
Submitted:
10 January 2024
Posted:
11 January 2024
You are already at the latest version
Abstract
The persistent threat of cancer remains a significant hurdle for global health, prompting the exploration of innovative approaches in the quest for successful therapeutic interventions. Cyclin-dependent kinase 9 (CDK9), a central player in transcription regulation and cell cycle progression, has emerged as a promising target to combat cancer. Its pivotal role in oncogenic pathways and the pressing need for novel cancer treatments has propelled CDK9 into the spotlight of drug discovery efforts. This article presents a comprehensive study that connects a multidisciplinary approach, combining computational methodologies, experimental validation, and the transformative PROTAC (Proteolysis-Targeting Chimera) technology. By uniting these diverse techniques, we aim to identify, characterize, and optimize a new class of degraders targeting CDK9. We explore these compounds for kinase inhibitory properties and potential to induce targeted protein degradation, offering a novel and potentially more sustainable approach to cancer therapy. This cohesive strategy expresses the interface between computational predictions and experimental insights, with the ultimate goal of advancing the development of effective anticancer therapeutics targeting CDK9.
Keywords:
cancer
; protein degradation
; degraders
; drug discovery
; PROTAC
; CDK9
1. Introduction
Cancer is one of the most overwhelming challenges to global public health, with an ever-evolving landscape of complexities requiring incessant innovative and effective therapeutic solutions. Affecting people of all ages, it is the second most common cause of death globally. As many of us accept this challenge and keep investigating different perspectives, more attention is falling on Cyclin-dependent kinase 9 (CDK9), a decisive regulator of transcription and cell cycle progression, as a promising path for cancer therapy [1,2,3,4].
CDK9, a member of the Cyclin-dependent kinase (CDK) family with its regulatory partner cyclin T1(CCNT1), forms the positive transcription extension factor b(P-TEFb) complex [5]. This complex coordinates the phosphorylation of RNA polymerase II and the liberation of paused RNA polymerase II, facilitating proficient transcriptional elongation of genes critical to cell cycle control and dodging apoptosis [6,7]. There is also evidence that CDK9-driven phosphorylation has been known to promote many cancers [8].
CDK9 drives vital oncogenic pathways and its frequent overexpression across various cancer types, emphasizing its significance as a prime target for cancer therapy [9,10]. The outlook of overriding CDK9 assures disruption of the transcriptional programs that fuel cancer evolution, positioning it as a compelling scheme in pursuing novel anticancer therapeutics [11]. We have a massive demand for a comprehensive and innovative approach to decode this promise into tangible clinical benefits. Several CDK9 inhibitors have recently been designed through molecular modeling techniques and showed good antitumoral activity in-vitro [12].
Traditional drug discovery methods repeatedly revolve around developing small molecules designed to inhibit the enzymatic activity of a target protein [13]. These approaches have yielded prominent successes with different kinase inhibitors, but they may not fully exploit the manifold functions of a target protein. While addressing this limitation and endeavoring for enhanced therapeutic efficacy, this article presents a comprehensive study that harmonizes computational methodologies, experimental validation, and the groundbreaking realm of Proteolysis-Targeting Chimeras (PROTACs) [14]. This approach aims to identify and exemplify the novel CDK9 degraders and explore the new territory of targeted protein degradation, steering a significant shift in cancer drug discovery.
Our study of the intersection of computational predictions, experimental insights, and PROTAC-mediated protein degradation seeks to bridge the considerable gap between target identification and the clinical development of effective therapies. In the upcoming sections, we will accurately explain the computational and experimental methodologies employed in our investigation and highlight their interactive potential to drive the field of CDK9-targeted cancer therapeutics to new heights.
2. Results
2.1. Computational Results
2.1.1. Docking Study
The outcomes of the molecular docking analysis involving CDK9 and Therabene's proprietary compounds (TB003, TB008, and TB0016)1 are documented in Table 1. The docking results elucidate the optimal binding sites of the ligands with the receptor, highlighting the species of the most stable binding determined by the binding affinity (Figure 1a). Notably, the docking performance of CDK9 surpasses that of other cyclin-dependent kinases (CDKs), as evidenced by a superior Area Under the Curve (AUC) value. Examining the AUC performance in Figure 1b reveals that CDK9 outperforms CDK2. This outstanding AUC performance of CDK9 can be anticipated to extend to other CDKs. The data thus underscores the efficacy of CDK9 in ligand binding, suggesting a notable advantage over its CDK counterparts based on our comprehensive analysis of molecular docking results.
2.1.2. ADMET Analysis
The physicochemical properties of the compounds and their PROTAC, which serve as the descriptors to evaluate their bioactivity, are shown in Table 2. TB003 exhibits high lipophilicity with a LogP value of 5.09, signifying a solid affinity for lipid environments. However, its low absorption and poorly soluble nature with a LogS of -6.29 suggest potential challenges in bioavailability. In contrast, TB008 demonstrates moderate lipophilicity (LogP 2.75), coupled with high absorption and good solubility (LogS -3.83). TB0016 shares similar attributes with TB008, displaying moderate lipophilicity (LogP 2.55), high absorption, and moderately soluble characteristics (LogS -4.28). The PROTAC versions of TB003 and TB008 also exhibit high lipophilicity, but their low absorption and moderately to poorly soluble nature could pose formulation challenges in drug development. These compound profiles underscore the importance of balancing lipophilicity, absorption, and solubility for optimal drug design and delivery. The PROTAC version of TB0016 is currently being made.
In summary, all drugs, including precursors and PROTACs, display LogP values that fall well within the desirable range for therapeutic compounds. As expected, PROTAC molecules, however, are less soluble than the precursor compounds, and also, because of size, the PROTACs are not as well absorbed.
2.1.3. MD Simulations
The molecular dynamics (MD) simulations conducted for each complex yielded valuable insights into the intricate molecular interactions between the target and our selected compounds. This comprehensive analysis delves into the nuances of the complexes' stability and dynamics under scrutiny. In addition to the simulations of CDK9 alone, the complexes subjected to MD simulations include CDK9-TB003, CDK9-TB008, and CDK9-TB0016, allowing for a detailed exploration of their behavior at the molecular level. These simulations show how these complexes evolve, unraveling essential information about their structural stability and dynamic behavior.
a) RMSD Analysis
To investigate the structural conformations of CDK9, CDK9-TB003, CDK9-TB008, and CDK9-TB0016 at the molecular level, the analysis focused on calculating Root Mean Square Deviations (RMSD) values. The mean RMSD values for CDK9, CDK9-TB003, CDK9-TB008, and CDK9-TB0016 were quantified as 0.29 nm, 0.30 nm, 0.28 nm, and 0.27 nm, respectively. These outcomes suggest minimal variations in the structural configurations of the complexes following binding events.
The RMSD plot, depicted in Figure 2a, visually represents the sustained stability observed throughout the simulation. The plot demonstrates a consistent trajectory, indicating that the complexes maintain high structural integrity over the simulation period. This implies that the binding of the ligands to CDK9 does not induce significant structural deviations.
Furthermore, the probability distribution analysis, as illustrated in Figure 2b, corroborates the stability of the complexes by revealing a lack of significant shifts. The probability distribution graph provides additional evidence supporting the notion that the structural integrity of each complex remains reasonably stable throughout the simulation, further substantiating the robustness of the molecular systems under investigation.
a) RMSF Analysis
Examining dynamic characteristics and flexibility within the complex employs Root Mean Square Fluctuation (RMSF) analysis, as shown in Figure 3a. The results reveal that residual fluctuations consistently display stability and remain minimized throughout the simulation duration. Notably, subtle changes are observable within the residues ranging from 250 to 300, positioned distally from the binding site at the protein's terminus.
The probability distribution plot of RMSF, as illustrated in Figure 3b, reinforces the system's stability by indicating an absence of significant shifts. This graphical representation further underscores the system's overall strength, emphasizing the minimal impact of fluctuations on the dynamic behavior of the complex.
a) Radius of Gyration Analysis
The examination considered the structure's intricacy and overall configuration before and after ligand binding, utilizing the gyration radius (Rg) metric throughout the simulation. The findings indicate that the protein's stability remained predominantly unaltered by the binding event, as evidenced by the consistent Rg values observed throughout the simulation duration, as illustrated in Figure 4a. This sustained stability highlights the robustness of the protein structure, implying that the ligand binding did not elicit notable structural disruptions. The probability distribution plot (Figure 4b) further supports the comparable stability of the system, revealing no significant shifts in Rg values.
a) Analysis of Solvent-Accessible Surface Area (SASA)
We conducted an in-depth examination of the protein's interaction with its surrounding solvent by computing the solvent-accessible surface area (SASA) for CDK9 and CDK9-TB complexes. The calculated average SASA values for CDK9, CDK9-TB003, CDK9-TB008, and CDK9-TB0016 were determined to be 163.98 nm2, 161.00 nm2, 163.68 nm2, and 160.62 nm2, respectively, as depicted in Figure 5a. This analysis offers valuable insights into the extent of protein exposure to the solvent in each complex.
Moreover, the probability distribution plot (Figure 5b) further reinforces that the systems, including CDK9, CDK9-TB003, CDK9-TB008, and CDK9-TB0016, remained stable throughout the simulation. This stability is a critical factor in understanding the structural integrity and behavior of these complexes, providing additional confidence in the reliability of the obtained results.
a) Hydrogen Bonds Dynamics
Preserving hydrogen bond stability during simulations is imperative for upholding the structural conformation of the protein and its complexes. In the context of CDK9 and its interactions with TB003, TB008, and TB0016, the mean count of intramolecular hydrogen bonds was consistently recorded at 198, 202, 197, and 203, respectively. This unwavering count emphasizes the stability of hydrogen bonding interactions between CDK9 and the CDK9-TB compounds throughout the simulation, as portrayed in Figure 6a. Notably, the slight elevation in the number of hydrogen bonds can be ascribed to the increased complexity inherent in the CDK9-TB complexes. The probability distribution plot of the number of hydrogen bonds further substantiates the system's stability during the simulation, as depicted in Figure 6b.
The intermolecular hydrogen bonds observed between CDK9 and TB compounds exhibit variability, extending up to five, yet with a noticeable consistency within the 1-3 bond range. This consistency reflects minimal fluctuations throughout the simulation, as illustrated in Figure 7a and further elucidated in its distribution plot, Figure 7b. The findings affirm that the ligands consistently maintained their initial binding positions from the docking phase throughout the simulation, corroborating the sustained stability of the hydrogen bonding interactions within the CDK9-TB complexes.
2.2. Experimental Results
2.2.1. Assay
The table presented below illustrates the notable effectiveness of both TB003 and TB008 in inhibiting the kinase activity of the CDK9/Cyclin T complex. Conversely, TB0016 exhibited no discernible inhibitory properties (Table 3).
Notably, TB003 and TB008 emerge as potent CDK9 degraders, with IC50 values of 5nM and 3.5nM, respectively. In contrast, TB0016, identified as a CDK9 ligand, exhibits no significant potency with an IC50 greater than 1µM. Further emphasizing the selectivity of TB003 and TB008 for CDK9, the fold selectivity values against other CDK family members, such as CDK7, CDK5, CDK4, CDK2, and CDK1, are provided. These compounds, suitable for administration via IP/Oral routes, present promising prospects for targeted therapeutic interventions.
2.2.2. CDK9 Degradation by Western Blot
To demonstrate that TB003 degrades CDK9 selectively, the Malme -3M cell line was grown to confluency in 35mm culture dishes and then treated with TB003 at 1µM for varying periods. The cell lysates from each time point, containing approximately 1-2 mg/mL of protein, were then analyzed by Western blot. An image of the Western blot is shown in Figure 8.
To mitigate variations in protein load, the Western blot was also stained for the light chain of myosin as a control. Notably, the myosin light chain appears consistently comparable across the lanes. However, the CDK9 band exhibits a diminishing intensity with increasing treatment time. This observation suggests that as cells are exposed to the degrader for longer durations, there is a proportional decrease in the intensity of the CDK9 band, providing compelling evidence for the specific degradation of CDK9 induced by TB003. The dynamic changes in CDK9 intensity, as evidenced by the Western blot, reinforce the temporal relationship between TB003 treatment and the degradation of the target protein. TB008 has not been tested by Western blot analysis.
2.2.3. Cell viability
TB003 and TB008 have also been evaluated for cell growth inhibition and killing using two cell lines, NCI-H358, a non-small cell lung carcinoma-derived cell line, and MIA-PaCa-2, a pancreatic carcinoma-derived cell line. For this study, both drugs were added to cells at an initial concentration of 10µM, then serially diluted (1:3) down to 0.2nM. Results are presented in Figure 9.
In the context of anti-proliferative efficacy and cell-killing potential, both TB003 and TB008 exhibited notable effectiveness when tested against the MIA-PaCa-2 and NCI H-358 cell lines. A direct comparison with standard chemotherapeutic agents, such as Paclitaxel and Oxaliplatin, revealed that TB003 and TB008, boasting EC50 values in the low nanomolar range, surpassed the potency of these reference compounds. Notably, both degraders demonstrated robust anti-proliferative properties, but TB008 outperformed in cell-killing efficacy, as evidenced by a cell viability signal approaching zero at elevated drug concentrations. These findings underscore the potential of TB003 and TB008 as highly effective agents in impeding cell growth and inducing cell death, positioning them as promising candidates for further therapeutic development.
3. Discussions
The comprehensive approach presented in this study combines computational methodologies, experimental validation, and PROTAC technology to identify, characterize, and optimize a new class of CDK9 degraders for cancer therapy. The study begins by emphasizing the pivotal role of CDK9 in transcription regulation and cell cycle progression, positioning it as a promising target for cancer treatment. Integrating computational predictions and experimental insights seeks to bridge the gap between target identification and the clinical development of effective therapies.
The computational results, encompassing molecular docking, ADMET analysis, and MD simulations, provide valuable insights into the binding affinities, physicochemical properties, and dynamic behavior of the CDK9-TB compounds. Notably, CDK9 exhibits superior docking performance compared to other CDKs, emphasizing its efficacy in ligand binding. The MD simulations reveal the stability of the complexes, with minimal structural variations and consistent interactions between the ligands and CDK9. ADMET analysis sheds light on the pharmacological behavior of the compounds, emphasizing the importance of balancing lipophilicity, absorption, and solubility for optimal drug design.
Experimental results further validate the efficacy of TB003 and TB008 as potent CDK9 degraders, with notable IC50 values and selectivity against other CDK family members. Western blot analysis demonstrates the specific degradation of CDK9 induced by TB003, reinforcing its potential as a targeted therapeutic agent. Cell viability assays highlight the anti-proliferative and cell-killing efficacy of TB003 and TB008, surpassing the potency of standard chemotherapeutic agents. In contrast to the consistency of the computational findings among the three molecules, TB0016 does not exhibit strong inhibitory effects in the CDK9 kinase assay. Therefore, a thorough examination of TB0016 is essential for a more comprehensive investigation, including assessments such as binding activity, cell viability, Western blot analysis, and exploring alternative binding interactions with CDK9. This disparity underscores the need for careful consideration and a more profound analysis when interpreting computational and molecular docking results.
4. Materials and Methods
4.1. Computational Methods
4.1.1. Compound Selection and Analysis
Candidate inhibitors targeting CDK9 were chosen through an extensive review of chemical databases and relevant literature. A subset of these inhibitors, already present in our in-house inventory and previously subjected to testing, remains actively under consideration for ongoing research initiatives [7,15,16]. Using Openbabel to expand our repertoire, a comprehensive database search was conducted using a similarity search approach [17]. Enamine library [18], comprising a diverse array of chemical compounds, was used for this. Approximately 10,000 combinations were scrutinized, subjected to rigorous filtration, and systematically sorted to identify compounds with potential inclusion in our study. This systematic approach not only broadens the scope of our investigation but also ensures that the selected inhibitors exhibit a range of structural and chemical characteristics, enriching the diversity of our research dataset.
4.1.2. Target Preparation
The structural examination of CDK9 involved utilizing tools for structural modeling, visualization, and refinement, as outlined in references [19,20,21]. To ensure a robust foundation, UniProt (P50750) was employed to identify potential CDK9 structures, with a particular emphasis on selecting the most optimal structure (PDB ID: 3BLR). This selection was based on criteria such as resolution, the absence of missing residues, and alignment with other available systems, ensuring a high-quality structural template for subsequent analyses [20,22].
Furthermore, an in-depth investigation was conducted using X-ray data to identify the existing ligand-binding pocket. Additionally, the exploration of potential alternative active sites was undertaken by applying CAVIAR [23]. This comprehensive structural analysis provides a reliable structural framework for CDK9 and lays the groundwork for a nuanced understanding of its ligand interactions and potential regulatory sites.
4.1.3. Molecular Docking
The molecular docking of compound collections and CDK9 was executed through AutoDock-Vina, employing Smina and the latest version of Vina (version 1.2.5) for docking and scoring methods, as described in references [24,25,26]. The compounds resulting from the docking process were then meticulously arranged based on their binding affinity, providing a systematic ranking of their potential interactions with CDK9. To ensure the accuracy of the docking simulations, the receptor and ligands were meticulously prepared using AutoDock tools [24]. In assessing the performance of CDK9 with its prospective novel inhibitors, a comparative analysis was conducted against other cyclin-dependent kinases (CDKs) utilizing the DUDE dataset for docking and scoring [27]. Among the identified compounds, the one with the highest binding affinity was selected for further investigation, designated as TB0016. This meticulous selection ensures that the combination chosen for in-depth study exhibits the most favorable binding characteristics with CDK9.
Moreover, in addition to TB0016, two previously identified compounds under examination by Therabene, namely TB003 and TB008, were also included in our study. All the compounds analyzed in our research are collectively referred to as TB compounds. This comprehensive approach involving diverse compounds, each selected based on their docking performance, enriches the breadth and depth of our investigation into potential inhibitors for CDK9.
4.1.3. ADMET Properties
The selected compound from the docking (TB0016) and two other compounds (TB003 and TB008) were analyzed with SwissADME [28] to study the ADMET properties (Absorption, Distribution, Metabolism, Elimination and Toxicity). Integrating these properties is expected to enhance the overall understanding of the compound's pharmacokinetic properties, aiding in the early identification and mitigation of potential safety concerns.
4.1.4. Molecular Dynamics Calculations
To assess the stability of the chosen compounds identified through library screening and docking within the CDK9 complex, a comprehensive evaluation was conducted using Molecular Dynamics (MD) simulations facilitated by the GROMACS package [29,30]. These simulations were carried out in a box, explicitly including water and ions, and employed the CHARMM force field to represent molecular interactions accurately [31]. The ligand's topology was meticulously prepared using the external tool CGenFF [32].
During the simulation, the system underwent position restraint, employing a 1000 kJ mol-1 nm-2 force constant. The simulation plan included an initial energy minimization with 10,000 steps, each with a minimization step size 0.01. Subsequently, NVT (canonical ensemble) and NPT (isothermal-isobaric ensemble) equilibration steps were conducted, each lasting one nanosecond, with a time step of 2 femtoseconds. The final production run extended over 50 nanoseconds, providing a prolonged timeframe for observing and assessing the stability of the compounds within the CDK9 complex. This MD simulation approach enhances our understanding of the selected compounds' dynamic behavior and structural stability during the simulated timespan.
4.2. Experimental Methods
4.2.1. Chemical Compounds
TB003 and TB008 are proprietary PROTAC structures; briefly, the constructs of these two molecules comprise two functional moieties, one that binds to the target protein, CDK9, and the other that binds to cereblon E3 ligase. The two moieties are linked through an alkyl chain or polyethylene glycol linker. These molecules range in size between 750 g/mole to 950 g/mole and are relatively hydrophobic. The structure of TB0016 is also proprietary; while the TB0016 degrader precursor has been synthesized and subjected to some biochemical characterization, the TB0016 degrader has yet to be synthesized.
4.2.2. Biochemical Assays
a) Adapta Assay
The Adapta universal kinase assay operates through two phases: a kinase reaction phase and an ADP detection phase. During the kinase reaction phase, all components necessary for the reaction are introduced into the well, allowing incubation for 60 minutes. Following the response, a detection solution is applied, comprising a europium-labeled anti-ADP antibody, an Alexa Fluor™ 647 labeled ADP tracer, and EDTA to halt the kinase reaction. ADP generated by the kinase reaction displaces the Alexa Fluor 647 labeled ADP tracer from the antibody, reducing the TR-FRET signal. In the presence of a kinase inhibitor, the formation of ADP is diminished, and the ensuing interaction between the intact antibody and tracer leads to a heightened TR-FRET signal.
The assay procedure entails using a multi-well plate. It involves the following steps: Initially, 100 nL of CDK inhibitors (concentrations ranging from 0.1nM to 1µM) in an aqueous solution with up to 1% DMSO are added to the wells. Subsequently, 2.4 µL of 30 mM HEPES (pH 7.5), 2.5 µL of a 4X ATP solution in water, and 5 µL of a 2X Kinase Mixture in 50 mM HEPES (pH 7.5), 0.01% BRIJ-35, ten mM MgCl2, and one mM EGTA are introduced into each well. The plate is then shaken for 30 seconds and centrifuged at 1000 x g for 1 minute. The reaction mixture undergoes incubation at room temperature for 60 minutes. Subsequently, 5 µL of a detection mix containing 30 mM EDTA, 6 nM Eu-anti-ADP antibody, and ADP tracer is added to each well. After shaking for 30 seconds and centrifugation, the mixture is equilibrated at room temperature for 60 minutes. Finally, the assay is read on a fluorescence plate reader, and the data are analyzed. This detailed assay procedure facilitates the assessment of CDK inhibitors' inhibitory effects on kinase activity, providing a systematic approach for experimentation and ensuring the generation of reliable data for subsequent analysis.
a) Z-Lyte Assay
The Z-LYTE biochemical assay utilizes a coupled-enzyme format with a fluorescence-based approach. Its methodology relies on the differential sensitivity of phosphorylated and non-phosphorylated peptide substrates to proteolytic cleavage. The peptide substrate, labeled with fluorophores at each end, constitutes a FRET pair. In the primary reaction, the Kinase transfers the gamma-phosphate of ATP to a specific tyrosine, serine, or threonine residue in the synthetic FRET-peptide. Subsequently, a site-specific protease recognizes and cleaves any non-phosphorylated FRET-peptide in the secondary response, as phosphorylation prevents peptide cleavage. Cleavage disrupts FRET between the donor (coumarin) and acceptor (fluorescein) fluorophores on the FRET-peptide, while uncleaved, phosphorylated FRET-peptide maintains the FRET signal. A ratiometric method, determining the Emission Ratio by calculating the ratio of coumarin emission (445nm) to FRET (fluorescein) emission (520nm) after exciting the donor fluorophore at 400 nm, is employed for quantifying reaction progress.
The assay procedure involves introducing inhibitory test samples (100 nL) with concentrations ranging from 0.1nM to 1µM in water with up to 1% DMSO onto a multi-well plate. Subsequently, 2.4 µL of kinase buffer, 5 µL of a 2X Peptide (4µM)/Kinase (2ng) Mixture, and 2.5 µL of 4X ATP (40µM) are successively added to each well. Following a 30-second shaking period, the reaction mixture undergoes incubation at room temperature for 60 minutes. Afterward, 5 µL of a proprietary Development Reagent Solution is added, and the plate is shaken for another 30 seconds. The reaction is performed at room temperature for 60 minutes before being read on a fluorescence plate reader. The resulting data are then analyzed to evaluate the inhibitory effects of the test samples on kinase activity.
4.2.3. CDK9 Degradation with TB003
To demonstrate that TB003 works as a degrader, the 3M cells were grown to confluency in 35 mm x 12mm cell culture dishes using IMDM media with 20% bovine serum. Cells were treated with and without 50nM TB003 for 60 minutes at 37C. After incubation, the cells were washed with ice-cold PBS and lysed with cell lysis buffer containing 1X protease cocktail. After removing cellular and nuclear debris by centrifugation at 13,000rpm for 10 minutes, a 75µL aliquot was mixed with 4X reducing Laemmli sample buffer and loaded onto a 4-12% Bis-Tris gel. The gel was run at 125V for 60-90 minutes and then electro-transferred onto PVDF. After transfer, the membrane was blocked with 2% milk extract and developed using a combination of rabbit anti-CDK9 and anti-myosin light chain monoclonal antibodies, used at 1:5000 dilution, followed by goat anti-rabbit antibody conjugated with HRP at 1:10,000 dilution. Bands on the membrane were visualized by chemiluminescence using an ECL reagent [33,34].
4.2.4. Effect of Degrader on Cell Viability
The cell lines used in this study were purchased from ATCC (https://www.atcc.org) and maintained under standard conditions at 37°C with a 5% CO2 atmosphere. NCI-H-358 cells were cultivated in 1X RPMI 1640 medium (Corning), while MIA-PaCa2 cells were cultured using 1X DMEM (ATCC). Both culture media were supplemented with 10% fetal bovine serum (Gibco) and 1X Penicillin/Streptomycin (Gibco). For culturing the Melma-3M cells, IMDM media fortified with 20% fetal bovine serum, and 1X Penicillin/Streptomycin was used. To prevent overgrowth, cells were regularly split using 0.25% trypsin (Thermo-Fisher) before reaching full confluence, and the culture media were refreshed every 3-4 days.
This rigorous cell culture protocol ensured optimal conditions for assessing the degrader's impact on cell viability. The choice of diverse cell lines and the meticulous cultivation practices contribute to the findings' reliability and relevance in elucidating the degrader's effects on cellular viability.
5. Conclusions
In summary, this integrated approach successfully identifies and characterizes a novel class of CDK9 degraders with promising anticancer properties. The study combines computational and experimental techniques, providing a thorough understanding of the molecular interactions, pharmacological behavior, and therapeutic potential of the CDK9-TB compounds. TB003 and TB008 emerge as promising candidates, demonstrating not only potent CDK9 inhibition but also effective induction of targeted protein degradation and robust anti-proliferative effects. The findings underscore the potential of these compounds as lead candidates for further therapeutic development in pursuing innovative and effective cancer treatments targeting CDK9. The positive computational results for TB0016 necessitate a deeper exploration of the biochemical and biological properties of TB0016 in its interactions with CDK9 and the development of its corresponding PROTAC structure. This study contributes to the evolving landscape of cancer drug discovery by presenting a holistic and multidisciplinary approach to identifying and optimizing novel therapeutic agents.
Author Contributions
MK conducted computational analyses and authored the manuscript, while MD played a key role in the experimental aspects, contributed to manuscript writing, and provided supervision throughout the project. All authors thoroughly reviewed and gave their approval to the final manuscript.
Funding
This research received no external funding.
Acknowledgments
We thank the Mansfield Bio-Incubator for providing laboratory space.
Conflicts of Interest
Author Mahesh Koirala and Mario DiPaola were employed by the company Therabene Inc. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Tai, W.; Mahato, R.; Cheng, K. The role of HER2 in cancer therapy and targeted drug delivery. J. Control. Release 2010, 146, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Bondeson, D.P.; Mares, A.; Smith, I.E.D.; Ko, E.; Campos, S.; Miah, A.H.; Mulholland, K.E.; Routly, N.; Buckley, D.L.; Gustafson, J.L.; et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat. Chem. Biol. 2015, 11, 611–617. [Google Scholar] [CrossRef]
- Garriga, J.; Graña, X. CDK9 inhibition strategy defines distinct sets of target genes. BMC Res. Notes 2014, 7, 301–301. [Google Scholar] [CrossRef] [PubMed]
- Sajjadi-Dokht, M.; Mohamad, T.A.M.; Rahman, H.S.; Maashi, M.S.; Danshina, S.; Shomali, N.; Solali, S.; Marofi, F.; Zeinalzadeh, E.; Akbari, M.; et al. MicroRNAs and JAK/STAT3 signaling: A new promising therapeutic axis in blood cancers. Genes Dis. 2021, 9, 849–867. [Google Scholar] [CrossRef]
- Zhou, Q.; Li, T.; Price, D.H. RNA Polymerase II Elongation Control. Annu. Rev. Biochem. 2012, 81, 119–143. [Google Scholar] [CrossRef]
- Ott, M.; Geyer, M.; Zhou, Q. The Control of HIV Transcription: Keeping RNA Polymerase II on Track. Cell Host Microbe 2011, 10, 426–435. [Google Scholar] [CrossRef] [PubMed]
- Blachly, J.S.; Byrd, J.C.; Grever, M. Cyclin-dependent kinase inhibitors for the treatment of chronic lymphocytic leukemia. Semin. Oncol. 2016, 43, 265–273. [Google Scholar] [CrossRef]
- Blake, D.R.; Vaseva, A.V.; Hodge, R.G.; Kline, M.P.; Gilbert, T.S.K.; Tyagi, V.; Huang, D.; Whiten, G.C.; Larson, J.E.; Wang, X.; et al. Application of a MYC degradation screen identifies sensitivity to CDK9 inhibitors in KRAS-mutant pancreatic cancer. Sci. Signal. 2019, 12. [Google Scholar] [CrossRef]
- Lyu, J.; Yang, E.J.; Zhang, B.; Wu, C.; Pardeshi, L.; Shi, C.; Mou, P.K.; Liu, Y.; Tan, K.; Shim, J.S. Synthetic lethality of RB1 and aurora A is driven by stathmin-mediated disruption of microtubule dynamics. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Knight, Z.A.; Shokat, K.M. Chemical Genetics: Where Genetics and Pharmacology Meet. Cell 2007, 128, 425–430. [Google Scholar] [CrossRef]
- Lu, H.; et al. Compensatory induction of MYC expression by sustained CDK9 inhibition via a BRD4-dependent mechanism. Elife 2015, 4, e06535. [Google Scholar] [CrossRef] [PubMed]
- Morales, F.; Giordano, A. Overview of CDK9 as a target in cancer research. Cell Cycle 2016, 15, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Rahaman, M.H.; Kumarasiri, M.; Mekonnen, L.B.; Yu, M.; Diab, S.; Albrecht, H.; Milne, R.W.; Wang, S. Targeting CDK9: A promising therapeutic opportunity in prostate cancer. Endocrine-Related Cancer 2016, 23, T211–T226. [Google Scholar] [CrossRef] [PubMed]
- Burslem, G.M.; Crews, C.M. Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell 2020, 181, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Qin, Z.; Tian, Y.; Wang, J.; Xu, C.; Li, Z.; Bian, J. Recent Developments in the Biology and Medicinal Chemistry of CDK9 Inhibitors: An Update. J. Med. Chem. 2020, 63, 13228–13257. [Google Scholar] [CrossRef] [PubMed]
- Olson, C.M.; Jiang, B.; Erb, M.A.; Liang, Y.; Doctor, Z.M.; Zhang, Z.; Zhang, T.; Kwiatkowski, N.; Boukhali, M.; Green, J.L.; et al. Pharmacological perturbation of CDK9 using selective CDK9 inhibition or degradation. Nat. Chem. Biol. 2018, 14, 163–170. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Yang, J.W.; Hoffmann, S.; List, B. Asymmetric Enamine Catalysis. Chem. Rev. 2007, 107, 5471–5569. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- DeLano, W.L. Pymol: An open-source molecular graphics tool. CCP4 Newsl. Protein Crystallogr 2002, 40, 82–92. [Google Scholar]
- Consortium, U. UniProt: A hub for protein information. Nucleic acids research 2015, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Marchand, J.-R.; Pirard, B.; Ertl, P.; Sirockin, F. CAVIAR: A method for automatic cavity detection, description and decomposition into subcavities. J. Comput. Mol. Des. 2021, 35, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Morris, G.M.; Forli, S. Using AutoDock 4 and AutoDock vina with AutoDockTools: A tutorial. The Scripps Research Institute Molecular Graphics Laboratory 2012, 10550, 1000. [Google Scholar]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE 2016, 11, e0155183. [Google Scholar] [CrossRef] [PubMed]
- Masters, L.; Eagon, S.; Heying, M. Evaluation of consensus scoring methods for AutoDock Vina, smina and idock. J. Mol. Graph. Model. 2020, 96, 107532. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Van Der Spoel, D.; Van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Shukla, R.; Tripathi, T. Molecular dynamics simulation of protein and protein–ligand complexes. Computer-aided drug design 2020, 133–161. [Google Scholar]
- Huang, J.; Mackerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; MacKerell, A.D. Automation of the CHARMM General Force Field (CGenFF) I: Bond Perception and Atom Typing. J. Chem. Inf. Model. 2012, 52, 3144–3154. [Google Scholar] [CrossRef] [PubMed]
- Bjørkøy, G.; et al. Monitoring autophagic degradation of p62/SQSTM1. Methods in enzymology 2009, 452, 181–197. [Google Scholar] [PubMed]
- Jang, J.; To, C.; De Clercq, D.J.H.; Park, E.; Ponthier, C.M.; Shin, B.H.; Mushajiang, M.; Nowak, R.P.; Fischer, E.S.; Eck, M.J.; et al. Mutant-Selective Allosteric EGFR Degraders are Effective Against a Broad Range of Drug-Resistant Mutations. Angew. Chem. 2020, 132, 14589–14597. [Google Scholar] [CrossRef]
Figure 1.
Docking and scoring results: a) docked pose of TB compounds to CDK9 (red: TB003; blue: TB008 & green: TB0016); b) AUC values of docking and scoring CDK9 (left) & CDK2(right).
Figure 1.
Docking and scoring results: a) docked pose of TB compounds to CDK9 (red: TB003; blue: TB008 & green: TB0016); b) AUC values of docking and scoring CDK9 (left) & CDK2(right).
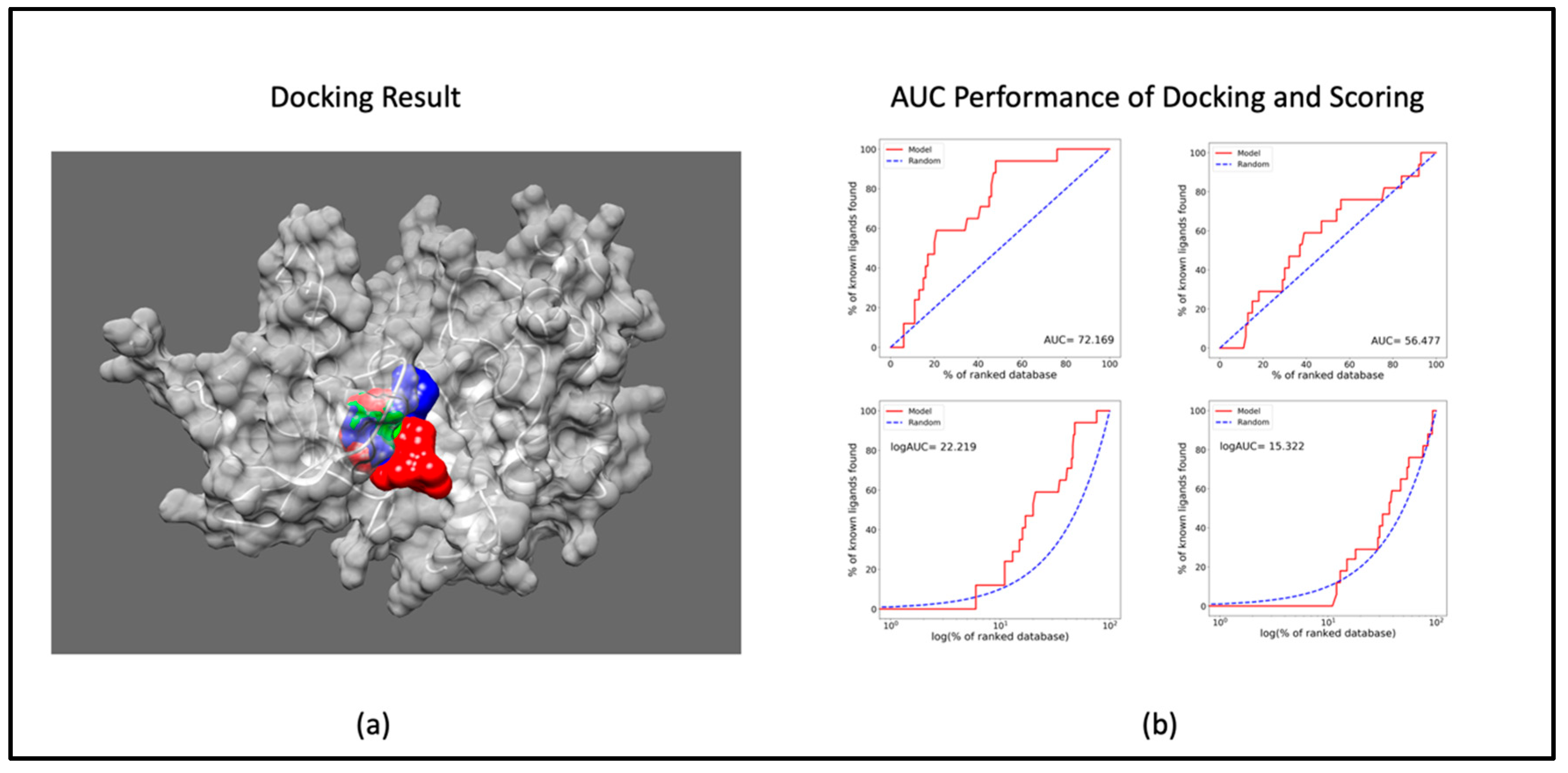
Figure 2.
RMSD analysis: a) RMSD plot of CDK9 and CDK9-TB compounds; b) probability distribution of RMSD.
Figure 2.
RMSD analysis: a) RMSD plot of CDK9 and CDK9-TB compounds; b) probability distribution of RMSD.
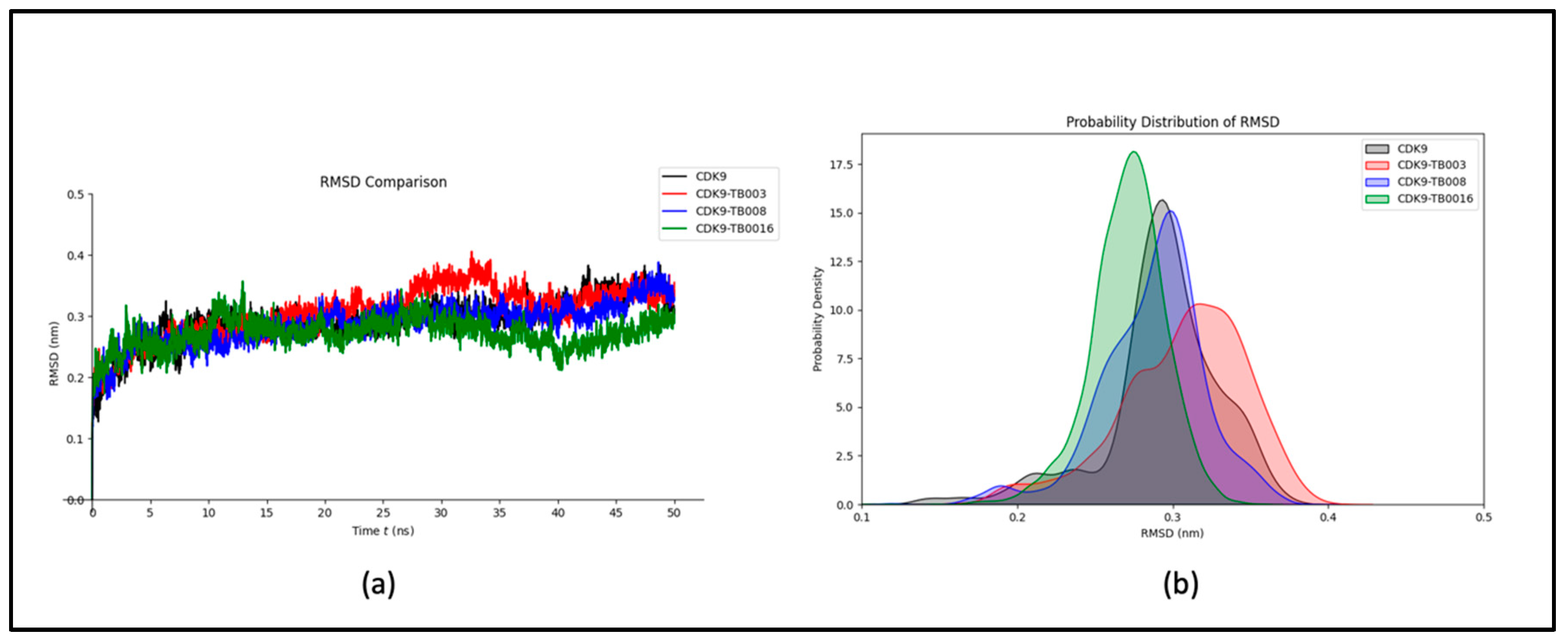
Figure 3.
RMSF analysis: a) RMSF fluctuations of CDK9 and CDK9-TB compounds, b) probability distribution of RMSF.
Figure 3.
RMSF analysis: a) RMSF fluctuations of CDK9 and CDK9-TB compounds, b) probability distribution of RMSF.
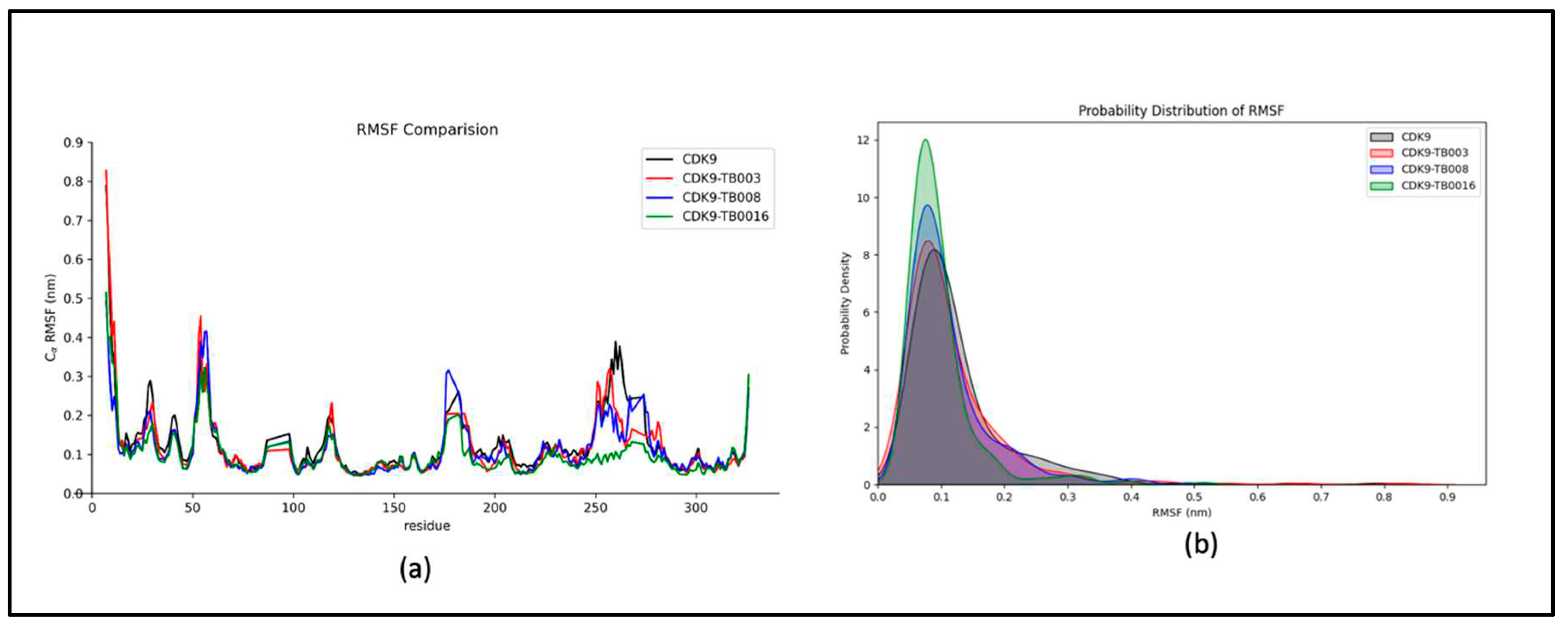
Figure 4.
The radius of gyration of CDK9 and CDK9-TB compounds: a) plot of Rg over time; b) probability distribution of Rg.
Figure 4.
The radius of gyration of CDK9 and CDK9-TB compounds: a) plot of Rg over time; b) probability distribution of Rg.
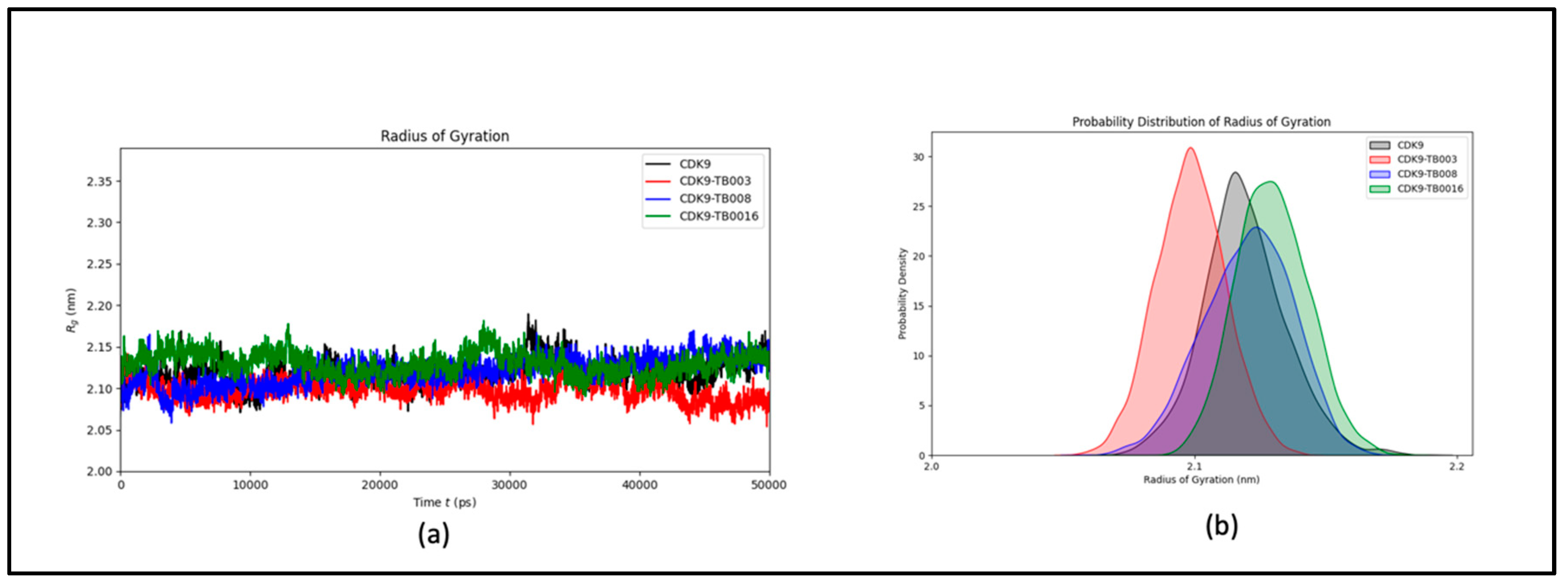
Figure 5.
Solvent-accessible surface area (SASA) analysis: a) SASA of CDK9 and CDK9-TB compounds; b) Probability distribution of SASA.
Figure 5.
Solvent-accessible surface area (SASA) analysis: a) SASA of CDK9 and CDK9-TB compounds; b) Probability distribution of SASA.
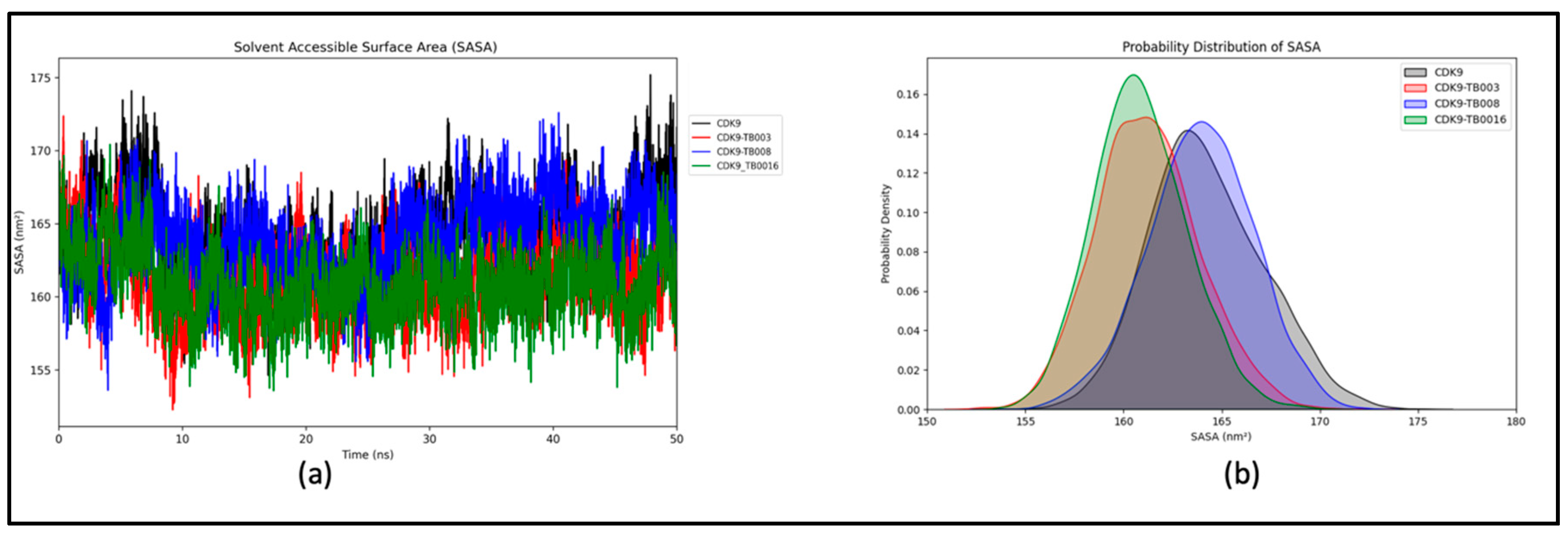
Figure 6.
Intramolecular hydrogen bonds dynamics: a) number of hydrogen bonds over time; b) probability distribution of intramolecular hydrogen bond.
Figure 6.
Intramolecular hydrogen bonds dynamics: a) number of hydrogen bonds over time; b) probability distribution of intramolecular hydrogen bond.
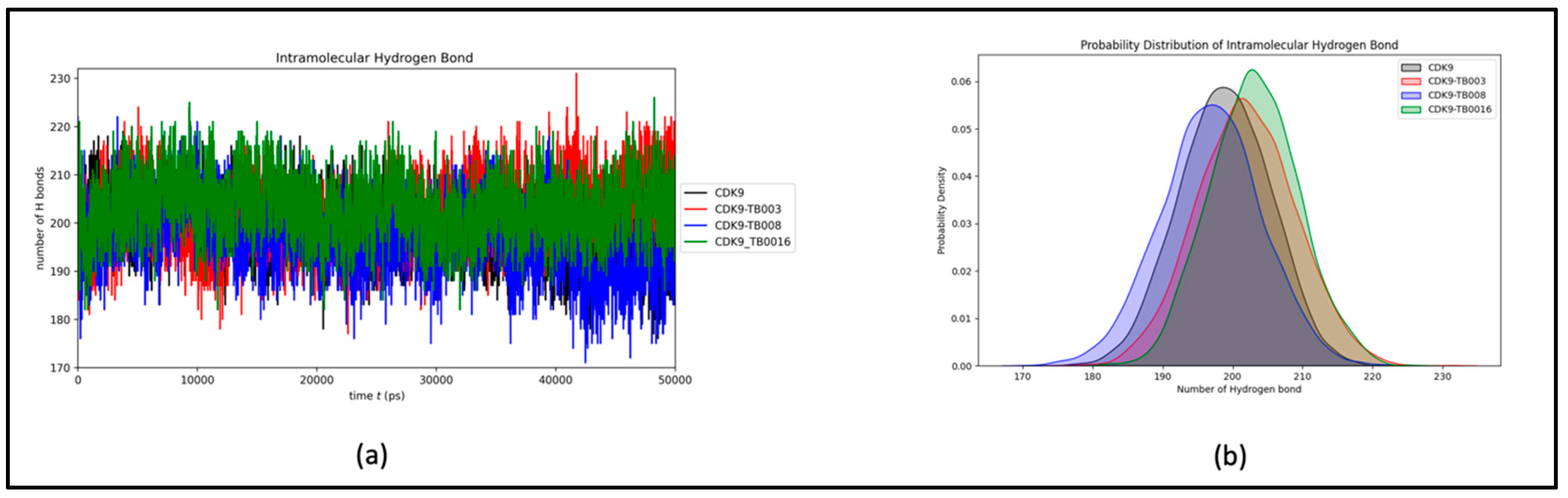
Figure 7.
Intermolecular hydrogen bond dynamics: a) number of hydrogen bonds between CDK9 and TB compounds; b) probability distribution of intermolecular hydrogen bond.
Figure 7.
Intermolecular hydrogen bond dynamics: a) number of hydrogen bonds between CDK9 and TB compounds; b) probability distribution of intermolecular hydrogen bond.
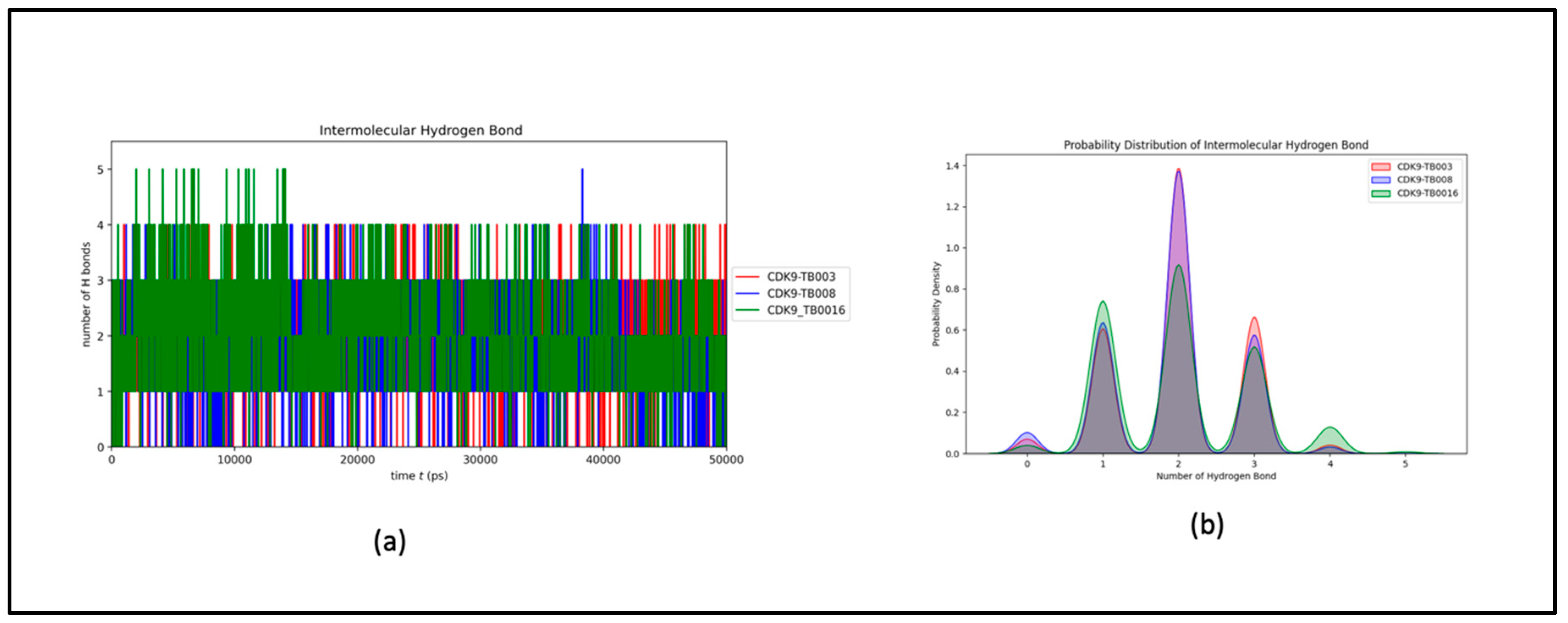
Figure 8.
Western blot of 3M cells treated with TB003: Approximately a 30µL volume of each lysate was loaded into the gel in the following order – MW markers lane 1, control lane 3, 2-hour control lane 4, 4-hour control lane 5, 12-hour control lane 7, 2-hour treatments lane 9, 4-hour treatment lane 10, 8-hour treatment lane 11 and 12-hour treatment lane 12.
Figure 8.
Western blot of 3M cells treated with TB003: Approximately a 30µL volume of each lysate was loaded into the gel in the following order – MW markers lane 1, control lane 3, 2-hour control lane 4, 4-hour control lane 5, 12-hour control lane 7, 2-hour treatments lane 9, 4-hour treatment lane 10, 8-hour treatment lane 11 and 12-hour treatment lane 12.
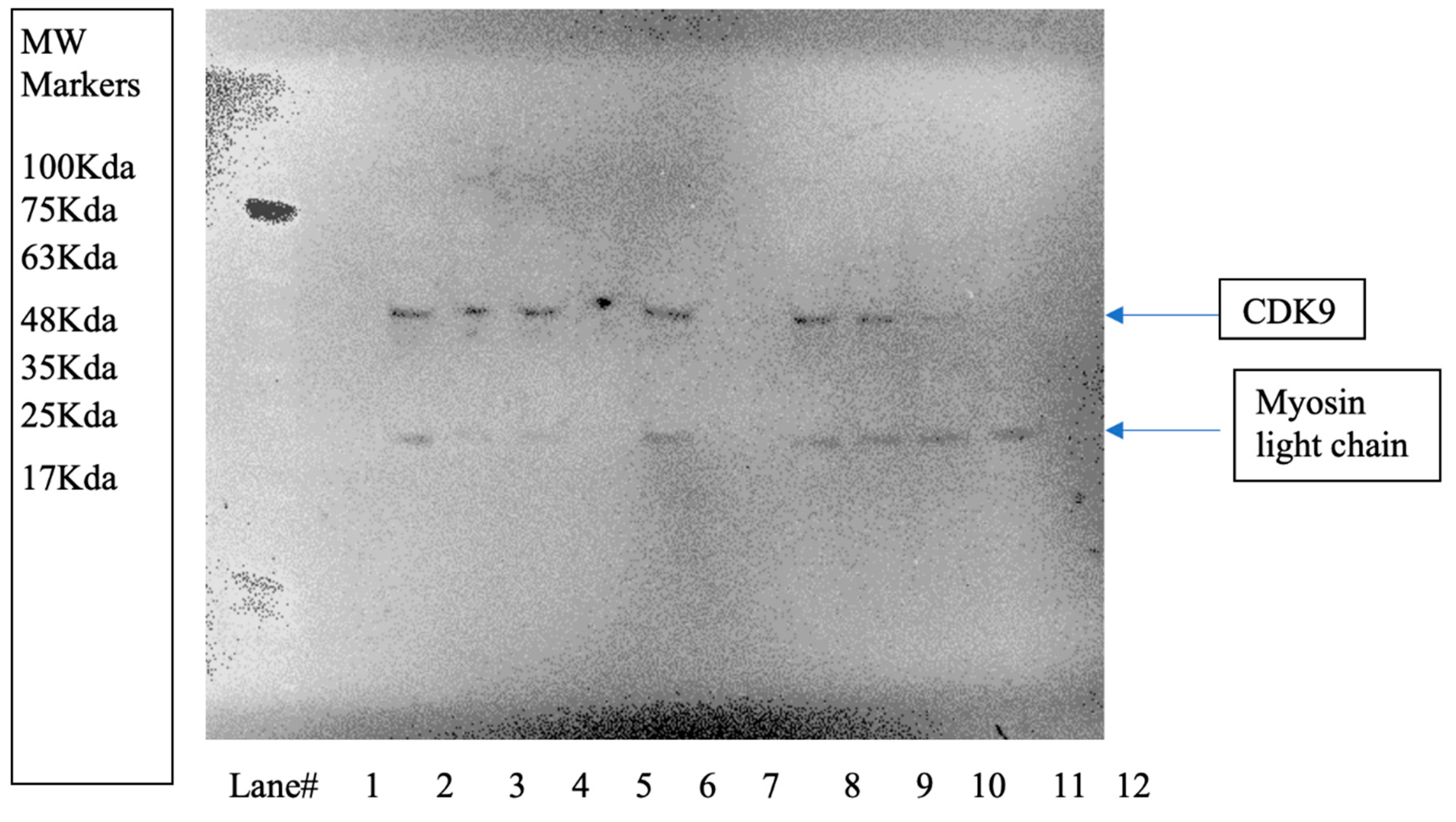
Figure 9.
Cell viability results: a) MIA-PaCa-2; b) NCI H-358.
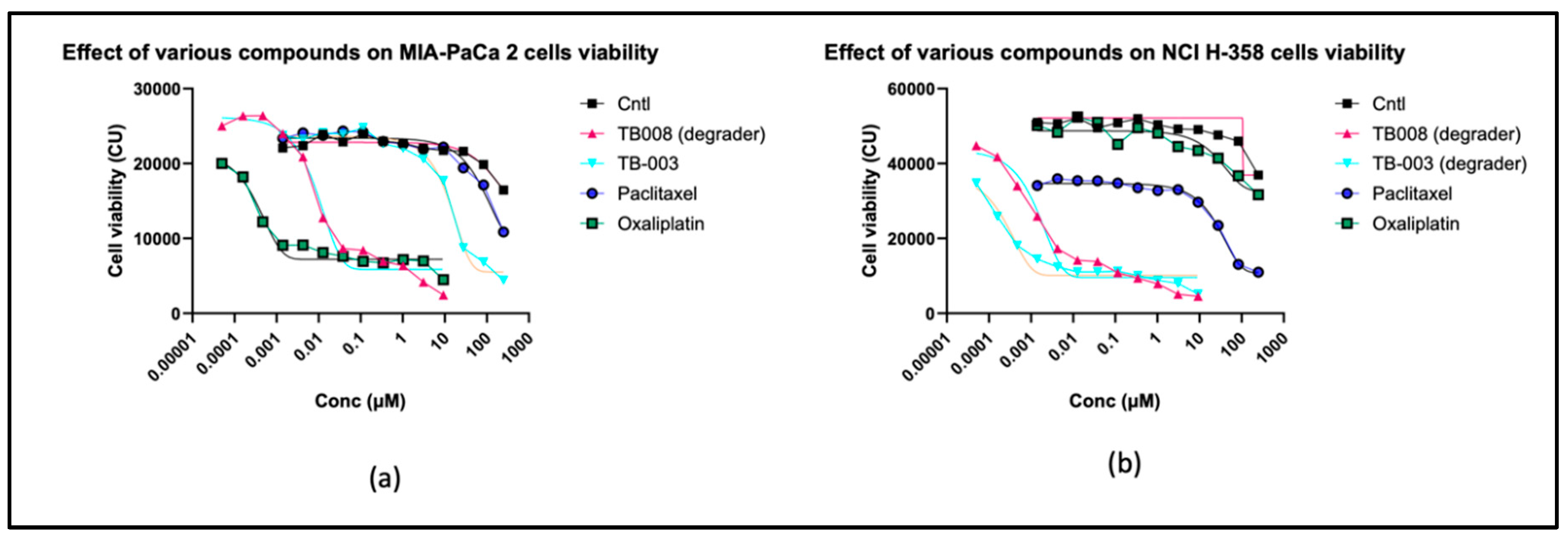

Table 1.
Ligands and CDK9 kinase binding affinity with different docking methods1.
| Ligand Name | Binding affinity (kcal/mol) | Docking Method |
|---|---|---|
| TB003 | -7.8 | Vina |
| TB003 | -8.4 | Smina |
| TB008 | -9.6 | Vina |
| TB008 | -9.8 | Smina |
| TB0016 | -9.7 | Vina |
| TB0016 | -8.8 | Smina |
1: Structural details are not revealed due to ongoing patenting activities.
Table 2.
Evaluation of Pharmacological behavior of compounds.
| Compound | Lipophilicity (LogP) | Absorption | Solubility (LogS) |
|---|---|---|---|
| TB003 | 5.09 (High) | Low | -6.29 (Poorly Soluble) |
| TB008 | 2.75 (Moderate) | High | -3.83 (Soluble) |
| TB0016 | 2.55 (Moderate) | High | -4.28 (Moderately Soluble) |
| TB003 PROTAC | 4.28 (High) | Low | -6.65 (Poorly Soluble) |
| TB008 PROTAC | 3.19 (High) | Low | -5.59 (Moderately Soluble) |
Table 3.
Summary of CDK9 kinase inhibition results2.
| Compound | TB003 | TB0016 | TB008 | |
|---|---|---|---|---|
| Modality | CDK9 degrader | CDK9 ligand | CDK9 degrader | |
| Potency (IC50) | 5nM | > 1µM | 3.5nM | |
| Fold selectivity CDK9 vs other CDK family members | CDK7 | >200 | > 1µM | >500 |
| CDK5 | NT | > 1µM | >500 | |
| CDK4 | NT | > 1µM | >500 | |
| CDK2 | >200 | > 1µM | >500 | |
| CDK1 | >200 | > 1µM | >500 | |
| Route of administration | IP/Oral | IP/Oral | IP/Oral | |
2: This study was done at Thermo-Fisher by the SelectScreen screening and profiling services division.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



