
Submitted:
02 January 2024
Posted:
18 January 2024
You are already at the latest version
Abstract
Bacteria can colonize and develop biofilm on a large variety of biotic and abiotic materials, it is accountable for nosocomial infections and chronic illness and consequently, it is a serious economic and public health problem. Staphylococcus epidermidis, thanks to its ability to form biofilm and colonize biomaterials, represents the most frequent causative agent involved in bio-film-associated infections of medical devices. Due to the biofilm presence, S. epidermidis infections are extremely difficult to treat and the research of new molecules able to interfere with its biofilm formation has a remarkable interest. In the present work, the attention was focused on Pseudomonas sp. TAE6080, an Antarctic marine bacterium able to produce and secrete an effective antibiofilm compound. The molecule responsible for this activity was purified by an activity-guided approach and it resulted to be a periplasmic protein, named Cold-Azurin. The Cold-Azurin was recombinantly produced in E. coli and purified. The recombinant protein was able to impair S. epidermidis attachment to the polystyrene surface and effectively prevent biofilm formation.
Keywords:
Staphylococcus epidermidis
; antibiofilm
; Azurin
; cold-adapted bacteria
1. Introduction
Current advances in surgical and medical science are increasing the use of indwelling medical devices, however, their implantation and in vivo use are compromised by their susceptibility to microbial colonization causing half of the problems in healthcare-associated infections (HAIs) [1,2,3]. Their surface and long-term use can be threatened by the adhesion and proliferation of microorganisms, which can interact and form biofilms exposing the body to a risk of permanent colonization and device potential replacement [4]. Staphylococci are the most common etiological agents of medical device infections in the US and Europe [5]. Among them, Staphylococcus epidermidis represents one of the leading species of contamination, due to its normal presence on human skin; it can colonize numerous sites throughout the body, and it is now the most common non-aureus Staphylococci (NAS) species associated with infections of indwelling medical devices, endocarditis and neonatal infections [6]. The ability of S. epidermidis to form biofilm on indwelling medical devices is central in the biomedical infection process and represents its major virulence determinant, rendering S. epidermidis a successful nosocomial pathogen stem [7]. It is noteworthy that currently, more than 70% of S. epidermidis healthcare-associated is methicillin-resistant [8] which is of concern and limits the options for effective antimicrobial treatment.
Currently, the treatment of staphylococcal biofilm infections is an expensive and significant challenge and the phenotypic heterogeneity within the biofilm population associated to the reduced antibiotic susceptibility, is a major obstacle for successful antimicrobial therapy. It is established that the biofilm confers competitive advantages to staphylococci, including a 10 to 1000-fold increase in resistance to antibiotics compared to planktonic cells [9]. The extracellular polymeric matrix of biofilm (EPS) protects the embedded bacteria hindering antibiotic penetration and makes difficult for the drug to reach the inner layer of the biofilm [10]. Additionally, the presence in biofilm of cells with different metabolic features promotes the emergence of tolerant and persister cells which exhibit physiological characteristics that make S. epidermidis cells resilient to high antibiotic concentrations [5]. Notably, extrinsic factors also play a role in the modulation of antibiotic susceptibility in staphylococci, for example, mixed biofilms have been shown to confer to S. epidermidis an enhanced tolerance toward vancomycin and other antimicrobials [11]. As a result, antimicrobial therapy often fails, and innovative approaches are needed to prevent bacterial adhesion and biofilm formation in the medical setting. The antibiofilm strategies do not aim to inhibit bacterial growth and cell division, but instead target molecules and pathways involved in the formation and maturation of biofilms without necessarily killing biofilm-associated cells. This latter should allow the development of narrow-spectrum agents, which will have low or no influence on commensal microbiota [1], moreover, an approach that targets biofilm without affecting bacterial vitality avoids the rapid appearance of escape mutants. Therefore, the use of antibiofilm molecules that are active against different stages of biofilm development represents a promising option.
Microorganisms able to survive in extreme environments, such as Antarctica, can be a promising source of new antibiofilm agents. Antarctic marine bacteria apply various survival strategies to persist in harsh conditions, and the reduction of competing microorganisms is one of these approaches. Due to the key role of biofilm in bacteria fitness in extreme conditions [12] the production of antibiofilm molecules could reduce the competitors’ survival. Indeed, several papers report that marine Antarctic bacteria produce and secrete anti-biofilm molecules [13,14,15,16,17,18].
In this work, we investigated the ability of the Antarctic marine bacterium Pseudomonas sp. TAE6080 [19] to produce compounds effective against S. epidermidis biofilm formation and we identified the molecule responsible for this activity.
The antibiofilm molecule is a protein named Cold-Azurin. The results reported in this paper sustain once again the great potential of Antarctic bacteria as producers of bioactive molecules.
2. Results
2.1. Production of Anti-Biofilm and Anti-Adhesive Molecules Active against S. epidermidis Biofilm
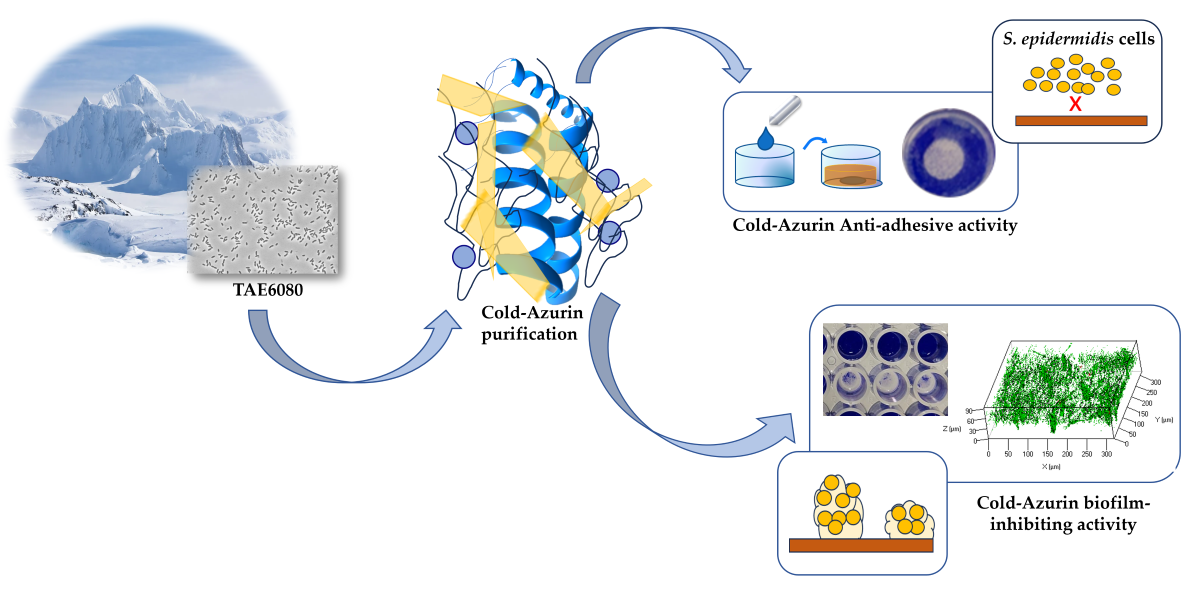
Pseudomonas sp. TAE6080 is able to produce and secrete molecule/s able to interfere with the biofilm formation of S. epidermidis RP62A [19]. Firstly, the ability of this Polar bacterium to also produce anti-adhesive compound was evaluated. The anti-adhesive and anti-biofilm activities of Pseudomonas sp. TAE6080 cell-free supernatant (SN TAE6080) were tested against the reference strain S. epidermidis RP62A [19] and against S. epidermidis O-47 [20] an agr-mutant considered a strong biofilm producer [21].
SN TAE6080 anti-adhesive capability was tested using the surface coating assay by evaluating the ability of the coated surfaces to avoid staphylococcal biofilm formation (Figure 1A). The capability of SN TAE6080 to inhibit the biofilm formation of S. epidermidis was evaluated by adding the supernatant to the staphylococcal growth medium at the beginning of the cultivation (Figure 1B). As reported in Figure 1A, SN TAE6080 showed anti-adhesive activity against both S. epidermidis strains, moreover it was able to impair the biofilm formation of the two strains (Figure 1B).
To ascertain more information about the chemical nature of the anti-adhesive and/or anti-biofilm compound/s, SN TAE6080 was treated with proteinase K, and the activity of the treated samples was compared to the activity of untreated samples. The treatments affected both anti-adhesive (Figure 1C) and biofilm-inhibiting activity (Figure 1D), suggesting that the activities could be due to a peptide and/or proteins. To have more information about the molecular weight of the active compound/s, the SN TAE6080 was concentrated ten-fold by ultrafiltration with membranes of 30kDa MWCO and the anti-adhesive and anti-biofilm activity of the retentate fraction (SNC) and the permeate fraction was determined by the coating assay against S. epidermidis RP62A and S. epidermidis O-47. The results demonstrated that only SNC can inhibit the initial attachment and the biofilm formation (data not shown).
2.2. Purification of anti-adhesive and antibiofilm molecule
To have amount of cell-free supernatant sufficient for the purification, a scale-up of Pseudomonas sp. TAE6080 growth in a bioreactor was set up. The Antarctic bacterium was grown in a 3 L stirred tank fermenter at 15 °C in synthetic G medium. The growth was followed for 72 h till the stationary phase, and the supernatant (SN TAE6080) was collected, separated from the cells, and ultrafiltered with membranes of 30kDa MWCO as described above. To purify the active molecule, SNC was subjected to adsorption chromatography on Amberlite XAD-2, a polystyrene resin, exploiting the reported ability of the anti-adhesive molecules to adhere to this material. Upon SNC loading, the column was extensively washed with G medium, while the elution steps were performed with methanol. The collected chromatographic fractions were tested by the surface coating assay to evaluate their anti-adhesive properties against S. epidermidis RP62A (data not shown) and O-47 (Figure 2A). As shown in Figure 2A the fraction eluted with methanol (named E), dried and resuspended in G medium, displays a strong antibiofilm activity by interfering with S. epidermidis adhesion on the entire well surface. This activity was confirmed also by biofilm inhibiting assay (data not shown). Given the putative proteinaceous nature of the anti-adhesive molecule/s, the fractions were analyzed by SDS-PAGE. The electrophoresis analysis demonstrated that the eluted fraction (E) had a less complex protein profile compared to the unbound (Ub) and wash fraction (W), and that the E fraction was enriched in two proteins (Figure 2B).
2.3. Cold Azurin Identification
SDS analysis of the active fraction obtained after the proposed purification protocol, showed the presence of two main protein bands, which were subsequently in situ hydrolyzed and peptide mixtures thus obtained were analyzed by LC-MSMS for proteins identifications as described in the Materials and Methods section. Our results led to the identification of two Pseudomonas sp. TAE6080 proteins namely a flagellin [20] (FliC the filament core of flagellum) with a molecular weight of about 60kDa (Figure 2B) and a 15kDa (Figure 2B) protein displaying a good similarity to the Azurin [21] from Pseudomonas aeruginosa PAO1 (Figure S1)., both of them were identified with a 35% of sequence coverage ( Figure 2C and 2D). The two proteins present in the active fraction and identified by MS, have usually a different cell localization. Indeed, Azurin is a periplasmic protein while the flagellin is a secreted protein. We took advantage from this difference to understand which one is the protein responsible for anti-biofilm and anti-adhesive activity.
Given the periplasmic nature of the Azurin, the outer membrane permeabilization by cold osmotic shock was performed on TAE6080 cell-pellet. The TAE6080 periplasmic extract named for simplicity OS, presumably enriched with cold-Azurin (Figure 3A), was analysed for anti-adhesive and anti-biofilm activity (Figure 3B and C) against S. epidermidis strains and proved to be able to impair the biofilm formation (Figure 3C) of both staphylococci and to have strong anti-adhesive activity (Figure 3B). Pseudomonas sp. TAE6080 periplasmic extract (OS) was more active against S.epidermidis RP62A while the cell-free supernatant resulted to be more efficient against S.epidermidis O-47 biofilm, the presence of some Pseudomonas sp. TAE6080 periplasmic proteins could be responsible of the recorded different behaviour, future experiments will be dedicated to clarify this point.
2.4. Recombinant Cold Azurin production and purification
Although the anti-biofilm and anti-adhesive activity of TAE6080 periplasmic extract (OS) is a strong clue that the protein responsible for the reported activity is the Cold-Azurin, we produce the cold-adapted protein in E. coli cells to confirm our results. As reported in Materials and Methods section, Cold-Azurin gene was cloned into pET28b(+) vector under the control of an inducible lac operon using E. coli BL21DE3 as host for the heterologous production. We chose to express the protein at 28 °C to not eventually compromise the correct folding of the rCold-Azurin. Figure S2 reports the electrophoretic profiles of the periplasmic fractions of recombinant cells induced or non-induced with IPTG. rCold-Azurin was produced at very large amount only when the inducer was added to the medium (+IPTG). The E.coli periplasmic extract containing the recombinant rCold-Azurin was biologically active as demonstrated by the coating assay on S. epidermidis O-47 strain and by biofilm inhibiting assay (Figure S2).
To purify the rCold-Azurin, the same protocol used for the purification of native protein was applied. The periplasmic fraction from recombinant E. coli BL21DE3 induced cells, was subjected to adsorption chromatography (Amberlite XAD-2) and the elution step was performed with methanol. After the dry, the eluted fraction was suspended in a small volume of PBS and the chromatography fractions were analyzed by SDS-PAGE, demonstrating the presence of only rCold-Azurin in the eluted sample (Figure 4A). The purified rCold-Azurin activity was evaluated and the recombinant protein showed strong anti-biofilm (Figure 4B) and anti-adhesive capability (Figure 4C) against both S. epidermidis strains.
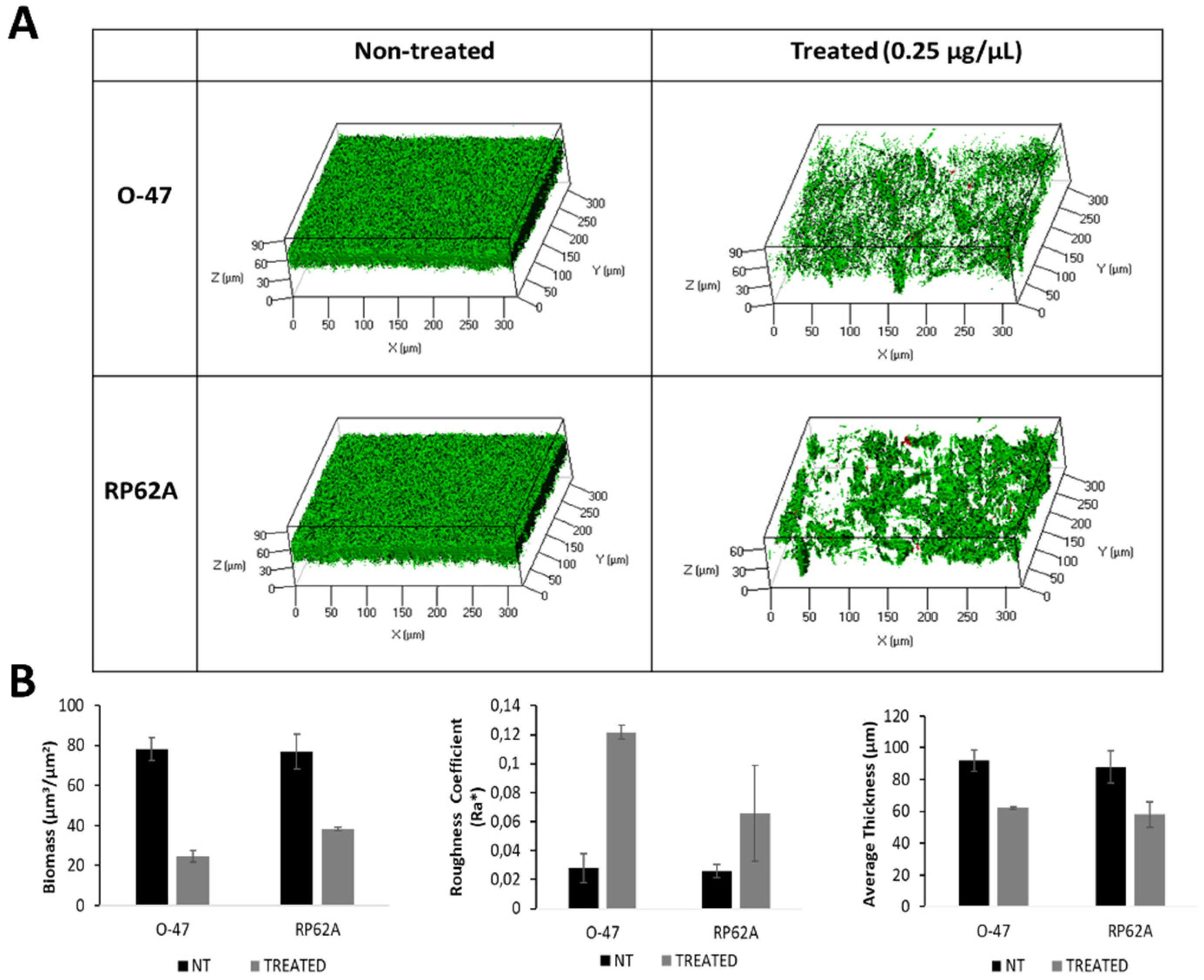
The effect of rCold-Azurin on both S. epidermidis strains biofilms was further explored by confocal laser scanning microscopy (CLSM). The biofilm structure and cell integrity were analyzed by LIVE/DEAD® Biofilm Viability Kit. As shown (Figure 5A), the CLMS analysis confirmed that rCold-Azurin can reduce S. epidermidis biofilm formation without affecting cell viability. The CLSM image stack data on treated biofilms were analysed using the COMSTAT image analysis software package [22] to evaluate the different variables describing the biofilm structure. As expected, the values of the biomass and the average thickness of the biofilm obtained in the presence of rCold-Azurin were lower if compared with the values obtained without the antibiofilm protein, while an increased roughness coefficient is observed for the treated sample (Figure 5B). This dimensionless factor grants a measure of the thickness variation of a biofilm, and thus it is used as an indirect indicator of biofilm heterogeneity. The assays revealed that the treatment resulted in an unstructured and non-homogeneus biofilm compared to untreated biofilm.
3. Discussion
In a previously published paper [19] we report the ability of Pseudomonas sp. TAE6080 to produce antibiofilm molecule/s capable of inhibiting the formation of S. epidermidis RP62A biofilm. In this paper we demonstrated that the bacterium produces and secretes molecule/s able to impair the biofilm formation of another S. epidermidis strain, S. epidermidis O-47, and to interfere also with the attachment to polystyrene surface of both strains. The observation that Pseudomonas sp. TAE6080 cell-free supernatant interfered with the surface adhesion and with biofilm formation suggested the presence of two different molecules involved in, or the presence of a single molecule capable to work as anti-adhesive and antibiofilm compound. To clarify this point and collect physico-chemical information, the cell-free supernatant was treated with proteinase K or subject to ultrafiltration and then tested against the two pathogens. The results demonstrated the involvement of a protein/s with molecular weight higher than 30 kDa. The purification strategy for the Pseudomonas sp. TAE6080 anti-adhesive molecule/s was based on the ability of the protein/s to bind polystyrene. As expected, the fraction eluted from the polystyrene chromatographic resin with methanol (E) had the ability to interfere with S. epidermidis adhesion and, surprisingly, it presented good anti-biofilm activity. These results indicated that the bacterium produces protein/s that impairs S. epidermidis attachment to the polystyrene surface and effectively prevents biofilm formation. The E fraction analysis by SDS-PAGE revealed the presence of two proteins identified by LC-MSMS , a flagellin and a 15kDa protein similar to the Azurin [23] from P. aeruginosa PAO1 that was named Cold-Azurin.
Several proteins resulted to be a good candidate for biofilm treating and prevention [24,25] but generally are hydrolytic enzymes, in our case the two potential antibiofilm proteins are known to be involved in different biological processes. The Flagellin protomers are secreted by the flagellar secretion apparatus and are arranged as a multimer to form a long filament [20]. Azurin is a low molecular weight, blue, copper-containing protein mainly present in the periplasmic space of different Gram-negative bacteria [26]. A possible antibiofilm activity of flagellin seems to be counterintuitive and the involvement of Azurin in the prevention of biofilm is not so obvious. Therefore, we awarded the anti-biofilm activity to the Cold-Azurin by testing the activity of the periplasmic extract of Pseudomonas sp. TAE6080 and evaluating the activity of the recombinant Cold-Azurin produced in E. coli. Both samples were active against S. epidermidis biofilm formation and displayed good anti-adhesive capability. Moreover, the CLSM analyses on S. epidermidis treated biofilm revealed that the purified rCold-Azurin, not only reduced the biofilm biomass but deeply modified the S. epidermidis biofilm structure without affecting cell viability.
These results allowed us to attribute the biological activity of the Pseudomonas sp. TAE6080 cell-free supernatant to the Cold-Azurin.
Azurin acts in respiratory electron transport chain in some bacteria and in Pseudomonas aeruginosa PAO1 it is involved in denitrification and protection against oxidative stress [21,27,28]. Indeed, Azurin allows single-electron transfer between enzymes associated with the cytochrome chain by undergoing oxidation-reduction between Cu(I) and Cu(II), moreover it supports oxidative deamination of primary amines by passing electrons from aromatic amine dehydrogenase to cytochrome oxidase, as well as from some c-type cytochromes to nitrite reductases [23]. In attempt to speculate that the antibiofilm activity of Cold-Azurin could be related to the Cu(2+)/Cu(+) reduction potentials of type-1 copper site [29] we tested the putative antibiofilm activity of poxA1b laccase [30] a well know blue copper-protein oxidase (Figure S3). The obtained results showed the absence of anti-biofilm activity due to the laccase; thus the molecular mechanism responsible for the activity of Cold-Azurin is not related to its redox property.
Unrelated to its electron-transfer property, Azurin has been found to be active against different agents of human diseases such as malaria, AIDS, and cancer [31,32]. Moreover, this protein inhibit the attachment and invasion of different pathogenic bacteria to host cells [33]. This versatile protein is able to interact with unrelated targets such as the surface protein MSP1-19 [34] of the malarial parasite Plasmodium falciparum, the HIV-protein gp120 [34], the ephrin receptor EphB2 [35], and the tumor suppressor protein p53 [36].
Fialho and coworker suggested that the Azurin’s promiscuity in targeting multiple proteins is related with its three-dimensional structure [31]. Azurin is a member of a family cupredoxins [35] and members of this family demonstrate structural features similar to the immunoglobulin variable domains [31,37]. Indeed, although Azurin and the immunoglobulins have a low sequence identity, their similarity is based on the presence of invariant super secondary substructures common to cupredoxins and immunoglobulins [31]. The hypothesis proposed by Fialho and coworkers [31] is that the Azurin is used by the bacterium as a multitarget weapon to avoid the entry of competitors and to eliminate foreign invaders in the host, in this way the bacterium preserves its own survival. This behavior is exactly the job of immune system made by immunoglobulins.
In this view, the reported antibiofilm action of Cold-Azurin can be interpreted as an additional strategy to reduce the presence of potential competitors and the activity could be related to the Cold-Azurin ability to interact with specific proteins required for biofilm formation. Different S. epidermidis proteins have an important role in the biofilm establishment [38,39] [38,39]. For example, the A domain of the accumulation associated protein Aap can promote adhesion to unconditioned biomaterial [40,41], small basic protein (Sbp) proteins can foster S. epidermidis biofilm formation [42] and the surface protein AtlE [43], a bi-functional adhesin/autolysin abundant in the cell wall of S. epidermidis, has a key role in S. epidermidis biofilm formation [44]. Therefore, further experiments are necessary to assess if the target of Cold-Azurin action is one of the proteins involved in S. epidermidis biofilm development.
The present study paves the way to the use of Cold-Azurin as a possible antibiofilm agent against S. epidermidis biofilm. Moreover, the studies on the use of Azurin from P. aeruginosa in different human pathologies [31,33,45,46], that demonstrated also its biocompatibility [32,47,48], are encouraging for the future use Cold-Azurin in the treatment of human infection in combination with conventional antibiotics.
4. Materials and Methods
4.1. Bacterial Strains and Culture Conditions
Bacterial strains used in this work were Pseudomonas sp. TAE6080, collected in 1992 from seawater near French Antarctic Station Dumont d’Urville, Terre Adélie (66°40′ S; 140° 01′ E); S. epidermidis O-47 [49] isolated from clinical septic arthritis; S. epidermidis RP62A reference strain [50] isolated from an infected catheter (ATCC collection no. 35984).
Pseudomonas sp. TAE6080 was grown in synthetic medium G (D-Gluconic acid sodium 10 g L−1, NaCl 10 g L−1; NH4NO3 1 g L−1; KH2PO·7H2O 1 g L−1; MgSO4·7H2O 200 mg L−1; FeSO4·7H2O 5 mg L−1; CaCl2·2H2O 5 mg L−1) [19] in planktonic conditions at 15 °C under vigorous agitation (250 rpm) for 72 h of growth. The cell-free supernatant was separated from the pellet by centrifugation (7000 rpm at 4 °C for 30 min), sterilized by filtration through membranes with a pore diameter of 0.22 μm and stored at 4 °C until use. The cell pellet was stored at –20 °C until use.
Staphylococci were grown at 37 °C in Brain Heart Infusion broth (BHI, Oxoid, Basingstoke, UK), and biofilm formation was assessed in static conditions while planktonic cultures were performed under agitation (180 rpm).
All strains were maintained at −80 °C in cryovials with 20% of glycerol.
4.2. TAE6080 cell-free supernatant preparation
The cell-free supernatant (SN TAE6080) was concentrated 10-fold with Amicon Ultrafiltration cell equipped with a 30 kDa cut-off PES Millipore Ultrafiltration Disc (Merck KGaA, Darmstadt, Germany). Then, retentate fraction (SNC) was collected.
4.3. Surface coating assay
For the surface coating assay, a volume of 5 μL of the tested sample was deposited onto the center of a well of a 24-well tissue-culture-treated polystyrene microtiter plate. The plate was incubated at room temperature to allow complete evaporation of the liquid in sterile conditions. The wells were then filled with 1 mL of S. epidermidis RP62A or S. epidermidis O-47 cultures in exponential growth phase diluted in BHI with a final concentration of about 0.1 and 0.001 OD600nm respectively and incubated at 37 °C in static condition. After 24 h, wells were rinsed with water and stained with 1 mL of 0.1% crystal violet. Stained biofilms were rinsed with water and dried, after that the wells were photographed.
4.4. Biofilm Inhibiting Assay
The quantification of in vitro biofilm production was based on the method described by Ricciardelli and coworkers [51]. For staphylococcal biofilm formation in the presence of Pseudomonas sp. TAE6080 cell-free supernatant (SN TAE6080), the wells of a sterile 96-well flat-bottomed polystyrene plate were filled with S. epidermidis RP62A or S. epidermidis O-47 cultures in exponential growth phase diluted in BHI 2x with a final concentration of about 0.1 and 0.001 OD600nm, respectively. Each well was filled with 100 μL of bacterial cultures and 100 μL of the cell-free supernatant. In this way, the sample was used diluted 1:2 with a final concentration of 50%. As control, the first row was filled with 100 μL of bacterial cultures and 100 μL of G medium (untreated bacteria). The plates were incubated aerobically for 24 h at 37 °C. Biofilm formation was measured using crystal violet staining. After incubation, planktonic cells were gently removed; and wells were washed three times with sterile PBS and thoroughly dried. Each well was then stained with 0.1% crystal violet and incubated for 15 min at room temperature, rinsed twice with double-distilled water, and thoroughly dried. The dye bound to adherent cells was solubilized with 20% (v/v) glacial acetic acid and 80% (v/v) ethanol. After 30 min of incubation at room temperature, the OD590nm was measured to quantify the total biomass of biofilm formed in each well. Each data point was composed of six independent samples.
SN TAE6080 was subjected to proteinase K treatment. The anti-biofilm activity of treated and untreated supernatant was evaluated using the microtiter plate assay against S. epidermidis strains as previously described. Each data point was composed of five independent samples.
For the assay in the presence of Pseudomonas sp. TAE6080 periplasmic extract by osmotic shock method (named OS), the wells of a sterile 96-well flat-bottomed polystyrene plate were filled with S. epidermidis RP62A or S. epidermidis O-47 cultures in exponential growth phase diluted in BHI with a final concentration of about 0.1 and 0.001 OD600nm, respectively. Each well was filled with 180 μL of cultures and 20 μL of periplasmic extract. In this way, the sample was used diluted 1:10 with a final concentration of 10%. As control, the first row was filled with 180 μL of cultures and 20 μL of buffer used for the periplasmic protein extraction (untreated bacteria). The plates were incubated aerobically for 24 h at 37 °C. Biofilm formation was measured as previously described. Each data point was composed of four independent samples.
For the assay with rCold-Azurin: the wells of a sterile 96-well flat-bottomed polystyrene plate were filled with 200 μL of S. epidermidis RP62A or S. epidermidis O-47 cultures in exponential growth phase diluted in BHI with a final concentration of about 0.1 and 0.001 OD600nm, respectively. The plate was incubated at 37 °C for 24 h in the absence and in the presence of rCold-Azurin (0.25 µg µL-1). Biofilm formation was measured as previously described. Each data point was composed of four independent samples.
4.5. Proteinase K treatment
To analyse the proteinaceous nature of active compound/s, proteinase K (Sigma Aldrich, St Louis, MO) was added to the sample at a final concentration of 2 mg mL−1 and the reaction was incubated for 2 h at 37 °C. To exclude an effect due to the temperature, the sample was incubated without proteinase K for 2 h at 37 °C.
4.6. Large-scale growth
Pseudomonas sp. TAE6080 bacterial culture was grown in G medium in a Stirred Tank Reactor 3 L fermenter (Applikon, Schiedam, The Netherlands) connected to an ADI-1030 Bio Controller with a working volume of 1 L. The bioreactor was equipped with the standard pH, pO2, level- and temperature sensors for bioprocess monitoring. The culture was carried out at 15 °C for 72 h in aerobic conditions (30% dissolved oxygen). Supernatant was recovered by centrifugation at 7000 rpm. Then, it was sterilized by filtration through membranes with a pore diameter of 0.22 µm and stored at 4 °C until use.
4.7. Adsorption chromatography
The primary enrichment of the active compound/s was achieved by adsorption chromatography on a polystyrene resin (Amberlite XAD-2; Rohm and Haas, Philadelphia, Pa.). The resin (3 g) was placed in a glass column (10 cm by 1 cm). The column was equilibrated with G medium and then 10 mL of retentate fraction (SNC) was applied at a flow rate of approximately 1 mL min−1. The column was then washed with 3-bed volumes of G medium. The elution of the active compound/s was subsequently carried out with methanol. Fractions of 15 mL volume were collected. The fraction eluted with methanol was recovered, dried, to obtain more concentrated samples, and resuspended in a small volume of G medium. Each chromatographic fraction was analyzed by surface coating assay against S. epidermidis O-47 and S. epidermidis RP62A.
For the rCold-Azurin purification with Amberlite XAD-2, the resin (8 g) was as placed in polypropylene column (12 cm by 1.5 cm) Econo-Pac®. The column was equilibrated with 5mM MgSO4 and then 5 mL of periplasmic extract from recombinant BL21DE3 induced cells was applied at a flow rate of approximately 1 mL min−1. The column was then washed with 3-bed volumes of 5mM MgSO4. The elution of the rCold-Azurin was subsequently carried out with methanol. Fractions of 10 mL volume were collected. The fraction eluted with methanol was recovered, dried, to obtain more concentrated samples, and resuspended in a small volume (about 0.3mL) of PBS buffer. The ativity of purified protein was analyzed by surface coating assay or anti-biofilm assay against S. epidermidis O-47 and S. epidermidis RP62A.
4.8. SDS-PAGE
Protein samples (prepared in Laemmli buffer 4x followed by boiling at 95 °C) were separated on SDS-PAGE gels. The gels were stained with colloidal Coomassie or silver nitrate staining, the protein sizes were determined by comparing the migration of the protein band to a molecular mass standard (Unstained Protein Molecular Weight Marker, Thermo Fisher Scientific Waltham, MA, USA). To analyse total proteins (tot), 1 OD600nm of liquid cultures were harvested at the end of the growth, centrifuged at 13000 rpm for 10 min at 4 °C, and the pellet (about 75 mg) was solubilized in 60 μL of Laemmli Sample buffer 4x. Then, the sample was boiled 95 °C for 20 min quickly cooled on ice for 5 min and finally centrifuged at 13,000 rpm for 5 min at RT. 2 μL of sample was analyzed by SDS-PAGE.
4.9. In-Situ Hydrolysis, LC-MS/MS Analysis and Protein Identification
Mono-dimensional SDS-PAGE gel was stained with Coomassie Brilliant Blue, the band approximately at 60 kDa and 15kDa were excised and de-stained with 100 µL of 0.1 M ammonium bicarbonate (AMBIC) and 130 µL of acetonitrile (ACN) and subsequently subjected to in-situ hydrolysis with 0.1 µg µL−1 trypsin mM in AMBIC for 18 h at 37 °C. The hydrolysis was stopped by adding acetonitrile and 0,1% formic acid. The sample was then filtered and dried in a vacuum centrifuge.
The peptide mixtures thus obtained were directly analyzed by LTQ Orbitrap XL™ Hybrid Ion Trap-Orbitrap Mass Spectrometer (Thermo Fisher Scientific, Bremen, Germany). C-18 reverse phase capillary column 75 μm × 10 cm (Thermo Fisher Scientific) was performed using a flow rate of 300 nL min−1, with a gradient from eluent A (0.2% formic acid in 2% acetonitrile) to eluent B (0.2% formic acid in 95% acetonitrile). The following gradient conditions were used: t = 0 min, 5% solvent B; t = 10 min, 5% solvent B; t = 90 MIN, 50% solvent B; t = 100 min, 80% solvent B; t = 105 min, 100% solvent B; t = 115 min, 100% solvent B; t = 120 min; 5% solvent B. Peptide analysis was performed using the data-dependent acquisition of one MS scan followed by CID fragmentation of the five most abundant ions.
For the MS scans, the scan range was set to 400–1800 m/z at a resolution of 60,000, and the automatic gain control (AGC) target was set to 1 × 106. For the MS/MS scans, the resolution was set to 15,000, the AGC target was set to 1 × 105, the precursor isolation width was 2 Da, and the maximum injection time was set to 500 ms. The CID normalized collision energy was 35%. Data were acquired by Xcalibur™ software (Thermo Fisher Scientific).
In-house Mascot software (version 2.4.0) was used as a search engine to identify proteins. The TAE6080 (2644 sequences; 878,869 residues) proteins database was used for proteins identification.
The software returns a list of proteins associated with a probability index (score), calculated as −10 × Log P, where P is the probability that the observed event is a random one. Proteins are considered as identified if a minimum number of 2 peptides reach the calculated threshold score.
4.10. Pseudomonas sp. TAE6080 periplasmic protein extraction
Osmotic shock. A cell pellet of about 30g was resuspended in 5mL of 30 mM Tris-HCl, pH 7.8 and 20% sucrose, 1x EDTA-free protease inhibitor (Roche). After 30 min of incubation at room temperature, the cells were centrifuged (7500 rpm for 20 min at 4 °C) and the supernatant containing the highest amount of active protein was carefully recovered and named OS. The pellet was then resuspended in 5 mL of ice-cold 0.5 mM MgCl2 and gently shaken for 10 min in an ice bath. The suspension was centrifuged (7500 rpm for 20 min at 4 °C). After centrifugation, the soluble fraction was carefully transferred into a new tube and the pellet, containing the cytoplasmatic fraction, was suspended in 30 mL lysis buffer (50 mM Tris-HCl, pH 7.8, 500 mM NaCl, one tablet of EDTA-free Complete Ultra protease inhibitor (Roche, Mannheim, Germany). The cells were mechanically lysed by a French Press at 2 Kbar for two consecutive cycles. The obtained lysate was centrifuged (6’500 rpm for 1 h at 4° C) to separate the soluble and insoluble protein fractions, then the soluble fraction (Sol) was analyzed on SDS-PAGE.
4.11. Cold-Azurin recombinant protein production
For the production of the recombinant protein, azurin gene was PCR amplified and cloned into the commercial expression vector pET28b (+). The resulting construct was indicated as pET28b-Azu.The azurin gene was amplified by forward primer Azu-NdeI: 5’- CCCTGGATCCGAGATTCATATGTTTGCC -3’ and reverse primer Azu-BamHI: 5’- CGATGAAGGATCCCGCGGTCTTGAG -3’. The primers were designed based on Pseudomonas TAE6080 sequence (GenBank, under the accession number JAHIDY000000000) which encompasses the whole sequence of azurin gene and harbor the restriction sites of NdeI and BamHI enzymes, respectively. The PCR was performed with initial denaturation (98°C, 30 s), 25 cycles of denaturation (98°C, 10 s), annealing (56 °C, 30 s) and extension (72°C, 30 s), and final extension (72°C, 2 min). The products were electrophoresed on 1,5 % agarose gel, purified by High Pure PCR Product Purification kit (Roche, Mannheim, Germany) and sequenced.
The cloning of azurin gene in pET28b vector was performed by double digestion of purified PCR product by 20 U/µL BamHI-NdeI, (Thermo Fisher Scientific, New York, USA). The pET8b vector was also double digested with the same restriction enzymes. The digested fragments were purified using High Pure PCR Product Purification kit (Roche, Mannheim, Germany) and incubated at 16°C for overnight with T7 ligation enzyme (Thermo Fisher Scientific, New York, USA). The pET28b-Azu construct was transformed into the E. coli BL21DE3 competent cells, plated on Luria-Bertani (LB) agar (Sigma aldrich, Steinheim, Germany) in the presence of 50 µg/mL of kanamycin (Sigma aldrich, Steinheim, Germany) and incubated overnight at 37 °C.
The transformed E. coli BL21DE3 cells containing pET28b-Azu construct were subjected to protein expression and production. Temperature and induction condition for rCold-Azurin expression were determined and optimized.
Briefly, the transformed colonies were inoculated into 10 mL of LB broth without antibiotic at 28 °C, 180 rpm. After 1 h, 10 mL of inoculum were diluted in 100 mL of fresh LB medium containing 50 µg/mL of kanamycin and 5 µg/ml CuSO4 until reaching the optical density of 0.5 at 600 nm (about 2 h) at 28 °C, 180 rpm. The expression was induced by addition of isopropyl-1-thio-β-D-galactopyranoside (IPTG) 2 mM to the culture medium and incubated for 16-20 h. After the incubation, cells were harvested by centrifugation at 4000 rpm at 4°C.
The bacterial pellet was washed with 150 mM of ice-cold phosphate buffer at pH 7, resuspended in buffer A (30 mM Tris–HCl pH 8, 20% sucrose, 1 mM EDTA pH 8) and incubated at room temperature for 20 min. The shocked cells were collected by centrifugation at 8000 rpm at 4°C and resuspended in ice-cold 5 mM MgSO4. After incubation at 4°C for 20 min and centrifugation at 13,000 rpm, the supernatant (periplasmic fractions) containing the recombinant Azurin was collected and stored at -20°C until use. The protein content was determined by Bradford assay.
4.12. Confocal microscopy
The activity of purified rCold-Azurin against staphylococcal biofilms was evaluated by Confocal Laser Scanning Microscopy (CLSM). Biofilms were formed on NuncTM Lab-Tek® 8-well Chamber Slides (n◦17744; Thermo Scientific, Ottawa, ON, Canada). Briefly, the wells of the chamber slide were filled with 300 μL of S. epidermidis RP62A or S. epidermidis O-47 cultures in exponential growth phase diluted in BHI with a final concentration of approximately 0.1 and 0.001 OD600nm, respectively. The culture was incubated at 37 °C for 24 h in the absence (control) and in the presence of protein (0.25 µg µL-1) to assess its antibiofilm activity and its influence on cell viability. The biofilm cell viability was determined by the FilmTracer™ LIVE/DEAD® Biofilm Viability Kit (Molecular Probes, Invitrogen, Carlsbad, CA, USA), following the manufacturer’s instructions. After rinsing with filter-sterilized PBS, each well of the chamber slide was filled with 300 µL of working solution of fluorescent stains, containing SYTO®9 green-fluorescent nucleic acid stain (10 µM) and propidium iodide, the red-fluorescent nucleic acid stain (60 µM), and incubated for 20–30 min at room temperature, protected from light. All excess stain was removed by rinsing gently with filter-sterilized PBS. All microscopic observations and image acquisitions were performed with a confocal laser scanning microscope (LSM700-Zeiss, Jena, Germany) equipped with an Ar laser (488 nm) and a He-Ne laser (555 nm). Images were obtained using a 20x/0.8 objective. The excitation/emission maxima for these dyes are 480/500 nm for SYTO®9 and 490/635 nm for PI. Z-stacks were obtained by driving the microscope to a point just out of focus on both the top and bottom of the biofilms. Images were recorded as a series of tif files with a file-depth of 16 bits. The COMSTAT software package [22] was used to determine biomasses (μm3 μm−2), average thicknesses (µm), and roughness coefficient (Ra*). For each condition, two independent biofilm samples were used.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: Comparison of Pseudomonas sp TAE6080 and Pseudomonas aeruginosa PAO1 Azurina aminoacidic sequences, Figure S2: Protein profile and anti-adhesive activity of periplasmic fraction from recombinant E. Coli BL21DE3. Figure S3: Antibiofilm activity of poxA1b laccase.
Author Contributions
Conceptualization, D.C. and E.P.; methodology, D.A., A.C., R.P., M.T.; validation, D.C.; formal analysis, R.P., E.P; investigation, D.A., A.C.; resources, M.A., L.S..; data curation, E.P., ML.T,M.T.; writing—original draft preparation, E.P.; writing—review and editing, R.P.,D.C., ML.T.; visualization, D.C., R.P..; supervision, E.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Abbreviation
| SN TAE6080 | Pseudomonas sp. TAE6080 cell-free supernatant |
| G | Pseudomonas sp. TAE6080 culture medium |
| SNC | Retentate fraction obtained after the ultrafiltration of TAE6080 cell-free supernatant |
| Ub | Unbound fraction obtained from adsorption chromatography, |
| W | Fractions obtained from adsorption chromatography during the washing steps |
| E or P | Fractions obtained from adsorption chromatography elution step with methanol |
| OS | Pseudomonas sp. TAE6080 periplasmic extract by osmotic shock method |
| Sol | soluble fraction obtained after TAE6080 cell-pellet lysis |
References
- Dickey, S.W.; Cheung, G.Y.C.; Otto, M. Different Drugs for Bad Bugs: Antivirulence Strategies in the Age of Antibiotic Resistance. Nat. Rev. Drug Discov. 2017, 16, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Moriarty, T.F.; Kuehl, R.; Coenye, T.; Metsemakers, W.J.; Morgenstern, M.; Schwarz, E.M.; Riool, M.; Zaat, S.A.J.; Khana, N.; Kates, S.L.; et al. Orthopaedic Device-Related Infection: Current and Future Interventions for Improved Prevention and Treatment. EFORT Open Rev. 2016, 1, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Gilmore, B.F.; Carson, L. Bioactive Biomaterials for Controlling Biofilms; Woodhead Publishing Limited, 2015; ISBN 9780857097224. [Google Scholar] [CrossRef]
- Pietrocola, G.; Campoccia, D.; Motta, C.; Montanaro, L.; Arciola, C.R.; Speziale, P. Colonization and Infection of Indwelling Medical Devices by Staphylococcus Aureus with an Emphasis on Orthopedic Implants. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Kranjec, C.; Angeles, D.M.; Mårli, M.T.; Fernández, L.; García, P.; Kjos, M.; Diep, D.B. Staphylococcal Biofilms: Challenges and Novel Therapeutic Perspectives. Antibiotics 2021, 10, 1–30. [Google Scholar] [CrossRef]
- Calvo, T.D.; Tejera, N.; McNamara, I.; Langridge, G.C.; Wain, J.; Poolman, M.; Singh, D. Genome-Scale Metabolic Modelling Approach to Understand the Metabolism of the Opportunistic Human Pathogen Staphylococcus Epidermidis RP62A. Metabolites 2022, 12. [Google Scholar] [CrossRef]
- Oliveira, F.; Rohde, H.; Vilanova, M.; Cerca, N. Fighting Staphylococcus Epidermidis Biofilm-Associated Infections: Can Iron Be the Key to Success? Front. Cell. Infect. Microbiol. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Saffari, F.; Gurram, B. Molecular and Phenotypic Characterization of Multidrug-Resistant Clones of Staphylococcus Epidermidis in Iranian Hospitals: Clonal Relatedness to Healthcare-Associated Methicillin-... 2016. [Google Scholar] [CrossRef]
- Høiby, N.; Bjarnsholt, T.; Givskov, M.; Molin, S.; Ciofu, O. International Journal of Antimicrobial Agents Antibiotic Resistance of Bacterial Biofilms. 2010, 35, 322–332. [Google Scholar] [CrossRef]
- Araújo, G.R. d. S.; Viana, N.B.; Gómez, F.; Pontes, B.; Frases, S. The Mechanical Properties of Microbial Surfaces and Biofilms. Cell Surf. 2019, 5. [Google Scholar] [CrossRef]
- Adam, B.; Baillie, G.S.; Douglas, L.J. Mixed Species Biofilms of Candida Albicans and Staphylococcus Epidermidis. 2002, 51, 344–349. [Google Scholar] [CrossRef]
- Parrilli, E.; Luisa, M.; Gennaro, T. Biofilm as an Adaptation Strategy to Extreme Conditions. Rend. Lincei. Sci. Fis. e Nat. 2022. [Google Scholar] [CrossRef]
- Artini, M.; Papa, R.; Vrenna, G.; Lauro, C.; Ricciardelli, A.; Casillo, A.; Corsaro, M.M.; Tutino, M.L.; Parrilli, E.; Selan, L. Cold-Adapted Bacterial Extracts as a Source of Anti-Infective and Antimicrobial Compounds against Staphylococcus Aureus. Future Microbiol. 2019, 14, 1369–1382. [Google Scholar] [CrossRef] [PubMed]
- Papa, R.; Selan, L.; Parrilli, E.; Tilotta, M.; Sannino, F.; Feller, G.; Tutino, M.L.; Artini, M. Anti-Biofilm Activities from Marine Cold Adapted Bacteria against Staphylococci and Pseudomonas Aeruginosa. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef]
- Casillo, A.; Papa, R.; Ricciardelli, A.; Sannino, F.; Ziaco, M.; Tilotta, M.; Selan, L.; Marino, G.; Corsaro, M.M.; Tutino, M.L.; et al. Anti-Biofilm Activity of a Long-Chain Fatty Aldehyde from Antarctic Pseudoalteromonas Haloplanktis TAC125 against Staphylococcus Epidermidis Biofilm. Front. Cell. Infect. Microbiol. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Riccardi, C.; D’Angelo, C.; Calvanese, M.; Ricciardelli, A.; Tutino, M.L.; Parrilli, E.; Fondi, M. Genome Analysis of a New Biosurfactants Source: The Antarctic Bacterium Psychrobacter Sp. TAE2020. Mar. Genomics 2022, 61, 1–5. [Google Scholar] [CrossRef]
- D’Angelo, C.; Casillo, A.; Melchiorre, C.; Lauro, C.; Corsaro, M.M.; Carpentieri, A.; Tutino, M.L.; Parrilli, E. CATASAN Is a New Anti-Biofilm Agent Produced by the Marine Antarctic Bacterium Psychrobacter Sp. TAE2020. Mar. Drugs 2022, 20, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Papa, R.; Vrenna, G.; D’angelo, C.; Casillo, A.; Relucenti, M.; Donfrancesco, O.; Corsaro, M.M.; Fiscarelli, E.V.; Assanti, V.T.G.; Tutino, M.L.; et al. Anti-Virulence Activity of the Cell-Free Supernatant of the Antarctic Bacterium Psychrobacter Sp. Tae2020 against Pseudomonas Aeruginosa Clinical Isolates from Cystic Fibrosis Patients. Antibiotics 2021. [Google Scholar] [CrossRef]
- Riccardi, C.; D’Angelo, C.; Calvanese, M.; Ricciardelli, A.; Sellitto, A.; Giurato, G.; Tutino, M.L.; Weisz, A.; Parrilli, E.; Fondi, M. Whole-Genome Sequencing of Pseudomonas Sp. TAE6080, a Strain Capable of Inhibiting Staphylococcus Epidermidis Biofilm. Mar. Genomics 2021. [Google Scholar] [CrossRef] [PubMed]
- Mondino, S.; San Martin, F.; Buschiazzo, A. 3D Cryo-EM Imaging of Bacterial Flagella: Novel Structural and Mechanistic Insights into Cell Motility. J. Biol. Chem. 2022, 298, 102105. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Seib, K.L.; Edwards, J.L.; Apicella, M.A.; McEwan, A.G.; Jennings, M.P. Azurin of Pathogenic Neisseria Spp. Is Involved in Defense against Hydrogen Peroxide and Survival within Cervical Epithelial Cells. Infect. Immun. 2005, 73, 8444–8448. [Google Scholar] [CrossRef]
- Heydorn, A.; Nielsen, A.T.; Hentzer, M.; Sternberg, C.; Givskov, M.; Ersboll, B.K.; Molin, S. Quantification of Biofilm Structures by the Novel Computer Program COMSTAT. Microbiology 2000, 146, 2395–2407. [Google Scholar] [CrossRef]
- van de KAMP, M.; SILVESTRINI, M.C.; BRUNORI, M.; Van BEEUMEN, J.; HALI, F.C.; CANTERS, G.W. Involvement of the Hydrophobic Patch of Azurin in the Electron-transfer Reactions with Cytochrome C551 and Nitrite Reductase. Eur. J. Biochem. 1990, 194, 109–118. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, Y.; Breslawec, A.P.; Liang, T.; Deng, Z.; Kuperman, L.L.; Yu, Q. Strategy to Combat Biofilms: A Focus on Biofilm Dispersal Enzymes. npj Biofilms Microbiomes 2023, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Artini, M.; Cicatiello, P.; Ricciardelli, A.; Papa, R.; Selan, L.; Dardano, P.; Tilotta, M.; Vrenna, G.; Tutino, M.L.; Giardina, P.; et al. Hydrophobin Coating Prevents Staphylococcus Epidermidis Biofilm Formation on Different Surfaces. Biofouling 2017, 33, 601–611. [Google Scholar] [CrossRef] [PubMed]
- Savelieff, M.G.; Lu, Y. CuA Centers and Their Biosynthetic Models in Azurin. J. Biol. Inorg. Chem. 2010, 15, 461–483. [Google Scholar] [CrossRef] [PubMed]
- Pinho, D.; Besson, S.; Brondino, C.D.; Pereira, E.; De Castro, B.; Moura, I. Two Azurins with Unusual Redox and Spectroscopic Properties Isolated from the Pseudomonas Chlororaphis Strains DSM 50083T and DSM 50135. J. Inorg. Biochem. 2004, 98, 276–286. [Google Scholar] [CrossRef]
- Vijgenboom, E.; Busch, J.E.; Canters, G.W. In Vivo Studies Disprove an Obligatory Role of Azurin in Denitrification in Pseudomonas Aeruginosa and Show That Azu Expression Is under Control of RpoS and ANR. Microbiology 1997, 143, 2853–2863. [Google Scholar] [CrossRef]
- Li, H.; Webb, S.P.; Ivanic, J.; Jensen, J.H. Determinants of the Relative Reduction Potentials of Type-1 Copper Sites in Proteins. J. Am. Chem. Soc. 2004, 126, 8010–8019. [Google Scholar] [CrossRef]
- Palmieri, G.; Giardina, P.; Bianco, C.; Scaloni, A.; Capasso, A.; Sannia, G. A Novel White Laccase from Pleurotus Ostreatus. J. Biol. Chem. 1997, 272, 31301–31307. [Google Scholar] [CrossRef]
- Fialho, A.M.; Stevens, F.J.; Das Gupta, T.K.; Chakrabarty, A.M. Beyond Host-Pathogen Interactions: Microbial Defense Strategy in the Host Environment. Curr. Opin. Biotechnol. 2007, 18, 279–286. [Google Scholar] [CrossRef]
- Pang, X.; Yuan, C.; Sun, R.; Wang, K.; Tang, B. Revealing the Underestimated Anticancer Effect of Azurin by Mechanical Unfolding. ACS Biomater. Sci. Eng. 2021, 7, 4809–4818. [Google Scholar] [CrossRef]
- Bakhshi, B.; Barzelighi, H.M.; Daraei, B. The Anti-Adhesive and Anti-Invasive Effects of Recombinant Azurin on the Interaction between Enteric Pathogens (Invasive/Non-Invasive) and Caco-2 Cells. Microb. Pathog. 2020, 147, 104246. [Google Scholar] [CrossRef]
- Chaudhari, A.; Fialho, A.M.; Ratner, D.; Gupta, P.; Hong, C.S.; Kahali, S.; Yamada, T.; Haldar, K.; Murphy, S.; Cho, W.; et al. Azurin, Plasmodium Falciparum Malaria and HIV/AIDS: Inhibition of Parasitic and Viral Growth by Azurin. Cell Cycle 2006, 5, 1642–1648. [Google Scholar] [CrossRef]
- Chaudhari, A.; Mahfouz, M.; Fialho, A.M.; Yamada, T.; Granja, A.T.; Zhu, Y.; Hashimoto, W.; Schlarb-Ridley, B.; Cho, W.; Das Gupta, T.K.; et al. Cupredoxin-Cancer Interrelationship: Azurin Binding with EphB2, Interference in EphB2 Tyrosine Phosphorylation, and Inhibition of Cancer Growth. Biochemistry 2007, 46, 1799–1810. [Google Scholar] [CrossRef]
- Hu, J.; Jiang, W.; Zuo, J.; Shi, D.; Chen, X.; Yang, X.; Zhang, W.; Ma, L.; Liu, Z.; Xing, Q. Structural Basis of Bacterial Effector Protein Azurin Targeting Tumor Suppressor P53 and Inhibiting Its Ubiquitination. Commun. Biol. 2023, 6, 1–10. [Google Scholar] [CrossRef]
- Stevens, F.J.; Kuemmel, C.; Babnigg, G.; Collart, F.R. Efficient Recognition of Protein Fold at Low Sequence Identity by Conservative Application of Psi-BLAST: Application. J. Mol. Recognit. 2005, 18, 150–157. [Google Scholar] [CrossRef]
- Foster, T.J. Surface Proteins of Staphylococcus Epidermidis. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Peña, S.; Martínez-García, S.; Rodríguez-Martínez, S.; Cancino-Diaz, M.E.; Cancino-Diaz, J.C. Overview of Staphylococcus Epidermidis Cell Wall-Anchored Proteins: Potential Targets to Inhibit Biofilm Formation. Mol. Biol. Rep. 2020, 47, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Büttner, H.; Mack, D.; Rohde, H. Structural Basis of Staphylococcus Epidermidis Biofilm Formation: Mechanisms and Molecular Interactions. Front. Cell. Infect. Microbiol. 2015, 5, 1–15. [Google Scholar] [CrossRef]
- Yarawsky, A.E.; Johns, S.L.; Schuck, P.; Herr, A.B. The Biofilm Adhesion Protein Aap from Staphylococcus Epidermidis Forms Zinc-Dependent Amyloid Fibers. J. Biol. Chem. 2020, 295, 4411–4427. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jiang, J.; Gao, Y.; Sun, Y.; Dai, J.; Wu, Y.; Qu, D.; Ma, G.; Fang, X. Staphylococcus Epidermidis Small Basic Protein (Sbp) Forms Amyloid Fibrils, Consistent with Its Function as a Scaffolding Protein in Biofilms. J. Biol. Chem. 2018, 293, 14296–14311. [Google Scholar] [CrossRef]
- Wang, B.; Zhan, Q.; Xiao, Y.; Xu, Y.; Zhao, H.; Rao, L.; Wang, X.; Zhang, J.; Shen, L.; Zhou, Y.; et al. Mupirocin Enhances the Biofilm Formation of Staphylococcus Epidermidis in an AtlE-Dependent Manner. Int. J. Antimicrob. Agents 2023, 62, 106904. [Google Scholar] [CrossRef]
- Ortega-Peña, S.; Martínez-García, S.; Rodríguez-Martínez, S.; Cancino-Diaz, M.E.; Cancino-Diaz, J.C. Overview of Staphylococcus Epidermidis Cell Wall-Anchored Proteins: Potential Targets to Inhibit Biofilm Formation. Mol. Biol. Rep. 2020, 47, 771–784. [Google Scholar] [CrossRef]
- Bhardwaj, R.; Mishra, P. Engineered Recombinant EGFP-Azurin Theranostic Nanosystem for Targeted Therapy of Prostate Cancer. Mol. Pharm. 2023. [Google Scholar] [CrossRef] [PubMed]
- Al-Hazmi, N.E.; Naguib, D.M. Microbial Azurin Immobilized on Nano-Chitosan as Anticancer and Antibacterial Agent Against Gastrointestinal Cancers and Related Bacteria. J. Gastrointest. Cancer 2022, 53, 537–542. [Google Scholar] [CrossRef] [PubMed]
- Quinteros, M.A.; Bonilla, J.O.; Alborés, S. V.; Villegas, L.B.; Páez, P.L. Biogenic Nanoparticles: Synthesis, Stability and Biocompatibility Mediated by Proteins of Pseudomonas Aeruginosa. Colloids Surfaces B Biointerfaces 2019, 184, 110517. [Google Scholar] [CrossRef] [PubMed]
- Punj, V.; Das Gupta, T.K.; Chakrabarty, A.M. Bacterial Cupredoxin Azurin and Its Interactions with the Tumor Suppressor Protein P53. Biochem. Biophys. Res. Commun. 2003, 312, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Raue, S.; Fan, S.H.; Rosenstein, R.; Zabel, S.; Luqman, A.; Nieselt, K.; Götz, F. The Genome of Staphylococcus Epidermidis O47. Front. Microbiol. 2020, 11, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Gowrishankar, S.; Pandian, S.K. Modulation of Staphylococcus Epidermidis (RP62A) Extracellular Polymeric Layer by Marine Cyclic Dipeptide-Cyclo(L-Leucyl-L-Prolyl) Thwarts Biofilm Formation. Biochim. Biophys. Acta - Biomembr. 2017, 1859, 1254–1262. [Google Scholar] [CrossRef] [PubMed]
- Ricciardelli, A.; Casillo, A.; Papa, R.; Monti, D.M.; Imbimbo, P.; Vrenna, G.; Artini, M.; Selan, L.; Corsaro, M.M.; Tutino, M.L.; et al. Pentadecanal Inspired Molecules as New Anti-Biofilm Agents against Staphylococcus Epidermidis. Biofouling 2018, 34, 1110–1120. [Google Scholar] [CrossRef]
Figure 1.
(A) Biofilm formation by S. epidermidis O-47 and S. epidermidis RP62A in polystyrene 24-wells microtiter plate wells coated with G medium (Control) and SN TAE6080 obtained after 72 h of growth. (B) Effect of SN TAE6080 on S. epidermidis biofilm formation. Each data point represents the mean ± SD of six independent samples. The results are expressed as the percentage of biofilm formed in the presence of SN TAE6080 compared to untreated bacteria (100%). Biofilm formation was considered unaffected in the range of 90–100% (C) Biofilm formation by S. epidermidis O-47 and S. epidermidis RP62A in polystyrene 24-wells microtiter plate wells coated with SN TAE6080 treated with proteinase K or SN TAE6080 incubated at 37 °C without proteinase K (D) Effect of SN TAE6080 treated with proteinase K on biofilm formation. Each data point represents the mean ± SD of five independent samples. The results are expressed as the percentage of biofilm formed in the presence of SN TAE6080 compared to untreated bacteria (100%). Biofilm formation was considered unaffected in the range of 90–100%.
Figure 1.
(A) Biofilm formation by S. epidermidis O-47 and S. epidermidis RP62A in polystyrene 24-wells microtiter plate wells coated with G medium (Control) and SN TAE6080 obtained after 72 h of growth. (B) Effect of SN TAE6080 on S. epidermidis biofilm formation. Each data point represents the mean ± SD of six independent samples. The results are expressed as the percentage of biofilm formed in the presence of SN TAE6080 compared to untreated bacteria (100%). Biofilm formation was considered unaffected in the range of 90–100% (C) Biofilm formation by S. epidermidis O-47 and S. epidermidis RP62A in polystyrene 24-wells microtiter plate wells coated with SN TAE6080 treated with proteinase K or SN TAE6080 incubated at 37 °C without proteinase K (D) Effect of SN TAE6080 treated with proteinase K on biofilm formation. Each data point represents the mean ± SD of five independent samples. The results are expressed as the percentage of biofilm formed in the presence of SN TAE6080 compared to untreated bacteria (100%). Biofilm formation was considered unaffected in the range of 90–100%.
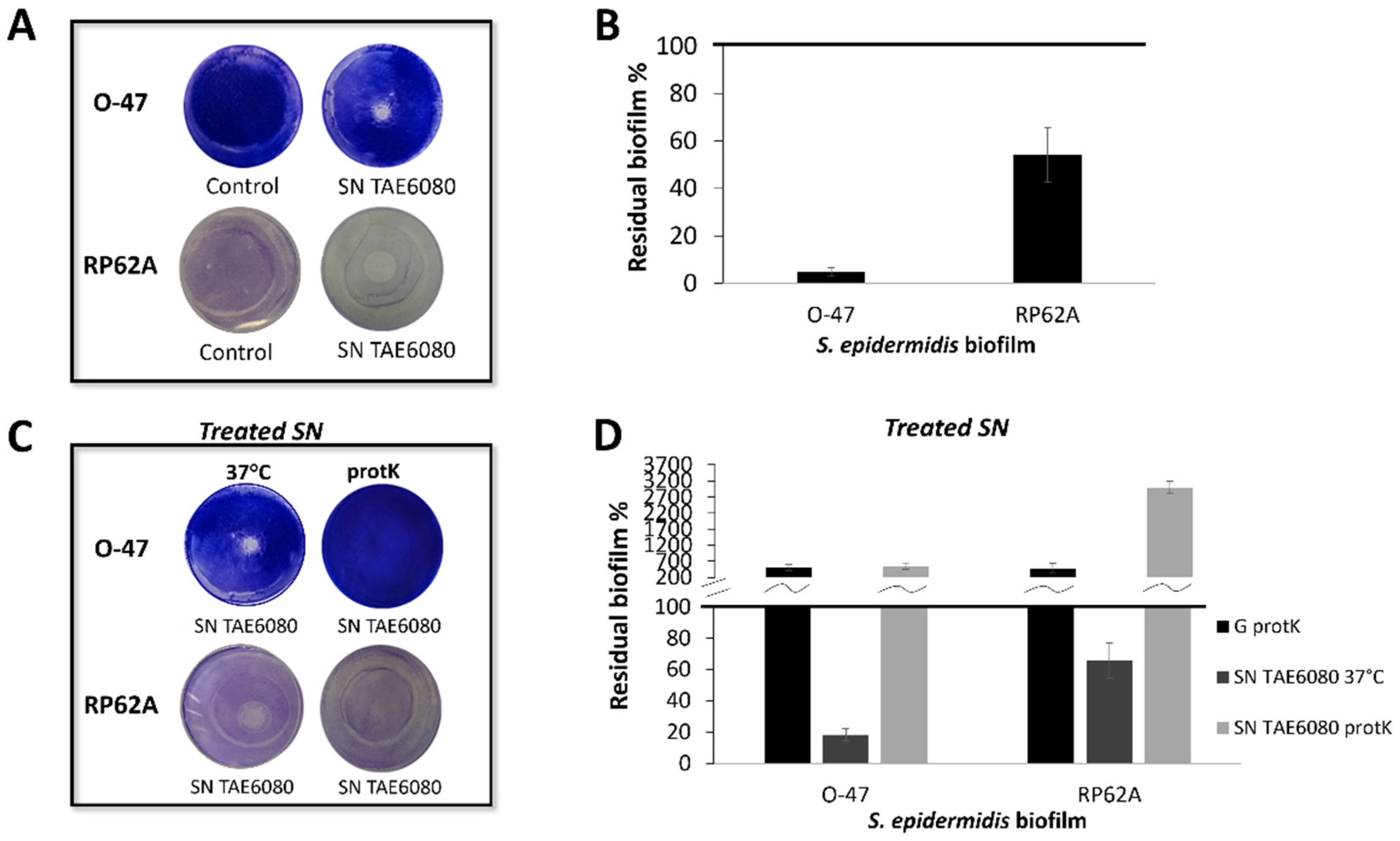
Figure 2.
(A) Biofilm formation by S. epidermidis O-47 in polystyrene 24-wells microtiter plate wells coated with G medium (NT) and fractions obtained from adsorption chromatography: Ub: unbound fraction, W: fraction obtained during the washing step with G medium, E: fraction eluted with methanol.(B) SDS-PAGE 15% stained with Comassie blue, protein profile of the chromatography fractions, SNC: load sample on chromatography, Ub: unbound fraction, W: fraction obtained during the washing step, E: fraction eluted with methanol, M: molecular weight marker.(C) Flagellin protein sequence coverage obtained by LCMSMS analysis (D) Azurin protein sequence coverage obtained by LCMSMS analysis.
Figure 2.
(A) Biofilm formation by S. epidermidis O-47 in polystyrene 24-wells microtiter plate wells coated with G medium (NT) and fractions obtained from adsorption chromatography: Ub: unbound fraction, W: fraction obtained during the washing step with G medium, E: fraction eluted with methanol.(B) SDS-PAGE 15% stained with Comassie blue, protein profile of the chromatography fractions, SNC: load sample on chromatography, Ub: unbound fraction, W: fraction obtained during the washing step, E: fraction eluted with methanol, M: molecular weight marker.(C) Flagellin protein sequence coverage obtained by LCMSMS analysis (D) Azurin protein sequence coverage obtained by LCMSMS analysis.
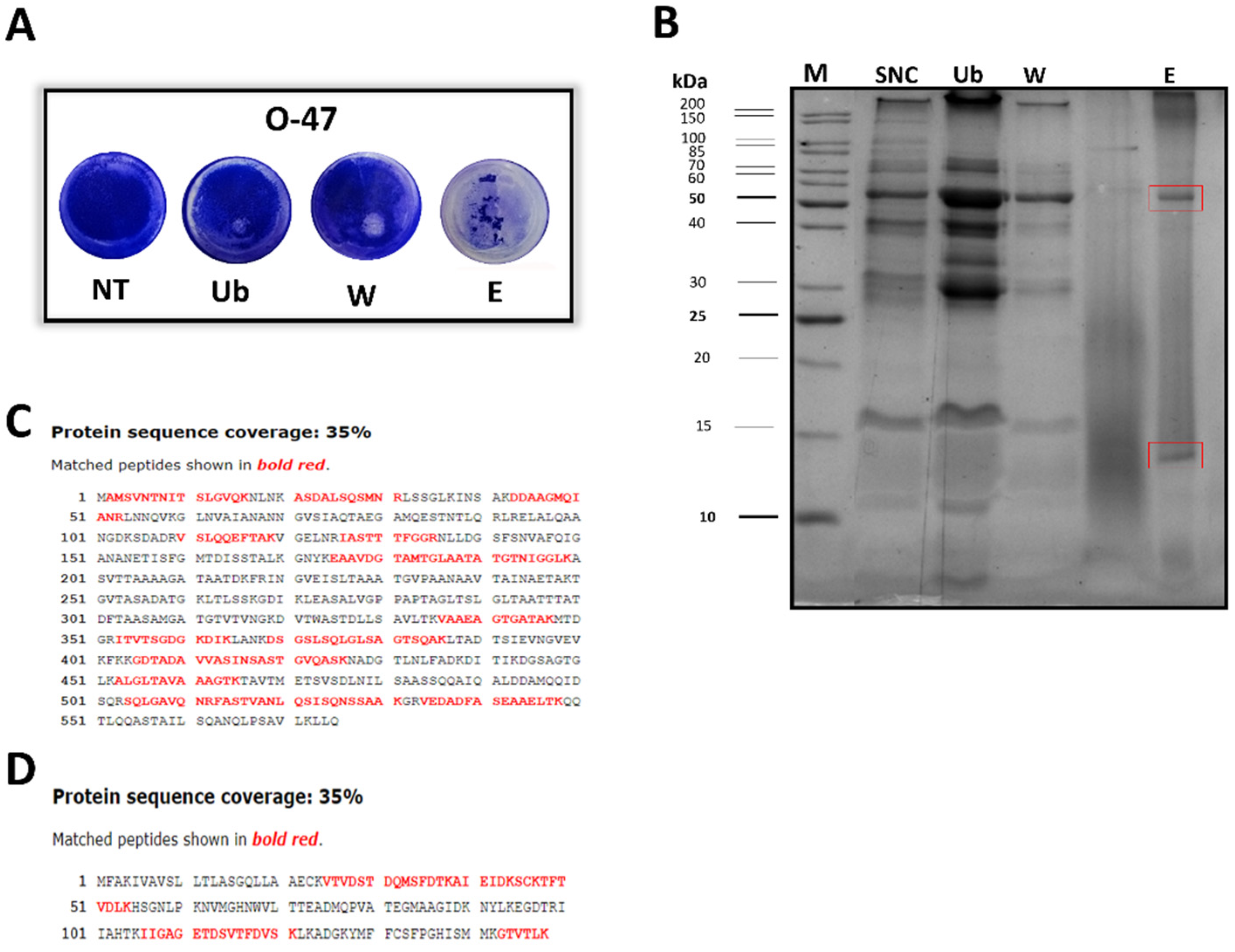
Figure 3.
(A) SDS-PAGE 15% stained with silver staining , protein profile of cell pellet before osmotic shock (tot), soluble post lysis (Sol) and TAE6080 periplasmic fraction (OS), M: molecular weight marker. (B) Biofilm formation by S. epidermidis O-47 in polystyrene 24-wells microtiter plate wells coated with OS buffer (NT) or TAE6080 periplasmic fraction (OS). (C) Effect of TAE6080 periplasmic fraction (OS) on S. epidermidis biofilm formation. Each data point represents the mean ± SD of four independent samples. The results are expressed as the percentage of biofilm formed in the presence of TAE6080 periplasmic fraction compared to untreated bacteria (100%). Biofilm formation was considered unaffected in the range of 90–100%.
Figure 3.
(A) SDS-PAGE 15% stained with silver staining , protein profile of cell pellet before osmotic shock (tot), soluble post lysis (Sol) and TAE6080 periplasmic fraction (OS), M: molecular weight marker. (B) Biofilm formation by S. epidermidis O-47 in polystyrene 24-wells microtiter plate wells coated with OS buffer (NT) or TAE6080 periplasmic fraction (OS). (C) Effect of TAE6080 periplasmic fraction (OS) on S. epidermidis biofilm formation. Each data point represents the mean ± SD of four independent samples. The results are expressed as the percentage of biofilm formed in the presence of TAE6080 periplasmic fraction compared to untreated bacteria (100%). Biofilm formation was considered unaffected in the range of 90–100%.
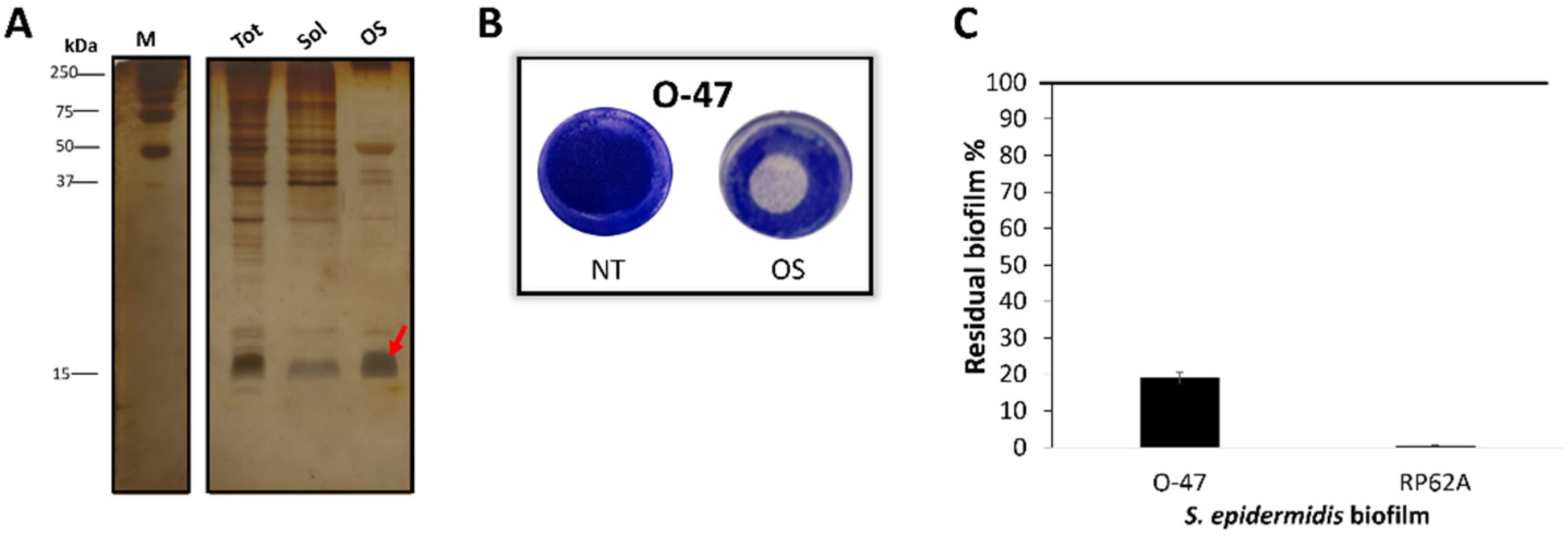
Figure 4.
Figure 4. (A) SDS-PAGE 15% stained with Comassie blue, protein profile of the chromatography fractions, M: molecular weight marker, Load: periplasmic extract fraction from recombinant induced BL21DE3 cells, Ub: unbound fraction, W: fractions obtained during the washing step with MgSO4 (5mM), P: eluted fraction with methanol. (B) Effect of r-Cold-Azurin (0.25µg/µL) eluted from adsorption chromatography on S. epidermidis biofilm formation. Each data point represents the mean ± SD of four independent samples. The results are expressed as the percentage of biofilm formed in the presence of r-Cold-Azurin compared to untreated bacteria (100%). Biofilm formation was considered unaffected in the range of 90–100%. (C) Biofilm formation by S. epidermidis O-47 or RP62A in polystyrene 24-wells microtiter plate wells coated with PBS (NT) or rCold-Azurin.
Figure 4.
Figure 4. (A) SDS-PAGE 15% stained with Comassie blue, protein profile of the chromatography fractions, M: molecular weight marker, Load: periplasmic extract fraction from recombinant induced BL21DE3 cells, Ub: unbound fraction, W: fractions obtained during the washing step with MgSO4 (5mM), P: eluted fraction with methanol. (B) Effect of r-Cold-Azurin (0.25µg/µL) eluted from adsorption chromatography on S. epidermidis biofilm formation. Each data point represents the mean ± SD of four independent samples. The results are expressed as the percentage of biofilm formed in the presence of r-Cold-Azurin compared to untreated bacteria (100%). Biofilm formation was considered unaffected in the range of 90–100%. (C) Biofilm formation by S. epidermidis O-47 or RP62A in polystyrene 24-wells microtiter plate wells coated with PBS (NT) or rCold-Azurin.
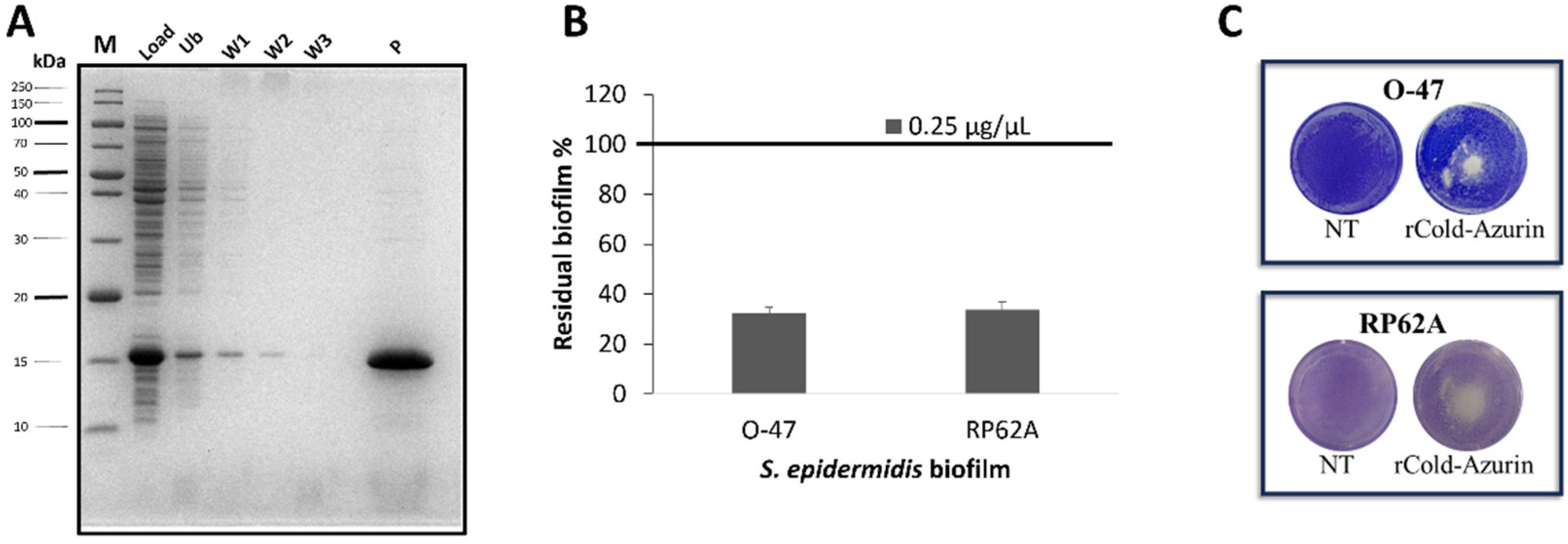
Figure 5.
Figure 5. (A) CLSM analysis of S. epidermidis biofilms formed in the absence (non-treated) and presence (treated) of rCold-Azurin (0.25µg/µL) (B) COMSTAT quantitative analysis of Biomass (µm3/µm2), Roughness Coefficient (Ra*) and Average thickness (µm) of untreated (NT) or treated (rCold-Azurin) S. epidermidis biofilms.
Figure 5.
Figure 5. (A) CLSM analysis of S. epidermidis biofilms formed in the absence (non-treated) and presence (treated) of rCold-Azurin (0.25µg/µL) (B) COMSTAT quantitative analysis of Biomass (µm3/µm2), Roughness Coefficient (Ra*) and Average thickness (µm) of untreated (NT) or treated (rCold-Azurin) S. epidermidis biofilms.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



