
Submitted:
10 January 2024
Posted:
10 January 2024
You are already at the latest version
Abstract
Background
Corpus callosum abnormalities (CCA) are midline developmental brain malformations and are usually associated with a wide spectrum of other neurological and non-neurological abnormalities. The study aims to highlight the diagnostic role of fetal MRI to characterize heterogeneous corpus callosum abnormalities using the latest classification system. It also helps to identify associated anomalies, which have prognostic implications on the postnatal outcome.
Methods
In this study, retrospective data from antenatal women who underwent fetal MRI between January 2014 and July 2023 at Rush University Medical Center were evaluated for CCA and classified based on structural morphology. Patients were further assessed for associated neurological and non-neurological anomalies.
Results
The most frequent class of CCA was complete agenesis (79.1%), followed by hypoplasia (12.5%), dysplasia (4.2%) and hypoplasia with dysplasia (4.2%). Amongst them, 17% had isolated CCA, while the majority (83%) had complex forms of CCA associated with other CNS and non-CNS anomalies. Out of the complex CCA cases, 58% were associated with other CNS anomalies, while 8% were associated with non-CNS anomalies. 17% of cases had both.
Conclusion
The use of fetal MRI is valuable in the classification of abnormalities of the corpus callosum after the confirmation of a diagnosis suspected on prenatal ultrasound. This technique is an invaluable method for distinguishing between isolated and complex forms of CCA, especially in the cases of apparent isolated CCA. The use of diffusion-weighted imaging or diffusion tensor imaging in fetal neuroimaging is expected to provide further insights into white matter abnormalities in fetuses diagnosed with CCA in the future.
Keywords:
fetal magnetic resonance imaging
; corpus callosum abnormality
; central nervous system anomalies
; probst bundles
; agenesis
; hypoplasia
; neuroradiology
Introduction
The corpus callosum (CC) serves as the primary cerebral commissure in the supratentorial region of the brain [1]. There are two smaller interhemispheric fissure connections next to the CC, namely, the anterior commissure and hippocampal commissure. The corpus callosum primarily consists of white matter fibers connecting the two cerebral hemispheres at the midline. There are more than 200-250 million myelinated axons crossing the midline in the corpus callosum during brain development, establishing crucial connections between the hemispheres [2].
During the sixth gestational week (GW), the commissural plate begins to differentiate, followed by the crossing of pioneer axons [3]. As corpus callosum forms between the eighth and fourteenth weeks of gestation, callosal precursors and cortical fibers develop in bilateral cerebral hemispheres. Anatomically, it consists of four components: the rostrum, the genu, the body, and the splenium. The genu is the first part of the corpus callosum to exhibit crossed fibers, followed by body and the splenium, and the rostrum was considered the last part of the corpus callosum to show crossed fibers [4]. However, the advanced techniques like diffusion tensor neuroimaging (DTI) could identify the rostrum at 15 weeks of gestational age along with the anterior part of body and the genu. Posterior part of the body and splenium develops in the later part of the gestation and completes by 18 to 19 gestational weeks, highlighting the importance of gestational age in describing the corpus callosal abnormalities [5,6,7].
An intricate and tightly regulated sequence of developmental processes culminate in the corpus callosum during gestation and into adulthood [8]. If any of these events are disrupted, it can result in corpus callosal abnormalities (CCA), which is a rare condition. In accordance with data from the National Organization for Disorders of the Corpus Callosum, a person in every 2053 experiences symptoms associated with abnormalities in the corpus callosum. Its frequency is between 0.5 and 70 per 10000, and its prevalence among children with developmental disorders is about 230 to 600 per 10000 (2.3%) [9,10,11,12]. It is found in one in every 19000 autopsies [13]. The incidence is higher among boys than girls [14]. Corpus callosum (CC) abnormalities can occur alone; however, they are more commonly occur with a wide range of other abnormalities of the central nervous system or malformations of other organ systems [15].
Heterogeneous terminologies are used in the classification of corpus callosal abnormalities. Different classification systems are based on either anatomical or functional basis. Barkovich et al. previously divided CCA into dysgenesis (a defective development) and hypogenesis (an incomplete formation) [16]. Another classification by Santo et al. subdivided CCA into complete agenesis and partial agenesis grouping hypogenesis and dysgenesis into partial agenesis [17]. They further classified them into isolated versus complex CCA. Witelson et al. used DTI and classified CCA on functional basis [18]. Al Hashim et al. used embryology based functional anatomical classification system and categorized into complete CCA, anterior CCA, posterior CCA and complete hypoplasia [19]. Hanna et al. updated the previous classification of CCA by subdividing them into complete agenesis, hypoplasia without dysplasia, hypoplasia with dysplasia and dysplasia [20]. The term partial agenesis was not used in this classification. This classification provides valuable information for future studies regarding the role of genetic mechanisms in CCA [21].
Complete agenesis has two types. In type 1, axons are present, but the commissural fibers that develop by crossing the midline are absent. Probst bundles are those uncrossed fibers that run along the superior and medial area of the bilateral lateral ventricles [22,23]. In type II, axons are absent and thus, Probst bundles are not seen [24]. Hypoplasia refers to uniformly thin CC or underdeveloped posterior corpus callosum with intact morphology. In callosal hypoplasia, sigmoid bundles represent a heterotopic connection between one hemisphere's frontal lobe and the other hemisphere's occipital lobe [25]. Dysplasia refers to defective development of CC, which has an abnormal shape [26].
As development starts anteriorly and progresses posteriorly, earlier the callosal development is arrested, the smaller the genu and anterior body is formed [27]. In contrast, with the sequential arrest of growth of corpus callosum in congenital hypogenesis, acquired defects can affect any of the parts of corpus callosum [27]. Number of reasons are incremented in causing insults to the developing brain including infectious diseases (Toxoplasmosis, Rubella, Cytomegalovirus, and Herpes), toxic /metabolic (like alcohol) and chromosomal/genetic factors or vascular disorders [28]. Many syndromes are associated sporadically with CCA like Aicardi syndrome, Shappiro syndrome, Anderman syndrome, and Acrocallosal syndromes [29,30,31,32,33,34].
The diagnosis of CCA by fetal ultrasound has a high false-positive rate and its range varies from 0% to 20% depending upon the operator [17]. Fetal magnetic resonance imaging (MRI) for evaluating fetal CC is considered superior because of its multiplanar capabilities and spatial resolution [26]. A fetal MRI not only confirms CCA, but also determines its structural morphology more accurately while detecting coexisting anomalies like gyration anomalies and heterotopia not visualized on prenatal ultrasound [17]. A systematic review of fetal MRI detected additional abnormalities in 22.5% of cases compared to ultrasonography in prenatal cases [35]. Poor postnatal neurodevelopmental outcome is even more common to occur in fetuses with CCA associated with other CNS anomalies [36]; hence, MRI should be performed as part of CCA assessment, particularly in fetuses with apparently isolated CCA. With the advent of ultra-fast T2 imaging techniques, fetal MRI allows direct visualization of CCA on a midline sagittal image even without volume imaging or multiplanar reconstruction.
In conclusion, understanding the intricate development and potential callosal abnormalities is essential for unraveling the complexities of cerebral connectivity. With various classification systems shedding light on the diverse manifestations of corpus callosal abnormalities (CCA), and advanced imaging techniques like fetal MRI enhancing diagnostic precision, our study endeavors to contribute to this knowledge landscape. By adopting the refined classification system proposed by Hanna et al. we aim to categorize 24 individuals with callosal anomalies on fetal MRI, unravel the nuances of these variations, and their associated neurological and non-neurological abnormalities [20]. Till date, very few studies used this latest classification to characterize neuroimaging findings in fetuses with CCA.
Methods
Study Population and Design
In this study, retrospective data from antenatal women who underwent fetal MRI between January 2014 and July 2023 at Rush University Medical Center were analyzed for CCA. The fetal maternal unit referred all these cases to the diagnostic neuroradiology department for fetal MRI upon detecting corpus callosal anomalies on prenatal ultrasound. The study received approval from the Institutional Review Board, and explicit consent was waived off for the study. The study included fetuses with abnormalities of the corpus callosum detected on fetal MRI and suspected related congenital anomalies that were not adequately assessed on ultrasound. A number of participants were excluded from the study, including those with MRI contraindications (pacemaker, metal foreign body or aneurysm clip), poor image quality, those without the abnormal corpus callosum of the MRI, those with secondary causes of callosal abnormalities ( examples include, congenital hydrocephalus, Chiari malformations, intracranial hemorrhage, metabolic disorders, infection, or toxins), those without genetic analysis, those with disagreement among neuroradiologists about CCA diagnosis were excluded from the study. A total of 140 fetal MRIs were analyzed for CCA, 30 fetal MRI’s had abnormal corpus callosum, but six cases were not included because they met exclusion criteria. In total, 24 antenatal women participated in the study.
Data Collection and Management
Chart reviews were performed and the following data were extracted from the medical records: maternal age at delivery, maternal comorbid diagnoses, number of gestations, maternal race/ethnicity, mean weeks of gestation (GW) while undergoing fetal MRI, route of delivery, known teratogenic exposures during pregnancy, amniocentesis results if performed, birth GA (completed gestational weeks), sex, and length of stay (LOS) in NICU (Neonatal intensive care unit). The pregnancy outcomes were categorized as termination of pregnancy, spontaneous intrauterine fetal demise, inborn live birth or death in neonatal period. In this study, clinical information was obtained from the pediatric neurology and genetics clinic.
Fetal Magnetic Resonance Imaging
At the study institute, fetal MR scans were conducted using an Espree 1.5-T MR scanner manufactured by Siemens Medical Solutions in Erlangen, Germany. The scans were performed without any sedation. Imaging was performed using HASTE pulse sequence. The parameters include Time to echo (TE): 120 ms; repetition time (TR): 4300 milliseconds (ms); field of view (FOV): 23 (phase) × 23 (frequency) cm; slice thickness (ST): 3 mm; interslice spacing (IS): zero, matrix: 256 (phase) × 180 (frequency) in axial, sagittal, and coronal projections. This T2-weighted MR sequence uses half-Fourier single-shot turbine spin echo (abbreviated HASTE) to acquire images in one second.
To ensure unbiased evaluation, two independent board-certified neuroradiologists (S.E.B and M.K with over 15 years' experience) reviewed the fetal MR brain images of participants without knowledge of the clinical history. All cases were categorized according to the proposed classification system. Throughout the study, all authors remained blinded to any identifiable information, and strict confidentiality was maintained regarding protected health information. The final diagnosis was arrived by their consensus. Neuroradiologists could establish concordance in 21 cases (92%). The non-concordance was noted in the diagnosis of one case in each class of hypoplasia with dysplasia, dysplasia, and complete agenesis. The interobserver agreement statistics showed kappa statistics between 0.86 to 1 for the presence of abnormal callosum, and between 0.79 to 1 for the presence of other brain anomalies.
Classification of CCA on Fetal MRI
Patients with CCA are further subclassified into complete agenesis, hypoplasia without dysplasia, dysplasia, hypoplasia with dysplasia [20]. Complete agenesis of corpus callosum was defined as the absence of corpus callosum in mid-sagittal plane. The term hypoplasia refers to a uniformly thin or underdeveloped posterior part of the corpus callosum with intact morphology. Dysplasia refers to defective CC development, which has an abnormal shape (Figure 1). The normative indices for the corpus callosal length for fetuses between 18-22 weeks were derived from the study by Parazzini et al. [37], while between 22-37 weeks were derived from the study by Garel et al. [38]. The presence of ventriculomegaly, colpocephaly, Probst bundles and absent septum pellucidum were considered secondary to abnormal corpus callosum rather than associated CNS anomalies. Fetal ventriculomegaly is subclassified into mild form (10-12mm in transverse diameter), moderate form (12-15mm) and severe form (>15mm) [39]. We further classified CCA into associated CNS anomalies, and non-CNS anomalies. We reviewed fetal brain MRI images for the following associated CNS abnormalities: hypoplastic or dysplastic brainstem, Dandy-Walker complex and other posterior fossa anomalies, hydrocephalus, interhemispheric cysts, optic nerve anomalies, septal anomalies, migrational anomalies. Non-central nervous system abnormalities were reviewed separately by a pediatric body radiologist. The anterior commissure and hippocampal commissure are not studied for the classification system used in this study.
Statistical Analysis
Data were obtained and entered into Microsoft Excel. Parametric tests were conducted because the data were normally distributed. Means and their corresponding standard deviations are analyzed for the quantitative variables. Frequencies were used as percentages to represent the qualitative variables.
Results
24 fetuses with abnormal corpus callosum on fetal MRI were recruited for final analyses. Twenty-three of the cases involved singleton pregnancies, and one involved a twin pregnancy. Antenatal women in the study range in age between 16 to 41 years, with a mean age of 30 years (standard deviation ± 7). Gestational age of the fetuses was between 19 to 36 weeks, with a mean age of 26 WG (standard deviation ± 5 days). In one case, the parents were consanguineous, and in all other cases, it was sporadic. There were 55% (13/24) male fetuses and 45% (11/24) female fetuses among the participants. Genetic abnormalities were identified in seven (28%) cases, including trisomy 8 (12%), trisomy 18 (4%), 14q pathogenic deletion (4%), 17q12 pathogenic deletion (4%) and 46 XX plus 47XX+marker chromosome mosaicism (4%) (Figure 2).
Two antenatal women (8%) terminated their pregnancies after fetal MRIs were performed, while the majority of women (91.6%) continued their pregnancy until delivery. Of them, two had stillbirth, and the remaining 20 had live-born babies, who were admitted to the neonatal intensive care unit. 12.6% (3/20) of liveborn infants admitted with CCA died before NICU discharge. The maternal and the birth characteristics of the study population are described in Table 1.
We classified these 24 fetuses based on the type of corpus callosal structural abnormality found on the fetal MRI midline sagittal image. 19 patients had complete agenesis (79.1%), three patients had hypoplasia and (12.5 %) and one patient each had dysplasia (4.2 %) and hypoplasia with dysplasia (4.2 %) respectively (Figure 3).
Individuals within each subclass of CCA displayed a range of abnormalities, both CNS and non-CNS. Out of 24 cases, 4/24 of cases (17 %) had isolated CCA (Figure 5) and 20/24 (83%) cases had complex form of CCA with associated CNS and/or non-CNS anomalies (Figure 4).
Colpocephaly is seen in 13/24 cases (54%) (Figure 5). Of them, 12 cases had complete agenesis and one case had hypoplasia. Absent septum pellucidum is seen in 5/24 cases (20.8%) (Figure 6), 4 cases had complete agenesis and one case had hypoplasia. Probst bundles are identified only in isolated form of CCA and accounts for 3/19 cases (15.7%) of complete agenesis and 3/4 cases of isolated form of CCA (Figure 7). Probst bundles are not seen in other subclasses of CCA Ventriculomegaly is seen in 7/24 cases (29%), 5 cases had complete agenesis, 1 case each had hypoplasia and hypoplasia with dysplasia.
Associated CNS anomalies includes 9/20 cases of interhemispheric cysts seen in 8 cases of complete agenesis class and 1 case of hypoplasia class. Out of 9 cases of IHC, 2 IHC was communicating with lateral ventricles. 4/20 (20%) cases had migration abnormalities such as gray matter heterotopia (Figure 8) in 3 cases of complete agenesis and hemimegalencephaly with schizencephaly in a case of dysplasia. 2 cases of cerebral cysts (2/20) including arachnoid cyst in a case of complete agenesis class and germinolytic cyst in a case of dysplasia. 3/20 cases of posterior fossa cysts, likely Blake's pouch cysts, 2 in complete agenesis class and 1 in hypoplasia class. 2 cases of vermian hypoplasia (2/18) noted, one each case in complete agenesis and hypoplasia class (Figure 9). A case of complete agenesis class had cerebellar hypoplasia and brain stem hypoplasia with a normal vermis. A case of complete agenesis class had neural tube defect in the form of sphenoidal meningoencephalocele. A case of dysplasia class had diffuse thinning of white matter.
Regarding non-neurological anomalies in complex form of CCA, 2 cases had non-CNS anomalies without other CNS anomalies, a case had nasal dermoid cyst while another case had T12 hemivertebra and thoracic bodies fusion. A total of 4 cases had both CNS and non-CNS anomalies, 1 of the cases had severe hydrocephalus, hemimegalencephaly, germinolytic cyst and facial dysmorphism secondary to fused eyes. Second case had heterotopia and hydroureteronephrosis, and another case had sphenoidal meningomyelocele and cleft lip with a cleft palate, last case had interhemispheric cyst, arachnoid cyst and thick nuchal fold, hydronephrosis (Table 2).
Figure 5.
Fetal MRI images of a 31 gestational weeks aged fetus with complete agenesis of the corpus callosum. a) A T2-weighted sequence image in sagittal plane shows that the corpus callosum is not visible (red arrow). b) A T2-weighted sequence image in axial plane shows a “teardrop”-like dilation in the posterior horn of bilateral lateral ventricles suggestive of colpocephaly (white arrow) and absence of corpus callosum and septum pellucidum (red arrow) in the midline. c) A T2-weighted axial image shows a small interhemispheric cyst in the midline. (arrow). No Probst bundles are observed.
Figure 5.
Fetal MRI images of a 31 gestational weeks aged fetus with complete agenesis of the corpus callosum. a) A T2-weighted sequence image in sagittal plane shows that the corpus callosum is not visible (red arrow). b) A T2-weighted sequence image in axial plane shows a “teardrop”-like dilation in the posterior horn of bilateral lateral ventricles suggestive of colpocephaly (white arrow) and absence of corpus callosum and septum pellucidum (red arrow) in the midline. c) A T2-weighted axial image shows a small interhemispheric cyst in the midline. (arrow). No Probst bundles are observed.
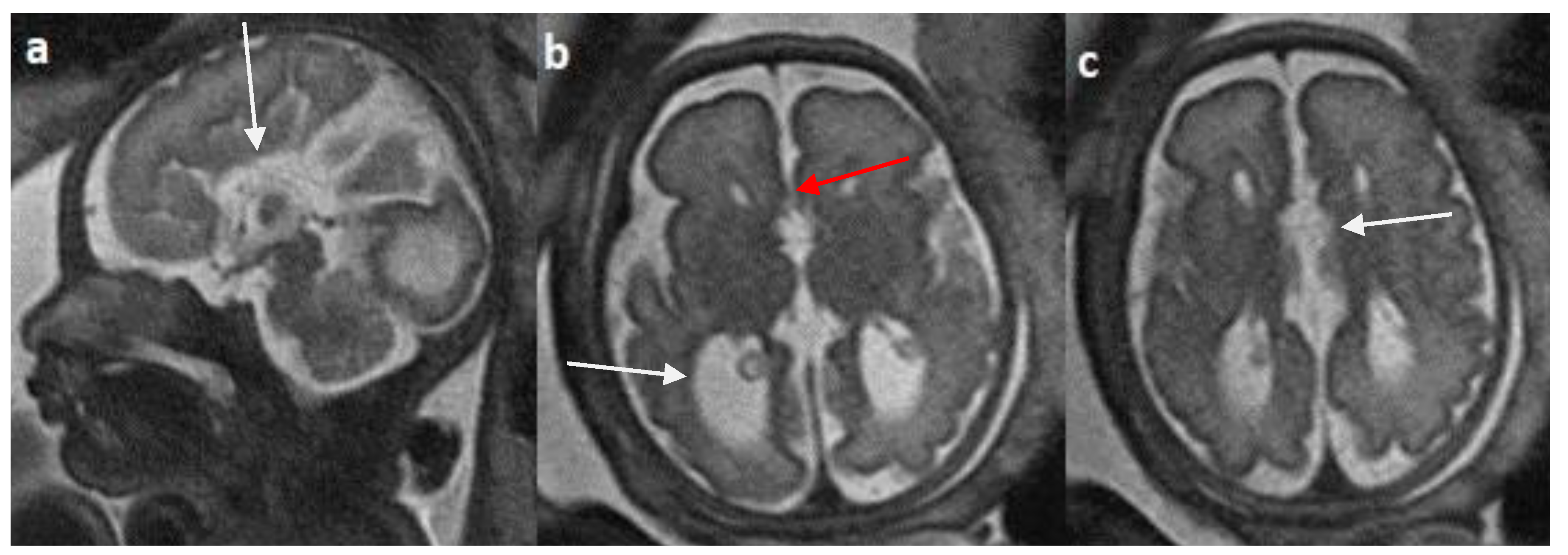
Figure 6.
Fetal MRI images of a 29 gestational weeks aged fetus in a case of hypoplasia with dysplasia of the corpus callosum a) A T2-weighted sequence in axial plane showing severe dilatation of the lateral ventricles, particularly at the body and atria, with thinning of the brain parenchyma in the posterior parieto-occipital region An associated absent cavum septum pellucidum was noted (white arrow).b) A T2-weighted sequence in mid sagittal plane shows thin anterior corpus callosum with dysplasia. Additionally, hypoplasia of the inferior vermis is noted (white arrow).
Figure 6.
Fetal MRI images of a 29 gestational weeks aged fetus in a case of hypoplasia with dysplasia of the corpus callosum a) A T2-weighted sequence in axial plane showing severe dilatation of the lateral ventricles, particularly at the body and atria, with thinning of the brain parenchyma in the posterior parieto-occipital region An associated absent cavum septum pellucidum was noted (white arrow).b) A T2-weighted sequence in mid sagittal plane shows thin anterior corpus callosum with dysplasia. Additionally, hypoplasia of the inferior vermis is noted (white arrow).
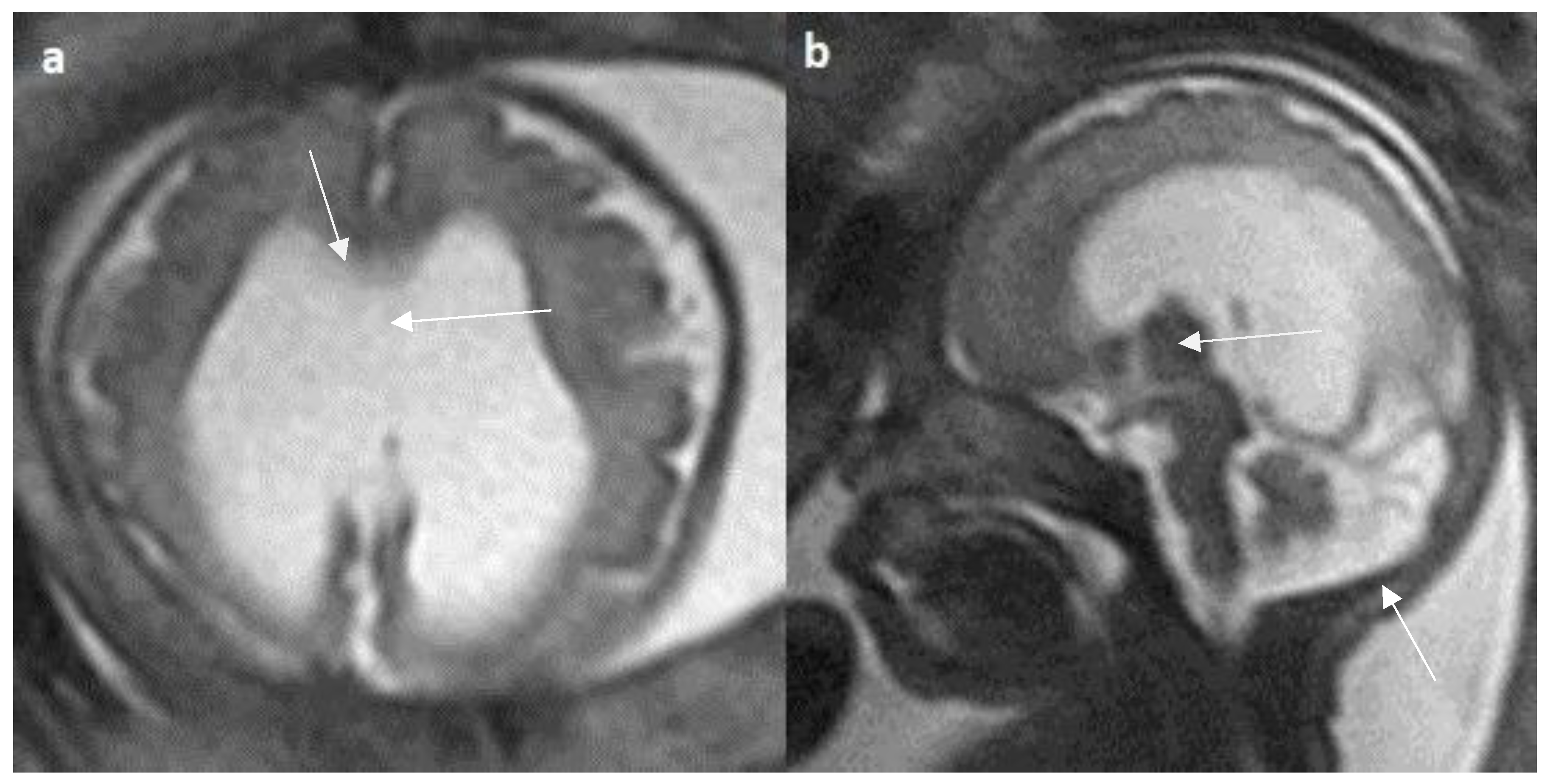
Figure 7.
Fetal MRI images of a 20 gestational weeks aged fetus with complete agenesis of the corpus callosum. A T2-weighted sequence image in axial plane showing non-decussating anterior-posterior white matter tracts known as Probst bundles medial to the lateral ventricles (arrow). A T2- weighted sequence coronal image showing Probst bundles indenting superomedial margins of lateral ventricles. Probst bundles are seen with complete agenesis of the corpus callosum.
Figure 7.
Fetal MRI images of a 20 gestational weeks aged fetus with complete agenesis of the corpus callosum. A T2-weighted sequence image in axial plane showing non-decussating anterior-posterior white matter tracts known as Probst bundles medial to the lateral ventricles (arrow). A T2- weighted sequence coronal image showing Probst bundles indenting superomedial margins of lateral ventricles. Probst bundles are seen with complete agenesis of the corpus callosum.
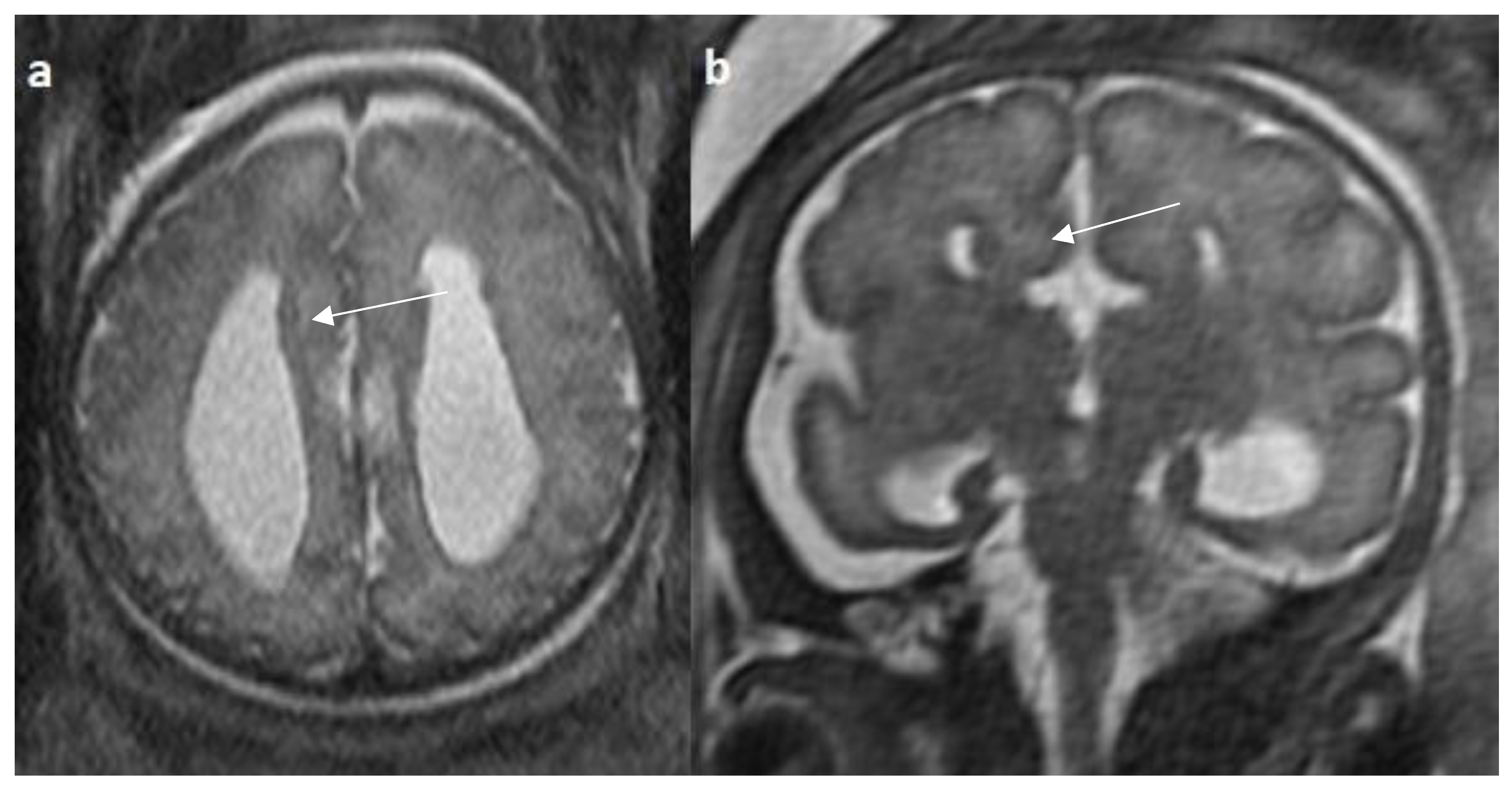
Figure 8.
Fetal MRI images of a 34 gestational weeks aged fetus, with complete agenesis of the corpus callosum and gray matter heterotopia. a) A T2-weighted sequence image in mid sagittal plane showing complete absence of the corpus callosum with secondary changes of ventriculomegaly. b) A T2-weighted axial sequence image showing associated subependymal heterotopia (white arrow) along the occipital horns of the bilateral lateral ventricles with moderate dilatation of lateral and third ventricles.
Figure 8.
Fetal MRI images of a 34 gestational weeks aged fetus, with complete agenesis of the corpus callosum and gray matter heterotopia. a) A T2-weighted sequence image in mid sagittal plane showing complete absence of the corpus callosum with secondary changes of ventriculomegaly. b) A T2-weighted axial sequence image showing associated subependymal heterotopia (white arrow) along the occipital horns of the bilateral lateral ventricles with moderate dilatation of lateral and third ventricles.
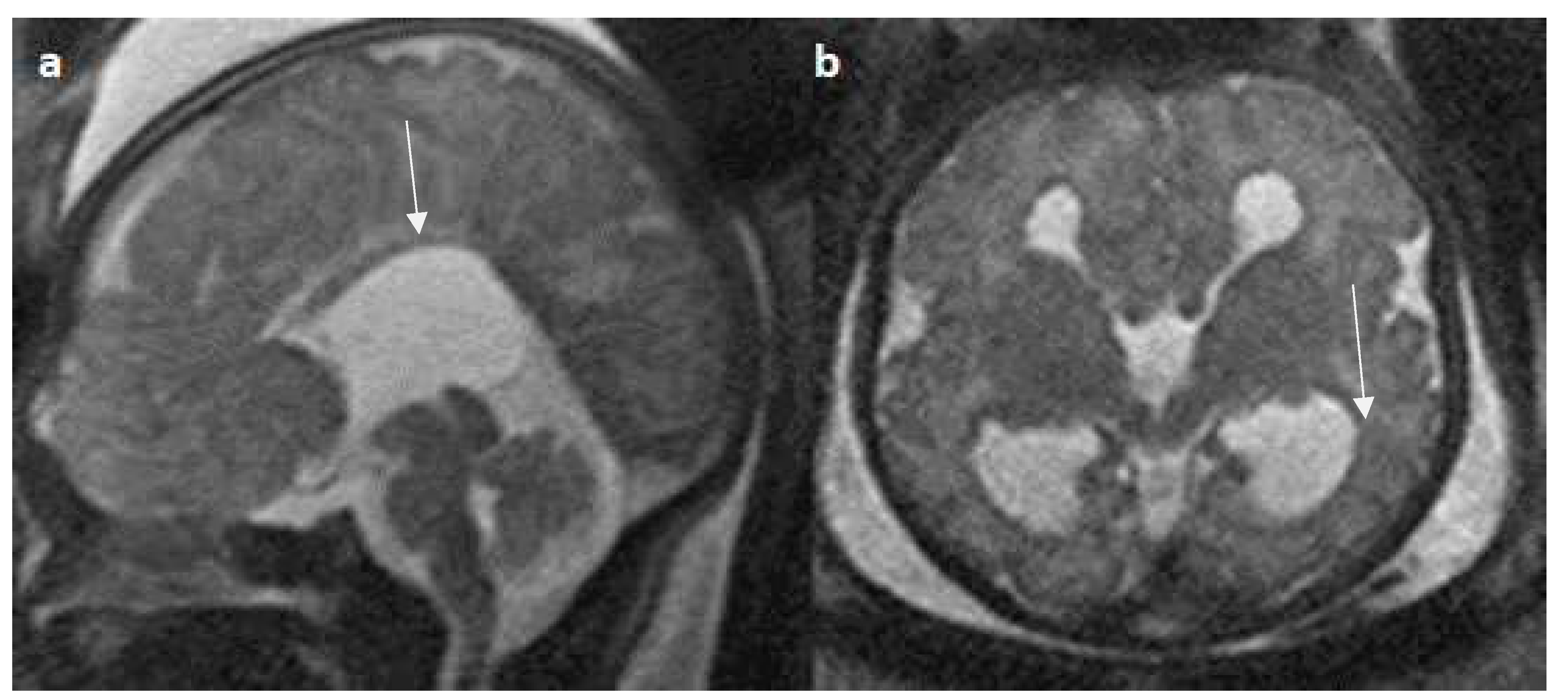
Figure 9.
Fetal MRI images of a 20 gestational weeks aged fetus with complete agenesis of the corpus callosum. a) A T2-weighted sequence image in axial plane shows non-visualization of the corpus callosum, suggestive of complete agenesis. b) A T2 weighted sequence axial image showing a well-defined extra axial fluid in the interhemispheric fissure. A lack of the septum pellucidum was also observed. c) The coronal plane shows an “steer horn” sign (arrow) in the anterior horn of the bilateral lateral ventricles and the absence of the corpus callosum and septum pellucidum in the midline region.
Figure 9.
Fetal MRI images of a 20 gestational weeks aged fetus with complete agenesis of the corpus callosum. a) A T2-weighted sequence image in axial plane shows non-visualization of the corpus callosum, suggestive of complete agenesis. b) A T2 weighted sequence axial image showing a well-defined extra axial fluid in the interhemispheric fissure. A lack of the septum pellucidum was also observed. c) The coronal plane shows an “steer horn” sign (arrow) in the anterior horn of the bilateral lateral ventricles and the absence of the corpus callosum and septum pellucidum in the midline region.
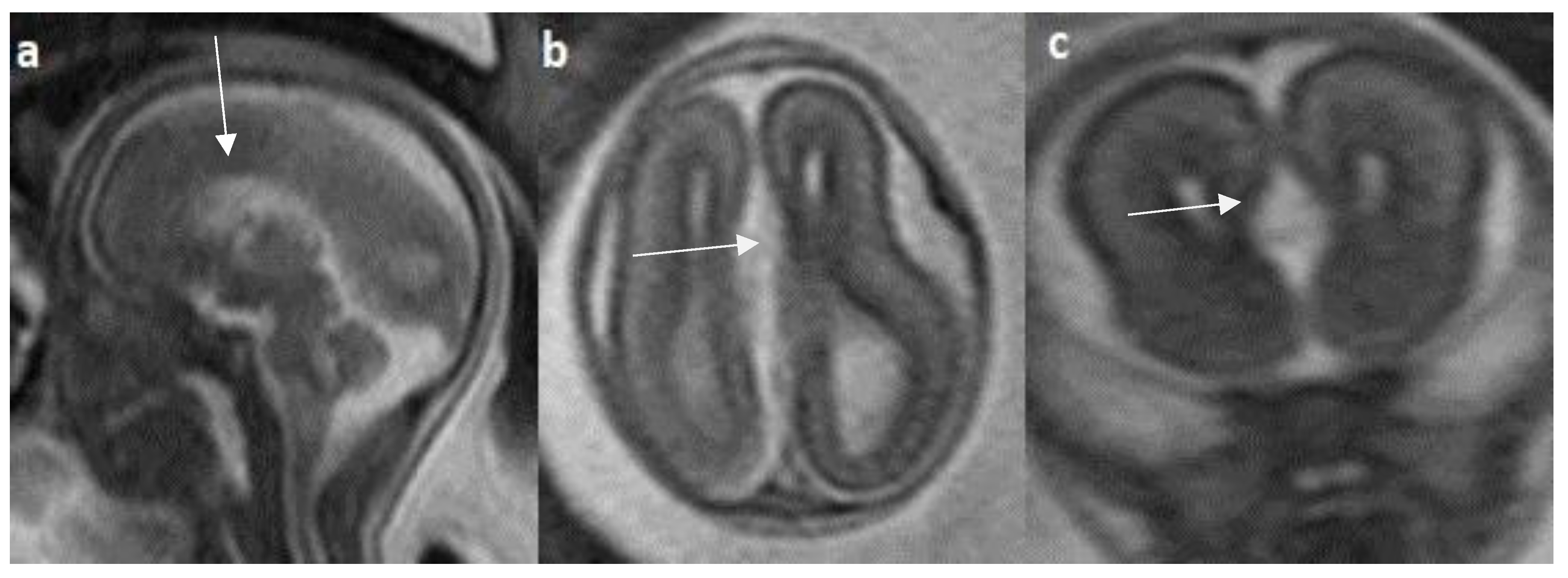
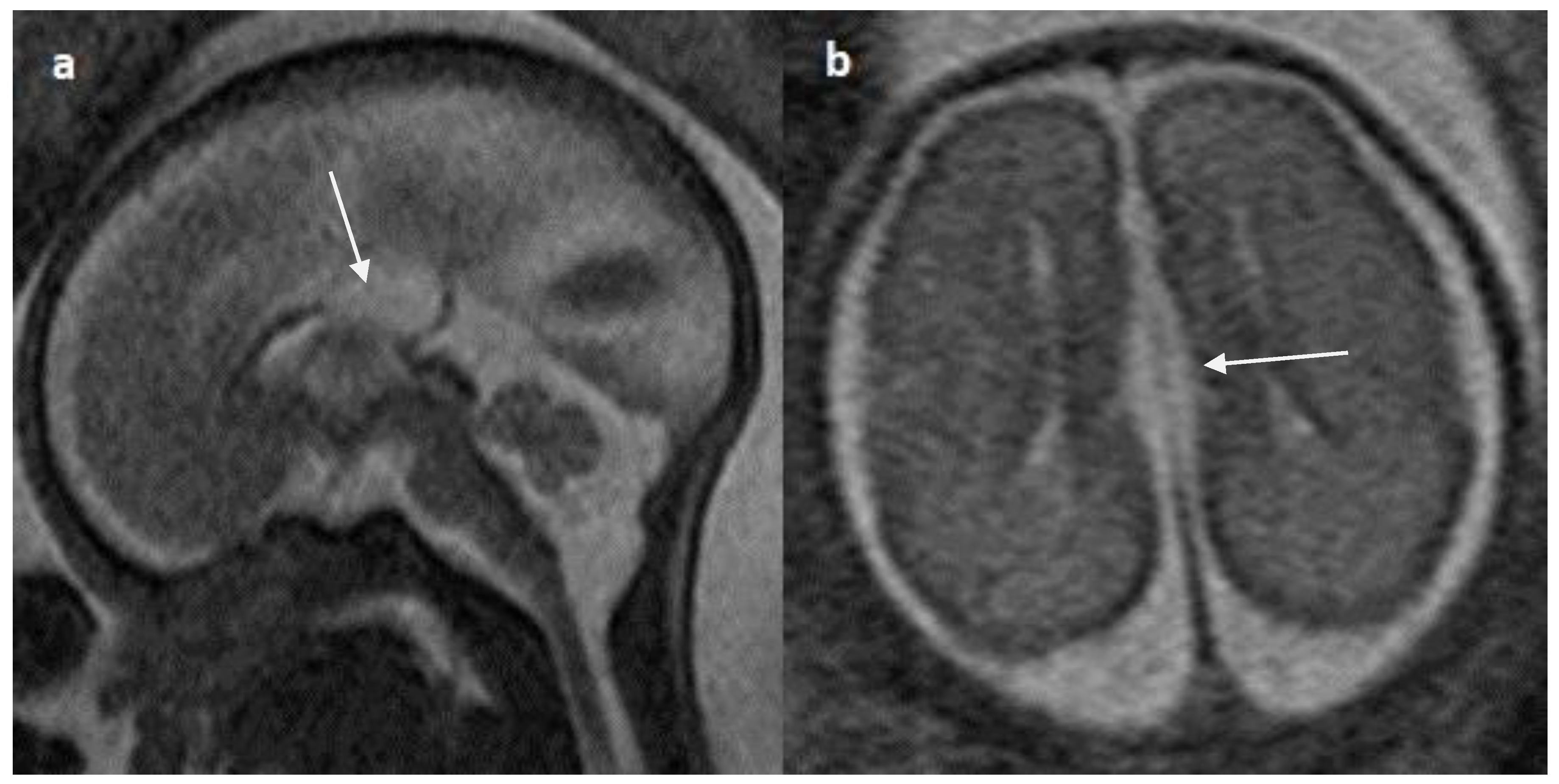
Figure 10.
Fetal MRI images of a 26 gestational weeks aged fetus with hypoplasia of the corpus callosum. a) A T2-weighted sequence in mid sagittal plane image showing normal anterior part of corpus callosum with absent posterior part (arrow), suggestive of hypoplasia of posterior region with anterior remnant, rest of the brain parenchyma in this section is normal. b) A T2-weighted sequence in axial plane showing a small midline interhemispheric fluid collection/cyst without communication with the lateral ventricles.
Figure 10.
Fetal MRI images of a 26 gestational weeks aged fetus with hypoplasia of the corpus callosum. a) A T2-weighted sequence in mid sagittal plane image showing normal anterior part of corpus callosum with absent posterior part (arrow), suggestive of hypoplasia of posterior region with anterior remnant, rest of the brain parenchyma in this section is normal. b) A T2-weighted sequence in axial plane showing a small midline interhemispheric fluid collection/cyst without communication with the lateral ventricles.
Discussion
The study included 16 to 41-year-old pregnant women, with a mean age of 30 ± 7 years similar to study done by Yeh et al. [40]. In this study population of 24 fetuses diagnosed with CCA, the male to female ratio is 1.2:1. Yeh et al. and Manganaro et al. found similar results for corpus callosal abnormalities in prenatally diagnosed fetuses [40,41]. Jeret et al. reported similar results in postnatally diagnosed fetuses [12]. The development of corpus callosum completes by around 18 to 19 WG when detected in prenatal imaging [28]. Therefore, the gestational age in our study ranged from 19 to 36 weeks, with a mean of 26 (standard deviation ± 5 days) consistent with the study of Tang et al. [42]. Sporadic cases accounted for 23 of 24 cases, with only one case resulting from consanguinity, which is consistent with other studies on prenatally detected CCA [43]. Current evidence suggests that both acquired and genetic mechanisms play an important role in the etiology of CCA [41]. Roughly 30% of the study participants had identifiable diagnoses, including cases of single-gene disorders, mosaicism, copy number variants, trisomy’s. Many genetic disorders have been linked to CCA including trisomy’s, gene deletion syndromes, copy number variants, and X-linked disorders [19,44]. The genetic variants noted in the study have also been identified in many other studies [45,46,47,48,49]. The trisomy 8 mosaicism is the most common (13%) genetic abnormality detected in our study. We detected 4/24 cases (25%) of syndromic corpus callosal agenesis of which 3 patients had trisomy 8 mosaicism and one patient had trisomy 18. The percentage of clinical syndromic diagnosis is similar to study done by Al-Hashim et al. (22%), Bedeshi et al. (33%) and Schell- Apasik et al. (32%) [19,36,50]. 2 cases had single gene disorder with pathogenic gene deletion at 14q24.3-32 and 17q12. Till date only few cases were reported with 14q24.3-32 gene deletion in cases with corpus callosum agenesis [47]. 17q12 pathogenic deletion was identified in few studies analyzing genetic variants in corpus callosal agenesis [48,49]. A total of (29%) cases had identifiable genetic cause similar to the study conducted by Al-Hashim et al. [19]. Regarding the disposition of pregnancy, 2 cases underwent the termination of pregnancy (abortion), 2 cases had still birth, while most of them continued their pregnancy until 37th WG. These findings can be comparable to study conducted by Ibrahim et al. [43]
The diagnosis of corpus callosal abnormalities in the axial planes in fetal MRI allows suspicion of the pathology while mid sagittal plane is necessary to make the diagnosis of CCA without the need for 3D reconstruction techniques or volumetric analysis. Fetal movement poses a great challenge in the collection of corpus callosum information, however visualization in the mid sagittal plane of the brain is possible by ultra-fast localization using axial and coronal planes [51]. In the light of high prevalence of complex form of CCA over isolated form, fetal MRI is advantageous and provide more information about cortical morphology especially when done after 27 weeks of gestation. Moreover, in nearly 83% of patients, fetal MRI identified abnormalities not revealed by antenatal ultrasonography [42]. In addition, fetal MRI can suggest a possible etiology for CCA such as developmental malformations can give a clue for a genetic syndrome and disruptive changes in the fetal brain parenchyma may suggest an acquired or metabolic abnormality associated with CCA [51]. Furthermore, by utilizing fetal MRI we can detect subtle other associated CNS abnormalities that may indicate postnatal neurodevelopmental outcomes [52]. Fetal MRI has a better contrast resolution, multiplanar capability and allows direct visualization of the corpus callosum [26]. A simple system-based classification based on fetal MRI midline sagittal image is the first step in the evaluation of CCA, however, future studies may use advanced imaging techniques like functional MRI or fractional anisotropy for precise anatomic subclassification [20]. The use of machine learning models in conjunction with genetic and clinical data to analyze prenatal brain multi-parametric MRI data may improve diagnosis and postnatal outcome monitoring [53]. Multidisciplinary collaborative teams are involved including genetics, obstetrics, perinatology, pediatric surgery, neuro surgery and neurology in the management of CCA [54].
Our study used the Hanna et al. classification to describe heterogeneity in CCA morphology that could describe our entire cohort [20]. Recent studies of fetal MRI have used this classification including a study done by El Ameen et al. in 2019 and Manor et al. in 2020 [55,26]. The study done by El Ameen et al. used this classification to evaluate the role of DTI and fiber tractography in children with CCA and also studied the clinico-radiological correlation [55]. Manor et al. described fetal MRI in CCA in his pictorial essay based on this classification [26]. The previous classification systems based on prenatal ultrasonography divided CCA broadly into complete and partial agenesis, and thus did not account for heterogeneity in callosal morphologies. The extensive spectrum of morphological description of CCA may throw light into the time of insult, etiology, and may have prognostic implications.
In the present study, the classification of CCA was done into complete, hypoplasia, dysplasia, and hypoplasia with dysplasia. The largest subgroup consisted of individuals with complete agenesis (79.1%), which is similar to the findings reported by Ruland et al. in which they reported the majority percentage of complete agenesis cases (76%) [56]. These findings are in line with study conducted by Ibrahim et al. in which they diagnosed 77.7% cases of complete agenesis [43]. Correspondingly, Al-Hashim et al. reported in their study with the largest subset of patients with complete agenesis (52%) [19]. The complete agenesis group exhibited a higher prevalence of ventriculomegaly, colpocephaly, and Probst bundles. Ventriculomegaly was not considered as an additional finding, as it can be seen with corpus callosal agenesis. In our study colpocephaly (58.3%) is more frequent in cases of complete agenesis than in hypoplasia or dysplasia group similar to study done by Neal et al. [21]. Colpocephaly occurs due to a decrease in white matter in the occipital cortex, resulting in the subsequent enlargement of the posterior horns of the lateral ventricles [1,57]. On the other hand, in the group with hypoplasia or dysplasia, the presence of intact myelinated callosal tracts is likely responsible for preserving the structural integrity, which in turn prevents the enlargement of the posterior horns of the lateral ventricles to certain extent. Probst bundles were observed in cases of complete agenesis of the corpus callosum but were not found in cases of hypoplasia, consistent with the findings reported by Hett et al. [1]. Furthermore, Probst bundles are found in 3/4 (75%) cases of isolated CCA and not seen in complex form of CCA, similar to findings reported by Byrd et al. [23]. However, the identification of Probst bundles on real-time fetal MRI can be challenging, possibly explaining the lower percentage of Probst bundles in the study cohort compared to post-natal research studies. Cavum septum pellucidum is considered present, even if incomplete or present in the form of remnants. This fact probably explains the smaller number of absent CSP cases in our cohort. Most cases of CSP are identified in cases of complete agenesis. These findings are in contrast with study done by Neal et al. who reported that septal anomalies are most common in hypoplasia class [21].
The complex form of CCA can be further classified as associated with neurological, non-neurological anomalies or both. Out of 24 patients in our analysis, only 4 (16%) had isolated CCA; the remaining 20 (83.3%) cases were linked to other abnormalities. This is consistent with study done by Ruland et al. that found that 71% of cases were non-isolated forms of CCA and 29% of cases were isolated forms. [56]. Our study results are also in line with similar work reported by Ibrahim et al. in which they revealed that 18.5% had isolated CCA and 74% were non-isolated form [43]. Manganaro et al. conducted a study in which they discovered that fetal MRI diagnosed 26.9% isolated CCA, with the majority of the cohort (about 73.1%) being associated with other abnormalities [42]. This maximized prevalence of non-isolated CCA over isolated CCA is an important predictor of postnatal prognosis according to recent data on autopsy studies [58,59,60].
Associated CNS anomalies were seen in 14/20 cases (70 %) which is in line with studies conducted by Al-Hashim et al. [19]. Interhemispheric cysts were the most frequent related neurological findings in around 9/20 cases (45%) which correlates well with study done by Ibrahim et al. who reported that IHC is the most common neurological finding representing upto 37.5% [43]. In another study done by Manganaro et al. it was found that around 19.7% of cases had IHC [42]. Additionally, these findings are similar to study done by Byrd et al. with 35/79 cases (44%) having associated IHC [23]. However, in another study done by Hetts et al. only 14% cases of corpus callosal agenesis had IHC [1]. Our study had (20%) cases of migrational anomalies, which is in accordance with the study by Hetts et al. (25%) and Byrd et al. (23%) [1,23]. In our study between CCA classes, migrational anomalies were more common in the complete agenesis group in comparison with hypoplasia group similar to Hetts et al. who reported migrational anomalies in 29% of cases with agenesis of corpus callosum and 21% of cases with hypogenesis [1].
In our study, vermian hypoplasia represented up to 10% of posterior fossa anomalies, which was lesser when compared to research by Manganaro et al. and Ibrahim et al. that found up to 30% of related cerebellar malformations [42, 43]. Furthermore, Byrd et al. reported that 7 % of cases had vermian hypoplasia [23]. 5% of our cases had Dandy walker variant with abnormal and distorted medulla, which is lower than study done by Neal et al. and Byrd et al. [21,23]. Neural tube defects are noted in 5% of cases which is in line with study done by Byrd et al. [23] and lower when compared to study done by Ibrahim et al [43].
6/24 cases (25%) of the total cohort had associated non-CNS anomalies, which is similar to study done by Taylor et al. [61]. The associated only non-neurological anomalies were found in two cases, in which one case had T12 hemivertebra with thoracic vertebral bodies fusion but suspected to have Aicardi syndrome, and another case had nasal dermoid cyst, which is a rare syndrome known as fronto-nasal dermoid cysts and agenesis of corpus callosum [62].
The cranio-facial abnormalities were the most common non-neurological anomalies detected in the study. This according to the studies done by Jeret et al. and Bedeshi et al. [12,36]. 4 cases (20%) showed combined neurological and non-neurological anomalies, which is higher than the study done by Ibrahim et al. (10%) [43]. Of these, first case had facial dysmorphism including frontal bossing secondary to severe hydrocephalus with associated fused eyes. Second case includes non-neurological anomaly of cleft lip with a cleft palate with associated sphenoidal meningomyelocele as neurological anomaly. The neurological anomalies associated with this case include hemimegalencephaly and germinolytic cyst. Another case had included non-neurological anomalies diagnosed as hydroureteronephrosis, which showed a neurological anomaly of heterotopia. The last case had trisomy 8 showed associated thick nuchal fold with an interhemispheric cyst. The study done by Ibrahim et al. reported 2 cases of combined anomalies, one case had bilateral enlarged cystic kidneys and posterior fossa cyst. Another case had bilateral club foot with large interhemispheric cyst and posterior fossa anomalies [43].
This study highlights the importance of diagnosing CCA abnormalities with fetal MRI, which has implications in the genetic counselling of parents of future pregnancy risks. Consequently, greater research on the significance of fetal MRI is needed, as behavioral and cognitive issues may not be recognized until school age, as corpus callosal abnormalities have been associated to autism and schizophrenia. Future studies using Diffusion-weighted and Diffusion tensor imaging will provide more insight into white matter abnormalities in fetuses with CCA.
The study is limited by a retrospective study design. Relation of various CNS and non-CNS abnormalities with different CCA subtypes could not be analyzed owing to the small sample size. The study included only fetuses diagnosed with CCA by prenatal ultrasound. Fetal ultrasonography is operator dependent and diagnosis depends on the obstetrician's judgment. Fetal MR imaging done at later gestational ages has better sensitivity, though clinically impractical. It is therefore important to perform fetal MRI at a consistent gestational age in order to maximize the study's accuracy. Replications of the study at different institutions may yield different results. As this study was conducted at a tertiary health care referral center, more proportion of fetuses are seen with associated anomalies, which makes our results less generalizable.
Conclusion
The prevalence of complex forms of CCA is higher than that of isolated CCA. Fetal MRI is recommended in the characterization of CCA, as in many instances of apparent isolated CCA It is the main method to differentiate between isolated CCAs and complex CCAs. It provides additional information about the type and severity of CCA, detects associated anomalies, and helps to comprehend the underlying mechanisms. The study emphasizes that in many instances a single midline sagittal MRI image is adequate in identifying an abnormal corpus callosum. However, in cases where the conclusion is unclear, it is advisable to examine the fetal CC in all three planes to reduce interpretation errors. Inclusion of the CCA subclasses' classification accurately describes the heterogeneity of callosal morphology and their associated prognosis. It is recommended that the classification scheme should include the nomenclature of Probst's bundles, which is commonly seen in sporadic cases of complete callosal agenesis and has significance in the post-natal outcome. We recommend that it is best a practice standard to include clinical genetics review, CMA testing in patients with both isolated and non-isolated forms of CCA. The integration of morphological classification combined with clinical and genetic information using deep machine learning techniques allows for further understanding of an individual with CCA. Future studies should use functional and tractography-based imaging techniques for early detection of white matter abnormalities, Probst tracts, and other faulty fibers in fetuses with CCA.
Funding
Supported by Colonel Robert R McCormick Professorship of Diagnostic Imaging Fund at Rush University Medical Center (The Activity Number is 1233-161-84), No. 8410152-03.
Conflicts of Interest
None.
References
- Hetts, S.W., et al., Anomalies of the corpus callosum: an MR analysis of the phenotypic spectrum of associated malformations. AJR Am J Roentgenol, 2006. 187(5): p. 1343-8. [CrossRef]
- Fitsiori, A., et al., The corpus callosum: white matter or terra incognita. Br J Radiol, 2011. 84(997): p. 5-18. [CrossRef]
- Richards, L.J., C. Plachez, and T. Ren, Mechanisms regulating the development of the corpus callosum and its agenesis in mouse and human. Clin Genet, 2004. 66(4): p. 276-89. [CrossRef]
- Kier EL, Truwit CL. The normal and abnormal genu of the corpus callosum: an evolutionary, embryologic, anatomic, and MR analysis. AJNR Am J Neuroradiol 1996; 17(9): 1631-1641.
- Ren T, Anderson A, Shen WB, et al. Imaging, anatomical, and molecular analysis of callosal formation in the developing human fetal brain. Anat Rec A Discov Mol Cell Evol Biol 2006; 288(2): 191-204. [CrossRef]
- Georgy, B.A., J.R. Hesselink, and T.L. Jernigan, MR imaging of the corpus callosum. AJR m J Roentgenol, 1993. 160(5): p. 949-55. [CrossRef]
- Lee, S.K., et al., Diffusion-tensor MR imaging and fiber tractography: a new method of describing aberrant fiber connections in developmental CNS anomalies. Radiographics, 2005. 25(1): p. 53-65; discussion 66-8. [CrossRef]
- Rakic, P. and P.I. Yakovlev, Development of the corpus callosum and cavum septi in man. J Comp Neurol, 1968. 132(1): p. 45-72. [CrossRef]
- Paul LK, Brown WS, Adolphs R, et al. Agenesis of the corpus callosum: genetic, development and functional aspects of connectivity. Nat Rev Neurosci. 2007;8(4):287-299. [CrossRef]
- Edwards TJ, Sherr EH, Barkovich AJ, Richards LJ. Clinical, genetic and imaging findings identify new causes for corpus callosum development syndromes. Brain. 2014 Jun;137(Pt 6):1579-613. [CrossRef]
- Glass HC, Shaw GM, Ma C, Sherr EH. Agenesis of the corpus callosum in California 1983-2003: a population-based study. Am J Med Genet A. 2008;146A(19):2495-2500. [CrossRef]
- Jeret, J.S., et al., Frequency of agenesis of the corpus callosum in the developmentally disabled population as determined by computerized tomography. Pediatr Neurosci, 1985. 12(2): p. 101-3. [CrossRef]
- Grogono, J.L., Children with agenesis of the corpus callosum. Dev Med Child Neurol, 1968. 10(5): p. 613-6. [CrossRef]
- Nagwa, S., et al., Imaging features of complete agenesis of corpus callosum in a 3-year-old child. Sudan J Paediatr, 2018. 18(2): p. 69-71. [CrossRef]
- Hofman, J., et al., Corpus Callosum Agenesis: An Insight into the Etiology and Spectrum of Symptoms. Brain Sci, 2020. 10(9). [CrossRef]
- Barkovich AJ. Pediatric Neuroimaging. Philadelphia: Lippincott Williams & Wilkins; 2005.
- Santo S, D'Antonio F, Homfray T, Rich P, Pilu G, Bhide A, Thilaganathan B, Papageorghiou AT. Counseling in fetal medicine: agenesis of the corpus callosum. Ultrasound Obstet Gynecol. 2012 Nov;40(5):513-21. [CrossRef]
- Witelson, S.F., 1989. Hand and sex differences in the isthmus and genu of the human corpus callosum. A postmortem morphological study. Brain. 1989;112 ( Pt 3):799-835. [CrossRef]
- Al-Hashim AH, Blaser S, Raybaud C, et al. Corpus callosum abnormalities: neuroradiological and clinical correlations. Dev Med Child Neurol. 2016;58(5):475-484. [CrossRef]
- Hanna RM, Marsh SE, Swistun D, Al-Gazali L, Zaki MS, Abdel-Salam GM, et al. Distinguishing 3 classes of corpus callosal abnormalities in consanguineous families. Neurology. 2011;76(4):373-82. [CrossRef]
- Neal, J.B., Filippi, C.G. & Mayeux, R. Morphometric variability of neuroimaging features in Children with Agenesis of the Corpus Callosum. BMC Neurol. 2015;15:116. [CrossRef]
- Matter Remodeling in a Dog With Corpus Callosum Hypoplasia and Dysplasia. Front Vet Sci, 2018. 5: p. 260. [CrossRef]
- Byrd SE, Radkowski MA, Flannery A, McLone DG. The clinical and radiological evaluation of absence of the corpus callosum. Eur J Radiol 1990;10:65-73. [CrossRef]
- Volpe P, Campobasso G, De Robertis V, et al. Disorders of prosencephalic development. Prenat Diagn 2009; 29(4): 340-354. [CrossRef]
- Chaudhari BP, Ho ML. Congenital Brain Malformations: An Integrated Diagnostic Approach. Semin Pediatr Neurol 2022;42:100973. [CrossRef]
- Manor C, Rangasami R, Suresh I, Suresh S. Magnetic Resonance Imaging Findings in Fetal Corpus Callosal Developmental Abnormalities: A Pictorial Essay. J Pediatr Neurosci. 2020 Oct-Dec;15(4):352-357. [CrossRef]
- Hyun Yoo J, Hunter J. Imaging spectrum of pediatric corpus callosal pathology: a pictorial review. J Neuroimaging. 2013 Apr;23(2):281-95. [CrossRef]
- Glenn, O.A., Goldstein, R.B., Li, K.C., Young, S.J., Norton, M.E., Busse, R.F., Goldberg, J.D. and Barkovich, A.J., Fetal Magnetic Resonance Imaging in the Evaluation of Fetuses Referred for Sonographically Suspected Abnormalities of the Corpus Callosum. Journal of Ultrasound in Medicine. 2005; 24: 791-804. [CrossRef]
- Vergani P, Ghidini A, Strobelt N, Locatelli A, Mariani S, Bertalero C, Cavallone M: Prognostic indicators in the prenatal diagnosis of agenesis of the corpus callosum. Am J Obstet Gynecol 1994;170:753–758. [CrossRef]
- Gupta JK, Lilford RJ: Assessment and management of fetal agenesis of the corpus callosum. Prenatal Diagn 1995;15:301–312. [CrossRef]
- Serur D, Jeret JS, Wisniewski K: Agenesis of the corpus callosum: Clinical, neuroradiological and cytogenetic studies. Neuropaediatrics 1988;19:87–91. [CrossRef]
- Cohen MM Jr, Kreiborg S: Agenesis of the corpus callosum: Its associated anomalies and syndromes with special reference to the Apert syndrome. Neurosurg Clin North Am 1991;2:565– 568. [CrossRef]
- Geoffrey G: Other syndromes frequently associated with callosal agenesis; in Lassonde M, Jeeves MA (eds): Callosal Agenesis: A Natural Split Brain? New York, Plenum Press, 1994, pp 55–62. [CrossRef]
- Aicardi J, Chevrie JJ: The Aicardi syndrome; in Lassonde M, Jeeves MA (eds): Callosal Agenesis: A 2Natural Split Brain? New York, Plenum Press, 1994, pp 7–15. [CrossRef]
- Sotiriadis A, Makrydimas G. Neurodevelopment after prenatal diagnosis of isolated agenesis of the corpus callosum: an integrative review. Am J Obstet Gynecol 2012; 206: 337.e1–5. [CrossRef]
- Bedeschi MF, Bonaglia MC, Grasso R, Pellegri A, Garghentino RR, Battaglia MA, Panarisi AM, Di Rocco M, Balottin U, Bresolin N, Bassi MT, Borgatti R. Agenesis of the corpus callosum: clinical and genetic study in 63 young patients. Pediatr Neurol. 2006 Mar;34(3):186-93. [CrossRef]
- Parazzini C, Righini A, Rustico M, Consonni D, Triulzi F. Prenatal magnetic resonance imaging: brain normal linear biometric values below 24 gestational weeks. Neuroradiology. 2008;50(10):877–883. [CrossRef]
- Garel C (2004) MRI of the fetal brain: normal development and cerebral pathologies, pathology of the midline cap. Springer, Berlin:131–149. [CrossRef]
- Sira LB, Kozyrev DA, Bashat DB, Constantini S, Roth J, Shiran S.Fetalventriculomegaly and hydrocephalus-What shouldn’t be missed on imaging?. Neurol India.2021;69(supplement): S298–304. [CrossRef]
- Yeh HR, Park HK, Kim HJ, Ko TS, Won HS, Lee MY, Shim JY, Yum MS. Neurodevelopmental outcomes in children with prenatally diagnosed corpus callosal abnormalities. Brain Dev. 2018 Sep;40(8):634-641. [CrossRef]
- Manganaro, L., Bernardo, S., DeVito, C., Antonelli, A., Marchionni, E., Vinci, V., Saldari, M., Di Meglio, L., Giancotti, A., Silvestri, E., Catalano, C., and Pizzuti, A. Role of fetal MRI in the evaluation of isolated and non-isolated corpus callosum dysgenesis: results of a cross-sectional study. Prenat Diagn, 2017;37: 244–252. [CrossRef]
- Tang PH, Bartha AI, Norton ME, Barkovich AJ, Sherr EH, Glenn OA. Agenesis of the corpus callosum: an MR imaging analysis of associated abnormalities in the fetus. AJNR Am J Neuroradiol. 2009. [CrossRef]
- Ibrahim and Emad-Eldin Egyptian Journal of Radiology and Nuclear Medicine. 2020; 51:75. [CrossRef]
- Richards LJ, Plachez C, Ren T. Mechanisms regulating the development of the corpus callosum and its agenesis in mouse and human. Clin Genet 2004;66:276 –289. [CrossRef]
- Settimo, C.; Bonanno, L.; Tresoldi, M.; Muratore, R.; Cucinotta, F.; Tripodi, E.; Piccolo, A.; Anchesi, S.; Impallomeni, C. Early and Innovative Rehabilitation in Warkany Syndrome 2 Associated with Agenesis of the Corpus Callosum: A Case Report. Children 2022, 9, 722. [CrossRef]
- Cereda A, Carey JC. The trisomy 18 syndrome. Orphanet J Rare Dis. 2012 Oct 23;7:81. [CrossRef]
- Nicita F, Di Giacomo M, Palumbo O, Ferri E, Maiorani D, Vigevano F, Carella M, Capuano A. Neurological features of 14q24-q32 interstitial deletion: report of a new case. Mol Cytogenet. 2015 Nov 24;8:93. [CrossRef]
- Ru Li, Fang Fu, Yong-Ling Zhang, Dong-Zhi Li, Can Liao. Prenatal diagnosis of 17q12 duplication and deletion syndrome in two fetuses with congenital anomalies. Taiwanese Journal of Obstetrics and Gynecology. 2014;53(4):579-582. [CrossRef]
- Kamath A, Linden SC, Evans FM, Hall J, Jose SF, Spillane SA, Hardie ADR, Morgan SM, Pilz DT. Chromosome 17q12 duplications: Further delineation of the range of psychiatric and clinical phenotypes. Am J Med Genet B Neuropsychiatr Genet. 2018 Jul;177(5):520-528. [CrossRef]
- Schell-Apacik CC, Wagner K, Bihler M, Ertl-Wagner B, Heinrich U, Klopocki E, Kalscheuer VM, Muenke M, von Voss H. Agenesis and dysgenesis of the corpus callosum: clinical, genetic and neuroimaging findings in a series of 41 patients. Am J Med Genet A. 2008 Oct 1;146A(19):2501-11. [CrossRef]
- Li X, Wang Q. Magnetic Resonance Imaging (MRI) Diagnosis of Fetal Corpus Callosum Abnormalities and Follow-up Analysis. J Child Neurol. 2021 Oct;36(11):1017-1026. [CrossRef]
- Sztriha L. Spectrum of corpus callosum agenesis. Pediatr Neurol. 2005;32(2):94–101. [CrossRef]
- Vahedifard F, Adepoju JO, Supanich M, Ai HA, Liu X, Kocak M, Marathu KK, Byrd SE. Review of deep learning and artificial intelligence models in fetal brain magnetic resonance imaging. World J Clin Cases. 2023 Jun 6;11(16):3725-3735. [CrossRef]
- Da Silva NA, Vassallo J, Sarian LO, Cognard CH, Sevely A. Magnetic resonance imaging of the fetal brain at 3 Tesla. Preliminary experience from a single series. Medicine.2018;97(40):1–6. [CrossRef]
- El Ameen NF, Ibrahim MA, Mouner SM. Fiber Tractography and Diffusion Tensor Imaging in Children with Agenesis and Dysgenesis of Corpus Callosum: A Clinico-Radiological Correlation. Int J Pediatr 2019; 7(8): 9903-15. [CrossRef]
- Rüland AM, Berg C, Gembruch U, Geipel A. Prenatal diagnosis of anomalies of the corpus callosum over a 13-year period. Ultraschall Med. 2016;37(6):598–603 15. [CrossRef]
- Barkovich AJ, Norman D. Anomalies of the corpus callosum: correlation with further anomalies of the brain. AJR Am J Roentgenol. 1988 Jul;151(1):171-9. [CrossRef]
- Kidron D, Shapira D, Ben Sira L et al. Agenesis of the corpus callosum. An autopsy study in fetuses. Virchows Arch 2016;468(2):219-30. [CrossRef]
- Kitova TT, Kitov B, Milkov D, Gaigi S. Postnatally diagnosed agenesis of corpus callosum in fetuses. Fetal Pediatr Pathol 2014;33(4):239-43. [CrossRef]
- Alby C, Malan V, Boutaud L et al. Clinical, genetic and neuropathological findings in a series of 138 fetuses with a corpus callosum malformation. Birth Defects Res A Clin Mol Teratol. 2016;106(1):36-46. [CrossRef]
- Taylor M, David AS. Agenesis of the corpus callosum: a United Kingdom series of 56 cases. J Neurol Neurosurg Psychiatry. 1998 Jan;64(1):131-4. [CrossRef]
- Jacobson, I., Jeeves, M.A. (1994). A New Syndrome: Familial Fronto-Nasal Dermoid Cysts with Agenesis of the Corpus Callosum. In: Lassonde, M., Jeeves, M.A. (eds) Callosal Agenesis. Advances in Behavioral Biology, vol 42. Springer, Boston, MA. [CrossRef]
Figure 1.
Schematic representative diagram showing structural morphology of normal corpus callosum and corpus callosal abnormalities on the midline sagittal fetal MRI. A Normal corpus callosum showing rostrum, genu, body and splenium. Corpus callosal abnormalities are divided into subclasses: B, Hypoplasia without dysplasia – uniform thinning with the intact structure of corpus callosum having all the four parts. C, Hypoplasia of posterior region with anterior remnant- Absent posterior corpus callosum. D, Dysplasia without hypoplasia- this represents as structurally abnormal corpus callosum with no thinning. E, Hypoplasia with dysplasia – striped type- lack of the structurally distinct genu and splenium with uniform thin stripe corpus callosum. F, Hypoplasia with dysplasia – kinked type- hypoplasia and kinking of anterior or posterior aspect. Complete, agenesis- Absence of corpus callosum at fetal MRI.
Figure 1.
Schematic representative diagram showing structural morphology of normal corpus callosum and corpus callosal abnormalities on the midline sagittal fetal MRI. A Normal corpus callosum showing rostrum, genu, body and splenium. Corpus callosal abnormalities are divided into subclasses: B, Hypoplasia without dysplasia – uniform thinning with the intact structure of corpus callosum having all the four parts. C, Hypoplasia of posterior region with anterior remnant- Absent posterior corpus callosum. D, Dysplasia without hypoplasia- this represents as structurally abnormal corpus callosum with no thinning. E, Hypoplasia with dysplasia – striped type- lack of the structurally distinct genu and splenium with uniform thin stripe corpus callosum. F, Hypoplasia with dysplasia – kinked type- hypoplasia and kinking of anterior or posterior aspect. Complete, agenesis- Absence of corpus callosum at fetal MRI.
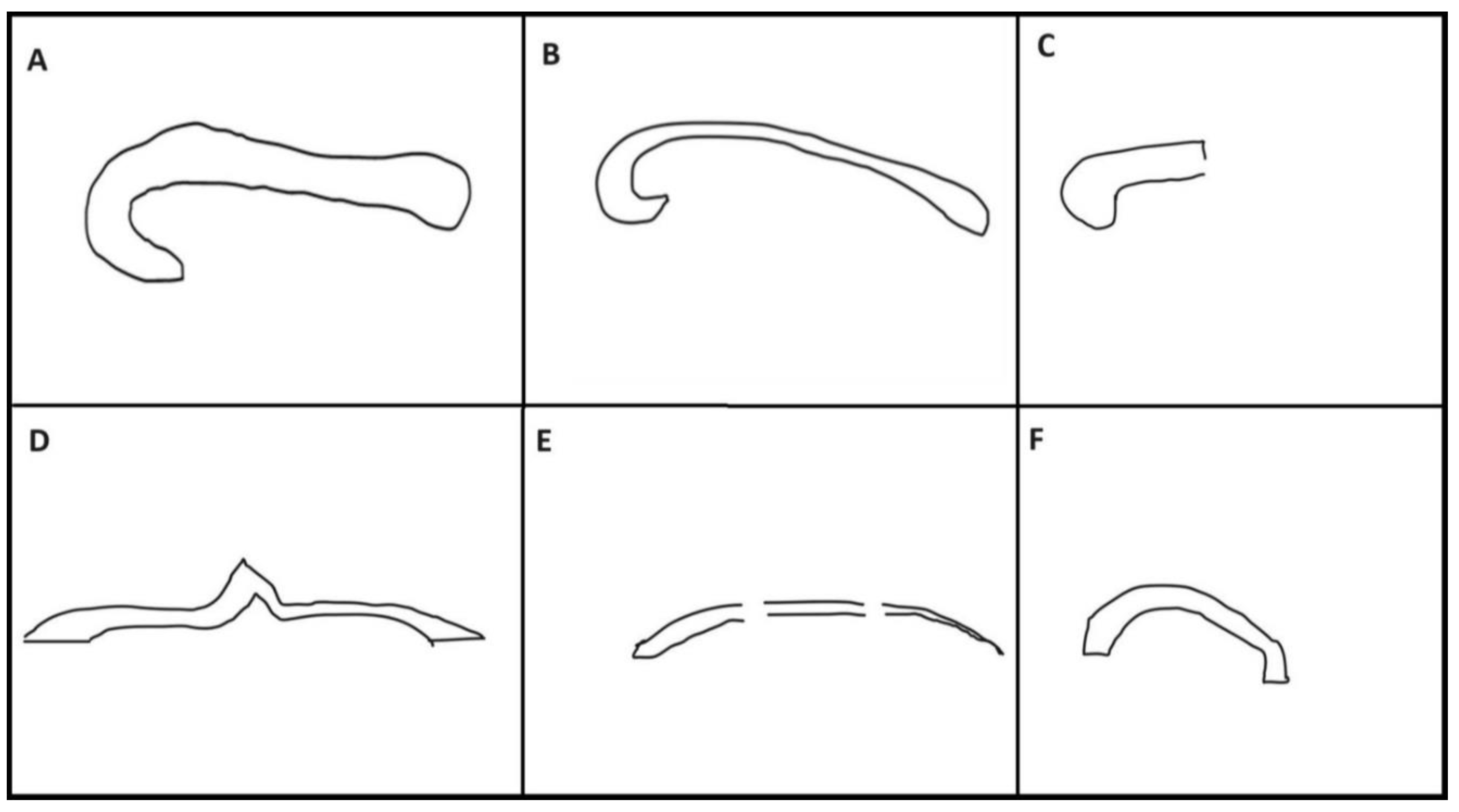
Figure 2.
Illustrative chart showing genetic analyses results of the study population. sSMCs, small supernumerary marker chromosomes.
Figure 2.
Illustrative chart showing genetic analyses results of the study population. sSMCs, small supernumerary marker chromosomes.
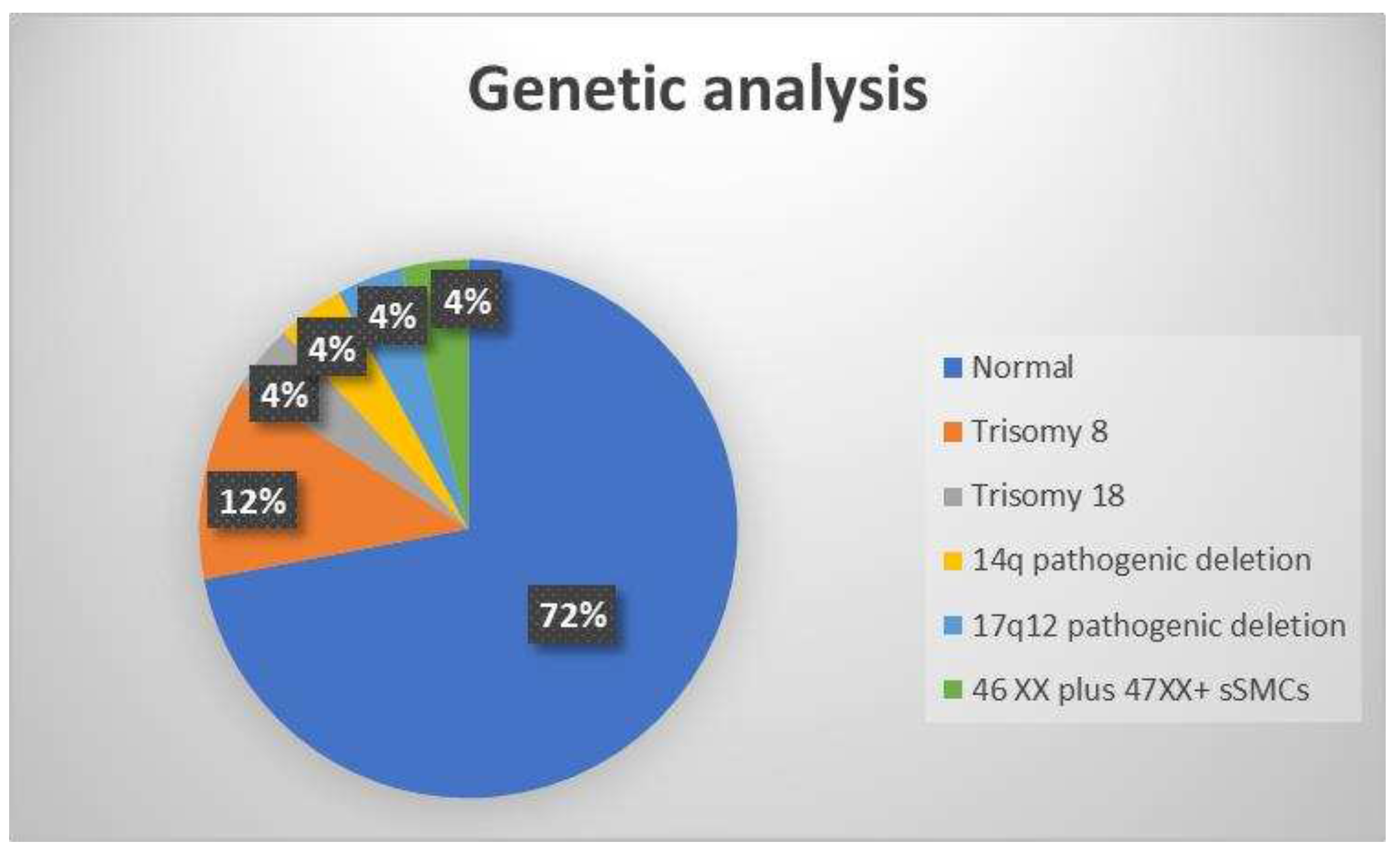
Figure 3.
Illustrative chart showing frequencies of various corpus callosal abnormalities detected on fetal MRI.
Figure 3.
Illustrative chart showing frequencies of various corpus callosal abnormalities detected on fetal MRI.
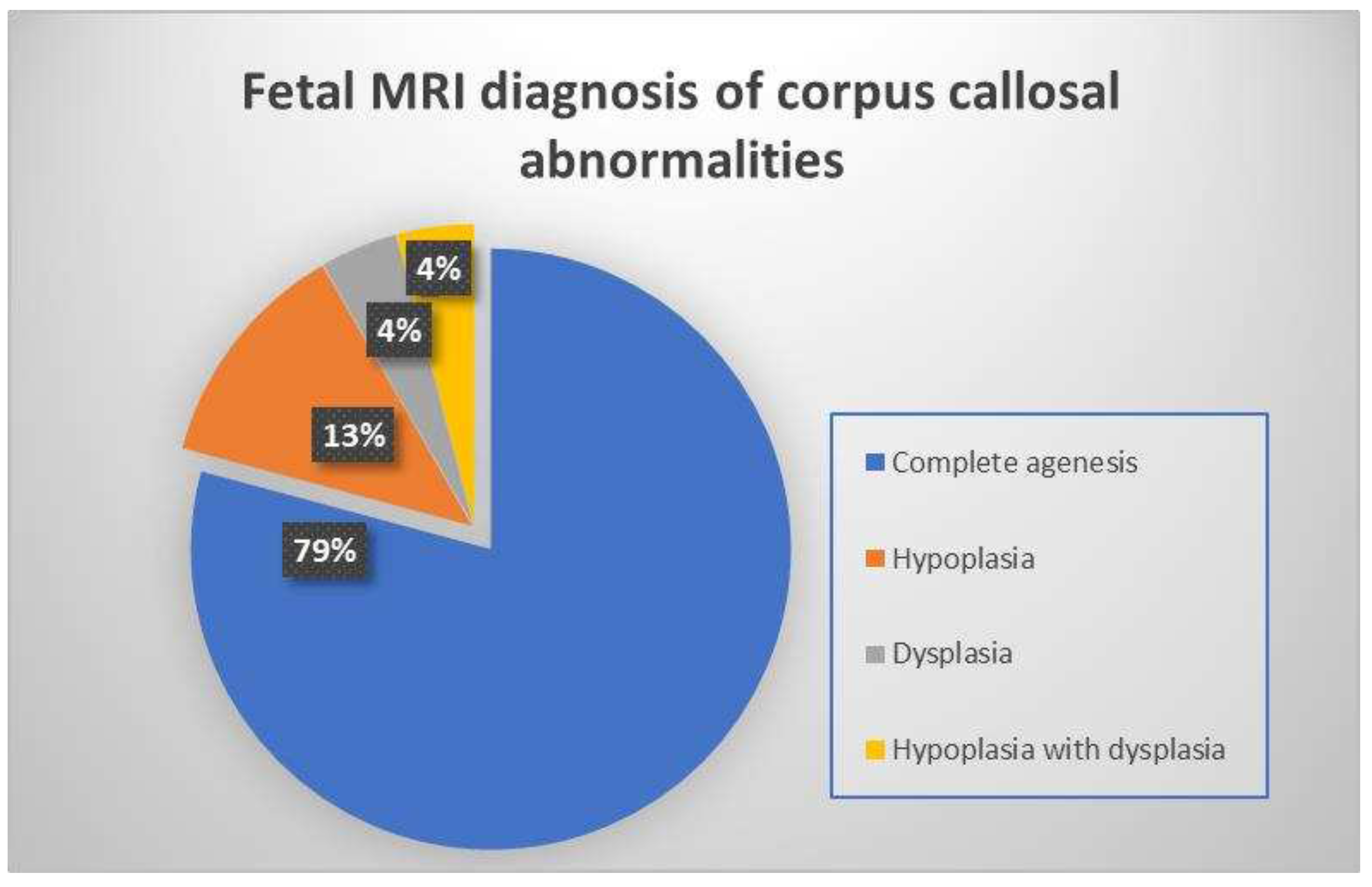
Figure 4.
Fetal MRI showing various anomalies associated with abnormal corpus callosum.
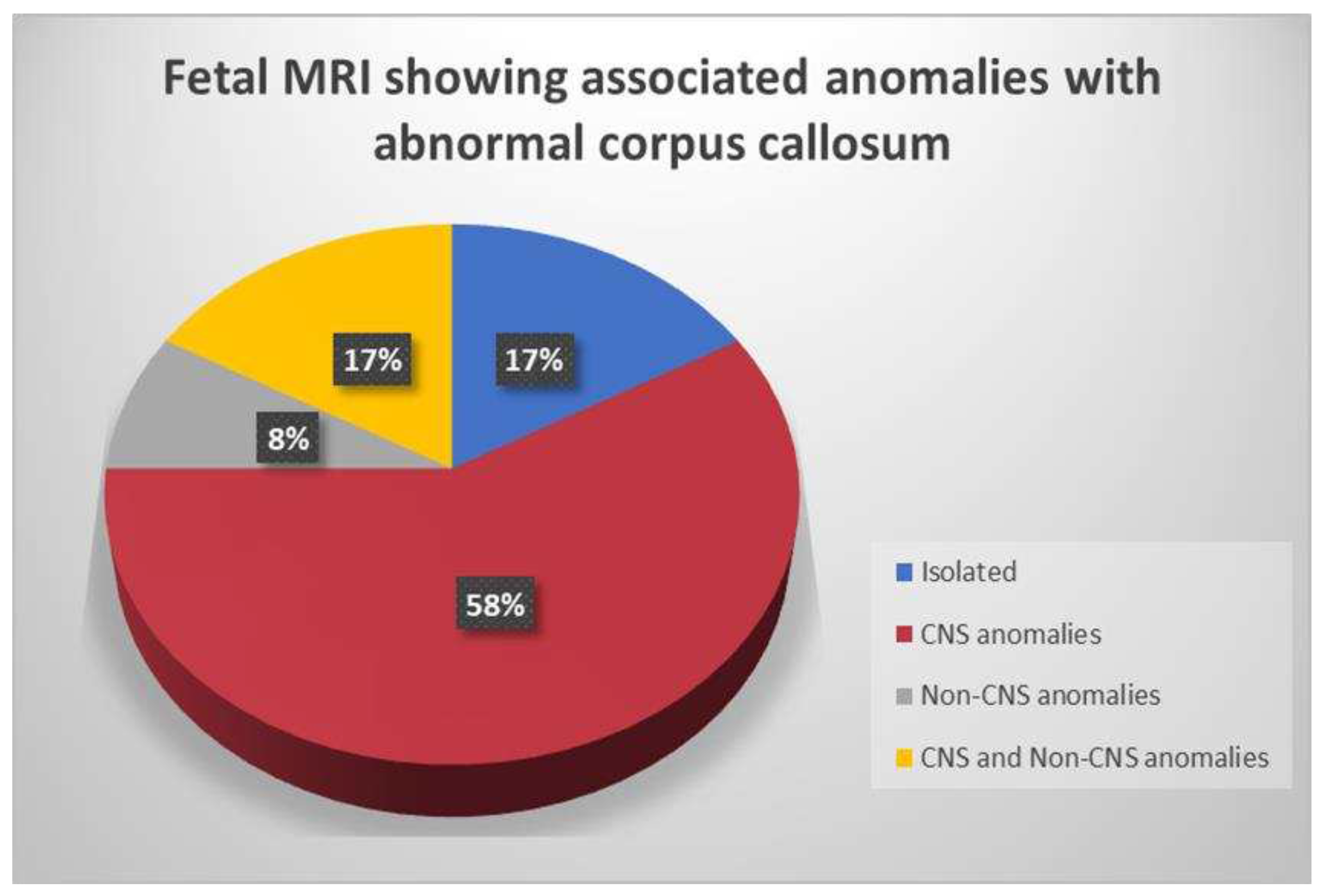

Table 1.
Maternal and birth characteristics of the study population.
| Fetuses with Corpus Callosal Abnormalities (N = 24) | Mean + SD or N (%) |
| Mean maternal age at delivery (years) | 30 + 7 |
| Mean WG (weeks) at the time of fetal MRI | 26 + 5 |
| Maternal comorbid diagnoses Preeclampsia Gestational or chronic hypertension Gestational diabetes Maternal cancer Teen pregnancy Elderly pregnancy |
5 (21%) 3 (13%) 3 (13%) 1 (4%) 4 (17%) 5 (21%) |
| Amniocentesis Normal karyotype and CMA Abnormal results |
17 (71%) 7 (29%) |
| Birth Weight (g) | 2947 ± 951 |
| Birth WG (week) | 36 + 5 |
| Male | 13 (55 %) |
| Disposition Inborn live birth MTP Stillbirths |
20 (83%) 2 (8%) 2 (8 %) |
SD, standard deviation; N, number; MRI, magnetic resonance imaging; WG, weeks of gestation; g, grams; NICU, neonatal intensive care unit; MTP, medical termination of pregnancy; CMA: chromosomal microarray analyses.
Table 2.
Fetal MRI characteristics of 24 patients with corpus callosal abnormalities, including neurological, non-neurological anomalies, and genetic analysis.
Table 2.
Fetal MRI characteristics of 24 patients with corpus callosal abnormalities, including neurological, non-neurological anomalies, and genetic analysis.
| No. | WG/Sex | Delivery History | CCA Subclass | Associated CNS Anomaly | Associated Non-CNS Anomaly | Genetic Analysis |
| 1 | 36/M | Full term | Complete Agenesis | colpocephaly | 46XY | |
| 2 | 34/F | Full term | Complete Agenesis | ventriculomegaly, heterotopia | 46XX | |
| 3 | 24/M | Full term | Complete Agenesis | IHC, Arachnoid cyst, colpocephaly, ventriculomegaly | Thick nuchal fold, hydronephrosis, | Trisomy 8 |
| 4 | 22/F | Full term | Complete Agenesis | colpocephaly, ventriculomegaly | 46XX | |
| 5 | 20/M | Preterm | Complete Agenesis | IHC, absent septum pellucidum, blake pouch cyst,vermian hypoplasia | Trisomy 18 | |
| 6 | 23/M | Preterm | Complete Agenesis | IHC, colpocephaly | 46 XY | |
| 7 | 31/F | N/A | Complete Agenesis | IHC, colpocephaly | 46 XY | |
| 8 | 22/F | Full term | Complete Agenesis | Heterotopia | 46 XX | |
| 9 | 25/M | Full term | Complete Agenesis | IHC, colpocephaly | 8q partial trisomy syndrome | |
| 10 | 20/M | Full term | Complete Agenesis | IHC | 46 XY | |
| 11 | 24/M | Full term | Complete Agenesis | Large IHC with incorporation of left ventricle into cyst | 46 XY | |
| 12 | 19/F | N/A | Complete Agenesis | Dandy walker variant with distorted medulla, cerebellar hypoplasia | 46XX,47XX+sSMCs | |
| 13 | 27/F | Full term | Complete Agenesis | Sphenoidal meningoencephalocele | Cleft lip, cleft palate | 46XX del 17q12 |
| 14 | 25/M | Full term | Complete Agenesis | Colpocephaly | Nasal dermoid cyst | 46 XY |
| 15 | 29/M (twin) | Full term | Complete Agenesis | IHC, colpocephaly | 46 XY | |
| 16 | 26/M | N/A | Complete Agenesis | IHC, colpocephaly, ventriculomegaly | 46 XY | |
| 17 | 33/M | Full term | Complete Agenesis | Colpocephaly, Absent septum pellucidum, ventriculomegaly | T12 hemivertebrae, thoracic bodies fusion | 46 XY |
| 18 | 27/F | Full term | Complete Agenesis | Colpocephaly, Absent septum pellucidum, ventriculomegaly | 46 XX | |
| 19 | 28/M | Preterm | Complete Agenesis | Colpocephaly, Absent septum pellucidum, heterotopia | Renal anomaly (hydroureteronephrosis) | Trisomy 8 mosaicism |
| 20 | 27/F | Full term | Hypoplasia of splenium | Colpocephaly | 46 XX | |
| 21 | 26/M | N/A | Hypoplasia of splenium | IHC, blake pouch cyst | 46 XY | |
| 22 | 29/F | Preterm | Complete Hypoplasia | Absent septum pellucidum, ventriculomegaly,vermian hypoplasia, aqueductal stenosis | 46 XX del 14q24.3 and q32.1. |
|
| 23 | 22/F | Preterm | Dysplasia | Hemimegalencephaly, germinolytic cyst, schizencephaly | Facial dysmorphism with fused eyes | 46 XX |
| 24 | 26/F | Preterm | Hypoplasia with Dysplasia | Ventriculomegaly, Reduced white matter | 46 XX |
CNS, central nervous system; N/A, not available; CCA, corpus callosal abnormalities; WG, weeks of gestation; IHC, interhemispheric cyst; sSMCs, small supernumerary marker chromosomes.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



