
Submitted:
17 January 2024
Posted:
18 January 2024
You are already at the latest version
Abstract
Acquired immunodeficiency syndrome (AIDS) is an enormous global health threat stemming from human immunodeficiency virus (HIV-1) infection. Up to now, the tremendous advances in combination antiretroviral therapy (cART) have shifted HIV-1 infection from a fatal illness into a manageable chronic disorder. However, the presence of latent reservoirs, multifaceted nature of HIV-1, drug resistance, severe off-target effects, poor adherence, and high cost restrict the efficacy of current cART targeting the distinct stages of the virus life cycle. Therefore, there is an unmet need for discovery of new therapeutics that not only bypass the limitations of the current therapy but also protect the body health at the same time. The main goal for complete HIV-1 eradication is purging latently infected cells from the patients’ bodies. A potential strategy called “lock-in apoptosis” targeting the budding phase of life cycle of virus leading to susceptibility to apoptosis of HIV-1 infected cells for elimination of HIV-1 reservoirs and ultimately for a complete eradication. The current work intends to present the main advantages and disadvantages of United States Food and Drug Administration (FDA) approved anti-HIV-1 drugs as well as plausible strategies for the design and development of more anti-HIV-1 compounds with better potency, favorable pharmacokinetic profiles, and improved safety issues.
Keywords:
HIV-1
; AIDS
; FDA
; Combination antiretroviral therapy
; latent reservoirs
; apoptosis
; drug resistance
; anti-HIV-1 drug design and discovery
1. Introduction
Human immunodeficiency virus (HIV), an enveloped virus with a diploid, positive-sense and single-stranded RNA, belongs to the Lentivirus genus within the Orthoretrovirinae subfamily of Retroviridae family. HIV-1 and HIV-2 are two main types of HIV and HIV-1 is responsible for the majority of infections across the globe [1,2,3,4]. Acquired immunodeficiency syndrome (AIDS), stemming from mainly HIV-1, is one of the most serious hurdles throughout the world. The last updated World Health Organization (WHO) report indicated that globally about 39.0 million people acquired HIV-1 and 630.000 people died from HIV-1-dependent problems at the end of 2022 [5]. Besides, HIV-1/AIDS provokes opportunistic infections including tuberculosis, cryptococcal infection, histoplasmosis, and severe bacterial infections; hepatitis B, and C co-infections [6,7,8]; and comorbidities such as cardiovascular, kidney, and liver disorders and cancer [9]. In the near future, COVID-19 and monkeypox virus infections could be possible challenges for clinicians and people with HIV-1 [10,11].
The typical flu symptoms may appear at 2-6 weeks after acute HIV-1 infection, then it can remain silent for years without any symptoms and testing is the only solution to diagnose HIV-1. The “eclipse period” or “window period” is known as the time length between exposure and confidential identification of infection. It is hard to predict this period due to obscurity of the exact time of the exposure, albeit to, recent HIV-1 testing technologies applied in clinic are able to identify some parameters, including viral DNA, capsid (CA) protein, HIV-1 antibodies and antibody/antigen in combination [12,13,14].
HIV-1 is capable of converting its RNA into DNA through reverse transcriptase or RNA-dependent DNA polymerase enzyme and subsequent integrating its viral DNA into the host cell DNA through integrase enzyme. HIV-1 incorporates nine genes: structural (gag, pol, and env), regulatory (tat, rev), and accessory (vif, vpr, vpu, nef) genes. The group specific antigen (gag) gene encodes for Gag polyprotein precursor (Pr55Gag: Gag), whereas the Gag-Pol polyprotein precursor is expressed via ribosomal frameshifting between the gag and pol genes. Finally, the env gene encodes for the viral envelope glycoproteins. All these proteins orchestrate the viral cell cycle of HIV-1 leading to redundancy in infected host cells, mainly CD4+ T cell lymphocytes [15,16,17,18,19,20,21].
The stages of HIV-1 cell cycle are: (i) attachment, fusion and entry: The attachment and fusion between HIV-1 envelope and the cell surface of CD4+ cell occur through viral glycoproteins 120 and 41 (gp120 and gp41), the subunits of Env glycoprotein, and the host chemokine receptors (CCR5 or CXCR4) located on the surface of CD4+ cell, respectively. Then, HIV-1 enters into the CD4+ cell; (ii) revers transcription of viral RNA: the viral RNA is transcribed to viral DNA by reverse transcriptase enzyme, located within the CA, which occludes exposure of the viral genome to host proteins. This process is known to start in the cytoplasm. Recent studies [22] have reported that cDNA synthesis is ended inside the nucleus (iii) integration: the HIV-1 DNA is integrated into cellular DNA by integrase enzyme for its further replication, transcription, and translation into viral proteins; (iv) assembly, budding, release and maturation: 1) Gag proteins move to the plasma membrane and form hexameric subunits and assemble into immature lattice, 2) RNA dimers are recruited to assembly region and Env trimers are included in the budding particles, 3) membrane sequestration and release of virus particle from cell surface driven by the endosomal sorting complexes required for transport (ESCRT) and the Gag-p6 domain, 4) subsequent viral protease-mediated cleavage of Gag and Gag-Pol polyprotein to constitute mature structural and viral proteins, self-assembly and generation of mature CA protein and final formation of mature particle, which is able to infect other immune cells (Figure 1) [23,24,25,26,27,28].
After the discovery of the first anti-HIV-1 drug zidovudine in 1987, numerous agents acting as inhibitors of reverse transcriptase, integrase, entry (attachment, fusion, post-attachment), protease, and CA or pharmacokinetic enhancers have rendered the impact of HIV-1/AIDS from devastating fatal disorder into a manageable chronic infection [29,30,31,32]. The combination use of these agents entitled as combination antiretroviral therapy (cART, ART, formerly highly active antiretroviral therapy (HAART), cocktail therapy) marked a watershed moment to ameliorate the prognosis of the illness [33,34,35]. The three-drug regimens with dual nucleoside analogue reverse transcriptase inhibitors (NRTIs) backbone, and a core drug from integrase strand transfer inhibitors (INSTIs), boosted protease inhibitors, attachment inhibitor, fusion inhibitor or non-nucleoside reverse transcriptase inhibitor (NNRTI) has become the mainstay for the initial HIV-1 treatment [36,37,38,39]. Besides, the once-daily fixed-dose three drug combination in a single tablet regimen is one of the milestones for HIV-1/AIDS treatment facilitating reduced pill burden and dosing frequency, and improved adherence [40,41].
The major obstacles of the current therapy are persistent latent reservoirs, adherence challenges, drug resistance, and limited treatment options for multi-class resistance, drug-drug interactions, adverse effects such as cardiovascular events, insulin resistance and type II diabetes, renal dysfunction, hepatotoxicity, lipodystrophy, gastrointestinal toxicities (nausea, vomiting and diarrhea), rash, chronic pain, and central nervous system (CNS) toxicities, limited availability of drug in poor countries, a high-cost of the lifetime treatment regimen [42,43,44,45,46]. There are also other factors influencing the HIV-1 treatment in particular continued viral transmission among people who cannot reach testing or treatment due to some barriers such as stigma, discrimination, lack of confidentiality or gender-based disadvantages [13,47,48]. The mother-to-child transmission during pregnancy and breastfeeding is also another point that caution is required for implementation of cART to enhance safety of mothers with HIV-1 infection and their exposed fetuses and children [49].
The current treatment is lifelong, which paves the way for HIV-1 drug resistance and poor medication compliance. Therefore, to develop new inhibitors with novel mechanism of action is crucial. This review focuses on evaluation of pearls and pitfalls, and updates in the use of approved anti-HIV-1 drugs and bona fide approaches for HIV-1 eradication.
2. United States Food and Drug Administration (FDA) Approved Drugs for HIV-1 Treatment
2.1. Reverse Transcriptase Inhibitors
Reverse transcriptase enzyme acts two distinct activities as DNA polymerase, which catalyses the synthesis of a double-stranded proviral DNA from a single-stranded viral RNA and as endonuclease, which exerts RNase H activity hydrolysing the RNA strand in an RNA/DNA hybrid. HIV-1 reverse transcriptase enzyme incorporates, 66 kDa (p66) and 51 kDa (p51) subunits. The p66 subunit takes after the closed right hand with palm, fingers, thumb and connection subdomains and contains the polymerase and the RNase H active sites [50,51,52,53].
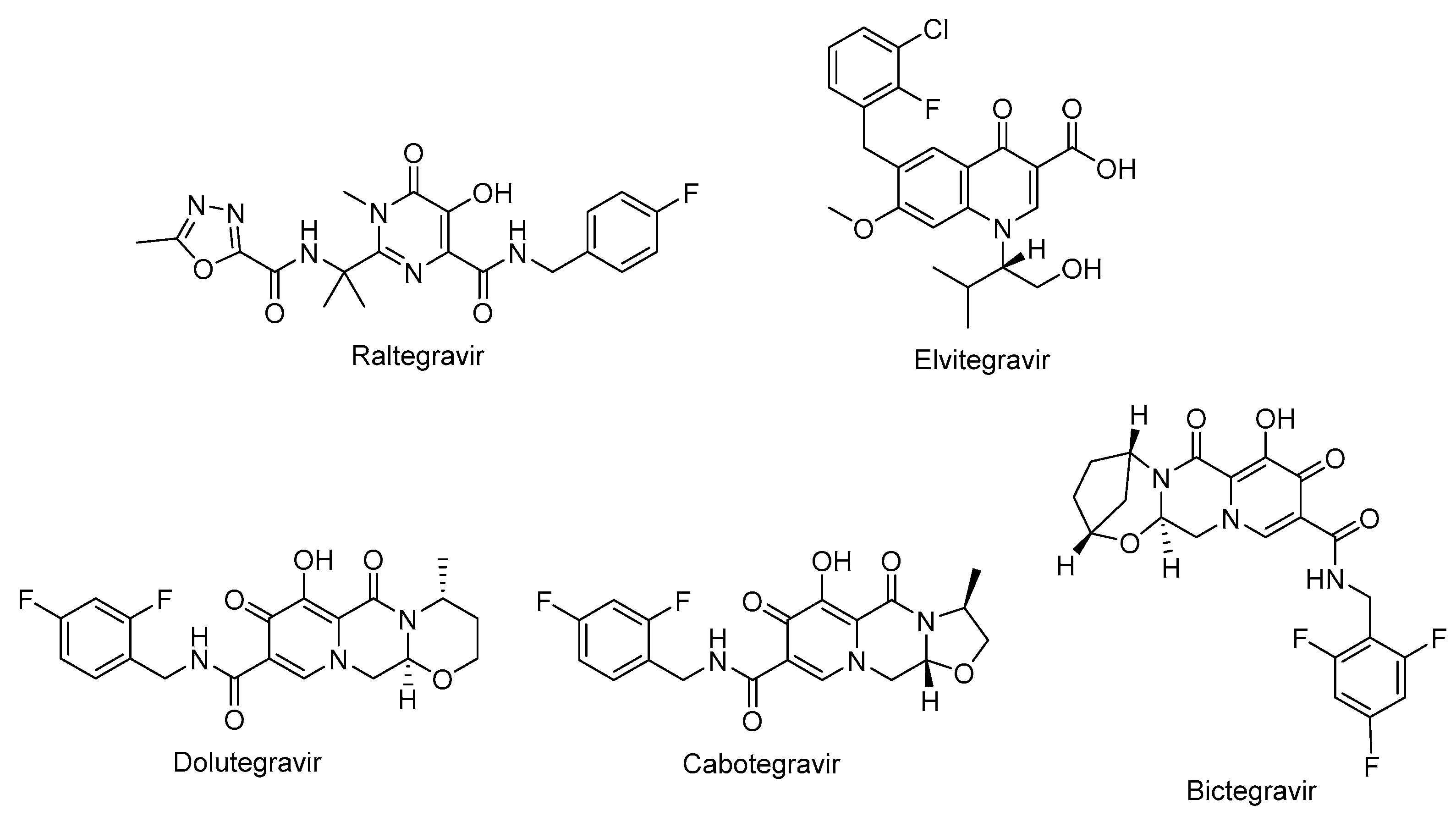
The non-nucleoside reverse transcriptase inhibitor (NNRTI) site is linked with the palm subdomain to a great extent and is far from the polymerase active site, thus they serve as non-competitive inhibitors on the contrary to nucleoside reverse transcriptase inhibitors (NRTIs), which compete at the active site. Therefore, non-nucleoside reverse transcriptase inhibitors (NNRTIs) constitute compounds possessing diverse chemical structures, whereas NRTIs are pyrimidine/purine (nucleoside) or nucleotide analogues. The first-generation NNRTIs, nevirapine (Viramune®) (Figure 2) and delavirdine (Rescriptor®) (Figure 2)) are prone to be affected by single point mutations. Delavirdine was discontinued due to severe adverse effects and nevirapine, a derivative of dipyridodiazepinone, is not recommended anymore due to side effects such as hepatotoxicity and rash though it is a potent and easy to take antiretroviral drug [54]. On the other hand, efavirenz (Sustiva®) (Figure 2) is found more stronger to the drug resistance mutations but side effects such as neuropsychiatric disorders and poor live function were manifested. Efavirenz was one of the first options appropriate for once-daily use [55,56]. Etravirine (Intelence®) (Figure 2), rilpivirine (Edurant®) (Figure 2), diarylpyrimidine derivatives, and doravirine (Pifeltro®) (Figure 2) are second-generation inhibitors. Etravirine is an alternative option for HIV-1-infected people with first-generation NNRTI resistance but its bitter taste and twice-daily dosing impede its clinical use [57]. Rilpivirine was demonstrated to be potent with less off-target effects in HIV-1 infected patients compared to efavirenz [58].
Rilpivirine containing regimens are expedient in various terms such as single tablet formulation with tenofovir and emtricitabine (NRTIs). Rilpivirine is a component of co-formulations (Complera®, Odefsey®) with emtricitabine (Figure 2) along with tenofovir diproxil fumarate (Figure 2) or tenovofir alafenamide (Figure 2) [59,60]. Since the bioavailability of rilpivirine is boosted under acidic conditions, it must not be administered with proton pump inhibitors concomitantly. Cytochrome p450 family 3 subfamily A (CYP3A) enzyme inducers such as anticonvulsants (carbamazepine, phenobarbital and phenytoin) and antimycobacterial agents (rifampicin) may cause the lower anti-HIV-1 activity [58]. Doravirine, a pyridone derivative, exhibited a broad spectrum of antiviral activity towards clinically relevant mutant viruses. It was also found beneficial with improved pharmacokinetic parameters for alternative use in patients with the advantages of less propensity for resistance and toxicity. Doravirine is on the market as a fixed dose combination tablet with lamivudine and tenofovir disoproxil fumarate (Delstrigo®). This combination requires attention for patients with HIV-1-hepatitis B co-infection [61,62,63,64].
HIV-1 NRTIs mimic and compete with natural deoxynucleotide triphosphates (such as dTTP, dCTP, dGTP and dATP) for incorporation at the polymerase active site. Most approved NRTIs with a missing hydroxy group at the 3′ position are incorporated into proviral DNA as chain terminators by reverse transcriptase since they are substrates for reverse transcriptase, which convert them to the corresponding 5′-triphosphates. Nucleoside analogues of NRTIs are phosphorylated to achieve their active di- or tri-phosphate anabolites by host enzymes within a cell. However, tenofovir disoproxil fumarate (Figure 2), a nucleotide analogue, is found in a monophosphate form and two additional phosphorylation steps are required to run into its active form. Since the common structure of NRTIs is a trigger for the development of resistance to NRTIs, they are used in combination with other NRTIs and NNRTIs rather than as a single agent nowadays [30,65,66,67,68].
NRTIs are the key backbones of combination regimens according to the recent guidelines. Zidovudine (Retrovir®) (Figure 2), the first approved anti-HIV-1 drug, and stavudine (Zerit®) (Figure 2) are thymidine analogues. The exploration of zidovudine as an anti-HIV agent is also the first example of drug repositioning in the history of medicinal chemistry as it was initially developed as an anticancer agent [69]. The use of stavudine is not recommended by WHO as it increases the lipoatrophy risk along with other side effects. Lamivudine (Epivir®) (Figure 2), zalcitabine (Hivid®) (Figure 2) and emtricitabine (Emtriva®) (Figure 2) are cytosine analogues. Renal dysfunction is a challenging point for the use of lamivudine and the dosage must be adjusted for patients with renal impairment. Lamivudine was licenced for the treatment of HIV-1 and hepatitis B virus infections in 1995 and 1998, respectively. Lamivudine, a key component in HIV-1 treatment, is highly recommended in almost all first-line and a majority of second-line combination regimens thanks to its remarkable efficacy and safety profile. The combinations of tenofovir disoproxil fumarate with lamivudine (Cimduo®) or emtricitabine (Truvada®) are also recommended as a first-line treatment for patients coinfected with HIV-1 and hepatitis B virus [70].
Emtricitabine received FDA approval in 2003 for use in anti-HIV-1 treatment. Emtricitabine, the most commonly prescribed anti-HIV-1 drug, is a fluorinated derivative of lamivudine, and they share the same sugar configuration [71]. Zalcitabine is not recommended for use in many countries due to high mitochondrial toxicity. Didanosine (Videx®) (Figure 2) and abacavir (Ziagen®) (Figure 2) are adenosine and guanosine analogues, respectively. Didanosine is not prescribed as it has toxicity, efficacy, formulation and drug interactions issues. Abacavir has a higher risk of cardiovascular diseases. Therefore, abacavir is also available in coformulated tablets that contain other antiretrovirals such as lamivudine (Kivexa®, Epzicom®), lamivudine plus dolutegravir (INSTI) (Triumeq®) and lamivudine plus zidovudine (Trizivir®) [72]. Given that tenofovir displays poor cellular absorption and oral bioavailability owing to the negative charges on the phosphonate moiety, the fumarate salts of tenofovir disoproxil (Viread®) and tenofovir alafenamide (Figure 2) are prepared as tenofovir prodrugs. Tenofovir alafenamide (Vemlidy®), which was developed to deal with the renal and bone toxicity of tenofovir disoproxil fumarate, has superior potential and safety properties over other NRTIs. Moreover, HIV-1 treatment guidelines mainly recommend its combination with emtricitabine. On the other hand, tenofovir disoproxil fumarate/emtricitabine is the recommended oral pre-exposure prophylaxis regimen for all populations at risk [65,73,74,75,76].
2.2. Protease Inhibitors
Protease enzymes drive the catalysis of the hydrolysis of polypeptide bonds. The HIV-1 protease is a homodimeric aspartyl protease, which can cleave HIV-1 precursor protein or polyprotein involving structural proteins (Gag proteins: matrix (MA), CA, nucleocapsid (NC), p6, and enzyme products), and viral enzymes (Pol proteins: protease, reverse transcriptase, RNase H, and integrase). The Gag and Gag-Pol viral polyprotein are associated with efficient viral assembly, genome packaging, formation of immature viral particles, budding, release from the cell, and maturation in order to infect new host cells. The timing of the release of HIV-1 protease from the Gag-Pol polyprotein is crucial for accomplished maturation process and consequently HIV-1 infectivity [77,78,79,80,81].
The active site of protease is a catalytic Asp-Ser-Gly triad that enables the nucleophilic attack of water onto the scissile amide bond of natural substrate for successful cleavage. The protease inhibitors, non-cleavable transition state isosteres, can compete with the natural substrate by a functional group acting as a scissile amide bond mimetic [82,83]. The protease inhibitors were initially developed based on a “structure-informed” strategy aiming at HIV-1 protease inhibition and successive inhibition of HIV-1 replication [84]. The first protease inhibitors, including saquinavir (Invirase®) (Figure 3), ritonavir (Norvir®) (Figure 3), indinavir (Crixivan®) (Figure 5), nelfinavir (Viracept®) (Figure 3), amprenavir (Agenerase®) (Figure 3), and fosamprenavir (Lexiva®) (Figure 3) shared some similar structural properties and binding pattern resulting in possible cross-resistance and common severe side effects. Some pharmacokinetic challenges were also encountered with first HIV-1 protease inhibitors such as poor oral bioavailability, extensively binding to plasma proteins, and rapid elimination [85]. The second-generation of protease inhibitors, including lopinavir (Kaletra®), atazanavir (Reyataz®), tipranavir (Aptivus®), and darunavir (Prezista®) are currently recommended in use as second-line regimens for patients with HIV-1, who have provided no benefit from the first-line therapy with integrase and reverse transcriptase inhibitors. They are available in a fixed dose regimen in combination with emtricitabine and tenofovir [86].
Lopinavir (Figure 3), a combination with ritonavir, was approved by the FDA in 2004 for the treatment of HIV infections. Ritonavir promotes the increased bioavailability of lopinavir as a pharmacoenhancer. Atazanavir (Figure 3), a hydroxyethyl hydrazine aza-peptide inhibitor, is a well-tolerated effective treatment for HIV-1 patients with an extended half-life, less side effect and resistance features [83]. Atazanavir is also reported for alleviating competitive bilirubin binding, and relative possible risk of cardiovascular disorders [87]. Tipranavir (Figure 3) is a sulfonamide-based dihydropyrone revealing a different binding profile than the other HIV-1 protease inhibitors making it stronger for the development of resistance. Tipranavir was found effective for both naïve and highly experienced antiretroviral treatment patients but its therapeutic effect is limited due to some harsh unwanted effects, including intracranial hemorrhage and hepatotoxicity [88,89]. Darunavir (Figure 3) is a small molecule that is capable of impeding the dimerization of HIV protease and its catalytic activity. Darunavir is effective for the treatment of both naïve and experienced HIV-1-infected patients. Darunavir has a high genetic barrier to resistance development and favorable safety profile [90,91].
2.3. Fusion Inhibitors
For the entry process, all enveloped viruses perform a multistep fusion machinery that their lipid bilayers and host cell membrane reunite. The HIV-1 Env glycoprotein (gp160) is cleaved to form a gp120 and gp411 heterodimer. The three surface gp120 subunits that are non-covalently bound to three transmembrane gp41 subunits constitute a functional trimeric envelope spike on the virion surface, which is essential for host cell recognition and membrane fusion. In the attachment process, gp120 attaches to the primary receptor CD4 antigen on the host cell. This attachment results in structural changes of gp120 and it interacts with a co-receptor (CCR5 or CXCR4). Co-receptor binding triggers the fusion mediated by gp41. During the fusion, the gp41 forms a stable six-helix bundle involving N-terminal heptad repeat (NHR) coiled coils and 3 C-terminal heptad repeat (CHR) helices packing into the hydrophobic NHR grooves as antiparallel [92,93,94,95,96,97].
The HIV-1 entry inhibitors were developed based on three categories: gp120-CD4 binding inhibitors, gp120-co-receptor binding inhibitors and fusion inhibitors. Enfuvirtide (Fuzeon®), the first approved peptide HIV-1 inhibitor, acts as a HIV-1 fusion inhibitor binding to a region of the gp41 of HIV-1. Enfuvirtide was found effective in clinical trials against resistant HIV-1 strains. The disadvantages of the treatment with enfuvirtide are short half-life that needs long-term application, uncomfortable subcutaneous administration that cause injection-site reactions, and high prices [98,99].
Maraviroc (Selzentry®) (Figure 4) is the first-in-class and only FDA approved drug as an inhibitor of CCR5 for the treatment of R5-tropic HIV-1 infected patients. It was discovered as the end product of a high-throughput screening and medicinal chemistry program. Maraviroc was found to be effective at disrupting gp120-CCR5 binding and subsequently membrane fusion events necessary for HIV-1 entry into cells. Maraviroc showed potent anti-HIV-1 activity, low level of resistance, favorable pharmacological properties, and mild-to-moderate hepatic and renal disorders [100,101,102,103].
Ibalizumab (Trogarzo®) is the first humanized IgG4 monoclonal antibody and CD4-mediated post-attachment inhibitor for the treatment of HIV-1 infection. Ibalizumab shows its effects binding to D2 immunoglobulin domain of cell-surface glycoprotein CD4. Ibalizumab, in combination with other antiretroviral(s), was approved by FDA in 2018 for the treatment of adults with multidrug resistant HIV-1 infection. Ibalizumab is a valuable option with a high potency and a favorable pharmacokinetic profile notwithstanding the necessity of additional studies and long-term post-marketing data [104,105,106,107,108].
Fostemsavir (Rukobia®) (Figure 4) was approved by FDA in 2020 for the treatment of multi-drug resistant HIV-1 infection of heavily treatment-experienced adults whose previous HIV-1 therapy was unsuccessful owing to resistance, intolerance, or safety considerations. Fostemsavir, a phosphonooxymethyl prodrug of temsavir, is converted to its active metabolite by alkaline phosphatase. Temsavir prevents attachment and subsequent entry into host cell via binding to gp120 and holding gp120 in the conformational state, and hampering the initial interaction with surface receptors on CD4+ cells. Fostemsavir showed good efficacy and a safety profile in patients with multidrug-resistant HIV-1 infection. Besides, it revealed no in vitro cross-resistance with other classes of entry inhibitors such as ibalizumab, maraviroc or enfuvirtide. Fostemsavir can be also used for HIV-1 CCR5/CXCR4 tropism. The possible drug-drug interactions can be occurred during the coadministration with strong CYP3A inducers [109,110,111,112,113].
2.4. HIV-1 Integrase Inhibitors
The HIV-1 intasome is a massive nucleoprotein complex containing the integrase protein and the ends of the viral DNA obtained by reverse transcription. Integration process is crucial for viral replication that HIV-1 integrase strand-transfer inhibitors (INSTIs) target the intasomes to halt the integration and subsequent replication processes by binding to catalytic site of integrase. This catalytic site of integrase is engaged with covalent bonds with the phosphodiester backbone of DNA through divalent cations, metals such as Mg+2, required for integrase catalytic reactions. The INSTIs demonstrate familiar mode of action, whereas they generally possess distinct pharmacokinetic profiles, resulting in different dose frequency, combinations and drug-drug interactions. INSTIs are current recommended components of frontline and drug-switch cART formulations [114,115,116,117].
Raltegravir (Isentress®) (Figure 5) and elvitegravir (Vitekta®) (Figure 5) are first-generation INSTIs. Raltegravir is the first approved INSTI blocking the formation of the covalent bonds. Patients with HIV-1 have experienced a well-tolerated, safe and potent antiretroviral treatment with raltegravir-based regimens. One of the advantages of raltegravir is glucuronidation metabolism instead of hepatic metabolism leading to less observed drug-drug interactions [118,119]. Elvitegravir, was then designed and obtained bearing a coplanar monoketo acid motif in 4-quinolone-3-carbocyclic acid as a bioisostere of a diketo acid motif of which raltegravir also contains a structural similar moiety, with a pyrimidinone carboxamide [120]. A fixed-dose combination of elvitegravir with cobicistat, emtricitabine and tenofovir disoproxil fumarate (Stribild®) and tenofovir alafenamide instead of tenofovir disoproxil fumarate (Genyova®) were approved for the treatment of HIV-1 infection. Dolutegravir (Tivicay®) (Figure 5), cabotegravir (Vocabria®) (Figure 5), and bictegravir (Figure 5), are second-generation INSTIs. Based on the reports of current guidelines, the combinations of bictegravir, dolutegravir, or raltegravir plus two NRTIs are recommended as initial therapies [74]. Dolutegravir and cabotegravir are bicyclic carbamoyl pyridone analogs using a two-metal chelation model of the integrase catalytic active site [121]. Dolutegravir exhibited a significant activity against HIV-1 isolates with a higher barrier to resistance development compared to raltegravir and elvitegravir. Dolutegravir has several advantages, including favourable pharmacokinetic properties such as long half-life, once-daily dosing without a need of pharmacokinetic enhancer or commitment to meal time. The fixed-dose combination of dolutegravir with lamivudine in single-tablet regimen (Dovato®) is confirmed to be effective and well-tolerated for adolescents and adults with HIV-1 infection [122,123,124]. Dolutegravir is also highly recommended by WHO in the initial therapy combined with NRTI or NNRTI and for use in pregnant women and women with child-bearing potential in second-line treatment. Dolutegravir plus either tenofovir disoproxil fumarate/emtricitabine or tenofovir alafenamide/ emtricitabine is recommended as a safe option during pregnancy [74,114,125].
Figure 5.
The chemical structures of FDA-approved first and second generation of INSTIs.
The oral tablet form of dolutegravir is available in the market and other long-acting injectables, implants, nasal and vaginal formulations were found beneficial for HIV-1 treatment in preclinical studies [126]. Cabotegravir received approval from the FDA for HIV-1 pre-exposure prophylaxis as the first long-acting injectable medication. It is announced that it is safe and effective for people carrying important HIV-1 infection risk [127,128,129,130]. Bictegravir, structurally deriven from dolutegravir, specially inhibited HIV-1 integrase with an IC50 value of 7.5 nM compared to dolutegravir and elvitegravir with IC50 values of 7.4 nM and 8.4 nM, respectively [131,132]. The advantage of bictegravir over cabotegravir and dolutegravir is integrated into fixed-dose combinations for higher potency and less drug resistance [133].
2.5. CA Inhibitor
Lenacapavir (GSK-6207) (Sunlenca®) (Figure 6), developed by Gilead Sciences Inc., was approved by FDA (2022) for clinical use in combination with other antiretroviral(s) in heavily treatment-experienced patients with multidrug-resistant HIV-1 infection. Lenacapavir (GSK-6207), an indazole derivative, is a long-acting first-in class approved CA inhibitor. Based on its high lipid solubility and permeability, lenacapavir can be administered as orally with an up to weekly dosing interval or bi-annual subcutaneous injectable solution. As a CA inhibitor, lenacapavir selectively binds to the hexamer subunits of the HIV-1 CA protein, which is generated by the cleavage of the Gag by viral protease. Upon targeting CA protein, lenacapavir enables the inhibition of HIV-1 assembly, appropriate viral CA generation and nuclear import of viral DNA [134,135,136,137,138,139].
Lenacapavir showed a potent in vitro anti-HIV-1 activity against HIV-1 infected MT-4 cells with a mean half-maximum effective concentration (EC50) 105 pmol/L. Lenacapavir manifested a well-tolerated and safe profile with injection site reactions, the most frequent off-target event, in clinical trials. In a phase 1b study, substantial antiviral activity was observed with lenacapavir. In phase 3 CAPELLA trial (NCT04150068), lenacapavir was found to reduce HIV-1 viral load in patients with multidrug-resistant infection. A phase 2 study sponsored by Gilead Sciences, Inc. is also ongoing in virologically suppressed people with HIV-1 for investigating the safety and efficacy of lenacapavir in combination with islatravir (an investigational drug and a first potential drug of a new class entitled nucleoside reverse transcriptase translocation inhibitors (NRTTIs)) (Figure 6) (NCT05052996) [140,141,142,143,144,145].
No cross resistance is reported for lenacapavir with approved antiretroviral drugs. The concomitant use of lenacapavir with strong inhibitors of CYP3A and P-glycoprotein is contraindicated due to the possible low plasma levels of lenacapavir and consequent antiviral activity loss and the emergence of viral resistance [141,143,145].
2.6. Pharmacokinetic Enhancers
In particular, HIV-1 protease inhibitors are prone to metabolize rapidly by CYP3A enzyme predominantly, which can lead to low systemic exposure. Ritonavir, a strong approved HIV protease inhibitor, was found to boost the pharmacokinetics of CYP3A substrate antiviral drugs, including elvitegravir and maraviroc. After discovery of CYP3A inhibitory potency of ritonavir, the strategy of enhancing pharmacokinetic profile of antiretroviral agents was followed due to the drawbacks of ritonavir use such as lipodystrophy, hyperlipidaemia and insulin resistance, and other undesired drug interactions. Therefore, researchers optimized the chemical structure of ritonavir starting with the removal of the key hydroxyl group and following intensive structure-activity relationship studies and obtained cobicistat (Figure 7), which is more soluble than ritonavir, making coformulation easier. Cobicistat revealed no activity against primary HIV-1 isolates and HIV-1 protease, whereas cobicistat selectively inhibited CYP3A enzyme with minimal cytotoxicity. Cobicistat (Tybost®) received its approval by FDA in 2014 for the treatment of HIV-1 infection. Cobicistat is a pharmacoenhancer of the HIV-1 protease inhibitors atazanavir and darunavir in adults with HIV-1 infection. A fixed-dose combination of cobicistat with darunavir, emtricitabine, and tenofovir alafenamide (Symtuza®) is the first protease-inhibitor-based single-tablet regimen approved by FDA. Cobicistat is also available in two drugs in one pill combinations with darunavir (Prezcobix®), and atazanavir (Evotaz®). The serum creatinine levels may be augmented with cobicistat use, so caution is required with patients showing kidney problems [90,91,146,147,148,149].
2.7. Standard Two-Drug, Three-Drug, and Four-Drug Regimens and Fixed-Dose Combinations as cART Options
Functional antagonism, boosted drug toxicity, and synergistic/additive effects are major possible consequences of combination therapy. Combination therapy has the potential to cause adverse effects due to drug-drug interactions, whereas the decrease of individual drug doses in synergistic drug combinations leads to reduced drug toxicities. To target multiple pathways responsible for the disease using different class of potential drugs contributes to increase in desired activity [41].
The administration of fixed dose combination therapies and single tablet regimens for HIV-1 treatment have enabled dramatic improvements such as attenuation the pill burden, prevention of drug resistance, and improved patient compliance. The combination of lamivudine and zidovudine (Combivir®) is the first fixed dose FDA approved combination therapy containing two NRTIs (in 1997) for HIV-1 infected people followed by emtricitabine and tenofovir disoproxil fumarate and, abacavir sulfate and lamivudine [150,151]. On the other hand, the combination of dolutegravir and rilpivirine (Juluca®) is the first (in 2017) FDA approved fixed-dose two-drug single-tablet regimen from two different classes of cART (INSTIs and NNRTIs, respectively) for the treatment of adults with HIV-1 infection [152,153].
The introduction of protease inhibitors to combination therapy made a huge impact for the management of HIV-1 infection. The current guidelines recommend three-drug combinations with dual NRTIs backbone, and a core drug from boosted protease inhibitors such as darunavir/ritonavir, integrase inhibitors or NNRTIs for the initial HIV-1 treatment. Bictegravir, dolutegravir, elvitegravir, raltegravir (from INSTIs), and rilpivirine, efavirenz (from NNRTIs) are usually in the frame for initial three-drug regimen. Tenofovir disoproxil fumarate plus emtricitabine (in fixed-dose combination) and abacavir plus lamivudine (in fixed-dose combination) are the mostly preferred NRTIs as a backbone [36,38]. The triple-drug regimen of bictegravir, emtricitabine and tenofovir alafenamide (Biktarvy®) received approval from the FDA in 2018 for the treatment of HIV-1 infected adults. The weight gain, which could result in high risk of diabetes and cardiovascular disorders, was observed as a disadvantage of this treatment regimen. As HIV-1 and tuberculosis coinfection was frequently encountered, this regimen was found to be safe to be administered along with isoniazid and rifapentine on the contrary of rifampicin as it induced CYP3A and P-glycoprotein significantly that could lead to drug-drug interactions [133,154,155].
However, three-drug regimen is also questioned due to the toxic effects and cross-resistance stemming from the NRTI class. Therefore, dual-therapy regimens stand out as a plausible alternative as a first-line therapy in treatment-naive patients, who cannot tolerate three-drug combination therapy mainly due to toxicity issues of tenofovir disoproxil fumarate and abacavir. On the other hand, four-drug regimens were considered to get benefit from the superior features of INSTIs and HIV-1 protease inhibitors, including swift viral suppression and higher genetic barrier to resistance, respectively. However, no significant differences in safety or efficacy as well as toxicity or adherence were observed with four-drug regimen compared to three-drug regimen [36,38,39,151,156,157,158,159].
Long-acting formulations such as implants, rings and nanoformulations are also future hallmarks of anti-HIV-1 drug development. The co-packaged cabotegravir (extended-release injectable suspension) and rilpivirine (extended-release injectable suspension) (Cabenuva®) is the first FDA-approved injectable, complete regimen for adults with HIV-1 infection [160,161].
3. The Other Anti-Gag Compounds
The HIV-1 Gag coordinates all major steps of assembly of viral particles and it is the only viral protein required to initiate and complete the budding, and release of virus-like particles (VLPs) from the plasma membrane [162,163]. The recent discovery and approval of lenacapavir pointed out that targeting Gag leads to the inhibition of HIV-1 assembly, budding, release and maturation steps and consequent infection of other immune cells.
3.1. MA Inhibitors
The HIV-1 Gag incorporates MA, CA, NC, and p6 domains, in addition to two spacer peptides, SP1 and SP2, which are located at the CA/NC and the NC/p6 junctions, respectively (Figure 8). The Gag-Gag (multimerization), Gag-membrane, and Gag-RNA interactions are crucial for assembly, mediated by CA, MA and NC domains, respectively. The minority of Gag molecules lead to assembly of a large number of Gag molecules recruiting a single viral RNA dimer to the plasma membrane. The MA domain anchors Gag into the plasma membrane through electrostatic and hydrophobic interactions but binding is mediated predominantly by dynamic, electrostatic interactions. The MA domain is myristoylated on its N terminus and forms ionic interactions with acidic polar heads of phosphatidylinositol-(4,5)-bisphosphate (PI(4,5)P2) (Figure 9) through its highly basic region (conserved stretch of lysine and arginine residues). The MA domain is also capable of binding RNA just like NC domain. The specificity of MA binding to PI(4,5)P2 might be attributed to MA-RNA binding because this binding prevents Gag from interacting with other lipid interfaces until it reaches to the plasma membrane. It was also reported that MA presented a hexamer-of-trimers arrangement for Env incorporation by interacting with the cytoplasmic tail of the Env gp41 protein and some residual mutations hinder this trimeric formation and subsequent Env incorporation for successful virus particle assembly (a stable immature Gag lattice) [162,163,164,165,166,167,168,169,170,171,172,173,174].
The NC domain, comprised of two zinc-finger motifs, contributes to Gag multimerization, binds signal Ψ (ΨRNA) specifically, packages genomic viral RNA. The p6 domain deploys the cellular endosomal sorting complex required for ESCRT machinery, a key component for the release of the budding viral particle from the cell surface [175,176,177,178].
Our research group developed a new surface plasmon resonance (SPR)-based technique in 2010 to detect the binding affinity between Gag/MA binding and various phosphoinositide derivatives containing a different number of phosphates and their regioisomers with or without an acyl chain. We identified that both the divalent phosphate groups and the acyl chains of PI(4,5)P2 [164] were essential for strong binding to MA [179]. Then, we carried out SPR analysis of the MA binding of highly phosphorylated inositol phosphates and confirmed that inositol hexakisphosphate (IP6) (Figure 9) bound to MA 10-fold strongly than IP3 (Kd= 272 µM) (Figure 9) with a dissociation constants (Kd) value of 25.7 µM and comparable to PI(4,5)P2 derivative (1) (Kd= 16.9 µM) (Figure 9) pointing out the importance of the presence of more phosphate groups in the inositol ring. Strikingly, the dissociation constant of IP6 was found concordant with the PI(4,5)P2 derivative due to absence of the diacylglycerol moiety in its chemical structure. This outcome encouraged us to design new phosphoinositides, carrying diacylglycerol moiety in IP6 framework. Among new phosphoinositides, compound 2 (later DL-Heptanoylphosphatidyl Inositol Pentakisphosphate, DL-HIPPO) revealed remarkable affinity (Kd = 0.25 µM), which was 70-fold and 100-fold stronger than 1 and IP6, respectively [180]. In continuous of our work, potent isomer L-HIPPO (Figure 9) was separated initially and Kd for MA binding of L-HIPPO was found 0.18 ± 0.08 μM [181]. Then, we recorded diffraction patterns of MA-IP6 microcrystals beyond 3.5 Å resolution using X-ray free-electron laser (XFEL) study [182]. We elucidated three high-resolution crystal structures of the MA domain in complex with IP6 molecules (PDB IDs: 7E1I, 7E1J, and 7E1K) [183] by both synchrotron cryo X-ray crystallography and ambient-temperature Serial Femtosecond X-ray crystallography (SFX) at an XFEL. We determined the important residues in IP6 binding and confirmed that the binding site of MA-IP6 is distinct from the PI(4,5)P2-binding site and IP6 was found important in the oligomerization of MA [183]. Afterwards, we carried out computational studies for new rationally designed L-HIPPO derivatives via Maestro software in the MA domain (PDB IDs: 7E1I, 7E1J, and 7E1K) and revealed that benzene-inserted compounds presented a more favorable binding profile than L-HIPPO. Therefore, we applied the same docking procedure to a large library of aromatic group carrying L-HIPPO derivatives and identified 3,4-dihydroxyphenyl and 3-methoxy-4-hydroxyphenyl carrying compounds as the most potent L-HIPPO derivatives for MA binding with optimum pharmacokinetic properties [184].
In other studies, Zentner et al. 2013 [185], developed compound 7 (Figure 10) using virtual screening method. They further reported that this compound showed potential anti-HIV-1 activity with IC50 values of 7.5–15.6 µM. It was also determined that compound 7 directly interacted with HIV-1 MA competing with PI(4,5)P2 for MA binding and blocking the generation of new viruses.
Alfadhli et al. 2013 [186], targeted the MA-RNA binding since RNA binding might protect MA interacting with other cellular membranes before Gag delivery to the cell surface. They determined four compounds, including three thiadiazolanes (TD 1-3) (Figure 10), that compete with RNA for MA binding. Although thiadiazolanes were found to halt HIV-1 replication, they suffered from toxicity.
3.2. Maturation Inhibitors
The viral protease cleaves Gag and Gag-Pol polyprotein into their mature subunits over the course of maturation process leading to dramatic structural rearrangements within the particle. In the final maturation step, the cleavage of six-helix bundle structure by protease sorts out CA from CA-SP1 cleavage site and disrupts the immature lattice and frees CA. Then, CA monomers build up hexameric and pentameric complex of mature core, which is crucial for maturation and infection. The CA monomers incorporates the N-terminal (CA-NTD) and the C-terminal (CA-CTD) domains, which are separated by a short flexible linker. IP6 interacts with the six-helix bundle structure and stabilizes the immature Gag lattice. IP6 is also very crucial for assembly of CA into the mature fullerene-like cone lattice [187,188,189,190,191,192].
To develop maturation inhibitors, which target the cleavage of the SP1 peptide from the CA-SP1 maturation intermediate, has been a bona fide approach to HIV eradication [193].
Fujioka et al. 1994 [194], isolated betulinic acid (Figure 11) and platanic acid (Figure 11) from Syzigium claviflorum and reported that both of them exhibited HIV-1 replication inhibitory activity against H9 lymphocyte cells. They also reported that dihydrobetulinic acid inhibited HIV replication in same cells with EC50 and IC50 values of 0.9 µM and 13 µM, respectively. Then, the same research group prepared betulinic acid and dihydrobetulinic acid derivatives and reported that 3-O-(3′,3′-dimethylsuccinyl)betulinic acid (later then called as DBS, YK-FH312, PA-457, Bevirimat) (Figure 11) exhibited the most potent anti-HIV activity in acutely infected H9 lymphocytes [195]. Kanamoto et al. 2001 [196], demonstrated the virus-induced cytopathic effects of bevirimat in HIV-1IIIB-infected MT-4 cells with an EC50 value of 0.011 μg/mL, a CC50 value of 14.03 μg/mL. Further mechanistic effects suggested that the formation of viral proteins continued though the virion could not be released pointing out that bevirimat affected the viral maturation. On the other hand, Li et al. 2003 [197], presented that bevirimat inhibited replication of wild-type and drug-resistant HIV-1 isolates along with inhibition of conversion of CA precursor to mature CA. Subsequent research studies also confirmed that bevirimat prevented the cleavage of SP1 from the C-terminus of CA leading to defective core condensation and inhibition of maturation [198,199,200,201,202]. Although phase I and II studies of bevirimat indicated that bevirimat posed a well-tolerated profile and alleviated viral load in a dose-dependent manner without drug resistance mutations [203], later study evaluating baseline susceptibility to bevirimat found that diminished bevirimat sensitivity was correlated with naturally occurring polymorphisms at 6-8 positions in Gag SP1 [204]. Another bevirimat derivative was prepared and evaluated for anti-HIV effects. Compound 16 (Figure 11) was found more effective with a higher hydrosolubility compared to bevirimat. They also characterized a direct interaction of compound 16 and CA-SP1-NC domain [205]. Dang et al. 2013 [206], synthesized new bevirimat analogues to cope with the resistance issues of bevirimat and evaluated towards bevirimat resistant HIV-1 variants. Compound 6 (Figure 11) was defined as the most potential analogue against wild-type virus and the bevirimat resistant NL4-3/V370A variant with IC50 values of 0.01 μM and 0.16 μM, respectively. Another bevirimat analogue GSK3532795 (BMS-955176) (Figure 11) was developed as a potent of CA/SP1 cleavage inhibitor, revealing a wide range of antiviral effects including V370A- and ΔV370-containing polymorphic viruses with a low serum binding [207,208,209]. In a phase IIa trial, GSK3532795 was generally detected safe and well-tolerated and demonstrated >1 log10 reduction in viral RNA [210]. The gastrointestinal intolerability and treatment-development resistance were detected with GSK3532795 associated with NRTI backbone in a randomized phase IIb trial [211]. Chroback et al. 2019 [212], synthesized and investigated phosphate and phosphonate analogues of bevirimat for anti-HIV-1 activity. According to the results, compound 14a (Figure 11) demonstrated similar and more selective anti-HIV-1 activity compared with bevirimat (IC50= 0.03 ± 0.009 μM; Selectivity Index (SI)= 967) with an IC50 value of 0.02 ± 0.01 μM and SI value of 3450). They confirmed by computational studies that the phosphonate group contributed to strong interactions of compound 14a in CTD of CA-SP1. Dicker et al. 2022 [213], developed GSK3640254 (GSK’254) (Figure 11) through a medicinal chemistry approach. They showed that GSK’254 displayed remarkable anti-HIV-1 effects towards a panel of HIV-1 clinical isolates, with a mean EC50 value of 9 nM. In phase I studies, GSK’254 was confirmed to possess a favorable clinical potential alone or in combination with tenofovir alafenamide/emtricitabine or dolutegravir. In phase IIa clinical study, GSK’254 blocked cleavage of p25 in a range of polymorphic HIV-1 Gag VLPs. Phase IIb trials are currently ongoing for GSK’254 (NCT04493216 and NCT04900038).
N’-(3-chloro-4-methylphenyl)-N-{2-[({5-[(dimethylamino)-methyl]-2-furyl}-methyl)-sulfanyl]ethyl}urea) (CAP-1) (Figure 12) decreased infectivity in latent infected U1 cultures and MAGI cells. Besides, it was reported that CAP-1 inhibited no early phase events, while it inhibited late-phase viral event [214]. In the continuation of the research to determine the mechanism of CAP-1 inhibition, a combination of X-ray crystallography and NMR spectroscopy were performed indicating the displacement of Phe32 in CAP-1 binding put emphasis on the significant role for a Phe32 conformational change during normal CA assembly [215].
During the journey to exploration and optimization of new benzodiazepines and benzimidazoles, researchers reached to compounds with potent antiviral activity against wild-type and drug-resistant HIV-1 and both series of inhibitors was able to bind to the N-terminal domain of CA. Then authors reported the binding of a novel CA-assembly inhibitor targeting an authentic inhibitory site on CA-NTD. The use of compound 1 (Figure 12) as a tool presented to enable ternary co-crystallizations with CA-NTD [216,217,218,219,220].
In another study, a CA assembly inhibitor (CAI), a 12-mer peptide (sequence: IT FEDLLDYYGP-amide), was found to be embedded into a conserved hydrophobic groove and changed the CA dimer interface (CAI binding site) indicating a new target for anti-HIV-1 drug discovery. This peptide is identified as the first known immature HIV-1 assembly inhibitor [221]. Authors later then expanded the study and designated i,i + 7 stapled peptides and identified NYAD-36 (sequence: Ac-ISF-R8-ELLDYY-S5-ESGS-amide), NYAD-66 (sequence: Ac-ISF-R8-ELLDYY-S5-ED-amide), and NYAD−67 (sequence: Ac-ISF-R8-EWLQAY-S5-EDE-amide) as three potent inhibitors, which could bind to CA robustly and collapsed the formation of mature-like particles [222].
Another new small-molecule inhibitor that targeted virion maturation was introduced from a HIV-1 antiviral screen. PF-46396 (Figure 12), a lead molecule, exhibited potential anti-HIV-1 activity. This compound inhibited the processing of CA-SP1 giving rise to the aggregation of CA/SP1 precursor proteins and maturation inhibition [223]. The same research group then reported PF-3450074 (PF74) (Figure 12) to be effective against all strains of HIV-1 tested with median EC50 values of 0.207 µM (range 0.113 to 0.362 µM). A co-crystal structure of PF-74 revealed a new binding site on HIV-1 CA. Moreover, PF-74 in vitro enhanced the rate of HIV-1 CA multimerization [224]. Dostálková et al. 2020 [225], reported a series of modifications of PF74 derivatives. They obtained compound D10 (Figure 12) with a modified indole moiety to benzimidazole moiety exerting in vitro stabilization activity in higher levels compared to the original PF74 molecule. Researchers continue further modifications to decrease the D10 cytotoxicity.
Yant et al. 2019 [226], reported GS-CA1 (Figure 12) as a novel small-molecule CA inhibitor, which showed remarkable and selective anti-HIV effect with an EC50 value of 240±40 pM and CC50 >50 µM against MT-4 cells acutely infected with HIV-1IIIB. Further mechanistic studies indicated that GS-CA1 directly interacted with CA and affected CA mediated nuclear import of viral DNA. Moreover, GS-CA1 presented favorable metabolic stability and low solubility to function sustained drug release in mice.
4. Strategies for Eradication of HIV-1
Recent advancements in HIV treatment are: sustained-release antiretroviral formulations such as subcutaneous implants, vaginal rings, and nanotherapies for long-lasting treatment [156]; gene editing technologies including the use of CRISPR/Cas9, clustered regularly interspaced short palindromic repeat (CRISPR)-associated nuclease 9 (Cas9) [227]; immunological approaches such as active immunization with vaccines and passive immunization with broadly neutralizing antibodies for controlling HIV-1 pandemic globally [228]; chimeric antigen receptor cell technology for prolonged remission of the reactivated latent viral reservoirs [229]; and the rational drug-design with structural optimization, combinatorial chemistry together with high-throughput screening and virtual screening methods for the rapid discovery of potent anti-HIV-1 lead [230,231]. Accordingly, a promising new strategy hints that premature protease activation leads to pyroptotic killing of infected cells for the eradication of the latent reservoir due to toxicity of HIV-1 protease [81]. Despite these revolutionary advances improving the efficacy and safety of HIV-1/AIDS treatment, there is still no definitive cure in the world hitherto and patients with HIV-1/AIDS need lifelong cART [232,233,234].
4.1. “Shock and Kill (Kick and Kill)” and “Block and Lock”
The remaining latent reservoirs are host cells containing integrated proviral DNA in the genome and can replicate competent viral particles. Various cells harbouring latent reservoir can circulate in blood after HIV infection, causing different types of diseases and HIV-1 development. During cART, the major HIV cellular reservoirs are memory CD4+ T cell whose longevity is very long. The HIV-1 reservoirs are mainly observed in gastrointestinal mucosa, CNS, lymph nodes, and genital tract, where viral replication become autonomous and undetectable [235,236,237,238,239].
There is no definite characterization of latent reservoirs, which makes the elimination of the cells harbouring the latent reservoir compelling. Clonal expansion known as latently infected cell proliferation is also need to be addressed as it is considered one of the main reasons of HIV-1 persistence during cART. Although several approaches such as latency reversal, latency silencing, immunotherapy with broadly neutralizing antibodies and HIV-1-specific chimeric antigen receptor-T cells or gene editing technologies that disrupt the provirus have been proposed to eliminate HIV latency, these studies encounter several high risks and limitations, so there is no broad consensus on the methodology yet [240,241,242,243,244].
The “shock and kill” (Figure 13) therapy targets the removal of latent proviral reservoirs with the activation of transcription of dormant provirus using pharmaceutical agents (shock) and elimination of the latently infected cells with cART, intrinsic cell death mechanisms, immune responses, such as activation of CD8+ T cells, and/or HIV-1-cytolysis (kill). Latency-reversing agents (LRAs), mainly functioning as either protein kinase C (PKC) activators, histone deacetylase inhibitors (HDACis), toll-like receptor (TLR) agonists or PI3K/Akt agonists, have been introduced for this purpose, but none of them have reached to the ultimate success. On the other hand, “block and lock” (Figure 13) strategy aiming the HIV-1 eradication through the inhibition of HIV transcription utilizing latency promoting agents (LPAs), has been proposed alternatively. The “block and lock” strategy also proposes destroying cellular epigenetic regulators using small interfering RNAs (siRNAs) and maintains HIV-1 latency. Briefly, “block and lock” strategy can target both replication competent and incompetent virus making it noteworthy because replication-deficient viruses can produce viral transcripts and toxic proteins. “Block and lock” strategy could be promising for a functional HIV-1 remission or therapy enabling long-lasting HIV-1 suppression even independent from cART use. Although a few agents such as Tat or Rev inhibitors were found to supress the provirus expression, no clinical data have been reported so far [245,246,247,248,249,250].
4.2. A New Approach: “Lock-in Apoptosis” and Compound L-HIPPO
The early clinical trials about afore mentioned strategies failed to reach a scalable solution for curing HIV-1. Our research group came up with a novel strategy called as “lock-in and apoptosis” (Figure 13) referring to L-HIPPO (L-Heptanoylphosphatidyl Inositol Pentakisphosphate) (Figure 9), which was synthesized by our research group as a potential Gag MA targeted anti-HIV-1 compound. The “killing process” is a problem in “shock and kill”, and this strategy may overcome this problem. Based on this strategy, L-HIPPO was able to circumvent the budding of provirus and facilitate to lock the virus into the HIV-1-infected host cell leading eliminating secretion of new viral particles, a build-up of viral products and ultimately to HIV-1-specific host-cell apoptosis [181].
As mentioned above, the apoptosis induction of L-HIPPO with a carrier (L-HIPPO-α-CDE complex) was determined using fluorescence-activated cell sorting (FACS) and microscopic analysis. Results indicated that HeLa cells and Jurkat T cells transfected with pNL4-3/Gag Venus underwent apoptosis in the presence of L-HIPPO-α-CDE complex without showing toxicity [181].
4.3. Apoptosis and HIV Latency
Apoptosis, programmed cell death, has already become an emerging concept in HIV-1 latency elimination. HIV-1 protease is also well-known to cleave host proteins in addition to HIV-1 polyprotein at a post-integration step. HIV-1 protease can induce apoptosis by affecting more than one cell death pathway such as cleaving anti-apoptotic factor Bcl-2, pro-caspase 8, and the breast carcinoma-associated protein 3 (BCA3) (Figure 14). The cleavage of caspase 8 leads to the production of casp8p41 fragment, which can induce apoptosis in a caspase 9- and Bak/Bax-dependent manner leading to cytochrome c release from mitochondria and activation of caspases 3 and 7. It is noteworthy that the HIV-1 protease cleavage site of pro-caspase-8 is different than the typical cleavage site of protease highlighting that there is low possibility of correlated mutation [251,252,253,254,255].
Cummins et al. 2021 [256], conducted a trial searching for once weekly oral ixazomib (Figure 14), FDA approved proteasome inhibitor, in ART-suppressed, HIV-1 carrying adults. They showed the safety of ixazomib for 24 weeks in HIV-1 infected persons. Latency reversal and decline in HIV-1 reservoir size were ex vivo and in vivo observed.
5. Conclusions and Future Perspectives
HIV-1/AIDS pandemic continues to grow at an alarming rate worldwide. In state-of-the-art progress in cART has made a dramatic change in the life quality and survival of people living with HIV-1. However, the presence of latent reservoirs, the development of drug resistance, nonadherence and serious side effects have posed the greatest challenges in HIV-1 treatment. It is imperative that HIV-1 carrying patients should administer lifelong multi-drug therapy. All the available anti-HIV-1 drugs only decrease the HIV-1 activity in infected cells without definite elimination. If therapy is interrupted, latent reservoirs can reignite new rounds of infection, destroy the immune system of patient and cause eventual death. In particular deciphering the precise mechanisms of HIV-1 latency will allow to develop new anti-HIV-1 compounds with durable efficacy, tolerability and safety.
A new approach proposed by our research group “lock-in and apoptosis” pertains to L-HIPPO, a potential anti-HIV-1 compound, is able to block the budding of offspring virus after the “kick” process and facilitates the virus to be stuck in an HIV-1-infected host cell, which subsequently undergoes apoptosis along with the virus, leading to the definite HIV elimination from the body. Intense research efforts and several modern modalities have been devoted to finding more effective anti-HIV drugs with well-matched pharmacokinetic profile, metabolic stability, improved adherence, and high barrier to resistance, along with eliminating the latent reservoirs, the insurmountable problem over the years. Up to now, there is no antiretroviral agent that targets the budding phase driven by Gag. At this point, L-HIPPO might be a game-changer filling gaps and accelerating HIV-1 cure.
Abner et al. 2019 [246], once proposed that the additional use of ixazomib could rekindle the hope to be the first true dual-functioning agent and for our continuation research, ixazomib might contribute to apoptotic effects of L-HIPPO and rapidly elimination of latent reservoirs.
This review aims to interrogate the positive and negative features of available approved drugs and alternative curative treatment strategies oriented with a purpose of developing novel potent anti-HIV-1 drug candidates with superior properties.
Author Contributions
Conceptualization, B.S. and H.C.; methodology, B.S. and H.C.; software, B.S. and H.C.; validation, B.S. and H.C.; formal analysis, B.S. and H.C.; investigation, B.S. and H.C.; resources, B.S. and H.C.; data curation, B.S. and H.C.; writing—original draft preparation, B.S. and H.C.; writing—review and editing, B.S., M.O., M.F., and H.C.; visualization, B.S. and H.C.; supervision, H.C.; project administration, H.C.; funding acquisition, H.C. All authors have read and agreed to the published version of the manuscript.
Funding
This project has received funding from the European Union’s Horizon Europe research and innovation programme under the Marie Skłodowska-Curie grant agreement No: 10106939.

Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Authors would like to dedicate this manuscript to the memory of Fevzi Sever and Nizar Turker.
Conflicts of Interest
The authors declare no conflict of interest with Science Farm Ltd.
References
- German Advisory Committee Blood (Arbeitskreis Blut), Subgroup ‘Assessment of Pathogens Transmissible by Blood’. Human Immunodeficiency Virus (HIV). Transfus Med Hemother 2016, 43, 203-22. [CrossRef]
- Bbosa, N.; Kaleebu P, Ssemwanga D. HIV subtype diversity worldwide. Curr Opin HIV AIDS 2019, 14, 153-160. [CrossRef]
- Kwon, D.; Han, M.J.; Seo, K.W.; Kang, K.S. Combinatorial Strategies for Long-term Control of HIV Infection. AIDS Rev 2020, 22, 175-182. [CrossRef]
- Moranguinho, I.; Taveira, N.; Bártolo, I. Antiretroviral Treatment of HIV-2 Infection: Available Drugs Resistance Pathways, and Promising New Compounds. Int J Mol Sci 2023, 24, 5905. [CrossRef]
- 5. World Health Organization, HIV and AIDS. https://www.who.int/news-room/fact-sheets/detail/hiv-aids, (accessed on 13 July 2023).
- Summers, N.A.; Armstrong, W.S. Management of Advanced HIV Disease. Infect Dis Clin North Am 2019, 33, 743-767. [CrossRef]
- Foka, F.E.T.; Mufhandu, H.T. Current ARTs, Virologic Failure, and Implications for AIDS Management: A Systematic Review. Viruses 2023, 15, 1732. [CrossRef]
- Lehman, A.; Ellis, J.; Nalintya, E.; Bahr, N.C.; Loyse, A.; Rajasingham, R. Advanced HIV disease: A review of diagnostic and prophylactic strategies. HIV Med 2023, 24, 859-876. [CrossRef]
- Shafran, S.D.; Di Perri, G.; Esser, S.; Lelièvre, J.D.; Parczewski, M. Planning HIV therapy to prevent future comorbidities: patient years for tenofovir alafenamide. HIV Med 2019, 20, 1-16. [CrossRef]
- Siliciano, J.D.; Siliciano, R.F. In Vivo Dynamics of the Latent Reservoir for HIV-1: New Insights and Implications for Cure. Annu Rev Pathol 2022, 17, 271-294. [CrossRef]
- Gandhi, R.T.; Bedimo, R.; Hoy, J.F.; Landovitz, R.J.; Smith, D.M.; Eaton, E.F.; Lehmann, C.; Springer, S.A.; Sax, P.E.; Thompson, M.A.; Benson, C.A.; Buchbinder, S.P.; Del Rio, C.; Eron, J.J. Jr.; Günthard, H.F.; Molina, J.M.; Jacobsen, D.M.; Saag, M.S. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2022 Recommendations of the International Antiviral Society-USA Panel. JAMA 2023, 329, 63-84. [CrossRef]
- Konrad, B.P.; Taylor, D.; Conway, J.M.; Ogilvie, G.S.; Coombs, D. On the duration of the period between exposure to HIV and detectable infection. Epidemics 2017, 20, 73-83. [CrossRef]
- Bekker, L.G.; Beyrer, C.; Mgodi, N.; Lewin, S.R.; Delany-Moretlwe, S.; Taiwo, B.; Masters, M.C.; Lazarus, J.V. HIV infection. Nat Rev Dis Primers 2023, 9, 42. [CrossRef]
- van Marle, G.; Church, D.L.; van der Meer, F.; Gill, M.J. Combating the HIV reservoirs. Biotechnol Genet Eng Rev 2018, 34, 76-89. [CrossRef]
- Park, J.; Morrow, C.D. Overexpression of the gag-pol precursor from human immunodeficiency virus type 1 proviral genomes results in efficient proteolytic processing in the absence of virion production. J Virol 1991, 65, 5111-5117. [CrossRef]
- Hamard-Peron, E.; Muriaux, D. Retroviral matrix and lipids, the intimate interaction. Retrovirology 2011, 8, 15. [CrossRef]
- Mattei, S.; Schur, F.K.; Briggs, J.A. Retrovirus maturation-an extraordinary structural transformation. Curr Opin Virol 2016, 18, 27-35. [CrossRef]
- Nagata, S.; Imai, J.; Makino, G.; Tomita, M.; Kanai, A. Evolutionary Analysis of HIV-1 Pol Proteins Reveals Representative Residues for Viral Subtype Differentiation. Front Microbiol 2017, 8, 2151. [CrossRef]
- Klingler, J.; Anton, H.; Réal, E.; Zeiger, M.; Moog, C., Mély, Y.; Boutant, E. How HIV-1 Gag Manipulates Its Host Cell Proteins: A Focus on Interactors of the Nucleocapsid Domain. Viruses 2020, 12, 888. [CrossRef]
- Azimi, F.C.; Lee, J.E. Structural perspectives on HIV-1 Vif and APOBEC3 restriction factor interactions. Protein Sci 2020, 29, 391-406. [CrossRef]
- Faust, T.B.; Binning, J.M.; Gross, J.D.; Frankel, A.D. Making Sense of Multifunctional Proteins: Human Immunodeficiency Virus Type 1 Accessory and Regulatory Proteins and Connections to Transcription. Annu Rev Virol 2017, 4, 241-260. [CrossRef]
- Müller, T.G.; Zila, V.; Peters, K.; Schifferdecker, S.; Stanic, M.; Lucic, B.; Laketa, V.; Lusic, M.; Müller, B.; Kräusslich, H.G. HIV-1 uncoating by release of viral cDNA from capsid-like structures in the nucleus of infected cells. Elife 2021, 10, e64776. [CrossRef]
- Alkhatib, G. The biology of CCR5 and CXCR4. Curr Opin HIV AIDS 2009, 4, 96-103. [CrossRef]
- Sundquist, W.I.; Kräusslich, H.G. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med 2012, 2, a006924. [CrossRef]
- Meng, B.; Ip, N.C.; Prestwood, L.J.; Abbink, T.E.; Lever, A.M. Evidence that the endosomal sorting complex required for transport-II (ESCRT-II) is required for efficient human immunodeficiency virus-1 (HIV-1) production. Retrovirology 2015, 12, 72. [CrossRef]
- Kleinpeter, A.B.; Freed, E.O. HIV-1 Maturation: Lessons Learned from Inhibitors. Viruses 2020, 12, 940. [CrossRef]
- Tompa, D.R.; Immanuel, A.; Srikanth, S.; Kadhirvel, S. Trends and strategies to combat viral infections: A review on FDA approved antiviral drugs. Int J Biol Macromol 2021, 172, 524-541. [CrossRef]
- Gupta, M.; Pak, A.J.; Voth, G.A. Critical mechanistic features of HIV-1 viral capsid assembly. Sci Adv 2023, 9, eadd7434. [CrossRef]
- Atta, M.G.; De Seigneux, S.; Lucas, G.M. Clinical Pharmacology in HIV Therapy. Clin J Am Soc Nephrol 2019, 14, 435-444. [CrossRef]
- Maeda, K.; Das, D.; Kobayakawa, T.; Tamamura, H.; Takeuchi, H. Discovery and Development of Anti-HIV Therapeutic Agents: Progress Towards Improved HIV Medication. Curr Top Med Chem 2019, 19, 1621-1649. [CrossRef]
- Deng, C.; Yan, H.; Wang, J.; Liu, B.; Liu, K.; Shi, Y. The anti-HIV potential of imidazole, oxazole and thiazole hybrids: A mini-review. Arab J Chem 2022, 15, 104242. [CrossRef]
- Vanangamudi, M.; Kurup, S.; Namasivayam, V. Non-nucleoside reverse transcriptase inhibitors (NNRTIs): A brief overview of clinically approved drugs and combination regimens. Curr Opin Pharmacol 2020, 54, 179-187. [CrossRef]
- Ghosn, J.; Taiwo, B.; Seedat, S.; Autran, B.; Katlama, C. HIV. Lancet 2018, 392, 685-697. [CrossRef]
- García-Deltoro, M. Rapid Initiation of Antiretroviral Therapy after HIV Diagnosis. AIDS Rev 2019, 21, 55-64. [CrossRef]
- Phanuphak, N.; Gulick, R.M. HIV treatment and prevention 2019: Current standards of care. Curr Opin HIV AIDS 2020, 15, 4-12. [CrossRef]
- Achhra, A.C.; Mwasakifwa, G.; Amin, J.; Boyd, M.A. Efficacy and safety of contemporary dual-drug antiretroviral regimens as first-line treatment or as a simplification strategy: a systematic review and meta-analysis. Lancet HIV 2016, 3, e351-e360. [CrossRef]
- Orkin, C.; Llibre, J.M.; Gallien, S.; Antinori, A.; Behrens, G.; Carr, A. Nucleoside reverse transcriptase inhibitor-reducing strategies in HIV treatment: assessing the evidence. HIV Med 2018, 19, 18-32. [CrossRef]
- Zhang, K.; Zhang, Y.; Liu, X.; Li, A.; Gao, M.; Hou, J.; Guo, C.; Zhang, T.; Wu, H.; Chen, G.; Huang, X. Three-Drug Regimens Containing Integrase Inhibitor Show Good Efficacy and Safety in Treatment-Naive Patients With HIV-1: A Bayesian Analysis. Front Pharmacol 2021, 12, 603068. [CrossRef]
- Burns, J.E.; Stöhr, W.; Kinloch-De Loes, S.; Fox, J.; Clarke, A.; Nelson, M.; Thornhill, J.; Babiker, A.; Frater, J.; Pett, S.L.; Fidler, S. Tolerability of four-drug antiretroviral combination therapy in primary HIV-1 infection. HIV Med 2021, 22, 770-774. [CrossRef]
- Flexner, C. Modern Human Immunodeficiency Virus Therapy: Progress and Prospects. Clin Pharmacol Ther 2019, 105, 61-70. [CrossRef]
- Shyr, Z.A.; Cheng, Y.S.; Lo, D.C.; Zheng, W. Drug combination therapy for emerging viral diseases. Drug Discov Today 2021, 26, 2367-2376. [CrossRef]
- Esté, J.A.; Cihlar, T. Current status and challenges of antiretroviral research and therapy. Antiviral Res 2010, 85, 25-33. [CrossRef]
- Hawkins, T. Understanding and managing the adverse effects of antiretroviral therapy. Antiviral Res 2010, 85, 201-209. [CrossRef]
- Addis, D.R.; DeBerry, J.J.; Aggarwal, S. Chronic Pain in HIV. Mol Pain 2020, 16, 1744806920927276. [CrossRef]
- Cambou, M.C.; Landovitz, R.J. Novel Antiretroviral Agents. Curr HIV/AIDS Rep 2020, 17, 118-124. [CrossRef]
- Chatzidimitriou, D.; Tsotridou, E.; Grigoropoulos, P.; Skoura, L. HIV-1: Towards understanding the nature and quantifying the latent reservoir. Acta Virol 2020, 64, 3-9. [CrossRef]
- Dubrocq, G.; Rakhmanina, N. Antiretroviral therapy interruptions: impact on HIV treatment and transmission. HIV AIDS (Auckl) 2018, 10, 91-101. [CrossRef]
- Kalidasan, V.; Theva Das, K. Lessons Learned From Failures and Success Stories of HIV Breakthroughs: Are We Getting Closer to an HIV Cure? Front Microbiol 2020, 11, 46. [CrossRef]
- Bailey, H.; Zash, R.; Rasi, V.; Thorne, C. HIV treatment in pregnancy. Lancet HIV 2018, 5, e457-e467. [CrossRef]
- Singh, K.; Marchand, B.; Kirby, K.A.; Michailidis, E.; Sarafianos, S.G. Structural Aspects of Drug Resistance and Inhibition of HIV-1 Reverse Transcriptase. Viruses 2010, 2, 606-638. [CrossRef]
- Cilento, M.E.; Kirby, K.A.; Sarafianos, S.G. Avoiding Drug Resistance in HIV Reverse Transcriptase. Chem Rev 2021, 121, 3271-3296. [CrossRef]
- Deng, C.; Yan, H.; Wang, J.; Liu, K.; Liu, B.; Shi, Y. Current scenario on non-nucleoside reverse transcriptase inhibitors (2018-present). Arab J Chem 2022, 15, 104378. [CrossRef]
- Wang, Z.; Cherukupalli, S.; Xie, M.; Wang, W.; Jiang, X.; Jia, R.; Pannecouque, C.; De Clercq, E.; Kang, D.; Zhan, P.; Liu, X. Contemporary Medicinal Chemistry Strategies for the Discovery and Development of Novel HIV-1 Non-nucleoside Reverse Transcriptase Inhibitors. J Med Chem 2022, 65, 3729-3757. [CrossRef]
- Podzamczer, D.; Fumero, E. The role of nevirapine in the treatment of HIV-1 disease. Expert Opin Pharmacother 2001, 2, 2065-78. [CrossRef]
- Ren, J.; Stammers, D.K. Structural basis for drug resistance mechanisms for non-nucleoside inhibitors of HIV reverse transcriptase. Virus Res 2008, 134, 157-70. [CrossRef]
- Costa, B.; Vale, N. Efavirenz: History, Development and Future. Biomolecules 2022, 13, 88. [CrossRef]
- Havens, J.P.; Podany, A.T.; Scarsi, K.K.; Fletcher, C.V. Clinical Pharmacokinetics and Pharmacodynamics of Etravirine: An Updated Review. Clin Pharmacokinet 2020, 59, 137-154. [CrossRef]
- Sharma, M.; Saravolatz, L.D. Rilpivirine: a new non-nucleoside reverse transcriptase inhibitor. J Antimicrob Chemother 2013, 68, 250-256. [CrossRef]
- Ferretti, F.; Boffito, M. Rilpivirine long-acting for the prevention and treatment of HIV infection. Curr Opin HIV AIDS 2018, 13, 300-307. [CrossRef]
- Mu, Y.; Pham, M.; Podany, A.T.; Cory, T.J. Evaluating emtricitabine + rilpivirine + tenofovir alafenamide in combination for the treatment of HIV-infection. Expert Opin Pharmacother 2020, 21, 389-397. [CrossRef]
- Colombier, M.A.; Molina, J.M. Doravirine: a review. Curr Opin HIV AIDS 2018, 13, 308-314. [CrossRef]
- Deeks, E.D. Doravirine: First Global Approval. Drugs 2018, 78, 1643-1650. [CrossRef]
- Pham, H.T.; Xiao, M.A.; Principe, M.A.; Wong, A.; Mesplède, T. Pharmaceutical, clinical, and resistance information on doravirine, a novel non-nucleoside reverse transcriptase inhibitor for the treatment of HIV-1 infection. Drugs Context 2020, 9, 2019-11-4. [CrossRef]
- Shin, Y.H.; Park, C.M.; Yoon, C.H. An Overview of Human Immunodeficiency Virus-1 Antiretroviral Drugs: General Principles and Current Status. Infect Chemother 2021, 53, 29-45. [CrossRef]
- Wonganan, P.; Limpanasithikul, W.; Jianmongkol, S.; Kerr, S.J.; Ruxrungtham, K. Pharmacokinetics of nucleoside/nucleotide reverse transcriptase inhibitors for the treatment and prevention of HIV infection. Expert Opin Drug Metab Toxicol 2020, 16, 551-564. [CrossRef]
- Yoshida, Y.; Honma, M.; Kimura, Y.; Abe, H. Structure, Synthesis and Inhibition Mechanism of Nucleoside Analogues as HIV-1 Reverse Transcriptase Inhibitors (NRTIs). ChemMedChem 2021, 16, 743-766. [CrossRef]
- Fraley, A.W.; Chen, D.; Johnson, K.; McLaughlin, L.W. An HIV reverse transcriptase-selective nucleoside chain terminator. J Am Chem Soc 2003, 125, 616-617. [CrossRef]
- Holec, A.D.; Mandal, S.; Prathipati, P.K.; Destache, C.J. Nucleotide Reverse Transcriptase Inhibitors: A Thorough Review, Present Status and Future Perspective as HIV Therapeutics. Curr HIV Res 2017, 15, 411-421. [CrossRef]
- Bianco, M.D.C.A.D.; Inacio Leite, D.; Silva Castelo Branco, F.; Boechat, N.; Uliassi, E.; Bolognesi, M.L.; Bastos, M.M. The Use of Zidovudine Pharmacophore in Multi-Target-Directed Ligands for AIDS Therapy. Molecules 2022, 27, 8502. [CrossRef]
- Quercia, R.; Perno, C.F.; Koteff, J.; Moore, K.; McCoig, C.; St Clair, M.; Kuritzkes, D. Twenty-Five Years of Lamivudine: Current and Future Use for the Treatment of HIV-1 Infection. J Acquir Immune Defic Syndr 2018, 78, 125-135. [CrossRef]
- Cahn, P. Emtricitabine: a new nucleoside analogue for once-daily antiretroviral therapy. Expert Opin Investig Drugs 2004, 13, 55-68. [CrossRef]
- Cusato, J.; Allegra, S.; Nicolò, A.; Calcagno, A.; D’Avolio, A. Precision medicine for HIV: where are we? Pharmacogenomics 2018, 19, 145-165. [CrossRef]
- Piliero, P.J. Pharmacokinetic properties of nucleoside/nucleotide reverse transcriptase inhibitors. J Acquir Immune Defic Syndr 2004, 37 Suppl 1, S2-S12. [CrossRef]
- Saag, M.S.; Gandhi, R.T.; Hoy, J.F.; Landovitz, R.J.; Thompson, M.A.; Sax, P.E.; Smith, D.M.; Benson, C.A.; Buchbinder, S.P.; Del Rio, C.; Eron, J.J. Jr.; Fätkenheuer, G.; Günthard, H.F.; Molina, J.M.; Jacobsen, D.M.; Volberding, P.A. Antiretroviral Drugs for Treatment and Prevention of HIV Infection in Adults: 2020 Recommendations of the International Antiviral Society-USA Panel. JAMA 2020, 324, 1651-1669. [CrossRef]
- Li, G.; Wang, Y.; De Clercq, E. Approved HIV reverse transcriptase inhibitors in the past decade. Acta Pharm Sin B 2022, 12, 1567-1590. [CrossRef]
- Schinazi, R.F.; Patel, D.; Ehteshami, M. The best backbone for HIV prevention, treatment, and elimination: Emtricitabine+tenofovir. Antivir Ther 2022, 27, 13596535211067599. [CrossRef]
- Louis, J.M.; Ishima, R.; Torchia, D.A.; Weber, I.T. HIV-1 protease: structure, dynamics, and inhibition. Adv Pharmacol 2007, 55, 261-298. [CrossRef]
- Huang, L.; Chen, C. Understanding HIV-1 protease autoprocessing for novel therapeutic development. Future Med Chem 2013, 5, 1215-1229. [CrossRef]
- Konvalinka, J.; Kräusslich, H.G.; Müller, B. Retroviral proteases and their roles in virion maturation. Virology 2015, 479-480, 403-417. [CrossRef]
- Majerová, T.; Konvalinka, J. Viral proteases as therapeutic targets. Mol Aspects Med 2022, 88, 101159. [CrossRef]
- Tabler, C.O.; Wegman, S.J.; Chen, J.; Shroff, H.; Alhusaini, N.; Tilton, J.C. The HIV-1 Viral Protease Is Activated during Assembly and Budding Prior to Particle Release. J Virol 2022, 96, e0219821. [CrossRef]
- Brik, A.; Wong, C.H. HIV-1 protease: mechanism and drug discovery. Org Biomol Chem 2003, 1, 5-14. [CrossRef]
- Ghosh, A.K.; Osswald, H.L.; Prato, G. Recent Progress in the Development of HIV-1 Protease Inhibitors for the Treatment of HIV/AIDS. J Med Chem 2016, 59, 5172-5208. [CrossRef]
- Eron, J.J. HIV-1 Protease Inhibitors. Clin Infect Dis 2000, 30, S160–S170. [CrossRef]
- Lv, Z.; Chu, Y.; Wang, Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS (Auckl) 2015, 7, 95-104. [CrossRef]
- Weber, I.T.; Wang, Y.F.; Harrison, R.W. HIV Protease: Historical Perspective and Current Research. Viruses 2021, 13, 839. [CrossRef]
- Nascimento, A.L.C.S.; Fernandes, R.P.; Quijia, C.; Araujo, V.H.S.; Pereira, J.; Garcia, J.S.; Trevisan, M.G.; Chorilli, M. Pharmacokinetic Parameters of HIV-1 Protease Inhibitors. ChemMedChem 2020, 15, 1018-1029. [CrossRef]
- Chan-Tack, K.M.; Struble, K.A.; Birnkrant, D.B. Intracranial hemorrhage and liver-associated deaths associated with tipranavir/ritonavir: review of cases from the FDA’s Adverse Event Reporting System. AIDS Patient Care STDS 2008, 22, 843-850. [CrossRef]
- Orman, J.S.; Perry, C.M. Tipranavir: a review of its use in the management of HIV infection. Drugs 2008, 68, 1435-1463. [CrossRef]
- Spagnuolo, V.; Castagna, A.; Lazzarin, A. Darunavir for the treatment of HIV infection. Expert Opin Pharmacother 2018, 19, 1149-1163. [CrossRef]
- Squillace, N.; Bozzi, G.; Colella, E.; Gori, A.; Bandera, A. Darunavir-cobicistat-emtricitabine-tenofovir alafenamide: safety and efficacy of a protease inhibitor in the modern era. Drug Des Devel Ther 2018, 12, 3635-3643. [CrossRef]
- Doms, R.W.; Moore, J.P. HIV-1 membrane fusion: targets of opportunity. J Cell Biol 2000, 151, F9-14. [CrossRef]
- Cai, L.; Jiang, S. Development of peptide and small-molecule HIV-1 fusion inhibitors that target gp41. ChemMedChem 2010, 5, 1813-1824. [CrossRef]
- Falkenhagen, A.; Joshi, S. HIV Entry and Its Inhibition by Bifunctional Antiviral Proteins. Mol Ther Nucleic Acids 2018, 13, 347-364. [CrossRef]
- Wang, C; Cheng, S.; Zhang, Y.; Ding, Y.; Chong, H.; Xing, H.; Jiang, S.; Li, X.; Ma, L. Long-Acting HIV-1 Fusion Inhibitory Peptides and their Mechanisms of Action. Viruses 2019, 11, 811. [CrossRef]
- Xiao, T.; Cai, Y.; Chen, B. HIV-1 Entry and Membrane Fusion Inhibitors. Viruses 2021, 13, 735. [CrossRef]
- Negi, G.; Sharma, A.; Dey, M.; Dhanawat, G.; Parveen, N. Membrane attachment and fusion of HIV-1, influenza A, and SARS-CoV-2: resolving the mechanisms with biophysical methods. Biophys Rev 2022, 14, 1109-1140. [CrossRef]
- Matthews, T.; Salgo, M.; Greenberg, M.; Chung, J.; DeMasi, R.; Bolognesi, D. Enfuvirtide: The first therapy to inhibit the entry of HIV-1 into host CD4 lymphocytes. Nat Rev Drug Discov 2004, 3, 215-225. [CrossRef]
- Pang, W.; Tam, S.C.; Zheng, Y.T. Current peptide HIV type-1 fusion inhibitors. Antivir Chem Chemother 2009, 20, 1-18. [CrossRef]
- Dorr, P.; Westby, M.; Dobbs, S.; Griffin, P.; Irvine, B.; Macartney, M.; Mori, J.; Rickett, G.; Smith-Burchnell, C.; Napier, C.; Webster, R.; Armour, D.; Price, D.; Stammen, B.; Wood, A.; Perros, M. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother 2005, 49, 4721-4732. [CrossRef]
- Tan, Q.; Zhu, Y.; Li, J.; Chen, Z.; Han, G.W.; Kufareva, I.; Li, T.; Ma, L.; Fenalti, G.; Li, J.; Zhang, W.; Xie, X.; Yang, H.; Jiang, H.; Cherezov, V.; Liu, H.; Stevens, R.C.; Zhao, Q.; Wu, B. Structure of the CCR5 chemokine receptor-HIV entry inhibitor maraviroc complex. Science 2013, 341, 1387-1390. [CrossRef]
- Kesavardhana, S.; Varadarajan, R. Stabilizing the native trimer of HIV-1 Env by destabilizing the heterodimeric interface of the gp41 postfusion six-helix bundle. J Virol 2014, 88, 9590-9604. [CrossRef]
- Woollard, S.M.; Kanmogne, G.D. Maraviroc: a review of its use in HIV infection and beyond. Drug Des Devel Ther 2015, 9, 5447-5468. [CrossRef]
- Freeman, M.M.; Seaman, M.S.; Rits-Volloch, S.; Hong, X.; Kao, C.Y.; Ho, D.D.; Chen, B. Crystal structure of HIV-1 primary receptor CD4 in complex with a potent antiviral antibody. Structure 2010, 18, 1632-1641. [CrossRef]
- Markham, A. Ibalizumab: First Global Approval. Drugs 2018, 78, 781-785. [CrossRef]
- Beccari, M.V.; Mogle, B.T.; Sidman, E.F.; Mastro, K.A.; Asiago-Reddy, E.; Kufel, W.D. Ibalizumab, a Novel Monoclonal Antibody for the Management of Multidrug-Resistant HIV-1 Infection. Antimicrob Agents Chemother 2019, 63, e00110-19. [CrossRef]
- Blair, H.A. Ibalizumab: A Review in Multidrug-Resistant HIV-1 Infection. Drugs 2020, 80, 189-196. [CrossRef]
- Chahine, E.B.; Durham, S.H. Ibalizumab: The First Monoclonal Antibody for the Treatment of HIV-1 Infection. Ann Pharmacother 2021, 55, 230-239. [CrossRef]
- Kozal, M.; Aberg, J.; Pialoux, G.; Cahn, P.; Thompson, M.; Molina, J.M.; Grinsztejn, B.; Diaz, R.; Castagna, A.; Kumar, P.; Latiff, G.; DeJesus, E.; Gummel, M.; Gartland, M.; Pierce, A.; Ackerman, P.; Llamoso, C.; Lataillade, M.; BRIGHTE Trial Team. Fostemsavir in Adults with Multidrug-Resistant HIV-1 Infection. N Engl J Med 2020, 382, 1232-1243. [CrossRef]
- Markham, A. Fostemsavir: First Approval. Drugs 2020, 80, 1485-1490. [CrossRef]
- Seval, N.; Frank, C.; Kozal, M. Fostemsavir for the treatment of HIV. Expert Rev Anti Infect Ther 2021, 19, 961-966. [CrossRef]
- Yuan, S.; Luo, Y.Q.; Zuo, J.H.; Liu, H.; Li, F.; Yu, B. New drug approvals for 2020: Synthesis and clinical applications. Eur J Med Chem 2021, 215, 113284. [CrossRef]
- Muccini, C.; Canetti, D.; Castagna, A.; Spagnuolo, V. Efficacy and Safety Profile of Fostemsavir for the Treatment of People with Human Immunodeficiency Virus-1 (HIV-1): Current Evidence and Place in Therapy. Drug Des Devel Ther 2022, 16, 297-304. [CrossRef]
- Krishnan, L.; Li, X.; Naraharisetty, H.L.; Hare, S.; Cherepanov, P.; Engelman, A. Structure-based modeling of the functional HIV-1 intasome and its inhibition. Proc Natl Acad Sci U S A 2010, 107, 15910-15915. [CrossRef]
- Engelman, A.N. Multifaceted HIV integrase functionalities and therapeutic strategies for their inhibition. J Biol Chem 2019, 294, 15137-15157. [CrossRef]
- Passos, D.O.; Li, M.; Jóźwik, I.K.; Zhao, X.Z.; Santos-Martins, D.; Yang, R.; Smith, S.J.; Jeon, Y.; Forli, S.; Hughes, S.H.; Burke, T.R. Jr.; Craigie, R.; Lyumkis, D. Structural basis for strand-transfer inhibitor binding to HIV intasomes. Science 2020, 367(6479), 810-814. [CrossRef]
- Lu, C.H.; Bednarczyk, E.M.; Catanzaro, L.M.; Shon, A.; Xu, J.C.; Ma, Q. Pharmacokinetic drug interactions of integrase strand transfer inhibitors. Curr Res Pharmacol Drug Discov 2021, 2, 100044. [CrossRef]
- Hicks, C.; Gulick, R.M. Raltegravir: the first HIV type 1 integrase inhibitor. Clin Infect Dis 2009, 48, 931-939. [CrossRef]
- Scarsi, K.K.; Havens, J.P.; Podany, A.T.; Avedissian, S.N.; Fletcher, C.V. HIV-1 Integrase Inhibitors: A Comparative Review of Efficacy and Safety. Drugs 2020, 80, 1649-1676. [CrossRef]
- Shimura, K.; Kodama, E.N. Elvitegravir: a new HIV integrase inhibitor. Antivir Chem Chemother 2009, 20, 79-85. [CrossRef]
- Johns, B.A.; Kawasuji, T.; Weatherhead, J.G.; Taishi, T.; Temelkoff, D.P.; Yoshida, H.; Akiyama, T.; Taoda, Y.; Murai, H.; Kiyama, R.; Fuji, M.; Tanimoto, N.; Jeffrey, J.; Foster, S.A.; Yoshinaga, T.; Seki, T.; Kobayashi, M.; Sato, A.; Johnson, M.N.; Garvey, E.P.; Fujiwara, T. Carbamoyl pyridone HIV-1 integrase inhibitors 3. A diastereomeric approach to chiral nonracemic tricyclic ring systems and the discovery of dolutegravir (S/GSK1349572) and (S/GSK1265744). J Med Chem 2013, 56, 5901-5916. [CrossRef]
- Fantauzzi, A.; Mezzaroma, I. Dolutegravir: clinical efficacy and role in HIV therapy. Ther Adv Chronic Dis 2014, 5, 164-177. [CrossRef]
- Scott, L.J. Dolutegravir/Lamivudine Single-Tablet Regimen: A Review in HIV-1 Infection. Drugs 2020, 80, 61-72. [CrossRef]
- Santevecchi, B.A.; Miller, S.; Childs-Kean, L.M. Doing More with Less: Review of Dolutegravir-Lamivudine, a Novel Single-Tablet Regimen for Antiretroviral-Naïve Adults With HIV-1 Infection. Ann Pharmacother 2020, 54, 1252-1259. [CrossRef]
- Max, B. Update on HIV integrase inhibitors for the treatment of HIV-1 infection. Future Virol 2019, 14, 693–709. [CrossRef]
- Sarode, I.M.; Jindal, A.B. Current status of dolutegravir delivery systems for the treatment of HIV-1 infection. J Drug Deliv Sci Technol 2022, 76, 103802. [CrossRef]
- Stellbrink, H.J.; Hoffmann, C. Cabotegravir: its potential for antiretroviral therapy and preexposure prophylaxis. Curr Opin HIV AIDS 2018, 13, 334-340. [CrossRef]
- Canetti, D.; Spagnuolo, V. An evaluation of cabotegravir for HIV treatment and prevention. Expert Opin Pharmacother 2021, 22, 403-414. [CrossRef]
- Prather, C.; Jeon, C. Cabotegravir: The first long-acting injectable for HIV pre-exposure prophylaxis. Am J Health Syst Pharm 2022, 79, 1898-1905. [CrossRef]
- Durham, S.H.; Milam, A.; Waer, D.; Chahine, E.B. Cabotegravir: The First Long-Acting Injectable for HIV Preexposure Prophylaxis. Ann Pharmacother 2023, 57, 306-316. [CrossRef]
- Liao, C.; Wang, Q. Authentic HIV-1 integrase inhibitors for the treatment of HIV-1/AIDS. In Privileged Scaffolds in Drug Discovery, Yu, B.; Li, N.; Fu, C., Eds.; Publisher: Academic Press, 2007; 154-196. doi. 10.1016/B978-0-443-18611-0.00026-7.
- Tsiang, M.; Jones, G.S.; Goldsmith, J.; Mulato, A.; Hansen, D.; Kan, E.; Tsai, L.; Bam, R.A.; Stepan, G.; Stray, K.M.; Niedziela-Majka, A.; Yant, S.R.; Yu, H.; Kukolj, G.; Cihlar, T.; Lazerwith, S.E.; White, K.L.; Jin, H. Antiviral Activity of Bictegravir (GS-9883), a Novel Potent HIV-1 Integrase Strand Transfer Inhibitor with an Improved Resistance Profile. Antimicrob Agents Chemother 2016, 60, 7086-7097. [CrossRef]
- De Clercq, E.; Zhang, Z.; Huang, J.; Zhang, M.; Li, G. Biktarvy for the treatment of HIV infection: Progress and prospects. Biochem Pharmacol 2023, 217, 115862. [CrossRef]
- Link, J.O.; Rhee, M.S.; Tse, W.C.; Zheng, J.; Somoza, J.R.; Rowe, W.; Begley, R.; Chiu, A.; Mulato, A.; Hansen, D.; Singer, E.; Tsai, L.K.; Bam, R.A.; Chou, C.H.; Canales, E.; Brizgys, G.; Zhang, J.R.; Li, J.; Graupe, M.; Morganelli, P.; Liu, Q.; Wu, Q.; Halcomb, R.L.; Saito, R.D.; Schroeder, S.D.; Lazerwith, S.E.; Bondy, S.; Jin, D.; Hung, M.; Novikov, N.; Liu, X.; Villaseñor, A.G.; Cannizzaro, C.E.; Hu, E.Y.; Anderson, R.L.; Appleby, T.C.; Lu, B.; Mwangi, J.; Liclican, A.; Niedziela-Majka, A.; Papalia, G.A.; Wong, M.H.; Leavitt, S.A.; Xu, Y.; Koditek, D.; Stepan, G.J.; Yu, H.; Pagratis, N.; Clancy, S.; Ahmadyar, S.; Cai, T.Z.; Sellers, S.; Wolckenhauer, S.A.; Ling, J.; Callebaut, C.; Margot, N.; Ram, R.R.; Liu, Y.P.; Hyland, R.; Sinclair, G.I.; Ruane, P.J.; Crofoot, G.E.; McDonald, C.K.; Brainard, D.M.; Lad, L.; Swaminathan, S.; Sundquist, W.I.; Sakowicz, R.; Chester, A.E.; Lee, W.E.; Daar, E.S.; Yant, S.R.; Cihlar, T. Clinical targeting of HIV capsid protein with a long-acting small molecule. Nature 2020, 584, 614-618. [CrossRef]
- Marcelin, A.G.; Charpentier, C.; Jary, A.; Perrier, M.; Margot, N.; Callebaut, C.; Calvez, V.; Descamps, D. Frequency of capsid substitutions associated with GS-6207 in vitro resistance in HIV-1 from antiretroviral-naive and -experienced patients. J Antimicrob Chemother 2020, 75, 1588-1590. [CrossRef]
- Bekerman, E.; Yant, S.R.; VanderVeen, L.; Hansen, D.; Lu, B.; Rowe, W.; Wang, K.; Callebaut, C. Long-acting lenacapavir acts as an effective preexposure prophylaxis in a rectal SHIV challenge macaque model. J Clin Invest 2023, 133, e167818. [CrossRef]
- Dzinamarira, T.; Almehmadi, M.; Alsaiari, A.A.; Allahyani, M.; Aljuaid, A.; Alsharif, A.; Khan, A.; Kamal, M.; Rabaan, A.A.; Alfaraj, A.H.; AlShehail, B.M.; Alotaibi, N.; AlShehail, S.M.; Imran, M. Highlights on the Development, Related Patents, and Prospects of Lenacapavir: The First-in-Class HIV-1 Capsid Inhibitor for the Treatment of Multi-Drug-Resistant HIV-1 Infection. Medicina (Kaunas) 2023, 59, 1041. [CrossRef]
- Subramanian, R.; Tang, J.; Zheng, J.; Lu, B.; Wang, K.; Yant, S.R.; Stepan, G.J.; Mulato, A.; Yu, H.; Schroeder, S.; Shaik, N.; Singh, R.; Wolckenhauer, S.; Chester, A.; Tse, W.C.; Chiu, A.; Rhee, M.; Cihlar, T.; Rowe, W.; Smith, B.J. Lenacapavir: A Novel, Potent, and Selective First-in-Class Inhibitor of HIV-1 Capsid Function Exhibits Optimal Pharmacokinetic Properties for a Long-Acting Injectable Antiretroviral Agent. Mol Pharm 2023, 20, 6213-6225. [CrossRef]
- Hitchcock, A.M.; Kufel, W.D.; Dwyer, K.A.M.; Sidman, E.F. Lenacapavir: A novel injectable HIV-1 capsid inhibitor. Int J Antimicrob Agents 2023, 63, 107009. [CrossRef]
- McFadden, W.M.; Snyder, A.A.; Kirby, K.A.; Tedbury, P.R.; Raj, M.; Wang, Z.; Sarafianos, S.G. Rotten to the core: antivirals targeting the HIV-1 capsid core. Retrovirology 2021, 18, 41. [CrossRef]
- Dvory-Sobol, H.; Shaik, N.; Callebaut, C.; Rhee, M.S. Lenacapavir: a first-in-class HIV-1 capsid inhibitor. Curr Opin HIV AIDS 2022, 17, 15-21. [CrossRef]
- Menéndez-Arias, L.; Delgado, R. Update and latest advances in antiretroviral therapy. Trends Pharmacol Sci 2022, 43, 16-29. [CrossRef]
- Paik, J. Lenacapavir: First Approval. Drugs 2022, 82, 1499-1504. [CrossRef]
- Segal-Maurer, S.; DeJesus, E.; Stellbrink, H.J.; Castagna, A.; Richmond, G.J.; Sinclair, G.I.; Siripassorn, K.; Ruane, P.J.; Berhe, M.; Wang, H.; Margot, N.A.; Dvory-Sobol, H.; Hyland, R.H.; Brainard, D.M.; Rhee, M.S.; Baeten, J.M.; Molina, J.M.; CAPELLA Study Investigators. Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection. N Engl J Med 2022, 386, 1793-1803. [CrossRef]
- Tailor, M.W.; Chahine, E.B.; Koren, D.; Sherman, E.M. Lenacapavir: A Novel Long-Acting Capsid Inhibitor for HIV. Ann Pharmacother 2023, 10600280231171375, (in press). [CrossRef]
- Xu, L.; Liu, H.; Murray, B.P.; Callebaut, C.; Lee, M.S.; Hong, A.; Strickley, R.G.; Tsai, L.K.; Stray, K.M.; Wang, Y.; Rhodes, G.R.; Desai, M.C. Cobicistat (GS-9350): A Potent and Selective Inhibitor of Human CYP3A as a Novel Pharmacoenhancer. ACS Med Chem Lett 2010, 1, 209-213. [CrossRef]
- Nathan, B.; Bayley, J.; Waters, L.; Post, F.A. Cobicistat: a Novel Pharmacoenhancer for Co-Formulation with HIV Protease and Integrase Inhibitors. Infect Dis Ther 2013, 2, 111-122. [CrossRef]
- Deeks, E.D. Cobicistat: a review of its use as a pharmacokinetic enhancer of atazanavir and darunavir in patients with HIV-1 infection. Drugs 2014, 74, 195-206. [CrossRef]
- Niu, Z.X.; Wang, Y.T.; Zhang, S.N.; Li, Y.; Chen, X.B.; Wang, S.Q.; Liu, H.M. Application and synthesis of thiazole ring in clinically approved drugs. Eur J Med Chem 2023, 250, 115172. [CrossRef]
- Choudhary, M.C.; Mellors, J.W. The transformation of HIV therapy: One pill once a day. Antivir Ther 2022, 27, 13596535211062396. [CrossRef]
- Portsmouth, S.D.; Scott, C.J. The renaissance of fixed dose combinations: Combivir. Ther Clin Risk Manag 2007, 3, 579-583.
- Blair, H.A. Dolutegravir/Rilpivirine: A Review in HIV-1 Infection. Drugs 2018, 78, 1741-1750. [CrossRef]
- Hester, E.K.; Astle, K. Dolutegravir-Rilpivirine, Dual Antiretroviral Therapy for the Treatment of HIV-1 Infection. Ann Pharmacother 2019, 53, 860-866. [CrossRef]
- Markham, A. Bictegravir: First Global Approval. Drugs 2018, 78, 601-606. [CrossRef]
- Pham, H.T.; Mesplède, T. Bictegravir in a fixed-dose tablet with emtricitabine and tenofovir alafenamide for the treatment of HIV infection: pharmacology and clinical implications. Expert Opin Pharmacother 2019, 20, 385-397. [CrossRef]
- Sang, Y.; Ding, L.; Zhuang, C.; Chen, F. Design strategies for long-acting anti-HIV pharmaceuticals. Curr Opin Pharmacol 2020, 54, 158-165. [CrossRef]
- Gulick, R.M.; Ribaudo, H.J.; Shikuma, C.M.; Lalama, C.; Schackman, B.R.; Meyer, W.A.; Acosta, E.P.; Schouten, J.; Squires, K.E.; Pilcher, C.D.; Murphy, R.L.; Koletar, S.L.; Carlson, M.; Reichman, R.C.; Bastow, B.; Klingman, K.L.; Kuritzkes, D.R.; AIDS Clinical Trials Group (ACTG) A5095 Study Team. Three- vs four-drug antiretroviral regimens for the initial treatment of HIV-1 infection: a randomized controlled trial. JAMA 2006, 296, 769-781. [CrossRef]
- Moreno, S.; Perno, C.F.; Mallon, P.W.; Behrens, G.; Corbeau, P.; Routy, J.P.; Darcis, G. Two-drug vs. three-drug combinations for HIV-1: Do we have enough data to make the switch? HIV Med 2019, 20, 2-12. [CrossRef]
- Waters, L.; Church, H. Two drugs regimens for HIV. Curr Opin Infect Dis 2020, 33, 28-33. [CrossRef]
- Bares, S.H.; Scarsi, K.K. A new paradigm for antiretroviral delivery: long-acting cabotegravir and rilpivirine for the treatment and prevention of HIV. Curr Opin HIV AIDS 2022, 17, 22-31. [CrossRef]
- Brizzi, M.; Pérez, S.E.; Michienzi, S.M.; Badowski, M.E. Long-acting injectable antiretroviral therapy: will it change the future of HIV treatment? Ther Adv Infect Dis 2023, 10, 20499361221149773. [CrossRef]
- Tedbury, P.R.; Novikova, M.; Alfadhli, A.; Hikichi, Y.; Kagiampakis, I.; KewalRamani, V.N.; Barklis, E.; Freed, E.O. HIV-1 Matrix Trimerization-Impaired Mutants Are Rescued by Matrix Substitutions That Enhance Envelope Glycoprotein Incorporation. J Virol 2019, 94, e01526-19. [CrossRef]
- Bou-Nader, C.; Muecksch, F.; Brown, J.B.; Gordon, J.M.; York, A.; Peng, C.; Ghirlando, R.; Summers, M.F.; Bieniasz, P.D.; Zhang, J. HIV-1 matrix-tRNA complex structure reveals basis for host control of Gag localization. Cell Host Microbe 2021, 29, 1421-1436.e7. [CrossRef]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) bisphosphate regulates HIV-1 Gag targeting to the plasma membrane. Proc Natl Acad Sci USA 2004, 101, 14889-14894. [CrossRef]
- Larson, D.R.; Ma, Y.M.; Vogt, V.M.; Webb, W.W. Direct measurement of Gag-Gag interaction during retrovirus assembly with FRET and fluorescence correlation spectroscopy. J Cell Biol 2003, 162, 1233-44. [CrossRef]
- Kutluay, S.B.; Zang, T.; Blanco-Melo, D.; Powell, C.; Jannain, D.; Errando, M.; Bieniasz, P.D. Global changes in the RNA binding specificity of HIV-1 gag regulate virion genesis. Cell 2014, 159, 1096-1109. [CrossRef]
- Dilley, K.A.; Nikolaitchik, O.A.; Galli, A.; Burdick, R.C.; Levine, L.; Li, K.; Rein, A.; Pathak, V.K.; Hu, W.S. Interactions between HIV-1 Gag and Viral RNA Genome Enhance Virion Assembly. J Virol 2017, 91, e02319-16. [CrossRef]
- Mercredi, P.Y.; Bucca, N.; Loeliger, B.; Gaines, C.R.; Mehta, M.; Bhargava, P.; Tedbury, P.R.; Charlier, L.; Floquet, N.; Muriaux, D.; Favard, C.; Sanders, C.R.; Freed, E.O.; Marchant, J.; Summers, M.F. Structural and Molecular Determinants of Membrane Binding by the HIV-1 Matrix Protein. J Mol Biol 2016, 428, 1637-1655. [CrossRef]
- Tedbury, P.R.; Novikova, M., Ablan, S.D.; Freed, E.O. Biochemical evidence of a role for matrix trimerization in HIV-1 envelope glycoprotein incorporation. Proc Natl Acad Sci U S A 2016, 113, E182-190. [CrossRef]
- Tran, R.J.; Lalonde, M.S.; Sly, K.L.; Conboy, J.C. Mechanistic Investigation of HIV-1 Gag Association with Lipid Membranes. J Phys Chem B 2019, 123, 22, 4673-4687. [CrossRef]
- Monje-Galvan, V.; Voth, G.A. Binding mechanism of the matrix domain of HIV-1 gag on lipid membranes. Elife 2020, 9, e58621. [CrossRef]
- Murphy, R.E.; Saad, J.S. The Interplay between HIV-1 Gag Binding to the Plasma Membrane and Env Incorporation. Viruses 2020, 12, 548. [CrossRef]
- Sumner, C.; Kotani, O.; Liu, S.; Musier-Forsyth, K.; Sato, H.; Ono, A. Molecular Determinants in tRNA D-arm Required for Inhibition of HIV-1 Gag Membrane Binding. J Mol Biol 2022, 434, 167390. [CrossRef]
- Socas, L.B.P.; Ambroggio, E.E. HIV-1 Gag specificity for PIP2 is regulated by macromolecular electric properties of both protein and membrane local environments. Biochim Biophys Acta Biomembr 2023, 1865, 184157. [CrossRef]
- Tomasini, M.D.; Johnson, D.S.; Mincer, J.S.; Simon, S.M. Modeling the dynamics and kinetics of HIV-1 Gag during viral assembly. PLoS One 2018, 13, e0196133. [CrossRef]
- Novikova, M.; Adams, L.J.; Fontana, J.; Gres, A.T.; Balasubramaniam, M.; Winkler, D.C.; Kudchodkar, S.B.; Soheilian, F.; Sarafianos, S.G.; Steven, A.C.; Freed, E.O. Identification of a Structural Element in HIV-1 Gag Required for Virus Particle Assembly and Maturation. mBio 2018, 9, e01567-18. [CrossRef]
- Chen, S.; Xu, J.; Liu, M.; Rao, A.L.N.; Zandi, R.; Gill, S.S.; Mohideen, U. Investigation of HIV-1 Gag binding with RNAs and lipids using Atomic Force Microscopy. PLoS One 2020, 15, e0228036. [CrossRef]
- Roos, W.H. High-speed AFM reveals the dynamics of virus budding. Biophys J 2022, 121, 4022-4023. [CrossRef]
- Anraku, K.; Fukuda, R.; Takamune, N.; Misumi, S.; Okamoto, Y.; Otsuka, M.; Fujita, M. Highly sensitive analysis of the interaction between HIV-1 Gag and phosphoinositide derivatives based on surface plasmon resonance. Biochemistry 2010, 49, 5109–5116. [CrossRef]
- Tateishi, H.; Anraku, K.; Koga, R.; Okamoto, Y.; Fujita, M.; Otsuka, M. Design and synthesis of lipid-coupled inositol 1,2,3,4,5,6-hexakisphosphate derivatives exhibiting high-affinity binding for the HIV-1 MA domain. Org Biomol Chem 2014, 12, 5006. [CrossRef]
- Tateishi, H.; Monde, K.; Anraku, K.; Koga, R.; Hayashi, Y.; Ciftci, H.I.; DeMirci, H.; Higashi, T.; Motoyama, K.; Arima, H.; Otsuka, M.; Fujita, M. A clue to unprecedented strategy to HIV eradication: “Lock-in and apoptosis”. Sci. Rep 2017, 7, 8957. [CrossRef]
- Ciftci, H.I.; Sierra, R.G.; Yoon, C.H.; Su, Z.; Tateishi, H.; Koga, R.; Kotaro, K.; Yumoto, F.; Senda, T.; Liang, M.; Wakatsuki, S.; Otsuka, M.; Fujita, M.; DeMirci, H. Serial Femtosecond X-Ray Diffraction of HIV-1 Gag MA-IP6 Microcrystals at Ambient Temperature. Int J Mol Sci 2019, 20, 1675. [CrossRef]
- Ciftci, H.; Tateishi, H.; Koiwai, K.; Koga, R.; Anraku, K.; Monde, K.; Dağ, Ç.; Destan, E.; Yuksel, B.; Ayan, E.; Yildirim, G.; Yigin, M.; Ertem, F.B.; Shafiei, A.; Guven, O.; Besler, S.O.; Sierra, R.G.; Yoon, C.H.; Su, Z.; Liang, M.; Acar, B.; Haliloglu, T.; Otsuka, M.; Yumoto, F.; Fujita, M.; Senda, T.; DeMirci, H. Structural insight into host plasma membrane association and assembly of HIV-1 matrix protein. Sci Rep 2021, 11, 15819. [CrossRef]
- Ciftci, H.; Sever, B.; Ayan, E.; Can, M.; DeMirci, H.; Otsuka, M.; TuYuN, A.F.; Tateishi, H.; Fujita, M. Identification of New L-Heptanoylphosphatidyl Inositol Pentakisphosphate Derivatives Targeting the Interaction with HIV-1 Gag by Molecular Modelling Studies. Pharmaceuticals (Basel) 2022, 15, 1255. [CrossRef]
- Zentner, I.; Sierra, L.J.; Fraser, A.K.; Maciunas, L.; Mankowski, M.K.; Vinnik, A.; Fedichev, P.; Ptak, R.G.; Martín-García, J.; Cocklin, S. Identification of a small-molecule inhibitor of HIV-1 assembly that targets the phosphatidylinositol (4,5)-bisphosphate binding site of the HIV-1 matrix protein. ChemMedChem 2013, 8, 426-432. [CrossRef]
- Alfadhli, A.; McNett, H.; Eccles, J.; Tsagli, S.; Noviello, C.; Sloan, R.; López, C.S.; Peyton, D.H.; Barklis, E. Analysis of small molecule ligands targeting the HIV-1 matrix protein-RNA binding site. J Biol Chem 2013, 288, 666-76. [CrossRef]
- Mattei, S.; Anders, M.; Konvalinka, J.; Kräusslich, H.G.; Briggs, J.A.; Müller, B. Induced maturation of human immunodeficiency virus. J Virol 2014, 88, 13722-13731. [CrossRef]
- Campbell, S.; Fisher, R.J.; Towler, E.M.; Fox, S.; Issaq, H.J.; Wolfe, T.; Phillips, L.R.; Rein, A. Modulation of HIV-like particle assembly in vitro by inositol phosphates. Proc Natl Acad Sci U S A 2001, 98, 10875-9. [CrossRef]
- Ricaña, C.L.; Dick, R.A. Inositol Phosphates and Retroviral Assembly: A Cellular Perspective. Viruses 2021, 13, 2516. [CrossRef]
- Poston, D.; Zang, T.; Bieniasz, P. Derivation and characterization of an HIV-1 mutant that rescues IP6 binding deficiency. Retrovirology 2021, 18, 25. [CrossRef]
- Pak, A.J.; Gupta, M.; Yeager, M.; Voth, G.A. Inositol Hexakisphosphate (IP6) Accelerates Immature HIV-1 Gag Protein Assembly toward Kinetically Trapped Morphologies. J Am Chem Soc 2022, 144, 10417-10428. [CrossRef]
- Kleinpeter, A.B.; Zhu, Y.; Mallery, D.L.; Ablan, S.D.; Chen, L.; Hardenbrook, N.; Saiardi, A.; James, L.C.; Zhang, P.; Freed, E.O. The Effect of Inositol Hexakisphosphate on HIV-1 Particle Production and Infectivity can be Modulated by Mutations that Affect the Stability of the Immature Gag Lattice. J Mol Biol 2023, 435, 168037. [CrossRef]
- Perilla, J.R.; Hadden-Perilla, J.A.; Gronenborn, A.M.; Polenova, T. Integrative structural biology of HIV-1 capsid protein assemblies: combining experiment and computation. Curr Opin Virol 2021, 48, 57-64. [CrossRef]
- Fujioka, T.; Kashiwada, Y.; Kilkuskie, R.E.; Cosentino, L.M.; Ballas, L.M.; Jiang, J.B.; Janzen, W.P.; Chen, I.S.; Lee, K.H. Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J Nat Prod 1994, 57, 243-247. [CrossRef]
- Kashiwada, Y.; Hashimoto, F.; Cosentino, L.M.; Chen, C.H.; Garrett, P.E.; Lee, K.H. Betulinic acid and dihydrobetulinic acid derivatives as potent anti-HIV agents. J Med Chem 1996, 39, 1016-7. [CrossRef]
- Kanamoto, T.; Kashiwada, Y.; Kanbara, K.; Gotoh, K.; Yoshimori, M.; Goto, T.; Sano, K.; Nakashima, H. Anti-human immunodeficiency virus activity of YK-FH312 (a betulinic acid derivative), a novel compound blocking viral maturation. Antimicrob Agents Chemother 2001, 45, 1225-1230. [CrossRef]
- Li, F.; Goila-Gaur, R.; Salzwedel, K.; Kilgore, N.R.; Reddick, M.; Matallana, C.; Castillo, A.; Zoumplis, D.; Martin, D.E.; Orenstein, J.M.; Allaway, G.P.; Freed, E.O.; Wild, C.T. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc Natl Acad Sci U S A 2003, 100, 13555-13560. [CrossRef]
- Adamson, C.S.; Ablan, S.D.; Boeras, I.; Goila-Gaur, R.; Soheilian, F.; Nagashima, K.; Li, F.; Salzwedel, K.; Sakalian, M.; Wild, C.T.; Freed, E.O. In vitro resistance to the human immunodeficiency virus type 1 maturation inhibitor PA-457 (Bevirimat). J Virol 2006, 80, 10957-10971. [CrossRef]
- Li, F.; Zoumplis, D.; Matallana, C.; Kilgore, N.R.; Reddick, M.; Yunus, A.S.; Adamson, C.S.; Salzwedel, K.; Martin, D.E.; Allaway, G.P.; Freed, E.O.; Wild, C.T. Determinants of activity of the HIV-1 maturation inhibitor PA-457. Virology 2006, 356, 217-224. [CrossRef]
- Sakalian, M.; McMurtrey, C.P.; Deeg, F.J.; Maloy, C.W.; Li, F.; Wild, C.T.; Salzwedel, K. 3-O-(3’,3’-dimethysuccinyl) betulinic acid inhibits maturation of the human immunodeficiency virus type 1 Gag precursor assembled in vitro. J Virol 2006, 80, 5716-22. [CrossRef]
- Zhou, J.; Chen, C.H.; Aiken, C. Human immunodeficiency virus type 1 resistance to the small molecule maturation inhibitor 3-O-(3’,3’-dimethylsuccinyl)-betulinic acid is conferred by a variety of single amino acid substitutions at the CA-SP1 cleavage site in Gag. J Virol 2006, 80, 12095-101. [CrossRef]
- Stoddart, C.A.; Joshi, P.; Sloan, B.; Bare, J.C.; Smith, P.C.; Allaway, G.P.; Wild, C.T.; Martin, D.E. Potent activity of the HIV-1 maturation inhibitor bevirimat in SCID-hu Thy/Liv mice. PLoS One 2007, 2, e1251. [CrossRef]
- Smith, P.F.; Ogundele, A.; Forrest, A.; Wilton, J.; Salzwedel, K.; Doto, J.; Allaway, G.P.; Martin, D.E. Phase I and II study of the safety, virologic effect, and pharmacokinetics/pharmacodynamics of single-dose 3-o-(3’,3’-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob Agents Chemother 2007, 51, 3574-3581. [CrossRef]
- Van Baelen, K.; Salzwedel, K.; Rondelez, E.; Van Eygen, V.; De Vos, S.; Verheyen, A.; Steegen, K.; Verlinden, Y.; Allaway, G.P.; Stuyver, L.J. Susceptibility of human immunodeficiency virus type 1 to the maturation inhibitor bevirimat is modulated by baseline polymorphisms in Gag spacer peptide 1. Antimicrob Agents Chemother 2009, 53, 2185-2188. [CrossRef]
- Coric, P.; Turcaud, S.; Souquet, F.; Briant, L.; Gay, B.; Royer, J.; Chazal, N.; Bouaziz, S. Synthesis and biological evaluation of a new derivative of bevirimat that targets the Gag CA-SP1 cleavage site. Eur J Med Chem 2013, 62, 453-465. [CrossRef]
- Dang, Z.; Ho, P.; Zhu, L.; Qian, K.; Lee, K.H., Huang, L.; Chen, C.H. New betulinic acid derivatives for bevirimat-resistant human immunodeficiency virus type-1. J Med Chem 2013, 56, 2029-2037. [CrossRef]
- Nowicka-Sans, B.; Protack, T.; Lin, Z.; Li, Z.; Zhang, S.; Sun, Y.; Samanta, H.; Terry, B.; Liu, Z.; Chen, Y.; Sin, N.; Sit, S.Y.; Swidorski, J.J.; Chen, J.; Venables, B.L.; Healy, M.; Meanwell, N.A.; Cockett, M.; Hanumegowda, U.; Regueiro-Ren, A.; Krystal, M.; Dicker, I.B. Identification and Characterization of BMS-955176, a Second-Generation HIV-1 Maturation Inhibitor with Improved Potency, Antiviral Spectrum, and Gag Polymorphic Coverage. Antimicrob Agents Chemother 2016, 60, 3956-3969. [CrossRef]
- Regueiro-Ren, A.; Liu, Z.; Chen, Y.; Sin, N.; Sit, S.Y.; Swidorski, J.J.; Chen, J.; Venables, B.L.; Zhu, J.; Nowicka-Sans, B.; Protack, T.; Lin, Z.; Terry, B.; Samanta, H.; Zhang, S.; Li, Z.; Beno, B.R.; Huang, X.S.; Rahematpura, S.; Parker, D.D.; Haskell, R.; Jenkins, S.; Santone, K.S.; Cockett, M.I.; Krystal, M.; Meanwell, N.A.; Hanumegowda, U.; Dicker, I.B. Discovery of BMS-955176, a Second Generation HIV-1 Maturation Inhibitor with Broad Spectrum Antiviral Activity. ACS Med Chem Lett 2016, 7, 568-572. [CrossRef]
- Regueiro-Ren, A.; Dicker, I.B.; Hanumegowda, U.; Meanwell, N.A. Second Generation Inhibitors of HIV-1 Maturation. ACS Med Chem Lett 2019, 10, 287-294. [CrossRef]
- Hwang, C.; Schürmann, D.; Sobotha, C.; Boffito, M.; Sevinsky, H.; Ray, N.; Ravindran, P.; Xiao, H.; Keicher, C.; Hüser, A.; Krystal, M.; Dicker, I.B.; Grasela, D.; Lataillade, M. Antiviral Activity, Safety, and Exposure-Response Relationships of GSK3532795, a Second-Generation Human Immunodeficiency Virus Type 1 Maturation Inhibitor, Administered as Monotherapy or in Combination With Atazanavir With or Without Ritonavir in a Phase 2a Randomized, Dose-Ranging, Controlled Trial (AI468002). Clin Infect Dis 2017, 65, 442-452. [CrossRef]
- Morales-Ramirez, J.; Bogner, J.R.; Molina, J.M.; Lombaard, J.; Dicker, I.B.; Stock, D.A.; DeGrosky, M.; Gartland, M.; Pene Dumitrescu, T.; Min, S.; Llamoso, C.; Joshi, S.R.; Lataillade, M. Safety, efficacy, and dose response of the maturation inhibitor GSK3532795 (formerly known as BMS-955176) plus tenofovir/emtricitabine once daily in treatment-naive HIV-1-infected adults: Week 24 primary analysis from a randomized Phase IIb trial. PLoS One 2018, 13, e0205368. [CrossRef]
- Chrobak, E.; Marciniec, K.; Dąbrowska, A.; Pęcak, P.; Bębenek, E.; Kadela-Tomanek, M.; Bak, A.; Jastrzębska, M.; Boryczka, S. New Phosphorus Analogs of Bevirimat: Synthesis, Evaluation of Anti-HIV-1 Activity and Molecular Docking Study. Int J Mol Sci 2019, 20, 5209. [CrossRef]
- Dicker, I.; Jeffrey, J.L., Protack, T.; Lin, Z.; Cockett, M.; Chen, Y.; Sit, S.Y.; Gartland, M.; Meanwell, N.A.; Regueiro-Ren, A.; Drexler, D.; Cantone, J.; McAuliffe, B.; Krystal, M. GSK3640254 Is a Novel HIV-1 Maturation Inhibitor with an Optimized Virology Profile. Antimicrob Agents Chemother 2022, 66, e0187621. [CrossRef]
- Tang, C.; Loeliger, E.; Kinde, I.; Kyere, S.; Mayo, K.; Barklis, E.; Sun, Y.; Huang, M.; Summers, M.F. Antiviral inhibition of the HIV-1 capsid protein. J Mol Biol 2003, 327, 1013-1020. [CrossRef]
- Kelly, B.N.; Kyere, S.; Kinde, I.; Tang, C.; Howard, B.R.; Robinson, H.; Sundquist, W.I.; Summers, M.F.; Hill, C.P. Structure of the antiviral assembly inhibitor CAP-1 complex with the HIV-1 CA protein. J Mol Biol 2007, 373, 355-366. [CrossRef]
- Fader, L.D.; Bethell, R.; Bonneau, P.; Bös, M.; Bousquet, Y.; Cordingley, M.G.; Coulombe, R.; Deroy, P.; Faucher, A.M.; Gagnon, A.; Goudreau, N.; Grand-Maître, C.; Guse, I.; Hucke, O.; Kawai, S.H.; Lacoste, J.E.; Landry, S.; Lemke, C.T.; Malenfant, E.; Mason, S.; Morin, S.; O’Meara, J.; Simoneau, B.; Titolo, S.; Yoakim, C. Discovery of a 1,5-dihydrobenzo[b][1,4]diazepine-2,4-dione series of inhibitors of HIV-1 capsid assembly. Bioorg Med Chem Lett 2011, 21, 398-404. [CrossRef]
- Lemke, C.T.; Titolo, S.; von Schwedler, U.; Goudreau, N.; Mercier, J.F.; Wardrop, E.; Faucher, A.M.; Coulombe, R.; Banik, S.S.; Fader, L.; Gagnon, A.; Kawai, S.H.; Rancourt, J.; Tremblay, M.; Yoakim, C.; Simoneau, B.; Archambault, J.; Sundquist, W.I.; Mason, S.W. Distinct effects of two HIV-1 capsid assembly inhibitor families that bind the same site within the N-terminal domain of the viral CA protein. J Virol 2012, 86, 6643-6655. [CrossRef]
- Lemke, C.T.; Titolo, S.; Goudreau, N.; Faucher, A.M.; Mason, S.W.; Bonneau, P. A novel inhibitor-binding site on the HIV-1 capsid N-terminal domain leads to improved crystallization via compound-mediated dimerization. Acta Crystallogr D Biol Crystallogr 2013, 69, 1115-1123. [CrossRef]
- Tremblay, M.; Bonneau, P.; Bousquet, Y.; DeRoy, P.; Duan, J.; Duplessis, M.; Gagnon, A.; Garneau, M.; Goudreau, N.; Guse, I.; Hucke, O.; Kawai, S.H.; Lemke, C.T.; Mason, S.W.; Simoneau, B.; Surprenant, S.; Titolo, S.; Yoakim, C. Inhibition of HIV-1 capsid assembly: optimization of the antiviral potency by site selective modifications at N1, C2 and C16 of a 5-(5-furan-2-yl-pyrazol-1-yl)-1H-benzimidazole scaffold. Bioorg Med Chem Lett 2012, 22, 7512-7517. [CrossRef]
- Goudreau, N.; Lemke, C.T.; Faucher, A.M.; Grand-Maître, C.; Goulet, S.; Lacoste, J.E.; Rancourt, J.; Malenfant, E.; Mercier, J.F.; Titolo, S.; Mason, S.W. Novel inhibitor binding site discovery on HIV-1 capsid N-terminal domain by NMR and X-ray crystallography. ACS Chem Biol 2013, 8, 1074-1082. [CrossRef]
- Sticht, J.; Humbert, M.; Findlow, S.; Bodem, J.; Müller, B.; Dietrich, U.; Werner, J.; Kräusslich, H.G. A peptide inhibitor of HIV-1 assembly in vitro. Nat Struct Mol Biol 2005, 12, 671-677. [CrossRef]
- Zhang, H.; Curreli, F.; Waheed, A.A.; Mercredi, P.Y.; Mehta, M.; Bhargava, P.; Scacalossi, D.; Tong, X.; Lee, S.; Cooper, A.; Summers, M.F.; Freed, E.O.; Debnath, A.K. Dual-acting stapled peptides target both HIV-1 entry and assembly. Retrovirology 2013, 10, 136. [CrossRef]
- Blair, W.S.; Cao, J.; Fok-Seang, J.; Griffin, P.; Isaacson, J.; Jackson, R.L.; Murray, E.; Patick, A.K.; Peng, Q.; Perros, M.; Pickford, C.; Wu, H.; Butler, S.L. New small-molecule inhibitor class targeting human immunodeficiency virus type 1 virion maturation. Antimicrob Agents Chemother 2009, 53, 5080-5087. [CrossRef]
- Blair, W.S.; Pickford, C.; Irving, S.L.; Brown, D.G.; Anderson, M.; Bazin, R.; Cao, J.; Ciaramella, G.; Isaacson, J.; Jackson, L.; Hunt, R.; Kjerrstrom, A.; Nieman, J.A.; Patick, A.K.; Perros, M.; Scott, A.D.; Whitby, K.; Wu, H.; Butler, S.L. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog 2010, 6, e1001220. [CrossRef]
- Dostálková, A.; Škach, K.; Kaufman, F.; Křížová, I.; Hadravová, R.; Flegel, M.; Ruml, T.; Hrabal, R.; Rumlová, M. PF74 and Its Novel Derivatives Stabilize Hexameric Lattice of HIV-1 Mature-Like Particles. Molecules 2020, 25, 1895. [CrossRef]
- Yant, S.R.; Mulato, A.; Hansen, D.; Tse, W.C.; Niedziela-Majka, A.; Zhang, J.R., Stepan, G.J.; Jin, D.; Wong, M.H.; Perreira, J.M.; Singer, E.; Papalia, G.A.; Hu, E.Y.; Zheng, J.; Lu, B.; Schroeder, S.D.; Chou, K.; Ahmadyar, S.; Liclican, A.; Yu, H.; Novikov, N.; Paoli, E.; Gonik, D.; Ram, R.R.; Hung, M.; McDougall, W.M.; Brass, A.L.; Sundquist, W.I.; Cihlar, T.; Link, J.O. A highly potent long-acting small-molecule HIV-1 capsid inhibitor with efficacy in a humanized mouse model. Nat Med 2019, 25, 1377-1384. [CrossRef]
- Zhang, Z.; Hou, W.; Chen, S. Updates on CRISPR-based gene editing in HIV-1/AIDS therapy. Virol Sin 2022, 37, 1-10. [CrossRef]
- Stephenson, K.E.; Wagh, K.; Korber, B.; Barouch, D.H. Vaccines and Broadly Neutralizing Antibodies for HIV-1 Prevention. Annu Rev Immunol 2020, 38, 673-703. [CrossRef]
- Choudhary, M.C.; Cyktor, J.C.; Riddler, S.A. Advances in HIV-1-specific chimeric antigen receptor cells to target the HIV-1 reservoir. J Virus Erad 2022, 8, 100073. [CrossRef]
- Sun, L.; Zhang, X.; Xu, S.; Huang, T.; Song, S.; Cherukupalli, S.; Zhan, P.; Liu, X. An insight on medicinal aspects of novel HIV-1 capsid protein inhibitors. Eur J Med Chem 2021, 217, 113380. [CrossRef]
- Zhang, L.; Wei, F.; Zhang, J.; Liu, C.; López-Carrobles, N.; Liu, X.; Menéndez-Arias, L.; Zhan, P. Current medicinal chemistry strategies in the discovery of novel HIV-1 ribonuclease H inhibitors. Eur J Med Chem 2022, 243, 114760. [CrossRef]
- Passaes, C.P.; Sáez-Cirión, A. HIV cure research: advances and prospects. Virology 2014, 454-455, 340-52. [CrossRef]
- Dick, A.; Cocklin, S. Recent Advances in HIV-1 Gag Inhibitor Design and Development. Molecules 2020, 25, 1687. [CrossRef]
- Saeb, S.; Wallet, C.; Rohr, O.; Schwartz, C.; Loustau, T. Targeting and eradicating latent CNS reservoirs of HIV-1: Original strategies and new models. Biochem Pharmacol 2023, 214, 115679. [CrossRef]
- García, M.; Buzón, M.J.; Benito, J.M.; Rallón, N. Peering into the HIV reservoir. Rev Med Virol 2018, 28, e1981. [CrossRef]
- Dufour, C.; Gantner, P.; Fromentin, R.; Chomont, N. The multifaceted nature of HIV latency. J Clin Invest 2020, 130, 3381-3390. [CrossRef]
- Devanathan, A.S.; Cottrell, M.L. Pharmacology of HIV Cure: Site of Action. Clin Pharmacol Ther 2021, 109, 841-855. [CrossRef]
- Chen, J.; Zhou, T.; Zhang, Y.; Luo, S.; Chen, H.; Chen, D.; Li, C.; Li, W. The reservoir of latent HIV. Front Cell Infect Microbiol 2022, 12, 945956. [CrossRef]
- Ta, T.M.; Malik, S.; Anderson, E.M.; Jones, A.D.; Perchik, J.; Freylikh, M.; Sardo, L.; Klase, Z.A.; Izumi, T. Insights Into Persistent HIV-1 Infection and Functional Cure: Novel Capabilities and Strategies. Front Microbiol 2022, 13, 862270. [CrossRef]
- Yang, H.; Wallace, Z.; Dorrell, L. Therapeutic Targeting of HIV Reservoirs: How to Give T Cells a New Direction. Front Immunol 2018, 9, 2861. [CrossRef]
- Elsheikh, M.M.; Tang, Y.; Li, D.; Jiang, G. Deep latency: A new insight into a functional HIV cure. EBioMedicine 2019, 45, 624-629. [CrossRef]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519-530. [CrossRef]
- Ward, A.R.; Mota, T.M.; Jones, R.B. Immunological approaches to HIV cure. Semin Immunol 2021, 51, 101412. [CrossRef]
- Bai, R.; Lv, S.; Wu, H.; Dai, L. Insights into the HIV-1 Latent Reservoir and Strategies to Cure HIV-1 Infection. Dis Markers 2022, 2022, 6952286. [CrossRef]
- Andersen, R.J.; Ntie-Kang, F.; Tietjen, I. Natural product-derived compounds in HIV suppression, remission, and eradication strategies. Antiviral Res 2018, 158, 63-77. [CrossRef]
- Abner, E.; Jordan, A. HIV “shock and kill” therapy: In need of revision. Antiviral Res 2019, 166, 19-34. [CrossRef]
- Atkins, A.J.; Allen, A.G.; Dampier, W.; Haddad, E.K.; Nonnemacher, M.R.; Wigdahl, B. HIV-1 cure strategies: why CRISPR? Expert Opin Biol Ther 2021, 21, 781-793. [CrossRef]
- Thomas, J.; Ruggiero, A.; Paxton, W.A.; Pollakis, G. Measuring the Success of HIV-1 Cure Strategies. Front Cell Infect Microbiol 2020, 10, 134. [CrossRef]
- Kim, Y.S. Long-Acting Injectable Antiretroviral Agents for HIV Treatment and Prevention. Infect Chemother 2021, 53, 686-695. [CrossRef]
- Kula-Pacurar, A.; Rodari, A.; Darcis, G.; Van Lint, C. Shocking HIV-1 with immunomodulatory latency reversing agents. Semin Immunol 2021, 51, 101478. [CrossRef]
- Rumlová, M.; Křížová, I.; Keprová, A.; Hadravová, R.; Doležal, M.; Strohalmová, K.; Pichová, I.; Hájek, M.; Ruml, T. HIV-1 protease-induced apoptosis. Retrovirology 2014, 11, 37. [CrossRef]
- Centazzo, M.; Manganaro, L.; Alvisi, G. Cellular Targets of HIV-1 Protease: Just the Tip of the Iceberg? Viruses 2023, 15, 712. [CrossRef]
- Kim, Y.; Anderson, J.L.; Lewin, S.R. Getting the “Kill” into “Shock and Kill”: Strategies to Eliminate Latent HIV. Cell Host Microbe 2018, 23, 14-26. [CrossRef]
- Kim, J.G.; Shan, L. Beyond Inhibition: A Novel Strategy of Targeting HIV-1 Protease to Eliminate Viral Reservoirs. Viruses 2022, 14, 1179. [CrossRef]
- Tanaka, K.; Kim, Y.; Roche, M.; Lewin, S.R. The role of latency reversal in HIV cure strategies. J Med Primatol 2022, 51, 278-283. [CrossRef]
- Cummins, N.W.; Baker, J.; Chakraborty, R.; Dean, P.G.; Garcia-Rivera, E.; Krogman, A.; Kumar, S.; Kuzmichev, Y.V.; Laird, G.M.; Landay, A.; Lichterfeld, M.; Mahmood, M.; Martinson, J.; Maynes, M.; Natesampillai, S.; Rajkumar, V.; Rassadkina, Y.; Ritter, K.D.; Rivera, C.G.; Rizza, S.A.; Subramanian, K.; Tande, A.J.; Wonderlich, E.R.; Whitaker, J.A.; Zeuli, J.; Badley, A.D. Single center, open label dose escalating trial evaluating once weekly oral ixazomib in ART-suppressed, HIV positive adults and effects on HIV reservoir size in vivo. EClinicalMedicine 2021, 42, 101225. [CrossRef]
Figure 1.
HIV-1 life cycle. Myr-MA: Myristoylated-Matrix, CA: Capsid, NC: Nucleocapsid, PR: Protease, RT: Reverse Transcriptase, IN: Integrase, ESCRT: Endosomal sorting complexes required for transport. Image created with BioRender.com.
Figure 1.
HIV-1 life cycle. Myr-MA: Myristoylated-Matrix, CA: Capsid, NC: Nucleocapsid, PR: Protease, RT: Reverse Transcriptase, IN: Integrase, ESCRT: Endosomal sorting complexes required for transport. Image created with BioRender.com.
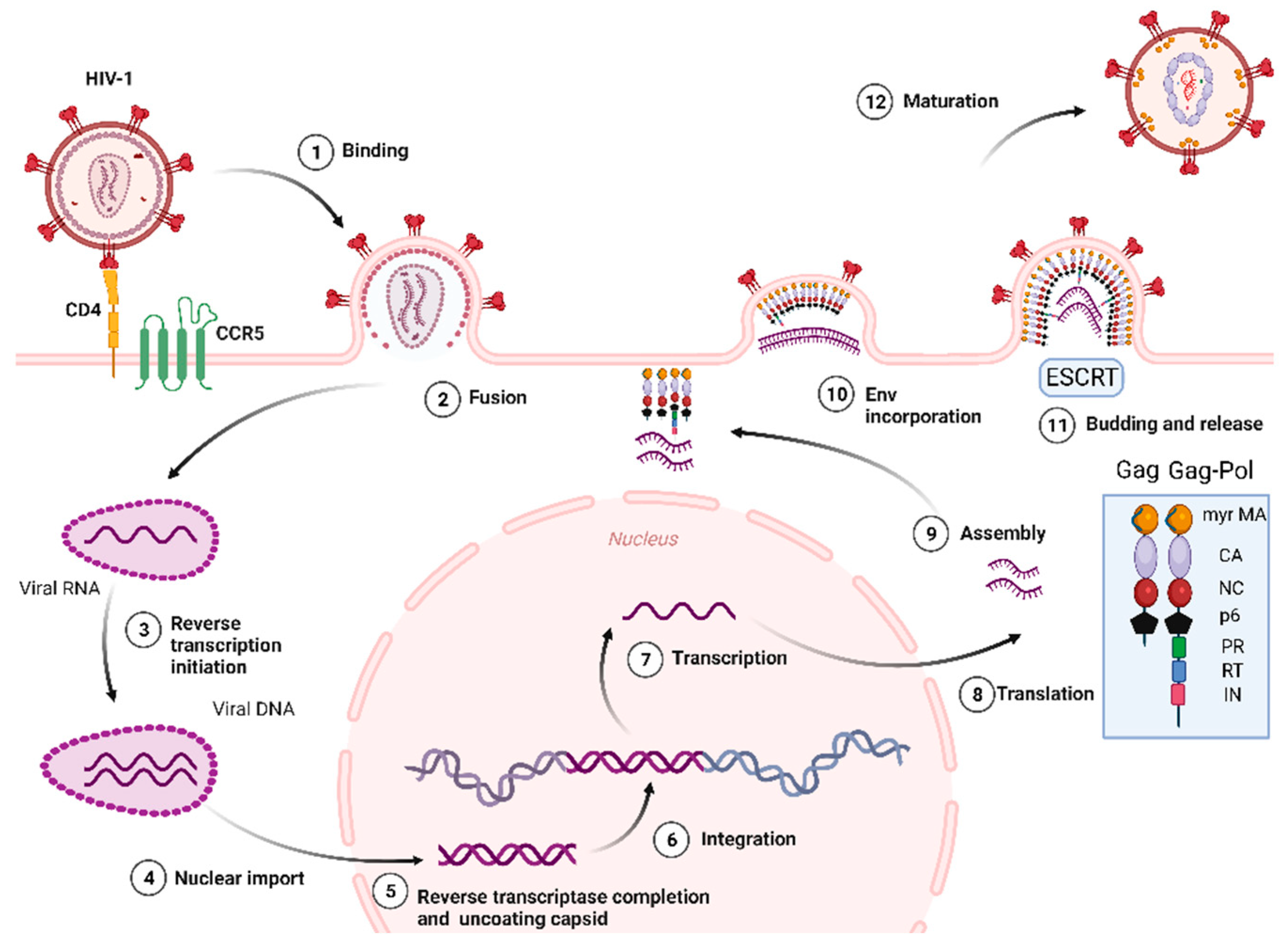
Figure 2.
The chemical structures of FDA-approved NNRTIs (A) and NRTIs (B).
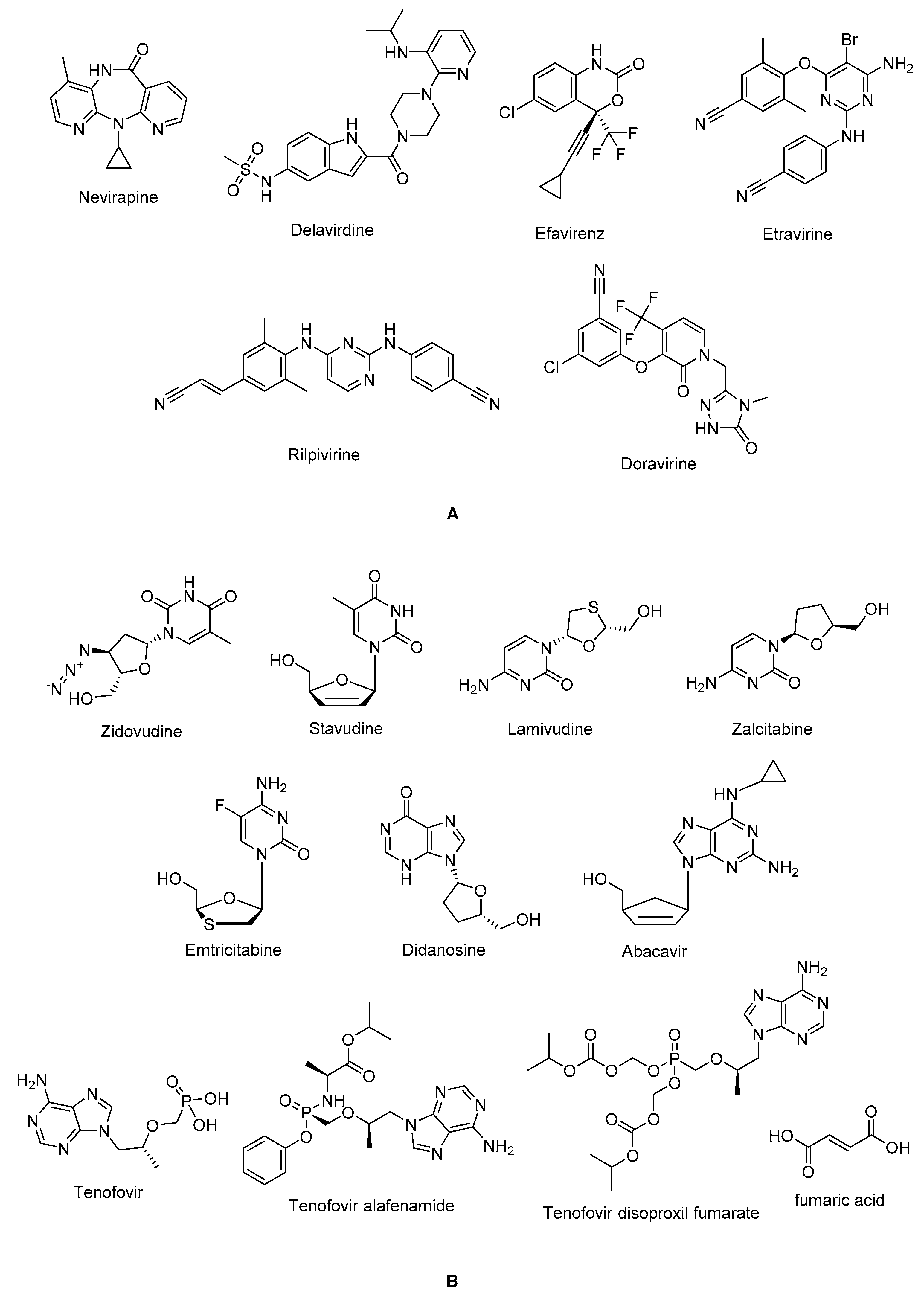
Figure 3.
The chemical structures of FDA-approved first and second generation of protease inhibitors.
Figure 3.
The chemical structures of FDA-approved first and second generation of protease inhibitors.
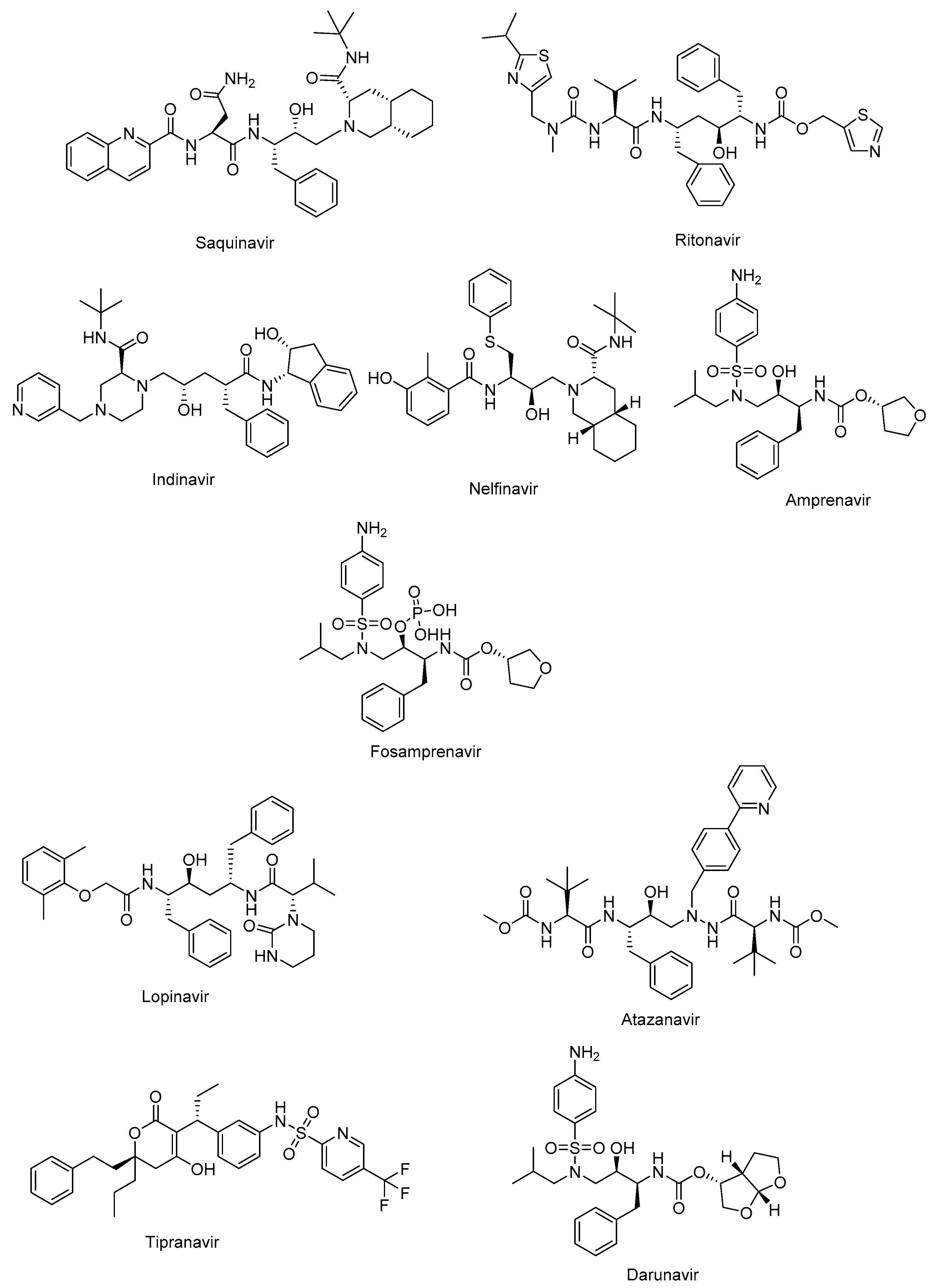
Figure 4.
The chemical structures of maraviroc (CCR5 Antagonist) and fostemsavir (Attachment Inhibitor).
Figure 4.
The chemical structures of maraviroc (CCR5 Antagonist) and fostemsavir (Attachment Inhibitor).
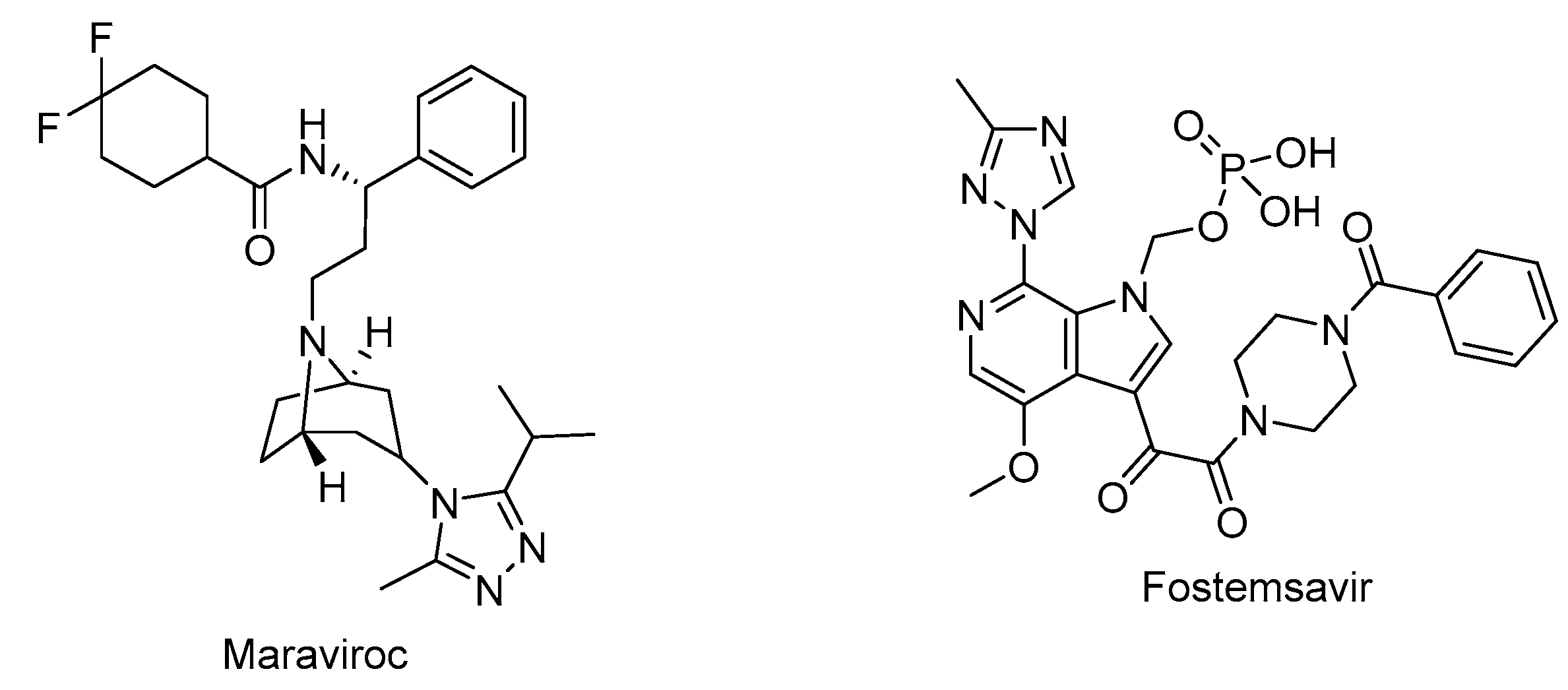
Figure 6.
The chemical structures of lenacapavir (CA inhibitor) and islatravir (NRTTI).
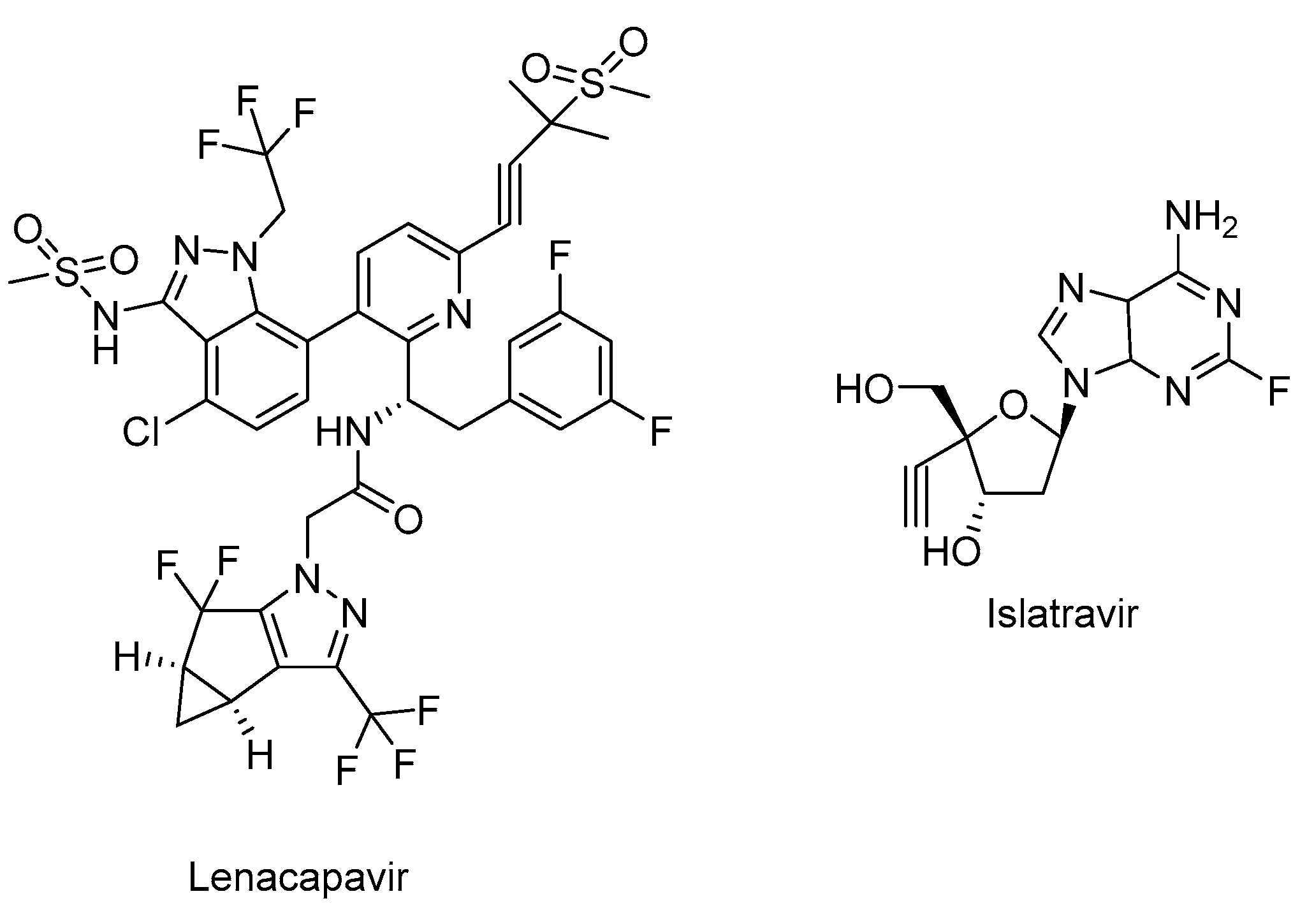
Figure 7.
The chemical structure of cobicistat.
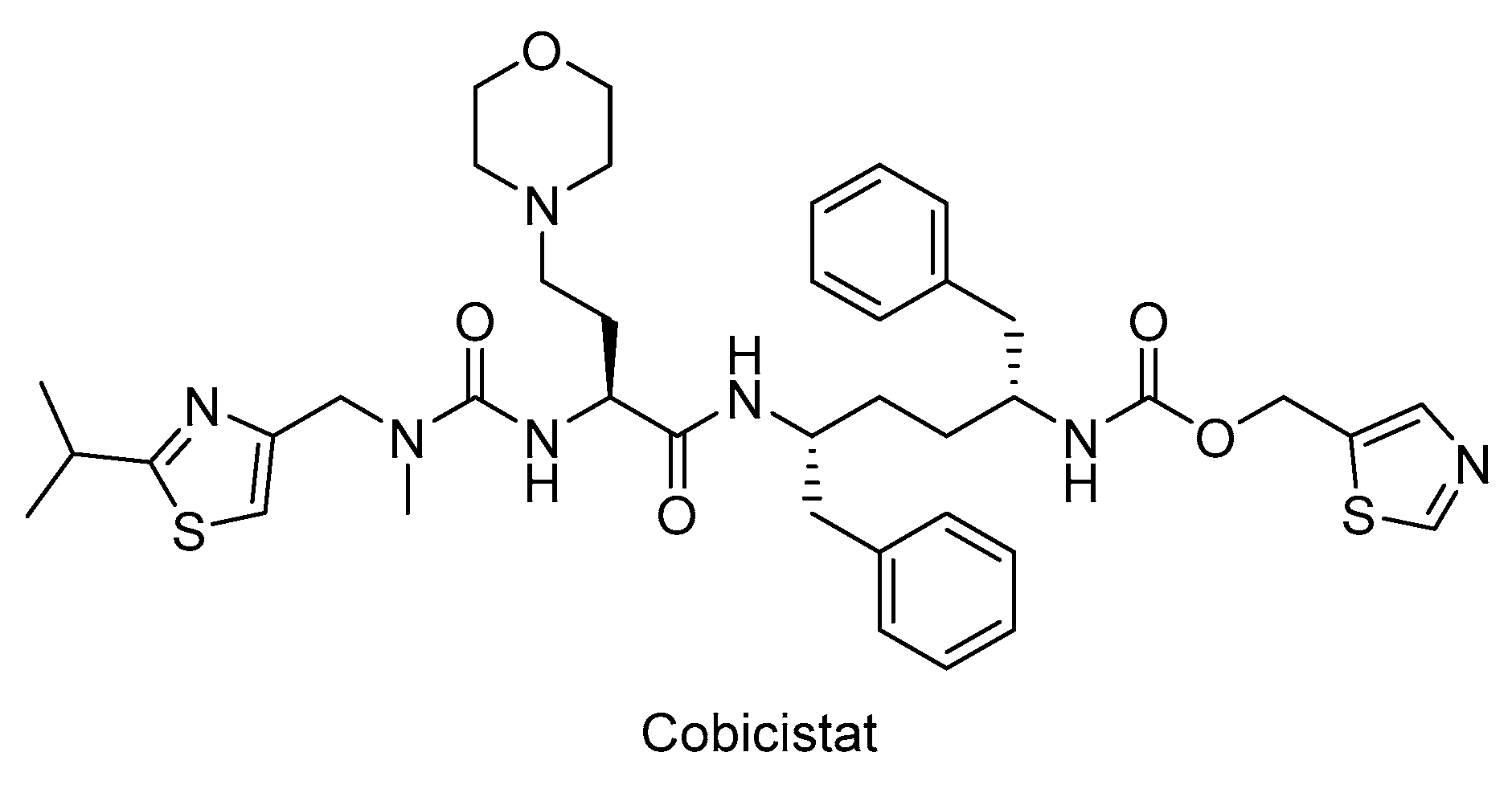
Figure 8.
Schematic illustration of HIV-1 Gag. HBR: Highly basic region, MA: Matrix, CA: Capsid, NC: Nucleocapsid, ESCRT: ESCRT: Endosomal sorting complexes required for transport, PI(4,5)P2: Phosphatidylinositol-(4,5)-bisphosphate [19]. Image created with BioRender.com.
Figure 8.
Schematic illustration of HIV-1 Gag. HBR: Highly basic region, MA: Matrix, CA: Capsid, NC: Nucleocapsid, ESCRT: ESCRT: Endosomal sorting complexes required for transport, PI(4,5)P2: Phosphatidylinositol-(4,5)-bisphosphate [19]. Image created with BioRender.com.
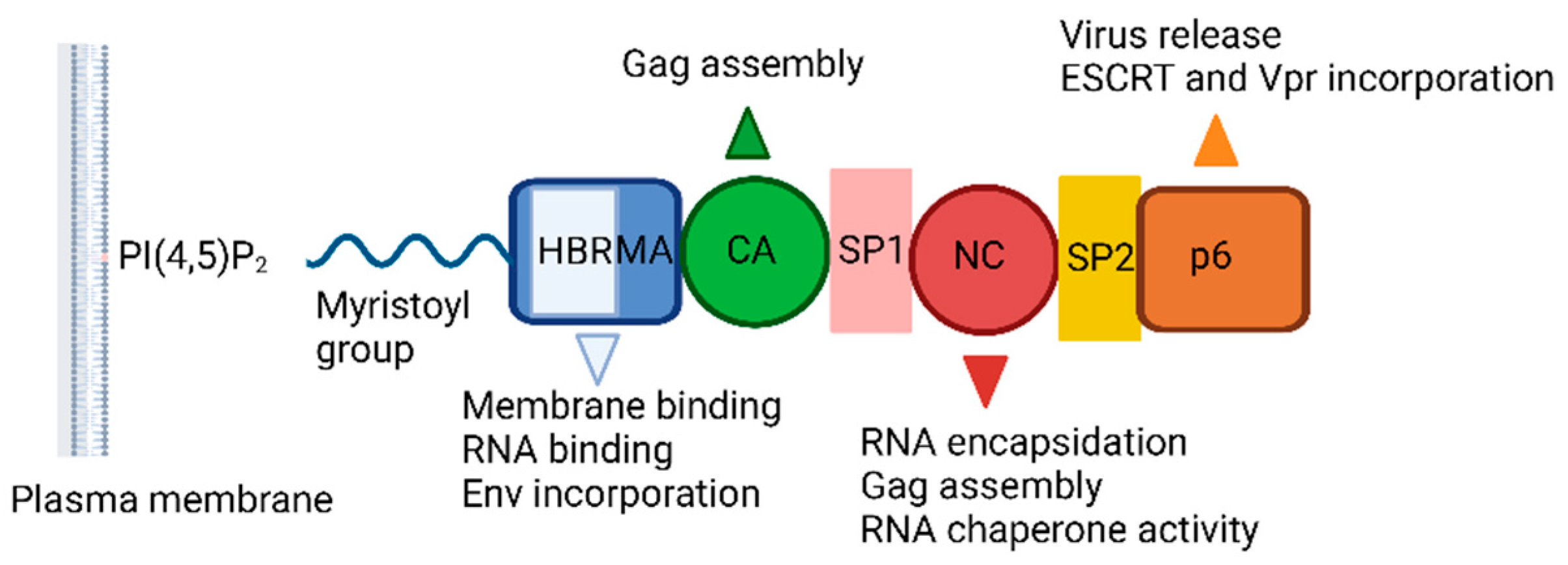
Figure 9.
The chemical structures of IP3, IP6, PI(4,5)P2, PI(4,5)P2 derivative and L-HIPPO.
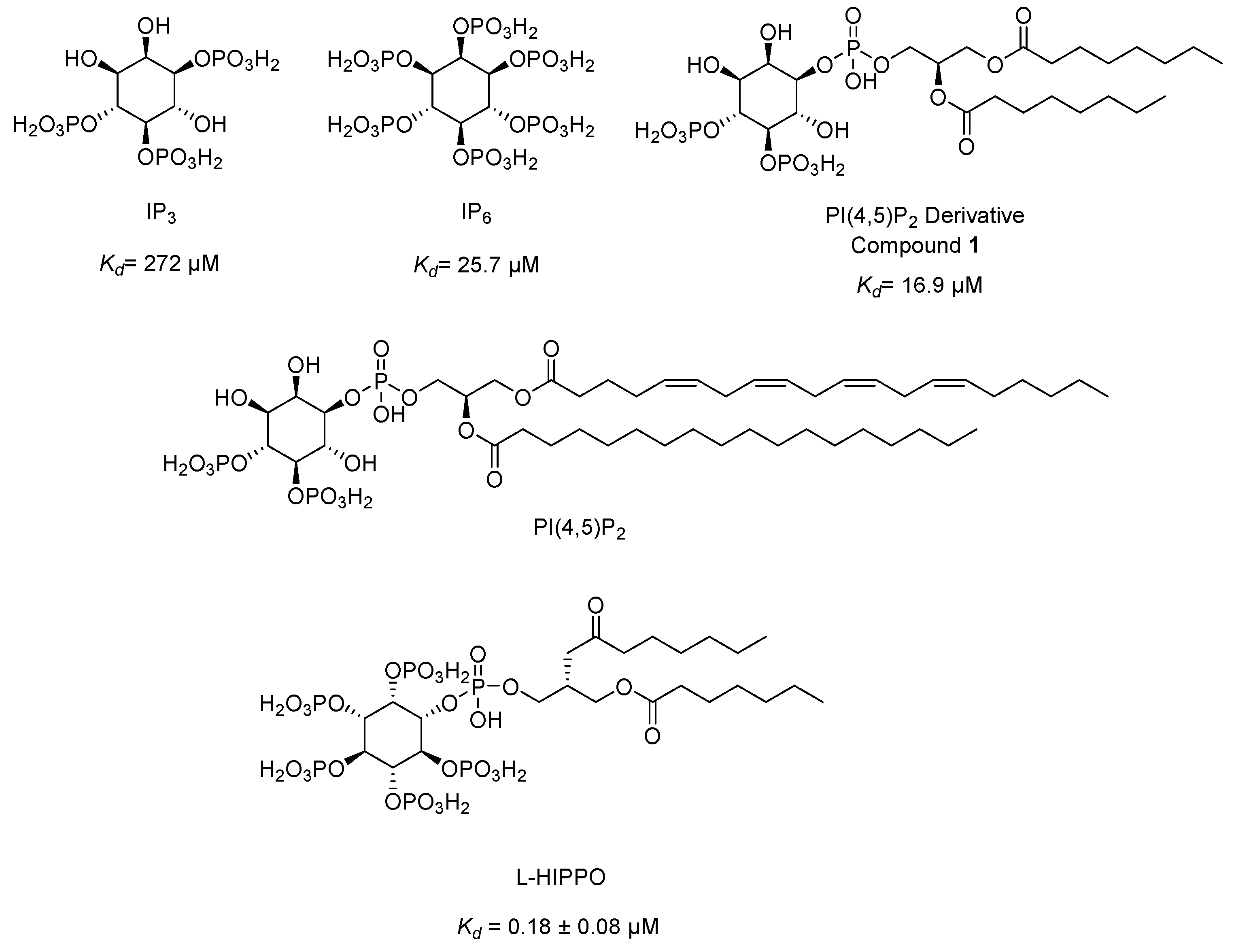
Figure 10.
The chemical structures of compounds 7 and TD 1-3.
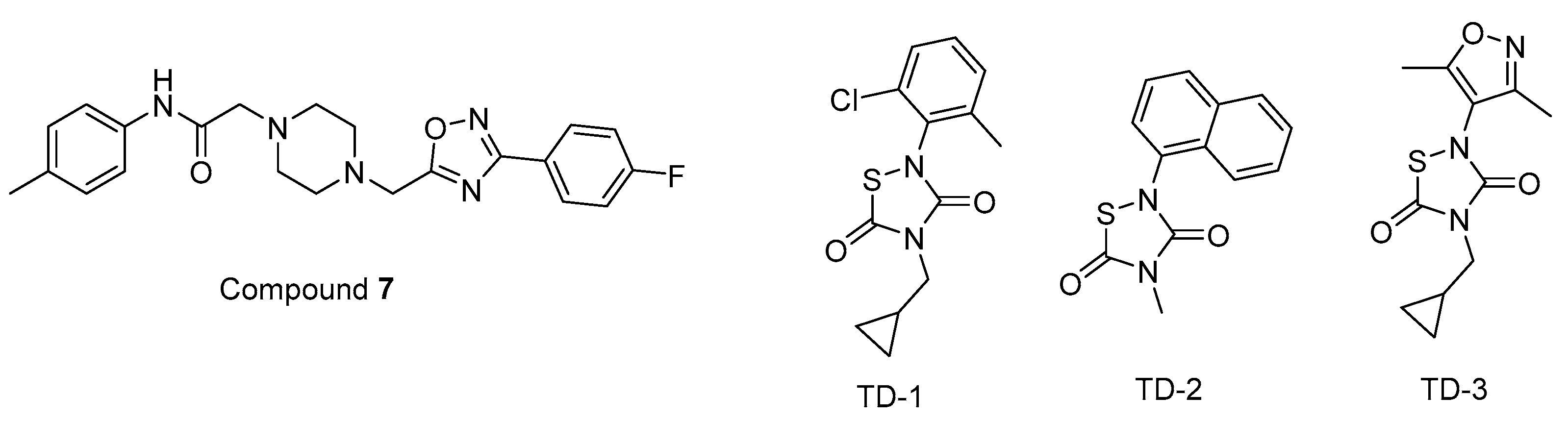
Figure 11.
The chemical structures of bevirimat and its potential analogues as anti-HIV-1 agents.
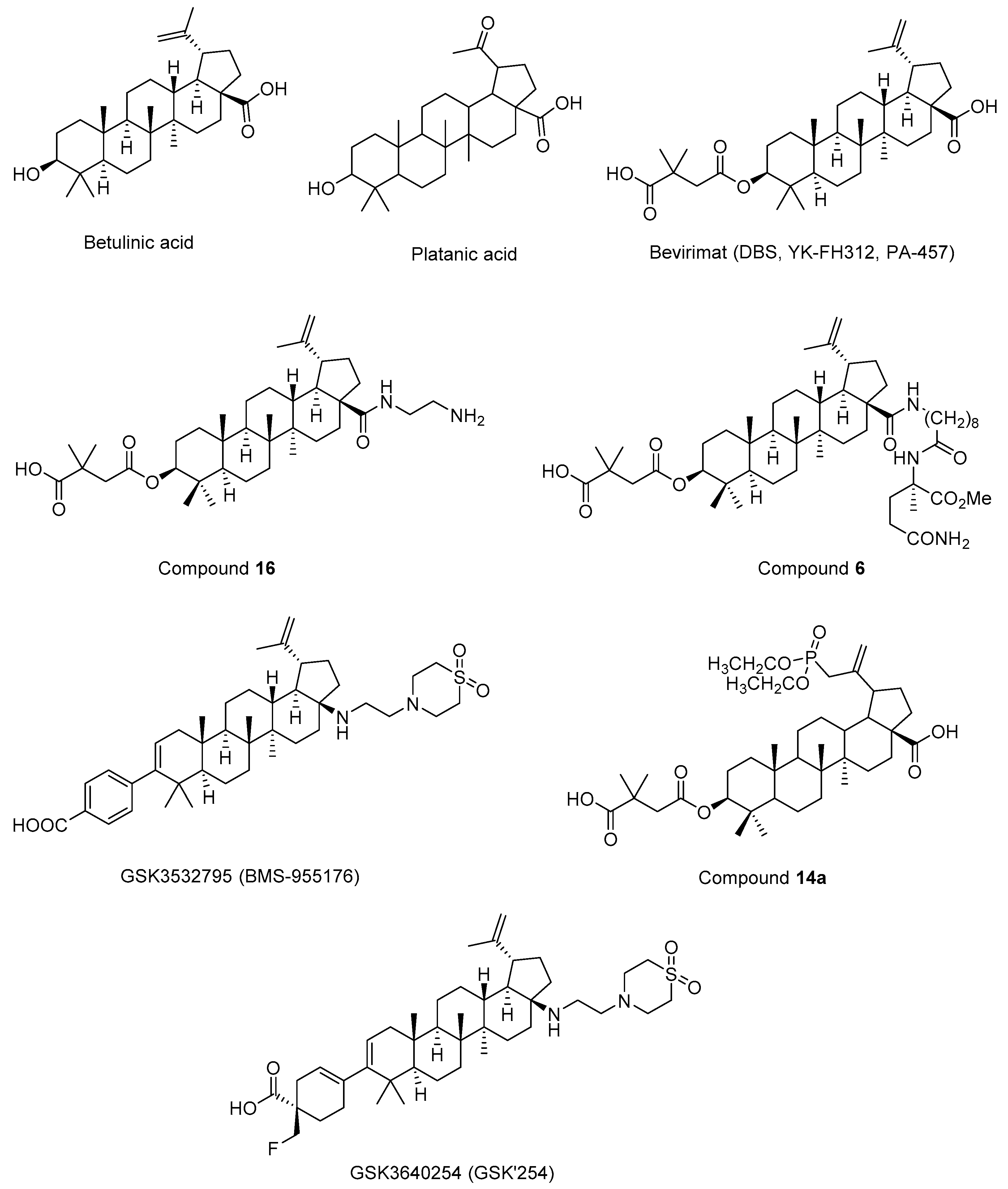
Figure 12.
The chemical structures of potential maturation inhibitors.
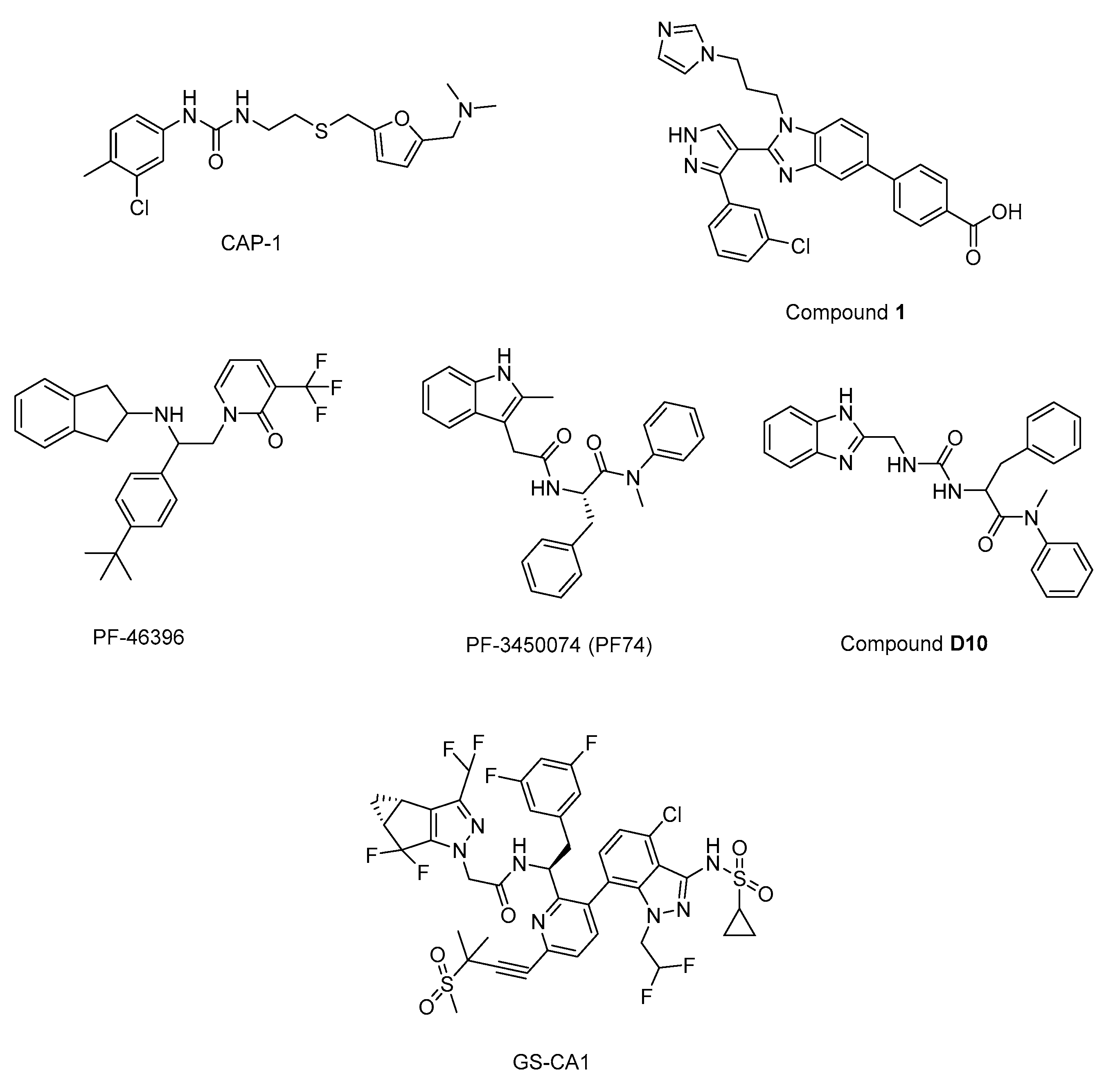
Figure 13.
Different strategies for HIV-1 cure. A) Shock and kill, B) Block and lock, C) Lock-in apoptosis. LRAs: Latency-reversing agents, latency promoting agents (LPAs), L-HIPPO: L-Heptanoylphosphatidyl Inositol Pentakisphosphate [248]. Image created with BioRender.com.
Figure 13.
Different strategies for HIV-1 cure. A) Shock and kill, B) Block and lock, C) Lock-in apoptosis. LRAs: Latency-reversing agents, latency promoting agents (LPAs), L-HIPPO: L-Heptanoylphosphatidyl Inositol Pentakisphosphate [248]. Image created with BioRender.com.

Figure 14.
The cell death pathways affected by HIV-1 protease and FDA approved proteasome inhibitor ixazomib. BCA3: breast carcinoma-associated protein 3.
Figure 14.
The cell death pathways affected by HIV-1 protease and FDA approved proteasome inhibitor ixazomib. BCA3: breast carcinoma-associated protein 3.
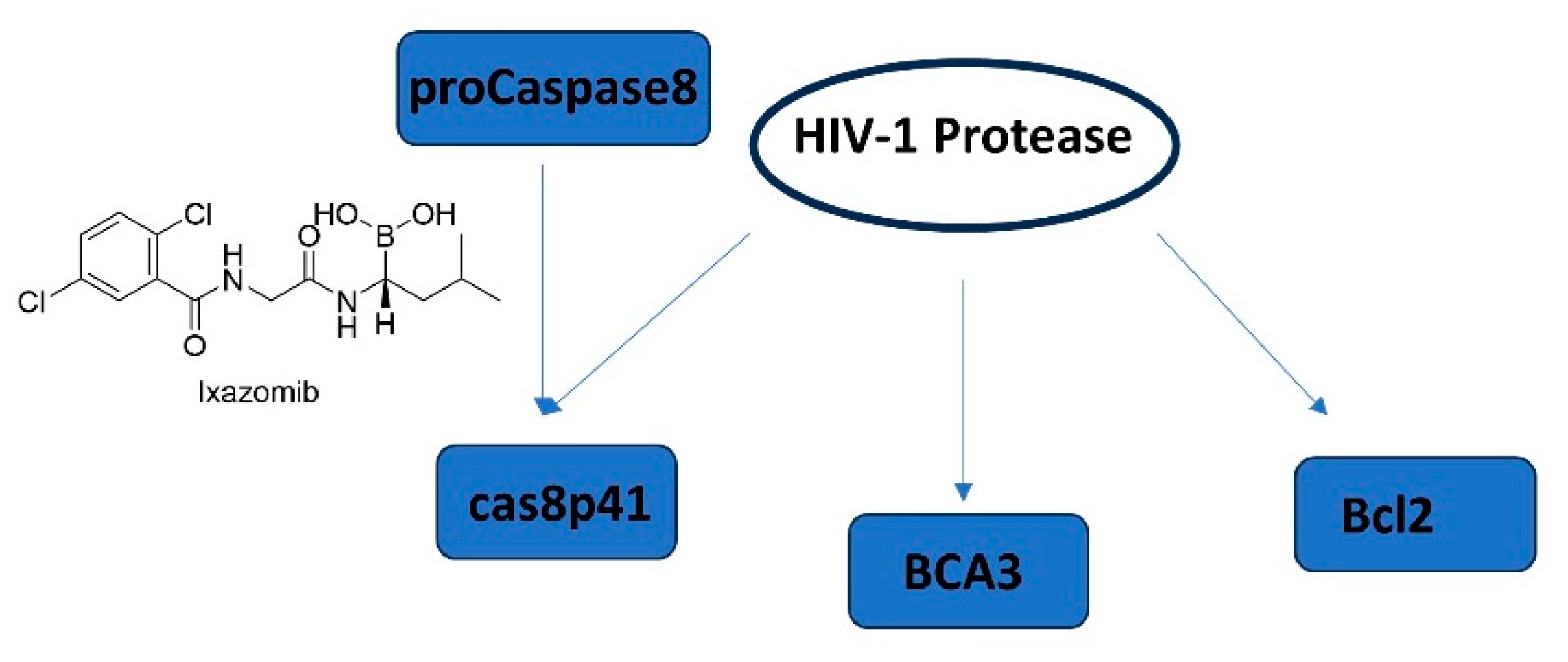
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



