
Submitted:
11 January 2024
Posted:
12 January 2024
You are already at the latest version
Abstract
Antimalarial drug resistance has become a real public health problem despite WHO measures. New sequencing technologies make it possible to investigate genomic variations associated with resistant phenotypes at the genome-wide scale. Based on the use of hemisynthetic nanopores, the PromethION technology from Oxford Nanopore Technologies can produce long-read sequences, in contrast to previous short-read technologies used as gold-standard to sequence Plasmodium. Two clones of P.falciparum (Pf3D7 and PfW2) were sequenced in long-read using the PromethION sequencer from Oxford Nanopore Technologies without genomic amplification. This made it possible to create a processing analysis pipeline for human Plasmodium with ONT Fastq only. De novo assembly revealed N50 lengths of 18,488 kb and 17,502 kb for the Pf3D7 and PfW2, respectively. The genome size was estimated at 23,235,407 base pairs for the Pf3D7 clone and 21,712,038 base pairs for the PfW2 clone. The average genome coverage depth was estimated at 787X and 653X for the Pf3D7 and PfW2 clones, respectively. This study proposes an assembly processing pipeline for the human Plasmodium genome using software’s adapted to large ONT data and the high AT percentage of Plasmodium. This search provides all the parameters, which have been optimized, for use with the software selected in the pipeline.
Keywords:
Genome assembly
; Plasmodium falciparum
; Long-read sequencing
; Nanopore
; PromethION
; Pf3D7
; PfW2
1. Introduction
Despite intensive efforts to eradicate malaria and the development of new combination therapies, it remains endemic in eighty-four countries. According to the 2023 report published by the World Health Organization (WHO), there were 248 million cases of malaria and 609,000 deaths in 2022 [1], compared to 229 million cases and 409,000 deaths in 2019. Since the 2000s, malaria cases have declined due to the development and use of rapid diagnostic tests, impregnated bed nets and new antimalarial drugs, including artemisinin-based combinations such as dihydroartemisinin-piperaquine, artemether-lumefantrine, and artesunate-amodiaquine [1]. However, Plasmodium falciparum has developed resistance to these new drugs, leading to therapeutic ineffectiveness and clinical failure, which constitutes a major public health problem [2,3]. The development of genetic monitoring of parasite genomic variations is one of the most reliable approaches to assess susceptibility decreases and genetic polymorphisms [4].
The genomic knowledge of Plasmodium falciparum has evolved since the first genomic sequencing of the Plasmodium falciparum 3D7 clone took place in 2002 [5]. This clone was provided from the NF54 isolate, recovered in the Netherlands from a malaria case airport, by limiting dilution and is widely used in in vitro studies as a reference clone [6]. The whole genome shotgun sequencing identified a genomic size of 22,853,764 base pairs coding for 5268 genes with 3465 hypothetical proteins. Fourteen chromosomes and two organelles were identified: the apicoplast and the mitochondria. The proportion of nucleotides (A+T) has been estimated at about 80.6% [7]. In 2019, the genome size was updated to 23,292,622 bp with 5280 genes and 1776 hypothetical proteins[5]. By 2023, 5389 proteins had been annotated and 1626 proteins had been annotated as hypothetical proteins for a genome size around 23.33 Mb [8,9]. This sequenced genome is also the only reference genome used in genomics studies. PfW2 was cloned form the Indochina III/CDC isolate, originally derived from Laotian patient who failed chloroquine therapy [10] and no reference genomes are available for this clone. It is also used as reference clone in in vitro studies for its resistance to chloroquine.
Several sequencing tools have been developed, including Illumina sequencers (San Diego, CA, US), that allow read sequencing between 150 and 300 base pairs [11]. Short-read sequencing with Illumina sequencers has become the reference for Plasmodium falciparum genomic studies [12]. However, short-read sequencing can be problematic for de novo assembly, due to genome size and the many repeated regions. Pacbio sequencers have also been used for Plasmodium sequencing, but the gold standard remains illumina technology.
Nanopore technology was introduced in 2014, with the MinION sequencer [13]. This technology allows for long-read sequencing with higher read depth when using the PromethION sequencer [11,13], and also enables high-throughput real-time analysis with a shorter processing time [14].
Since this implementation, moreover, few genomic studies have been published on Plasmodium falciparum due to the accessibility and price of sequencers but also due to the richness of the genome in (A+T) which leads to a higher error rate [14]. These studies focus on nanopore sequencers, such as the MinION, to explore resistance on identified genes, listed as causing antimalarial drug resistance [15,16,17,18].
In addition, most existing Plasmodium pipelines are suitable for illumina short reads like for example GATK4 [19] or pipelines are specialized in other, smaller microorganisms, and incompatible with Plasmodium genomics.
The aim of this study was to sequence, without any genomics amplification, Plasmodium falciparum clones 3D7 and W2 with the ONT technology with the PromethION sequencer in order to de novo assemble their whole genomes by assembling a Plasmodium specific analysis pipeline. The aim was also to demonstrate the feasibility of nanopore whole genome sequencing, considering the high AT richness of Plasmodium, which generally hinders sequencing.
2. Materials and Methods
2.1. Plasmodium falciparum Laboratory Cultures
The two clones (Pf3D7, PfW2) used in this study were obtained from the Malaria Research and Reference Reagent Resource Center (MRA-102 and MRA-157) (BEI resources, Manassas, VA, US).
The laboratory parasitic cultures were maintained in 4.5 ml of RPMI medium (Invitrogen, Paisley, UK) buffered supplemented in 10% of human serum (EFS, France) in 500 µL of human blood (A+, EFS, France). Cultures were maintained under a controlled atmosphere: 37°C, 5% CO2 and 10% O2. Cultures were maintained at high parasitaemia (>80% ring stage) to obtain an adequate amount of genetic material for DNA extraction. The RPMI medium was prepared with RPMI 1640 MEDIUM W/L-Glutamine and 26 ml of Hepes buffer (1M), 26 ml of sodium bicarbonate (7.5%), 3.2 ml of neomycin (10 mg/ml) and 1 ml of hypoxanthine (500 mg/L), orotique acid (250 mg/L) and L-Glutamine. Then 20 mL of 10% D-glucose was added and, finally, was adjusted to a volume of 1 L with ultrapure water.
2.2. DNA Extraction
DNA from each sample was extracted using a QIAamp DNA blood Mini Kit (Qiagen, Hilden. Germany). Briefly, 200 µl of ATL buffer and 40 µl of proteinase k were mixed with 200 µl of each sample prior to a four-hour long incubation at 56°C with agitation not exceeding 1200 rpm for sequencing. Each sample was supplemented with 200 µL of ethanol before adding wash buffer. DNA was eluted in 60 µL of ultrapure water and 1µL was quantified using the Qubit dsDNA high sensitivity kit (Thermo Fisher Scientific) following the manufacturer’s recommendation. DNA fragmentation was controlled with 9 µL of DNA on 0.7% agarose gel prepared with TBE 1x.
2.3. Whole Genome Library
The whole genome sequencing of Plasmodium falciparum clones was performed using the PromethION sequencer. After DNA extraction, without any amplification, a maximum of 1 µg of the DNA template was prepared using the ligation sequencing kit 110 (LSK110) according to manufacturing’s instructions with the following minor modifications. The elution buffer was provided by Qiagen (Qiagen, Hilden. Germany) and the Short Fragment Buffer (SFB) from LSK 110 kit was used to keep all DNA fragments. The ratio of AMPure XP beads (Beckman Coulter) used for DNA purification was adapted at 1:1 for each step where beads were used. The sequencing was performed for 24 hours on R9.4.1 (FLO-PRO002) flow cells with sequencer default parameters and the “super high accuracy basecalling” was selected.
The sequencing time had to be adapted according to the sequencing library quality and the number of reads required at the end of sequencing. Sequencing reports are in additional file S1 and S2.
2.4. Data Analysis
Bioinformatics analysis was performed on the fastq output data from the sequencer according to the pipeline presented in Figure 1 (bash script is in additional file S3). Data quality control was verified using the NanoPlot software (v1.32.1) [20] and data were filtered using the Filtlong tool (v0.2.1) [21] to keep high-quality long reads only (https://github.com/rrwick/Filtlong). The Filtlong parameters were minimum length 5000, keep percent 90, and target bases 20,000,000,000 for Pf3D7 and minimum length 2000, keep percent 90, and target bases 20,000,000,000 for PfW2. These parameters were adjusted according to the sequencing results. Parameters were set up to keep long reads.
2.4.1. Genome Consensus of the Plasmodium falciparum 3D7 Clone
For the Plasmodium falciparum 3D7 clone, long reads were assembled using Flye (v2.8.1) [22] specifying the genome size around 23 Mb and the “--asm-coverage” parameter to 35. The contig quality was checked using Quast (v5.0.2) [23] against the reference genome. Chromosomes were assembled manually using the Plasmodium falciparum 3D7 reference genome with the help of Bedtools (v2.30.0) [24] to polish the contigs. The apicoplast and the mitochondria were assembled in totality with Flye. Flye was chosen because he had excellent results on various organism and is versatile. Also, it has been design to build assembly with error prone reads and is based on k-mer and the graph theory [22,25].
2.4.2. Genome Consensus of the Plasmodium falciparum W2 Clone
For the Plasmodium falciparum W2 clone, long reads were also assembled using Flye (v2.8.1) [22], specifying the genome size at around 23 Mb but the "--asm-coverage" parameter was 300. This parameter was high to allow genome assembly. Since no reference genome was available for this clone, a consensus was created with Minimap2 (v.2.17) (“skeleton genome”) [26,27] (Figure 1).
The raw reads were mapped to the Plasmodium falciparum 3D7 reference genome, and the consensus was extracted using the CLC Genomics Workbench 7.5 (Qiagen) software. This process was performed to facilitate contig repair for chromosome assembly because of the high variability between the two clones. Chromosomes were assembled manually using the CLC consensus genome and Bedtools (v2.30.0) [24]. The apicoplast and the mitochondria were assembled in totality with Flye. This last step could be changed with the GreenHill software [28]. It should be noted that we had already repaired our genome using the technique described here, so we didn’t use the GreenHill software.
2.4.3. Final Consensus of the Two Clones
The assembly was refined for both clones. One refining event (also known as a “polishing step”) was associated with the following six sub-steps: firstly, minimap2 [26,27] was used to map filtered reads to the newly generated assembly resulting in a SAM file. Secondly, the Samtools sort function was used to compress SAM files into the BAM binary format. Thirdly, Bcftools mpileup and, fourthly, Bcftools call were used to identify all variants specifying the “--ploidy 1” parameter and to gather them inside a VCF file [29]. Bcftools view [29] was then used to filter the VCF file on the DP4 parameter with the filtering command: “((DP4[1] + DP4[2]) < (DP4[3] + DP4[4]) && (DP4[3] + DP4[4] > 40)”. Bcftools consensus was used to polish the assembly – that one assembly given to minimap2 in the first sub-step – from the filtered VCF file and to generate a new consensus assembly. To proceed to another refinement step, this consensus assembly could then be given to Minimap2 with the same filtered reads; otherwise, the refining process would be stopped at that point and the resulting consensus would be called the final consensus. That final consensus was, thus, a refined assembly assimilated to a complete genome sequence. This process was performed eleven times for Pf3D7 and twenty times for PfW2.
2.4.5. Genome Annotation
Genome annotations of both clones were performed using BUSCO (v5.4.6) and Metaeuk [30]. The data were uploaded to the Galaxy web platform, and we used the public server at usegalaxy.eu to analyse the data [31]. The BUSCO Plasmodium dataset includes 3642 genes from 23 species of Plasmodium [32]. BUSCO was run in eukaryotic mode.
A Venn diagram was created with BUSCO results and an R script. (All BUSCO data are provided in additional files S3, S4 and S5 and the R script is in the additional file S6).
Whole genomes annotation were also performed with Companion for compared with BUSCO [33]
2.4.6. Apicoplast and Mitochondria Annotation
2.5. Plasmodium falciparum 3D7 Reference Genome Variant Caller
The Filtlong-filtered Nanopore Fastq data was mapped to the Pf3D7 reference genome with the Minimap2 software [26,27]. The variant caller ran using bcftools mpileup (v1.13), bcftools call and bcftools view (v1.13) [29], specifying filter parameters on quality and on the DP4 parameter with the command “((DP4[1] + DP4[2]) < (DP4[3] + DP4[4]) && (DP4[3] + DP4[4] > 40)”. Bcftools stats were then used to create VCFs statistics, and the plot was generated with a R script (in additional file S8).
The same pipeline was used to identify the difference between the Pf3D7 reference and the PfW2 reads sequenced.
3. Results
3.1. Long-Read Sequencing Results
Long-read sequencing with PromethION initially produced reads with an estimated N50 length of 9731 kb for Pf3D7 and 16,305 kb for PfW2 (Table 1). The analyses were performed using NanoPlot from the raw fastq and the filtered fastq with Filtlong. Filtering made it possible to increase the quality score from 14.4 to 17 for the Pf3D7 clone and from 13.9 to 14.5 for the PfW2 clone. For both clones, sequencing was performed for 24 hours, resulting in over 12 million reads for Pf3D7 and over two million reads for PfW2. After filtering, only 1,238,210 reads were retained for Pf3D7 and 1,296,307 for PfW2. The assembly was thus carried out with reads having N50 lengths of 18,448 kb for the Pf3D7 clone and 17,502 kb for the PfW2 clone (Table 1).
3.2. Plasmodium falciparum 3D7 De Novo Assembly
For Pf3D7, the assembly resulted in a total genome length of 23,477,924 base pairs distributed in 32 fragments. The average genome coverage depth was estimated to 787X by Flye (Table 2). Quast estimated the genome size to be 23,330,137 base-pair long, with 48 reported mis-assembly events on Flye out. The genome mapped with the Pf3D7 reference showed up to 99.94% similarity.
After de novo assembly, chromosomes were assembled manually from fragments using the Pf3D7 reference genome as a model and Bedtools. The chromosome quality was then assessed using Quast, ensuring errors were kept to a minimum. The last consensus sequence was obtained after multiple polishing steps involving VCF files. Ultimately, Quast estimated this genome size to 23,235,407 bp with 24 mis-assemblies and the newly built genome fraction mapping with the Pf3D7 reference showed 99.348% similarity.
A final quality check was performed by mapping the filtered reads to both our new Pf3D7 genome and the older reference genome. This revealed that, although similar, the alignment rate was slightly lower than with the reference genome (94.20% identity compared to 94.18% identity, a difference which corresponds to 46,471 bp). This result is discussed in the discussion part.
3.3. Plasmodium falciparum W2 De Novo Assembly
The PfW2 genome assembly resulted in a length of 23,302,768 base pairs distributed in 31 fragments and two scaffolds. The average genome coverage depth was estimated to 653X by Flye (Table 2). Quast results on PfW2 assembly showed that the differences between the two strains were too high to use the Pf3D7 reference genome. A “skeleton” genome was therefore created from the fastq file and the Pf3D7 reference genome, with the help of the CLC Genomics wb7 software. This step was essential to improvement the genome. Chromosomes were assembled manually from fragments using the “skeleton” genome and Bedtools (method described in section number 2). Again, the last consensus sequence was obtained after multiple polishing steps involving the VCF file. The genome length was then estimated to be 21,712,038 bp long (Table 3). A final quality check was performed by mapping the filtered reads to our PfW2 genome. It revealed that 99.14% reads mapped to the genome.
3.4. Genome Depth and Length
The consensus apicoplast and mitochondria for both genomes have similar lengths. However, PfW2 chromosomes lengths are still shorter than that of Pf3D7 (Table 3). Differences in genome size can be explained by the major deletions observed. This result is discussed in discussion part.

Table 3.
Chromosome length and average assembly depth for Pf3D7 and PfW2.
| Pf3D7 | PfW2 | |||
|---|---|---|---|---|
| length | average depth | length | average depth | |
| chromosome 1 | 638,193 | 754.4 ± 64.6 | 621,378 | 946 ± 1041.9 |
| chromosome 2 | 940,408 | 764 ± 95.5 | 942,789 | 731 ± 487.8 |
| chromosome 3 | 1,056,079 | 768.3 ± 56.3 | 955,041 | 669.7 ± 381 |
| chromosome 4 | 1,195,288 | 808.9 ± 161.5 | 969,939 | 749.4 ± 545.6 |
| chromosome 5 | 1,338,524 | 774 ± 81.4 | 1,353,934 | 705.9 ± 264.2 |
| chromosome 6 | 1,412,742 | 784 ± 81.7 | 1,294,864 | 669.7 ± 223.7 |
| chromosome 7 | 1,438,736 | 779.7 ± 59.8 | 1,272,765 | 685.7± 366.4 |
| chromosome 8 | 1,445,520 | 762 ± 68.1 | 1,320,440 | 660.6 ± 334.2 |
| chromosome 9 | 1,534,997 | 765 ± 59.8 | 1,420,162 | 632.8 ± 144.6 |
| chromosome 10 | 1,716,863 | 760.8 ± 56.2 | 1,528,816 | 627.5 ± 62.8 |
| chromosome 11 | 2,029,548 | 769.6 ± 73.7 | 1,894,263 | 620.9 ± 59.5 |
| chromosome 12 | 2,257,511 | 774.8 ± 109.7 | 2,097,388 | 635.9 ± 123.6 |
| chromosome 13 | 2,913,737 | 779.7 ± 126 | 2,786,778 | 624.8 ± 58.8 |
| chromosome 14 | 3,277,058 | 776.9 ± 91.4 | 3,213,288 | 630.5 ± 98.5 |
| apicoplast | 34,237 | 465.8 ± 113.4 | 34,226 | 348.2 ± 80.6 |
| mitochondria | 5966 | 7922.1 ± 151.9 | 5967 | 7823.6 ± 251.4 |
| total length | 23,235,407 | 21,712,038 | ||
3.5. Genome Annotation
For the assembled Pf3D7 consensus sequence, BUSCO identified 2925 complete genes out of the 3642 in the database. Three hundred and five genes were fragmented and 412 were missing (Figure 3). In comparison, for the Pf3D7 reference genome, BUSCO identified 3587 genes out of the 3642 including 2 fragmented genes and 53 missing genes (Figure 4).
For the PfW2 consensus sequence, 2595 complete genes were identified out of the 3642 in its database: 404 genes were fragmented and 643 are missing (Figure 4).
The BUSCO results were compared with one another to identify which genes were common to the clones and the Pf3D7 reference. The Venn diagram shows that 2853 genes are shared between the three clones and 228 genes are present only in the reference. Five genes are present only in the Pf3D7 sequenced and two genes are present only in the PfW2 sequenced (in chromosome 3). Three hundred and sixty-eight genes are shared only by the Pf3D7 clone and the Pf3D7 reference and 140 genes are shared only by the Pf3D7 reference and the PfW2 (Figure 5). All BUSCO data are presented in additional file S3-S5.
The Companion software was also used to compared BUSCO annotation. For Pf3D7, 3477 genes were identified out of 5562 genes of the database and for PfW2 is 1567 genes out of 5562 of the databases. For both, the P.falciparum 3D7 database reference was used. 2535 and 4060 pseudogenes were identified for Pf3D7 and PfW2 respectively. This result is discussed in the discussion part.
3.7. Apicoplast and Mitochondria Annotations
Apicoplast and mitochondria were annotated using the Prokka software in Galaxy. Prokka was choose because hit’s a software specialized in the annotation of prokaryotic genome and can manage circular genome. The two organelles are highly similar to prokaryote genome.
Thirty CDS, 33 tRNA and four rRNA were identified for the newly assembled Pf3D7 apicoplast. As for PfW2, 29 CDS, 33 tRNA and four rRNA were identified.
Three CDS were identified within the mitochondria. These CDS are shared by the three studied Plasmodium.
3.8. Genomic Variability of the Pf3D7 Clone against the Pf3D7 Reference Genome
The sequenced Pf3D7 clone reads were mapped to the reference genome and revealed a very high variability throughout the genome. The variant call format revealed 3719 variants between the reference Pf3D7 genome and the Pf3D7 sequenced reads (Figure 6A), and most variations are in chromosome 12. Besides, most observed substitutions are A>T, C>T and G>A (Figure 6B). For the observed variants, quality was compared with the sequencing depth. Chromosome 12 had a good average quality but low depth. As for the apicoplast, both parameters were low. Other chromosomes shared a high mean depth (between 600X and 700X) and a high average quality (≥ 80) (Figure 6C).
No mutations were observed inside the mitochondria.
3.9. Genomic Variability of the PfW2 Clone against the Pf3D7 Reference Genome
The PfW2 reads sequenced were mapped onto the Pf3D7 reference genome and after a variant call, 100,000 variants were identified between them which represent 0.42%.
4. Discussion
In this study, a bioinformatics assembly pipeline was developed for the human parasite Plasmodium falciparum using only nanopore long-read sequencing from library preparation to de novo genome assembly. The selected software packages were chosen for their compatibility with Nanopore fastq and their ability to analyse Plasmodium falciparum data. Each software parameter is specified for more practical use.
This pipeline enabled the de novo assembly of two whole genomes with a substantial sequencing depth never-archived before. This sequencing depth is necessary to compensate for the sequencing errors of nanopore technology. However, the errors generated during sequencing cannot be ignored and are mainly due to the high AT content (80.6%) of Plasmodium and its sequenced genome size (23 Mb) [7]. Besides, in this study the LSK110 ligation kit was used to prepare the sequencing library and Oxford Nanopore Technologies now recommends using the LSK114 kit ligation chemistry, which would create even fewer sequencing errors. We were also confronted with the bioinformatic limitations of genome repair. Some base pairs in the consensus genome did not match the sequenced reads, despite the many polishing steps performed to generate the consensus. These sequencing errors were highlighted with the VCF files and are in the order of 0.0004% over the whole genome, which remains very low. We shall provide some insight about its potential cause in our discussion of bioinformatics issues. Finally, these errors are counterbalanced by the sequencing depth obtained for the two strains sequenced here, enabling the creation of a robust genome.
One of the main limitations of Oxford Nanopore Technologies is the quantity of DNA required. ONT recommends loading 1 µg of DNA into PromethION Flow Cells. For clinical samples, this protocol therefore needs to be adapted by using filtration columns to remove human DNA according to the protocol presented by Coppée et al. [35], followed by a whole genome amplification for small parasitaemia [36]. This protocol was used to sequenced Plasmodium clinical sample by nanopore adaptive sampling which shows that the technology can be adapted to clinical isolates with very low parasitaemia [37].
With regards to bioinformatics assembly, high peak and low depth regions (Figure 1 and Figure 2) at the start and end of chromosome assemblies correspond respectively to an underestimation or overestimation of repeated-region repeat number. It is a known pattern associated with the presence of telomeres. Such a result was, therefore, expected. Other patterns of sudden increases and falls in depth alongside a chromosome may correspond to a mis-assembly pattern due to incorrect number of repeats for repeated regions. This might be especially true if the depth of this specific region is a multiple of the surrounding region depth.
We can observe that the Pf3D7 and PfW2 clones both have regions that have higher depth of coverage than others. This is particularly visible in the apicoplast. This observation could result from a repeated region with a length or number of repeats which was underestimated by Flye during the assembly. Indeed, the resolution of extensively large, repeated regions is one of the major troubleshooting of de novo assembly. Despite long-read sequencing technology being devised to bypass repeated regions, some regions are still too wide for all repetitions to be encompassed within long reads.
As mentioned above, assemblies were carried out with N50 lengths of 18,448 kb and 17,502 kb for each of the Pf3D7 and PfW2 clones. In Pf3D7, the higher depth apicoplast region corresponds approximately to the 23,750–31,250 bp segment, hence a 7500 base-long segment. This is supposedly shorter than the N50 length, so that, the assembly should have been resolved. However, as the depth in this particular segment is twice as high as the other surrounding regions within the apicoplast, it is most likely that the segment should have been a two-time repeated segment. Hence, the length ranges from 7500 to 15,000 base pairs long, nearing the N50 length. If the assembly software was lacking reads for the resolution of this particular regions – which seems to be the case at the end of the segment, considering the far below average depth (see Figure 2) – it could have opted for an inappropriate alternative assembly, although algorithmically correct assembly.
This type of reasoning can be applied to other regions within the genome. A repeated-region repeat underestimation is also most likely what happened for the ninth chromosome of clone PfW2, as some regions have a near-stagnant depth of 1000X while the majority of the chromosome has a depth of 500X. Hence, some regions should have been duplicated (rather than truncated during the assembly). In contrast, another region is far less represented. As it is enclosed between two underestimated regions (i.e., these regions are missing a copy inside the final assembly), it is most likely that this region was present only in one of the supposed duplicates.
In addition, we can observe that some peaks are very high in other chromosomes. This could indicate a small specific segment in which number of repeats has been greatly underestimated during assembly.
Inside the VCF file, and despite the numerous polishing steps of the assembled genomes, there was a point up to which the mutation did not decrease. This could be explained by the underestimated repetition number of repeated regions. Indeed, for a repeated region that should have been duplicated (but has only one copy inside the assembly), the mapped reads would report around 50% the current base and around 50% another base, which would then be written inside the VCF file for the next polishing step. Each subsequent polishing steps would then only correspond to a switch between the other of the two bases. This would continue indefinitely.
Despite these mis-assembly events, the produced de novo assemblies are still of use. They present at least one copy of the underestimated regions, thus enabling read mapping. Indeed, the alignment of the trimmed reads to the final assembly reached 94.20% for Pf3D7 and 99.14% for PfW2. It could thus be used to compare genomes in reference to these novel assemblies and more modern strains. It may also support exploratory RNA-seq studies and other omics studies.
Unfortunately, we did not used the ILRA pipeline to improve long-read assemblies because the research was published in July 2023 and we assembled our genomes before the publication [38]. Manual repair steps can be replaced by the ILRA pipeline. To develop their pipelines, Ruiz et al used Plasmodium reads sequenced with PacBio technology, and also used two fungal genomes sequenced with ONT technology.
Finally, in this study, the genomic variability of Plasmodium falciparum clone 3D7 was highlighted. This variability may be the result of culture adaptation, which would potentially explain why BUSCO has not identified all the genes. However, it could also be due to a lack of data inside the BUSCO database. A comparison between BUSCO and Companion was also carried out in order to identify the best software for annotation. Both software packages showed limitations in annotation, which could indicate either too much in vitro genomic variability. In fact, many stop codons were found by visualising the genome using IGV software. According to Claessens et al. [39], the in vitro variability has already been observed. Thus, it would most likely be due to laboratory culture adaptation rather than that of an insufficient BUSCO or Companion database. Comparison of the two clones revealed a difference of 100,000 base pairs. This difference can be explained by culture adaptability, also by sequencing errors due to the technology employed as well as genetic drift of the clone.
In future, this pipeline could also produce a hybrid genome with the addition of short reads, produced for example, by Illumina. Using both techniques would improve the robustness of the sequenced genome in a complementary way. Hybrid genome assembly can be useful for clinical isolates presenting therapeutic failures unexplained by currently known molecular markers. Nanopore technology also makes it possible to study DNA methylation profiles, which would enable the study of resistances that are epigenetic rather than genetic [40].
5. Conclusions
This study proposes an assembly processing pipeline from biology to bioinformatics analysis for the human Plasmodium genome. It enables the assembly of complex Plasmodium genomes (80.6% AT) using exclusively ONT long-reads without any genomic amplification. This pipeline is useful for the analysis of clinical isolates, but first filtration should be necessary in laboratory.
However, it might still need to be optimized and adapted to each clinical isolate, meaning that the software parameters and thresholds should be overhauled. In perspective of this work, we could make a hybrid genome composed of illumina and ONT reads.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Additional file 1: Bash script assembly pipeline. This script provides the command lines that can be used in an Ubuntu terminal and the parameters used for each software. Additional file 2: R script for chromosome depth coverage. This R script is used to produce figures 2 and 3 at chromosome depth. Additional file 3: BUSCO annotation result for Pf3D7 assembly. The results were obtained from the Galaxy platform with the Pf3D7 consensus assembly. Additional file 4: BUSCO annotation result for PfW2 assembly. The results were obtained from the Galaxy platform with the PfW2 consensus assembly. Additional file 5: BUSCO annotation result for Pf3D7 reference genome.The results were obtained from the Galaxy platform with the Pf3D7 reference genome. Additional file 6: R script for BUSCO comparison and Ven diagram creation. This R script was used to produce figure 4 by comparing BUSCO data obtained for the three ge-nomes. Additional file 7: R script for VCF analysis. This script was used to produce figure 6.
Author Contributions
Conceptualization, Océane Delandre and Bruno Pradines; Formal analysis, Océane Delandre; Funding acquisition, Bruno Pradines; Investigation, Océane Delandre, Ombeline Lamer, Jean-Marie Loreau, Nasserdine Papa Mze, Isabelle Fonta, Joel Mosnier and Nicolas Gomez; Methodology, Océane Delandre and Ombeline Lamer; Project administration, Bruno Pradines; Resources, Bruno Pradines; Supervision, Bruno Pradines; Validation, Océane Delandre and Bruno Pradines; Writing – original draft, Océane Delandre, Ombeline Lamer and Bruno Pradines; Writing – review & editing, Océane Delandre, Ombeline Lamer, Emilie Javelle and Bruno Pradines. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the Direction Générale de l’Armement [grant no. NBC-2-B-2120].
Institutional Review Board Statement
Not applicable
Informed Consent Statement
Not applicable
Data Availability Statement
This project was registered under the BioProject accession number PRJNA987860 , and the SAMN36493565 (Pf3D7) and SAMN36493566 (PfW2) Biosample accession numbers. The genomes have been deposited in NCBI (987860[BioProject] - Nucleotide - NCBI (nih.gov) (link available on January 2024). Sequencing report, BUSCO annotation results, R scripts, and bash script used in the pipeline are in the additional files.
Acknowledgments
The authors would like to thank the IHU genomics platform (Marseille, France) for access to sequencers and for access to the CLC Genomicswb7 software.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- WHO. World malaria report, 2023.
- Amaratunga C, Witkowski B, Khim N, Menard D, Fairhurst RM. Artemisinin resistance in Plasmodium falciparum. Lancet Infect Dis. 2014;14:449–50.
- Menard, D.; Dondorp, A. Antimalarial Drug Resistance: A Threat to Malaria Elimination. Cold Spring Harb. Perspect. Med. 2017, 7, a025619, . [CrossRef]
- Haldar K, Bhattacharjee S, Safeukui I. Drug resistance in Plasmodium. Nat Rev Microbiol. 2018;16:156–70.
- Böhme, U.; Otto, T.D.; Sanders, M.J.; Newbold, C.I.; Berriman, M. Progression of the canonical reference malaria parasite genome from 2002–2019. Wellcome Open Res. 2019, 4, 58, . [CrossRef]
- Delemarre-van de Waal HA, de Waal FC. [A 2d patient with tropical malaria contracted in a natural way in the Netherlands]. Ned Tijdschr Geneeskd. 1981;125:375–7.
- Gardner, M.J.; Hall, N.; Fung, E.; White, O.; Berriman, M.; Hyman, R.W.; Carlton, J.M.; Pain, A.; Nelson, K.E.; Bowman, S.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511, . [CrossRef]
- Bahl, A.; Brunk, B.; Crabtree, J.; Fraunholz, M.J.; Gajria, B.; Grant, G.R.; Ginsburg, H.; Gupta, D.; Kissinger, J.C.; Labo, P.; et al. PlasmoDB: the Plasmodium genome resource. A database integrating experimental and computational data. Nucleic Acids Res. 2003, 31, 212–215, . [CrossRef]
- Data Set Plasmodium falciparum 3D7 Genome Sequence and Annotation [Internet]. [cited 2023 Jul 21]. Available from: https://plasmodb.org/plasmo/app/record/dataset/DS_1d17c1883c.
- Oduola, A.M.; Weatherly, N.F.; Bowdre, J.H.; Desjardins, R.E. Plasmodium falciparum: Cloning by single-erythrocyte micromanipulation and heterogeneity in vitro. Exp. Parasitol. 1988, 66, 86–95, . [CrossRef]
- Garrido-Cardenas, J.A.; Garcia-Maroto, F.; Alvarez-Bermejo, J.A.; Manzano-Agugliaro, F. DNA Sequencing Sensors: An Overview. Sensors 2017, 17, 588, . [CrossRef]
- LE Roch, K.G.; Chung, D.D.; Ponts, N. Genomics and integrated systems biology in Plasmodium falciparum: a path to malaria control and eradication. Parasite Immunol. 2011, 34, 50–60, . [CrossRef]
- Wang Y, Zhao Y, Bollas A, Wang Y, Au KF. Nanopore sequencing technology, bioinformatics and applications. Nat Biotechnol. 2021;39:1348–65.
- Akoniyon, O.P.; Adewumi, T.S.; Maharaj, L.; Oyegoke, O.O.; Roux, A.; Adeleke, M.A.; Maharaj, R.; Okpeku, M. Whole Genome Sequencing Contributions and Challenges in Disease Reduction Focused on Malaria. Biology 2022, 11, 587, . [CrossRef]
- Cesare M de, Mwenda M, Jeffreys AE, Chirwa J, Drakeley C, Schneider K, et al. Flexible and cost-effective genomic surveillance of P. falciparum malaria with targeted nanopore sequencing [Internet]. bioRxiv; 2023 [cited 2023 Mar 7]. p. 2023.02.06.527333. Available from: https://www.biorxiv.org/content/10.1101/2023.02.06.527333v1.
- Girgis ST, Adika E, Nenyewodey FE, Jnr DKS, Ngoi JM, Bandoh K, et al. Nanopore sequencing for real-time genomic surveillance of Plasmodium falciparum [Internet]. bioRxiv; 2022 [cited 2023 Mar 7]. p. 2022.12.20.521122. Available from: https://www.biorxiv.org/content/10.1101/2022.12.20.521122v1.
- Runtuwene, L.R.; Tuda, J.S.B.; Mongan, A.E.; Makalowski, W.; Frith, M.C.; Imwong, M.; Srisutham, S.; Thi, L.A.N.; Tuan, N.N.; Eshita, Y.; et al. Nanopore sequencing of drug-resistance-associated genes in malaria parasites, Plasmodium falciparum. Sci. Rep. 2018, 8, 8286, . [CrossRef]
- Sabin, S.; Jones, S.; Patel, D.; Subramaniam, G.; Kelley, J.; Aidoo, M.; Talundzic, E. Portable and cost-effective genetic detection and characterization of Plasmodium falciparum hrp2 using the MinION sequencer. Sci. Rep. 2023, 13, 1–11, . [CrossRef]
- Niaré, K.; Greenhouse, B.; Bailey, J.A. An optimized GATK4 pipeline for Plasmodium falciparum whole genome sequencing variant calling and analysis. Malar. J. 2023, 22, 1–11, . [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669, . [CrossRef]
- Wick R. rrwick/Filtlong [Internet]. 2023 [cited 2023 Feb 9]. Available from: https://github.com/rrwick/Filtlong.
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546, . [CrossRef]
- Mikheenko, A.; Prjibelski, A.; Saveliev, V.; Antipov, D.; Gurevich, A. Versatile genome assembly evaluation with QUAST-LG. Bioinformatics 2018, 34, i142–i150, . [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842, . [CrossRef]
- Lin, Y.; Yuan, J.; Kolmogorov, M.; Shen, M.W.; Chaisson, M.; Pevzner, P.A. Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl. Acad. Sci. 2016, 113, 201604560–E8405, . [CrossRef]
- Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 2021, 37, 4572–4574, . [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100, . [CrossRef]
- Ouchi, S.; Kajitani, R.; Itoh, T. GreenHill: a de novo chromosome-level scaffolding and phasing tool using Hi-C. Genome Biol. 2023, 24, 162, . [CrossRef]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993, . [CrossRef]
- Karin EL, Mirdita M, Söding J. MetaEuk—sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome [Internet]. 2020 [cited 2023 Jul 25];8. Available from: https://link.springer.com/epdf/10.1186/s40168-020-00808-x.
- The Galaxy Community; Afgan, E.; Nekrutenko, A.; Blankenberg, D.; Goecks, J.; Schatz, M.C.; E Ostrovsky, A.; Mahmoud, A.; Lonie, A.J.; Syme, A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351, . [CrossRef]
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2.
- Steinbiss, S.; Silva-Franco, F.; Brunk, B.; Foth, B.; Hertz-Fowler, C.; Berriman, M.; Otto, T.D. Companion: a web server for annotation and analysis of parasite genomes. Nucleic Acids Res. 2016, 44, W29–W34, . [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; A Simão, F.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654, . [CrossRef]
- Coppée, R.; Mama, A.; Sarrasin, V.; Kamaliddin, C.; Adoux, L.; Palazzo, L.; Ndam, N.T.; Letourneur, F.; Ariey, F.; Houzé, S.; et al. 5WBF: a low-cost and straightforward whole blood filtration method suitable for whole-genome sequencing of Plasmodium falciparum clinical isolates. Malar. J. 2022, 21, 1–13, . [CrossRef]
- Oyola, S.O.; Ariani, C.V.; Hamilton, W.L.; Kekre, M.; Amenga-Etego, L.N.; Ghansah, A.; Rutledge, G.G.; Redmond, S.; Manske, M.; Jyothi, D.; et al. Whole genome sequencing of Plasmodium falciparum from dried blood spots using selective whole genome amplification. Malar. J. 2016, 15, 1–12, . [CrossRef]
- De Meulenaere, K.; Cuypers, W.L.; Gauglitz, J.M.; Guetens, P.; Rosanas-Urgell, A.; Laukens, K.; Cuypers, B. Selective whole-genome sequencing of Plasmodium parasites directly from blood samples by nanopore adaptive sampling. mBio 2023, 15, e0196723, . [CrossRef]
- Ruiz, J.L.; Reimering, S.; Escobar-Prieto, J.D.; Brancucci, N.M.B.; Echeverry, D.F.; I Abdi, A.; Marti, M.; Gómez-Díaz, E.; Otto, T.D. From contigs towards chromosomes: automatic improvement of long read assemblies (ILRA). Briefings Bioinform. 2023, 24, . [CrossRef]
- Claessens, A.; Stewart, L.B.; Drury, E.; Ahouidi, A.D.; Amambua-Ngwa, A.; Diakite, M.; Kwiatkowski, D.P.; Awandare, G.A.; Conway, D.J. Genomic variation during culture adaptation of genetically complex Plasmodium falciparum clinical isolates. Microb. Genom. 2023, 9, 001009, . [CrossRef]
- Shim J, Kim Y, Humphreys GI, Nardulli AM, Kosari F, Vasmatzis G, et al. Nanopore-Based Assay for Detection of Methylation in Double-Stranded DNA Fragments [Internet]. ACS Publications. American Chemical Society; 2015 [cited 2023 Jul 17]. Available from: https://pubs.acs.org/doi/pdf/10.1021/nn5045596.
Figure 1.
Assembly processing pipeline of Plasmodium falciparum Oxford Nanopore long reads sequencing. (Bash script is in additional file 1). Raw assembly (Sky blue); optional, non automized assembly optimisation step (Grey); Refinement processing (Green): The step should be processed as long as it reduces the reported mutations number (a repetition corresponds to following the green dotted arrow pathway). Quality control (Orange): to help set the parameters and thresholds. Optional skeleton build (Navy Blue): if no satisfying reference exist for your strain.
Figure 1.
Assembly processing pipeline of Plasmodium falciparum Oxford Nanopore long reads sequencing. (Bash script is in additional file 1). Raw assembly (Sky blue); optional, non automized assembly optimisation step (Grey); Refinement processing (Green): The step should be processed as long as it reduces the reported mutations number (a repetition corresponds to following the green dotted arrow pathway). Quality control (Orange): to help set the parameters and thresholds. Optional skeleton build (Navy Blue): if no satisfying reference exist for your strain.
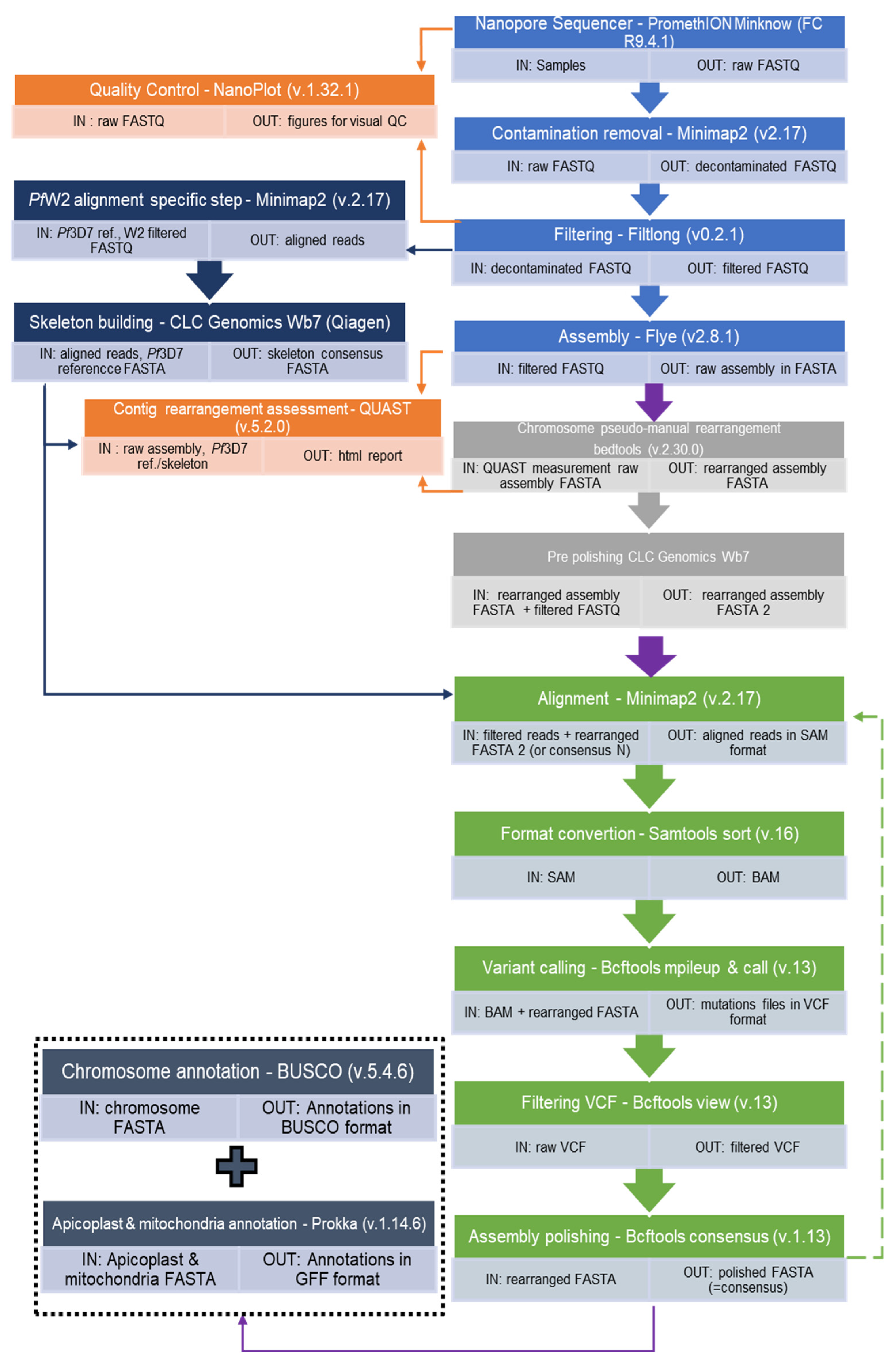
Figure 2.
Sequencing depth of Plasmodium falciparum 3D7 clone genome assembly for the 14 chromosomes and the apicoplast and the mitochondria. The figure was realized with Samtools depth and R script (in additional file S2).The mean depth per chromosomes : 3D7_chromosome_1 (754.4 ± 64.6), 3D7_chromosome_2 (764 ± 95.5), 3D7_chromosome_3 (768.3 ± 56.3), 3D7_chromosome_4 (808.9 ± 161.5), 3D7_chromosome_5 (774 ± 81.4), 3D7_chromosome_6 (784 ± 81.7), 3D7_chromosome_7 (779.7 ± 59.8), 3D7_chromosome_8 (762 ± 68.1), 3D7_chromosome_9 (765 ± 59.8), 3D7_chromosome_10 (760.8 ± 56.2), 3D7_chromosome_11 (769.6 ± 73.7), 3D7_chromosome_12 (774.8 ± 109.7), 3D7_chromosome_13 (779.7 ± 126), 3D7_chromosome_14 (776.9 ± 91.4), 3D7_apicoplaste (465.8 ± 113.4), 3D7_mitochondria (7922.1 ± 151.9).
Figure 2.
Sequencing depth of Plasmodium falciparum 3D7 clone genome assembly for the 14 chromosomes and the apicoplast and the mitochondria. The figure was realized with Samtools depth and R script (in additional file S2).The mean depth per chromosomes : 3D7_chromosome_1 (754.4 ± 64.6), 3D7_chromosome_2 (764 ± 95.5), 3D7_chromosome_3 (768.3 ± 56.3), 3D7_chromosome_4 (808.9 ± 161.5), 3D7_chromosome_5 (774 ± 81.4), 3D7_chromosome_6 (784 ± 81.7), 3D7_chromosome_7 (779.7 ± 59.8), 3D7_chromosome_8 (762 ± 68.1), 3D7_chromosome_9 (765 ± 59.8), 3D7_chromosome_10 (760.8 ± 56.2), 3D7_chromosome_11 (769.6 ± 73.7), 3D7_chromosome_12 (774.8 ± 109.7), 3D7_chromosome_13 (779.7 ± 126), 3D7_chromosome_14 (776.9 ± 91.4), 3D7_apicoplaste (465.8 ± 113.4), 3D7_mitochondria (7922.1 ± 151.9).
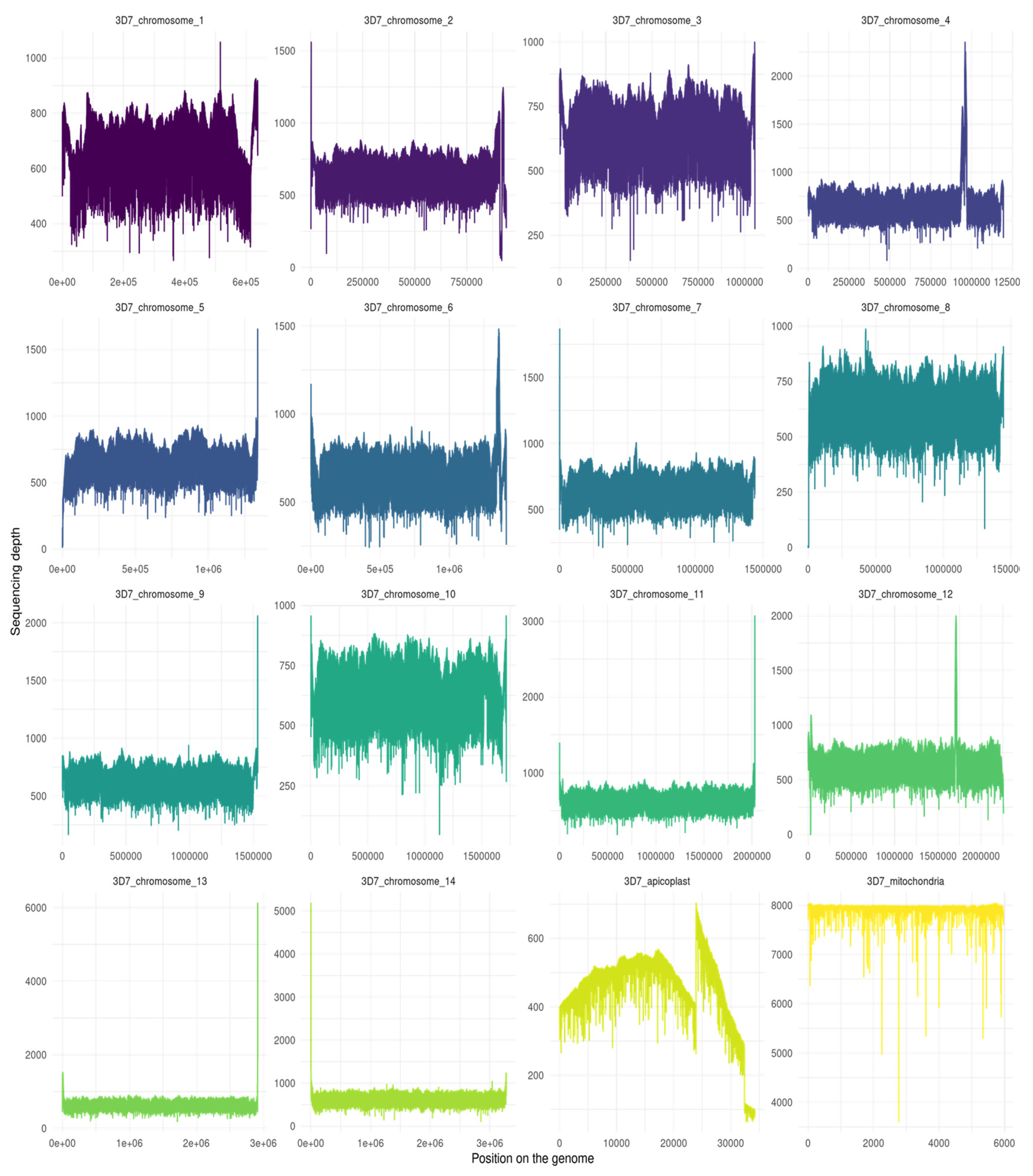
Figure 3.
Sequencing depth of Plasmodium falciparum W2 clone genome assembly for the 14 chromosomes and the apicoplast and the mitochondria. The figure was realized with Samtools depth and R script (in additional file S2). The mean depth per chromosome : PfW2_chromosome_1 (946 ± 1041.9), PfW2_chromosome_2 (731 ± 487.8), PfW2_chromosome_3 (669.7 ± 381), PfW2_chromosome_4 (749.4 ± 545.6), PfW2_chromosome_5 (705.9 ± 264.2), PfW2_chromosome_6 (669.7 ± 223.7), PfW2_chromosome_7 (685.7± 366.4), PfW2_chromosome_8 (660.6 ± 334.2), PfW2_chromosome_9 (632.8 ± 144.6), PfW2_chromosome_10 (627.5 ± 62.8), PfW2_chromosome_11 (620.9 ± 59.5), PfW2_chromosome_12 (635.9 ± 123.6), PfW2_chromosome_13 (624.8 ± 58.8), PfW2_chromosome_14 (630.5 ± 98.5), PfW2_apicoplaste (348.2 ± 80.6), PfW2_mitochondria (7823.6 ± 251.4).
Figure 3.
Sequencing depth of Plasmodium falciparum W2 clone genome assembly for the 14 chromosomes and the apicoplast and the mitochondria. The figure was realized with Samtools depth and R script (in additional file S2). The mean depth per chromosome : PfW2_chromosome_1 (946 ± 1041.9), PfW2_chromosome_2 (731 ± 487.8), PfW2_chromosome_3 (669.7 ± 381), PfW2_chromosome_4 (749.4 ± 545.6), PfW2_chromosome_5 (705.9 ± 264.2), PfW2_chromosome_6 (669.7 ± 223.7), PfW2_chromosome_7 (685.7± 366.4), PfW2_chromosome_8 (660.6 ± 334.2), PfW2_chromosome_9 (632.8 ± 144.6), PfW2_chromosome_10 (627.5 ± 62.8), PfW2_chromosome_11 (620.9 ± 59.5), PfW2_chromosome_12 (635.9 ± 123.6), PfW2_chromosome_13 (624.8 ± 58.8), PfW2_chromosome_14 (630.5 ± 98.5), PfW2_apicoplaste (348.2 ± 80.6), PfW2_mitochondria (7823.6 ± 251.4).
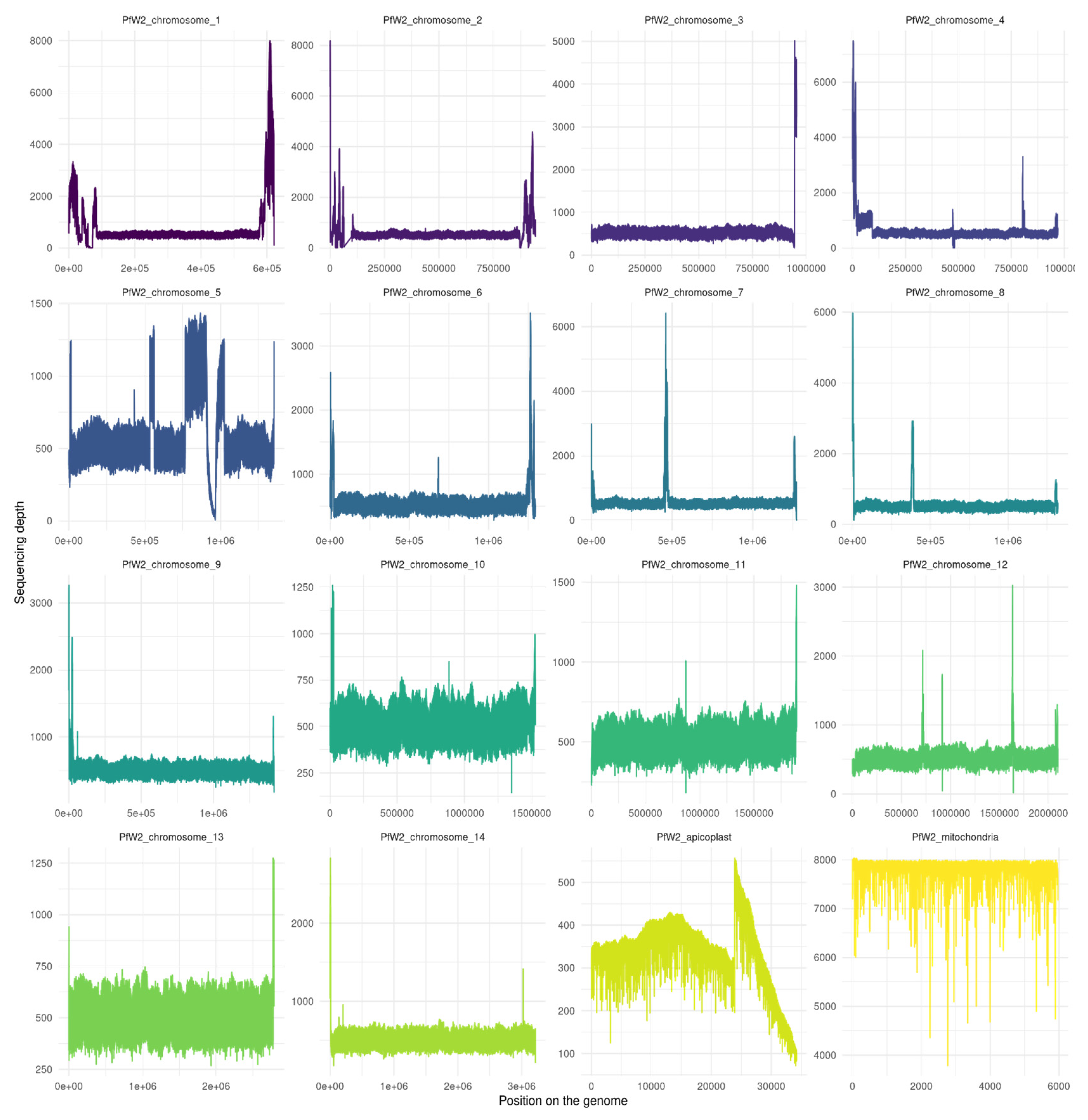
Figure 4.
BUSCO completeness results. Pf3D7, PfW2 consensus genomes and the Pf3D7 reference genome are used. The three genomes were annotated with BUSCO and Metaeuk on Galaxy platform [31]. The figure was realized with the BUSCO.py script [32].
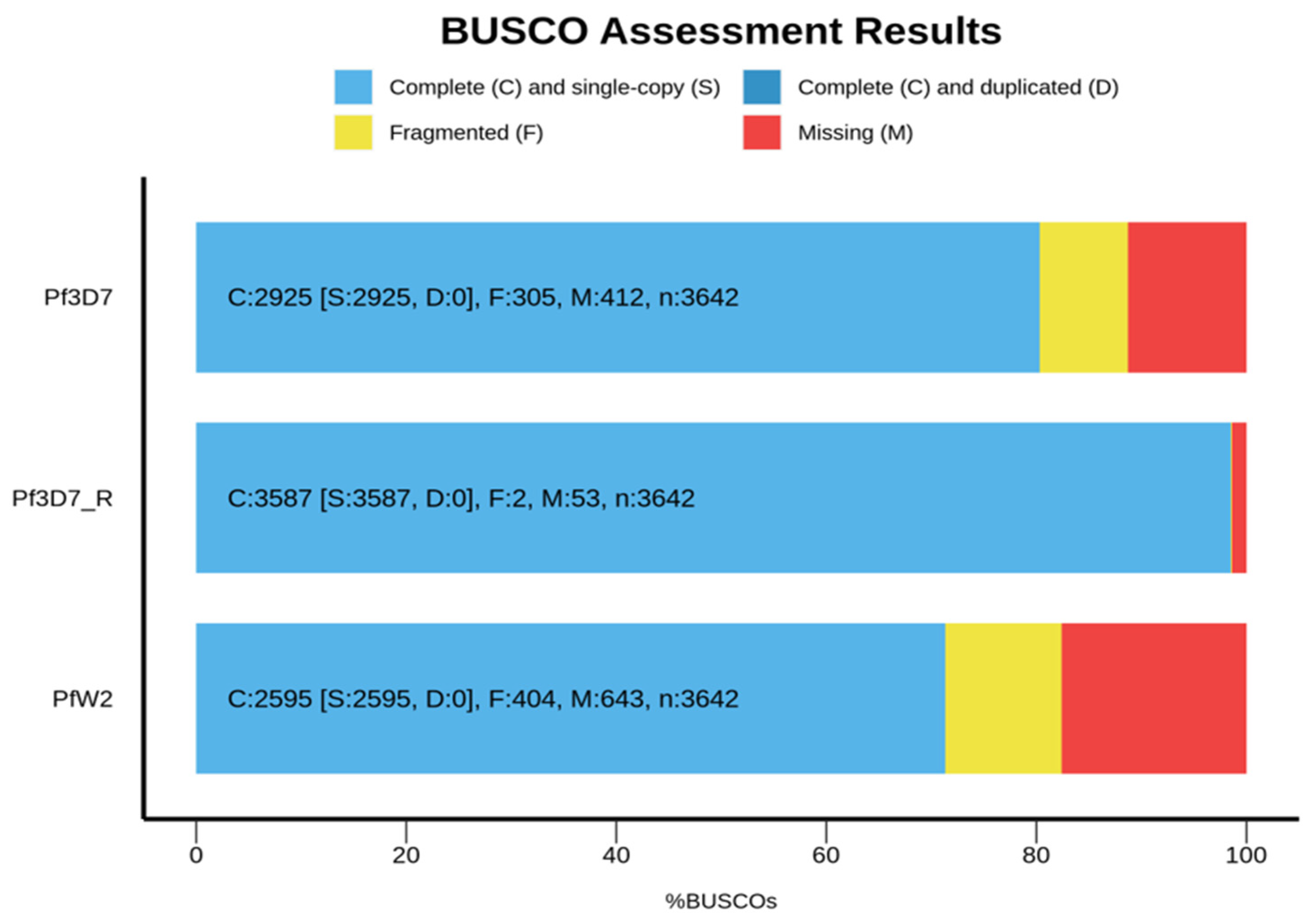
Figure 5.
Venn diagram showing the number distribution of shared genes between the three Plasmodium falciparum clones. The Venn diagram shows the genes shared by the three strains, whether fragmented or complete. Based exclusively on BUSCO data. The Pf3D7r is the reference genome. Missing genes were not represented. (R script is in additional file S6).
Figure 5.
Venn diagram showing the number distribution of shared genes between the three Plasmodium falciparum clones. The Venn diagram shows the genes shared by the three strains, whether fragmented or complete. Based exclusively on BUSCO data. The Pf3D7r is the reference genome. Missing genes were not represented. (R script is in additional file S6).
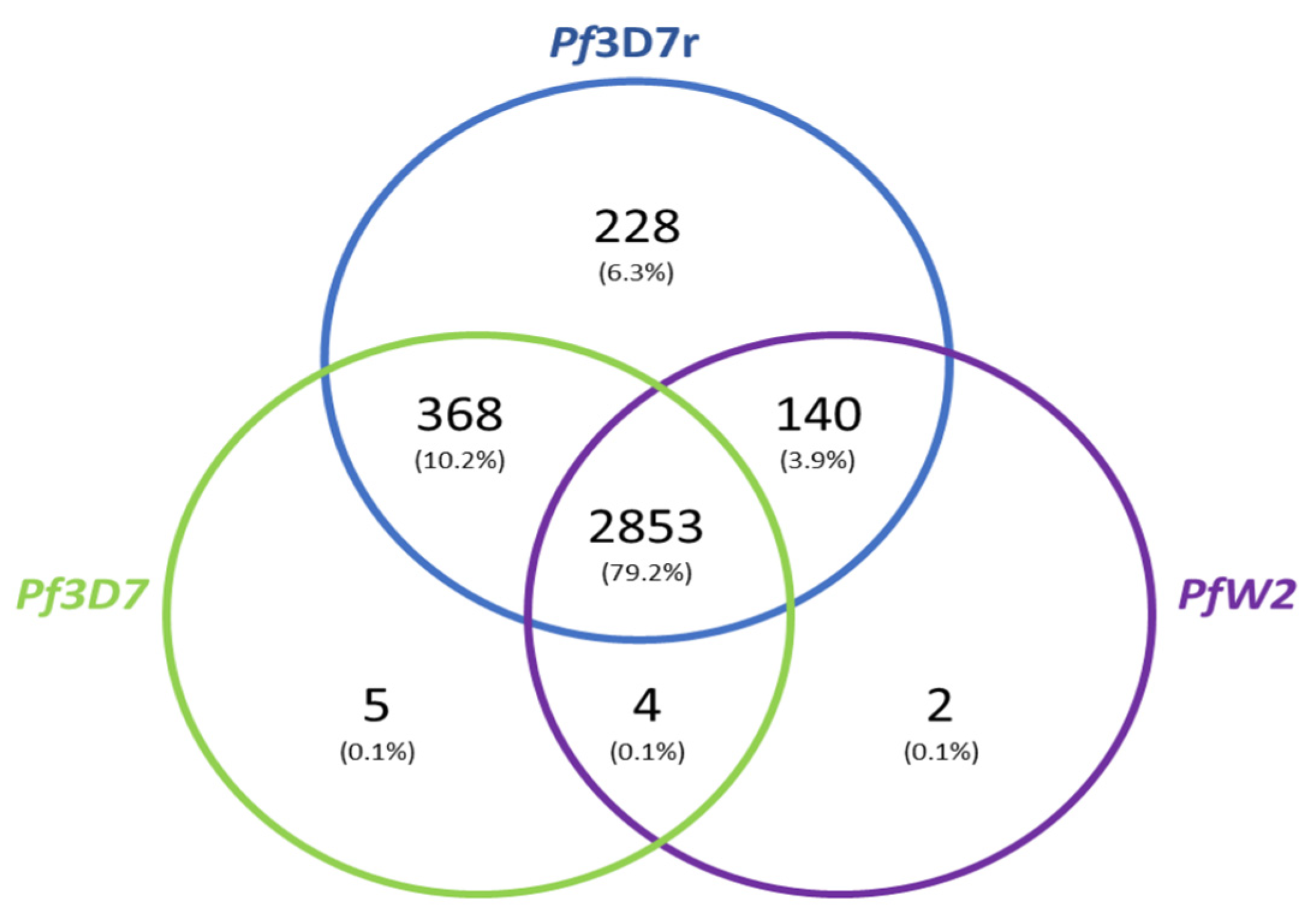
Figure 6.
Variability between the Pf3D7 clone sequencing and the Pf3D7 reference genome. A. Variants observed chromosome by chromosome. The plot was generated with the filtered VCF file (R script is in additional file S7). Each number (1-14) corresponds to a chromosome (1-14) and “A” to the apicoplast. No variant was observed in the mitochondria. B. Number of SNP substitution for whole genome of Pf3D7 genome reference C. Mean quality versus mean depths for the variants observed.
Figure 6.
Variability between the Pf3D7 clone sequencing and the Pf3D7 reference genome. A. Variants observed chromosome by chromosome. The plot was generated with the filtered VCF file (R script is in additional file S7). Each number (1-14) corresponds to a chromosome (1-14) and “A” to the apicoplast. No variant was observed in the mitochondria. B. Number of SNP substitution for whole genome of Pf3D7 genome reference C. Mean quality versus mean depths for the variants observed.
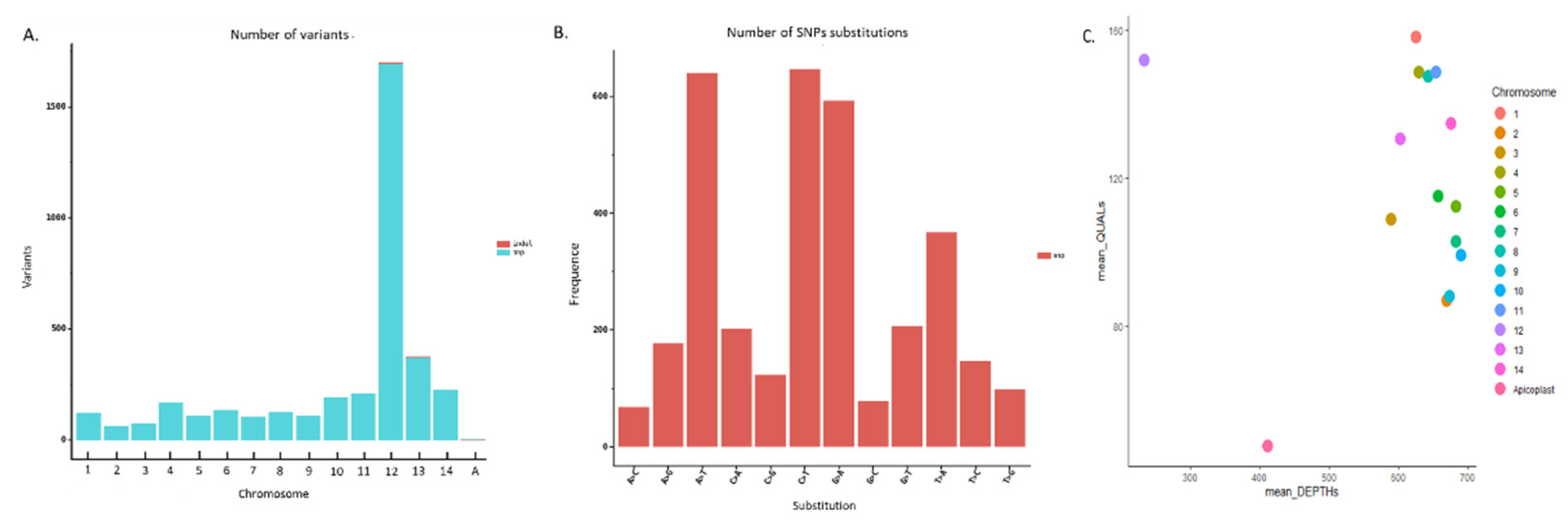
Table 1.
NanoPlot data for Pf3D7 and PfW2 clones. NanoPlot analyses were performed on the fastq files from the sequencer output and from the fastq files filtered by the Filtlong software (filter parameters are in the method section). (Pf3D7: Plasmodium falciparum 3D7 clone; PfW2: Plasmodium falciparum W2 clone; *data are in kb).
Table 1.
NanoPlot data for Pf3D7 and PfW2 clones. NanoPlot analyses were performed on the fastq files from the sequencer output and from the fastq files filtered by the Filtlong software (filter parameters are in the method section). (Pf3D7: Plasmodium falciparum 3D7 clone; PfW2: Plasmodium falciparum W2 clone; *data are in kb).
| Nanoplot data | Pf3D7 reads | Pf3D7 filtered reads | PfW2 reads | PfW2 filtered reads |
|---|---|---|---|---|
| Mean read length* | 3440.2 | 15,843.6 | 7701.3 | 11,600.2 |
| Mean read quality | 14.4 | 17.0 | 13.9 | 14.5 |
| Median read length* | 1,037.0 | 12,363.0 | 4,068.0 | 7,903.0 |
| Median read quality | 14.1 | 17.1 | 14.0 | 14.6 |
| Number of reads | 12,854,191.0 | 1,238,210.0 | 2,192,635.0 | 1,296,307.0 |
| Read length N50* | 9731 | 18,448 | 16,305 | 17,502 |
| Total bases | 44,221,405,060.0 | 19,617,651,842.0 | 16,886,156,110.0 | 15,037,370,421.0 |
Table 2.
De novo assembly data’s for Pf3D7 and PfW2 clones. De novo assembly were performed using the Flye software (all parameters are in the method section).
Table 2.
De novo assembly data’s for Pf3D7 and PfW2 clones. De novo assembly were performed using the Flye software (all parameters are in the method section).
| Pf3D7 | PfW2 | |
|---|---|---|
| Total length | 23,477,924 | 23,302,768 |
| Fragments | 32 | 31 |
| Fragments N50 | 1,265,374 | 1,700,513 |
| Largest fragments | 3,284,512 | 3,249,547 |
| Scaffolds | 0 | 2 |
| Mean coverage | 787 | 653 |
| N50 (Kb) | 18,488 | 17,502 |
| N90 | 8461 | 5277 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



