
Submitted:
08 January 2024
Posted:
09 January 2024
You are already at the latest version
Abstract
Malaria, leishmaniasis, and African trypanosomiasis are protozoan diseases that constitute a major global health problem, especially in developing countries; however the development of drug resistance coupled with the toxicity of current treatments have hindered their management. The implication of certain enzymes (dihydrofolate reductase, DHFR) or proteins (potassium channels) in the pathogenesis of these protozoan diseases is undeniable. In this study, a series of three DHFR inhibitors (6-5 fused heterocyclic derivatives X, Y and Z) and one K+ channel blocker (E4031) were screened for their inhibitory effect on Leishmania donovani, Plasmodium falciparum, and Trypanosoma brucei. Resazurin assay was used to assess antitrypanosomal and antileishmanial activities of test compounds, whereas the antimalarial activity was appraised through SYBR Green I test. Moreover, cytotoxicity of test compounds was evaluated on Vero, Raw 264.7 and HepG-2 cells using the resazurin-based test, whilst their pharmacokinetic properties were predicted using the online tool pkCSM. As a result, compound Y revealed a selective (selectivity index range: 2.69 - >61.4; Vero, Raw 264.7 and HepG-2 cells) and broad-spectrum antiprotozoal activity against L. donovani promastigotes (IC50: 12.4 µM) and amastigotes (IC50: 4.28 µM), P. falciparum (IC50: 0.028 µM), and T. brucei brucei (IC50: 0.81 µM). In addition, compound X inhibited the growth P. falciparum (IC50: 0.0052 µM) and T. brucei brucei (IC50: 6.49 µM). In silico screening of the active antiprotozoal compounds revealed positive drug-likeness scores as none of Lipinski's rule of five’s criteria was violated by these compounds. However, in-depth pharmacokinetic and mechanistic studies are warranted to support discovery of novel antiprotozoal agents against malaria, leishmaniasis, and African trypanosomiasis by repurposing K+ channel blocker and DHFR inhibitors.
Keywords:
Protozoan diseases
; Drug repurposing
; Cytotoxicity
; Dihydrofolate reductase inhibitors
; Potassium (K+) channel blocker
; ADME properties
1. Introduction
Parasitic diseases represent an overwhelming health problem for impoverished populations living in developing countries with poor sanitary conditions. Previous reports have shown that the three most important protozoan diseases have an estimated disability-adjusted life year (DALY) of approximately one million [1]. These diseases include malaria, leishmaniasis and trypanosomiasis, which are caused by vector-borne protozoan parasites, including Plasmodium, Leishmania and Trypanosoma species, respectively. In malaria, for instance, the convergence of the COVID-19 pandemic and escalating medication resistance has propelled this pathological condition into the realm of the world’s deadliest diseases [2], with an estimated 249 million cases and 608 000 malaria deaths in 85 countries in 2022. In 2022, the high rates of malaria burden was endorsed by the World Health Organization (WHO)’s African region, with 94% of malaria cases (233 million) and 95% (580 000) of malaria deaths [3]. Core activities for the management of malaria include vector control and treatment of patients with appropriate antimalarial drugs, all impaired by the endless development of mosquito and Plasmodium spp. resistant strains and vaccine administration [2]. However, the vaccine approach has shown little success as the world’s first RTS, and the S vaccine offers only 30% protection [4]. Leishmaniasis and trypanosomiasis are neglected tropical diseases (NTDs) that are fatal if left untreated [5]. Leishmaniasis is endemic in Africa, Asia, the Americas, and the Mediterranean region [6], where it can develop into 3 main clinical forms according to the involved Leishmania species. These include cutaneous and mucocutaneous (Leishmania major, L. tropica and L. braziliensis, etc.) and the most severe visceral (L. donovani, and L. infantum, etc.) forms [7]. Annually, 700 000 to 1 million people are newly infected with leishmaniasis, with 20 000 to 30 000 deaths [7]. The treatment options for leishmaniasis include pentavalent antimonials, sodium stibogluconate (pentostam), and meglumine antimoniate (glucantime) as first-line drugs and pentamidine, amphotericin B, paromomycin and miltefosine as second-line treatments [8]. African trypanosomiasis, which affects both humans (human African trypanosomiasis) and animals (animal African trypanosomiasis) is a health concern in endemic areas. Human African trypanosomiasis (HAT) threatens millions of people in 36 sub-Saharan African countries, with 55 million people at risk of being infected [9]. With the multiple control strategies initiated, an important decline in the number of new cases from approximately 40 000 in 1998 to 663 in 2020 has been recorded. However, important efforts should be made to completely fulfil the WHO’s initiative toward the eradication of sleeping sickness by 2030 [9]. Trypanosoma brucei gambiense and T. brucei rhodesiense are the main pathogens responsible for HAT. Animal African trypanosomiasis (AAT) is among the most important diseases in cattle with severe economic consequences [10]. Indeed, the disease was reported to cause 3 million deaths in cattle [11]. AAT is mainly caused by three species of the genus Trypanosoma, including T. brucei gambiense, T. vivax, and T. congolense. Pentamidine, eflornithine, nifurtimox, fexinidazole, suramin, and melarsoprol are among the current treatments of HAT, whereas homidium bromide, diminazene aceturate, and suramin are the main drugs for AAT [9]. However, these drugs reveal poor efficacy, unacceptable toxicity, and drug resistance as the main limitations that make them inappropriate treatments [10, 12]. The currently developed acoziborole drug, which showed 95% success rate and effectiveness during phases 2 and 3 trials against HAT is noteworthy; however, recent development of Trypanosoma resistance to this drug has been identified [13]. Thus, there is a crucial need to search for alternative treatments against Trypanosoma infections. In spite of the recent advances in research to control these infectious diseases, they remain prevalent, supporting the need to search for effective and safe treatments against malaria, leishmaniasis and trypanosomiasis. The natural origin of antimalarial drugs is undeniable as quinine and artemisinin are some of the foremost examples that were respectively identified from cinchona bark [14] and Artemisia annua [15]. Moreover, synthesis and structural modifications of natural product scaffolds have provided a number of active principles for antileishmanial (miltefosine) [16, 17] and antitrypanosomal (fexinidazole) [9, 18] drug development. The mechanistic basis of antiprotozoal action of these active principles revealed inhibition of a number of enzymes, such as dihydrofolate reductase, which are crucial for the survival and virulence of Plasmodium [19], Leishmania and Trypanosoma [20] species. According to reported studies, potassium channels are integral membrane proteins that are intricately involved in the maintenance of vital parameters, including cellular membrane potential and cell volume in malaria parasites [21]. Noteworthy, the inhibitory effect of glibenclamide [22] and a number of halogenated glucose analogs [23] vis-à-vis K+ channels is reported in Leishmania sp. and Trypanosoma brucei, respectively. Thus, it is not unreasonable to speculate that compounds or drugs that inhibit dihydrofolate reductase or K+ channels can afford potentially active antiprotozoal leads against malaria, leishmaniasis, and African trypanosomiasis. Indeed, target-based drug discovery and drug repositioning can afford potentially active compounds that can serve as scaffolds for drug development, thereby reducing the overall cost and time frame used in the traditional drug discovery process [24]. Based on the foregoing, this study aims to investigate the antiprotozoal activity of a series of 3 heterocyclic potential DHFR inhibitors (compound X, Y and Z) (Figure 1) and 1 potassium channel blocker (E4031) (Figure 1, Table 1) against Plasmodium, Leishmania and Trypanosoma parasites.
2. Materials and Methods
2.1. Parasites and culture
Three parasites, including P. falciparum strain 3D7, L. donovani 1S (MHOM/SD/62/1S) promastigotes and T. brucei bloodstream trypomastigotes (subsp. brucei, Strain Lister 427 VSG 221), which were used in this study were received as a kind gift from BEI resources (https://www.beiresources.org/).
The axenic promastigote forms of L. donovani were cultured at 28°C in M199 culture medium (Sigma Aldrich) supplemented with 1% streptomycin/penicillin (Sigma Aldrich) and 10% heat-inactivated fetal bovine serum (FBS) (Sigma Aldrich).
Plasmodium falciparum parasites were continuously maintained in Petri dishes containing complete RPMI 1640 medium [RPMI 1640 (Sigma Aldrich) supplemented with 0.5% Albumax II (Gibco), 0.2% sodium bicarbonate (Sigma Aldrich), 1% hypoxanthine 100X (Gibco), 25 mM HEPES and 0.04% gentamicin (Sigma Aldrich)] with fresh O+ erythrocytes obtained from healthy human volunteers, and suspended at 4% hematocrit (v/v), followed by an incubation at 37°C in a 5% CO2 humidified atmosphere [28]. The culture medium was renewed daily, and parasitemia was monitored by microscopy through 10% Giemsa-stained thin blood smears. Two days prior to the assay, parasites were synchronized at the ring stage by serial treatment with 5% sorbitol.
The bloodstream form of T. brucei subsp. brucei was grown and maintained in vented flasks containing standard HMI-9 (Hirumi’s modified Iscove’s medium 9) supplemented with 10% heat-inactivated FBS. Parasites were subsequently incubated at 37°C in a 5% CO2 atmosphere and examined by inverted microscopy every 72 h to monitor parasite density. Next, the cells were passaged by transferring 250 µL of the medium containing parasites into 4.750 mL of fresh medium in a new sterile vented flask [29].
2.2. Mammalian cell lines’ culture
Three cell lines were used to appraise the cytotoxicity of the test compounds: the African green monkey kidney cell line (Vero; ATCC CRL-1586), and the macrophage murine leukemia (RAW 264.7) cell lines (obtained from American Type Culture Collection, ATCC, Manassas, VA, USA), as well as the human hepatoma HepG-2 cells (Acc 85011430) (procured from Sigma Aldrich). Vero and Raw264.7 cells were maintained in T-25 vented cap culture flasks containing complete Dulbecco's Modified Eagle Medium (DMEM) [(10% (v/v) heat inactivated fetal bovine serum (HIFBS); 1% (v/v) nonessential amino acids mixture, and 1% penicillin/streptomycin], followed by an incubation at 37°C in a 5% CO2 humidified atmosphere. Vero and Raw cells were passaged with trypsin-EDTA (ethylenediaminetetraacetic acid) (Gibco, 0.25%) up to approximately 80-90% confluence. HepG-2 cells were grown and maintained in Eagle’s minimum essential medium supplemented with 10% (v/v) HIFBS, 2.2 g/mL sodium bicarbonate, 1% penicillin/streptomycin, and 1% sodium pyruvate at 37°C under the same conditions. Cells were passaged every 72 hours to retain a cell density between 1 × 105 and 3 × 105 cells/mL in a 25 cm2 culture flask.
2.3. Antiparasitic assays
2.3.1. Antikinetoplastid activity
Assay of Trypanosoma brucei bloodstream and Leishmania donovani promastigote inhibition
Resazurin assay was used to determine the antitrypanosomal and antileishmanial activities of compounds in a 96-well microplate using modified protocols as reported by Bowling et al. [30] and Siqueira-Neto et al. [31], respectively. Either 2 x 105 trypanosomes or 4 × 105 Leishmania parasites per mL were seeded with various compounds (concentrations’ range: 10 to 0.016 μM). The final concentration of dimethylsulfoxide (DMSO) was kept at 0.1%, and a vehicle control was used in all assays. Pentamidine (1-0.0016 µM) and amphotericin B (10-0.16 µg/mL) were respectively used as positive controls for Trypanosoma and Leishmania. After a 24-hr incubation of Leishmania plates, 1 mg/mL resazurin dye (prepared in Dulbecco’s phosphate buffered saline) was added, followed by an incubation for 48 h in darkness. Following 68 h incubation period for trypanosomes at 37°C and 5% CO2, 10 µL of resazurin solution (0.15 mg/mL) was added to each well, and plates were subsequently incubated for an extra 4 h. For both tests, the fluorescence was then read using a Tecan Infinite F200 reader using excitation and emission wavelengths of 530 and 590 nm, respectively.
Antiamastigote assay
Compounds that displayed activity against the promastigote forms were further screened against the intracellular amastigotes of L. donovani as reported by Jain et al. [32] with slight modifications (by using Raw 264.7 macrophages as host cells in lieu of THP-1 human acute monocytic leukemia cells). In brief, an exponentially growing Raw 264.7 macrophages (4 × 103 cells/well) were seeded in sterile flat bottomed 96-well plates, followed by an incubation for 6 h at 37°C under 5% CO2 to allow adhesion. Next, plates were washed with sterile PBS to discard nonadherent cells and further incubated for 24 h at 37°C in a 5% CO2 atmosphere with M199 medium containing Leishmania metacyclic promastigotes (4 × 105 cells) at 1:10 macrophage/promastigote ratio. Thereafter, 3-4 successive washes with phosphate buffer saline (PBS) were applied to carefully remove the noninternalized promastigote forms. A solution containing 10% FBS, fresh M199 medium, and test compounds (final assay concentrations range: 10 to 0.016 µM) was prepared and incubated for 48 h at 37°C under 5% CO2 atmosphere. Next, 0.05% SDS (sodium dodecyl sulfate) was introduced into plates for 30 seconds for controlled lysis, followed by the addition of complete M199 (10% FBS) to stop macrophage lysis. Afterwards, resazurin (250 μg/mL) was introduced into each well, followed by plates’ incubation for 24 h. Then, the fluorescence of the preparation was read using a microplate reader (TECAN-Infinite M200, Tecan Austria GmbH, Grödig Flachgau, Austria) at λexcitation and λemission of 530 and 590 nm, respectively.
2.3.2. Antiplasmodial assay
SYBR Green I-based fluorescence method was used to assess the antiplasmodial activity of the selected compounds [33]. Briefly, 10 µL of each compound was introduced into a 96-well assay plate in duplicate and serially diluted to reach final concentrations ranging from 10 to 0.016 µM. Artemisinin (10 μM) and chloroquine (10 μM) were used as positive controls (0% growth), whereas 0.1% DMSO (v/v) was used as a negative control (100% growth). Next, 90 µL of parasitized red blood cells at 1% hematocrit and 2% parasitemia were dispensed into each well, followed by an incubation of the preparation for 72 hours. To evaluate the parasite growth, an immediate check for the presence or absence of trophozoites in the plates was achieved. For this purpose, cells were lysed by effecting freeze‒thaw cycles, then 50 µL of the thawed culture in each well was gently mixed with 50 µl of SYBR Green I lysis buffer [0.2 μL of 10,000×SYBR Green I (Invitrogen) per mL of lysis buffer {Tris (20 mM; pH 7.5), saponin (0.008%; wt/v), EDTA (5 mM), and Triton X-100 (0.08%; v/v)}]. Plates were further incubated for an additional 60 minutes in darkness at room temperature, and fluorescence was measured using a microplate reader (TECAN-Infinite M200, Tecan Austria GmbH, Grödig Flachgau, Austria) at excitation and emission wavelengths of 485 and 530 nm, respectively.
2.4. Cytotoxicity assay
The resazurin based-colorimetric method [30] was used to assess the cytotoxic effects of the active antiprotozoal compounds against two human mammalian cells, viz. Raw 264.7 and Vero cells, as well as the human hepatoma HepG-2 cells. In brief, cells were seeded at 104 cells/well (100 µL) in 96-well culture plates and incubated overnight to allow cell adherence. After renewing the culture medium, 10 µL of serially diluted compound solutions (concentration range: 50-0.08 µM) were added in duplicate. Plates were then incubated at 37°C for 48 h in a 5% CO2 humidified atmosphere. Wells containing 10% DMSO (v/v) and podophyllotoxin (20 µM) were regarded as positive controls, while those containing only cultured cells were considered to have 100% growth. Next, ten microliters of a stock solution of resazurin [0.15 mg/mL, Dulbecco's Phosphate-Buffered Saline (DPBS)] was added to each well, gently mixed, and subsequently incubated for an additional 4 h. Fluorescence was read using a microplate reader (TECAN-Infinite M200, Tecan Austria GmbH, Grödig Flachgau, Austria) with excitation and emission wavelengths of 530 and 590 nm, respectively.
2.5. In silico prediction of physicochemical and pharmacokinetic properties
The studied compounds were subjected to in silico studies for their profile’s prediction in terms of absorption, distribution, metabolism, and excretion [34]. The chemical structures of test compounds were drawn using ChemBio2D Draw, whereas their SMILES codes were produced, and further used as the main material to predict the pharmacokinetic properties by running the pkCSM online tool (https://biosig.lab.uq.edu.au/pkcsm/prediction; accessed on 13th August 2022).
2.6. Data analysis
Each experiment was carried out in duplicate and repeated twice. The growth inhibition percentages for each test compound were determined using Microsoft Excel software and then used for dose‒response curve plotting (inhibitory percentage versus log10 [drug concentration]) to deduce the half-maximal inhibitory concentration (IC50) or the half-cytotoxic concentration (CC50) using GraphPad Prism 8.0 software. Compounds’ selectivity indices were calculated for each test sample as follows: SI=CC50 cells/IC50 parasites.
3. Results and discussion
Whole-cell phenotypic screening of compounds with known inhibitory effects against validated therapeutic targets is called target-based drug discovery. This approach includes target identification and validation using a number of tests, such as in silico prediction, biochemical and genetic analyses to identify proteins or enzymes that are crucial for the survival and virulence of the parasites [35]. In this regard, several targets, including dihydrofolate reductase have been validated as important components involved in the pathogenesis of Plasmodium, Leishmania and Trypanosoma species [36]. In fact, dihydrofolate reductase aids in the replication of several microorganisms by reducing dihydrofolate into tetrahydrofolate for DNA synthesis [37, 38]. Moreover, the implication of K+ channels in the survival and virulence of these parasites is noteworthy. According to the literature, a number of anticancer drugs, such as methotrexate exert their activity through inhibition of DHFR [39, 40]. Furthermore, the two antimalarial drugs cycloguanil and pyrimethamine, as well as a codified antimalarial compound (P218) were also found to inhibit DHFR enzyme [41]. Unlike the other most popular antifolate agents, such as trimethoprim, cycloguanil, and pyrimethamine that exhibited weak inhibition of Leishmania major DHFR, methotrexate inhibited this enzyme (L. major DHFR) in a nanomolar range (IC50: 5 nM). The whole cell phenotypic screening of methotrexate against L. major revealed IC50 value of 0.3 µM, confirming that this compound might have exerted antileishmanial activity through DHFR inhibition [42, 43]. Recently, Dize et al. revealed the inhibitory potential of MMV675968 derivatives bearing the quinazoline scaffold on the Trypanosoma brucei brucei DHFR together with their potent antitrypanosomal activity (IC50 range: 45-60 nM) [44]. On the other hand, potassium channels are among the proteins, which are found in the membrane of parasites, including Plasmodium, Leishmania and Trypanosoma. A number of authors have described the importance of K+ channels in the survival of these parasites [45,46,47,48]. Several synthetic compounds, such as clofazimine derivatives that revealed antiprotozoal activity against Leishmania and Plasmodium species were found to inhibit K+ channels [49]. Moreover, Waller et al. evaluated the antiplasmodial activity of several known K+ channel blockers, including quinidine, clotrimazole, haloperidol, charybdotoxin, bicuculline methiodide, tubocurarine chloride, trifluoperazine hydrochloride, and verruculogen, and the results showed inhibition of Plasmodium falciparum 3D7 with IC50 values stretching from 0.046 to 187.86 µM [45].
In this study, the antiprotozoal activity of a series of DHFR inhibitors (compound X, Y, and Z) and the potassium channel blocker E4031 was evaluated against Plasmodium, Leishmania and Trypanosoma species. As a result, compounds X, Y, and Z, and E4031 showed different degrees of antiprotozoal activity extending from poorly (IC50S>10 µM) to highly active (IC50S< 10 µM) [50, 51]. Against T. brucei, compounds X and Y exhibited IC50 values of 6.49 and 0.81 µM, respectively, vs pentamidine (IC50 value: 0.006 µM), whereas these compounds revealed IC50 values of 0.0052 and 0.028 µM, when tested against Plasmodium falciparum 3D7 with high selectivity in Raw cells (SI: 366.6 and 1179 for compounds X and Y, respectively), compared to the value obtained with artemisinin (IC50: 0.03 µM) (Table 2). In addition, compound Y showed IC50 values of 12.47 and 4.28 µM (SI: >2.69; Vero and Raw cells), when tested against L. donovani promastigotes and amastigotes, respectively, vs amphotericin B (IC50 values: 20 and 247.81 µM, respectively). Furthermore, compound Z and E4031 showed IC50 values > 10 µM when tested against T. brucei, promastigotes of L. donovani and P. falciparum 3D7, and were predicted to be poorly active compounds. Although numerous authors have reported the anticancer and antifolate activities of compounds X, Y, and Z [25, 27], the antiprotozoal activity of these compounds has not yet been unveiled. Compounds X, Y and Z are structurally similar compounds, in which one amino group of the pyrimidine moiety is replaced by an OH group in compound Z. This structural modification might have contributed for the decrease in the observed antiprotozoal activity of compound Z. Although there is reported evidence that the presence of OH groups tend to increase the antiprotozoal activity of bioactive compounds [52], other reports highlight a different opinion about this matter [53]. Compounds’ selection based on their positive drug-likeness scores increases the likelihood to identify potential drug candidates with favorable pharmacokinetics, efficacy, and safety. As realigning pharmacokinetic studies early in the discovery phase can assist in selecting an ideal drug candidate, active antiprotozoal compounds were subjected to in silico screening using the pkCSM online tool. Regarding the in silico examination of physicochemical properties, molecular weight (441.488, 442.472, 442.476 and 415.559) ClogP (lipophilicity; octanol-water partition coefficient) (1.741, 1.865, 1.221 and 3.289), rotatable bonds (10, 10, 10 and 8), hydrogen bond acceptors (HBA) (7, 7, 8 and 5), hydrogen bond donors (HBD) (5, 5, 6 and 1) and Topological Polar Surface Area (TPSA) (184.604, 184.058, 183.789, and 172.949) (Table 3) of compounds X, Y, and Z and E4031 were respectively predicted. Notably, none of these physicochemical parameters were violated according to criteria by the Lipinski's rule of five, which states that a molecule has an increased chance of being directly bioavailable when it obeys to the conditions of having (i) no more than five hydrogen bond donors, (ii) ten hydrogen bond acceptors, (iii) a molecular weight under 500, and (iv) a LogP number under 5 [54]. In fact, all tested compounds (X, Y, Z and E4031) qualify as orally active compounds as they abide by the "Lipinski's rule of five" criteria [lipophilicity (LogP) < 5, HBD ≤ 5, HBA ≤ 10, molecular weight (g/mol) ≤ 500 g/mol, and flexibility (rotatable bonds): < 10] [55, 56]. Other rulesets for drug-likeness, including the Veber [flexibility (rotatable bonds): < 10; HBD ≤ 5; HBA ≤ 10; except for TPSA (140 Å2)], Ghose filter (-0.4<Log P<+ 5.6; HBD ≤ 5 and HBA ≤ 10; MW<500 g/mol), Muegge (HBD ≤ 5, except for E4031 and MW<500 g/mol) and Egan (-0.4<Log P<+5.6; MW<500 g/mol and HBA ≤ 10)] rules showed favorable drug-like characteristics [55, 56]. Furthermore, in silico tests on ADME (absorption, distribution, metabolism and excretion) revealed poor permeability of compounds X, Y and Z, and E4031 across the skin, blood brain barrier and central nervous system. Nevertheless, in vitro and in vivo pharmacokinetic studies need to be thoroughly investigated for the successful utilization of these compounds as scaffolds in antiprotozoal drug discovery.
In this study, the antiprotozoal activity of a series of DHFR inhibitors (compound X, Y, and Z) and the potassium channel blocker E4031 was evaluated against Plasmodium, Leishmania and Trypanosoma species. As a result, compounds X, Y, and Z, and E4031 showed different degrees of antiprotozoal activity extending from poorly (IC50S>10 µM) to highly active (IC50S< 10 µM) [50, 51]. Against T. brucei, compounds X and Y exhibited IC50 values of 6.49 and 0.81 µM, respectively, vs pentamidine (IC50 value: 0.006 µM), whereas these compounds revealed IC50 values of 0.0052 and 0.028 µM, when tested against Plasmodium falciparum 3D7 with high selectivity in Raw cells (SI: 366.6 and 1179 for compounds X and Y, respectively), compared to the value obtained with artemisinin (IC50: 0.03 µM) (Table 2). In addition, compound Y showed IC50 values of 12.47 and 4.28 µM (SI: >2.69; Vero and Raw cells), when tested against L. donovani promastigotes and amastigotes, respectively, vs amphotericin B (IC50 values: 20 and 247.81 µM, respectively). Furthermore, compound Z and E4031 showed IC50 values > 10 µM when tested against T. brucei, promastigotes of L. donovani and P. falciparum 3D7, and were predicted to be poorly active compounds. Although numerous authors have reported the anticancer and antifolate activities of compounds X, Y, and Z [25, 27], the antiprotozoal activity of these compounds has not yet been unveiled. Compounds X, Y and Z are structurally similar compounds, in which one amino group of the pyrimidine moiety is replaced by an OH group in compound Z. This structural modification might have contributed for the decrease in the observed antiprotozoal activity of compound Z. Although there is reported evidence that the presence of OH groups tend to increase the antiprotozoal activity of bioactive compounds [52], other reports highlight a different opinion about this matter [53]. Compounds’ selection based on their positive drug-likeness scores increases the likelihood to identify potential drug candidates with favorable pharmacokinetics, efficacy, and safety. As realigning pharmacokinetic studies early in the discovery phase can assist in selecting an ideal drug candidate, active antiprotozoal compounds were subjected to in silico screening using the pkCSM online tool. Regarding the in silico examination of physicochemical properties, molecular weight (441.488, 442.472, 442.476 and 415.559) ClogP (lipophilicity; octanol-water partition coefficient) (1.741, 1.865, 1.221 and 3.289), rotatable bonds (10, 10, 10 and 8), hydrogen bond acceptors (HBA) (7, 7, 8 and 5), hydrogen bond donors (HBD) (5, 5, 6 and 1) and Topological Polar Surface Area (TPSA) (184.604, 184.058, 183.789, and 172.949) (Table 3) of compounds X, Y, and Z and E4031 were respectively predicted. Notably, none of these physicochemical parameters were violated according to criteria by the Lipinski's rule of five, which states that a molecule has an increased chance of being directly bioavailable when it obeys to the conditions of having (i) no more than five hydrogen bond donors, (ii) ten hydrogen bond acceptors, (iii) a molecular weight under 500, and (iv) a LogP number under 5 [54]. In fact, all tested compounds (X, Y, Z and E4031) qualify as orally active compounds as they abide by the "Lipinski's rule of five" criteria [lipophilicity (LogP) < 5, HBD ≤ 5, HBA ≤ 10, molecular weight (g/mol) ≤ 500 g/mol, and flexibility (rotatable bonds): < 10] [55, 56]. Other rulesets for drug-likeness, including the Veber [flexibility (rotatable bonds): < 10; HBD ≤ 5; HBA ≤ 10; except for TPSA (140 Å2)], Ghose filter (-0.4<Log P<+ 5.6; HBD ≤ 5 and HBA ≤ 10; MW<500 g/mol), Muegge (HBD ≤ 5, except for E4031 and MW<500 g/mol) and Egan (-0.4<Log P<+5.6; MW<500 g/mol and HBA ≤ 10)] rules showed favorable drug-like characteristics [55, 56]. Furthermore, in silico tests on ADME (absorption, distribution, metabolism and excretion) revealed poor permeability of compounds X, Y and Z, and E4031 across the skin, blood brain barrier and central nervous system. Nevertheless, in vitro and in vivo pharmacokinetic studies need to be thoroughly investigated for the successful utilization of these compounds as scaffolds in antiprotozoal drug discovery.
4. Conclusions
We report that a group composed of three DHFR inhibitors (6-5 fused ring heterocyclic derivatives) and a K+ channel blocker (E4031) exhibit antiprotozoal activity on three parasite strains namely Leishmania donovani, Trypanosoma brucei, and Plasmodium falciparum 3D7, without cytotoxicity to the human mammalian cells Vero and Raw, as well as HepG-2 cells. In silico screening of the active antiprotozoal compounds using the pkCSM online tool revealed positive drug-likeness scores as none of the physicochemical parameters of these compounds were violated according to criteria by Lipinski's rule of five, which states that a molecule has an increased probability of being directly bioavailable if it has no more than five hydrogen bond donors, ten hydrogen bond acceptors, a molecular weight under 500, and a LogP number under 5. Nonetheless, in-depth in vitro and in vivo pharmacokinetic and antiprotozoal mechanistic studies are warranted to support discovery of novel antiprotozoal agents against malaria, leishmaniasis, and African trypanosomiasis by repurposing K+ channel blocker and DHFR inhibitors.
Author Contributions
“Conceptualization, F.F.B.; methodology, D.D., M.B.T.T., C.A.N.N., K.H., and R.K.; software, D.D., M.B.T.T., C.A.N.N. and R.K.; validation, E.A.M.K., L.R.T.Y., B.P.K., P.V.T.F. and F.F.B; formal analysis, K.H., B.P.K., E.A.M.K., L.R.T.Y., and P.V.T.F.; investigation, K.H., D.D., M.B.T.T., C.A.N.N. and R.K.; resources, B.P.K., E.A.M.K., L.R.T.Y., P.V.T.F. and F.F.B; data curation, D.D., M.B.T.T., C.A.N.N. and R.K.; writing—original draft preparation, D.D., M.B.T.T., C.A.N.N., E.A.M.K., L.R.T.Y., K.H. and R.K.; writing—review and editing, E.A.M.K., L.R.T.Y., P.V.T.F., B.P.K. and F.F.B; visualization, E.A.M.K., L.R.T.Y., P.V.T.F. and B.P.K.; supervision, F.F.B.; project administration, F.F.B.; funding acquisition, F.F.B. All authors have read and agreed to the published version of the manuscript.”.
Funding
This research was funded by the Grand Challenges Africa programme [GCA/DD/rnd3/006] awarded to FFB. Grand Challenges Africa, which is an AAS (African Academy of Sciences)’ programme is implemented through the Alliance for Accelerating Excellence in Science in Africa (AESA) platform, an initiative of AAS and the African Union Development Agency-New Partnership for Africa's Development (AUDA-NEPAD). For this work, GC Africa’s support was also empowered by the Bill & Melinda Gates Foundation (BMGF), Medicines for Malaria Venture (MMV), and Drug Discovery and Development Centre of University of Cape Town (H3D), and the APC was funded by the authors.
Institutional Review Board Statement
NA.
Informed Consent Statement
NA.
Data Availability Statement
NA.
Acknowledgments
The authors acknowledge Eisai Co., Ltd. for support, designing and supplying the 6-5 fused ring heterocyclic antifolates (Compound-X, Compound-Y and Compound-Z) and the potassium channel blocker (E4031).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Murray, C.J.L.; Vos, T.; Lozano, R.; Naghavi, M.; Flaxman, A.D.; Michaud, C.; Ezzati, M.; Shibuya, K.; Salomon, J.A.; Abdalla, S.; Aboyans, V.; Abraham, J.; Ackerman, I.; Aggarwal, R.; Ahn, S.Y.; Ali, M.K.; AlMazroa, M.A.; Alvarado, M.; Anderson, H.R.; Lopez, A.D. and Collaborators. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: A systematic analysis for the Global Burden of Disease Study 2010. The Lancet, 2012, 380 (9859), 2197-2223. [CrossRef]
- The World Health Organization (WHO). (2023a). Malaria. https://www.who.int/news-room/fact-sheets/detail/malaria.
- The World Health Organization (WHO) (2022). World Malaria Report 2022. https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2022.
- The World Health Organization (WHO) (2021). WHO recommends groundbreaking malaria vaccine for children at risk. https://www.who.int/news/item/06-10-2021-who-recommends-groundbreaking-malaria-vaccine-for-children-at-risk.
- The World Health Organization (WHO) (2010). First WHO report on neglected tropical diseases: working to overcome the global impact of neglected tropical diseases. https://www.who.int/publications/i/item/9789241564090.
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. (2017). Leishmaniasis: A review. F1000 Research, 6 (May), 1-15.
- The World Health Organization (WHO) (2023b). Leishmaniasis. https://www.who.int/news-room/fact-sheets/detail/leishmaniasis.
- Silva-Jardim, I.; Thiemann, O.H.; Anibal, F.F. (2014). Leishmaniasis and Chagas disease chemotherapy: A critical review. J. Braz. Chem. Soc. 25 (10), 1810-1823. [CrossRef]
- The World Health Organization (WHO) (2023c). Trypanosomiasis (Sleeping sickness). https://www.who.int/news-room/fact-sheets/detail/trypanosomiasis-human-african-(sleeping-sickness).
- Giordani, F.; Morrison, L.J.; Rowan, T.G.; De Koning, H.P.; Barrett, M.P. The animal trypanosomiases and their chemotherapy: A review. Parasitology, 2016, 143 (14), 1862-1889. [CrossRef]
- Food and Agriculture Organization (FAO) (2023). Programme Against Africa, Trypanosomiasis. https://www.fao.org/paat/the-programme/the-disease/en/.
- Fairlamb, A.H. Chemotherapy of human African trypanosomiasis: Current and future prospects. Trends Parasitol. 2003, 19 (11), 488-494. [CrossRef]
- Betu Kumeso, V.K.; Kalonji, W.M.; Rembry, S.; Mordt, O.V.; Tete, D.N.; Prêtre, A.; Delhomme, S.; Kyhi, M.I.W.; Camara, M.; Catusse, J.; Schneitter, S.; Nusbaumer, M.; Miaka, E.M.; Mbembo, H.M.; Mayawula, J.M.; Camara, M..; Massa, F.A.; Badibabi, L.K.; Bonama, A.K.; Lukula, P.K.; Kalonji, S.M.; Philemon, P.M.; Nganyonyi, R.M.; Mankiara, H.E.; Nguba, A.A.A.; Muanza, V.K.; Nasandhel, E.M.; Bambuwu, A.F.N.; Scherrer, B.; Strub-Wourgaft, N.; Tarral, A. Efficacy and safety of acoziborole in patients with human African trypanosomiasis caused by Trypanosoma brucei gambiense: a multicenter, open-label, single-arm, phase 2/3 trial. Lancet Infect. Dis. 2023, 23 (4), 463-470.
- Christensen, S.B. Natural products that changed society. Biomedicines 2021, 9(5), 472. [CrossRef]
- Ma, N.; Zhang, Z.; Liao, F.; Jiang, T.; Tu, Y. The birth of artemisinin. Pharmacol. Ther. 2020, 216:107658. [CrossRef]
- Dorlo, T.P.C.; Balasegaram, M.; Beijnen, J.H.; de Vries, P.J. Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J. Antimicrob. Chemother. 2012, 67 (11), 2576-2597. [CrossRef]
- Pinto-Martinez, A.K.; Rodriguez-Durán, J.; Serrano-Martin, X.; Hernandez-Rodriguez, V.; Benaim, G. Mechanism of action of miltefosine on Leishmania donovani involves the impairment of acidocalcisome function and the activation of the sphingosine-dependent plasma membrane Ca2+ channel. Antimicrob. Agents Chemother. 2017, 62(1), e01614-17. [CrossRef]
- Torreele, E.; Bourdin Trunz, B.; Tweats, D.; Kaiser, M.; Brun, R.; Mazué, G.; Bray, M.A.; Pécoul, B. Fexinidazole--a new oral nitroimidazole drug candidate entering clinical development for the treatment of sleeping sickness. PLoS Negl. Trop. Dis. 2010, 4(12):e923, 1-15. [CrossRef]
- Bilsland, E.; van Vliet, L.; Williams, K.; Feltham, J.; Carrasco, M.P.; Fotoran, W.L.; Cubillos, E.F.G.; Wunderlich, G.; Grøtli, M.; Hollfelder, F.; Jackson, V.; King, R.D.; Oliver, S.G. 2018. Plasmodium dihydrofolate reductase is a second enzyme target for the antimalarial action of triclosan. Sci. Rep. 2018, 8:1038, 1-8. [CrossRef]
- Shamshad, H.; Bakri, R.; Mirza, A.Z. Dihydrofolate reductase, thymidylate synthase, and serine hydroxy methyltransferase: successful targets against some infectious diseases. Mol. Biol. Rep. 2022, 49(7), 6659-6691. [CrossRef]
- Ellekvist, P.; Mlambo, G.; Kumar, N.; Klaerke, D.A. Functional characterization of malaria parasites deficient in the K+ channel Kch2. Biochem. Biophys. Res. Commun. 2017, 493 (1) 690-696. [CrossRef]
- Serrano-Martín, X.; Payares, G.; Mendoza-León, A. Glibenclamide, a blocker of K+(ATP) channels, shows antileishmanial activity in experimental murine cutaneous leishmaniasis. Antimicrob. Agents Chemother. 2006, 50 (12) 4214-4216. [CrossRef]
- Schmidt, R.S.; Macedo, J.P.; Steinmann, M.E.; Salgado, A.G.; Butikofer, P.; Sigel, E.; Rentsch, D.; Maser, P. Transporters of Trypanosoma brucei-phylogeny, physiology, pharmacology. The FEBS Journal. 2018, 285, 1012-1023.
- Müller, J.; Hemphill, A. Drug target identification in protozoan parasites Drug target identification in protozoan parasites. Expert Opin. Drug Discov. 2016, 11(8), 815-824.
- Kotake, Y.; Iijima, A.; Yoshimatau, K.; Tamai, N.; Ozawa, Y.; Koyanagi, N.; Kitoh, K.; Nomura, H. Synthesis and antitumor activities of novel 6-5 fused ring heterocycle antifolates: N-[4-[o-(2-amino-4-substituted-6,7-dihydrocyclopenta[~pyrimidin-5-yl)alkyl]benzoyl]-L-glutamic Acids. J. Med. Chem., 1994, 37(11), 1616-1624.
- Kotake, Y.; Okauchi, T.; Iuima, A.; Yoshimatau, K.; Nomura, H. Novel 6-5 fused ring heterocycle antifolates with potent antitumor activity: bridge modifications and heterocyclic Benzoyl isosters of 2,4-diamino-6.7-difydro-5H-cyclopenta[d]pyrimidine antifolate. Chem. Pharm. Bull. 1995, 43 (5), 829-841. [CrossRef]
- Mcguire, J.J.; Bergoltz, V.V.; Heitzman, K.J.; Haile, W.H.; Russell, C.A., Bolanowska, E. Novel 6,5-fused ring heterocyclic antifolates : Biochemical and biological characterization. Cancer Res. 1994, 54, 2673-2680.
- Trager, W.; Jensen, J.B. Human malaria parasites in continuous culture. Science, 1976, 193 (4254), 673-675. [CrossRef]
- Hirumi, H.; Hirumi, K. Continuous cultivation of Trypanosoma brucei blood stream forms in a medium containing a low concentration of serum protein without feeder cell layers. J. Parasitol. 1989, 75 (6), 985-989. [CrossRef]
- Bowling, T.; Mercer, L.; Don, R.; Jacobs, R.; Nare, B. Application of a resazurin-based high-throughput screening assay for the identification and progression of new treatments for human african trypanosomiasis. Int. J. Parasitol. Drugs Drug Resist. 2012, 2, 262-270. [CrossRef]
- Siqueira-Neto, J.L.; Song, O.R.; Oh, H.; Sohn, J.H.; Yang, G.; Nam, J.; Jang, J.; Cechetto, J.; Lee, C. B.; Moon, S.; Genovesio, A.; Chatelain, E.; Christophe, T.; Freitas-Junior, L.H. Antileishmanial high-throughput drug screening reveals drug candidates with new scaffolds. PLoS Negl. Trop. Dis. 2010, 4 (5), 1-9. [CrossRef]
- Jain, S.K.; Sahu, R.; Walker, L.A.; Tekwani, B.L. A parasite rescue and transformation assay for antileishmanial screening against intracellular Leishmania donovani amastigotes in THP1 human acute monocytic leukemia cell line. Journal of Visualized Experiments (JoVE) 2012, 70, 1-14.
- Smilkstein, M.; Sriwilaijaroen, N.; Kelly, J.X.; Wilairat, P.; Riscoe, M. Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother. 2004, 48 (5), 1803-1806. [CrossRef]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58 (9), 4066-4072. [CrossRef]
- Andrade, E.L.; Bento, A.F.; Cavalli, J.; Oliveira, S.K.; Freitas, C.S.; Marcon, R.; Schwanke, R.C.; Siqueira, J.M., Calixto, J.B. Non-clinical studies required for new drug development – Part I : early in silico and in vitro studies , new target discovery and validation , proof of principles and robustness of animal studies. Braz. J. Med. Biol. Res. 2016, 49, 1-9.
- Gangjee, A.; Kurup, S.; Namjoshi, O. Dihydrofolate reductase as a target for chemotherapy in parasites. Curr. Pharm. Des. 2007, 13(6), 609-639. [CrossRef]
- Sharma, M.; Chauhan, M. Dihydrofolate reductase as a therapeutic target for infectious diseases : opportunities and challenges. Future Med. Chem. 2012, 4 (10), 1335-1365.
- Choudhury, A.A.K.; Vinayagam, S.; Adhikari, N.; Ghosh, S.K.; Sattu, K. Microwave synthesis and antimalarial screening of novel 4-amino benzoic acid (PABA)-substituted pyrimidine derivatives as Plasmodium falciparum dihydrofolate reductase inhibitors. 3 Biotech. 2022, 12(8), 170. [CrossRef]
- Raimondi, M.V.; Randazzo, O.; La Franca, M.; Barone, G.; Vignoni, E.; Rossi, D.; Collina, S. DHFR inhibitors: Reading the past for discovering novel anticancer agents. Molecules. 2019, 24(6) 1140. [CrossRef]
- Wróbel, A.; Drozdowska, D. Recent design and structure-activity relationship studies on the modifications of DHFR inhibitors as anticancer agents. Curr. Med. Chem. 2021, 28(5), 910-939. [CrossRef]
- Yuthavong, Y.; Tarnchompoo, B.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Charman, S.A.; McLennan, D.N.; White, K.L.; Vivas, L.; Bongard, E.; Thongphanchang, C.; Taweechai, S.; Vanichtanankul, J.; Rattanajak, R.; Arwon, U.; Fantauzzi, P.; Yuvaniyama, J.; Charman, W.N.; Matthews, D. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc. Natl. Acad. Sci. U S A. 2012, 109 (42), 16823-16828. [CrossRef]
- Gilbert, I.H. Inhibitors of dihydrofolate reductase in leishmania and trypanosomes. Biochim. Biophys Acta Mol. Basis Dis. 2002, 1587 (2-3), 249-257. [CrossRef]
- Teixeira, B.V.F.; Teles, A.L.B.; Silva, S.G.D.; Brito, C.C.B.; Freitas, H.F.; Pires, A.B.L.; Froes, T.Q.; Castilho, M.S. Dual and selective inhibitors of pteridine reductase 1 (PTR1) and dihydrofolate reductase-thymidylate synthase (DHFR-TS) from Leishmania chagasi. J. Enzyme Inhib. Med. Chem. 2019, 34(1), 1439-1450. [CrossRef]
- Dize, D.; Tata, R.B.; Keumoe, R.; Kouipou Toghueo, R.M.; Tchatat, M.B.; Njanpa, C.N.; Tchuenguia, V.C.; Yamthe, L.T.; Fokou, P.V.T.; Laleu, B.; Duffy, J.; Bishop, O.T.; Boyom, F.F. Preliminary structure–activity relationship study of the MMV pathogen box compound MMV675968 (2,4-diaminoquinazoline) unveils novel inhibitors of Trypanosoma brucei brucei. Molecules, 2022, 27 (19), 1-38.
- Waller, K.L.; Kim, K.; McDonald, T.V. Plasmodium falciparum: Growth response to potassium channel blocking compounds. Exp. Parasitol. 2008, 120 (3), 280-285. [CrossRef]
- Mosimann, M.; Goshima, S.; Wenzler, T.; Lu, A.; Uozumi, N.; Ma, P. (2010). A Trk/HKT-Type K+ transporter from Trypanosoma brucei. Eukaryot. Cell. 2010, 9 (4), 539-546.
- Kuang, Q.; Purhonen, P.; Hebert, H. Structure of potassium channels. Cell. Mol. Life Sci. 2015, 72, 3677-3693. [CrossRef]
- Paul, A.; Mubashra; Singh, S. Identification of a novel calcium activated potassium channel from Leishmania donovani and in silico predictions of its antigenic features. Acta Trop. 2021, 220, 105922. [CrossRef]
- Barteselli, A.; Casagrande, M.; Basilico, N.; Parapini, S.; Rusconi, C.M.; Tonelli, M.; Boido, V.; Taramelli, D.; Sparatore, F.; Sparatore, A. Clofazimine analogs with antileishmanial and antiplasmodial activity. Bioorg. Med. Chem. 2015, 23 (1), 55-65. [CrossRef]
- Boniface, P.K.; Ferreira, E.I. Flavonoids as efficient scaffolds: Recent trends for malaria, leishmaniasis, Chagas disease, and dengue. Phytother. Res. 2019, 33 (10), 2473-2517. [CrossRef]
- Tchatat Tali, M.B.; Boniface, P.K.; Jean Claude, T.; Fabrice, F.F. 2023. Current developments on the antimalarial, antileishmanial, and antitrypanosomal potential and mechanisms of action of Terminalia spp. S. Afr. J. Bot. 2023, 156, 309-333.
- Pérez-Silanes, S.; Berrade, L.; García-Sánchez, R.N.; Mendoza, A.; Galiano, S.; Pérez-Solórzano, B.M.; Nogal-Ruiz, J.J.; Martínez-Fernández, A.R.; Aldana, I.; Monge, A. New 1-aryl-3-substituted propanol derivatives as antimalarial agents. Molecules. 2009, 14(10), 4120-4135. [CrossRef]
- Ravindar, L.; Hasbullah, S.A. ; Rakesh, K.P. ; Hassan, N.I. Recent developments in antimalarial activities of 4-aminoquinoline derivatives. Eur. J. Med. Chem. 2023, 256, 115458. [CrossRef]
- Chen, X.; Li. H.; Tian. L.; Li, Q.; Luo, J.; Zhang, Y. Analysis of the physicochemical properties of Acaricides based on Lipinski's Rule of five. J. Comput. Biol. 2020, 27(9), 1397-1406.
- Rai, M.; Singh, A.V.; Paudel, N.; Kanase, A.; Falletta, E.; Kerkar, P.; Heyda, J.; Barghash, R.F.; Pratap Singh, S.; Soos, M. Herbal concoction unveiled: A computational analysis of phytochemicals' pharmacokinetic and toxicological profiles using novel approach methodologies (NAMs). Curr. Res. Toxicol. 2023, 5:100118. [CrossRef]
- Guenfoud, F.; Khaoua, O.; Cherak, Z.; Loucif, L.; Boussebaa, W.; Benbellat, N.; Laabassi, M.; Mosset, P. 2024. Synthesis, antimicrobial, DFT, and in silico pharmacokinetic profiling of nitroaldol quinoline derivatives: A comprehensive exploration for designing potential oral antibacterial agents targeting DNA-gyrase. J. Mol. Struct. 1300, 137293. [CrossRef]
Figure 1.
Structures of the 6,5-fused ring heterocyclic antifolates (X, Y, Z) and the potassium channel blocker (E4031).
Figure 1.
Structures of the 6,5-fused ring heterocyclic antifolates (X, Y, Z) and the potassium channel blocker (E4031).
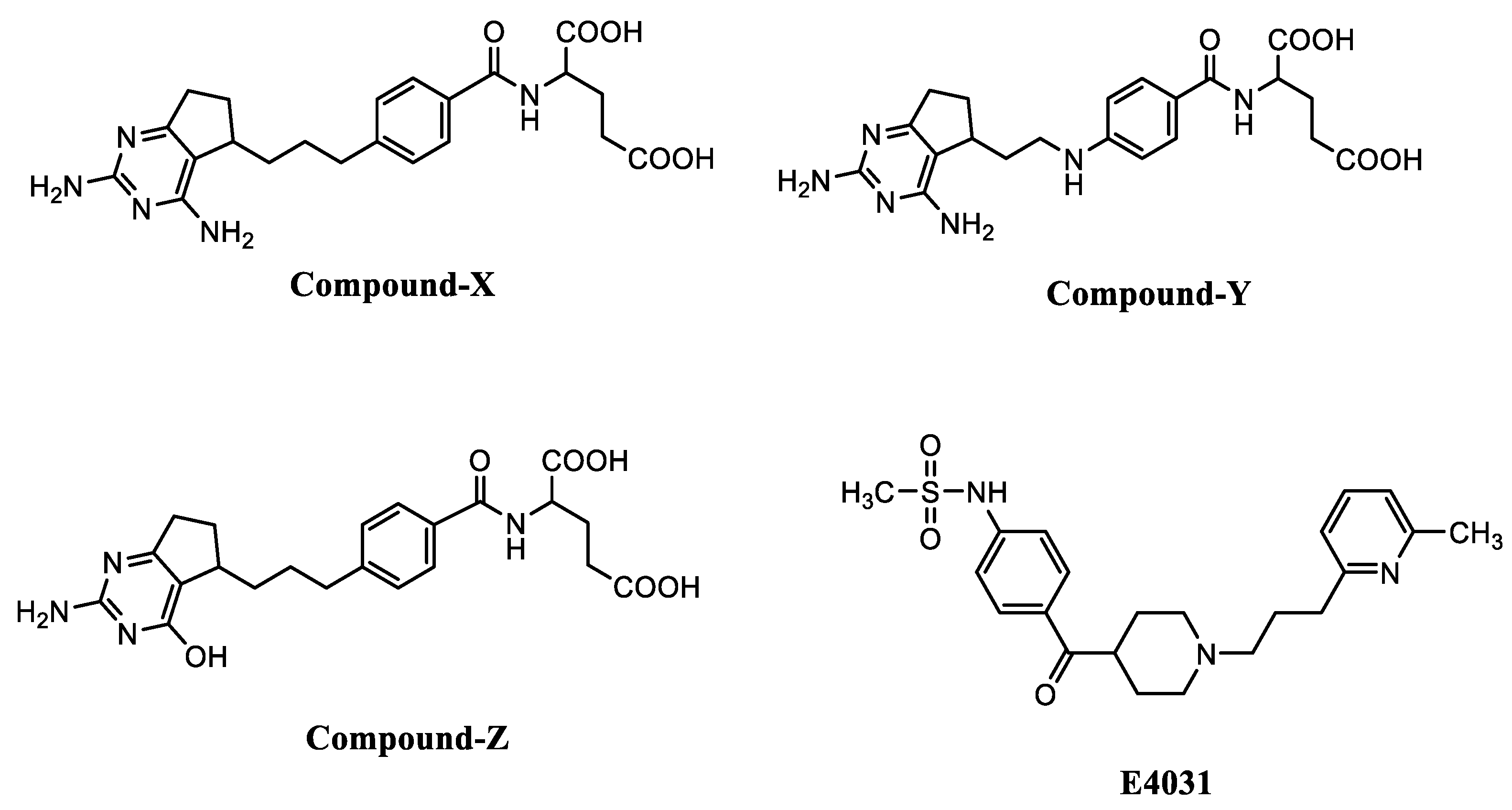

Table 1.
Reported activities of the antifolate compounds against DHFR enzymes from various sources.
| Compounds | *IC50 (nM)_DHFR | References | ||
|---|---|---|---|---|
| Bovine liver | P388 | CCRF-CEM | ||
| X | 2.5 | 7.1 | 0.6 | [25, 26, 27] |
| Y | 5.9 | - | 0.8 | [26,27] |
| Z | 60 000 | - | - | [25] |
* Median inhibitory concentration (IC50) of the antifolates on DHFR from bovine liver, P388 (leukaemia cells) and CCRF-CEM (human T-lymphoblastic leukaemia cells).
Table 2.
Antiparasitic and cytotoxic activities of the 6,5-fused ring heterocyclic antifolates and the potassium channel blocker.
Table 2.
Antiparasitic and cytotoxic activities of the 6,5-fused ring heterocyclic antifolates and the potassium channel blocker.
| Compounds ID | IC50±SD (µM) | CC 50± SD (µM) | SI | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| T. b. brucei |
L. donovani prom |
L. donovani ama |
Pf_3D7 | ||||||||||||
| T. b. brucei |
L. donovani Prom |
L. donovani ama |
Pf_3D7 | Raw264.7 | Vero | HepG-2 | Raw264.7 | Vero HepG-2 |
Raw264.7 | Vero/ HepG-2 |
Raw264.7 | Vero/ HepG-2 |
Raw264.7 | Vero/ HepG-2 |
|
| Compound-X | 6.49±0.4 | ˃10 | NT | 0.0052 | 1.91±0.09 | ˃50 | ˃50 | 0.29 | ˃7.7 | 366.6 | ˃9596 | ||||
| Compound-Y | 0.81±0.00 | 12.47±3.04 | 4.28±0.12 | 0.028 | 33.58±5.5 | ˃50 | ˃50 | 41 | ˃61.4 | 2.69 | ˃4 | 7.85 | ˃11.7 | 1179 | ˃1756 |
| Compound-Z | ˃10 | ˃10 | NT | ˃10 | ˃50 | ˃50 | ˃50 | - | - | - | - | - | - | - | - |
| E4031 | ˃10 | ˃10 | NT | ˃10 | ˃50 | ˃50 | ˃50 | - | - | - | - | - | - | - | - |
| Pentamidine | 0.006±0.0 | - | - | - | - | - | - | - | - | - | - | - | - | - | - |
| Artemisinin | - | - | - | 0.03±0.004 | - | - | - | - | - | - | - | - | - | - | - |
| Amphotericin B | - | 20±1.63 | 247.812±24 | ND | - | - | - | - | - | - | - | - | - | - | - |
The IC50s: half inhibitory concentration are mean values obtained from two replicates on Plasmodium falciparum, Trypanosoma brucei brucei, Leishmania donovani promastigotes and intracellular amastigotes. CC50: half cytotoxic concentration on Vero, Raw264.7 and HepG-2 cells. SI (selectivity index). Positive controls: amphotericin B for Leishmania donovani, Pentamidine for Trypanosoma brucei brucei and Artemisinin for Plasmodium falciparum.
Table 3.
Predicted physicochemical and ADME properties of the test compounds.
| Compounds | Compound-X | Compound-Y | Compound-Z | E4031 | |
|---|---|---|---|---|---|
| Physicochemical properties | MW | 441.488 | 442.472 | 442.476 | 415.559 |
| ClogP | 1.7415 | 1.8649 | 1.2208 | 3.28902 | |
| Rotatable Bonds | 10 | 10 | 10 | 8 | |
| Acceptors | 7 | 7 | 8 | 5 | |
| Donors | 5 | 5 | 6 | 1 | |
| TPSA | 184.604 | 184.058 | 183.789 | 172.949 | |
| Absorption | Water solubility (log mol/L) | -2.907 | -2.911 | -2.903 | -4.414 |
| Caco2 permeability (log Papp) | -0.822 | -0.758 | -0.797 | 1.093 | |
| Intestinal absorption (%) | 28.627 | 25.758 | 22.436 | 92.885 | |
| Skin Permeability (log Kp) | -2.735 | -2.735 | -2.735 | -3.35 | |
| P-gp substrate | Yes | Yes | Yes | Yes | |
| P-gp I inhibitor | No | No | No | No | |
| P-gp II inhibitor | No | No | No | Yes | |
| Distribution | VDss (human) (log L/kg) | -0.62 | -0.25 | -0.224 | 0.677 |
| Fraction unbound | 0.282 | 0.204 | 0.369 | 0.286 | |
| BBB permeability (log BB) | -1.544 | -1.67 | -1.649 | -0.448 | |
| CNS permeability (log PS) | -3.528 | -3.545 | -3.654 | -2.916 | |
| Metabolism | CYP2D6 substrate | No | No | Yes | No |
| CYP3A4 substrate | No | No | No | Yes | |
| CYP1A2 inhibitior | No | No | No | No | |
| CYP2C19 inhibitior | No | No | No | No | |
| CYP2C9 inhibitior | No | No | No | No | |
| CYP2D6 inhibitior | No | No | No | No | |
| CYP3A4 inhibitior | No | No | No | Yes | |
| Excretion | Total Clearance (log ml/min/kg) | 0.689 | 0.722 | 0.481 | 0.744 |
| Renal OCT2 substrate | No | No | No | No | |
BBB: blood‒brain barrier; CLogP o/w: lipophilicity (recommended value: LogP <5); CYP1A2: cytochrome P450 family 1 subfamily A member 2; CYP3A4: cytochrome P450 family 3 subfamily A member 4; CYP2C9: cytochrome P450 family 2 subfamily C member 9; CYP2C19: cytochrome P450 family 2 subfamily C member 19; CYP2D6: cytochrome P450 family 2 subfamily D member 6; Lipinski (criteria: MW≤500, LogP≤5, N or O ≤ 10, NH or OH≤5); MW: Molecular weight of the compounds; OCT2: organic cation transporter 2; P-gp: P-glycoprotein substrate; TPSA: topological polar surface area.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



