
Submitted:
10 January 2024
Posted:
11 January 2024
You are already at the latest version
Abstract
Background: Atypical teratoid/rhabdoid tumor (AT/RT) is an uncommon and aggressive pediatric central nervous system neoplasm. However, a universal clinical consensus or reliable prognostic evaluation system for this malignancy is lacking. Our study aimed to develop a risk model based on comprehensive clinical data to assist in clinical decision-making. Methods: We conducted a retrospective study by examining data from the Surveillance, Epidemiology, and End Results (SEER) repository, spanning 2000 to 2019. The external validation cohort was sourced from the Children's Hospital Affiliated to Chongqing Medical University, China. To discern independent factors affecting overall survival (OS) and cancer-specific survival (CSS), we applied Least Absolute Shrinkage and Selection Operator (LASSO) and Random Forest (RF) regression analyses. Based on these factors, we structured nomogram survival predictions and initiated a dynamic online risk-evaluation system. To contrast survival outcomes among diverse treatments, we used propensity score matching (PSM) methodology. Molecular data with the most common mutations in AT/RT were extracted from the Catalogue of Somatic Mutations in Cancer (COSMIC) database. Results: The annual incidence of AT/RT showed an increasing trend (APC, 2.86%; 95% CI:0.75-5.01). Our prognostic study included 316 SEER database participants and 27 external validation patients. The entire group had a median OS of 18 months (range 11.5 to 24 months) and median CSS of 21 months (range 11.7 to 29.2). Evaluations involving C-statistics, DCA, and ROC analysis underscored the distinctive capabilities of our prediction model. An analysis via PSM highlighted that individuals undergoing triple therapy (integrating surgery, radiotherapy, and chemotherapy) had discernibly enhanced OS and CSS. The most common mutations of AT/RT identified in the COSMIC database were SMARCB1, BRAF, SMARCA4, NF2, and NRAS. Conclusions: In this study, we devised a predictive model that effectively gauges the prognosis of AT/RT and briefly analyzed its genomic features, which might offer a valuable tool to address existing clinical challenges.
Keywords:
Atypical teratoid/rhabdoid tumor
; LASSO
; Random Forest
; SEER
; COSMIC
; Risk prognostic model
; Genomic Landscape.
1. Introduction
An Atypical teratoid/rhabdoid tumor, commonly referred to as AT/RT, is an exceptionally aggressive form of central nervous system (CNS) tumor known for its prognostic outlook [1,2]. Notably, it predominantly targets children below the age of 3 years, constituting less than 5% of all pediatric CNS tumors. However, this percentage rises to 20% when focusing solely on the subgroup of children under the age of three [3]. Although there are some histopathological similarities between AT/RT and other embryonal tumors of the CNS, it was only in 1996 that AT/RT was acknowledged as a unique tumor [4]. In 2016, the World Health Organization (WHO) categorized it as a grade IV embryonal malignancy of the CNS [5]. Delving into its genetic underpinnings, a significant number of affected children exhibit mutations in genes associated with chromatin alterations. This includes, but is not limited to, the SMARCB1 (INI-1) gene on chromosome 22q11.2 and the SMARCA4 (BRG1) gene on chromosome 19p13.2 [6,7]. These genetic changes are pivotal for diagnostic assessment of AT/RT. Clinically, patients often experience symptoms such as vomiting, gait imbalance, and recurrent seizures, with the disease advancing at an alarming rate [8]. The median survival duration typically hovers around a mere year [9].
The infrequency of AT/RT and its diagnostic intricacies mean that prior investigations of this ailment have largely focused on individual case analyses and modest retrospective evaluations [10]. There is an existing void concerning a robust prognostic staging framework and authoritative guidance for the most effective therapeutic approaches. Recent studies have underscored that conventional treatments, including surgical interventions, chemotherapy, and radiation therapy, continue to be the predominant therapeutic choices, even though their survival enhancements remain limited [11]. An appraisal of prolonged survival data from extant cases underlines the effectiveness of a tripartite approach, amalgamating surgical procedures with chemotherapy and radiation therapy, in increasing patient longevity [12]. Additionally, post-surgical localized radiation combined with systemic chemotherapy is indispensable, considering the pronounced invasiveness and propensity for metastasis of the tumor [13]. Evidence suggests that high-dose alkylating agent chemotherapy, along with intrathecal chemotherapy, constitutes potent systemic therapeutic avenues, markedly elevating survival rates in pediatric cohorts [14]. While radiation offers amplified tumor containment postoperatively, its repercussions on neural development in children, potentially instigating enduring neurocognitive impairments, spur contention in its clinical application [15].
Amid prevailing clinical uncertainties, our research endeavors to conceive and corroborate a dynamic assessment tool for survival risks. This tool draws upon comprehensive demographic data and integrates clinical and genomic characteristics. The objective is not only to surpass the shortcomings intrinsic to current prognostic frameworks but also to enrich the foundation upon which clinicians base their decisions.
2. Materials and Methods
2.1. Study design and selection criteria
This study adhered to the Transparent Reporting of a Multivariable Prediction Model for Individual Prognosis or Diagnosis (TRIPOD) reporting guidelines for prognostic studies. A comprehensive workflow is shown in Figure 1. Data retrieval was facilitated using the SEER* Stat software (version 8.4.1), accessing the most updated iteration of the SEER database.
Incidence data were acquired using the Incidence-SEER 22 Regs Research Limited-Field Data, Nov 2022 Sub (2000-2020) and the incidence rates were adjusted relative to the age of the standard American population as of 2000. Complete follow-up and treatment data were collected from the Incidence-SEER 17 Regs Research Plus Data, Nov 2021 Sub (2000-2019), and the criteria employed during the screening phase were detailed as follows. First, we only considered patients identified with an AT/RT diagnosis, bearing the International Classification of Diseases for Oncology, Third Edition (ICD-O-3) code 9508/3, between the years 2000 and 2019. The patient’s medical record should indicate the sole primary tumor. Second, the documented survival duration for patients was at least 1 month. Third, the dataset for each patient should encompass comprehensive follow-up data. Lastly, patient data had to incorporate critical information elements: vital status, duration of survival, demographics (including age, sex, and race), combined summary stage, CS tumor dimensions, and primary therapeutic interventions. For external validation, we enrolled 27 patients with AT/RT treated at the Children’s Hospital Affiliated to Chongqing Medical University from January 2018 to October 2023. Molecular data with common mutations in AT/RT were extracted from the Catalogue of Somatic Mutations in the Cancer (COSMIC) database. STRING database was used to collect and integrate potential protein interactions (PPI).
2.2. Statistical analysis
Age-adjusted incidence rates were computed as per 100,00 individuals using the SEER statistic, and annual percentage changes (APCs) were also determined. Data sourced from the SEER database were randomly divided into two distinct sets: a training cohort and a validation cohort, at proportions of 70% and 30%, respectively. Categorical variables were assessed by tabulating their frequencies and expressed as percentages, and the chi-square test was then applied for their evaluation. To map out survival trends, the Kaplan-Meier technique was used, and any disparities among these curves were discerned using the log-rank test. To identify factors that had a significant influence on overall survival (OS) and cancer-specific survival (CSS), our approach focused on applying Least Absolute Shrinkage and Selection Operator (LASSO) and Random Forest (RF) regression analysis.
To assess model discrimination, we evaluated the area under the time-dependent ROC and utilized the C-index. Calibration plots were designed to compare the predicted survival rates with actual outcomes. To ascertain the predictive power of our system against SEER stage, we relied on both DCA and time-dependent ROC. Individualized risk scores were determined by leveraging the formulated nomograms. This led to the categorization of patients into groups with higher or lower risks using the Surv_Cutpoint function to identify the best cut-off values for OS and CSS. Visual heatmaps highlighted the associations between risk factors and spread of clinical features across different risk categories for OS and CSS. Sankey diagrams were crafted for every variable within the concluding risk category, thereby enriching the clinical applicability of our framework. To ensure a meticulous comparison of survival rates across various treatments, we integrated Propensity Score Matching (PSM) analysis with a match tolerance/caliper of 0.02. The top 20 mutated genes derived from the COSMIC database were utilized for subsequent PPI network analysis (Confidence score > 0.7) and imported into Cytoscape software (v3.8.2) for visualization. For biological process and pathway enrichment analyses, the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) analyses were performed using the R clusterProfiler package. Our analytical methods hinged on SPSS 26.0 and R software (version 4.1.1), all findings were deemed significant at P values less than 0.05.
3. Results
3.1. Epidemiological Characteristics Analysis
The incidence of AT/RT consistently increased between 2000 and 2020, with an APC of 2.86% (95% CI:0.75-5.01; P < 0.05) (Figure 2A). Distribution analysis showed that regardless of gender differences, children younger than three years old accounted for the vast majority of the population (Figure 2B).
3.2. Clinical characteristics of patients
Our study incorporated data from 316 individuals diagnosed with AT/RT gathered from the SEER 17 Regs Research Plus database between 2000 and 2019. For analytical purposes, this patient cohort was divided in a 7:3 ratio, assigning 221 individuals to the training set and the remaining 95 to the validation set. We assessed the clinical characteristics to discern any potential disparities between the two subsets. Notably, the distribution did not indicate any marked discrepancies (P>0.05) in demographic or clinical factors. Table 1 presents the demographic and clinical data. Key observations include the fact that a significant proportion of the study participants were infants aged under 3 years (n=248, 78.5%), predominantly of Caucasian descent (n=241, 76.3%), having primary tumors located intracranially (n=299, 94.6%), and hailing from households with lower incomes (n=221, 69.9%). In terms of disease progression, as classified by the SEER system, most patients presented with localized tumors at diagnosis (n=189, 59.8%), followed by those with regional (n=61, 19.3%) and metastatic disease (n=66, 20.9%). Treatment modalities revealed that gross total resection/subtotal resection (GTR/STR) was performed in 94.0% of the cases, chemotherapy in 81.0%, and radiotherapy in 46.8%. The median survival span for all patients in the database was 18 months (range: 11.5-24.5) with a median cancer-specific survival of 21 months (range: 11.7-29.2). The training set exhibited a median OS of 19 months (range: 12.3-25.6) and CSS of 22 months (range: 12.6-31.3). For the validation set, these measures were 17 months (range: 10.8-23.2) and 20 months (range: 9.0-30.1), respectively. Moreover, 27 patients with AT/RT treated at the Children’s Hospital Affiliated to Chongqing Medical University were included in the external validation. This external set had a median OS of 10 months (range:5.8-14.2), and demographic and clinical details are provided in Supplementary Table S1.
3.3. Prognostic factors selection and model construction
Before delving into machine learning algorithm screening, we first evaluated the potential collinearity between all scrutinized parameters using Spearman correlation analysis, as shown in Figure 3A. To identify the best coefficient for each prognostic determinant, we employed the LASSO and RF algorithms, which ensured the circumvention of overfitting during the selection of important variables [16]. LASSO regression was performed by minimizing the partial probability deviation and generating coefficient curves from a logarithmic (lambda) series (Figure 3C,E). Guided by the requisite standards for Lasso-Cox regression and adopting a 10-fold cross-validation, the algorithm discerned six pivotal clinical parameters (age, SEER stage, tumor size, surgical interventions, chemotherapeutic approaches, and radiological treatments) with significance as standalone predictors in both the OS and CSS frameworks (Figure 3B,D). In the RF algorithms, by increasing the number of random forests, the out-of-bag (OOB) error rate gradually decreases (Figure 3G,I), allowing for the determination of the importance index of each parameter in both OS and CSS (Figure 3H,J). Furthermore, the TOP 6 common parameters identified by both algorithms (Figure 3F) were selected as the final predictor variables for the model. Finally, we synthesized these selected prognostic markers into forest diagrams (Figure 4A,B) to devise nomogram-driven prognostic models for OS and CSS (Figure 4C,D).
3.4. Dynamic web version survival model
To support researchers and clinicians, our team launched digital versions of our nomograms designed to assess OS and CSS in patients with AT/RT. These tools are accessible at the following URLs: https://atrtapp.shinyapps.io/shinyNomoforATRTinOS/ and https://atrtapp.shinyapps.io/shinyNomoforATRTinCSS/ (Figure 4E,F).
3.5. Internal and external multidimensional validation of models
The nomogram showed notable capabilities in forecasting OS for intervals of 1, 2, and 3 years. Both the training (0.815) and validation (0.801) cohorts registered C-index values that outshone those of the SEER stage method, which scored 0.648 and 0.656, respectively. Furthermore, when considering 1-, 2-, and 3-year CSS projections, our model surpassed the SEER-stage method, yielding C-index scores of 0.809 and 0.661 for the training set and 0.778 and 0.653 for the validation set. In the evaluations against the SEER-stage method, our nomograms consistently achieved a time-dependent AUC above 0.8, underscoring their enhanced forecasting process (Supplementary Figure S1). The calibration curves displayed a close match between the forecasted and actual survival rates. The presented models precisely forecasted OS and CSS for all mentioned durations in both cohorts (Supplementary Figures S2 and S3). Decision curve assessments for the OS and CSS models confirmed their elevated clinical relevance and forecasting competency for the stated durations, as illustrated by the expansive range of optimal threshold probabilities (Supplementary Figure S4). Moreover, in the external validation cohort, metrics such as the calibration curve, time-dependent ROC, DCA curve, and risk stratification analysis unequivocally showed the robustness and superiority of the model (Supplementary Figure S5).
3.6. Risk Stratification and Sankey Diagram Based on the Model
Using the Surv_miner R package, we established an optimal threshold to segregate patients into high-risk and low-risk categories concerning OS and CSS, with scores of 135 and 155, respectively. There was a pronounced divergence in survival trajectories among these risk groups (P<0.001), underscoring the relevance of our nomogram and the stratification approach (Figure 5A–D). Furthermore, we leveraged heat maps to visualize variations in clinical features among the OS (Figure 5E) and CSS (Figure 5F) designated risk brackets. The model’s practical utility in clinical settings can be enhanced by illustrating a Sankey diagram that delineates the progression of each factor and its culmination into a designated risk category. As displayed in Figure 6A,B, this visual tool elucidates the influence of individual variables on the resulting risk classification.
3.7. Optimal treatment strategy analysis
To examine how diverse treatments influence patient prognosis, PSM analysis was applied to mitigate the influence of confounding factors [17]. The results for both the pre- and post-PSM are presented in Supplementary Table S2. Prior to the matched assessment, triple therapy indicated more favorable OS and CSS outcomes than SR/SC. The comparative median survival periods were 10 months versus 91 months and 11 months versus 91 months. Notably, the 5-year OS for triple-therapy reached 57.1% and 58.7% for CSS, in contrast to SR/SC, which stood at 26.2% for OS and 29.3% for CSS (Figure 6C,D). Post-matching, the edge triple therapy persisted in terms of OS and CSS. The median survival intervals were 10 months juxtaposed at 93 months and 11 months compared with 93 months. Furthermore, the 5-year SR/SC survival rates were 26.2% (OS) and 29.3% (CSS), while for triple-therapy, they were 55.5% (OS) and 57.2% (CSS) (Figure 6E,F).
3.8. Genetic Mutations and GO/KEGG analysis
The AT/RT genetic mutation data were extracted from COSMIC (https://cancer.sanger.ac.uk/cosmic, accessed on December 30, 2023) version GRCh38 COSMIC v99. In total, 17650 cases of CNS tumors were evaluated for genetic mutations in the database. In the subselection category, all brain sites were selected for data extraction. For histological selection, only AT/RT cases were selected, and a final total of 194 cases were analyzed for genetic mutations. The top 20 genes that were mutated in AT/RT were SMARCB1 45% (in all samples tested = 269), BRAF 8% (73), SMARCA4 8% (26), NF2 2% (45), NRAS 2% (43), TP53 2% (43), KRAS 2% (43), MSH2 2% (43), IDH2 3% (31), KAT6B 4% (24), GATA2 4% (24), FANCD2 4% (24), SPEN 4% (24), ZFHX3 4% (24), NCOR2 4% (24), BCL11B 4% (24), ERBB4 4% (24), AFF3 4% (24), MAF 4% (24), and MDM4 4% (24) (Figure 7A). An overview of the mutation types and PPI network are shown in Figure 7B,C. We performed GO and KEGG analyses of these genes. Biological process analysis showed that the top 20 genes were enriched in RNA transcription, transcription factor complex, DNA-binding transcription factor, and ErbB signaling pathways (Figure 7D). Supplementary Table S3 summarizes the details of the GO functions and KEGG pathways of the top 20 genes for co-expression enrichment analysis.
4. Discussion
AT/RT, recognized as a profoundly malignant CNS tumor, has a particularly poor prognosis, marked by an unusual level of aggressiveness [1,2,3,18]. Given the rarity of AT/RT instances, adequate research poses a challenge. The SEER database is a reputable repository of U.S. cancer statistics, making it instrumental in probing uncommon tumors [19,20]. Through analysis, we engaged a sizable cohort of AT/RT individuals from SEER (n=316) and an external validation cohort from Chongqing, China (n=27), subsequently discerning six clinical determinants linked to OS and CSS. Additionally, we used the COSMIC database to analyze the genomic variation characteristics of AT/RT. To the best of our knowledge, this inquiry marks a seminal SEER- and COSMIC-driven exploration to formulate predictive frameworks for specific survival to AT/RT. To amplify the real-world applicability of our findings, we incorporated web-enabled prognostic tools and devised a visual representation of Sankey to facilitate risk-based clinical decisions.
The actual incidence of AT/RT might have been underestimated owing to gaps in prior knowledge. Our epidemiological survey demonstrated a noticeable increase in the incidence of AT/RT over the past two decades. Consequently, it is imperative to prioritize and enhance AT/RT-related management in the future. Research indicates that AT/RT primarily targets infants younger than three years [3], with age being a pivotal determinant of patient outcomes. Our analysis confirmed that a significant fraction of the patient population aged < 3 years (n=248, 78.5%) demonstrated inferior prognostic outcomes. While tumor dimensions serve as a pivotal influencer on the outcomes of diverse solid tumors [21] by mirroring the tumor’s reach, our findings are consistent with clinical anecdotes, establishing that an expanded tumor size (≥ 4 cm) negatively affects both OS and CSS. However, a universally recognized staging forecast system for AT/RT remains elusive. The SEER database employs a novel approach that uses a combined summary stage [22]. This unique categorization delineates cancer progression from its inception into localized, regional, or distant stages, aiming to streamline clinical reference. Leveraging this SEER stage in our study, we found that a significant proportion of patients (n=127, 40.2%) had already progressed beyond the localized phase upon initial identification.
For aggressive brain tumors, achieving maximal safe resection is the accepted standard for surgical management [23]. Our research further investigates how medical interventions tie with patient outcomes, underscoring the pivotal role that surgery holds in bolstering OS and CSS for AT/RT treatments. However, some studies have questioned the pronounced survival benefit of complete surgical intervention over other treatment modalities, suggesting that adjunct postoperative therapies might hold more weight [24,25]. The efficacy of high-dose chemotherapy in managing AT/RT has undergone rigorous examination, with prevalent regimens including the American COG’s CCG9933 and German GPOH’s HIT program [26,27]. Athale et al.’s meta-analysis postulated a potential survival advantage with intrathecal chemotherapy, especially for those unsuitable for craniospinal radiotherapy [28]. Furthermore, conjoining postoperative local radiotherapy with craniospinal radiotherapy demonstrates remarkable potential in enhancing local disease control and prolonging survival [9]. Given the adverse neurotoxic side effects inherent to traditional radiotherapy and the looming threat of secondary malignancies, judicious consideration is imperative when choosing surgical strategies, orchestrating radiotherapy and chemotherapy sequences, and calibrating dosages for AT/RT patients under 3 years. Recent advancements have highlighted the potential of proton therapy to augment survival by fine-tuning radiation responses [29]. COG’s current investigation into AT/RT posits that radiation for children under 3 Gy should be primarily tumor-focused, and recommends tapering the dosage for whole brain and spinal cord radiotherapy in metastatic cases from 30 Gy to 24 Gy [14]. Recognizing the ambiguity surrounding current treatments, our exploration gravitates towards triple therapy’s promise in extending survival durations. Post-PSM evaluations indicated that, even after equalizing confounding elements between cohorts, the triple-therapy recipients outperformed their counterparts in OS and CSS, who underwent surgery accompanied by chemotherapy/radiotherapy. This finding is consistent with the results of previous studies and once again emphasizes the superiority of triple therapy, but ignores side effects such as radiotoxicity [30,31]. Hence, forward-focused clinical trials are essential to corroborate the efficacy and safety of this therapeutic approach.
However, the origin of AT/RT remains unclear. Based on the genetic and DNA methylation status and transcriptome profiles, AT/RTs are further divided into three distinct molecular subgroups: ATRT-SHH, ATRT-TYR, and ATRT-MYC [32]. Both previous literature and our analysis suggest that mutations or absences in SMARCB1(INI-1) play a pivotal role in its development and progression [6,33]. INI-1 is ubiquitously expressed in the nucleus of normal cells and is considered a tumor suppressor gene. Studies have shown that INI-1 is a core component of the switch/sucrose-non-fermentable (SWI/SNF) chromatin remodeling complex, which regulates gene expression important for lineage specification and maintenance of stem cell pluripotency [34,35]. The GO/KEGG analysis based on the top 20 mutated gene sets also revealed that the biological properties of AT/RT are related to RNA transcription regulation, transcription factor complex formation, RNA-DNA specific binding and ErbB signaling pathway. Several studies showed that lapatinib has a good inhibitory effect on AT/RT by targeting the EGFR-ErbB2 signaling pathway [36,37]. Therefore, conducting an in-depth genomic analysis of AT/RT patients is necessary as it will provide potential therapeutic targets for this disease.
In the current investigation, the LASSO and RF regression methodologies were used to craft a model aimed at assessing survival threats and the genomic landscape used to clarify the underlying pathogenesis. Despite its strengths, our study has some limitations. Given the retrospective nature of the analysis, we bypassed patients who were absent from the SEER registry, potentially introducing a sampling bias. The SEER datasets also did not provide exhaustive details concerning pivotal clinical aspects such as performance status, specific chemotherapy regimens, number of cycles, radiation dosages, and subsequent lines of therapy. The absence of metrics, such as disease progression-free survival and recurrence survival, in the SEER database could also pose limitations to the broader utilization of the model. Genomics analysis needs to be more in-depth, such as exploration of epigenetic changes and functional mechanisms.
5. Conclusions
We meticulously examined patient data from the SEER database spanning the years 2000 to 2019 and the genetic mutation characteristics of the patients in the COSMIC database. Our study identified the clinical determinants of prognosis in patients with AT/RT and mapped the genetic mutation landscape. The prediction model that we devised may offer a valuable tool to address existing clinical challenges. Additionally, analysis based on mutational genomics will facilitate the research of molecular targeted drugs. As clinical understanding deepens and genomic profiling expands, precise and efficient combination therapies are expected to drive this disease toward a positive direction.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Aishun Jin and Menglei Wang had full access to all the data in the study and took responsibility for the integrity of the data and accuracy of the data analysis. Concept and design: S. Chen and Y. He. Acquisition, analysis, and interpretation of data: S. Chen, Y. He, J. Liu and R. Wu. Drafting of the manuscript: S. Chen and Y. He. Critical revision of the manuscript for intellectual content: A. Jin. Statistical analysis: S. Chen, Y. He, J. Liu, R. Wu, and M. Wang. Obtained funding: A. Jin. Administrative, technical, and material support: M. Wang and A. Jin. All authors helped with the preparation of the manuscript, and read and approved the final manuscript.
Funding
This study was supported by a grant from the Chongqing Medical University Professor Scientific Research Start-up Fund (R1047).
Institutional Review Board Statement
This study was conducted in accordance with the Declaration of Helsinki (revised in 2013) and approved by the Ethics Committee of the Children’s Hospital Affiliated to Chongqing Medical University, China (Approval No. 2023-013).
Informed Consent Statement
Since individual information was removed from all SEER and COSMIC databases, informed consent from patients and approval by the institutional review board were waived.
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its additional information files. Further inquiries can be made at https://github.com/SihaoChen95/ATRT
Acknowledgments
We are immensely grateful to all investigators involved in this study.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
| AT/RT | Atypical teratoid/rhabdoid tumor |
| COSMIC | Catalogue of Somatic Mutations in Cancer |
| SEER | Surveillance, Epidemiology, and End Results |
| APCs | Annual percentage changes |
| LASSO | Least Absolute Shrinkage and Selection Operator |
| RF | Random Forest |
| CNS | Central nervous system |
| GTR/STR | Gross total resection/subtotal resection |
| PSM | Propensity Score Matching |
| APCs | Annual percentage changes |
| GO/KEGG | Gene Ontology and Kyoto Encyclopedia of Genes and Genomes |
References
- Nesvick CL, Nageswara Rao AA, Raghunathan A, et al. Case-based review: atypical teratoid/rhabdoid tumor. Neurooncol Pract. 2019 May;6(3):163-178. [CrossRef]
- Ma XJ, Li D, Wang L, et al. Overall Survival of Primary Intracranial Atypical Teratoid Rhabdoid Tumor Following Multimodal Treatment: A Pooled Analysis of Individual Patient Data. Neurosurg Rev. 2020 Feb;43(1):281-292. [CrossRef]
- Park M, Han JW, Hahn SM, et al. Atypical Teratoid/Rhabdoid Tumor of the Central Nervous System in Children under the Age of 3 Years. Cancer Res Treat. 2021 Apr;53(2):378-388. [CrossRef]
- Rorke LB, Packer RJ, Biegel JA. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: definition of an entity. J Neurosurg. 1996 Jul;85(1):56-65. [CrossRef]
- Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016 Jun;131(6):803-20. [CrossRef]
- Biegel JA, Tan L, Zhang F, et al. Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res. 2002 Nov;8(11):3461-7.
- Holdhof D, Johann PD, Spohn M, et al. Atypical teratoid/rhabdoid tumors (ATRTs) with SMARCA4 mutation are molecularly distinct from SMARCB1-deficient cases. Acta Neuropathol. 2021 Feb;141(2):291-301. [CrossRef]
- Rorke LB, Packer R, Biegel J. Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol. 1995;24(1):21-8. [CrossRef]
- Buscariollo DL, Park HS, Roberts KB, et al. Survival outcomes in atypical teratoid rhabdoid tumor for patients undergoing radiotherapy in a Surveillance, Epidemiology, and End Results analysis. Cancer. 2012 Sep 1;118(17):4212-9. [CrossRef]
- Broggi G, Gianno F, Shemy DT, et al. Atypical teratoid/rhabdoid tumor in adults: a systematic review of the literature with meta-analysis and additional reports of 4 cases. J Neurooncol. 2022 Mar;157(1):1-14. [CrossRef]
- Lafay-Cousin L, Strother D. Current treatment approaches for infants with malignant central nervous system tumors. Oncologist. 2009 Apr;14(4):433-44. [CrossRef]
- Almalki MH, Alrogi A, Al-Rabie A, et al. Atypical Teratoid/Rhabdoid Tumor of the Sellar Region in an Adult With Long Survival: Case Report and Review of the Literature. J Clin Med Res. 2017 Mar;9(3):216-220. [CrossRef]
- Yang WC, Yen HJ, Liang ML, et al. Role of early and aggressive post-operative radiation therapy in improving outcome for pediatric central nervous system atypical teratoid/rhabdoid tumor. Childs Nerv Syst. 2019 Jun;35(6):1013-1020. [CrossRef]
- Reddy AT, Strother DR, Judkins AR, et al. Efficacy of High-Dose Chemotherapy and Three-Dimensional Conformal Radiation for Atypical Teratoid/Rhabdoid Tumor: A Report From the Children’s Oncology Group Trial ACNS0333. J Clin Oncol. 2020 Apr 10;38(11):1175-1185. [CrossRef]
- Squire SE, Chan MD, Marcus KJ. Atypical teratoid/rhabdoid tumor: the controversy behind radiation therapy. J Neurooncol. 2007 Jan;81(1):97-111. [CrossRef]
- Wang W, Liu W. Integration of gene interaction information into a reweighted Lasso-Cox model for accurate survival prediction. Bioinformatics. 2020 Dec 16:btaa1046. [CrossRef]
- West SG, Cham H, Thoemmes F, et al. Propensity scores as a basis for equating groups: basic principles and application in clinical treatment outcome research. J Consult Clin Psychol. 2014 Oct;82(5):906-19. [CrossRef]
- Hilden JM, Meerbaum S, Burger P, et al. Central nervous system atypical teratoid/rhabdoid tumor: results of therapy in children enrolled in a registry. J Clin Oncol. 2004 Jul 15;22(14):2877-84. [CrossRef]
- Liang W, Li L, Wang M, et al. Non-inferior efficacy of non-surgical treatment to surgical treatment in patients with nonmetastatic head and neck rhabdomyosarcoma: a SEER-based study. Clin Transl Oncol. 2023 Jun;25(6):1779-1792. [CrossRef]
- Tang N, Chen ZY, Yang Z, et al. Development and verification of prognostic nomogram for ampullary carcinoma based on the SEER database. Front Oncol. 2023 May 29;13:1197626. [CrossRef]
- Dai W, Mo S, Xiang W, et al. The Critical Role of Tumor Size in Predicting Prognosis for T1 Colon Cancer. Oncologist. 2020 Mar;25(3):244-251. [CrossRef]
- Wu XC, Yu Q, Andrews PA, et al. Comparisons of directly coded SEER Summary Stage 2000 and Collaborative Staging Derived SEER Summary Stage 2000. J Registry Manag. 2010 Winter;37(4):137-40.
- Hatoum R, Chen JS, Lavergne P, et al. Extent of Tumor Resection and Survival in Pediatric Patients With High-Grade Gliomas: A Systematic Review and Meta-analysis. JAMA Netw Open. 2022 Aug 1;5(8):e2226551. [CrossRef]
- Fischer-Valuck BW, Chen I, Srivastava AJ, et al. Assessment of the treatment approach and survival outcomes in a modern cohort of patients with atypical teratoid rhabdoid tumors using the National Cancer Database. Cancer. 2017 Feb 15;123(4):682-687. [CrossRef]
- Elsayad K, Kriz J, Samhouri L, et al. Long-term survival following additive radiotherapy in patients with atypical teratoid rhabdoid tumors. Strahlenther Onkol. 2016 Aug;192(8):569-81. English. [CrossRef]
- MacDonald TJ, Arenson EB, Ater J, et al. Phase II study of high-dose chemotherapy before radiation in children with newly diagnosed high-grade astrocytoma: final analysis of Children’s Cancer Group Study 9933. Cancer. 2005 Dec 15;104(12):2862-71. [CrossRef]
- von Hoff K, Hinkes B, Dannenmann-Stern E, et al. Frequency, risk-factors and survival of children with atypical teratoid rhabdoid tumors (AT/RT) of the CNS diagnosed between 1988 and 2004, and registered to the German HIT database. Pediatr Blood Cancer. 2011 Dec 1;57(6):978-85. [CrossRef]
- Slavc I, Chocholous M, Leiss U, et al. Atypical teratoid rhabdoid tumor: improved long-term survival with an intensive multimodal therapy and delayed radiotherapy. The Medical University of Vienna Experience 1992-2012. Cancer Med. 2014 Feb;3(1):91-100. [CrossRef]
- De Amorim Bernstein K, Sethi R, Trofimov A, et al. Early clinical outcomes using proton radiation for children with central nervous system atypical teratoid rhabdoid tumors. Int J Radiat Oncol Biol Phys. 2013 May 1;86(1):114-20. [CrossRef]
- Quinn TJ, Almahariq MF, Siddiqui ZA, et al. Trimodality therapy for atypical teratoid/rhabdoid tumor is associated with improved overall survival: A surveillance, epidemiology, and end results analysis. Pediatr Blood Cancer. 2019 Dec;66(12):e27969. [CrossRef]
- Bhutada AS, Adhikari S, Cuoco JA, et al. Survival Benefit from Multimodal Treatment for Patients with Atypical Teratoid Rhabdoid Tumor in a Surveillance, Epidemiology and End Result Database Analysis. Oncology. 2023 Aug 25. [CrossRef]
- Ho B, Johann PD, Grabovska Y, et al. Molecular subgrouping of atypical teratoid/rhabdoid tumors-a reinvestigation and current consensus. Neuro Oncol. 2020 May 15;22(5):613-624. [CrossRef]
- Cacciotti C, Fleming A, Ramaswamy V. Advances in the molecular classification of pediatric brain tumors: a guide to the galaxy. J Pathol. 2020 Jul;251(3):249-261. [CrossRef]
- Duan Z, Yao K, Yang S, et al. Primary adult sellar SMARCB1/INI1-deficient tumor represents a subtype of atypical teratoid/rhabdoid tumor. Mod Pathol. 2022 Dec;35(12):1910-1920. [CrossRef]
- Valencia AM, Collings CK, Dao HT, et al. Recurrent SMARCB1 Mutations Reveal a Nucleosome Acidic Patch Interaction Site That Potentiates mSWI/SNF Complex Chromatin Remodeling. Cell. 2019 Nov 27;179(6):1342-1356.e23. [CrossRef]
- Singh A, Lun X, Jayanthan A, et al. Profiling pathway-specific novel therapeutics in preclinical assessment for central nervous system atypical teratoid rhabdoid tumors (CNS ATRT): favorable activity of targeting EGFR- ErbB2 signaling with lapatinib. Mol Oncol. 2013 Jun;7(3):497-512. [CrossRef]
- Sredni ST, Patel K, D’Almeida Costa F, et al. Activation of ErbB2- ErbB3 signaling pathway supports potential therapeutic activity of ErbB inhibitors in AT/RT. J Neurooncol. 2014 May;118(1):201-3. [CrossRef]
Figure 1.
Study design and the workflow diagram.
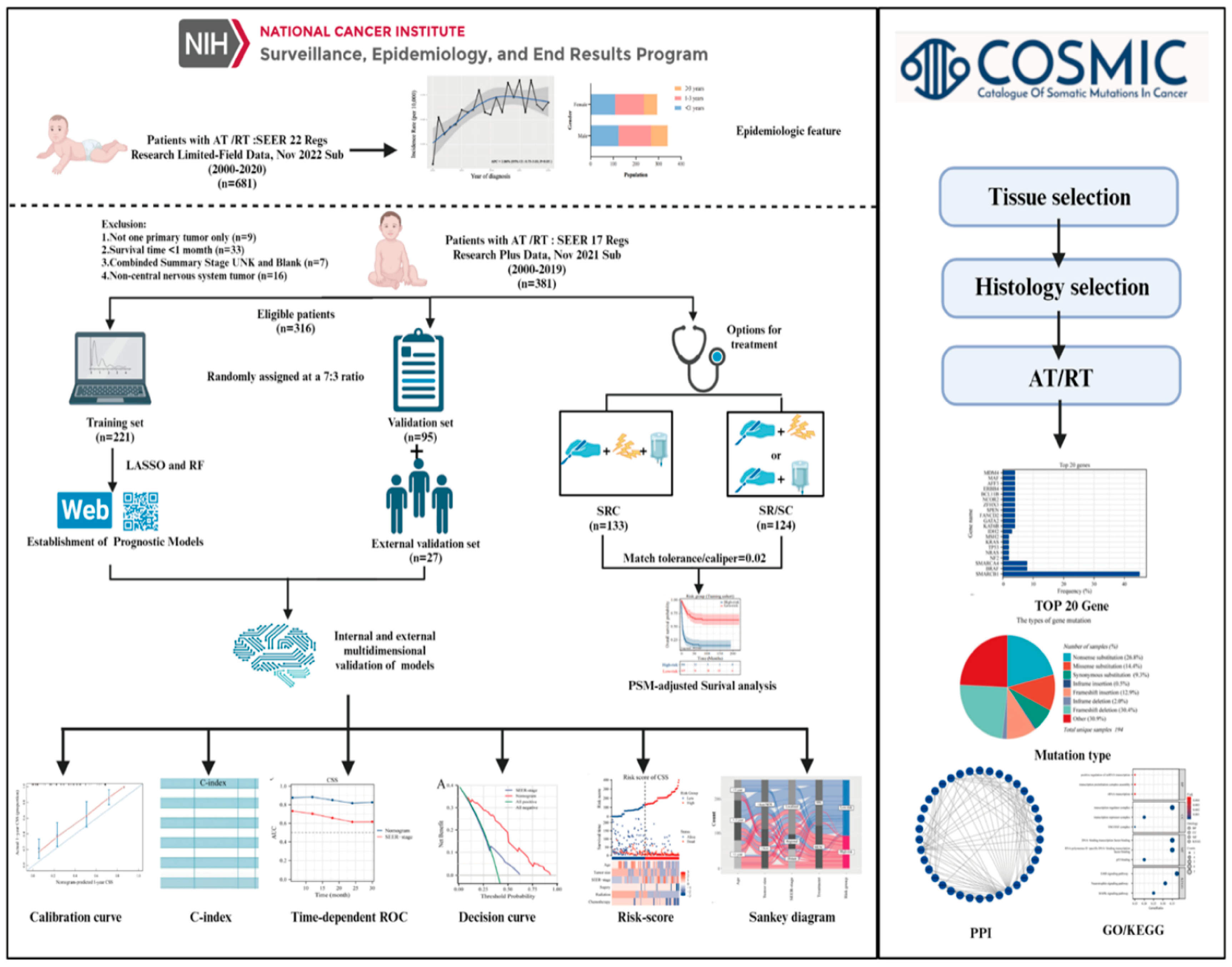
Figure 2.
Trends and characteristics of AT/RT. (A) Incidence of AT/RT from 2000 to 2020. (B) Age and gender distribution.
Figure 2.
Trends and characteristics of AT/RT. (A) Incidence of AT/RT from 2000 to 2020. (B) Age and gender distribution.
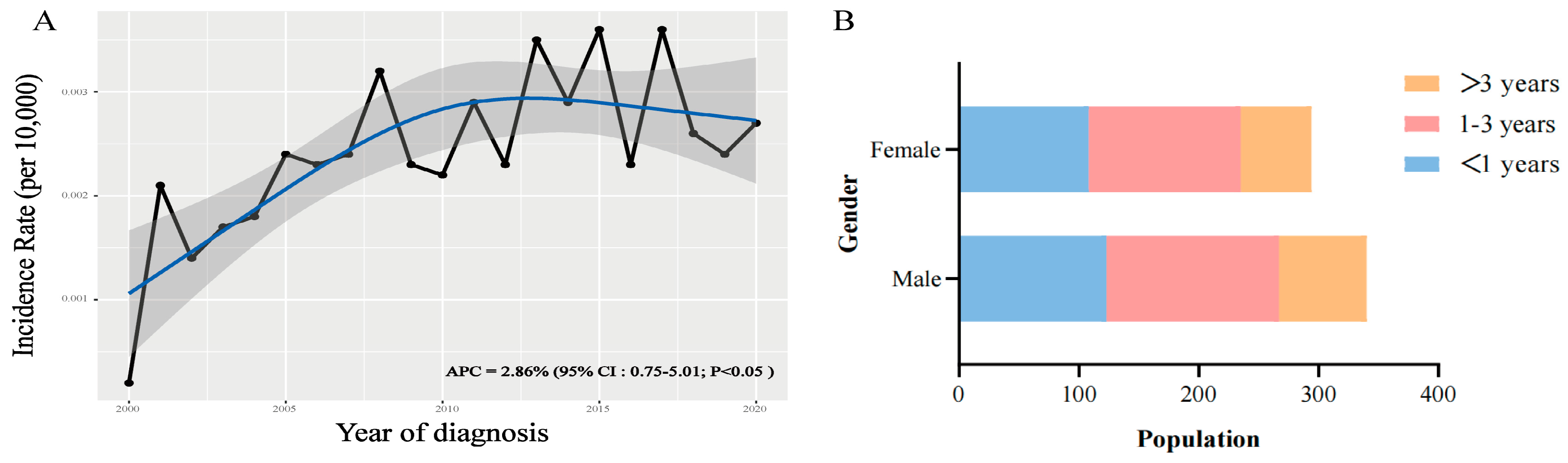
Figure 3.
Correlation analysis, LASSO and RF regression model in both OS and CSS. The results of correlation analysis between all included variables (A). Selection of tuning parameter (λ) for the LASSO model (C,E), 10-fold cross-validation (B,D) . The OBB error rate of Random forest (G,I) and variable importance ranking (H,J). Intersection variables of the two algorithms (F).
Figure 3.
Correlation analysis, LASSO and RF regression model in both OS and CSS. The results of correlation analysis between all included variables (A). Selection of tuning parameter (λ) for the LASSO model (C,E), 10-fold cross-validation (B,D) . The OBB error rate of Random forest (G,I) and variable importance ranking (H,J). Intersection variables of the two algorithms (F).
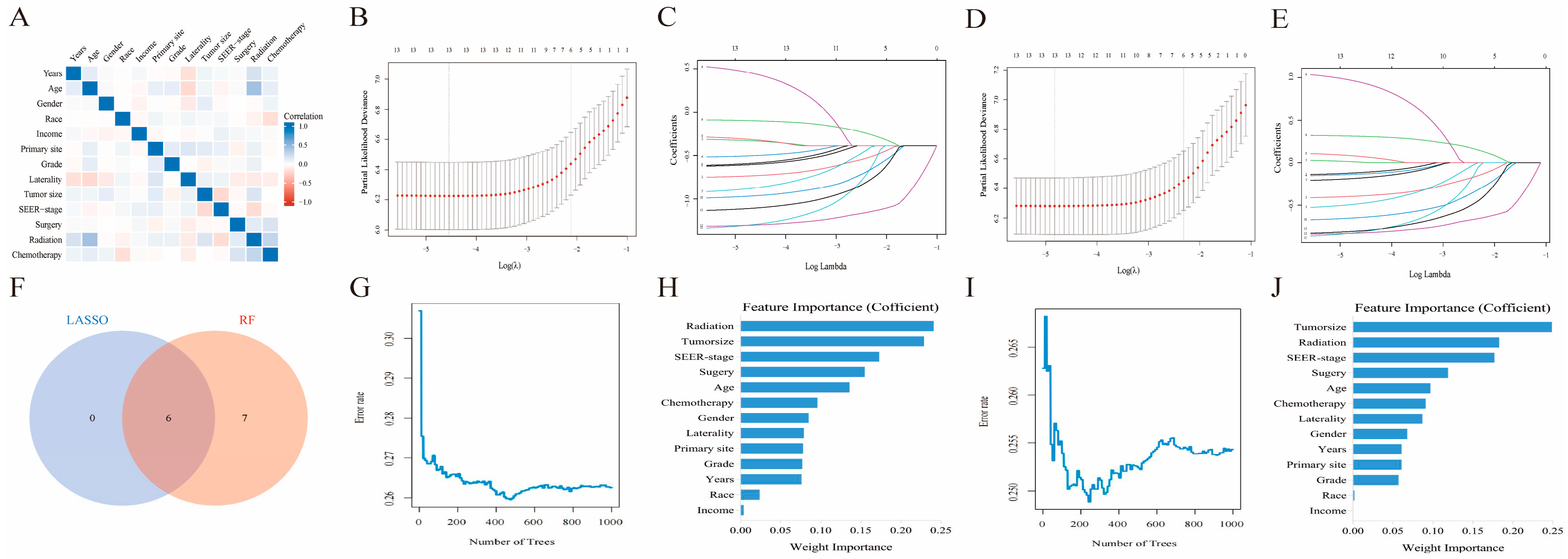
Figure 4.
Th forest plot and Nomogram. Forest plot demonstrating the model for OS (A) and CSS (B) in the training cohort and the Nomogram for predicting 1-, 2- and 3-year OS (C) and CSS (D). Dynamic survival risk system for OS (A) and CSS (B) in AT/RT patients.
Figure 4.
Th forest plot and Nomogram. Forest plot demonstrating the model for OS (A) and CSS (B) in the training cohort and the Nomogram for predicting 1-, 2- and 3-year OS (C) and CSS (D). Dynamic survival risk system for OS (A) and CSS (B) in AT/RT patients.
Figure 5.
Overview of risk-stratification system according to risk points calculated by the model. OS and CSS analysis of patients with AT/RT in the training cohort (A,B) and validation cohort (C,D). The distribution of clinicopathological features in different risk groups for OS (E) and CSS (F).
Figure 5.
Overview of risk-stratification system according to risk points calculated by the model. OS and CSS analysis of patients with AT/RT in the training cohort (A,B) and validation cohort (C,D). The distribution of clinicopathological features in different risk groups for OS (E) and CSS (F).
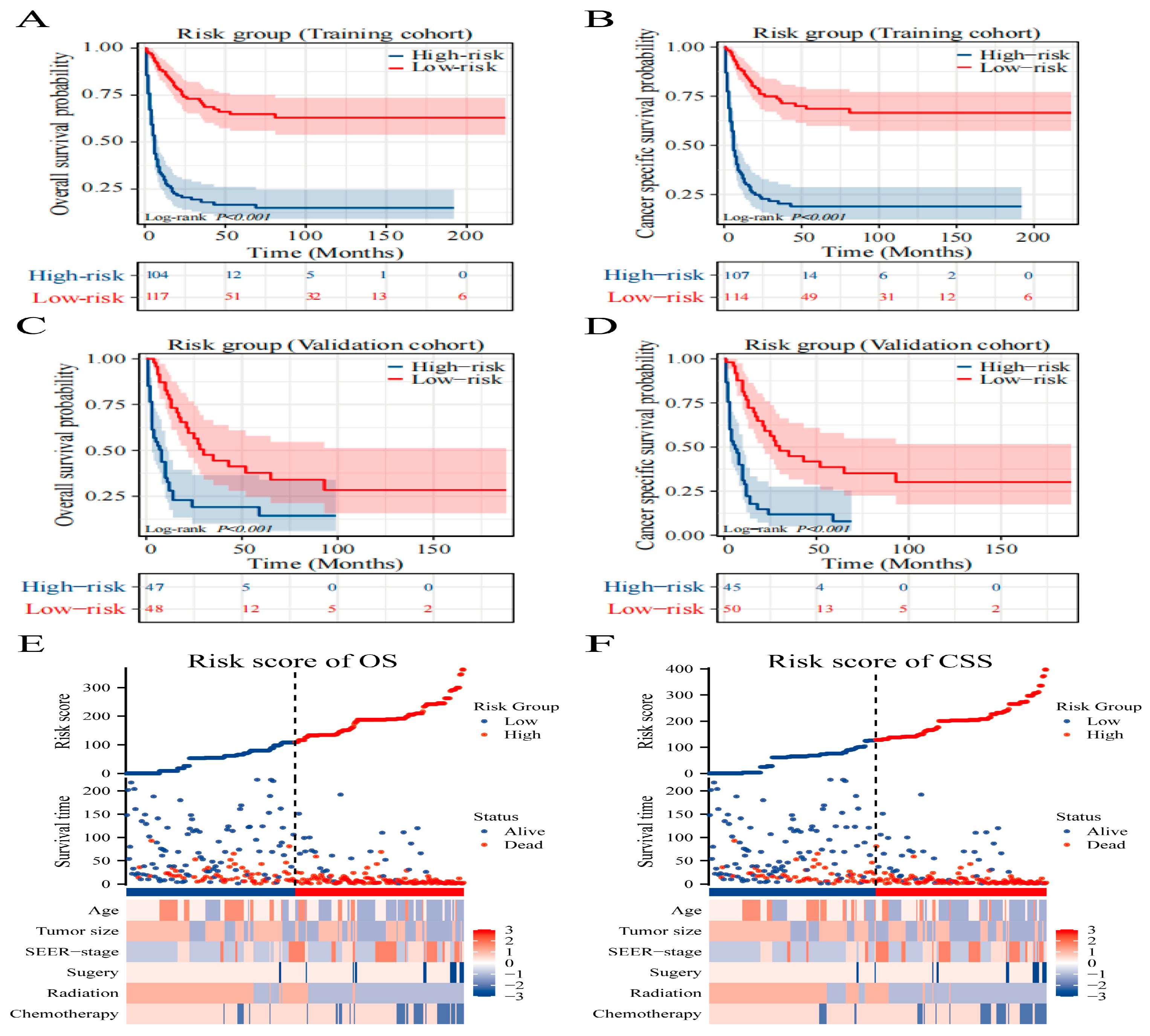
Figure 6.
The Sankey diagram and PSM analysis. Sankey diagram of each predictor feature and risk grouping of OS (A) and CSS (B); The survival curves of two groups before and after matching analysis in OS (C,E) and CSS (D,F).
Figure 6.
The Sankey diagram and PSM analysis. Sankey diagram of each predictor feature and risk grouping of OS (A) and CSS (B); The survival curves of two groups before and after matching analysis in OS (C,E) and CSS (D,F).
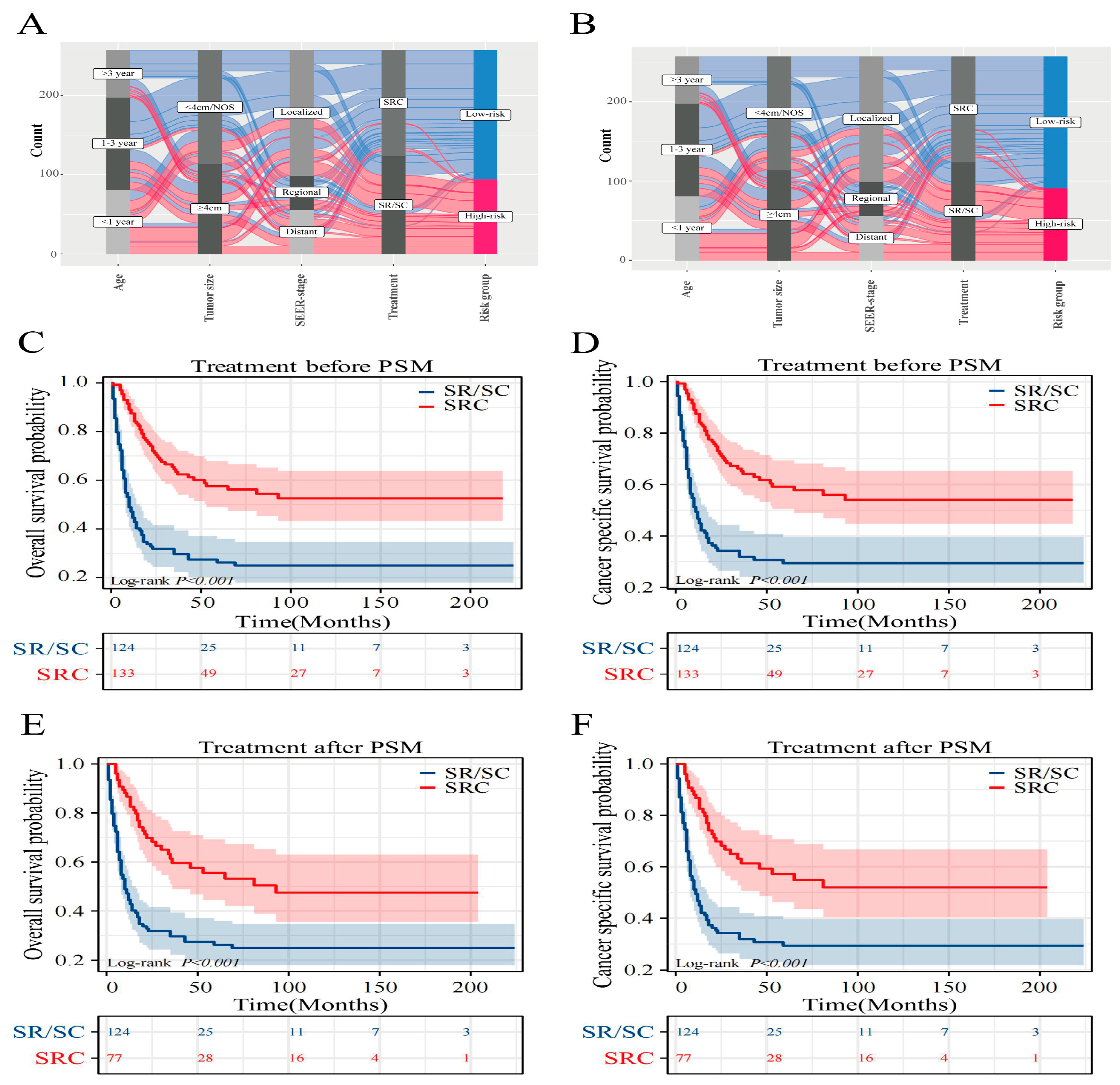
Figure 7.
The Genomic Landscape, PPI and GO/KEGG analysis in AT/RT. (A) The top 20 mutated genes. (B) An overview of the types of mutation observed. (C) PPI network of the top 20 mutated genes. (D) The GO/KEGG enrichment analysis of top 20 genes.
Figure 7.
The Genomic Landscape, PPI and GO/KEGG analysis in AT/RT. (A) The top 20 mutated genes. (B) An overview of the types of mutation observed. (C) PPI network of the top 20 mutated genes. (D) The GO/KEGG enrichment analysis of top 20 genes.
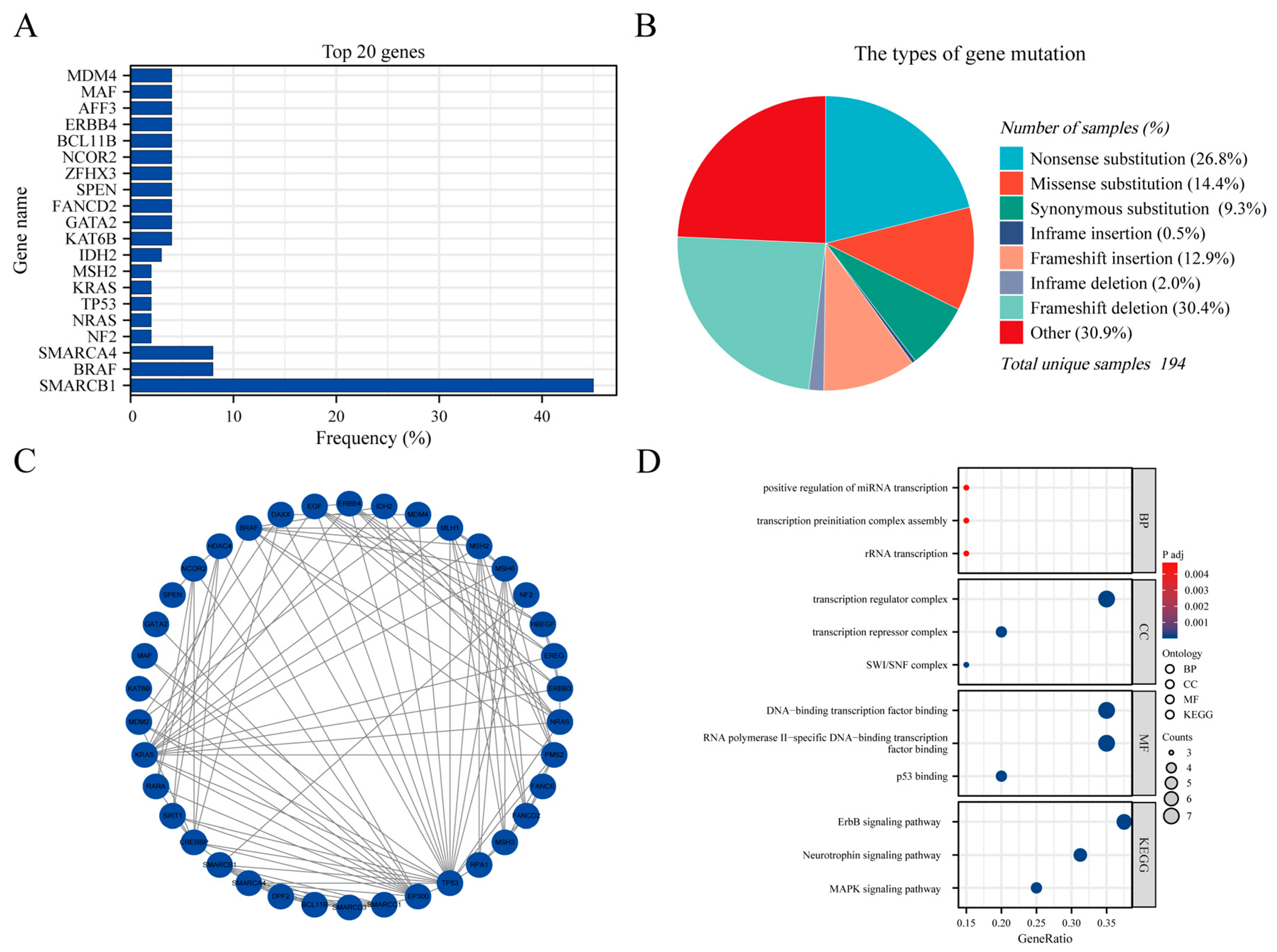

Table 1.
Characteristics of patients with AT/RT in the training and validation group.
| Characteristics | Total (n=316) |
Training group (n=221) |
Validation group (n=95) |
P value |
|---|---|---|---|---|
| no.(%) | no.(%) | no.(%) | ||
| Years of diagnosis | 0.361 | |||
| 2000-2009 | 137(43.4%) | 100 (45.2%) | 37 (38.9%) | |
| 2010-2019 | 179(56.6%) | 121 (54.8%) | 58 (61.1%) | |
| Age | 0.171 | |||
| <1 year | 114(36.1%) | 87 (39.4%) | 27 (28.4%) | |
| 1-3 year | 134(42.4%) | 88 (39.8%) | 46 (48.4%) | |
| >3 year | 68(21.5%) | 46 (20.8%) | 22 (23.2%) | |
| Gender | 0.786 | |||
| Male | 165(52.2%) | 117 (52.9%) | 48 (50.5%) | |
| Female | 151(47.8%) | 104 (47.1%) | 47 (49.5%) | |
| Race | 0.295 | |||
| White | 241(76.3%) | 173 (78.3%) | 68 (71.6%) | |
| Black | 41(13.0%) | 28 (12.7%) | 13 (13.7%) | |
| Others | 34(10.7%) | 20 (9%) | 14 (14.7%) | |
| Household income | 0.987 | |||
| <75000$ | 221(69.9%) | 154 (69.7%) | 67 (70.5%) | |
| ≥75000$ | 95(30.1%) | 67 (30.3%) | 28 (29.5%) | |
| Grade | 0.893 | |||
| Unknown | 253(80.1%) | 176 (79.6%) | 77 (81.1%) | |
| III-IV | 63(19.9%) | 45 (20.4%) | 18 (18.9%) | |
| Primary site | 0.194 | |||
| Intracranial | 299(94.6%) | 212 (95.9%) | 87 (91.6%) | |
| Spinal cord | 17(5.4%) | 9 (4.1%) | 8 (8.4%) | |
| Laterality | 0.674 | |||
| Left | 51(16.1%) | 35 (15.8%) | 16 (16.8%) | |
| Right | 55(17.4%) | 36 (16.3%) | 19 (20%) | |
| Others | 210(66.5%) | 150 (67.9%) | 60 (63.2%) | |
| Tumor size | 0.152 | |||
| <4cm/NOS | 174(55.1%) | 128 (57.9%) | 46 (48.4%) | |
| ≥4cm | 142(44.9%) | 93 (42.1%) | 49 (51.6%) | |
| SEER-stage | 0.348 | |||
| Localized | 189(59.8%) | 136 (61.5%) | 53 (55.8%) | |
| Regional | 61(19.3%) | 38 (17.2%) | 23 (24.2%) | |
| Distant | 66(20.9%) | 47 (21.3%) | 19 (20.0%) | |
| Surgery | 0.999 | |||
| GTR/STR | 297(94.0%) | 208 (94.1%) | 89 (93.7%) | |
| No/Unknown | 19(6.0%) | 13 (5.9%) | 6 (6.3%) | |
| Chemotherapy | 0.647 | |||
| Yes | 256(81.0%) | 181 (81.9%) | 75 (78.9%) | |
| No/Unknown | 60(19.0%) | 40 (18.1%) | 20 (21.1%) | |
| Radiation | 0.999 | |||
| Yes | 148(46.8%) | 103 (46.6%) | 45 (47.4%) | |
| No/Unknown | 168(53.2%) | 118 (53.4%) | 50 (52.6%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



