
Submitted:
17 January 2024
Posted:
17 January 2024
You are already at the latest version
Abstract
Prostate cancer (PC) is a common malignancy of elderly men, characterized by great heterogeneity in its clinical course, ranging from an indolent to a highly aggressive disease. The aggressive variant prostate cancer (AVPC) clinically shows an atypical pattern of disease progression, similar to that of small cell PC (SCPC), and also shares the chemo-responsiveness of SCPC. The term AVPC does not describe a specific histologic subtype of PC but rather the group of tumors that, irrespective of morphology, show an aggressive clinical course, dictated by androgen receptor (AR) indifference. AR indifference represents an adaptive response to androgen deprivation therapy (ADT), driven by epithelial plasticity, an inherent ability of tumor cells to adapt to their environment by changing their phenotypic characteristics in a bi-directional way. The molecular profile of AVPC entails combined alterations in tumor suppressor genes retinoblastoma protein 1 (RB1), tumor protein 53 (TP53) and phosphatase and tensin homolog (PTEN). The understanding of the biologic heterogeneity of castration-resistant PC (CRPC) and the need to identify the subset of patients that would potentially benefit from specific therapies necessitate the development of prognostic and predictive biomarkers. This review aims to discuss the possible pathophysiologic mechanisms of AVPC development and the potential use of emerging tissue-based biomarkers in clinical practice.
Keywords:
AVPC
; biomarkers
; NEPC
; transdifferentiation
; ADT
; tumor suppressors
; DDR
; epigenetic regulation
1. Introduction
At the initial stages of oncogenesis, prostate cancer (PC) is an androgen receptor (AR)-driven disease [1], treated systematically with androgen ablation. Eventually, a castrate resistant state is reached where disease progresses despite castrate levels of androgen [2]. In most cases, resistance continues to involve AR-dependent mechanisms, temporarily counteracted by new androgen-targeting drugs, enzalutamide (a potent AR inhibitor) and abiraterone (an androgen biosynthesis inhibitor) [3]. However, a subset of castration-resistant PCs (CRPCs) evolves into AR-indifferent disease with inherent resistance to androgen deprivation therapy (ADT) [4]. The aggressive variant prostate cancer (AVPC) is the current umbrella term for the clinically virulent AR-independent tumors, encompassing neuroendocrine PC (NEPC) and double negative PC (DNPC) [5]. NEPC, in the vast majority of cases, emerges after treatment (t-NEPC) of a prostate adenocarcinoma, the prototype of PC (from here on referred as PCA), rather than de novo [6]. It is estimated that at least 20-30% of metastatic CRPCs (mCRPCs) progress to NEPC. NEPC represents the extreme end of AR independence with lineage switching, acquisition of NE markers and activation of alternative survival mechanisms [7]. DNPC, on the other hand, are tumors lacking both AR and neuroendocrine (NE) markers expression that possibly correspond to a transitional state from AR-dependent disease to NEPC [8]. It appears that biologic heterogeneity even within the AVPC group of tumors [9] is a real challenge in the attempt to accurately define and recognize these tumors. This is further exacerbated by the paucity of diagnostically valuable biopsy samples in the context of advanced, metastatic disease [10]. However, the need to identify AVPC patients is becoming increasingly relevant considering 1) the expected rise in AVPC incidence by the wider implementation of new, efficient antiandrogens in clinical practice [11] and 2) evidence of benefit from chemotherapy combinations for this subset of patients [12,13]. In this review, we intend to describe biomarkers emerging from current knowledge regarding AVPC molecular pathogenesis that could identify high-risk PC patients and guide therapeutic decisions.
2. AVPC Definition
AVPC was conceptualized after the clinical observation that a subset of CRPCs followed a non-conventional clinical course, reminiscent of that of small cell PC (SCPC), with predominantly visceral rather than bony metastases, lytic rather than osteoblastic bone disease and relatively low PSA levels. The hypothesis that these tumors in addition to clinical features also show similar biology and chemo-responsiveness to SCPC was tested with a phase II clinic trial of first-line carboplatin-docetaxel and salvage etoposide-cisplatin. Eligibility criteria for patients’ selection were the following: 1) SCPC histology, 2) exclusively metastatic visceral spread, 3) predominantly lytic bone metastases, 4) bulky (≥5 cm) lymph node mass or bulky (≥5 cm) mass in prostate/pelvis with Gleason score (GS) ≥8, 5) low prostate-specific antigen (PSA) (≤10 ng/mL) at first presentation (before ADT) or at symptomatic progression during ADT despite high volume (≥20) bone metastases, 6) positive immunohistochemistry (IHC) for NE markers (chromogranin A or synaptophysin) or abnormally elevated serum NE markers [chromogranin A or gastrin-releasing peptide) at initial presentation or progression together with non-otherwise explained serum LDH and/or CEA ≥ 2 X upper normal value and/or malignant hypercalcemia, 7) short interval period (≤6 months) between ADT initiation and AR-independent progression. Except from patients with a histologic diagnosis of SCPC, all others were required to have undergone ADT and have progressed during treatment or have an unsatisfactory response. This set of aggressive features was used to define clinicopathologic AVPC (AVPC-c) [12]. The term “anaplastic PC” that was previously used to describe patients with similar virulent features was abandoned on the grounds that the term “anaplasia” from a pathologist’s viewpoint implies cellular pleiomorphism [14]. A high percentage of patients fulfilling at least one of the AVPC-c criteria responded, although briefly, to platinum-based combination therapy. Overall survival (OS) was significantly shortened in relation to the number of fulfilled criteria [12]. Subsequent clinical trials reinforced the notion of platinum efficiency in AVPC. The addition of carboplatin to cabazitaxel was selectively beneficial for AVPC compared to non-AVPC patients [13]. In another study, platinum-based therapy could counterbalance the poorer prognosis of AVPC by achieving similar progression free survival (PFS) and OS scores with conventional CRPC [15].
The molecular signature of AVPC was investigated in AVPC tissue samples and patient derived xenografts (PDX). Molecular AVPC (AVPC-m) is defined by combined loss of tumor suppressors retinoblastoma protein 1 (RB1), tumor protein 53 (TP53) and/or phosphatase and tensin homolog (PTEN) (≥2/3), as this molecular profile was more frequent in AVPC-c (48,3%) compared to unselected CRPC patients (26%). Morphology did not predict the underlying molecular changes [16].
AVPC spans a spectrum of histological appearances from poorly-differentiated adenocarcinoma, to mixed NEPC-PCA to pure NEPC, either SCPC or large cell neuroendocrine prostate carcinoma (LCPC) [14] (Figure 1). Unusual features such as aberrant squamous differentiation have also been described [17]. AVPC with PCA histology usually shows a typical GS 5 architectural configuration with nested or solid growth without lumina formation and with prominent nucleoli [18]. SCPC is histologically distinctive with high nuclear to cytoplasmic ratio, nuclear molding, indistinct cell borders, lack of nucleoli, prominent apoptotic and mitotic activity and necrosis. Positivity for least one NE marker [synaptophysin, chromogranin A, CD56 or the newer NE marker insulinoma-associated protein 1 (INSM1)] is confirmatory (Figure 1). LCPC is rare, characterized by large nests of cells with peripheral palisading, prominent nucleoli and NE markers expression [14]. Histologic diagnosis of NEPC in the context of prior therapy is by definition conclusive of AVPC. The challenge lies in predicting which cases of morphologically conventional PC would have an aggressive behavior or benefit from specific treatments.
3. NE and AR-Signaling Markers
Taking into consideration that a subset of AVPC show positivity for NE markers and that elevated serum chromogranin A has been previously reported to have an unfavorable prognostic role in CRPC patients [19,20] it seems reasonable to consider NE markers as possible biomarkers for AVPC detection. NE cells are normally scattered in small numbers in the basal cell layer of prostate acini [14]. Focal expression of NE markers is seen in 10-100% of usual PCAs [14] and although it has been associated with poorly differentiated tumors, it does not represent an independent prognostic factor [21,22]. NE cells in PCA do not proliferate but they secrete peptides that may boost the survival of adjacent cancer cells [23]. The expansion of NE cells that occurs after prolonged exposure to ADT has been hypothesized to confer resistance to therapy due to the delivery of alternative, AR-independent survival signals [24]. Serum or tissue chromogranin A or synaptophysin expression did not show, however, a strong correlation with progression free survival (PFS) or OS in AVPC-c patients. Expression of NE markers may represent an epiphenomenon rather than a driver of aggressive disease [12]. It has also been repeatedly shown that serum NE markers alone cannot predict response to chemotherapy [25,26,27,28].
Downregulation of AR and AR-regulated genes, such as PSA, transmembrane serin protease 2 (TMRSS2) and NK3 homeobox 1 (NKX3.1), along with loss of AR expression, in SCPC is consistent with AR-independent growth [29,30,31] (Figure 1). Reduced AR staining (<10%) is also observed in 36% of AVPC [16]. In the contrary, frequent intense AR expression has also been reported in therapy-related SCPC (t-SCPC). Rates of intense AR staining did not differ significantly between t-SCPC and castration resistant PCA (75% and 87% respectively), despite lower AR transcriptional activity in t-SCPC. Epigenetic regulation may explain this discordance [32]. “Atypical” SCPCs with retained AR signaling activity have been also described by other groups [33]. In AR-positive AVPC cases, downstream molecules of AR signaling were co-expressed [16]. In addition, AR staining does not necessarily predict response to ADT [34]. Levels of AR expression and AR transcriptional scores between t-NEPC and castration resistant PCA patients significantly overlap [35]. Apparently, there is a spectrum of AR expression and activity in CRPC that cannot reliably distinguish AVPC patients. AR-independent resistance mechanisms in AVPC are implied by the paucity of AR splice variants [16,35] and the absence of AR activating mutations and amplification in t-NEPC compared to castrate resistance PCA [35].
Transcriptome analysis of mCRPC samples revealed an inverse relationship between AR and NE signature. Low AR signaling and high NE transcript levels were mostly assigned to tumors with a NE histology. Notably, some cases of low AR and high NE scores as well as some of intermediate AR and NE scores corresponded to adenocarcinomas with distinctive atypical nuclei, possibly in transition to NEPC [36].
DNPC are tumors with an AR-/NE- phenotype with an increase in their prevalence from 5% to 20% of PC after the introduction of the new antiandrogens into clinical practice. Experimental models reveal that these tumors develop from clones that have resisted strong AR inhibition and rely on alternative survival pathways, namely fibroblast growth factor receptor (FGFR) – mitogen-activated protein kinase (MAPK) signaling [8]. Labrecque et al. classified mCRPC into 5 phenotypes based on IHC and transcriptional expression of AR and NE markers: 1) AR+(high)/NE- (ARPC), 2) AR+(low)/NE- (ARLPC), 3) AR+/NE+ (amphicrine), 4) AR-/NE- (DNPC), 5) AR-/NE+ (SCPC). Each phenotype is enriched for different biological properties with DNPC and SCPC sharing a high metastatic potential. These phenotypes are dynamically related. Most importantly, DNPC has an intrinsic ability to transform to SCPC and may embody a transitional state to NEPC [37] (Figure 2). Squamous differentiation, previously reported in AVPC [17], was seen in a subset of DNPC. The above evidence discloses the complementary role of AR signaling and NE profile analysis in understanding the heterogenous biology of CRPC.
4. Tumor Suppressors RB1, TP53 and PTEN
Loss of at least two of the tumor suppressors RB1, TP53 and PTEN characterizes half of the AVPC-c cases [16] and is associated with clinically significant benefit from chemotherapy. Indeed, combined defects in at least two of the three genes, termed as unfavorable genomics [13] is included in the NCCN criteria for adding cabazitaxel to carboplatin [38].
Immunohistochemical detection of defects in two of these three molecules predicted improved responses to chemotherapy [13] and accurately categorized cases as AVPC-m. A cut-off value of 10% was used to define gene loss by IHC (≤10% for RB and PTEN and ≥10% for p53, the latter due to nuclear accumulation of the mutated protein) and intensity of staining was considered to achieve optimal correlations with loss-of-function transcriptional scores. Intense (2+3+) staining was used to define abnormal RB1 and p53 expression, while any staining (1+-3+) defined abnormal PTEN expression [39]. In contrast to TP53 missense mutations that lead to p53 nuclear accumulation and, thus, intense p53 staining by IHC (Figure 1), nonsense, frameshift and indel alterations were not easily detected by IHC due to the low basal p53 expression in normal prostate and p53-wild type PC [40]. Next-generation sequencing (NGS) remained a valuable tool for identifying TP53 alterations [39], albeit IHC proved to be just as, if not more, accurate as NGS.
A large number of clinical and preclinical data suggest that the use of RB1, p53 and PTEN as biomarkers of AVPC in clinical practice is not only accurate and feasible but also biologically relevant. RB1 deletions and microdeletions represent a well-established genetic event in SCPC [30,35,41] resulting in almost universal absence of protein expression by IHC [30,41]. RB1 allelic loss is extremely rare in primary PC but relatively common in mCRPC [41,42]. The retained RB1 expression in these mCRPC cases along with its loss in NEPC, suggests a driver role of RB1 protein loss in NEPC, further highlighted by the frequently observed concurrent loss of RB1 expression in the adenocarcinoma component in mixed PCA-NEPC [30,41]. Evidence supports a common origin of the two components in mixed tumors [43,44] and NEPC emergence through transdifferentiation of PCA under the pressure of ADT [35,45]. Loss of RB1 protein seems to predict transition to NEPC as it precedes the morphologic shift of PCA to NEPC [30]. Furthermore, presence of RB1 alterations has been identified as a poor prognostic factor in mCRPC [36]. Functionally, RB1 loss relieves inhibition of E2F target genes and not only enables cell cycle progression but also deregulates AR expression [42,46].
RB1 alterations tend to be mutually exclusive with AR alterations [36] but frequently coincide with TP53 mutations [16,35,36]. Concurrent RB1 and TP53 alterations are seen in 74% of mCRPC with a NE phenotype in contrast to 5% of primary PC and 39% of mCRPC-Adeno [47]. Combined RB1 and TP53 inactivation is prevalent in poorly differentiated NE tumors [48] and has been reported to induce small cell lung cancer [49]. In experimental models of PC, while knockout of either RB1 or TP53 failed to give rise to invasive carcinomas [50,51,52,53], simultaneous inactivation of both molecules resulted in poorly differentiated PCs with co-expression of luminal and NE markers, de novo ADT resistance and visceral metastatic spread [53,54], features reminiscent of AVPC. In transgenic mouse models of prostate adenocarcinoma (TRAMP), SV40 large T antigen, which binds and inactivates RB1 and p53, induced NE-like, metastatic tumors [48,55]. While other oncogenic events can initiate poorly differentiated PC from prostate basal cells, additional defects in RB1 and/or TP53 are required for SCPC emergence [56]. RB1 and p53 act as guardians of lineage commitment [57,58,59,60,61], so their inactivation enables lineage plasticity and reprogramming of PC cells. In the LNCaP/AR and CWR22Pc-ER models, concurrent RB1 and p53 loss conferred AR-independent resistance to enzalutamide with expression of basal and neuroendocrine markers and suppression of luminal markers. This transition was mediated by the transcription factor (TF) SRY-Box transcription factor 2 (SOX2) [47], previously found to induce pluripotency [62], and was rapidly reversible after return of RB1 and p53 to baseline levels. This suggests a bidirectional, universal passage of tumor cells from a dedifferentiated, flexible state, marked by loss of luminal markers [47] and is consistent with the theory of DNPC being a transitional state (Figure 2). RB1 and p53 loss-of-function alterations may determine transcriptionally and epigenetically the conversion to NEPC. This dual inactivation has a synergic effect in increasing chromatin accessibility in genomic regions crucial for neuronal differentiation and, accordingly, decreasing accessibility to genes related with PCA and epithelial differentiation [56]. An additional mechanism of synergy may be the unhindered cell proliferation by the “double hit” in cell cycle checkpoints. RB1 regulates transition from G1 to S phase, so inactivation of RB1 leads to uncontrollable cell cycle progression that would be otherwise preventable by a functional p53 [53].
TP53 alterations are frequent in AVPC and SCLC [16,41,63] and mostly include deleterious mutations that, also, extend the protein’s half-life resulting in nuclear accumulation, detected by IHC [39,41]. In most studies, TP53 alterations are encountered in <10% of primary PC [45,64] but are significantly enriched in advanced disease [45,64,65,66,67,68]. TP53 defects are an independent poor prognostic factor among CRPC patients [40,69,70] and are associated with rapid acquisition of resistance to abiraterone or enzalutamide [54,69,70]. p53 abnormal staining, when found in the initial prostate biopsy or prostatectomy specimen, could be used as an early predictor of aggressive progression and resistance to enzalutamide or abiraterone [40,68,69]. TP53 knockout however does not suffice to induce resistance to enzalutamide [47]. Certainly, TP53 could not be independently used as a specific biomarker for AVPC [41].
In contrast to TP53 and RB1 alterations, PTEN deletion is a recurrent genomic alteration of primary PC, documented decades ago [71,72]. PTEN loss is capable of PC initiation [73,74,75] and is frequently hemizygous [76,77], consistent with the identification of PTEN as a happloinsufficient gene [78]. Interestingly, bi-allelic PTEN deletion induces a p53-mediated program of cellular senescence that restrains tumorigenesis [52,79,80]. Homozygous PTEN deletion is more common in CRPC [66,81] where it is often coupled with TP53 loss [45], resulting in dysregulated senescent response and lethal tumor progression [52]. The detrimental effect of combined PTEN and TP53 loss was validated with the PBCre4+:Ptenfl/fl:TP53fl/fl model. The aggressive biology of tumors in this model was attributed not only to decreased senescence but also to the induction and increased plasticity of prostate stem cells [82]. Loss of PTEN and TP53 leads to histologically diverse tumors [45,82] that are phenotypically and molecularly related to human CRPC-NE and have intrinsic resistance to abiraterone [45].
In PTEN-deficient tumors, RB1 deletion facilitates visceral metastatic spread and lineage plasticity, as evidenced by the heterogenous cellular composition of the emerging tumors with variable expression of cytokeratin, AR and synaptophysin. The addition of TP53 loss resulted in rapidly metastasizing, lethal tumors with de novo ADT-resistance. Both PBCre4+:Ptenfl/fl:Rb1fl/fl and PBCre4+:Ptenfl/fl:Rb1fl/fl:Trp53 fl/fl models recapitulate the molecular signature of human NEPC [54].
It becomes therefore apparent that combined defects in tumor suppressors potentiate lineage plasticity and the acquisition of ADT resistance and other clinical and molecular features of AVPC. The combination of tumor suppressor gene alterations as biomarkers achieves higher specificity than either of these biomarkers alone (Table 1). The value of tumor suppressors as prognostic biomarkers in PC was highlighted by a recent study declaring the superiority of tumor suppressors alterations detection compared to traditional prognostic parameters in early prediction of aggressive disease course [83].
5. Oncogenes MYCN and AURKA
N-myc proto-oncogene protein (N-Myc), encoded by MYCN Proto-Oncogene, BHLH Transcription Factor (MYCN), is a transcription factor essential for brain development that is not expressed in normal prostate epithelium. MYCN is amplified in neural, hematologic and other tumors, including 40% of NEPC and a small percentage of CRPP-Adeno [29,86,87,88]. MYCN amplification has been linked with poor prognosis in both NEPC and CRPC-Adeno [87] and the timing of its occurrence coincides with metastatic spread [35]. N-Myc-driven AVPCs have been reported as poor responders to docetaxel [9], the standard of care for mCRPC [89]. N-Myc downregulates AR expression and directly suppresses AR-targeted genes, providing a mechanism for abrogating AR dependence [29,87,90]. A role of N-Myc in lineage determination, through epigenetic reprogramming, has also been suggested. Following castration, N-Myc signature shifts and is enriched with neural lineage and stem cell programs. N-Myc also biases bivalent H3K27me3 and H3K4me3 histone marks toward a neural lineage gene activation [90] and directly upregulates NE markers [29]. N-Myc signature is, thus, consistent with the NEPC molecular program [87] and could be used to predict CRPC patients at risk of developing NEPC [90].
Supporting its role in lineage plasticity, N-Myc overexpression, combined with activated AKT serine/threonine kinase 1 (AKT) or PTEN loss, induces invasive tumors with adenocarcinoma, NEPC, mixed or aberrant phenotypes [87,91] with prevalence of NEPC after prolonged castration [91]. These findings suggest that primary PC with MYCN amplification may have inherent plasticity and the potential to develop into NEPC after ADT [91]. N-Myc overexpression in conjunction with RB1 and PTEN loss leads to aggressive, NE-like tumors. The lineage switch is accompanied by alterations in chromatin accessibility and redirection of N-Myc binding to NE-associated genes [92], further supporting N-Myc’s role in conferring lineage plasticity properties to the tumor cells.
In PC, MYCN amplification is nearly always concurrent with amplification of Aurora kinase A (AURKA) [29], a serine/threonine kinase that regulates mitotic division and has oncogenic functions in various malignancies [93]. AURKA amplification has been detected in prostate intraepithelial neoplasia (PIN) lesions and may represent an early oncogenic event in PC [94]. AURKA seems to also mediate resistance in CRPC and its expression is analogous to disease progression [95]. Other mechanisms of AURKA overexpression, besides amplification, may be involved in PC oncogenesis [29,30]. N-MYC and AURKA stabilize each other by forming a complex [29,87,96] and cooperate in the induction of NE features in PC [29]. Combined AURKA and MYCN amplification has been also identified in NE carcinomas of other sites [97]. As expected, both MYCN and AURKA amplification in primary PC predict the transformation to t-NEPC [29,97], independently from other factors such as tumor stage, PSA levels or GS. MYCN and AURKA amplifications arise early and may even be present in GS 6 tumors that could be otherwise dealt with active surveillance [97].
The use of MYCN and AURKA as biomarkers, apart from risk stratification purposes, could have therapeutic implications [97]. Although clinical trials of AURKA inhibitors either in unselected CRPC or in AVPC patients did not show significant clinical efficacy [85,98], rare responders exhibited AURKA and MYCN overexpression [85]. N-Myc defies direct therapeutic targeting because firstly, it is an intrinsically disordered protein [99] and secondly, its structure lacks “druggable” pockets [100]. However, some AURKA inhibitors that alter AURKA conformation, destabilize N-Myc too [100], [101]. The sensitivity of MYCN amplification to poly (ADP-ribose) polymerase 1 (PARP1) inhibition in neuroblastoma [102] and the identification of MYCN-PARP1/2-DNA damage repair (DDR) pathway as a driver of transition to NEPC provide the rational for combining AURKA inhibitors with PARP1 inhibitors. More specifically, N-Myc has been identified as a direct transcriptional activator of PARP1 and PARP2, that in turn regulate the expression of DDR-related genes [103]. Compounds that indirectly target MYCN, by disrupting its heretodimerization with myc-associated factor X (MAX) for example, have been developed [104], [105]. Recently, a dual inhibitor of N-Myc and AURKA effectively constrained cellular growth in cell lines of PC and NEPC [106].
Diagnostically, MYCN and AURKA amplifications, although highly specific, are detected in only 20% and 25% of AVPC patients respectively (Table 1). Another limitation is the lack of established criteria for defining AURKA overexpression by IHC [16]. The use of IHC as a surrogate of MYCN amplification remains also widely unexplored, besides some evidence showing nearly complete agreement between N-Myc IHC and florescence in situ hybridization (FISH) [107].
6. DNA Damage Repair (DDR) Pathway
A significant percentage (20-30%) of mCRPC harbor germline or somatic DDR alterations, associated with adverse prognosis [108,109,110], among which Breast cancer 2 (BRCA2) defects are the most prevalent [66,111]. BRCA1/2 defects are responsible for defective homologous recombination (HR) of DNA double-strands [112]. A synthetic lethality approach of PARP inhibition in HR-defective PCs has been proven effective and PARP inhibitors Olaparib and Rucaparib have been approved by FDA in this context [113,114,115,116]. Furthermore, the presence of HR and especially BRCA2 defects is associated with responses to platinum-based chemotherapy [15,117,118,119,120].
Although alterations in mismatch repair (MMR) proteins are uncommon in PC [66,121], they have been correlated with disease progression [121] and an hypermutated phenotype of advanced PC [122,123]. The detection of MMR germline mutations in a minority of these cases suggests that a subset of MMR-deficient PCs is associated with Lynch syndrome [121,124]. MMR defects and microsatellite instability (MSI) predict response to Programmed cell death 1 (PD-1) blockage [121] in contrast to modest response rates in unselected mCRPC patients [125].
The efficacy of PARP inhibition and anti-PD-1 therapy for AVPC patients is currently investigated (NCT04592237). Screening for DDR defects in AVPC could be predictive of sensitivity to novel therapies. DDR alterations were reported to be mutually exclusive with the histologic diagnosis of t-SCPC [32]. Conflicting results show MutS homolog 2 (MSH2) loss in 5% of NEPC [126] and approximately 30% incidence of BRCA2 alterations in AVPC-c [85] (Table 1), in line with the association of BRCA2 mutations with low PSA levels in metastatic PC [127]. DDR defects are biologically related to the AVPC phenotype. The frequently observed BRCA2 and RB1 co-deletion [65,128] drives aggressive behavior and acquisition of ADT resistance and an epithelial-to-mesenchymal transition (EMT)-like state in PC [128]. Regardless of the presence of DDR alterations, AVPC seems to have inherent genomic instability [16]. p53 and PTEN defects accelerate the cell cycle and do not allow enough time for DDR [15,129]. AURKA can also impair DDR [130]. It is yet unclear if predictive biomarkers could further specify which patients of the AVPC group would benefit from therapies associated with DDR deficiency.
7. Gene Expression Profiles, Epigenetic Regulators and Proneural Transciption Factors
NEPC displays a downregulation of AR-mediated and epithelial differentiation genes and overexpress NE lineage, EMT, cell cycle and E2F target genes [29,30,32,33,35,54,131,132,133,134] (Figure 2). Paternally expressed gene 10 (PEG10), a gene of placental development with inherent oncogenic properties, is reactivated in NEPC and represents an attractive biomarker for early NEPC detection and a potential therapeutic target due to the absence of expression in normal adult tissues [135]. The upregulation of mitotic and proneural genes has been validated in AVPC [16]. Gene expression classifiers predictive of NEPC have been developed by different research groups [32,33,35,132,133,134]. The advantage of these classifiers as biomarkers stems from their implementation on the limited sampled metastatic tissue that is preclusive of extensive IHC studies. The 70-gene NEPC classifier developed by Beltran et al. accurately recognizes NEPC. However, in 20% of cases, elevated scores corresponded to adenocarcinomas, hypothesized to represent tumors in transition to NEPC, or predisposed for NEPC transformation [36], or simply highlighting the fact that morphology alone is not fully predictive of the behavior of the neoplasm.
The differential gene expression of NEPC is epigenetically regulated [35]. Several studies have highlighted diverse epigenetic programs between AVPC and non-AVPC CRPCs. Similar to gene expression, an absolute correlation between AVPC-related epigenome and NEPC morphology has not been seen. For example, the DNA methylation pattern of NEPC can be shared by cases with a morphological diagnosis of PCA but with AVPC clinical features [35], suggesting that epigenetic features may help distinguish AVPC irrespectively of morphology.
It has been postulated that epigenetic alterations are responsible for the lineage plasticity of the AVPC phenotype. RB1 and TP53 loss, frequent molecular events in AVPC, enable epigenetic reprogramming by SOX2 and Enhancer of zeste homolog 2 (EZH2) and the adoption of a stem cell-like program, permissive of lineage switching [54]. EZH2 is a histone methyltransferase, member of the Polycomb Repressor Complex 2 (PRC2), which suppresses the transcription of developmental regulators and maintains stemness [136,137] dictating poor outcomes in PC [138]. EZH2 has been associated with AR independence and has been proposed as a prognostic biomarker in PC [139]. More recently, EZH2 upregulation has been particularly associated with t-NEPC [29,35,140,141] and has been found essential for the transformation of PCAs into NEPCs [140]. EZH2 synergizes with N-Myc in the induction of NEPC transcriptional program [87] and is a central downstream mediator of multiple pathogenetic events in NEPC [140]. The oncogenic role of EZH2 is indicative of the benefit of EZH2 inhibitors in combinational therapies [140,142,143,144]. EZH2 expression could be predictive of response to such therapies [142]. In pre-clinical studies, EZH2 inhibitors demonstrate anti-tumor effect on PC and preferentially NEPC cell lines [35,145] and re-sensitize NEPC cells to ADT [54]. Clinical trials are currently conducted in order to investigate the efficacy of EZH2 inhibitors alone or in combination with abiraterone, enzalutamide or the PARP inhibitor Talazoparib in CRPC patients (NCT04179864, NCT03480646, NCT04846478, NCT03460977). In addition to EZH2, Clermont et al. identified Chromobox 2 (CBX2), a member of the PRC1, as a frequently overexpressed epigenetic regulator in NEPC and developed a “neuroendocrine-associated repression signature” (NEARS). This signature showed an enrichment for polycomb group (PcG)-silenced genes and was associated with patients’ prognosis [146]. The significance of PcG proteins in AVPC has also been highlighted by the role of PRC1 in forming an immunosuppressive and angiogenetic microenvironment favoring metastases in DNPC [147].
Other epigenetic regulators besides PcG proteins have been implicated in AVPC pathogenesis and could be potential biomarkers and therapeutic targets. DNA methyltransferases (DNMTs) are upregulated in NEPC [35,146,148] and synergize with PcG proteins [146,149] in the induction of a stem cell-like chromatin state [148], that renders tumor suppressor genes vulnerable to silencing [150]. Similar to EZH2 inhibitors, DNA hypomethylating agents restore AR dependence in PC cells [151]. DEK, a DNA topology modulator, is overexpressed in NEPC [32,35,152] and regulates neural genes and genes associated with proliferative and migratory potential. DEK expression in a small percentage of hormone-naïve PCs is an independent poor prognostic factor and could therefore be informative of a propensity towards NEPC transformation [152]. Heterochromatin protein 1a (HP1a) expression is an early and persistent event in NEPC development that drives the NE phenotype after castration, via repression of AR and (RE)-1 silencing TF (REST). HP1a is part of a heterochromatin gene signature that distinguishes NEPC from PCA [153].
Noncoding RNAs (ncRNAs) are regulatory molecules of gene expression with an emerging role in determining tumor phenotypes including NEPC transformation [154]. Several microRNAs (miRNAs), short ncRNAs with a post-transcriptional regulatory function, have been identified as drivers of NE differentiation and candidate diagnostic biomarkers of NEPC [10,155,156,157]. The observation of the altered miRNA expression profile in NEPC [158,159] led to the development of a miRNA-based classifier that distinguishes CRPC-NEPC from CRPC-Adeno. The practical advantage of this classifier compared to gene-expression and mRNA-based classifiers is the stability of miRNAs in formalin-fixed tissues [159]. AR-negative CRPC/NEPC transcriptome is also characterized by a distinct compilation of long ncRNAs (lncRNAs) with diagnostic and prognostic value [160,161]. These lncRNAs embody an additional epigenetic mechanism facilitating lineage plasticity and NEPC induction, partly via interacting with PRC2 [149,160,161].
Transcription factors’ aberrations have also been shown in AVPCs. Loss of REST repression on neural lineage genes has been reported in NEPC and mixed PCA-NEPC [131]. The defect in REST function is, at least in part, mediated by alternative splicing of the respective mRNA by serine/arginine repetitive matrix 4 (SRRM4) [37,162,163]. The alternative splicing fingerprint of NEPC includes a number of SRRM4-regulated genes. SRRM4 emerges as a master regulator that in AR-depleted conditions orchestrates the transcriptional and epigenetic modifications needed for transformation into NEPC [162]. REST downregulation induces NE markers expression [162,163,164] but results in amphicrine tumors with AR co-expression and probably sustained sensitivity to ADT. Additional expression of proneural transcription factors (TFs) is required for definite conversion to NEPC [37,163]. In AVPC PDX models, REST presence did not prevent the expression of proneural TFs, however the possibility of inactive REST splice variants could not be excluded [16].
Proneural TFs reported to mediate transition to NEPC include SOX2, Achaete-scute homolog 1 (ASCL1), POU domain, class 3, transcription factor 2 (POU3F2/BRN2), POU3F4/BRN4, Forkhead box protein A1 (FOXA1), INSM1 and Neurogenic differentiation 1 (NEUROD1) [32,47,92,165,166,167,168,169,170,171]. ASCL1 upregulation is an early occurrence following enzalutamide treatment [166] (Figure 2) and can predict aggressive disease [172]. ASCL1 remodels the chromatin architecture and directs PC identity towards a neuronal and stem cell fate, acting in concert with EZH2 to promote lineage plasticity [166]. BRN2, encoded by the AR-repressed gene POU3F2, is required for terminal and SOX2-mediated NE differentiation. BRN2 overexpression is observed not only in NEPC but also in PCA with low serum PSA and probably signifies and is predictive of the transition to an AR-independent state after ADT [167]. BRN4 has been recently identified as an inducible TF upon ADT that cooperates with BRN2 in the initiation of the NEPC program. In fact, BRN2 and BRN4, excreted in the form of extracellular vesicles, could serve as predictive serum biomarkers of NEPC [168]. FOXA1 is a TF that mediates prostate development but remains essential in NEPC as it relocates to NE regulatory elements [169]. INSM1 emerges as a highly specific marker of NEPC [170,173,174] and its expression coincides with Yes-associated protein (YAP) silencing; thus, these two markers could be complementary in the prediction of NEPC [170]. Interestingly, NEUROD1 and ASCL1 expression characterize distinct coexisting subpopulations in NEPC [171]. Wang et al. described a time-dependent expression of TFs and NE genes during transition to NEPC. ASCL1, as a pioneer factor, governs the initial oncogenic phase but is lost during the late phase of NE differentiation [175]. Expression of TFs SOX2 and POU3F2 and terminal NE markers (chromogranin A and B, enolase 2) are late events in the transdifferentiation process [135,175] (Figure 2). The spatial and temporal heterogeneity of proneural TF expression probably explains why none of those TFs was consistently expressed in all AVPC samples [16].
8. Conclusions
A downside of potent AR inhibition in PC is that it drives, via selective pressure mechanisms, the emergence of AR-indifferent tumors. The need for biomarkers capable of predicting this transdifferentiation process is imperative. The concurrence of tumor suppressors alterations is a well-characterized but not uniformly present feature of AVPC. Given the similar biological background of AVPC and NEPC, the current understanding of NEPC pathogenesis could prove useful in broadening the scope of available biomarkers. Epigenetic modulations in particular could not only be predictive of an AR-independent path but also be therapeutically exploited to restore AR dependence. Hypothesizing that at least a part of AVPCs is in a transitional state towards overt NEPC, the sequence of biomarkers expression during NEPC progression should be clarified in order to select early predictors. The validation of these biomarkers in clinical studies would establish them as valuable tools in risk stratification and clinical decision making in the management of CRPC.
Author Contributions
Conceptualization, V.T.; writing—original draft preparation, O.K.; writing—review and editing, V.T., V.B. and K.G.; visualization, O.K. and V.T.; supervision, V.T.; funding acquisition, V.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded in part by the Ronald and Rita McAulay Foundation (#82690) (V.T.).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Weischenfeldt, J.; Simon, R. Integrative genomic analyses reveal an androgen-driven somatic alteration landscape in early-onset prostate cancer. Cancer cell 2013, 23, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Halabi, S. Design and end points of clinical trials for patients with progressive prostate cancer and castrate levels of testosterone: Recommendations of the Prostate Cancer Clinical Trials Working Group. J. Clin. Oncol. 2008, 26, 1148–1159. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekar, T.; Yang, J.C. Mechanisms of resistance in castration-resistant prostate cancer (CRPC). Transl. Androl. Urol. 2015, 4, 365–380. [Google Scholar] [CrossRef]
- Berchuck, J.E.; Viscuse, P.V. Clinical considerations for the management of androgen indifferent prostate cancer. Prostate Cancer Prostatic Dis. 2021, 24, 623–637. [Google Scholar] [CrossRef] [PubMed]
- Laudato, S.; Aparicio, A. Clonal Evolution and Epithelial Plasticity in the Emergence of AR-Independent Prostate Carcinoma. Trends Cancer 2019, 5, 440–455. [Google Scholar] [CrossRef] [PubMed]
- Terry, S.; & Beltran, H. The many faces of neuroendocrine differentiation in prostate cancer progression. Front. Oncol. 2014, 4, 60. [CrossRef]
- Aggarwal, R.; Zhang, T. Neuroendocrine prostate cancer: Subtypes, biology, and clinical outcomes. J. Natl. Compr. Canc. Netw. 2014, 12, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Coleman, I.M. Androgen Receptor Pathway-Independent Prostate Cancer Is Sustained through FGF Signaling. Cancer cell 2017, 32, 474–489.e6. [Google Scholar] [CrossRef]
- Han, H.; Lee, H.H. Prostate epithelial genes define therapy-relevant prostate cancer molecular subtype. Prostate Cancer Prostatic Dis. 2021, 24, 1080–1092. [Google Scholar] [CrossRef]
- Li, Z.; Sun, Y. p53 Mutation Directs AURKA Overexpression via miR-25 and FBXW7 in Prostatic Small Cell Neuroendocrine Carcinoma. Mol. Cancer. Res. 2015, 13, 584–591. [Google Scholar] [CrossRef]
- Akamatsu, S.; Inoue, T. Clinical and molecular features of treatment-related neuroendocrine prostate cancer. Int. J. Urol. 2018, 25, 345–351. [Google Scholar] [CrossRef]
- Aparicio, A.M.; Harzstark, A.L. Platinum-based chemotherapy for variant castrate-resistant prostate cancer. Clin. Cancer Res. 2013, 19, 3621–3630. [Google Scholar] [CrossRef]
- Corn, P.G.; Heath, E.I. Cabazitaxel plus carboplatin for the treatment of men with metastatic castration-resistant prostate cancers: A randomised, open-label, phase 1-2 trial. Lancet. Oncol. 2019, 20, 1432–1443. [Google Scholar] [CrossRef]
- Epstein, J.I.; Amin, M.B. Proposed morphologic classification of prostate cancer with neuroendocrine differentiation. Am. J. Surg. Pathol. 2014, 38, 756–767. [Google Scholar] [CrossRef] [PubMed]
- Slootbeek, P.H.J.; Duizer, M.L. Impact of DNA damage repair defects and aggressive variant features on response to carboplatin-based chemotherapy in metastatic castration-resistant prostate cancer. Int. J. Cancer 2021, 148, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, A.M.; Shen, L. Combined Tumor Suppressor Defects Characterize Clinically Defined Aggressive Variant Prostate Cancers. Clin. Cancer Res. 2016, 22, 1520–1530. [Google Scholar] [CrossRef] [PubMed]
- Manucha, V.; Henegan, J. Clinicopathologic Diagnostic Approach to Aggressive Variant Prostate Cancer. Arch. Pathol. Lab. Med. 2020, 144, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Montironi, R.; Cimadamore, A. Morphologic, Molecular and Clinical Features of Aggressive Variant Prostate Cancer. Cells 2020, 9, 1073. [Google Scholar] [CrossRef] [PubMed]
- Taplin, M.E.; George, D.J. Prognostic significance of plasma chromogranin a levels in patients with hormone-refractory prostate cancer treated in Cancer and Leukemia Group B 9480 study. Urology 2015, 66, 386–391. [Google Scholar] [CrossRef] [PubMed]
- Berruti, A.; Mosca, A. Independent prognostic role of circulating chromogranin A in prostate cancer patients with hormone-refractory disease. Endocr. Relat. Cancer 2005, 12, 109–117. [Google Scholar] [CrossRef] [PubMed]
- Speights, V.O., Jr.; Cohen, M.K. Neuroendocrine stains and proliferative indices of prostatic adenocarcinomas in transurethral resection samples. Br. J. Urol. 1997, 80, 281–286. [Google Scholar] [CrossRef]
- Pruneri, G.; Galli, S. Chromogranin A and B and secretogranin II in prostatic adenocarcinomas: Neuroendocrine expression in patients untreated and treated with androgen deprivation therapy. Prostate 1998, 34, 113–120. [Google Scholar] [CrossRef]
- Bonkhoff, H.; Wernert, N. Relation of endocrine-paracrine cells to cell proliferation in normal, hyperplastic, and neoplastic human prostate. Prostate 199, 19, 91–98. [CrossRef]
- Hirano, D.; Okada, Y. Neuroendocrine differentiation in hormone refractory prostate cancer following androgen deprivation therapy. Eur. Urol. 2014, 45, 586–592. [Google Scholar] [CrossRef]
- Steineck, G.; Reuter, V. Cytotoxic treatment of aggressive prostate tumors with or without neuroendocrine elements. Acta Oncol. 2002, 41, 668–674. [Google Scholar] [CrossRef]
- Culine, S.; El Demery, M. Docetaxel and cisplatin in patients with metastatic androgen independent prostate cancer and circulating neuroendocrine markers. J. Urol. 2007, 178, 844–848. [Google Scholar] [CrossRef]
- Loriot, Y.; Massard, C. Combining carboplatin and etoposide in docetaxel-pretreated patients with castration-resistant prostate cancer: A prospective study evaluating also neuroendocrine features. Ann. Oncol. 2009, 20, 703–708. [Google Scholar] [CrossRef]
- Fléchon, A.; Pouessel, D. Phase II study of carboplatin and etoposide in patients with anaplastic progressive metastatic castration-resistant prostate cancer (mCRPC) with or without neuroendocrine differentiation: Results of the French Genito-Urinary Tumor Group (GETUG) P01 trial. Ann. Oncol. 2011, 22, 2476–2481. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Rickman, D.S. Molecular characterization of neuroendocrine prostate cancer and identification of new drug targets. Cancer Discov. 2011, 1, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Tzelepi, V.; Zhang, J. Modeling a lethal prostate cancer variant with small-cell carcinoma features. Clin. Cancer. Res. 2012, 18, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Epstein, J.I. Small cell carcinoma of the prostate. A morphologic and immunohistochemical study of 95 cases. Am. J. Surg. Pathol. 2008, 32, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Huang, J. Clinical and Genomic Characterization of Treatment-Emergent Small-Cell Neuroendocrine Prostate Cancer: A Multi-institutional Prospective Study. J. Clin. Oncol. 2018, 36, 2492–2503. [Google Scholar] [CrossRef] [PubMed]
- Tsai, H.K.; Lehrer, J. Gene expression signatures of neuroendocrine prostate cancer and primary small cell prostatic carcinoma. BMC Cancer 2017, 17, 759. [Google Scholar] [CrossRef]
- Shah, R.B.; Mehra, R. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004, 64, 9209–9216. [Google Scholar] [CrossRef]
- Beltran, H.; Prandi, D. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med. 2016, 22, 298–305. [Google Scholar] [CrossRef]
- Abida, W.; Cytra, J. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. 2019, 116, 11428–11436. [Google Scholar] [CrossRef]
- Labrecque, M.P.; Coleman, I.M. Molecular profiling stratifies diverse phenotypes of treatment-refractory metastatic castration-resistant prostate cancer. J. Clin. Invest. 2019, 129, 4492–4505. [Google Scholar] [CrossRef]
- Schaeffer, E.M.; Srinivas, S. Prostate Cancer, Version 4.2023, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Canc. Netw. 2023, 21, 1067–1096. [Google Scholar] [CrossRef]
- Soundararajan, R.; Viscuse, P. Genotype-to-Phenotype Associations in the Aggressive Variant Prostate Cancer Molecular Profile (AVPC-m) Components. Cancers 2022, 14, 3233. [Google Scholar] [CrossRef] [PubMed]
- Guedes, L.B.; Almutairi, F. Analytic, Preanalytic, and Clinical Validation of p53 IHC for Detection of TP53 Missense Mutation in Prostate Cancer. Clin. Canc. Res. 2017, 23, 4693–4703. [Google Scholar] [CrossRef] [PubMed]
- Tan, H.L.; Sood, A. Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin. Cancer Res. 2014, 20, 890–903. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Yeow, W.S. The retinoblastoma tumor suppressor controls androgen signaling and human prostate cancer progression. J. Clin. Invest. 2010, 120, 4478–4492. [Google Scholar] [CrossRef] [PubMed]
- Williamson, S.R.; Zhang, S. ERG-TMPRSS2 rearrangement is shared by concurrent prostatic adenocarcinoma and prostatic small cell carcinoma and absent in small cell carcinoma of the urinary bladder: Evidence supporting monoclonal origin. Mod. Pathol. 2011, 24, 1120–1127. [Google Scholar] [CrossRef]
- Hansel, D.E.; Nakayama, M. Shared TP53 gene mutation in morphologically and phenotypically distinct concurrent primary small cell neuroendocrine carcinoma and adenocarcinoma of the prostate. Prostate 2009, 69, 603–609. [Google Scholar] [CrossRef]
- Zou, M.; Toivanen, R. Transdifferentiation as a Mechanism of Treatment Resistance in a Mouse Model of Castration-Resistant Prostate Cancer. Cancer Disc. 2017, 7, 736–749. [Google Scholar] [CrossRef]
- Frolov, M.V.; Dyson, N.J. Molecular mechanisms of E2F-dependent activation and pRB-mediated repression. J. Cell Sci. 2004, 117, 2173–2181. [Google Scholar] [CrossRef]
- Mu, P.; Zhang, Z. SOX2 promotes lineage plasticity and antiandrogen resistance in TP53- and RB1-deficient prostate cancer. Science 2017, 355, 84–88. [Google Scholar] [CrossRef]
- Rickman, D.S.; Beltran, H. Biology and evolution of poorly differentiated neuroendocrine tumors. Nat. Med. 2017, 23, 1–10. [Google Scholar] [CrossRef]
- Meuwissen, R.; Linn, S.C. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Maddison, L.A.; Sutherland, B.W. Conditional deletion of Rb causes early stage prostate cancer. Cancer Res. 2004, 64, 6018–6025. [Google Scholar] [CrossRef] [PubMed]
- Elgavish, A.; Wood, P.A. Transgenic mouse with human mutant p53 expression in the prostate epithelium. Prostate 2004, 61, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Trotman, L.C. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature 2005, 436, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Flesken-Nikitin, A. Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res. 2006, 66, 7889–7898. [Google Scholar] [CrossRef]
- Ku, S.Y.; Rosario, S. Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 2017, 355, 78–83. [Google Scholar] [CrossRef]
- Masumori, N.; Thomas, T.Z. A probasin-large T antigen transgenic mouse line develops prostate adenocarcinoma and neuroendocrine carcinoma with metastatic potential. Cancer Res. 2011, 61, 2239–2249. [Google Scholar]
- Park, J.W.; Lee, J.K. Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 2018, 362, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Calo, E.; Quintero-Estades, J.A. Rb regulates fate choice and lineage commitment in vivo. Nature 2010, 466, 1110–1114. [Google Scholar] [CrossRef] [PubMed]
- Tschaharganeh, D.F.; Xue, W. p53-dependent Nestin regulation links tumor suppression to cellular plasticity in liver cancer. Cell 2014, 158, 579–592. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, T.; Suzuki, J. Linking the p53 tumour suppressor pathway to somatic cell reprogramming. Nature 2019, 460, 1140–1144. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Lu, C. Multiple roles of p53-related pathways in somatic cell reprogramming and stem cell differentiation. Cancer Res. 2012, 72, 5635–5645. [Google Scholar] [CrossRef] [PubMed]
- Marión, R.M.; Strati, K. A p53-mediated DNA damage response limits reprogramming to ensure iPS cell genomic integrity. Nature 2009, 460, 1149–1153. [Google Scholar] [CrossRef]
- Picanço-Castro, V.; Russo-Carbolante, E. Pluripotent reprogramming of fibroblasts by lentiviral mediated insertion of SOX2, C-MYC, and TCL-1A. Stem Cells Dev. 2011, 20, 169–180. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sun, Y. Pathogenesis of prostatic small cell carcinoma involves the inactivation of the P53 pathway. Endocr. Relat. Cancer 2012, 19, 321–331. [Google Scholar] [CrossRef]
- Bookstein, R.; MacGrogan, D. p53 is mutated in a subset of advanced-stage prostate cancers. Cancer Res. 1993, 53, 3369–3373. [Google Scholar]
- Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Yelensky, R. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur. Urol. 2013, 63, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Maughan, B.L.; Guedes, L.B. p53 status in the primary tumor predicts efficacy of subsequent abiraterone and enzalutamide in castration-resistant prostate cancer. Prostate Cancer Prostatic Dis. 2018, 21, 260–268. [Google Scholar] [CrossRef]
- Annala, M.; Vandekerkhove, G. Circulating Tumor DNA Genomics Correlate with Resistance to Abiraterone and Enzalutamide in Prostate Cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef] [PubMed]
- Cairns, P.; Okami, K. Frequent inactivation of PTEN/MMAC1 in primary prostate cancer. Cancer Res. 1997, 57, 4997–5000. [Google Scholar] [PubMed]
- Li, J.; Yen, C. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Wang, S.; Garcia, A.J. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc. Natl. Acad. Sci. USA 2006, 103, 1480–1485. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, J. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Backman, S.A.; Ghazarian, D. Early onset of neoplasia in the prostate and skin of mice with tissue-specific deletion of Pten. Proc. Natl. Acad. Sci. USA 2004, 101, 1725–1730. [Google Scholar] [CrossRef] [PubMed]
- Feilotter, H.E.; Nagai, M.A. Analysis of PTEN and the 10q23 region in primary prostate carcinomas. Oncogene 1998, 16, 1743–1748. [Google Scholar] [CrossRef] [PubMed]
- Müller, M.; Rink, K. PTEN/MMAC1 mutations in prostate cancer. Prostate Cancer Prostatic Dis. 2000, 3, S32. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, A.; Carracedo, A. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458. [Google Scholar] [CrossRef]
- Alimonti, A.; Nardella, C. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin. Invest. 2010, 120, 681–693. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.R.; Chen, M. The functions and regulation of the PTEN tumour suppressor: New modes and prospects. Nat. Rev. Mol. Cell Biol. 2018, 19, 547–562. [Google Scholar] [CrossRef]
- Sircar, K.; Yoshimoto, M. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J. Pathol. 2009, 218, 505–513. [Google Scholar] [CrossRef]
- Martin, P.; Liu, Y.N. Prostate epithelial Pten/TP53 loss leads to transformation of multipotential progenitors and epithelial to mesenchymal transition. Am. J. Pathol. 2011, 179, 422–435. [Google Scholar] [CrossRef]
- Velez, M.G.; Kosiorek, H.E. Differential impact of tumor suppressor gene (TP53, PTEN, RB1) alterations and treatment outcomes in metastatic, hormone-sensitive prostate cancer. Prostate Cancer Prostatic Dis. 2022, 25, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Young, F.P.; Becker, T.M. Biomarkers of Castrate Resistance in Prostate Cancer: Androgen Receptor Amplification and T877A Mutation Detection by Multiplex Droplet Digital PCR. J. Clin. Med. 2022, 11, 257. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Oromendia, C. A Phase II Trial of the Aurora Kinase A Inhibitor Alisertib for Patients with Castration-resistant and Neuroendocrine Prostate Cancer: Efficacy and Biomarkers. Clin. Cancer Res. 2019, 25, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Strieder, V.; Lutz, W. Regulation of N-myc expression in development and disease. Cancer Lett. 2002, 180, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Dardenne, E.; Beltran, H. N-Myc Induces an EZH2-Mediated Transcriptional Program Driving Neuroendocrine Prostate Cancer. Cancer Cell 2016, 30, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Rickman, D.S.; Schulte, J.H. The Expanding World of N-MYC-Driven Tumors. Cancer Discov. 2018, 8, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Tangen, C.M. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. N. Engl. J. Med. 2004, 351, 1513–1520. [Google Scholar] [CrossRef]
- Berger, A.; Brady, N.J. N-Myc-mediated epigenetic reprogramming drives lineage plasticity in advanced prostate cancer. J. Clin. Invest. 2019, 129, 3924–3940. [Google Scholar] [CrossRef]
- Lee, J.K.; Phillips, J.W. N-Myc Drives Neuroendocrine Prostate Cancer Initiated from Human Prostate Epithelial Cells. Cancer Cell 2016, 29, 536–547. [Google Scholar] [CrossRef]
- Brady, N.J.; Bagadion, A.M. Temporal evolution of cellular heterogeneity during the progression to advanced AR-negative prostate cancer. Nat. Commun. 2021, 12, 3372. [Google Scholar] [CrossRef]
- Nikonova, A.S.; Astsaturov, I. Aurora A kinase (AURKA) in normal and pathological cell division. Cell. Mol. Life Sci. 2013, 70, 661–687. [Google Scholar] [CrossRef]
- Buschhorn, H.M.; Klein, R.R. Aurora-A over-expression in high-grade PIN lesions and prostate cancer. Prostate 2005, 64, 341–346. [Google Scholar] [CrossRef]
- Jones, D.; Noble, M. Aurora A regulates expression of AR-V7 in models of castrate resistant prostate cancer. Sci. Rep. 2017, 7, 40957. [Google Scholar] [CrossRef]
- Otto, T.; Horn, S. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer Cell 2009, 15, 67–78. [Google Scholar] [CrossRef]
- Mosquera, J.M.; Beltran, H. Concurrent AURKA and MYCN gene amplifications are harbingers of lethal treatment-related neuroendocrine prostate cancer. Neoplasia 2013, 15, 1–10. [Google Scholar] [CrossRef]
- Meulenbeld, H.J.; Bleuse, J.P. Randomized phase II study of danusertib in patients with metastatic castration-resistant prostate cancer after docetaxel failure. BJU Int. 2013, 111, 44–52. [Google Scholar] [CrossRef]
- Kumar, D.; Sharma, N. Therapeutic Interventions of Cancers Using Intrinsically Disordered Proteins as Drug Targets: C-Myc as Model System. Cancer Inform. 2017, 16, 1176935117699408. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, W.C.; Meyerowitz, J.G. Drugging MYCN through an allosteric transition in Aurora kinase A. Cancer Cell 2014, 26, 414–427. [Google Scholar] [CrossRef] [PubMed]
- Richards, M.W.; Burgess, S.G. Structural basis of N-Myc binding by Aurora-A and its destabilization by kinase inhibitors. Proc. Natl. Acad. Sci. USA 2016, 113, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- Hallett, R.M.; Seong, A.B. Transcript signatures that predict outcome and identify targetable pathways in MYCN-amplified neuroblastoma. Mol. Oncol. 2016, 10, 1461–1472. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, B. Targeting the MYCN-PARP-DNA Damage Response Pathway in Neuroendocrine Prostate Cancer. Clin. Cancer Res. 2018, 24, 696–707. [Google Scholar] [CrossRef]
- Liu, Z.; Chen, S.S. Targeting MYCN in Pediatric and Adult Cancers. Front. Oncol. 2021, 10, 623679. [Google Scholar] [CrossRef]
- Carabet, L.A.; Rennie, P.S. Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. Int. J. Mol. Sci. 2018, 20, 120. [Google Scholar] [CrossRef]
- Ton, A.T.; Singh, K. Dual-Inhibitors of N-Myc and AURKA as Potential Therapy for Neuroendocrine Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 8277. [Google Scholar] [CrossRef] [PubMed]
- Santiago, T.; Tarek, N. Correlation Between MYCN Gene Status and MYCN Protein Expression in Neuroblastoma: A Pilot Study To Propose the Use of MYCN Immunohistochemistry in Limited-Resource Areas. J. Glob. Oncol. 2019, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Narod, S.A.; Neuhausen, S. Rapid progression of prostate cancer in men with a BRCA2 mutation. Br. J. Cancer 2018, 99, 371–374. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Goh, C. Germline BRCA mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J. Clin. Oncol. 2013, 31, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Castro, E.; Goh, C. Effect of BRCA Mutations on Metastatic Relapse and Cause-specific Survival After Radical Treatment for Localised Prostate Cancer. Eur. Urol. 2015, 68, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Nombela, P.; Lozano, R. BRCA2 and Other DDR Genes in Prostate Cancer. Cancers 2019, 11, 352. [Google Scholar] [CrossRef]
- Gottipati, P.; Vischioni, B. Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 2010, 70, 5389–5398. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.J.; Tutt, A.N. Synthetic lethality and cancer therapy: Lessons learned from the development of PARP inhibitors. Annu. Rev. Med. 2015, 66, 455–470. [Google Scholar] [CrossRef]
- de Bono, J.; Mateo, J. Olaparib for Metastatic Castration-Resistant Prostate Cancer. N. Eng. J. Med. 2020, 382, 2091–2102. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Porta, N. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet. Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Nizialek, E.; Antonarakis, E.S. PARP Inhibitors in Metastatic Prostate Cancer: Evidence to Date. Cancer Manag. Res. 2020, 12, 8105–8114. [Google Scholar] [CrossRef]
- Zafeiriou, Z.; Bianchini, D. Genomic Analysis of Three Metastatic Prostate Cancer Patients with Exceptional Responses to Carboplatin Indicating Different Types of DNA Repair Deficiency. Eur. Urol. 2019, 75, 184–192. [Google Scholar] [CrossRef] [PubMed]
- Mota, J.M.; Barnett, E. Platinum-Based Chemotherapy in Metastatic Prostate Cancer With DNA Repair Gene Alterations. JCO Precis. Oncol. 2020, 4, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Spisák, S. The association between germline BRCA2 variants and sensitivity to platinum-based chemotherapy among men with metastatic prostate cancer. Cancer 2017, 123, 3532–3539. [Google Scholar] [CrossRef]
- Cheng, H.H.; Pritchard, C.C. Biallelic Inactivation of BRCA2 in Platinum-sensitive Metastatic Castration-resistant Prostate Cancer. Eur. Urol. 2016, 69, 992–995. [Google Scholar] [CrossRef]
- Abida, W.; Cheng, M.L. Analysis of the Prevalence of Microsatellite Instability in Prostate Cancer and Response to Immune Checkpoint Blockade. JAMA Oncol. 2019, 5, 471–478. [Google Scholar] [CrossRef]
- Kumar, A.; White, T.A. Exome sequencing identifies a spectrum of mutation frequencies in advanced and lethal prostate cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 17087–17092. [Google Scholar] [CrossRef]
- Pritchard, C.C.; Morrissey, C. MSH2 and MSH6 mutations in hypermutated microsatellite unstable advanced prostate cancer. Nat. Commun. 2014, 5, 4988. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Joost, P. Frequent mismatch-repair defects link prostate cancer to Lynch syndrome. BMC Urol. 2016, 16, 15. [Google Scholar] [CrossRef]
- Antonarakis, E.S.; Piulats, J.M. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Guedes, L.B.; Antonarakis, E.S. MSH2 Loss in Primary Prostate Cancer. Clin. Cancer Res. 2017, 23, 6863–6874. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Park, C.K. Characteristics of BRCA2 Mutated Prostate Cancer at Presentation. Int. J. Mol. Sci. 2022, 23, 13426. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, G.; Armenia, J. Significance of BRCA2 and RB1 Co-loss in Aggressive Prostate Cancer Progression. Clin. Cancer Res. 2020, 26, 2047–2064. [Google Scholar] [CrossRef] [PubMed]
- Mansour, W.Y.; Tennstedt, P. Loss of PTEN-assisted G2/M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Sci. Rep. 2018, 8, 3947. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sun, H. Aurora-A: A potential DNA repair modulator. Tumour Biol. 2014, 35, 2831–2836. [Google Scholar] [CrossRef]
- Lapuk, A.V.; Wu, C. From sequence to molecular pathology, and a mechanism driving the neuroendocrine phenotype in prostate cancer. J. Pathol. 2012, 227, 286–297. [Google Scholar] [CrossRef]
- Cheng, S.; Yu, X. Bioinformatics analyses of publicly available NEPCa datasets. Am. J. Clin. Exp. Urol. 2019, 7, 327–340. [Google Scholar]
- Ostano, P.; Mello-Grand, M. Gene Expression Signature Predictive of Neuroendocrine Transformation in Prostate Adenocarcinoma. Int. J. Mol. Sci. 2020, 21, 1078. [Google Scholar] [CrossRef]
- Alshalalfa, M.; Liu, Y. Characterization of transcriptomic signature of primary prostate cancer analogous to prostatic small cell neuroendocrine carcinoma. Int. J. Cancer 2019, 145, 3453–3461. [Google Scholar] [CrossRef]
- Akamatsu, S.; Wyatt, A.W. The Placental Gene PEG10 Promotes Progression of Neuroendocrine Prostate Cancer. Cell Rep. 2015, 12, 922–936. [Google Scholar] [CrossRef]
- Lee, T.I.; Jenner, R.G. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell 2016, 125, 301–313. [Google Scholar] [CrossRef]
- Flora, P.; Dalal, G. Polycomb Repressive Complex(es) and Their Role in Adult Stem Cells. Genes 2021, 12, 1485. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Yu, J. A polycomb repression signature in metastatic prostate cancer predicts cancer outcome. Cancer Res. 2007, 67, 10657–10663. [Google Scholar] [CrossRef] [PubMed]
- Karanikolas, B.D.; Figueiredo, M.L. Comprehensive evaluation of the role of EZH2 in the growth, invasion, and aggression of a panel of prostate cancer cell lines. Prostate 2010, 70, 675–688. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Al-Muftah, M.A. Targeting Wnt/EZH2/microRNA-708 signaling pathway inhibits neuroendocrine differentiation in prostate cancer. Cell Death Discov. 2019, 5, 139. [Google Scholar] [CrossRef]
- Zhang, Y.; Zheng, D. Androgen deprivation promotes neuroendocrine differentiation and angiogenesis through CREB-EZH2-TSP1 pathway in prostate cancers. Nat. Commun. 2018, 9, 4080. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.S.; Schaarschuch, K. Top2a identifies and provides epigenetic rationale for novel combination therapeutic strategies for aggressive prostate cancer. Oncotarget 2015, 6, 3136–3146. [Google Scholar] [CrossRef]
- Wee, Z.N.; Li, Z. EZH2-mediated inactivation of IFN-γ-JAK-STAT1 signaling is an effective therapeutic target in MYC-driven prostate cancer. Cell Rep. 2014, 8, 204–216. [Google Scholar] [CrossRef]
- Schade, A.E.; Kuzmickas, R. Combating castration-resistant prostate cancer by co-targeting the epigenetic regulators EZH2 and HDAC. PLoS Biol. 2023, 21, e3002038. [Google Scholar] [CrossRef]
- Crea, F.; Hurt, E.M. Pharmacologic disruption of Polycomb Repressive Complex 2 inhibits tumorigenicity and tumor progression in prostate cancer. Mol. Cancer 2011, 10, 40. [Google Scholar] [CrossRef]
- Clermont, P.L.; Lin, D. Polycomb-mediated silencing in neuroendocrine prostate cancer. Clin. Epigenetics 2015, 7, 40. [Google Scholar] [CrossRef]
- Su, W.; Han, H.H. The Polycomb Repressor Complex 1 Drives Double-Negative Prostate Cancer Metastasis by Coordinating Stemness and Immune Suppression. Cancer Cell 2019, 36, 139–155.e10. [Google Scholar] [CrossRef]
- Smith, B.A.; Balanis, N.G. A Human Adult Stem Cell Signature Marks Aggressive Variants across Epithelial Cancers. Cell Rep. 2018, 24, 3353–3366.e5. [Google Scholar] [CrossRef] [PubMed]
- Xiang, S.; Zou, P. HOTAIR-mediated reciprocal regulation of EZH2 and DNMT1 contribute to polyphyllin I-inhibited growth of castration-resistant prostate cancer cells in vitro and in vivo. Biochim. Biophys. Acta. Gen. Subj. 2018, 1862, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Ohm, J.E.; McGarvey, K.M. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007, 39, 237–242. [Google Scholar] [CrossRef]
- Gravina, G.L.; Festuccia, C. Chronic azacitidine treatment results in differentiating effects, sensitizes against bicalutamide in androgen-independent prostate cancer cells. Prostate 2008, 68, 793–801. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Dong, X. Identification of DEK as a potential therapeutic target for neuroendocrine prostate cancer. Oncotarget 2015, 6, 1806–1820. [Google Scholar] [CrossRef] [PubMed]
- Ci, X.; Hao, J. Heterochromatin Protein 1α Mediates Development and Aggressiveness of Neuroendocrine Prostate Cancer. Cancer Res. 2018, 78, 2691–2704. [Google Scholar] [CrossRef]
- Clermont, P.L.; Ci, X. Treatment-emergent neuroendocrine prostate cancer: Molecularly driven clinical guidelines. Int. J. Encodr. Oncol. 2019, 6, IJE20. [Google Scholar] [CrossRef]
- Dang, Q.; Li, L. Anti-androgen enzalutamide enhances prostate cancer neuroendocrine (NE) differentiation via altering the infiltrated mast cells → androgen receptor (AR) → miRNA32 signals. Mol. Oncol. 2015, 9, 1241–1251. [Google Scholar] [CrossRef]
- Ding, M.; Lin, B. A dual yet opposite growth-regulating function of miR-204 and its target XRN1 in prostate adenocarcinoma cells and neuroendocrine-like prostate cancer cells. Oncotarget 2015, 6, 7686–7700. [Google Scholar] [CrossRef] [PubMed]
- Nam, R.K.; Benatar, T. MicroRNA-652 induces NED in LNCaP and EMT in PC3 prostate cancer cells. Oncotarget 2018, 9, 19159–19176. [Google Scholar] [CrossRef] [PubMed]
- Dankert, J.T.; Wiesehöfer, M. The deregulation of miR-17/CCND1 axis during neuroendocrine transdifferentiation of LNCaP prostate cancer cells. PLoS ONE 2018, 13, e0200472. [Google Scholar] [CrossRef] [PubMed]
- Bhagirath, D.; Liston, M. MicroRNA determinants of neuroendocrine differentiation in metastatic castration-resistant prostate cancer. Oncogene 2020, 39, 7209–7223. [Google Scholar] [CrossRef]
- Ramnarine, V.R.; Alshalalfa, M. The long noncoding RNA landscape of neuroendocrine prostate cancer and its clinical implications. Gigascience 2018, 7, giy050. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Ramnarine, V.R. The long noncoding RNA H19 regulates tumor plasticity in neuroendocrine prostate cancer. Nat. Commun. 2021, 12, 7349. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Donmez, N. SRRM4 Drives Neuroendocrine Transdifferentiation of Prostate Adenocarcinoma Under Androgen Receptor Pathway Inhibition. Eur. Urol. 2017, 71, 68–78. [Google Scholar] [CrossRef]
- Zhang, X.; Coleman, I.M. SRRM4 Expression and the Loss of REST Activity May Promote the Emergence of the Neuroendocrine Phenotype in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 4698–4708. [Google Scholar] [CrossRef]
- Svensson, C.; Ceder, J. REST mediates androgen receptor actions on gene repression and predicts early recurrence of prostate cancer. Nucleic Acids Res. 2014, 42, 999–1015. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, A.; Tzelepi, V. Neuroendocrine prostate cancer xenografts with large-cell and small-cell features derived from a single patient’s tumor: Morphological, immunohistochemical, and gene expression profiles. Prostate 2011, 71, 846–856. [Google Scholar] [CrossRef] [PubMed]
- Nouruzi, S.; Ganguli, D. ASCL1 activates neuronal stem cell-like lineage programming through remodeling of the chromatin landscape in prostate cancer. Nat. Commun. 2022, 13, 2282. [Google Scholar] [CrossRef]
- Bishop, J.L.; Thaper, D. The Master Neural Transcription Factor BRN2 Is an Androgen Receptor-Suppressed Driver of Neuroendocrine Differentiation in Prostate Cancer. Cancer Discov. 2017, 7, 54–71. [Google Scholar] [CrossRef]
- Bhagirath, D.; Yang, T.L. BRN4 Is a Novel Driver of Neuroendocrine Differentiation in Castration-Resistant Prostate Cancer and Is Selectively Released in Extracellular Vesicles with BRN2. Clin. Cancer Res. 2019, 25, 6532–6545. [Google Scholar] [CrossRef]
- Baca, S.C.; Takeda, D.Y. Reprogramming of the FOXA1 cistrome in treatment-emergent neuroendocrine prostate cancer. Nat. Commun. 2021, 12, 1979. [Google Scholar] [CrossRef]
- Asrani, K.; Torres, A.F. Reciprocal YAP1 loss and INSM1 expression in neuroendocrine prostate cancer. J. Pathol. 2021, 255, 425–437. [Google Scholar] [CrossRef] [PubMed]
- Cejas, P.; Xie, Y. Subtype heterogeneity and epigenetic convergence in neuroendocrine prostate cancer. Nat. Commun. 2021, 12, 5775. [Google Scholar] [CrossRef]
- Vias, M.; Massie, C.E. Pro-neural transcription factors as cancer markers. BMC Med. Genomics 2018, 1, 17. [Google Scholar] [CrossRef]
- Xin, Z.; Zhang, Y. Insulinoma-associated protein 1 is a novel sensitive and specific marker for small cell carcinoma of the prostate. Hum. Pathol. 2018, 79, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.F.; Yang, C. Expression of novel neuroendocrine marker insulinoma-associated protein 1 (INSM1) in genitourinary high-grade neuroendocrine carcinomas: An immunohistochemical study with specificity analysis and comparison to chromogranin, synaptophysin, and CD56. Pathol. Res. Pract. 2020, 216, 152993. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, T. Single-cell transcriptional regulation and genetic evolution of neuroendocrine prostate cancer. iScience 2022, 25, 104576. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Representative histologic images from two AVPC cases (a,b) with mixed adenocarcinoma and small cell carcinoma. (a) The two components are admixed. AR is expressed in the adenocarcinoma component and chromogranin A is expressed in the small cell carcinoma; (b) The two components are separate within the tumor, as demonstrated by the intense expression of the neuroendocrine marker CD56 only in the small cell carcinoma. Their distinctive morphology and immunohistochemical profiles are depicted in the right panel, yet, both show intense p53 expression, consistent with TP53 mutation. [AR=Androgen receptor, AVPC=Aggressive variant prostate cancer, TP53=Tumor protein 53].
Figure 1.
Representative histologic images from two AVPC cases (a,b) with mixed adenocarcinoma and small cell carcinoma. (a) The two components are admixed. AR is expressed in the adenocarcinoma component and chromogranin A is expressed in the small cell carcinoma; (b) The two components are separate within the tumor, as demonstrated by the intense expression of the neuroendocrine marker CD56 only in the small cell carcinoma. Their distinctive morphology and immunohistochemical profiles are depicted in the right panel, yet, both show intense p53 expression, consistent with TP53 mutation. [AR=Androgen receptor, AVPC=Aggressive variant prostate cancer, TP53=Tumor protein 53].
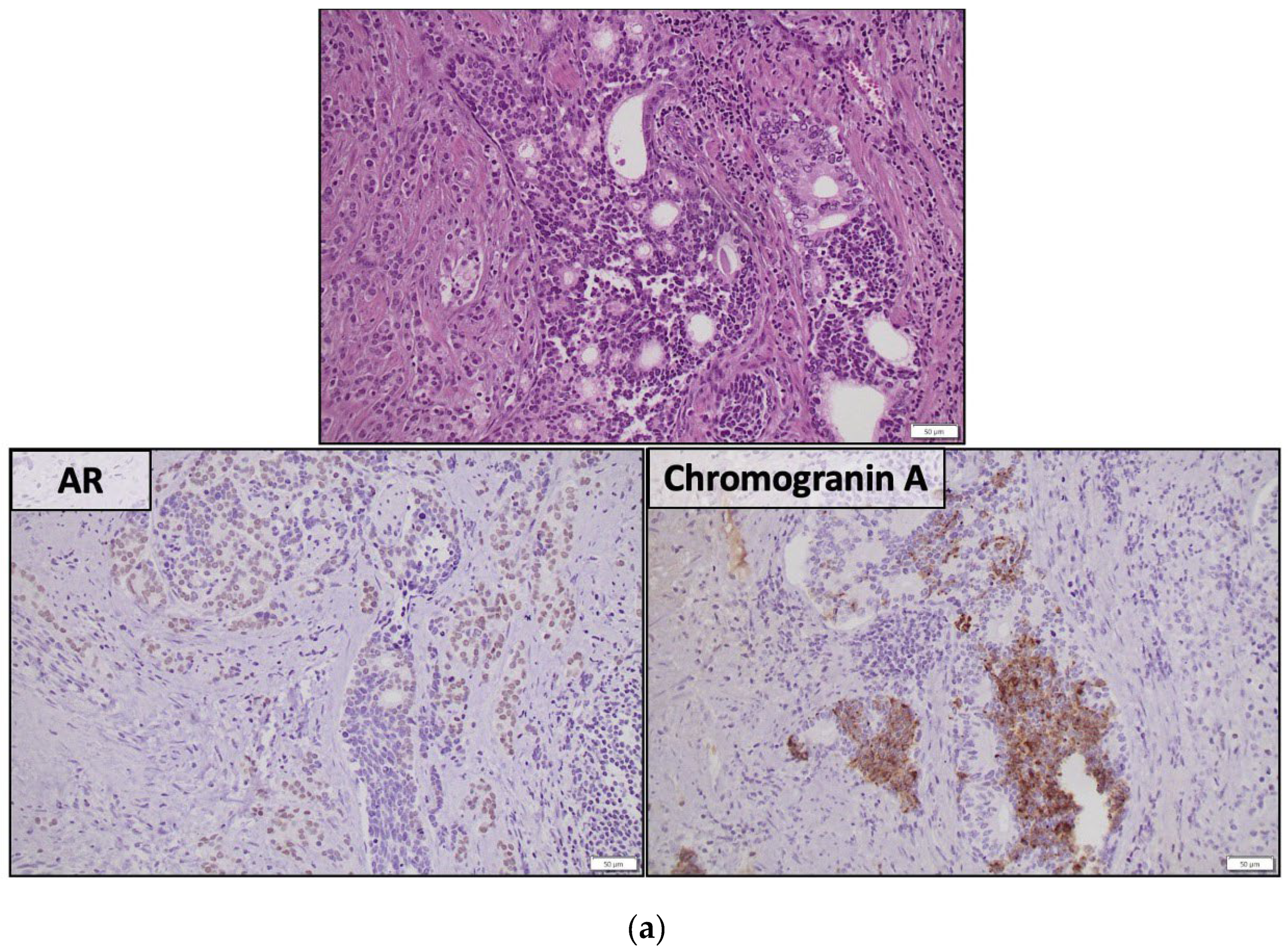
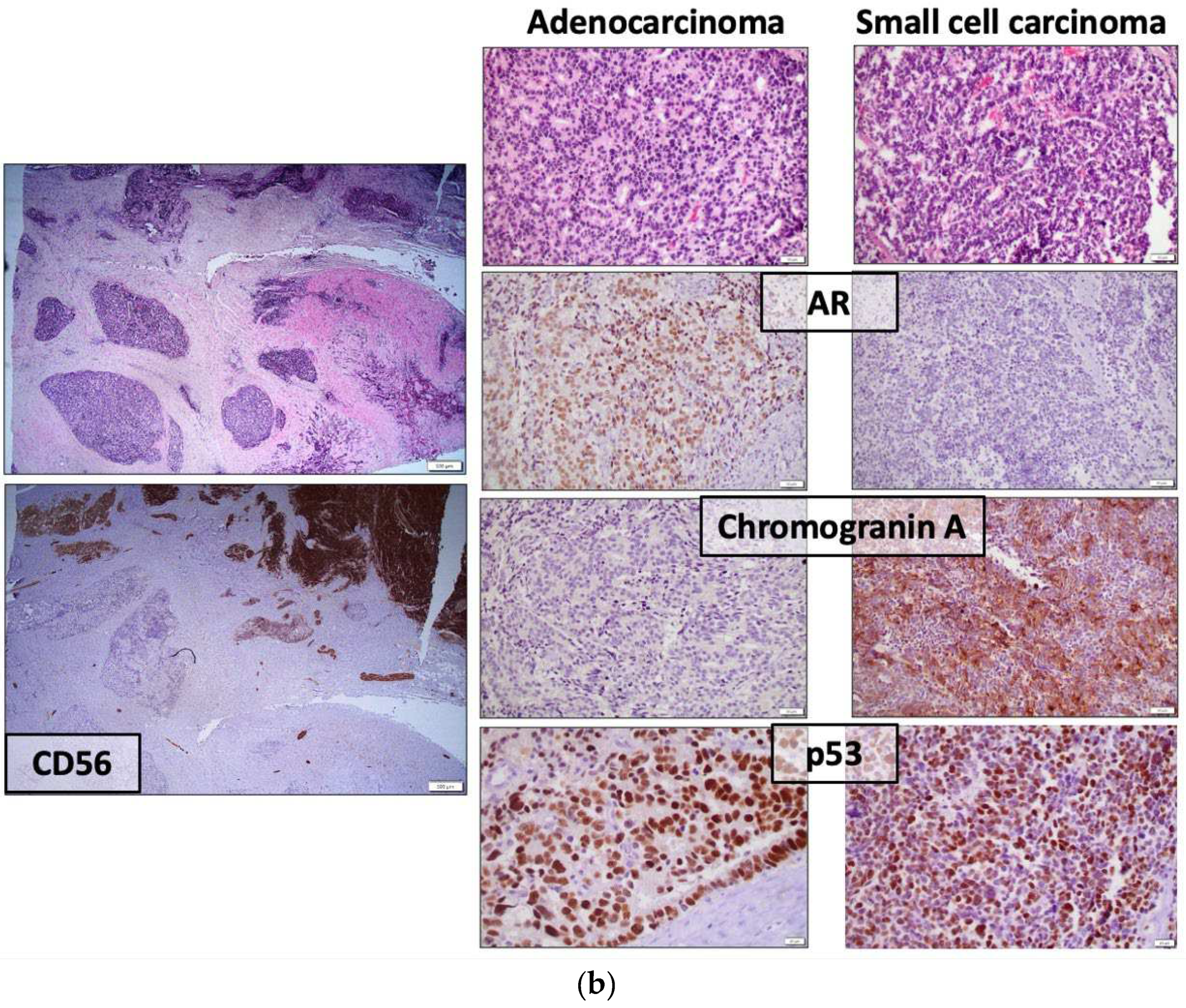
Figure 2.
Schematic representation of the transition of PCA to NEPC with the accompanying molecular and phenotypic changes. During transition to NEPC, PCA under antiandrogen therapy switches from an AR-dependent to an AR-independent state. An intermediate phase of double negativity for AR and NE markers indicates transient reversal to an undifferentiated, stem-cell-like state. The acquisition of a mesenchymal program marks EMT. On the molecular level, combined alterations of tumor suppressors and recruitment of pioneer epigenetic regulators initiate AVPC emergence. Additional epigenetic events potentiate NE differentiation with the expression of late proneural TFs and NE markers. [AR=Androgen receptor, ASCL1=Achaete-scute homolog 1, AVPC=Aggressive variant prostate cancer, BRN2 (POU3F2)=POU domain class 3 transcription factor 2, DNPC=Double negative prostate cancer, EMT=Epithelial-to-mesenchymal transition, NE=Neuroendocrine (markers), NEPC=Neuroendocrine prostate cancer, PCA=Prostate adenocarcinoma, PTEN=Phosphatase and tensin homolog, SOX2=SRY-Box transcription factor 2, RB1=Retinoblastoma protein 1, TP53=Tumor protein 53].
Figure 2.
Schematic representation of the transition of PCA to NEPC with the accompanying molecular and phenotypic changes. During transition to NEPC, PCA under antiandrogen therapy switches from an AR-dependent to an AR-independent state. An intermediate phase of double negativity for AR and NE markers indicates transient reversal to an undifferentiated, stem-cell-like state. The acquisition of a mesenchymal program marks EMT. On the molecular level, combined alterations of tumor suppressors and recruitment of pioneer epigenetic regulators initiate AVPC emergence. Additional epigenetic events potentiate NE differentiation with the expression of late proneural TFs and NE markers. [AR=Androgen receptor, ASCL1=Achaete-scute homolog 1, AVPC=Aggressive variant prostate cancer, BRN2 (POU3F2)=POU domain class 3 transcription factor 2, DNPC=Double negative prostate cancer, EMT=Epithelial-to-mesenchymal transition, NE=Neuroendocrine (markers), NEPC=Neuroendocrine prostate cancer, PCA=Prostate adenocarcinoma, PTEN=Phosphatase and tensin homolog, SOX2=SRY-Box transcription factor 2, RB1=Retinoblastoma protein 1, TP53=Tumor protein 53].
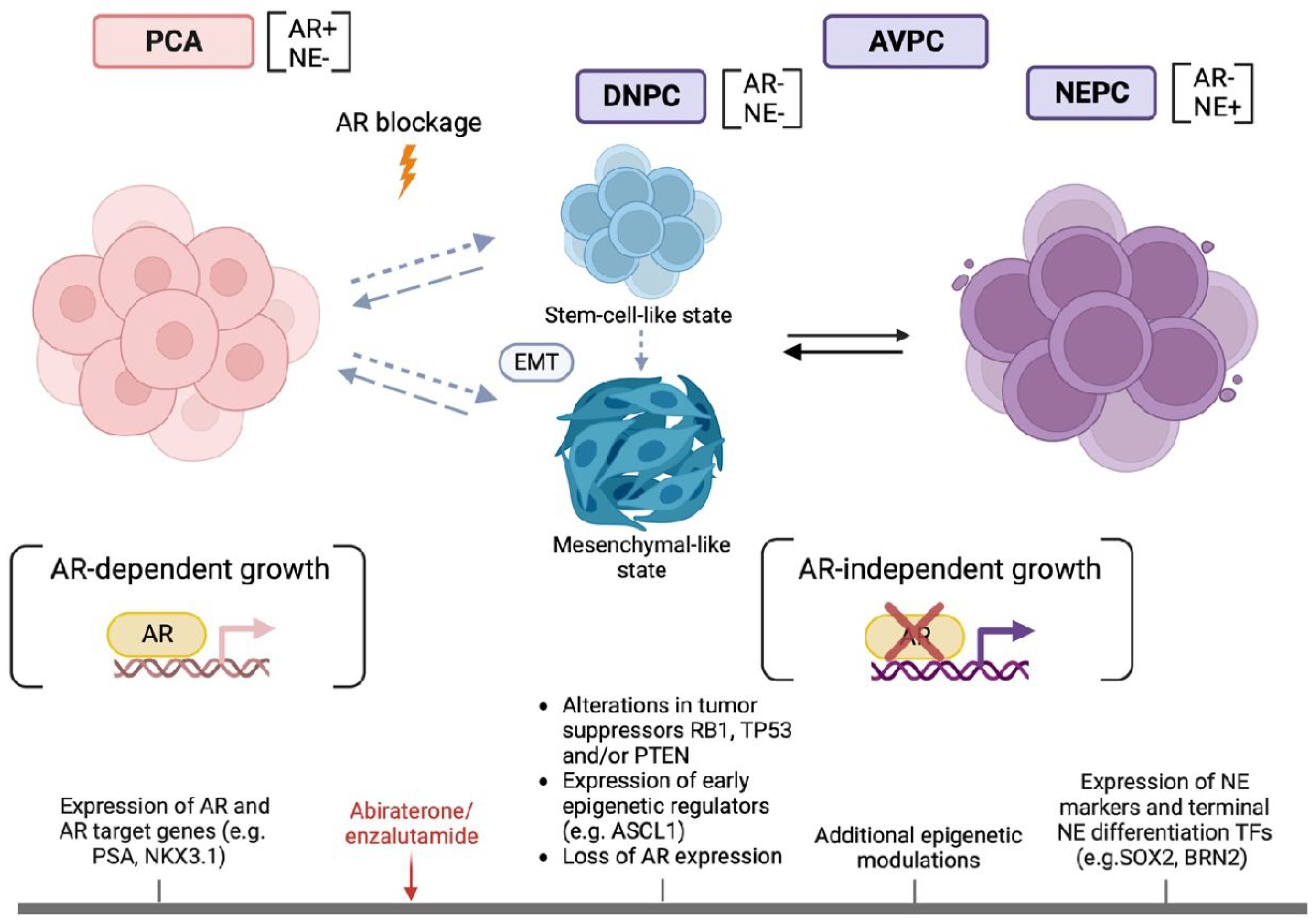

Table 1.
Candidate tissue-based biomarkers for AVPC detection with their respective sensitivity and specificity values.
Table 1.
Candidate tissue-based biomarkers for AVPC detection with their respective sensitivity and specificity values.
| Candidate Tissue-Based AVPC biomarkers | Method | Evaluation | Sensitivity | Specificity |
|---|---|---|---|---|
| Chromogranin and/or synaptophysin | IHC | Any extent of positive staining | 57% [12] | 0–90% [14] 1 |
| AR |
IHC Copy number analysis |
Reduced (<10%) or weak (1+) staining Absence of copy number gain |
36% [16] 80% [16] |
87% [32] 2 30–50% [84] 3 |
| RB1 | IHC | Reduced (<10%) staining | 61% [16] | 26–93% [41,42] 4 |
| Copy number analysis | Copy number loss | 54% [16] | 72% [68] 5 | |
| p53 | IHC | ≥10% staining | 41% [16] | ≈ 60% 6 |
| PTEN | Copy number analysis | Copy number loss | 48% [16] | 23% [81] 7 |
| RB1, TP53 and/or PTEN (≥2/3) | DNA sequencing8 | Combined alterations | 48% [16] | 74% [16] 9 |
| MYCN | Copy number analysis | Copy number gain | 20% [16] | 96% [29] 10 |
| AURKA | Copy number analysis | Copy number gain | 25% [16] | 95% [29] 11 |
| BRCA2 | DNA sequencing | Mutation or deletion | 29% [85] | 87% [66] 12 |
1,10,11 compared to unselected PCA 2,12 compared to unselected mCRPC 4compared to unselected CRPC (26% specificity) [42] or PCA in general (93% specificity) [41] 3,5,7,9 compared to unselected CRPC 6 estimated by considering p53 IHC as a surrogate for TP53 mutations in unselected CRPC [66,68] 8 IHC with a standardized evaluation approach is acceptable [39] [AURKA=Aurora kinase A, AR=Androgen receptor, BRCA2=Breast cancer 2, IHC=Immunohistochemistry, MYCN=MYCN Proto-Oncogene, BHLH Transcription Factor, PTEN=Phosphatase and tensin homolog, RB1=Retinoblastoma protein 1, TP53=Tumor pro..tein 53]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



