
Submitted:
09 January 2024
Posted:
10 January 2024
You are already at the latest version
Abstract
Microbiomes, comprised of diverse microbial species and viruses, play pivotal roles in human health, environmental processes, and biotechnological applications and interact with themselves, their environment, and hosts via metabolites and signaling molecules. Our understanding of microbiomes is still limited and hampered by their complexity. A concept improving this understanding is systems biology, which focuses on the holistic description of biological systems utilizing experimental and computational methods. An important set of such experimental methods are metaomics methods which analyze microbiomes and output lists of molecular features. These lists of data are integrated, interpreted, and compiled into computational microbiome models, to predict, optimize, and control microbiome behavior. There exists a gap in understanding between microbiologists and modelers/bioinformaticians, stemming from a lack of interdisciplinary knowledge. This knowledge gap hinders the establishment of computational models in microbiome analysis. This review aims to bridge this gap and is tailored for microbiologists, researchers new to microbiome modeling, and bioinformaticians. To achieve this goal, it provides an interdisciplinary overview of microbiome modeling, starting with fundamental knowledge of microbiomes, metagenomics methods, and modeling formalisms. Furthermore, the review explains model building, examples of microbiome model applications for prediction, optimization, and control. It concludes with guidelines, software, and repositories for modeling. Each section provides entry-level information, serving as a valuable resource for comprehending and navigating the complex landscape of microbiome research and modeling.
Keywords:
systems microbiology
; microbial ecology
; omics data integration
; metaproteomics
; genome-scale modeling
; constraint-based modeling
; boolean modeling
; bioinformatics
Figure 1.
Graphical abstract
1. Introduction
Most habitats on earth are populated by microbiomes consisting of various microbial species and viruses 1. Due to their ubiquity and versatility, microbiomes are essential for human life, development, and health [1,2]. The human microbiome can, for instance, increase cancer risk and progression by promoting local chronic inflammation, the release of free radicals, or the induction of pro-inflammatory cytokines [3]. The intestinal microbiomes of livestock ferment feed that is indigestible for humans. Products from livestock such as meat or milk are valuable protein sources but cause 30% of the global anthropogenic methane emission at the same time [4]. Similar microbiomes as in livestock degrade organic waste and renewables in anaerobic digesters to methane, which can be used for the production of renewable electric energy. In Germany, electricity from biogas covered about 5.8% of the electricity demand 2 and contributed 10% to the prevented greenhouse gas emissions in 2022 3. Lastly, microbiomes play a major role in nutrient cycling and are important for soil fertility and plant growth [5]. These examples demonstrate how important microbiomes are for human health, biotechnology, and the environment.
Despite their importance, member species of most natural microbiomes are unknown [6,7] and their behavior is not fully understood [1]. The reason for the lack of knowledge is the complexity of molecular interactions between microbiome members and their environment/hosts. These molecular interactions concern the processing of cellular energy and biomass (i.e., metabolism), as well as cellular signaling and regulation.
Parts of the missing knowledge on microbiomes can be uncovered by metaomics methods. These analytical methods identify and quantify genes, transcripts, proteins, and metabolites in microbiomes [8,9,10] analyzing many samples and molecules in a relatively short time, thus branded as high throughput. Making sense of the high throughput of metaomics data requires bioinformatics for automated data integration and analysis [10,11,12].
Metaomics data analysis results in mechanistic knowledge, which can be used to construct mathematical models of microbiomes [13,14,15,16,17]. Microbiome models indeed enable microbiome prediction, optimization, and control of microbiomes. Model predictions estimate the properties or the behavior of microbiomes under certain conditions and can support or falsify hypotheses and help understand microbiomes. Microbiome optimization comprises the identification of process conditions or interventions shifting specific process parameters and microbiome performance to a specified goal. For example, models determine optimal conditions for producing chemical compounds [18] or determine drug targets for growth inhibition of pathogens [19]. Lastly, microbiome models aid the control of dynamic processes towards a specified outcome. For instance, in biotechnological processes, model outputs are used to regulate the production of chemical compounds or biogas [20,21,22].
The outlined principle of collecting high-throughput data on a biological system, integrating data, analyzing data, and building predictive models is termed systems biology [23]. Although microbiome research, omics analysis, and systems biology have been reviewed independently several times, there is a lack of holistic and interdisciplinary training at the interface between microbiome research, omics analysis, and computational methods. Since this knowledge is the key to the successful application of systems biology on microbiomes, this review focuses on bridging all three fields starting from the basics and targeting the following four aspects:
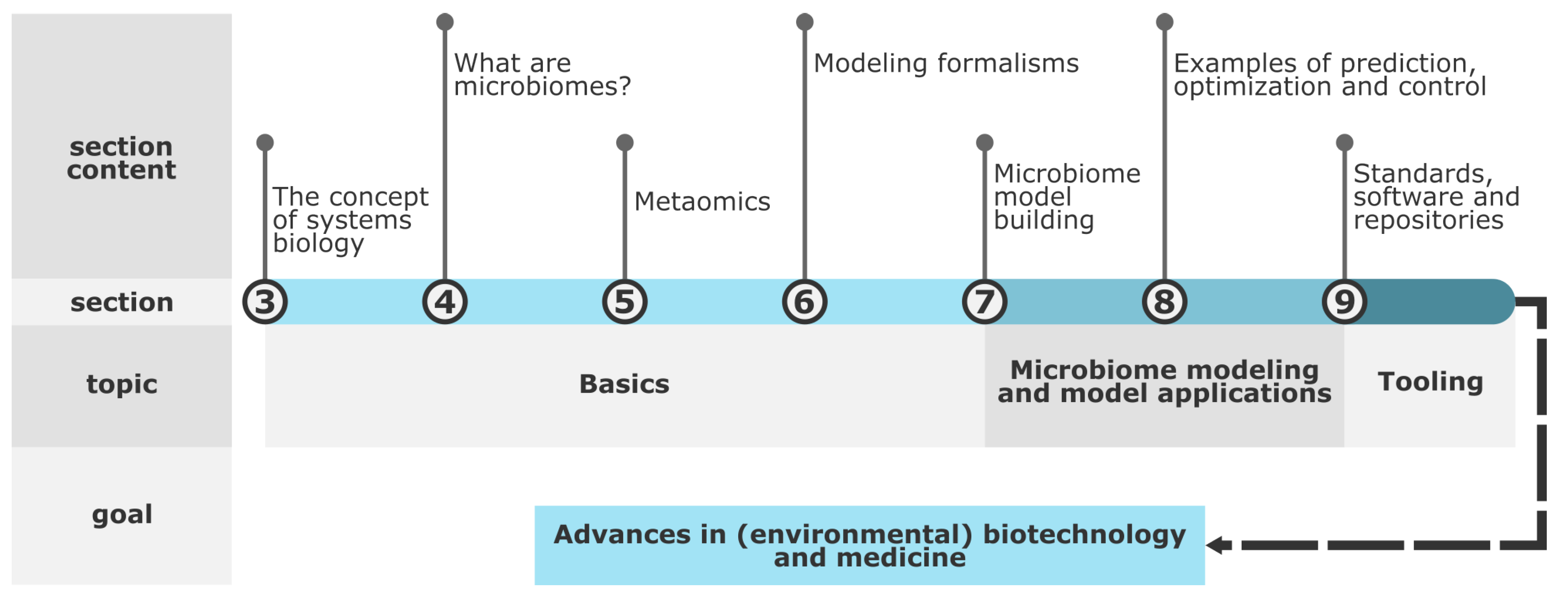
- First, the manuscript gives an interdisciplinary overview of microbiome modeling. To this end, the concept of systems biology (Section 3), microbiome properties (Section 4), metaomics methods (Section 5), and mathematical modeling (Section 6) are explained. In addition, mechanistic model building (Section 7) and their role in predicting, optimizing, and controlling microbiomes (Section 8) are covered. Finally, an overview of guidelines, software, and repositories for microbiome modeling is provided (Section 9).
- Second, metaomics and its peculiarities are explained. Metaomics methods based on liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS) allow the determination of cellular phenotypes. Because it is difficult to cover every meta-omics method, metaproteomics is presented as an exemplary technology. In addition, extensive references to other omics technologies for microbiome analytics are given.
- Third, modeling concepts from metabolic modeling, as well as modeling of signaling and regulation are discussed. While metabolic models are standardized and can reach the scale of genomes [24], modeling of signaling and regulation is less uniform. To fully understand the interference of microbiomes and their hosts, highly resolved models of signaling and regulation are required. The building process of both model types is compared and formalisms that could facilitate genome-scale modeling of signaling and regulation are described.
- Fourth, guidelines facilitating reusability and reproducibility are introduced.
The main sections of the manuscript are mostly independent of each other for readers exclusively interested in one topic. Moreover, at the end of each section, a summary of the section’s contents is included.
2. Methods
2.1. Targeted Literature Research Strategy
This review addresses metaomics methods, metabolism, signaling, and regulation, microbiome modeling, and guidelines for improving the reuse of microbiome models. A Python script was used to extract relevant papers from the respective fields, which queries the PubMed API [25], obtains a list of articles, and determines the most cited references across these articles (the used queries are listed in Table 1). The script was inspired by an available project (https://github.com/paulamartingonzalez/Targeted_Literature_Reviews_via_webscraping) and is available on our GitHub repository (https://github.com/voidsailor/targeted_literature_search, https://zenodo.org/doi/10.5281/zenodo.10402352).
The parameter for the initial number of papers was always set to 100. Afterwards, the most cited papers were extracted from the references of these initial 100 and ordered by node degree of the reference network. Starting with the highest-ranked articles, the best-fitting articles were selected for the respective sections. The generated output files can be found in the supplementary files (Table S1). Additionally, we added further references discovered while reading the fetched set of articles.
3. The Concept of Systems Biology
Systems biology can be defined in different ways [23]. In this review, it is defined as the combination of experimental and computational methods for collecting, integrating, and analyzing data to obtain a holistic view of biological systems and to predict, optimize, and control these systems. Systems biology of microbiomes and their hosts comprise four key aspects: Collecting data by experimental methods, integrating data, analyzing data, and building predictive models.
The first aspect of systems biology is to collect data on a complex biological system. These data can be generated by omics methods, which identify and quantify molecules such as metabolites, proteins, RNA, and DNA [23]. Metaomics methods analyze microbiomes and their environment (Section 5). Apart from (meta)omics, other methods are employed, for example, determination of biomass composition or growth rates.
Secondly, different data types and data sets from (meta)omics measurements and other sources need to be integrated, [23], to relate molecular information from every "ome" and permit a holistic view of cellular physiology. This requires a systematization of molecules, which is facilitated by biochemical databases. For each type of molecule, biochemical databases assign unique identifiers. Identifiers enable the labeling of measured molecules in meta-omics data sets. Different types of measurements are then connected following the molecular organization of cells. For example, genes link to their protein products, which link to enzymatic reactions, which link to metabolites. Additionally, (meta)omics data need to be annotated with metadata providing information on samples (e.g. origin, patient group, used lab workflow).
Thirdly, meaning needs to be extracted from meta-omics data. To this end, “classical” methods from statistics reveal group differences, patterns, and correlations [26,27,28]. Other statistical methods such as network analyses and pathway enrichment additionally provide biological contexts for meta-omics data [29,30,31]. Data visualization facilitates comprehension of metaomics data and communication of analysis results [27,32].
Lastly, knowledge from (meta)omics data can be used to build and refine computational models of microbiomes (Section 6 and Section 7). These models make predictions on microbiome properties and can simulate microbiome behavior, thereby facilitating the generation and validation of scientific hypotheses. Additionally, model predictions guide experiments and reduce experimental efforts (Section 8), for example, by predicting the most promising interventions for metabolic engineering as done by Bekiaris and Klamt [33]. Data produced in model-guided experiments can be used for model validation or refinement in case of successful or incorrect predictions respectively. Lastly, models are based on assumptions defining conditions under which they are applicable and/or that simplify the model structure (e.g., time invariance, metabolic steady state, homogeneity of cell populations). Eventually, the cycle of experimentation, model building, prediction, and model refinement leads to an increase in knowledge of microbiomes.
3.1. Summary – Section 3
Systems biology tries to resolve molecular mechanisms of biological systems and predicts system behavior. Collecting metaomics data, integrating data, analyzing data, and building predictive models are core aspects of systems biology on microbiomes.
4. What Are Microbiomes?
Native microbiomes are heterogeneous communities of microorganisms living in the same habitat or host. Microbiomes are complex biological systems and their composition and behavior emerge from molecular interactions (Figure 2). This section describes these molecular interactions (Section 4.1), emerging microbiome characteristics (Section 4.2), and considerations for cultivating microbiomes (Section 4.3).
4.1. Microorganisms and Hosts Interact via Metabolism and Signaling
Microorganisms exchange molecules with their inanimate environment, their hosts, and other microorganisms. These molecular interactions directly affect two systems [34]: Metabolism and signaling. Cellular metabolism constitutes the uptake, conversion, and excretion of chemical compounds, termed metabolites, by networks of enzymatic reactions. These reactions generate energy and building blocks for cellular maintenance and growth [35].
Cellular signaling detects and processes different stimuli (e.g., pH, osmolarity, temperature, signaling molecules). Cells receive signals with membrane-bound or intracellular receptor proteins. Receptors detect stimuli and transduce signals via cascades of sequentially activated proteins and small molecules (2nd messengers) [35]. Terminal molecular signals cause cellular responses (e.g., cellular shape changes [36]) or activate gene expression with transcription factors [37]. Activated genes regulate metabolism and signaling by expressing regulatory RNAs, enzymes, and signaling proteins. Additionally, genes regulate other genes by expressing transcription factors forming gene regulatory networks. These networks encode biological programs that correspond to behaviors or phenotypes [38,39,40] and can be considered as the third cellular system apart from metabolism and signaling [34].
Metabolism, signaling, and gene regulation are dynamic systems with inputs and outputs. For example, stimuli and activated transcription factors could be considered as respective inputs and outputs of signaling networks. Inputs and outputs are connected by regulated molecular interactions. Protein activity, for example, is regulated by protein expression, protein degradation, or post-translational modifications [41]. Especially relevant for signaling and gene regulation are feed-forward and feed-back loops formed by molecular interactions. These motifs determine dynamic system behaviors such as signal amplification or oscillation [42]. Signaling and gene regulation also possess stable or dynamic activation patterns sustaining cellular phenotypes. Resultingly, signaling can operate on a broad timescale ranging from milliseconds to hours [43].
For the remainder of this review, signaling, and gene regulation are described collectively as “signaling and regulation”. Similar modeling formalisms can be applied for signaling and gene regulation, as both are closely connected and often considered together for modeling. One could further resolve these main cellular systems into networks of transcripts [44] or regulatory RNAs [45]. However, these are not discussed for the sake of simplicity.
4.2. Microbiome Characteristics
Microbiomes are heterogeneous in their taxonomic and molecular compositions. They contain hundreds to thousands of species spanning all domains of life (i.e., Archaea, Bacteria, Eukaryotes, and Viruses). Microbiome members vary greatly in their size spanning several orders of magnitude [2]. Species also vary in their elemental and macro-molecular biomass composition. The estimated elemental composition of Escherichia coli, for example, is [46]. E. coli’s macro molecular dry weight composition consists of 50-55% proteins, 7-9% lipids, 20% RNA, and 3% DNA [47]. However, these values vary depending on the growth conditions and even differ across strains of the same species.
The composition of microbiomes is shaped by interactions among microorganisms and their hosts. Investigating these interactions is termed microbial ecology. Species do, for instance, compete for the same carbon and energy resources (competition), produce growth-inhibiting metabolites or antimicrobials (ammensalism), nurture other species (commensalism), and couple themselves tightly to the metabolism of other microorganisms (syntrophy) [48,49,50].
Many microbial interactions are related to metabolism. Exchanging metabolites allows division of labor, meaning that microbiomes contain specialists carrying out dedicated functions. In biogas-producing microbiomes, for example, hydrolyzing species and primary fermenters break down polymers into small organic molecules and hydrogen that can be used subsequently by methane-producing species [51]. Division of labor makes microbiomes more flexible and robust in comparison to individual species because cellular resources do not need to be allocated across different metabolic tasks [49]. When analyzing microbiomes, taxonomic profiles are thus of high interest, because the presence of one species may indicate a specific metabolic process [52]. Turnbaugh et al. [53], for instance, studied gut microbiomes of obese patients. They showed that relative changes in two dominant taxa could indicate greater energy harvesting potential of the microbiome.
In addition to metabolism, inter-cellular signaling influences microbiomes and their hosts. Signaling can occur across microorganisms. Quorum Sensing is an example of signaling in microbial populations. In quorum sensing, microorganisms respond with biofilm formation if the concentration of a signaling molecule exceeds a threshold [54]. Furthermore, microbiomes and their hosts communicate via molecular signaling: Plant roots secrete attractants or antimicrobial molecules to establish specific microbiomes, gut microbiomes might directly stimulate host neurons, influence host development, or interfere with host signaling [55,56,57,58].
Another important characteristic is the spatial and temporal variability of microbiomes. For example, human microbiomes from different body sites differ in their composition due to different physical conditions (e.g., the pH value) [59]. Because physical conditions depend on space and time, taxonomic profiles, generation times and adaption times vary across these dimensions as well. Depending on these conditions, microorganisms can live free-floating, as aggregates, or attached to surfaces in biofilms [60]. The type of organization determines mass transfer of molecules across the microbial population. Microorganisms at the surface of a biofilm can, for example, consume available oxygen completely and create anaerobic conditions inside the biofilm [61]. Additionally, large inter-individual variations of cells can exist inside the same population [62]. Furthermore, cellular density varies depending on the environment (e.g., 106 cells in 1 m3 air or 1011-1012 per mL in the colon) [63].
4.3. Culturing Microorganisms to Model Microbiomes
Most microbial species are still uncharacterized [6,7,64,65]. Out of the estimated 0.8–1.6 million prokaryotic species (based on operational taxonomic units) [66], about 0.57 million have sequenced genomes (NCBI, date of access: November 21, 2023, https://www.ncbi.nlm.nih.gov/genome/browse#!/prokaryotes/), but less than 10% are available as isolate from the German Collection of Microorganisms and Cell Cultures (https://www.dsmz.de/, date of access: November 21, 2023, 26,609 bacterial and 627 archaeal strains).
Characterizing unknown microorganisms requires cultivation-based studies to determine the functions of their genes [67]. However, many species are difficult to grow in enriched or axenic cultures (i.e., single-species cultures), due to unknown nutritional requirements, or because they can only survive in synthrophies [7]. Ongoing efforts optimize media and culture conditions for axenic cultures [67]. Furthermore, synthrophic species have been successfully grown and characterized in co-cultures with their interaction partners [67]. The resulting resources on characterized prokaryotic species are collected in databases such as BacDive [68].
Further growth experiments in axenic lab cultures are usually required to determine parameters and data for microbiome modeling (Section 6). Such cultures can provide enough material to determine cellular dry weight, macro-molecular biomass composition, ATP-maintenance coefficients, metabolic fluxes [69,70,71,72] or analyze biomolecules by “classical” omics methods [73]. It is beneficial to plan experiments with modeling assumptions in mind. For example, metabolic modeling (Section 6) assumes constant cellular metabolite concentrations and growth rate. Therefore, cultivation in continuously stirred tank processes is suitable to determine parameters for metabolic modeling, because process parameters remain constant [74].
Lab cultures of reduced microbiomes (i.e., two to ten species) allow investigation of species interactions under controlled conditions. Reduced cultures are used to mimic the functional composition of more complex microbiomes, for example, biogas-producing microbiomes [75,76], or the human gut microbiome [77]. It is also possible to inoculate lab cultures with samples from native microbiomes [78].
In many instances, microbiomes need to be analyzed in their native environments because native microbiomes and lab-cultured microbiomes may differ in their phenotypes and interactions. Mesocosm experiments are a compromise between the native environment and controlled conditions. In such experiments, organisms are subjected to environments similar to their native environment, but specific conditions can be altered [79,80]. It is also possible to sample native microbiomes in situ. Metaomics methods facilitate the analysis of biomolecules from complex or native microbiomes.
Due to the diversity and complexity of microbiomes, the role and contribution of an individual species to the microbiome cannot be deduced from growth or process parameters alone. Dissecting individual species activities is possible with metaomics methods. Metaomics data are resolved by taxonomy and are essential to model microbiomes (Section 5).
4.4. Summary – Section 4
Microbiomes are complex and heterogeneous biological systems interacting with their environment and hosts. The taxonomic and functional microbiome composition is shaped by metabolism and signaling and varies over time and space. Many microbiome members are uncharacterized because they are difficult or impossible to culture axenically. However, culturing individual microorganisms and microbiomes is necessary to generate the data required for microbiome modeling. If culturing is infeasible, in situ microbiome samples from mesocosms and native environments can be analyzed by metaomics.
5. Meta-Omics Create Inventory Lists of Microbiomes
Omics methods identify and quantify genes (genomics), transcripts (transcriptomics), proteins (proteomics), and metabolites (metabolomics) in biological systems. Metaomics refers to omics methods for (native) microbiomes and is applicable for in situ samples. Metaomics differ from “classical” omics because they need to deal with the high complexity and heterogeneity of samples [81].
Every omics method analyzes one distinct layer of molecular information. Integrating these layers is required to extract meaning from raw data. For example, genomic sequences are required to identify corresponding proteins from raw proteomics data (Section 5.1). Data integration is also required for statistical analysis and interpretation of outputs from (meta)omics methods. These outputs are usually lists of genes or molecules and their abundances (i.e., quantities). Pathway enrichment analysis, for example, integrates gene lists and information on metabolic pathways. Firstly, genes are grouped by their encoded metabolic pathway and then analyzed by a statistical test identifying pathways whose genes are significantly enriched [30]. The described procedure of pathway enrichment applies only to one omics layer which is integrated with information from a database. Multiomics in contrast applies two or more omics methods to the same system at the same time. Multi-omics provide a holistic insight into the analyzed system rather than just one “ome” but are more expensive, and require specific experimental considerations and analysis methods (see Arıkan and Muth [28] for a comprehensive and recent review).
This section introduces the general workflow of one metaomics method, namely metaproteomics (Section 5.1). Metaproteomics was chosen as an example because it utilizes similar methods as metabolomics, namely liquid-chromatography coupled to tandem mass spectrometry (LC-MS/MS). Secondly, it faces similar challenges as metagenomics, i.e., assigning functions and taxonomies to sequence information. Thirdly, it is underrepresented in microbiome modeling. For example, only one study uses metaproteomic data for reconstructing metabolic microbiome models [14]. In addition, taxonomic and functional protein annotation is discussed (Section 5.2), and other (meta)omics typically integrated with metaproteomics are introduced (Section 5.3). Furthermore, a list of references on not discussed omics methods with relevance for microbiome modeling is presented.
5.1. The Metaproteomics Workflow
Metaproteomics is the identification and quantification of proteins from microbiomes and their hosts, based on LC-MS/MS [82,83]. It can unravel the taxonomic and functional microbiome composition, decipher the expression of metabolic pathways, investigate microbiome interactions, and determine substrate usage [84]. Furthermore, protein abundances are used as estimates for enzymatic activities for microbiome analysis [82] and modeling [85]. However, it needs to be considered that protein activity is attenuated by temperature, pH value, metabolite concentrations, and post-translational modifications.
The metaproteomics workflow (Figure 3) starts in the laboratory with sample preparation. Firstly, cell lysis, extraction, and purification of proteins are performed. For each sample, the total protein concentration is determined for later sample normalization. Subsequently, proteins are purified and can optionally be separated by SDS-PAGE. The purified (and separated) proteins are then digested into peptides using the enzyme trypsin. Another separation of tryptic peptides by (two-dimensional) liquid chromatography (LC) can follow. Separation generally reduces sample complexity and can improve the resolution of subsequent analysis by mass spectrometry [86].
The last step of sample preparation separates peptides by LC, which is coupled to tandem mass spectrometry (MS/MS) [10]. During MS/MS, peptides are ionized and separated by their ratio of mass to charge (m/z ratio). In the next step, the mass spectrometer selects specific peptide ions. The selection of peptide ions is carried out for the most abundant peptide ions (data-dependent acquisition, DDA) or all peptide ions (data-independent acquisition, DIA) [87,88]. Lastly, the selected ions are fragmented, and recorded on a detector, resulting in a mass spectrum. The mass spectrum contains signal peaks of peptide ions or peptide ion fragments over their m/z ratio.
Proteins can be identified by comparing a recorded mass spectrum against libraries of reference spectra obtained from actual measurements. Alternatively, proteins are identified by searching the mass spectrum in a protein sequence database. To this end, the protein database is digested and fragmented in silico and searched using algorithms such as Mascot or X!Tandem [89,90]. Database searches result in assignments of the mass spectrum to peptides, termed peptide-to-spectrum match (PSM). PSMs are evaluated by a probability-based score, usually considering the PSM with the highest score as correct. A second search against a decoy database is conducted to exclude random PSMs. This search calculates the false discovery rate (FDR), a metric that filters out low confidence PSMs [91]. Protein identification is done using bioinformatic software such as the MetaProteomeAnalyzer or Galaxy [92,93]. Even though metaproteomics workflows differ, it could be shown in a ring trial that all applied workflows provide similar protein identifications [94].
Metaprotein abundance is quantified relative or absolute using the number of detected spectra (spectral counts), or the signal intensities of peptide ions. Relative, label-free quantification compares abundances of all metaproteins across samples [10,95]. Relative, label-based quantification introduces unique molecular labels (e.g., isotopic labels, TMT [96]) to the proteins of distinct samples. Subsequently, samples are pooled (i.e., multiplexed) and subjected to sample preparation. During MS/MS analysis, specific signals from the molecular labels are used for relative protein quantification. TMT labels, for example, emit unique reporter ions during MS/MS proportional to the abundances of labeled proteins and are used to infer relative abundances [96]. The advantages of label-based quantification are higher accuracy and reduced analysis times [96]. The output of relative quantification is a list of protein fold changes compared to a reference sample.
Absolute protein quantification determines absolute protein concentrations, for example, in mmol/mL. To this end, a protein standard containing a mixture of proteins with known concentrations (e.g., UPS2) is added to a sample before tryptic digestion [97,98]. Another method is the absolute quantification (AQUA) strategy, where the standard contains isotopically labeled peptides also occurring in the sample [99]. To quantify all proteins, present in the sample, a calibration curve can be created from abundances of standard peptides and used to interpolate concentrations of other proteins [97] (see Section 6.1 for a similar example of a calibration curve). It has also been shown that fractions of the total protein content can be estimated from peptide signal intensities without using a protein standard [95,97].
Identifying and quantifying proteins in complex in situ samples is a big challenge. Soil samples, sludge from wastewater treatment plants, or biogas plants contain large amounts of impurities (e.g., minerals, humic substances) [51,100]. These impurities must be removed during sample preparation since they disturb total protein quantification, protein separation, and LC-MS/MS measurements. However, extensive sample preparation could cause a bias in protein quantification, leading to more considerable deviations from the actual protein concentration. An approach to control this bias is to add internal standards to samples [101]. Furthermore, identified proteins are biased by the used reference database. If a protein sequence is not included in the database, it cannot be identified. However, including more protein sequences in a database may result in more false positive identifications. Another challenge is the identification and quantification of homologous proteins from different species. Homologous proteins can only be uniquely identified if they have unique peptides. If no unique peptides can be detected for homologous proteins, unique identification is impossible. In such cases, proteins can be assigned to protein groups (metaproteins) [10,102]. Resultingly, quantification of metaproteins can sometimes only be performed for protein groups.
Guidelines for sample preparation, metaomics study design, and reporting of data are described in several articles [103,104]. Developments in data integration, data processing, and standardization in (meta)proteomics are coordinated by platforms such as Elixir https://elixir-europe.org/communities/proteomics [105] or the metaproteomics initiative (https://metaproteomics.org/) [106].
5.2. Taxonomic and Functional Annotation of Protein Groups in Meta-Proteomics
Taxonomic and functional annotation of identified proteins is of interest to figure out which species perform what kind of function in a microbiome. Protein identity can be inferred from a reference database (Section 5.1), which can be derived from public protein databases (e.g., UniProt/SwissProt [107]) or constructed from metagenome assembled genomes (MAG, Section 5.3) [10,108]. Protein taxonomy and function are usually annotated in reference databases as well and can be adopted for newly identified proteins. Notably, taxonomic and functional profiling by metaproteomics is always biased towards the used reference database.
Protein taxonomy is related to the organism expressing the protein and generally employs the NCBI taxonomy [109] or the GTDB taxonomy [110]. The taxonomy for a protein group (Section 5.1) is defined by the lowest common ancestor, sometimes extended by the expression profile of all matching peptides, the sequence similarity, or taxonomic constraints [92,111,112,113,114]. Alternatively, researchers focus only on unique peptides [115] or on marker proteins such as ribosomal proteins for taxonomic profiling [116].
Functional protein annotation is done according to the systematics of biochemical databases. For example, the UniProt database assigns unique identifiers to protein sequences (https://www.uniprot.org/help/accession_numbers) [107], the BRENDA database [117] uses enzyme commission (EC) numbers to assign functional ontologies for metabolic enzymes, and the gene ontology knowledgebase provides general terms for cellular processes [118,119]. Database entries provide cross-references to other functional identifiers, associated molecules, and pathways [120,121,122]. Functional annotation of protein groups poses a similar challenge to taxonomic annotation. On one hand, homologous proteins from different species will have similar functions. On the other hand, large proteins, particularly from eukaryotic cells, contain several functional domains. Grouping these multi-domain proteins may result in an unspecific protein grouping and overestimation of functions.
The selection of the best approach for the taxonomic (and functional) assignment of peptides depends on the microbiome complexity, quality of the reference database, and microbiome stability. Currently, the Critical Assessment of Meta-Proteome Interpretation (CAMPI3) is conducted and benchmarks different bioinformatic workflows for protein assignment (https://metaproteomics.org/campi/campi3/).
5.3. Other Omics and Experimental Methods for Microbiome Analysis
The metaproteomics analysis and following data analyses benefit from other metaomics methods, such as metagenomics. Whole metagenome shotgun sequencing (WGS) is a metagenomics method that aims to determine genomic sequences in a microbiome [12]. WGS methods process snippets of sequenced DNA (i.e., reads) for functional or taxonomic microbiome profiling. Reads can also be assembled into longer contiguous sequences (contigs), which can be used to predict genes, as well as associated gene taxonomy and function. Predicted and annotated genes can be used for the de novo reconstruction of genomes (i.e., MAGs) for unknown organisms [12,123]. However, such genomes can be incomplete or contain genes from different organisms.
Functional and taxonomic annotation can be performed for reads or contigs. Taxonomy can be classified by 16s ribosomal RNA marker genes or by searches against reference databases [12]. As for proteins, gene taxonomies are assigned according to NCBI [109] or GTDB [124]. Functional annotations can be retrieved from reference databases or, for de novo assembled genomes, from homology searches against databases for functional ontologies or protein families [12]. Another method for taxonomic profiling is amplicon sequencing, which relies on the quantification of strain-specific ribosomal RNA [12].
Microbiome composition can furthermore be investigated using flow cytometry. Flow cytometry sorts and counts cells according to cellular features or chemical labels. Sorted cells can also be subjected to further (omics) analyses or cultivation [125,126]. Lastly, microscopic observation gives clues about present species and is necessary to determine physical properties of microorganisms (e.g., shape and cell sizes) [127].
Metabolic activity is another key feature apart from the taxonomic and functional composition of microbiomes. Expression of metabolic enzymes is indicated by metaproteomics, but enzyme activity does not necessarily correlate with enzyme abundance. Metabolomics quantifies metabolite pools and hints at metabolic activity [128]. The metabolome generally comprises molecules below 1500 Da and is dynamic. Sampling and sample preparation protocols thus aim to preserve the metabolome state, for example, by minimizing enzyme activity and reducing chemical reactivity [129]. Metabolomics utilizes LC or gas chromatography (GC) coupled to mass spectrometers or nuclear magnetic resonance devices (NMR) to identify and quantify metabolites [130]. It is possible to quantify metabolites for the complete microbiome or the microbiome medium. Determining metabolite pools of individual cells requires single-cell methods. Alternatively, chemically, or isotopically labeled substrates can be added to the medium to measure the incorporation of metabolites into biomass, which indicates metabolic activity [126].
The mentioned technologies allow for top-down analyses of microbiomes and their expressed and active metabolic functions. Mechanistic models with molecular resolution (Section 6) can be reconstructed (Section 7) from annotated (meta)genomes and refined, validated, and integrated with metaproteomics and metabolomics data. Microbiome modeling is not limited to these data types and can exploit other omics and experimental methods depending on the utilized modeling framework and research question (Section 6.3). Koch et al. [76] for example, used the measured community growth rate to constrain a microbiome model and validated predicted biogas production yields with values determined from measurements. A (non-exhaustive) list of data types/methods useful for microbiome modeling and corresponding references is provided (Table 2).
5.4. Summary – Section 5
Omics and metaomics methods identify and quantify genes, transcripts, proteins, and metabolites in individual organisms or microbiomes respectively. Metaproteomics is based on LC-MS/MS and advantageous over metagenomics because it determines expressed cellular functions. Annotating metaproteins by taxonomy illuminates the origin of these functions. Metaproteomics relies on other technologies such as metagenomics providing reference databases for protein identification and is supplemented by metabolomics to determine metabolic activity. Microbiome modeling can exploit other data types apart from metaproteomics and metagenomics.
6. Mathematical Models Are Formalisms to Describe Biological Mechanisms
Models aim to capture real-world phenomena by mathematical expressions and can be used to describe biological systems in time and space. An important part of model expressions are parameters, which quantify the properties of a biological system (e.g., the ATP maintenance coefficient [71]). The value of models lies in their capability to integrate and compile knowledge and complement newly generated experimental data. Beyond compiling knowledge, they can make predictions, generate, and validate hypotheses. Making predictions is usually cheaper than performing experiments and at the same time, model predictions can guide the design of experiments by making experiments more targeted.
Choosing an appropriate model structure depends on many factors, for example, the research question, required mechanistic resolution, available data, and available knowledge (Figure 4). This section explains choices for mathematical models that are often applied for signaling and regulation and metabolic models. Aspects of mechanistic and statistical modeling (Section 6.1), model scale (Section 6.2), and mechanistic mathematical frameworks (Section 6.3) are introduced. More information on modeling of biological systems and formalisms that were not considered can be found in [34,150,151].
6.1. Statistical Models and Mechanistic Models
The first decision for a model framework can be made between statistical models and mechanistic models (Figure 4). Statistical models comprise a heterogeneous group of model frameworks (part of which are machine learning models) applied to detect patterns in data, classification, or regression. These models generally capture relations between one or more input and output variables of a biological system from data [152]. Assumptions on the structure (i.e., distribution, dependencies) of input and output data determines the chosen model framework [153]. The available data are utilized to adjust model parameters in a procedure termed model training. The lack of mechanistic information is a disadvantage of statistical models because there is no information on the causal connection between input and output variables, models can be biased towards the structure of training data, and their range of validity is often limited [153]. Statistical modeling is, for example, applied in meta-proteomics to improve protein identification [154], predict disease states from meta-genomes [155], or for the detection of potential disease biomarkers [156] and biomarker panels [157].
A simple example of statistical modeling is the generation of a calibration curve for protein quantification. The chosen model framework is a linear equation containing the y-interception and slope as parameters. The input variable is the absorption value of a colorimetric protein assay [158] and the output is the protein concentration. A dataset for model training is generated from a dilution series of a standard protein. This dataset contains absorptions assigned to known protein concentrations and is used to train model parameters by linear regression. Using the trained model, the total protein concentration in unknown samples can be determined from protein assays [158].
Contrary to statistical models are mechanistic models, which represent physiological processes in (more or less resolved) detail [153]. Constructing mechanistic models often follows a bottom-up approach in the sense that an overarching process is reconstructed from its parts [152]. Overall, this necessitates less data than statistical models but requires knowledge of the components of a biological system. The great advantage of mechanistic models is their display of causality. Additionally, model entities and model parameters can be integrated with (meta-)omics measurements.
Hereafter, mechanistic modeling will be the focus of this review, because it is suited for microbiomes. Microbiome models integrate omics data (Section 7), represent reusable knowledgebases, and give insight into microbiome physiology. Nevertheless, statistical modeling is of equal value, and combining both formalisms can be advantageous [20,153,159,160].
6.2. Scales of Mechanistic Models
The second decision requires modelers to set a model resolution (Figure 4). Mechanistic models cover different scales of detail, distinguishable into sub-cellular scale describing molecular interactions within cells, the cellular scale characterizing molecular interactions between cells, and the macroscopic scale representing the function of entire technical systems, environments, or complex organisms [150]. Combining several scales results in multi-scale models, which usually describe macroscopic processes based on molecular effects [79].
It is possible to reconstruct macroscopic processes from molecular interactions but many research questions do not require models of molecular resolution. Additionally, non-available data, lack of knowledge, or the required effort prevent the creation of such models. Thus, models are often tuned to the scale of interest, for example, cellular, or macroscopic scale models often incorporate lesser mechanistic detail.
6.3. Mathematical Modeling Frameworks
Modelers are now confronted with a variety of mathematical frameworks (Figure 4). Choosing an appropriate mathematical framework is decided by the chosen scale, the biological system of interest, and available data.
6.3.1. Graphs
Graphs are mathematical representations of networks and describe the structure of networks (i.e., network topology) [161]. In the context of systems biology, graphs capture interactions (edges) between biological entities (nodes). Nodes could be molecules, functional modules, or cells. Edges can be undirected indicating associations (e.g., protein A interacts with protein B), or directed to indicate a directed flow of mass (e.g., metabolite A is catalyzed by reaction Y to metabolite B) or information (e.g., protein A activates protein B). A graph can be expressed as an adjacency matrix containing one row and column for each node with matrix entries representing the occurrence and the type of an interaction [42,161].
The analysis of graphs gives information on the organization of biological networks, for example, whether the network has a modular organization [161]. Metrics such as node degree (number of edges connected to a node) and betweenness centrality (number of paths going through a node/edge) can respectively highlight molecular hubs or potential metabolic bottlenecks [161]. Furthermore, for networks representing signal flow, paths (routes between input and output) and feed-forward or feed-back loops can be uncovered to obtain insight into the dynamic behaviors of networks [42,161]. More information on biological graphs can be obtained from an introduction on protein-protein interaction networks (https://doi.org/10.6019/tol.networks_t.2016.00001.1) and articles by Samaga and Klamt [42], Koutrouli et al. [161].
Graphs represent knowledgebases compiling information from various resources [161,162]. Graph databases exploit this flexible framework by attaching information to nodes and edges [163]. Furthermore, systems of differential equations, Boolean, and constraint-based models inhere a graph structure, because they represent interactions among parts of biological systems [161]. Graph methods can thus be applied to more advanced mathematical frameworks.
6.3.2. Boolean Models
Boolean models commonly represent cellular signaling and gene regulation [161,164]. They are based on a set of variables having a (binary) activation state (e.g., zero or one). Accordingly, these variables can correspond to genes or signaling molecules. Activation states are updated by Boolean functions linking all activating/inhibiting interactions from other variables. Boolean models can be visualized as directed graphs, representing Boolean variables as nodes and activating/inhibiting interactions as edges [164,165].
Boolean models are qualitative, meaning they do not capture molecular quantities but can represent relations and "on" or "off" states. They can be applied when parameters are difficult to determine, for example, kinetic parameters for models based on differential equations (Section 6.3.3) [34]. Typical analyses of Boolean models investigate dynamic input-output behaviors or steady states [42]. Dynamic simulations require a time-scale separation of fast and slow processes because Boolean models are updated in discrete time steps. More information on Boolean models can be found in [42,164,165,166].
6.3.3. Models Based on Differential Equations
Ordinary differential equations (ODEs) express changes in biological entities (e.g., chemical compounds, cells) over time. Biological entities are represented by quantitative state variables, for example, corresponding to molecular or biomass concentrations. A differential equation contains terms describing rates of production and consumption of the corresponding variable (e.g., the ODE for growth rate may contain terms for production of biomass and loss of biomass due to cell death). Each rate term can depend on parameters that are functions of environmental features, for example, a chemical reaction rate r depends on the temperature-dependent reaction rate constant k, and the concentrations of its substrates , (Equation (1)).
Essentially, differential equations can be applied to model any type of network exhibiting a dynamic behavior on any scale. Metabolic networks, for example, consist of biochemical reactions. The change in concentration of each metabolite, i.e., the reaction rate, can be expressed by an ODE. This includes the sum of reaction rates of all reactions producing and consuming the compound times the corresponding stoichiometric coefficients. The resulting system of ODEs can be characterized by a stoichiometric matrix N representing metabolites as rows, reactions as columns, and each entry as the stoichiometry of compound i in reaction j (Figure 5). Additionally, this matrix is multiplied by the (concentration- and temperature-dependent) vector of reaction rates r [167] (Equation (2)).
Like Boolean models, state variables can be simulated over time. Simulation of differential equations requires numerical integration, which is computationally more expensive than the discrete simulation scheme of Boolean models. Both model types require initial conditions for state variables (e.g., metabolite concentrations) from which the evolution of the dynamic system is determined.
The system can evolve from its initial state into a steady state in which state variables remain constant over time or oscillate. Depending on the initial conditions, the system may run into different steady states. The number and properties of steady states are determined by model structure and parameter values and may correspond to specific phenotypes. Apart from steady-state analyses, input-output behaviors can be analyzed. Specific to ODE models are sensitivity analysis (identification of parameters and initial conditions with the highest impact on the system output) and bifurcation analysis (investigation of changes in network behavior dependent on parameter changes) [168]. Further information on quantitative dynamic modeling is explained by Palsson [169] and Novere [151].
6.3.4. Constraint-Based Metabolic Models
Kinetic parameters in metabolic models, such as reaction rate constants from the model described in Equation (1), are often not available or imprecise [170]. To mitigate this issue, constraint-based modeling assumes that metabolism quickly reaches a steady state. For microbiomes, a steady state can be assumed if a continuous supply of substrates is available to the system, for example, in continuous cultivation or during the exponential growth phase in batch cultures [74,171]. In the steady state, metabolite concentrations are constant over time, which simplifies Equation (2) into a system of linear algebraic equations [167] (Equation (3)).
Because metabolite concentrations are assumed constant, only metabolic fluxes can be calculated from Equation (3). A solution to Equation (3) is termed flux distribution. For larger networks, there is usually no unique flux distribution, meaning the system is under-determined [172]. The under-determined system thus has a solution space containing multiple possible flux distributions [167].
Flux balance analysis (FBA) is a method, which determines a flux distribution fulfilling a specific biological objective and additional constraints. To this end, upper and lower limits for reaction rates are set as constraints (e.g. restriction of oxygen uptake in anaerobic systems) and an objective function is defined. The objective function usually represents a biological objective, for example, biomass growth. To represent the biomass objective in the model, a biomass reaction abstracting all anabolic reactions is introduced (see Orth et al. [173] for an example biomass reaction for E. coli). The resulting optimization problem can be solved by linear optimization, which determines a global optimum for the objective function [167]. However, the flux distribution fulfilling the optimum is not necessarily unique.
Flux variability analysis (FVA) can be used to explore the limits of the solution space, i.e., investigate the set of all possible flux distributions. FVA performs FBA for each reaction to find its minimal and maximal values [174]. Incorporating omics data into models is a way to reduce the size of the solution space and achieve predictions for specific biological contexts (Section 7.2.2).
The core structure of constraint-based models and methods has been extended for a wide spectrum of scenarios, for example, alternative objectives, multiple objectives, metabolic regulation, and explicit biosynthesis of enzymes. More information is available in reviews by Terzer et al. [167], Lewis et al. [175], Bordbar et al. [176].
6.3.5. Rule-Based Models and the Rxncon Language
Signaling proteins can have multiple sites for post-translational modifications, which determine their activity. The combination of possible modifications results in many possible microstates. Modeling each microstate explicitly, for example, by ODEs would result in models infeasible for simulation in terms of required parameters and computation power [177,178].
Rule-based models can define molecular reactions in a more scalable fashion than Boolean models or ODEs. In a rule-based model, molecules are defined as objects that are modified by reaction rules. A molecule could, for example, be a protein with a specific phosphorylation site. A reaction rule could then describe the procedure of phosphorylation which applies to all proteins having this phosphorylation site. Rule-based models are supported, for example, by the BioNetGen language (BNGL) [179].
The reaction contingency language (rxncon) is related to rule-based modeling [180]. Analogous to rule-based models, a rxncon model contains molecules and elemental reactions. Additionally, it contains contingencies, which are conditions that permit elemental reactions (e.g., activation of a kinase) [178].
Rxncon and BNGL are rather languages than actual mathematical models and are not ready for simulation [179,180]. BNGL requires kinetic parameters and can be compiled into ODEs [179]. However, these parameters are difficult to obtain, and ODE models can only be small or coarse-grained due to the combinatorial explosion of microstates. Rxncon on the other hand, can be compiled into Boolean models which can simulate large-scale networks without the need for kinetic parameters [180].
6.3.6. Combining Model Formalisms
Theoretically, every cellular process could be modeled by ODEs, but models of metabolism, signaling, and regulation commonly employ specific modeling frameworks. Cellular processes are not isolated, and assumptions like steady state, or homogeneity do not always apply in reality. Thus, integration of different modeling frameworks is necessary if one wants to consider these circumstances [34]. Multi-scale models (Section 6.2), for example, integrate different frameworks because frameworks for molecular processes may not capture processes on higher scales [181].
Generally, there is no standard way of integrating modeling formalisms. Implementing an integrated framework thus depends on available knowledge of cellular processes and the modeler’s creativity. Mahadevan et al. [182], for example, implemented dynamic FBA by combining a constraint-based model with differential equations into a dynamic optimization problem. Another example by Orth et al. [173] combines metabolism and transcriptional regulation by integrating a constraint-based model with Boolean rules representing enzyme availability. In this example, reactions can be constrained to carry no flux, if the corresponding transcriptional rule evaluates to “FALSE”.
Fully understanding the interactions between microbiome and host will require a combined model of all cellular processes. Such models are termed whole-cell models and are currently limited to microorganisms such as Mycoplasma genitalium or E. coli [183]. Modeling microbiomes and hosts at the whole cell resolution are far away, but some of the presented principles and modeling frameworks will potentially facilitate this enterprise.
6.4. Summary – Section 6
Models constitute different mathematical frameworks fitting distinct problems. The availability of large datasets or the need to represent mechanistic information determines the choice between statistical or mechanistic models. The model scale is determined by the scale of the problem to be investigated. For mechanistic models, plenty of mathematical frameworks are available such as graphs, Boolean models, differential equations, and constraint-based models. Different modeling frameworks can be connected and will facilitate the development of models that can capture all cellular processes in microbiomes and their hosts.
7. Building and Adjusting Models to (meta)omics Data
This section explains model building and model adjustment to (meta)omics data. These two procedures can be seen as data integration workflows. Firstly, model building incorporates and connects existing knowledge in the mechanistic structure of models. Secondly, adjusting an unspecific model to data “embeds” these data into the model. Additionally, constructed models enable data integration as well, because they provide a framework relating the parts of a biological system. If model parts correspond to biological entities, they can be annotated with database identifiers, which facilitate the integration of model predictions and data from (meta)omics experiments.
Hereafter explained are network reconstruction and model building (Section 7.1), parameter estimation, contextualization, and model reduction with emphasis on genome-scale models (Section 7.2, see Figure 4 for an overview). Reviews by García-Jiménez et al. [18], Papin et al. [43], Feist et al. [184], Gu et al. [185], Heinken et al. [186], Garza et al. [187], do Rosario Martins Conde et al. [188] are recommended for further information on network reconstruction, microbiome modeling, and the related multi-tissue modeling.
Figure 6.
Overview of model building (including model validation), adjustment, and related challenges. Reconstruction of metabolic microbiome metabolism is in the most advanced state, compared to other modeling formalisms. One bottleneck is the lack of annotated genomes and biomass composition data for uncommon species. Modeling signaling and regulation suffers from a general lack of understanding of the biological processes and there is usually no (semi-)automated procedure to create models as in constraint-based modeling. Additionally, standards for modeling and exchange formats are less established and there are no databases containing standard model components. All model types commonly lack model annotation. However, even if annotated with identifiers, these identifiers can be ambiguous. Many simulation results cannot be reproduced due to insufficient reporting. Lastly, using software packages for (microbiome) modeling usually requires programming experience or software is not available anymore.
Figure 6.
Overview of model building (including model validation), adjustment, and related challenges. Reconstruction of metabolic microbiome metabolism is in the most advanced state, compared to other modeling formalisms. One bottleneck is the lack of annotated genomes and biomass composition data for uncommon species. Modeling signaling and regulation suffers from a general lack of understanding of the biological processes and there is usually no (semi-)automated procedure to create models as in constraint-based modeling. Additionally, standards for modeling and exchange formats are less established and there are no databases containing standard model components. All model types commonly lack model annotation. However, even if annotated with identifiers, these identifiers can be ambiguous. Many simulation results cannot be reproduced due to insufficient reporting. Lastly, using software packages for (microbiome) modeling usually requires programming experience or software is not available anymore.
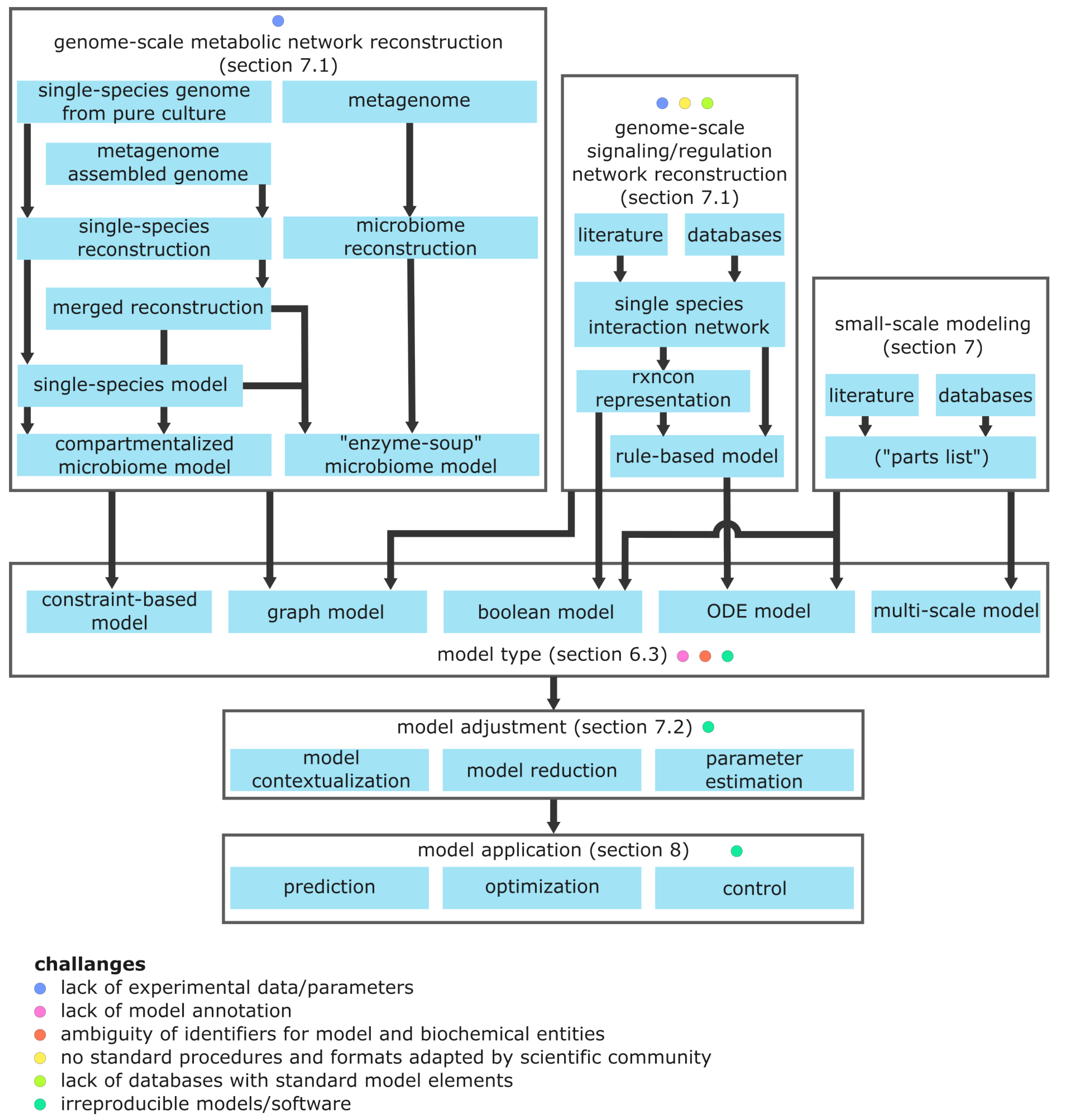
7.1. Reconstruction of Genome-Scale Biological Networks
Different strategies exist to build computational models. Firstly, a research question should be defined to establish a goal for modeling (e.g., finding the optimal substrate for maximal growth of E. coli) and to choose a modeling framework (Section 6.3). Secondly, literature and databases need to be scanned to extract all involved entities, their relationships, and the character of relationships (e.g., directionality, activation, inhibition) [151]. The resulting list of entities and relations can then be formulated as graphs, Boolean rules, or mathematical equations utilizing available software (Section 9) [151]. Subsequently, the model is adjusted to experimental data (Section 7.2) and ready to make predictions. Model predictions are validated on experimental data and the model structure and parameters are adjusted if predictions and data do not match. This results in an iterative model development cycle [151].
Conducting model development manually is a tedious approach, but feasible for small models for specific contexts. A complementary, partly automated approach utilizes annotated genomes to create inventory lists for signaling, regulatory, or metabolic networks termed network reconstruction [184]. Network reconstructions serve as knowledgebases and are used to build mechanistic models with molecular resolution. Because all relevant gene products are considered, corresponding reconstructions and models are termed genome-scale. Genome-scale models essentially have the same resolution as (meta)omics data. Due to the combinatorial explosion of possible microstates and the non-availability of kinetic parameters (Section 6.3.5), it is currently impossible to build genome-scale ODE models. The alternative modeling frameworks for genome-scale modeling are constraint-based models and Boolean models.
Hereafter, the reconstruction of genome-scale models for metabolism, signaling, and regulation are explained. Some more information on reconstructing other network types and building kinetic models can be found in [151,184,189].
7.1.1. Reconstruction of Single Species Metabolic Networks
Reconstruction of single species metabolic networks is a standardized four-step procedure [24] and results in a simulatable constraint-based model (a detailed description is given by Thiele and Palsson [24], Orth et al. [173]):
- Draft Reconstruction: Starting point is a whole genome sequence of an organism. The genome is annotated, i.e., genes are linked to transcribed enzymes and transport proteins, which are associated with metabolic reactions. Biochemical databases (e.g., KEGG) can be used to annotate known genes. Genes and reactions are connected in Boolean expressions named gene protein reaction rules. These describe enzymatic subunits required to perform a reaction and facilitate in silico gene knockout analyses. The resulting “parts list” of genes and reactions is generated automatically and represents a draft reconstruction that needs further refinement.
- Refinement: Errors within the reconstruction, such as wrong stoichiometries, wrong cofactor usage, or falsely assigned reactions need to be resolved. This step often requires manual curation linked to extensive literature research and mining of organism-specific databases. Furthermore, processes such as non-growth associated maintenance, recovery of reducing agents, and biomass synthesis (i.e., cell growth) are typically lumped into respective model reactions and added to the reconstruction. For example, substrates of the biomass reaction (Section 6.3.4) are macromolecules or their precursors, whose stoichiometries are determined experimentally from the organism’s macromolecular composition (Section 4.2) [70] or adapted from other organisms. Beck et al. [69] reviewed and evaluated several lab procedures to obtain the macromolecular composition.
- Mathematical model implementation: Thirdly, the network reconstruction is converted to a constraint-based model, which involves creating the stoichiometric matrix, defining compartments, and specifying reaction directionalities.
- Model validation and refinement: The fourth step is a loop of model validation and refinement. The computational model is used to diminish flaws in the reconstruction, for example, missing pathways, or unreachable reactions and metabolites. Furthermore, model constraints can be fine-tuned to biological data (e.g., maximal uptake rates, growth, and non-growth-associated maintenance coefficients). It is ensured that biomass precursors can be synthesized and that the model reproduces relevant growth conditions. The primary reconstruction may contain network gaps, which can be closed by automated, optimization-based gap-filling algorithms. These gap-filling algorithms aim, for example, to identify a minimum set of reactions from a biochemical database enabling the model to simulate growth for different growth media [11]. Another gap-filling approach searches for reactions that support growth, biomass precursor synthesis, utilization of specified alternative energy sources, and metabolite production based on high genetic evidence [190]. After gap-filling, the model might contain blocked reactions (reactions unable to carry any flux), which can be identified using FVA (Section 6.3.4). Manual curation resolves these errors, for example, by adding further reactions. Growth and knockout screenings are used to validate the model output. Lastly, basic model properties, for example, the stoichiometric balance of reactions can be validated with the MEMOTE software [191].
7.1.2. Reconstruction of Microbiome Metabolism
Two approaches exist to build constraint-based models of microbiomes. The “enzyme-soup” approach combines all biochemical reactions of the microbiome into a unified model. This can be done by identifying all microbiome members and merging their models [192] or by performing the previously described reconstruction process based on the metagenome or metaproteome [14]. “Enzyme-soup” models are utilized to investigate shifts in metabolic network topology [14,193], to predict active metabolic pathways, species contributions to metabolic functions [14], and interactions between microbiome and environment [192].
The second approach is based on model compartmentalization and can simulate species interactions. In constraint-based models, compartmentalization is implemented by creating compartment-specific metabolites. Transport reactions “shuttle” the same chemical compound by transforming it from one compartment to the other (e.g., the transformation of extracellular glucose to cytosolic glucose) [194]. Interactions across species are resolved by treating each species as an individual compartment and placing it in an exchange compartment corresponding to the microbiome medium [76,195]. The exchange compartment connects species compartments by transport reactions, which indicate species interactions [196,197]. Additionally, the contribution of biomasses from microbiome members to the total microbiome biomass is incorporated to account for microbiome growth [76,195,198].
A compartmentalized model can be built from available single-species models, but these are often not available for less characterized species part of microbiomes. In such cases, metabolic models can be reconstructed from MAGs (Section 5.3). Although metagenomic sequences are prone to errors and may be incomplete [132,133], the model building pipelines CarveMe [15] and gapseq [190] generate simulatable models. Both pipelines are based on “carving out” reactions that are not supported by metagenomic data from a universal model. The metaGEM pipeline by [134] provides a complete workflow to build models from raw metagenomic reads. MetaGEM uses CarveMe and can additionally estimate taxonomic microbiome composition and growth rates.
Automatically generated network reconstructions and models usually require manual curation. Recent studies have performed large-scale curations for the human gut microbiome by propagating refinements across multiple reconstructions [199,200]. Refinements can be done for individual species, but because metaomics data are usually available at the community level, it is worthwhile to refine the complete community model. Henry et al. [192], for instance, showed that merging reconstructions before gap-filling (step 2 of the reconstruction process) resulted in more correctly predicted interactions for a simple community model Henry et al. [192]. This indicates that models reconstructed and refined for single microbiome members may not mirror the “true” behavior in the microbiome.
Other metaomics data apart from metagenomics are useful in microbiome model reconstruction. Metabolomics data, for example, quantify enzyme activities, carbon utilization, fermentation products, and nutrient requirements. Metabolomics data can be retrieved in situ [201] and are already used in model validation [190,199,200]. Metaproteomics data seem to be a blank spot in microbiome reconstruction because only one reference making use of it could be identified [14]. Metaproteomics data could be utilized for model validation by comparing the occurrence of a metaprotein with the predicted activity of related model reactions or by comparing pathway mappings [202] with predicted pathway activities [203,204].
A challenge of microbiome modeling is the construction of biomass reactions for microbiome members. Adopting the biomass reaction from model organisms or “universal” biomass reactions is common [14,15,190], but biomass composition can vary between different strains and growth conditions influencing quantitative model predictions [69,70]. Single-cell and Flow cytometry based techniques could be of use to determine the total biomass and its macro-molecular composition for microbiome members to create biomass reactions [126,205]. Yet, biomass reactions are not always necessary to analyze species interactions. For example, graph-based identification of potential metabolic interactions is independent of biomass reaction or objective function and feasible for qualitative predictions [197].
Analysis methods for constraint-based microbiome models comprise graph-based analyses, optimization-based approaches, dynamic analyses, and spatiotemporal analyses [18,195,206]. Optimization-based approaches typically extend the linear optimization problem from FBA (Section 6.3.4) by additional constraints for microbiome composition and microbiome growth. The extended optimization problems are not necessarily linear and commonly solved by iterative optimization runs. For example, a first optimization could determine the maximal microbiome growth rate, which is incorporated into the second optimization run to determine the growth rates of microbiome members [196,198]. Koch et al. [76] simplified their optimization by assuming balanced growth (i.e., all species have the same specific growth rate) and fixing parameters such as microbiome composition (e.g., from experimental data) or microbiome growth rate (e.g., to dilution rate of a continuous bioreactor). The resulting optimization problem becomes linear and can be analyzed by FBA [76]. Optimization-based approaches are generally used to simulate metabolic fluxes, microbiome interactions, microbiome composition, or growth rates using compartmentalized microbiome models [206]. Overviews on all analysis methods are given in [18,195] and an evaluation of methods and software can be found in [206].
7.1.3. Reconstruction of Signaling Networks
Signaling and regulation networks are more complex than metabolic networks: They can operate on different timescales (from ms to hours), involve heterogeneous molecules (e.g. 2nd messengers), and contain multiple modification sites. As stated in Section 6.3.5, it is infeasible to model every possible state of signaling molecules explicitly. This problem can be mitigated by creating less resolved models [41], for example, by considering one activation state per protein [207]. However, this circumstance and the lack of standardized and automated protocols for constructing and annotating coarse-grained models complicates the integration of omics data.
The rxncon framework (Section 6.3.5) provides some benefits that could foster the standardized reconstruction of genome-scale models for host signaling and regulation (Section 7.1.4). Analogous to the reconstruction of metabolic models, rxncon models are reconstructed in four steps [208]:
- Draft Reconstruction: Networks are reconstructed in the context of macroscopic behaviors in response to stimuli. Firstly, the inputs and outputs of interest are defined, which helps to restrict the scope of the reconstruction. Secondly, the molecules propagating signals from input to output are identified. In addition, information on the sequence of interactions should be collected [208]. Data on molecular interactions are determined experimentally [144] or predicted [209,210] and are available in interaction databases such as String [162] and scientific literature [42,208]. In non-specific interaction networks (e.g., if retrieved from a database), algorithms can determine potential connections between inputs and outputs [211]. For well-investigated processes, existing signaling networks are available in pathway databases [120,122,212,213,214] and can serve as templates [42]. The result of the first step is an interaction network specific to the defined scope.
- Rxncon model implementation: Firstly, the elemental reactions, involved molecules, and resulting states need to be defined. Secondly, the sequence of signaling events is implemented by defining the contingencies (i.e., conditions) for elemental reactions. Information from expression arrays, knock-out screenings, (meta)omics analyses, databases, and literature provide the required information [41,43,208]. The result of this step is a rxncon model, which is comparable to a metabolic network in the reconstruction state. It represents an interaction network with causal relationships and thus could be analyzed by graph methods [42].
- Boolean model implementation and
- Model validation: Rxncon models can be compiled into Boolean models, which can be validated on experimental data (e.g., reproduction of input-output behavior or activation of internal nodes). If model predictions are not consistent with data, model building is re-iterated from the first or second steps. Additionally, it is possible to compile a rxncon model into a rule-based model and subsequently to an ODE model [208].
The complete process relies on manual curation and Romers et al. [208] even suggest revisiting original data from primary literature. This effort is worthwhile because the resulting rxncon models include detailed mechanistic information and model elements can be annotated and integrated with high-throughput data (e.g., phosphorylation sites with phosphoproteomic data). Furthermore, models are modular [178,180], implying that elements from existing rxncon models could be used to compose new ones, which is common practice in constraint-based modeling [215,216]. This would result in a more standardized and accelerated approach to creating models for cellular signaling. Additionally, the illustrated workflow has been proven practical by reconstructing and simulating a genome-scale model for signaling of the yeast cell cycle [178].
7.1.4. A Perspective for Reconstruction of Signaling in Microbiomes
Currently, there is no mechanistic model of microbiome or host signaling comparable to the size or resolution of genome-scale metabolic models. As a comparison, genome-scale metabolic modeling was initiated between 1999 and 2004 ([176]). If the recently published yeast model [178] is considered the first genome-scale model of signaling, genome-scale modeling of signaling and regulation is behind by more than 15 years.
Prospectively there is much to catch up on: Standard workflows and formats would need to be established, more reference reconstructions of single species or tissue networks need to be available and databases such as BiGG or ModelSEED [215,216] need to be created for reusable model elements to facilitate automation of the reconstruction process. Even with these tools at hand, retrieving experimental data for signaling and regulation is a bottleneck for modeling. Mechanistic models of signaling and regulation will likely stay qualitative or semi-qualitative for the next years, simply because kinetic parameters are not available [42,178].
The majority of known interactions in microbiomes occur by metabolite exchanges and are already covered by constraint-based models. Modeling of signaling systems in microbiomes such as quorum sensing could potentially be of interest, especially due to its relevance for microbiome engineering [217]. However, interactions of microbial metabolites and host signaling are of major importance for many diseases [58] and will potentially be the focus of future research. Ultimately, the goal is to connect models of metabolism and signaling for microbiomes and hosts on the genome-scale, to integrate models and (meta)omics data.
7.2. Parameter Estimation, Model Contextualization and Model Reduction
The previous sections introduced different aspects of model building; this section aims to illustrate how to tune them to experimental data (parameter estimation), adapt them to specific scenarios (contextualization), or reduce them to essential parts (reduction).
7.2.1. Parameter Estimation
Parameters are the set screws for tuning the behavior of a model. Determining these parameters from experimental data is a challenge. One parameter property is identifiability describing whether a parameter can be (uniquely) determined if all required data would be available. Because biological data are noisy, identifiability also assesses the uncertainty of a parameter value, usually based on the number of measurements and their variance [218].
The Michaelis-Menten kinetics, for example, is a simple model applicable to many enzyme reactions (Equation (4)) [219].
The model calculates a reaction rate v from the substrate concentration and contains enzyme parameters representing the maximal reaction rate and the Michaelis constant .
To determine parameters, model predictions need to be compared to experimental data. In the Michaelis-Menten example, the initial predictions are generated using arbitrary parameter values. The experimental data would be generated in an enzyme assay and comprise measurements of the reaction rate v for a range of substrate concentrations .
The difference between prediction and experimental value (i.e., the error or residual) determines a metric quantifying the disagreement, for example, the root mean squared error [218,220]. Optimization-based approaches iteratively adjust the parameters to minimize this disagreement. In the Michaelis-Menten model, and would be adjusted until the disagreement between prediction and data is minimal. For large and non-linear model equations there can exist multiple sets of parameters satisfying the objective of a minimal disagreement (i.e., local optima) [218]. The described process is also termed parameter fitting or model training and is employed similarly in statistical modeling.
Dynamic systems can be described by differential equations, which predict evolutions of state variables (e.g., metabolite concentrations) over time utilizing numerical integration. Accordingly, model parameters of ODEs are fitted (and validated) on time series data [168]. Additionally, initial conditions are necessary for numerical integration [168]. For example, investigating the dynamics of a receptor system during its stimulation would require the quantities of all signaling molecules (i.e., the state variables) before stimulation as an initial condition. It is usually impossible to measure all initial state variables, but unknown initial conditions can be optimized by fitting model predictions to time-series data [20]. More information on parameter estimation in ODE models can be found in Ashyraliyev et al. [218].
Boolean models do not contain any parameters but require initial conditions for dynamic analysis. Deducing a binary state from continuous data demands a threshold value. This can be determined experimentally by assessing the respective signaling compound’s activity or by comparing phenotypes (e.g., inactive in wild-type individuals, active in disease) [221]. Romers et al. [180] used an approach independent of experimental data. They initialized all model elements to an “off-state” and let the model evolve into a steady state, which was used as a new initial condition to analyze dynamic behavior after stimulation [180].
Parameters in “default” constraint-based models are metabolite stoichiometries and rate constraints. Both parameter types can be adjusted by model training as described above [33] but are usually determined directly from experimental data. Reaction stoichiometries are configured during network reconstruction according to biochemical knowledge, similar to stoichiometries in the biomass reaction derived from the macromolecular biomass composition. Biomass composition is specific to culture conditions and should be adjusted to each simulated condition to facilitate accurate predictions. Rate constraints of metabolic reactions can be fixed to measured metabolic fluxes [72,74,141]. If constraints should be “loose” they can be set to maximal rates (e.g., measured maximal uptake rates of metabolites). Lastly, models may contain reactions abstracting biological processes. A good example is the ATP-maintenance reaction describing the “drainage” of ATP for biomass maintenance. Its lower rate boundary is the ATP-maintenance coefficient quantifying this ATP requirement [173]. Similar to biomass composition, ATP maintenance may greatly influence model predictions [75]. Such coefficients can be estimated from growth experiments and are subject to growth conditions as well [71,149].
7.2.2. Contextualization
Genome-scale reconstructions contain all possible biochemical processes encoded by the genome. However, most processes are regulated on the gene or post-translational level and are only active in specific conditions [173,184]. Contextualization is a variant of data integration that adjusts a model to experimental data so that it reflects a specific biological scenario such as a growth condition or a tissue type. Contextualizing a model for growth on a specific substrate, for example, could be done by introducing measured reaction rates and a biomass reaction for this scenario or removing inactive metabolic reactions from the model.
Contextualization is useful because contextualized models are less general and may exclude implausible predictions. Most computational models are not on the genome-scale and thus inherently context-specific, because they were created with a specific biological context in mind. Constraint-based models currently represent the only available type of genome-scale model, thus only examples for this model framework are covered. As soon as more genome-scale signaling models become available, they will also require contextualization. Potentially, many methods targeting constraint-based models could become relevant for signaling and regulation.
The input for contextualization methods is a genome-scale model, (meta)omics data, information from biochemical databases, and mechanistic knowledge. (Meta)omics data are mapped to model elements and used to knock out (switch-based) not-supported metabolic reactions or constrain them (valve-based) [222]. Contextualization is (semi-)automated and requires annotation of model elements with standard database identifiers to facilitate data mapping.
An example of switch-based contextualization is tINIT [223,224], which scores enzymes and metabolites according to transcriptomic, proteomic, and metabolomics abundance data. Afterwards, it extracts a sub-network that includes reactions supported by the data and excludes reactions with low evidence. Additionally, metabolic functions that should be included in the output model can be specified. The output model contains fewer reactions than the original model.
A second example are methods such as GECKO [85] and sMOMENT [33]. These impose protein allocation constraints on the input model by adding reactions describing the availability of enzymes. Total protein content, absolute proteomic abundances of enzymes, and values for enzyme turnover ( values) are used to constrain the limits for enzyme usage. In addition to metabolic fluxes, enzyme-constrained models can also predict enzyme usage. Generated output models contain more reactions than the input.
Switch- and valve-based methods generate output models in standard formats, that can perform standard analyses, which does not apply to all contextualization methods (e.g. [225,226]). More information is available in reviews by Opdam et al. [227], Kerkhoven [228]. The introduced methods are tailored to single-species models, but contextualization has also significance for microbiome modeling. One example is the investigation of metabolic interactions, which depend on the potential to exchange metabolites across cells. Less probable interactions can be ruled out by contextualization and could make quantitative predictions more accurate.
Contextualization could be applied in microbiome modeling in two ways: Firstly, single species models can be contextualized with metaomics data before being assembled to the microbiome model and secondly, contextualization could be applied to the readily assembled microbiome model. Machado et al. [15] and Zimmermann et al. [190] essentially apply the first approach in their “model carving” methods gapseq and CarveME. Like tINIT, a universal, non-specific microbiome model can be contextualized with metagenomic data. Metatranscriptomic and –proteomic data could be applied to exclude non-expressed metabolic reactions. Relatively quantified molecular abundances could be applicable in tINIT-like methods and usable to compare microbiomes across conditions. Creating enzyme-constrained models from metaproteomic data poses some difficulties because absolute quantification of metaproteins is not very reliable (Section 5.1). Furthermore, a strategy to handle metaproteins that cannot be classified on the species level (i.e., protein groups) would be required (Section 5.2). Optionally, uniquely identifiable proteins could be used to impose at least some protein constraints. Another problem is that required values are not available or less accurate for enzymes of less characterized species. Innovations in machine-learning based prediction from protein sequences could alleviate this issue [160]. Lastly, model size needs to be considered because microbiomes may contain several hundreds of species. Microbiome models can thus become very large, which can cause long calculation times for analyses [76,229]. Enzyme constraints bloat the number of model elements [33] and could be less preferential in contrast to tINIT-like methods, which reduce model sizes [223,224].
The second strategy of contextualizing assembled microbiome models could apply to unified microbiome models. Tobalina et al. [14] created their context-specific model from metaproteomic data in a bottom-up manner. Creating a unified microbiome model from metagenomic data and “carving” it with expression data could be another viable approach. Contextualization of compartmentalized microbiome models would require the extension of existing algorithms for single-species models. Microbiome model contextualization would only be beneficial if the model was assembled before gap-filling as done by Henry et al. [192]. This approach may result in more interactions across microbiome member models that could be adjusted by contextualization.
7.2.3. Model Reduction
Together with the advantages of genome-scale models come disadvantages related to large model sizes and insufficient performance of model analyses. Furthermore, it is difficult to comprehend a model with more than 13,000 metabolic reactions such as the general Human1 model [230]. A step beyond contextualization is the reduction of such large models to a minimal size while preserving key qualities of its template [229]. Potential applications of reduced models are, for example, education, tool benchmarking [173], kinetic modeling [229], multi-scale modeling, construction of microbiome models containing many species [76] and model predictive control (MPC) (Section 8.3.3).
Model reduction can be performed manually. Orth et al. [173], for example, derived a reduced model representing the central metabolic functions of E. coli from a genome-scale template. Automated reduction methods are preferred, because they require less manual work, and updates in templates could be propagated automatically to reduced models [229]. Erdrich et al. [231] developed an algorithm that uses a template model, mandatory reactions, metabolites, and phenotypes as input. It removes unprotected model elements in the first step and subsequently compresses the pruned model by lumping together reactions while preserving phenotypes of the template [231]. Another approach by Koch et al. [76] reduces compartmentalized community models. The authors first determined conversions of microbial substrates to products (net conversions) for single species models and reduced these models to exclusively represent these conversions. The reduced models were then assembled into a microbiome model and can be utilized to analyze species interactions and microbiome composition.
Model reduction is also of interest for rule-based models, mainly to reduce model sizes when compiled into ODE models. Borisov et al. [177], for example, determined protein domains that are modified independently of their scaffold protein (i.e., the protein inheriting all domains). These domains can be modeled independently, thereby reducing the number of possible microstate combinations. An algorithm by Danos et al. [232] extracts a reduced differential equation model from a rule-based template. Their reduced models provide solutions that are linear combinations of the solutions from the uncompressed ODE model.
Machine learning provides capabilities to reduce any model type to a black box model. Black box models only consider inputs and outputs of a biological system, while omitting mechanistic information. Wagner and Schlüter [233], for example, used a deep neural network to reduce a macroscopic model for the biogas process (Section 8.1). To this end, they trained the neural network on simulation data of the original model and could reproduce new simulation results with high accuracy. The resulting black box model was then used with an MPC to control methane production.
7.3. Summary – Section 7
Mechanistic models integrate biological data and represent knowledgebases. They can be reconstructed coarse-grained and with specific applications in mind or on the scale of complete genomes. The reconstruction process of genome-scale metabolic models is a standardized process applicable to building complete microbiome models, while genome-scale reconstruction of signaling networks is still in its infancy and restricted to intracellular processes. During model building, model parameters quantifying specific parts of a system are fitted to experimental data. Furthermore, the structure of a “general-purpose” genome-scale model can be contextualized to better represent a specific biological scenario. It is also possible to reduce models to a minimal size while preserving key features of the template model, which is mainly relevant to improving the performance of model analyses and simulations. Lastly, metatranscriptomic and metaproteomic data are infrequently exploited in these approaches.
8. Examples of Model-Based Microbiome Prediction, Optimization, and Control
This section approaches the last aspect of systems biology as defined in Section 3, namely the application of computational models to predict (Section 8.1), optimize (Section 8.2) and control (Section 8.3) microbiomes. These terms are differentiated because model predictions are applied to different extents. Firstly, predictions are mere explanations of a biological system that are used to build or validate a hypothesis. Secondly, optimization applies model predictions to a microbiome to achieve a desired outcome. Thirdly, control applies model predictions to determine strategies for achieving a desired dynamic microbiome behavior. The subsequent sections contain examples of how models are applied according to these three categories. Due to many other reviews containing microbiome modeling examples [18,185,186,187,234], this section was limited to a few examples on gut microbiomes, drug discovery, and from biotechnology.
8.1. Predicting and Understanding Microbiomes
The human gut microbiome is of major importance for human well-being but is also involved in diseases like inflammatory bowel disease (IBD). IBD has two subtypes, Crohn’s disease (CD) and ulcerative colitis (UC), and is characterized by chronic inflammation of the gastrointestinal tract. Further hallmarks are alterations of interactions between the immune system and microbiome, also influencing microbial interactions and microbiome composition [235].
Aden et al. [16] investigated the microbiome of IBD patients during treatment with anti-inflammatory anti-TNF. They acquired taxonomic microbiome profiles from amplicon sequencing of fecal samples and built patient-specific constraint-based microbiome models containing the detected species. The authors found that IBD microbiomes with fewer predicted metabolic interactions might reduce therapeutic success. Furthermore, the microbiome models identified butyrate metabolism as a significantly altered pathway and predicted a restoration of metabolic exchanges in remitting microbiomes.
Similarly, Marcelino et al. [17] performed a meta-study evaluating metabolic interactions in diseased gut microbiomes. They aimed to identify disease-specific disruptions of metabolite exchanges. The authors reconstructed microbiome models from fecal metagenomes and simulated microbiome growth. Based on simulated metabolic fluxes, they determined the capability to exchange metabolites across species for healthy and disease samples. They found important metabolites, such as thiamin and short-chain fatty acid precursors, to be significantly altered between healthy and diseased samples. Furthermore, they predicted metabolites that were previously shown to be disease-related, including known biomarkers for disease progression. In a case study for CD, the authors investigated the causes of altered metabolic exchanges of , which can cause gut inflammation. Resultingly, the authors determined a disbalance in -producing and consuming species to be the origin of altered exchanges.
Another aspect of IBD is the modulation of host signaling by the microbiome. Andrighetti et al. [209] aimed to find microbial proteins that could interfere with cellular signaling and modulate gene expression in humans. To this end, they developed a workflow integrating metaproteomic data and several other data resources. In their workflow, putative interactions between metaproteins and human receptor proteins are predicted, embedded into a graph model of signaling, and propagated to the affected genes. The authors applied their approach to CD and proposed microbial metaproteins that could modulate the expression of genes involved in autophagy pathways. A similar approach by Zhou et al. [210] aims to predict interferences with host signaling by microbial metaproteins homologous to endogenous effector proteins. This method elucidated the role of non-annotated metaproteins and indicated that metaproteins might modulate known drug targets in humans.
This section is closed with an example from biotechnology. The anaerobic digestion model 1 (ADM1) is a macroscopic process model routinely applied to monitor biogas production. ADM1 describes the step-wise degradation of complex organic matter to biogas (CO2 and CH4) by microbial processes utilizing differential and algebraic equations [236]. The model includes biochemical reactions for conversion of organic matter and physicochemical reactions (e.g., ion association/dissociation, gas-liquid transfer). Seven biochemical reactions modeling the degradation of key compounds are linked to the accumulation of microbial biomass. ADM1 was established as a standard to simulate and benchmark degradation processes and was enhanced extensively [237]. Weinrich et al. [238], for instance, extended the ADM1 model with genome-scale metabolic models of a methanogenic (i.e., biogas-producing) microorganism. The resulting multi-scale model reproduced simulation results of the standard AMD1 model and predicted metabolic fluxes on the sub-cellular scale. Weinrich et al. [238] proposed that such models will facilitate the integration and interpretation of time-resolved metaomics data from biogas plants, estimate process yields, determine interventions for process optimization, and identify signals indicating reactor breakdowns.
8.2. Optimizing Microbiomes
Model-based optimization of microbiome behavior is established differently across scientific domains. Many examples can be found for biotechnological applications: García-Jiménez et al. [18] listed six categories for model-based optimization in biotechnology, namely the production of chemicals, pathway distribution (i.e., division of labor), microbiome stability, medium composition, spatial organization, and flexible optimization goals. Additionally, the degradation of pollutants can be added to this list [239]. Model-based optimization in therapeutic contexts focuses on microbiome composition, microbiome host interactions [240,241], and drug target discovery [19]. However, computational approaches targeting microbiomes for therapy development are still in their infancy [242].
An example of optimization of microbiome composition and host-microbiome interactions is represented by Stein et al. [240]. It is known that the gut microbiome modulates the host immune system, and has an important role in host immune system development. Stein et al. [240] investigated the modulation of regulatory T-cells (Treg) by the microbiome, which can have anti-inflammatory effects in diseases such as UC. They studied different combinations of Clostridia strains to determine microbiome compositions that would stably colonize germ-free mice and maximally stimulate Tregs. To this end, they trained macroscopic ODE models on time-resolved microbial abundance data and population counts for Tregs. Using their model, they predicted promising strain combinations and validated these microbiomes in germ-free mice. Their approach demonstrates the potential of model predictions to design therapeutic microbiomes.
The application of a host-microbe model for drug target discovery is demonstrated by Curran et al. [19]. They aimed to inhibit the growth of the parasitic nematode Brugia malayi, which causes the tropical disease lymphatic filariasis. B. malayi lives in endosymbiotic relationship with bacteria of the genus Wolbachia. These bacteria are vital for the nematode’s fitness and thus a target for antibiotic therapy. To identify antibiotic targets in Wolbachia, Curran et al. [19] built a compartmentalized constraint-based model for nematode and bacterium. Moreover, they predicted the use of different metabolic pathways and performed in silico knock-out experiments. These experiments revealed growth-essential gene products suitable as drug targets. In experiments, three of these targets could be successfully inhibited by known antibiotics.
An example of optimized degradation of pollutants can be found in Xu et al. [239]. Using constraint-based microbiome models, they investigated the effect of the pesticide atrazine on interactions in soil microbiomes. They utilized metagenomic data to identify member species in soil samples and reconstructed microbiome models based on available reference genomes. A dynamic version of FBA was used for analysis and could predict an improved performance if the main degrading species grows in a microbiome compared to growth in isolation. Furthermore, supplementation with glucose resulted in increased degradation. These results were verified by growth experiments [239].
8.3. Controlling Microbiomes
Control refers to the regulation of a dynamic system to achieve a desired dynamic behavior of the system. Interventions to control a system are termed control strategies and can be applied to steer the behavior of microbiomes.
Controlling native microbiomes dynamically is difficult because environmental factors may dominate microbiome behavior and measuring native samples can be difficult. However, in biotechnological production processes the influence of environmental factors is minimized allowing for microbiome control. This section briefly introduces the concept of closed-loop control, discusses elements of closed-loop control concerning microbiomes, and emphasizes model-based control strategies for microbiomes with examples from biotechnology. Further information on this topic can be found in the review by Lee and Steel [243].
8.3.1. The Concept of Closed-Loop Control
Control strategies can follow a feedback structure (Figure 7) allowing it to affect a dynamic system, such as a microbiome. The system has a measurable output that should be controlled, for example, the concentration of a metabolite. The output response is affected by the system input, for example, the concentration of a specific nutrient. As the system is dynamic, the output may change over time. To validate that the output has a desired value, the output is compared regularly to a reference value. The difference between the measured output and the reference is the error. The error is fed back into a controller, which computes a system input according to a control algorithm. The controller tries to maintain a low error. If the error increases, the controller steers the system input to reduce the mismatch between output and reference. Because the controller closes the loop to the system, this feedback structure is named closed-loop control.
8.3.2. System Inputs and System Outputs of Microbiomes
Nutrient concentration was a previous example of an input for a microbiome, but any environmental factor can be altered to influence microbiome output. This includes pH, level of oxygen, temperature, or salinity. Additionally, population sizes of individual species can be targeted by the input. Population size can be increased by the expression of growth-inducing genes [244] or by directly adding a species to the community [245]. On the other hand, the population size can be decreased by initiating cell death [246], by introducing antibiotics, or targeted bacteriophages [247].
The control output is the response of the system to the input. Several methods exist to measure the output depending on factors such as the complexity of the community, the control goal, the measurement frequency, the economic cost, or the duration of measurements. Process parameters such as the pH value or oxygen concentration are easy, cheap, and quick to measure but do not give any insight into the microbiome. Other methods that are applicable on-line (i.e., "during cultivation") are flow cytometry or metabolomics. Flow cytometry can distinguish different strains using universal dyes, thereby giving an insight into microbiome composition [248]. GC-based metabolomics can be applied to measure gaseous metabolites during cultivation [249]. Metaproteomics in contrast are less suited for on-line measurements due to the extensive sample preparation but can resolve expressed enzymes.
8.3.3. Control Algorithms and Model-Based Control
The control algorithm determines how the controller steers the system inputs. The selection of the control algorithm depends on the system and the control goals. One of the most straightforward approaches is PID (Proportional, Integral, Derivative) control. A PID controller consists of three adjustable parameters for corrections based on the proportional, the integral, and the derivative term of the error value. Due to its simple structure, PID control is easy to implement without much knowledge of the system. However, the performance of the controller depends on the chosen parameters. Controller parameters can be tuned using a mathematical model of the system. This results in a more accurate parameter set without the need for extensive experiments. Bensmann et al. [250], for example, performed a comprehensive simulation study of biogas plants. They used an extended version of the ADM1 model to propose and test a PI (i.e., PID without the derivative term) feed-back control for the biological methanation of hydrogen.
Model predictive control (MPC) is an advanced control strategy for complex control goals or cases where multiple inputs need to be controlled. MPC is an optimal control strategy and, therefore, aims to optimize a given objective function, such as the population growth of a microbiome member. For the optimization, MPC uses a model of the system to predict the future system behavior over a finite time interval. Xue et al. [22], for example, used nonlinear MPC to control the anaerobic digestion process in biogas plants. For this, the authors used a reduced version of the AMD1 model. Because many state variables of the anaerobic digestion process are immeasurable, these values need to be estimated. To this end, the authors applied an estimation algorithm termed Unscented Kalman Filtering, which determines parameters based on available measurements [22,251].
MPC has also been applied in cybergenetic control. Cybergenetics regulates gene activity in genetically engineered microorganisms by external stimuli, such as light. In this way, metabolic functions or growth can be controlled. Espinel-Ríos et al. [20] performed cybergenetic simulation studies in which they optimized nianigrin production in a co-culture of engineered E. coli and yeast. Furthermore, the authors simulated a scenario where some process parameters were unknown. They trained a model using machine learning to estimate these parameters and adopted MPC to improve the predictions of this model. The same authors also implemented cybergenetic MPC for a lactate-producing E. coli culture in a bioreactor [21]. Here, a dynamic constraint-based model with protein resource allocation was used to control the expression of ATPase by light. This approach could also be extended to synthetic microbiomes, as stated by the authors.
MPC can also employ machine learning models, as highlighted by Wagner and Schlüter [233]. They discussed that mechanistic understanding of the controlled system is not relevant for controlling production processes. The authors demonstrated that a deep neural network trained on simulated data could accomplish similar precision as the ADM1 model. Additionally, the neural network could be used with MPC to control methane production. This approach could also be applied to train machine learning models with experimental data.
8.4. Summary - Section 8
The knowledge compiled in computational models can be applied to predict and understand processes in microbiomes and their hosts. Furthermore, model predictions are applied to optimize microbiomes for several biotechnological and therapeutic purposes. Thirdly, microbiome computational models aid microbiome control. Furthermore explained were the concept of feedback control and how models are applied to tune parameters of control algorithms, in simulation studies, or model predictive control.
9. Microbiome Modeling Requires Standards, Software and Repositories
Standards facilitate the reuse of data, models, and simulation results. This section describes the concept of FAIR (findable, accessible, interoperable, and reusable) guidelines for research data and expands to the standards of the modeling community. Furthermore, software tools and repositories used in the modeling domain are introduced. More information on standards in systems biology is given in articles by Waltemath and Wolkenhauer [252] and Stanford et al. [253].
9.1. FAIR Data
Biological data are generated at a high pace and good data management is required to facilitate the reuse and integration of data. In 2016, the FAIR guidelines were published to improve existing issues in research data management and stewardship [254]. These principles apply to research data, as well as algorithms, software, and workflows [254]. Additionally, FAIR guidelines apply to metadata, which is information associated with the “actual” data or software. Metadata describes, for example, the subject of research, data origin, or time of generation. Finding, retrieving, and integrating big amounts of data, for example, to build genome-scale models requires automation. Hence, another motivation for having FAIR data and software is to provide minimal requirements facilitating automation.
Four main principles are covered by FAIR (explanations are taken from Boeckhout et al. [255])):
- Findability (“Datasets should be described, identified and registered or indexed in a clear and unequivocal manner”
- Accessibility (“Datasets should be accessible through a clearly defined access procedure, ideally using automated means. Metadata should always remain accessible.”)
- Interoperability (“Data and metadata are conceptualized, expressed and structured using common, published standards”)
- Reusability (“Characteristics of data and their provenance are described in detail according to domain-relevant community standards, with clear and accessible conditions for use”)
FAIR is highly relevant for research, but factors such as incomplete metadata and insufficient reporting of parameters and initial conditions hamper the reusability of biological and biomedical data [256] or computational models [257].
FAIRDOM (https://fair-dom.org/about) is a consortium supporting scientific communities in implementing FAIR guidelines. They provide FAIRDOMHub [258], a web-based repository to publish scientific data, protocols, and models, as well as FAIRsharing (https://fairsharing.org/), a web tool for searching community guidelines and scientific databases.
9.2. Initiatives and Community Guidelines
While FAIRDOM is a more general consortium, the COmputational Modeling in BIology Network (COMBINE) is an initiative establishing standards on the level of the modeling community [259,260]. COMBINE coordinates standards for exchange formats and modeling languages (e.g., SBML, see below) and organizes regular community meetings [259]. Another initiative cooperating with COMBINE is the Consortium for Logical Models and Tools (CoLoMoTo) [261]. CoLoMoTo has similar aims as COMBINE but specializes in logical modeling (including Boolean modeling).
COMBINE supports guidelines for metadata on model elements and simulation experiments. Model elements usually represent biological entities or relations between them (e.g., in chemical formulas) and their meaning can be described with metadata. Metadata links model entities to unique identifiers for biological entities. The association of model entities and metadata is termed model annotation, which is important for data integration (Section 3) [262,263]. MIRIAM (Minimum information requested in the annotation of biochemical models) provides guidelines for these annotations aiming to improve model reusability. It specifies model documentation, correspondence between models and articles, utilization of machine-readable exchange formats, and the quality of model annotations [263].
MIASE (Minimum Information About a Simulation Experiment) is complementary to MIRIAM and provides guidelines facilitating the reproduction of simulation experiments [264]. MIASE-compliant reporting includes the specification and definition of used models, precise descriptions of simulation steps, and descriptions of the analysis of simulation data (e.g., post-processing steps) [264].
9.3. Languages for Modeling and Exchange Formats
The interoperability principle in FAIR specifies the use of formal languages to express knowledge [254]. Systems biology has adopted this principle to describe model structures and simulation experiments.
The systems biology markup language (SBML) is a widely used standard in the metabolic modeling community [265] and one of the languages maintained by COMBINE. It builds on the extensible markup language (XML) and describes model structures while being agnostic to any software or analysis method [266]. A metabolic model, for example, is represented by semantic elements describing biological entities (reactions, metabolites, gene products, compartments) and default parameters. These semantic elements are organized hierarchically, and specific information is assigned by element attributes. An important aspect of SBML is the use of systems biology ontology (SBO) terms to characterize model elements (e.g., mathematical expressions, metadata, or physical entities) [266].
SBML is a modeling language and exchange file format at the same time. Furthermore, it allows the implementation of MIRIAM guidelines by providing means for model annotation, fostering the reusability of models. For annotation, the resource description framework (RDF) is utilized, supporting references to multiple (biochemical) databases [266]. Additionally, the current SBML version 3 is designed in a modular manner, providing extensions to the core language for the representation of constraint-based, ODE, logical, and rule-based models, as well as means to store network layout information [267].
Rxncon is another language introduced earlier (Section 6.3.5) and is dedicated to describing the structure of signaling and regulatory networks [178,208]. The language can represent biological entities, supports an iterative model-building cycle, and compiles biological knowledge into standardized knowledgebases [208]. Models in rxncon language are built and stored in the SBtab spreadsheet format [178,268]. SBtab files can be exchanged and are compliant with MIRIAM, as metadata on model elements could be added to model spreadsheets [268]. However, rxncon defines SBtab columns for annotation less strictly compared to SBML [208,266] (SBML has specific RDF tags containing uniform resource identifiers, whereas rxncon does not have such specifications). Models in SBtab rely on rxncon software to be compiled into executable Boolean or rule-based models and are thus not software agnostic. A drawback of rxncon is that it is purely qualitative and kinetic parameters need to be added manually when transformed to a quantitative rule-based model [208]. However, as it is dedicated to model genome-scale signaling networks, it will be hard to retrieve all those parameters experimentally anyway.
To our knowledge, rxncon is the only language dedicated to supporting genome-scale signaling models. However, it is not established in the modeling community, as it is not part of COMBINE or CoLoMoTo and appears less often in publications than SBML (querying PubMed for “SBML” and “rxncon” resulted in 478 and 7 hits respectively; date of access: November 29, 2023).
9.4. Software
Reconstruction and modeling are linked to using programming languages or a command line. Python and Matlab are the most common languages and incorporate features for reconstruction and modeling by importing available software packages. Additionally, a few applications with (web-based) graphical user interfaces (GUI) are available.
According to a community survey from 2019 [265], the most used platforms for constraint-based modeling are COBRA and COBRApy, which are implemented for the programming languages Matlab and Python, respectively [270,271]. Both software packages provide core functionalities, standard analysis methods, and advanced methods for data integration, in the case of COBRA. Additionally, packages for network reconstruction and model curation [15,190,272], data integration [33,85,223,224] and advanced simulations [273] are available. In addition, a variety of tools for microbiome modeling exists, of which 24 have been evaluated for usability and reproducibility in a recent publication [206]. The most used pipeline with web-based GUI according to Carey et al. [265] is KBase [274], which allows composing modeling and simulation workflows from workflow modules. A software package/application with a user interface helping to implement MIRIAM guidelines in genome-scale metabolic models is MEMOTE [191]. MEMOTE facilitates quality control for annotations and model consistency and provides a framework to set up version-controlled repositories for model development.
A list of tools for modeling and simulation of Boolean models is available on the CoLoMoTo website (http://www.colomoto.org/software/). Featured software and languages are, for example, PyBoolNet (Python) [275], CellNetAnalyzer (Matlab) [276] and BoolNet (R) [277]. Rxncon comes with software packages based on Python, containing functions to compile rxncon models into executable Boolean or rule-based models [208]. For simulation, rxncon depends on the BoolNet package. An example of a web-based application for signaling and regulation is the Cell Collective fostering model building, simulation, analysis, and integration of knowledge [278].
Building and simulating ODE models is natively supported in Matlab but does not adhere to any modeling language. The Simbiology Toolbox is a proprietary Matlab plugin focused on dynamical pharmacological modeling (https://www.mathworks.com/products/simbiology.html), which can interact with SBML files. CellDesigner is a free software providing a GUI and utilizing the Systems Biology Graphical Notation (SBGN) for “drawing” biological process models [279]. It allows for dynamic simulations if kinetic equations are provided, supports MIRIAM-compliant annotation, and is SBML-compatible. Open-source packages for dynamical modeling are the GUI-based OpenCOR [280] and COPASI [281], as well as Python-based PySceS [282] and PySB [283]. Another notable software is Morpheus, which facilitates the modeling of multiscale and multicellular systems [284].
9.5. Repositories
Repositories are platforms to store and share data or models. They are accessible through websites or programmatically via application programming interfaces (API). Repositories for experimental data are important for network reconstruction, validation, refinement, and contextualization of models. A list of biochemical databases for model annotation is, for example, available in the supplementary of Lieven et al. [191]. Other resources can be found on the FAIRSharing platform, which indexes domain-specific databases, for example, STRING for protein-protein interaction networks [162], BacDive for growth screenings [68], Sabio-RK [285] and BRENDA [117] for enzyme constants or MGnify for microbiome sequence analysis and storage [286].
Models are published in dedicated repositories or on GitHub (e.g., https://github.com/SysBioChalmers/Human-GEM), an online platform for version-controlled projects commonly used in software development. BioModels is one of the biggest dedicated model repositories. It contains different model types, models are partly curated, and provides a version control system [287]. BiGG is a fully curated repository providing constraint-based models and model elements [215]. Model elements are aligned to a common namespace (i.e., a naming scheme) and contain cross-references to biochemical databases. MetaNetX is another database for constraint-based models, which collects its entries from various resources (including BiGG) and aims to unify models under the MNXref namespace [288].
The list of explicit microbiome models in public repositories is short. Except for BioModels, all mentioned repositories contain single-species models. Using the keywords “microbiome” and “microbial community” in BioModels resulted in six models representing more than one species (date of access: August 4, 2023, Table S2). However, a common strategy for metabolic models is to make models of individual species available and share the code to assemble microbiome models (e.g., Heinken et al. [200] and Ankrah et al. [289]).
9.6. Remarks on Languages and Software for Community Modeling
Even though several initiatives and standards are set up, modeling is not FAIR. A survey among 89 members of the constraint-based modeling community showed that only 56% were aware of MIRIAM [265], which is in accordance with Lieven et al. [191], who demonstrated that many constraint-based models lack annotation or semantic SBO identifiers. MIASE was familiar to less than 25% of constraint-based modelers, pointing out potential issues in reporting simulation experiments. This hypothesis applies at least to kinetic models, as shown by Tiwari et al. [257]. They tried to reproduce 455 kinetic models from the BioModels repository, which was possible for only 49% based on information from respective publications. The main reasons for irreproducibility were inconsistencies in model structure, as well as insufficient reporting of initial values and parameters.
Kim et al. [290] showed that irreproducibility also occurs for bioinformatics software: Conflicts of operating systems, dependency issues, and poor documentation are common examples researchers must face when using foreign code [290]. Additionally, researchers without advanced training in programming or bioinformatics will quickly surrender, as resolving these issues requires some debugging experience. A resolution to this issue could be the use of lightweight software containers [291]. Such containers are isolated from the hosting system and run their own operating system, preinstalled dependencies, and configurations, allowing to share containerized software (https://docs.docker.com/get-started/) [291].
Reusability ultimately affects microbiome modeling, because microbiome models can consist of individual sub-models (from third parties) that need to be reusable. Even if sub-models are annotated, metabolite identifiers can be ambiguous [292]. Furthermore, there is no standard namespace for model elements, and merging models from different sources can be problematic if no common identifiers or annotations are included (i.e., if the models use different namespaces) [293]. To alleviate this problem, MNXref aims to provide a common namespace by connecting several database references to unique identifiers usable for model annotation [288]. Additionally, connecting different species models requires annotation of model taxonomies.
The MEMOTE software tests for annotations from a list of identifiers that should be ideally included in a model (Table S3). Based on own experience and [191,294], the recommended set of identifiers for minimal annotation includes:
(Meta)omics data should include the respective identifiers to facilitate data integration. Following the suggested set of minimal annotations, metabolomic data should include InChI, ChEBI, and MNXref identifiers, and genomic, transcriptomic, or proteomic data should include EC numbers, MNXref identifiers, and UniProt Accessions.
Carey et al. [265] pointed out that community standards are inherently lagging behind new analysis methods. This could also be a reason that most available genome-scale community models need to be assembled from their member species and require the original code to assemble microbiome models. Nevertheless, SBML can represent compartmentalized metabolic community models, but there is still a lack of standards for other model types, e.g., individual-based models [297]. A future solution could be the addition of new SBML extensions to keep up [265].
Prospectively, it will take further time and effort to assimilate guidelines into the modeling community and minimize reproducibility issues. Giving more incentives by rewarding model annotation, stricter requirements by journals, providing user-friendly annotation tools, peer-reviewing models and software, and coordinating standardization efforts are examples of potential large-scale solutions to the problem [256,257,265,298].
9.7. Summary - Section 9
Findability, Accessibility, Interoperability, and Reusability are principles that facilitate the management and stewardship of data, software, and algorithms. Initiatives such as FAIRDOM, COMBINE, and CoLoMoTo promote these principles and coordinate efforts for standardization in the modeling community. This results in established languages/exchange formats for models and simulation experiments, such as SBML and SED-ML, as well as guidelines for model annotation and reporting of analyses (i.e., MIRIAM and MIASE).
10. Discussion
The holistic approach of systems biology paves the way to understanding microbiomes. Every aspect of systems biology, i.e., measuring metaomics data, data integration, data analysis, and modeling is linked with a vast amount of challenges and options. Only specialists can overview the challenges and options in their research area. At the same time, it is counterproductive to study them in isolation from other areas. Thus, researchers with interdisciplinary education are needed to mediate between specialists and keep the flow of systems biology running. This was the intention behind the four goals for this review.
Improvements in methods and technology of metaomics will provide standardized workflows and more reliable data. Better data will direct the application of metaomics towards routine diagnostics and monitoring of technical processes [10,28,299], but will also be profitable for microbiome model quality. Furthermore, technologies such as single-cell omics will be applied more frequently [126] and could provide a better separation of taxa for subsequent metaomics analyses. With the lower taxonomic complexity of microbiomes, challenges such as taxonomic and functional annotation of metaproteins could be alleviated. Flow cytometric separation of cells and subsequent cultivation and observation could also contribute to the characterization of microbiome members [126]. Another alley of interest is the parallel measurement of different "omes" over time by applying time-resolved multi-omics to microbiomes. This would provide more mechanistic insight into the dynamics of microbiomes and facilitate the development of dynamic models integrating metabolism, signaling, and gene regulation.
The examples introduced in Section 8 show the potential of microbiome modeling for medical research and biotechnology, but modeling is not fully established in the standard workflow of metaomics data analysis. A potential reason for this could be the lack of accessibility as modeling mostly relies on bioinformatics experience. Furthermore, there is a lack of standardization even for bioinformatics workflows for data analysis, which is slowly counteracted by initiatives and ring trials such as CAMPI3 (https://metaproteomics.org/campi/campi3/). The cooperation of lab experts and bioinformaticians/modelers is one solution to establishing modeling and has already been realized by many research groups. The second option is to provide user-friendly software for modeling, such as KBase [274]. A drawback of such software is that it takes time to implement new features. For example, KBase is focused on processing genomic data but has currently no features for handling metaproteomic data. In the future, web-based software for modeling and data repositories could be merged into organism or process-specific hubs. These hubs could provide reference (meta)omics datasets, for example, from healthy and diseased individuals. Additionally, workflows could be provided to integrate these reference datasets with newly generated data and computational models.
The realization of guidelines such as FAIR facilitates a landscape of data and model repositories and available software for microbiome modeling. Nevertheless, standards are not fully established in modeling communities and many are unaware of their existence. As a result, many models are not reusable for data integration because of missing or not unified annotations and simulation results are not reproducible. In addition, standards naturally are behind emerging analysis methods, which ultimately affect microbiome modeling. Therefore, it is often the case that original code from publications needs to be executed. However, software is affected by irreproducibility as well. Containerizing software for modeling or implementing web applications are short-term perspectives to make microbiome modeling easily accessible for researchers. In the long run, standards need to be assimilated by scientific communities, which could be facilitated by repositories and journals giving incentives for the usage of standards, as well as peer-reviewing of models and software.
Prospectively, microbiome modeling will develop new ways to exploit metaomics data. Metaproteomics data, for example, could be used to contextualize microbiome models. For example, achieving reliable absolute quantification of metaproteins could provide data to model protein resource allocation in microbiomes. Alternatively, algorithms could be developed to deal with protein groups and exploit relative metaproteomic abundances. Resultingly, studies similar to the example by Marcelino et al. [17] (Section 8) could employ metaproteomic data to obtain more precise predictions of microbiome interactions in diseases or biotechnological processes.
An obstacle in kinetic modeling of biological systems is the non-availability of parameters. Statistical modeling could help us determine unknown model parameters from available information. Li et al. [160] for example, predicted values for enzymes from substrate structures and protein sequences based on machine learning. They used these predicted values to create enzyme-constrained metabolic models and could achieve reasonable results. Another example is hybrid modeling, such as employed by Espinel-Ríos et al. [20]. A hybrid model consists of a mechanistic part coupled to a statistical model (e.g., a neural network), which predicts an uncertain term or variable of the mechanistic model. Hybrid modeling could be applied in instances where some parts of a molecular mechanism are unknown, but sufficient training data are available.
Metabolic modeling approaches whole body modeling of humans as studies by Thiele et al. [300,301] show. Thiele et al. [301] built a metabolic model accounting for every human organ including the microbiome and could predict known inter-organ metabolic cycles and energy use. Microbiomes also interact with human cellular signaling, meaning the next step could be the extension of human tissue models by cellular signaling and regulation. Signaling and regulation are currently a blank spot in host-microbiome modeling, but modeling formalisms such as rxncon were showcased, which could facilitate genome-scale modeling of these processes. As whole-body genome-scale models become very large (over 80,000 reactions in Harvey/Harvetta [301] vs. 4,131 reactions in Yeast8 [302]), model reduction techniques will be used to increase the performance of simulations. While the models by Thiele et al. [301] are restricted to constraint-based modeling, a multi-scale modeling approach would be required to capture the dynamics of physiological processes in humans (reviewed in Thiele et al. [300]). Such advanced whole-body models will improve the understanding of microbiome-host interactions in well-being and diseases. Ultimately, these models could find application in diagnostics and personalized medicine [301].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
EL: Conceptualization, Data curation, Investigation, Methodology, Project administration, Software, Writing – original draft; LK: Investigation, Writing – original draft, Writing – review & editing; JK: Conceptualization, Writing – review & editing; DB: Conceptualization, Writing – review & editing; RH: Conceptualization, Investigation, Project administration, Supervision, Writing – original draft, Writing – review & editing
Data Availability Statement
Scripts and generated output files are available on GitHub (https://github.com/voidsailor/automated_literature_search) and Zenodo (https://zenodo.org/doi/10.5281/zenodo.10402352).
Acknowledgments
We thank Maximilian Wolf for proofreading. We thank our colleagues, friends, and family members whose engaging discussions sparked ideas and contributed to the development of this manuscript. We also appreciate their interest and encouragement throughout the process.
Conflicts of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nature Medicine 2018, 24, 392–400. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D. Human gut microbiome: hopes, threats and promises. Gut 2018, 67, 1716–1725. [Google Scholar] [CrossRef]
- Helmink, B.A.; Khan, M.A.W.; Hermann, A.; Gopalakrishnan, V.; Wargo, J.A. The microbiome, cancer, and cancer therapy. Nature Medicine 2019, 25, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Jackson, R.B.; Saunois, M.; Bousquet, P.; Canadell, J.G.; Poulter, B.; Stavert, A.R.; Bergamaschi, P.; Niwa, Y.; Segers, A.; Tsuruta, A. Increasing anthropogenic methane emissions arise equally from agricultural and fossil fuel sources. Environmental Research Letters 2020, 15, 071002. [Google Scholar] [CrossRef]
- Naylor, D.; Sadler, N.; Bhattacharjee, A.; Graham, E.B.; Anderton, C.R.; McClure, R.; Lipton, M.; Hofmockel, K.S.; Jansson, J.K. Soil Microbiomes Under Climate Change and Implications for Carbon Cycling. Annual Review of Environment and Resources 2020, 45, 29–59. [Google Scholar] [CrossRef]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiological Reviews 1995, 59, 143–169. [Google Scholar] [CrossRef]
- Wade, W. Unculturable bacteria–the uncharacterized organisms that cause oral infections. JRSM 2002, 95, 81–83. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; Mende, D.R.; Li, J.; Xu, J.; Li, S.; Li, D.; Cao, J.; Wang, B.; Liang, H.; Zheng, H.; Xie, Y.; Tap, J.; Lepage, P.; Bertalan, M.; Batto, J.M.; Hansen, T.; Le Paslier, D.; Linneberg, A.; Nielsen, H.B.; Pelletier, E.; Renault, P.; Sicheritz-Ponten, T.; Turner, K.; Zhu, H.; Yu, C.; Li, S.; Jian, M.; Zhou, Y.; Li, Y.; Zhang, X.; Li, S.; Qin, N.; Yang, H.; Wang, J.; Brunak, S.; Doré, J.; Guarner, F.; Kristiansen, K.; Pedersen, O.; Parkhill, J.; Weissenbach, J.; Bork, P.; Ehrlich, S.D.; Wang, J. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Aguiar-Pulido, V.; Huang, W.; Suarez-Ulloa, V.; Cickovski, T.; Mathee, K.; Narasimhan, G. Metagenomics, Metatranscriptomics, and Metabolomics Approaches for Microbiome Analysis: Supplementary Issue: Bioinformatics Methods and Applications for Big Metagenomics Data. Evolutionary Bioinformatics 2016, 12s1, EBO.S36436. [Google Scholar] [CrossRef]
- Heyer, R.; Schallert, K.; Zoun, R.; Becher, B.; Saake, G.; Benndorf, D. Challenges and perspectives of metaproteomic data analysis. Journal of Biotechnology 2017, 261, 24–36. [Google Scholar] [CrossRef]
- Henry, C.S.; DeJongh, M.; Best, A.A.; Frybarger, P.M.; Linsay, B.; Stevens, R.L. High-throughput generation, optimization and analysis of genome-scale metabolic models. Nature Biotechnology 2010, 28, 977–982. [Google Scholar] [CrossRef] [PubMed]
- Jünemann, S.; Kleinbölting, N.; Jaenicke, S.; Henke, C.; Hassa, J.; Nelkner, J.; Stolze, Y.; Albaum, S.P.; Schlüter, A.; Goesmann, A.; Sczyrba, A.; Stoye, J. Bioinformatics for NGS-based metagenomics and the application to biogas research. Journal of Biotechnology 2017, 261, 10–23. [Google Scholar] [CrossRef]
- Faust, K.; Raes, J. Microbial interactions: from networks to models. Nature Reviews Microbiology 2012, 10, 538–550. [Google Scholar] [CrossRef]
- Tobalina, L.; Bargiela, R.; Pey, J.; Herbst, F.A.; Lores, I.; Rojo, D.; Barbas, C.; Peláez, A.I.; Sánchez, J.; von Bergen, M.; Seifert, J.; Ferrer, M.; Planes, F.J. Context-specific metabolic network reconstruction of a naphthalene-degrading bacterial community guided by metaproteomic data. Bioinformatics 2015, 31, 1771–1779. [Google Scholar] [CrossRef]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Research 2018, 46, 7542–7553. [Google Scholar] [CrossRef]
- Aden, K.; Rehman, A.; Waschina, S.; Pan, W.H.; Walker, A.; Lucio, M.; Nunez, A.M.; Bharti, R.; Zimmerman, J.; Bethge, J.; Schulte, B.; Schulte, D.; Franke, A.; Nikolaus, S.; Schroeder, J.O.; Vandeputte, D.; Raes, J.; Szymczak, S.; Waetzig, G.H.; Zeuner, R.; Schmitt-Kopplin, P.; Kaleta, C.; Schreiber, S.; Rosenstiel, P. Metabolic Functions of Gut Microbes Associate With Efficacy of Tumor Necrosis Factor Antagonists in Patients With Inflammatory Bowel Diseases. Gastroenterology 2019, 157, 1279–1292.e11. [Google Scholar] [CrossRef] [PubMed]
- Marcelino, V.R.; Welsh, C.; Diener, C.; Gulliver, E.L.; Rutten, E.L.; Young, R.B.; Giles, E.M.; Gibbons, S.M.; Greening, C.; Forster, S.C. Disease-specific loss of microbial cross-feeding interactions in the human gut. Nature Communications 2023, 14. [Google Scholar] [CrossRef]
- García-Jiménez, B.; Torres-Bacete, J.; Nogales, J. Metabolic modelling approaches for describing and engineering microbial communities. Computational and Structural Biotechnology Journal 2021, 19, 226–246. [Google Scholar] [CrossRef] [PubMed]
- Curran, D.M.; Grote, A.; Nursimulu, N.; Geber, A.; Voronin, D.; Jones, D.R.; Ghedin, E.; Parkinson, J. Modeling the metabolic interplay between a parasitic worm and its bacterial endosymbiont allows the identification of novel drug targets. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Espinel-Ríos, S.; Bettenbrock, K.; Klamt, S.; Avalos, J.L.; Findeisen, R. Machine learning-supported cybergenetic modeling, optimization and control for synthetic microbial communities. In Computer Aided Chemical Engineering; Elsevier, 2023; pp. 2601–2606. [Google Scholar] [CrossRef]
- Espinel-Ríos, S.; Morabito, B.; Pohlodek, J.; Bettenbrock, K.; Klamt, S.; Findeisen, R. Toward a modeling, optimization, and predictive control framework for fed-batch metabolic cybergenetics. Biotechnology and Bioengineering 2023. [Google Scholar] [CrossRef]
- Xue, L.; Li, D.; Xi, Y. Nonlinear model predictive control of anaerobic digestion process based on reduced ADM1. In Proceedings of the 2015 10th Asian Control Conference (ASCC); IEEE, 2015; pp. 1–6. [Google Scholar] [CrossRef]
- Veenstra, T.D. Omics in Systems Biology: Current Progress and Future Outlook. PROTEOMICS 2021, 21, 2000235. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Palsson, B.Ø. A protocol for generating a high-quality genome-scale metabolic reconstruction. Nature Protocols 2010, 5, 93–121. [Google Scholar] [CrossRef]
- Sayers, E. Entrez programming utilities help. 2009; 17. Available online: http://www.ncbi.nlm.nih.gov/books/NBK25499.
- Bartel, J.; Krumsiek, J.; Theis, F.J. STATISTICAL METHODS FOR THE ANALYSIS OF HIGH-THROUGHPUT METABOLOMICS DATA. Computational and Structural Biotechnology Journal 2013, 4, e201301009. [Google Scholar] [CrossRef] [PubMed]
- Yamada, R.; Okada, D.; Wang, J.; Basak, T.; Koyama, S. Interpretation of omics data analyses. Journal of Human Genetics 2020, 66, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Arıkan, M.; Muth, T. Integrated multi-omics analyses of microbial communities: a review of the current state and future directions. Molecular Omics 2023. [Google Scholar] [CrossRef]
- Jiang, D.; Armour, C.R.; Hu, C.; Mei, M.; Tian, C.; Sharpton, T.J.; Jiang, Y. Microbiome Multi-Omics Network Analysis: Statistical Considerations, Limitations, and Opportunities. Frontiers in Genetics 2019, 10. [Google Scholar] [CrossRef]
- Reimand, J.; Isserlin, R.; Voisin, V.; Kucera, M.; Tannus-Lopes, C.; Rostamianfar, A.; Wadi, L.; Meyer, M.; Wong, J.; Xu, C.; Merico, D.; Bader, G.D. Pathway enrichment analysis and visualization of omics data using g:Profiler, GSEA, Cytoscape and EnrichmentMap. Nature Protocols 2019, 14, 482–517. [Google Scholar] [CrossRef]
- Salvato, F.; Hettich, R.L.; Kleiner, M. Five key aspects of metaproteomics as a tool to understand functional interactions in host-associated microbiomes. PLOS Pathogens 2021, 17, e1009245. [Google Scholar] [CrossRef] [PubMed]
- Gehlenborg, N.; O’Donoghue, S.I.; Baliga, N.S.; Goesmann, A.; Hibbs, M.A.; Kitano, H.; Kohlbacher, O.; Neuweger, H.; Schneider, R.; Tenenbaum, D.; Gavin, A.C. Visualization of omics data for systems biology. Nature Methods 2010, 7, S56–S68. [Google Scholar] [CrossRef]
- Bekiaris, P.S.; Klamt, S. Automatic construction of metabolic models with enzyme constraints. BMC Bioinformatics 2020, 21. [Google Scholar] [CrossRef]
- Machado, D.; Costa, R.S.; Rocha, M.; Ferreira, E.C.; Tidor, B.; Rocha, I. Modeling formalisms in Systems Biology. AMB Express 2011, 1, 45. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Der Stoffwechsel: Konzepte und Grundmuster. In Stryer Biochemie; Springer: Berlin/Heidelberg, Germany, 2013; pp. 431–455. [Google Scholar] [CrossRef]
- Huang, J.; Zhang, P.; Solari, F.A.; Sickmann, A.; Garcia, A.; Jurk, K.; Heemskerk, J.W.M. Molecular Proteomics and Signalling of Human Platelets in Health and Disease. International Journal of Molecular Sciences 2021, 22, 9860. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Signaltransduktionswege. In Stryer Biochemie; Springer: Berlin/Heidelberg, Germany, 2013; pp. 404–430. [Google Scholar] [CrossRef]
- Davidson, E.; Levin, M. Gene regulatory networks. Proceedings of the National Academy of Sciences 2005, 102, 4935–4935. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Kontrolle der Genexpression bei Prokaryoten. In Stryer Biochemie; Springer: Berlin/Heidelberg, Germany, 2013; pp. 933–933. [Google Scholar] [CrossRef]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Kontrolle der Genexpression bei Eukaryoten. In Stryer Biochemie; Springer: Berlin/Heidelberg, Germany, 2013; pp. 949–969. [Google Scholar] [CrossRef]
- Terfve, C.; Saez-Rodriguez, J. Modeling Signaling Networks Using High-throughput Phospho-proteomics. In Advances in Experimental Medicine and Biology; Springer: New York, 2011; pp. 19–57. [Google Scholar] [CrossRef]
- Samaga, R.; Klamt, S. Modeling approaches for qualitative and semi-quantitative analysis of cellular signaling networks. Cell Communication and Signaling 2013, 11, 43. [Google Scholar] [CrossRef] [PubMed]
- Papin, J.A.; Hunter, T.; Palsson, B.O.; Subramaniam, S. Reconstruction of cellular signalling networks and analysis of their properties. Nature Reviews Molecular Cell Biology 2005, 6, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Sorrells, T.; Johnson, A. Making Sense of Transcription Networks. Cell 2015, 161, 714–723. [Google Scholar] [CrossRef] [PubMed]
- Panni, S.; Lovering, R.C.; Porras, P.; Orchard, S. Non-coding RNA regulatory networks. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 2020, 1863, 194417. [Google Scholar] [CrossRef] [PubMed]
- Popovic, M. Thermodynamic properties of microorganisms: determination and analysis of enthalpy, entropy, and Gibbs free energy of biomass, cells and colonies of 32 microorganism species. Heliyon 2019, 5, e01950. [Google Scholar] [CrossRef]
- Feijó Delgado, F.; Cermak, N.; Hecht, V.C.; Son, S.; Li, Y.; Knudsen, S.M.; Olcum, S.; Higgins, J.M.; Chen, J.; Grover, W.H.; Manalis, S.R. Intracellular Water Exchange for Measuring the Dry Mass, Water Mass and Changes in Chemical Composition of Living Cells. PLoS ONE 2013, 8, e67590. [Google Scholar] [CrossRef] [PubMed]
- Großkopf, T.; Soyer, O.S. Synthetic microbial communities. Current Opinion in Microbiology 2014, 18, 72–77. [Google Scholar] [CrossRef]
- Roell, G.W.; Zha, J.; Carr, R.R.; Koffas, M.A.; Fong, S.S.; Tang, Y.J. Engineering microbial consortia by division of labor. Microbial Cell Factories 2019, 18. [Google Scholar] [CrossRef] [PubMed]
- Schink, B. Energetics of syntrophic cooperation in methanogenic degradation. Microbiology and Molecular Biology Reviews 1997, 61, 262–280. [Google Scholar] [CrossRef]
- Heyer, R.; Kohrs, F.; Reichl, U.; Benndorf, D. Metaproteomics of complex microbial communities in biogas plants. Microbial Biotechnology 2015, 8, 749–763. [Google Scholar] [CrossRef]
- Muth, T.; Renard, B.Y.; Martens, L. Metaproteomic data analysis at a glance: advances in computational microbial community proteomics. Expert Review of Proteomics 2016, 13, 757–769. [Google Scholar] [CrossRef]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Solano, C.; Echeverz, M.; Lasa, I. Biofilm dispersion and quorum sensing. Current Opinion in Microbiology 2014, 18, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Quiza, L.; St-Arnaud, M.; Yergeau, E. Harnessing phytomicrobiome signaling for rhizosphere microbiome engineering. Frontiers in Plant Science 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Jameson, K.; Olson, C.; Kazmi, S.; Hsiao, E. Toward Understanding Microbiome-Neuronal Signaling. Molecular Cell 2020, 78, 577–583. [Google Scholar] [CrossRef]
- Shin, S.C.; Kim, S.H.; You, H.; Kim, B.; Kim, A.C.; Lee, K.A.; Yoon, J.H.; Ryu, J.H.; Lee, W.J. Drosophila Microbiome Modulates Host Developmental and Metabolic Homeostasis via Insulin Signaling. Science 2011, 334, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Segre, J.A. Signaling in Host-Associated Microbial Communities. Cell 2016, 164, 1288–1300. [Google Scholar] [CrossRef]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.M. Non-surface Attached Bacterial Aggregates: A Ubiquitous Third Lifestyle. Frontiers in Microbiology 2020, 11. [Google Scholar] [CrossRef]
- Rani, S.A.; Pitts, B.; Beyenal, H.; Veluchamy, R.A.; Lewandowski, Z.; Davison, W.M.; Buckingham-Meyer, K.; Stewart, P.S. Spatial Patterns of DNA Replication, Protein Synthesis, and Oxygen Concentration within Bacterial Biofilms Reveal Diverse Physiological States. Journal of Bacteriology 2007, 189, 4223–4233. [Google Scholar] [CrossRef]
- Kreft, J.U.; Plugge, C.M.; Prats, C.; Leveau, J.H.J.; Zhang, W.; Hellweger, F.L. From Genes to Ecosystems in Microbiology: Modeling Approaches and the Importance of Individuality. Frontiers in Microbiology 2017, 8. [Google Scholar] [CrossRef]
- Blum, W.E.; Zechmeister-Boltenstern, S.; Keiblinger, K.M. Does Soil Contribute to the Human Gut Microbiome? Microorganisms 2019, 7, 287. [Google Scholar] [CrossRef]
- Pasolli, E.; Asnicar, F.; Manara, S.; Zolfo, M.; Karcher, N.; Armanini, F.; Beghini, F.; Manghi, P.; Tett, A.; Ghensi, P.; Collado, M.C.; Rice, B.L.; DuLong, C.; Morgan, X.C.; Golden, C.D.; Quince, C.; Huttenhower, C.; Segata, N. Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle. Cell 2019, 176, 649–662.e20. [Google Scholar] [CrossRef] [PubMed]
- Almeida, A.; Mitchell, A.L.; Boland, M.; Forster, S.C.; Gloor, G.B.; Tarkowska, A.; Lawley, T.D.; Finn, R.D. A new genomic blueprint of the human gut microbiota. Nature 2019, 568, 499–504. [Google Scholar] [CrossRef]
- Louca, S.; Mazel, F.; Doebeli, M.; Parfrey, L.W. A census-based estimate of Earth’s bacterial and archaeal diversity. PLOS Biology 2019, 17, e3000106. [Google Scholar] [CrossRef] [PubMed]
- Overmann, J.; Abt, B.; Sikorski, J. Present and Future of Culturing Bacteria. Annual Review of Microbiology 2017, 71, 711–730. [Google Scholar] [CrossRef] [PubMed]
- Reimer, L.C.; Sardà Carbasse, J.; Koblitz, J.; Ebeling, C.; Podstawka, A.; Overmann, J. BacDive in 2022: the knowledge base for standardized bacterial and archaeal data. Nucleic Acids Research 2021, 50, D741–D746. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.; Hunt, K.; Carlson, R. Measuring Cellular Biomass Composition for Computational Biology Applications. Processes 2018, 6, 38. [Google Scholar] [CrossRef]
- Lachance, J.C.; Lloyd, C.J.; Monk, J.M.; Yang, L.; Sastry, A.V.; Seif, Y.; Palsson, B.O.; Rodrigue, S.; Feist, A.M.; King, Z.A.; Jacques, P.É. BOFdat: Generating biomass objective functions for genome-scale metabolic models from experimental data. PLOS Computational Biology 2019, 15, e1006971. [Google Scholar] [CrossRef]
- Vos, T.; Hakkaart, X.D.V.; de Hulster, E.A.F.; van Maris, A.J.A.; Pronk, J.T.; Daran-Lapujade, P. Maintenance-energy requirements and robustness of Saccharomyces cerevisiae at aerobic near-zero specific growth rates. Microbial Cell Factories 2016, 15. [Google Scholar] [CrossRef]
- Zamboni, N.; Fendt, S.M.; Rühl, M.; Sauer, U. 13C-based metabolic flux analysis. Nature Protocols 2009, 4, 878–892. [Google Scholar] [CrossRef]
- Palazzotto, E.; Weber, T. Omics and multi-omics approaches to study the biosynthesis of secondary metabolites in microorganisms. Current Opinion in Microbiology 2018, 45, 109–116. [Google Scholar] [CrossRef]
- Winter, G.; Krömer, J.O. Fluxomics - connecting ‘omics analysis and phenotypes. Environmental Microbiology 2013, 15, 1901–1916. [Google Scholar] [CrossRef]
- Koch, S.; Benndorf, D.; Fronk, K.; Reichl, U.; Klamt, S. Predicting compositions of microbial communities from stoichiometric models with applications for the biogas process. Biotechnology for Biofuels 2016, 9. [Google Scholar] [CrossRef]
- Koch, S.; Kohrs, F.; Lahmann, P.; Bissinger, T.; Wendschuh, S.; Benndorf, D.; Reichl, U.; Klamt, S. RedCom: A strategy for reduced metabolic modeling of complex microbial communities and its application for analyzing experimental datasets from anaerobic digestion. PLOS Computational Biology 2019, 15, e1006759. [Google Scholar] [CrossRef] [PubMed]
- Schäpe, S.S.; Krause, J.L.; Engelmann, B.; Fritz-Wallace, K.; Schattenberg, F.; Liu, Z.; Müller, S.; Jehmlich, N.; Rolle-Kampczyk, U.; Herberth, G.; von Bergen, M. The Simplified Human Intestinal Microbiota (SIHUMIx) Shows High Structural and Functional Resistance against Changing Transit Times in In Vitro Bioreactors. Microorganisms 2019, 7, 641. [Google Scholar] [CrossRef] [PubMed]
- Hanreich, A.; Schimpf, U.; Zakrzewski, M.; Schlüter, A.; Benndorf, D.; Heyer, R.; Rapp, E.; Pühler, A.; Reichl, U.; Klocke, M. Metagenome and metaproteome analyses of microbial communities in mesophilic biogas-producing anaerobic batch fermentations indicate concerted plant carbohydrate degradation. Systematic and Applied Microbiology 2013, 36, 330–338. [Google Scholar] [CrossRef]
- Lui, L.M.; Majumder, E.L.W.; Smith, H.J.; Carlson, H.K.; von Netzer, F.; Fields, M.W.; Stahl, D.A.; Zhou, J.; Hazen, T.C.; Baliga, N.S.; Adams, P.D.; Arkin, A.P. Mechanism Across Scales: A Holistic Modeling Framework Integrating Laboratory and Field Studies for Microbial Ecology. Frontiers in Microbiology 2021, 12. [Google Scholar] [CrossRef]
- Petersen, C.; Hamerich, I.K.; Adair, K.L.; Griem-Krey, H.; Torres Oliva, M.; Hoeppner, M.P.; Bohannan, B.J.M.; Schulenburg, H. Host and microbiome jointly contribute to environmental adaptation. The ISME Journal 2023, 17, 1953–1965. [Google Scholar] [CrossRef] [PubMed]
- Muth, T.; Benndorf, D.; Reichl, U.; Rapp, E.; Martens, L. Searching for a needle in a stack of needles: challenges in metaproteomics data analysis. Mol. BioSyst. 2013, 9, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Wilmes, P.; Bond, P.L. Metaproteomics: studying functional gene expression in microbial ecosystems. Trends in Microbiology 2006, 14, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Valera, F. Environmental genomics, the big picture? FEMS Microbiology Letters 2004, 231, 153–158. [Google Scholar] [CrossRef]
- Kleiner, M. Metaproteomics: Much More than Measuring Gene Expression in Microbial Communities. 2019; 4. [Google Scholar] [CrossRef]
- Domenzain, I.; Sánchez, B.; Anton, M.; Kerkhoven, E.J.; Millán-Oropeza, A.; Henry, C.; Siewers, V.; Morrissey, J.P.; Sonnenschein, N.; Nielsen, J. Reconstruction of a catalogue of genome-scale metabolic models with enzymatic constraints using GECKO 2.0. Nature Communications 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Kohrs, F.; Heyer, R.; Magnussen, A.; Benndorf, D.; Muth, T.; Behne, A.; Rapp, E.; Kausmann, R.; Heiermann, M.; Klocke, M.; Reichl, U. Sample prefractionation with liquid isoelectric focusing enables in depth microbial metaproteome analysis of mesophilic and thermophilic biogas plants. Anaerobe 2014, 29, 59–67. [Google Scholar] [CrossRef] [PubMed]
- Aakko, J.; Pietilä, S.; Suomi, T.; Mahmoudian, M.; Toivonen, R.; Kouvonen, P.; Rokka, A.; Hänninen, A.; Elo, L.L. Data-Independent Acquisition Mass Spectrometry in Metaproteomics of Gut Microbiota—Implementation and Computational Analysis. Journal of Proteome Research 2019, 19, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Pietilä, S.; Suomi, T.; Elo, L.L. Introducing untargeted data-independent acquisition for metaproteomics of complex microbial samples. ISME Communications 2022, 2. [Google Scholar] [CrossRef]
- Perkins, D.N.; Pappin, D.J.C.; Creasy, D.M.; Cottrell, J.S. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 1999, 20, 3551–3567. [Google Scholar] [CrossRef]
- Craig, R.; Beavis, R.C. TANDEM: matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef] [PubMed]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature Methods 2007, 4, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Muth, T.; Behne, A.; Heyer, R.; Kohrs, F.; Benndorf, D.; Hoffmann, M.; Lehtevä, M.; Reichl, U.; Martens, L.; Rapp, E. The MetaProteomeAnalyzer: A Powerful Open-Source Software Suite for Metaproteomics Data Analysis and Interpretation. Journal of Proteome Research 2015, 14, 1557–1565. [Google Scholar] [CrossRef]
- Afgan, E.; Nekrutenko, A.; Grüning, B.A.; Blankenberg, D.; Goecks, J.; Schatz, M.C.; Ostrovsky, A.E.; Mahmoud, A.; Lonie, A.J.; Syme, A.; Fouilloux, A.; Bretaudeau, A.; Nekrutenko, A.; Kumar, A.; Eschenlauer, A.C.; DeSanto, A.D.; Guerler, A.; Serrano-Solano, B.; Batut, B.; Grüning, B.A.; Langhorst, B.W.; Carr, B.; Raubenolt, B.A.; Hyde, C.J.; Bromhead, C.J.; Barnett, C.B.; Royaux, C.; Gallardo, C.; Blankenberg, D.; Fornika, D.J.; Baker, D.; Bouvier, D.; Clements, D.; de Lima Morais, D.A.; Tabernero, D.L.; Lariviere, D.; Nasr, E.; Afgan, E.; Zambelli, F.; Heyl, F.; Psomopoulos, F.; Coppens, F.; Price, G.R.; Cuccuru, G.; Corguillé, G.L.; Kuster, G.V.; Akbulut, G.G.; Rasche, H.; Hotz, H.R.; Eguinoa, I.; Makunin, I.; Ranawaka, I.J.; Taylor, J.P.; Joshi, J.; Hillman-Jackson, J.; Goecks, J.; Chilton, J.M.; Kamali, K.; Suderman, K.; Poterlowicz, K.; Yvan, L.B.; Lopez-Delisle, L.; Sargent, L.; Bassetti, M.E.; Tangaro, M.A.; van den Beek, M.; Čech, M.; Bernt, M.; Fahrner, M.; Tekman, M.; Föll, M.C.; Schatz, M.C.; Crusoe, M.R.; Roncoroni, M.; Kucher, N.; Coraor, N.; Stoler, N.; Rhodes, N.; Soranzo, N.; Pinter, N.; Goonasekera, N.A.; Moreno, P.A.; Videm, P.; Melanie, P.; Mandreoli, P.; Jagtap, P.D.; Gu, Q.; Weber, R.J.M.; Lazarus, R.; Vorderman, R.H.P.; Hiltemann, S.; Golitsynskiy, S.; Garg, S.; Bray, S.A.; Gladman, S.L.; Leo, S.; Mehta, S.P.; Griffin, T.J.; Jalili, V.; Yves, V.; Wen, V.; Nagampalli, V.K.; Bacon, W.A.; de Koning, W.; Maier, W.; Briggs, P.J. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Research 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Bossche, T.V.D.; Kunath, B.J.; Schallert, K.; Schäpe, S.S.; Abraham, P.E.; Armengaud, J.; Arntzen, M.Ø.; Bassignani, A.; Benndorf, D.; Fuchs, S.; Giannone, R.J.; Griffin, T.J.; Hagen, L.H.; Halder, R.; Henry, C.; Hettich, R.L.; Heyer, R.; Jagtap, P.; Jehmlich, N.; Jensen, M.; Juste, C.; Kleiner, M.; Langella, O.; Lehmann, T.; Leith, E.; May, P.; Mesuere, B.; Miotello, G.; Peters, S.L.; Pible, O.; Queiros, P.T.; Reichl, U.; Renard, B.Y.; Schiebenhoefer, H.; Sczyrba, A.; Tanca, A.; Trappe, K.; Trezzi, J.P.; Uzzau, S.; Verschaffelt, P.; von Bergen, M.; Wilmes, P.; Wolf, M.; Martens, L.; Muth, T. Critical Assessment of MetaProteome Investigation (CAMPI): a multi-laboratory comparison of established workflows. Nature Communications 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Delogu, F.; Kunath, B.J.; Evans, P.N.; Arntzen, M.Ø; Hvidsten, T.R.; Pope, P.B. Integration of absolute multi-omics reveals dynamic protein-to-RNA ratios and metabolic interplay within mixed-domain microbiomes. Nature Communications 2020, 11. [Google Scholar] [CrossRef]
- Sivanich, M.K.; Gu, T.; Tabang, D.N.; Li, L. Recent advances in isobaric labeling and applications in quantitative proteomics 2022. 22. [CrossRef]
- Ahrné, E.; Molzahn, L.; Glatter, T.; Schmidt, A. Critical assessment of proteome-wide label-free absolute abundance estimation strategies. PROTEOMICS 2013, 13, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, B.J.; Zhang, C.; Nilsson, A.; Lahtvee, P.J.; Kerkhoven, E.J.; Nielsen, J. Improving the phenotype predictions of a yeast genome-scale metabolic model by incorporating enzymatic constraints. Molecular Systems Biology 2017, 13, 935. [Google Scholar] [CrossRef] [PubMed]
- Kirkpatrick, D.S.; Gerber, S.A.; Gygi, S.P. The absolute quantification strategy: a general procedure for the quantification of proteins and post-translational modifications. Methods 2005, 35, 265–273. [Google Scholar] [CrossRef]
- Starke, R.; Jehmlich, N.; Bastida, F. Using proteins to study how microbes contribute to soil ecosystem services: The current state and future perspectives of soil metaproteomics. Journal of Proteomics 2019, 198, 50–58. [Google Scholar] [CrossRef]
- Pratt, J.M.; Simpson, D.M.; Doherty, M.K.; Rivers, J.; Gaskell, S.J.; Beynon, R.J. Multiplexed absolute quantification for proteomics using concatenated signature peptides encoded by QconCAT genes. Nature protocols 2006, 1, 1029–1043. [Google Scholar] [CrossRef] [PubMed]
- Schallert, K.; Verschaffelt, P.; Mesuere, B.; Benndorf, D.; Martens, L.; Bossche, T.V.D. Pout2Prot: An Efficient Tool to Create Protein (Sub)groups from Percolator Output Files. Journal of Proteome Research 2022, 21, 1175–1180. [Google Scholar] [CrossRef]
- Zhang, X.; Figeys, D. Perspective and Guidelines for Metaproteomics in Microbiome Studies. Journal of Proteome Research 2019, 18, 2370–2380. [Google Scholar] [CrossRef] [PubMed]
- Mirzayi, C.; Renson, A.; Furlanello, C.; Sansone, S.A.; Zohra, F.; Elsafoury, S.; Geistlinger, L.; Kasselman, L.J.; Eckenrode, K.; van de Wijgert, J.; Loughman, A.; Marques, F.Z.; MacIntyre, D.A.; Arumugam, M.; Azhar, R.; Beghini, F.; Bergstrom, K.; Bhatt, A.; Bisanz, J.E.; Braun, J.; Bravo, H.C.; Buck, G.A.; Bushman, F.; Casero, D.; Clarke, G.; Collado, M.C.; Cotter, P.D.; Cryan, J.F.; Demmer, R.T.; Devkota, S.; Elinav, E.; Escobar, J.S.; Fettweis, J.; Finn, R.D.; Fodor, A.A.; Forslund, S.; Franke, A.; Furlanello, C.; Gilbert, J.; Grice, E.; Haibe-Kains, B.; Handley, S.; Herd, P.; Holmes, S.; Jacobs, J.P.; Karstens, L.; Knight, R.; Knights, D.; Koren, O.; Kwon, D.S.; Langille, M.; Lindsay, B.; McGovern, D.; McHardy, A.C.; McWeeney, S.; Mueller, N.T.; Nezi, L.; Olm, M.; Palm, N.; Pasolli, E.; Raes, J.; Redinbo, M.R.; Rühlemann, M.; Sartor, R.B.; Schloss, P.D.; Schriml, L.; Segal, E.; Shardell, M.; Sharpton, T.; Smirnova, E.; Sokol, H.; Sonnenburg, J.L.; Srinivasan, S.; Thingholm, L.B.; Turnbaugh, P.J.; Upadhyay, V.; Walls, R.L.; Wilmes, P.; Yamada, T.; Zeller, G.; Zhang, M.; Zhao, N.; Zhao, L.; Bao, W.; Culhane, A.; Devanarayan, V.; Dopazo, J.; Fan, X.; Fischer, M.; Jones, W.; Kusko, R.; Mason, C.E.; Mercer, T.R.; Sansone, S.A.; Scherer, A.; Shi, L.; Thakkar, S.; Tong, W.; Wolfinger, R.; Hunter, C.; Segata, N.; Huttenhower, C.; Dowd, J.B.; Jones, H.E.; Waldron, L. Reporting guidelines for human microbiome research: the STORMS checklist. Nature Medicine 2021, 27, 1885–1892. [Google Scholar] [CrossRef]
- Vizcaíno, J.A.; Walzer, M.; Jiménez, R.C.; Bittremieux, W.; Bouyssié, D.; Carapito, C.; Corrales, F.; Ferro, M.; Heck, A.J.; Horvatovich, P.; Hubalek, M.; Lane, L.; Laukens, K.; Levander, F.; Lisacek, F.; Novak, P.; Palmblad, M.; Piovesan, D.; Pühler, A.; Schwämmle, V.; Valkenborg, D.; van Rijswijk, M.; Vondrasek, J.; Eisenacher, M.; Martens, L.; Kohlbacher, O. A community proposal to integrate proteomics activities in ELIXIR. F1000Research 2017, 6, 875. [Google Scholar] [CrossRef]
- Van Den Bossche, T.; Arntzen, M.Ø; Becher, D.; Benndorf, D.; Eijsink, V.G.H.; Henry, C.; Jagtap, P.D.; Jehmlich, N.; Juste, C.; Kunath, B.J.; Mesuere, B.; Muth, T.; Pope, P.B.; Seifert, J.; Tanca, A.; Uzzau, S.; Wilmes, P.; Hettich, R.L.; Armengaud, J. The Metaproteomics Initiative: a coordinated approach for propelling the functional characterization of microbiomes. Microbiome 2021, 9. [Google Scholar] [CrossRef]
- Bateman, A.; Martin, M.J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; Cukura, A.; Denny, P.; Dogan, T.; Ebenezer, T.; Fan, J.; Garmiri, P.; da Costa Gonzales, L.J.; Hatton-Ellis, E.; Hussein, A.; Ignatchenko, A.; Insana, G.; Ishtiaq, R.; Joshi, V.; Jyothi, D.; Kandasaamy, S.; Lock, A.; Luciani, A.; Lugaric, M.; Luo, J.; Lussi, Y.; MacDougall, A.; Madeira, F.; Mahmoudy, M.; Mishra, A.; Moulang, K.; Nightingale, A.; Pundir, S.; Qi, G.; Raj, S.; Raposo, P.; Rice, D.L.; Saidi, R.; Santos, R.; Speretta, E.; Stephenson, J.; Totoo, P.; Turner, E.; Tyagi, N.; Vasudev, P.; Warner, K.; Watkins, X.; Zaru, R.; Zellner, H.; Bridge, A.J.; Aimo, L.; Argoud-Puy, G.; Auchincloss, A.H.; Axelsen, K.B.; Bansal, P.; Baratin, D.; Batista Neto, T.M.; Blatter, M.C.; Bolleman, J.T.; Boutet, E.; Breuza, L.; Gil, B.C.; Casals-Casas, C.; Echioukh, K.C.; Coudert, E.; Cuche, B.; de Castro, E.; Estreicher, A.; Famiglietti, M.L.; Feuermann, M.; Gasteiger, E.; Gaudet, P.; Gehant, S.; Gerritsen, V.; Gos, A.; Gruaz, N.; Hulo, C.; Hyka-Nouspikel, N.; Jungo, F.; Kerhornou, A.; Le Mercier, P.; Lieberherr, D.; Masson, P.; Morgat, A.; Muthukrishnan, V.; Paesano, S.; Pedruzzi, I.; Pilbout, S.; Pourcel, L.; Poux, S.; Pozzato, M.; Pruess, M.; Redaschi, N.; Rivoire, C.; Sigrist, C.J.A.; Sonesson, K.; Sundaram, S.; Wu, C.H.; Arighi, C.N.; Arminski, L.; Chen, C.; Chen, Y.; Huang, H.; Laiho, K.; McGarvey, P.; Natale, D.A.; Ross, K.; Vinayaka, C.R.; Wang, Q.; Wang, Y.; Zhang, J. UniProt: the Universal Protein Knowledgebase in 2023. Nucleic Acids Research 2022, 51, D523–D531. [Google Scholar] [CrossRef]
- Schiebenhoefer, H.; Bossche, T.V.D.; Fuchs, S.; Renard, B.Y.; Muth, T.; Martens, L. Challenges and promise at the interface of metaproteomics and genomics: an overview of recent progress in metaproteogenomic data analysis. Expert Review of Proteomics 2019, 16, 375–390. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Ciufo, S.; Domrachev, M.; Hotton, C.L.; Kannan, S.; Khovanskaya, R.; Leipe, D.; Mcveigh, R.; O’Neill, K.; Robbertse, B.; Sharma, S.; Soussov, V.; Sullivan, J.P.; Sun, L.; Turner, S.; Karsch-Mizrachi, I. NCBI Taxonomy: a comprehensive update on curation, resources and tools. Database 2020, 2020. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Research 2021, 50, D785–D794. [Google Scholar] [CrossRef]
- Mesuere, B.; der Jeugt, F.V.; Willems, T.; Naessens, T.; Devreese, B.; Martens, L.; Dawyndt, P. High-throughput metaproteomics data analysis with Unipept: A tutorial. Journal of Proteomics 2018, 171, 11–22. [Google Scholar] [CrossRef]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Research 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Penzlin, A.; Lindner, M.S.; Doellinger, J.; Dabrowski, P.W.; Nitsche, A.; Renard, B.Y. Pipasic: similarity and expression correction for strain-level identification and quantification in metaproteomics. Bioinformatics 2014, 30, i149–i156. [Google Scholar] [CrossRef] [PubMed]
- Schiebenhoefer, H.; Schallert, K.; Renard, B.Y.; Trappe, K.; Schmid, E.; Benndorf, D.; Riedel, K.; Muth, T.; Fuchs, S. A complete and flexible workflow for metaproteomics data analysis based on MetaProteomeAnalyzer and Prophane. Nature Protocols 2020, 15, 3212–3239. [Google Scholar] [CrossRef]
- Mesuere, B.; Van der Jeugt, F.; Devreese, B.; Vandamme, P.; Dawyndt, P. The unique peptidome: Taxon-specific tryptic peptides as biomarkers for targeted metaproteomics. PROTEOMICS 2016, 16, 2313–2318. [Google Scholar] [CrossRef] [PubMed]
- Starke, R.; Fiore-Donno, A.M.; White, R.A.; Parente Fernandes, M.L.; Martinović, T.; Bastida, F.; Delgado-Baquerizo, M.; Jehmlich, N. Biomarker metaproteomics for relative taxa abundances across soil organisms. Soil Biology and Biochemistry 2022, 175, 108861. [Google Scholar] [CrossRef]
- Chang, A.; Jeske, L.; Ulbrich, S.; Hofmann, J.; Koblitz, J.; Schomburg, I.; Neumann-Schaal, M.; Jahn, D.; Schomburg, D. BRENDA, the ELIXIR core data resource in 2021: new developments and updates. Nucleic Acids Research 2020, 49, D498–D508. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; Harris, M.A.; Hill, D.P.; Issel-Tarver, L.; Kasarskis, A.; Lewis, S.; Matese, J.C.; Richardson, J.E.; Ringwald, M.; Rubin, G.M.; Sherlock, G. Gene Ontology: tool for the unification of biology. Nature Genetics 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Aleksander, S.A.; Balhoff, J.; Carbon, S.; Cherry, J.M.; Drabkin, H.J.; Ebert, D.; Feuermann, M.; Gaudet, P.; Harris, N.L.; et al. The Gene Ontology knowledgebase in 2023. Genetics 2023, 224, iyad031. [Google Scholar] [PubMed]
- Kanehisa, M.; Furumichi, M.; Sato, Y.; Kawashima, M.; Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucleic Acids Research 2022. [Google Scholar] [CrossRef]
- Karp, P.D.; Billington, R.; Caspi, R.; Fulcher, C.A.; Latendresse, M.; Kothari, A.; Keseler, I.M.; Krummenacker, M.; Midford, P.E.; Ong, Q.; Ong, W.K.; Paley, S.M.; Subhraveti, P. The BioCyc collection of microbial genomes and metabolic pathways. Briefings in Bioinformatics 2017, 20, 1085–1093. [Google Scholar] [CrossRef]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; Deng, C.; Varusai, T.; Ragueneau, E.; Haider, Y.; May, B.; Shamovsky, V.; Weiser, J.; Brunson, T.; Sanati, N.; Beckman, L.; Shao, X.; Fabregat, A.; Sidiropoulos, K.; Murillo, J.; Viteri, G.; Cook, J.; Shorser, S.; Bader, G.; Demir, E.; Sander, C.; Haw, R.; Wu, G.; Stein, L.; Hermjakob, H.; D’Eustachio, P. The reactome pathway knowledgebase 2022. Nucleic Acids Research 2021, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chowdhury, D.; Zhang, Z.; Cheung, W.K.; Lu, A.; Bian, Z.; Zhang, L. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. 2021, 19, 6301–6314. [CrossRef]
- Parks, D.H.; Rinke, C.; Chuvochina, M.; Chaumeil, P.A.; Woodcroft, B.J.; Evans, P.N.; Hugenholtz, P.; Tyson, G.W. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nature Microbiology 2017, 2, 1533–1542. [Google Scholar] [CrossRef] [PubMed]
- Props, R.; Kerckhof, F.M.; Rubbens, P.; Vrieze, J.D.; Sanabria, E.H.; Waegeman, W.; Monsieurs, P.; Hammes, F.; Boon, N. Absolute quantification of microbial taxon abundances. The ISME Journal 2016, 11, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Hatzenpichler, R.; Krukenberg, V.; Spietz, R.L.; Jay, Z.J. Next-generation physiology approaches to study microbiome function at single cell level. Nature Reviews Microbiology 2020, 18, 241–256. [Google Scholar] [CrossRef] [PubMed]
- Cesar, S.; Huang, K.C. Thinking big: the tunability of bacterial cell size. FEMS Microbiology Reviews 2017, 41, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Bauermeister, A.; Mannochio-Russo, H.; Costa-Lotufo, L.V.; Jarmusch, A.K.; Dorrestein, P.C. Mass spectrometry-based metabolomics in microbiome investigations. Nature Reviews Microbiology 2021, 20, 143–160. [Google Scholar] [CrossRef]
- Liu, X.; Locasale, J.W. Metabolomics: A Primer. Trends in Biochemical Sciences 2017, 42, 274–284. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Sun, H.; Wang, P.; Han, Y.; Wang, X. Modern analytical techniques in metabolomics analysis. The Analyst 2012, 137, 293–300. [Google Scholar] [CrossRef]
- Bragg, L.; Tyson, G.W. Metagenomics Using Next-Generation Sequencing. In Methods in Molecular Biology; Humana Press, 2014; pp. 183–201. [Google Scholar] [CrossRef]
- Segata, N.; Boernigen, D.; Tickle, T.L.; Morgan, X.C.; Garrett, W.S.; Huttenhower, C. Computational meta’omics for microbial community studies. Molecular Systems Biology 2013, 9, 666. [Google Scholar] [CrossRef]
- Frioux, C.; Singh, D.; Korcsmaros, T.; Hildebrand, F. From bag-of-genes to bag-of-genomes: metabolic modelling of communities in the era of metagenome-assembled genomes. Computational and Structural Biotechnology Journal 2020, 18, 1722–1734. [Google Scholar] [CrossRef] [PubMed]
- Zorrilla, F.; Buric, F.; Patil, K.R.; Zelezniak, A. metaGEM: reconstruction of genome scale metabolic models directly from metagenomes. Nucleic Acids Research 2021, 49, e126–e126. [Google Scholar] [CrossRef] [PubMed]
- Bashiardes, S.; Zilberman-Schapira, G.; Elinav, E. Use of Metatranscriptomics in Microbiome Research. Bioinformatics and Biology Insights 2016, 10, BBI.S34610. [Google Scholar] [CrossRef] [PubMed]
- Gifford, S.M.; Sharma, S.; Rinta-Kanto, J.M.; Moran, M.A. Quantitative analysis of a deeply sequenced marine microbial metatranscriptome. The ISME Journal 2010, 5, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Gosalbes, M.J.; Durbán, A.; Pignatelli, M.; Abellan, J.J.; Jiménez-Hernández, N.; Pérez-Cobas, A.E.; Latorre, A.; Moya, A. Metatranscriptomic Approach to Analyze the Functional Human Gut Microbiota. PLoS ONE 2011, 6, e17447. [Google Scholar] [CrossRef] [PubMed]
- Mijakovic, I.; Macek, B. Impact of phosphoproteomics on studies of bacterial physiology. FEMS Microbiology Reviews 2012, 36, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Mashego, M.R.; Rumbold, K.; Mey, M.D.; Vandamme, E.; Soetaert, W.; Heijnen, J.J. Microbial metabolomics: past, present and future methodologies. Biotechnology Letters 2006, 29, 1–16. [Google Scholar] [CrossRef]
- Stitt, M.; Gibon, Y. Why measure enzyme activities in the era of systems biology? Trends in Plant Science 2014, 19, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Wiechert, W. 13C Metabolic Flux Analysis. Metabolic Engineering 2001, 3, 195–206. [Google Scholar] [CrossRef]
- Wang, D.; Bodovitz, S. Single cell analysis: the new frontier in ‘omics’. Trends in Biotechnology 2010, 28, 281–290. [Google Scholar] [CrossRef]
- Duncan, K.D.; Fyrestam, J.; Lanekoff, I. Advances in mass spectrometry based single-cell metabolomics. The Analyst 2019, 144, 782–793. [Google Scholar] [CrossRef]
- Zhou, M.; Li, Q.; Wang, R. Current Experimental Methods for Characterizing Protein-Protein Interactions. ChemMedChem 2016, 11, 738–756. [Google Scholar] [CrossRef]
- Maier, R.M.; Pepper, I.L. Bacterial Growth. In Environmental Microbiology; Elsevier, 2015; pp. 37–56. [Google Scholar] [CrossRef]
- Oh, Y.K.; Palsson, B.O.; Park, S.M.; Schilling, C.H.; Mahadevan, R. Genome-scale Reconstruction of Metabolic Network in Bacillus subtilis Based on High-throughput Phenotyping and Gene Essentiality Data. Journal of Biological Chemistry 2007, 282, 28791–28799. [Google Scholar] [CrossRef]
- Noble, J.E.; Knight, A.E.; Reason, A.J.; Matola, A.D.; Bailey, M.J.A. A Comparison of Protein Quantitation Assays for Biopharmaceutical Applications. Molecular Biotechnology 2007, 37, 99–111. [Google Scholar] [CrossRef]
- Noble, J.E.; Bailey, M.J. Chapter 8 Quantitation of Protein. In Methods in Enzymology; Elsevier, 2009; pp. 73–95. [Google Scholar] [CrossRef]
- Stouthamer, A.; Bettenhaussen, C. Utilization of energy for growth and maintenance in continuous and batch cultures of microorganisms. Biochimica et Biophysica Acta (BBA) - Reviews on Bioenergetics 1973, 301, 53–70. [Google Scholar] [CrossRef]
- Motta, S.; Pappalardo, F. Mathematical modeling of biological systems. Briefings in Bioinformatics 2012, 14, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Novère, N.L. Quantitative and logic modelling of molecular and gene networks. Nature Reviews Genetics 2015, 16, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Bruggeman, F.J.; Westerhoff, H.V. The nature of systems biology. Trends in Microbiology 2007, 15, 45–50. [Google Scholar] [CrossRef]
- Baker, R.E.; Peña, J.M.; Jayamohan, J.; Jérusalem, A. Mechanistic models versus machine learning, a fight worth fighting for the biological community? Biology Letters 2018, 14, 20170660. [Google Scholar] [CrossRef] [PubMed]
- Bouwmeester, R.; Gabriels, R.; Bossche, T.V.D.; Martens, L.; Degroeve, S. The Age of Data-Driven Proteomics: How Machine Learning Enables Novel Workflows. PROTEOMICS 2020, 20, 1900351. [Google Scholar] [CrossRef]
- Pasolli, E.; Truong, D.T.; Malik, F.; Waldron, L.; Segata, N. Machine Learning Meta-analysis of Large Metagenomic Datasets: Tools and Biological Insights. PLOS Computational Biology 2016, 12, e1004977. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Mou, M.; Wang, Y.; Luo, Y.; Zhu, F. MetaFS: Performance assessment of biomarker discovery in metaproteomics. Briefings in Bioinformatics 2020, 22. [Google Scholar] [CrossRef]
- Sydor, S.; Dandyk, C.; Schwerdt, J.; Manka, P.; Benndorf, D.; Lehmann, T.; Schallert, K.; Wolf, M.; Reichl, U.; Canbay, A.; Bechmann, L.P.; Heyer, R. Discovering Biomarkers for Non-Alcoholic Steatohepatitis Patients with and without Hepatocellular Carcinoma Using Fecal Metaproteomics. International Journal of Molecular Sciences 2022, 23, 8841. [Google Scholar] [CrossRef]
- Ninfa, A.J.; Ballou, D.P.; Benore, M. Fundamental laboratory approaches for biochemistry and biotechnology; John Wiley & Sons, 2009. [Google Scholar]
- Suthers, P.F.; Foster, C.J.; Sarkar, D.; Wang, L.; Maranas, C.D. Recent advances in constraint and machine learning-based metabolic modeling by leveraging stoichiometric balances, thermodynamic feasibility and kinetic law formalisms. Metabolic Engineering 2021, 63, 13–33. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Yuan, L.; Lu, H.; Li, G.; Chen, Y.; Engqvist, M.K.M.; Kerkhoven, E.J.; Nielsen, J. Deep learning-based kcat prediction enables improved enzyme-constrained model reconstruction. Nature Catalysis 2022, 5, 662–672. [Google Scholar] [CrossRef]
- Koutrouli, M.; Karatzas, E.; Paez-Espino, D.; Pavlopoulos, G.A. A Guide to Conquer the Biological Network Era Using Graph Theory. Frontiers in Bioengineering and Biotechnology 2020, 8. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; Jensen, L.J.; von Mering, C. The STRING database in 2021: customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Research 2020, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Walke, D.; Micheel, D.; Schallert, K.; Muth, T.; Broneske, D.; Saake, G.; Heyer, R. The importance of graph databases and graph learning for clinical applications. Database 2023, 2023, baad045. [Google Scholar] [CrossRef] [PubMed]
- Barbuti, R.; Gori, R.; Milazzo, P.; Nasti, L. A survey of gene regulatory networks modelling methods: from differential equations, to Boolean and qualitative bioinspired models. Journal of Membrane Computing 2020, 2, 207–226. [Google Scholar] [CrossRef]
- Wang, R.S.; Saadatpour, A.; Albert, R. Boolean modeling in systems biology: an overview of methodology and applications. Physical Biology 2012, 9, 055001. [Google Scholar] [CrossRef] [PubMed]
- Karlebach, G.; Shamir, R. Modelling and analysis of gene regulatory networks. Nature Reviews Molecular Cell Biology 2008, 9, 770–780. [Google Scholar] [CrossRef]
- Terzer, M.; Maynard, N.D.; Covert, M.W.; Stelling, J. Genome-scale metabolic networks. WIREs Systems Biology and Medicine 2009, 1, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Aldridge, B.B.; Burke, J.M.; Lauffenburger, D.A.; Sorger, P.K. Physicochemical modelling of cell signalling pathways. Nature Cell Biology 2006, 8, 1195–1203. [Google Scholar] [CrossRef] [PubMed]
- Palsson, B.Ø. Systems biology: simulation of dynamic network states; Cambridge University Press, 2011. [Google Scholar]
- Reed, J.L.; Palsson, B.Ø. Thirteen Years of Building Constraint-Based In Silico Models of Escherichia coli. Journal of Bacteriology 2003, 185, 2692–2699. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.S.; Ibarra, R.U.; Palsson, B.O. In silico predictions of Escherichia coli metabolic capabilities are consistent with experimental data. Nature Biotechnology 2001, 19, 125–130. [Google Scholar] [CrossRef]
- Bonarius, H.P.; Schmid, G.; Tramper, J. Flux analysis of underdetermined metabolic networks: the quest for the missing constraints. Trends in Biotechnology 1997, 15, 308–314. [Google Scholar] [CrossRef]
- Orth, J.D.; Fleming, R.M.T.; Palsson, B.Ø. Reconstruction and Use of Microbial Metabolic Networks: the Core Escherichia coli Metabolic Model as an Educational Guide. EcoSal Plus 2010, 4. [Google Scholar] [CrossRef]
- Gudmundsson, S.; Thiele, I. Computationally efficient flux variability analysis. BMC Bioinformatics 2010, 11. [Google Scholar] [CrossRef]
- Lewis, N.E.; Nagarajan, H.; Palsson, B.O. Constraining the metabolic genotype–phenotype relationship using a phylogeny of in silico methods. Nature Reviews Microbiology 2012, 10, 291–305. [Google Scholar] [CrossRef]
- Bordbar, A.; Monk, J.M.; King, Z.A.; Palsson, B.O. Constraint-based models predict metabolic and associated cellular functions. Nature Reviews Genetics 2014, 15, 107–120. [Google Scholar] [CrossRef]
- Borisov, N.M.; Chistopolsky, A.S.; Faeder, J.R.; Kholodenko, B.N. Domain-oriented reduction of rule-based network models. IET systems biology 2008, 2, 342–351. [Google Scholar] [CrossRef]
- Münzner, U.; Klipp, E.; Krantz, M. A comprehensive, mechanistically detailed, and executable model of the cell division cycle in Saccharomyces cerevisiae. Nature Communications 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Faeder, J.R.; Blinov, M.L.; Hlavacek, W.S. Rule-Based Modeling of Biochemical Systems with BioNetGen. In Methods in Molecular Biology; Humana Press, 2009; pp. 113–167. [Google Scholar] [CrossRef]
- Romers, J.; Thieme, S.; Münzner, U.; Krantz, M. A scalable method for parameter-free simulation and validation of mechanistic cellular signal transduction network models. npj Systems Biology and Applications 2020, 6. [Google Scholar] [CrossRef]
- Dada, J.O.; Mendes, P. Multi-scale modelling and simulation in systems biology. Integrative Biology 2011, 3, 86. [Google Scholar] [CrossRef] [PubMed]
- Mahadevan, R.; Edwards, J.S.; Doyle, F.J. Dynamic Flux Balance Analysis of Diauxic Growth in Escherichia coli. Biophysical Journal 2002, 83, 1331–1340. [Google Scholar] [CrossRef]
- Sun, G.; Ahn-Horst, T.A.; Covert, M.W. The E. coli Whole-Cell Modeling Project. EcoSal Plus 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Feist, A.M.; Herrgård, M.J.; Thiele, I.; Reed, J.L.; Palsson, B.Ø. Reconstruction of biochemical networks in microorganisms. Nature Reviews Microbiology 2008, 7, 129–143. [Google Scholar] [CrossRef]
- Gu, C.; Kim, G.B.; Kim, W.J.; Kim, H.U.; Lee, S.Y. Current status and applications of genome-scale metabolic models. Genome Biology 2019, 20. [Google Scholar] [CrossRef]
- Heinken, A.; Basile, A.; Thiele, I. Advances in constraint-based modelling of microbial communities. Current Opinion in Systems Biology 2021, 27, 100346. [Google Scholar] [CrossRef]
- Garza, D.R.; Gonze, D.; Zafeiropoulos, H.; Liu, B.; Faust, K. Metabolic models of human gut microbiota: Advances and challenges. Cell Systems 2023, 14, 109–121. [Google Scholar] [CrossRef]
- do Rosario Martins Conde, P.; Sauter, T.; Pfau, T. Constraint Based Modeling Going Multicellular. Frontiers in Molecular Biosciences 2016, 3. [Google Scholar] [CrossRef]
- Mendes, P.; Hoops, S.; Sahle, S.; Gauges, R.; Dada, J.; Kummer, U. Computational Modeling of Biochemical Networks Using COPASI. In Systems Biology; Humana Press, 2009; pp. 17–59. [Google Scholar] [CrossRef]
- Zimmermann, J.; Kaleta, C.; Waschina, S. gapseq: informed prediction of bacterial metabolic pathways and reconstruction of accurate metabolic models. Genome Biology 2021, 22. [Google Scholar] [CrossRef] [PubMed]
- Lieven, C.; Beber, M.E.; Olivier, B.G.; Bergmann, F.T.; Ataman, M.; Babaei, P.; Bartell, J.A.; Blank, L.M.; Chauhan, S.; Correia, K.; Diener, C.; Dräger, A.; Ebert, B.E.; Edirisinghe, J.N.; Faria, J.P.; Feist, A.M.; Fengos, G.; Fleming, R.M.T.; García-Jiménez, B.; Hatzimanikatis, V.; van Helvoirt, W.; Henry, C.S.; Hermjakob, H.; Herrgård, M.J.; Kaafarani, A.; Kim, H.U.; King, Z.; Klamt, S.; Klipp, E.; Koehorst, J.J.; König, M.; Lakshmanan, M.; Lee, D.Y.; Lee, S.Y.; Lee, S.; Lewis, N.E.; Liu, F.; Ma, H.; Machado, D.; Mahadevan, R.; Maia, P.; Mardinoglu, A.; Medlock, G.L.; Monk, J.M.; Nielsen, J.; Nielsen, L.K.; Nogales, J.; Nookaew, I.; Palsson, B.O.; Papin, J.A.; Patil, K.R.; Poolman, M.; Price, N.D.; Resendis-Antonio, O.; Richelle, A.; Rocha, I.; Sánchez, B.J.; Schaap, P.J.; Sheriff, R.S.M.; Shoaie, S.; Sonnenschein, N.; Teusink, B.; Vilaça, P.; Vik, J.O.; Wodke, J.A.H.; Xavier, J.C.; Yuan, Q.; Zakhartsev, M.; Zhang, C. MEMOTE for standardized genome-scale metabolic model testing. Nature Biotechnology 2020, 38, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.S.; Bernstein, H.C.; Weisenhorn, P.; Taylor, R.C.; Lee, J.Y.; Zucker, J.; Song, H.S. Microbial Community Metabolic Modeling: A Community Data-Driven Network Reconstruction. Journal of Cellular Physiology 2016, 231, 2339–2345. [Google Scholar] [CrossRef] [PubMed]
- Greenblum, S.; Turnbaugh, P.J.; Borenstein, E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proceedings of the National Academy of Sciences 2011, 109, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Klitgord, N.; Segre, D. The importance of compartmentalization in metabolic flux models: yeast as an ecosystem of organelles 2010. pp. 41–55.
- Biggs, M.B.; Medlock, G.L.; Kolling, G.L.; Papin, J.A. Metabolic network modeling of microbial communities. WIREs Systems Biology and Medicine 2015, 7, 317–334. [Google Scholar] [CrossRef] [PubMed]
- Zomorrodi, A.R.; Maranas, C.D. OptCom: A Multi-Level Optimization Framework for the Metabolic Modeling and Analysis of Microbial Communities. PLoS Computational Biology 2012, 8, e1002363. [Google Scholar] [CrossRef]
- Zelezniak, A.; Andrejev, S.; Ponomarova, O.; Mende, D.R.; Bork, P.; Patil, K.R. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proceedings of the National Academy of Sciences 2015, 112, 6449–6454. [Google Scholar] [CrossRef] [PubMed]
- Diener, C.; Gibbons, S.M.; Resendis-Antonio, O. MICOM: Metagenome-Scale Modeling To Infer Metabolic Interactions in the Gut Microbiota. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Magnúsdóttir, S.; Heinken, A.; Kutt, L.; Ravcheev, D.A.; Bauer, E.; Noronha, A.; Greenhalgh, K.; Jäger, C.; Baginska, J.; Wilmes, P.; Fleming, R.M.T.; Thiele, I. Generation of genome-scale metabolic reconstructions for 773 members of the human gut microbiota. Nature Biotechnology 2016, 35, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Heinken, A.; Hertel, J.; Acharya, G.; Ravcheev, D.A.; Nyga, M.; Okpala, O.E.; Hogan, M.; Magnúsdóttir, S.; Martinelli, F.; Nap, B.; Preciat, G.; Edirisinghe, J.N.; Henry, C.S.; Fleming, R.M.T.; Thiele, I. Genome-scale metabolic reconstruction of 7,302 human microorganisms for personalized medicine. Nature Biotechnology 2023. [Google Scholar] [CrossRef] [PubMed]
- Geier, B.; Sogin, E.M.; Michellod, D.; Janda, M.; Kompauer, M.; Spengler, B.; Dubilier, N.; Liebeke, M. Spatial metabolomics of in situ host–microbe interactions at the micrometre scale. Nature Microbiology 2020, 5, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Walke, D.; Schallert, K.; Ramesh, P.; Benndorf, D.; Lange, E.; Reichl, U.; Heyer, R. MPA_Pathway_Tool: User-Friendly, Automatic Assignment of Microbial Community Data on Metabolic Pathways. International Journal of Molecular Sciences 2021, 22, 10992. [Google Scholar] [CrossRef]
- Li, L.; Figeys, D. Proteomics and Metaproteomics Add Functional, Taxonomic and Biomass Dimensions to Modeling the Ecosystem at the Mucosal-luminal Interface. Molecular Cellular Proteomics 2020, 19, 1409–1417. [Google Scholar] [CrossRef] [PubMed]
- Rosario, D.; Boren, J.; Uhlen, M.; Proctor, G.; Aarsland, D.; Mardinoglu, A.; Shoaie, S. Systems Biology Approaches to Understand the Host–Microbiome Interactions in Neurodegenerative Diseases. Frontiers in Neuroscience 2020, 14. [Google Scholar] [CrossRef] [PubMed]
- Cermak, N.; Becker, J.W.; Knudsen, S.M.; Chisholm, S.W.; Manalis, S.R.; Polz, M.F. Direct single-cell biomass estimates for marine bacteria via Archimedes’ principle. The ISME Journal 2016, 11, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Scott, W.T.; Benito-Vaquerizo, S.; Zimmermann, J.; Bajić, D.; Heinken, A.; Suarez-Diez, M.; Schaap, P.J. A structured evaluation of genome-scale constraint-based modeling tools for microbial consortia. PLOS Computational Biology 2023, 19, e1011363. [Google Scholar] [CrossRef] [PubMed]
- Saez-Rodriguez, J.; Simeoni, L.; Lindquist, J.A.; Hemenway, R.; Bommhardt, U.; Arndt, B.; Haus, U.U.; Weismantel, R.; Gilles, E.D.; Klamt, S.; Schraven, B. A Logical Model Provides Insights into T Cell Receptor Signaling. PLoS Computational Biology 2007, 3, e163. [Google Scholar] [CrossRef] [PubMed]
- Romers, J.; Thieme, S.; Münzner, U.; Krantz, M. Using rxncon to develop rule based models. Modeling Biomolecular Site Dynamics: Methods and Protocols 2018. [Google Scholar] [CrossRef]
- Andrighetti, T.; Bohar, B.; Lemke, N.; Sudhakar, P.; Korcsmaros, T. MicrobioLink: An Integrated Computational Pipeline to Infer Functional Effects of Microbiome–Host Interactions. Cells 2020, 9, 1278. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Beltrán, J.F.; Brito, I.L. Host-microbiome protein-protein interactions capture disease-relevant pathways. Genome Biology 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Ritz, A.; Poirel, C.L.; Tegge, A.N.; Sharp, N.; Simmons, K.; Powell, A.; Kale, S.D.; Murali, T. Pathways on demand: automated reconstruction of human signaling networks. npj Systems Biology and Applications 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Türei, D.; Korcsmáros, T.; Saez-Rodriguez, J. OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nature Methods 2016, 13, 966–967. [Google Scholar] [CrossRef]
- Kandasamy, K.; Mohan, S.; Raju, R.; Keerthikumar, S.; Kumar, G.S.S.; Venugopal, A.K.; Telikicherla, D.; Navarro, D.J.; Mathivanan, S.; Pecquet, C.; Gollapudi, S.K.; Tattikota, S.G.; Mohan, S.; Padhukasahasram, H.; Subbannayya, Y.; Goel, R.; Jacob, H.K.C.; Zhong, J.; Sekhar, R.; Nanjappa, V.; Balakrishnan, L.; Subbaiah, R.; l Ramachandra, Y.; Rahiman, A.; s Keshava Prasad, T.; Lin, J.X.; Houtman, J.C.D.; Desiderio, S.; Renauld, J.C.; Constantinescu, S.; Ohara, O.; Hirano, T.; Kubo, M.; Singh, S.; Khatri, P.; Draghici, S.; Bader, G.D.; Sander, C.; Leonard, W.J.; Pandey, A. NetPath: a public resource of curated signal transduction pathways. Genome Biology 2010, 11, R3. [Google Scholar] [CrossRef] [PubMed]
- Martens, M.; Ammar, A.; Riutta, A.; Waagmeester, A.; Slenter, D.N.; Hanspers, K.; Miller, R.A.; Digles, D.; Lopes, E.N.; Ehrhart, F.; Dupuis, L.J.; Winckers, L.A.; Coort, S.L.; Willighagen, E.L.; Evelo, C.T.; Pico, A.R.; Kutmon, M. WikiPathways: connecting communities. Nucleic Acids Research 2020, 49, D613–D621. [Google Scholar] [CrossRef] [PubMed]
- King, Z.A.; Lu, J.; Dräger, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Research 2015, 44, D515–D522. [Google Scholar] [CrossRef]
- Seaver, S.M.D.; Liu, F.; Zhang, Q.; Jeffryes, J.; Faria, J.P.; Edirisinghe, J.N.; Mundy, M.; Chia, N.; Noor, E.; Beber, M.E.; Best, A.A.; DeJongh, M.; Kimbrel, J.A.; D’haeseleer, P.; McCorkle, S.R.; Bolton, J.R.; Pearson, E.; Canon, S.; Wood-Charlson, E.M.; Cottingham, R.W.; Arkin, A.P.; Henry, C.S. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Research 2020, 49, D575–D588. [Google Scholar] [CrossRef]
- Ronda, C.; Wang, H.H. Engineering temporal dynamics in microbial communities. Current Opinion in Microbiology 2022, 65, 47–55. [Google Scholar] [CrossRef]
- Ashyraliyev, M.; Fomekong-Nanfack, Y.; Kaandorp, J.A.; Blom, J.G. Systems biology: parameter estimation for biochemical models. FEBS Journal 2009, 276, 886–902. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.M.; Tymoczko, J.L.; Gatto, G.J.; Stryer, L. Enzyme: Grundlegende Konzepte und Kinetik. In Stryer Biochemie; Springer: Berlin/Heidelberg, Germany, 2017; pp. 255–297. [Google Scholar] [CrossRef]
- Choi, K.; Medley, J.K.; König, M.; Stocking, K.; Smith, L.; Gu, S.; Sauro, H.M. Tellurium: An extensible python-based modeling environment for systems and synthetic biology. Biosystems 2018, 171, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Albert, R.; Thakar, J. Boolean modeling: a logic-based dynamic approach for understanding signaling and regulatory networks and for making useful predictions. WIREs Systems Biology and Medicine 2014, 6, 353–369. [Google Scholar] [CrossRef] [PubMed]
- Hyduke, D.R.; Lewis, N.E.; Palsson, B.Ø. Analysis of omics data with genome-scale models of metabolism. Mol. BioSyst. 2013, 9, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Bordel, S.; Mardinoglu, A.; Pornputtapong, N.; Nookaew, I.; Nielsen, J. Reconstruction of Genome-Scale Active Metabolic Networks for 69 Human Cell Types and 16 Cancer Types Using INIT. PLoS Computational Biology 2012, 8, e1002518. [Google Scholar] [CrossRef] [PubMed]
- Agren, R.; Mardinoglu, A.; Asplund, A.; Kampf, C.; Uhlen, M.; Nielsen, J. Identification of anticancer drugs for hepatocellular carcinoma through personalized genome-scale metabolic modeling. Molecular Systems Biology 2014, 10. [Google Scholar] [CrossRef]
- Yizhak, K.; Benyamini, T.; Liebermeister, W.; Ruppin, E.; Shlomi, T. Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 2010, 26, i255–i260. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Reed, J.L. Integrating proteomic or transcriptomic data into metabolic models using linear bound flux balance analysis. Bioinformatics 2018, 34, 3882–3888. [Google Scholar] [CrossRef] [PubMed]
- Opdam, S.; Richelle, A.; Kellman, B.; Li, S.; Zielinski, D.C.; Lewis, N.E. A Systematic Evaluation of Methods for Tailoring Genome-Scale Metabolic Models. Cell Systems 2017, 4, 318–329.e6. [Google Scholar] [CrossRef]
- Kerkhoven, E.J. Advances in constraint-based models: methods for improved predictive power based on resource allocation constraints. Current Opinion in Microbiology 2022, 68, 102168. [Google Scholar] [CrossRef]
- Hädicke, O.; Klamt, S. EColiCore2: a reference network model of the central metabolism of Escherichia coli and relationships to its genome-scale parent model. Scientific Reports 2017, 7. [Google Scholar] [CrossRef]
- Robinson, J.L.; Kocabaş, P.; Wang, H.; Cholley, P.E.; Cook, D.; Nilsson, A.; Anton, M.; Ferreira, R.; Domenzain, I.; Billa, V.; Limeta, A.; Hedin, A.; Gustafsson, J.; Kerkhoven, E.J.; Svensson, L.T.; Palsson, B.O.; Mardinoglu, A.; Hansson, L.; Uhlén, M.; Nielsen, J. An atlas of human metabolism. Science Signaling 2020, 13. [Google Scholar] [CrossRef]
- Erdrich, P.; Steuer, R.; Klamt, S. An algorithm for the reduction of genome-scale metabolic network models to meaningful core models. BMC Systems Biology 2015, 9. [Google Scholar] [CrossRef] [PubMed]
- Danos, V.; Feret, J.; Fontana, W.; Harmer, R.; Krivine, J. Abstracting the Differential Semantics of Rule-Based Models: Exact and Automated Model Reduction. 2010 25th Annual IEEE Symposium on Logic in Computer Science. IEEE, 2010. [CrossRef]
- Wagner, D.; Schlüter, W. Vorhersage und Regelung der Methanproduktion durch maschinelles Lernen. Proceedings ASIM SST 2020. ARGESIM Publisher Vienna, 2020, Vol. 25. [CrossRef]
- Eng, A.; Borenstein, E. Microbial community design: methods, applications, and opportunities. Current Opinion in Biotechnology 2019, 58, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Morgan, X.C.; Tickle, T.L.; Sokol, H.; Gevers, D.; Devaney, K.L.; Ward, D.V.; Reyes, J.A.; Shah, S.A.; LeLeiko, N.; Snapper, S.B.; Bousvaros, A.; Korzenik, J.; Sands, B.E.; Xavier, R.J.; Huttenhower, C. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biology 2012, 13, R79. [Google Scholar] [CrossRef]
- Batstone, D.; Keller, J.; Angelidaki, I.; Kalyuzhnyi, S.; Pavlostathis, S.; Rozzi, A.; Sanders, W.; Siegrist, H.; Vavilin, V. The IWA Anaerobic Digestion Model No 1 (ADM1). Water Science and Technology 2002, 45, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Batstone, D.; Keller, J.; Steyer, J. A review of ADM1 extensions, applications, and analysis: 2002–2005. Water Science and Technology 2006, 54, 1–10. [Google Scholar] [CrossRef]
- Weinrich, S.; Koch, S.; Bonk, F.; Popp, D.; Benndorf, D.; Klamt, S.; Centler, F. Augmenting Biogas Process Modeling by Resolving Intracellular Metabolic Activity. Frontiers in Microbiology 2019, 10. [Google Scholar] [CrossRef]
- Xu, X.; Zarecki, R.; Medina, S.; Ofaim, S.; Liu, X.; Chen, C.; Hu, S.; Brom, D.; Gat, D.; Porob, S.; Eizenberg, H.; Ronen, Z.; Jiang, J.; Freilich, S. Modeling microbial communities from atrazine contaminated soils promotes the development of biostimulation solutions. The ISME Journal 2018, 13, 494–508. [Google Scholar] [CrossRef] [PubMed]
- Stein, R.R.; Tanoue, T.; Szabady, R.L.; Bhattarai, S.K.; Olle, B.; Norman, J.M.; Suda, W.; Oshima, K.; Hattori, M.; Gerber, G.K.; Sander, C.; Honda, K.; Bucci, V. Computer-guided design of optimal microbial consortia for immune system modulation. eLife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- van Leeuwen, P.T.; Brul, S.; Zhang, J.; Wortel, M.T. Synthetic microbial communities (SynComs) of the human gut: design, assembly, and applications. FEMS Microbiology Reviews 2023, 47. [Google Scholar] [CrossRef]
- Sorbara, M.T.; Pamer, E.G. Microbiome-based therapeutics. Nature Reviews Microbiology 2022, 20, 365–380. [Google Scholar] [CrossRef]
- Lee, T.A.; Steel, H. Cybergenetic control of microbial community composition. Frontiers in Bioengineering and Biotechnology 2022, 10, 1873. [Google Scholar] [CrossRef]
- Gutiérrez Mena, J.; Kumar, S.; Khammash, M. Dynamic cybergenetic control of bacterial co-culture composition via optogenetic feedback. Nature Communications 2022, 13. [Google Scholar] [CrossRef]
- Aditya, C.; Bertaux, F.; Batt, G.; Ruess, J. A light tunable differentiation system for the creation and control of consortia in yeast. Nature Communications 2021, 12. [Google Scholar] [CrossRef] [PubMed]
- Scott, S.R.; Hasty, J. Quorum Sensing Communication Modules for Microbial Consortia. ACS Synthetic Biology 2016, 5, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.K.; Collins, J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proceedings of the National Academy of Sciences 2007, 104, 11197–11202. [Google Scholar] [CrossRef]
- Buysschaert, B.; Kerckhof, F.; Vandamme, P.; De Baets, B.; Boon, N. Flow cytometric fingerprinting for microbial strain discrimination and physiological characterization. Cytometry Part A 2017, 93, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Khesali Aghtaei, H.; Püttker, S.; Maus, I.; Heyer, R.; Huang, L.; Sczyrba, A.; Reichl, U.; Benndorf, D. Adaptation of a microbial community to demand-oriented biological methanation. Biotechnology for Biofuels and Bioproducts 2022, 15. [Google Scholar] [CrossRef] [PubMed]
- Bensmann, A.; Hanke-Rauschenbach, R.; Heyer, R.; Kohrs, F.; Benndorf, D.; Reichl, U.; Sundmacher, K. Biological methanation of hydrogen within biogas plants: A model-based feasibility study. Applied Energy 2014, 134, 413–425. [Google Scholar] [CrossRef]
- Simon, D. Kalman filtering. Embedded systems programming 2001, 14, 72–79. [Google Scholar]
- Waltemath, D.; Wolkenhauer, O. How Modeling Standards, Software, and Initiatives Support Reproducibility in Systems Biology and Systems Medicine. IEEE Transactions on Biomedical Engineering 2016, 63, 1999–2006. [Google Scholar] [CrossRef]
- Stanford, N.J.; Scharm, M.; Dobson, P.D.; Golebiewski, M.; Hucka, M.; Kothamachu, V.B.; Nickerson, D.; Owen, S.; Pahle, J.; Wittig, U.; Waltemath, D.; Goble, C.; Mendes, P.; Snoep, J. Data Management in Computational Systems Biology: Exploring Standards, Tools, Databases, and Packaging Best Practices. In Methods in Molecular Biology; Springer: New York, 2019; pp. 285–314. [Google Scholar] [CrossRef]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; Bouwman, J.; Brookes, A.J.; Clark, T.; Crosas, M.; Dillo, I.; Dumon, O.; Edmunds, S.; Evelo, C.T.; Finkers, R.; Gonzalez-Beltran, A.; Gray, A.J.; Groth, P.; Goble, C.; Grethe, J.S.; Heringa, J.; ’t Hoen, P.A.; Hooft, R.; Kuhn, T.; Kok, R.; Kok, J.; Lusher, S.J.; Martone, M.E.; Mons, A.; Packer, A.L.; Persson, B.; Rocca-Serra, P.; Roos, M.; van Schaik, R.; Sansone, S.A.; Schultes, E.; Sengstag, T.; Slater, T.; Strawn, G.; Swertz, M.A.; Thompson, M.; van der Lei, J.; van Mulligen, E.; Velterop, J.; Waagmeester, A.; Wittenburg, P.; Wolstencroft, K.; Zhao, J.; Mons, B. The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data 2016, 3. [Google Scholar] [CrossRef]
- Boeckhout, M.; Zielhuis, G.A.; Bredenoord, A.L. The FAIR guiding principles for data stewardship: fair enough? European Journal of Human Genetics 2018, 26, 931–936. [Google Scholar] [CrossRef]
- Hughes, L.D.; Tsueng, G.; DiGiovanna, J.; Horvath, T.D.; Rasmussen, L.V.; Savidge, T.C.; Stoeger, T.; Turkarslan, S.; Wu, Q.; Wu, C.; Su, A.I.; and, L.P. Addressing barriers in FAIR data practices for biomedical data. Scientific Data 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, K.; Kananathan, S.; Roberts, M.G.; Meyer, J.P.; Shohan, M.U.S.; Xavier, A.; Maire, M.; Zyoud, A.; Men, J.; Ng, S.; Nguyen, T.V.N.; Glont, M.; Hermjakob, H.; Malik-Sheriff, R.S. Reproducibility in systems biology modelling. Molecular Systems Biology 2021, 17. [Google Scholar] [CrossRef]
- Wolstencroft, K.; Krebs, O.; Snoep, J.L.; Stanford, N.J.; Bacall, F.; Golebiewski, M.; Kuzyakiv, R.; Nguyen, Q.; Owen, S.; Soiland-Reyes, S.; Straszewski, J.; van Niekerk, D.D.; Williams, A.R.; Malmström, L.; Rinn, B.; Müller, W.; Goble, C. FAIRDOMHub: a repository and collaboration environment for sharing systems biology research. Nucleic Acids Research 2016, 45, D404–D407. [Google Scholar] [CrossRef] [PubMed]
- Hucka, M.; Nickerson, D.P.; Bader, G.D.; Bergmann, F.T.; Cooper, J.; Demir, E.; Garny, A.; Golebiewski, M.; Myers, C.J.; Schreiber, F.; Waltemath, D.; Novère, N.L. Promoting Coordinated Development of Community-Based Information Standards for Modeling in Biology: The COMBINE Initiative. Frontiers in Bioengineering and Biotechnology 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Waltemath, D.; Golebiewski, M.; Blinov, M.L.; Gleeson, P.; Hermjakob, H.; Hucka, M.; Inau, E.T.; Keating, S.M.; König, M.; Krebs, O.; Malik-Sheriff, R.S.; Nickerson, D.; Oberortner, E.; Sauro, H.M.; Schreiber, F.; Smith, L.; Stefan, M.I.; Wittig, U.; Myers, C.J. The first 10 years of the international coordination network for standards in systems and synthetic biology (COMBINE). Journal of Integrative Bioinformatics 2020, 17. [Google Scholar] [CrossRef] [PubMed]
- Naldi, A.; Monteiro, P.T.; Müssel, C.; Kestler, H.A.; Thieffry, D.; Xenarios, I.; Saez-Rodriguez, J.; Helikar, T.; and, C.C. Cooperative development of logical modelling standards and tools with CoLoMoTo. Bioinformatics 2015, 31, 1154–1159. [Google Scholar] [CrossRef] [PubMed]
- Tatka, L.T.; Smith, L.P.; Hellerstein, J.L.; Sauro, H.M. Adapting modeling and simulation credibility standards to computational systems biology. Journal of Translational Medicine 2023, 21. [Google Scholar] [CrossRef] [PubMed]
- Novère, N.L.; Finney, A.; Hucka, M.; Bhalla, U.S.; Campagne, F.; Collado-Vides, J.; Crampin, E.J.; Halstead, M.; Klipp, E.; Mendes, P.; Nielsen, P.; Sauro, H.; Shapiro, B.; Snoep, J.L.; Spence, H.D.; Wanner, B.L. Minimum information requested in the annotation of biochemical models (MIRIAM). Nature Biotechnology 2005, 23, 1509–1515. [Google Scholar] [CrossRef]
- Waltemath, D.; Adams, R.; Beard, D.A.; Bergmann, F.T.; Bhalla, U.S.; Britten, R.; Chelliah, V.; Cooling, M.T.; Cooper, J.; Crampin, E.J.; Garny, A.; Hoops, S.; Hucka, M.; Hunter, P.; Klipp, E.; Laibe, C.; Miller, A.K.; Moraru, I.; Nickerson, D.; Nielsen, P.; Nikolski, M.; Sahle, S.; Sauro, H.M.; Schmidt, H.; Snoep, J.L.; Tolle, D.; Wolkenhauer, O.; Novère, N.L. Minimum Information About a Simulation Experiment (MIASE). PLoS Computational Biology 2011, 7, e1001122. [Google Scholar] [CrossRef]
- Carey, M.A.; Dräger, A.; Beber, M.E.; Papin, J.A.; Yurkovich, J.T. Community standards to facilitate development and address challenges in metabolic modeling. Molecular Systems Biology 2020, 16. [Google Scholar] [CrossRef] [PubMed]
- Hucka, M.; Bergmann, F.T.; Chaouiya, C.; Dräger, A.; Hoops, S.; Keating, S.M.; König, M.; Novère, N.L.; Myers, C.J.; Olivier, B.G.; Sahle, S.; Schaff, J.C.; Sheriff, R.; Smith, L.P.; Waltemath, D.; Wilkinson, D.J.; Zhang, F. The Systems Biology Markup Language (SBML): Language Specification for Level 3 Version 2 Core Release 2. Journal of Integrative Bioinformatics 2019, 16. [Google Scholar] [CrossRef] [PubMed]
- Keating, S.M.; Waltemath, D.; König, M.; Zhang, F.; Dräger, A.; Chaouiya, C.; Bergmann, F.T.; Finney, A.; Gillespie, C.S.; Helikar, T.; Hoops, S.; Malik-Sheriff, R.S.; Moodie, S.L.; Moraru, I.I.; Myers, C.J.; Naldi, A.; Olivier, B.G.; Sahle, S.; Schaff, J.C.; Smith, L.P.; Swat, M.J.; Thieffry, D.; Watanabe, L.; Wilkinson, D.J.; Blinov, M.L.; Begley, K.; Faeder, J.R.; Gómez, H.F.; Hamm, T.M.; Inagaki, Y.; Liebermeister, W.; Lister, A.L.; Lucio, D.; Mjolsness, E.; Proctor, C.J.; Raman, K.; Rodriguez, N.; Shaffer, C.A.; Shapiro, B.E.; Stelling, J.; Swainston, N.; Tanimura, N.; Wagner, J.; Meier-Schellersheim, M.; Sauro, H.M.; Palsson, B.; Bolouri, H.; Kitano, H.; Funahashi, A.; Hermjakob, H.; Doyle, J.C.; Hucka, M.; Adams, R.R.; Allen, N.A.; Angermann, B.R.; Antoniotti, M.; Bader, G.D.; Červený, J.; Courtot, M.; Cox, C.D.; Pezze, P.D.; Demir, E.; Denney, W.S.; Dharuri, H.; Dorier, J.; Drasdo, D.; Ebrahim, A.; Eichner, J.; Elf, J.; Endler, L.; Evelo, C.T.; Flamm, C.; Fleming, R.M.; Fröhlich, M.; Glont, M.; Gonçalves, E.; Golebiewski, M.; Grabski, H.; Gutteridge, A.; Hachmeister, D.; Harris, L.A.; Heavner, B.D.; Henkel, R.; Hlavacek, W.S.; Hu, B.; Hyduke, D.R.; de Jong, H.; Juty, N.; Karp, P.D.; Karr, J.R.; Kell, D.B.; Keller, R.; Kiselev, I.; Klamt, S.; Klipp, E.; Knüpfer, C.; Kolpakov, F.; Krause, F.; Kutmon, M.; Laibe, C.; Lawless, C.; Li, L.; Loew, L.M.; Machne, R.; Matsuoka, Y.; Mendes, P.; Mi, H.; Mittag, F.; Monteiro, P.T.; Natarajan, K.N.; Nielsen, P.M.; Nguyen, T.; Palmisano, A.; Pettit, J.B.; Pfau, T.; Phair, R.D.; Radivoyevitch, T.; Rohwer, J.M.; Ruebenacker, O.A.; Saez-Rodriguez, J.; Scharm, M.; Schmidt, H.; Schreiber, F.; Schubert, M.; Schulte, R.; Sealfon, S.C.; Smallbone, K.; Soliman, S.; Stefan, M.I.; Sullivan, D.P.; Takahashi, K.; Teusink, B.; Tolnay, D.; Vazirabad, I.; von Kamp, A.; Wittig, U.; Wrzodek, C.; Wrzodek, F.; Xenarios, I.; Zhukova, A.; and, J.Z. SBML Level 3: an extensible format for the exchange and reuse of biological models. Molecular Systems Biology 2020, 16. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, T.; Hahn, J.; Bergmann, F.T.; Noor, E.; Klipp, E.; Liebermeister, W. SBtab: a flexible table format for data exchange in systems biology. Bioinformatics 2016, 32, 2559–2561. [Google Scholar] [CrossRef]
- Köhn, D.; Novère, N.L. SED-ML – An XML Format for the Implementation of the MIASE Guidelines. In Computational Methods in Systems Biology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 176–190. [Google Scholar] [CrossRef]
- Heirendt, L.; Arreckx, S.; Pfau, T.; Mendoza, S.N.; Richelle, A.; Heinken, A.; Haraldsdóttir, H.S.; Wachowiak, J.; Keating, S.M.; Vlasov, V.; Magnusdóttir, S.; Ng, C.Y.; Preciat, G.; Žagare, A.; Chan, S.H.J.; Aurich, M.K.; Clancy, C.M.; Modamio, J.; Sauls, J.T.; Noronha, A.; Bordbar, A.; Cousins, B.; Assal, D.C.E.; Valcarcel, L.V.; Apaolaza, I.; Ghaderi, S.; Ahookhosh, M.; Guebila, M.B.; Kostromins, A.; Sompairac, N.; Le, H.M.; Ma, D.; Sun, Y.; Wang, L.; Yurkovich, J.T.; Oliveira, M.A.P.; Vuong, P.T.; Assal, L.P.E.; Kuperstein, I.; Zinovyev, A.; Hinton, H.S.; Bryant, W.A.; Artacho, F.J.A.; Planes, F.J.; Stalidzans, E.; Maass, A.; Vempala, S.; Hucka, M.; Saunders, M.A.; Maranas, C.D.; Lewis, N.E.; Sauter, T.; Palsson, B.Ø.; Thiele, I.; Fleming, R.M.T. Creation and analysis of biochemical constraint-based models using the COBRA Toolbox v.3.0. Nature Protocols 2019, 14, 639–702. [Google Scholar] [CrossRef]
- Ebrahim, A.; Lerman, J.A.; Palsson, B.O.; Hyduke, D.R. COBRApy: COnstraints-Based Reconstruction and Analysis for Python. BMC Systems Biology 2013, 7. [Google Scholar] [CrossRef]
- Camborda, S.; Weder, J.N.; Töpfer, N. CobraMod: a pathway-centric curation tool for constraint-based metabolic models. Bioinformatics 2022, 38, 2654–2656. [Google Scholar] [CrossRef]
- Bauer, E.; Zimmermann, J.; Baldini, F.; Thiele, I.; Kaleta, C. BacArena: Individual-based metabolic modeling of heterogeneous microbes in complex communities. PLOS Computational Biology 2017, 13, e1005544. [Google Scholar] [CrossRef]
- Arkin, A.P.; Cottingham, R.W.; Henry, C.S.; Harris, N.L.; Stevens, R.L.; Maslov, S.; Dehal, P.; Ware, D.; Perez, F.; Canon, S.; Sneddon, M.W.; Henderson, M.L.; Riehl, W.J.; Murphy-Olson, D.; Chan, S.Y.; Kamimura, R.T.; Kumari, S.; Drake, M.M.; Brettin, T.S.; Glass, E.M.; Chivian, D.; Gunter, D.; Weston, D.J.; Allen, B.H.; Baumohl, J.; Best, A.A.; Bowen, B.; Brenner, S.E.; Bun, C.C.; Chandonia, J.M.; Chia, J.M.; Colasanti, R.; Conrad, N.; Davis, J.J.; Davison, B.H.; DeJongh, M.; Devoid, S.; Dietrich, E.; Dubchak, I.; Edirisinghe, J.N.; Fang, G.; Faria, J.P.; Frybarger, P.M.; Gerlach, W.; Gerstein, M.; Greiner, A.; Gurtowski, J.; Haun, H.L.; He, F.; Jain, R.; Joachimiak, M.P.; Keegan, K.P.; Kondo, S.; Kumar, V.; Land, M.L.; Meyer, F.; Mills, M.; Novichkov, P.S.; Oh, T.; Olsen, G.J.; Olson, R.; Parrello, B.; Pasternak, S.; Pearson, E.; Poon, S.S.; Price, G.A.; Ramakrishnan, S.; Ranjan, P.; Ronald, P.C.; Schatz, M.C.; Seaver, S.M.D.; Shukla, M.; Sutormin, R.A.; Syed, M.H.; Thomason, J.; Tintle, N.L.; Wang, D.; Xia, F.; Yoo, H.; Yoo, S.; Yu, D. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nature Biotechnology 2018, 36, 566–569. [Google Scholar] [CrossRef]
- Klarner, H.; Streck, A.; Siebert, H. PyBoolNet: a python package for the generation, analysis and visualization of boolean networks. Bioinformatics 2016, 33, 770–772. [Google Scholar] [CrossRef]
- von Kamp, A.; Thiele, S.; Hädicke, O.; Klamt, S. Use of CellNetAnalyzer in biotechnology and metabolic engineering. Journal of Biotechnology 2017, 261, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Müssel, C.; Hopfensitz, M.; Kestler, H.A. BoolNet—an R package for generation, reconstruction and analysis of Boolean networks. Bioinformatics 2010, 26, 1378–1380. [Google Scholar] [CrossRef] [PubMed]
- Helikar, T.; Kowal, B.; McClenathan, S.; Bruckner, M.; Rowley, T.; Madrahimov, A.; Wicks, B.; Shrestha, M.; Limbu, K.; Rogers, J.A. The Cell Collective: Toward an open and collaborative approach to systems biology. BMC Systems Biology 2012, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Funahashi, A.; Ghosh, S.; Kitano, H. Modeling and Simulation Using CellDesigner. In Transcription Factor Regulatory Networks; Springer: New York, 2014; pp. 121–145. [Google Scholar] [CrossRef]
- Garny, A.; Hunter, P.J. OpenCOR: a modular and interoperable approach to computational biology. Frontiers in Physiology 2015, 6. [Google Scholar] [CrossRef]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI—a COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [PubMed]
- Olivier, B.G.; Rohwer, J.M.; Hofmeyr, J.H.S. Modelling cellular systems with PySCeS. Bioinformatics 2004, 21, 560–561. [Google Scholar] [CrossRef] [PubMed]
- Lopez, C.F.; Muhlich, J.L.; Bachman, J.A.; Sorger, P.K. Programming biological models in Python using PySB. Molecular Systems Biology 2013, 9. [Google Scholar] [CrossRef] [PubMed]
- Starruß, J.; de Back, W.; Brusch, L.; Deutsch, A. Morpheus: a user-friendly modeling environment for multiscale and multicellular systems biology. Bioinformatics 2014, 30, 1331–1332. [Google Scholar] [CrossRef]
- Wittig, U.; Rey, M.; Weidemann, A.; Kania, R.; Müller, W. SABIO-RK: an updated resource for manually curated biochemical reaction kinetics. Nucleic Acids Research 2017, 46, D656–D660. [Google Scholar] [CrossRef]
- Mitchell, A.L.; Almeida, A.; Beracochea, M.; Boland, M.; Burgin, J.; Cochrane, G.; Crusoe, M.R.; Kale, V.; Potter, S.C.; Richardson, L.J.; Sakharova, E.; Scheremetjew, M.; Korobeynikov, A.; Shlemov, A.; Kunyavskaya, O.; Lapidus, A.; Finn, R.D. MGnify: the microbiome analysis resource in 2020. Nucleic Acids Research 2019. [Google Scholar] [CrossRef] [PubMed]
- Malik-Sheriff, R.S.; Glont, M.; Nguyen, T.V.N.; Tiwari, K.; Roberts, M.G.; Xavier, A.; Vu, M.T.; Men, J.; Maire, M.; Kananathan, S.; Fairbanks, E.L.; Meyer, J.P.; Arankalle, C.; Varusai, T.M.; Knight-Schrijver, V.; Li, L.; Dueñas-Roca, C.; Dass, G.; Keating, S.M.; Park, Y.M.; Buso, N.; Rodriguez, N.; Hucka, M.; Hermjakob, H. BioModels—15 years of sharing computational models in life science. Nucleic Acids Research 2019. [Google Scholar] [CrossRef] [PubMed]
- Moretti, S.; Tran, V.D.T.; Mehl, F.; Ibberson, M.; Pagni, M. MetaNetX/MNXref: unified namespace for metabolites and biochemical reactions in the context of metabolic models. Nucleic Acids Research 2020, 49, D570–D574. [Google Scholar] [CrossRef]
- Ankrah, N.Y.D.; Barker, B.E.; Song, J.; Wu, C.; McMullen, J.G.; Douglas, A.E. Predicted Metabolic Function of the Gut Microbiota of Drosophila melanogaster. mSystems 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Poline, J.B.; Dumas, G. Experimenting with reproducibility: a case study of robustness in bioinformatics. GigaScience 2018, 7. [Google Scholar] [CrossRef]
- Boettiger, C. An introduction to Docker for reproducible research. ACM SIGOPS Operating Systems Review 2015, 49, 71–79. [Google Scholar] [CrossRef]
- Pham, N.; van Heck, R.; van Dam, J.; Schaap, P.; Saccenti, E.; Suarez-Diez, M. Consistency, Inconsistency, and Ambiguity of Metabolite Names in Biochemical Databases Used for Genome-Scale Metabolic Modelling. Metabolites 2019, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Chindelevitch, L.; Stanley, S.; Hung, D.; Regev, A.; Berger, B. MetaMerge: scaling up genome-scale metabolic reconstructions, with application to Mycobacterium tuberculosis. Genome Biology 2012, 13, R6. [Google Scholar] [CrossRef]
- Ravikrishnan, A.; Raman, K. Critical assessment of genome-scale metabolic networks: the need for a unified standard. Briefings in Bioinformatics 2015, 16, 1057–1068. [Google Scholar] [CrossRef]
- Goodman, J.M.; Pletnev, I.; Thiessen, P.; Bolton, E.; Heller, S.R. InChI version 1.06: now more than 99.99. Journal of Cheminformatics 2021, 13. [Google Scholar] [CrossRef]
- Hastings, J.; Owen, G.; Dekker, A.; Ennis, M.; Kale, N.; Muthukrishnan, V.; Turner, S.; Swainston, N.; Mendes, P.; Steinbeck, C. ChEBI in 2016: Improved services and an expanding collection of metabolites. Nucleic Acids Research 2015, 44, D1214–D1219. [Google Scholar] [CrossRef]
- Vieira, L.S.; Laubenbacher, R.C. Computational models in systems biology: standards, dissemination, and best practices. Current Opinion in Biotechnology 2022, 75, 102702. [Google Scholar] [CrossRef]
- Papin, J.A.; Gabhann, F.M.; Sauro, H.M.; Nickerson, D.; Rampadarath, A. Improving reproducibility in computational biology research. PLOS Computational Biology 2020, 16, e1007881. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Schallert, K.; Knipper, L.; Sickmann, A.; Sczyrba, A.; Benndorf, D.; Heyer, R. Advances in the clinical use of metaproteomics. Expert Review of Proteomics 2023, 20, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Thiele, I.; Clancy, C.M.; Heinken, A.; Fleming, R.M. Quantitative systems pharmacology and the personalized drug–microbiota–diet axis. Current Opinion in Systems Biology 2017, 4, 43–52. [Google Scholar] [CrossRef]
- Thiele, I.; Sahoo, S.; Heinken, A.; Hertel, J.; Heirendt, L.; Aurich, M.K.; Fleming, R.M. Personalized whole-body models integrate metabolism, physiology, and the gut microbiome. Molecular Systems Biology 2020, 16. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Li, F.; Sánchez, B.J.; Zhu, Z.; Li, G.; Domenzain, I.; Marcišauskas, S.; Anton, P.M.; Lappa, D.; Lieven, C.; Beber, M.E.; Sonnenschein, N.; Kerkhoven, E.J.; Nielsen, J. A consensus S. cerevisiae metabolic model Yeast8 and its ecosystem for comprehensively probing cellular metabolism. Nature Communications 2019, 10. [Google Scholar] [CrossRef] [PubMed]
| 1 | With the term "microbiomes", we include the terms "microbial community" or "microbiota" and refer to any community of microorganisms living with or without a eukaryotic host. |
| 2 |
https://www.destatis.de/DE/Presse/Pressemitteilungen/2023/03/PD23_090_43312.html, date of access: December 13, 2023 |
| 3 |
https://www.umweltbundesamt.de/daten/energie/erneuerbare-energien-vermiedene-treibhausgase#stromerzeugung, date of access: December 13, 2023 |
Figure 2.
Characteristics of microbiomes with relevance for their general understanding and modeling.
Figure 2.
Characteristics of microbiomes with relevance for their general understanding and modeling.
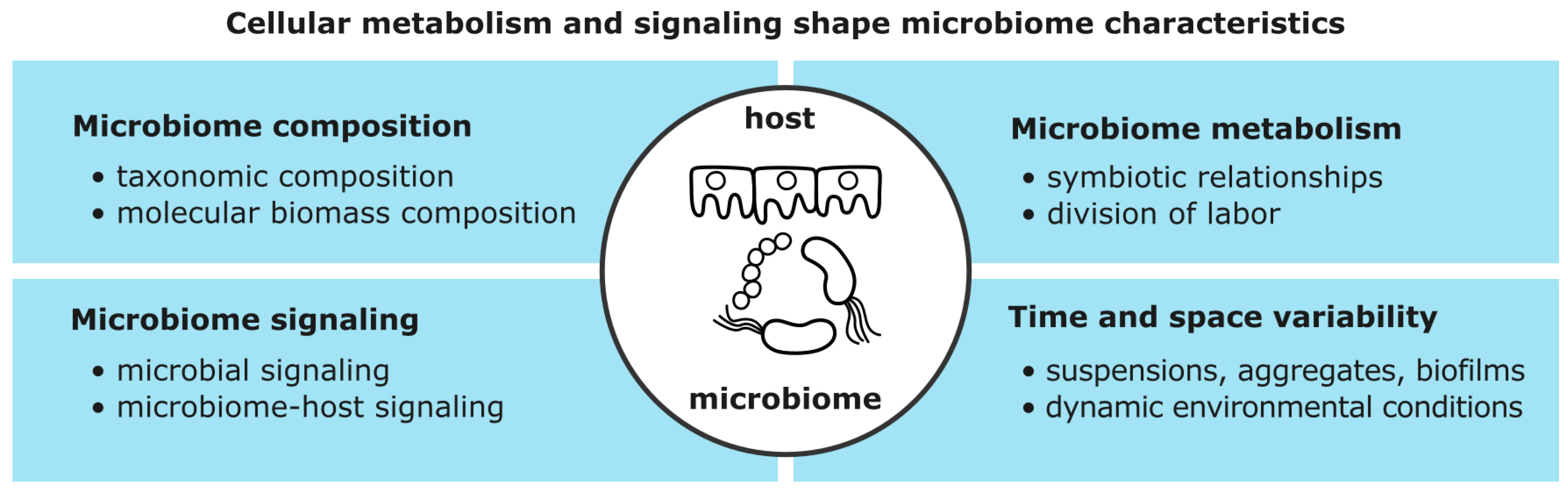
Figure 3.
(Meta)omics provide several workflows for investigating single species and microbiomes. Microbiome samples include microorganisms, host cells, and related molecules. Samples are prepared and measured according to standardized protocols. Data from measurements require bioinformatics workflows for their analysis. Metaproteomic analyses are integrated with meta-genomics and rely on functionally and taxonomically annotated gene and protein sequence data from public repositories. Information obtained from (meta)proteomics, (meta)genomics, and other omics measurements are integrated with microbiome modeling. Grey background - omics workflow(s), Arrow – flow of information, black background – omics method/modeling, blue background – workflow step, orange background – workflow step for absolute protein quantification, box with round edges - output data.
Figure 3.
(Meta)omics provide several workflows for investigating single species and microbiomes. Microbiome samples include microorganisms, host cells, and related molecules. Samples are prepared and measured according to standardized protocols. Data from measurements require bioinformatics workflows for their analysis. Metaproteomic analyses are integrated with meta-genomics and rely on functionally and taxonomically annotated gene and protein sequence data from public repositories. Information obtained from (meta)proteomics, (meta)genomics, and other omics measurements are integrated with microbiome modeling. Grey background - omics workflow(s), Arrow – flow of information, black background – omics method/modeling, blue background – workflow step, orange background – workflow step for absolute protein quantification, box with round edges - output data.
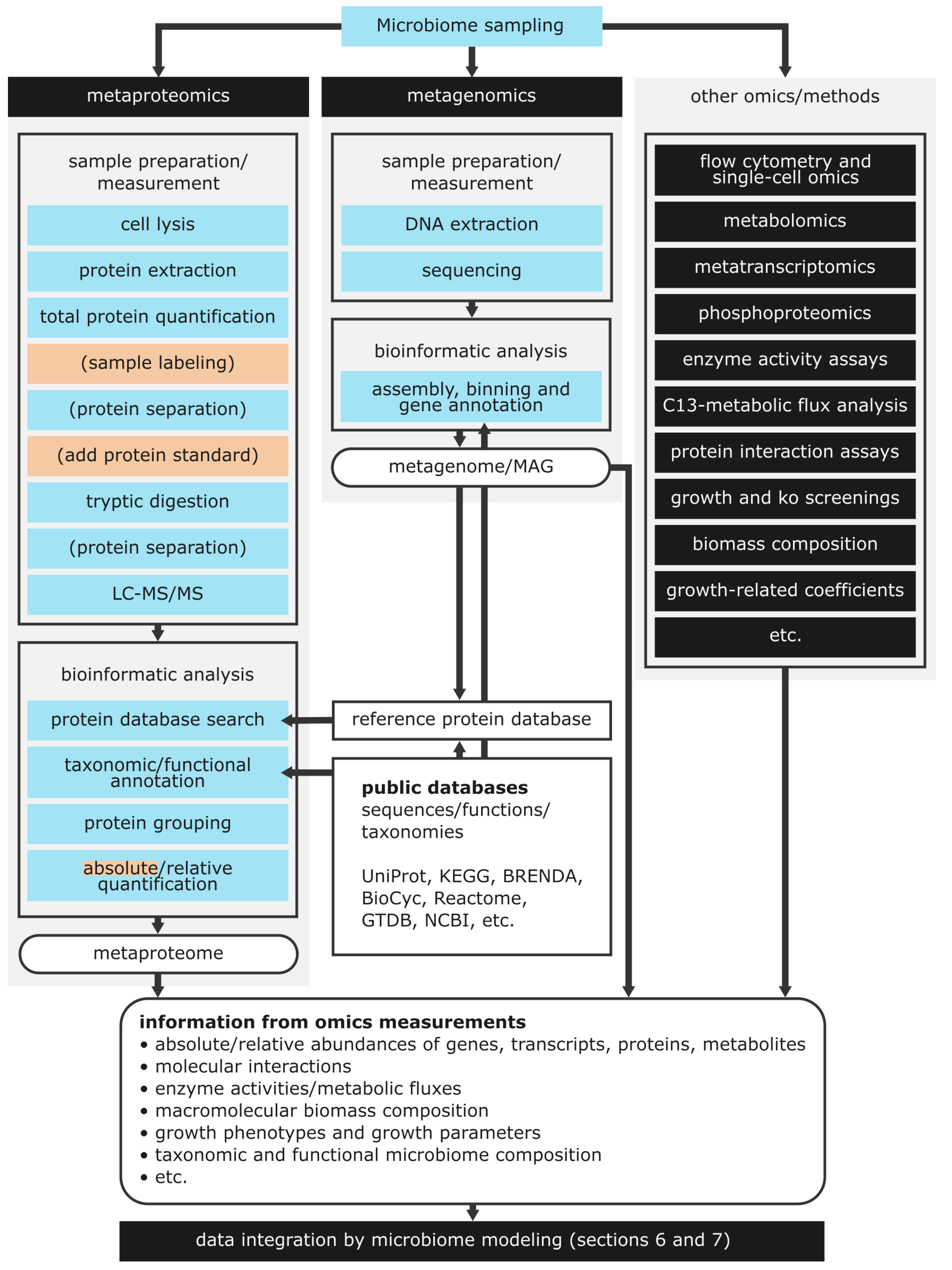
Figure 4.
Decision tree for basic model types. Deciding factors are, for example, the level of mechanistic detail, model scale, biological process, dynamics, and availability of data. All model types, except for multi-scale models, can be expressed as networks and thus analyzed by graph analysis methods. Combined models are applied to join different modeling formalisms, which is always required when models span different spatial scales.
Figure 4.
Decision tree for basic model types. Deciding factors are, for example, the level of mechanistic detail, model scale, biological process, dynamics, and availability of data. All model types, except for multi-scale models, can be expressed as networks and thus analyzed by graph analysis methods. Combined models are applied to join different modeling formalisms, which is always required when models span different spatial scales.
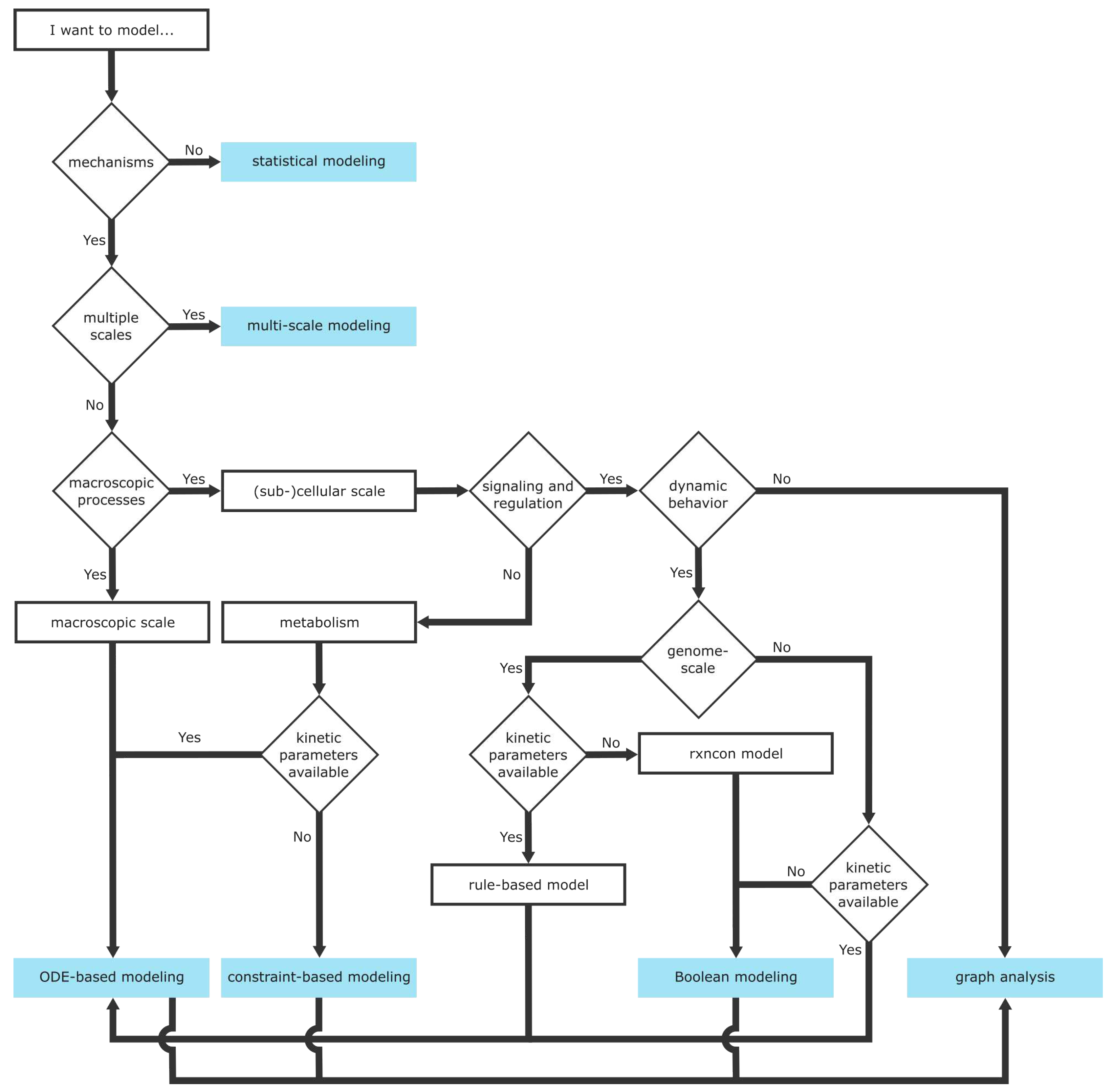
Figure 5.
Stoichiometric matrix. Matrix rows correspond to metabolites, matrix columns correspond to reactions. Matrix entries correspond to the stoichiometries of the respective metabolite in the respective reaction.
Figure 5.
Stoichiometric matrix. Matrix rows correspond to metabolites, matrix columns correspond to reactions. Matrix entries correspond to the stoichiometries of the respective metabolite in the respective reaction.
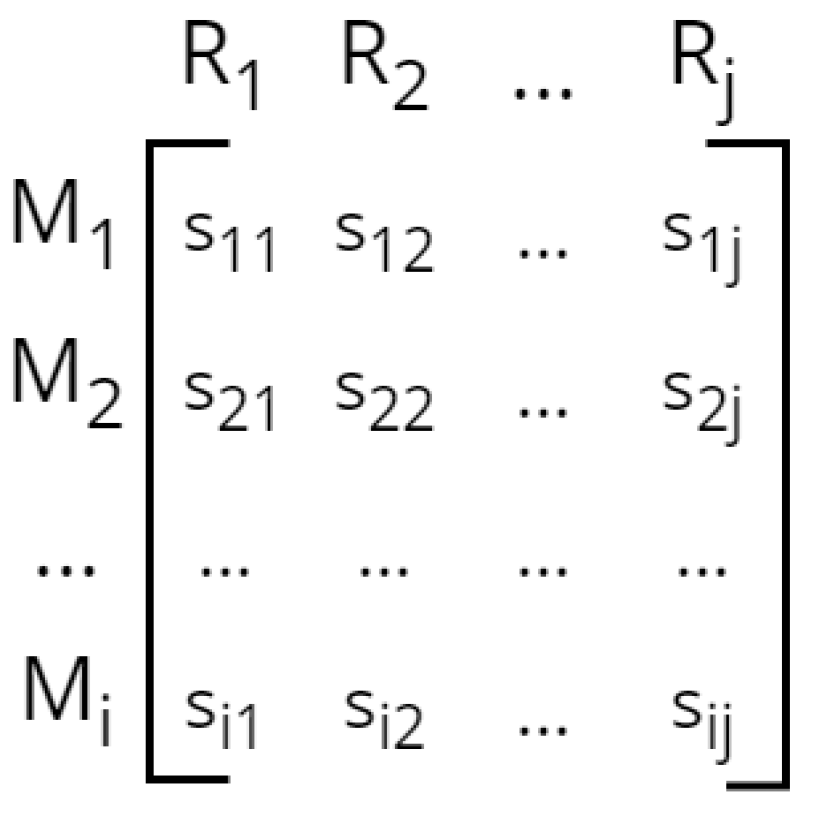
Figure 7.
Block diagram of a closed-loop control with feedback. The controller computes an action that affects the microbiome based on a control algorithm. The action is applied to the microbiome, which reacts with a measurable output. The output is compared with the desired reference value. The difference between both values is the error, which is fed back into the controller.
Figure 7.
Block diagram of a closed-loop control with feedback. The controller computes an action that affects the microbiome based on a control algorithm. The action is applied to the microbiome, which reacts with a measurable output. The output is compared with the desired reference value. The difference between both values is the error, which is fed back into the controller.

Table 1.
Pubmed queries.
| section | Script query | date | Number of hits | Topics |
|---|---|---|---|---|
| 4 | (microbiome) AND (microbial community) | November 7, 2023 | 4465 | Role and properties of microbiomes |
| 5 | (metaproteomics) OR (metagenomics) OR (metaomics) | November 7, 2023 | 4200 | Metaomics methods, metaproteomics, bioinformatic challenges |
| 6 | (computational model) AND ((metabolism) OR (regulation) OR (signaling)) | November 7, 2023 | 3163 | model types, modeling approaches applicable to metabolism and signaling |
| 7.1 | (biological network reconstruction) AND ((microbiome) OR (microbial community)) | November 7, 2023 | 6531 | Reconstruction of metabolic and signaling networks |
| 7.2 | (computational model) AND ((parameter estimation) OR (contextualization) OR (reduction)) | November 7, 2023 | 1035 | parameter estimation, context-specific models, reduction of model size |
| 8.1 and 8.2 | (computational modeling) AND ((microbiome) OR (microbial community)) | November 7, 2023 | 1035 | examples of prediction, optimization |
| 8.3 | (control algorithm) AND ((microbiome) OR (microbial community)) | November 7, 2023 | 4313 | microbiome control |
| 9 | (network modeling) AND (guidelines OR software OR repository) | November 7, 2023 | 1671 | FAIR, initiatives, standards, languages, software, repositories |
Table 2.
List of references to other (metaomics) methods that can be used in microbiome modeling.
| Data type/Method | References |
|---|---|
| WGS/amplicon | Bragg and Tyson [131], |
| Segata et al. [132], | |
| Frioux et al. [133], | |
| Jünemann et al. [12], | |
| Zorrilla et al. [134] | |
| metatranscriptomics | Bashiardes et al. [135], |
| Gifford et al. [136], | |
| Gosalbes et al. [137] | |
| phosphoproteomics | Terfve and Saez-Rodriguez [41], |
| Mijakovic and Macek [138] | |
| metabolomics | Mashego et al. [139], |
| Zhang et al. [130], | |
| Liu and Locasale [129], | |
| Bauermeister et al. [128] | |
| enzyme activity assays | Stitt and Gibon [140] |
| C13-metabolic flux analysis | Winter and Krömer [74], |
| Wiechert [141], | |
| Zamboni et al. [72] | |
| single-cell omics | Wang and Bodovitz [142], |
| Hatzenpichler et al. [126], | |
| Duncan et al. [143] | |
| protein interaction data | Zhou et al. [144] |
| growth screenings | Maier and Pepper [145], |
| Oh et al. [146] | |
| knock out screenings and gene essentiality data | Oh et al. [146] |
| biomass composition | Beck et al. [69], |
| Lachance et al. [70] | |
| total protein content | Noble et al. [147], |
| Noble and Bailey [148] | |
| maintenance coefficients | Stouthamer and Bettenhaussen [149], |
| Vos et al. [71] | |
| microscopy | Cesar and Huang [127] |
| flow cytometry | Hatzenpichler et al. [126], |
| Props et al. [125] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



