Submitted:
08 January 2024
Posted:
09 January 2024
You are already at the latest version
Abstract
To obtain a cost-effective and high-performance composite material of polybutylene adipate-terephthalate (PBAT), for this study we selected microcrystalline cellulose, which is inexpensive and easily available, as the reinforcing medium. Hexadecyl trimethoxysilane (HDTMS) containing a long carbon chain was chosen to silanize the microcrystalline cellulose (MCC) to obtain silanized cellulose (SG). Three types of SGs with different degrees of silanization were obtained by controlling the reaction ratio. Characterization of these three types of SGs was conducted using FTIR, TEM, and water absorption analysis. Subsequently, PBAT/SG composites were prepared through a sol-gel method, and the effects of these three types of SGs on the thermal stability, compatibility, mechanical properties, and dynamic thermomechanical properties of PBAT were evaluated. Furthermore, the mechanism behind enhancing the mechanical properties of the composite was analyzed. The results demonstrated successful synthesis of SG. As the reaction ratio between HDTMS and MCC increased, the nanoparticle size increased, while the water absorption decreased significantly. After SG was added to the PBAT composites, the yield stress increased while maintaining good thermal stability. Both the SEM and DMA results indicated good compatibility of the PBAT/SG composites. Analysis of the mechanical properties revealed that the tensile strength initially increased and then decreased with increasing blending ratio for all three composites tested; among them, the PBAT/SG2 composites exhibit superior performance, with a maximum tensile strength reaching 22 MPa at an 85/15 blending ratio—nearly 30% higher than that of pure PBAT alone. The addition of SG significantly improved the strength of the PBAT, and we speculated as to the underlying mechanism involved. This study provides a new idea for the industrial-scale development of degradable polyesters with low cost and good mechanical properties.
Keywords:
silanized cellulose
; sol-gel
; compatibility
; PBAT
1. Introduction
With the increasing severity of environmental pollution and the growing consciousness of environmental issues, an increasing amount of attention is being directed toward the advancement and utilization of biodegradable materials. Biodegradable materials, also known as "green plastics," can undergo degradation into water and carbon dioxide through microbial activity under natural or compost conditions[1,2]. Among the various biodegradable materials, polyadipate-butylene terephthalate (PBAT) is the most extensively employed and promising option. PBAT is flexible and rigidly soluble in aromatic polyesters, making it a highly suitable substitute for conventional plastics[3,4,5].
However, the high production cost and low mechanical properties of PBAT have emerged as significant obstacles impeding its widespread application as a potential substitute for polyethylene and other plastics[6]. In recent years, numerous studies have focused on enhancing the mechanical properties of PBAT. The commonly employed approach involves blending PBAT with high-strength substances, including high-strength polymers, rigid inorganic particles, and cellulose[7,8,9,10].
Among the many polymers blended with PBAT, polylactic acid (PLA) has become one of the most popular high-strength polymers due to its excellent mechanical and processing properties[11]. Yeh[12] prepared a PBAT/PLA composite material by melt blending and reported that when the PBAT content was low, the PBAT could be evenly dispersed in the PLA matrix, and the tensile strength of the composite improved. When the content of PBAT is above 5%, phase separation of the composite system also occurs, and the tensile strength decreases obviously. To improve the compatibility of PBAT and PLA, Chen[13] used a small amount (0.5-3 wt %) of epoxy-functional styryl-acrylic oligomer (ESA) as a high-efficiency crosslinking agent to prepare supertoughened and mechanically robust PLA/PBAT blends by dynamic vulcanization. When ESA was added to the surface, the PBAT phase had a strong interfacial adhesion force, which gave rise to the highest impact toughness and ductility while maintaining high strength.
The inorganic particles used to improve the strength of PBAT include carbon nanotubes and CaCO3. The mechanical properties of carbon nanotubes (CNTs) are excellent, with a theoretical strength of 150 GPa and a density of only 1/6 that of steel. The use of CNTs to modify polymers has obvious advantages[14]. Zhao[15] added carbon nanotubes (CNTs) with different contents to PBAT/PLA blends to form branched-chain carbon nanotube copolymers using multifunctional epoxy oligomers (ADRs) as reaction compatibilizers. The results showed that the addition of CNTs and ADR improved both the strength and toughness of the samples. The impact strength was 35.3 kJ/m2, approximately 7 times that of the PLA/PBAT blend, and the tensile strength increased from 33.6 MPa to 42.8 MPa. The performance of the PLA/PBAT blends comodified with ADR and CNTs was significantly better than that of the PLA/PBAT blends comodified with ADR or CNTs. CaCO3 has strong toughening and strengthening effects and can significantly improve the bending strength and bending modulus of materials[16] and enhance their thermal stability. Liu[17] designed and prepared PBAT/CaCO3 composite films with PBAT as the resin matrix and calcium carbonate (CaCO3) as the filler by using a twin screw extruder and a single screw extrusion blow molding machine. The results showed that the size and content of the CaCO3 particles significantly influence the tensile properties of the composites. The addition of unmodified CaCO3 reduces the tensile properties of the composites by more than 30%. The modification of CaCO3 by the titanate coupling agent 201 (TC-2) improved the overall performance of the PBAT/CaCO3 composite film. When the addition of TC-2 was 1%, the maximum tensile strength of the film was 20.55 MPa, the water vapor permeability of the composite was reduced by 27.99%, and the water vapor permeability coefficient was reduced by 43.19%.
Cellulose has unique properties, such as low density, high toughness, high strength[18,19], and complete degradation. In addition, cellulose comes from a wide range of sources and is inexpensive, so the price of cellulose-based composites is relatively low[20,21]. However, cellulose also has several drawbacks, mainly because it is a polyhydroxyl compound that absorbs water, and if it is directly blended with hydrophobic polyester, an incompatible phenomenon will occur[22,23]. As a result, the properties of composite materials cannot be optimized, which restricts the use of cellulose to a certain extent. Giri[24] prepared microcrystalline cellulose (MCC) from wheat straw as a raw material and prepared the composite material by melting composites with PBAT in different proportions. The results showed that the composite material could maintain good mechanical properties when the MFC filling amount was low, and when the MFC filling amount exceeded 10%, the composite material could maintain good mechanical properties. Due to the aggregation of MFC and poor interfacial bonding, the PBAT composite cracks prematurely and leads to fracture when subjected to external forces. Hou[25] used octadecylamine (ODA) to graft nanocellulose (CNF) to improve its compatibility with the PBAT matrix. PBAT composites containing 1 wt% CNFs were prepared by the masterbatch premixing method to avoid CNF aggregation during extrusion. The results showed that the tensile strength of the CNF(OCNF)/PBAT fused extrusion composite was 17.2% greater than that of the PBAT polymer without affecting the thermal stability of the PBAT.
At present, there have been many studies dedicated to increasing the mechanical properties of PBATs, but there are several problems that remain to be addressed, such as limited strength improvement and poor compatibility of blended materials. To reduce the cost of the material, improve the compatibility of PBAT and additives, and further improve the mechanical properties of the material, for this study we selected inexpensive and easily available microcrystalline cellulose as the strengthening medium and selected hexadecyl trimethoxysilane, which contains a long carbon chain, to silanize the microcrystalline cellulose to obtain silanized cellulose (SG). PBAT/SG composites were then blended with PBAT to prepare PBAT/SG composites, and the thermal stability, compatibility and mechanical properties of the composites were further studied through thermogravimetric testing, scanning analysis, dynamic thermomechanical property analysis and mechanical property analysis to explore the mechanism of enhancing the mechanical properties of the composites. This approach provides a new industrial approach for further study of the blending of degradable polyesters.
2. Materials and Methods
2.1 Sources of the Materials
PBAT, Injection molding grade, Xinjiang Changji Lanshan Tunhe Co., Ltd.; Microcrystalline cellulose, Column chromatography, Shanghai Hengxin Chemical Reagent Co., Ltd.; Sodium hydroxide, Analytical pure, Tianjin Zhiyuan Chemical Reagent Co., Ltd..; Thiourea, Analytical pure, Tianjin Shengmiao Chemical Reagent Co., LTD.; Cetyltrimethoxysilane, Analytically pure, Shanghai Aladdin Co., Ltd..; Anhydrous ethanol, Analytically pure, Tianjin Yongsheng Fine Chemical Co., Ltd.. Concentrated hydrochloric acid, analytically pure, Tianjin Kemeng Chemical Plant.
2.2 Synthesis of SG
Three grams of microcrystalline cellulose (MCC) was added to 100 g of alkali solution (NaOH) mixed with sodium hydroxide (NaOH) and thiourea (CH4N2S). CH4N2S:H2O = 9.5:4.5:86 (w/w/w)) was uniformly stirred for 100 min to form a homogeneous solution, and the solution was frozen in a refrigerator at -20 °C for 24 h. After thawing, 0.1 g of cetyltrimethoxy-silane (HDTMS) was added, and the mixture was stirred evenly to form a sol. Then, 15 mL of 4 mol/L HCl was added until a gel was formed. Silanized cellulose 1 (SG1) was obtained by aging the gel at room temperature for 10 h, washing it with distilled water to a neutral pH and drying it to constant weight in a drying oven at 100 °C. The preparation methods for silanized cellulose 2 (SG2) and silanized cellulose 3 (SG3) were the same as above, except that the amounts of HDTMS added were 0.9 g and 1.5 g, respectively.
2.3 Preparation of PBAT/SG composites
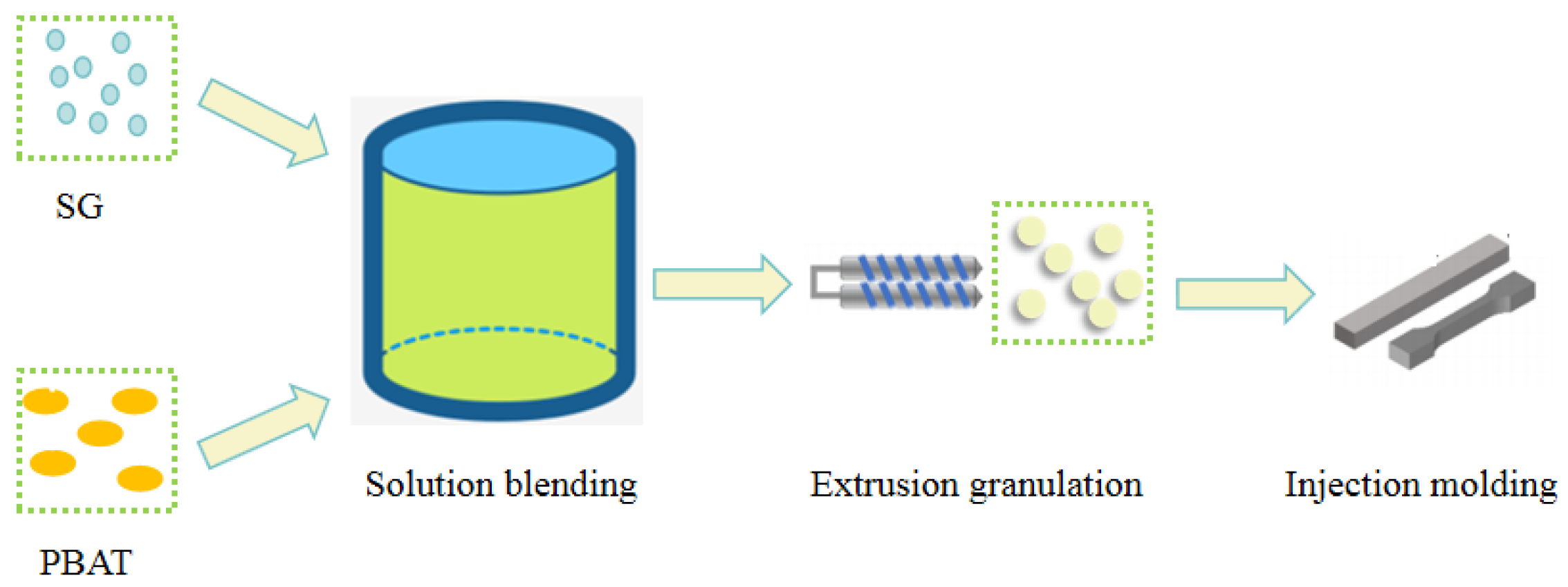
SG was ground into 200-300 mesh powder and dried with PBAT in a blast drying oven at 70 °C for 12 h. The solution blending method[26,27] was used to mix SG and PBAT evenly; that is, PBAT was first added to the chloroform solution and evenly stirred until all was dissolved. Then, SG was added, and the mixture was continuously stirred until SG was evenly dispersed in the PBAT solution. After the solvent volatilized completely, the composite material was extruded on a twin screw extruder at 130 °C for granulation. Reinjection into standard splines The process flow chart is as follows:
Figure 1.
Preparation process for the PBAT/SG composites.
2.4 Measurements and characterizations
Before the performance test, the sample was treated to eliminate the residual internal stress of the composite material and remove moisture. The method was as follows: the composite material was placed in a blast box at 80 °C and dried for 12 hours to fully remove moisture and eliminate internal residual stress. The composite material was then stored in a sealed bag to prevent moisture absorption.
Fourier transform infrared (FTIR) spectroscopy: The potassium bromide tablet method was used to observe the chemical composition of the SGs in the range of 4000~400 cm-1 to determine whether silanization modification occurred in the microcrystalline cellulose.
Transmission electron microscopy (TEM): SGs were dispersed in anhydrous ethanol and observed on a Hitachi H-600 transmission electron microscope at an acceleration voltage of 100 kV to determine the size and dispersion of the silica in the SGs. TEM was used to analyze the composite materials via the ultrathin slice method. Tensile properties: The tensile strength of the composite was tested according to GB/T1040-1992. The drawing rate was 10 mm/min, and the average value was measured 5 times for each sample.
Scanning electron microscopy (SEM): The composite material was soaked in liquid nitrogen for 40 min, followed by brittle fracture, gold spraying on its cross section, and observation of the cross section morphology of the composite material by scanning electron microscopy at an accelerating voltage of 15 kV.
Thermogravimetric analysis (TGA): A sample of the PBAT/SG composite material was cut into small pieces of approximately 5 mg, and the temperature was increased to 1000 °C at a heating rate of 10 °C/min under a nitrogen atmosphere. The heating process data were recorded, and thermogravimetric analysis of the composite material was performed.
Dynamic thermal analysis (DMA): A Q800 V7.5 DMA tester was used for dynamic thermal mechanical testing of the composite materials. The temperature range was -50-110 °C, the heating rate was 3 °C/min, and the instrument was operated in strain mode at a fixed frequency of 1 Hz. The whole test process was carried out in a N2 atmosphere.
3. Results and Discussions
3.1. Synthesis and characterization of SG
(1) Sol-gel reaction
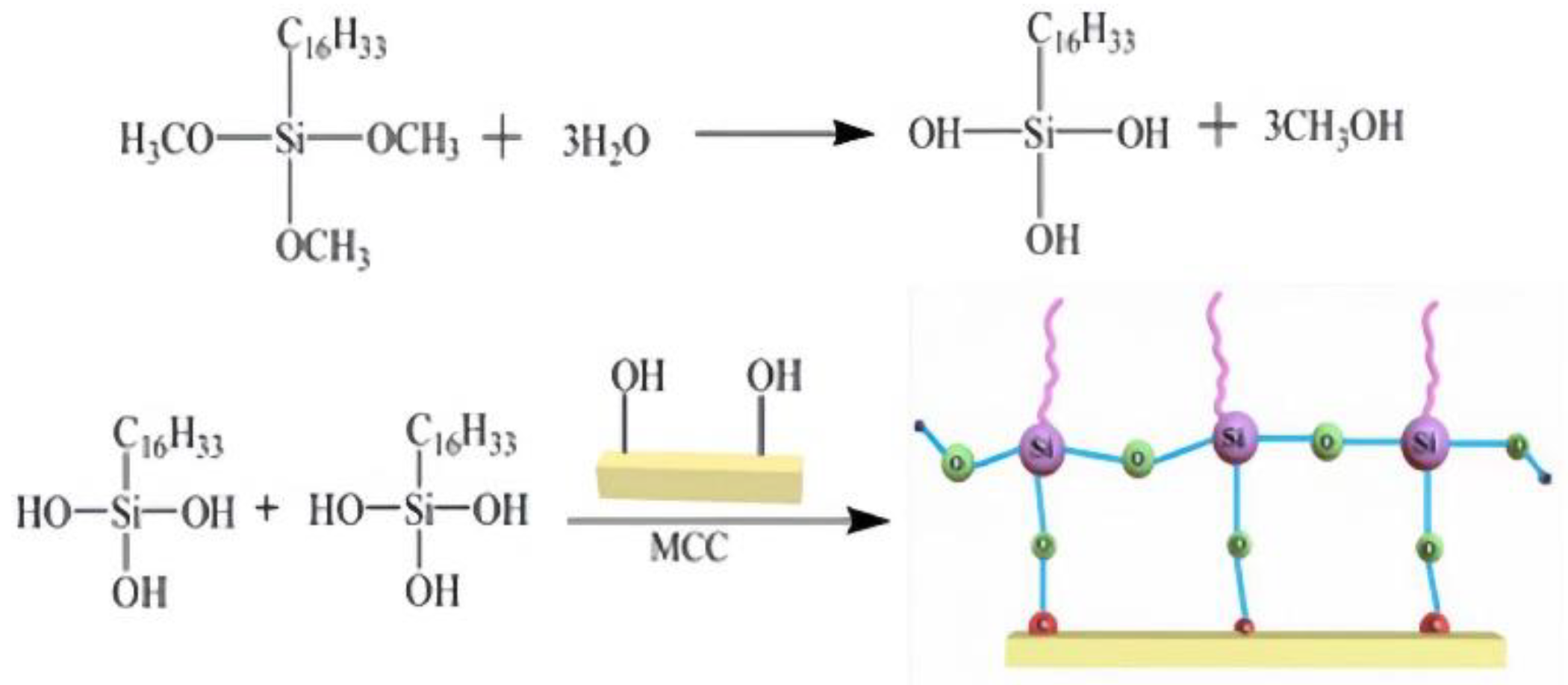
SG is prepared by the sol‒gel method, which includes the hydrolysis and condensation of HDTMS in cellulose solution. When HDTMS is added to an alkali solution of cellulose, hydrolysis occurs, the three methoxy groups of HDTMS are hydrolyzed into hydroxyl groups, and methanol is obtained as a byproduct. Methanol helps HDTMS dissolve in water[28] and therefore facilitates the hydrolysis reaction. When HCl was added to the solution, the condensation rate increased. After hydrolysis, part of the hydroxyl group of HDTMS condenses with the hydroxyl group of MCC to form a Si-O-C covalent bond, and part of the hydroxy group is self-condensed to form a polysiloxane network structure. The condensation reaction of HDTMS and MCC results in cross-linking between microcrystalline cellulose, resulting in the hydrophobicity of silanized cellulose.
Figure 2.
Synthetic route of SG.
(2) IR and TEM analysis of SGs
As seen from the infrared spectrum of MCC (Figure 3A(a)), the peak attributed to the stretching vibration of -OH groups ranged from 3600 to 3200 cm-1. The strong absorption peak near 1056 cm-1 is the characteristic peak of C-O-C in hemiacetal in pyran sugar. The absorption peak of C-H is at approximately 2898 cm-1. As the ratio of HDTMS to MCC increases, the absorption peak attributed to OH in SG becomes increasingly weaker (Figure 3A(b-d)), mainly because MCC condensation occurs during the silanization of cellulose and possibly because Si-OH, like the OH in MCC[29], can also form intramolecular hydrogen bonds in its own chains. Compared with that of MCC, the absorption peak band corresponding to C-H is more intense at 2840-2930 cm-1 for SG. With the increase in the reaction proportion of HDTMS and MCC, the corresponding peak intensity also significantly increases[30], which further proves that the sol-gel reaction introduces alkyl chains. However, it is difficult to determine Si-O-Si and Si-O-C on SG in the spectrum[31] because the absorption peaks of Si-O-Si and Si-O-C overlap near 1100-1150 cm-1. However, the increase in the intensity of the SG absorption peak near 1100-1150 cm-1 also indirectly proves that the sol-gel reaction occurred. The TEM image in Figure 3(C-E) shows that after the sol-gel reaction, SiO2 nanoparticles are formed in the SG, and the size of the nanoparticles increases with increasing reaction proportion of HDTMS and MCC, whose sizes are 11 nm, 17 nm and 21 nm, respectively.
(3) Water absorption of SG
The wettability of solids is usually measured by the water contact angle, but due to the uneven surface of SGs, it is difficult to detect the baseline water droplets[32]. Therefore, in this study, the degree of silanization and hydrophobicity of SGs were measured by the amount of water absorbed. Before silanization, MCC has high water absorption capacity and can be almost completely wetted by water. With increasing reaction ratio of HDTMS to MCC, the water absorption rate decreased significantly, and the water absorption rate of SG3 was only 0.54% (Table 1). When HDTMS modifies MCC, on the one hand, the hydroxy groups of cellulose and silanol undergo dehydration, which causes the long hydrophobic carbon chain of HDTMS to bind to MCC; on the other hand, the intramolecular condensation reaction results in the formation of a network structure, which also reduces the water absorption of SG.
3.2. Performance analysis of composite materials
3.2.1. IR and TEM analysis of composite materials
Samples of PBAT/SG blend materials were tested by FTIR and TEM to analyze the changes in the composite groups after blending PBAT and SG and the dispersion of SG in PBAT. The results are shown in Figure 4.
PBAT belongs to the polyester family, and the stretching vibration peak of C-O near 1111 cm-1 and the stretching vibration peak of C=O near 1728 cm-1 are the characteristic peaks of esters[33]. C-H asymmetric and symmetric bending vibration absorption peaks are found at approximately 1457 cm-1 and 1392 cm-1, respectively. The stretching vibration peak of -OH was near 3436 cm-1 (Figure 4a).
Compared with those of pure PBAT, the positions of the characteristic absorption peaks of the composite material are basically unchanged, but the intensities of some absorption peaks change obviously. The strength of the peak attributed to the stretching vibration of the composite material becomes stronger near 1020 cm-1. This is because of the stretching vibration peaks of C-O-C and Si-O-C formed by SG near 1020 cm-1. The intensity of the C-H absorption peak at 2952 cm-1 was significantly greater than that of the other peaks, especially for PBAT/SG2 and PBAT/SG3. This was because SG2 and SG3 had the highest degree of silanization and contained more alkanes than SG1.
Figure 4(B-D) shows TEM images of PBAT/SG1, PBAT/SG2 and PBAT/SG3, respectively. The figure shows that the SG particle size gradually increases, but the dispersion in PBAT is relatively uniform.
3.2.2. Influence of the SG dosage on the mechanical properties of the composite materials
Figure 5 shows the stress‒strain curve of the PBAT/SG composites. Elastic deformation occurs when the strain is 33.9%. When the yield stage is reached, the yield stress of the composites is greater than that of the PBAT. The yield stress of PBAT/SG2 is the highest (10.7 MPa), followed by that of PBAT/SG3 and PBAT/SG1. After the yield stage, the deformation generated by the material is irreversible permanent deformation. When the stress reaches the highest point, the material is pulled off, and the maximum stress decreases in the order of PBAT/SG2 > PBAT/SG3 > PBAT/SG1 > PBAT. The elongation at break values of the PBAT, PBAT/SG1, PBAT/SG2 and PBAT/SG3 composites are 700.2%, 598.1%, 601.2% and 609.8%, respectively, indicating that the toughness of the composites is very good.
Figure 6 shows the influence of the amount of SG on the tensile properties of the composites. The figure shows that the tensile strength of PBAT/SG has no obvious improvement trend when the dosage of SG is low; this is mainly because when the amount of SG added is low (when the blend ratio is approximately 95/5), the continuous phase structure of the base resin PBAT is destroyed, causing defects in the composite system, resulting in stress concentration during tensile testing and a slight reduction in tensile strength. When the amount of SG continues to increase, the tensile strength of the composite improves to a certain extent, which is due to the interaction between SG and the PBAT matrix through hydrophobic and polar interactions, etc., which enhances the compatibility of the composite material, and the combination of inorganic silicon particles also enhances the tensile strength. However, when the amount of SG is too high, on the one hand, the enhanced interaction between SG and the PBAT matrix restricts the movement of PBAT chain segments; on the other hand, due to the grain distribution of SG, both the increase in the SG content and the trend toward phase separation increase, which causes the mechanical properties of the composite to decline.
In addition to the amount of SG, the tensile strength is also affected by the size and hydrophilicity of the SG. Among the three composites, PBAT/SG1 and PBAT/SG3 both reach the maximum tensile strength when the blending ratios of PBAT with SG1 and SG3 are 90/10, which are 16.4 MPa and 17.3 MPa, respectively. Compared with those of the previous two materials, the mechanical properties of PBAT/SG2 are better. When the blending ratio of PBAT to SG2 is 85/15, the tensile strength of the composite material reaches 22.0 MPa, which is 6.1 MPa greater than that of cellulose after blending with a silane coupling agent[34]. This is because SG1 has strong hydrophilicity and poor interface compatibility with PBAT, which cannot effectively improve the mechanical properties of PBAT. Although the hydrophobicity of SG3 is very good, due to the large particles, agglomeration easily occurs, and the mechanical properties of PBAT cannot be effectively improved. In contrast, SG2 is not only hydrophobic but also has a moderate and uniform particle size, which can effectively improve the mechanical properties of PBAT. The elongation at break of the three composites gradually decreased with increasing SG concentration. However, before the blending ratio reached 75/25, the elongation at break was the highest for PBAT/SG2, followed by that for PBAT/SG3, and that for PBAT/SG1 was the lowest. The elongation at break of PBAT/SG2 and PBAT/SG3 decreased rapidly, while the elongation at break of PBAT/SG1 did not decrease significantly. The introduction of SG isolates the interaction between PBAT molecular chains and increases the flexibility of molecular chains; on the other hand, silica plays a role as a filler, which limits the conformational change of molecular chains. Therefore, at a certain blending ratio, the hydrophobic effect of SG plays a leading role, and the good compatibility of the resulting material shows that the elongation at break of PBAT/SG2 is high. However, with a further increase in the SG proportion, the limiting effect of SG as a filler becomes more dominant, so the elongation at break decreases rapidly for the SG2 and SG3 series with larger particle sizes.
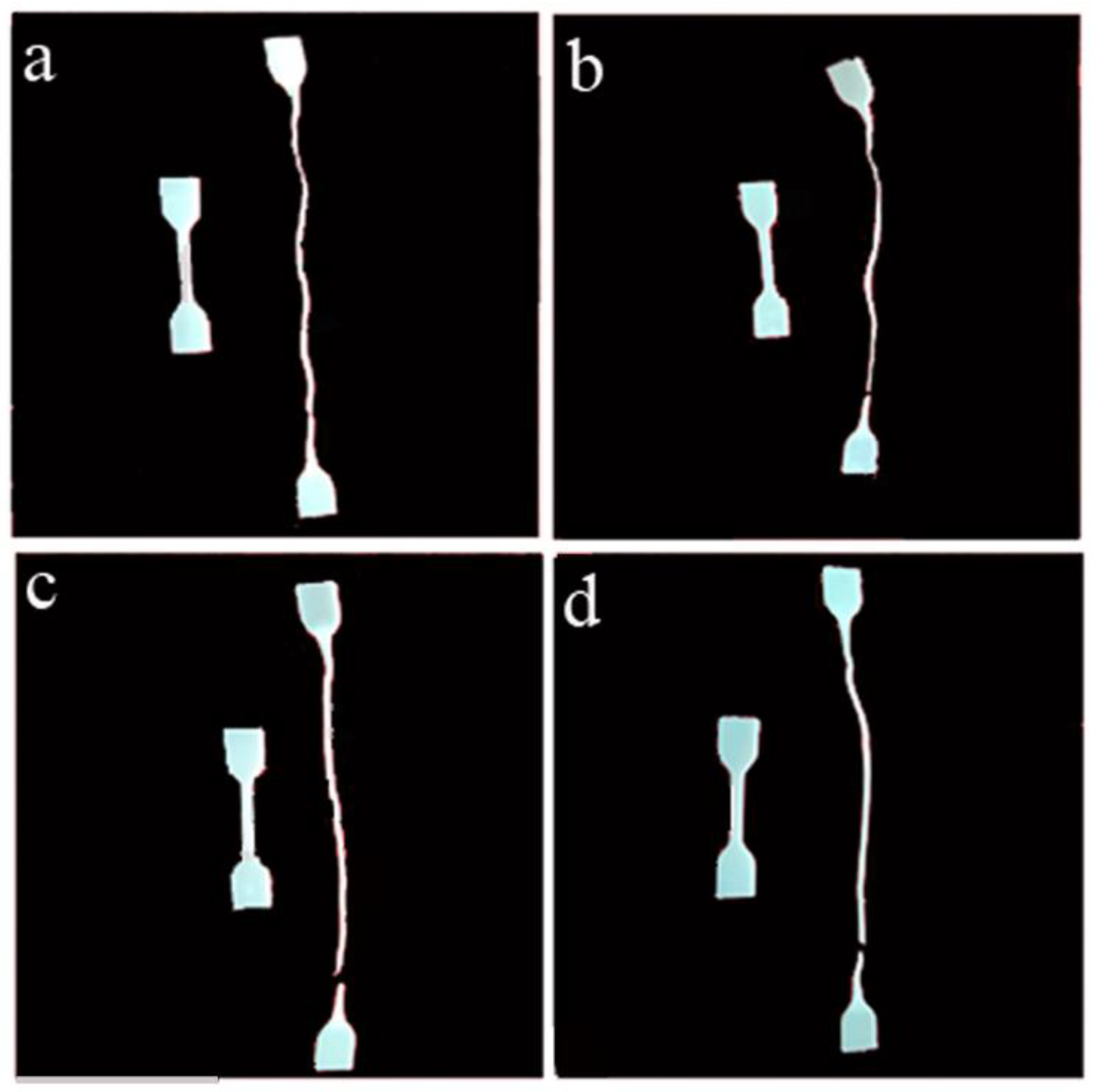
Figure 7.
Photos before and after break of (a) PBAT, PBAT/SG1 (90/10), (b) PBAT/SG2 (85/15), and (d) PBAT/SG3 (90/10).
Figure 7.
Photos before and after break of (a) PBAT, PBAT/SG1 (90/10), (b) PBAT/SG2 (85/15), and (d) PBAT/SG3 (90/10).
3.2.3. Thermal behavior analysis of different kinds of composite materials
The thermal properties of PBAT and its three composites are shown in Figure 8, and the thermal decomposition of PBAT and the PBAT/SG composites begins at approximately 320 °C. In contrast, the initial degradation temperature of the composite is slightly lower than that of PBAT, which may be caused by the low degradation temperature of the alkyl chain in SG. The weight loss of PBAT and its composite materials mainly occurs in the range of 350-430 °C. When the temperature reaches 1000 °C, almost no PBAT remains, while the residual amounts of PBAT/SG1, PBAT/SG2 and PBAT/SG3 are 0.91%, 3.2% and 2.9%, respectively, which are the SiO2 generated by SG oxidation at high temperature.
3.2.4. SEM analysis of different kinds of composite materials
The cross-sectional morphology of the composite was observed via SEM, and the compatibility of the two phases of the composite was further studied. Figure 9 shows the SEM image of the brittle liquid nitrogen section of PBAT and its three composites. The cross section of the PBAT is relatively flat, indicating that the PBAT underwent brittle fracture after being treated with liquid nitrogen (Figure 9a). There was little difference in the cross-sectional morphology of the three composites (Figure 9b-d). The cross sections of the PBAT/SG1, PBAT/SG2 and PBAT/SG3 composites are all flat, indicating that SG1, SG2 and SG3 have good dispersion in PBAT. This is because the silanization modification causes the hydrophilic hydroxyl group on the cellulose molecular chain to be replaced by a hydrophobic Si-O-C bond, which improves the interdependence of the two phases, so the surface of the composite material is flat. However, only the surface of the PBAT/SG3 composite contains insignificant particles because the SG3 particle size is 21 nm, which is larger than that of SG1 (11 nm) and SG2 (17 nm) and easy to separate from the matrix.
3.2.5. DMA analysis of different kinds of composite materials
The DMA curve of PBAT/SG provides information on not only the molecular chain movement of the composites but also the glass transition temperature (Tg) of the PBAT and composites. In addition, the modulus variation rule of composites can be obtained, which is highly important for determining the dynamic mechanical properties of composites.
Figure 10 shows the curves of the energy storage modulus, damping factor and loss modulus of the PBAT and its composite material with changing temperature. In the figure, the sharp decrease in the energy storage modulus and the peak damping factor correspond to the glass transition temperature of the material. The energy storage module scale is a measure of the elasticity (rigidity) of a material and describes the energy stored by elastic deformation during the deformation process. Figure 10A shows that the energy storage modulus of PBAT and the three other composites all decrease with increasing temperature. The glass transition temperature of PBAT is -20.7 °C, and the glass transition temperatures of the PBAT/SG1 and PBAT/SG3 composites increase slightly. The glass transition temperature of the PBAT/SG2 composites decreased slightly to -22.1 °C and -15.8 °C. The results indicated that the blending of SG and PBAT, on the one hand, had a certain plasticizing effect, promoted the movement of polymer molecular segments, and thus reduced the Tg. On the other hand, the introduction of rigid silica particles increases the Tg. Therefore, the blending ratio of SG and PBAT, as well as the size of the silica particles, will affect the Tg.
In addition, the Tg peak value of PBAT is large, indicating that the addition of SG reduces the internal friction of polymer molecules and the relative loss. In the blending system, the loss modulus of PBAT/SG2 is the largest, at 392.0 MPa, followed by that of PBAT/SG3, and the loss modulus of PBAT/SG1 is the smallest, at 292.8 MPa. PBAT has the highest energy storage modulus. After blending, the modulus of the composite materials is lower than that of PBAT. Among the three composite materials tested, PBAT/SG2 has the highest modulus, 2362.3 MPa, followed by PBAT/SG3 and PBAT/SG1, 1540.2 MPa and 1334.7 MPa, respectively. Since the energy storage modulus is a measure of the elasticity of the material, among the three composites, PBAT/SG2 has the best elasticity.
4. Conclusions
In this work, MCC and HDTMS with long carbon chains were silanized to obtain SGs, and three SGs with different silanization degrees were obtained by controlling the reaction ratio. By the sol-gel method, three SGs were mixed with PBAT at different blending ratios to prepare the PBAT/SG1, PBAT/SG2 and PBAT/SG3 composites. PBAT/SG composites have only one glass transition temperature, and the cross section of the composite is flat, which indicates that the compatibility of these two phases is good. The thermogravimetric test results showed that the yield stress of the composites increased while maintaining good thermal stability. The test results of the mechanical properties showed that the tensile strength of the three composites first increased and then decreased with increasing blending ratio. When the blending ratios of the PBAT/SG1, PBAT/SG2 and PBAT/SG3 composites are 90/10, 85/15 and 90/10, respectively, the tensile strength reaches the maximum value. The elongations at break were 16.4 MPa, 22.0 MPa and 17.3 MPa, while the elongations at break were 601.7%, 577.6% and 592.4%, respectively. Among the three composites, the composite with the best performance was PBAT/SG2. When the blending ratio is 85/15, the tensile strength is nearly 30% greater than that of pure PBAT, mainly because SG2 not only is hydrophobic but also has a moderate and uniform particle size coupled with good compatibility, which can effectively improve the mechanical properties of PBAT. The results obtained in this study help to further elucidate the relationship between the PBAT structure and performance and provide a feasible and efficient method for evaluating PBAT modifications.
Author Contributions
W. X. and M. W. contributed equally to this work. W. J. and Z. Y. developed the original idea and designed the experiments, Z. Y. directed the project; W. X. and M. W. performed the experiments; W. X. and Z. Y. co-wrote the manuscript. All the authors discussed the results and revised the manuscript.
Funding
This work was supported by the Natural Science Foundation of Changji University (Grant Nos. KYLK016).
Data Availability Statement
All data used to support the findings of this study are included within the article.
Conflicts of Interest
The authors declare no competing financial interest.
References
- Dufresne, A. Cellulose nanomaterial reinforced polymer nanocomposites. Curr Opin Colloin In. 2017, 29, 1-8. [CrossRef]
- Chaves, R.P.; Fechine, G.J.M. Thermo stabilisation of poly(butylene adipate-co- terephthalate). Polimeros. 2016, 26, 102-105. [CrossRef]
- Haider, T.P.; Völker, C.; Kramm, J.; Landfester, K.; Wurm, F. R. Plastics of the Future? The Impact of Biodegradable Polymers on the Environment and on Society. Angew. Chem. Inter. Ed. 2019, 58, 50-62. [CrossRef]
- Bruno, V.M.; Aline, S.; Gabriela, F.S.; Luana, M. R.; Fernanda, R.; Anderson, O. Influence of low contents of superhydrophilic MWCNT on the properties and cell viability of electrospun poly(butylene adipate-co-terephthalate) fibers. Mat Sci Eng C-Mater. 2016, 59, 782-791.
- Roldán-San Antonio, J.E.; Martín, M. Optimal Integrated Plant for Biodegradable Polymer Production. ACS Sustain Chem Eng. 2023, 11, 2172-2185. [CrossRef]
- Kargarzadeh, H.; Galeski, A.; Pawlak, A. PBAT green composites: Effects of kraft lignin particles on the morphological, thermal, crystalline, macro and micromechanical properties. Polymer. 2020, 203, 122748. [CrossRef]
- Liu, T.; Lian, X.; Li, L.; Peng, X.; Kuang, T. Facile fabrication of fully biodegradable and biorenewable poly (lactic acid)/poly (butylene adipate-co-terephthalate) in-situ nanofbrillar composites with high strength, good toughness and excellent heat resistance-ScienceDirect. Polym Degrad Stab. 2020, 171, 109044.
- Yang, K.; Liu, W.; Zhang, S.; Yu, W.; Shi, J.; Lin, Z.; Zheng, Q. Influence of the aggregated structures of layered double hydroxide nanoparticles on the degradation behavior of poly(butyleneadipate-co-terephthalate) composites. Appl Clay Sci. 2022, 230, 106713. [CrossRef]
- Caligiuri, V.; Tedeschi, G.; Palei, M.; Miscuglio, M.; MartinGarcia, B.; Guzman-Puyol, S.; Hedayati, M.K.; Kristensen, A.; Athanassiou, A.; Cingolani,R.; Sorger,V. J.; Salerno, M.; Bonaccorso, F.; Krahne, R.; Heredia-Guerrero, J.A. Biodegradable and Insoluble Cellulose Photonic Crystals and Metasurfaces. ACS Nano. 2020, 14, 9502-9511. [CrossRef]
- Shanker, R.; Ravi Anusuyadevi, P.; Gamage, S.; Hallberg, T.; Kariis, H.; Banerjee, D.; Svagan, A.J.; Jonsson, M.P. Structurally Colored Cellulose Nanocrystal Films as Transreflective Radiative Coolers. ACS Nano. 2022, 16, 10156-10162. [CrossRef]
- Han, D., Wang, H., Lu, T., Cao, L., Dai, Y., Cao, H., Yu, X. Scalable manufacturing green core-shell structure flame retardant,with enhanced mechanical and flame-retardant performances of polylactic acid. J Polym Environ. 2022, 30, 2516-2533. [CrossRef]
- Yeh, J.; Tsou, C.; Huang, C.; Chen. K.; Wu, C.; Chai, W. Compatible and crystallization properties of poly(Iactic acid)/poly(butylene adipate-co-terephthalate) blends. J Appl Polym Sci. 2010, 116, 680-687.
- Chen, X.; Zeng, Z., Ju, Y., Zhou, M., Bai, H., Fu, Q. Design of biodegradable PLA/PBAT blends with balanced toughness and strength via interfacial compatibilization and dynamic vulcanization. Polymer, 2023, 266 . [CrossRef]
- Wu, C. S. Antibacterial and static dissipating composites of poly(butyleneadipate-co-terephthalate) and multi-walled carbon nanotubes. Carbon. 2009, 47, 3091-3098.
- Zhao, X.; Yu, J.; Wang, X.; Huang, Z.; Zhou, W.; Peng, S. Strong synergistic toughenin and compatibilization enhancement of carbon nanotubes and multi-functional epoxy compatibilizer in high toughened polylactic acid (PLA)/poly (butylene adipate-co- terephthalate) (PBAT) blends. Int J Biol Macromol. 2023, 250, 126204.
- Jandas, P.J.; Mohanty, S.; Nayak, S.K. Thermal properties and cold crystallization kinetics of surface-treated banana fiber (BF)-reinforced poly(lactic acid) (PLA) nanocomposites. J Therm Anal Calorim. 2013, 114, 1265–1278. [CrossRef]
- Liu, Z,; Meng, F.; Tang, X.; Su, C.; Mu,Q.; Ju,G. Research on Properties of PBAT/CaCO3 Composite Films Modified with Titanate Coupling Agent. Polymers. 2023, 15, 2379. [CrossRef]
- Lo Re,G.; Engel, E.R.; Björn, L.; Sicairos, M.G.; Liebi, M.; Wahlberg, J.; Jonasson, K.; Larsson, P.A. Melt processable cellulose fibres engineered for replacing oil-based thermoplastics. Chem. Eng. J. 2023, 458, 141372. [CrossRef]
- Zhou, K.; Zhang, M.; Zhang, X.; Wang, T.; Wang, H.; Wang, Z.; Tang, X.; Bai, M.; Li, S.; Wang, Z.; Yue, M. cellulose reinforced polymer composite electrolyte for the wide-temperature-range solid lithium batteries. Chem. Eng. J. 2023, 464, 142537. [CrossRef]
- Malkapuram, R.; Kumar, V.; Negi, Y.S. Recent development in natural fiber reinforced polypropylene composites. J Reinf Plast Comp. 2009, 28, 1169-1189. [CrossRef]
- Mohanty, A.K.; Misra, M.; Hinrichsen, G. Biofibers, biodegradable polymers and biocomposites: An overview. Macromol Mater Eng. 2000, 276, 1-24.
- Arif, M.F.; Yusoff, P.S.; Ahmad, M.F. Effects of chemical treatment on oil palmempty fruit bunch reinforced high density polyethylene composites. J Reinf Plast Comp. 2010, 29, 2105–2118.
- Araujo, J.R.; Waldman, W.R.; De, M.A. Thermal properties of high densitypolyethylene composites with natural fibers: Coupling agent effect. Polym Degrad Stabil. 2008, 93, 1770-1775.
- Giri, J.; Lach, R.; Le, H.; Grellmann, W.; Saiter, J.; Henning, S.; Radusch H.; Adhikariet, R. Structural, thermal and mechanical properties of composites of poly (butylene adipate-co-terephthalate) with wheat straw microcrystalline cellulose. Polym Bull. 2020, 78, 4779-4795. [CrossRef]
- Hou, L.; Chen, J; Liu, J. Octadecylamine graft-modified cellulose nanofiber and its reinforcement to poly(butylene adipate-co-terephthalate) composites. Paper & Biomaterials. 2023, 7, 42-50.
- Sara, F.; Elena, M.; Cristina, M.; Pierluigi, M. Comparison of solution-blending and melt- intercalation for the preparation of poly(ethylene-co-acrylic acid)/organoclay nano- composites. Eur Polym J. 2008, 43, 1645-1659.
- Das, C.K.; Reddy, C.S. HLDPE/Organic Functionalized SiO2 Nanocomposites with Improved Thermal Stability and Mechanical Properties. Compos Interface. 2005, 11, 687-699. [CrossRef]
- Sankaraiah, S.; Lee, J.M.; Kim, J.H.; Choi, S.W. Preparation and characterization of surface-functionalized polysilsesquioxane hard spheres in aqueous medium. Macromolecules. 2008, 41, 6195-6204. [CrossRef]
- Liu J.; Huang W.; Xing Y.; Li, R.; Dai, J. Preparation of durable superhydrophobic surface by sol–gel method with water glass and citric acid. J Sol-Gel Sci Techn. 2011, 58, 18-23. [CrossRef]
- Abdelmouleh, M.; Boufi, S.; Belgacem, M.N.; Dufresne, A.; Gandini, A. Modificationof cellulose fibers with functionalized silanes: Effect of the fiber treatment on the mechanicalperformances of cellulose-thermoset composites. J Appl Polym Sci. 2005, 98, 974-984.
- Qu, P.; Zhou, Y.; Zhang, X.; Yao, S.; Zhang, L. Surface modification of cellulose nanofibrils for poly(lactic acid) composite application. J Appl Polym Sci. 2011, 125, 3084-3091. [CrossRef]
- Xu, L.; Zhang, W.; Xu, B.; Cai, Z. Fabrication of superhydrophobic cotton fabricsby silica hydrosol and hydrophobization. Appl Surf Sci. 2011, 257, 5491-5498.
- Banerjee, M.; Saraswatula, S.; Willows, L.G.; Woods, H.; Brettmann, B.Pharmaceutical crystallization in surface-modified nanocellulose organogels. J Matere Chem B. 2018, 6, 7317-7328. [CrossRef]
- Phosee, J.; Wittayakun, J.; Suppakarn, N. Mechanical properties and morphologies of rice husk silica (RHS)/poly(butylene adipate-co-terephthalate) (PBAT) composites: Effect of silane coupling agent. Advanced Materials Research. 2010, 978, 141-144.
Figure 3.
Figure 3. FTIR and TEM images: (A) FTIR spectra of MCC (a) SG with different ratios of HDTMS to MCC (a): 1:10 (b); 3:10 (c); 5:10 (d)); (B) enlarged FTIR spectra (A) in the range of 800 cm-1 to 1300 cm-1; (C-E) TEM image of SG with different ratios of HDTMS to MCC (1:10 (C); 3:10 (D); 5:10 (E)).
Figure 3.
Figure 3. FTIR and TEM images: (A) FTIR spectra of MCC (a) SG with different ratios of HDTMS to MCC (a): 1:10 (b); 3:10 (c); 5:10 (d)); (B) enlarged FTIR spectra (A) in the range of 800 cm-1 to 1300 cm-1; (C-E) TEM image of SG with different ratios of HDTMS to MCC (1:10 (C); 3:10 (D); 5:10 (E)).
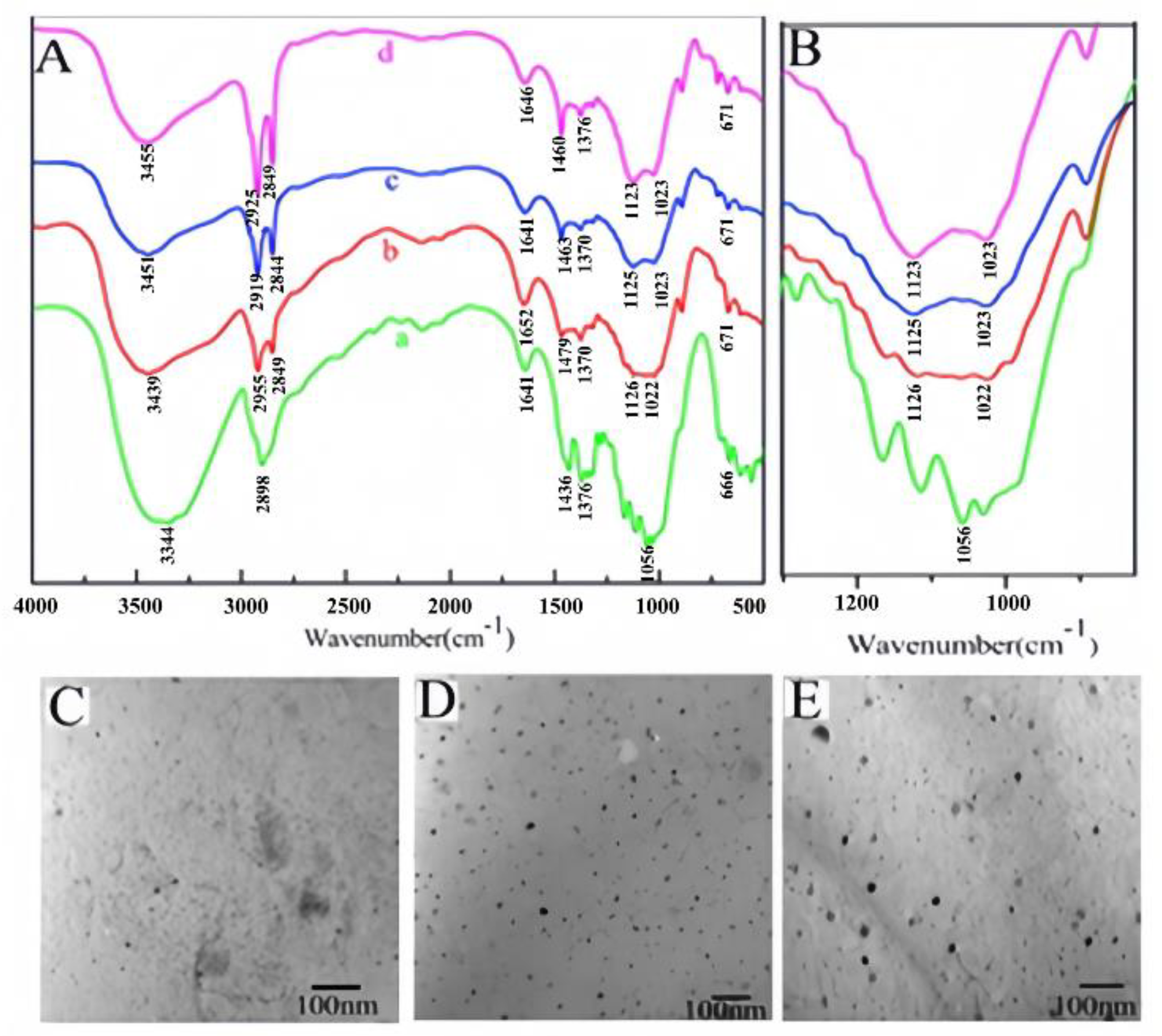
Figure 4.
IR and TEM images of (a) PBAT, (b and B) PBAT/SG1, (c and C) PBAT/SG2 and (d and D) PBAT/SG3.
Figure 4.
IR and TEM images of (a) PBAT, (b and B) PBAT/SG1, (c and C) PBAT/SG2 and (d and D) PBAT/SG3.
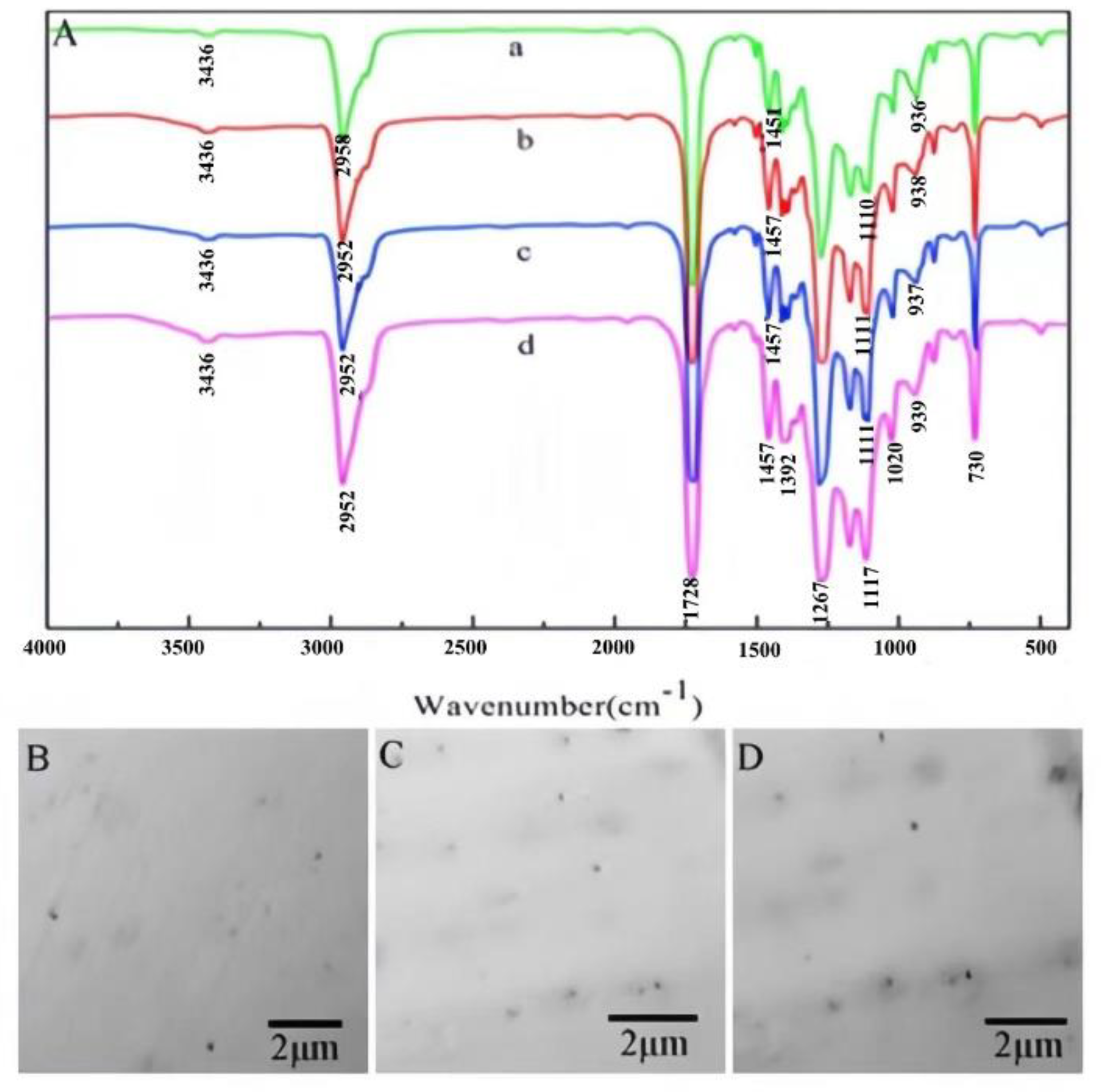
Figure 5.
Stress‒strain curves of the PBAT/SG composites.
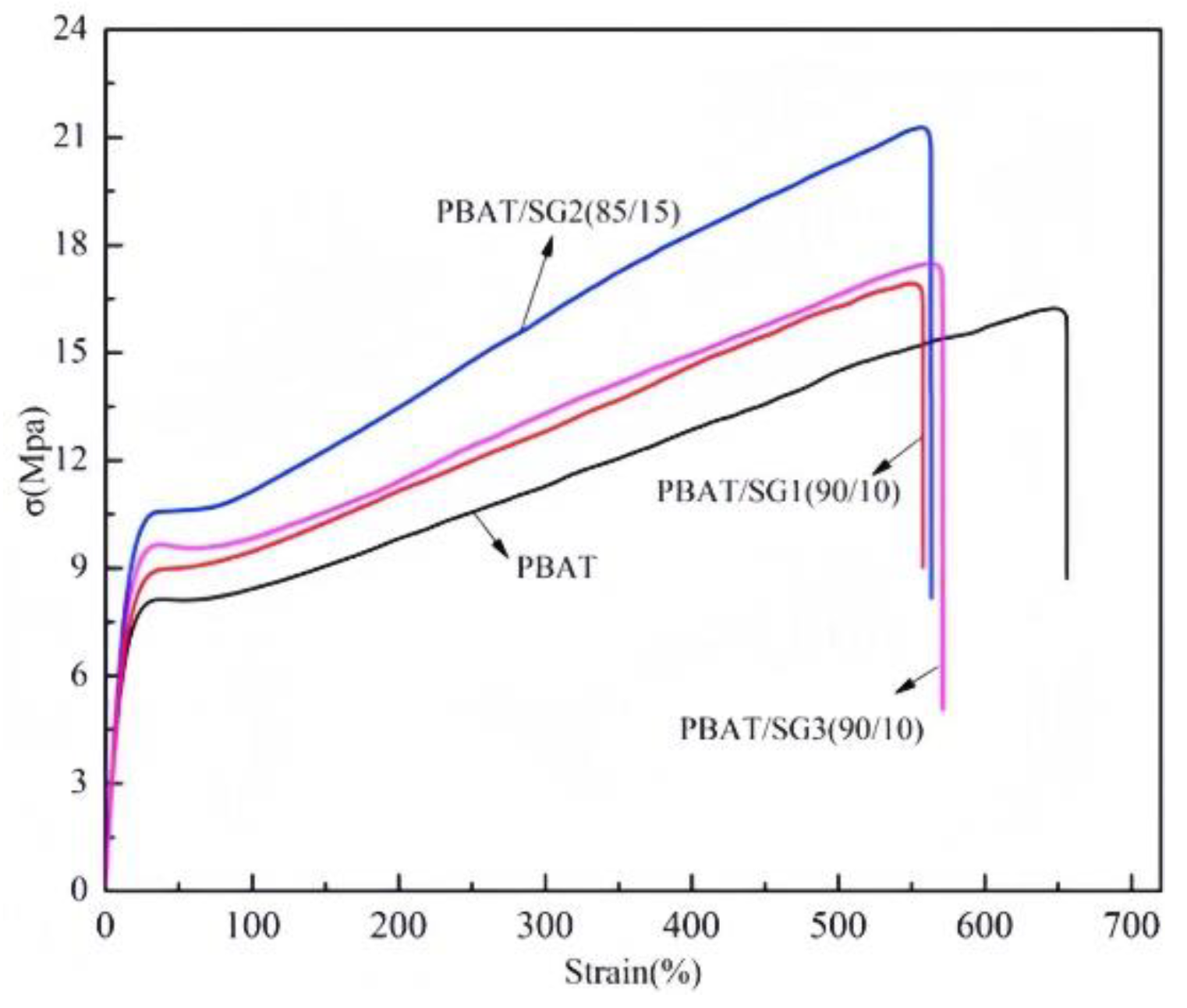
Figure 6.
Tensile strength and elongation at break of composites with different SG amounts.
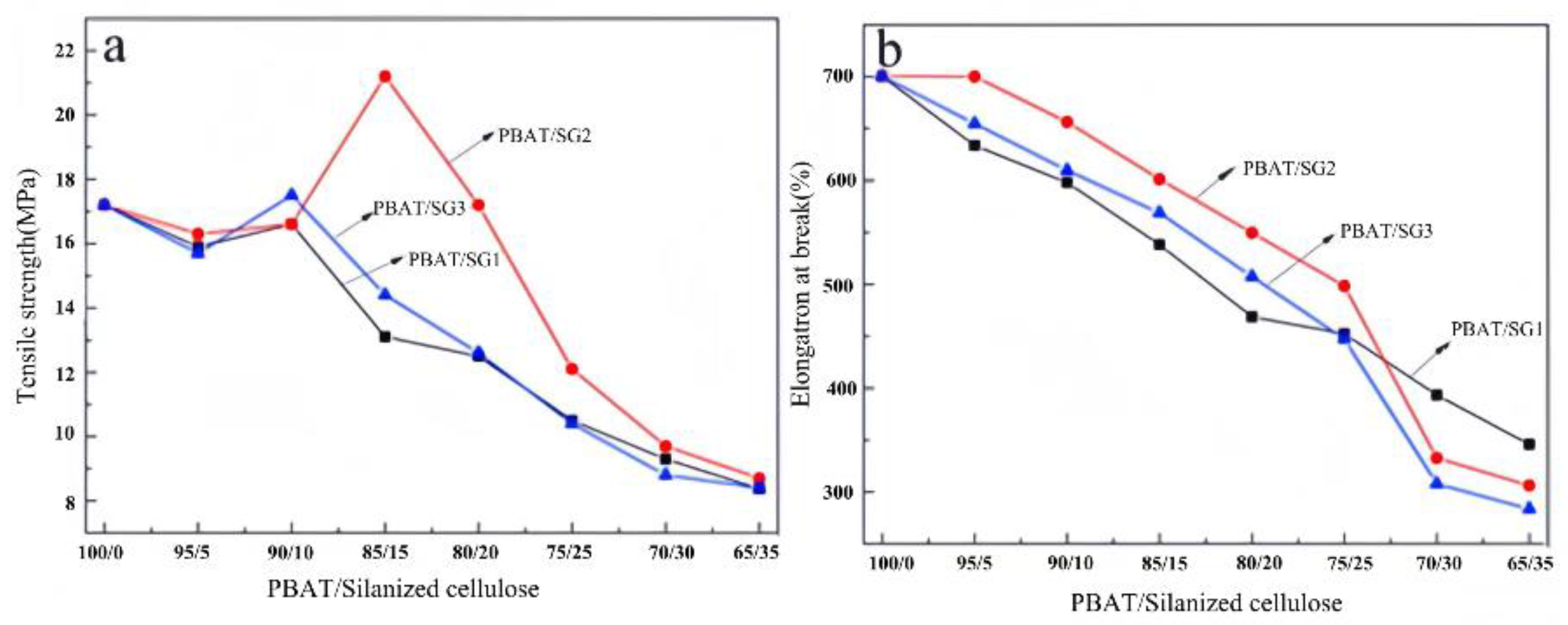
Figure 8.
TGA and DTG curves of (a) PBAT, (b) PBAT/SG1 (90/10), (c) PBAT/SG2 (85/15) and (d) PBAT/SG3 (90/10).
Figure 8.
TGA and DTG curves of (a) PBAT, (b) PBAT/SG1 (90/10), (c) PBAT/SG2 (85/15) and (d) PBAT/SG3 (90/10).
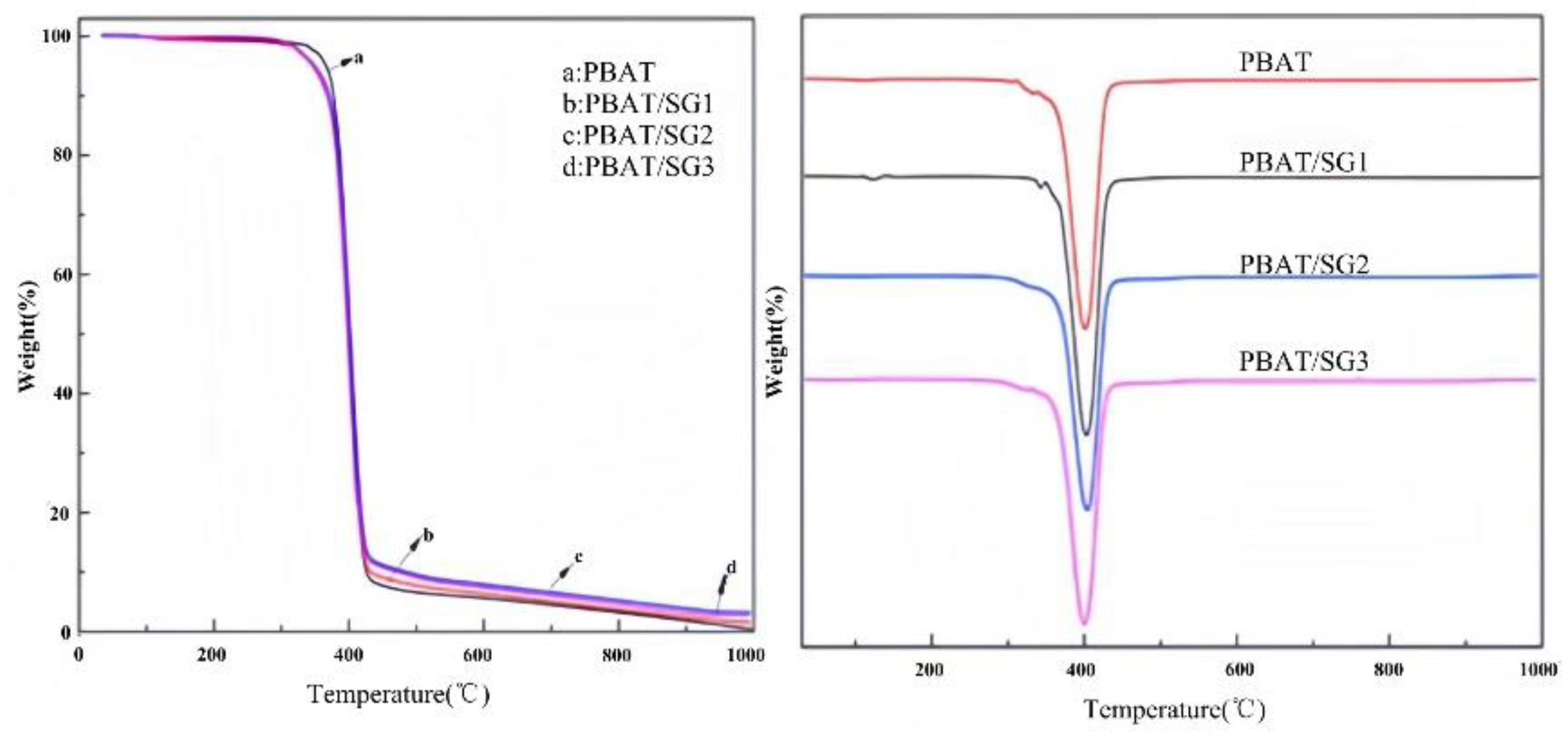
Figure 9.
SEM images of (a) PBAT, (b) PBAT/SG1 (90/10), (c) PBAT/SG2 (85/15) and (d) PBAT/SG3 (90/10).
Figure 9.
SEM images of (a) PBAT, (b) PBAT/SG1 (90/10), (c) PBAT/SG2 (85/15) and (d) PBAT/SG3 (90/10).

Figure 10.
Figure 10. DMA curves of PBAT and composites at different temperatures
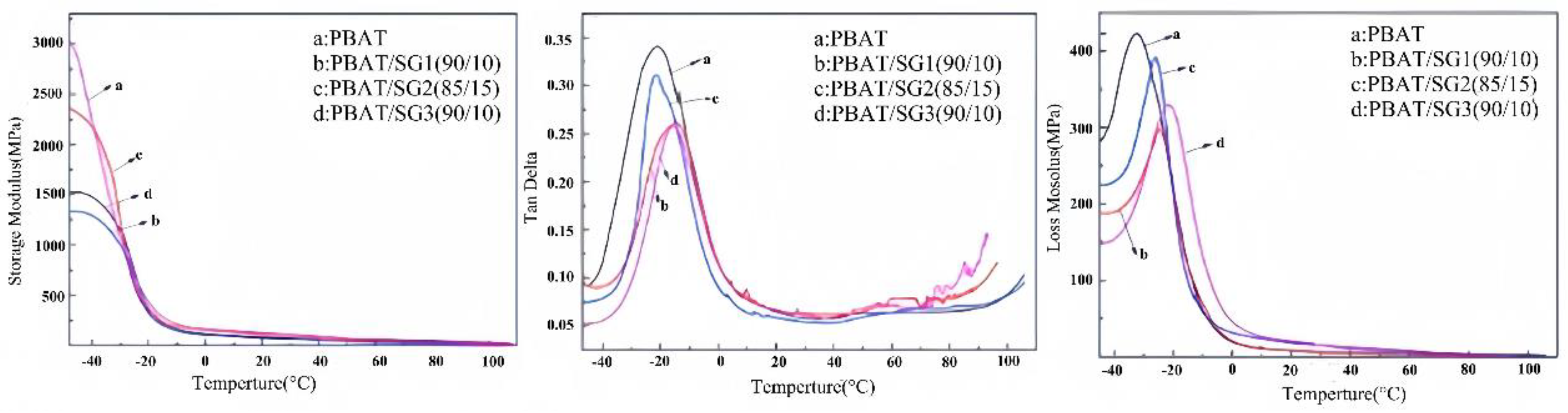

Table 1.
Water adsorption of MCC and SG with different mass ratios of HDTMS to MCC.
| Sample | Mass ratio of HDTMS to MCC | Water absorption (%) |
|---|---|---|
| MCC | - | 82.80 |
| SG1 | 1:10 | 65.59 |
| SG2 | 3:10 | 30.11 |
| SG3 | 5:10 | 0.54 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



