
Submitted:
12 January 2024
Posted:
12 January 2024
You are already at the latest version
Abstract
Newly designed pentacyclic benzimidazole derivatives, featuring amino or amido side chains, were synthesized to assess their in vitro antiproliferative activity. Additionally, we conducted investigations into their direct interaction with nucleic acids, aiming to uncover potential mechanisms of biological action. These compounds were synthesized using conventional organic synthesis methodologies, alongside photochemical and microwave-assisted reactions. Upon synthesis, the newly derived compounds underwent in vitro testing for their antiproliferative effects on various human cancer cell lines. Notably, derivatives 6 and 9 exhibited significant antiproliferative activity within the submicromolar concentration range. Our biological assessment highlighted that the biological activity was strongly influenced by both the position of the N atom on the quinoline moiety and the position and nature of the side chain on the pentacyclic skeleton. Findings from fluorescence, circular dichroism spectroscopy, and thermal melting assays indicated a mixed binding mode—comprising intercalation and the binding of aggregated compounds along the polynucleotide backbone—of these pentacyclic benzimidazoles with DNA and RNA.
Keywords:
antiproliferative activity
; amines
; amides
; benzimidazoles
; interaction with DNA
1. Introduction
Nowadays, nitrogen heterocycles play an essential role as fundamental structures in numerous synthetic and natural pharmacological compounds. They are frequently utilized in the strategic development of new biologically active molecules [1,2,3]. These compounds exhibit diverse biological characteristics, making them pivotal elements in constructing innovative therapeutic agents [4,5]. Recently, there has been a growing focus on producing fused, benzannulated benzimidazole derivatives due to their demonstrated significance in natural, medicinal, and environmental sciences [6,7]. Cyclic benzimidazoles possess a highly conjugated, flat chromophore with outstanding spectroscopic properties, which are not only vital in medicinal chemistry but also advantageous for various other fields [8]. Due to their structural resemblance to naturally occurring purines, benzimidazole derivatives possess the capability to readily engage with biomolecules such as DNA, RNA, or proteins within living systems [9]. Among the most commonly used classes of chemotherapeutic agents are those that directly interact with DNA [10]. Numerous critical intracellular processes, including transcription, regulation, and translation, rely on the interaction of small molecules with nucleic acid structures [11,12], thereby expanding the broad applications of small molecules in cancer therapy [13]. Aside from covalent bonding, various non-covalent reversible binding mechanisms exist, including intercalation, minor and major groove binding, electrostatic interactions with the nucleic acid backbone, among others [14].
Given that quinoline nuclei serve as crucial pharmacophores in medicinal chemistry, forming a structural component in a myriad of alkaloids and synthetic molecules with diverse biological functions [15,16,17], we have recently synthesized a series of compounds. These include tetracyclic quinoline-fused benzimidazoles (Figure 1a) [18,19,20], tetracyclic imidazo[4,5-b]pyridine derivatives (Figure 1b) [21,22], and pentacyclic benzo[b]thieno[2,3-b]pyrido-[1,2-a]benzimidazoles (Figure 1c) [23]. These compounds exhibited significant antiproliferative activity, indicating substantial potential for enhancing such frameworks to develop superior antitumor agents. Through comprehensive studies involving cytostatic assessment, analysis of DNA/RNA interactions, inhibition of topoisomerase I and II, and proteomic profiling, we confirmed the impact of the substituent type (amidino, amido, cyano) and its position on the tetracyclic and pentacyclic structures on their biological activity and mode of action.
Benzimidazo[1,2-a]quinolines and their heteroaromatic counterparts, featuring positively charged amidino substituents, exhibited notable selectivity toward colon carcinoma cells within submicromolar inhibitory concentration ranges, along with the ability to intercalate into double-stranded DNA or RNA [24,25]. Moreover, we synthesized 2-amino-, 5-amino-, and 2,5-diaminobenzimidazo[1,2-a]quinoline-6-carbonitriles, varying the lengths of secondary or tertiary amino chains linked to the tetracyclic framework, significantly influencing their anti-tumoral activity. Derivatives with N,N-dimethylaminopropyl substitutions displayed heightened antiproliferative activity, unequivocally establishing them as potent DNA-binders and intercalative agents. The nature and length of the amino side chain notably impacted cellular uptake and nuclear targeting [18,19,20,21,22,23]. Pentacyclic derivatives containing piperazinyl nuclei also demonstrated robust antiproliferative activity. Translocating amino side chains from position 3 to 6 marginally enhanced antitumoral activity. Conversely, for tetracyclic benzimidazoles, derivatives with amino side chains at position 2 exhibited superior activity [18,19,20].
Considering the significant biological promise of benzannulated benzimidazoles, we have devised and prepared novel pentacyclic derivatives to serve as potent and innovative antiproliferative agents. These compounds were modified with diverse amino and amido side chains situated at various positions within the skeleton. Furthermore, we explored the influence of the N atom position on the quinoline nuclei. Additionally, we investigated the DNA/RNA binding mechanisms of the most active compounds.
2. Results and Discussion
2.1. Chemistry
All targeted amino-substituted compounds at positions 6-9 and 19-22, as well as amido-substituted compounds at positions 25-26 and 29-30 in the pentacyclic benzimidazole derivatives, along with their corresponding intermediates, were synthesized using two outlined synthetic procedures in Scheme 1 and Scheme 2.
The synthesis of 7-amino substituted pentacyclic derivatives commenced from commercially available 2-chloro-3-quinolinecarbonyl chloride 1. This compound underwent condensation with 2-cyanomethylbenzimidazole 2, yielding the corresponding acyclic hydroxyl-substituted acrylonitrile derivative 3 with a good reaction yield of 76% [26]. Upon thermal cyclization with t-KOBu in DMF, the 7-oxo substituted pentacyclic derivative 4 was obtained with an 83% yield. Subsequently, derivative 4 was successfully transformed into the 7-chloro substituted derivative 5 (yield: 12%), serving as the primary precursor for synthesizing the 7-amino substituted targeted compounds 6-9. Using an uncatalyzed microwave-assisted amination method with the respective amines in acetonitrile at 170°C (800 W), the corresponding 7-amino-6-cyano substituted pentacyclic benzimidazoles 6-9 were prepared, yielding moderate results ranging from 83% to 32%. The successful substitution of chlorine with the amine side chain was confirmed through NMR spectra, revealing proton signals associated with amino groups, methine, methylene, and methyl groups within the aliphatic portion, observable in both the 1H (3.91-1.80 ppm) and 13C NMR spectra (54.5-19.6 ppm).
To synthesize the 11-amino substituted pentacyclic derivatives 19-22, the synthetic pathway initiated with commercially available 3-quinolinecarboxaldehyde 10. This compound underwent condensation with 2-cyanomethylbenzimidazole 2, resulting in the formation of corresponding acyclic acrylonitrile 13 with a moderate reaction yield of 76%. Through thermal cyclization in sulfolane at 300°C, the primary product obtained was the 11-fluoro substituted pentacyclic derivative 16, serving as the main precursor, synthesized with a good yield of 78%. Following the previously mentioned uncatalyzed microwave-assisted amination technique, the corresponding 11-amino substituted pentacyclic derivatives 19-22 were synthesized, yielding in the range of low to moderate (8-67%) yields.
Additionally, two distinct synthetic methods were employed for preparing the targeted 6-amido substituted pentacyclic derivatives. These methods differed in the positioning of the nitrogen atom within the quinoline moiety in the pentacyclic skeleton. To synthesize the 6-amido substituted benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridines 25-26, the previously mentioned synthesis commencing from chloro-substituted 3-quinolinecarboxaldehyde 11 was utilized to prepare acyclic 14 and the pentacyclic substituted 6-cyano substituted derivative 17.
Through acidic hydrolysis using concentrated sulfuric acid, the corresponding carboxylic acid 23 was successfully obtained with a commendable reaction yield of 97%. This carboxylic acid was then employed in the reaction with thionyl chloride to yield pentacyclic carbonyl chloride 24 [27]. Subsequent reactions with suitable amines in dichloromethane (DCM) resulted in the synthesis of targeted pentacyclic amides 25-26, although achieved in relatively low reaction yields.
For the synthesis of the alternative pentacyclic regioisomers, specifically the 7-amido substituted benzo[g]benzo[4,5]imidazo[1,2-a][1,5]-naphthyridines 29-30, the initial synthetic step involved preparing unsubstituted acyclic acrylonitrile 15 from quinoline-3-carbaldehyde, yielding a good reaction yield of 82%. The cyano-substituted cyclic derivative 18 was synthesized via photochemical cyclization in ethanol under irradiation for 4 hours, resulting in a 72% yield. Using the previously mentioned standard synthetic techniques, acidic hydrolysis and reaction with thionyl chloride, pentacyclic carbonyl chloride 28 was obtained with a moderate yield. The structural confirmation of both pentacyclic carbonyl chlorides 24 and 28 relied solely on IR spectroscopy TCL chromatograpy. Following the reaction with appropriate amines in dimethylformamide (DMF), 7-amido substituted benzo[g]benzo[4,5]imidazo[1,2-a][1,5]-naphthyridines 29-30 were obtained with moderate reaction yields. Structural verification of all synthesized compounds was carried out using 1H and 13C NMR spectroscopy, along with mass spectrometry (MS), high-resolution mass spectrometry (HRMS), elemental analysis, and UV/Vis spectroscopy. The structural characterization demonstrated that the observed chemical shifts in the 1H and 13C NMR spectra, as well as the H–H coupling constants, were consistent with the proposed structures. The formation of pentacyclic structures was confirmed by the disappearance of the signal associated with the proton of the NH group on the benzimidazole nuclei. Both photochemical and thermal cyclization processes were monitored using UV/Vis spectroscopy.
2.2. Biological activity
2.2.1. Antiproliferative activity in vitro
The in vitro antiproliferative potential of the synthesized pentacyclic benzimidazole derivatives, whether amino or amido substituted, was evaluated across a diverse panel of cancer cell lines, encompassing pancreatic adenocarcinoma (Capan-1), chronic myeloid leukemia (Hap-1), colorectal carcinoma (HCT-116), lung carcinoma (NCI-H460), acute lymphoblastic leukemia (DND-41), acute myeloid leukemia (HL-60), chronic myeloid leukemia (K-562), and non-Hodgkin lymphoma (Z-138). The obtained results are presented in Table 1, demonstrating the IC50 values (50% inhibitory concentration). Etoposide, a widely used chemotherapeutic and topoisomerase II inhibitor, was included as a reference compound. Comparatively, certain derivatives exhibited higher antiproliferative activity against specific cancer cell lines when juxtaposed with the reference molecule. Table 1 highlights several compounds demonstrating notable and selective antiproliferative effects, with inhibitory concentrations in the submicromolar range. While most derivatives displayed moderate activity, three compounds exhibited no antitumoral activity even at the highest tested dose (100 µM). Among these compounds, derivative 6, featuring an amino side chain at position 7 of the pentacyclic skeleton with N,N-dimethylaminopropyl substitution, showcased the most significant antiproliferative activity. It demonstrated submicromolar inhibitory concentrations (IC50 0.3 – 1.8 µM) against multiple cancer cell lines. This specific compound has been identified as a lead compound for further structural optimization aimed at enhancing both its activity and selectivity. Additionally, a structural analogue of derivative 6, namely compound 19, bearing an amino side chain at position 11 of the pentacyclic skeleton with N,N-dimethylaminopropyl substitution, displayed notably similar activity against K-562 and Z-138 cancer cells, also within the submicromolar range of inhibitory concentrations (IC50 0.4 µM and 0.6 µM, respectively).
Additionally, the 7-piperazinyl substituted pentacyclic derivative 9 displayed an overall IC50 range of 1.5 – 5.6 µM, while its structural analogue, compound 22, featuring piperazinyl nuclei at position 11 of the pentacyclic skeleton, exhibited IC50 values ranging from 1.2 – 8.4 µM.
Comparing the outcomes for these structural analogues, both bearing amino side chains at either position 7 or position 11 within the pentacyclic skeleton, indicates a slight enhancement in antiproliferative activity when the amino side chain is positioned at position 7. Conversely, compounds like the 7-piperidinyl substituted derivative 8 and the 11-fluor and 11-unsubstituted cyano substituted pentacyclic precursors 16 and 17 showed inactivity against the panel of cancer cell lines (IC50 > 100 µM).
Among the amido-substituted compounds, the carboxamide derivative 25, featuring an N,N-dimethylaminopropyl side chain, demonstrated the highest activity, exhibiting an IC50 range of 2.1 – 11.9 µM. Notably, this compound displayed increased antiproliferative activity against the Z-138 cancer cell line, with an IC50 value of 2.1 µM. Upon comparing the outcomes for both types of amido-substituted regioisomers, it was observed that the 6-amido-benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridines exhibited slightly higher activity compared to the 7-amido-benzo[g]benzo[4,5]imidazo[1,2-a] [1,5]naphthyridines.
In summary, it can be affirmed that the N,N-dimethylaminopropyl amino side chain positioned at C-7 of the pentacyclic skeleton notably enhances antiproliferative activity compared to analogues with an amino side chain at C-11, as well as cyano-substituted precursors or 6-amido/7-amido pentacyclic derivatives (Figure 2). Hence, both the position (C-7 or C-11) and the nature of the substituent (amino or amido) strongly influence the observed antiproliferative activity.
2.2.2. Binding with nucleic acids
Four compounds, namely 6, 9, 19, and 22, were selected based on their performance in the antitumoral evaluation for further investigation concerning their interaction with nucleic acids. These compounds were dissolved in DMSO at a concentration of c=2 × 10-3 mol dm-3. The absorbance measurements of their buffered aqueous solutions indicated a direct proportionality to their concentrations up to c=2 × 10-5 mol dm-3. This observation suggested that within this concentration range, the compounds did not form assemblies through intermolecular stacking interactions.
The absorption maxima and corresponding molar extinction coefficients (ε) were determined and are provided in Figure 3 and Table S1 (Figures S1-S4 in the Supporting Information, SI). Fluorimetric measurements were conducted in a spectral region devoid of emission and excitation spectral overlap (Figure S5-S8 in SI).
The investigation into the interaction of these compounds involved the use of calf thymus (ct)DNA, representing regular B-form DNA (with 58% AT base pairs and 42% GC base pairs), and AU homopoly-nucleotide (rArU), serving as a model for A-helical structure (RNA) [28,29,30]. Thermal melting (Tm) experiments provided crucial parameters, including the ΔTm value, representing the disparity between the Tm value of the free polynucleotide and the Tm of the complex formed with a small molecule [31].
Groove binding interactions can result in either substantial (positive ΔTm values) or minor stabilization or destabilization of DNA (negative ΔTm values). Conversely, positive ΔTm values are typically characteristic of intercalating small molecules. Upon evaluation, compounds 6, 9, 19, and 22 exhibited moderate stabilizing effects on both DNA (ctDNA) and RNA (rArU), as illustrated in Table 2, Figure 4, and Figures S15 and S16 in the Supporting Information (SI). Among these compounds, compound 22, which features a piperazine ring at C-11, demonstrated the most pronounced stabilizing effect on RNA.
Titrations with ctDNA and rArU resulted in fluorescence decrease of 6, 9, 19 and 22 (Figure 5, see Figures S9-S14 in SI).
The binding constants (Ka) for the complexes formed between the ligands and DNA/RNA were determined from the fluorimetric titration data using the Scatchard equation, and the calculated values are detailed in Table 2. Notably, compounds 19 and 22, both featuring an amino substituent (piperazine or dimethylaminopropylamino) at the C-11 position, displayed substantial binding affinities towards nucleic acids (both DNA and RNA). This increase in binding affinity was particularly pronounced for the AU homopolynucleotide (RNA).
Small fluorescence changes of derivatives 6 and 9 in the titration with ctDNA disabled the accurate calculation of binding constants (see Figures S11 and S12 in SI).
Circular dichroism (CD) spectroscopy serves as a valuable tool in monitoring alterations in nucleic acid conformation following the introduction of small molecules. It also offers insights into the interaction mode based on the relative orientation of the small molecule and the chiral axis of the nucleic acid [34]. When interacting with nucleic acids, achiral small molecules like 6, 9, 19, and 22 can induce a CD spectrum known as Induced Circular Dichroism (ICD). In the spectral region beyond 300 nm, where DNA exhibits no absorption (as depicted in Figure 6 and Figure S17 in the Supporting Information, SI), informative data regarding binding can be extracted. Upon the addition of compounds 6, 9, 19, and 22, a substantial reduction in CD intensity was observed in ctDNA at 275 nm and in RNA at 260 nm.
Compounds 6, 9, 19, and 22 exhibited the emergence of negative Induced Circular Dichroism (ICD) bands, which were less prominent at lower ratios (r [compound]/[nucleotide phosphate]) and became more pronounced at higher ratios (r > 0.2), as depicted in Figure 6 and Figure S17 in the Supporting Information (SI). The induced circular dichroism was notably more conspicuous for DNA compared to RNA.
Both the CD spectroscopy and thermal melting analysis collectively suggest that the anticipated mode of binding to nucleic acids at lower ratios (r [compound]/[nucleotide phosphate] < 0.2) involves intercalation. However, at higher ratios (r > 0.2), there appears to be a shift towards binding of aggregated compounds along the polynucleotide backbone [35].
2.2.3. Effects of pentacyclic benzimidazole derivatives on cancer cell cycle regulation
To further validate and explore the impact of the newly synthesized derivatives on cancer cell proliferation, we investigated the effect of derivatives 6 and 19 on cell cycle regulation. Our findings show that both derivatives dose dependently increase the percentage of cells in the G2/M phase following a 24-hour treatment with a concomitant reduction of cells in the G1 phase of the cell cycle. The same pattern can be seen for the reference drug doxorubicin (DOXO), an antineoplastic agent known to intercalate into nucleic acids. As shown in Figure 7, the effect on the cell cycle regulation was somewhat more pronounced in the NCI-H460 cell line (B) when compared to the Capan-1 cell line (A).
Alongside the G2/M arrest, an increasing percentage of cells in the sub-G1 phase was measured after treatment with derivatives 6 and 19. An increase in the percentage of cells in the sub-G1 phase in a cell cycle experiment is typically indicative of apoptotic cell death, since the sub-G1 phase represents a population of cells with DNA content lower than the G1 phase, often resulting from DNA fragmentation during apoptosis.
2.2.4. Effects of pentacyclic benzimidazole derivatives on caspase-3 and -7 activity
As suggested by the increased percentage of cells in the sub-G1 phase following exposure to derivatives 6 and 19, our objective was to examine the potential of these pentacyclic benzimidazole derivatives to induce apoptosis in various cancer cell types. Therefore, we investigated the dose dependent effects of 6 and 19 on pancreatic adenocarcinoma and lung carcinoma apoptosis.
As depicted in Figure 8, we observed an increase in caspase-3 and -7 activity in both cell lines after 24 hours of incubation with 10 µM of each derivative, while lower doses (1 and 0.1 µM) exhibited no discernible effect. Notably, derivative 6 exerted a more pronounced effect on the NCI-H460 cell line compared to derivative 19, whereas the opposite was true for the Capan-1 cell line. The reference drug doxorubicin (DOXO) was able to induce apoptosis even at a concentration of 1 µM, and this was seen in both cancer cell lines.
3. Materials and methods
3.1. Chemistry - General methods
Chemicals and solvents used in this study were procured from commercial suppliers such as Aldrich and Acros. Melting points were determined using SMP11 Bibby and Büchi 535 apparatus. NMR spectra were acquired in DMSO-d6 solutions, utilizing TMS as an internal standard. The 1H and 13C NMR spectra were recorded on Varian Gemini 300 or Varian Gemini 600 spectrometers, operating at 300, 600, 150, and 75 MHz, respectively. Chemical shifts were reported in ppm (δ) relative to TMS. To ensure the purity of compounds, TLC analysis was routinely conducted using Merck silica gel 60F-254 glass plates. In preparative photochemical experiments, irradiation was conducted at room temperature utilizing a water-cooled immersion well with an "Origin Hanau" 400 W high-pressure mercury arc lamp, employing Pyrex glass as a filter. High-resolution mass spectrometry (HRMS) analysis was performed using a mass spectrometer (MALDI TOF/TOF 4800 plus analyzer). Elemental analysis for carbon, hydrogen, and nitrogen was carried out using a Perkin-Elmer 2400 elemental analyzer. In cases where elemental analyses are represented by elemental symbols, the obtained analytical results were within 0.4% of the theoretical values.
3.2. Synthesis of 7-amino substituted benzo[g]benzo[4,5]imidazo[1,2-a][1,8]- -naphthyridine-6-carbonitriles
3.2.1. (E)-2-benzimidazolyl-1-(2-chloroquinolin-3-yl)-2-isocyanoethen-1-ol 3
A solution of 0.40 g (2.34 mmol) 2-cyanomethylbenzimidazole 2 and 0.60 g (2.45 mmol) 2-chloroquinoline-3-carbonyl chloride 1 in pyridine (6 mL) was refluxed for 1.5 h. The cooled mixture was poured into water (50 mL) and the resulting product was filtered off and recrystallized from ethanol to obtain a brown powder (1.24 g, 76%). m.p. >300 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 13.38 (s, 1H, OH), 12.62 (s, 1H, NHbenzim.), 9.30 (s, 1H, Harom.), 9.21–9.19 (m, 1H, Harom.), 8.34 (d, 1H, J = 8.61 Hz, Harom.), 8.30 (d, 1H, J = 8.52 Hz, Harom.), 8.00 (t, 1H, J = 7.65 Hz, Harom.), 7.72 (t, 1H, J = 7.12 Hz, Harom.), 7.58–7.55 (m, 1H, Harom.), 7.50–7.74 (m, 2H, Harom.); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = not enough soluble; Found: C, 65.60; H, 3.05; N, 16.36. Calc. for C19H11ClN4O: C, 65.81; H, 3.20; N, 16.16%.
3.2.2. 7-oxo-5,7-dihydrobenzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo-nitrile 4
A solution of 1.24 g (1.69 mmol) 3 and 1.20 g (11.00 mmol) t-KOBu in DMF (7 mL) was refluxed for 2 h. After cooling, the reaction mixture was evaporated under vacuum and dissolved in water (50 mL). Resulting product was filtered off and recrystallized from ethanol to obtain a light brown powder (0.93 g, 83%). m.p. >300 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 13.31 (bs, 1H, NH), 9.35 (s, 1H, Harom.), 8.47 (d, 1H, J = 7.92 Hz, Harom.), 8.22 (d, 1H, J = 7.91 Hz, Harom.), 8.01 (d, 1H, J = 8.53 Hz, Harom.), 7.88 (dt, 1H, J1 = 8.52 Hz, J2 = 1.42 Hz, Harom.), 7.57–7.48 (m, 3H, Harom.), 7.38–7.33 (m, 1H, Harom.); 13C NMR (DMSO-d6, 100 MHz): δ/ppm = not enough soluble; Found: C, 73.69; H, 3.15; N, 18.00. Calc. for C19H10N4O: C, 73.54; H, 3.25; N, 18.06%.
3.2.3. 7-chlorobenzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbonitrile 5
A solution of 0.93 g (2.98 mmol) 7-oxo-5,7-dihydrobenzo[g]benzo[4,5]imidazo[1,2-a] [1,8]naphthyridine-6-carbonitrile 4 and 0.35 g (1.70 mmol) PCl5 in POCl3 (17 mL) was refluxed for 2 h. After cooling, the reaction mixture was evaporated under vacuum and dissolved in water (10 ml). Resulting product was filtered off and washed with water to obtain a yellow powder (0.12 g, 12%). m.p. 293–295 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 9.46 (s, 1H, Harom.), 9.29 (bs, 1H, Harom.), 8.45 (d, 1H, J = 8.62 Hz, Harom.), 8.37 (d, 1H, J = 7.61 Hz, Harom.), 8.11 (t, 1H, J = 7.43 Hz, Harom.), 8.04 (d, 1H, J = 7.10 Hz, Harom.), 7.82 (t, 1H, J = 7.33 Hz, Harom.), 7.71–7.60 (m, 2H, Harom.); 13C NMR (DMSO-d6, 100 MHz): δ/ppm = 147.8, 145.0, 144.5, 143.9, 143.3, 139.5, 134.7, 131.3, 130.4, 128.2, 127.6, 126.2, 125.8, 125.4, 120.5, 117.3, 115.8, 113.8, 103.7; Found: C, 69.26; H, 2.68; N, 17.29. Calc. for C19H9ClN4: C, 69.42; H, 2.76; N, 17.04%; MS: m/z= 351.0410 ([M+Na]+).
3.2.4. General method for preparation of compounds 6–9
Compounds 6–9 were prepared using microwave irradiation, at optimized reaction time with power 800 W and 40 bar pressure, from compound 5 in acetonitrile (10 mL) with excess of added corresponding amine. After cooling, the reaction mixture was filtered off and resulting product was separated by column chromatography on SiO2 using dichloromethane/methanol as eluent.
3.2.4.1. 7-((3-N,N-(dimethylamino)propyl)amino)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]-naphthyridi- ne-6-carbonitrile 6
Compound 6 was prepared using above described method from 5 (0.06 g, 0.20 mmol) and N,N-dimethylamino-propyl-1-amine (0.14 mL, 1.44 mmol) after 4 h of irradiation to yield 0.05 g (65%) of yellow powder. m.p. 250–253 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 9.11 (s, 1H, Harom.), 8.92 (d, 1H, J = 7.14 Hz, Harom.), 8.67 (bs, 1H, NH), 8.11 (d, 1H, J = 8.40 Hz, Harom.), 7.99-7.93 (m, 1H, J = 8.10 Hz, Harom.), 7.93 (t, 1H, J = 8.16 Hz, Harom.), 7.69–7.63 (m, 2H, Harom.), 7.41–7.30 (m, 2H, Harom.), 3.91 (t, 2H, J = 6.36 Hz, CH2), 2.50–2.46 (m, 2H, CH2), 2.25 (s, 6H, CH3), 1.98–1.89 (m, 2H, CH2); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 150.4, 147.2, 144.7, 135.3, 133.5, 131.6, 129.5, 127.9, 127.1, 124.9, 124.8, 122.6, 118.5, 117.8, 116.4, 113.2, 57.2, 45.8 (2C), 44.0, 27.0;
Found: C, 73.27; H, 5.50; N, 21.23. Calc. for C24H22N6: C, 73.07; H, 5.62; N, 21.30%; MS: m/z= 395.1998 ([M+H]+).
3.2.4.2. 7-(N-isobutylamino)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo- nitrile 7
Compound 7 was prepared using above described method from 5 (0.06 g, 0.20 mmol) and isobutylamine (0.12 mL, 1.23 mmol) after 4 h of irradiation to yield 0.05 g (72%) of yellow powder. m.p. 296–297 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 9.51 (s, 1H, Harom.), 9.01 (dd, 1H, J1 = 6.71 Hz, J1 = 2.24 Hz, Harom.), 8.42 (t, 1H, J = 6.22 Hz, NHamin), 8.20 (d, 1H, J = 8.42 Hz, Harom.), 8.07 (d, 1H, J = 7.82 Hz, Harom.), 8.00–7.94 (m, 1H, Harom.), 7.72–7.67 (m, 2H, Harom.), 7.42–7.34 (m, 2H, Harom.), 3.70 (t, 2H, J = 6.62 Hz, CH2), 2.29–2.13 (m, 1H, CH), 1.05 (d, 6H, J = 6.62 Hz, CH3); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 150.0, 149.1, 146.9, 144.4, 144.3, 135.1, 132.9, 131.2, 128.9, 127.4, 126.5, 124.6, 124.3, 122.1, 118.0, 117.1, 115.9, 112.7, 51.5, 28.3, 19.6 (2C); Found: C, 75.39; H, 5.40; N, 19.21. Calc. for C23H19N4: C, 75.59; H, 5.24; N, 19.16%; MS: m/z= 366.1703 ([M+H]+).
3.2.4.3. 7-(piperidin-1-yl)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo- nitrile 8
Compound 8 was prepared using above described method from 5 (0.06 g, 0.20 mmol) and piperidine (0.13 mL, 1.27 mmol) after 4 h of irradiation to yield 0.06 g (87%) of yellow powder. m.p. >300 °C. 1H NMR (DMSO-d6, 400 MHz): δ/ppm = 9.19 (dd, J1 = 6.42 Hz, J2 = 2.93 Hz, 1H, Harom.), 9.05 (s, 1H, Harom.), 8.36 (d, J = 8.11 Hz, 1H, Harom.), 8.29 (d, J = 8.43 Hz, 1H, Harom.), 8.03 (t, J = 8.32 Hz, 1H, Harom.), 7.86 (dd, J1 = 6.32 Hz, J2 = 2.83 Hz, 1H, Harom.), 7.74 (t, J = 7.20 Hz, 1H, Harom.), 7.54–7.49 (m, 2H, Harom.), 3.72 (t, J = 5.21 Hz, 4H, CH2), 1.93 (bs, 4H, CH2), 1.80 (bs, 2H, CH2); 13C NMR (DMSO-d6, 100 MHz): δ/ppm = 158.5, 147.9, 147.4, 145.7, 144.2, 138.7, 133.7, 131.3, 130.4, 127.9, 126.9, 123.8, 119.3, 116.9, 116.4, 115.7, 54.4 (2C), 26.4 (2C), 24.1; Found: C, 76.20; H, 5.15; N, 18.65. Calc. for C24H19N5: C, 76.37; H, 5.07; N, 18.55%; MS: m/z= 400.1519 ([M+Na]+).
3.2.4.4. 7-(piperazin-1-yl)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo- nitrile 9
Compound 9 was prepared using above described method from 5 (0.06 g, 0.20 mmol) and piperazine (0.11 g, 1.27 mmol) after 4 h of irradiation to yield 0.02 g (32%) of yellow powder. m.p. 274–277 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 9.09–9.07 (m, 1H, Harom.), 8.98 (s, 1H, Harom.), 8.28 (d, 1H, J = 8.31 Hz, Harom.), 8.18 (d, 1H, J = 8.13 Hz, Harom.), 7.97 (t, 1H, J = 7.38 Hz, Harom.), 7.83–7.80 (m, 1H, Harom.), 7.68 (t, 1H, J = 7.42 Hz, Harom.), 7.47 (bs, 2H, Harom.), 3.64 (s, 4H, CH2), 3.08 (s, 4H, CH2); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 157.9, 147.9, 147.3, 145.5, 144.4, 138.6, 133.6, 131.4, 130.3, 127.8, 126.8, 125.3, 125.1, 123.7, 119.4, 116.8, 116.4, 115.3, 54.5 (2C), 46.7 (2C); Found: C, 73.10; H, 4.88; N, 22.00. Calc. for C23H18N6: C, 73.00; H, 4.79; N, 22.21%; MS: m/z= 401.1507 ([M+Na]+).
3.3. Synthesis of 11-amino substituted benzo[g]benzo[4,5]imidazo[1,2-a][1,8]- -naphthyridine-6-carbonitriles
3.3.1. (E)-2-(1H-benzo[d]imidazol-2-yl)-3-(2-chloro-7-fluoroquinolin-3-yl)acrylonitrile 13
Compound 13 was prepared from 2 (0.38 g, 2.39 mmol) and 2-chloro- 7-fluoroquinoline-3-carbaldehyde 10 (0.50 g, 2.39 mmol) and few drops of piperidine in absolute ethanol (7 mL) after refluxing for 2 h and recrystallization from ethanol to yield 0.43 (52%) of yellow powder. m.p. >300 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 13.43 (s, 1H, NHbenz.), 9.18 (s, 1H, Hetenil), 8.57 (s, 1H, Harom.), 8.34 (dd, 1H, J1 = 9.06 Hz, J2 = 6.24 Hz, Harom.), 7.87 (dd, 1H, J1 = 10.05 Hz, J2 = 2.37 Hz, Harom.), 7.76–7.69 (m, 3H, Harom.), 7.32–7.29 (m, 2H, Harom.); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 164.6 (d, J = 250.7 Hz), 162.9, 150.4, 148.8, 148.6, 146.8, 140.3 (2C), 139.8 (2C), 132.3 (d, J = 10.5 Hz), 125.9, 124.2, 119.2 (d, J = 25.2 Hz), 115.5, 112.6 (d, J = 21.4 Hz), 108.7; Found: C, 68.78; H, 3.40; N, 16.84. Calc. for C19H11ClN4: C, 68.99; H, 3.35; N, 16.94%.
3.3.2. 11-fluorobenzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbonitrile 16
Compound 13 (0.50 g, 1.60 mmol) was dissolved in 3 ml of sulfolane and reaction mixture was heated for 15 minutes at 280 °C. The cooled mixture was poured into water (15 mL) and the resulting product was filtered off and recrystallizated from ethanol (150 mL) to obtain a yellow powder (0.57 g, 78%). m.p. >300 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 9.16–9.13 (m, 1H, Harom.), 9.08 (s, 1H, Harom.), 8.77 (s, 1H, Harom.), 8.31 (dd, 1H, J1 = 6.39 Hz, J2 = 8.97 Hz, Harom.), 7.98–7.95 (m, 2H, Harom.), 7.66–7.56 (m, 3H, Harom.); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 164.8 (d, J = 253.6 Hz), 148.5, 145.9, 145.0, 143.4, 140.4, 140.1, 132.2 (d, J = 10.6 Hz), 130.7, 125.4, 124.5, 122.7, 119.9, 117.3 (d, J = 25.8 Hz), 116.9, 115.7, 114.8, 111.8 (d, J = 21.4 Hz), 102.4; Found: C, 73.27; H, 2.81; N, 17.78. Calc. for C19H9FN4: C, 73.07; H, 2.90; N, 17.94%; MS: m/z= 313.0882 ([M+H]+).
3.3.3. General method for preparation of compounds 19–22
Compounds 19–22 were prepared using microwave irradiation, at optimized reaction time with power 800 W and 40 bar pressure, from compound 16 in acetonitrile (10 mL) with excess of added corresponding amine. After cooling, the reaction mixture was filtered off and resulting product was separated by column chromatography on SiO2 using dichloromethane/methanol as eluent.
3.3.3.1. 11-((3-N,N-(dimethylamino)propyl)amino)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]-naphthyridine-6-carbonitrile 19
Compound 19 was prepared using above described method from 16 (0.07 g, 0.22 mmol) and N,N-dimethylamino-propyl-1-amine (0.18 mL, 1.56 mmol) after 3 h of irradiation to yield 0.04 g (50%) of yellow powder. m.p. 230–235 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 9.22–9.19 (m, 1H, Harom.), 8.58 (s, 2H, Harom.), 7.99–7.79 (m, 1H, Harom.), 7.74 (d, 1H, J = 9.01 Hz, Harom.), 7.58–7.54 (m, 2H, Harom.), 7.25 (t, 1H, J = 4.72 Hz, NHamin), 7.06 (d, 1H, J = 7.22 Hz, Harom.), 6.86 (s, 1H, Harom.), 3.26 (q, 2H, J = 5.64 Hz, CH2), 2.38 (t, 2H, J = 6.81 Hz, CH2), 2.20 (s, 6H, CH3), 1.87–1.75 (m, 2H, CH2); 13C NMR (DMSO-d6, 151 MHz): δ/ppm = 153.5, 150.8, 145.9, 145.6, 143.5, 140.2, 138.9, 130.6, 129.9, 124.9, 123.4, 119.3, 118.8, 116.9, 115.8, 111.1, 100.7, 97.1, 56.7, 45.2 (2C), 40.7, 26.3; Found: C, 73.17; H, 5.40; N, 21.45. Calc. for C24H22N6: C, 73.07; H, 5.62; N, 21.30%; MS: m/z= 395.1986 ([M+H]+).
3.3.3.2. 11-(N-isobutylamino)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbonitrile 20
Compound 20 was prepared using above described method from 16 (0.21 g, 0.66 mmol) and isobutylamine (0.468 mL, 4.71 mmol) after 3 h of irradiation to yield 0.12 g (54%) of yellow powder. m.p. 286–288 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 9.26–9.21 (m, 1H, NH), 8.60 (s, 2H, Harom), 7.96–7.91 (m, 1H, Harom.), 7.77 (d, 1H, J = 9.03 Hz, Harom.), 7.59–7.52 (m, 2H, Harom.), 7.26 (t, 1H, J = 5.32 Hz, Hamin), 7.15 (dd, 1H, J1 = 9.01 Hz, J2 = 2.06 Hz, Harom.), 6.89 (s, 1H, Harom.), 3.09 (t, 2H, J = 6.09 Hz, CH2), 2.06–1.92 (m, 2H, CH2), 1.04 (d, 6H, J = 6.63 Hz, CH3); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 154.4, 151.2, 146.7, 144.0, 140.6, 139.4, 131.2, 130.5, 125.5, 123.9, 120.0, 119.8, 119.4, 117.5, 116.2, 111.7, 101.6, 97.8, 50.8, 27.9, 20.8 (2C); Found: C, 75.36; H, 5.41; N, 19.22. Calc. for C23H19N5: C, 75.59; H, 5.24; N, 19.16%; MS: m/z= 366.1720 ([M+H]+).
3.3.3.3. 11-(piperidin-1-yl)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo-nitrile 21
Compound 21 was prepared using above described method from 16 (0.21 g, 0.66 mmol) and piperidine (0.47 mL, 4.68 mmol) after 3 h of irradiation to yield 0.062 g (8%) of yellow powder. m.p. >300 °C.
1H NMR (DMSO-d6, 300 MHz): δ/ppm = 9.43–9.38 (m, 1H, Harom.), 8.83 (s, 1H, Harom.), 8.76 (s, 1H, Harom.), 8.02 (d, 1H, J = 9.32 Hz, Harom.), 7.99–7.96 (m, 1H, Harom.), 7.64–7.57 (m, 3H, Harom.), 7.48 (s, 1H, Harom.), 3.65 (bs, 4H, CH2), 1.70 (bs, 6H, CH2); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 154.6, 150.8, 146.5, 144.1, 140.7, 139.7, 131.3, 130.8, 125.6, 124.2, 119.9, 119.5, 118.6, 117.7, 116.1, 112.8, 106.5, 98.7, 48.5 (2C), 25.6 (2C), 24.4; Found: C, 76.20; H, 5.17; N, 18.63. Calc. for C24H19N5: C, 76.37; H, 5.07; N, 18.55%; MS: m/z= 378.1720 ([M+H]+).
3.3.3.4. 11-(piperazin-1-yl)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbo-nitrile 22
Compound 22 was prepared using above described method from 16 (0.21 g, 0.66 mmol) and piperazine (0.30 g, 4.71 mmol) after 3 h of irradiation to yield 0.20 g (67%) of yellow powder. m.p. >300 °C. 1H NMR (DMSO-d6, 300 MHz): δ/ppm = 9.24 (d, 1H, J = 7.08 Hz, Harom.), 8.70 (s, 1H, Harom.), 8.63 (s, 1H, Harom.), 7.96–7.90 (m, 2H, Harom.), 7.58–7.48 (m, 3H, Harom.), 7.27 (s, 1H, Harom.), 3.52 (bs, 4H, CH2), 2.96 (s, 4H, CH2), 2.86 (s, 1H, NH); 13C NMR (DMSO-d6, 75 MHz): δ/ppm = 154.7, 150.4, 146.4, 145.9, 143.9, 140.8, 139.8, 131.2, 130.7, 125.6, 124.2, 119.9, 119.6, 118.4, 117.6, 116.2, 112.9, 106.6, 98.8, 47.9, 45.6 (2C), 43.8; Found: C, 73.13; H, 4.86; N, 22.01. Calc. for C23H18N6: C, 73.00; H, 4.79; N, 22.21%; MS: m/z= 379.1660 ([M+H]+).
3.4. Synthesis of amido substituted benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridines and benzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridines
3.4.1. General method for the synthesis of compounds 14 and 15
Solution of equimolar amounts of 2-cyanomethylbenzimidazole, corresponding aromatic aldehydes (10 or 12) and a few drops of piperidine in absolute ethanol, was refluxed for 2 h. Following reaction, the mixture was cooled to room temperature, the crude product was filtered off and recrystallized from ethanol.
3.4.1.1. (E)-2-(1H-benzo[d]imidazol-2-yl)-3-(2-chloroquinolin-3-yl)acrylonitrile 14
Compound 14 was prepared using above described method, from 2-cyano- methylbenzimidazole 2 (0.25 g, 1.56 mmol) and 2-chloroquinoline- 3-carbaldehyde 11 (0.30 g, 1.56 mmol) in absolute ethanol (6 mL) to yield 0.45 g (86%) of orange powder. m.p. 294–297 °C. 1H NMR (600 MHz, DMSO): δ/ppm = 13.38 (s, 1H, NHbenzimid.), 9.12 (s, 1H, Harom.), 8.56 (s, 1H, Harom.), 8.19 (d, J = 7.92 Hz, 1H, Harom.), 8.04 (d, J = 8.43 Hz, 1H, Harom.), 7.94 (t, J = 7.82 Hz, 1H, Harom.), 7.76 (t, J = 7.50 Hz, 1H, Harom.), 7.75 (bs, 1H, Harom.) 7.59 (bs, 1H, Harom.), 7.31 (bs, 1H, Harom.), 7.27 (bs, 1H, Harom.); 13C NMR (150 MHz, DMSO): δ/ppm = 147.1, 145.2, 145.1, 143.2, 141.0, 140.4, 133.4, 130.8, 129.3, 128.0, 126.8, 125.4, 125.3, 124.6, 119.9, 116.8, 116.3, 115.0, 102.3; Found: C, 69.05; H, 3.54; N, 16.70. Calc. for C19H11ClN4: C, 68.99; H, 3.35; N, 16.94%; MS: m/z= 331.0750 ([M+H]+).
3.4.1.2. (E)-2-(1H-benzo[d]imidazol-2-yl)-3-(quinolin-3-yl)acrylonitrile 15
Compound 15 was prepared using above described method, from 2-cyano- methylbenzimidazole 2 (0.30 g, 1.91 mmol) and quinoline-3-carbaldehyde 12 (0.30 g, 1.91 mmol) in absolute ethanol (6 mL) to yield 0.47 g (82%) of yellow powder. m.p. 290–292 °C. 1H NMR (600 MHz, DMSO): δ/ppm = 13.16 (s, 1H, NHbenzimid.), 9.34 (d, J = 2.22 Hz, 1H, Harom.), 8.98 (s, 1H, Harom.), 8.55 (s, 1H, Harom.), 8.14 (d, J = 7.82 Hz, 1H, Harom.), 8.11 (d, J = 8,34 Hz, 1H, Harom.), 7.91 (t, J = 8.33 Hz, 1H, Harom.), 7.73 (t, J = 7.98 Hz, 2H, Harom.), 7.61 (bs, 1H, Harom.), 7.29 (bs, 2H, Harom.); 13C NMR (75 MHz, DMSO): δ/ppm = 150.9, 148.5, 147.5, 142.6, 137.0, 132.2, 129.7, 129.3, 128.3, 127.3, 126.8, 124.4, 123.0, 119.9, 116.5, 112.2, 105.0; Found: C, 77.15; H, 4.20; N, 18.70. Calc. for C19H12N4: C, 77.01; H, 4.08; N, 18.91%; MS: m/z= 297.1143 ([M+H]+).
3.4.2. Benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbonitrile 17
Compound 17 (0.45 g, 1.35 mmol) was dissolved in sulfolane (2.5 mL) and reaction mixture was heated for 30 min at 280 °C. The cooled mixture was poured into water (10 mL) and the resulting product was filtered off and recrystallized from ethanol to obtain a yellow powder (0.39 g, 99%). m.p. 291–293 °C. 1H NMR (600 MHz, DMSO): δ/ppm = 9.30 (d, J = 7.62 Hz, 1H, Harom.), 9.16 (s, 1H, Harom.), 8.88 (s, 1H, Harom.), 8.31 (d, J = 8.43 Hz, 1H, Harom.), 8.26 (d, J = 8.02 Hz, 1H, Harom.), 8.04 (t, J = 7.12 Hz, 1H, Harom.), 8.01 (d, J = 8.04 Hz, 1H, Harom.), 7.74 (t, J = 7.43 Hz, 1H, Harom.), 7.64 (t, J = 7.51 Hz, 1H, Harom.), 7.60 (t, J = 7.42 Hz, 1H, Harom.); 13C NMR (75 MHz, DMSO): δ/ppm = 147.6, 143.8, 141.4, 140.8, 133.8, 131.2, 129.8, 128.4, 127.2, 125.8, 125.8, 125.0, 120.4, 117.3, 116.8, 115.5, 102.8; Found: C, 77.35; H, 3.56; N, 19.08. Calc. for C19H10N4: C, 77.54; H, 3.42; N, 19.04%; MS: m/z= 3295.0958([M+H]+).
3.4.3. Benzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridine-7-carbonitrile 18
An ethanolic solution (400 mL) of compound 15 (0.35 g, 1.18 mmol) was irradiated at room temperature, with 400 W high-pressure mercury lamp, using a Pyrex filter for 4 h. The solution was concentrated under reduced pressure and resulting product was filtered off to give 0.42 g (72%) of yellow powder. m.p. 243–246 °C. 1H NMR (600 MHz, DMSO): δ/ppm = 9.40 (d, J = 2.71 Hz, 1H, Harom.), 8.96 (d, J = 3.12 Hz 1H, Harom.), 8.80–8.77 (m, 1H, Harom.), 8.27 (d, J = 8.32 Hz, 1H, Harom.), 8.18 (dd, J1 = 8.41 Hz, J2 = 4.14 Hz, 1H, Harom.), 8.08 (d, J = 7.93 Hz, 1H, Harom.), 8.04 (t, J = 7.52 Hz, 1H, Harom.), 7.80 (t, J = 7.40 Hz, 1H, Harom.), 7.66 (t, J = 7.62 Hz, 1H, Harom.), 7.48 (t, J = 7.43 Hz, 1H, Harom.); 13C NMR (150 MHz, DMSO): δ/ppm = 150.7, 148.5, 146.8, 144.6, 138.6, 138.0, 132.5, 131.8, 129.6, 126.3, 125.9, 124.1, 121.8, 120.6, 116.4, 115.9, 115.2, 115.1, 101.9; Found: C, 77.68; H, 3.50; N, 18.82. Calc. for C19H10N4: C, 77.54; H, 3.42; N, 19.04%; MS: m/z= 295.0988 ([M+H]+).
3.4.4. General method for the synthesis of compounds 23 and 27
2 N solution of sulfuric acid and compounds 17 and 18 was refluxed for 24 h. Cooled reaction mixture was poured into ice, and resulting product was filtered off.
3.4.4.1. Benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carboxylic acid 23
Compound 23 was prepared using above described method, from 17 (0.42 g, 1.43 mmol) and 2 N aqueaus solution of sulfuric acid (4.09 mL) to yield 0.43 g (97%) of yellow powder. m.p. 287–290 °C. 1H NMR (300 MHz, DMSO): δ/ppm = 9.49 (s, 1H, Harom.), 9.46 (bs, 1H, Harom.), 9.16 (s, 1H, Harom.), 8.34 (d, J = 8.62 Hz, 1H, Harom.), 8.27 (d, J = 8.22 Hz, 1H, Harom.), 8.11–8.04 (m, 2H, Harom.), 7.79 (t, J = 7.43 Hz, 1H, Harom.), 7.78–7.73 (m, 2H, Harom.), 4.62 (bs, 1HCOOH); Found: C, 72.90; H, 3.65; N, 13.36. Calc. for C19H11N3O2: C, 72.84; H, 3.54; N, 13.51%.
3.4.4.2. Benzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridine-7-carboxylic acid 27
Compound 27 was prepared using above described method, from 18 (0.40 g, 1.36 mmol) and 2 N aqueaus solution of sulfuric acid (3.90 mL) to yield 0.28 g (68%) of yellow powder. m.p. 290–291 °C. 1H NMR (400 MHz, DMSO): δ/ppm = 9.79 (s, 1H, Harom.), 9.43 (s, 1H, Harom.), 8.81 (d, J = 7.92 Hz, 1H, Harom.), 8.39 (d, J = 7.63 Hz, 1H, Harom.), 8.34 (d, J = 8.61 Hz, 1H, Harom.), 8.22 (d, J = 8.11 Hz, 1H, Harom.), 8.17 (t, J = 8.32 Hz, 1H, Harom.), 7.93–7.86 (m, 2H, Harom.), 7.68 (t, J = 8.52 Hz, 1H, Harom.), 7.12 (bs, 1H, HCOOH); 13C NMR (101 MHz, DMSO): δ/ppm = 164.3, 152.0, 148.8, 145.0, 139.6, 138.4, 135.1, 134.0, 130.3, 123.0, 129.3, 127.1, 124.9, 124.4, 117.5, 117.3, 117.0, 116.8, 116.5; Found: C, 72.77; H, 3.46; N, 13.70. Calc. for C19H11N3O2: C, 72.84; H, 3.54; N, 13.51%.
3.4.5. General method for the synthesis of compounds 24 and 28
A mixture of corresponding carboxylic acids 23 and 27 and thionyl chloride in absolute toluene was refluxed for 22 h. Toluene and excess of thionyl chloride was removed under reduce pressure. The crude product was washed 3 times with absolute toluene to obtained powdered product.
3.4.5.1. Benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridine-6-carbonyl chloride 24
Compound 24 was prepared using above described method, from 23 (0.24 g, 0.80 mmol), absolute toluene (15 mL) and 0.58 mL (8.00 mmol) thionyl chloride to yield 0.19 g (72%) of yellow powder.
3.4.5.2. Benzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridine-7-carbonyl chloride 28
Compound 28 was prepared using above described method, from 27 (0.20 g, 0.67 mmol), absolute toluene (100 mL) and 0.48 mL (6.070 mmol) thionyl chloride to yield 0.17 g (77%) of yellow powder.
3.4.6. General method for the synthesis of compounds 25 and 26
A mixture of carbonyl chloride 24 and excess of corresponding amine in dry dichloromethane was stirred at room temperature for 2 h. The mixture was washed with 10 mL of 20% Na2CO3 and 10 mL water. After drying over MgSO4, the organic layer was concentrated at reduced pressure and resulting product was separated by column chromatography on SiO2 using dichloromethane/methanol as eluent.
3.4.6.1. N-(3-N,N-(dimethylamino)propyl)benzo[g]benzo[4,5]imidazo[1,2-a][1,8]- -naphthyridine-6-carboxamide 25
Compound 25 was prepared using above described method, from 24 (0.19 g, 0.57 mmol), dry dichloromethane (20 mL) and 0.19 mL (1.73 mmol) N,N-dimethyl- aminopropyl-1-amine to obtain 0.04 g (20%) of light orange powder. m.p. 208–212 °C. 1H NMR (400 MHz, DMSO): δ/ppm = 10.34 (t, J = 5.81 Hz, 1H, Hamide), 9.41 (d, J = 7.13 Hz, 1H, Harom.), 9.32 (s, 1H, Harom.), 8.81 (s, 1H, Harom.), 8.32 (d, J = 8.53 Hz, 1H, Harom.), 8.22 (d, J = 7.80 Hz, 1H, Harom.), 8.04–7.99 (m, 2H, Harom.), 7.74 (t, J = 7.12 Hz, 1H, Harom.), 7.68–7.61 (m, 2H, Harom.), 3.59 (q, 2H, CH2), 2.88 (t, J = 7.40 Hz, 2H, CH2), 2.57 (bs, 6H, CH3), 2.00– 1.93 (m, 2H, CH2); 13C NMR (101 MHz, DMSO): δ/ppm = 162.3, 147.3, 146.9, 145.9, 142.7, 141.4, 134.9, 133.2, 130.8, 129.5, 128.5, 127.0, 126.1, 125.8, 124.9, 121.4, 119.8, 117.6, 117.3, 55.8, 43.9 (2C), 37.3, 25.8; Found: C, 72.70; H, 5.90; N, 17.40. Calc. for C24H23N5O: C, 72.52; H, 5.83; N, 17.62%; MS: m/z= 398.1974 ([M+H]+).
3.4.6.2. Benzo[g]benzo[4,5]imidazo[1,2-a][1,8]naphthyridin-6-yl(piperidin-1-yl)metha- none 26
Compound 26 was prepared using above described method, from 24 (0.21 g, 0.65 mmol), dry dichloromethane (20 mL) and 0.19 mL (1.93 mmol) piperidine to obtain 0.02 g (9%) of light orange powder. m.p. 258–261 °C. 1H NMR (400 MHz, DMSO): δ/ppm = 9.42 (d, J = 7.61 Hz, 1H, Harom.), 9.11 (s, 1H, Harom.), 8.36 (d, J = 8.53 Hz, 1H, Harom.), 8.24 (d, J = 7.92 Hz, 1H, Harom.), 8.09 (s, 1H, Harom.), 8.02–7.98 (m, 2H, Harom.), 7.74 (t, J = 7.11 Hz, 1H, Harom.), 7.64 (t, J = 7.62 Hz, 1H, Harom.), 7.59 (t, J = 7.58 Hz 1H, Harom.), 3.74 (bs, 4H, CH2), 1.67 (s, 4H, CH2), 1.50 (bs, 2H, CH2); 13C NMR (101 MHz, DMSO): δ/ppm = 164.0, 146.6, 146.3, 145.8, 144.3, 138.7, 132.4, 131.3, 129.0, 128.4, 128.1, 128.1, 126.8, 126.0, 125.4, 124.5, 120.3, 117.9, 117.3, 48.1, 42.6, 26.5, 25.8, 24.5; Found: C, 75.50; H, 5.40; N, 14.84. Calc. for C24H20N4O: C, 75.77; H, 5.30; N, 14.73%; MS: m/z= 381.1719 ([M+H]+).
3.4.7. General method for the synthesis of compounds 29 and 30
A mixture of carboxylic acid 27, thionyl chloride and DMF in absolute toluene was stirred at room temperature for 10 minutes, after that the appropriate amine was added and heated at reflux. Toluene and excess of thionyl chloride was removed under reduce pressure. The crude product was washed 3 times with toluene to obtained powdered product. Resulting product was separated by column chromatography on SiO2 using dichloromethane/methanol as eluent.
3.4.7.1. N-isobutylbenzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridine-7-carboxamide 29
Compound 29 was prepared using above described method, from 27 (0.07 g, 0.23 mmol), thionyl chloride (0.03 mL, 0.46 mmol), absolute toluene (20 mL), 0.03 mL DMF and 0.09 mL (1.06 mmol) isobutyamine to obtain 0.03 g (40%) of yellow powder. m.p. 166–175 °C. 1H NMR (400 MHz, DMSO): δ/ppm = 10.45 (t, J = 5.80 Hz, 1H, HNH), 9.59 (s, 1H, Harom.), 8.93 (s, 1H, Harom.), 8.77 (d, J =8.42 Hz, 1H, Harom.), 8.28 (dd, J1 = 8.42, J2 = 0.91 Hz, 1H, Harom.), 8.19 (d, J = 8.54 Hz, 1H, Harom.), 8.06 – 7.99 (m, 2H, Harom.), 7.78 (t, J = 8.36 Hz, 1H, Harom.), 7.66 (t, J = 7.22 Hz, 1H, Harom.), 7.47 (t, J = 8.48 Hz, 1H, Harom.), 3.39 (t, J = 6.14 Hz, 2H, HCH2), 2.01 – 1.99 (m, 1H, HCH), 1.06 (d, J = 6.78 Hz, 6H, HCH3); 13C NMR (101 MHz, DMSO) δ/ppm = 162.0, 152.3, 148.5, 148.2, 144.0, 137.7, 132.9, 132.2, 131.6, 130.1, 126.7, 126.0, 124.4, 122.0, 121.5, 120.5, 116.9, 116.3, 116.3, 47.1, 29.1, 20.6.
3.4.7.2. N,N-dimethylbenzo[g]benzo[4,5]imidazo[1,2-a][1,5]naphthyridine-7-carbox-amide 30
Compound 30 was prepared using above described method, from 27 (0.20 g, 0.64 mmol), thionyl chloride (0.10 mL, 1.28 mmol), absolute toluene (50 mL), 0.10 mL DMF and 0.11 mL (3.19 mmol) methylamine to obtain 0.11 g (48%) of light yellow powder. m.p. 160–165 °C. 1H NMR (400 MHz, DMSO): δ/ppm = 9.46 (s, 1H, Harom.), 8.83 (d, J = 8.43 Hz, 1H, Harom.), 8.29 (d, J = 8.42 Hz, 1H, Harom.), 8.23 (s, 1H, Harom.), 8.22 (d, J = 7.20 Hz, 1H, Harom.), 8.06 (d, J = 7.89 Hz, 1H, Harom.), 8.00 (t, J = 8.31 Hz, 1H, Harom.), 7.81 (t, J = 8.29 Hz, 1H, Harom.), 7.63 (t, J = 8.12 Hz, 1H, Harom.), 7.47 (t, J = 8.42 Hz, 1H, Harom.), 3.16 (s, 3H, CH3), 2.99 (s, 3H, CH3); 13C NMR (101 MHz, DMSO): δ/ppm = 165.7, 151.5, 148.1, 147.2, 145.4, 136.9, 132.1, 131.6, 130.1, 128.1, 127.5, 126.1, 126.0, 124.2, 121.7, 121.0, 117.3, 116.6, 116.2, 38.6, 35.0; Found: C, 74.30; H, 4.58; N, 16.62. Calc. for C21H16N4O: C, 74.10; H, 4.74; N, 16.46%; MS: m/z= 341.1408 ([M+H]+).
3.5. Antiproliferative activity
3.5.1. Cell culture and reference compounds
Human cancer cell lines used in this manuscript, namely Capan-1, HCT-116, NCI-H460, LN-229, LS513, HL-60, K-562 and Z-138 were acquired from the American Type Culture Collection (ATCC, Manassas, VA, USA), while the DND-41 cell line was purchased from the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ Leibniz-Institut, Germany). Culture media were purchased from Gibco Life Technologies, USA, and supplemented with 10% fetal bovine serum (HyClone, GE Healthcare Life Sciences, USA). Etoposide and doxorubicin, which were used as reference inhibitors, were purchased from Selleckchem (Munich, Germany). Stock solutions were prepared in DMSO.
3.5.2. Proliferation assays
Adherent cell lines Hap-1, HCT-116, NCI-H460, and Capan-1 cells were seeded at a density between 500 and 1500 cells per well, in 384-well tissue culture plates (Greiner). After overnight incubation, cells were treated with seven different concentrations of the test compounds, ranging from 100 to 0.006µM. Suspension cell lines HL-60, K-562, Z-138, MM.1S and DND-41 were seeded at densities ranging from 2500 to 5500 cells per well in 384-well culture plates containing the test compounds at the same concentration points. Cells were incubated for 72 hours with compounds and were then analyzed using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) reagent (Promega) according to the manufacturer’s instructions. Absorbance of the samples was measured at 490 nm using a SpectraMax Plus 384 (Molecular Devices), and OD values were used to calculate the 50% inhibitory concentration (IC50). Compounds were tested in at least two independent experiments.
3.5.3. High content imaging analysis of cell cycle distribution
Capan-1 and NCI-H460 cells were seeded at 5000 cells per well in 96 well clear flat bottom tissue culture plates. After overnight incubation, the cells were treated with the test compounds at different concentrations for 24 h. Cells were then fixed with 4% PFA in PBS for 10 min, washed and the nuclei stained by adding a solution of 300 nM 4′,6-diamidino-2-phenylindole (DAPI, Molecular Probes). The plates were imaged on a CX5 High Content Screening device (ThermoFisher Scientific), using the Cell Cycle Analysis bio-application. A minimal of 500 cells was imaged for each well.
3.5.4. Caspase-3/7 Activity
Caspase activity was assayed by measuring light intensity using Caspase-Glo® 3/7 (Promega Corporation, Fitchburg, WI, USA) according to the manufacturer’s protocol. Briefly, Capan-1 and NCI-H460 cells were seeded at 5000 cells per well in white 96-well microplates and treated with the test compounds at different concentrations for 24 h. Caspase-Glo reagent was added and incubated at room temperature for 30 minutes. The caspase activity was then measured using the GloMax®-96 (Promega Corporation, Fitchburg, WI, USA) microplate luminometer, recording the mean of relative light units (RLU) per well. Consequently, the nuclei were stained by adding Hoechst 33342 (ThermoFisher Scientific) at a final concentration of 1 µg/mL, and after another 30 minute incubation period a CX5 High Content Screening device (ThermoFisher Scientific) was employed to measure the total amount of cells per well to facilitate the calculation of the apoptotic rate.
3.6. Binding with nucleic acids
The UV/Vis spectra were recorded on a Varian Cary 100 Bio spectrophotometer, CD spectra on JASCO J815 spectrophotometer and fluorescence spectra on a Varian Cary Eclipse spectrophotometer at 25oC using appropriate 1cm path quartz cuvettes. Calf thymus DNA, ctDNA and rArU were purchased in Sigma-Aldrich and dissolved in Na-cacodylate buffer, I=0.05 mol dm-3, pH=7.0. The ctDNA was additionally sonicated and filtered through a 0.45 mm filter [36]. DNA and RNA concentration was determined spectroscopically as the concentration of phosphates [37].
Spectrophotometric titrations were performed at pH=7.0 (I=0.05 mol dm-3, sodium cacodylate buffer) by adding portions of polynucleotide solution into the solution of the studied compound for fluorimetric experiments and CD experiments were done by adding portions of the compound stock solution into the solution of a polynucleotide. In fluorimetric experiments excitation wavelength of λexc= >300 nm was used to avoid the inner filter effect caused due to increasing absorbance of the polynucleotide. Emission was collected in the range λem=450–750 nm. Values for Ka were obtained by processing titration data using the Scatchard equation (Table 2), most of them have satisfactory correlation coefficients (>0.99). Thermal melting curves for DNA and their complexes with studied compounds were determined as previously described by following the absorption change at 260 nm as a function of temperature. The absorbance of the ligands was subtracted from every curve and the absorbance scale was normalized. Tm values are the midpoints of the transition curves determined from the maximum of the first derivative and checked graphically by the tangent method. The ΔTm values were calculated subtracting Tm of the free nucleic acid from Tm of the complex. Every ΔTm value here reported was the average of at least two measurements. The error in ΔTm is ±0.5oC.
4. Conclusion
This manuscript details the synthesis, structural characterization, assessment of anti-proliferative activity, and examination of DNA/RNA binding properties in pentacyclic benzimidazole derivatives containing amino or amido side chains. The placement of amino side chains was at positions 7 or 11 of the pentacyclic skeleton, whereas amido side chains were positioned at positions 6 or 7, with variations in the positioning of the N atom on the quinoline nuclei. The synthesized compounds underwent in vitro evaluation for their antiproliferative activity, and for the most promising compounds, investigations were carried out to elucidate their binding modes with DNA/RNA, aiming to uncover their mechanisms of biological action. Synthesis of the targeted compounds employed conventional organic synthesis techniques alongside environmentally friendly methods such as photochemical cyclization or microwave-assisted amination.
Only soluble compounds were subjected to in vitro testing for their antiproliferative effects on various human cancer cell lines. The most promising compound identified was the N,N-dimethylaminopropyl-substituted derivative 6, carrying an amino side chain at position 7 of the pentacyclic skeleton. This compound exhibited noteworthy inhibition of proliferation across multiple cancer cell lines, with inhibitory concentrations ranging from 0.3 to 0.6 µM. Additionally, structural analogue 19, featuring an amino side chain at position 11 of the pentacyclic skeleton, also demonstrated potent activity, particularly against K-562 and Z-138 cancer cells, with inhibitory concentrations ranging from 0.4 to 0.6 µM. Moreover, both the 7- and 11-piperazinyl-substituted pentacyclic derivatives, compounds 9 and 22, respectively, showcased robust and extensive antitumoral activity. In summary, the results obtained suggest that the substituent positioned at position 7 predominantly influenced the observed antiproliferative activities.
Thermal melting experiments, fluorescence, and circular dichroism spectroscopy collectively indicate a dual binding mode—comprising intercalation and the binding of aggregated compounds along the polynucleotide backbone—for the examined drugs concerning both DNA and RNA. The strength of these interactions with DNA/RNA was notably influenced by the positioning of the amino substituent on the pentacyclic ring. Specifically, derivatives 19 and 22, featuring an amino substituent (piperazine or N,N-dimethylaminopropylamino) at C-11, demonstrated enhanced binding affinities and thermal stabilization effects towards double-stranded DNA (ds-DNA) and double-stranded RNA (ds-RNA) in comparison to derivatives 6 and 9 with substituents at C-7 (as outlined in Table 2). The considerable binding affinities observed for compounds 19 and 22 suggest that direct interaction with DNA/RNA might represent their biological targets. Additional experiments to evaluate the effects of the most promising pentacyclic benzimidazole derivatives 6 and 19 on both cancer cell cycle regulation and caspase-3/7 activity yielded results paralleling those of the included reference drug doxorubicin, also confirming a similar mode of action by direct interaction with nucleic acids.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org; Spectroscopic characterization of pentacyclic benzimidazoles in aqueous solutions (Figures S1-S8); Interactions of pentacyclic benzimidazoles with ds-polynucleotides in neutral medium (pH=7.0): Fluorimetric titrations (Figures S9-S14); Thermal melting experiments (Figures S15-S16); Circular dichroism (CD) titrations (Figure S17); 1H and 13C NMR spectra of synthesized compounds (Figures S18-S59).
Author Contributions
Conceptualization, M.H.; methodology, N.P., M.K., L.P., I.F. and M.R.S.; software, L.P. and M.R.S.; formal analysis, N.P.; investigation, N.P., L.P., D.D. and M.R.S.; resources, M.R.S. and M.H.; data curation, L.P.; writing—original draft preparation, N.P., L.P., D.D., M.R.S. and M.H; writing—review and editing, L.P., D.D., M.R.S. and M.H; supervision, M.H.; All authors have read and agreed to the published version of the manuscript.
Funding
We greatly appreciate the financial support of the Croatian Science Foundation under the projects IP-2018-01-4379 and IP-2018-01-4694.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Additional data are available on request.
Acknowledgments
We greatly appreciate the financial support of the Croatian Science Foundation under the projects IP-2018-01-4379 and IP-2018-01-4694.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kumar, A.; Singh, A.K.; Singh, H.; Vijayan, V.; Kumar, D.; Naik, J.; Thareja, S.; Yadav, J.P.; Pathak, P.; Grishina, M.; et al. Nitrogen Containing Heterocycles as Anticancer Agents: A Medicinal Chemistry Perspective. Pharmaceuticals 2023, 16, 299. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Zadsirjan, V. Prescribed drugs containing nitrogen heterocycles: An overview. RSC Adv. 2020, 10, 44247. [Google Scholar] [CrossRef] [PubMed]
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.K.; Kaur, R.; Arora, R.; Saini, B.; Arora, S. Nitrogen-Containing Heterocycles as Anticancer Agents: An Overview. Anticancer Agents Med. Chem. 2020, 20, 2150–2168. [Google Scholar] [CrossRef] [PubMed]
- Nehra, B.; Mathew, B.; Chawla, P.A. A Medicinal Chemist’s Perspective Towards Structure Activity Relationship of Heterocycle Based Anticancer Agents. Curr. Top Med. Chem. 2022, 22, 493. [Google Scholar] [CrossRef]
- Vušak, D.; Perin, N.; Martin-Kleiner, I.; Kralj, M.; Karminski-Zamola, G.; Hranjec, M.; Bertoša, B. Synthesis and antiproliferative activity of amino-substituted benzimidazo[1,2-a]quinolines as mesylate salts designed by 3D-QSAR analysis. Mol. Divers. 2017, 21, 621. [Google Scholar] [CrossRef] [PubMed]
- Meščić Macan, A.; Perin, N.; Jakopec, S.; Mioč, M.; Radić Stojković, M.; Kralj, M.; Hranjec, M.; Raić-Malić, S. Synthesis, antiproliferative activity and DNA/RNA-binding properties of mono- and bis-(1,2,3-triazolyl)-appended benzimidazo[1,2-a]quinoline derivatives. Eur. J. Med. Chem. 2020, 185, 111845. [Google Scholar] [CrossRef] [PubMed]
- Perin, N.; Babić, D.; Kassal, P.; Čikoš, A.; Hranjec, M.; Vianello, R. Spectroscopic and Computational Study of the Protonation Equilibria of Amino-Substituted benzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazoles as Novel pH-Sensing Materials. Chemosensors 2022, 10, 21. [Google Scholar] [CrossRef]
- Akhtar, W.; Faraz Khan, M.; Verma, G.; Shaquiquzzaman, M.; Rizvi, M.A.; Mehdi, S.H.; Akhter, M.; Mumtaz Alam, M. Therapeutic evolution of benzimidazole derivatives in the last quinquennial period. Eur. J. Med. Chem. 2017, 126, 705–753. [Google Scholar] [CrossRef]
- Venugopal, S.; Sharma, V.; Mehra, A.; Singh, I.; Singh, G. DNA intercalators as anticancer agents. Chem Biol Drug Des. 2022, 100, 580. [Google Scholar] [CrossRef]
- Sharma, V.; Gupta, M.; Kumar, P.; Sharma, A. A Comprehensive Review on Fused Heterocyclic as DNA Intercalators: Promising Anticancer Agents. Curr Pharm Des. 2021, 27, 15. [Google Scholar] [CrossRef] [PubMed]
- Simon, Z.; Voiculetz, N.; Motoc, I. Specific Interaction and Biological Recognition Processes; CRC Press: Boca Raton, FL, USA, 1993. [Google Scholar]
- Xu, J.; Wang, G.A.; Gao, L.; Wu, L.; Lei, Q.; Deng, H.; Li, F. Enabling programmable dynamic DNA chemistry using small-molecule DNA binders. Nat Commun. 2023, 14, 4248. [Google Scholar] [CrossRef]
- Sischka, A.; Toensing, K.; Eckel, R.; Wilking, S.D.; Sewald, N.; Ros, R.; Anselmetti, D. Molecular mechanisms and kinetics between DNA and DNA binding ligands. Biophys. J. 2005, 88, 404–411. [Google Scholar] [CrossRef]
- Mekheimer, R.A.; Al-Sheikh, M.A.; Medrasi, H.Y.; Sadek, K.U. Advancements in the synthesis of fused tetracyclic quinoline derivatives. RSC Adv. 2020, 10, 19867. [Google Scholar] [CrossRef] [PubMed]
- Ajani, O.O.; Iyaye, K.T.; Ademosun, O.T. Recent advances in chemistry and therapeutic potential of functionalized quinoline motifs—a review. RSC Adv. 2022, 12, 18594–18614. [Google Scholar] [CrossRef] [PubMed]
- Shiro, T.; Fukaya, T.; Tobe, M. The chemistry and biological activity of heterocycle-fused quinoline derivatives: A review. Eur. J. Med. Chem. 2015, 97, 397–408. [Google Scholar] [CrossRef]
- Perin, N.; Martin-Kleiner, I.; Nhili, R.; Laine, W.; David-Cordonnier, M.-H.; Vugrek, O.; Karminski-Zamola, G.; Kralj, M.; Hranjec, M. Biological activity and DNA binding studies of 2-substituted benzimidazo[1,2-a]quinolines bearing different amino side chains. Med. Chem. Comm. 2013, 4, 1537–1550. [Google Scholar] [CrossRef]
- Perin, N.; Nhili, R.; Ester, K.; Laine, W.; Karminski-Zamola, G.; Kralj, M.; David-Cordonnier, M.-H.; Hranjec, M. Synthesis, antiproliferative activity and DNA binding properties of novel 5-aminobenzimidazo[1,2-a]quinoline-6-carbonitriles. Eur. J. Med. Chem. 2014, 80, 218–227. [Google Scholar] [CrossRef]
- Perin, N.; Nhili, R.; Cindrić, M.; Bertoša, B.; Vušak, D.; Martin-Kleiner, I.; Laine, W.; Karminski-Zamola, G.; Kralj, M.; David-Cordonnier, M.-H.; et al. Amino substituted benzimidazo[1,2-a]quinolines: Antiproliferative potency, 3D QSAR study and DNA binding properties. Eur. J. Med. Chem. 2016, 122, 530–545. [Google Scholar] [CrossRef]
- Lončar, B.; Perin, N.; Mioč, M.; Boček, I.; Grgić, L.; Kralj, M.; Tomić, S.; Radić Stojković, M.; Hranjec, M. Novel amino substituted tetracyclic imidazo[4,5-b]pyridine derivatives: Design, synthesis, antiproliferative activity and DNA/RNA binding study. Eur. J. Med. Chem. 2021, 217, 113342. [Google Scholar] [CrossRef]
- Hranjec, M.; Lučić, B.; Ratkaj, I.; Kraljević Pavelić, S.; Piantanida, I.; Pavelić, K.; Karminski-Zamola, G. Novel Imidazo[4,5-b]Pyridine and Triaza-Benzo[c]Fluorene Derivatives: Synthesis, Antiproliferative Activity and DNA Binding Studies. Eur. J. Med. Chem 2011, 46, 2748–2758. [Google Scholar] [CrossRef]
- Perin, N.; Bobanović, K.; Zlatar, I.; Jelić, D.; Kelava, V.; Koštrun, S.; Gabelica Marković, V.; Brajša, K.; Hranjec, M. Antiproliferative activity of amino substituted benzo[b]thieno[2,3-b]pyrido[1,2-a]benzimidazoles explored by 2D and 3D cell culture system. Eur. J. Med. Chem 2017, 125, 722–735. [Google Scholar] [CrossRef]
- Hranjec, M.; Piantanida, I.; Kralj, M.; Šuman, L.; Pavelić, K.; Karminski-Zamola, G. Novel amidino-substituted thienyl- and furyl-vinyl-benzimidazole derivatives and their photochemical conversion into corresponding diaza-cyclopenta[c]fluorenes. Synthesis, interactions with DNA and RNA and antitumor evaluation. Part 4. J. Med. Chem. 2008, 51, 4899–4910. [Google Scholar] [CrossRef]
- Brajša, K.; Vujasinović, I.; Jelić, D.; Trzun, M.; Zlatar, I.; Karminski-Zamola, G.; Hranjec, M. Antitumor activity of amidino-substituted benzimidazole and benzimidazo[1,2-a]quinoline derivatives tested in 2D and 3D cell culture systems. J. Enzym. Inhib. Med. Chem. 2016, 31, 1139–1145. [Google Scholar] [CrossRef] [PubMed]
- Perin, N.; Alić, J.; Liekens, S.; Van Aerschot, A.; Vervaeke, P.; Gadakh, B.; Hranjec, M. Different positions of amide side chains on the benzimidazo[1,2-a]quinoline skeleton strongly influenced biological activity. New J. Chem. 2018, 42, 7096–7104. [Google Scholar] [CrossRef]
- Cindrić, M.; Jambon, S.; Harej, A.; Depauw, S.; David-Cordonnier, M.-H.; Kraljević Pavelić, S.; Karminski-Zamola, G.; Hranjec, M. Novel amidino substituted benzimidazole and benzothiazole benzo[b]thieno-2-carboxamides exert strong antiproliferative and DNA binding properties. Eur. J. Med. Chem 2017, 136, 468–479. [Google Scholar] [CrossRef]
- Saenger, W. Principles of Nucleic Acid Structure; Springer: New York, NY, USA, 1984. [Google Scholar]
- Cantor, C.R. Techniques for the Study of Biological Structure and Function; W. H. Freeman: San Francisco, CA, USA, 1980. [Google Scholar]
- Neidle, S. Oxford Handbook of Nucleic acid Structure; Oxford University Press: Oxford, UK, 1999. [Google Scholar]
- Mergny, J.L.; Lacroix, L. Analysis of thermal melting curves. Oligonucleotides 2003, 13, 515–537. [Google Scholar] [CrossRef] [PubMed]
- Scatchard, G. The attractions of proteins for small molecules and ions. Ann. N. Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar] [CrossRef]
- McGhee, J.D.; von Hippel, P.H. Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974, 86, 469–489. [Google Scholar] [CrossRef]
- Berova, N.; Nakanishi, K.; Woody, R.W. Circular Dichroism Principles and Applications, 2nd ed.; Wiley-VCH: New York, NY, USA, 2000. [Google Scholar]
- Smidlehner, T.; Piantanida, I.; Pescitelli, G. Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids—Tutorial. Beilstein J. Org. Chem. 2017, 14, 84–105. [Google Scholar] [CrossRef]
- Chaires, J.B.; Dattagupta, N.; Crothers, D.M. Studies on Interaction of Anthracycline Antibiotics and Deoxyribonucleic-Acid—Equilibrium Binding-Studies on Interaction of Daunomycin with Deoxyribonucleic-Acid. Biochemistry 1982, 17, 3933–3940. [Google Scholar] [CrossRef]
- Chalikian, T.V.; Volker, J.; Plum, G.E.; Breslauer, K.J. A more unified picture for the thermodynamics of nucleic acid duplex melting: A characterization by calorimetric and volumetric techniques. Proc. Natl. Acad. Sci. USA 1999, 14, 7853–7858. [Google Scholar] [CrossRef]
Figure 1.
Previously synthesized benzimidazo[1,2-a]quinolines (a), tetracyclic imidazo[4,5-b]pyridines (b) and pentacyclic benzo[b]thieno[2,3-b]- pyrido[1,2-a]benzimidazoles (c) with promising biological potential.
Figure 1.
Previously synthesized benzimidazo[1,2-a]quinolines (a), tetracyclic imidazo[4,5-b]pyridines (b) and pentacyclic benzo[b]thieno[2,3-b]- pyrido[1,2-a]benzimidazoles (c) with promising biological potential.
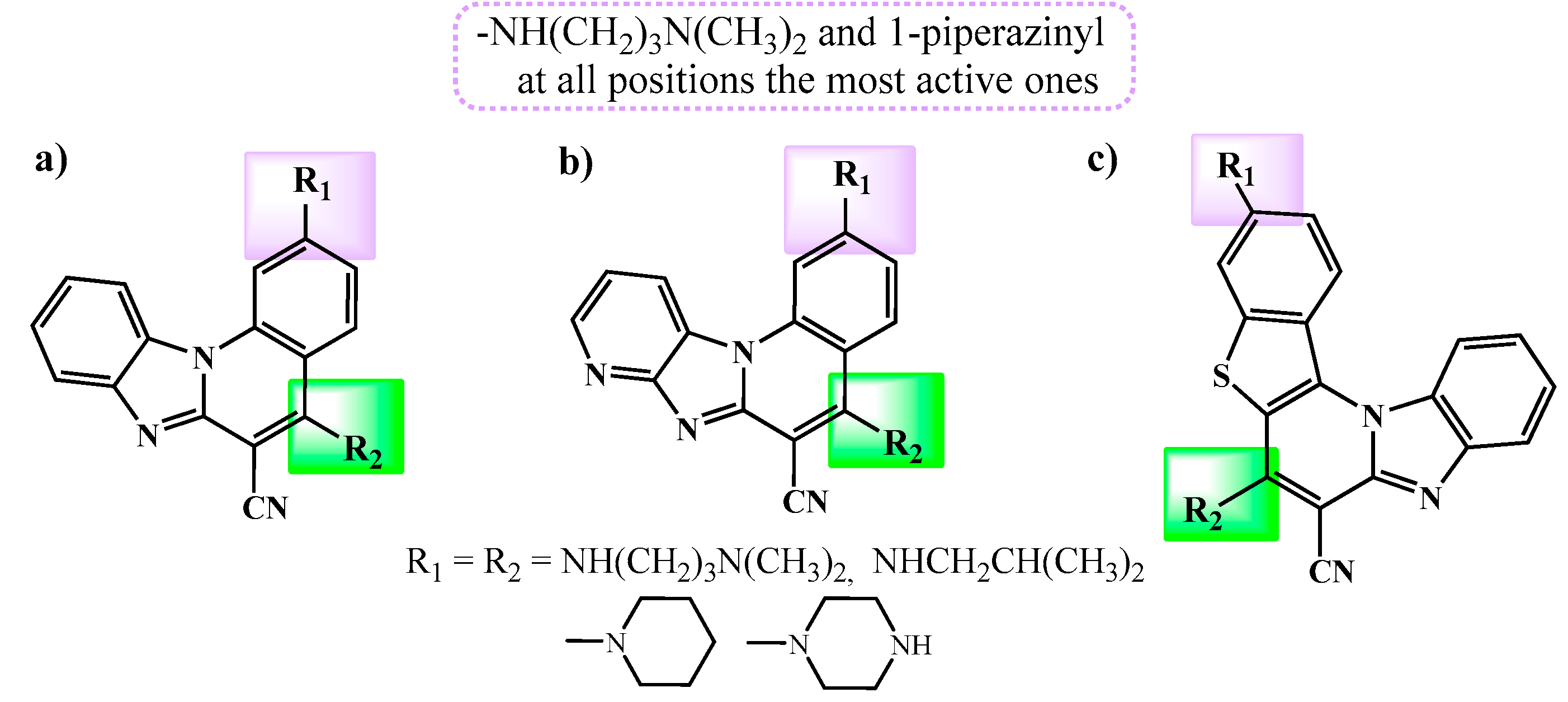
Scheme 1.
Synthesis of amino substituted pentacyclic benzimidazole derivatives 6-9.
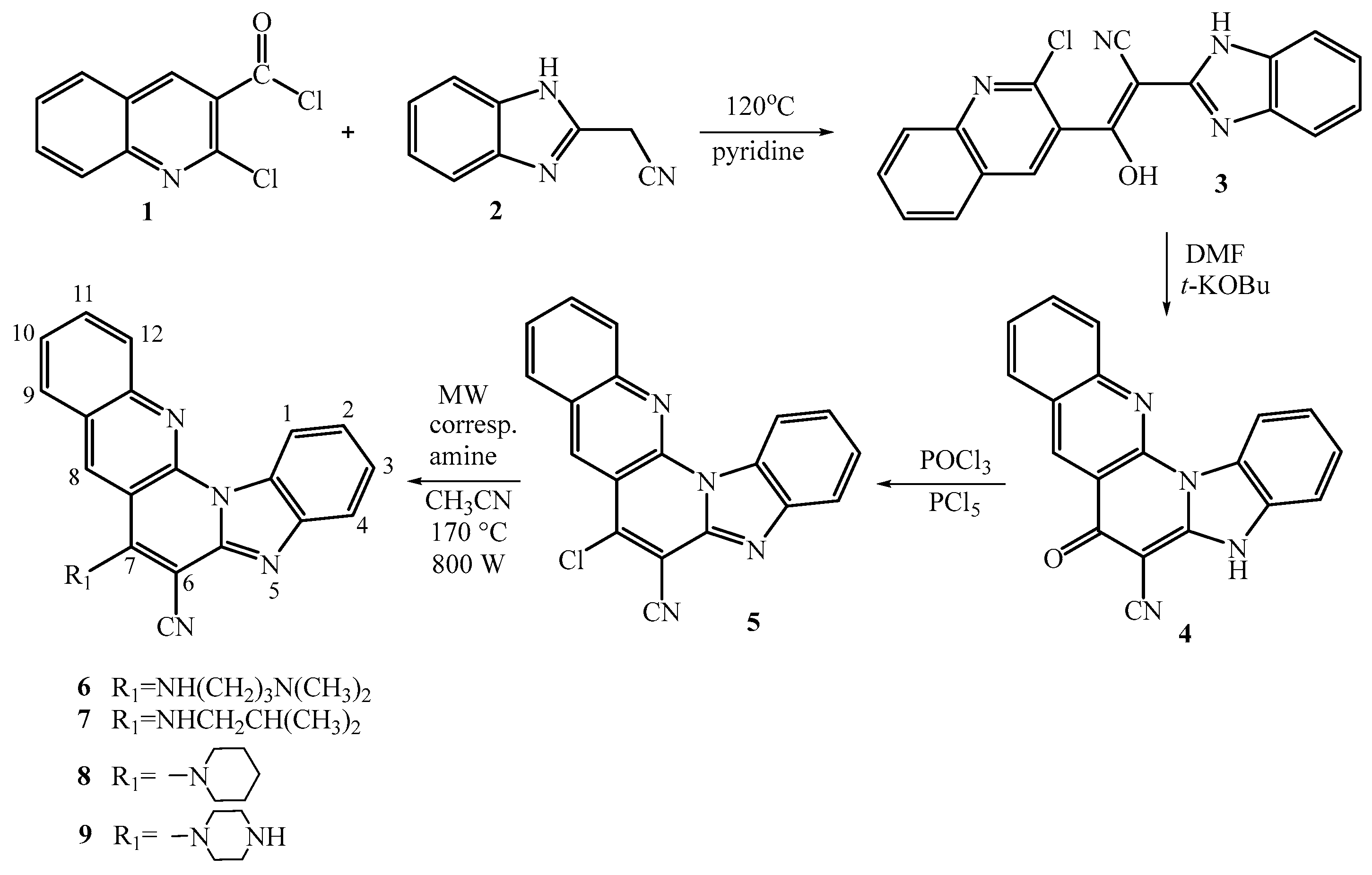
Scheme 2.
Synthesis of amino 19-22 and amido substituted 25-26 and 29-30 pentacyclic benzimidazole derivatives.
Scheme 2.
Synthesis of amino 19-22 and amido substituted 25-26 and 29-30 pentacyclic benzimidazole derivatives.
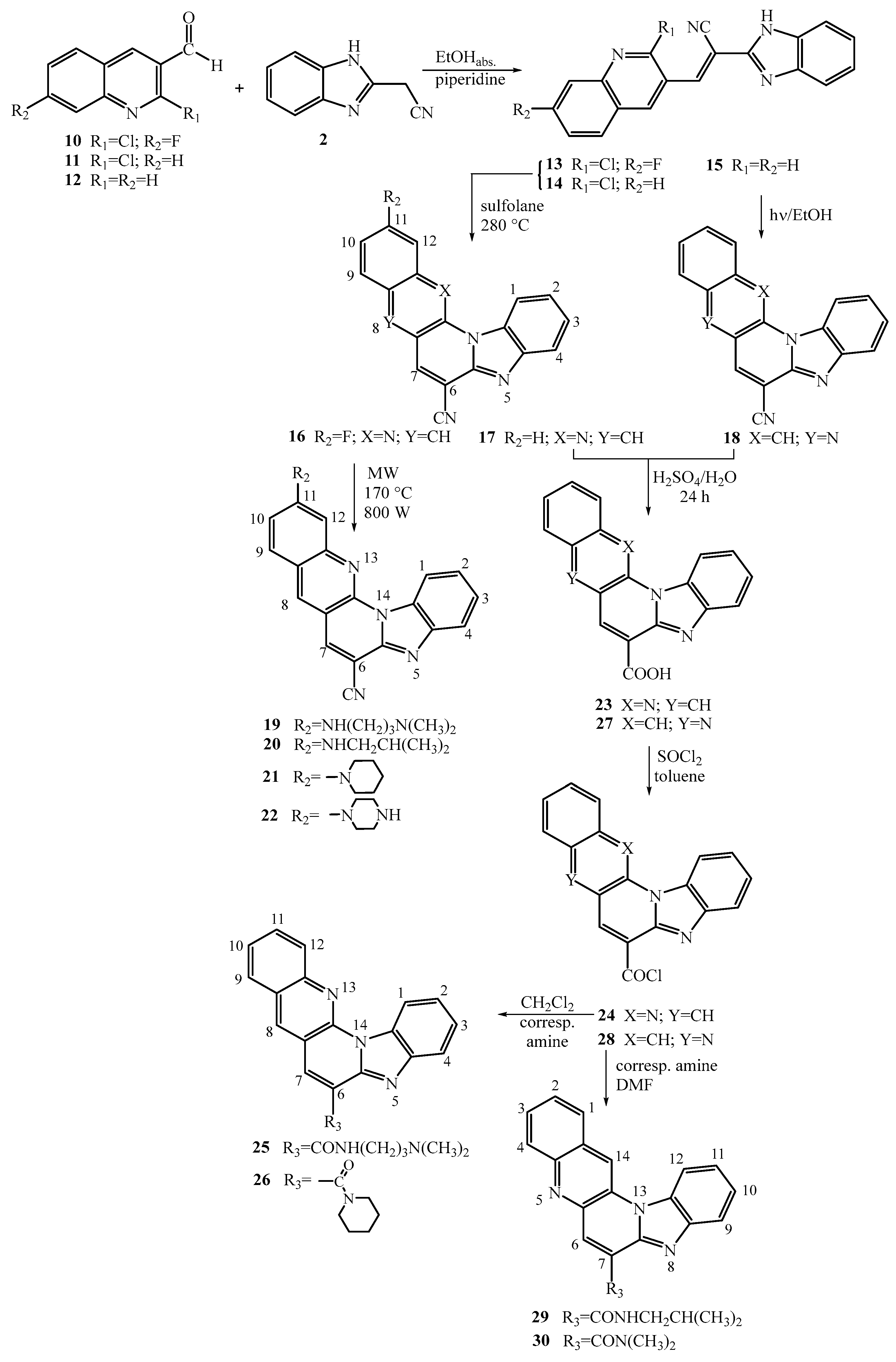
Figure 2.
Structure-activity relationship for antiproliferative activity.
Figure 3.
A) UV/Vis spectra of 6, 9, 19 and 22 (c = 2.0 × 10-5 mol dm-3) at pH=7.0, sodium cacodylate buffer, I=0.05 M; B) Normalized emission spectra of 6 (λexc=392nm), 9 (λexc=406nm), 19 (λexc=447nm) and 22 (λexc=436nm), and at concentration, c =1.0 × 10-6 mol dm-3; at pH=7.0, Na cacodylate buffer, I=0.05 mol dm-3.
Figure 3.
A) UV/Vis spectra of 6, 9, 19 and 22 (c = 2.0 × 10-5 mol dm-3) at pH=7.0, sodium cacodylate buffer, I=0.05 M; B) Normalized emission spectra of 6 (λexc=392nm), 9 (λexc=406nm), 19 (λexc=447nm) and 22 (λexc=436nm), and at concentration, c =1.0 × 10-6 mol dm-3; at pH=7.0, Na cacodylate buffer, I=0.05 mol dm-3.
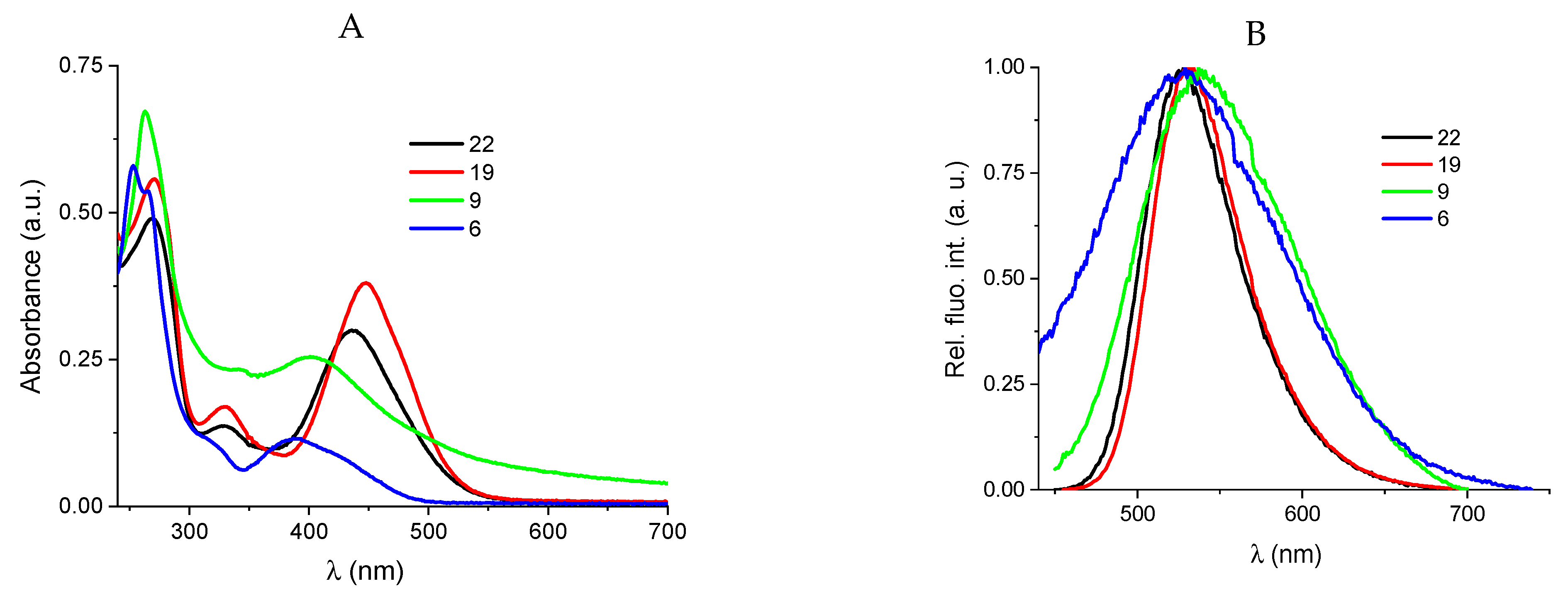
Figure 4.
Melting curve of RNA upon addition of compounds, with ratio, r ([compound/ [polynucleotide])=0.1 of 19 and 22 at pH=7.0 (buffer sodium cacodylate, I=0.05 mol dm-3).
Figure 4.
Melting curve of RNA upon addition of compounds, with ratio, r ([compound/ [polynucleotide])=0.1 of 19 and 22 at pH=7.0 (buffer sodium cacodylate, I=0.05 mol dm-3).
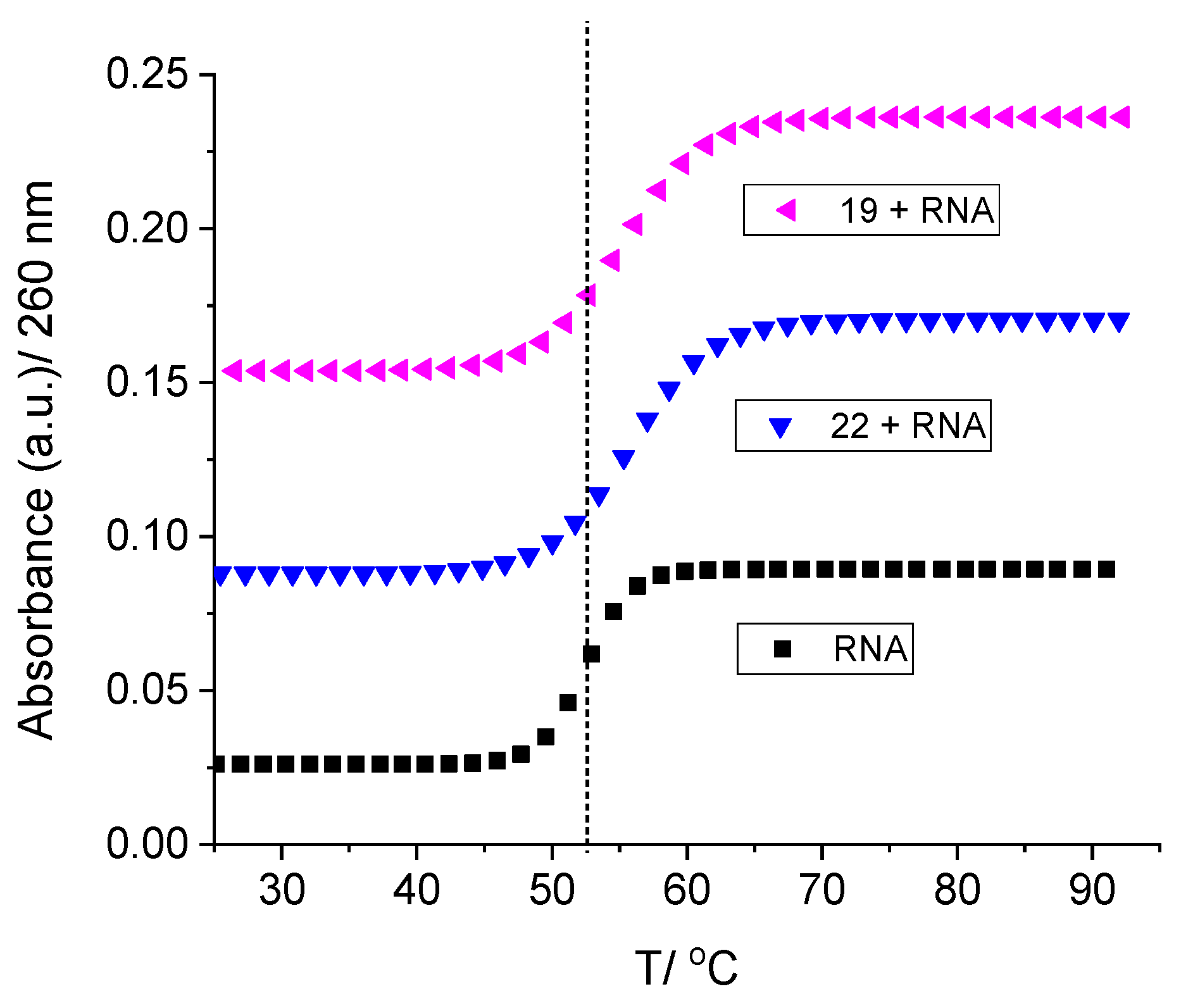
Figure 5.
Changes in fluorescence spectrum of compound 22 (c=1.0 × 10-6 mol dm-3, λexc=436 nm) upon titration with ctDNA (c=5.0 × 10-7 – 8 × 10-6 mol dm-3); Inset: Dependence of fluorescence of 22 at λem = 537 nm on c(ctDNA); (buffer sodium cacodylate, pH = 7.0, I = 0.05 mol dm-3).
Figure 5.
Changes in fluorescence spectrum of compound 22 (c=1.0 × 10-6 mol dm-3, λexc=436 nm) upon titration with ctDNA (c=5.0 × 10-7 – 8 × 10-6 mol dm-3); Inset: Dependence of fluorescence of 22 at λem = 537 nm on c(ctDNA); (buffer sodium cacodylate, pH = 7.0, I = 0.05 mol dm-3).

Figure 6.
CD spectra of ctDNA (c=3.0 × 10-5 mol dm-3) treated with 19 at different molar ratios r=[compound]/[nucleotide phosphate] indicated in the graph legend (pH=7.0, buffer sodium cacodylate, I=0.05 mol dm-3).
Figure 6.
CD spectra of ctDNA (c=3.0 × 10-5 mol dm-3) treated with 19 at different molar ratios r=[compound]/[nucleotide phosphate] indicated in the graph legend (pH=7.0, buffer sodium cacodylate, I=0.05 mol dm-3).
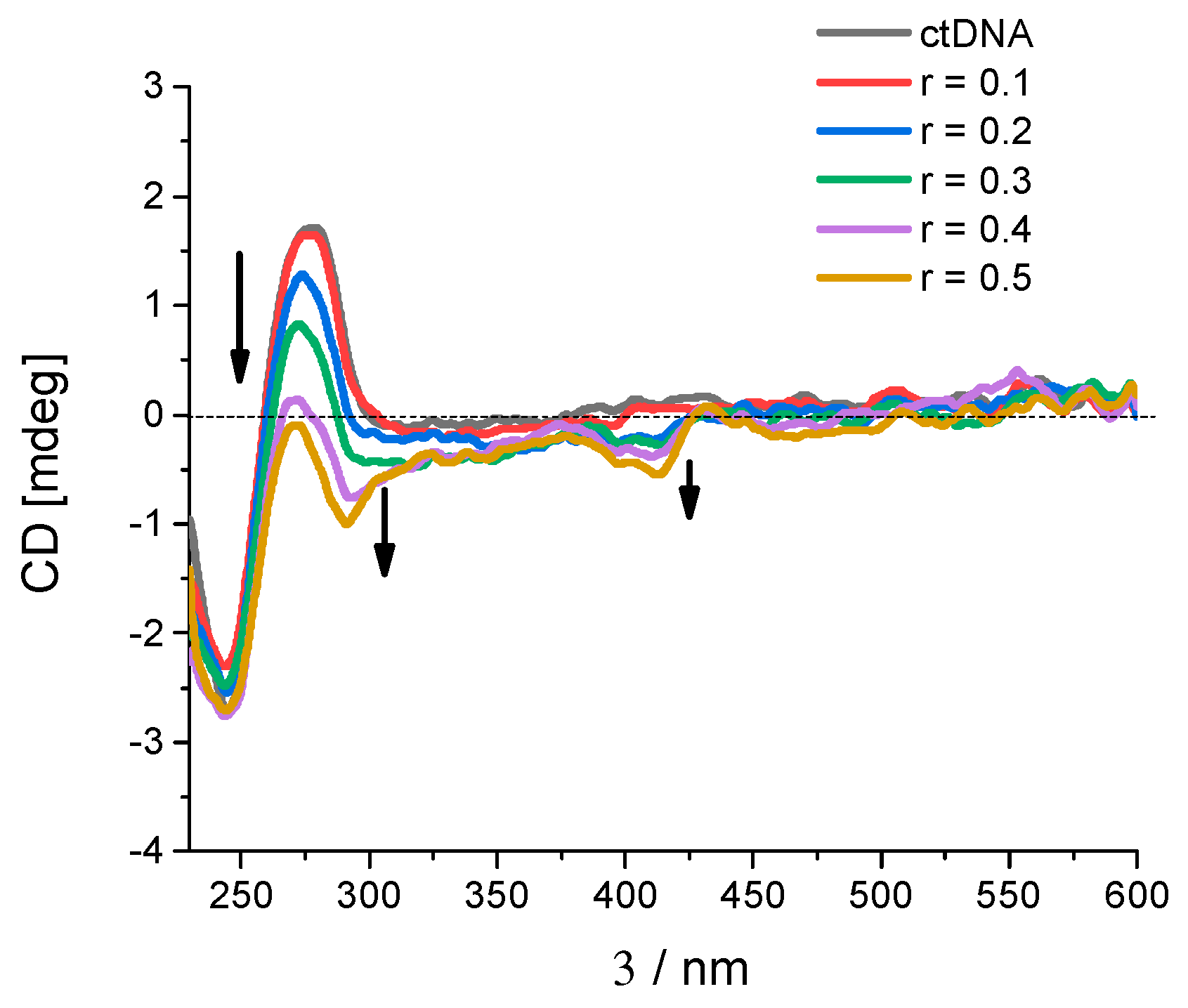
Figure 7.
Effect of the synthesized pentacyclic benzimidazole derivatives on cell cycle regulation: Cancer cells were treated with derivative 6 (left), 19 (middle) or doxorubicin (DOXO, right) at the indicated doses for 24 h, after which the percentage of cells in each phase of the cell cycle was assessed through high content imaging of Capan-1 cells (A) and NCI-H460 cells (B). The data represent the mean ± SD of two independent experiments conducted in duplicate. UTC: untreated control.
Figure 7.
Effect of the synthesized pentacyclic benzimidazole derivatives on cell cycle regulation: Cancer cells were treated with derivative 6 (left), 19 (middle) or doxorubicin (DOXO, right) at the indicated doses for 24 h, after which the percentage of cells in each phase of the cell cycle was assessed through high content imaging of Capan-1 cells (A) and NCI-H460 cells (B). The data represent the mean ± SD of two independent experiments conducted in duplicate. UTC: untreated control.
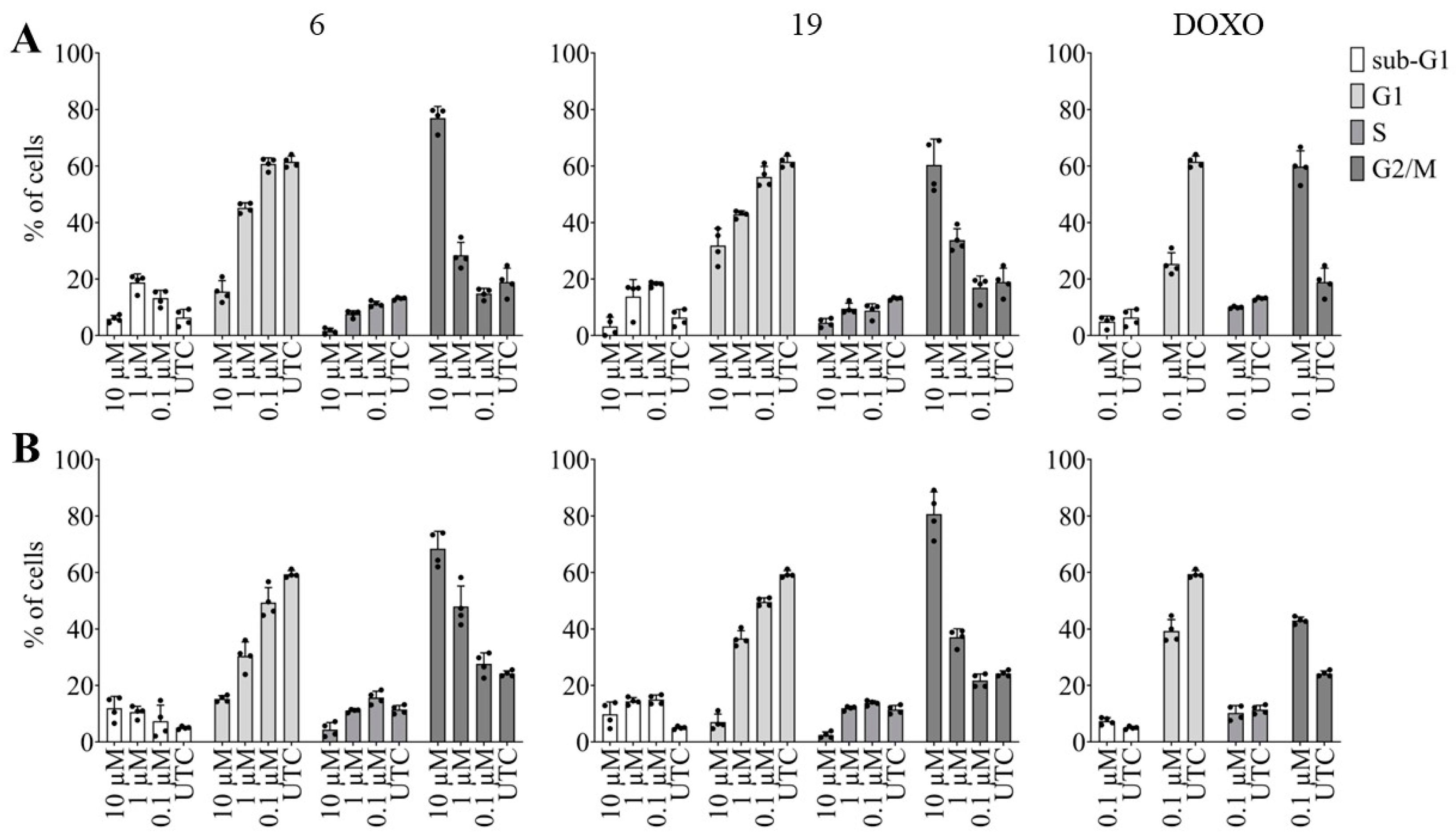
Figure 8.
Effect of the synthesized pentacyclic benzimidazole derivatives on caspase-3 and -7 activity: Cancer cells were treated with derivative 6 (left), 19 (middle) or doxorubicin (DOXO, right) at the indicated doses for 24 h, after which caspase-3 and -7 activity was measured using a Caspase-Glo 3/7 assay in Capan-1 cells (A) and NCI-H460 cells (B). The apoptotic rate was calculated and represented as the relative fold change to the untreated control cells (UTC). The data represent the mean ± SD of two independent experiments conducted in duplicate.
Figure 8.
Effect of the synthesized pentacyclic benzimidazole derivatives on caspase-3 and -7 activity: Cancer cells were treated with derivative 6 (left), 19 (middle) or doxorubicin (DOXO, right) at the indicated doses for 24 h, after which caspase-3 and -7 activity was measured using a Caspase-Glo 3/7 assay in Capan-1 cells (A) and NCI-H460 cells (B). The apoptotic rate was calculated and represented as the relative fold change to the untreated control cells (UTC). The data represent the mean ± SD of two independent experiments conducted in duplicate.


Table 1.
Antiproliferative activity of tested compounds in vitro.
| Cpd | IC50 /μM * | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cell line | |||||||||
| Capan-1 | HCT-116 | Hap-1 | NCI-H460 | DND-41 | HL-60 | K-562 | MM.1S | Z-138 | |
| 5 | 5.8 ±2.5 | 16.5 ±0.7 | 23.1 ±0.1 | 29.5 ±2.5 | 4.9 ±0.2 | 5.2 ±1.3 | 64.1 ±3.4 | 12.9 ±0.8 | 2.4 ±0.3 |
| 6 | 0.3 ±0.1 | 1.8 ±0.6 | 1.2 ±0.2 | 1.0 ±0.8 | 0.5 ±0.0 | 0.5 ±0.1 | 0.3 ±0.0 | 0.6 ±0.0 | 0.4 ±0.1 |
| 7 | ≥44.3 | >100 | 34.2 ±2.9 | 41.4 ±4.0 | ≥86.4 | 28.5 ±4.5 | >100 | >100 | 34.4 ±2.3 |
| 8 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 9 | 2.3 ±0.1 | 5.6 ±2.1 | 5.5 ±2.1 | 5.2 ±2.8 | 2.0 ±0.3 | 3.5 ±2.9 | 3.0 ±1.9 | 5.6 ±3.7 | 1.5 ±0.1 |
| 16 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 17 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 | >100 |
| 18 | 27.2 ±2.1 | 41.9 ±5.7 | 22.2 ±0.5 | 32.5 ±8.1 | 62.3 ±3.3 | 12.5 ±2.3 | 16.3 ±2.5 | >100 | 36.0 ±7.0 |
| 19 | 1.0 ±0.5 | 5.9 ±1.9 | 6.5 ±1.3 | 1.1 ±0.3 | 1.3 ±0.9 | 1.7 ±0.2 | 0.4 ±0.0 | 2.4 ±0.5 | 0.6 ±0.3 |
| 20 | >100 | ≥85.1 | >100 | 30.5 ±2.5 | >100 | >100 | >100 | >100 | >100 |
| 21 | 78.8 ±8.4 | >100 | >100 | 47.6 ±2.5 | >100 | >100 | >100 | >100 | >100 |
| 22 | 1.2 ±0.2 | 6.2 ±0.9 | 5.9 ±2.1 | 5.4 ±2.2 | 2.1 ±0.1 | 2.0 ±0.2 | 8.4 ±0.7 | 2.9 ±0.1 | 1.4 ±0.2 |
| 25 | 5.8 ±2.4 | 5.5 ±2.1 | 28.5 ±5.2 | 7.8 ±1.0 | 4.4 ±2.6 | 4.2 ±1.7 | 5.3 ±4.2 | 11.9 ±0.9 | 2.1 ±0.4 |
| 26 | 47.4 ±8.1 | >100 | 49.8 ±4.8 | ≥70.4 | >100 | >100 | >100 | >100 | 84.9 ±6.6 |
| 29 | ≥41.7 | ≥49.8 | 15.9 ±2.5 | 23.4 ±2.5 | >100 | ≥61.2 | >100 | >100 | 49.1 ±2.5 |
| 30 | 36.2 ±1.3 | 44.4 ±0.4 | 25.5 ±4.9 | 26.6 ±0.8 | ≥86.1 | 49.5 ±6.4 | >100 | ≥92.1 | 44.4 ±8.6 |
| Etoposide | 0.2 ±0.1 | 3.1 ±0.5 | 0.2 ±0.0 | 5.6 ±0.9 | 0.4 ±0.2 | 1.6 ±1.0 | 9.0 ±1.1 | 0.8 ±0.5 | 0.9 ±0.5 |
* Results are depicted as mean ±SD of two independent experiments.
Table 2.
Binding constants (logKa)a,b for complexes of ligand-DNA/RNA calculated from the fluorescence titrations and ΔTmc values (°C) of DNA/RNA upon addition of ratiod r=0.1 of 6, 9, 19 and 22 at pH 7.0 (sodium cacodylate buffer, I=0.05 mol dm-3).
Table 2.
Binding constants (logKa)a,b for complexes of ligand-DNA/RNA calculated from the fluorescence titrations and ΔTmc values (°C) of DNA/RNA upon addition of ratiod r=0.1 of 6, 9, 19 and 22 at pH 7.0 (sodium cacodylate buffer, I=0.05 mol dm-3).
| ctDNA | rArU | |||
|---|---|---|---|---|
| logKa | ΔTm / oC | logKa | ΔTm / oC | |
| 22 | 6.8 | 2.3 | 7.9 | 4.9 |
| 19 | 6.8 | 2.3 | 8.6 | 1.1 |
| 9 | -b | 1.6 | -e | -e |
| 6 | -b | 1.8 | -e | -e |
a Processing of titration data was performed using Scatchard equation [32,33]; correlation coefficients were >0.99 for most of the calculated K; b small fluorescence changes in titrations of 6 and 9 with DNA disabled accurate calculation of binding constants; c Error in ΔTm: ± 0.5°C; d r = [compound] / [polynucleotide]; e not determined.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



