
Submitted:
06 January 2024
Posted:
09 January 2024
You are already at the latest version
Abstract
The brain's unique characteristics make it exceptionally susceptible to oxidative stress, which arises from an imbalance between reactive oxygen species (ROS) production, reactive nitrogen species (RNS) production, and antioxidant defense mechanisms. This review explores the factors contributing to the brain vascular tone vulnerability in the presence of oxidative damage, which can be of clinical interest in critically ill patients or presenting acute brain injuries. The brain's high metabolic rate and inefficient electron transport chain in mitochondria lead to significant ROS generation. Moreover, non-replicating neuronal cells and low repair capacity increase susceptibility to oxidative insult. ROS can influence cerebral vascular tone and permeability, with a potential impact on cerebral autoregulation. Different ROS species, including superoxide and hydrogen peroxide, exhibit vasodilatory or vasoconstrictive effects on cerebral blood vessels. Reactive nitrogen species (RNS), particularly NO and peroxynitrite, also exert vasoactive effects. This review further investigates the neuroprotective effects of antioxidants, including superoxide dismutase (SOD), vitamin C, vitamin E, and the glutathione redox system. Various studies suggest that these antioxidants could be used as adjunct therapies to protect the cerebral vascular tone during high oxidative stress conditions. Nevertheless, more extensive research is required to comprehensively grasp the relationship between oxidative stress and cerebrovascular tone, as well as to explore the potential benefits of antioxidants as adjunctive therapies in critical illnesses and acute brain injuries.
Keywords:
Oxidants
; Reactive Oxygen Species
; Reactive Nitrogen Species
; antioxidants
; cerebrovascular tone
1. Introduction
Brain tissue is exceptionally susceptible to the harmful effects of oxidative stress due to its unique characteristics. Despite accounting for only 2% of the body weight, the brain consumes approximately 20% of the total basal oxygen [1,2]. This heightened oxygen consumption is essential for ATP production, which is necessary to maintain the brain's high metabolic rate. Unfortunately, the electron transport chain in the mitochondria, responsible for ATP synthesis, leads to the leakage of reactive oxygen species (ROS) [3]. Consequently, the brain cells ceaselessly generate a significant amount of ROS. Additionally, the brain comprises non-replicating neuronal cells, possesses a high cell surface-to-cytoplasm ratio, and has a relatively low repair capacity. These structural and functional features further contribute to its vulnerability to oxidative damage [4]. A summary of the main factors contributing to the brain tissue's high susceptibility to oxidative insults are described in Table 1.
The regulation of vascular tone in cerebral vessels is controlled by both muscular and endothelial mechanisms [5]. The former is regulated by myogenic mechanisms, which originate from vascular smooth muscle. The latter is regulated by endothelial factors, such as nitric oxide and endothelin, which can decrease or increase the vascular tone, respectively. In addition, local hormones, metabolic by-products, ROS and RNS, as well as drugs, can influence and contribute to cerebral vascular tone. Notably, ROS can arise from all the layers of the vascular wall and adjacent tissues through several mechanisms, and, among these, the NADPH-oxidase enzyme may be the primary source of ROS production from the vascular cells [6].
This widespread existence of ROS within and surrounding cerebral blood vessels aligns with their ability to influence the vascular tone and vascular permeability [7,8,9,10], and their effects may be greater in the cerebral vasculature than in any others [11,12]. Even cerebral autoregulation (CAR), a complex physiological mechanism that adjust cerebrovascular tone to maintain cerebral blood flow within desired ranges in response to changes in cerebral perfusion pressure, may be altered by ROS [13]. All these vascular effects may come from the direct activity of ROS on ion channels [14,15], through their interrelation with reactive nitrogen species (RNS), particularly nitric oxide (NO), or other vasoactive mechanisms still unknown.
The balance of redox reactions, given by ROS and RNS, may be of clinical interest especially in critically ill settings when high oxygen levels could be provided (thus increasing reactive species production), or in acute brain injuries when these species are more frequently produced [16,17]. In this review, we describe the primary ROS and RNS and their documented impact on cerebral vascular tone. Furthermore, since oxidative stress relies on oxidant species production and antioxidant defense mechanisms, we will also explore the main antioxidants and their effects on cerebral vascular tone.
2. Reactive Oxygen Species
The term Reactive Oxygen Species (ROS) refers to oxygen-containing chemical species that are unstable and highly reactive, capable of causing damage to various biological molecules. These species arise from the uptake of electrons from molecular oxygen, i.e. O2. Multiple sources contribute to their formation, first-of-all, the electron transport chain within the mitochondria, where, during cellular respiration, electrons are transferred through a series of protein complexes. When there's a leak or premature transfer of an electron to molecular oxygen, superoxide anion (O₂-) is formed. Other ways in which ROS are formed encompass, among others, metabolizing enzymes, NAD(P)H oxidases (NOS), ionizing radiation, the Fenton reaction [which can be simplified as Fe2+ + H2O2 → Fe3+ + OH− + HO•], cell uptake of microbes or xenobiotics.
Given their strong ability to react chemically with their environment, ROS are well known to lead to several pathological consequences [18]. Surprisingly, depending on the specific context, ROS can exhibit both protective and deleterious effects on the organism, the same molecule exerting opposing interferences on biological processes [19]. The impact of these effects relies on the interplay between the production of ROS and their clearance rates, wherein antioxidants and scavengers assume a critical role [20]. When this balance is disrupted, these reactive molecules can unopposedly interact with and damage vital cellular components such as DNA, proteins, and lipids. This molecular damage can compromise the normal function of cells, leading to tissue injury. This harmful state, characterized by the overwhelming presence of oxidative species and the resultant cellular and tissue damage, is termed "oxidative stress" [21].
The main mechanisms of the formation of ROS, further described later, are illustrated in Figure 1.
2.1. The main Reactive Oxygen Species
As previously mentioned, ROS originate from O2, a diradical, meaning it has two unpaired electrons in its molecular orbital. Despite being relatively stable under standard conditions, its diradical nature makes O2 more reactive than if it were a closed-shell molecule, and prone to participate in various chemical reactions, especially those involving electron transfer.
When O2 acquires an electron, primarily from the mitochondrial electron transport chain as earlier described, it gives rise to superoxide anion (O2-). This species carries both a negative electric charge and an unpaired electron, making it both an anion and a radical and it can be seen as the precursor to ROS and, as we will see later, it also plays a role in the formation of RNS. It is a free radical, i.e., a molecule that contains one free unpaired electron in its outer orbital. However, despite its name, O2- does not seem to act as a potent oxidizing agent. Instead, its detrimental effects stem from its secondary interactions, particularly when it reacts with nitric oxide (NO). Due to its low permeability, the actions of superoxide are primarily related to the compartment in which it is produced; however, it can also influence the surrounding cellular environment by crossing the cell's ion channels exerting its effects at a distance from the production point, even with a half-life, at normal pH, of about 5 seconds [22].
Hydrogen Peroxide (H2O2) is generated from O2- through a reaction called dismutation (i.e., a reaction in which the same species is both reduced and oxidized), in which 2 superoxide molecules are transformed in H2O2 and O2, catalyzed by the enzyme superoxide dismutase (SOD). H2O2 is not a free radical, and therefore it does not directly oxidize other molecules. However, it can move quickly through the cell membranes and, transforming into a potent compound (hydroxyl radical, described further in the text), it can exert the effect of ROS at different sites instead of its production site. Indeed, due to a non-enzymatic reaction that requires iron in its reduced state (Fe2+), known as the Fenton reaction, the most reactive compound known, hydroxyl radical (OH-), can be generated from H2O2. The OH- can also be formed through the reaction between O2- and nitric oxide (NO), and the Haber-Weiss reaction [which can be simplified as O2- + H2O2 → OH˙ + OH- + O2] [13,23]. Due to its tendency to react quickly with the first available molecule, this radical has a short half-life [20]. To prevent the continuation of the reaction chain, catalases can decompose H2O2 into water and O2, thereby preventing the formation of OH˙. Because of this cooperative capability, both SOD and catalase are classified as antioxidants.
The main ROS described, and their formation mechanisms, are schematically depicted in Figure 2.
2.2. The effects of ROS on cerebral vascular tone
In the brain, whose metabolism relies almost exclusively on the presence of oxygen, ROS appear to have a decisive influence on cerebral flow [13,24]. Indeed, the brain has a unique neurovascular coupling structure, facilitating functional and anatomical interactions among neurons, astrocytes, pericytes, microglia, and blood vessels, known as the neurovascular unit [25]. This unit allows selective adaptation of blood flow in different brain regions, influenced by ROS and nitric oxide (NO), which act on smooth muscle cells and pericytes [13,26]. For example, in animal models, ROS have been shown to have a role in microvascular reperfusion disturbances [27].
ROS can affect cerebral vascular smooth muscle tone in several biochemical pathways [28], potentially in a more pronounced way in the presence of cerebrovascular diseases [29]. The overall vasoactive effect varies based on 1) the specific type of molecule involved and 2) its concentration. For example, O2- can act as a direct endothelium-dependent vasodilator and a smooth-muscle vasoconstrictor through the inactivation of NO [30,31]. Indeed, O2-, as well as H2O2, can affect the vascular tone, causing vasodilation, by activating calcium-dependent potassium channels (Kca), which are the most common K+ channel expressed on cerebral arteriolar musculature, and which are sensitive to the redox state of the environment [13]. By contrast, O2- leads to vasoconstriction through a powerful inhibition of endothelial NO-mediated dilation. The equilibrium between these two opposite results depends on the net concentration of the reactive species. Indeed, findings from numerous in vitro and in vivo animal studies confirm that both O2- and H2O2 exhibit a biphasic impact on cerebral blood vessels, contingent upon their concentration. At low concentrations, O2- leads to vasodilation, while at high concentrations, it causes vasoconstriction. Similarly, H2O2, when present in very high concentrations, initially induces vasoconstriction, and later leads to vasodilation [30].
In addition to the aforementioned mechanisms, the modulation of cerebral arteriolar tone through chloride channels has also been suggested [32]. Moreover, O2- can react with arachidonic acid and other unsaturated fatty acids, to form isoprostanes. They are known as strong vasoconstrictors that can cause a reduction of cerebral blood flow. Thus O2- can indirectly further affect vascular tone and may be of pathological interest in traumatic brain injuries [13,30]. Finally, H2O2 would also act in some cases as a membrane depolarizer, exerting, with a hyperpolarization, a vasodilatory effect [13,30,31].
Finally, at different oxygenation levels, both the production of ROS and antioxidants exhibit distinct kinetics [33]. Therefore, it is conceivable to assume that the vasoactive activity of ROS, as well as RNS and antioxidants, may depend not only on the type of ROS and their concentration but also on a third feature, i.e., the kinetics of their production and degradation. Indeed, compensatory mechanisms might come into play when a stimulus becomes a new steady state. This can occur especially in pathological conditions, such as in acute brain injury or in critically ill patients, where elevated ROS levels may increment acutely, and persist for an extended period.
The vascular impacts of ROS on cerebrovascular tone are diverse and involve complex mechanisms. These also include interactions with NO and RNS, which will be further discussed later.
2.3. NADPH Oxidases
The nicotinamide adenine dinucleotide (phosphate) oxidases (NADH/NADPH oxidases, or Nox) are prominently involved in the enzymatic generation of ROS [34]. These membrane-bound enzymes produce superoxide radicals by utilizing NADH or NADPH as electron donors. In mammals, there exist seven isoforms of Nox. Among them, Nox1, Nox2, Nox4, and Nox5 are expressed in various tissues such as endothelium, vascular smooth muscle cells, fibroblasts, and perivascular adipocytes [35]. Under normal physiological conditions, vascular Nox exhibits a relatively low baseline activity. However, in response to various stimuli like cytokines, the enzyme activity can be augmented both in the short term and over an extended period. This response to stimuli disrupts vascular homeostasis, leading to pathological changes and associated health issues, such as increased blood pressure [36].
The involvement of NADH and NADPH reinforces the notion that the redox state is critical in influencing the tone of cerebral circulation. Indeed, interestingly, studies have shown that NADPH oxidase activity exhibits a significantly higher magnitude in cerebral arteries than in systemic arteries [12]. As electron acceptors, both NADH and NADPH facilitate the activity of Nox enzymes, leading to the production of O2- [24,37], which is in the end responsible for the impact on the vascular tone. Thus, ROS production derived from NADPH oxidase may contribute to vascular pathology while also to the maintenance of normal physiological vascular tone.
2.4. Additional viewpoints
ROS have more effects than expected, and they could work as signal molecule. In this way it is thought ROS play a role in the degradation of HIF-1α, as there is evidence of a negative correlation between these two [26]. HIF-1α, known as the hypoxia-inducible factor, is a cytosolic transcription factor that moves to the nucleus during hypoxia. It binds to HIF-β and activates the expression of genes containing hypoxia-responsive elements (HRE), which results in the production of hypoxia-related proteins such as erythropoietin (EPO). The production of ROS is implicated in the proposed "normobaric oxygen paradox" [38], where intermittent hyperoxic stimulation could paradoxically mimic a hypoxic-like condition, leading to the activation of HIF-1α [39]. Moreover, ROS production via NADPH oxidase, especially Nox2, may intervene in the evoked HIF-1α synthesis and stability [40]. These mechanisms may be especially relevant in brain cells, as they tend to increase ROS production under injured conditions. However, the existence of this phenomenon and its potential effects on the brain tissue have not been thoroughly investigated.
2.5. Regional Differences
It has been observed that the mechanisms that intervene in oxidative equilibrium may have differences between cerebral and systemic circulation [12]. Additionally, there may be differences in susceptibility to oxidative stress among cerebral microvessels in various brain regions [41]. This could account, for example, for the increased vulnerability of the hippocampus and cortex to oxidative stress and endothelial dysfunction compared to the cerebellum. These regional differences may also be influenced by the local availability of NO, which has been demonstrated in experimental animal models [42]. Therefore, it appears conceivable that even small changes in ROS and RNS concentrations can lead to clinical changes via their effect on the brain vasculature, especially during brain injuries and cerebrovascular diseases. Nevertheless, further human research is required to gain data and a deeper comprehension of these aspects, moving beyond theoretical discourses.
3. Reactive Nitrogen Species
Reactive Nitrogen Species (RNS) are a group of highly reactive nitrogen-containing molecules that are generated as by-products of nitrogen metabolism. These molecules are crucial in various physiological and pathological processes, including vascular control [43].
3.1. The formation of the main Reactive Nitrogen Species
Nitric Oxide (NO) is the most well-known and significant member of the RNS family. Also formerly known as endothelium-derived relaxing factor (EDRF), it is constantly generated in the body through the enzymatic action of Nitric Oxide Synthases (NOS) on L-Arginine and Oxygen. There are three distinct isoforms of NOS present in the body: iNOS (inducible), eNOS (endothelial), and nNOS (neuronal) [44,45]. NOS generates in loco NO, which readily exerts its biological activity. Although its half-life is limited to a few seconds, NO can react with hemoglobin, forming S-nitroso hemoglobin, or be converted to nitrite. S-nitroso hemoglobin (SNO-Hb) is a derivative of hemoglobin that has been modified by the addition of a nitric oxide (NO) group to a cysteine thiol group, in a reaction called S-nitrosylation.
Nitrite (NO2-) is a chemical compound that consists of one nitrogen atom and two oxygen atoms. It is an anion that can form salts such as sodium nitrite (NaNO2) or potassium nitrite (KNO2). SNO-Hb and NO2- salts act as NO reservoirs, which can be transported through systemic circulation and released under certain conditions to promote vasodilation and increase blood flow to tissues distant from the site of origin [46,47].
Other molecules can be categorized into RNS, produced as metabolites in NO decomposition or by reaction between other compounds such as CO2 and ROS. For example, peroxynitrite (ONOO-) is formed from the reaction between NO and superoxide ions, thus reducing NO concentrations [48]. Although evidence addresses NOS as responsible for nitroxyl (HNO) production, it is still unclear how this compound is synthesized in vivo [49,50]. Indeed, HNO can also be generated after the reduction of NO by mitochondrial cytochrome C, xanthine oxidase, ubiquinol, hemoglobin, and manganese superoxide dismutase (SOD) [50].
Nitrate (NO3-) is a chemical compound that consists of one nitrogen atom and three oxygen atoms. Similarly to NO2-, it carries a negative charge, and thus, it is found in salts chemical form. A significant amount of nitrate comes from leafy green vegetables. Salivary glands actively take up nitrate from circulation, resulting in saliva concentration 10-fold higher concentration than in plasma [51]. A certain quantity of NO3- is converted to NO2- by reductase enzymes in the salivary glands [51]. Finally, some nitrites are absorbed when ingested and become part of the "nitrate-nitrite-nitric oxide pathway" [52]. Kapil et al. have demonstrated how nitrate supplementation can lower blood pressure and that depleting oral microbiomes can jeopardize endogenous nitrate production, leading to hypertension [53]. There are other endogenous ways to synthesize nitrate and nitrite [52]. Over the years, many trials have established the conversion of NO to nitrite occurring naturally in vivo. This reaction has been linked to hypoxia, deoxyhemoglobin, and lower pH [54,55,56,57,58]. Furthermore, a significant increase in plasma nitrite was displayed during the administration of 80ppm iNO in a murine model [59]. With all these observations, nitrates and nitrites can be considered as of NO reservoir [60]. Indeed, while NO has a half-life limited to 2 ms, that of nitrite is much longer and can quickly spread all over the organism [52].
The formation of the main RNS described is schematically depicted in Figure 3.
3.2. The effects on cerebral vascular tone
3.2.1. Nitric Oxide
Since its recognition as the endothelium-derived relaxing factor [61,62], several studies have described NO as a strong vasodilator agent [44]. After synthesizing in the endothelium, NO increases cyclic guanosine monophosphate (cGMP) in smooth muscles, by stimulating soluble guanylyl cyclase. cGMP finally inhibits proteins responsible for contraction, causing vasorelaxation [44,63]. In adjunct to this mechanism, NO induces the reduction of intracellular calcium, mediated by ATPases that actively transport calcium inside the sarcoplasmic/endoplasmic reticulum [44,63]. Other pathways toward vasodilation include the reduction in 20-hydroxyeicosatetraenoic acid [64] and prostaglandins [65]. NO may be key in regulating cerebral blood flow (CBF) [66]. Indeed, in hyperoxia [67,68,69], hypoxia [67,68], and hypercapnia [70], it was found to be an essential mediator of CBF in animal models. Moreover, NO also has a leading role in coupling CBF and neuronal activity [71].
Several studies explored the role of NO in critical neurologic illnesses. Indeed, early stages of severe brain injuries recognize a depletion of NO, probably contributing to secondary brain damage [72]. By contrast, when present NO reduces injuries and has neuroprotective properties in ischemia/reperfusion animal models, both in stroke and cardiac arrest [72]. For this reason, a prospective trial has been performed, proving the feasibility of iNO administration in cardiac arrest, and showing promising results in terms of survival [73].
In subarachnoid hemorrhage, NOS inactivation is associated with alterations in microvascular density and hemostasis, leading to rebleeding, intracranial hypertension, and larger hemorrhage volume [74]. Conversely, NO reduction is also correlated with a lower grade of neuroinflammation and better neurobehavioral function in rats [75]. Studies in humans showed a correlation between elevated asymmetric dimethylarginine (which is an inhibitor of iNOS, and thus of the vasodilator molecule NO) with vasospasm and worse outcome [76]; another small human study found elevated levels of NO metabolites, especially in subjects with poorer outcomes [77]. Although results may be controversial, the literature suggests deleterious effects of NO depletion, supported by increased microthrombi formation and reduced cortical activity [72]. A pilot human trial established the safety of iNO administration and showed promising results in treating delayed cerebral ischemia [78].
Pathways in traumatic brain injury (TBI) appear more complex. Mechanical insults may induce up-regulation of iNOS and over-production of NO and ONOO-. The RNS stimulate the production of glutamate, which triggers nNOS. Nevertheless, NO plays an uncertain role in this disease, and its implications are still unknown [79]. Studies in newborn animal models are also contradictory. Some reports showed protective properties of NO by vasodilation, reducing ROS concentrations and scavenging radicals; others linked it to deleterious effects and greater brain injuries. These discrepancies might depend on the timing of production and concentrations of NO [80].
The brain controls blood flow by interacting with vessel diameter. Autoregulation ensures constant blood supply; changes in systemic blood pressure -if they occur within certain limits- induce vasodilation or vasoconstriction. Additionally, the brain increases blood flow in areas where neurons are more active through a mechanism named neurovascular coupling. Although other pathways and elements participate, NO plays a significant role in both of them. NOS inhibition (and thus lowering NO production) increases the lower limit of mean arterial blood pressure where autoregulation acts, reducing its efficiency [52,53,54]. Through neurovascular coupling, blood flow is directed to more active areas, where glutamate (the main excitatory neurotransmitter) induces calcium entry in neurons and activation of neuronal nNOS finally resulting in NO production and vasodilation [65].
Interestingly, CO2 may act as a regulator of NO. Indeed, the process of adjusting cerebral vascular tone in response to changes in arterial carbon dioxide partial pressure (PaCO2) is known as chemoregulation, which could be the main trigger of endothelial NO-release [81]. In humans, hypocapnia leads to vasoconstriction and reduced CBF, while hypercapnia and changes in perivascular pH result in vasodilation and increased CBF [81]. Cerebral endothelial cells and astrocytes have been shown to release NO under normocapnic conditions, while NO production increases during hypercapnia and decreases during hypocapnia, regardless of pH levels [82]. It has been proposed that these NO variations in response to PaCO2 are specific to NOS regulation and that administering exogenous NO may influence the CO2-dependent chemoregulation mechanism [82].
Finally, as an NO reservoir, the vasodilating effects of nitrites have long been investigated. Although some results failed to find any vasoactive activity [83], most studies confirmed that nitrite has vasorelaxant effects through the activation of guanylate cyclase [46,84,85,86,87]. >
In summary, NO stands as a potent vasodilator, playing diverse roles in the regulation of blood flow and brain protection. Its mechanism is intricate and involves numerous interactions. When it comes to brain injuries, a deficiency in NO could contribute to secondary brain damage, while NO possesses neuroprotective qualities in animal models of ischemia/reperfusion, including stroke. Interestingly, NO inhalation has been suggested as a neuroprotective intervention during cardiopulmonary resuscitation [88]. Consequently, extensive research is needed to gain a deeper understanding of its mechanisms and potential therapeutic effects.
3.2.2. Peroxynitrite
Peroxynitrite (ONOO-) has shown vasoactive properties, producing vasorelaxation of arterioles in vitro [89] and in vivo [20], although its potency seems very low, up to 50-fold less than that of NO [90]. The effect doesn't appear to involve the endothelium [91], and the vasoactive mechanism is still unclear. Some authors proposed that the effect is linked to the opening of ATP-sensitive potassium channels [20,92], by directly activating the channels or reducing ATP concentration by interfering with cellular metabolism [20,92,93]. Other mechanisms proposed to explain this vasodilating activity include elevation of cGMP levels, membrane hyperpolarization via K+ channel activation, activation of myosin phosphatase activity, and interference with cytosolic calcium movement and cellular membrane Ca2+ entry [89].
However, in contrast with all the above results, Daneva et al. induced an increase in ONOO- by upregulating iNOS in mice, reporting an increase in pulmonary arterial pressure by impeding calcium entry in smooth muscle cells of pulmonary arteries [94]. Similarly, Ottolini et al. investigated the same pathway in diet-obese mice, finding a link between obese hypertension and high ONOO- levels [95]. Finally, in vitro and animal studies [96,97] have observed that peroxynitrite may induce vasoconstriction in cerebral arteries. This effect is likely due to inhibiting the cerebral K+-dependent calcium channel. Interestingly, the addition of glutathione inhibited this cerebral vasoconstrictive effect.
Therefore, the net effects of peroxynitrite appear conflicting, and are not completely understood, probably being dose and time-dependent [98].
3.2.3. Nitroxyl
HNO has been proven to act as an endothelium-derived vasorelaxant [99,100,101,102] not susceptible to tolerance [100]. Moreover, HNO has multiple effects, including inhibiting platelet aggregation, limiting vascular smooth muscle proliferation, and interacting with metallo and thiol-containing proteins.
HNO promotes vasodilation by activating several molecular pathways [50,101,102,103]. These pathways include the stimulation of soluble guanylyl cyclase (sGC), leading to an increase in cGMP. From animal studies, it has been suggested that the sGC-cGMP pathway may be necessary and sufficient for HNO-induced vasodilation in vivo [104]. Others proposed that HNO may activate ATP-sensitive potassium (KATP) channels and voltage-dependent potassium (Kv) channels, resulting in the outflow of potassium ions. This causes the cell membrane to become hyperpolarized and leads to a reduction in intracellular calcium levels. Finally, from an elegant animal study, Eberhardt et al. suggested a vasodilation HNO mediated vasodilation via the transient receptor potential channel A1 (TRPA1) and calcitonin gene-related peptide (CGRP) [105]. This HNO-TRPA1-CGRP signaling pathway could be a crucial component in the neuroendocrine regulation of vascular tone, mediated by HNO.
4. Antioxidants
Antioxidants are substances that protect from the deleterious effects of free radicals. Their action, which can be enzymatic and non-enzymatic, can be further divided into two groups: 1) preventing the generation of ROS and blocking the generated ROS, and 2) repairing the damages the free radicals have caused. The main antioxidants are graphically depicted in Figure 4.
Reducing the level of antioxidants leads to an increase in oxidative stress, thus indirectly influencing the cerebrovascular tone. Indeed, a more oxidative cellular environment can contribute to an increase in vascular tone by reducing the activity of NO [106,107]. For this reason, antioxidant mechanisms are widely expressed in vascular cells and brain tissue.
4.1. Superoxide dismutase
Superoxide dismutase (SOD) is an enzymatic defense system catalyzing O2- into H2O2. It exists in 3 different forms: copper-zinc SOD (SOD1 or Cu-Zn SODI), manganese SOD (SOD2 or MnSOD), and extracellular SOD (SOD3 or ecSOD). They are one of the most important protective mechanisms against oxidative stress in the body, and decreased SOD levels may be associated with a worse outcome in acute ischemic stroke (AIS) patients, faster vascular damage, and blood-brain barrier breakdown [108]. It has been proposed as a marker of cardiovascular alterations in hypertensive and diabetic patients, as fluctuations in serum levels are linked to alterations in vascular structure and function [109].
SOD1 is predominantly found in the cytoplasm and nucleus of cells, where it helps neutralize superoxide radicals generated during normal metabolic processes. The copper and zinc ions are essential for its enzymatic activity. SOD2 is usually located in the mitochondria, thus representing a critical defense mechanism against oxidative damage to mitochondrial components. Finally, SOD3 is primarily found in the extracellular matrix and fluids, acting as a defense outside the cells, protecting from oxidative stress caused by superoxide radicals released from inflammatory cells. Moreover, it is active in the arterial wall, strategically between endothelium and smooth muscle, thus controlling reactivity to oxygen levels and vascular tone [110], by regulating nitric oxide. Indeed, from animal studies, SOD regulates in vivo both the basal tone and the vascular response to different pressure of oxygen [110]. In TBI patients, SOD levels may be reduced already during the first day and may persist low for 7 days [111]. Indeed, exogenous SOD has been proposed as a therapeutic adjunct in several conditions [112], and may reduce vasospasm after SAH [113]. The reduction of cerebral vasospasm in experimental SAH animal models obtained by administering SOD [114] underscores the significant involvement of superoxide anions and redox equilibrium in vascular regulation, particularly during pathological conditions.
4.2. Glutathione Redox System
Glutathione peroxidase (GPx) is an enzyme that reduces hydrogen peroxide to water via a tripeptide, called glutathione (GSH). GSH is the most common thiol, i.e., a molecule that contains a sulfhydryl group (SH), and which is present in the cytosol and its organelles. Composed of glutamine, cysteine, and glycine, glutathione works as an electron donor, passing from its reduced state (GSH), the active form, to an oxidate state (GGS). The enzyme glutathione reductase converts GSSG back into GSH, using NADPH as an electron donor. GPx has a central role in reducing hydrogen peroxide to water, keeping it away from the production of hydroxyl radicals. Moreover, GSH can directly and independently neutralize free radicals, acting as a non-enzymatic antioxidant. Indeed, it can donate electrons to free radicals, an activity called “scavenging”, oxidizing to form glutathione disulfide (GSSG).
This mechanism is one of the most important antioxidants in the body. Indeed, GSH is the most abundant intracellular antioxidant, as its concentration within the cytoplasm where it is produced is in mmol, where the ratio GSH:GSSG is normally maintained around 100:1 (but it can fall lower than 10:1 during oxidative stress). It can move into the extracellular fluids, but plasma levels are 1.000 less concentrated than intracellular levels. An exception is in the lining fluid in the lungs, where the concentration is 140 times more than the plasma.
Other molecules can also intervene in this system. For example, as already described, NADPH serves to maintain GSH reduced. The pentose phosphate pathway generates NADPH, and glucose-6-phosphate dehydrogenase (G6PD) is one of the enzymes in the pathway. Deficiency of this enzyme, which affects millions of people worldwide, can result in GSH deficiency, predisposing to oxidative injury. Thiamine is a cofactor in the way of the pentose phosphate. Indeed, evidence has been found that thiamine administration may alleviate oxidative stress, even in cases where it is not attributable to thiamine deficiency [115,116]. Finally, selenium is necessary for the glutathione peroxidase activity. Indeed, this enzyme contains selenium in the active site [117]. In sepsis and septic shock, selenium is abnormally low and, when infused, has been shown to reduce mortality [118]. This has also been reported by a recent meta-analysis, even if with a low quality of evidence [119].
Glutathione deficiency is a common finding in neurological disorders [120], and it may be related to several cardiovascular diseases [121]. Even in the brain vasculature, GSH may have protective effects. Despite apparently being the most convenient and safe method of GSH administration, oral GSH is not frequently utilized in clinical trials due to its limited effectiveness [122].
An alternative approach to increase GSH production involves employing N-acetylcysteine (NAC). NAC is smaller than GSH, can move into the cells, and acts as a cysteine precursor, a crucial factor in limiting GSH synthesis [118]. NAC administration presents various neuroprotective effects, such as a reduction in the size of cerebral stroke [123] and better outcomes in neurological deficits and disability [124]. Moreover, NAC can potentially reduce ischemia-reperfusion injury [125,126], and may enhance the vasodilator effects of other compounds, such as acetylcholine [127]. As shown with other antioxidants, NAC treatment may effectively reduce cerebral vasospasm in SAH animal models [128], potentially carrying significant clinical implications that will require clinical trials to determine efficacy, despite promising pre-clinical data.
4.3. Vitamin C
Vitamin C, also known as ascorbic acid (AA), is a crucial water-soluble antioxidant that plays multiple roles in the body, including acting as a cofactor for various enzymes and inhibiting the generation of ROS. Additionally, it directly scavenges ROS and RNS while repairing other oxidized scavengers. One fascinating aspect of ascorbic acid is its neuroprotective activity, and AA has been proposed as a therapeutical adjunct in several neurological conditions [129].
Indeed, the brain exhibits one of the highest ascorbic acid concentrations in the body, thanks to an active uptake from the bloodstream. The brain concentration of AA varies from 200–400 μM in the extracellular space to 10 mM intracellular, i.e., almost 100 times the concentration in the plasma [130,131].
The effect of AA on vessels started to be investigated more than forty years ago [132]. It has demonstrated that it has the potential to enhance impaired endothelium-dependent vasodilation in peripheral arteries in various conditions characterized by endothelial dysfunction [133]. However, the impact of vitamin C on peripheral and cerebral circulations may differ [134].
Although blood hyperoxia does not appear to be convincingly associated with cerebral vasospasm following a subarachnoid hemorrhage [135], animal studies have shown that intracisternal injected Oxy-Hb (oxyhemoglobin) may result in a concentration-dependent contraction of cerebral artery strips. However, the presence of ascorbic acid significantly modifies the biological activity of Oxy-Hb, suppressing its vasoconstrictor activity and minimizing its ability to reduce vasodilator actions [136].
The cerebrovascular effects of AA were recently evaluated in an elegant study by Mattos et al. [137], investigating the effects of isocapnic hyperoxia (IH) on cerebral blood flow (CBF) and metabolism. Ten male participants underwent a 10-minute IH trial with intravenous saline and AA infusion (3g). AA infusion inhibited ROS production and preserved NO bioavailability, as indicated by reduced ROS biomarkers and unchanged nitrite levels. The infusion also prevented regional and total CBF decline and restored the cerebral metabolic rate of oxygen (CMRO2) during IH. This study further confirms that ROS play a role in CBF regulation and metabolism, and antioxidants such as AA can have indirect vasoactive effects.
Considering all this, the use of ascorbic acid in protecting the cerebral vascular tone during high oxidative stress conditions, like those seen in critically ill patients or acute brain injuries, requires further study as there are few adverse reactions [129].
4.4. Vitamin E
Vitamin E is not a single molecule, but rather a group of fat-soluble substances with similar vitamin activity, which includes 4 tocopherols (alpha, beta, gamma, delta) and 4 tocotrienols (alpha, beta, gamma, delta). The biologically most active isoform among them is alpha-tocopherol [139]. These compounds protect the integrity of cellular membranes by shielding them from free radicals that can generate and by directly neutralizing superoxide and hydroxyl radicals. Vitamin E may have potential neuroprotective effects, particularly in neurodegenerative diseases, stemming from its activity in reducing neuroinflammation [140].
Besides its role as an antioxidant, vitamin E plays a significant role in various cellular processes, such as in the vascular vessels. For example, in patients with coronary spastic angina, administering alpha-tocopherol acetate at 300 mg/day restored flow-dependent vasodilation [141]. This improvement was accompanied by a reduction in levels of plasma lipid peroxidation substances and a decrease in anginal attacks.
Endothelial cells, when replete with vitamin E, are better equipped to prevent blood-cell components from adhering to their interior surface in blood vessels. Moreover, vitamin E enhances the expression of two enzymes that suppress arachidonic acid metabolism, leading to increased release of prostacyclin from the endothelium, promoting blood vessel dilation and inhibiting platelet aggregation [6]. Its administration can improve arteriolar compliance [142] and endothelium-dependent relaxation [143]. Furthermore, a meta-analysis indicates that vitamin E might provide protection in preventing ischemic stroke [144], but another recent one failed to show a benefit [145], concluding that further well-designed randomized controlled trials are required to reach a conclusive result.
4.5. Other antioxidants
Other antioxidants can be promising adjunct therapies in critically ill patients or those with brain injuries. For instance, catalase, which degrades hydrogen peroxide, when administered intracisternal in meningitis models demonstrated a reduction in the elevation of rCBF (regional cerebral blood flow), ICP (intracranial pressure), and brain water content, primarily caused by superoxide or its derivatives [146]. Indeed, hydrogen peroxide induces relaxation in cerebral arteries, regardless of the presence of intact endothelial cells. However, these relaxations were suppressed by catalase, while SOD (superoxide dismutase) had no impact, suggesting that the relaxations result from the direct effect of hydrogen peroxide [147]. Likewise, other than improving endothelial function [148], flavonoids may elicit favorable impacts on the vascular system, resulting in ameliorating cerebrovascular blood flow [149], potentially preventing vasospasm in SAH [150], and with therapeutic effects on ischemic stroke-induced models [151].
Vitamin D has demonstrated the potential to improve neurological outcomes in brain-injured patients with significant vitamin D deficiency [152]. Interestingly, its levels have shown a correlation with intracranial aneurysm rupture in patients with SAH [153]. Furthermore, it has been suggested that the vasoprotective effects induced by vitamin D may be attributed to a reduction in oxidative stress [154]. Nevertheless, its ability to effectively prevent oxidative stress remains still inconclusive, and its role as an antioxidant cannot currently be definitively established [155].
Finally, circulating uric acid may act as a significant water-soluble antioxidant, particularly against peroxynitrites (ONOO-) in hydrophilic settings. However, within the cells, uric acid may turn into a potent pro-oxidant, triggering oxidative stress cells and mitochondria and promoting the production of inflammatory cytokines [33], in a paradoxical dualism [156]. This exemplifies the difficulties in evaluating these data and underscores the need for high quality clinical studies.
5. Future perspectives
The regulation of vascular tone in cerebral vessels is a complex process that involves multiple factors and mechanisms [157], which include pressure-induced regulation, shear stress, local metabolism, and the control of the vascular diameter by neuronal activity. As described, the presence of ROS, RNS, and antioxidants can result in vasodilatory and vasoconstrictive effects in cerebral blood vessels, with a complex interplay activity [158]. In situations of intense hyperoxia, where there's a significant rise in O2-, the existing NO can be neutralized by O2-. The rate of this reaction matches the combined rates of other known superoxide degradation reactions [159]. The chemistry of NO suggests that its half-life is likely controlled by ROS, which increases in proportion to the partial pressure of oxygen in the brain, leading to an increase in arterial tone. Interestingly, during hyperbaric oxygenation in animal models, the vasodilating effect of superoxide dismutase when animals were pre-treated with a NO-synthase inhibitor, was not seen [160], suggesting that one cause of vasoconstriction in the brain during hyperoxia might be the neutralization of NO by O2-, impacting its natural ability to relax blood vessels.
Critically ill patients and those with acute brain injuries are particularly vulnerable to oxidative stress due to elevated oxygen levels [161] and increased ROS and RNS production [162]. In these patients, the balance of redox reactions becomes essential, making antioxidants potential therapeutic agents to counteract oxidative damage. Antioxidants have been considered as a potential adjunctive treatment for various critical illnesses and have notably been suggested as a potential therapeutic option for patients with sepsis [163]. Nevertheless, they have been studied and shown to be ineffective in a clinical study of sepsis [164]. In the context of patient-tailored therapy, it is plausible that also antioxidant therapy should be targeted toward those who truly require it, and it is conceivable that antioxidant levels should be evaluated in the intensive care unit, as most ICU patients experienced swift and severe deficiencies in antioxidants [165]. Future research should focus on how to assess antioxidant levels and identifying its most valuable indicators within this patient population.
Antioxidants can neutralize ROS and RNS, prevent their generation, and repair the damage caused by free radicals. For this reason, they have been proposed and tested to reduce the effects of reactive species on cerebral blood flow [137]. Indeed, even the mildest reduction in the regional cerebral blood may lead to cognitive dysfunction [166]. Therefore, utilizing antioxidants may potentially serve as a relatively safe supplementary therapy for specific critically ill patients or those with brain injuries. This may mitigate cognitive impairments in the former group and reduce secondary brain damage in the latter pending clinical trials. For example, ascorbic acid has demonstrated its ability to preserve NO bioavailability, prevent CBF decline, and protect the cerebral metabolic rate of oxygen during conditions of high oxidative stress [137]. N-acetylcysteine (NAC) administration has shown neuroprotective effects in reducing the size of cerebral stroke and improving neurological deficits [167].
While antioxidants hold promise for potential benefits, there is currently limited human evidence available. To our knowledge, as of now, there are no FDA-approved antioxidant therapies for TBI. Thus, further research is required to elucidate their precise mechanisms of action, optimal dosages, combinations of antioxidants, and potential interactions with other treatment modalities.
6. Conclusions
Understanding the complex interplay between ROS, RNS, antioxidants, and cerebral vascular tone is crucial for developing effective therapeutic strategies for neurological disorders. Antioxidants show promise as potential adjunct therapies to mitigate the harmful effects of oxidative stress and preserve cerebral blood flow in critically ill patients and acute brain injury cases. As research in this area continues to evolve, new opportunities may be unlocked to improve the prognosis and quality of life for patients with neurological conditions through targeted antioxidant interventions.
Author Contributions
Conceptualization, M.S.; methodology, M.S., C.B., F.S.T.; writing—original draft preparation, M.S., E.D.S., M.Z., S.M.S., I.W., C.B., F.S.T.; writing—review and editing, M.S., E.D.S., M.Z., S.M.S., I.W., C.B., F.S.T.; visualization, M.S.; supervision, I.W., C.B., F.S.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rink, C.; Khanna, S. Significance of Brain Tissue Oxygenation and the Arachidonic Acid Cascade in Stroke. Antioxid Redox Signal 2011, 14, 1889. [Google Scholar] [CrossRef] [PubMed]
- Clarke, D.D.; Sokoloff, L. Regulation of Cerebral Metabolic Rate. 1999.
- Turrens, J.F. Mitochondrial Formation of Reactive Oxygen Species. J Physiol 2003, 552, 335. [Google Scholar] [CrossRef] [PubMed]
- Salim, S. Oxidative Stress and the Central Nervous System. J Pharmacol Exp Ther 2017, 360, 201. [Google Scholar] [CrossRef] [PubMed]
- Cipolla, M.J. Regulation of Cerebrovascular Tone. 2009.
- Griendling, K.K.; Sorescu, D.; Ushio-Fukai, M. NAD(P)H Oxidase: Role in Cardiovascular Biology and Disease. Circ Res 2000, 86, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Staiculescu, M.C.; Foote, C.; Meininger, G.A.; Martinez-Lemus, L.A. The Role of Reactive Oxygen Species in Microvascular Remodeling. Int J Mol Sci 2014, 15, 23792. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Kim, S.R.; Park, S.J.; Park, H.S.; Min, K.H.; Lee, M.H.; Jin, S.M.; Jin, G.Y.; Yoo, W.H.; Lee, Y.C. Hydrogen Peroxide Induces Vascular Permeability via Regulation of Vascular Endothelial Growth Factor. Am J Respir Cell Mol Biol 2006, 35, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Horke, S.; Förstermann, U. Oxidative Stress in Vascular Disease and Its Pharmacological Prevention. Trends Pharmacol Sci 2013, 34, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Montezano, A.C.; Touyz, R.M. Reactive Oxygen Species, Vascular Noxs, and Hypertension: Focus on Translational and Clinical Research. Antioxid Redox Signal 2014, 20, 164–182. [Google Scholar] [CrossRef]
- Miller, A.A.; Drummond, G.R.; De Silva, T.M.; Mast, A.E.; Hickey, H.; Williams, J.P.; Broughton, B.R.S.; Sobey, C.G. NADPH Oxidase Activity Is Higher in Cerebral versus Systemic Arteries of Four Animal Species: Role of Nox2. Am J Physiol Heart Circ Physiol 2009, 296, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Drummond, G.R.; Schmidt, H.H.H.W.; Sobey, C.G. NADPH Oxidase Activity and Function Are Profoundly Greater in Cerebral versus Systemic Arteries. Circ Res 2005, 97, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Terashvili, M.; Pratt, P.F.; Gebremedhin, D.; Narayanan, J.; Harder, D.R. Reactive Oxygen Species Cerebral Autoregulation in Health and Disease. Pediatr Clin North Am 2006, 53, 1029. [Google Scholar] [CrossRef] [PubMed]
- Kitazono, T.; Faraci, F.M.; Taguchi, H.; Heistad, D.D. Role of Potassium Channels in Cerebral Blood Vessels. Stroke 1995, 26, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Gutterman, D.D.; Miura, H.; Liu, Y. Redox Modulation of Vascular Tone: Focus of Potassium Channel Mechanisms of Dilation. Arterioscler Thromb Vasc Biol 2005, 25, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Yang, J. The Role of Reactive Oxygen Species in Angiogenesis and Preventing Tissue Injury after Brain Ischemia. Microvasc Res 2019, 123, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Khatri, N.; Thakur, M.; Pareek, V.; Kumar, S.; Sharma, S.; Datusalia, A.K. Oxidative Stress: Major Threat in Traumatic Brain Injury. CNS Neurol Disord Drug Targets 2018, 17, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid Med Cell Longev 2017, 2017. [Google Scholar] [CrossRef]
- Leveque, C.; Mrakic-Sposta, S.; Lafère, P.; Vezzoli, A.; Germonpré, P.; Beer, A.; Mievis, S.; Virgili, F.; Lambrechts, K.; Theunissen, S.; et al. Oxidative Stress Response’s Kinetics after 60 Minutes at Different (30% or 100%) Normobaric Hyperoxia Exposures. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Wei, E.P.; Kontos, H.A.; Beckman, J.S. Mechanisms of Cerebral Vasodilation by Superoxide, Hydrogen Peroxide, and Peroxynitrite. Am J Physiol 1996, 271. [Google Scholar] [CrossRef]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid Med Cell Longev 2017, 2017. [Google Scholar] [CrossRef]
- Marklund, S. Spectrophotometric Study of Spontaneous Disproportionation of Superoxide Anion Radical and Sensitive Direct Assay for Superoxide Dismutase. Journal of Biological Chemistry 1976, 251, 7504–7507. [Google Scholar] [CrossRef]
- Beckman, J.S.; Beckman, T.W.; Chen, J.; Marshall, P.A.; Freeman, B.A. Apparent Hydroxyl Radical Production by Peroxynitrite: Implications for Endothelial Injury from Nitric Oxide and Superoxide. Proc Natl Acad Sci U S A 1990, 87, 1620–1624. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.A.; Drummond, G.R.; Sobey, C.G. Reactive Oxygen Species in the Cerebral Circulation: Are They All Bad? Antioxid Redox Signal 2006, 8, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.S.; Sherrington, C.S. On the Regulation of the Blood-Supply of the Brain. J Physiol 1890, 11, 85. [Google Scholar] [CrossRef] [PubMed]
- Watts, M.E.; Pocock, R.; Claudianos, C. Brain Energy and Oxygen Metabolism: Emerging Role in Normal Function and Disease. Front Mol Neurosci 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Liachenko, S.M.; Tang, P.; Chan, P.H. Faster Recovery of Cerebral Perfusion in SOD1-Overexpressed Rats after Cardiac Arrest and Resuscitation. Stroke 2009, 40, 2512–2518. [Google Scholar] [CrossRef]
- Faraci, F.M. Oxidative Stress: The Curse That Underlies Cerebral Vascular Dysfunction? Stroke 2005, 36, 186–188. [Google Scholar] [CrossRef] [PubMed]
- De Silva, T.M.; Faraci, F.M. Reactive Oxygen Species and the Regulation of Cerebral Vascular Tone. Oxidative Stress in Applied Basic Research and Clinical Practice 2017, 89–112. [Google Scholar] [CrossRef]
- Faraci, F.M. Reactive Oxygen Species: Influence on Cerebral Vascular Tone. J Appl Physiol (1985) 2006, 100, 739–743. [Google Scholar] [CrossRef] [PubMed]
- Kontos, H.A. Oxygen Radicals in Cerebral Ischemia. Stroke 2001, 32, 2712–2716. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.T.; Conway, M.A.; Knot, H.J.; Brayden, J.E. Chloride Channel Blockers Inhibit Myogenic Tone in Rat Cerebral Arteries. J Physiol 1997, 502 Pt 2, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Leveque, C.; Mrakic-Sposta, S.; Lafère, P.; Vezzoli, A.; Germonpré, P.; Beer, A.; Mievis, S.; Virgili, F.; Lambrechts, K.; Theunissen, S.; et al. Oxidative Stress Response’s Kinetics after 60 Minutes at Different (30% or 100%) Normobaric Hyperoxia Exposures. Int J Mol Sci 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Paravicini, T.M.; Chrissobolis, S.; Drummond, G.R.; Sobey, C.G. Increased NADPH-Oxidase Activity and Nox4 Expression During Chronic Hypertension Is Associated With Enhanced Cerebral Vasodilatation to NADPH In Vivo. Stroke 2004, 35, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Lassègue, B.; Clempus, R.E. Vascular NAD(P)H Oxidases: Specific Features, Expression, and Regulation. Am J Physiol Regul Integr Comp Physiol 2003, 285. [Google Scholar] [CrossRef] [PubMed]
- Konior, A.; Schramm, A.; Czesnikiewicz-Guzik, M.; Guzik, T.J. NADPH Oxidases in Vascular Pathology. Antioxid Redox Signal 2014, 20, 2794. [Google Scholar] [CrossRef] [PubMed]
- Didion, S.P.; Hathaway, C.A.; Faraci, F.M. Superoxide Levels and Function of Cerebral Blood Vessels after Inhibition of CuZn-SOD. Am J Physiol Heart Circ Physiol 2001, 281. [Google Scholar] [CrossRef] [PubMed]
- Balestra, C.; Germonpré, P. Hypoxia, a Multifaceted Phenomenon: The Example of the Normobaric Oxygen Paradox. Eur J Appl Physiol 2012, 112, 4173–4175. [Google Scholar] [CrossRef] [PubMed]
- Salvagno, M.; Coppalini, G.; Taccone, F.S.; Strapazzon, G.; Mrakic-Sposta, S.; Rocco, M.; Khalife, M.; Balestra, C. The Normobaric Oxygen Paradox-Hyperoxic Hypoxic Paradox: A Novel Expedient Strategy in Hematopoiesis Clinical Issues. Int J Mol Sci 2022, 24. [Google Scholar] [CrossRef] [PubMed]
- Nanduri, J.; Vaddi, D.R.; Khan, S.A.; Wang, N.; Makarenko, V.; Semenza, G.L.; Prabhakar, N.R. HIF-1α Activation by Intermittent Hypoxia Requires NADPH Oxidase Stimulation by Xanthine Oxidase. PLoS One 2015, 10, e0119762. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.A.; Santhanam, A.V.R.; D’Uscio, L. V.; Katusic, Z.S. Regional Heterogeneity of Cerebral Microvessels and Brain Susceptibility to Oxidative Stress. PLoS One 2015, 10, e0144062. [Google Scholar] [CrossRef]
- Mattos, J.D.; Campos, M.O.; Rocha, M.P.; Mansur, D.E.; Rocha, H.N.M.; Garcia, V.P.; Rocha, N.G.; Alvares, T.S.; Secher, N.H.; Nóbrega, A.C.L.; et al. Differential Vasomotor Responses to Isocapnic Hyperoxia: Cerebral versus Peripheral Circulation. Am J Physiol Regul Integr Comp Physiol 2020, 318, R182–R187. [Google Scholar] [CrossRef]
- Nathan, C. Specificity of a Third Kind: Reactive Oxygen and Nitrogen Intermediates in Cell Signaling. Journal of Clinical Investigation 2003, 111, 769. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Kampoli, A.-M.; Tentolouris Nikolaos Papageorgiou, C.; Stefanadis, C. The Role of Nitric Oxide on Endothelial Function. Curr Vasc Pharmacol 2012, 10, 4–18. [Google Scholar] [CrossRef] [PubMed]
- Tejero, J.; Shiva, S.; Gladwin, M.T. Sources of Vascular Nitric Oxide and Reactive Oxygen Species and Their Regulation. Physiol Rev 2019, 99, 311–379. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Raat, N.J.H.; Shiva, S.; Dezfulian, C.; Hogg, N.; Kim-Shapiro, D.B.; Patel, R.P. Nitrite as a Vascular Endocrine Nitric Oxide Reservoir That Contributes to Hypoxic Signaling, Cytoprotection, and Vasodilation. Am J Physiol Heart Circ Physiol 2006, 291. [Google Scholar] [CrossRef] [PubMed]
- Premont, R.T.; Reynolds, J.D.; Zhang, R.; Stamler, J.S. Role of Nitric Oxide Carried by Hemoglobin in Cardiovascular Physiology: Developments on a Three-Gas Respiratory Cycle. Circ Res 2020, 126, 129–158. [Google Scholar] [CrossRef] [PubMed]
- Piacenza, L.; Zeida, A.; Trujillo, M.; Radi, R. The Superoxide Radical Switch in the Biology of Nitric Oxide and Peroxynitrite. Physiol Rev 2022, 102, 1881–1906. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, S.; Koga, Y.; Fujita, M.; Suehiro, E.; Kaneda, K.; Oda, Y.; Ishihara, H.; Suzuki, M.; Tsuruta, R. Hyperoxemia during the Hyperacute Phase of Aneurysmal Subarachnoid Hemorrhage Is Associated with Delayed Cerebral Ischemia and Poor Outcome: A Retrospective Observational Study. J Neurosurg 2019, 134, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Irvine, J.C.; Ritchie, R.H.; Favaloro, J.L.; Andrews, K.L.; Widdop, R.E.; Kemp-Harper, B.K. Nitroxyl (HNO): The Cinderella of the Nitric Oxide Story. Trends Pharmacol Sci 2008, 29, 601–608. [Google Scholar] [CrossRef]
- Ahluwalia, A.; Gladwin, M.; Coleman, G.D.; Hord, N.; Howard, G.; Kim-Shapiro, D.B.; Lajous, M.; Larsen, F.J.; Lefer, D.J.; McClure, L.A.; et al. Dietary Nitrate and the Epidemiology of Cardiovascular Disease: Report From a National Heart, Lung, and Blood Institute Workshop. J Am Heart Assoc 2016, 5. [Google Scholar] [CrossRef] [PubMed]
- DeMartino, A.W.; Kim-Shapiro, D.B.; Patel, R.P.; Gladwin, M.T. Nitrite and Nitrate Chemical Biology and Signalling. Br J Pharmacol 2019, 176, 228–245. [Google Scholar] [CrossRef] [PubMed]
- Kapil, V.; Haydar, S.M.A.; Pearl, V.; Lundberg, J.O.; Weitzberg, E.; Ahluwalia, A. Physiological Role for Nitrate-Reducing Oral Bacteria in Blood Pressure Control. Free Radic Biol Med 2013, 55, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Cosby, K.; Partovi, K.S.; Crawford, J.H.; Patel, R.P.; Reiter, C.D.; Martyr, S.; Yang, B.K.; Waclawiw, M.A.; Zalos, G.; Xu, X.; et al. Nitrite Reduction to Nitric Oxide by Deoxyhemoglobin Vasodilates the Human Circulation. Nat Med 2003, 9, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.H.; Isbell, T.S.; Huang, Z.; Shiva, S.; Chacko, B.K.; Schechter, A.N.; Darley-Usmar, V.M.; Kerby, J.D.; Lang, J.D.; Kraus, D.; et al. Hypoxia, Red Blood Cells, and Nitrite Regulate NO-Dependent Hypoxic Vasodilation. Blood 2006, 107, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Dalsgaard, T.; Simonsen, U.; Fago, A. Nitrite-Dependent Vasodilation Is Facilitated by Hypoxia and Is Independent of Known NO-Generating Nitrite Reductase Activities. Am J Physiol Heart Circ Physiol 2007, 292. [Google Scholar] [CrossRef] [PubMed]
- Maher, A.R.; Milsom, A.B.; Gunaruwan, P.; Abozguia, K.; Ahmed, I.; Weaver, R.A.; Thomas, P.; Ashrafian, H.; Born, G.V.R.; James, P.E.; et al. Hypoxic Modulation of Exogenous Nitrite-Induced Vasodilation in Humans. Circulation 2008, 117, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Pinder, A.G.; Pittaway, E.; Morris, K.; James, P.E. Nitrite Directly Vasodilates Hypoxic Vasculature via Nitric Oxide-Dependent and -Independent Pathways. Br J Pharmacol 2009, 157, 1523. [Google Scholar] [CrossRef] [PubMed]
- Hataishi, R.; Rodrigues, A.C.; Neilan, T.G.; Morgan, J.G.; Buys, E.; Shiva, S.; Tambouret, R.; Jassal, D.S.; Raher, M.J.; Furutani, E.; et al. Inhaled Nitric Oxide Decreases Infarction Size and Improves Left Ventricular Function in a Murine Model of Myocardial Ischemia-Reperfusion Injury. Am J Physiol Heart Circ Physiol 2006, 291. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Hu, L.; Feng, X.; Wang, S. Nitrate and Nitrite in Health and Disease. Aging Dis 2018, 9, 938. [Google Scholar] [CrossRef] [PubMed]
- Stamler, J.S.; Singel, D.J.; Loscalzo, J. Biochemistry of Nitric Oxide and Its Redox-Activated Forms. Science 1992, 258, 1898–1902. [Google Scholar] [CrossRef] [PubMed]
- Palmer, R.M.J.; Ferrige, A.G.; Moncada, S. Nitric Oxide Release Accounts for the Biological Activity of Endothelium-Derived Relaxing Factor. Nature 1987, 327, 524–526. [Google Scholar] [CrossRef]
- Cohen, R.A.; Adachi, T. Nitric-Oxide-Induced Vasodilatation: Regulation by Physiologic s-Glutathiolation and Pathologic Oxidation of the Sarcoplasmic Endoplasmic Reticulum Calcium ATPase. Trends Cardiovasc Med 2006, 16, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.W.; Falck, J.R.; Okamoto, H.; Harder, D.R.; Roman, R.J. Role of CGMP versus 20-HETE in the Vasodilator Response to Nitric Oxide in Rat Cerebral Arteries. Am J Physiol Heart Circ Physiol 2000, 279. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; MacVicar, B.A.; Newman, E.A. Glial and Neuronal Control of Brain Blood Flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef]
- Iadecola, C.; Pelligrino, D.A.; Moskowitz, M.A.; Lassen, N.A. Nitric Oxide Synthase Inhibition and Cerebrovascular Regulation. J Cereb Blood Flow Metab 1994, 14, 175–192. [Google Scholar] [CrossRef]
- Matsuura, T.; Kanno, I. Effect of Nitric Oxide Synthase Inhibitor on the Local Cerebral Blood Flow Evoked by Rat Somatosensory Stimulation under Hyperoxia. Comparative Biochemistry and Physiology - A Molecular and Integrative Physiology 2002, 131, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Takuwa, H.; Matsuura, T.; Bakalova, R.; Obata, T.; Kanno, I. Contribution of Nitric Oxide to Cerebral Blood Flow Regulation under Hypoxia in Rats. J Physiol Sci 2010, 60, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, I.T.; Oury, T.D.; Crapo, J.D.; Piantadosi, C.A. Regulation of the Brain’s Vascular Responses to Oxygen. Circ Res 2002, 91, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Iadecola, C. Does Nitric Oxide Mediate the Increases in Cerebral Blood Flow Elicited by Hypercapnia? Proc Natl Acad Sci U S A 1992, 89, 3913. [Google Scholar] [CrossRef] [PubMed]
- Hoiland, R.L.; Macleod, D.B.; Stacey, B.S.; Caldwell, H.G.; Howe, C.A.; Carr, J.M.J.R.; Tymko, M.M.; Nowak-flu, D.; Coombs, G.B.; Patrician, A.; et al. Hemoglobin and Cerebral Hypoxic Vasodilation in Humans: Evidence for Nitric Oxide-Dependent and S-Nitrosothiol Mediated Signal Transduction. J Cereb Blood Flow Metab 2023, 0, 271678X231169579–271678X231169579. [Google Scholar] [CrossRef] [PubMed]
- Garry, P.S.; Ezra, M.; Rowland, M.J.; Westbrook, J.; Pattinson, K.T.S. The Role of the Nitric Oxide Pathway in Brain Injury and Its Treatment--from Bench to Bedside. Exp Neurol 2015, 263, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Patel, J.K.; Schoenfeld, E.; Hou, W.; Singer, A.; Rakowski, E.; Ahmad, S.; Patel, R.; Parikh, P.B.; Smaldone, G. Inhaled Nitric Oxide in Adults with In-Hospital Cardiac Arrest: A Feasibility Study. Nitric Oxide 2021, 115, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Lenz, I.J.; Plesnila, N.; Terpolilli, N.A. Role of Endothelial Nitric Oxide Synthase for Early Brain Injury after Subarachnoid Hemorrhage in Mice. J Cereb Blood Flow Metab 2021, 41, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Qu, W.; Cheng, Y.; Peng, W.; Wu, Y.; Rui, T.; Luo, C.; Zhang, J. Targeting INOS Alleviates Early Brain Injury After Experimental Subarachnoid Hemorrhage via Promoting Ferroptosis of M1 Microglia and Reducing Neuroinflammation. Mol Neurobiol 2022, 59, 3124–3139. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Wu, W.; Liu, M.; Zhang, X.; Zhang, Q.R.; Ni, L.; Hang, C.H. Increased Cerebrospinal Fluid Concentrations of Asymmetric Dimethylarginine Correlate with Adverse Clinical Outcome in Subarachnoid Hemorrhage Patients. J Clin Neurosci 2014, 21, 1404–1408. [Google Scholar] [CrossRef] [PubMed]
- Ng, W.H.; Moochhala, S.; Yeo, T.T.; Ong, P.L.; Ng, P.Y. Nitric Oxide and Subarachnoid Hemorrhage: Elevated Level in Cerebrospinal Fluid and Their Implications. Neurosurgery 2001, 49, 622–627. [Google Scholar] [CrossRef] [PubMed]
- Fung, C.; Z’Graggen, W.J.; Jakob, S.M.; Gralla, J.; Haenggi, M.; Rothen, H.U.; Mordasini, P.; Lensch, M.; Söll, N.; Terpolilli, N.; et al. Inhaled Nitric Oxide Treatment for Aneurysmal SAH Patients With Delayed Cerebral Ischemia. Front Neurol 2022, 13, 817072. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, A. V.; Bahrami, S.; Redl, H.; Szabo, C. Alterations in Nitric Oxide Homeostasis during Traumatic Brain Injury. Biochim Biophys Acta Mol Basis Dis 2017, 1863, 2627–2632. [Google Scholar] [CrossRef] [PubMed]
- Angelis, D.; Savani, R.; Chalak, L. Nitric Oxide and the Brain. Part 2: Effects Following Neonatal Brain Injury-Friend or Foe? Pediatr Res 2021, 89, 746–752. [Google Scholar] [CrossRef] [PubMed]
- Lavi, S.; Gaitini, D.; Milloul, V.; Jacob, G. Impaired Cerebral CO2 Vasoreactivity: Association with Endothelial Dysfunction. Am J Physiol Heart Circ Physiol 2006, 291. [Google Scholar] [CrossRef]
- Fathi, A.R.; Yang, C.; Bakhtian, K.D.; Qi, M.; Lonser, R.R.; Pluta, R.M. Carbon Dioxide Influence on Nitric Oxide Production in Endothelial Cells and Astrocytes: Cellular Mechanisms. Brain Res 2011, 1386, 50. [Google Scholar] [CrossRef] [PubMed]
- Lauer, T.; Preik, M.; Rassaf, T.; Strauer, B.E.; Deussen, A.; Feelisch, M.; Kelm, M. Plasma Nitrite Rather than Nitrate Reflects Regional Endothelial Nitric Oxide Synthase Activity but Lacks Intrinsic Vasodilator Action. Proc Natl Acad Sci U S A 2001, 98, 12814–12819. [Google Scholar] [CrossRef]
- Ignarro, L.J.; Gruetter, C.S. Requirement of Thiols for Activation of Coronary Arterial Guanylate Cyclase by Glyceryl Trinitrate and Sodium Nitrite: Possible Involvement of S-Nitrosothiols. Biochim Biophys Acta 1980, 631, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Ignarro, L.J.; Byrns, R.E.; Buga, G.M.; Wood, K.S. Endothelium-Derived Relaxing Factor from Pulmonary Artery and Vein Possesses Pharmacologic and Chemical Properties Identical to Those of Nitric Oxide Radical. Circ Res 1987, 61, 866–879. [Google Scholar] [CrossRef] [PubMed]
- Gladwin, M.T.; Shelhamer, J.H.; Schechter, A.N.; Pease-Fye, M.E.; Waclawiw, M.A.; Panza, J.A.; Ognibene, F.P.; Cannon, R.O. Role of Circulating Nitrite and S-Nitrosohemoglobin in the Regulation of Regional Blood Flow in Humans. Proc Natl Acad Sci U S A 2000, 97, 11482–11487. [Google Scholar] [CrossRef] [PubMed]
- Furchgott, R.; Bhadrakom, S. Reactions of Strips of Rabbit Aorta to Epinephrine, Isopropylarterenol, Sodium Nitrite and Other Drugs - PubMed. J Pharmacol Exp Ther. 1953, 2, 129–143. [Google Scholar]
- Brücken, A.; Derwall, M.; Bleilevens, C.; Stoppe, C.; Götzenich, A.; Gaisa, N.T.; Weis, J.; Nolte, K.W.; Rossaint, R.; Ichinose, F.; et al. Brief Inhalation of Nitric Oxide Increases Resuscitation Success and Improves 7-Day-Survival after Cardiac Arrest in Rats: A Randomized Controlled Animal Study. Crit Care 2015, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, W.; Altura, B.T.; Altura, B.M. Peroxynitrite-Induced Relaxation in Isolated Canine Cerebral Arteries and Mechanisms of Action. Toxicol Appl Pharmacol 2004, 196, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Pritchard, K.A.; Kaminski, P.M.; Fayngersh, R.P.; Hintze, T.H.; Wolin, M.S. Involvement of Nitric Oxide and Nitrosothiols in Relaxation of Pulmonary Arteries to Peroxynitrite. Am J Physiol 1994, 266. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Beckman, J.S.; Ku, D.D. Peroxynitrite, a Product of Superoxide and Nitric Oxide, Produces Coronary Vasorelaxation in Dogs - PubMed. J Pharmacol Exp Ther. 1994, 268, 1114–1121. [Google Scholar] [PubMed]
- Wei, E.P.; Kontos, H.A.; Beckman, J.S.; Faraci, F.M. Antioxidants Inhibit ATP-Sensitive Potassium Channels in Cerebral Arterioles. Stroke 1998, 29, 817–823. [Google Scholar] [CrossRef] [PubMed]
- Nossaman, B.D.; Kadowitz, P.J. Potential Benefits of Peroxynitrite. Open Pharmacol J 2008, 2, 31. [Google Scholar] [CrossRef] [PubMed]
- Daneva, Z.; Marziano, C.; Ottolini, M.; Chen, Y.L.; Baker, T.M.; Kuppusamy, M.; Zhang, A.; Ta, H.Q.; Reagan, C.E.; Mihalek, A.D.; et al. Caveolar Peroxynitrite Formation Impairs Endothelial TRPV4 Channels and Elevates Pulmonary Arterial Pressure in Pulmonary Hypertension. Proc Natl Acad Sci U S A 2021, 118, e2023130118–e2023130118. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, M.; Hong, K.; Cope, E.L.; Daneva, Z.; Delalio, L.J.; Sokolowski, J.D.; Marziano, C.; Nguyen, N.Y.; Altschmied, J.; Haendeler, J.; et al. Local Peroxynitrite Impairs Endothelial Transient Receptor Potential Vanilloid 4 Channels and Elevates Blood Pressure in Obesity. Circulation 2020, 141, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- Brzezinska, A.K.; Gebremedhin, D.; Chilian, W.M.; Kalyanaraman, B.; Elliott, S.J. Peroxynitrite Reversibly Inhibits Ca(2+)-Activated K(+) Channels in Rat Cerebral Artery Smooth Muscle Cells. Am J Physiol Heart Circ Physiol 2000, 278. [Google Scholar] [CrossRef] [PubMed]
- Elliott, S.J.; Lacey, D.J.; Chilian, W.M.; Brzezinska, A.K. Peroxynitrite Is a Contractile Agonist of Cerebral Artery Smooth Muscle Cells. Am J Physiol 1998, 275. [Google Scholar] [CrossRef] [PubMed]
- Ronson, R.S.; Nakamura, M.; Vinten-Johansen, J. The Cardiovascular Effects and Implications of Peroxynitrite. Cardiovasc Res 1999, 44, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.L.; Irvine, J.C.; Tare, M.; Apostolopoulos, J.; Favaloro, J.L.; Triggle, C.R.; Kemp-Harper, B.K. A Role for Nitroxyl (HNO) as an Endothelium-Derived Relaxing and Hyperpolarizing Factor in Resistance Arteries. Br J Pharmacol 2009, 157, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.L.; Lumsden, N.G.; Farry, J.; Jefferis, A.M.; Kemp-Harper, B.K.; Chin-Dusting, J.P.F. Nitroxyl: A Vasodilator of Human Vessels That Is Not Susceptible to Tolerance. Clin Sci (Lond) 2015, 129, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Paolocci, N.; Jackson, M.I.; Lopez, B.E.; Miranda, K.; Tocchetti, C.G.; Wink, D.A.; Hobbs, A.J.; Fukuto, J.M. The Pharmacology of Nitroxyl (HNO) and Its Therapeutic Potential: Not Just the Janus Face of NO. Pharmacol Ther 2007, 113, 442. [Google Scholar] [CrossRef] [PubMed]
- Favaloro, J.L.; Kemp-Harper, B.K. The Nitroxyl Anion (HNO) Is a Potent Dilator of Rat Coronary Vasculature. Cardiovasc Res 2007, 73, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Kemp-Harper, B.K.; Horowitz, J.D.; Ritchie, R.H. Therapeutic Potential of Nitroxyl (HNO) Donors in the Management of Acute Decompensated Heart Failure. Drugs 2016, 76, 1337–1348. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Groneberg, D.; Sikka, G.; Hori, D.; Ranek, M.J.; Nakamura, T.; Takimoto, E.; Paolocci, N.; Berkowitz, D.E.; Friebe, A.; et al. Soluble Guanylate Cyclase Is Required for Systemic Vasodilation But Not Positive Inotropy Induced by Nitroxyl in the Mouse. 2014. [CrossRef]
- Eberhardt, M.; Dux, M.; Namer, B.; Miljkovic, J.; Cordasic, N.; Will, C.; Kichko, T.I.; De La Roche, J.; Fischer, M.; Suárez, S.A.; et al. H2S and NO Cooperatively Regulate Vascular Tone by Activating a Neuroendocrine HNO–TRPA1–CGRP Signalling Pathway. Nat Commun 2014. [Google Scholar] [CrossRef]
- Senoner, T.; Dichtl, W. Oxidative Stress in Cardiovascular Diseases: Still a Therapeutic Target? Nutrients 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Gryglewski, R.J.; Palmer, R.M.J.; Moncada, S. Superoxide Anion Is Involved in the Breakdown of Endothelium-Derived Vascular Relaxing Factor. Nature 1986, 320, 454–456. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.S.; Liang, J.H.; Yang, M.J.; Ren, Y.R.; Cheng, D.H.; Wu, Q.H.; He, Y.; Yin, J. Low Serum Superoxide Dismutase Is Associated With a High Risk of Cognitive Impairment After Mild Acute Ischemic Stroke. Front Aging Neurosci 2022, 14, 834114. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Marcos, M.A.; Blázquez-Medela, A.M.; Gamella-Pozuelo, L.; Recio-Rodriguez, J.I.; García-Ortiz, L.; Martínez-Salgado, C. Serum Superoxide Dismutase Is Associated with Vascular Structure and Function in Hypertensive and Diabetic Patients. Oxid Med Cell Longev 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, I.T.; Oury, T.D.; Crapo, J.D.; Piantadosi, C.A. Regulation of the Brain’s Vascular Responses to Oxygen. Circ Res 2002, 91, 1031–1037. [Google Scholar] [CrossRef] [PubMed]
- Fesharaki-Zadeh, A. Oxidative Stress in Traumatic Brain Injury. International Journal of Molecular Sciences 2022, 23, 13000. [Google Scholar] [CrossRef] [PubMed]
- Younus, H. Therapeutic Potentials of Superoxide Dismutase. Int J Health Sci (Qassim) 2018, 12, 88. [Google Scholar] [PubMed]
- McGirt, M.J.; Parra, A.; Sheng, H.; Higuchi, Y.; Oury, T.D.; Laskowitz, D.T.; Pearlstein, R.D.; Warner, D.S. Attenuation of Cerebral Vasospasm after Subarachnoid Hemorrhage in Mice Overexpressing Extracellular Superoxide Dismutase. Stroke 2002, 33, 2317–2323. [Google Scholar] [CrossRef]
- Aladag, M.A.; Turkoz, Y.; Sahna, E.; Parlakpinar, H.; Gul, M. The Attenuation of Vasospasm by Using a Sod Mimetic after Experimental Subarachnoidal Haemorrhage in Rats. Acta Neurochir (Wien) 2003, 145, 673–677. [Google Scholar] [CrossRef] [PubMed]
- Hazell, A.S.; Faim, S.; Wertheimer, G.; Silva, V.R.; Marques, C.S. The Impact of Oxidative Stress in Thiamine Deficiency: A Multifactorial Targeting Issue. Neurochem Int 2013, 62, 796–802. [Google Scholar] [CrossRef] [PubMed]
- Bozic, I.; Lavrnja, I. Thiamine and Benfotiamine: Focus on Their Therapeutic Potential. Heliyon 2023, 9, e21839. [Google Scholar] [CrossRef]
- Burk, R.F.; Hill, K.E. Glutathione Peroxidases. Comprehensive Toxicology, Second Edition 2010, 4, 229–242. [Google Scholar] [CrossRef]
- Angstwurm, M.W.A.; Engelmann, L.; Zimmermann, T.; Lehmann, C.; Spes, C.H.; Abel, P.; Strauß, R.; Meier-Hellmann, A.; Insel, R.; Radke, J.; et al. Selenium in Intensive Care (SIC): Results of a Prospective Randomized, Placebo-Controlled, Multiple-Center Study in Patients with Severe Systemic Inflammatory Response Syndrome, Sepsis, and Septic Shock. Crit Care Med 2007, 35, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Jaff, S.; Zeraattalab-Motlagh, S.; Amiri Khosroshahi, R.; Gubari, M.; Mohammadi, H.; Djafarian, K. The Effect of Selenium Therapy in Critically Ill Patients: An Umbrella Review of Systematic Reviews and Meta-Analysis of Randomized Controlled Trials. Eur J Med Res 2023, 28, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K. Glutathione in the Brain. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Matuz-Mares, D.; Riveros-Rosas, H.; Vázquez-Meza, H.; Vilchis-Landeros, M.M. Glutathione Participation in the Prevention of Cardiovascular Diseases. Antioxidants 2021, 10. [Google Scholar] [CrossRef]
- Schmitt, B.; Vicenzi, M.; Garrel, C.; Denis, F.M. Effects of N-Acetylcysteine, Oral Glutathione (GSH) and a Novel Sublingual Form of GSH on Oxidative Stress Markers: A Comparative Crossover Study. Redox Biol 2015, 6, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Uemura, T.; Watanabe, K.; Ko, K.; Higashi, K.; Kogure, N.; Kitajima, M.; Takayama, H.; Takao, K.; Sugita, Y.; Sakamoto, A.; et al. Protective Effects of Brain Infarction by N-Acetylcysteine Derivatives. Stroke 2018, 49, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Sabetghadam, M.; Mazdeh, M.; Abolfathi, P.; Mohammadi, Y.; Mehrpooya, M. Evidence for a Beneficial Effect of Oral N-Acetylcysteine on Functional Outcomes and Inflammatory Biomarkers in Patients with Acute Ischemic Stroke. Neuropsychiatr Dis Treat 2020, 16, 1265. [Google Scholar] [CrossRef] [PubMed]
- Olesen, H.Ø.; Pors, S.E.; Jensen, L.B.; Grønning, A.P.; Lemser, C.E.; Nguyen Heimbürger, M.T.H.; Mamsen, L.S.; Getreu, N.; Christensen, S.T.; Andersen, C.Y.; et al. N-Acetylcysteine Protects Ovarian Follicles from Ischemia-Reperfusion Injury in Xenotransplanted Human Ovarian Tissue. Hum Reprod 2021, 36, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Cayuela, N.C.; Koike, M.K.; Jacysyn, J. de F.; Rasslan, R.; Cerqueira, A.R.A.; Costa, S.K.P.; Diniz-Júnior, J.A.P.; Utiyama, E.M.; Montero, E.F. de S. N-Acetylcysteine Reduced Ischemia and Reperfusion Damage Associated with Steatohepatitis in Mice. Int J Mol Sci 2020, 21, 1–19. [Google Scholar] [CrossRef]
- Raghu, G.; Berk, M.; Campochiaro, P.A.; Jaeschke, H.; Marenzi, G.; Richeldi, L.; Wen, F.-Q.; Nicoletti, F.; Calverley, P.M.A. The Multifaceted Therapeutic Role of N-Acetylcysteine (NAC) in Disorders Characterized by Oxidative Stress. Curr Neuropharmacol 2021, 19, 1202. [Google Scholar] [CrossRef] [PubMed]
- Güney, O.; Erdi, F.; Esen, H.; Kiyici, A.; Kocaogullar, Y. N-Acetylcysteine Prevents Vasospasm after Subarachnoid Hemorrhage. World Neurosurg 2010, 73, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Ballaz, S.J.; Rebec, G. V. Neurobiology of Vitamin C: Expanding the Focus from Antioxidant to Endogenous Neuromodulator. Pharmacol Res 2019, 146, 104321. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin C Function in the Brain: Vital Role of the Ascorbate Transporter SVCT2. Free Radic Biol Med 2009, 46, 719–730. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate Regulation and Its Neuroprotective Role in the Brain. Trends Neurosci 2000, 23, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, M.; Dobrev, D.; Himmel, H.M.; Ravens, U.; Kirch, W. Ascorbic Acid–Induced Modulation of Venous Tone in Humans. Hypertension 2001, 37, 949–954. [Google Scholar] [CrossRef] [PubMed]
- May, J.M. How Does Ascorbic Acid Prevent Endothelial Dysfunction? Free Radic Biol Med 2000, 28, 1421–1429. [Google Scholar] [CrossRef] [PubMed]
- Barak, O.F.; Caljkusic, K.; Hoiland, R.L.; Ainslie, P.N.; Thom, S.R.; Yang, M.; Jovanov, P.; Dujic, Z. Differential Influence of Vitamin C on the Peripheral and Cerebral Circulation after Diving and Exposure to Hyperoxia. Am J Physiol Regul Integr Comp Physiol 2018, 315, R759–R767. [Google Scholar] [CrossRef] [PubMed]
- Salvagno, M.; Taccone, F.S.; Bogossian, E.G. Tissue Hyperoxia Is Not a Risk Factor for Cerebral Vasospasm Following Subarachnoid Hemorrhage. J Neurol Sci 2023, 445. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, M.; Kodama, N.; Toda, N. Suppression of the Cerebral Vasospastic Actions of Oxyhemoglobin by Ascorbic Acid. Neurosurgery 1991, 28, 33. [Google Scholar] [CrossRef] [PubMed]
- Mattos, J.D.; Campos, M.O.; Rocha, M.P.; Mansur, D.E.; Rocha, H.N.M.; Garcia, V.P.; Batista, G.; Alvares, T.S.; Oliveira, G. V.; Souza, M. V.; et al. Human Brain Blood Flow and Metabolism during Isocapnic Hyperoxia: The Role of Reactive Oxygen Species. Journal of Physiology 2019, 597, 741–755. [Google Scholar] [CrossRef]
- Abdullah, M.; Jamil, R.T.; Attia, F.N. Vitamin C (Ascorbic Acid). Encyclopedia of Toxicology: Third Edition 2023, 962–963. [CrossRef]
- Engin, K.N. Alpha-Tocopherol: Looking beyond an Antioxidant. Mol Vis 2009, 15, 855. [Google Scholar] [PubMed]
- La Torre, M.E.; Villano, I.; Monda, M.; Messina, A.; Cibelli, G.; Valenzano, A.; Pisanelli, D.; Panaro, M.A.; Tartaglia, N.; Ambrosi, A.; et al. Role of Vitamin E and the Orexin System in Neuroprotection. Brain Sci 2021, 11, 1098. [Google Scholar] [CrossRef] [PubMed]
- Motoyama, T.; Kawano, H.; Kugiyama, K.; Hirashima, O.; Ohgushi, M.; Tsunoda, R.; Moriyama, Y.; Miyao, Y.; Yoshimura, M.; Ogawa, H.; et al. Vitamin E Administration Improves Impairment of Endothelium-Dependent Vasodilation in Patients with Coronary Spastic Angina. J Am Coll Cardiol 1998, 32, 1672–1679. [Google Scholar] [CrossRef]
- Rasool, A.H.G.; Rahman, A.R.A.; Yuen, K.H.; Wong, A.R. Arterial Compliance and Vitamin E Blood Levels with a Self Emulsifying Preparation of Tocotrienol Rich Vitamin E. Arch Pharm Res 2008, 31, 1212–1217. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, T.; Ylä Herttuala, S.; Wild, E.; Luoma, J.; Drexler, H. Effect of Vitamin E on Endothelial Vasodilator Function in Patients with Hypercholesterolemia, Chronic Smoking or Both. J Am Coll Cardiol 1999, 33, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Wang, L.; Ning, S.; Liu, Z.; Lin, H.; Chen, S.; Zhu, J. Vitamin E Intake and Risk of Stroke: A Meta-Analysis. British Journal of Nutrition 2018, 120, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Loh, H.C.; Lim, R.; Lee, K.W.; Ooi, C.Y.; Chuan, D.R.; Looi, I.; Kah Hay, Y.; Abdul Karim Khan, N. Effects of Vitamin E on Stroke: A Systematic Review with Meta-Analysis and Trial Sequential Analysis. Stroke Vasc Neurol 2021, 6, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Pfister, H.W.; Ködel, U.; Dirnagl, U.; Haberl, R.L.; Ruckdeschel, G.; Einhäupl, K.M. Effect of Catalase on Regional Cerebral Blood Flow and Brain Edema during the Early Phase of Experimental Pneumococcal Meningitis. J Infect Dis 1992, 166, 1442–1445. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Katusic, Z.S.; Wei, E.P. Mechanisms of Cerebral Arterial Relaxations to Hydrogen Peroxide. Stroke 2000, 31, 2224–2230. [Google Scholar] [CrossRef] [PubMed]
- Bondonno, N.P.; Bondonno, C.P.; Blekkenhorst, L.C.; Considine, M.J.; Maghzal, G.; Stocker, R.; Woodman, R.J.; Ward, N.C.; Hodgson, J.M.; Croft, K.D. Flavonoid-Rich Apple Improves Endothelial Function in Individuals at Risk for Cardiovascular Disease: A Randomized Controlled Clinical Trial. Mol Nutr Food Res 2018, 62. [Google Scholar] [CrossRef] [PubMed]
- Vauzour, D.; Vafeiadou, K.; Rodriguez-Mateos, A.; Rendeiro, C.; Spencer, J.P.E. The Neuroprotective Potential of Flavonoids: A Multiplicity of Effects. Genes Nutr 2008, 3, 115–126. [Google Scholar] [CrossRef]
- Gül, Ş.; Aydoğmuş, E.; Bahadir, B.; Büyükuysal, Ç.; Güven, B. Neuroprotective Effects of Quercetin on Cerebral Vasospasm Following Experimental Subarachnoid Haemorrhage in Rats. Turk J Med Sci 2020, 50, 1106. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Ma, J.; Yang, F.; Li, S.; Ma, W.; Chang, X.; Yang, L. Neuroprotective Effects of Quercetin on Ischemic Stroke: A Literature Review. Front Pharmacol 2022, 13, 854249. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Jeong, S.W.; Kim, M.Y.; Park, J.B.; Kim, M.S. The Effect of Vitamin D Supplementation in Patients with Acute Traumatic Brain Injury. World Neurosurg 2019, 126, e1421–e1426. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Yuan, X.; Li, D.; Fan, F.; Guo, X.; Xu, Y.; Guan, S. Vitamin D Level Is Associated with Rupture of Intracranial Aneurysm in Patients with Subarachnoid Hemorrhage. Front Neurol 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Della Nera, G.; Sabatino, L.; Gaggini, M.; Gorini, F.; Vassalle, C. Vitamin D Determinants, Status, and Antioxidant/Anti-Inflammatory-Related Effects in Cardiovascular Risk and Disease: Not the Last Word in the Controversy. Antioxidants 2023, 12, 948. [Google Scholar] [CrossRef] [PubMed]
- Tagliaferri, S.; Porri, D.; De Giuseppe, R.; Manuelli, M.; Alessio, F.; Cena, H. The Controversial Role of Vitamin D as an Antioxidant: Results from Randomised Controlled Trials. Nutr Res Rev 2019, 32, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Sautin, Y.Y.; Johnson, R.J. URIC ACID: THE OXIDANT–ANTIOXIDANT PARADOX. Nucleosides Nucleotides Nucleic Acids 2008, 27, 608. [Google Scholar] [CrossRef] [PubMed]
- Halvorson, B.; Frisbee, J.; Halvorson, B.; Frisbee, J. Cerebral Vascular Tone Regulation: Integration and Impact of Disease. Basic and Clinical Understanding of Microcirculation 2020. [Google Scholar] [CrossRef]
- Hsieh, H.J.; Liu, C.A.; Huang, B.; Tseng, A.H.; Wang, D.L. Shear-Induced Endothelial Mechanotransduction: The Interplay between Reactive Oxygen Species (ROS) and Nitric Oxide (NO) and the Pathophysiological Implications. Journal of Biomedical Science 2014 21:1 2014, 21, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Demchenko, I.T.; Boso, A.E.; O’Neill, T.J.; Bennett, P.B.; Piantadosi, C.A. Nitric Oxide and Cerebral Blood Flow Responses to Hyperbaric Oxygen. J Appl Physiol 2000, 88, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Zhilyaev, S.Y.; Moskvin, A.N.; Platonova, T.F.; Gutsaeva, D.R.; Churilina, I. V.; Demchenko, I.T. Hyperoxic Vasoconstriction in the Brain Is Mediated by Inactivation of Nitric Oxide by Superoxide Anions. Neurosci Behav Physiol 2003, 33, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Kraft, F.; Andel, H.; Gamper, J.; Markstaller, K.; Ullrich, R.; Klein, K.U. Incidence of Hyperoxia and Related In-Hospital Mortality in Critically Ill Patients: A Retrospective Data Analysis. Acta Anaesthesiol Scand 2018, 62, 347–356. [Google Scholar] [CrossRef] [PubMed]
- Angeloni, C.; Prata, C.; Vieceli Dalla Sega, F.; Piperno, R.; Hrelia, S. Traumatic Brain Injury and NADPH Oxidase: A Deep Relationship. Oxid Med Cell Longev 2015, 2015. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Yin, L.; Song, Y.; Zhang, M.; Lu, Y.; Wang, S. The Use of Hydrocortisone, Ascorbic Acid and Thiamine in Patients with Sepsis and Septic Shock - A Systematic Review. J Pharm Pract 2023, 36. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, F.; Masse, M.-H.; Menard, J.; Sprague, S.; Pinto, R.; Heyland, D.K.; Cook, D.J.; Battista, M.-C.; Day, A.G.; Guyatt, G.H.; et al. Intravenous Vitamin C in Adults with Sepsis in the Intensive Care Unit. New England Journal of Medicine 2022, 386, 2387–2398. [Google Scholar] [CrossRef] [PubMed]
- Margaritelis, N. V.; Paschalis, V.; Theodorou, A.A.; Vassiliou, V.; Kyparos, A.; Nikolaidis, M.G. Rapid Decreases of Key Antioxidant Molecules in Critically Ill Patients: A Personalized Approach. Clinical Nutrition 2020, 39, 1146–1154. [Google Scholar] [CrossRef] [PubMed]
- Bowton, D.L.; Bertels, N.H.; Prough, D.S.; Stump, D.A. Cerebral Blood Flow Is Reduced in Patients with Sepsis Syndrome. Crit Care Med 1989, 17, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Sabetghadam, M.; Mazdeh, M.; Abolfathi, P.; Mohammadi, Y.; Mehrpooya, M. Evidence for a Beneficial Effect of Oral N-Acetylcysteine on Functional Outcomes and Inflammatory Biomarkers in Patients with Acute Ischemic Stroke. Neuropsychiatr Dis Treat 2020, 16, 1265. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The main pathways of the formation of ROS. ROS are formed through various pathways, contributing to their accumulation, which can have beneficial and detrimental effects on cellular processes. 1) Microbes and Xenobiotics: microbes can produce ROS as part of their metabolism or defense mechanism, xenobiotics can be metabolized to produce ROS; 2) NOX: it is an enzyme that produces superoxide by transferring electrons from NADPH to molecular oxygen; 3) Radiation: ionizing radiation, such as X-rays or gamma rays, can cause the ionization of water molecules, leading to the formation of hydroxyl radicals; 4) Metabolizing Enzymes: some enzymes involved can produce ROS as by-products of their reactions; 5) Fenton Reaction: a chemical reaction in which iron (Fe2+) reacts with hydrogen peroxide to produce hydroxyl radicals; 6) Peroxisomes: organelles that contain enzymes involved in a variety of metabolic processes, including the breakdown of fatty acids and the detoxification of harmful substances, during which they can produce ROS; they also have catalase, an enzyme that breaks down hydrogen peroxide into water and oxygen; if the production of ROS exceeds the capacity of catalase to break it down, ROS can accumulate; 7) Endoplasmic Reticulum (ER) Stress and Unfolded Protein Response: the ER is an organelle in the cell that is responsible for the synthesis, folding, and modification of proteins; when the ER becomes overwhelmed with misfolded proteins, it triggers a response known as ER stress, caused by various factors, including nutrient deprivation, hypoxia, and changes in calcium levels; the accumulation of misfolded proteins in the ER can cause oxidative stress; moreover, in response to ER stress, the cell activates the Unfolded Protein Response, a signaling pathway that aims to restore ER homeostasis by halting protein synthesis, increasing the production of chaperone proteins that assist in protein folding, and enhancing the degradation of misfolded proteins; thus, the demand for energy increases, leading to an increase in mitochondrial activity; 8) Oxidative Phosphorylation in the mitochondria: electrons are transferred through the electron transport chain to produce ATP; some electrons may leak and react with oxygen to form superoxide. ROS: Reactive Oxygen Species; NOX: NADPH Oxidase; ER: Endoplasmic Reticulum.
Figure 1.
The main pathways of the formation of ROS. ROS are formed through various pathways, contributing to their accumulation, which can have beneficial and detrimental effects on cellular processes. 1) Microbes and Xenobiotics: microbes can produce ROS as part of their metabolism or defense mechanism, xenobiotics can be metabolized to produce ROS; 2) NOX: it is an enzyme that produces superoxide by transferring electrons from NADPH to molecular oxygen; 3) Radiation: ionizing radiation, such as X-rays or gamma rays, can cause the ionization of water molecules, leading to the formation of hydroxyl radicals; 4) Metabolizing Enzymes: some enzymes involved can produce ROS as by-products of their reactions; 5) Fenton Reaction: a chemical reaction in which iron (Fe2+) reacts with hydrogen peroxide to produce hydroxyl radicals; 6) Peroxisomes: organelles that contain enzymes involved in a variety of metabolic processes, including the breakdown of fatty acids and the detoxification of harmful substances, during which they can produce ROS; they also have catalase, an enzyme that breaks down hydrogen peroxide into water and oxygen; if the production of ROS exceeds the capacity of catalase to break it down, ROS can accumulate; 7) Endoplasmic Reticulum (ER) Stress and Unfolded Protein Response: the ER is an organelle in the cell that is responsible for the synthesis, folding, and modification of proteins; when the ER becomes overwhelmed with misfolded proteins, it triggers a response known as ER stress, caused by various factors, including nutrient deprivation, hypoxia, and changes in calcium levels; the accumulation of misfolded proteins in the ER can cause oxidative stress; moreover, in response to ER stress, the cell activates the Unfolded Protein Response, a signaling pathway that aims to restore ER homeostasis by halting protein synthesis, increasing the production of chaperone proteins that assist in protein folding, and enhancing the degradation of misfolded proteins; thus, the demand for energy increases, leading to an increase in mitochondrial activity; 8) Oxidative Phosphorylation in the mitochondria: electrons are transferred through the electron transport chain to produce ATP; some electrons may leak and react with oxygen to form superoxide. ROS: Reactive Oxygen Species; NOX: NADPH Oxidase; ER: Endoplasmic Reticulum.
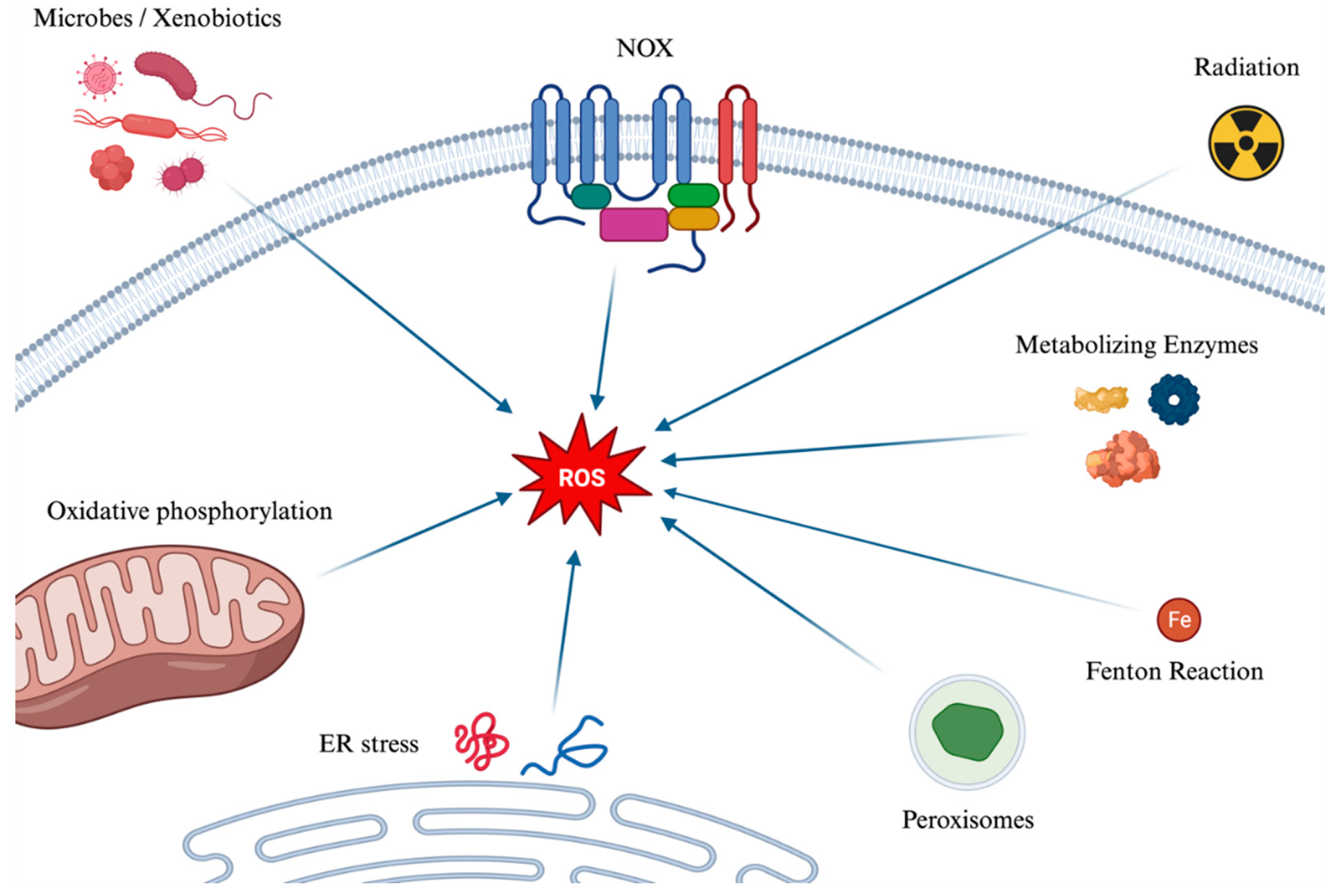
Figure 2.
Reactive Oxygen Species (ROS): their structure and formation. The process begins with molecular oxygen (O2), which has two unpaired electrons in the outer orbital. Thus, it can acquire an electron to form the superoxide anion (O2-), which is a radical (i.e., with an unpaired electron). Superoxide dismutase (SOD) converts superoxide into hydrogen peroxide (H2O2) and oxygen. Catalase can further break down H2O2 into water and oxygen. In the presence of transition metals, the Haber-Weiss reaction generates hydroxyl radicals (OH˙) and hydroxyl ions (OH-) from superoxide and H2O2. The Fenton reaction also produces hydroxyl radicals and hydroxyl ions by reducing H2O2 in the presence of iron ions. O: Oxygen atom; H: Hydrogen atom; H2O: water molecule; O2˙: superoxide anion; H2O2: Hydrogen Peroxide; OH˙: Hydroxyl Radical; OH-: Hydroxyl Ion; NO: Nitric Oxide; Fe2+: iron in reduced state; Fe3+: iron in oxidized state; SOD: superoxide dismutase.
Figure 2.
Reactive Oxygen Species (ROS): their structure and formation. The process begins with molecular oxygen (O2), which has two unpaired electrons in the outer orbital. Thus, it can acquire an electron to form the superoxide anion (O2-), which is a radical (i.e., with an unpaired electron). Superoxide dismutase (SOD) converts superoxide into hydrogen peroxide (H2O2) and oxygen. Catalase can further break down H2O2 into water and oxygen. In the presence of transition metals, the Haber-Weiss reaction generates hydroxyl radicals (OH˙) and hydroxyl ions (OH-) from superoxide and H2O2. The Fenton reaction also produces hydroxyl radicals and hydroxyl ions by reducing H2O2 in the presence of iron ions. O: Oxygen atom; H: Hydrogen atom; H2O: water molecule; O2˙: superoxide anion; H2O2: Hydrogen Peroxide; OH˙: Hydroxyl Radical; OH-: Hydroxyl Ion; NO: Nitric Oxide; Fe2+: iron in reduced state; Fe3+: iron in oxidized state; SOD: superoxide dismutase.
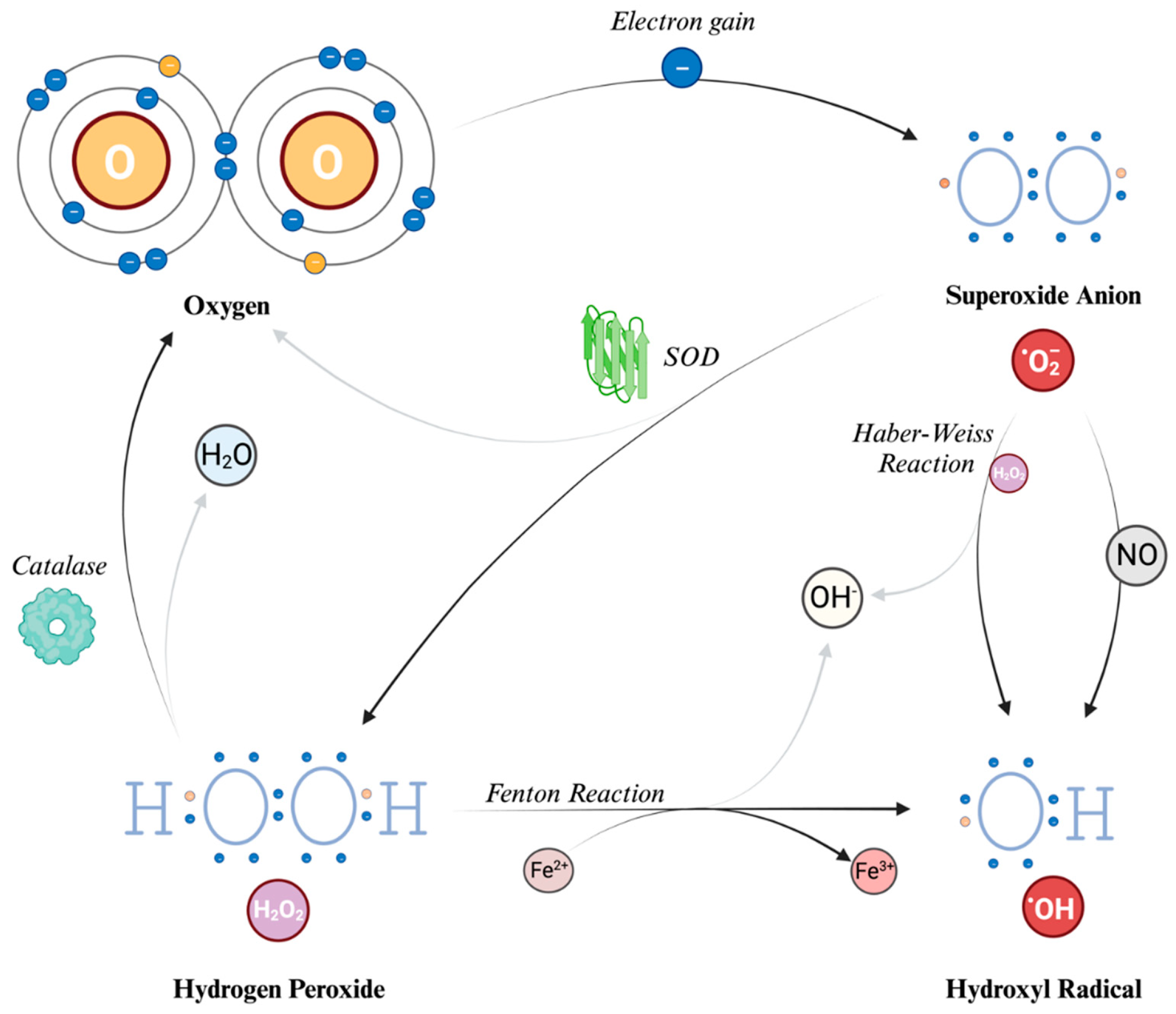
Figure 3.
Overview of the main reactive nitrogen species (RNS) and their formation pathways. The figure illustrates the generation of nitric oxide (NO), a small, gaseous molecule that acts as a signaling molecule in the body. It is produced by the enzyme nitric oxide synthase (NOS) from the amino acid L-arginine and molecular oxygen. Through S-nitrosylation, a NO group is added to the thiol group of a cysteine residue, for example, Hb. Otherwise, NO can be transformed into salts or esters of nitrous acid, containing the anion nitrite (NO2-) which can be converted back to NO under certain conditions. Nitrates are taken into the body from external sources, such as food or supplements. They can be converted into nitrites (and thus potentially into NO). This process is known as the nitrate-nitrite-NO pathway and is an alternative source of NO, especially when the enzymatic production of NO is impaired. Peroxynitrite is a highly reactive nitrogen species formed by NO with O2−. Finally, Nitroxyl (HNO) is a reactive nitrogen species isoelectronic with NO, formed by the reduction of NO or the decomposition of certain NO donors. O2: molecular Oxygen; NO: Nitric Oxide; NOS: Nitric Oxide Synthase; N: Nitrogen atom; Hb: Hemoglobin; NO2-: Nitrite; NO3-: Nitrate; ONOO-: Peroxynitrite; HNO: Nitroxyl.
Figure 3.
Overview of the main reactive nitrogen species (RNS) and their formation pathways. The figure illustrates the generation of nitric oxide (NO), a small, gaseous molecule that acts as a signaling molecule in the body. It is produced by the enzyme nitric oxide synthase (NOS) from the amino acid L-arginine and molecular oxygen. Through S-nitrosylation, a NO group is added to the thiol group of a cysteine residue, for example, Hb. Otherwise, NO can be transformed into salts or esters of nitrous acid, containing the anion nitrite (NO2-) which can be converted back to NO under certain conditions. Nitrates are taken into the body from external sources, such as food or supplements. They can be converted into nitrites (and thus potentially into NO). This process is known as the nitrate-nitrite-NO pathway and is an alternative source of NO, especially when the enzymatic production of NO is impaired. Peroxynitrite is a highly reactive nitrogen species formed by NO with O2−. Finally, Nitroxyl (HNO) is a reactive nitrogen species isoelectronic with NO, formed by the reduction of NO or the decomposition of certain NO donors. O2: molecular Oxygen; NO: Nitric Oxide; NOS: Nitric Oxide Synthase; N: Nitrogen atom; Hb: Hemoglobin; NO2-: Nitrite; NO3-: Nitrate; ONOO-: Peroxynitrite; HNO: Nitroxyl.
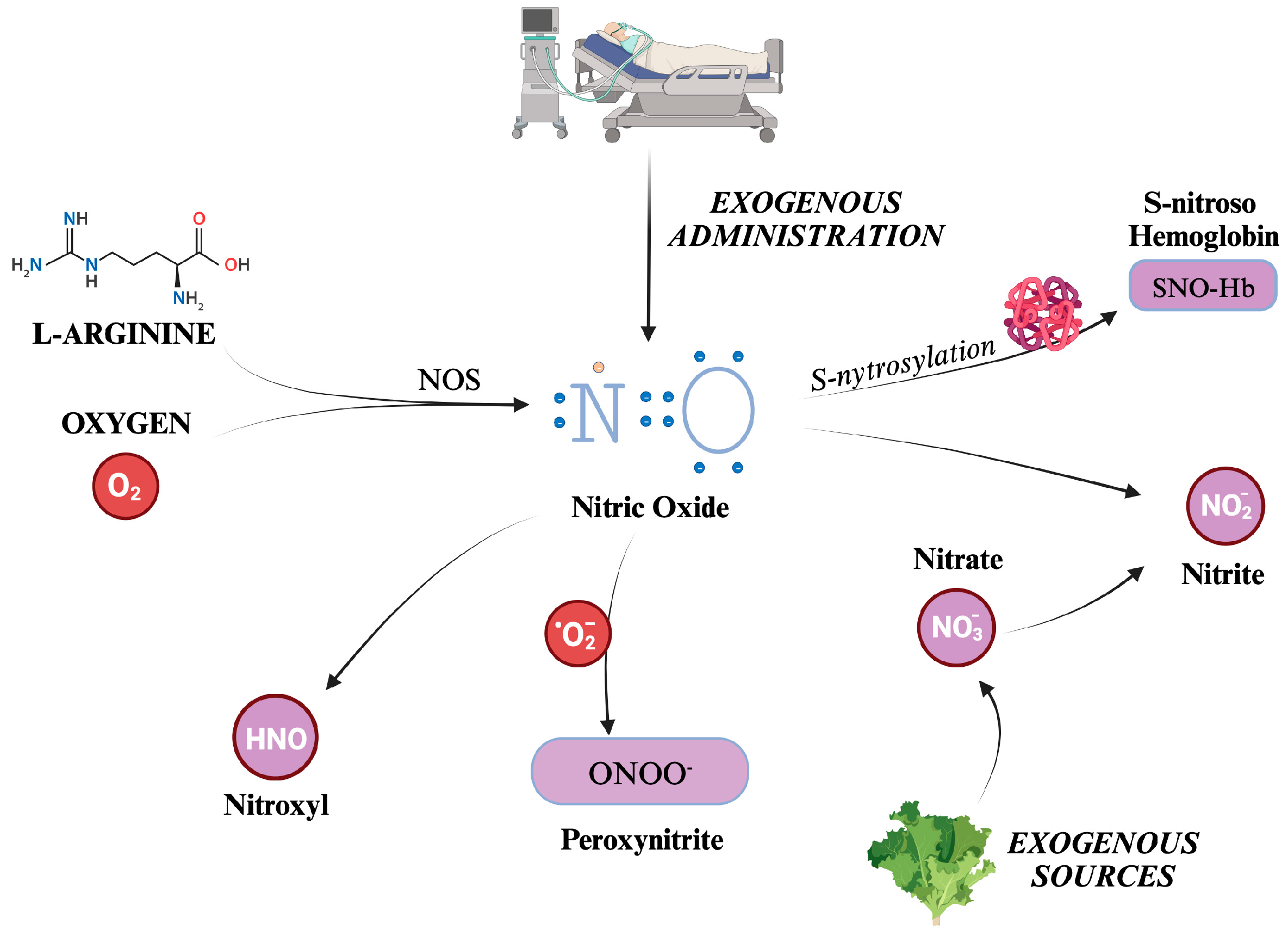
Figure 4.
Classification of the main antioxidants into enzymatic and non-enzymatic. Enzymatic antioxidants include enzymes that help to catalyze the breakdown of reactive oxygen species (ROS). Glutathione Peroxidase (GPx) catalyzes the reduction of hydrogen peroxide to water using glutathione as a reducing agent. Catalase catalyzes the decomposition of hydrogen peroxide into water and oxygen, it is found in the peroxisomes of nearly all aerobic. Superoxide Dismutase (SOD) intervenes in the dismutation of superoxide into oxygen and hydrogen peroxide. Non-enzymatic antioxidants directly neutralize free radicals and prevent oxidative damage. Vitamin C (Ascorbic Acid) is a water-soluble vitamin that acts as a potent antioxidant by directly scavenging free radicals and regenerating other antioxidants, such as vitamin E. Vitamin E (Tocopherols and Tocotrienols) is a group of fat-soluble vitamins that act as antioxidants by scavenging lipid peroxyl radicals and preventing the propagation of lipid peroxidation. Thus, it is important for maintaining the integrity of cell membranes and protecting against oxidative damage. Finally, glutathione (GSH) is a tripeptide composed of cysteine, glycine, and glutamate, that acts both independently and in conjunction with GPx. SOD: Superoxide Dismutase; GPx: Glutathione Peroxidase; GSH: Glutathione.
Figure 4.
Classification of the main antioxidants into enzymatic and non-enzymatic. Enzymatic antioxidants include enzymes that help to catalyze the breakdown of reactive oxygen species (ROS). Glutathione Peroxidase (GPx) catalyzes the reduction of hydrogen peroxide to water using glutathione as a reducing agent. Catalase catalyzes the decomposition of hydrogen peroxide into water and oxygen, it is found in the peroxisomes of nearly all aerobic. Superoxide Dismutase (SOD) intervenes in the dismutation of superoxide into oxygen and hydrogen peroxide. Non-enzymatic antioxidants directly neutralize free radicals and prevent oxidative damage. Vitamin C (Ascorbic Acid) is a water-soluble vitamin that acts as a potent antioxidant by directly scavenging free radicals and regenerating other antioxidants, such as vitamin E. Vitamin E (Tocopherols and Tocotrienols) is a group of fat-soluble vitamins that act as antioxidants by scavenging lipid peroxyl radicals and preventing the propagation of lipid peroxidation. Thus, it is important for maintaining the integrity of cell membranes and protecting against oxidative damage. Finally, glutathione (GSH) is a tripeptide composed of cysteine, glycine, and glutamate, that acts both independently and in conjunction with GPx. SOD: Superoxide Dismutase; GPx: Glutathione Peroxidase; GSH: Glutathione.
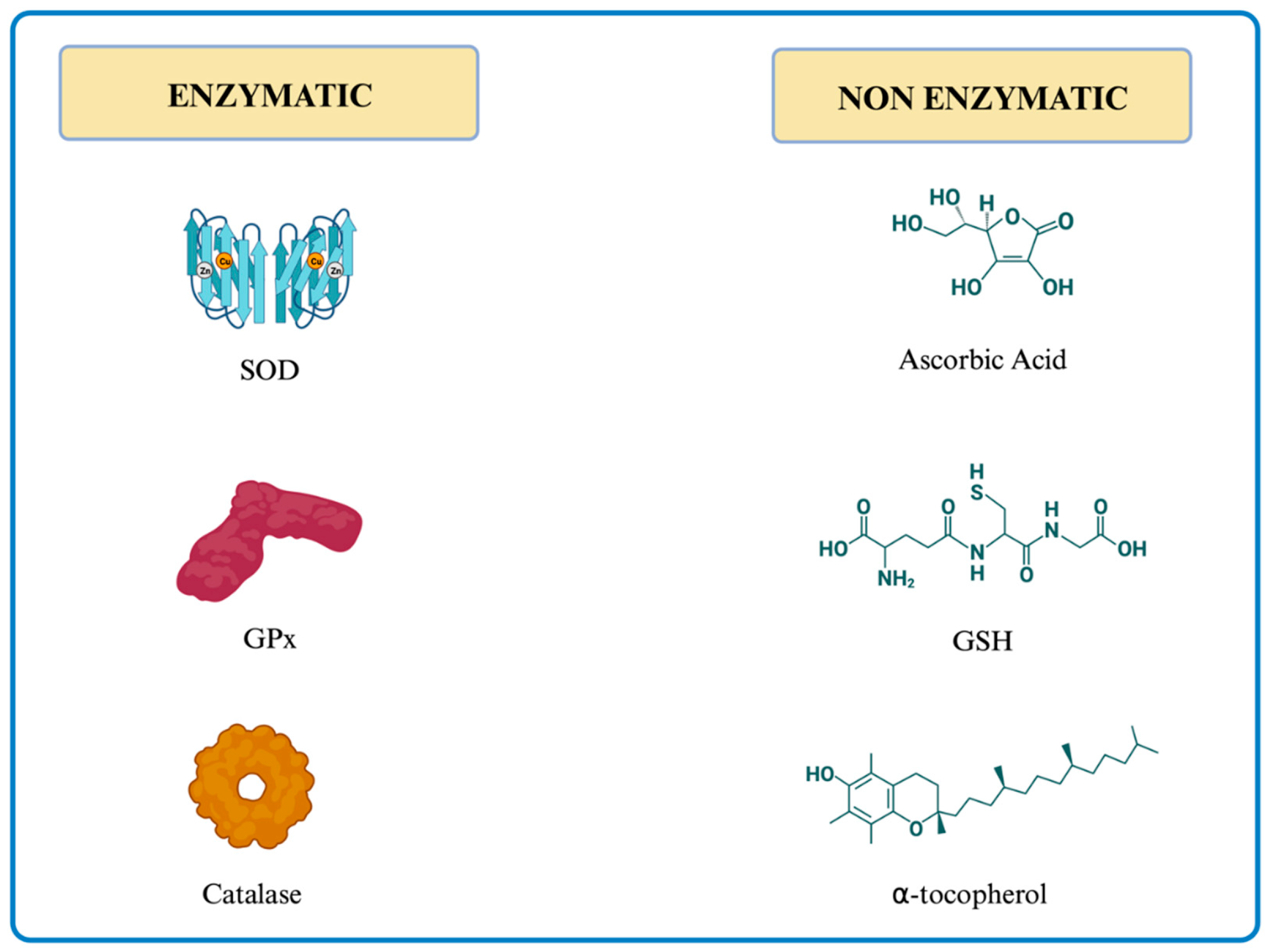

Table 1.
Factors contributing to the heightened vulnerability of brain tissue to oxidative insults.
| Factors of high susceptibility and vulnerability of the brain tissue |
|---|
| Intense production or ROS |
| High energy requirements |
| Neurodegeneration |
| Non – replicating neuronal cells |
| High ratio of membrane surface to cytoplasm |
| Weak antioxidant capacity |
| High lipid content |
| Low repair capacity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



