
Submitted:
12 January 2024
Posted:
15 January 2024
You are already at the latest version
Abstract
Objective: The pleiotropic effect of hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (statins) is responsible for potent defense against inflammatory response. This study evaluated the inhibitory effects of HMG-CoA reductase inhibitors on monosodium urate (MSU)-induced inflammatory response through the regulation of interleukin-37 (IL-37) expression.
Methods: Serum was collected from patients with gout (n = 40) and from healthy controls (n = 30). The mRNA and protein expression of target molecules IL-1b, IL-37, caspase-1, and Smad3 was measured in THP-1 cells stimulated with MSU, atorvastatin, or rosuvastatin using real-time quantitative polymerase chain reaction and Western blot assay. Transfection with IL-1b or Smad3 siRNA in THP-1 cells was used to verify the pharmaceutical effect of statins in uric acid-induced inflammation.
Results: Serum IL-37 level in gout patients was significantly higher than in controls (p < 0.001) and was associated with serum uric acid level (r = 0.382, p = 0.008). THP-1 cells stimulated with MSU markedly induced IL-37 mRNA expression and transition of IL-37 from cytoplasm to nucleus. Recombinant IL-37 treatment dose-dependently inhibited activation of caspase-1 and IL-1b in MSU-induced inflammation. Atorvastatin and rosuvastatin attenuated caspase-1 activation and mature IL-1b expression but augmented translocation of IL-37 from cytoplasm to nucleus. Atorvastatin and rosuvastatin induced phosphorylation of Smad3 in THP-1 cells treated with MSU crystals. Statins potently attenuated translocation of IL-37 from cytoplasm to nucleus in THP-1 cells transfected with Smad3 siRNA compared to cells with negative control siRNA.
Conclusion: This study revealed that statins inhibit MSU-induced inflammatory response through augmentation of phosphorylated Smad3-mediated IL-37 expression in THP-1 cells.
Keywords:
Monosodium urate
; Interleukin-37
; Statin
; Smad3
; Caspase-1
1. Introduction
Gout is a crystal-induced inflammatory disease triggered by excessive deposition of monosodium urate (MSU) in intraarticular and periarticular structures and other body organs [1,2]. Recently, it has been recognized that the innate immune system mediated through activation of NLRP3 inflammasome triggered by MSU crystals is involved in the pathogenesis of gouty arthritis [3,4]. The NLRP3 inflammasome is an intracellular multi-protein signaling platform that leads to proteolytic cleavage of pro-interleukin-1 (pro-IL-1) into mature IL-1, a proinflammatory cytokine mainly responsible for uric acid-induced inflammation. Various molecules negatively regulate the NLRP3 inflammasome activation such as A20, nitric oxide, carbon monoxide, and aryl hydrocarbon receptor [5,6], suggesting that identification of novel negative regulators of NLRP3 inflammasome activation might offer therapeutic options for control aberrant uric acid-induced inflammation.
IL-37, one member of IL-1 cytokine family, is a novel anti-inflammatory cytokine that primarily reduces inflammatory response by suppressing production of proinflammatory cytokines and attenuating the expression of transcriptional cytokines [7,8,9,10]. IL-37 is upregulated by inflammatory stimuli including various proinflammatory cytokines, TLR ligands, or lipopolysaccharide via diverse signal transduction pathway. Clinically, IL-37 plays an important role in the mechanisms that protect against aberrant immune/inflammatory responses in chronic inflammatory and/or autoimmune diseases and against progression and development of diverse tumors. Anti-inflammatory effects of IL-37 have been extensively identified in diverse autoimmune and inflammatory rheumatic diseases such as rheumatoid arthritis (RA) [11,12], systemic lupus erythematosus (SLE) [13], osteoarthritis (OA) [14], and ankylosing spondylitis (AS) [15] through suppression of proinflammatory cytokines or inflammatory cells. Consistently, IL-37 expression in serum and peripheral blood mononuclear cells (PBMCs) in gout patients was also significantly higher than in healthy controls [16,17]. In addition, other experimental studies showed that IL-37 exerted a potent anti-inflammatory effect by reducing release of proinflammatory cytokines in uric acid-induced inflammation [18,19].
Hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors, generally referred to as “statins”, are inhibitors of cholesterol biosynthesis [20,21]. Accumulating evidence also proposes that HMG-CoA reductase inhibitors have some favorable cholesterol-independent or pleiotropic properties such as improvement for endothelial dysfunction, abnormal glucose metabolism, and aberrant inflammation. IL-1 is a critical proinflammatory cytokine that is responsible for the development of uric acid-induced sterile inflammatory disease like gout [3,4]. Some studies demonstrated that variable HMG-CoA reductase inhibitors markedly suppressed mature IL-1 release generated during MSU-induced inflammatory response [22,23]. Data on whether HMG-CoA reductase inhibitors affect the immunomodulatory effect of IL-37 are insufficient, although some studies demonstrated that IL-37 could be implicated in the pathogenesis of uric acid-induced inflammation [16,17,18,19]. Shaoyuan et al. demonstrated that atorvastatin treatment significantly attenuated IL-37 and Smad3 that were increased in the atherosclerotic plaque in rabbit models [18]. There is a lack of data on the relationship between HMG-CoA reductase inhibitors and IL-37 expression in the pathogenesis of gout. Thus, this study investigated whether the therapeutic effect of HMG-CoA reductase inhibitors is related to the regulation of IL-37 expression in uric acid-induced inflammation.
2. Materials and methods
2.1. Study population and collecting blood samples
This study consecutively enrolled 40 male patients who met the classification criteria for diagnosis of gout proposed by the American College of Rheumatology/European League Against Rheumatism collaborative initiative [24]. An additional 30 healthy male subjects were recruited as controls who were age-matched and without a history of gout. Subjects with other forms of inflammatory arthritis including RA, SLE, psoriatic arthritis, OA, and AS were excluded from both gout and control groups by review of medical records and by subject interview if needed. At the time of enrollment, we collected some laboratory data such as serum uric acid (mg/dL), erythrocyte sedimentation rate (ESR, mm/hr), and C-reactive protein (CRP, mg/L) in gout patients.
2.2. Cell culture
Human monocytic cell line, THP-1 cells were grown in RPMI1640 (Gibco, BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA) and 1% antibiotics (100 units/mL penicillin and 100 g/mL streptomycin). THP-1 cells were differentiated into macrophages by 24 h incubation with 100 nM phorbol 12-myristate 13-acetate (PMA) prior to stimulation.
2.3. Chemicals and antibodies
Atorvastatin, rosuvastatin, p-nitrophenylphosphate, and phorbol 12-myristate 13-acetate (PMA) were purchased from Sigma-Aldrich (St Louis, MO, USA). Human recombinant IL-37 protein was purchased from PeproTech (Rocky Hill, NJ, USA). MSU crystals were prepared as previously described in our previous study [25].
Antibodies were purchased from the following companies: Caspase-1 and Smad3 (Ser423/425) (Abcam, Cambridge, UK), IL-1, anti-cleaved IL-1 and phospho-Smad3 (Ser423/425) (Cell Signaling, Danvers, MA, USA), IL-37 (R&D Systems, Minneapolis, MN, USA), and -actin were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
2.4. Enzyme-linked immunosorbent assay (ELISA)
The concentration of human IL-37 was measured by ELISA kit according to assay instructions (R&D Systems, Minneapolis, MN, USA). For this measurement, 96-well plates were coated with 100 L of IL-37 captured antibody and were incubated overnight at room temperature. Plates were washed 3 times in washing buffer (0.05% Tween 20 in phosphate buffered saline [PBS]) and blocked with 300 L per well of 1% bovine serum albumin (BSA) in PBS for 1h at room temperature. Serums and standards were added and then the plates were incubated for 2 h at room temperature. After washing, detection antibody was added and incubated for 2 h at room temperature. The plates were incubated with biotin-conjugated detection antibody followed by horse radish peroxidase-labelled streptavidin. The absorbance values of the serums were measured at 450 nm using an ELISA plate reader (BMG Lab Technologies, Offenburg, Germany).
2.5. Quantitative real time-polymerase chain reaction (qRT-PCR)
Cells were seeded on 60 mm culture dishes at a density of 8 x 105 cells/dish, were pre-treated with the indicated concentrations of atorvastatin and rosuvastatin for 24 h, and then stimulated with MSU (0.2 mg/mL) for 24 h. Total RNA was extracted from cells using Trizol reagent (Gibco BRL, Grand Island, NY, USA) according to the manufacturer’s instruction. A 1 g sample of total RNAs was used for complementary DNA (cDNA) synthesis using ReverTra Ace-α-1 (Toyobo, Osaka, Japan) followed by incubation at 37 °C for 15 min, 50 °C for 5 min and 98 °C for 5 min.
The gene expression was measured by quantitative PCR with SYBR Green Master Mix (Toyobo, Tokyo, Japan) and a Mini Option TM Real-time PCR system (Bio-Rad, Hercules, CA, USA). For PCR amplification, 20 L containing 2 L cDNA, 10 µl of SYBR® Green Realtime PCR Master Mix, 10 pmol/L of each primer, and 6.4 µl of distilled water was analyzed. The reaction was carried out at an initial denaturation at 95 °C for 15 min, followed by 40 cycles of 9 °C for 5 s, 58-63 °C for 30 s, and 72 °C for 15 s.
Primer sequences were as follows: IL-37 (Forward) 5’-CGG CCC TTC ATC TTT TAT AGG-3’ and (Reverse) 5’-TTT ATC TGT CAC CCC AAC AGG-3’, NLRP3 (Forward) 5’- GAT CTT CGC TGC GAT CAA CA-3’ and (Reverse) 5’-GGG ATT CGA AAC ACG TGC ATT A-3’, Caspase-1 (Forward) 5’- GCC TGT TCC TGT GAT GTG GAG-3’ and (Reverse) TGC CCA CAG ACA TTC ATA CAG TTT C-3’, IL-1 (Forward) 5’- CCA CAG ACC TTC CAG GAG AAT-3’ and (Reverse) 5’- GTG CAC ATA AGC CTC GTT ATC C-3’, Elastase (Forward) 5’- CCC TCA CGA GAG TGC AGA CGT T-3’ and (Reverse) 5’- CGT AAA CTT CTT GCT CAA CGA CAT C-3’, cathepsin S (Forward) 5’- GCC TGT GCC TAT CAC CTC TTA T-3’ and (Reverse) 5’- CCT TCT CTG TCT GTC TCC TGG T-3’, and -actin (Forward) 5’- CCT GAC TGA CTA CCT CAT GAA GG-3’ and (Reverse) 5’- CGT AGC ACA GCT TCT CCT TA-3’. All reactions were run in triplicate, and relative expression of each gene was analyzed using the 2–ΔΔCt method.
2.6. Transfection of siRNA
Transfection was performed using Lipofectamine RNAiMAX transfection reagent (Invitrogen, Waltham, MA, USA) following the manufacturer’s protocol. Cells were seeded at 4 x 105 cells / well in a 24-well plate and transiently transfected with human IL-1 or Smad3 siRNA and negative control (NC) siRNA (Invitrogen, Waltham, MA, USA).
Briefly, siRNA was diluted in Opti-mem media (Life Technologies, Darmstadt, Germany) and mixed with 1 L Lipofectamine RNAiMAX transfection reagent diluted in Opti-MEM media and incubated for 10 min at room temperature. Transfection complexes were added for 48 h in 24 well plates. Next, cells were pretreated with statins for 24 h, treatment with MSU (0.2 mg/mL) for 24 h, and harvested for subsequent experiments.
2.7. Western blotting
Cells (2 x 106) were seeded on 100-mm culture dishes, pretreated with the indicated concentrations of atorvastatin and rosuvastatin for 24 h, and stimulated with MSU (0.2 mg/mL) for 24 h. Total proteins were lysed in lysis buffer (Bio-Rad, Hercules, CA, USA) supplemented with protease inhibitor (Roche, Diagnostics, Mannheim, Germany) for 15 min on ice. The cell lysates were clarified by centrifugation at 15,000 rpm for 10 min at 4 °C, and protein concentrations were determined by a PierceTM BCA Protein Assay Kit (Thermo Fisher, Waltham, MA, USA).
Cytoplasmic and nuclear proteins were extracted using the NE-PER™ Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher, Waltham, MA, USA) according to the manufacturer’s instructions. Equal amounts of total proteins were separated on 10-12 % SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad, Hercules, CA, USA) by electrophoresis.
The membranes were blocked in 5% BSA (BD Bioscience, San Francisco, CA, USA) and probed with appropriate dilutions of primary antibodies followed by horseradish peroxidase (HRP)-conjugated secondary antibodies. Immunoreactive protein detection was performed with the ECL chemiluminescence kit (Thermo Fisher, Waltham, MA, USA). Densitometry was analyzed and quantified with Quantity One software (Bio-Rad, Hercules, CA, USA).
2.8. Statistical analysis
Data are described as the mean ± standard error of the mean. The difference of serum IL-37 levels between gout patients and controls was evaluated using the nonparametric Mann-Whiney U test. Correlation of IL-37 level with serum uric acid, ESR, and CRP in patients with gout was assessed by linear regression analysis. The statistical differences for target genes of caspase-1, IL-1, and IL-37 mRNA and protein expression between cells treated with each statin or not were measured by Mann-Whitney U test. Statistical significance was considered at a p value of less than 0.05. The statistical analyses and generation of plots illustrated in figures were performed by GraphPad Prism version 5.04 (San Diego, CA, USA).
3. Results
3.1. IL-37 expression in MSU-induced inflammation
We compared serum IL-37 level between gout patients (n = 40) and controls (n = 30), measured by ELISA method. Gout patients showed higher serum IL-37 level, compared to controls (p < 0.001) (Figure 1A). We assessed whether MSU crystals can stimulate IL-37 expression in THP-1 cells. Stimulation with MSU at concentrations of 0.1 and 0.2 mg/mL in THP-1 cells gradually induced IL-37 mRNA expression in a dose-dependent manner (*p < 0.05 and **p < 0.01 compared to unstimulated cells, respectively) (Figure 1B). Consistently, IL-37 protein expression in cytoplasm of THP-1 cells treated with MSU was reduced in a dose-dependent manner, whereas nuclear IL-37 expression was gradually increased. Positive correlation was noted between serum IL-37 level and serum uric acid in patients with gout (r = 0.382, p = 0.008) (Figure 1C). However, acute phase reactants including ESR and CRP in gout patients were not related with serum IL-37 level (p > 0.05 of both). Considering the anti-inflammatory effect of IL-37 in uric acid-induced inflammation, THP-1 cells transfected with IL-1 siRNA significantly suppressed IL-37 mRNA expression compared to cells with NC siRNA (Figure 1D). In addition, THP-1 cells stimulated with recombinant IL-37 dose-dependently suppressed activation of caspase-1 and IL-1 in MSU-induced inflammation (Figure 1E). This suggests that IL-37 expression could be tightly regulated by IL-1 in uric acid-induced inflammation.
3.2. Effect of statins on caspase-1, IL-1, and IL-37 expression in MSU-induced inflammatory response
We evaluated the effects of HMG-CoA reductase inhibitors including atorvastatin and rosuvastatin on caspase-1, IL-1, and IL-37 expression in THP-1 cells treated with 0.2 mg/mL of MSU crystals. Atorvastatin suppressed caspase-1 and IL-1 mRNA expression at doses of 5.0 and/or 10.0 M but increased IL-37 mRNA expression in THP-1 cells treated with MSU at the same dosages (*p < 0.05 and **p < 0.01 compared to cells without atorvastatin treatment) (Figure 2A). Similarly, rosuvastatin at dose of 3.0 or 5.0M in uric acid-induced inflammation inhibited caspase-1 and IL-1 mRNA expression, whereas IL-37 mRNA expression in THP-1 cells stimulated with MSU was increased by rosuvastatin at a dose of 5.0M (*p < 0.05 and **p < 0.01 compared to cells without rosuvastatin treatment) (Figure 2A).
Consistently, increased expression of caspase-1 and IL-1 proteins in THP-1 cells stimulated with MSU alone was dose-dependently attenuated in cells treated with atorvastatin of 1.0, 5.0, or 10.0 M (Figure 2B). Atorvastatin treatment markedly increased IL-37 expression in the nucleus of cells stimulated with MSU crystals compared to cells stimulated with MSU alone. However, IL-37 expression in cytoplasm was dose-dependently reduced by atorvastatin. In addition, rosuvastatin treatment significantly suppressed activation of caspase-1 and IL-1 protein from the inactive forms of these proteins in a dose-dependent manner (Figure 2B). Translocation of IL-37 from cytoplasm into nucleus was augmented by treatment with rosuvastatin under stimulation of MSU.
3.3. Effects of statins on interactions with Smad3 and IL-37 in MSU-induced inflammation
Mature IL-37 binds to phosphorylated Smad3 to lead to generation of an IL-37/Smad3 complex in the cytoplasm that then moves into the nucleus. Finally, IL-37 binding to Smad3 results in up- or down-regulation of diverse proinflammatory or anti-inflammatory gene expression (10). Here, it was determined whether HMG-CoA reductase inhibitors regulated phosphorylation of Smad3 in the condition of MSU crystal-induced stimulation in THP-1 cells. We found that atorvastatin and rosuvastatin significantly induced phosphorylation of Smad3 in dose-dependent manner (Figure 3A). This suggests that HMG-CoA reductase inhibitors augment generation of IL-37/Smad3 complex in uric acid-inflammation. In the assessment for the effect of statins on Smad3-dependent IL-37 expression, IL-37 mRNA expression stimulated by atorvastatin and rosuvastatin in cells transfected with Smad3 siRNA was downregulated rather than in those with NC siRNA (Figure 3B). Cells transfected with Smad3 siRNA under stimulation with MSU alone showed lower IL-37 mRNA expression than did those with NC siRNA. In addition, stimulation with atorvastatin or rosuvastatin markedly decreased translocation of IL-37 from cytoplasm to nucleus in THP-1 cells transfected with Smad3 siRNA under stimulation with MSU (Figure 3C).
3.4. Effect of statins on proteolytic activity in the process of IL-37 activation
This study demonstrated that HMG-CoA reductase inhibitors under stimulation with MSU significantly induced IL-37 expression, while down-regulating caspase-1 expression. Thus, we evaluated whether two different HMG-CoA reductase inhibitors may influence biological activity of other proteolytic enzymes including neutrophil elastase and cathepsin S during cleavage of precursor IL-37 into mature IL-37. We found that stimulation with MSU markedly induced two different proteases in THP-1 cells, but that two HMG-CoA reductase inhibitors, atorvastatin and rosuvastatin, did not have any stimulatory or inhibitory effect on mRNA and protein expression of these enzymes (Figure 4A and 4B). It suggests that IL-37 is activated by proteases other than caspase-1 in uric acid-induced inflammation, and that these enzymes are not affected by HMG-CoA reductase inhibitors in the process of IL-37 activation.
4. Discussion
The regulation of proinflammatory cytokines has a crucial role in the inflammatory reaction to MSU crystals in the pathogenesis of gout [1,2]. Recent studies have implicated IL-1 as being generally considered a key proinflammatory cytokine, leading to the development of acute gout attack. Several clinical studies demonstrated that statin use could provide greater benefits on uric acid homeostasis, the risk of gout, and mortality in gout patients [26,27,28], although action mechanisms of statins for uric acid-induced inflammatory responses have not been determined. Recently, HMG-CoA reductase inhibitors including atorvastatin, simvastatin, and mevastatin inhibited activation of MSU-induced NLRP3 inflammasome by upregulation of peroxisome proliferator-activated receptor-γ (PPAR-), resulting in anti-inflammatory therapeutic action in gout [22]. THP-1 cells pretreated with pitavastatin led to reduction of MSU-stimulated IL-1 production and secretion through blocking caspase-1 activation [23]. To investigate the anti-inflammatory mechanism of statin in MSU-induced inflammation, we found that HMG-CoA reductase inhibitors including atorvastatin and rosuvastatin potently suppressed release of IL-1 through up-regulation of the IL-37/Smad3 complex in THP-1 cells.
Growing evidence has well demonstrated that IL-37 is implicated in the pathogenesis of chronic autoimmune and inflammatory rheumatic diseases including RA, SLE, OA, and AS [11,12,13,14,15]. Results from these clinical studies consistently showed that IL-37 level in serum or PBMC was significantly higher than in healthy controls. IL-37 level was found to be closely linked with disease activity markers such as disease activity score of 28 joints (DAS28) for RA, SLE disease activity index (SLEDAI) score and autoantibodies for SLE, visual analog scale (VAS) for OA, and Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) for AS. The main mechanism of action of IL-37 is related to reducing proinflammatory cytokines and inflammatory cells including tumor necrosis factor- (TNF-, IL-1, IL-6, IL-17, IL-23, and Th17 cells or increasing anti-inflammatory cytokines such as IL-10 through modulating inflammatory signal pathways. In addition, patients with gout, especially those with an active gouty attack, had significantly higher IL-37 level in serum, PBMC, and synovial tissue, compared to healthy controls [16,17,18]. These studies verified that IL-37 level was significantly associated with ESR, CRP, and the presence of tophi, which implicated much higher levels of this cytokine in active gout than in inactive gout. These results are consistent with the finding of this study that serum IL-37 level in gout was significantly higher than in controls. We also found serum IL-37 level to be closely associated with serum uric acid level but not with ESR and CRP in gout. In contrast, Ding et al. identified no correlation between IL-37 level and serum uric acid [17]. It has been well recognized that the main mechanism of IL-37 for limiting the inflammatory response caused by uric acid in gout is alleviation of the release of proinflammatory cytokines and chemokines [16,17,18]. In the DNA sequencing of IL-37 using molecular inversion probe resequencing in gout patients, detection of p.(N182S)(rs752113534) carrier status was identified as a potent genetic variant favoring the risk of gout [29]. Recently, Zhao et al. demonstrated novel opinion that IL-37 enhanced the shaping of macrophages with non-inflammatory phagocytic activity [19]. Thus, IL-37 is considered a potential biomarker for disease activity and severity and as a pathogenic target molecule in inflammatory diseases.
Considering the biological activity of intracellular IL-37, diverse inflammatory stimuli and cytokines lead to production of precursor IL-37 and activation of caspase-1, which in turn trigger cleavage of pro-IL-37 into mature IL-37. IL-37, as a transcription factor, binds to phosphorylated activated Smad3 [7,8,9,10]. The IL-37/Smad3 complex generated in the cytoplasm translocates to the nucleus to regulate gene transcription, reducing secretion of proinflammatory cytokine and chemokines. It is considered that the binding to IL-37 and phosphorylation of Smad3 are essential in the IL-37 activation. Some debates have demonstrated that HMG-CoA reductase inhibitors could regulate Smad3 phosphorylation in the TGF- /Smad signal pathway in various cellular conditions. Accumulation of IL-37 and Smad3 in the macrophage-derived foam cells in the atherosclerotic plaque was significantly inhibited by atorvastatin treatment [30]. This supports regulation of biological activity of IL-37 by blocking Smad3 phosphorylation by statin treatment. Liu et al. found that IL-37 potently limited the uric acid-induced innate inflammatory response through augmentation of Smad3 and IL-1R8 expression, blocking activation of NLRP3 inflammasome and activating SOCS in human synovial cells and THP-1 cells [18]. Consistently, we also found that two HMG-CoA reductase inhibitors, atorvastatin and rosuvastatin, activated Smad3 phosphorylation. It suggests that atorvastatin and rosuvastatin might contribute to greater translocation of cytoplasm to nucleus of IL-37/Smad3 complex.
Regarding the effect of anti-inflammatory drugs on IL-37 level, IL-37 expression together with TNF-, IL-6, and IL-17A during use of disease modifying anti-rheumatic drugs (DMARDs), especially drug-responders, in RA was significantly decreased rather than before initiation of DMARDs [11]. Similarly, mRNA expression of IL-37 and plasma IL-37 level along with those of other inflammatory cytokines including interferon- and IL-6 was reduced after steroid (1 mg/kg) treatment, compared to pre-treatment [13]. It is presumed that treatment with DMARDs and steroid suppressed production and release of proinflammatory cytokines that can stimulate IL-37. Overexpression of IL-37 using transfection with pcDNA3.1-IL-37b or recombinant human IL-37 treatment under stimulation with MSU crystals inhibited production of multiple inflammatory cytokines and chemokines including IL-1, IL-6, IL-8, CCL2, and TNF-, suggesting that augmentation of IL-37 expression effectively controls excessive gouty inflammation [16,18]. For up-regulation of IL-37 level that plays a crucial role in limiting diverse inflammatory response, some anti-inflammatory medicines including Tripterygium wilfordii Glycosides, chloroquine, and rapamycin induce IL-37 expression. These pharmaceuticals have been verified in in vitro and in vivo studies through upregulation of ERK1/2, p38 MAPK, and NF-B/AP-1 signal transduction [31,32]. Consistently, we found that atorvastatin and rosuvastatin significantly induced IL-37 mRNA and protein expression in THP-1 cells treated with MSU crystals. Based on these observations, IL-37 is considered an effective therapeutic agent for inflammatory rheumatic disorders.
Several proteases including caspase-1, caspase-8, neutrophil enolase, granzyme B, cathepsin S, and cathepsin G are involved in proteolytic processing of IL-1 family cytokines [33]. In the biological activity of IL-37, caspase-1 is primarily involved in cleavage from pro-IL-37 to mature IL-37 and in nuclear translocation of IL-37 [7,8]. In addition to caspase-1, IL-37 could be cleaved by additional proteases including neutrophil enolase and cathepsin S [26,34]. Active caspase-1 is well recognized to cleave pro-IL-1 to produce mature IL-1 in the process of NLRP3 inflammasome activation, that is considered a pathogenic mechanism in the pathogenesis of gout [33]. In this study to determine the role of statins in regulating IL-37 expression in the MSU-induced inflammatory response, we found that statins inhibited the activation of caspase-1. In other words, statins can be expected to block the activation of IL-1 and IL-37, but processing of IL-37 activation was not limited. It suggests that IL-37 activation is affected by proteases other than caspase-1. In this study, we found that neutrophil enolase was suppressed by only 10 M of atorvastatin and 5 M of rosuvastatin, but cathepsin S was not inhibited by the two statins. Based on our results, it is presumed that IL-37 activation by statins is related to activation of Smad3 phosphorylation, independent of caspase-1.
HMG-CoA reductase inhibitors are classified into two categories of hydrophilic statins (rosuvastatin and pravastatin) and lipophilic statins (simvastatin, atorvastatin, lovastatin, and pitavastatin) based on water- or lipid solubility, respectively [35]. The possible superiority of either lipophilic or hydrophilic statins considering anti-inflammatory potency has not been determined. Chang et al. demonstrated that articular chondrocytes treated with simvastatin or pravastatin with hyaluronic acid showed down-regulated release of proinflammatory cytokines and inflammatory mediators, while hydrophilic statin resulted in better overcome [36]. Regarding potential risk of statins and incident RA, long-term lipophilic statin use (> 365 days) lead to lower possibility of development of RA [37]. But there was no association between hydrophilic statins or short-term use and the risk of RA. Consistently, lipophilic statins showed anti-inflammatory action on MSU-induced NLRP3 inflammasome activation [22]. Atorvastatin and rosuvastatin used in this study equally suppressed MSU-induced inflammation through augmentation of IL-37 production.
5. Conclusions
This study found that two different HMG-CoA reductase inhibitors, atorvastatin and rosuvastatin, have anti-inflammatory effects through augmentation of IL-37 expression due to increased phosphorylation of Smad3 in MSU-induced inflammation in THP-1 cells (Figure 5). HMG-CoA reductase inhibitors are suggested as being responsible for potent negative regulation of uric acid-induced inflammation. IL-37 is considered an anti-inflammatory cytokine to suppress uric acid-induced inflammation. Our finding provides evidence that recombinant IL-37 treatment or pharmacological agents that amplify the IL-37 expression might be considered as a therapeutic strategy to control acute uric acid-induced inflammation.
Author Contributions
Conceptualization, K.-Y.P., J.-W.K., and S.-K.K.; Investigation, K.-Y.P., J.-Y.C., J.-W.K., B.K., and S.-K.K; Writing-Original Draft Preparation, S.-K.K.; Writing-Review & Editing, S.-K.K. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the grant of Daegu Catholic University Medical Center (2023).
Institutional Review Board Statement
This study was approved by Institutional Review Board at Daegu Catholic University Medical Center (IRB No. CR-21-187-L).
Informed Consent Statement
Patients and controls provided informed consent at the time of study enrollment.
Data Availability Statement
The data underlying this article will be shared on reasonable request to the corresponding author.
Acknowledgments
None.
Conflicts of Interest
There are no financial and non-financial conflicts of interest for any of the authors regarding specific financial interests that are relevant to the work conducted or reported in this manuscript.
References
- Wu, M.; Tian, Y.; Wang, Q.; Guo, C. Gout: a disease involved with complicated immunoinflammatory responses: a narrative review. Clin. Rheumatol. 2020, 39, 2849–2859. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.K. The Mechanism of the NLRP3 Inflammasome Activation and Pathogenic Implication in the Pathogenesis of Gout. J. Rheum. Dis. 2022, 29, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Pétrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006, 440, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-. Mol. Cell. 2002, 10, 417–426. [Google Scholar] [CrossRef] [PubMed]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Akbal, A.; Dernst, A.; Lovotti, M.; Mangan, M.S.J.; McManus, R.M.; Latz, E. How location and cellular signaling combine to activate the NLRP3 inflammasome. Cell. Mol Immunol. 2022, 19, 1201–1214. [Google Scholar] [CrossRef]
- Nold, M.F.; Nold-Petry, C.A.; Zepp, J.A.; Palmer, B.E.; Bufler, P.; Dinarello, C.A. IL-37 is a fundamental inhibitor of innate immunity. Nat. Immunol. 2010, 11, 1014–1022. [Google Scholar] [CrossRef]
- Jia, H.; Liu, J.; Han, B. Reviews of Interleukin-37: Functions, Receptors, and Roles in Diseases. Biomed. Res. Int. 2018, 2018, 3058640. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Quan, Y.; Yue, Y.; Heng, X.; Che, F. Interleukin-37: A crucial cytokine with multiple roles in disease and potentially clinical therapy. Oncol. Lett. 2018, 15, 4711–4719. [Google Scholar] [CrossRef]
- Su, Z.; Tao, X. Current Understanding of IL-37 in Human Health and Disease. Front. Immunol. 2021, 12, 696605. [Google Scholar] [CrossRef]
- Zhao, P.W.; Jiang, W.G.; Wang, L.; Jiang, Z.Y.; Shan, Y.X.; Jiang, Y.F. Plasma levels of IL-37 and correlation with TNF-α, IL-17A, and disease activity during DMARD treatment of rheumatoid arthritis. PLoS. One. 2014, 9, e95346. [Google Scholar] [CrossRef]
- Xia, T.; Zheng, X.F.; Qian, B.H.; Fang, H.; Wang, J.J.; Zhang, L.L.; Pang, Y.F.; Zhang, J.; Wei, X.Q.; Xia, Z.F.; et al. Plasma Interleukin-37 Is Elevated in Patients with Rheumatoid Arthritis: Its Correlation with Disease Activity and Th1/Th2/Th17-Related Cytokines. Dis. Markers. 2015, 2015, 795043. [Google Scholar] [CrossRef]
- Song, L.; Qiu, F.; Fan, Y.; Ding, F.; Liu, H.; Shu, Q.; Liu, W.; Li, X. Glucocorticoid regulates interleukin-37 in systemic lupus erythematosus. J. Clin. Immunol. 2013, 33, 111–117. [Google Scholar] [CrossRef]
- Ding, L.; Hong, X.; Sun, B.; Huang, Q.; Wang, X.; Liu, X.; Li, L.; Huang, Z.; Liu, D. IL-37 is associated with osteoarthritis disease activity and suppresses proinflammatory cytokines production in synovial cells. Sci. Rep. 2017, 7, 11601. [Google Scholar] [CrossRef]
- Chen, B.; Huang, K.; Ye, L.; Li, Y.; Zhang, J.; Zhang, J.; Fan, X.; Liu, X.; Li, L.; Sun, J.; et al. Interleukin-37 is increased in ankylosing spondylitis patients and associated with disease activity. J. Transl. Med. 2015, 13, 36. [Google Scholar] [CrossRef]
- Zeng, M.; Dang, W.; Chen, B.; Qing, Y.; Xie, W.; Zhao, M.; Zhou, J. IL-37 inhibits the production of pro-inflammatory cytokines in MSU crystal-induced inflammatory response. Clin. Rheumatol. 2016, 35, 2251–2258. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Li, H.; Sun, B.; Wang, T.; Meng, S.; Huang, Q.; Hong, X.; Liu, D. Elevated interleukin-37 associated with tophus and pro-inflammatory mediators in Chinese gout patients. Cytokine. 2021, 141, 155468. [Google Scholar] [CrossRef]
- Liu, L.; Xue, Y.; Zhu, Y.; Xuan, D.; Yang, X.; Liang, M.; Wang, J.; Zhu, X.; Zhang, J.; Zou, H. Interleukin 37 limits monosodium urate crystal-induced innate immune responses in human and murine models of gout. Arthritis. Res. Ther. 2016, 18, 268. [Google Scholar] [CrossRef]
- Zhao, L.; Zhao, T.; Yang, X.; Cao, L.; Xu, R.; Liu, J.; Lin, C.; Yu, Y.; Xuan, D.; Zhu, X.; et al. IL-37 blocks gouty inflammation by shaping macrophages into a non-inflammatory phagocytic phenotype. Rheumatology. (Oxford). 2022, 61, 3841–3853. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, S.I.; Muniyappa, R.; Francisco, R.; Sowers, J.R. Clinical review 145: Pleiotropic effects of statins: lipid reduction and beyond. J. Clin. Endocrinol. Metab. 2002, 87, 1451–1458. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 89–118. [Google Scholar] [CrossRef]
- Kim, S.K.; Choe, J.Y.; Kim, J.W.; Park, K.Y. HMG-CoA Reductase Inhibitors Suppress Monosodium Urate-Induced NLRP3 Inflammasome Activation through Peroxisome Proliferator-Activated Receptor- Activation in THP-1 Cells. Pharmaceuticals. (Basel). 2023, 16, 522. [Google Scholar] [CrossRef]
- Cui, H.; Soga, K.; Tamehiro, N.; Adachi, R.; Hachisuka, A.; Hirose, A.; Kondo, K.; Nishimaki-Mogami, T. Statins repress needle-like carbon nanotube- or cholesterol crystal-stimulated IL-1β production by inhibiting the uptake of crystals by macrophages. Biochem. Pharmacol. 2021, 188, 114580. [Google Scholar] [CrossRef]
- Neogi, T.; Jansen, T.L.; Dalbeth, N.; Fransen, J.; Schumacher, H.R.; Berendsen, D.; Brown, M.; Choi, H.; Edwards, N.L.; Janssens, H.J.; et al. 2015 Gout Classification Criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis. Rheumatol. 2015, 67, 2557–2568. [Google Scholar] [CrossRef]
- Choe, J.Y.; Jung, H.Y.; Park, K.Y.; Kim, S.K. Enhanced p62 expression through impaired proteasomal degradation is involved in caspase-1 activation in monosodium urate crystal-induced interleukin-1b expression. Rheumatology. (Oxford). 2014, 53, 1043–1053. [Google Scholar] [CrossRef]
- Bulau, A.M.; Nold, M.F.; Li, S.; Nold-Petry, C.A.; Fink, M.; Mansell, A.; Schwerd, T.; Hong, J.; Rubartelli, A.; Dinarello, C.A.; et al. Role of caspase-1 in nuclear translocation of IL-37, release of the cytokine, and IL-37 inhibition of innate immune responses. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 2650–2655. [Google Scholar] [CrossRef]
- Milionis, H.J.; Kakafika, A.I.; Tsouli, S.G.; Athyros, V.G.; Bairaktari, E.T.; Seferiadis, K.I.; Elisaf, M.S. Effects of statin treatment on uric acid homeostasis in patients with primary hyperlipidemia. Am. Heart. J. 2004, 148, 635–640. [Google Scholar] [CrossRef]
- Lin, G.L.; Lin, H.C.; Lin, H.L.; Keller, J.J.; Wang, L.H. Association between statin use and the risk of gout in patients with hyperlipidemia: A population-based cohort study. Front. Pharmacol. 2023, 14, 1096999. [Google Scholar] [CrossRef]
- Klück, V.; van Deuren, R.C.; Cavalli, G.; Shaukat, A.; Arts, P.; Cleophas, M.C.; Crișan, T.O.; Tausche, A.K.; Riches, P.; Dalbeth, N.; et al. Rare genetic variants in interleukin-37 link this anti-inflammatory cytokine to the pathogenesis and treatment of gout. Ann. Rheum. Dis. 2020, 79, 536–544. [Google Scholar] [CrossRef] [PubMed]
- Shaoyuan, C.; Ming, D.; Yulang, H.; Hongcheng, F. Increased IL-37 in Atherosclerotic Disease could be Suppressed by Atorvastatin Therapy. Scand. J. Immunol. 2015, 82, 328–336. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Li, R.; He, S.; He, L.; Zhao, H.; Deng, X.; Chen, Z. Tripterygium wilfordii Glycosides Upregulate the New Anti-Inflammatory Cytokine IL-37 through ERK1/2 and p38 MAPK Signal Pathways. Evid. Based. Complement. Alternat. Med. 2017, 2017, 9148523. [Google Scholar] [CrossRef]
- Shi, X.; Lai, C.; Zhao, L.; Zhang, M.; Liu, X.; Peng, S.; Guo, W.; Xu, Q.; Chen, S.; Chen, G.X. Chloroquine and Rapamycin Augment Interleukin-37 Expression via the LC3, ERK, and AP-1 Axis in the Presence of Lipopolysaccharides. J. Immunol. Res. 2020, 2020, 6457879. [Google Scholar] [CrossRef]
- Afonina, I.S.; Müller, C.; Martin, S.J.; Beyaert, R. Proteolytic Processing of Interleukin-1 Family Cytokines: Variations on a Common Theme. Immunity. 2015, 42, 991–1004. [Google Scholar] [CrossRef]
- Sullivan, G.P.; Davidovich, P.; Muñoz-Wolf, N.; Ward, R.W.; Hernandez Santana, Y.E.; Clancy, D.M.; Gorman, A.; Najda, Z.; Turk, B.; Walsh, P.T.; et al. Myeloid cell-derived proteases produce a proinflammatory form of IL-37 that signals via IL-36 receptor engagement. Sci. Immunol. 2022, 7, eade5728. [Google Scholar] [CrossRef]
- Climent, E.; Benaiges, D.; Pedro-Botet, J. Hydrophilic or Lipophilic Statins. Front. Cardiovasc. Med. 2021, 8, 687585. [Google Scholar] [CrossRef]
- Chang, C.H.; Hsu, Y.M.; Chen, Y.C.; Lin, F.H.; Sadhasivam, S.; Loo, S.T.; Savitha, S. Anti-inflammatory effects of hydrophilic and lipophilic statins with hyaluronic acid against LPS-induced inflammation in porcine articular chondrocytes. J. Orthop. Res. 2014, 32, 557–565. [Google Scholar] [CrossRef]
- Kwon, M.J.; Kim, J.H.; Kim, J.H.; Park, H.R.; Kim, N.Y.; Hong, S.; Choi, H.G. Incident Rheumatoid Arthritis Following Statin Use: From the View of a National Cohort Study in Korea. J. Pers. Med. 2022, 12, 559. [Google Scholar] [CrossRef]
Figure 1.
IL-37 expression in gout and uric acid-induced inflammation. A) Comparison of serum IL-37 level between gout patients (n=40) and controls (n=30). B) Expression of IL-37 mRNA and IL-37 protein in cytoplasm and nucleus in THP-1 cells treated with MSU crystals. C) Correlation of IL-37 level with uric acid, ESR, and CRP in serum in gout patients. D) Expression of IL-37 mRNA and IL-37 protein in cytoplasm and nucleus in THP-1 cells transfected with IL-1 siRNA under stimulation with MSU crystals. E) Expression of active caspase-1 and IL-1 in THP-1 cells stimulated with recombinant IL-37. *p < 0.05 and **p < 0.01. Values presented as mean ± SEM of three independent experiments. The representative images are illustrated after three independent experiments. Abbreviation: MSU, monosodium urate; IL-37, interleukin-37; IL-1, interleukin-1; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein.
Figure 1.
IL-37 expression in gout and uric acid-induced inflammation. A) Comparison of serum IL-37 level between gout patients (n=40) and controls (n=30). B) Expression of IL-37 mRNA and IL-37 protein in cytoplasm and nucleus in THP-1 cells treated with MSU crystals. C) Correlation of IL-37 level with uric acid, ESR, and CRP in serum in gout patients. D) Expression of IL-37 mRNA and IL-37 protein in cytoplasm and nucleus in THP-1 cells transfected with IL-1 siRNA under stimulation with MSU crystals. E) Expression of active caspase-1 and IL-1 in THP-1 cells stimulated with recombinant IL-37. *p < 0.05 and **p < 0.01. Values presented as mean ± SEM of three independent experiments. The representative images are illustrated after three independent experiments. Abbreviation: MSU, monosodium urate; IL-37, interleukin-37; IL-1, interleukin-1; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein.
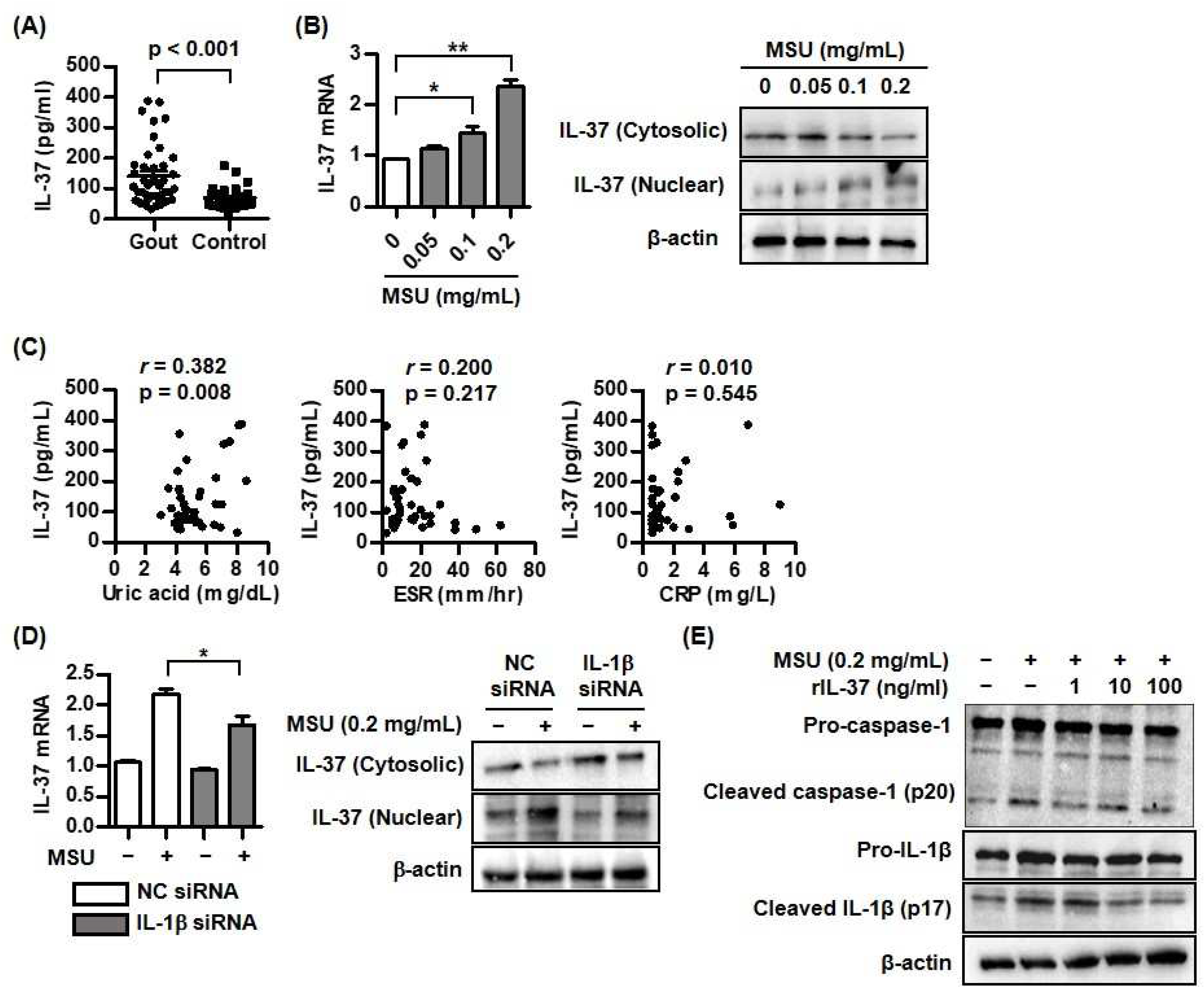
Figure 2.
Changes of caspase-1, IL-1, and IL-37 mRNA and protein expression by atorvastatin or rosuvastatin in THP-1 cells treated with MSU crystals. A) Expression of IL-37 mRNA after addition of atorvastatin or rosuvastatin in THP-1 cells treated with MSU crystals. B) Expression of cleaved caspase-1, cleaved IL-1, and IL-37 protein in cytoplasm and nucleus after addition of atorvastatin or rosuvastatin in THP-1 cells treated with MSU crystals. *p < 0.05 and **p < 0.01. Values presented as mean ± SEM of three independent experiments. The representative images are illustrated after three independent experiments. Abbreviation: MSU, monosodium urate; IL-37, interleukin-37; IL-1, interleukin-1.
Figure 2.
Changes of caspase-1, IL-1, and IL-37 mRNA and protein expression by atorvastatin or rosuvastatin in THP-1 cells treated with MSU crystals. A) Expression of IL-37 mRNA after addition of atorvastatin or rosuvastatin in THP-1 cells treated with MSU crystals. B) Expression of cleaved caspase-1, cleaved IL-1, and IL-37 protein in cytoplasm and nucleus after addition of atorvastatin or rosuvastatin in THP-1 cells treated with MSU crystals. *p < 0.05 and **p < 0.01. Values presented as mean ± SEM of three independent experiments. The representative images are illustrated after three independent experiments. Abbreviation: MSU, monosodium urate; IL-37, interleukin-37; IL-1, interleukin-1.
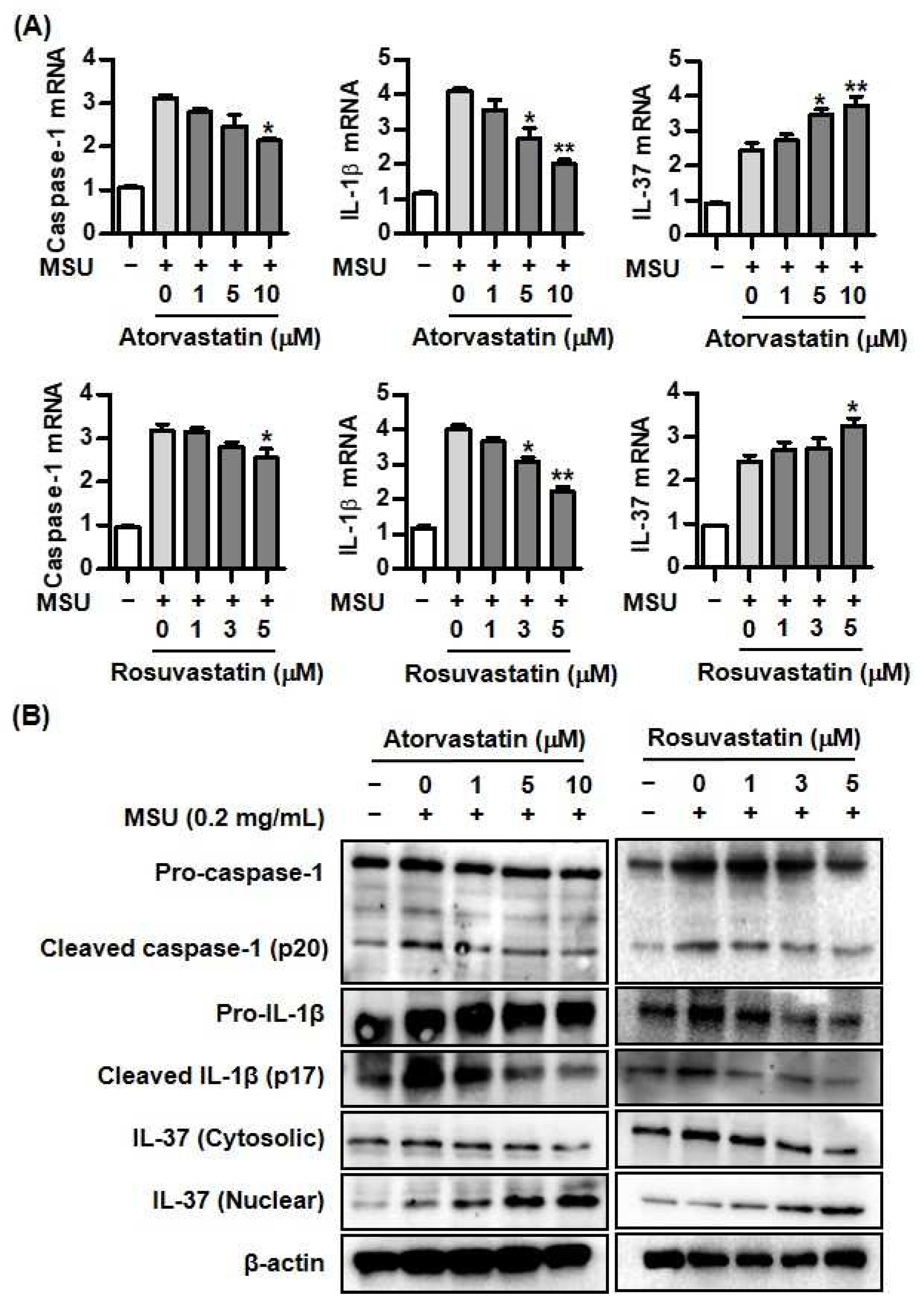
Figure 3.
Changes of Smad3 and IL-37 expression by atorvastatin or rosuvastatin in MSU-induced inflammation. A) Expression of phosphorylation of Smad3 after addition of atorvastatin or rosuvastatin in THP-1 cells treated with MSU crystals. B) Comparison of IL-37 mRNA expression between cells transfected with NC siRNA and Smad3 siRNA under stimulation with MSU crystals. C) Comparison of IL-37 protein expression in cytoplasm and nucleus between cells transfected with NC siRNA and Smad3 siRNA under stimulation with MSU crystals. *p < 0.05. Values presented as mean ± SEM of three independent experiments. The representative images are illustrated after three independent experiments. Abbreviation: MSU, monosodium urate; IL-37, interleukin-37.
Figure 3.
Changes of Smad3 and IL-37 expression by atorvastatin or rosuvastatin in MSU-induced inflammation. A) Expression of phosphorylation of Smad3 after addition of atorvastatin or rosuvastatin in THP-1 cells treated with MSU crystals. B) Comparison of IL-37 mRNA expression between cells transfected with NC siRNA and Smad3 siRNA under stimulation with MSU crystals. C) Comparison of IL-37 protein expression in cytoplasm and nucleus between cells transfected with NC siRNA and Smad3 siRNA under stimulation with MSU crystals. *p < 0.05. Values presented as mean ± SEM of three independent experiments. The representative images are illustrated after three independent experiments. Abbreviation: MSU, monosodium urate; IL-37, interleukin-37.
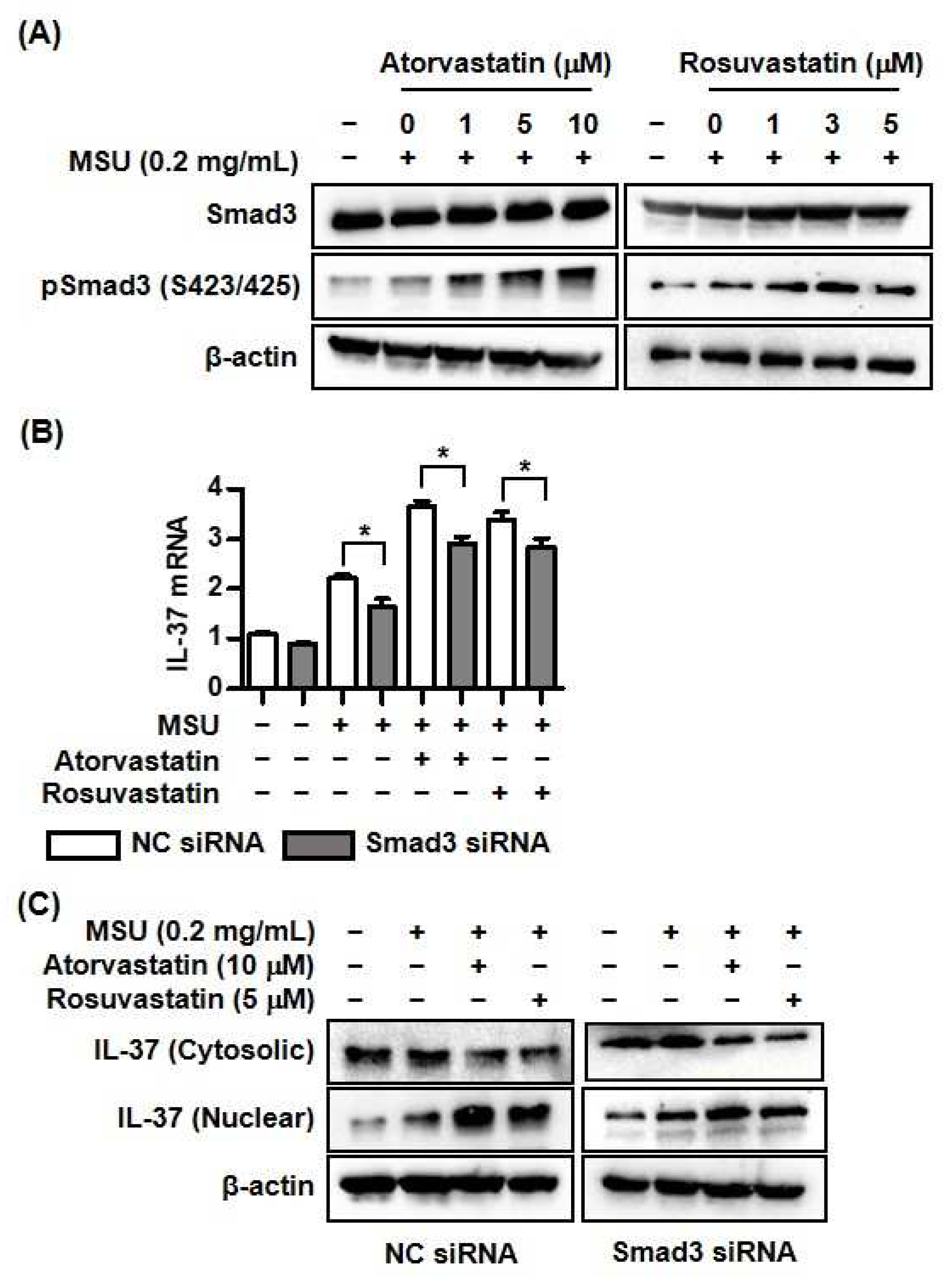
Figure 4.
Changes of proteolytic enzymes by atorvastatin or rosuvastatin in THP-1 cells. A and B) Expression of elastase and cathepsin S mRNA and protein by atorvastatin or rosuvastatin in THP-1 cells treated with MSU crystals. Values presented as mean ± SEM of three independent experiments. The representative images are illustrated after three independent experiments. Abbreviation: MSU, monosodium urate.
Figure 4.
Changes of proteolytic enzymes by atorvastatin or rosuvastatin in THP-1 cells. A and B) Expression of elastase and cathepsin S mRNA and protein by atorvastatin or rosuvastatin in THP-1 cells treated with MSU crystals. Values presented as mean ± SEM of three independent experiments. The representative images are illustrated after three independent experiments. Abbreviation: MSU, monosodium urate.
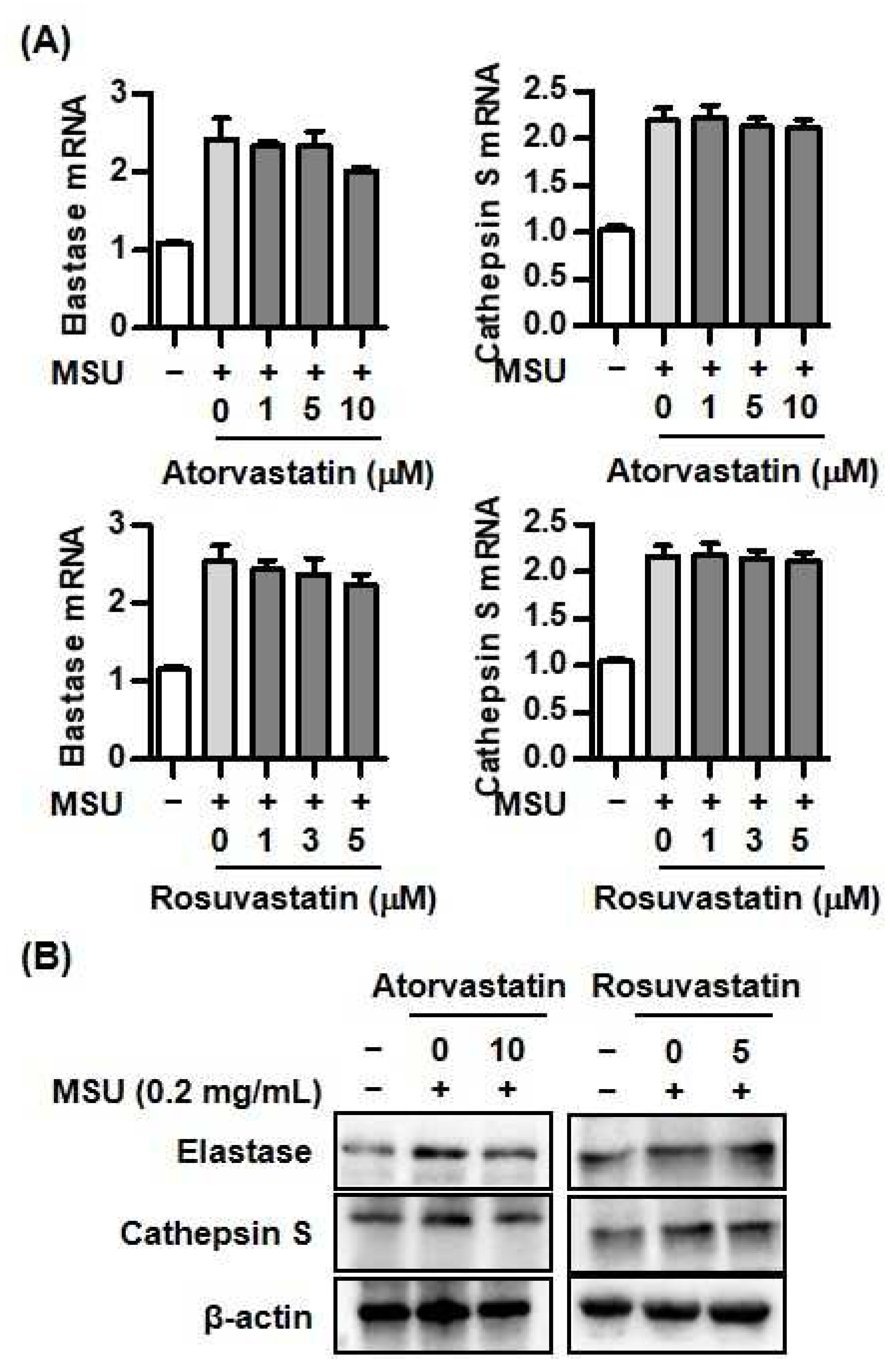
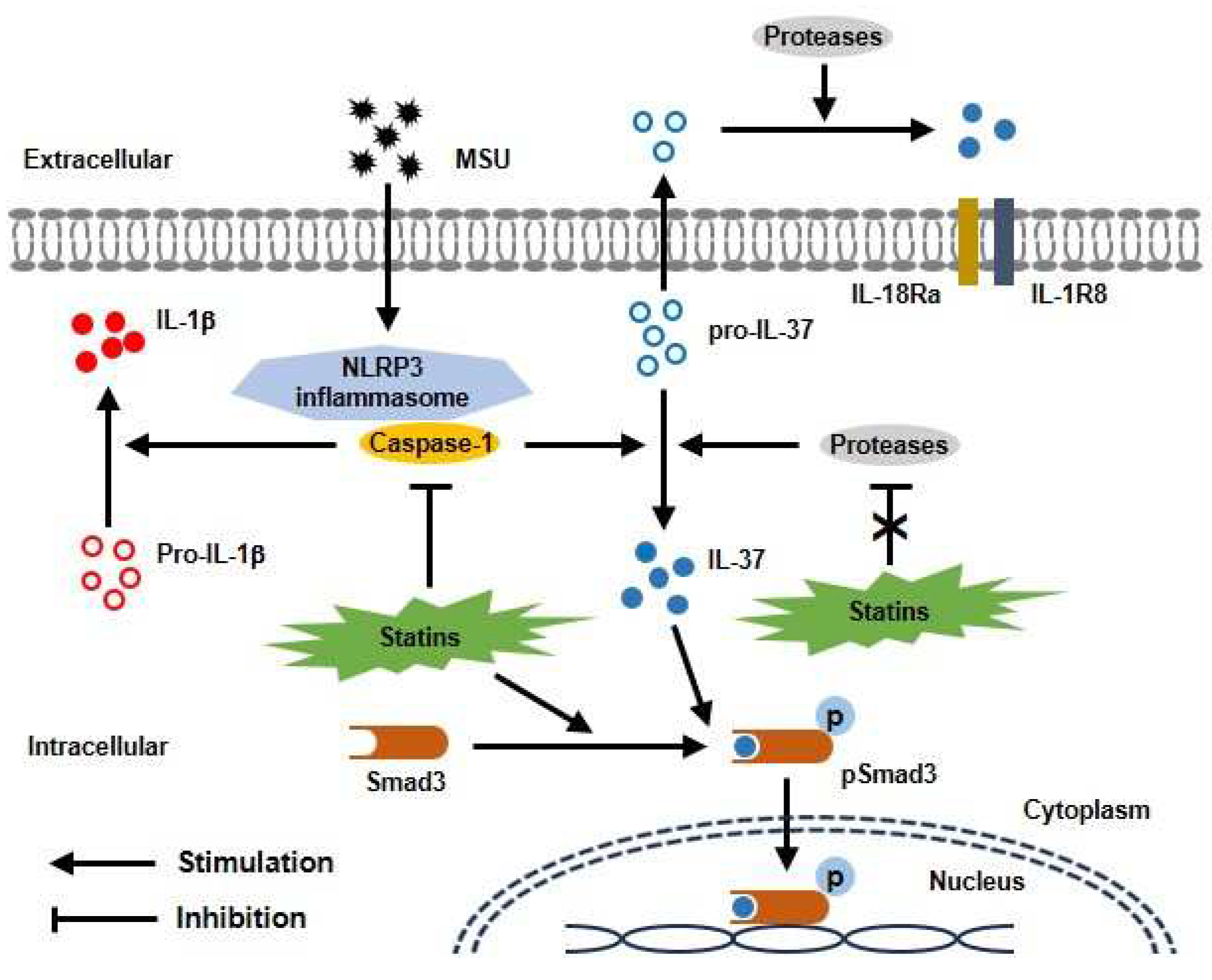
Figure 5.
Proposed model for protective effect of statins through interaction with Smad3 and IL-37 in uric acid-induced inflammation. MSU crystals induce activation of caspase-1 in NLRP3 inflammasome, then leading to conversion of mature IL-37 from pro-IL-37. Statins block caspase-1 activation, but not other proteases in THP-1 cells treated with MSU crystals. In addition, statins activate phosphorylation of Smad3. Binding with activated IL-37 and Smad3 in cytoplasm is translocated into nucleus, then stimulates transcription of anti-inflammatory molecules.
Figure 5.
Proposed model for protective effect of statins through interaction with Smad3 and IL-37 in uric acid-induced inflammation. MSU crystals induce activation of caspase-1 in NLRP3 inflammasome, then leading to conversion of mature IL-37 from pro-IL-37. Statins block caspase-1 activation, but not other proteases in THP-1 cells treated with MSU crystals. In addition, statins activate phosphorylation of Smad3. Binding with activated IL-37 and Smad3 in cytoplasm is translocated into nucleus, then stimulates transcription of anti-inflammatory molecules.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



