
Submitted:
10 January 2024
Posted:
10 January 2024
You are already at the latest version
Abstract
Four models of Ce(III) complexes with four ligands based on a 1,2,3-triazole derivatives were studied at four DFT levels. The effect of different ligands on the atomic charge value on the cerium ion was analyzed. Several relationships were clearly established between this atomic charge and that of the surrounding atoms, as well as with several geometric parameters of the ligand and with the molecular properties of the Ce(III) complex. The experimental IR and Raman spectra of the newly obtained Ce(III) complex with the (pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylate ligand were interpreted based on their comparison to the theoretical scaled ones using the scaling equations determined by two procedures and four DFT levels. Therefore, the structure predicted for the synthesized Ce(III) complex was clearly characterized and confirmed. The potential antioxidant action of Ce(III) complex was elucidated and compared with other Ln(III) complexes in order to find out the differences in their biological activity profiles.
Keywords:
cerium(III) complex
; 1
; 2
; 3-triazoles
; scaling
; vibrational analysis
; antioxidant activity
1. Introduction
Organometallic compounds with rare earth elements are extensively used in modern medicine [1,2], especially in cancer therapy, because of their molecular properties and low toxicities. Many studies have been conducted on them, revealing multiple possible applications. The authors, however, have not yet discovered reports that include triazoles as ligands in coordination interactions with rare earth elements. For the first time, in a previous manuscript, we have carried out this type of study using lanthanum(III) as a coordination ion [3]. In the present manuscript, several cerium complexes with a 1,2,3-triazole derivatives as ligands have been analyzed.
Cerium complexes have been reported to have important pharmaceutical properties. For example, the cerium-humic acid complex has bacteriostatic potency inhibiting the growth of several pathogenic bacterial strains [4], cerium-curcumin and cerium-quercetin complexes are toxic against both breast and melanoma cancer cells in photodynamic therapy (PDT) [5], cerium-ofloxacin and 2,2´-bipyridine complexes have antimicrobial and anticancer activities against breast and colon cell lines [6] and azamacrocyclic cerium complexes promote the hydrolysis of the phosphodiester bond of supercoiled DNA [7].
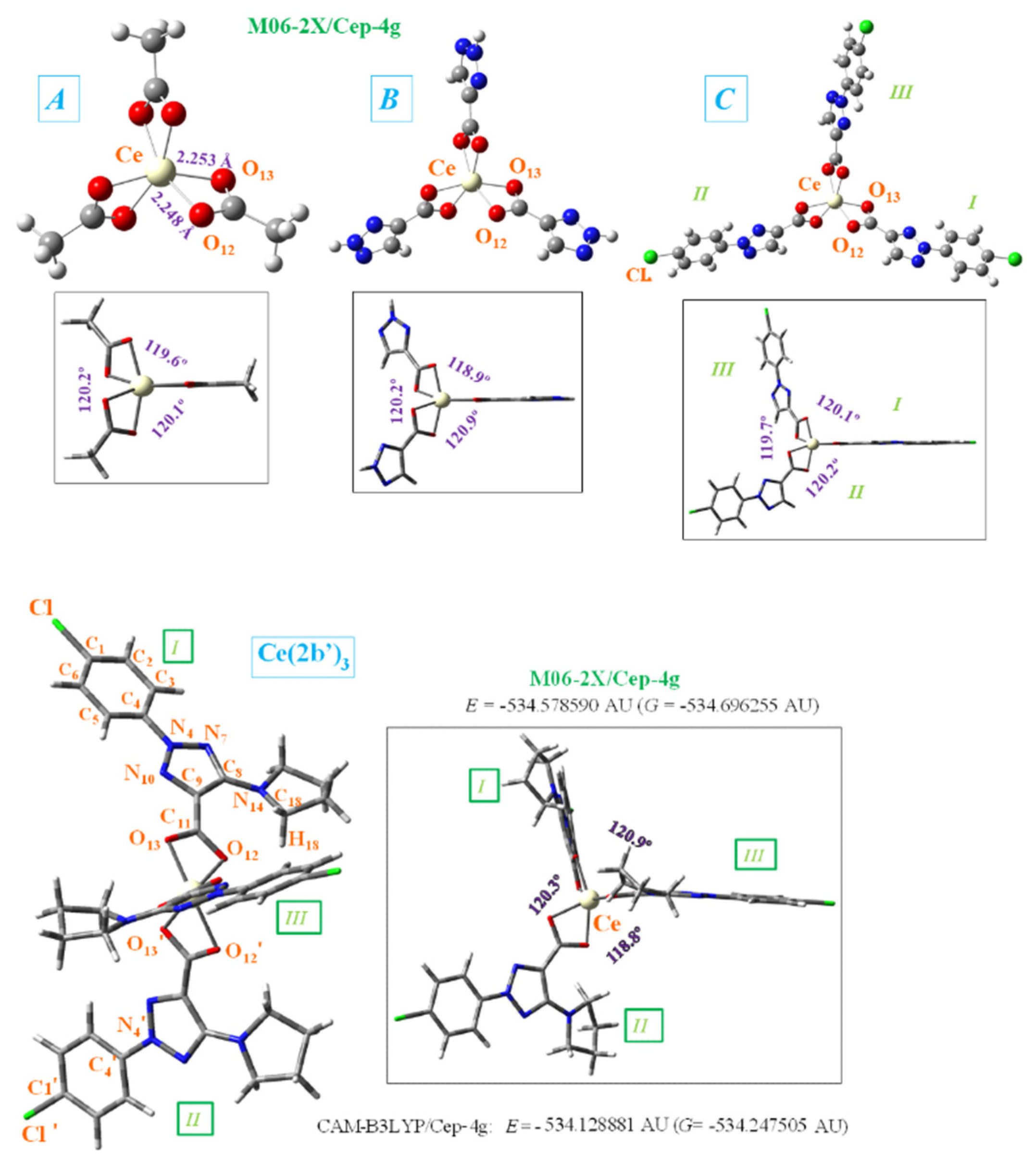
Among the four cerium complexes studied here, only that with the ligand 2-(4-chlorophenyl)-5-(pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylate sodium salt), reported by us earlier [8], has been synthesized. The ligand has been labeled as 2b (sodium salt) and 2b’ (its anionic form)[8] and this notation will be used here. The current study describes the synthesis, structural investigation and biological properties of new a cerium complex of the triazole ligand Ce(2b’)3. Due to the typical Ce+3 oxidation state, the synthetized complex can be assumed to be Ce(2b’)3. The cerium cation is coordinated through the carboxylate groups, linked in a three-dimensional coordination complex, Figure 1. This arrangement is similar to the one associated with the respective lanthanum(III) complex [3] as well as complexes of other carboxylic acid derivatives [9]. To reveal the influence of individual structural fragments on the features of the geometry and electronic structure of the Ce(2b’)3 complex, three simpler complexes A, B and C were visualized and used as models for quantum mechanical chemical calculations.
1,2,3-triazole derivatives are widely synthesized and studied due to their remarkable biological potentials [10,11]. The 2b’ ligand with its 1,2,3-triazole ring is expected to conserve these properties. In addition, the ligand has a strong-accepting COO- group that could provide the interaction [12] with the aminoacids of the MMP-2 metalloproteinase receptor of cancer cells, and it also has pyrrolidine and aryl rings that provide the complex liposolubility, thus facilitating cell membrane permeation.
In order to elucidate the metal–ligand binding mode and confirm the molecular structure proposed of the newly synthesized Ce(2b’)3 coordination complex, a thorough theoretical study was carried out. The rationale behind the chosen approach is that a single crystal structure of this complex, suitable for X-ray, cannot be obtained because of its low solubility. Therefore, the present study has the following objectives: i) to analyze the influence of various ligands on the charge and molecular properties of the Ce(III) complex; ii) To elucidate the impact of the different ligands on the properties of their respective complexes; iii) To assign the experimental IR and Raman spectra by comparison with the theoretical ones using accurate scaling procedures, and therefore to confirm the structures of the compounds.
The reported study is a continuation of our work with lanthanide(III) complexes with a number of biologically active ligands. The elucidation of the biological activity of the obtained Ln(III) complexes is a practical upgrade of the applied theoretical approaches and physicochemical methodologies for the characterization of this class of compounds, which enables the output of useful structure-activity correlations. The lanthanide(III) complexes studied have exhibited pronounced bioactivity, with the complexes invariably showing significantly higher potential than the initial ligands and inorganic salts [2,3]. Their influence on the levels of reactive species (RS), generated by various model systems has been studied, confirming the antioxidant capacity of the lanthanide complexes. The information obtained on the interaction of Ln(III) complexes with the model systems studied could serve to clarify the mechanisms of action in future experiments with these compounds in vitro and in vivo.
2. Results
The N-2-aryl-triazole ligands, including 2b, were prepared according to procedures reported in the literature [3,8,12]. The complex Ce(2b)3 was synthesized by the interaction of triazole sodium salt 2b with Ce(NO3)3·6H2O at a molar ratio of 3:1 in water solution (Scheme 1) with an yield (85%) and characterized by elemental analysis and vibrational spectroscopy.
Elemental analysis of Ce(III) complex of 2-(4-chlorophenyl)-5-(pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylic acid: (% calculated/found): Ce(2b’)3 ● 2H2O: C: 44.61/45.02; H: 4.00/3.78; N: 16.01/15.68; H2O: 3.43/3.98; Ce: 13.35/12.95, where 2b’ = C13H12N4O2Cl-.
The mode of bonding of the ligand to Ce(III) ions was elucidated by recording the IR and Raman spectra of the complex as compared with those of the free ligand and the theoretical predictions. The vibrational fundamentals from the IR and Raman spectra were analyzed by comparing these modes with those from the literature in combination with the results of our DFT calculations (i.e., harmonic vibrational wavenumbers and their Raman scattering activities) for the ligand and for the Ce(III) complex.
2.1. Molecular Structure of the Cerium Compex
The Cerium(III) ion, similar to other lanthanide ions, appears to coordinate well to oxygen atoms rather than nitrogen atoms [9]. That would be due to the large flexibility of the carboxylic oxygens, which facilitates bonding to the cerium(III) ion. Therefore, the starting geometry to be optimized was that with the Ce(III) ion coordinated through the COO- group with three ligands. In order to study the geometry structure of this coordination, four types of ligands were utilized, from a simpler form with methyl carboxylate (A-complex) through two “intermediate” forms to the 2b’ ligand (complex Ce(2b’)3), Figure 1. The structure incorporating 1,2,3-triazole-4-carboxylate ligand was labeled as B-complex, while that with 2-(4-chlorophenyl)- 2H-1,2,3-triazole-4-carboxylate ligand was labeled as C-complex. For simplicity, in the bottom of Figure 1 the total energy (E), which includes the ZPE (zero-point vibrational energy) correction, and the Gibbs energy (G) value were only shown for Ce(2b’)3 complex and with the CAM-B3LYP and M06-2X methods. Because the optimized structure by the B3LYP method appears noticeably distorted, it has been included in Figure S1 (Supplementary Material). That distortion is due to the lack of long range interactions of B3LYP for stabilizing the complex to a symmetric arrangement. The study of the geometry and atomic charges in the coordination of the Ce(III) ion with different ligands is in view of the potentially different bio-profiles of these complexes for their further application as possible anticancer and antioxidant agent.
In the complex formation with the cerium ion, the CO bonds of the ligands are lengthened as compared to the free form or in a dimer form, which gives rise to a slight shortening of the C9-C11 bond length. This feature in the triazole ring leads to a decrease of the N7=C8 and C9=N10 double bond character (an increase of their bond lengths) and shortening of the neighbor N4-N10 and C8-N14 bonds. It is noted that a lengthening of these bond lengths leads to more rotated triazole substituents, and thus they can interact more easily with other ligands, especially with the pyrrolidine ring. Therefore, with the cerium binding, the triazole ring bonds and angles are slightly modified, which consequently results in modification of their molecular properties.
The different ligands have little impact on the almost symmetric arrangement with the Ce(III) ion, Figure 1. Therefore, these ligands appear placed at angles (C11···Ce···C′11 and C′11···Ce···C′′11) very close to 120.0° by all methods and complexes, and with very little rotation with C9-C11···C′11-C′9 and C′9-C′11···C′′11-C′′9 torsional angles close to 0°, with the exception of the calculated values in Ce(2b’)3 complex by M06-2X method, -18.0° and 16.7°, respectively. As expected, the main differences appear in the coordination distances between the carboxylate oxygens and Ce(III) ion, which significantly affects the neighboring O-C11 (slightly, for example 1.434, 1.434, 1.436, 1, 437Å) and (and more prominently: 1.657, 1.587, 1,583, 1.576 Å) C11-C9 bond lengths. These differences can be observed in Table 1, which includes several selected optimized geometrical parameters in one of the ligands (labeled as I) at three DFT levels. The notation used for labeling the atoms is from that reported on 2b’ ligand [8]. Large differences also appear among the three DFT levels used, with the values by CAM-B3LYP closer to M06-2X than to B3LYP.
The optimized structure of the aryl ring in the C- and Ce(2b’)3 complexes is full planar at the three DFT levels used, as it is expected. It is almost coplanar with the triazole ring, with a C5-C4-N4-N10 torsional angle of ca. -0.4° in C-complex and slightly larger by effect of the pyrrolidine ring in Ce(2b’)3 complex, -4.7° by B3LYP and -2.3° by M06-2X. Similar values are also observed in the isolated ligands of C- and Ce(2b’)3 complexes, which indicate the very small effect of the Ce(III) ion in this coplanarity.
The triazole ring is also full planar in B- and C-complexes at all three DFT levels, and with torsional angle values lower than 0.5° in Ce(2b’)3 complex. By contrast, the pyrrolidine substituent appears out-of-plane, as it is found in cyclopyrrol, and out-of-coplanarity with the triazole ring plane, C9-C8-N14-C18 = -20.2° by M06-2X and 16.6° by CAM-B3LYP. However, this value is noticeably lower than that calculated in the 2b ligand alone at M06-2X/cep-4g level, -40.3°. That can be explained by the strong intramolecular H-bond O12···H18 between the carboxylate oxygen O12 and the pyrrolidine hydrogen H18, 1.711 Å, vs. 2.605 Å in Ce(2b‘)3 complex. This H-bond forces the rotation of the pyrrolidine ring.
In the Ce(2b’)3 complex, C11 carbon atom appears slightly rotated related to the triazole ring plane, being this rotation slightly smaller with O12 than with O13. Therefore, the torsional angle C8-C9-C=O12 with O12 has a smaller value of -1.7° by M06-2X (4.8° by CAM-B3LYP) vs that with O13, N10-C9-C=O13 of -8.2° (8.9° by CAM-B3LYP). This difference is due to a weak intramolecular H-bond O12···H18, 2.605 Å by M06-2X (2.332 Å by CAM-B3LYP), but it has not been observed in B- and C-complexes with lack of the pyrrolidine ring. Therefore, in these B- and C- complexes, the carboxylate group appears coplanar to the triazole ring plane, and with the same N10-C9-C=O13 and C8-C9-C=O12 torsional angle values of -0.2°. This intramolecular H-bond in the Ce(2b’)3 complex is also the main responsible of the difference in the Ce-O12 and Ce-O13 coordination distance values, and especially in the C-O12 and C-O13 bond lengths that are different in the Ce(2b’)3 complex, but are the same in A-, B- and C-complexes.
The different ligands also have little influence on the calculated bond angles, such as C=O12-Ce (90.3° by M06-2X in B-complex and 90.4° in Ce(2b’)3 complex), the C=O13-Ce angle (91.0° in B-complex and 91.6° in Ce(2b)3 complex), as well as the OCO, O12-Ce-O′13 and O13-Ce-O′12 angles, with differences lower than 2°, Table 1.
Significant differences are observed if we compare complex Ce(2b’)3 and La(2b’)3, published previously [[3]]. For example, the lengths of the C-O and C-C bonds surrounding the metal cation in the complex with cerium are noticeably shorter (by about 7-13% M06-2X), whereas the La3+-O bonds are noticeably shorter (on 10-11%). These differences in the spatial structures of the complexes may affect their behavior in biological systems and the magnitude or selectivity of their biological effects.
2.2. APT Atomic Charges and Relationships Established
In the complexes under theoretical study, the oxygen atoms have the highest negative charge, as expected, appearing therefore as the most reactive. Due to their high reactivity, it is also expected that they will have a key role in the H-bonding of these complexes to the aminoacids of the cancer cells proteins. N4 and N14 nitrogen atoms also have a large negative charge, although lower than that of the oxygen atoms and therefore, these atoms are also expected to participate in the biological activity of the synthesized complexes under study. N7 nitrogen has also negative charge, but very small, and in N10 it is positive but small. Thus, these nitrogen atoms are not expected to be involved in interaction with biological targets.
C8 and C11 carbon atoms have a high positive charge due to the fact that they appear bonded to highly negatively charged atoms. In the calculations, the cerium(III) ion appears positively charged around 3 - 4e depending on the DFT method used and complex studied, Table 2. Therefore, it is smaller by B3LYP (between 2.3e and 3.7e) and larger by CAM-B3LYP and M06-2X methods, between 2.4e and 4.5e. Both CAM-B3LYP and M06-2X methods have overall similar results. From A- to Ce(2b’)3 complexes the cerium charge is increased. That could be be explained by a better charge distribution on the cerium ion in larger ligands than in smaller ones, such as in A-complex.
The charge of the Ce(III) ion has a large influence in the bond lengths and atomic charges on the ligand atoms in the A-, B-, C- and Ce(2b’)3 complexes and therefore, several relationships can be well established at the B3LYP/Cep-4g, CAM-B3LYP/Cep-4g and M06-2X/Cep-4g levels, Figure 2. An increase in the positive calculated atomic APT charge on the cerium atom appears well linear related to an increment in the negative charge on O12 and O13 atoms, Figure 2a and 2b. The magnitude of the electronic charge lost by the Ce(III) ion is almost the same that was transferred to both O12 and O13 atoms. Large ligands appear to facilitate this negative electron transfer. In this transfer a large change when the triazole ring is inserted in the A-complex (B-complex), and a serious increase when the aryl ring is inserted (C-complex) are noted. The addition of the pyrrolidine ring increments this electron transfer a little. B3LYP differs in this point. The values by CAM-B3LYP and M06-2X methods are very close, differing somewhat to those calculated by B3LYP. These differences with B3LYP are increased in the remaining Figure 2c–e. Moreover, B3LYP shows large discrepancies in the calculated values for the Ce(2b)3 complex, and for this reason the results obtained by B3LYP were not discussed in the present manuscript.
The increment in the negative charge on the oxygen atoms leads to a similar increase in the positive charge on the C11 atom, Figure 2c. Consequently, the atomic charge on C9 is negative and increased, Figure 2d. These features show the facility for electron transfer in the present type of complexes.
The charge variation on the cerium ion was also related to the C9-C11 bond length, Figure 2e. Although this relation is not linear, there is a relationship between both parameters. In general, the charge variation on the ligand atoms leads to changes in the geometrical parameters. In Figure S2 we show several relations with the increase in the cerium charge, leading to an increase in the Ce-O12 and C=O12 bond lengths and a decrease in Ce-O13. The relationships plotted in Figure 2 have been also observed with the lanthanum ion [3]. As an example, the relationship calculated even with the better Lanl2dz basis set between the atomic APT charge on the lanthanum ion and the atomic charge on O12 is shown in Figure 2f.a demonstrated increasing both the positive charge value on central cerium cation and atom C11, and negative charge calculated for C9, O12 and O13 in comparison with La-cations and the same atoms in complex La(2b’).
2.3. Molecular Properties
Several parameters, such as rotational constants, heat capacity at constant volume, dipole moments, molecular orbitals and other global chemical descriptors in the A-, B-, C- and Ce(2b’)3 complexes have been determined and collected in Table 2. The large symmetry obtained in the optimized complexes leads to values of the rotational constant similar in the three (A, B, C) directions. Very small differences are observed in its calculation by the four DFT methods used. Its value decreases with the increment of the system complexity, i.e. from A- to Ce(2b’)3 complexes.
The Constant Volume Heat Capacity (Cv) value differs little between the DFT methods used, being slightly higher with CAM-B3LYP than with M06-2X, with the exception of A-complex. As it is expected, its value remarkably increases with the system complexity. Entropy (S) values also increase with the system complexity although in less amount.
The dipole moment values indicate an almost null water solubility in A- and B-complexes, but remarkable increase when the aryl and pyrrolidine rings are inserted in the ligands, the C- and Ce(2b’)3 complexes. In these last complexes, the calculated value by M06-2X is almost half that by the other three DFT methods.
The HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) values have been also determined. Their values slightly decreased with the system complexity, and they appear linear relationship with the atomic charge on the Ce ion, with the exception of the Ce(2b’)3 complex values, Figure 3a,b.
M06-2X and CAM-B3LYP methods calculate almost the same HOMO energy orbital value, but differing largely in the LUMO value. With these computed energies, the global chemical reactivity descriptors [13,14] were determined, which facilitates a better understanding of the stability and reactivity of the four Ce(III) complexes studied in the present manuscript. The energy gap (Eg) between HOMO and LUMO frontier orbitals (HOMO–LUMO gap) appears as one of the meaningful characteristics of molecules, and it facilitates the characterization of its chemical reactivity and kinetic stability. By all methods, its value was linearly related to the Ce atomic charge (Figure 3c) and it decreases with the complexity of the system, with the exception of Ce(2b’)3 complex that has a similar value to the C-complex. A high Eg shows that the molecule (or complex system) is less polarizable, which is in general related to a low chemical reactivity and hence to a high kinetic stability. Since the Eg values decrease with the system complexity, and the lowest value appears in the C-complex, it means that this C-complex is more reactive than Ce(2b’)3, the A-complex being the most stable one. The pyrrolidine ligand appears to slightly reduce the reactivity of the Ce(2b’)3 complex, but it gives liposolubility to the complex that is necessary to cross the cell membrane.
The low Eg values obtained in the Ce(2b’)3 complex with the four DFT methods used, as well as in the C-complex, reveal a noticeable chemical reactivity of them and therefore, small energies are required for excitation.
With the HOMO and LUMO energies, several global chemical reactivity descriptors were calculated according to the well-known formulas:
Due to the large reactivity of the Ce(2b’)3 complex, its computed ionization potential (IP) value appears slightly lower with the four DFT methods used. It is noted that the calculations by CAM-B3LYP and M06-2X methods lead to the same value, 0.390 eV, remaining very close to those determined by B3LYP and D3-B3LYP methods. Compared to the others Ce(III) complexes, these values are the lowest, increasing with the complexity decrease (the Ce atomic charge decrease).
The electron affinity (EA) appears to be noticeably lower than IP, and it slightly increases in the complexes as complexity decreases, as expected. The M06-2X method predicts higher values than the CAM-B3LYP method. The low electronegativity (χ) observed is in agreement with neutral complexes and also it slightly increases with the complexity decrease. The chemical hardness (η) and global softness (S) values indicate the opposition of a system to an alteration in its number of electrons. Their values are low in our complexes and also, they slightly increase with the complexity decrease. When the values of η are low, the system (or the complex system in our case) is named soft, while when they are high, the system is identified as hard. Therefore, according to the low values obtained in our complexes, they are soft systems, with small gap, and with an easily modified electron density.
In addition, the significant difference should be noted in the global chemical reactivity descriptors of the complex Ce(2b’)3 and the same complex with La3+ (La(2b’)3). For instance, all leveles rezults showed an increase in the dipole moment of the cerium complex. Such a discrepancy in properties can lead to different biological behavior and both qualitative and quantitative differences in their interaction with biological targets.
2.4. Vibrational Analysis
A detailed vibrational analysis was carried out on the newly synthetized Ce(2b’)3 complex. One of the goals of the present manuscript was to confirm the molecular structure proposed for this synthetized complex through a detailed analysis and comparison of the calculated and experimental IR and Raman spectra. For this task, a first theoretical-experimental comparison of the full IR spectra in the 3750-400 cm-1 range has been carried out and it is shown in Figure 4S with the scaled values obtained by the four DFT levels used. The same comparison but with the Raman spectra and in the 3750-0 cm-1 range is included in Figure 5S. The identification and characterization of all vibrations in the 3500-400 cm-1 range is collected in Table 1S (Supplementary Material Section) for the calculations with the CAM-B3LYP method, while the results with the M06-2X method are included in Table 2S. All values correspond to the most stable conformer, with the arrangement of the ligands such as shown in Figure 1. Because these tables are too long, a resume is included in Table 4 including only the frequencies with high IR or Raman intensities, or those characteristics of the complex. This Table 4 is divided in two parts corresponding to the results with the CAM-B3LYP and M06-2X methods.

Table 4.
Calculated, scaled and experimental wavenumbers (ν, cm-1) in the Ce(2b)3 complex by CAM-B3LYP and M06-2X methods. Relative infrared intensity (A) in %, relative Raman intensity (S) in %, and Raman depolarization ratios for plane (DP) and unpolarized incident light (DU). For each vibration of the tetramer, the wavenumber with the highest IR intensity is indicated in bold type and that with the highest Raman intensity is indicated in italic type. The relative IR and Raman intensities were shown only for these wavenumbers. DP and DU values were from most intense Raman line. The number of the ring mode corresponds to Wilson’s notation [14].
Table 4.
Calculated, scaled and experimental wavenumbers (ν, cm-1) in the Ce(2b)3 complex by CAM-B3LYP and M06-2X methods. Relative infrared intensity (A) in %, relative Raman intensity (S) in %, and Raman depolarization ratios for plane (DP) and unpolarized incident light (DU). For each vibration of the tetramer, the wavenumber with the highest IR intensity is indicated in bold type and that with the highest Raman intensity is indicated in italic type. The relative IR and Raman intensities were shown only for these wavenumbers. DP and DU values were from most intense Raman line. The number of the ring mode corresponds to Wilson’s notation [14].
| Calculated by CAM-B3LYP | scaled | Experimental | Characterization by CAM-B3LYP | ||||||
|---|---|---|---|---|---|---|---|---|---|
| ν | A | S | DP | DU | LSE | PSE | IR | Raman | |
|
3324, 3323, 3323 3312, 3312, 3312 3303, 3303, 3303 3211, 3211, 3211 1664, 1660, 1660 1644, 1644, 1644 1635, 1635, 1635 1558, 1525, 1525 1475, 1464, 1464 1385, 1383, 1383 1362, 1362, 1362 1347, 1339, 1339 1337, 1336, 1336 1306, 1305, 1305 1303, 1301, 1301 1259, 1248, 1248 1238, 1234, 1233 1216, 1215, 1215 1189, 1188, 1188 1168, 1168, 1168 1112, 1109, 1109 1011, 1010, 1010 956, 956, 956 932, 931, 931 887, 887, 887 840, 839, 839 742, 742, 742 713, 709, 709 567, 557, 557 555, 554, 554 474, 474, 473 344, 344, 343 |
47 4 13 5 56 0 0 97 100 7 2 26 7 6 3 40 41 3 1 2 9 4 0 1 0 4 9 4 12 4 16 3 |
1 3 0 3 35 0 92 100 60 7 1 12 16 1 19 3 7 3 0 1 2 1 0 0 4 22 0 11 2 1 2 1 |
0.70 0.64 0.75 0.01 0.09 0.22 0.18 0.75 0.04 0.04 0.75 0.10 0.05 0.75 0.75 0.35 0.75 0.75 0.75 0.75 0.75 0.75 0.73 0.09 0.02 0.04 0.75 0.07 0.75 0.75 0.05 0.75 |
0.82 0.78 0.86 0.02 0.16 0.36 0.31 0.86 0.08 0.07 0.86 0.18 0.09 0.86 0.86 0.52 0.86 0.86 0.86 0.86 0.86 0.86 0.84 0.16 0.04 0.08 0.86 0.13 0.86 0.86 0.10 0.86 |
3057 3048 3040 2960 1623 1605 1598 1503 1450 1380 1362 1342 1339 1312 1309 1263 1250 1234 1211 1194 1143 1057 1011 989 951 909 826 797 666 663 594 481 |
3043 3034 3028 2957 1671 1653 1645 1546 1491 1417 1398 1377 1374 1345 1342 1293 1279 1262 1237 1219 1164 1071 1020 996 954 908 815 783 636 633 554 426 |
2968.1 s 2872.1 m 1577.2 br, vs 1500.1 s 1484.4 vs 1418.0 w 1398.3 m 1372.2 vs 1343.1 s 1301.8 vs 1285.1 s 1246.8 m 1218.1 m 1178.3 m 1091.2 vs 1011.9 m 969.1 vs 914.3 w 829.9 vs 804.8 m 654 m 647.1 m 509.0 m 466.9 m |
1595.0 vs 1504.5 s 1376.8 vs 1171.5 w 1089.9 m 1012.8 w 970.2 s |
20b, ν(C5-H) in aryl (100) 7b, ν(C6-H) in aryl (100) 20a, ν(C2-H) in aryl (100) νs(C-H) in C15H2 in pyrrolidine (100) ν(C8-N14) (45) + νs(CC) (34) 8b, ν(C=C) in aryl (89) 8a, ν(C=C) in aryl (82) ν(C9-C11)+νs(COO)+ν(C4N)+19a,ν(CC,CH) 19a,ν(CC,CH)+ν(C9-C11)+ν(triazol) νas(NNN, CN) +19a,ν(CC)(35)+νs(COO) 19b, ν(CC)(85) ν(triazol)+νas(CO12)+3,δ(CH)+δ(pyrrolidine) νs(COO) (49) + ν(triazol) + δ(pyrrolidine) δ(C-H) in pyrrolidine +νas(COO) νas(CO12) + 3,δ(C-H) aryl + δ(triazol) νas(CO13) + ν(C-N) triazol + Γ(pyrrolidine) νas(CO13) + ν(C-N) triazol + Γ(pyrrolidine) νas(CO12) + ν(C-N) triazol + δ(C-H) in aryl δs(C-H) in pyrrolidine 3, δ(C-H) in aryl νs(COO) + ν(NCCN) + γs(CC,CH) νs(COO) + ν(triazol) + 18a, δ(CC,CH) γas(CC,CH) in pyrrolidine νas(triazol) + νs(COO) + γ(CC,CH) γ(CC) pyrrolidine +νas(NNN) νas(NNN) + 12, δ(CCC) in aryl 17b, γ(C-H) in aryl ν(COO) + ν(triazol) + γ(CC) pyrrolidine Γ(triazol) +δ(COO) + 4,γ(CCC) δ(COO)+ r(triazol) + 4,γCCC) δ(COO) + δ(triazol) + δ(CC) in aryl δ(COO) + δ(triazol) + ν(aryl,C-Cl) |
| Calculated by M06-2X | scaled | Experimental | Characterization by M06-2X | ||||
| ν | A | S | LSE | PSE | IR | Raman | |
| 3326, 3326, 3325 3314, 3314, 3313 3308, 3308, 3307 3223, 3223, 3223 1702, 1700, 1698 1652, 1651, 1651 1646, 1646, 1645 1584, 1550, 1549 1493, 1482, 1481 1407, 1405, 1405 1378, 1378, 1378 1358, 1357, 1352 1345, 1344, 1344 1330, 1325, 1325 1309, 1308, 1308 1283, 1274, 1269 1242, 1242, 1241 1184, 1184, 1183 1168, 1166, 1166 1134, 1132, 1131 1119, 1119, 1118 1027, 1025, 1025 959, 958, 958 945, 944, 944 851, 851, 851 751, 750, 750 725, 724, 723 575, 573, 573 572, 565, 564 478, 476, 475 372, 370, 363 |
37 3 15 6 46 0 1 100 94 5 2 28 8 2 0 81 1 1 5 8 1 4 0 3 2 7 3 0 11 16 4 |
1 1 0 5 20 1 100 73 98 10 0 9 19 14 4 11 4 0 1 1 1 1 1 1 19 0 10 1 2 2 1 |
3050 3039 3034 2961 1649 1607 1603 1520 1461 1395 1372 1349 1343 1326 1312 1278 1255 1205 1189 1159 1149 1068 1010 998 918 831 808 680 671 594 498 |
3036 3027 3022 2957 1699 1655 1651 1565 1503 1434 1409 1386 1378 1361 1345 1309 1285 1231 1214 1182 1170 1083 1019 1006 918 821 795 652 641 554 443 |
2968.1 s 2872.1 m 1577.2 br, vs 1500.1 s 1484.4 vs 1418.0 w 1398.3 m 1372.2 vs 1343.1 s 1301.8 vs 1285.1 s 1246.8 m 1218.1 m 1178.3 m 1091.2 vs 1011.9 m 969.1 vs 914.3 w 829.9 vs 804.8 m 654 m 647.1 m 509.0 m 466.9 m |
1595.0 vs 1504.5 s 1376.8 vs 1171.5 w 1089.9 m 1012.8 w 970.2 s |
7b, ν(C5-H) in aryl (100) 20b, ν(C6-H) in aryl (100) 20a, ν(C2-H) in aryl (100) νs(C-H) in C15H2 pyrrolidine (100) ν(C8-N14) + νs(NNN-NC) in triazol 8b, ν(C=C) in aryl (93) 8a, ν(C=C) in aryl (93) ν(C9-C11)+νs(COO)+νs(CCN) triazol + ν(C4-N4) 19a, ν(CC,CH) + ν(C4-N4) +ν(C9-C11) νas(NNN) in triazol + 19a,ν(CC) in aryl 19b, ν(CC,CH) in aryl νas(COO) + νs(C9-N10) triazol + 19b, ν(CC,CH) in aryl νs(NNN) + νas(COO) +19a,ν(CC) + δ(CC) pyrrolidine νas(COO)+νas(triazol) +νas(CNC) pyrrolidine+ 19a,ν(CC) δs(C-H) in pyrrolidine + νs(NNN) ν(CO13)(45)+ν(CN) triazol (40)+ δs(CC,CH) pyrrolidine (12) νas(CO12) +νs(C9NN) triazol + δs(CC,CH) in pyrrolidine γas(C-H) in pyrrolidine 3, δ(CH) + γas(C-H) in pyrrolidine + νs(NNN) νs(COO)+ ν(triazol)+δas(C-H) in pyrrolidine γas(C-H) in pyrrolidine + νs(triazol) νs(NNN) + 18a, δ(C-H) in aryl δas(CC, CH) in pyrrolidine νas(NC9-C8) + δas(CC, CH) in pyrrolidine νas(NNN)(36) + 12, δ(CCC) in aryl 17b, γ (C-H) in aryl δas(COO) + γ(CC, CH) pyrrolidine +δ(triazol) γ(triazol) + γas(COO) + 8b, δ(CCC) in aryl γas(COO) + γ(triazol) δas(COO) + δ(triazol) + δ(CC) in aryl δas(COO) + δ(triazol) + δ(CC) in aryl |
2.4.1. General Comparison of the IR and Raman Spectra
In a first comparison of the scaled IR spectra with the experimental one plotted in Figure 4S the following has been observed:
- (i)
- A good accordance between the scaled theoretical spectra with the experimental one has been found, in particular, the scaled strongest vibrations have their correspondence in the experimental spectrum. This feature confirms the scaling carried out on the calculated wavenumbers, therefore the theoretical methods used appear appropriate. Thus, in general, the assignments proposed could be considered true, identifying most of the computed modes in their normal ranges.
- (ii)
- A very broad and strong band centered at 3400.8 cm-1 has been observed in the experimental spectrum that by its position, it can be only assigned to the O-H stretching ν(O-H) mode, corresponding to water molecules strongly H-bonded to the three 2b’ ligands of Ce(2b’)3 complex. These water molecules were not included in our optimized theoretical complex, but in previous studies with lanthanum(III) ion we noted that these water molecules only slightly affect the carboxylate group, the other groups remaining unaffected [3,9]. This hydration appears due to the spatial arrangement of these ligands in the complex, which leaves cavities that can be occupied by water molecules. As it is expected, this band is not observed in the Raman spectrum.
- (iii)
- Another broad but very strong experimental IR band is observed at 1577.2 cm-1. Its large broadening can be interpreted as a result of the additional contribution of the in-plane bending δ(O-H) mode of these hydrated water molecules to the main assignment of this band corresponding to the C8-N14 and C-C stretching, Table 4.
- (iv)
- A noticeable resemblance between the scaled spectra obtained by the D3-B3LYP, CAM-B3LYP and M06-2X methods that include long-range correction has been observed, while that by B3LYP differs noticeably. As compared to the experimental spectrum, the two best methods are CAM-B3LYP and M06-2X and for this reason, their spectra were analyzed in detail and included in Table 4. In this analysis, the scaled wavenumbers by CAM-B3LYP appear slightly better than by M06-2X, that is why it has been mainly utilized in the experimental spectra assignment.
- (v)
- Coordination of the 2b’ ligands to Ce(III) ion remarkably modified the IR and Raman spectra appearing distinct of that found with 2b ligand anion alone [12].
The same analysis has been carried out with the Raman spectra, Figure 5S. In this case, the scaled spectra show large differences among the DFT methods. CAM-B3LYP and M06-2X methods again seem to be better with bands showing good agreement with the experimental ones. Unfortunately, the experimental spectrum appears with a noticeable background noise, which complicate the detection and further analysis of all weak lines, making this comparison difficult. The appearance of a broad Raman line with medium intensity at 73.1 cm-1 was observed in the experimental spectrum, which has not been detected in the theoretical spectra. Due to this feature, it was not assigned in Table 4.
2.4.2. Specific Comparison of the IR and Raman Spectra
For a comprehensive, specific and better comparison of the distinct scaled and experimental frequencies, the spectra were divided in three ranges. In the IR spectra, these ranges were from 3750-2600 cm-1 (Figure 4), from 1800-1000 cm-1 (Figure 5) and from 1000-400 cm-1 (Figure 6). In the Raman spectrum the comparison was only carried out in the 1800-800 cm-1 range, Figure 7, in which the Raman lines can be clearly identified, because of the noticeable background noise of the experimental spectrum. In these figures, for simplicity, only the assignment of strong and characteristic vibrational modes was included.
For this purpose, a resume with the most characteristic frequencies determined in the Ce(2b’)3 complex with the CAM-B3LYP and M06-2X methods is shown in Table 4. The first column collects the three computed frequencies for each mode related to the three 2b’ ligands of the complex. Of these three frequencies, that with higher computed IR intensity was typed in bold style, whereas that with higher Raman intensity was typed in italic style. This notation was not used if the intensity of these three wavenumbers is similar, or very weak. The second and third columns list the relative IR and Raman intensities (%) corresponding to the frequencies of the first column typed in bold style and in italic style, respectively. These relative intensities were determined by normalizing each computed data to the strongest intensity of the spectrum. In the Table with the CAM-B3LYP method two columns are included, the fourth and fifth, with the Raman depolarization ratios for plane (DP) and unpolarized incident light (DU), respectively. The values included correspond to the wavenumbers typed in italic type in the first column. The scaled wavenumbers by the LSE or PSE procedures were listed in the next two columns. For simplicity, only the wavenumbers typed in bold type are included as scaled values in these two columns. The observed experimental IR and Raman bands with their corresponding intensities were collected in the next two columns, respectively. Finally, the last column shows the principal characterization of the computed wavenumbers determined with the CAM-B3LYP (first part of the Table) and M06-2X (second part of the Table) methods. The % contribution (PEDs) of the distinct modes to a calculated wavenumber was added only in few cases.
The main purpose of this vibrational study is the identification and characterization of the synthetized Ce(2b’)3 coordination complex. For this reason, the principal interest was focused on the strongest IR and Raman bands, as well as on the most characteristic modes to confirm the structure intended and optimized in Figure 1. Since the spectra with the CAM-B3LYP method were the best, their frequencies have been mainly used for discussion. In specific cases, the scaled values obtained with the M06-2X method were also discussed. For simplicity, the scaled values with the PSE scaling procedure were mainly used in the discussion due to their slightly better accordance to the experimental results than those with the LSE procedure. The analysis and assignment of the different vibrational modes have been carried out under the following sections: (i) The COO- group modes, (ii) the triazole ring modes, and (iii) the aryl ring modes. Due to the low quality of the Raman spectra, the vibrational modes with the Ce(III) ion were not detected in the experimental spectrum and therefore they were not discussed in the present work.
2.4.2.1. The Carboxylate COO- Group Modes
In solid state samples [15,16], the asymmetric νas(COO-) mode of the carboxylate group appears as a strong IR band in the 1600-1560 cm-1 range, while the νs(COO-) mode appears at lower wavenumbers. In 2b ligand alone, the νas(COO-) stretching has been scaled at 1710 cm-1 with very strong IR intensity [12]. Nevertheless, in the Ce(2b’)3 complex a noticeable red shift is expected to lower values due to the remarkably lengthened the C-O bonds to make the six O-Ce coordination bonds. In our complex and by both DFT methods, the symmetric mode has been clearly characterized first at higher frequencies than the asymmetric one, in clear contradiction with that expected. The spectrum of the free ligand is complicated due to the intermolecular H-bonding, including the carboxylic group. That is why the calculations show that the intermolecular H-bonding in the ligand and the Ce(III) complex produce similar ν(COO-) frequency changes.
Therefore, with the CAM-B3LYP method, a large contribution of the symmetric νs(COO-) mode was identified in the calculated wavenumber at 1525 cm-1 (scaled at 1503 cm-1 by the LSE procedure) with very strong IR intensity, the second stronger of the spectrum, in very good agreement with the experimental strong IR band at 1500.1 cm-1 and with the strong Raman line at 1504.5 cm-1. This feature was also found with the M06-2X method, appearing scaled at 1520 cm-1 by LSE which is in good accordance with the experimental spectrum.
A large contribution of this symmetric νs(COO-) mode was also found in the calculated wavenumber at 1336 cm-1 (scaled at 1374 cm-1 by the PSE procedure), and in agreement with that found in the 1420-1400 cm-1 range in solid state samples of related compounds [15,16].
A large contribution of the asymmetric stretching vibration νas(COO-) was clearly identified by CAM-B3LYP in the scaled wavenumber by PSE at 1377 cm-1 with strong IR intensity and medium Raman intensity, in excellent agreement with the very strong bands at 1372.3 (IR) and 1376.8 cm-1 (Raman). The M06-2X method confirms this assignment with scaled wavenumbers at 1386 and 1378 cm-1. Asymmetric stretching vibrations νas(CO12) and νas(CO13) were also identified in the scaled wavenumbers by CAM-B3LYP at 1342, 1293, 1279 and 1262 cm-1 which are in good agreement with the experimental very strong IR band at 1301.8 cm-1 and with the strong band at 1285.1 cm-1. The M06-2X method confirms these assignments in the scaled values at 1309 and 1285 cm-1, respectively.
2.4.2.2. Triazole Ring Modes
The geometry and vibrational frequencies of the 1,2,3-triazole ring in different triazoles have already been studied [17,18,19]. Our computations and experimental results are in good agreement with them. For simplicity, the analysis of the different vibrations has been only focused in the assignment of the strongest bands.
NNN modes: The νas(NNN) stretching was characterized by CAM-B3LYP as strongly coupled with the 19a aryl mode in the scaled frequency with weak IR intensity at 1417 cm-1, being in very good accordance with the weak experimental IR band at 1418.0 cm-1. The results by M06-2X are in agreement with this assignment with a scaled wavenumber at 1434 cm-1 also with weak IR intensity. Due to the large background noise of the experimental Raman spectrum, the weak lines predicted for this mode were not clearly identified.
Other triazole ring stretching modes with asymmetric character and strongly coupled with other modes were identified in the scaled wavenumbers at 996 and 908 cm-1, and related to the very strong experimental IR band at 969.1 cm-1 and to the weak band at 914.3 cm-1, respectively. The Raman line detected at 1012.8 cm-1 was also assigned to this mode.
The symmetric stretching νs(NNN) mode was identified at lower wavenumbers and strongly coupled with the asymmetric νas(COO-) one, as well as with other ring modes. It was predicted by CAM-B3LYP with medium-strong IR intensity at 1377 cm-1 and corresponds to the very strong IR band at 1372.2 cm-1. The very strong experimental Raman line observed at 1376.8 cm-1 can be assigned to this mode or to the neighbor scaled wavenumber at 1374 cm-1. Similar scaled wavenumber 1378 cm-1 was calculated by the M06-2X method. In 2b ligand alone (isolated state), it has been scaled at 1360 cm-1 and assigned to the experimental IR band at 1340.5 cm-1 [20].
C8-N14 modes: The stretching mode has been scaled by the LSE procedure at 1623 cm-1 with strong IR intensity and corresponds to the very strong and broad band observed in the experimental IR spectrum at 1577.2 cm-1. This difference in the wavenumbers and the broadening of the experimental IR band can be due to the contribution of the δ(O-H) mode corresponding to the hydrated water molecules. This assignment was in accordance to that found by M06-2X in the scaled wavenumber at 1649 cm-1 by the LSE procedure. This stretching mode was identified in isolated 2b ligand in the scaled wavenumber at 1556 cm-1 [12], being in good agreement with the experimental bands with medium intensity at 1543.9 cm-1 (IR) and 1550.6 cm-1 (Raman).
2.4.2.3. Aryl Ring Modes
For the assignments of the ring modes the Varsanyi notation [21] for a 1,4-disubstituted benzene derivative was followed. Therefore, the substituent modes of the aryl ring for stretching vibrations correspond to 7a and 13 modes, for in-plane vibrations to 9b and 15 modes, and for out-of-plane vibrations to 10b and 11 modes. In particular, the C4-N4 bond is represented by 13, 15 and 10b modes, while the C-Cl bond is represented by 7a, 9b and 11 modes. In a chloro-substituted benzene derivative [20], mode 7a ν(C-Cl) was found at 359 cm-1, mode 9b δ(C-Cl) at 310 cm-1 and mode 11 γ(C-Cl) at 93 cm-1.
The assignments of many of the remaining ring modes are clear in Table 1-SUP and do not need further analysis, thus interest was directed only to the strongest vibrations in order to confirm the structure of the synthesized complex.
The aromatic C-H stretching modes appear mainly in the 3200-2950 cm-1 region theoretically as almost pure modes (100% PED) with weak or very weak IR and Raman intensities. Thus, only mode 20b with a scaled value by the PSE procedure at 3043 cm-1 correlated to the experimental IR band with strong intensity at 2968.1 cm-1. This wavenumber is exactly the same as the one found in an isolated 2b ligand [12], which can be explained by the negligible effect of the Ce(III) ion on the far aryl ring.
The aromatic C-C stretching vibrations, modes 8a and 8b, appear as nearly pure modes but with %PED around 90%. Through the CAM-B3LYP method, mode 8a was scaled by the LSE procedure at 1598 cm-1 with practically null IR intensity, but being the second vibration with the highest Raman intensity, which is in excellent agreement with the very strong Raman line at 1595.0 cm-1. Similar scaled wavenumber at 1603 cm-1 is predicted by M06-2X. Mode 8b was calculated by both DFT methods with practically null IR and Raman intensities and therefore, it has not been detected in the experimental spectra.
Mode 19a was calculated by both DFT methods with very strong IR and Raman intensities at 1491 cm-1 being in excellent agreement with the experimental strong IR band at 1484.4 cm-1. Mode 19b was scaled at 1398 cm-1 with weak IR and very weak Raman intensities, which is in agreement with the experimental IR band with medium intensity detected at 1398.3 cm-1.
2.5. Free Radical-Scavenging Activity
The radical-scavenging activity of the sodium salt 2b and its La(III) complex – La(2b’)3, have previously been reported[3]. For comparison’s sake, results, obtained after testing in the model systems reported herein, will be presented together with data for the novel Ce(III) complex.
2.5.1. Impact of Ce(2b’)3 on 2-Deoxyribose Degradation
The impact of the Ce(III) complex on 2-deoxyribose degradation as a result of UV-induced water radiolysis are presented in Figure 8:
The observed effect with Ce(2b’)3 is concentration-dependent, as is the case with the previously reported 2b and La(2b’)3. At concentrations below 1·10-5 M the activity of the complex is very mild (RSA<5%). At 1·10-5 M and higher the cerium complex is more active than the ligand at the same concentration. At 3·10-5 M Ce(2b’)3 and La(2b’)3 have similar activity (RSA=46.31±3.72% and 53.59±4.99% respectively). The observed impact of the coordination center in this model system seemed very mild, both complexes exhibiting similar activity within the tested range of molarities.
2.5.2. Impact of Ce(2b’)3 on a model system, containing the stable radical DPPH●
The ability of Ce(2b’)3 to exchange hydrogen with DPPH● is presented in Figure 9.
Previous experiments have shown that the ligand 2b and the complex La(2b’)3 manifest very mild activity in this model system - RSA<5% at even the highest concentration with the effect decreasing to zero at 3·10-6 M and lower. The case of Ce(2b’)3 is very similar. At 3·10-5 M this complex has RSA=5.17±0.53%. The effect decreases in a concentration dependent manner to 1.50±0.41% at 3·10-6. DPPH● is scavenged by way of HAT, similar to OH●[22], however its relatively large size may be implicated in the observed low activity in this model system, compared to the 2-deoxyribose degradation model. Ce(2b’)3 is more active that 2b at three times higher molarity. That may be due to the impact of Ce(III) on the distribution of electron density within the coordinated ligands, yielding active hydrogen atoms that could interact with DPPH●.
2.5.3. Impact of Ce(2b’)3 on a model system, containing the stable radical ABTS●+
The ability of the novel complex to participate in electron-exchange reactions with ABTS●+ is presented in Figure 10:
A concentration-dependent impact is observed with RSA increasing from 0.24±0.44% at 1·10-6 M to 23.36±1.12% at 3·10-5 M. At the same molarities, the activity of Ce(2b’)3 is higher than that of 2b and even the lanthanum(III) counterpart at 3·10-6 M or more. The activity of the Ce(III) complex tends to be higher than that of 2b at three times higher concentration. At the highest tested molarity (3·10-5 M) Ce(2b’)3 has RSA=23.36±1.12%, compared to 9.32±1.01% for La(2b’)3 (3·10-5 M) and 16.92±0.67% for the ligand 2b (1·10-4 M). Unlike La(III), complexation of 2b with Ce(III) seems to promote electron-exchange with ABTS●+, rather that suppress it.
2.5.4. Impact of Ce(2b’)3 on MTT-formazan transformation, triggered by Fenton reaction-derived hydroxyl radicals.
The Fenton reaction is a chemical process with clear clinical significance[23]. It involves transition metal-catalyzed production of hydroxyl radicals from H2O2. OH● are highly reactive species that tend to attack molecular sites incorporating conjugated double bonds, causing molecular fragmentation, lipid peroxidation and generation of malondialdehyde-like products. Since both the Fenton reaction and water radiolysis produce OH●, similar activity in both model systems would also be expected. This, however, is not the case for Ce(2b’)3, much in line with previously published observations with 2b and La(2b’)3. Results are presented in Figure 11.
Previous observations demonstrate that in this model system the ligand 2b is inert at 3·10-5 M or less. At 1·10-4 M it actually increases MTT-formazan formation to a significant degree (RSA= -114.67±71.87%) – a sign of pro-oxidant action. At 3·10-5 M Ce(2b’)3 also seems to act as a pro-oxidant (RSA= -79.41±4.06%), similar to the previously reported La(2b’)3 (RSA=- 68.14±22.57%). This effect decreases in a concentration-dependent manner. At 1·10-5 M Ce(2b’)3 seems to behave as a slightly more potent prooxidant, compared to La(2b’)3 (RSA= -39.36±3.31% and 8.99±10.92% respectively). Ce(2b’)3 seems to act as a more potent prooxidant than 2b at concentrations 3·10-5 M and lower. At 3·10-6 M and 1·10-5 M it increases MTT-formazan transformation to a greater extent than the ligand 2b at three times greater molarity.
3. Discussion and Conclusions
New example of rare-earth metal ion complex with biological active ligand was synthesized and their structure, electronic properties, and antioxidant activity were carefully studied by the experimantal methods and quantum-mechanical calculation. The composition and structure of the tri-coordinated complex of Ce3+ and 2-(4-chlorophenyl)-5-(pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylate was determined using elemental analysis and spectral data. The structural features of Ce(2b’)3 and global chemical reactivity descriptors were revealed based on quantum mechanical calculations performed at the at the CAM-B3LYP/Cep-4g and M06-2X/Cep-4g levels in comparison with three models of cerium complexes obtaind by the simplification of the structure of a triazole ligand and also with lantanum complex bearing the same ligand.
The next text may be the same as the previous one, but so many conclusions are not good. I think that points 3 - 9 should be shortened as it was in previous paper about La-complex.
The most important findings are as follows:
- 1)
- In the complex formation, the CO bonds of the ligands are lengthened as compared to the free form, which gives rise to a slight shortening of the C9-C11 bond length, to a decrease of the N7=C8 and C9=N10 double bond character, and to more rotated triazole substituents.
- 2)
- The influence of different ligands on the charge and molecular properties of the Ce(III) complex was analyzed. Thus, the cerium charge is increased from the smallest A-complex to the largest Ce(2b’)3 complex due to the better charge distribution on the cerium ion in larger systems. The pyrrolidine ring slightly increments this electron transfer.
- 3)
- New relationships with different ligands were established to find better properties. In particular, the charge on the cerium ion has a large influence in the bond lengths and atomic charges on the ligand atoms in all complexes. An increase in the positive calculated charge of the Ce(III) ion appears well linear associated with an increment in the negative charge of O12 and O13 atoms. The magnitude of the electronic charge lost by the Ce(III) ion is almost the same that was transferred to both O12 and O13 atoms. Large ligands appear to facilitate this negative electron transfer.
- 4)
- Global chemical reactivity descriptors were calculated. The HOMO–LUMO gap value linearly decreases with the system complexity (higher Ce atomic charge), with the exception of Ce(2b’)3 complex which has a similar value to C-complex. Its low energy value indicates a large chemical reactivity and small excitation energies to the manifold of excited states.
- 5)
- The pyrrolidine ligand slightly reduces the reactivity of the Ce(2b’)3 complex, but it gives liposolubility to the complex that would be necessary to cross the cell membrane.
- 6)
- The dipole moment value indicates an almost null water solubility(insolubility) in A- and B-complexes, but remarkable increase when the aryl and pyrrolidine rings are inserted in the ligands (C and Ce(2b’)3 complexes). It is not so significant.
- 7)
- New scaling equations for the D3-B3LYP, CAM-B3LYP and M06-2X DFT methods have been utilized to improve the computed IR and Raman spectra. The noticeable agreement with the experimental ones, raised in both frequencies and intensities, corroborates the spatial orientation of the ligands in the synthesized Ce(2b’)3 complex. The best correlations have been found with CAM-B3LYP method.
- 8)
- The complex Ce(2b’)3 scavenges hydroxyl radicals, generated by UV-infuced water radiolysis. Comparison with the analogous La(2b’)3 shows that in this particular model system the impact of the coordination center, Ce(III) or La(III), on activity is very mild.
- 9)
- In line with previous observations on 2b and La(2b’)3, the ability of Ce(2b’)3 to participate in HAT with DPPH● is very limited. These results, skarkly contrasting with the ones, derived from the 2-deoxyribose degradation assay, suggest that the low DPPH-scavenging activity may be due to steric hindrance, rather than low hydrogen-donating capacity of the complex.
- 10)
- Coordination with Ce(III) seems to improve the ligand’s ability to participate in SET, as observed in the ABTS●+ assay. Ce(2b’)3 seems to scavenge the stable radical much more actively, compared to its La(III) counterpart (almost threefold higher RSA values). In this case we can comfortably deduce that the electron-exchanging capacity is significantly impacted by the type of metal ion, coordinated with 2b.
- 11)
- Ce(2b’)3 reaffirms previous observations that 2b and its lanthanide complexes tend to behave as prooxidants in presence of the clinically significant Fenton reaction. A concentration-dependent increase in MTT-formazan formation was observed with Ce(2b’)3, similar to La(2b’)3 .
Structural and electronic characterization of the studied triazole complexes, together with the new relationships established in quantum-mechanical calculations and findings revealed in biological tests, show that this combination of study methods could help in the synthesis of novel lanthanide(III) complexes with possible antioxidant or anticancer activities.
4. Materials and Methods
4.1. Experimental Details
The compounds used for the synthesis of Ce(III) complex were from Merck company, p.a.grade: Ce(NO3)3·6H2O. The sodium salt 2b [8] was used as a ligand. The complex was synthesized through the interaction between cerium(III) inorganic salt and the ligand. The synthesis was carried out by adding an aqueous solution of La(III) nitrate to an aqueous solution of the ligand in an amount equal to a metal:ligand molar ratio of 1:3. The reaction mixture was stirred with an electromagnetic stirrer at room temperature for one hour. As a result, the precipitate of the complex was obtained, which was filtered, washed with water, and dried in a desiccator to constant weight. The resulting complex was slightly soluble in water, methanol, and ethanol, but soluble in dimethyl sulfoxide (DMSO).
The chemical composition of the newly obtained Ce(III) complex was characterized by elemental analysis. The binding mode in the Ce(III) complex was confirmed by means of vibrational spectroscopy. The synthesized Ce(2b’)3 complex was investigated as KBr pellet at room temperature for the IR spectrum, which was recorded in the 3900-400 cm–1 range using a FTIR IFS25 Bruker spectrophotometer. The Raman spectrum was registered in the 4000-0 cm−1 range using a Horiba Jobin Yvon, model LabRam, equipped with 1800 grooves/mm holographic grating.
Radical-scavenging assays: The in vitro scavenging activity of the sodium salt of the ligand (2b) and its La(III) complex in presence of several free radical-generating model systems has already been discussed in a previous work[3]. Herein the behavior of 2b and its cerium(III) complex on RS, generated by four model systems is revealed and compared to that of the respective lanthanum(III) complex.
4.2. Materials
All materials used in the RS-generating model systems were pro analysis grade SIGMA-ALDRICH (Sigma-Aldrich Chemie GmbH, Taufkirchen, Germany). Bi-distilled water and 95% ethanol were utilized for solution preparation. The Deoxyribose Degradation Assay required the following solutions and reagents: 50mM K-Na-phosphate saline solution (PBS), pH= 7.45; water solution of deoxyribose, 6.0 mM; 1% water solution of thiobarbituric acid (TBA); 3% water solution of trichloroacetic acid (TCA). The Fenton reaction MTT assay involved: 3 mg/mL water solution of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT); Fenton reagent, containing FeCl3, H2O2 and Na2-EDTA; 4 mg/mL water solution of ascorbic acid. Participation in hydrogen atom transfer (HAT) reactions was elucidated according to well-established protocols [24,25], using a 0,6 mM solution of 2,2-diphenyl-1-picrylhydrazyl radical (DPPH●) in ethanol. The ability of the tested compounds to participate in single electron transfer (SET) reactions was established using the 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic) free radical (ABTS) assay, as described by Erel et al [26,27]. The ABTS assay employed the following reagents: 0.4 mM acetate buffer, pH=5.8; 0.03 mmol acetate buffer solution, pH = 3.6 with ABTS and H2O2 in order to produce the stable radical-ion ABTS●+.
4.3. Methods
4.3.1. Deoxyribose Degradation Assay
A modified protocol, utilizing UV-induced water radiolysis, [28] was applied to generate hydroxyl radicals (OH●) [29]. Degradation of 2-deoxyribose to malondialdehyde(MDA)-like products is detected with the aid of the TBA assay. Every sample is comprised of the following solutions: 0.5 mL 2-deoxyribose, solution of the investigated compound and PBS up to 5.0 mL. Control samples did not include the investigated compounds. After irradiation with an UV lamp (220-400 nm, 30 min), 1.0 mL from each 5.0 mL volume was withdrawn. Consequently, to each 1.0 mL volume was added 0.6 mL TCA and 0.6 mL TBA, followed by cultivation in water bath (100 ̊С, 30 min). Absorbance was measured at λ=532 nm. The degree of 2-deoxyribose degradation is presented as Radical-Scavenging Activity (RSA):
4.3.2. Fenton Reaction MTT Assay
The reduction of MTT to formazan by Fenton-generated OH● was investigated. Ascorbic acid increases OH● production in the model system. Formazan production is registered as increase in absorbance at λ= 578 nm, using the kinetic function of the apparatus with 10 seconds lag time and 600 seconds measuring time. Sample composition (2.0 mL total volume) is as follows (Table 5):
Results are presented as Radicals-scavenging activity (RSA), calculated with the formula:
where: Аcontrol is the absorbance at 578 nm in presence of the Fenton reagent, but in absence of the investigated compound. Аsample is the absorbance at 578 nm in presence of the Fenton reagent together with the investigated compound. Аblank is the absorbance at 578 nm in absence of the Fenton reagent, but in presence of the investigated compound.
4.3.3. DPPH Assay
The assay was performed according to literature data [24,25,30]. DPPH● absorbance at λ=517 nm was measured in three types of samples – “blank”, “control” and “sample” (Table 6).
Total volume of each sample is 2.0 mL. Changes in absorbance were measured with the kinetic function of the apparatus: 10 seconds lag time, 600 seconds measuring time. Results are presented as RSA:
4.3.4. ABTS Assay
Two reagents are prepared, as described by Erel [26] – Reagent 1 (R1) and Reagent 2 (R2). R1 is Na-acetate buffer with pH=5.8. R2 includes ABTS, dissolved in Na-acetate buffer with pH=3.8, with the addition of H2O2 to produce the stable ABTS●+ radical-ion. 1.0 mL is the total volume of each sample. Absorbance was measured at 660 nm, using the kinetic function of the apparatus: 10 seconds lag time, 600 seconds measuring time. Results are presented as RSA – same as with the DPPH assay. Sample composition is as follows (Table 7).
4.4. Computational details
Four theoretical levels were used for the optimization of all Ce(III) complexes. Density Functional methods (DFT) [31] have been chosen for this purpose since they have provided accurate vibrational wavenumber values in biomolecules in good agreement with the experimental ones and in better accordance than those determined by MP2 method [32]. Among these DFT methods, the Minnesota M06-2X functional was the first chosen because it appears as one of the best meta-generalized gradient functionals to analyze dispersion–bound in large systems [33,34], especially in biomolecules with non-covalent weak interactions, as those included in the present work. Moreover, this method has also shown a large applicability in chemistry [35].
The B3LYP functional has been secondly chosen since it has yielded excellent results in the computation of the IR and Raman wavenumbers of biomolecules and is better than other DFT methods [32,36]. In particular, with the Ce(III) ion it performs slightly better than others hybrid functionals [37]. However, the B3LYP functional alone is not appropriate for reproduction of systems with non-covalent weak interactions like those associated with our complexes, therefore the spectra obtained differ from the experimental ones. For this reason, the D3-B3LYP and the CAM-B3LYP methods [38], which combine the hybrid qualities of B3LYP and the long-range correction presented by Tawada et al. [39] were also used to improve the calculated structural parameters. The CEP-4G basis set is the only one that appears available in Gaussian-16 program package for cerium [40]. Although it is a very small basis set, the vibration spectra obtained with this basis set can be well correlated to the experimental spectra, and therefore a good characterization of the synthesized Ce(2b’)3 complex was carried out. With these three DFT methods used, a charge +1 on the whole system was necessary to be included, even with the smaller Ce(III) complexes, labeled as A, B and C in Figure 1. With neutral charge 0 on the system, the Gaussian-16 program package does not run indicating an inconsistence for calculations in the ground state (multiplicity = 1). The authors deem that unusual since the three positive charges on Ce ion are compensated with three 2b’ anions. The authors propose that could be due to a default of the CEP-4G basis for the Ce ion. Despite that the effect on the organic ligands is very slight and their calculated spectra are in accordance to the experimental ones. Various Ce(III) and Ce(IV) complexes with organic molecules have been reported [41,42]. The UNIX version with standard parameters of this Gaussian-16 program was running in the Brigit super-computer of the Complutense University at Madrid.
All optimized complexes have shown positive wavenumbers, indicating a minimum in the potential energy surface. For this task, harmonic wavenumber computations have been performed at the same level of the corresponding optimization process. The Raman scattering activities (Si) obtained in the calculations have been transformed into relative Raman intensities (Ii) by the following relationship [43],
where νo corresponds to the wavenumber (cm-1) of exciting radiation and νi to the wavenumber of the ith normal mode. The remaining symbols, h, c, k and T correspond to Planck constant, speed of light, Boltzmann constant and absolute temperature, respectively. Finally, the symbol f is a suitable scale factor chosen to be common to all peak intensities.
4.4.1. Scaling the wavenumbers
The calculated wavenumbers by the theoretical DFT methods appear overestimated due to different reasons [6]. To correct this overestimation, different scaling procedures have been reported for each specific method and basis set [6,36], obtaining a noticeable improvement in the wavenumbers. Today, it is a normal procedure followed by the different authors to obtain an accurate assignment of the experimental bands. Among the different procedures available, the Linear Scaling Equation (LSE) and the Polynomic Scaling Equation procedures (PSE) appear as the most appropriate, and therefore they were used in the present manuscript. The specific equations utilized here for the four levels of computation were the following:
LSE procedure: νscal = 195.7 + 0.8706 νcal at B3LYP/Cep-4g level.
νscal = 194.0 + 0.8714 νcal at D3-B3LYP/Cep-4g level.
νscal = 184.0 + 0.8646 νcal at CAM-B3LYP/Cep-4g level.
νscal = 185.0 + 0.8613 νcal at M06-2X/Cep-4g level.
PSE procedure: νscal = 90.6 + 1.0305 νcal - 0.0000403· (νcal )2 at B3LYP/Cep-4g level
νscal = 92.7 + 1.0253 νcal - 0.0000388· (νcal )2 at D3-B3LYP/Cep-4g level
νscal = 78.7 + 1.0216 νcal - 0.0000390· (νcal )2 at CAM-B3LYP/Cep-4g level
νscal = 78.0 + 1,0204 νcal - 0.0000394· (νcal )2 at M06-2X/ Cep-4g level
Author Contributions
Conceptualization, I.K. and M.P.; methodology, I.K., N.B., M.P., N. H-A. and L.T.; software, M.P.; validation, I.K., N.B., M.P., N. H-A. and L.T.; formal analysis, M.P. and L.T.; investigation, M.P., N. H-A. and L.T.; resources, I.K., N.B., M.P., N. H-A. and L.T.; data curation, M.P. and L.T.; writing—original draft preparation, I.K., N.B., M.P. and L.T.; writing—review and editing, I.K., N.B., M.P. and L.T.; visualization, M.P., N.B. and L.T.; supervision, I.K. and M.P.; project administration, I.K., M.P. and L.T.; funding acquisition, M.P. and I.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study is financed by the European Union-NextGenerationEU, through the National Recovery and Resilience Plan of the Republic of Bulgaria, project No BG-RRP-2.004-0004-C01.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Lewandowski, W.; Kalinowska, M.; Lewandowska, H. The influence of metals on the electronic system of biologically important ligands. Spectroscopic study of benzoates, salicylates, nicotinates and isoorotates. Review. Journal of inorganic biochemistry 2005, 99, 1407–1423. [Google Scholar] [CrossRef]
- Goswami, A.K.; Kostova, I. Medicinal and Biological Inorganic Chemistry: Walter de Gruyter GmbH & Co KG; 2022; ISBN 1501516116.
- Alcolea Palafox, M.; Belskaya, N.P.; Todorov, L.T.; Kostova, I.P. Structural Study of a La (III) Complex of a 1, 2, 3-Triazole Ligand with Antioxidant Activity. Antioxidants 2023, 12, 1872. [Google Scholar] [CrossRef]
- Zhang, H.; Feng, J.; Zhu, W.; Liu, C.; Gu, J. Bacteriostatic effects of cerium-humic acid complex: An experimental study. Biological trace element research 2000, 73, 29–36. [Google Scholar] [CrossRef]
- Hosseinzadeh, R.; Khorsandi, K.; Esfahani, H.S.; Habibi, M.; Hosseinzadeh, G. Preparation of cerium-curcumin and cerium-quercetin complexes and their LEDs irradiation assisted anticancer effects on MDA-MB-231 and A375 cancer cell lines. Photodiagnosis and Photodynamic Therapy 2021, 34, 102326. [Google Scholar] [CrossRef]
- Abd El-Hamid, S.M.; Sadeek, S.A.; Zordok, W.A.; El-Shwiniy, W.H. Synthesis, spectroscopic studies, DFT calculations, cytotoxicity and antimicrobial activity of some metal complexes with ofloxacin and 2, 2′-bipyridine. Journal of Molecular Structure 2019, 1176, 422–433. [Google Scholar] [CrossRef]
- Feng, F.-M.; Cai, S.; Liu, F.-A.; Xie, J.-Q. Studies of DNA-binding and DNA-cutting mechanism of an azamacrocyclic cerium complex with carboxyl branch. Progress in Reaction Kinetics and Mechanism 2013, 38, 283–294. [Google Scholar] [CrossRef]
- Safronov, N.E.; Kostova, I.P.; Palafox, M.A.; Belskaya, N.P. Combined NMR Spectroscopy and Quantum-Chemical Calculations in Fluorescent 1, 2, 3-Triazole-4-carboxylic Acids Fine Structures Analysis. International Journal of Molecular Sciences 2023, 24, 8947. [Google Scholar] [CrossRef]
- Peica, N.; Kostova, I.; Kiefer, W. Theoretical and experimental studies on binding mode of 3, 5-pyrazoledicarboxylic acid in its new La (III) complex. Chemical physics 2006, 325, 411–421. [Google Scholar] [CrossRef]
- Alam, M.M. 1, 2, 3-Triazole hybrids as anticancer agents: A review. Archiv der Pharmazie 2022, 355, 2100158. [Google Scholar] [CrossRef]
- Hrimla, M.; Oubella, A.; Laamari, M.R.; Bahsis, L.; Ghaleb, A.; Auhmani, A.; et al. Click synthesis, anticancer activity, and molecular docking investigation of some functional 1, 2, 3-triazole derivatives. Biointerface Research in Applied Chemistry 2022, 12, 7633–7667. [Google Scholar]
- Palafox, M.A.; Belskaya, N.P.; Kostova, I.P. Study of the Molecular Architectures of 2-(4-Chlorophenyl)-5-(Pyrrolidin-1-Yl)-2H-1, 2, 3-Triazole-4-Carboxylic Acid as the Potential Anticancer Drug by Their Vibrational Spectra and Quantum Chemical Calculations. 2023.
- Mishra, V.R.; Sekar, N. Photostability of coumarin laser dyes-a mechanistic study using global and local reactivity descriptors. Journal of fluorescence 2017, 27, 1101–1108. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical hardness and density functional theory. Journal of Chemical Sciences 2005, 117, 369–377. [Google Scholar] [CrossRef]
- George, S. Infrared and Raman characteristic group frequencies: tables and charts. Wiley: Chichester 2001, 82, 85–87. [Google Scholar]
- Tammer, M.G. Sokrates: Infrared and Raman characteristic group frequencies: tables and charts: Wiley, Chichester, 2004. ISBN 0-470-09307-2, 347 pages, paperback; US $60. Springer; 2004.
- Aziz, S.G.; Elroby, S.A.; Alyoubi, A.; Osman, O.I.; Hilal, R. Experimental and theoretical assignment of the vibrational spectra of triazoles and benzotriazoles. Identification of IR marker bands and electric response properties. Journal of molecular modeling 2014, 20, 1–15. [Google Scholar]
- Törnkvist, C.; Bergman, J.; Liedberg, B. Geometry and vibrations of the 1, 2, 3-triazole anion. A theoretical and experimental study. The Journal of Physical Chemistry 1991, 95, 3119–3123. [Google Scholar] [CrossRef]
- El-Azhary, A.; Suter, H.; Kubelka, J. Experimental and theoretical investigation of the geometry and vibrational frequencies of 1, 2, 3-triazole, 1, 2, 4-triazole, and tetrazole anions. The Journal of Physical Chemistry A 1998, 102, 620–629. [Google Scholar] [CrossRef]
- Palafox, M.A.; Rastogi, V. Spectra and structure of benzonitriles and some of its simple derivatives. Asian Journal of Physics Vol 2013, 22, 1–30. [Google Scholar]
- Varsányi, G.; Láng, L.; Kovner, M.A.E.; Lempert, K. Assignment for vibrational spectra of seven hundred benzene derivatives. (No Title). 1974.
- Galano, A. Free radicals induced oxidative stress at a molecular level: The current status, challenges and perspectives of computational chemistry based protocols. Journal of the Mexican Chemical Society 2015, 59, 231–262. [Google Scholar] [CrossRef]
- Kell, D.B. Iron behaving badly: inappropriate iron chelation as a major contributor to the aetiology of vascular and other progressive inflammatory and degenerative diseases. BMC medical genomics 2009, 2, 1–79. [Google Scholar] [CrossRef]
- Kedare, S.B.; Singh, R. Genesis and development of DPPH method of antioxidant assay. Journal of food science and technology 2011, 48, 412–422. [Google Scholar] [CrossRef]
- Molyneux, P. The use of the stable free radical diphenylpicrylhydrazyl (DPPH) for estimating antioxidant activity. Songklanakarin J sci technol 2004, 26, 211–219. [Google Scholar]
- Erel, O. A novel automated direct measurement method for total antioxidant capacity using a new generation, more stable ABTS radical cation. Clinical biochemistry 2004, 37, 277–285. [Google Scholar] [CrossRef]
- Erel, O. A novel automated method to measure total antioxidant response against potent free radical reactions. Clinical biochemistry 2004, 37, 112–119. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.; Aruoma, O.I. The deoxyribose method: a simple “test-tube” assay for determination of rate constants for reactions of hydroxyl radicals. Analytical biochemistry 1987, 165, 215–219. [Google Scholar] [CrossRef]
- Burns, W.G.; Sims, H.E. Effect of radiation type in water radiolysis. Journal of the Chemical Society, Faraday Transactions 1: Physical Chemistry in Condensed Phases 1981, 77, 2803–2813. [Google Scholar] [CrossRef]
- Chrzczanowicz, J.; Gawron, A.; Zwolinska, A.; de Graft-Johnson, J.; Krajewski, W.; Krol, M.; et al. Simple method for determining human serum 2, 2-diphenyl-1-picryl-hydrazyl (DPPH) radical scavenging activity–possible application in clinical studies on dietary antioxidants. Clinical Chemical Laboratory Medicine 2008, 46, 342–349. [Google Scholar] [CrossRef]
- Seminario, J.M. Modern density functional theory: a tool for chemistry: Elsevier; 1995. ISBN 008053 6700.
- Palafox, M.A. DFT computations on vibrational spectra: Scaling procedures to improve the wavenumbers. Physical Sciences Reviews 2018, 3, 20170184. [Google Scholar] [CrossRef]
- Riley, K.E.; Hobza, P. Noncovalent interactions in biochemistry. Wiley Interdisciplinary Reviews: Computational Molecular Science 2011, 1, 3–17. [Google Scholar] [CrossRef]
- Riley, K.E.; Pitonák, M.; Jurecka, P.; Hobza, P. Stabilization and structure calculations for noncovalent interactions in extended molecular systems based on wave function and density functional theories. Chemical Reviews 2010, 110, 5023–5063. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Applications and validations of the Minnesota density functionals. Chemical Physics Letters 2011, 502, 1–13. [Google Scholar] [CrossRef]
- Alcolea, M.P. Scaling factors for the prediction of vibrational spectra. I. Benzene molecule. International Journal of Quantum Chemistry 2000, 77, 661–684. [Google Scholar] [CrossRef]
- Kullgren, J.; Castleton, C.W.; Müller, C.; Ramo, D.M.; Hermansson, K. B3LYP calculations of cerium oxides. The Journal of chemical physics 2010, 132. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chemical physics letters 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. The Journal of chemical physics 2004, 120, 8425–8433. [Google Scholar] [CrossRef]
- Frisch Me Trucks, G.; Schlegel, H.; Scuseria, G.; Robb, M.; Cheeseman, J.; et al. Gaussian 16, revision C. 01. Gaussian, Inc., Wallingford CT; 2016.
- Das, B.; Ghosh, K.; Baruah, J.B. Tris-dipicolinate Cerium Complexes Bearing Dications of Arginine, Histidine, and Ornithine. Synthesis and Reactivity in Inorganic, Metal-Organic, and Nano-Metal Chemistry 2014, 44, 251–257. [Google Scholar] [CrossRef]
- Levin, J.R.; Dorfner, W.L.; Dai, A.X.; Carroll, P.J.; Schelter, E.J. Density functional theory as a predictive tool for cerium redox properties in nonaqueous solvents. Inorganic Chemistry 2016, 55, 12651–12659. [Google Scholar] [CrossRef]
- Srivastav, G.; Yadav, B.; Yadav, R.K.; Yadav, R. DFT studies of molecular structures conformers and vibrational characteristics of sulfanilamide. Computational and Theoretical Chemistry 2019, 1167, 112588. [Google Scholar] [CrossRef]
Figure 1.
Front and lateral views of the optimized structure of four Ce(III) complexes at the CAM-B3LYP/Cep-4g and M06-2X/Cep-4g levels with the following ligands: methyl carboxylate (A-complex), 1,2,3-triazole-4-carboxylate (B-complex), 2-(4-chlorophenyl)-2H-1,2,3-triazole-4-carboxylate (C-complex), and 2-(4-chlorophenyl)-5-(pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylate (2b’ ligand) (Ce(2b’)3 complex). The labeling of the most characteristic atoms, the total energy of the system (E) including zero-point correction and the Gibbs free energy (G), as well as several bond length values are also included. 1 AU = 2625.5 kJ/mol:.
Figure 1.
Front and lateral views of the optimized structure of four Ce(III) complexes at the CAM-B3LYP/Cep-4g and M06-2X/Cep-4g levels with the following ligands: methyl carboxylate (A-complex), 1,2,3-triazole-4-carboxylate (B-complex), 2-(4-chlorophenyl)-2H-1,2,3-triazole-4-carboxylate (C-complex), and 2-(4-chlorophenyl)-5-(pyrrolidin-1-yl)-2H-1,2,3-triazole-4-carboxylate (2b’ ligand) (Ce(2b’)3 complex). The labeling of the most characteristic atoms, the total energy of the system (E) including zero-point correction and the Gibbs free energy (G), as well as several bond length values are also included. 1 AU = 2625.5 kJ/mol:.
Scheme 1.
Synthesis of the Ce(III) complex.
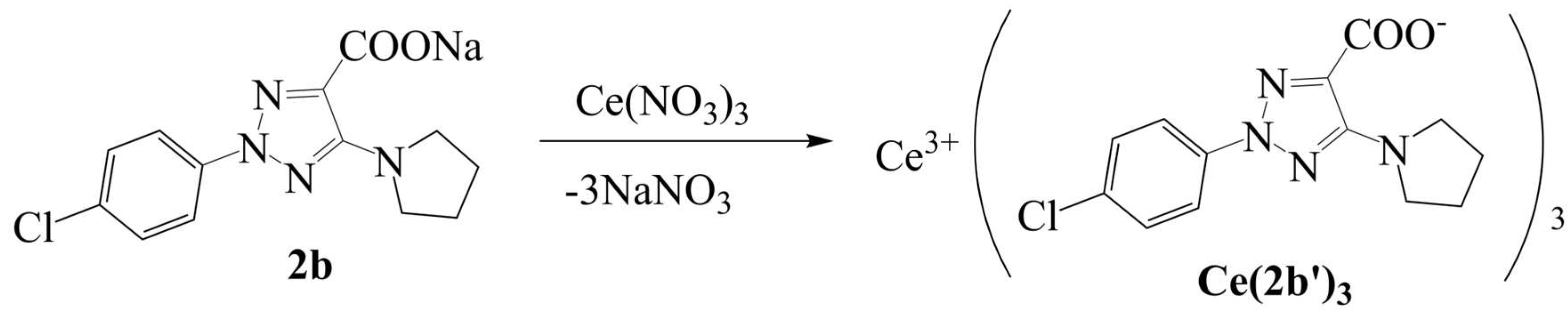
Figure 2.
Relationships established at the B3LYP/Cep-4g, CAM-B3LYP/Cep-4g and M06-2X/Cep-4g levels between the positive calculated atomic APT charge on the cerium ion in the A-, B-, C- and Ce(2b’)3 complexes with: (a) the atomic charge on O12, (b) the atomic charge on O13, (c) the atomic charge on C11, (d) the atomic charge on C9, (e) the C9-C11 bond length, and (f) relationship established at the CAM-B3LYP/Lanl2dz and M06-2X/Lanl2dz levels between the atomic APT charge on the lanthanum atom and the atomic charge on O12.
Figure 2.
Relationships established at the B3LYP/Cep-4g, CAM-B3LYP/Cep-4g and M06-2X/Cep-4g levels between the positive calculated atomic APT charge on the cerium ion in the A-, B-, C- and Ce(2b’)3 complexes with: (a) the atomic charge on O12, (b) the atomic charge on O13, (c) the atomic charge on C11, (d) the atomic charge on C9, (e) the C9-C11 bond length, and (f) relationship established at the CAM-B3LYP/Lanl2dz and M06-2X/Lanl2dz levels between the atomic APT charge on the lanthanum atom and the atomic charge on O12.
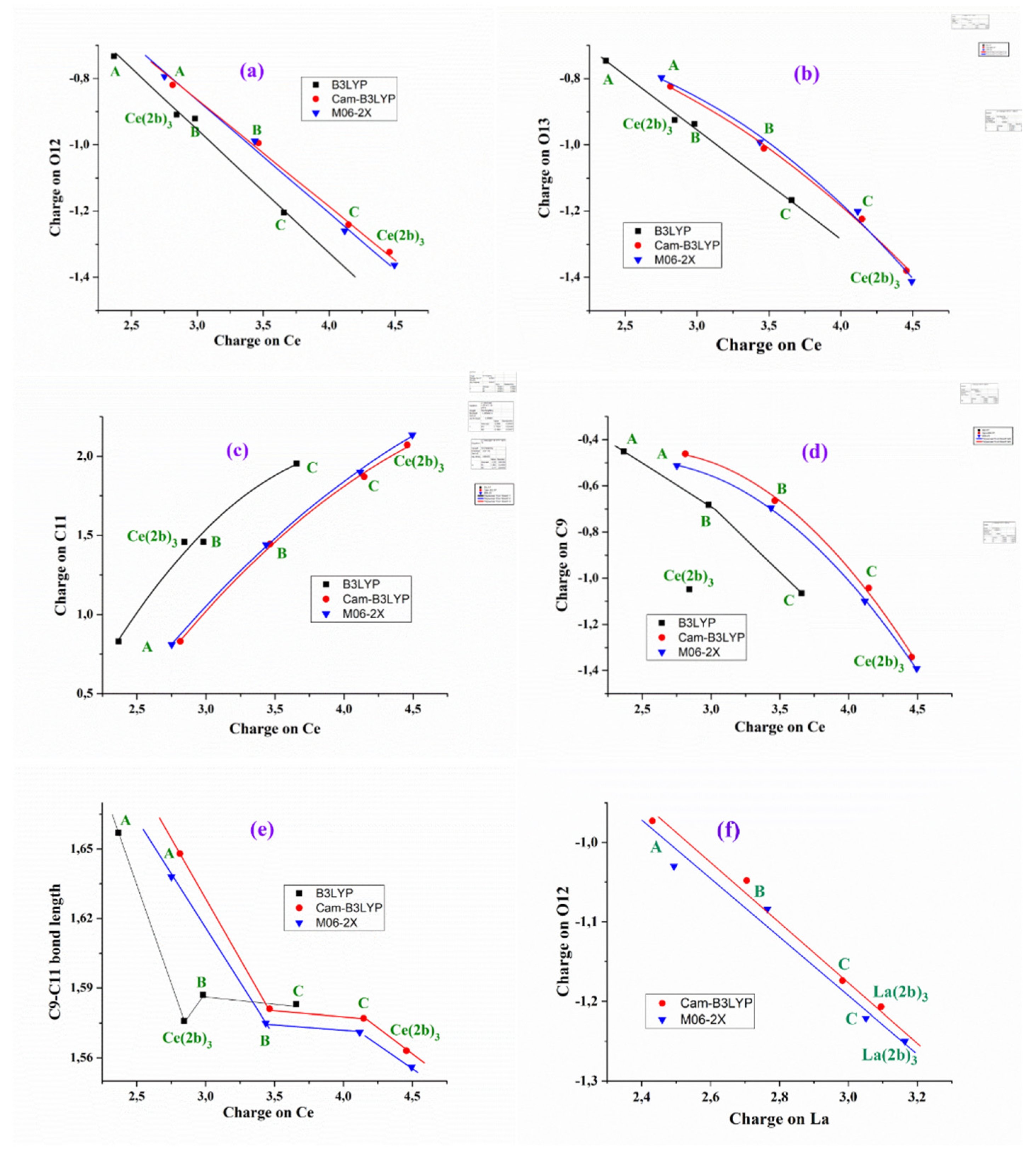
Figure 3.
Relationships at the B3LYP/Cep-4g, CAM-B3LYP/Cep-4g and M06-2X/Cep-4g levels between the positive calculated atomic APT charge on the cerium ion in the A-, B-, C- and Ce(2b’)3 complexes with: (a) the HOMO energy orbital. (b) the LUMO energy orbital. (c) the (HOMO-LUMO) energy difference.
Figure 3.
Relationships at the B3LYP/Cep-4g, CAM-B3LYP/Cep-4g and M06-2X/Cep-4g levels between the positive calculated atomic APT charge on the cerium ion in the A-, B-, C- and Ce(2b’)3 complexes with: (a) the HOMO energy orbital. (b) the LUMO energy orbital. (c) the (HOMO-LUMO) energy difference.
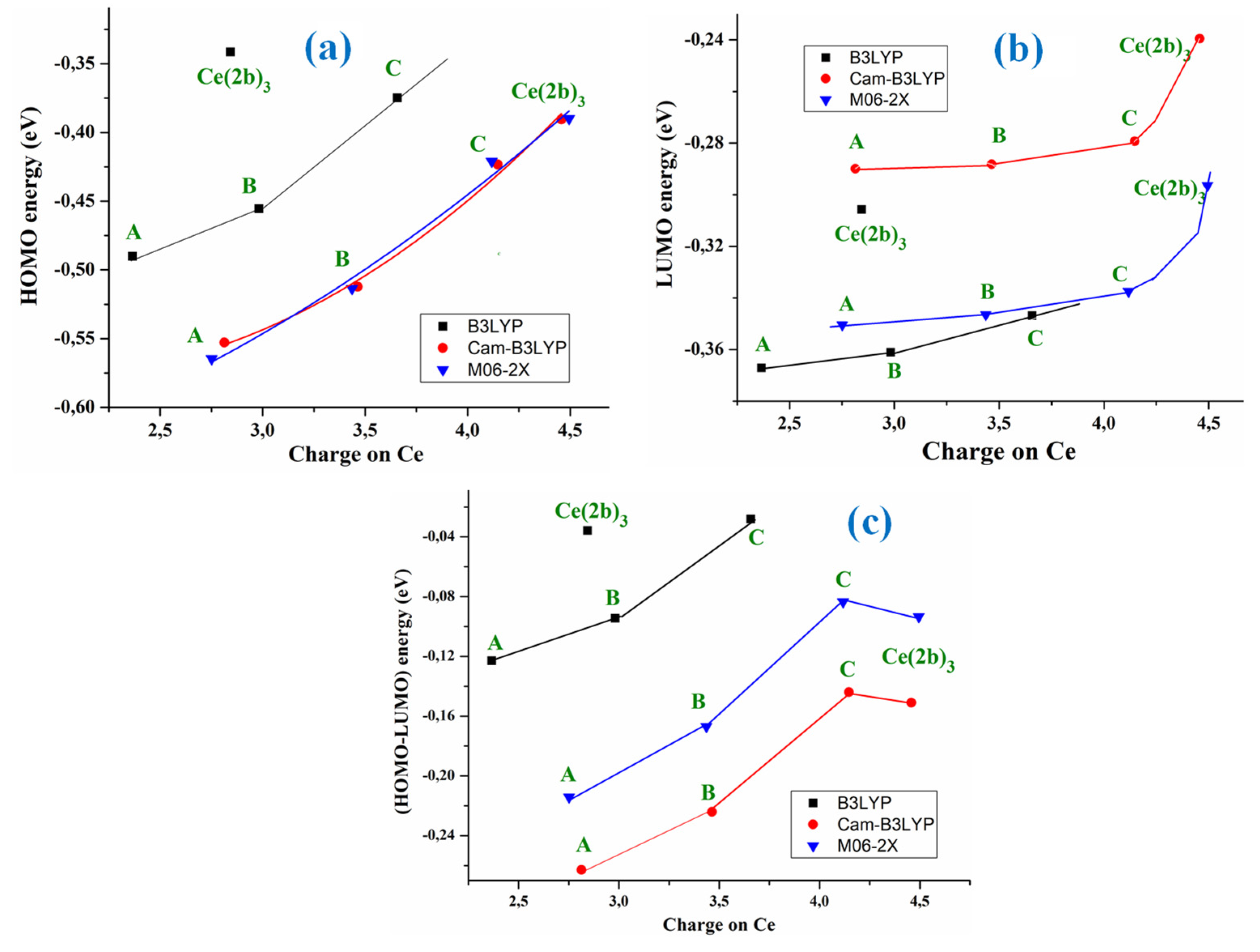
Figure 4.
Comparison of the scaled IR spectra at the B3LYP/Cep-4g, CAM-B3LYP/Cep-4g and M06-2X/Cep-4g levels by the PSE procedure with the experimental ones in the 3750-2600 cm-1 range.
Figure 4.
Comparison of the scaled IR spectra at the B3LYP/Cep-4g, CAM-B3LYP/Cep-4g and M06-2X/Cep-4g levels by the PSE procedure with the experimental ones in the 3750-2600 cm-1 range.
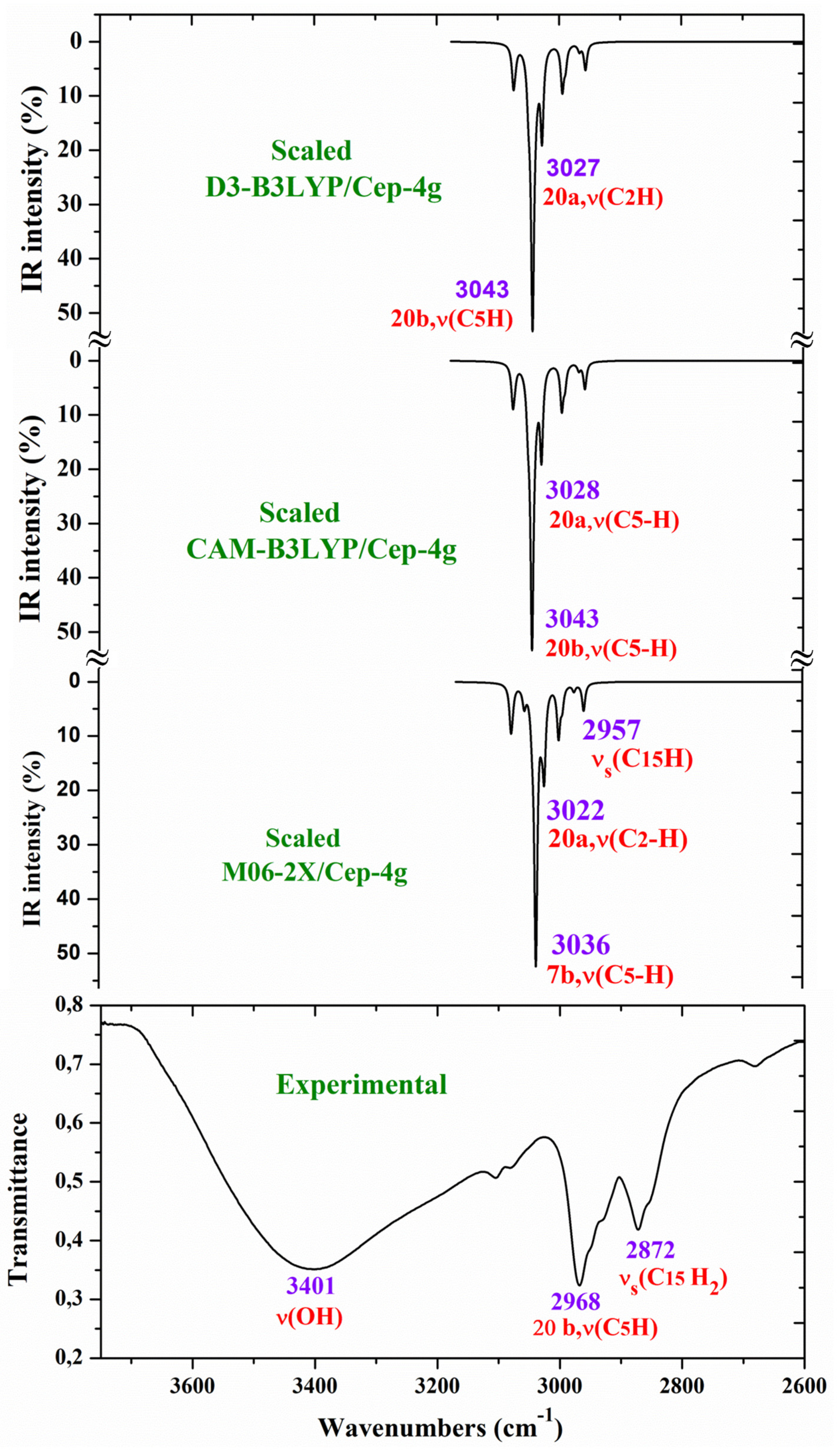
Figure 5.
Comparison of the scaled IR spectra at three different levels by the PSE procedure with the experimental ones in the 1800-1000 cm-1 range.
Figure 5.
Comparison of the scaled IR spectra at three different levels by the PSE procedure with the experimental ones in the 1800-1000 cm-1 range.
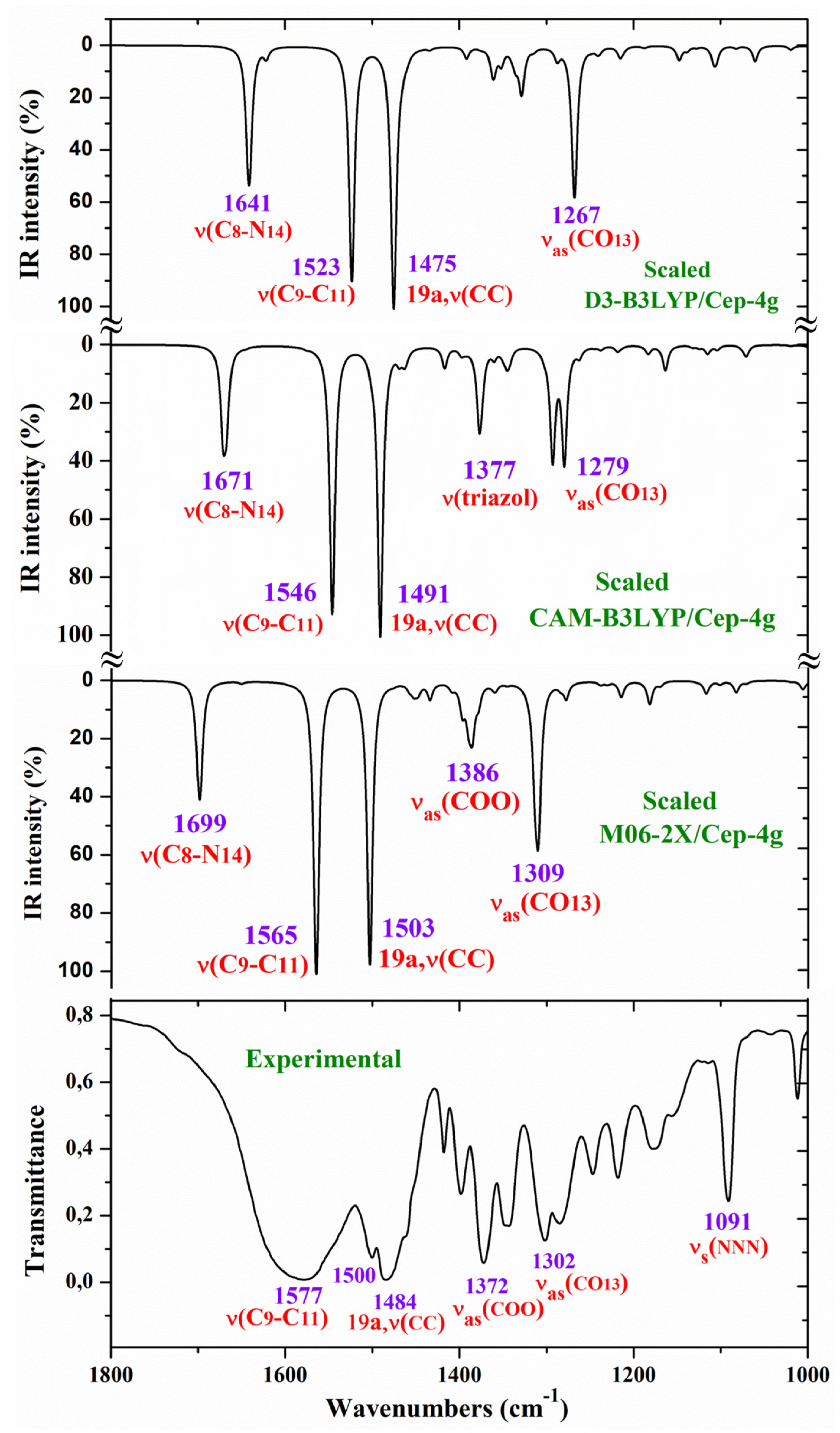
Figure 6.
Comparison of the scaled IR spectra at three different levels by the PSE procedure with the experimental ones in the 1000-400 cm-1 range.
Figure 6.
Comparison of the scaled IR spectra at three different levels by the PSE procedure with the experimental ones in the 1000-400 cm-1 range.
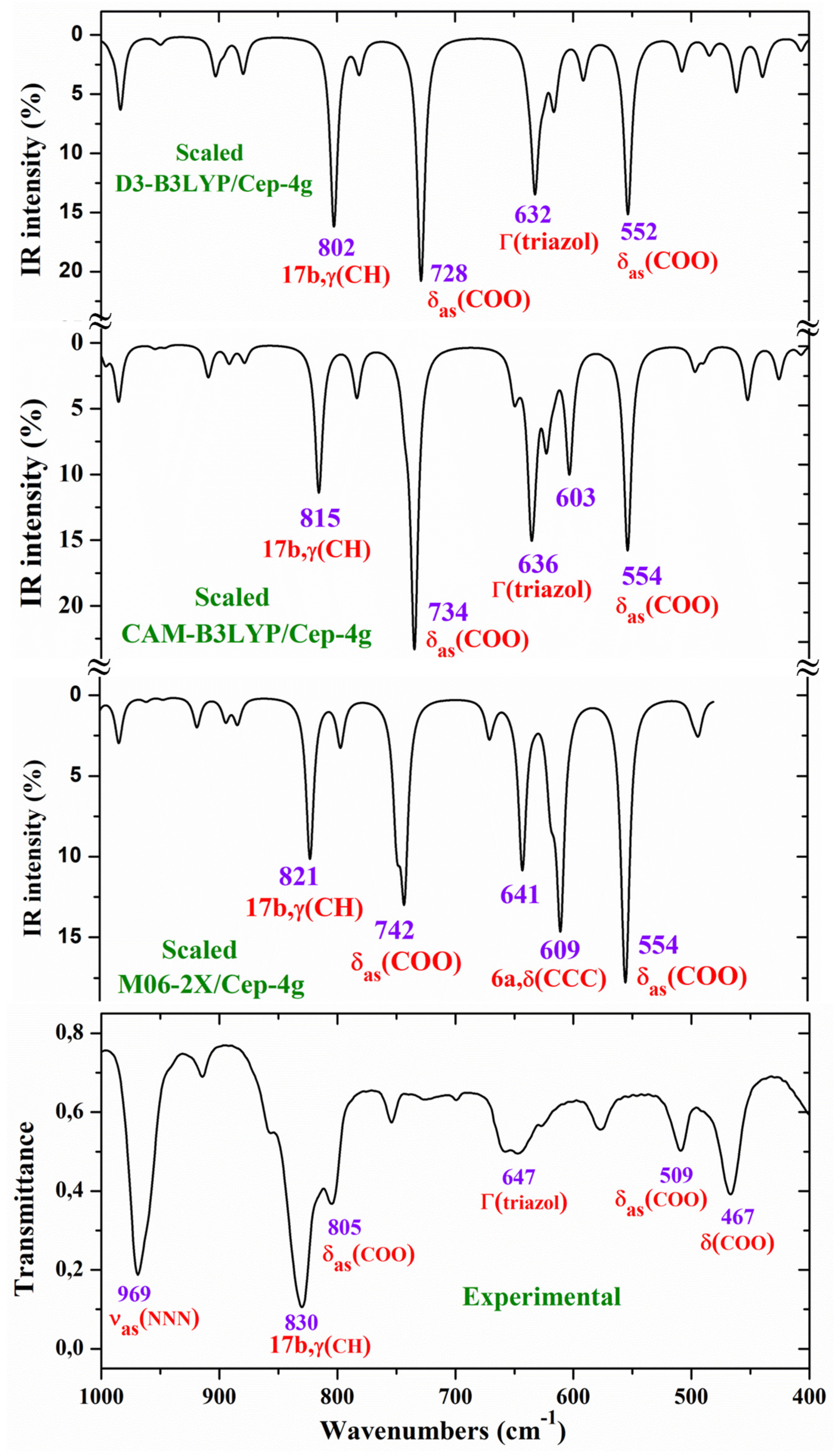
Figure 7.
Comparison of the scaled Raman spectrum at three different levels by the PSE procedure with the experimental one in the 1800-800 cm-1 range.
Figure 7.
Comparison of the scaled Raman spectrum at three different levels by the PSE procedure with the experimental one in the 1800-800 cm-1 range.
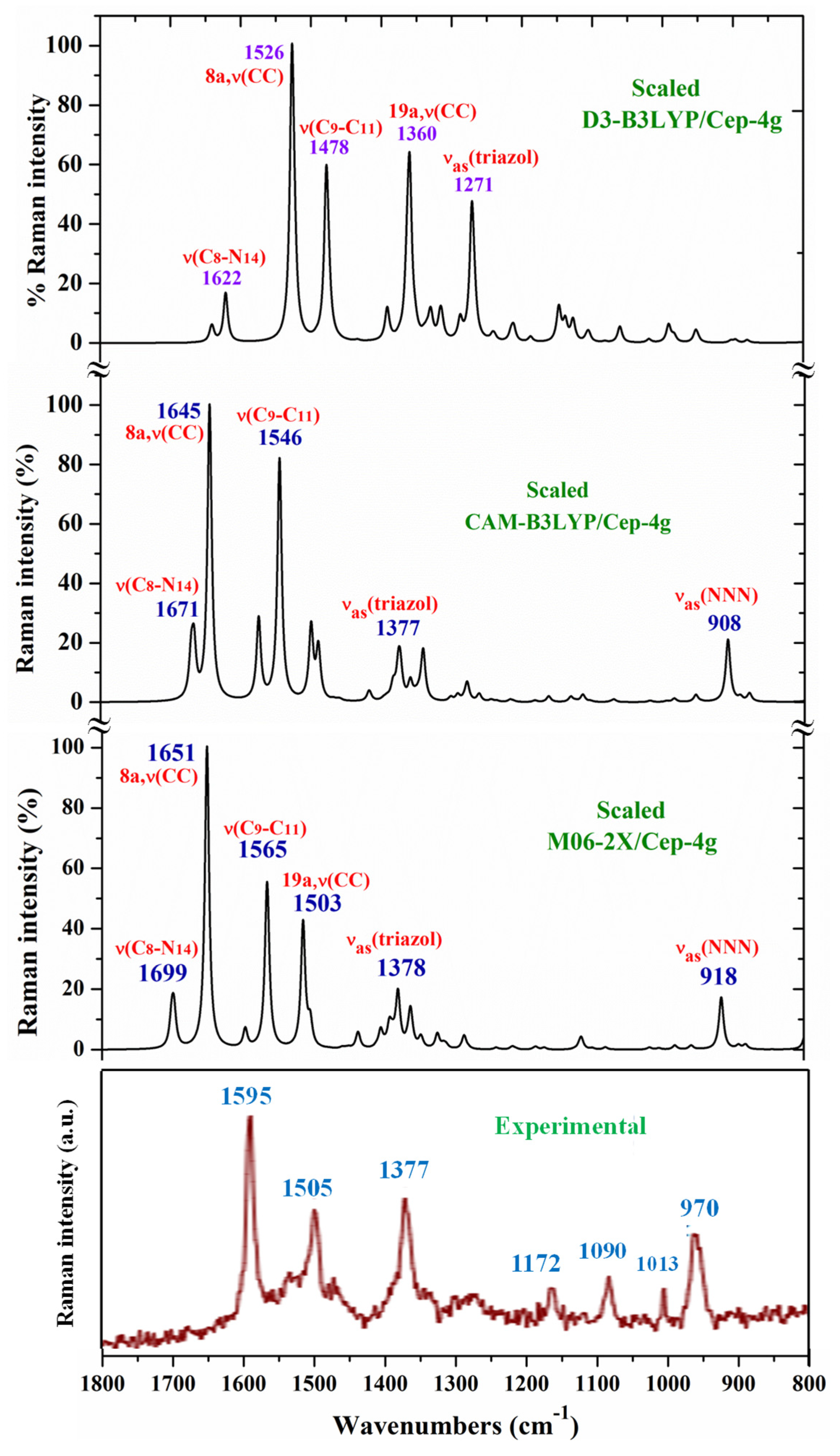
Figure 8.
Impact of 2b, Ce(2b’)3 and La(2b’)3 on 2-deoxyribose degradation. Data= Mean±StDev, p<0.05, N=3. Higher results mean higher scavenging activity.
Figure 8.
Impact of 2b, Ce(2b’)3 and La(2b’)3 on 2-deoxyribose degradation. Data= Mean±StDev, p<0.05, N=3. Higher results mean higher scavenging activity.
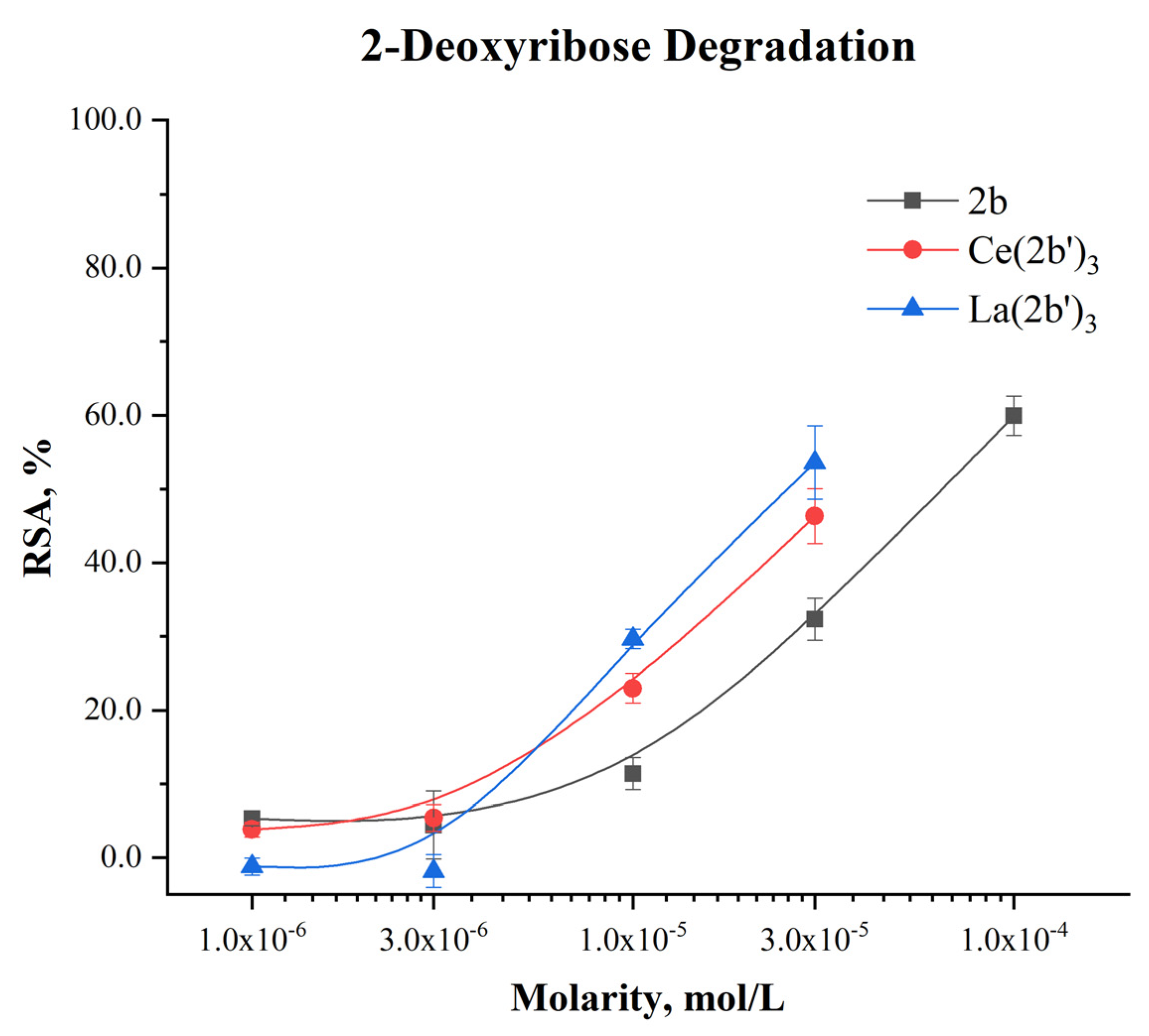
Figure 9.
Impact of 2b, La(2b’)3 and Ce(2b’)3 on DPPH●. Data= Mean±StDev, p<0.05, N=3. Higher result means higher scavenging activity.
Figure 9.
Impact of 2b, La(2b’)3 and Ce(2b’)3 on DPPH●. Data= Mean±StDev, p<0.05, N=3. Higher result means higher scavenging activity.
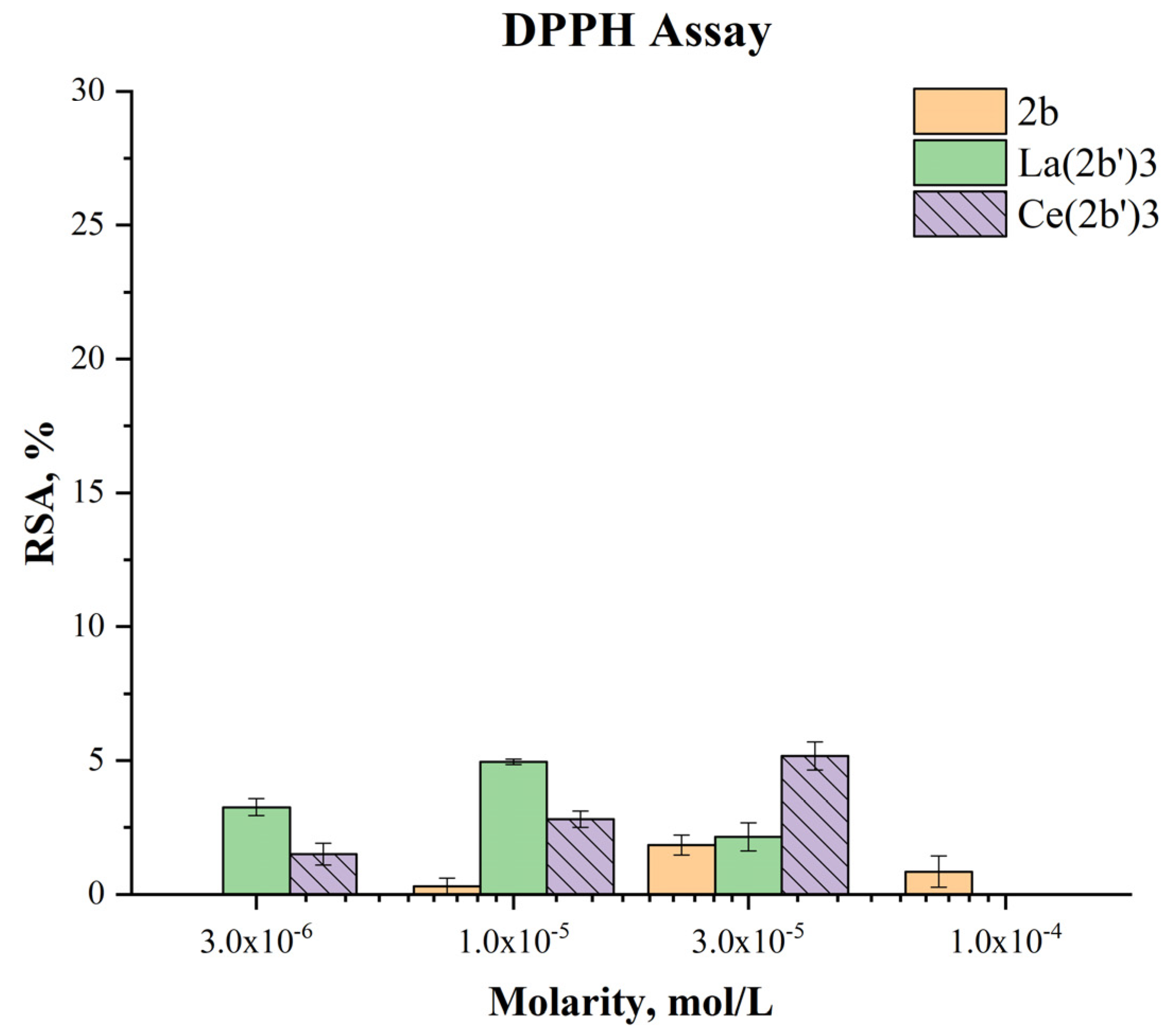
Figure 10.
Impact of 2b, La(2b’)3 and Ce(2b’)3 on DPPH●. Data= Mean±StDev, p<0.05, N=3. Higher result means higher scavenging activity.
Figure 10.
Impact of 2b, La(2b’)3 and Ce(2b’)3 on DPPH●. Data= Mean±StDev, p<0.05, N=3. Higher result means higher scavenging activity.
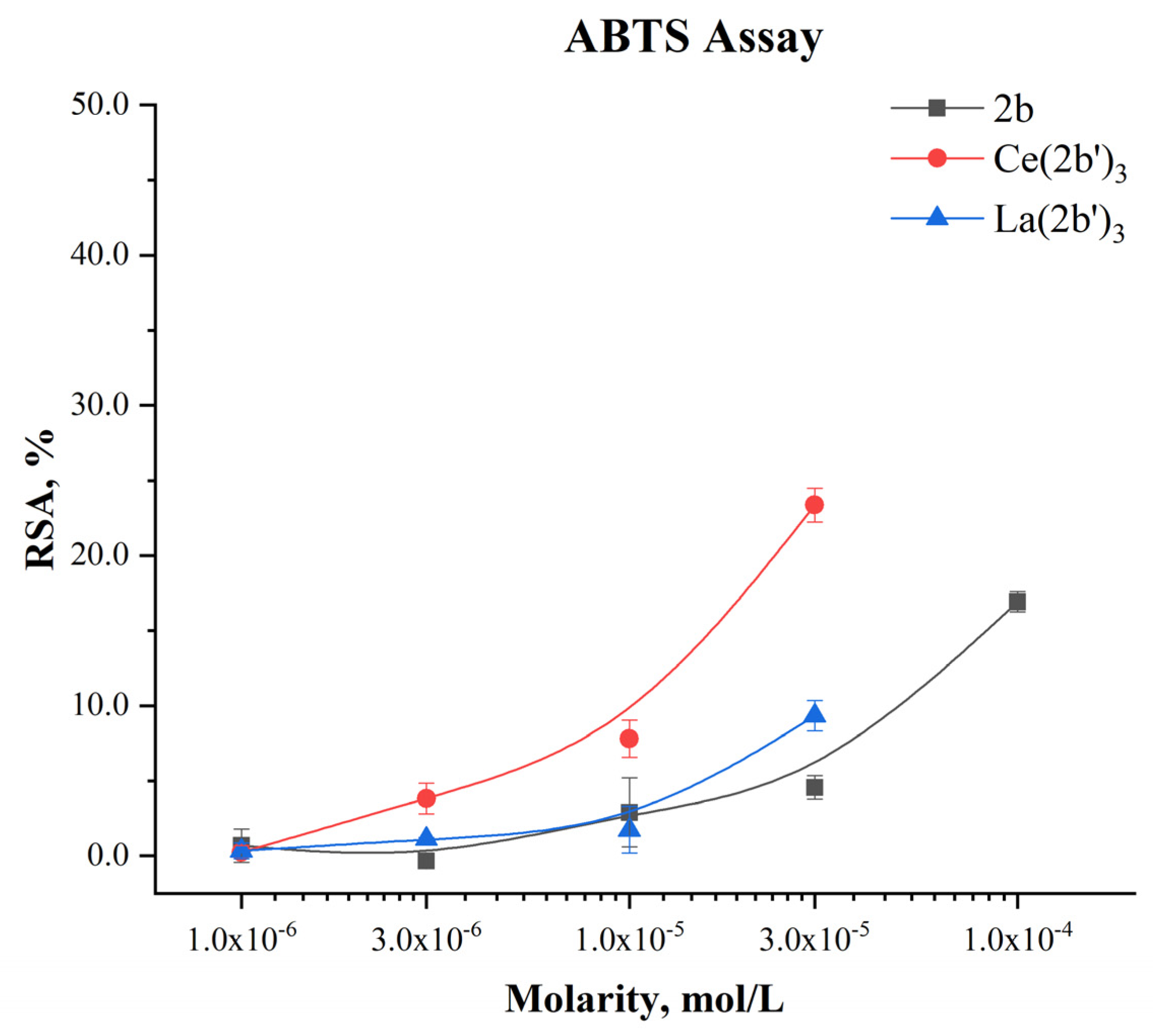
Figure 11.
Impact of 2b, La(2b’)3 and Ce(2b’)3 on MTT-Formazan transformation by Fenton-generated OH●. Data= Mean±StDev, p< 0.05, N=3. Higher result means higher scavenging activity.
Figure 11.
Impact of 2b, La(2b’)3 and Ce(2b’)3 on MTT-Formazan transformation by Fenton-generated OH●. Data= Mean±StDev, p< 0.05, N=3. Higher result means higher scavenging activity.
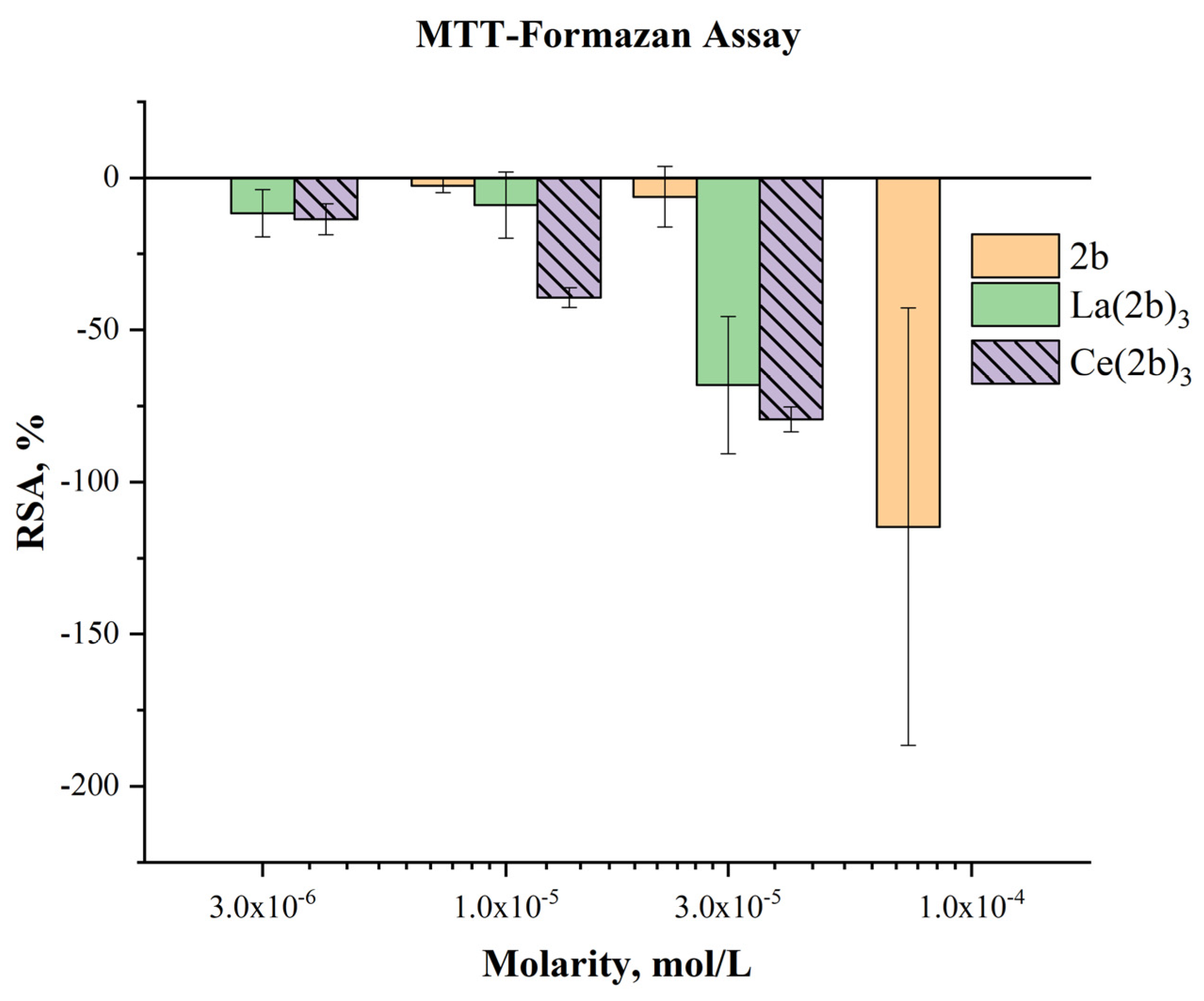
Table 1.
Several selected optimized geometrical parameters calculated with different DFT methods and the Cep-4g basis set in the Ce(III) complexes of Figure 1. Bond lengths (r) in Å, bond angles and dihedral angles (∠) in degrees. The data were from ligand I.
Table 1.
Several selected optimized geometrical parameters calculated with different DFT methods and the Cep-4g basis set in the Ce(III) complexes of Figure 1. Bond lengths (r) in Å, bond angles and dihedral angles (∠) in degrees. The data were from ligand I.
| A-complex | B-complex | C-complex | Ce-(2b’)3 | ||||||||||
| Parameters | B3LYP | CAM-B3LYP | M06-2X/ | B3LYP/ | CAM-B3LYP | M06-2X/ | B3LYP | CAM-B3LYP | M06-2X/ | B3LYP | D3-B3LYP | CAM-B3LYP | M06-2X/ |
| r(C9-C11) r(C=O12) r(C=O13) r(Ce-O12) r(Ce-O13) ∠(C9-C11=O12) ∠(O=C=O) ∠(C=O12-Ce) ∠(C=O13-Ce) ∠(O12-Ce-O′13) ∠(O13-Ce-O′12) ∠(C11-O12···O′12-C′11) ∠(C9-C11···C′11-C′9) ∠(C′9-C′11···C′′11-C′′9) ∠(C11···Ce··· C′11) ∠(C′11···Ce··· C′′11) |
1.657 1.434 1.434 2.287 2.286 123.7 112.5 92.3 92.3 153.9 102.2 57.6 -0.1 0.1 120.1 120.0 |
1.648 1.423 1.423 2.256 2.255 123.9 112.1 92.3 92.4 155.3 100.8 60.6 -0.2 0.3 120.2 119.6 |
1.638 1.413 1.412 2.248 2.253 123.4 113.2 91.9 91.7 100.5 102.1 -98.1 0.5 -0.5 119.6 120.2 |
1.587 1.434 1.435 2.290 2.282 122.2 114.2 90.9 91.2 154.3 102.7 56.9 0.2 2.1 120.2 119.0 |
1.581 1.422 1.423 2.260 2.251 122.4 113.8 91.0 91.4 155.5 101.2 60.5 0.0 2.6 120.2 119.0 |
1.575 1.410 1.414 2.261 2.240 122.0 114.8 90.3 91.0 100.5 100.1 -98.8 1.8 0.2 118.9 120.9 |
1.583 1.436 1.436 2.288 2.281 122.1 114.2 90,9 91.2 153.8 103.1 55.9 0.0 0.0 119,9 120.1 |
1.577 1.423 1.423 2.257 2.251 122.4 113.7 91.1 91.3 155.3 101.3 59.8 0.0 0.1 120.1 120.0 |
1.571 1.413 1.413 2.253 2.245 121.7 114.8 90.5 90.8 101.7 100.4 -99.1 2.7 1.0 120.1 119.7 |
1.576 1.437 1.442 2.304 2.275 125.9 113.7 90.9 91.9 152.4 104.5 56.4 -0.2 -0.1 120.0 119.8 |
1.570 1.437 1.441 2.292 2.272 125.4 113.9 90.8 91.5 153.3 103.4 59.6 -1.1 -1.1 119.9 119.8 |
1.563 1.425 1.433 2.264 2.231 126.6 112.7 91.1 92.2 154.8 101.6 61.7 0.0 -0.1 119.8 119.9 |
1.556 1.415 1.425 2.260 2.224 125.5 113.8 90.4 91.6 99.7 99.9 -103.5 -18.0 16.7 120.3 118.8 |
Table 2.
APT (Atomic Polar Tensor) charges calculated with different DFT methods and the Cep-4g basis set in the Ce(III) complexes of Figure 1. The values of ligand I are only shown.
Table 2.
APT (Atomic Polar Tensor) charges calculated with different DFT methods and the Cep-4g basis set in the Ce(III) complexes of Figure 1. The values of ligand I are only shown.
| A-complex | B-complex | C-complex | Ce-(2b’)3 | ||||||||||
| atom | B3LYP | CAM-B3LYP | M06-2X/ | B3LYP/ | CAM-B3LYP | M06-2X | B3LYP | CAM-B3LYP | M06-2X/ | B3LYP | D3-B3LYP | CAM-B3LYP | M06-2X/ |
|
Ce N4 N7 C8 C9 N10 C11 O12 O13 |
2.366 - - - -0.450 - 0.830 -0.733 -0.747 |
2.814 - - - -0.461 - 0.831 -0.819 -0.823 |
2,752 - - - -0.512 -- 0.810 -0.794 -0.797 |
2.982 -0.596 -0.013 -0.071 -0.682 0.253 1.459 -0.921 -0.937 |
3.464 -0.608 -0.018 -0.090 -0.664 0.262 1.446 -0.995 -1.011 |
3.436 -0.625 -0.031 -0.079 -0.695 0.276 1.441 -0.989 -0.992 |
3.657 -0.210 -0.111 0.021 -1.065 0.335 1.954 -1.205 -1.167 |
4.147 -0.440 -0.076 -0.093 -1.042 0.443 1.871 -1.241 -1.224 |
4.117 -0.383 -0.105 -0.072 -1.100 0.472 1.900 -1.260 -1.201 |
2.844 -0.058 -0.099 0.444 -1.048 0.105 1.458 -0.909 -0.925 |
3.056 -0.109 -0.109 0.453 -1.096 0.157 1.591 -0.974 -1.001 |
4.457 -0.466 -0.124 0.596 -1.342 0.411 2.071 -1.323 -1.380 |
4.494 -0.430 -0.133 0.610 -1.392 0.426 2.134 -1.363 -1.413 |
Table 3.
Molecular properties and global chemical reactivity descriptors (eV) calculated at different DFT levels in the Ce(III) complexes.
Table 3.
Molecular properties and global chemical reactivity descriptors (eV) calculated at different DFT levels in the Ce(III) complexes.
| Molecular properties | A-complex | B-complex | C-complex | Ce-(2b’)3 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CAM-B3LYP | M06-2X/ |
CAM-B3LYP | M06-2X | CAM-B3LYP | M06-2X/ | B3LYP | D3-B3LYP | CAM-B3LYP | M06-2X/ | |
| Rotational constants: A (GHz) B C |
0.575 0.572 0.328 |
0.582 0.582 0.331 |
0.133 0.129 0.070 |
0.135 0.131 0.071 |
0.023 0.023 0.013 |
0.028 0.020 0.012 |
0.018 0.018 0.011 |
0.018 0.018 0.011 |
0.018 0.018 0.011 |
0.019 0.017 0.011 |
| Cv (cal/mol·K) S (cal/mol·K) |
51.7 134.3 |
53.6 142.9 |
86.8 188.4 |
83.8 181.6 |
163.8 297.1 |
162.4 296.2 |
231.5 387.8 |
231.0 388.5 |
225.9 383.0 |
224.1 379.4 |
| Dipole moment (Debye) | 0.098 | 0.130 | 0.040 | 0.036 | 5.416 | 2.571 | 11.523 | 10.916 | 10.604 | 5.193 |
| HOMO LUMO Eg IP EA χ η S |
-0.553 -0.290 -0.263 0.553 0.290 0.421 0.131 0.066 |
-0.565 -0.350 -0.214 0.565 0.350 0.457 0.107 0.054 |
-0.512 -0.288 -0.224 0.512 0.288 0.400 0.112 0.056 |
-0.513 -0.346 -0.167 0.513 0.346 0.430 0.083 0.042 |
-0.423 -0.279 -0.144 0.423 0.279 0.351 0.072 0.036 |
-0.421 -0.337 -0.083 0.421 0.337 0.379 0.042 0.021 |
-0.342 -0.306 -0.107 0.342 0.306 0.324 0.018 0.009 |
-0.344 -0.305 -0.039 0.344 0.305 0.324 0.019 0.009 |
-0.390 -0.239 -0.151 0.390 0.239 0.315 0.076 0.038 |
-0.390 -0.296 -0.093 0.390 0.296 0.343 0.047 0.023 |
Table 5.
Fenton reaction-MTT assay sample composition.
| Control | Sample | Blank | |
|---|---|---|---|
| Tested compound | no | 200 μL | 200 μL |
| MTT | 200 μL | 200 μL | 200 μL |
| Fe2+/H2O2/Na2-EDTA | 100 μL | 100 μL | no |
| Ascorbic acid | 100 μL | 100 μL | no |
| Bi-destilled water | up to 2.0 mL | дo 2.0 mL | дo 2.0 mL |
Table 6.
DPPH assay sample composition.
| Blank | Control | Sample | |
|---|---|---|---|
| Tested compound | 200 μL | no | 200 μL |
| DPPH | no | 1800 μL | 1800 μL |
| Ethanol | 1800 μL | no | no |
| Bi-distilled water | no | 200 μL | no |
Table 7.
ABTS assay sample composition.
| Blank | Control | Sample | |
|---|---|---|---|
| Tested compound | 100 μL | no | 100 μL |
| R1 | 860 μL | 860 μL | 860 μL |
| R2 | no | 40 μL | 40 μL |
| Bi-destilled water | 40 μL | 100 μL | no |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



