
Submitted:
15 January 2024
Posted:
16 January 2024
You are already at the latest version
Abstract
Background: Medulloblastoma (MB) comprises four broad molecular subgroups - wingless (WNT), sonic hedgehog (SHH), Group 3, and Group 4 respectively with subgroup-specific developmental origins, unique genetic profiles, distinct clinico-demographic characteristics, and diverse clinical outcomes. WNT-MB has the best outcomes (5-year survival >90%) with contemporary multi-modality treatment prompting attempts at treatment de-intensification to reduce late toxicity. This is a retrospective audit of clinical outcomes in WNT-MB patients treated at a tertiary-care comprehensive cancer centre.
Methods: Patients with molecularly confirmed WNT-MB treated with maximal safe resection followed by post-operative standard-of-care risk-stratified adjuvant radio(chemo)therapy were identified retrospectively via electronic search of the neuro-oncology database. Data regarding clinico-demographic characteristics, histo-molecular features, treatment details, patterns of failure, and survival outcomes was retrieved from electronic medical records and/or hospital case files. Time-to-event outcomes were analyzed using Kaplan-Meier methods and compared with the log-rank test.
Results: Between 2004 to 2020, 74 patients with WNT-MB were registered in the neuro-oncology unit of the institute. Seven patients treated on a prospective clinical trial of therapy de-intensification were excluded leaving 67 patients that constitute the present study cohort. Median age at presentation was 12 years (inter-quartile range 9-18 years) with a male preponderance (2:1). Six patients (1 post-operative mortality and 5 without adequate details of treatment or outcomes) were excluded from the survival analysis which was restricted to 61 patients. At a median follow-up of 72 months, Kaplan-Meier estimates of 5-year progression-free survival and overall survival were 87.7% and 91.2% respectively. Traditional high-risk features such as large residual tumor (≥1.5cm2) and leptomeningeal metastases (M+) did not significantly impact upon survival in this molecularly-characterized WNT-MB cohort.
Conclusions: This retrospective clinical audit confirms excellent survival in WNT-MB patients treated with contemporary multi-modality therapy. Lack of prognostic impact of conventional high-risk features suggests the need for refined risk-stratification and potential de-intensification of therapy.
Keywords:
medulloblastoma
; multi-modality therapy
; relapse
; survival
; WNT
1. Introduction
Medulloblastoma (MB), the most common malignant tumor involving the brain and central nervous system (CNS) in children is now considered to be a heterogeneous disease comprising of four broad molecular subgroups - wingless (WNT), sonic hedgehog (SHH), Group 3, and Group 4 respectively with subgroup-specific developmental origins, unique genetic profiles, distinct clinico-demographic characteristics, and diverse clinical outcomes “[1,2,3,4]”. This led to the incorporation of molecular/genetic information in the World Health Organization (WHO) CNS tumor classification that recommended an integrated layered diagnosis including available molecular/genetic information in the WHO 2016 update “[5]”. The 5th edition of WHO classification of tumors involving the CNS (WHO CNS 5) “[6]” now classifies MB as molecularly-defined and histologically-defined groups to reflect this knowledge regarding the existence of clinical and biological heterogeneity. Molecularly-defined MB comprises of WNT-activated subgroup; SHH-activated subgroup which could be TP53-wildtype or TP53-mutant; and non-WNT/non-SHH subgroup. The WHO 2016 update “[5]” had classified MB into four histo-morphologic types labelled as classic; desmoplastic/nodular (D/N); medulloblastoma with extensive nodularity (MBEN); and large cell/anaplastic (LC/A). These histologic subtypes have now been combined into one section that describes them as morphologic patterns of an inclusive tumor type, MB-histologically defined in WHO CNS 5 “[6]”.
MB has a high propensity to spread throughout the craniospinal axis via cerebrospinal fluid (CSF) pathways with metastatic disease being identified on neuraxial staging in nearly one third of patients at initial diagnosis “[2,3]” necessitating treatment of the entire brain and spinal cord including its covering meninges for disease control. Contemporary management for non-infantile MB “[4,7,8]” comprises of maximal safe resection followed by post-operative risk-stratified adjuvant craniospinal irradiation (CSI) to a dose of 23.4-36Gy/13-20 fractions plus boost irradiation of the tumor-bed to a dose of 18-30.6Gy/10-17 fractions resulting in total primary site radiotherapy (RT) dose of 54Gy/30 fractions followed by 6-9 cycles of multi-agent adjuvant systemic chemotherapy “[4,7,9]”. Traditionally, children over the age of 3-years at diagnosis, with no/small residual tumor (<1.5cm2) and absence of metastatic disease (M0) were classified as average-risk disease “[10]” with >80% long-term survival “[3,4,5,6,7,8,9,10,11]”; while younger age (<3 years), large residual tumor (≥1.5cm2), and presence of leptomeningeal metastases (M+ disease) either alone or in combination were considered high-risk features “[10]” yielding much worse 5-year survival (30-60%) despite aggressive multi-modality therapy “[13,14]”. This traditional risk-stratification has been further refined by incorporating molecular/genetic information in the contemporary molecular era into low-risk, standard-risk, high-risk, and very high-risk categories with expected 5-year overall survival of >90%, 75-90%, 50-75%, and <50% respectively “[15]”.
Aggressive multi-modality treatment achieves good survival outcomes in MB but is associated with significant acute and late treatment-related toxicities including but not limited to neuro-cognitive impairment, neuro-psychological dysfunction, endocrinopathy particularly growth retardation, sensori-neural hearing loss (SNHL), vasculopathy specially cerebro-vascular accident (CVA), and second malignant neoplasm (SMN) “[16,17]”. Of all MBs, WNT-subgroup has the best outcomes (5-year survival >90%) particularly in children “[3,4,13,18]” making these long-term survivors more susceptible to dose-dependent treatment-related late morbidity prompting systematic attempts at de-intensification of therapy “[19]”. An appropriate risk-classification schema and optimal treatment regimen for WNT-MB is yet to be defined. For this, it is important to identify prognostic factors and assess patterns of failure to guide therapeutic decision-making for tailoring adjuvant therapy in WNT-subgroup MB.
2. Materials and Methods
Patients with molecularly-confirmed WNT-MB treated with maximal safe resection followed by post-operative standard-of-care risk-stratified adjuvant radio(chemo)therapy were identified retrospectively via electronic search of the neuro-oncology database.
Molecular subgrouping: Molecular subgroup assignment of MB was based on an inhouse developed assay combining differential expression of 12 protein-coding genes and 9 miRNAs using real-time reverse transcriptase polymerase chain reaction (RT-PCR) as described previously “[20]”. Briefly, RNA (1–2 mg) was reverse transcribed using random hexameric primers and M-MLV reverse transcriptase (Invitrogen). The primers for real-time PCR analysis were designed such that they corresponded to 2 adjacent exons and, wherever possible, were located at exon boundaries to avoid amplification of genomic DNA. The amplicon size was maintained below 75 –80 bp, so as to enable amplification of the fragmented RNA from formalin-fixed paraffin-embedded (FFPE) tissues. The expression was analyzed by SYBR Green PCR amplification assay on an Applied Biosystems 7900HT real-time PCR system using 10 ng cDNA per reaction for frozen tissues and 10-100 ng cDNA per reaction for FFPE tissues. For miRNA expression analysis, 50 ng RNA from fresh tissues and 50–200 ng RNA from FFPE tissues were reverse transcribed using multiplex RT primer pools and the Taqman MicroRNA Reverse Transcription Kit (Applied Biosystems) according to manufacturer’s instructions. The expression of each miRNA was analyzed by TaqMan real-time miRNA assay (Applied Biosystems) on the ABI 7900HT real-time PCR system using 10 ng cDNA from frozen tissues and 10–40 ng cDNA from FFPE tissues. The relative quantity (RQ) of each protein-coding gene/miRNA compared with GAPDH/RNU48 was determined by the comparative cycle threshold (Ct) method. Genes that were significantly differentially expressed in the 4 molecular subgroups were identified by Significance Analysis of Microarray (MeV, http://www.TM4.com) of expression profiling data previously obtained using Affymetrix Gene 1.0 ST array. The selection of 12 protein-coding marker genes for classification from the significantly differentially expressed genes was based on the standardized fold-change in expression of the gene in a particular subgroup. Concomitant over-expression of WIF1, DKK2, and MYC identified WNT-MB. Over-expression of HHIP, EYA1, and MYCN with under-expression of OTX2 served as markers for the SHH-subgroup. The over-expression of EOMES helped to identify Group 3 and Group 4 tumors, while higher expression of NPR3, MYC, and IMPG2 with lower expression of GRM8 and UNC5D helped to distinguish Group 3 from Group 4 tumors. Similar to gene-expression profiling, the differential expression of 9 selected miRNAs was used for subgroup assignment. WNT-activated tumors showed significant over-expression of miR-193a-3p, miR-224, miR-148a, miR-23b, and miR-365 compared with other subgroups. MiR-182 was found to be over-expressed in all WNT-MBs and in many Group 3 and some Group 4 MBs, while miR-204 was over-expressed in all WNT-MBs and in most Group 4 MBs. MiR-182, miR-135b, and miR-204 were found to be under-expressed in SHH-activated MBs. MiR-135b was found to be over-expressed in Group 3 and Group 4 tumors. MiR-592, a miRNA that is located within the GRM8 gene was over-expressed in Group 4 MB. This aforesaid assay had previously been successfully validated “[20]” against a set of 34 well-annotated FFPE MB samples with subgroup assignment based on the 22-gene set NanoString assay from the German Cancer Research Centre (DFKZ). In recent times (after 2017), confirmation of WNT-activation was further supplemented by testing for monosomy 6 (fluorescence in-situ hybridization), CTNNB1 mutation analysis (Sanger sequencing), and/or nuclear beta-catenin positivity (immunohistochemical staining) as orthogonal techniques.
Treatment & follow-up: Information regarding patient demographics, clinical features, histopathological features, molecular profiling, risk-stratification, treatment details, and outcomes were retrieved from hospital case files and/or electronic medical records as was appropriate. All patients underwent maximal safe resection followed by post-operative risk-stratified adjuvant radio(chemo)therapy. Risk-stratification after surgery was based on conventional criteria without upfront knowledge of the molecular subgroup. Children (≤16-years) with average-risk MB defined as residual tumor <1.5cm2 with no evidence of metastases (M0) were treated with CSI (23.4Gy) plus boost irradiation (30.6Gy) for total primary-site dose of 54Gy followed by 6-cycles of adjuvant systemic chemotherapy. For adolescents and young adults (AYA) over 16-years of age at initial diagnosis with average-risk MB, RT alone was considered and comprised of full-dose CSI (35-36Gy) plus boost (18-19.8Gy) for total primary-site dose of 54-54.8Gy without adjuvant chemotherapy. The presence of any high-risk features such as large residue (≥1.5cm2), metastatic disease (M+) or adverse histology (LC/A) mandated full-dose/extended-dose CSI (35-40Gy) plus boost irradiation of primary-site (14.4-19.8Gy) with or without boost (5.4-9Gy) to the metastatic deposits followed by 6-cycles of adjuvant systemic chemotherapy. Following completion of adjuvant radio(chemo)therapy, patients were followed up clinically at 3-4 monthly intervals for the first two years, 6-monthly intervals till 5-years, and annually thereafter with periodic surveillance magnetic resonance imaging (MRI) scans as per institutional policy.
Statistical analysis: Clinical and demographic variables were analyzed and summarized using descriptive statistics with measures of central tendency and dispersion being reported. Patterns of relapse were defined as local recurrence (in and around the surgical cavity/resected tumor-bed); metastatic disease either involving the leptomeningeal space outside the initial tumor-bed in the cranial and/or spinal leptomeninges or extra-neural metastases (ENM) involving the bones, lymph nodes, or bone marrow; or a combination of the above. Progression free survival (PFS) was defined as the time interval from the date of surgery till documented clinico-radiological progression, or death due to any cause, or last follow-up. Overall survival (OS) was defined from the date of surgery till death due to any cause or last documented follow-up. Median follow up of surviving patients was calculated by the reverse Kaplan-Meier method. Time-to-event outcomes were analyzed using the product-limit method of Kaplan Meier and presented as 5-year estimates with 95% confidence interval (CI). Univariate analysis of variables of known and/or presumed prognostic significance was done using the log-rank test after dichotomization at median values or cut-offs established from earlier literature as appropriate. Statistical analysis was performed using SPSS version 25.0 (IBM Corporation, Armonk, USA) and R Studio version 3.2.7 (R Corporation, Vienna). The study was duly reviewed and approved by the Institutional Ethics Committee (IEC) that functions in accordance with the Declaration of Helsinki. IEC also granted waiver of consent due to retrospective nature of the study with no/minimal risk to participants.
3. Results
Electronic search of the neuro-oncology database identified a total of 504 MB patients registered in the neuro-oncology unit of the institute between 2004 till 2020 of which 74 (14.6%) were diagnosed as having WNT-subgroup MB “[20]”. Seven patients who were treated on a prospective protocol of therapy de-intensification in WNT-MB “[21]” were excluded from the dataset leaving 67 patients which constitute the present study cohort.
Clinico-demographic features: Patient, disease, and treatment characteristics of the study cohort are summarized in Table 1. Median age of the study cohort was 12 years with an inter-quartile range (IQR) of 9-18 years and preponderance of male gender (2:1 ratio). Pediatric WNT-MB (defined as age ≤ 16 years) comprised 73.1% (n=49) of patients compared to 26.9% (n=18) of AYA WNT-MB (defined as age >16 years). All patients underwent maximal safe resection with gross total resection (GTR) achieved in 49% of patients. Classic histology was the most common histological subtype seen in 61.2% (n=41) patients. Metastatic disease status by CSF cytology and/or neuro-imaging was available in 62 patients with majority (91.9%, n=57) being non-metastatic at initial diagnosis. Presence of any one or more of the following adverse features such as large residual tumor (≥1.5cm2), metastatic disease (M+) and LC/A histology classified 16 (29.1%) patients as having high-risk disease and 39 (70.9%) patients as average-risk disease. All included patients were treated post-operatively with contemporary risk-stratified RT comprising of CSI plus boost irradiation with or without adjuvant systemic chemotherapy. The median dose of CSI was 35Gy (IQR: 23.4-35Gy) with median tumor-bed boost dose of 19.8Gy (IQR: 19.8-30.6Gy). Extended dose CSI (40Gy) and boost irradiation of metastatic deposits was also done at the discretion of the treating radiation oncologist. Most of the patients were treated with conformal techniques either three-dimensional conformal radiotherapy (3D-CRT) or intensity modulated radiation therapy (IMRT) using 6MV photons on modern linear accelerators including tomotherapy. Adjuvant systemic chemotherapy was delivered in 72.2% (n=39) patients whereas 27.8% (n=15) patients did not receive any chemotherapy after completion of RT. Chemotherapy was initiated 4-6 weeks after completion of RT after sufficient myelo-recovery defined as absolute neutrophil count (ANC) >1500/dl and platelet count >100000/dl. Adjuvant chemotherapy generally comprised of 6 cycles of cisplatin (75mg/m2 intravenously on d1 in alternate cycles 2,4,6), cyclophosphamide (1000mg/m2 intravenously on d1-d2 in cycles 1,3,5 and d2-d3 in cycles 2,4,6) and vincristine (1.5mg/m2 intravenously d1 & d8 in all 6 cycles) given at 4-weekly intervals with adequate hydration, forced saline diuresis, mesna prophylaxis and requisite dose modifications as appropriate (7). Two of 5 children with metastatic disease at initial diagnosis also received 1-year of maintenance chemotherapy post-completion of standard therapy using the modified combined oral metronomic bio-differentiating anti-angiogenic therapy (COMBAT) regimen comprising of temozolomide, etoposide, celecoxib, fenofibrate, and retinoic acid.
Patterns of failure, causes of death, and survival outcomes: Six patients (1 post-operative mortality and 5 without adequate details of treatment or outcomes) were excluded from the survival analysis which was restricted to 61 patients. Nine of the 61 included patients experienced an event of interest (relapse and/or death). Seven patients were detected with relapse on follow-up with leptomeningeal dissemination seen in 4 patients (including one with synchronous local recurrence), local tumor-bed recurrence in 3 patients (including one with synchronous neuraxial relapse), and isolated extra-neural metastases (lymph nodes, bones) in a single patient. Images from one such case scenario each of tumor-bed recurrence only, synchronous local recurrence with neuraxial failure, and isolated extra-neural (ENM) metastases from the study cohort are illustrated in Figure 1. Seven of 61 patients have died by the time of this analysis, six of recurrent/progressive disease and one due to chemotherapy-induced febrile neutropenia leading to septic shock and death. Clinico-demographic details, pattern of relapse, and outcomes of all these 9 patients experiencing an event are summarized in Table 2. Of the five WNT-MB patients who were treated with salvage therapy at relapse, two patients (W1: tumor-bed recurrence and W2: diffuse leptomeningeal metastases) achieved post-relapse survival of 55 and 29 months respectively, while the lone patient with ENM (W8) was alive with disease on salvage systemic chemotherapy at the time of this analysis. Two patients treated with re-excision alone without further re-irradiation or salvage chemotherapy succumbed to further progressive disease within 6-9 months of relapse. All the three WNT-MB patients offered best supportive care at relapse died of progressive disease within 3-months of first relapse. At a median follow-up of 72 months (IQR: 51-101 months) for the entire study cohort (N=61), the 5-year Kaplan-Meier estimates of PFS and OS were 87.7% (95%CI: 75.1-96.1%) and 91.2% (95%CI: 83.0-100%) respectively (Figure 2). Univariate analysis of various patient, disease, and treatment related factors did not identify any putative prognostic factor impacting upon PFS or OS (Table 3). Multi-variate analysis was considered inappropriate due to small number of events in the study cohort.
4. Discussion
The clinico-demographic characteristics of this large cohort of WNT-MB patients treated at an academic neuro-oncology unit of tertiary-care comprehensive cancer centre are largely in accordance with previously published literature with minor differences. The present study had more males with WNT-MB than females (2:1) possibly due to socio-cultural differences and patriarchal mindset prevalent in low-middle income country setting in South-East Asia compared to the fairly balanced gender ratio reported previously from high-income countries of the West “[18]”. Median age at diagnosis of the present study cohort was also slightly higher (12 years, IQR: 9-18 years) reflecting an increased representation of adult WNT-MB compared to an international reference cohort (median 10 years, IQR: 8-14.2 years) which was largely limited to the pediatric age group “[22]”.
Given the low prevalence of WNT-MB (constituting around 10% of all MBs) coupled with very low risk of failure in appropriately treated patients, prognostic factors impacting upon survival, patterns of relapse, and drivers of metastatic dissemination are relatively poorly understood. Nobre et al. “[22]”assembled a retrospective multi-institutional clinically annotated cohort of 93 WNT-pathway medulloblastoma patients using an integrated genomic approach. Fifteen patients with relapse were identified, 12 in metastatic compartment including one with ENM and 3 in the surgical cavity. Interestingly, 8 of 11 neuraxial relapses were in lateral ventricles (6 confined to frontal horns) leading to the hypothesis that ependymal lining of the lateral ventricles may be more conducive to homing of WNT-MB. Maintenance systemic chemotherapy (p=0.033), specifically lower cumulative dose of cyclophosphamide/ifosfamide (<12mg/m2) was reported to be associated with increased risk of relapse. It was proposed that the paracrine signals driven by mutant β-catenin protein induce a fenestrated tumor vasculature promoting accumulation of chemotherapeutic agents within the tumor-bed. The authors also reported that male gender (p=0.032) was associated with significantly increased risk of relapse in WNT-MB. Age at diagnosis, extent of resection, metastatic status at presentation, dose of CSI, and additional molecular/genetic alterations did not predict the risk of relapse in their study. In another cohort of 191 WNT-MB patients registered in the HIT database “[23]”, mutations in CTNNB1, APC, and TP53 were analyzed by DNA sequencing and chromosomal copy number aberrations by molecular inversion probe technology to identify the prognostic impact of TP53 mutations and other chromosomal aberrations in WNT-subgroup. Patients with tumors harboring TP53 mutation showed worse outcomes (5-year PFS: 68% vs 93%, p=0.001 and 5-year OS: 81% vs 95%, p=0.105) compared to TP53 wild-type tumors. Gain of OTX2 was associated with inferior survival outcomes (5-year PFS: 72% vs 93%, p=0.017 and 5-year OS:83% vs 97%, p=0.006). Multivariable Cox regression analysis identified both genetic alterations as independent prognostic markers for survival raising concerns regarding inclusion of such patients in ongoing prospective trials of therapy de-intensification.
The presence of intra-tumoral heterogeneity within the four broad molecular subgroups prompted several researchers to perform large-scale integrative clustering analysis combining DNA methylation and gene-expression profiling to identify further subtypes within each broad molecular subgroup “[24,25,26]” resulting in consensus definition of 12 subtypes of MB in second generation molecular subgrouping “[27]”. WNT-pathway MB typically demonstrate homogenous genome-wide expression patterns and methylation profiles; however, two molecular subtypes of WNT-activated MB have been identified “[27]” referred to as WNT-α and WNT-β that differ in age at diagnosis (median age of 10 vs 20 years), frequency of monosomy 6 (>85% vs <50%), histo-morphology (typically classic vs sometimes LC/A) and metastatic disease (absent vs occasionally present) respectively. Very rarely WNT-MB may harbor distinct genetic alterations typical of another molecular subgroup (such as SHH and non-WNT/non-SHH) in addition to WNT-activation referred to as hybrid molecular subtypes “[28]” indicating intra-tumoral heterogeneity with potential prognostic implications. MYC oncogenes are the most commonly amplified loci in MB “[29,30]” that are generally associated with non-WNT/non-SHH disease (particularly subgroup 3), LC/A histology, and metastatic dissemination making them known biomarkers of poor prognosis. Although overexpression of MYC can also be seen in WNT-subgroup MB with no detrimental impact on survival “[30]”, MYC-amplification has rarely been described “[31]” in WNT-activated tumors. It may be pertinent to note that increased MYC-signaling has been shown to accelerate tumor growth and promote metastases in a murine model of WNT-MB “[32]”.
Although MB largely remains a disease of childhood with much lower incidence in the AYA population, it is common perception that excellent survival outcomes achieved in the pediatric population may not be exactly mirrored in the AYA cohort “[33,34]”. However, contrary to popular belief, analysis of the Surveillance, Epidemiology, and End Results (SEER) database “[35]” from 1992-2013 reported comparable 2-year, 5-year, and 10-year survival outcomes between childhood (n=616) and adult MB (n=349). The first comprehensive molecular analysis adult MB “[36]” defined three broad molecular subgroups viz. WNT, SHH, and Group D (later reclassified as Group 4) with absence of Group 3 tumors. The authors reported worse prognosis of adult WNT-MB and Group 4 disease compared to corresponding subgroups of childhood MB; however, survival in SHH-subgroup MB was similar across both age groups. In another large multi-institutional dataset of adult MB “[37]”, there was no prognostic impact of molecular subgrouping with 5-year survival of 45%, 67%, 62% and 67% for WNT, SHH, Group 3, and Group 4 respectively. The largest integrative analysis of adult MB “[38]” also reported no statistically significant survival differences between the four broad molecular subgroups with 5-year PFS (95%CI) of 64.4% (48.0-86.5%), 61.9% (51.6-74.2%), 80.0% (51.6-100%), and 44.9% (28.6-70.7%) for WNT (n=30), SHH (n=112), Group 3 (n=6), and Group 4 (n=41) respectively. However, what stands out clearly is the substantially lower survival in adults with WNT-activated MB (5-year survival 45-70%) compared to benchmark outcomes in childhood WNT-MB (5-year survival >90%). However, this notion has recently been challenged with molecular subgrouping emerging as a significant prognostic factor in AYA-MB. In a large single-institutional dataset “[39]” of molecularly-characterized AYA-MB (≥15-years at initial diagnosis), the reported 5-year survival was 87.5%, 62.2%, and 50.1% for WNT (n=14), SHH (n=71), and non-WNT/non-SHH (n=21) subgroups respectively. A comparative analysis of pediatric versus AYA WNT-MB also reported similar survival outcomes “[40]” suggesting that age alone should not be used to intensify treatment in WNT-MB.
The time to recurrence (early vs delayed), pattern of failure (local, metastatic, or combined) and post-relapse survival in MB is largely dictated by disease biology and varies across the four broad molecular subgroups “[41,42,43]”. Time to relapse even within the WNT-subgroup has been variable across studies with both early as well as delayed relapses being reported “[22,34] “sometimes even beyond 10-years from initial diagnosis. Management of relapsed MB after appropriate and adequate upfront radio(chemo)therapy is not clearly defined with no universally acceptable standard-of-care salvage treatment “[44]”. A substantial and large proportion of these patients particularly with disseminated disease are offered best supportive care alone with only a small minority being treated with aggressive multi-modality salvage therapies including a combination of re-excision (isolated local relapse), re-irradiation, and systemic therapies that might include high-dose chemotherapy with autologous stem cell rescue and targeted therapy as appropriate. The prognosis of relapsed MB in patients previously treated with CSI in the upfront setting is typically poor and considered non-curative with <5% long-term survival despite aggressive salvage therapies “[44]”. However, it is now being increasingly appreciated that post-relapse outcomes might be somewhat subgroup-dependent with WNT-MB and Group 4 tumors demonstrating a more indolent clinical course with favorable outcomes compared to SHH-MB and Group 3 disease. This study also reported favorable outcomes in a subset of relapsed WNT-MB patients further raising the question whether these patients can be treated with upfront de-intensified therapy at initial diagnosis reserving treatments associated with high morbidity at the time of relapse. The patterns of failure and 5-year survival outcomes of appropriately treated WNT-MB patients in various molecularly-informed prospective cohort studies including randomized controlled trials are summarized in Table 4 “[12,13,42,45,46,47,48]” that reaffirms excellent prognosis and provides justification for ongoing global efforts towards de-escalation of therapy “[19]”. However, such an approach warrants caution as two prospective de-intensification studies had to be terminated prematurely due to unacceptably high risk of failures. The first of these “[21]” treated rigorously-defined low-risk WNT-MB patients with focal-only conformal RT to the index tumor-bed (54Gy) plus adjuvant systemic chemotherapy with omission of upfront CSI. Three of the first 7 patients accrued on the study were detected with neuraxial dissemination within 2-years of index diagnosis and although they were subsequently successfully salvaged in the short-term with aggressive multi-modality therapy including full-dose CSI and systemic chemotherapy, mature outcomes of salvage therapy remain to be reported. The second study “[49]” utilized a post-surgery primary chemotherapy approach eliminating RT completely in low-risk WNT-MB. Once again, three of the first 6 children on the study developed local recurrence and neuraxial dissemination shortly after completing chemotherapy leading to early closure due to safety concerns. Two of them were successfully salvaged with RT including CSI plus additional chemotherapy, but one child succumbed to further progressive disease at 35-months from initial diagnosis. Of the remaining 3 patients, 2 children proceeded to immediate RT after completion of primary chemotherapy to protect against early relapse, while the remaining child was switched mid-treatment to high-dose chemotherapy with autologous stem-cell rescue. Both these studies reinforce the need of RT, particularly CSI for effective disease control even in low-risk, favorable-biology WNT-pathway MB “[50]”.
Strengths & limitations: This study represents the largest descriptive analysis of WNT-MB treated with contemporary risk-stratified radio(chemo)therapy at a single institution anywhere in the world. Access to advanced molecular diagnostics for subgroup assignment and therapeutic decision-making in multi-disciplinary neuro-oncology clinic add further strength to the study. However, despite the above-mentioned strengths, several caveats and limitations remain. Retrospective nature of the study makes it susceptible to inherent biases that could potentially confound interpretation of results. Various platforms exist for robust molecular subgrouping of MB including immunohistochemistry panel, gene-expression analysis, microRNA profiling, and DNA methylation array. The study used combined gene-expression analysis and microRNA profiling for molecular subgroup assignment; however, DNA methylation which is considered the current gold-standard and method of choice for molecular classification of MB was not performed due to issues with availability, accessibility, and affordability. Although rare, the co-occurrence of additional genetic alterations to identify any hybrid molecular subtypes was not assessed in the study. Analysis of survival outcomes was restricted to 61 patients (after excluding 6 patients) which could be a potential source of bias. Follow-up duration though long (median of 72 months) may be considered inadequate to capture very delayed relapses and SMNs. Although therapeutic decision-making was largely based on discussion in a multi-disciplinary tumor board, patients may not have been treated uniformly over the long period of the study potentially impacting upon outcomes. Finally, lack of documented data on neuro-cognitive impairment, neuro-psychological dysfunction, SNHL, endocrinopathies, CVA, and resultant quality-of-life precludes assessment of the impact of treatment-related late toxicity in these long-term survivors.
5. Conclusions
This retrospective clinical audit confirms excellent survival in WNT-MB patients treated with contemporary multi-modality therapy comprising of maximal safe resection followed by risk-stratified appropriate radio(chemo)therapy. Lack of prognostic impact of conventional high-risk features suggests the need for refined risk-stratification and potential for de-intensification of therapy in WNT-MB.
Author Contributions
“Conceptualization, TG; methodology, TG, AC, & SM; formal analysis, AP, SM, & TG; investigation, NS, SE, AyS, & ArS; resources, TG, AM, & GC; data curation, SM, AD, AC, & TG; writing—original draft preparation, SM; writing—review and editing, TG, MP, & AC; supervision, TG, AC, SM; project administration, TG; funding acquisition, not applicable. All authors have read and agreed to the published version of the manuscript.”
Funding
This research received no external funding.
Institutional Review Board Statement
The study was reviewed by the Institutional Ethics Committee (IEC) of ACTREC, Tata Memorial Centre, Mumbai, India (IEC Project No. 900642) that granted approval on 03.07.2020 with waiver of consent.
Informed Consent Statement
The study was reviewed by Institutional Ethics Committee (IEC) of ACTREC, Tata Memorial Centre, Mumbai, India (IEC Project No. 900642) that granted approval on 03.07.2020 with waiver of consent.
Data Availability Statement
All the data generated from this study are vested with the Principal Investigator and corresponding author. Data are not publicly available due to privacy issues but can be made available in an anonymized format upon reasonable request to the corresponding author.
Acknowledgments
Brain Tumor Foundation (BTF) of India.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Taylor MD, Northcott PA, Korshunov A, Remke M, Cho YJ, Clifford SC, et al. Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol. 2012 Apr;123(4):465–72. [CrossRef]
- Gajjar A, Bowers DC, Karajannis MA, Leary S, Witt H, Gottardo NG. Pediatric Brain Tumors: Innovative Genomic Information Is Transforming the Diagnostic and Clinical Landscape. J Clin Oncol. 2015 Sep 20;33(27):2986–98. [CrossRef]
- Gupta T, Shirsat N, Jalali R. Molecular Subgrouping of Medulloblastoma: Impact Upon Research and Clinical Practice. Curr Pediatr Rev. 2015;11(2):106–19. [CrossRef]
- Northcott PA, Robinson GW, Kratz CP, Mabbott DJ, Pomeroy SL, Clifford SC, et al. Medulloblastoma. Nature Reviews Disease Primers [Internet]. 2019 Feb 14;5(1):11. Available from: https://doi.org/10.1038/s41572-019-0063-6. [CrossRef]
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016 Jun;131(6):803–20. [CrossRef]
- Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021 Aug 2;23(8):1231–51. [CrossRef]
- Gupta T, Sarkar C, Rajshekhar V, Chatterjee S, Shirsat N, Muzumdar D, et al. Indian Society of Neuro-Oncology consensus guidelines for the contemporary management of medulloblastoma. Neurol India. 2017 Apr;65(2):315–32. [CrossRef]
- Lazow MA, Palmer JD, Fouladi M, Salloum R. Medulloblastoma in the Modern Era: Review of Contemporary Trials, Molecular Advances, and Updates in Management. Neurotherapeutics. 2022 Oct;19(6):1733-51. [CrossRef]
- Mushtaq N, Ul Ain R, Hamid SA, Bouffet E. Evolution of Systemic Therapy in Medulloblastoma Including Irradiation-Sparing Approaches. Diagnostics [Internet]. 2023 Jan [cited 2023 Dec 18];13(24):3680. Available from: https://www.mdpi.com/2075-4418/13/24/3680. [CrossRef]
- Zeltzer PM, Boyett JM, Finlay JL, Albright AL, Rorke LB, Milstein JM, et al. Metastasis Stage, Adjuvant Treatment, and Residual Tumor Are Prognostic Factors for Medulloblastoma in Children: Conclusions From the Children’s Cancer Group 921 Randomized Phase III Study. JCO [Internet]. 1999 Mar [cited 2023 Dec 20];17(3):832–832. Available from: https://ascopubs.org/doi/10.1200/JCO.1999.17.3.832. [CrossRef]
- Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, et al. Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol. 2006 Sep 1;24(25):4202–8. [CrossRef]
- Michalski JM, Janss AJ, Vezina LG, Smith KS, Billups CA, Burger PC, et al. Children’s Oncology Group Phase III Trial of Reduced-Dose and Reduced-Volume Radiotherapy With Chemotherapy for Newly Diagnosed Average-Risk Medulloblastoma. J Clin Oncol. 2021 Aug 20;39(24):2685–97. [CrossRef]
- Gajjar A, Robinson GW, Smith KS, Lin T, Merchant TE, Chintagumpala M, et al. Outcomes by Clinical and Molecular Features in Children With Medulloblastoma Treated With Risk-Adapted Therapy: Results of an International Phase III Trial (SJMB03). J Clin Oncol. 2021 Mar 1;39(7):822–35. [CrossRef]
- Bouffet E. Management of high-risk medulloblastoma. Neurochirurgie. 2021 Feb;67(1):61–8. [CrossRef]
- Ramaswamy V, Remke M, Bouffet E, Bailey S, Clifford SC, Doz F, et al. Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathol. 2016 Jun;131(6):821–31. [CrossRef]
- Fossati P, Ricardi U, Orecchia R. Pediatric medulloblastoma: toxicity of current treatment and potential role of protontherapy. Cancer Treat Rev. 2009 Feb;35(1):79–96. [CrossRef]
- Salloum R, Chen Y, Yasui Y, Packer R, Leisenring W, Wells E, et al. Late Morbidity and Mortality Among Medulloblastoma Survivors Diagnosed Across Three Decades: A Report From the Childhood Cancer Survivor Study. J Clin Oncol. 2019 Mar 20;37(9):731–40. [CrossRef]
- Kool M, Korshunov A, Remke M, Jones DTW, Schlanstein M, Northcott PA, et al. Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 2012 Apr;123(4):473–84. [CrossRef]
- Thompson EM, Ashley D, Landi D. Current medulloblastoma subgroup specific clinical trials. Transl Pediatr. 2020 Apr;9(2):157–62. [CrossRef]
- Kunder R, Jalali R, Sridhar E, Moiyadi A, Goel N, Goel A, et al. Real-time PCR assay based on the differential expression of microRNAs and protein-coding genes for molecular classification of formalin-fixed paraffin embedded medulloblastomas. Neuro Oncol. 2013 Dec;15(12):1644–51. [CrossRef]
- Gupta T, Pervez S, Dasgupta A, Chatterjee A, Epari S, Chinnaswamy G, et al. Omission of Upfront Craniospinal Irradiation in Patients with Low-Risk WNT-Pathway Medulloblastoma Is Associated with Unacceptably High Risk of Neuraxial Failure. Clin Cancer Res. 2022 Oct 3;28(19):4180–5.
- Nobre L, Zapotocky M, Khan S, Fukuoka K, Fonseca A, McKeown T, et al. Pattern of Relapse and Treatment Response in WNT-Activated Medulloblastoma. Cell Rep Med. 2020 Jun 23;1(3):100038. [CrossRef]
- Goschzik T, Mynarek M, Doerner E, Schenk A, Spier I, Warmuth-Metz M, et al. Genetic alterations of TP53 and OTX2 indicate increased risk of relapse in WNT medulloblastomas. Acta Neuropathol. 2022 Dec;144(6):1143–56.
- Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, et al. Intertumoral Heterogeneity within Medulloblastoma Subgroups. Cancer Cell. 2017 Jun 12;31(6):737-754.e6. [CrossRef]
- Schwalbe EC, Lindsey JC, Nakjang S, Crosier S, Smith AJ, Hicks D, et al. Novel molecular subgroups for clinical classification and outcome prediction in childhood medulloblastoma: a cohort study. The Lancet Oncology [Internet]. 2017 Jul 1 [cited 2024 Jan 8];18(7):958–71. Available from: https://www.thelancet.com/journals/lanonc/article/PIIS1470-2045(17)30243-7/fulltext. [CrossRef]
- Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, et al. The whole-genome landscape of medulloblastoma subtypes. Nature [Internet]. 2017 Jul [cited 2021 Jun 26];547(7663):311–7. Available from: https://www.nature.com/articles/nature22973. [CrossRef]
- Hovestadt V, Ayrault O, Swartling FJ, Robinson GW, Pfister SM, Northcott PA. Medulloblastomics revisited: biological and clinical insights from thousands of patients. Nat Rev Cancer [Internet]. 2020 Jan [cited 2024 Jan 8];20(1):42–56. Available from: https://www.nature.com/articles/s41568-019-0223-8. [CrossRef]
- Helgager J, Pytel P, Vasudevaraja V, Lee EQ, Snuderl M, Iorgulescu JB, et al. WNT-Activated Medulloblastomas With Hybrid Molecular Subtypes. JCO Precis Oncol. 2020;4:PO.19.00332. [CrossRef]
- Ryan SL, Schwalbe EC, Cole M, Lu Y, Lusher ME, Megahed H, et al. MYC family amplification and clinical risk-factors interact to predict an extremely poor prognosis in childhood medulloblastoma. Acta Neuropathol [Internet]. 2012 Apr 1 [cited 2023 Dec 25];123(4):501–13. Available from: https://doi.org/10.1007/s00401-011-0923-y. [CrossRef]
- Park AK, Lee SJ, Phi JH, Wang KC, Kim DG, Cho BK, et al. Prognostic classification of pediatric medulloblastoma based on chromosome 17p loss, expression of MYCC and MYCN, and Wnt pathway activation. Neuro Oncol. 2012 Feb;14(2):203–14. [CrossRef]
- Green S, Hoover T, Doss D, Davidow K, Walter AW, Cottrell CE, et al. WNT-activated, MYC-amplified medulloblastoma displaying intratumoural heterogeneity. Neuropathology and Applied Neurobiology [Internet]. [cited 2023 Dec 25];n/a(n/a):e12945. Available from: https://onlinelibrary.wiley.com/doi/abs/10.1111/nan.12945. [CrossRef]
- Hartley R, Phoenix TN. MYC promotes aggressive growth and metastasis of a WNT-medulloblastoma mouse model. Dev Neurosci. 2023 Aug 5; [CrossRef]
- Korshunov A, Remke M, Werft W, Benner A, Ryzhova M, Witt H, et al. Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. J Clin Oncol. 2010 Jun 20;28(18):3054–60. [CrossRef]
- Wooley JR, Penas-Prado M. Pediatric versus Adult Medulloblastoma: Towards a Definition That Goes beyond Age. Cancers (Basel). 2021 Dec 16;13(24):6313.
- Li Q, Dai Z, Cao Y, Wang L. Comparing children and adults with medulloblastoma: a SEER based analysis. Oncotarget [Internet]. 2018 Jul 10 [cited 2024 Jan 8];9(53):30189–98. Available from: https://www.oncotarget.com/article/23773/. [CrossRef]
- Remke M, Hielscher T, Northcott PA, Witt H, Ryzhova M, Wittmann A, et al. Adult medulloblastoma comprises three major molecular variants. J Clin Oncol. 2011 Jul 1;29(19):2717–23. [CrossRef]
- Wong Wong GCH, Li KKW, Wang WW, Liu APY, Huang QJ, Chan AKY, et al. Clinical and mutational profiles of adult medulloblastoma groups. Acta Neuropathol Commun. 2020 Nov 10;8(1):191.
- Coltin H, Sundaresan L, Smith KS, Skowron P, Massimi L, Eberhart CG, et al. Subgroup and subtype-specific outcomes in adult medulloblastoma. Acta Neuropathol. 2021 Aug 18; [CrossRef]
- Patil R, Gupta T, Maitre M, Dasgupta A, Sahay A, Epari S, et al. Clinical Audit of Survival Outcomes and Prognostic Factors in Adolescents and Adults with Medulloblastoma. Journal of Adolescent and Young Adult Oncology [Internet]. 2021 Apr 23 [cited 2021 Oct 6]; Available from: https://www.liebertpub.com/doi/abs/10.1089/jayao.2021.0034. [CrossRef]
- Mani S, Chatterjee A, Dasgupta A, Shirsat N, Epari S, Chinnaswamy G, et al. WNT-pathway medulloblastoma: what constitutes low-risk and how low can one go? Oncotarget [Internet]. 2023 Jan 12 [cited 2023 Mar 7];14:105–10. Available from: https://www.oncotarget.com/article/28360/text/.
- Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol. 2013 Nov;14(12):1200–7. [CrossRef]
- Sabel M, Fleischhack G, Tippelt S, Gustafsson G, Doz F, Kortmann R, et al. Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. J Neurooncol. 2016 Sep;129(3):515–24. [CrossRef]
- Hill RM, Richardson S, Schwalbe EC, Hicks D, Lindsey JC, Crosier S, et al. Time, pattern, and outcome of medulloblastoma relapse and their association with tumour biology at diagnosis and therapy: a multicentre cohort study. Lancet Child Adolesc Health. 2020 Dec;4(12):865–74. [CrossRef]
- Hill RM, Plasschaert SLA, Timmermann B, Dufour C, Aquilina K, Avula S, et al. Relapsed Medulloblastoma in Pre-Irradiated Patients: Current Practice for Diagnostics and Treatment. Cancers [Internet]. 2022 Jan [cited 2024 Jan 8];14(1):126. Available from: https://www.mdpi.com/2072-6694/14/1/126. [CrossRef]
- Pietsch T, Schmidt R, Remke M, Korshunov A, Hovestadt V, Jones DTW, et al. Prognostic significance of clinical, histopathological, and molecular characteristics of medulloblastomas in the prospective HIT2000 multicenter clinical trial cohort. Acta Neuropathol. 2014 Jul;128(1):137–49.
- Von Bueren AO, Kortmann RD, von Hoff K, Friedrich C, Mynarek M, Müller K, et al. Treatment of Children and Adolescents With Metastatic Medulloblastoma and Prognostic Relevance of Clinical and Biologic Parameters. J Clin Oncol. 2016 Dec;34(34):4151–60. [CrossRef]
- Leary SES, Packer RJ, Li Y, Billups CA, Smith KS, Jaju A, et al. Efficacy of Carboplatin and Isotretinoin in Children With High-risk Medulloblastoma: A Randomized Clinical Trial From the Children’s Oncology Group. JAMA Oncol. 2021 Sep 1;7(9):1313–21.
- Dufour C, Foulon S, Geoffray A, Masliah-Planchon J, Figarella-Branger D, Bernier-Chastagner V, et al. Prognostic relevance of clinical and molecular risk factors in children with high-risk medulloblastoma treated in the phase II trial PNET HR+5. Neuro Oncol. 2021 Jul 1;23(7):1163–72. [CrossRef]
- Cohen KJ, Munjapara V, Aguilera D, Castellino RC, Stapleton SL, Landi D, et al. A Pilot Study Omitting Radiation in the Treatment of Children with Newly Diagnosed Wnt-Activated Medulloblastoma. Clin Cancer Res. 2023 Dec 15;29(24):5031–7. [CrossRef]
- Gottardo NG, Gajjar A. Verschlimmbesserung: Craniospinal Radiotherapy Is Essential in WNT Medulloblastoma Patients. Clinical Cancer Research [Internet]. 2023 Dec 15 [cited 2024 Jan 8];29(24):4996–8. Available from: https://doi.org/10.1158/1078-0432.CCR-23-2331. [CrossRef]
Figure 1.
Kaplan–Meier curves of progression-free survival (A) and overall survival (B) for WNT-pathway medulloblastoma in the study cohort.
Figure 1.
Kaplan–Meier curves of progression-free survival (A) and overall survival (B) for WNT-pathway medulloblastoma in the study cohort.
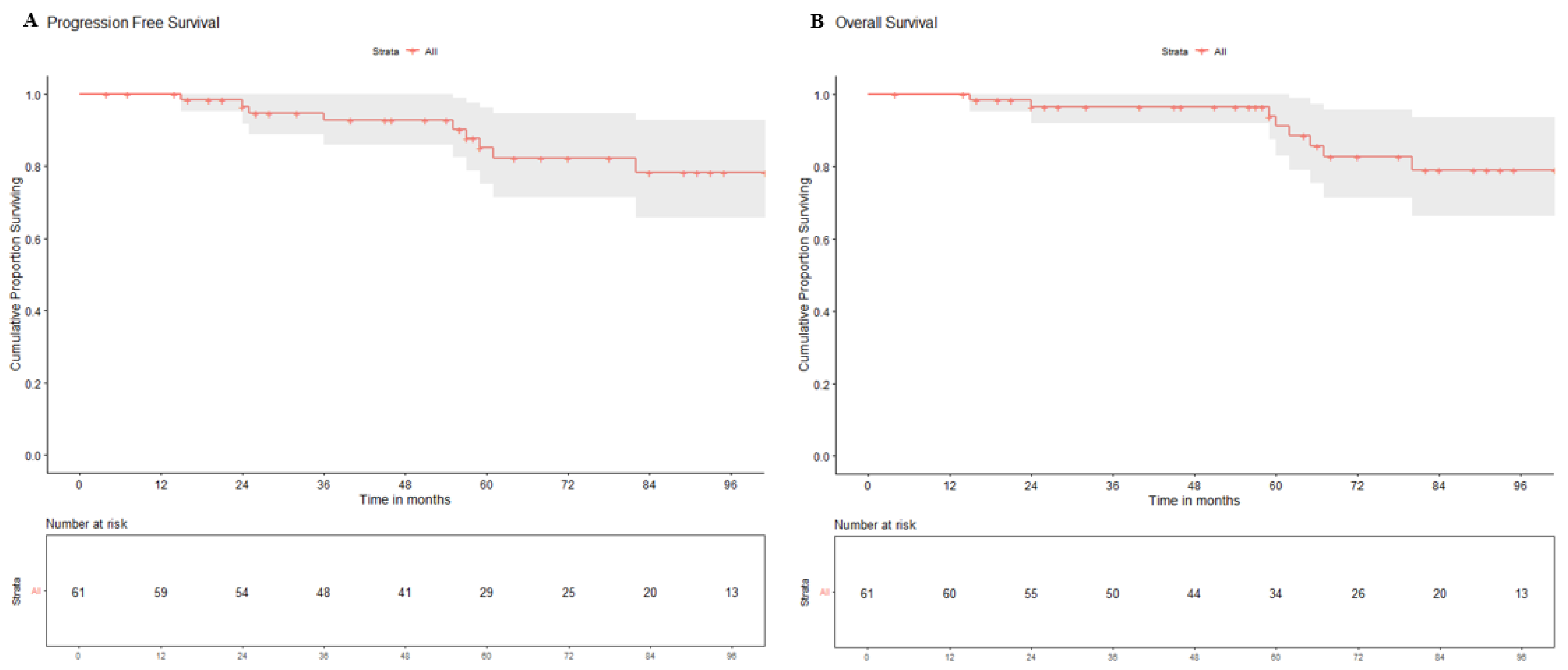
Figure 2.
Representative images of three patients from the study cohort with local recurrence in the tumor-bed (A), synchronous local relapse with spinal deposits (B), and extra-neural metastases (C) in WNT-subgroup medulloblastoma.
Figure 2.
Representative images of three patients from the study cohort with local recurrence in the tumor-bed (A), synchronous local relapse with spinal deposits (B), and extra-neural metastases (C) in WNT-subgroup medulloblastoma.
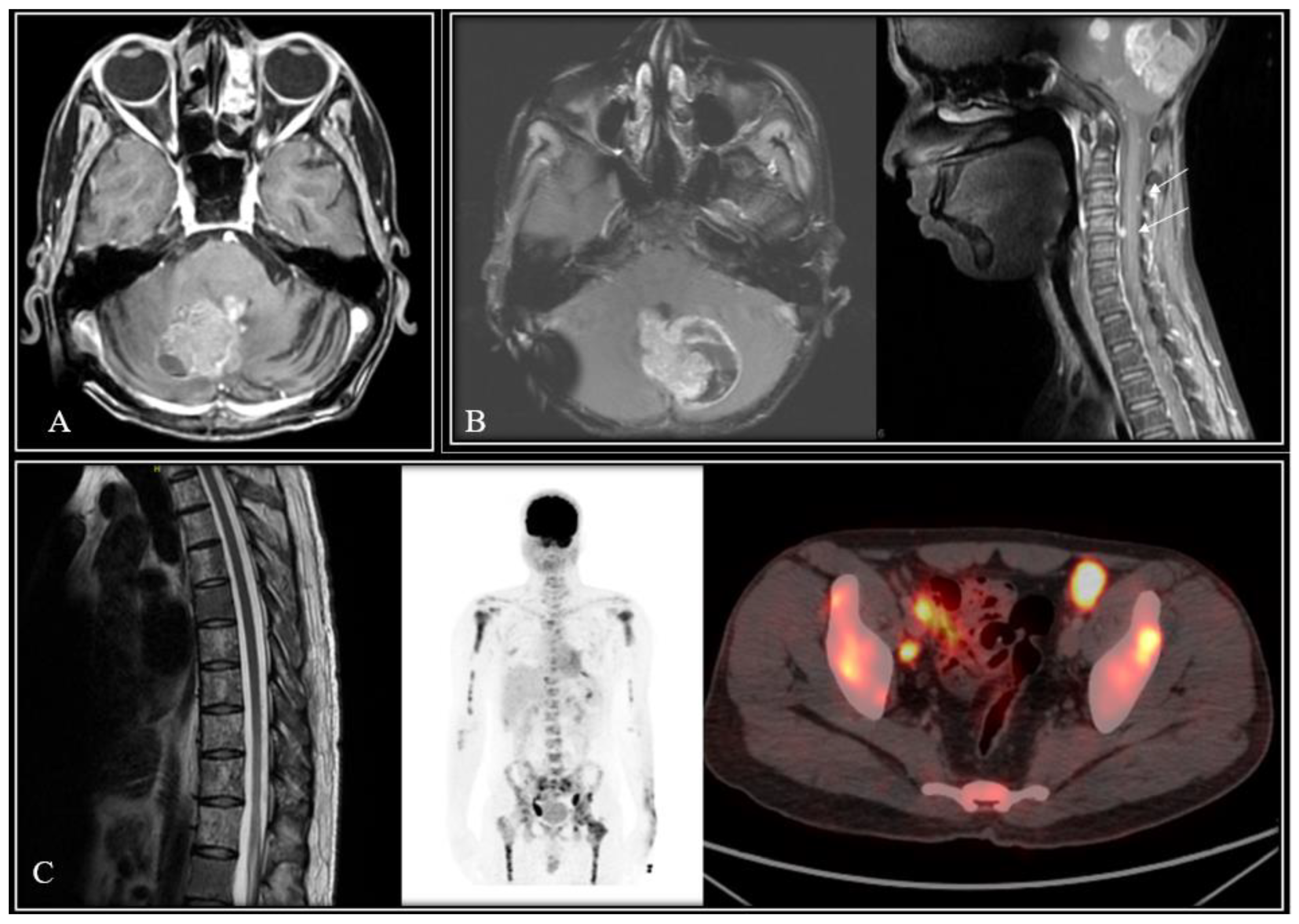

Table 1.
Patient, disease, and treatment characteristics of the study cohort (N=67).
| Characteristics | Number of patients (%) |
| Median age (inter-quartile range) at diagnosis | 12 years (9-18 years) |
| Gender Male Female |
44 (65.7%) 23 (34.3%) |
| Post-operative residual tumor (n=57) <1.5cm2 ≥1.5cm2 |
47 (82.4%) 10 (17.6%) |
| Metastastic status at diagnosis (n=62) Non-metastatic (M0) Metastatic disease (M+) |
57 (91.9%) 05 (08.1%) |
| Conventional risk-stratification (n=55) Average-risk High-risk |
39 (70.9%) 16 (29.1%) |
| Histological subtype Medulloblastoma (not otherwise specified) Classic Desmoplastic Large-cell/Anaplastic |
22 (32.8%) 41 (61.2%) 03 (04.5%) 01 (01.5%) |
| Time interval from surgery to adjuvant radiotherapy (n=40) ≤6 weeks >6 weeks |
17 (42.5%) 23 (57.5%) |
| Craniospinal irradiation dose (n=54) 23.4-26Gy$ 35-36Gy |
23 (42.6%) 31 (57.4%) |
| Craniospinal irradiation technique (n=39) Conventional radiotherapy Three-dimensional conformal radiotherapy Intensity modulated radiation therapy |
01 (02.6%) 16 (41.0%) 22 (56.4%) |
| Adjuvant systemic chemotherapy (n=54) Yes No |
39 (72.2%) 15 (27.8%) |
| Cumulative cyclophosphamide dose (n=39) ≤12mg/m2 >12mg/m2 |
11 (28.2%) 28 (71.8%) |
$One patient was planned for 23.4Gy craniospinal irradiation but defaulted after 14.4Gy/8 fractions.
Table 2.
Patterns of relapse and salvage therapy in WNT-pathway medulloblastoma experiencing an event (relape/death) in the study (n=9).
Table 2.
Patterns of relapse and salvage therapy in WNT-pathway medulloblastoma experiencing an event (relape/death) in the study (n=9).
| Sr No. |
Age (years) /Gender |
Stage | CSI dose at initial diagnosis | Pattern of first failure | PFS | Salvage therapy at relapse | Final Outcome | OS |
|---|---|---|---|---|---|---|---|---|
| W1 | 9/Male | Non-Metastatic | 36Gy/18fx | Tumor-bed recurrence | 25-months | Re-RT & chemotherapy | Died of disease | 80-months |
| W2 | 11/Male | Non-metastatic | 26Gy/13fx | Tumor-bed relapse plus metastases in brainstem, temporal lobe & spine | 37-months | Re-CSI (36Gy/36fx) & chemotherapy | Died of disease | 66-months |
| W3 | 13/Female | Non-metastatic | 35Gy/21fx | Leptomeningeal dissemination | 25-months | Best supportive care | Died of disease | 25-months |
| W4 | 10/Male | Non-metastatic | 14.4Gy/8fx (Incomplete RT) | Leptomeningeal dissemination |
61-months | Best supportive care | Died of disease | 62-months |
| W5 | 27/Male | Non-metastatic | 35Gy/21fx | Leptomeningeal dissemination | 58-months | Best supportive care | Died of disease | 60-months |
| W6 | 14/Male | Metastatic (frontal horn lesion) | 35Gy/21fx | No evidence of disease progression/failure | 15-months | Not applicable | Died of toxicity | 15-months |
| W7 | 9/Male | Non-metastatic | 36Gy/18fx | Tumor-bed recurrence | 56-months | Re-surgery | Died of disease | 60-months |
| W8 | 22/Male | Non-metastatic | 35Gy/21fx | Extra-neural metastases | 83-months | Chemotherapy | Alive with disease | Not applicable |
| W9 | 15/Male | Non-metastatic | Not known | Tumor-bed relapse plus metastases in frontal horn & multiple spinal metastases | 59-months | Re-surgery | Died of disease | 67-months |
WNT=wingless, CSI-craniospinal irradiation, RT=radiotherapy, PFS=progression-free survival, OS=overall survival, fx=fraction.
Table 3.
Univariate analysis of survival outcomes for WNT-pathway medulloblastoma in the study cohort (N=61).
Table 3.
Univariate analysis of survival outcomes for WNT-pathway medulloblastoma in the study cohort (N=61).
| Variables | Category | 5-year PFS (95%CI) | p-value | 5 years OS (95%CI) | p-value |
|---|---|---|---|---|---|
|
Gender |
Male | 86.0% (73.8-100%) | 0.480 | 92.6% (83.1-100%) | 0.440 |
| Female | 93.3% (81.5-100%) | 93.7% (82.6-100%) | |||
| Age at diagnosis | Child (≤16-years) | 83.5% (83.0-100%) | 0.722 | 89.5% (87.5-100%) | 0.323 |
| Adult (>16-years) | 90.0% (68.0-100%) | 90.9% (72.0-100%) | |||
|
Residual disease |
<1.5 cm2 | 84.5% (72.6-98.4%) | 0.250 | 90.7% (80.9-100%) | 0.260 |
| ≥1.5 cm2 | 100% (NE) | 100% (NE) | |||
|
Metastatic status |
Non-metastatic (M0) | 89.1% (79.1-100%) | 0.320 | 94.2% (86.4-100%) | 0.270 |
| Metastatic (M+) | 80.0% (51.6-100%) | 80.0% (51.6-100%) | |||
|
Risk-stratification |
Average-risk | 93.3% (81.5-100%) | 0.560 | 100% (NE) | 0.640 |
| High-risk | 81.2% (63.9-100%) | 87.9% (73.5-100%) | |||
|
Time interval (Surgery to RT) |
≤42 days | 87.3% (72.4-100%) | 0.500 | 94.1% (83.6-100%) | 0.440 |
| >42 days | 89.4% (76.7-100%) | 94.7% (85.2-100%) | |||
|
Dose of CSI |
Low-dose (14.4-26Gy) | 94.1% (83.0-100%) | 0.441 | 100% (NE) | 0.698 |
| High-dose (35-40Gy) | 81.0% (68.1-97.0%) | 84.6% (72.0-100%) | |||
|
Adjuvant chemotherapy |
No | 91.6% (77.2-100%) | 0.990 | 100% (NE) | 0.940 |
| Yes | 84.3% (70.6-100%) | 87.8% (75.2-100%) | |||
|
Cyclophosphamide dose |
<12gm/m2 | 92.8% (80.3-100%) | 0.970 | 100% (NE) | 0.970 |
| ≥12gm/m2 | 88.7% (77.4-100%) | 92.6% (83.2-100%) |
WNT=wingless, PFS=progression-free survival, OS=overall survival, CI=confidence interval, RT=radiotherapy, CSI=craniospinal irradiation, NE=not estimable.
Table 4.
Summary outcomes including patterns of relapse in WNT-MB patients treated adequately on molecularly-informed prospective cohort studies and randomized controlled trials.
Table 4.
Summary outcomes including patterns of relapse in WNT-MB patients treated adequately on molecularly-informed prospective cohort studies and randomized controlled trials.
| Trial Identity “[Ref]” & Registration |
Risk category | WNT-MB patients | WNT-MB failures | Patterns of relapse | 5-year EFS/PFS | 5-year OS | ||
|---|---|---|---|---|---|---|---|---|
| Local | Metastatic | Combined | ||||||
| HIT 2000 “[45]” NCT00303810 |
Non-metastatic (average-risk) |
15 | 0 | 0 | 0 | 0 | 100% | 100% |
| SIOP PNET-4 “[42]” NCT01351870 |
Average-risk | 58 | 8 | 2 | 4 | 2 | 91% | 95% |
| HIT 2000 “[46]” NCT00303810 |
Metastatic (high-risk) |
4 | 0 | 0 | 0 | 0 | 100% | 100% |
| COG ACNS 0331 “[12]” NCT00085735 |
Average-risk | 64 | 4 | 4 | 0 | 0 | 93.3% | 95.5% |
| SJMB-03 “[13]” NCT00085202 |
Average-risk & high-risk | 46 | 0 | 0 | 0 | 0 | 100% | 100% |
| COG ACNS 0332 “[47]” NCT00392327 |
High-risk | 14 | 1 | 0 | 0 | 1 | 92.9% | 100% |
| SIOP PNET 5 HR+ “[48]” NCT00936156 |
High-risk | 3 | 0 | 0 | 0 | 0 | 100% | 100% |
WNT=wingless; MB=medulloblastoma; EFS=event-free survival; PFS=progression-free survival; OS=overall survival.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



