
Submitted:
18 January 2024
Posted:
19 January 2024
You are already at the latest version
Abstract
The fertility rate and litter size of female pigs are critically affected by the expression of estrus. The objective of this study was to elucidate the regulatory mechanisms of estrus expression by analyzing the differential expression of genes and long intergenic non-coding RNAs (lincRNA), as well as the utilization of alternative polyadenylation (APA) sites, in the vulva and vagina during the estrus and diestrus stages of Large White and indigenous Chinese Mi gilts. Our study revealed that the number of differentially expressed genes (DEG) in the vulva was less than that in the vagina, and the DEGs in the vulva were enriched in pathways such as “neural” pathways and steroid hormone responses, including the “Calcium signaling pathway” and “Oxytocin signaling pathway”. The DEGs in the vagina were enriched in the “Metabolic pathways” and “VEGF signaling pathway”. Furthermore, 27 and 21 differentially expressed lincRNAs (DEL), whose target genes were enriched in the “Endocrine resistance” pathway, were identified in the vulva and vagina, respectively. Additionally, we observed that 63 and 618 transcripts of the 3'-untranslated region (3'-UTR) were lengthened during estrus in the vulva and vagina, respectively. Interestingly, the genes undergoing APA events in the vulva exhibited species-specific enrichment in neural or steroid-related pathways, whereas those in the vagina were enriched in apoptosis or autophagy-related pathways. Further bioinformatic analysis of these lengthened 3'-UTRs revealed the presence of multiple miRNAs binding sites and cytoplasmic polyadenylation element (CPE) regulatory elements. In particular, we identified more than 10 CPEs in the validated lengthened 3'-UTRs of the NFIX, PCNX4, CEP162 and ABHD2 genes using RT-qPCR. These findings demonstrated the involvement of APA and lincRNAs in the regulation of estrus expression in female pigs, providing new insights into the molecular mechanisms underlying estrus expression in pigs.
Keywords:
Alternative Polyadenylation
; Estrus
; LncRNA
; Pigs
1. Introduction
Extended and evident estrus expression is essential for timely insemination, whereas concealed or less evident estrus expression causes a decrease in conception rate [1]. The lack of estrus expression also leads to the culling of a considerable number of gilts from the breeding herd. This has emerged as one of the pivotal challenges in the pig farming industry that remains unresolved [2]. During estrus in female pigs, a series of signs are exhibited, such as vaginal mucus secretion, vascular ectasia, and pigment deposition in the vulva, which signal ovulation and prepare for mating [3,4]. Although earlier studies acknowledge these signs and our recent research confirms them [5,6,7], gaps in knowledge remain regarding the regulation of estrus expression changes throughout the estrus cycle.
In eukaryotes, there is a specific "CA" dinucleotide site at the 3'-untranslated regions (3’-UTRs) of pre-mRNA, known as the polyadenylation signal site (PAS), where cleavage occurs and approximately 100-200 adenosine acid nucleotides are added to a poly (A) tail to complete polyadenylation [8]. Genome-wide studies have shown that 60-70% of human genes have multiple polyadenylation sites [9,10]. Some polyadenylation sites have also been identified in pigs [11,12]. Alternative polyadenylation (APA) refers to the differential use of multiple PAS within a given pre-mRNA [13]. The use of alternative polyadenylation sites is an important mechanism for generating different isoforms of 3'-UTRs that regulate gene expression levels, mRNA stability, localization, and translation after transcription [14,15,16,17]. Different lengths of 3'-UTRs produce variable miRNA binding sites, with lengthened 3'-UTRs increasing the number of miRNA binding sites, thereby enhancing mRNA transcription inhibition [18]. The 3'-UTRs contain important elements such as hexameric PAS and cytoplasmic polyadenylation elements (CPE), which regulate cytoplasmic polyadenylation and interact with post-transcriptional factors to influence mRNA degradation and translation [19,20]. APA regulates gene expression and function in a cell- and tissue-specific manner [8,21]. It has been demonstrated that long non-coding RNAs (lncRNAs) exert impacts on pig traits [22], especially those related to pig reproductive traits [23]. We hypothesized that long intergenic non-coding RNAs (lincRNAs) and APA in the vaginal and vulva tissues may be involved in regulating the estrus expression of female pigs.
In the present study, vaginal and vulva tissues from European Large White gilts and indigenous Chinese Mi gilts were utilized to analyze mRNA, APA, and lincRNAs during different stages of estrus cycles. The expression characteristics and potential functions of these APA and lincRNAs sites provides new insights into the regulation of estrus expression in female pigs.
2. Materials and Methods
This study was approved by the Animal Care and Use Committee of Nanjing Agricultural University (SYXK2017-0027).
2.1. Sample collection
As described previously [24], the expression of estrus was observed in 30 Large White gilts and 30 Mi gilts at Yong Kang Agricultural Science and Technology Co., Ltd in Changzhou, Jiangsu Province, China. The samples were collected during the estrus and diestrus stages. Estrus stage: On the first day of gilt exhibiting standing reflex; Diestrus stage: On the 10th day of the third estrus. Three Large White and three Mi gilts were humanely euthanized at the estrus stage (designated as LE and ME) and diestrus stage (designated as LD and MD), respectively. Meticulous dissection of the vagina and vulva tissues was performed, and a total of 24 tissue samples were systematically collected. Subsequently, the collected samples were immediately preserved at a temperature of −80 °C until the initiation of RNA isolation.
2.2. Library preparation and sequencing
The total RNA was extracted from the vagina and vulva tissues using TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Agarose gel electrophoresis was performed to evaluate the samples for evidence of degradation and contamination. Subsequently, the RNA concentration and purity were evaluated using a Qubit RNA Assay Kit on a Qubit 2.0 Fluorometer (Life Technologies, CA, USA) and a NanoPhotometer® Spectrophotometer (IMPLEN, CA, USA). Following quality control, the samples were subjected to sequencing on the DNBSEQ platform (DNBSEQ Technology) provided by BGI (Shenzhen, China). The sequencing procedure involved the following steps: 1) Extraction of a specific quantity of total RNA samples, followed by the enrichment of mRNA containing a poly-A tail using oligo dT beads; 2) Fragmentation of mRNA molecules into small fragments; 3) Generation of first-strand cDNA from the fragmented mRNA using random primers; 4) Synthesis of the second-strand cDNA; 5) End-repair and 3' adenylation of the synthesized cDNA, followed by the ligation of adaptors to the ends of these 3' adenylated cDNA fragments; 6) PCR amplification and subsequent purification; 7) Quality control of the library; 8) Circularization of the library; 9) Amplification of the library to generate DNA nanoballs (DNBs); 10) Sequencing of the DNBs on the DNBSEQ platform (DNBSEQ Technology).
2.3. Transcriptional and assembly and lincRNAs identification
Twelve vagina and 12 vulva RNA-seq libraries were constructed separately. To obtain clean reads, the tools Trimmomatic [25] and SOAPnuke [26] were used for pre-processing. Then, the quality of the clean reads was evaluated using Q20, Q30, and GC content metrics. Only high-quality clean reads were selected for subsequent analysis. The selected clean reads were aligned against the pig reference genome (Sscrofa11.1) using HISAT2 aligner [27], and the bam files were obtained using Samtools [28] and employed for further analysis.
To identify the lincRNAs, we implemented the following steps: 1) filtering using gffcompare in StringTie was applied to transcripts categorized as 'U'; 2) transcripts with less than 2 exons and a length shorter than 200 bp were excluded; 3) the coding potential of transcripts was assessed using CPC2, Pfam, NCBI NR, and the UniRef 90 databases, resulting in the retention of transcripts incapable of encoding proteins; 4) transcripts expressed in at least one sample were retained as final candidate lincRNAs for further analysis. To explore the functions of lincRNAs, the target genes by cis-acting [29] of the lincRNAs were predicted. For cis-acting analysis, the protein-coding genes located within a range of 100 kb upstream and downstream of lincRNAs were identified by BEDTools.
2.4. Analysis of differential expression and function enrichment analysis
We aligned the genes and identified lincRNAs with the pig genome reference sequence (Sscrofa11.1) for the purpose of conducting differential expression analysis. To count the number of reads across each sample, we employed the "featureCounts" program. To identify the differentially expressed transcripts in the two tissues, including LE vs. ME, LD vs. LE, LD vs. MD, and MD vs. ME comparisons, we set a screening threshold of “adjusted P-value < 0.05 and |log2 (fold change)| > 1” using the “DESeq2” package [30]. Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of the genes were performed by the KOBAS database (http://kobas.cbi.pku.edu.cn/) [31]. A P-value threshold of less than 0.05 was considered statistically enrichment terms or pathways.
2.5. Analysis of alternative polyadenylation in the 3'-UTR
DaPars (https://github.com/ZhengXia/dapars) was used to analyze the use of APA sites in the vagina and vulva during the estrus and diestrus stages. First, the bam files were converted to wig files, and then the wig files of different groups were compared to obtain transcript subtypes with different 3'-UTR lengths. By calculating the percentage change of the usage index of distal poly(A) sites (PDUI), the usage of proximal and distal poly(A) sites was quantified. Finally, ΔPDUI (defined as the average PDUI of one group − the average PDUI of another group) was calculated to indicate the shortened or lengthened 3'-UTR length in the sample. An adjusted P-value less than 0.05 and |ΔPDUI| greater than 0.05 was considered statistically APA site [20].
2.6. Analysis of 3'-UTR sequences and functional elements
The IGV tool was used to analyze and visualize the length and abundance information of the 3'-UTR in each sample [32]. First, the reference genome (Sus_scrofa.Sscrofa11.1.dna.toplevel.fa) and annotation files were imported into IGV, followed by the import of bam files for each sample. Subsequently, the lengths and differences of the identified 3'-UTRs were examined. Then, we used an online program (http://genome.crg.es/CPE/server.html) to predict the types, numbers, and positions of CPE sites (CPENC, CPEC, HEXA and PBE) [33]. Miranda was used to analyze the binding sites of miRNAs in the extended sequences of 3'-UTRs [34], and the predicted results were visualized using Cytoscape (https://cytoscape.org).
2.7. RT-qPCR verification
Total cDNA was synthesized using a reverse transcriptase kit and subjected to RT-qPCR using SYBR Green Master Mix (Vazyme Biotech, Nanjing, China). The PCR reactions were performed in triplicate, and primers of the lengthened 3'-UTRs were designed using the Primer Premier 5 software (Table S1). The relative gene expression was determined using the 2−∆∆Ct method and normalized to the reference gene GAPDH. Expression data analysis was carried out using GraphPad Prism (version 8.0, San Diego, CA, USA). The t-test for independent samples was used to analyze statistical significance. Significant differences at P-value < 0.05. All reported results were presented as means ± standard error of the mean (SEM).
3. Results
3.1. Analysis of differentially expressed genes in the vulva and vagina
With optimal sequencing and alignment quality (Table S2), we identified 136 DEGs in the LDV vs. LEV comparison, including 21 upregulated genes and 115 downregulated genes (Figure 1A). Similarly, we have observed 826 DEGs in the LDY vs. LEY comparison, with 451 genes upregulated and 375 genes downregulated (Figure 1C). Notably, these DEGs in the vulva were significantly enriched in pathways such as the “Calcium signaling pathway”, “Wnt signaling pathway” and “Oxytocin signaling pathway” (Figure 1B). These DEGs in the vagina were significantly enriched in pathways such as “Metabolic pathways”, “PI3K−Akt signaling pathway” and “VEGF signaling pathway” (Figure 1D).
In Chinese Mi gilts, we detected 136 DEGs in the MDV vs. MEV comparison (Figure S1A), which were significantly enriched in pathways like the “Wnt signaling pathway” and “Toll−like receptor signaling pathway” (Figure S1B). Simultaneously, we also discovered 136 DEGs in the MDV vs. MEV comparison (Figure S1C), similarly showing significant enrichment in pathways such as “Metabolic pathways”, “Wnt signaling pathway” and “GnRH signaling pathway” (Figure S1D).
3.2. Analysis of differentially expressed lincRNAs in the vulva and vagina
To fully explore the lincRNAs that are associated with estrus expression in gilts, we have divided the sequencing samples into two cross-stage comparisons (LD vs. LE, MD vs. ME) and two cross-breed comparisons (LE vs. ME, LD vs. MD). Specifically, there were 0, 26, 2 and 2 DELs detected in LD vs. LE, LD vs. MD and MD vs. ME and LE vs. ME comparisons of the vulva, respectively (Figure 2A). Moreover, there were 10, 9, 1 and 2 DELs detected in LD vs. LE, LD vs. MD and MD vs. ME and LE vs. ME comparisons of the vagina, respectively (Figure 2B).
Among the four comparisons of the vulva, a total of 27 DELs were identified (Figure 2A), and 53 potential cis-acting target genes located within a range of 100 kb upstream and downstream of these lincRNAs were predicted (Figure 2C, Table S3). These target genes were found to be significantly enriched in pathways such as “Metabolic pathways”, “Oxidative phosphorylation” and “Hippo signaling pathway” (Figure S2A).Among the four comparisons of the vagina, a total of 21 DELs were identified (Figure 2B), and 36 potential cis-acting target genes of these lincRNAs were predicted (Figure 2D, Table S3). These target genes were found to be significantly enriched in pathways such as “N−Glycan biosynthesis”, “Endocrine resistance” and “mTOR signaling pathway” (Figure S2B).
3.3. Identification and analysis of APA sites in the estrus cycles
During the estrus and diestrus stages in Large White gilts, we found changes in the 3'-UTR length of 631 (Figure 3A) and 748 (Figure 3E) transcripts in the vulva and vagina, respectively. Principal component analysis (PCA) revealed separate clustering of samples from the vulva (Figure 3B) or vagina (Figure 3F) tissues at different estrus stages. Specifically, in the genes undergoing APA site in the vulva, there are 568 transcripts with shortened 3'-UTRs during estrus and 63 transcripts with lengthened 3'-UTRs (Figure 3C). Genes with lengthened 3'-UTRs during estrus were enriched in 11 pathways, including some interesting pathways “Neurotrophin signaling pathway”, “TNF signaling pathway”, and “Apoptosis”, while genes with shortened 3'-UTRs were enriched in 24 pathways, primarily pathways “Mitophagy-animal”, “Spliceosome”, and “Adherens junction” (Figure 3D). In the vagina, only 130 transcripts have shortened 3'-UTRs, while 618 transcripts have lengthened 3'-UTRs (Figure 3G). Genes with lengthened 3'-UTRs during estrus were enriched in 13 pathways, including pathways “Metabolic pathways”, “Autophagy-animal”, and “FoxO signaling pathway”, while genes with shortened 3'-UTRs were only enriched in “Endocytosis” pathway (Figure 3H).
In Mi gilts, we observed changes in the 3'-UTR length of 663 (Figure S3A) and 353 (Figure S3E) transcripts during the estrus cycle in the vulva and vagina, respectively. There are 179 (Figure S3C) and 151 (Figure S3G) transcripts with shortened 3'-UTRs in the vulva and vagina, respectively. Interestingly, the “Steroid biosynthesis” and “Cell adhesion molecules (CAMs)” pathways were enriched in the vulva (Figure S3D, Table S4); whereas the “HIF-1 signaling pathway” was enriched in the vagina (Figure S3H, Table S5).
Furthermore, we also conducted an analysis of cross-tissue APA events in these two pig breeds. while 2628 (Figure S4A) transcripts had different 3'-UTR lengths in the vulva vs. vagina in Large White gilts. Compared to the vagina, genes with lengthened 3'-UTRs in the vulva were enriched in 20 pathways, including pathways “Metabolic pathways”, “PI3K-Akt signaling pathway”, and “Spliceosome”, while genes with shortened 3'-UTRs were enriched in “Glycerophospholipid metabolism” and “Ether lipid metabolism” pathways (Figure S4D). In Mi gilts, a total of 1902 (Figure S3A) transcripts with different 3'-UTR lengths were identified in the vulva vs. vagina (Figure S4D). Compared to the vagina, the vulva of Mi gilts had 133 transcripts with shortened 3'-UTRs and 1769 with lengthened 3'-UTRs (Figure S3G). In addition, genes with 3'-UTR alterations in both the vulva and vagina also exhibited specific enrichment in the “Oocyte meiosis” pathway (Figure S3H, Table S6).
3.4. Visualization and validation of 3'-UTR expression abundance
To investigate the role of APA sites in the 3'-UTR, we characterized the expression abundance of the 3'-UTR in each sample. In the vulva, the heatmap revealed that the expression abundance of lengthened 3'-UTRs was lower during the diestrus stage and higher during the estrus stage, whereas the expression of shortened 3'-UTRs was higher during the diestrus stage and lower during the estrus stage (Figure 4A). In the vagina, the heatmap showed that the expression abundance of lengthened 3'-UTRs was higher during the estrus stage compared to the diestrus stage, while the expression of shortened 3'-UTRs was lower during the estrus stage (Figure 4B). Specifically, the expression of the TOMM40L gene shortened 3'-UTR was higher in the vulva during the diestrus stage compared to the estrus stage, while the expression of ABHD2 gene shortened 3'-UTR was higher in the vulva during the estrus stage (Figure 4C). Moreover, the expression of the POLA2 gene shortened 3'-UTR was higher in the vagina during the diestrus stage compared to the estrus stage, while the expression of the CEP162 gene shortened 3'-UTR was higher in the vagina during the estrus stage (Figure 4D). We designed primers specific to each gene located in the lengthened 3'-UTR and found that the relative expression results were consistent with the results obtained using IGV.
Moreover, when comparing the vulva, it was observed that the expression abundance of lengthened 3'-UTRs was higher in the vagina, whereas the expression of shortened 3'-UTRs was lower (Figure S5A). The expression of the NFIB gene shortened 3'-UTR was lower in the vagina compared to the vulva, while the expression of the PCNX4 gene shortened 3-UTR was higher in the vagina (Figure S5B).
3.5. Regulatory elements in lengthened 3'-UTR
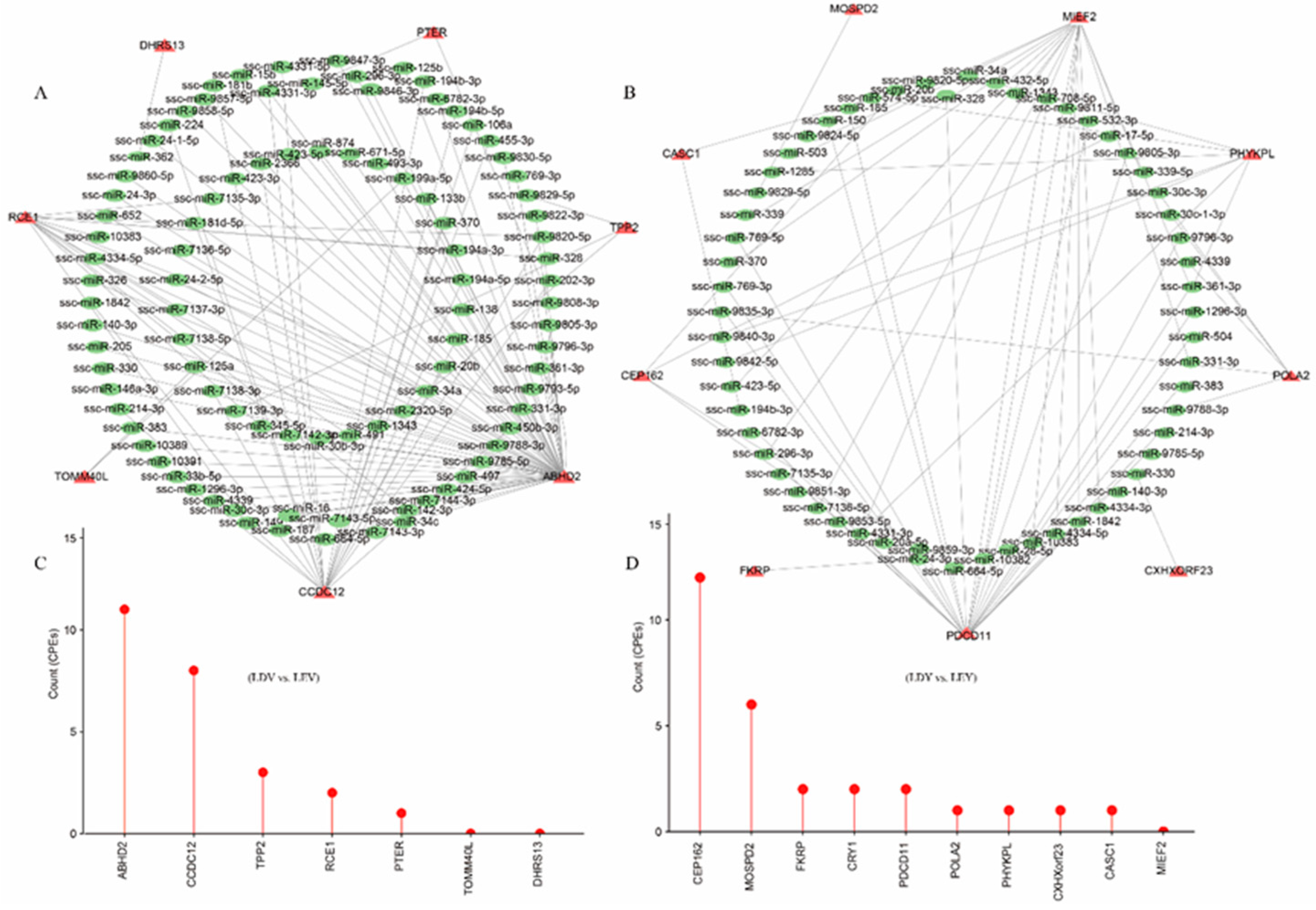
By filtering for significant transcripts with |ΔPDUI| ≥ 0.35, we obtained 7, 10, and 20 high-confidence transcripts in the LDV vs. LEV, LDY vs. LEY and LY vs. LV comparisons, respectively (Table S7). We analyzed the potential miRNA binding sites within these binding sequences. We found that among the 7 genes in the LDV vs. LEV comparison, a total of 95 miRNA binding sites were predicted in their lengthened 3'-UTRs (Figure 5A). Among the 10 genes in the LDY vs. LEY comparison, a total of 59 miRNA binding sites were predicted in their lengthened 3'-UTRs (Figure 5B). Furthermore, a total of 114 miRNA binding sites were predicted in the lengthened 3'-UTRs of the 10 genes in the LY vs. LV comparison (Figure S5C). These miRNAs could be one of the reasons for regulating the abundance of 3'-UTRs or mRNAs expression. Furthermore, we have also conducted predictions on the CPEs present in the lengthened 3'-UTRs of these genes (Table S8). Among them, 9 genes were found to harbor more than 10 CPEs, including the NFIX, PCNX4, CEP162 and ABHD2 genes. These findings provide important clues for further research on the transcriptional regulation of genes.
4. Discussion
The importance of estrus expression in sows during estrus is evident for pig reproduction [35,36]. During estrus, female pigs exhibit estrus signs, such as standing reflex, reddening and swelling of the vulva, and mucus discharge from the vulva [37]. Our previous studies have demonstrated differences in the color of the vulva and vaginal mucus between estrus and diestrus stages. However, the regulatory mechanisms underlying these changes are still lacking. We have conducted gene set enrichment analysis (GSEA) to analyze the functions of genes in the vulva and vaginal tissues. In this study, DEGs, lincRNAs, and APA were analyzed to further investigate the transcriptional changes in the vulva and vagina during the estrus cycle.
In the present study, the number of DEGs in the vulva is much lower than that in the vagina, which may be related to the vagina having more physiological functions than the vulva. Interestingly, we identified enrichment of the “Calcium signaling pathway”, “Oxytocin signaling pathway” and “Dopaminergic synapse” pathways in DEGs in the vulva, which may be related to the vulva's response to steroid hormones and neural regulation of estrus expression. In addition, the vagina is enriched with the “Metabolic pathways” and “VEGF signaling pathway”, this is because there are target genes of estrogen in the vagina, which may be involved in vaginal mucus secretion and metabolic functions [38]. LncRNAs have been increasingly found to play a role in the reproductive regulation of pigs [39,40]. In our earlier studies, lincRNAs in the ovaries during different estrus phases have been identified [5]. To comprehensively understand the regulation mechanism of the vulva and vagina during the estrus cycle, we characterized all DELs that could potentially be involved. The results indicate that the cis-acting target genes of these lincRNAs may be associated with estrus expression. For instance, the enrichment of target genes of DELs was observed in the “Endocrine resistance” pathway in the vagina.
Recent studies have unveiled the role of APA sites, a process involving selective splice-site usage, in regulating mRNA expression, translation, and localization of protein-coding genes [41,42]. As a transcriptional modulator, APA exerts its influence by directly influencing the length of a gene's 3'-UTR, thereby participating in the regulation of organismal homeostasis and phenotypic traits [21,43,44,45]. In this study, we employed RNA-seq to identify numerous APA events associated with estrus expression in the vulva and vagina tissues of gilts [46]. Remarkably, these APA events were observed not only in indigenous Chinese pig breeds but also in European pig breeds, suggesting their ubiquitous presence throughout different estrus cycles and possibly serving as a crucial modulatory mechanism of estrus expression in female pigs. In this study, our findings reveal that, regardless of whether in the vagina or vulva, the estrus stage induces APA in hundreds of genes, resulting in 3'-UTR elongation or shortening. Additionally, we have identified distinct stage specificity in APA sites [47]. By comparing APA sites in the vulva and vagina, we observe a greater tissue specificity, with a much larger number of genes involved in 3'-UTR modifications compared to genes involved in different estrus stages [48]. This suggests that APA-mediated regulation of gene expression may be exquisitely precise and sensitive.
Investigations on APA have revealed its implications in the exploration of numerous pathologies and traits [49,50]. In our current study, we have identified genes undergoing APA in the vulva of Large White gilts that are enriched in the “Neurotrophin signaling” pathway, whereas in Mi gilts, they are enriched in “Steroid synthesis” pathways. It is well-established that estrus expression in pigs is initiated by ovarian steroid hormones and further regulated through the hypothalamic-pituitary axis [51,52]. Therefore, the hypothalamic-pituitary-ovary (HPO) axis stands as the principal regulatory mechanism for the estrus cycle, while the vulva and vagina serve as carriers of estrus expression [53]. Consequently, the enrichment of genes related to neural and hormonal functions is to be expected, as these genes respond to the regulation of steroid hormones and neural activities. The 3'-UTRs of genes harbor numerous regulatory elements that are involved in mRNA translation, expression, localization, and more [54]. Through analysis of these lengthened 3'-UTRs, we have discovered multiple miRNA binding sites and CPE regulatory elements. These findings suggest that APA may be involved in the transcriptional regulation of mRNA to facilitate rapid responses in the vulva and vagina during estrus in gilts. Although we have quantitatively validated APA events for several representative genes, their involvement in the regulation of estrus expression remains unknown and represents a future direction for our research.
Taken together, we conducted a comprehensive analysis of the transcriptional differences, including protein-coding genes, lincRNAs, and APA, in the vulva and vagina of gilts at estrus and diestrus. These findings provide important insights into the transcriptional regulatory mechanisms of mRNA, as well as the involvement of lincRNAs and APA sites in the homeostasis and phenotypic traits. Further research on the functions and regulatory mechanisms of lincRNAs and APA across diverse biological processes promises to provide a more comprehensive understanding of the complexity and diversity of gene expression regulation. Our study provides new research perspectives for understanding the regulation of estrus expression in gilts.
5. Conclusions
In this study, we identified differentially expressed genes and lincRNA in the vulva and vagina at different stages of estrus. Differentially expressed genes during estrus in the vulva were enriched in the “Calcium signaling pathway” and “Oxytocin signaling pathway”, while those in the vagina were enriched in the “Metabolic pathways” and “VEGF signaling pathway”. The target genes of differentially expressed lincRNA were enriched in the “Endocrine resistance” pathway. Furthermore, genes undergoing APA in the vulva are enriched in “neural” or steroid-related pathways, while those in the vagina are enriched in apoptotic or autophagy-related pathways. Further analysis of this lengthened 3'-UTR revealed the presence of multiple miRNA binding sites and CPE regulatory elements. These findings demonstrated that lincRNAs and APA regulate functional genes involved in estrus expression in gilts in a stage-dependent manner, providing new insights into the molecular regulatory mechanisms of estrus expression in gilts.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S1: Analysis of Differentially Expressed genes of the Mi gilts; Figure S2: Functional analysis of lincRNA target genes; Figure S3: Identification and analysis of APA sites in the estrus cycles of the Mi gilts; Figure S4: Identification and analysis of cross-tissue APA events. Figure S5: Regulatory elements in lengthened 3'-UTRs of cross-tissue APA events. Table S1. Primer for the 3'-UTR of genes for RT-qPCR Table; S2: Summary of mapping in all RNA-seq samples; Table S3: Cis-acting target genes of differentially expressed lincRNAs; Table S4: Functional enrichment analysis of genes undergoing APA in the vulva during the estrus and diestrus stages; Table S5: Functional enrichment analysis of genes undergoing APA in the vagina during the estrus and diestrus stages; Table S6: Functional enrichment analysis of genes undergoing APA in the vulva and vagina; Table S7: The transcripts with |ΔPDUI| ≥ 0.35; Table S8: Overview of the CPEs present in the lengthened 3'-UTRs of these genes with |ΔPDUI| ≥ 0.35.
Author Contributions
Conceptualization, M.L. and B.Z.; data curation, M.L.; formal analysis, M.L and C.Z; funding acquisition, B.Z., W.A.; investigation, B.Z.; methodology, M.L., J.C., X.C., S.L. and B.Z.; project administration, B.Z.; software, M.L., C.Z., J.C., A.M.; supervision, M.L., C.Z., S.L., H.Y. and B.Z.; Writing—original draft, M.L.; writing—review and editing, W.A.; A.P.S. and B.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (NO. 32172786), the Key Project of Tarim University (TDZKZD202203) and the “JBGS” Project of Breeding Industry Revitalization in Jiangsu Province (JBGS[2021]101).
Institutional Review Board Statement
This study was approved by the Animal Care and Use Committee of Nanjing Agricultural University (SYXK Su 2017-0027).
Informed Consent Statement
Not applicable.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
We thank the Yong Kang Agricultural Science and Technology Co., Ltd for their participation.
Conflicts of Interest
The authors declare have no conflict of interest.
References
- Yang, S.B.; Zhou, X.L.; Pei, Y.; Wang, H.; He, K.; Zhao, A.Y. Identification of Differentially Expressed Genes in Porcine Ovaries at Proestrus and Estrus Stages Using RNA-Seq Technique. Biomed Research International 2018, 2018. [Google Scholar] [CrossRef]
- Verhoeven, S.; Chantziaras, I.; Bernaerdt, E.; Loicq, M.; Verhoeven, L.; Maes, D. The evaluation of an artificial intelligence system for estrus detection in sows. Porcine Health Manag 2023, 9. [Google Scholar] [CrossRef]
- Tang, L.T.; Ran, X.Q.; Mao, N.; Zhang, F.P.; Niu, X.; Ruan, Y.Q.; Yi, F.L.; Li, S.; Wang, J.F. Analysis of alternative splicing events by RNA sequencing in the ovaries of Xiang pig at estrous and diestrous. Theriogenology 2018, 119, 60–68. [Google Scholar] [CrossRef]
- Shade, K.A.; Stewart, K.R.; Johnson, J.S. Characterizing body temperature and movement differences at the onset of estrus in replacement gilts. Journal of Animal Science 2016, 94, 194–194. [Google Scholar] [CrossRef]
- Liu, M.; Xu, Q.; Zhao, J.; Guo, Y.; Zhang, C.; Chao, X.; Cheng, M.; Schinckel, A.P.; Zhou, B. Comprehensive Transcriptome Analysis of Follicles from Two Stages of the Estrus Cycle of Two Breeds Reveals the Roles of Long Intergenic Non-Coding RNAs in Gilts. Biology 2022, 11. [Google Scholar] [CrossRef]
- Glencorse, D.; Grupen, C.G.; Bathgate, R. Vaginal and vestibular electrical resistance as an alternative marker for optimum timing of artificial insemination with liquid-stored and frozen-thawed spermatozoa in sows. Sci Rep-Uk 2023, 13. [Google Scholar] [CrossRef]
- Yilma, T.; Sobiraj, A. Relationships Between Intra-Vaginal Electrical Impedance, Estrus Behavior, Plasma Levels of Ovarian Steroids and the Pre-Ovulatory Luteinizing Hormone Surge in the Prediction of the Optimal Insemination Time in Normal Cycling Pigs. Indian J Anim Res 2012, 46, 114–120. [Google Scholar]
- Cao, J.; Kuyumcu-Martinez, M.N. Alternative polyadenylation regulation in cardiac development and cardiovascular disease. Cardiovasc Res 2023, 119, 1324–1335. [Google Scholar] [CrossRef]
- Derti, A.; Garrett-Engele, P.; MacIsaac, K.D.; Stevens, R.C.; Sriram, S.; Chen, R.H.; Rohl, C.A.; Johnson, J.M.; Babak, T. A quantitative atlas of polyadenylation in five mammals. Genome Research 2012, 22, 1173–1183. [Google Scholar] [CrossRef]
- Ji, Z.; Luo, W.T.; Li, W.C.; Hoque, M.; Pan, Z.H.; Zhao, Y.; Tian, B. Transcriptional activity regulates alternative cleavage and polyadenylation. Mol Syst Biol 2011, 7. [Google Scholar] [CrossRef]
- Wang, Y.; Wu, Z.W.; Mou, Q.; Chen, L.; Fang, T.; Zhang, Y.Q.; Yin, Z.J.; Du, Z.Q.; Yang, C.X. Global 3'-UTRome of porcine immature Sertoli cells altered by acute heat stress. Theriogenology 2023, 196, 79–87. [Google Scholar] [CrossRef]
- Stotts, M.J.; Zhang, Y.Z.; Zhang, S.W.; Michal, J.J.; Velez, J.; Hans, B.; Maquivar, M.; Jiang, Z.H. Alternative polyadenylation events in epithelial cells sense endometritis progression in dairy cows. J Integr Agr 2023, 22, 1820–1832. [Google Scholar] [CrossRef]
- Ren, F.G.; Zhang, N.; Zhang, L.; Miller, E.; Pu, J.J. Alternative Polyadenylation: a new frontier in post transcriptional regulation. Biomark Res 2020, 8. [Google Scholar] [CrossRef]
- Cheng, L.C.; Zheng, D.H.; Zhang, Q.; Guvenek, A.; Cheng, H.; Tian, B. Alternative 3 ' UTRs play a widespread role in translation-independent mRNA association with the endoplasmic reticulum. Cell Reports 2021, 36. [Google Scholar] [CrossRef]
- Masamha, C.P.; Wagner, E.J. The contribution of alternative polyadenylation to the cancer phenotype. Carcinogenesis 2018, 39, 2–10. [Google Scholar] [CrossRef]
- Berkovits, B.D.; Mayr, C. Alternative 3 ' UTRs act as scaffolds to regulate membrane protein localization. Nature 2015, 522, 363–367. [Google Scholar] [CrossRef]
- Mayr, C.; Bartel, D.P. Widespread Shortening of 3 ' UTRs by Alternative Cleavage and Polyadenylation Activates Oncogenes in Cancer Cells. Cell 2009, 138, 673–684. [Google Scholar] [CrossRef]
- Rane, S.; Sayed, D.; Abdellatif, M. MicroRNA with a MacroFunction. Cell Cycle 2007, 6, 1850–1855. [Google Scholar] [CrossRef]
- Wang, R.J.; Nambiar, R.; Zheng, D.H.; Tian, B. PolyA_DB 3 catalogs cleavage and polyadenylation sites identified by deep sequencing in multiple genomes. Nucleic Acids Research 2018, 46, D315–D319. [Google Scholar] [CrossRef]
- Xia, Z.; Donehower, L.A.; Cooper, T.A.; Neilson, J.R.; Wheeler, D.A.; Wagner, E.J.; Li, W. Dynamic analyses of alternative polyadenylation from RNA-seq reveal a 3 '- UTR landscape across seven tumour types. Nat Commun 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Mittleman, B.E.; Pott, S.; Warland, S.; Zeng, T.; Mu, Z.P.; Kaur, M.; Gilad, Y.; Li, Y. Alternative polyadenylation mediates genetic regulation of gene expression. Elife 2020, 9. [Google Scholar] [CrossRef]
- Lv, W.; Jiang, W.; Luo, H.M.; Tong, Q.; Niu, X.Y.; Liu, X.; Miao, Y.; Wang, J.N.; Guo, Y.W.; Li, J.A.; et al. Long noncoding RNA lncMREF promotes myogenic differentiation and muscle regeneration by interacting with the Smarca5/p300 complex. Nucleic Acids Research 2022, 50, 10733–10755. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Ji, J.Z.; Cui, H.D.; Zhao, Q.; Ding, C.M.; Xu, C. Functional evaluation of LTR-derived lncRNAs in porcine oocytes and zygotes with RNA-seq and small RNA-seq. Frontiers in Genetics 2022, 13. [Google Scholar] [CrossRef] [PubMed]
- Chu, Q.; Zhou, B.; Xu, F.; Chen, R.; Shen, C.; Liang, T.; Li, Y.; Schinckel, A.P. Genome-wide differential mRNA expression profiles in follicles of two breeds and at two stages of estrus cycle of gilts. Sci Rep 2017, 7, 5052. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. GigaScience 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- Li, H. The Sequence Alignment/Map format and SAMtools. BIOINFORMATICS -OXFORD- 2009. [Google Scholar] [CrossRef] [PubMed]
- Bao, Z.; Yang, Z.; Huang, Z.; Zhou, Y.; Cui, Q.; Dong, D. LncRNADisease 2.0: an updated database of long non-coding RNA-associated diseases. Nucleic Acids Res 2019, 47, D1034–D1037. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014, 15, 550. [Google Scholar] [CrossRef]
- Bu, D.C.; Luo, H.T.; Huo, P.P.; Wang, Z.H.; Zhang, S.; He, Z.H.; Wu, Y.; Zhao, L.H.; Liu, J.J.; Guo, J.C.; et al. KOBAS-i: intelligent prioritization and exploratory visualization of biological functions for gene enrichment analysis. Nucleic Acids Research 2021, 49, W317–W325. [Google Scholar] [CrossRef] [PubMed]
- Thorvaldsdottir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Pique, M.; Lopez, J.M.; Foissac, S.; Guigo, R.; Mendez, R. A combinatorial code for CPE-mediated translational control. Cell 2008, 132, 434–448. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, V.; Bell, G.W.; Nam, J.W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Xue, H.X.; Chen, J.X.; Ding, Q.A.; Sun, Y.W.; Shen, M.X.; Liu, L.S.; Chen, X.D.; Zhou, J.Y. Automatic detection of sow posture and estrus based on convolutional neural network. Front Phys-Lausanne 2022, 10. [Google Scholar] [CrossRef]
- Niu, X.; Huang, Y.L.; Lu, H.; Li, S.; Huang, S.H.; Ran, X.Q.; Wang, J.F. CircRNAs in Xiang pig ovaries among diestrus and estrus stages. Porcine Health Manag 2022, 8. [Google Scholar] [CrossRef] [PubMed]
- Knauer, M.T.; Cassady, J.P.; Newcom, D.W.; See, M.T. Estimates of variance components for genetic correlations among swine estrus traits. Journal of Animal Science 2010, 88, 2913–2919. [Google Scholar] [CrossRef] [PubMed]
- Long, X.; Burke, K.A.; Bigsby, R.M.; Nephew, K.P. Effects of the xenoestrogen bisphenol A on expression of vascular endothelial growth factor (VEGF) in the rat. Exp Biol Med 2001, 226, 477–483. [Google Scholar] [CrossRef]
- Ni, Y.F.; Wu, F.; Chen, Q.Q.; Cai, J.F.; Hu, J.P.; Shen, J.C.; Zhang, J.Z. Long noncoding RNA and mRNA profiling of hypothalamic-pituitary-mammary gland axis in lactating sows under heat stress. Genomics 2020, 112, 3668–3676. [Google Scholar] [CrossRef]
- Li, M.X.; Liu, Y.; Xie, S.; Ma, L.P.; Zhao, Z.C.; Gong, H.B.; Sun, Y.S.; Huang, T. Transcriptome analysis reveals that long noncoding RNAs contribute to developmental differences between medium-sized ovarian follicles of Meishan and Duroc sows. Sci Rep-Uk 2021, 11. [Google Scholar] [CrossRef]
- Tian, B.; Hu, J.; Zhang, H.B.; Lutz, C.S. A large-scale analysis of mRNA polyadenylation of human and mouse genes. Nucleic Acids Research 2005, 33, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Di Giammartino, D.C.; Nishida, K.; Manley, J.L. Mechanisms and Consequences of Alternative Polyadenylation. Molecular Cell 2011, 43, 853–866. [Google Scholar] [CrossRef] [PubMed]
- Devany, E.; Park, J.Y.; Murphy, M.R.; Zakusilo, G.; Baquero, J.; Zhang, X.K.; Hoque, M.; Tian, B.; Kleiman, F.E. Intronic cleavage and polyadenylation regulates gene expression during DNA damage response through U1 snRNA. Cell Discov 2016, 2. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.T.; Zhan, D.D.; Qu, S.; Jiang, S.; Gan, W.H.; Qin, W.S.; Zheng, C.X.; Cheng, F.; Lu, Y.H.; Liu, M.W.; et al. Transcriptomics-proteomics Integration reveals alternative polyadenylation driving inflammation-related protein translation in patients with diabetic nephropathy. J Transl Med 2023, 21. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.Y.; Lin, S.B. mRNA alternative polyadenylation (APA) in regulation of gene expression and diseases. Genes Dis 2023, 10, 165–174. [Google Scholar] [CrossRef]
- Chen, M.L.; Ji, G.L.; Fu, H.J.; Lin, Q.M.; Ye, C.T.; Ye, W.B.; Su, Y.R.; Wu, X.H. A survey on identification and quantification of alternative polyadenylation sites from RNA-seq data. Brief Bioinform 2020, 21, 1261–1276. [Google Scholar] [CrossRef]
- Yan, J.; Marr, T.G. Computational analysis of 3 '-ends of ESTs shows four classes of alternative polyadenylation in human, mouse, and rat. Genome Research 2005, 15, 369–375. [Google Scholar] [CrossRef]
- MacDonald, C.C. Tissue-specific mechanisms of alternative polyadenylation: Testis, brain, and beyond (2018 update). Wires Rna 2019, 10. [Google Scholar] [CrossRef]
- Imada, E.L.; Wilks, C.; Langmead, B.; Marchionni, L. REPAC: analysis of alternative polyadenylation from RNA-sequencing data. Genome Biology 2023, 24. [Google Scholar] [CrossRef]
- Hao, Y.J.; Cai, T.; Liu, C.; Zhang, X.; Fu, X.D. Sequential Polyadenylation to Enable Alternative mRNA3' End Formation. Mol Cells 2023, 46, 57–64. [Google Scholar] [CrossRef]
- Li, X.; Su, J.; Lei, Z.H.; Zhao, Y.Y.; Jin, M.M.; Fang, R.; Zheng, L.C.; Jiao, Y. Gonadotropin-inhibitory hormone (GnIH) and its receptor in the female pig: cDNA cloning, expression in tissues and expression pattern in the reproductive axis during the estrous cycle. Peptides 2012, 36, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Soede, N.M.; Langendijk, P.; Kemp, B. Reproductive cycles in pigs. Animal Reproduction Science 2011, 124, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Gade, S.; Bennewitz, J.; Kirchner, K.; Looft, H.; Knap, P.W.; Thaller, G.; Kalm, E. A note on genetic parameters for estrus symptoms in sows. Applied Animal Behaviour Science 2008, 109, 406–409. [Google Scholar] [CrossRef]
- Tian, B.; Manley, J.L. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Bio 2017, 18, 18–30. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Analysis of differentially expressed genes of the Large White gilts. (A) A volcano plot displays differentially expressed genes in the LDV vs. LEV comparison. (B) KEGG enrichment analysis of differentially expressed genes in the LDV vs. LEV comparison. (C) A volcano plot displays differentially expressed genes in the LDY vs. LEY comparison. (D) KEGG enrichment analysis of differentially expressed genes in the LDY vs. LEY comparison. L: Large white gilts, M: Mi gilts, E: stage of estrus, D: stage of diestrus, Y: vagina, V: vulva. For instance, LEY: Samples were obtained from the vagina in Large White gilts during estrus.
Figure 1.
Analysis of differentially expressed genes of the Large White gilts. (A) A volcano plot displays differentially expressed genes in the LDV vs. LEV comparison. (B) KEGG enrichment analysis of differentially expressed genes in the LDV vs. LEV comparison. (C) A volcano plot displays differentially expressed genes in the LDY vs. LEY comparison. (D) KEGG enrichment analysis of differentially expressed genes in the LDY vs. LEY comparison. L: Large white gilts, M: Mi gilts, E: stage of estrus, D: stage of diestrus, Y: vagina, V: vulva. For instance, LEY: Samples were obtained from the vagina in Large White gilts during estrus.
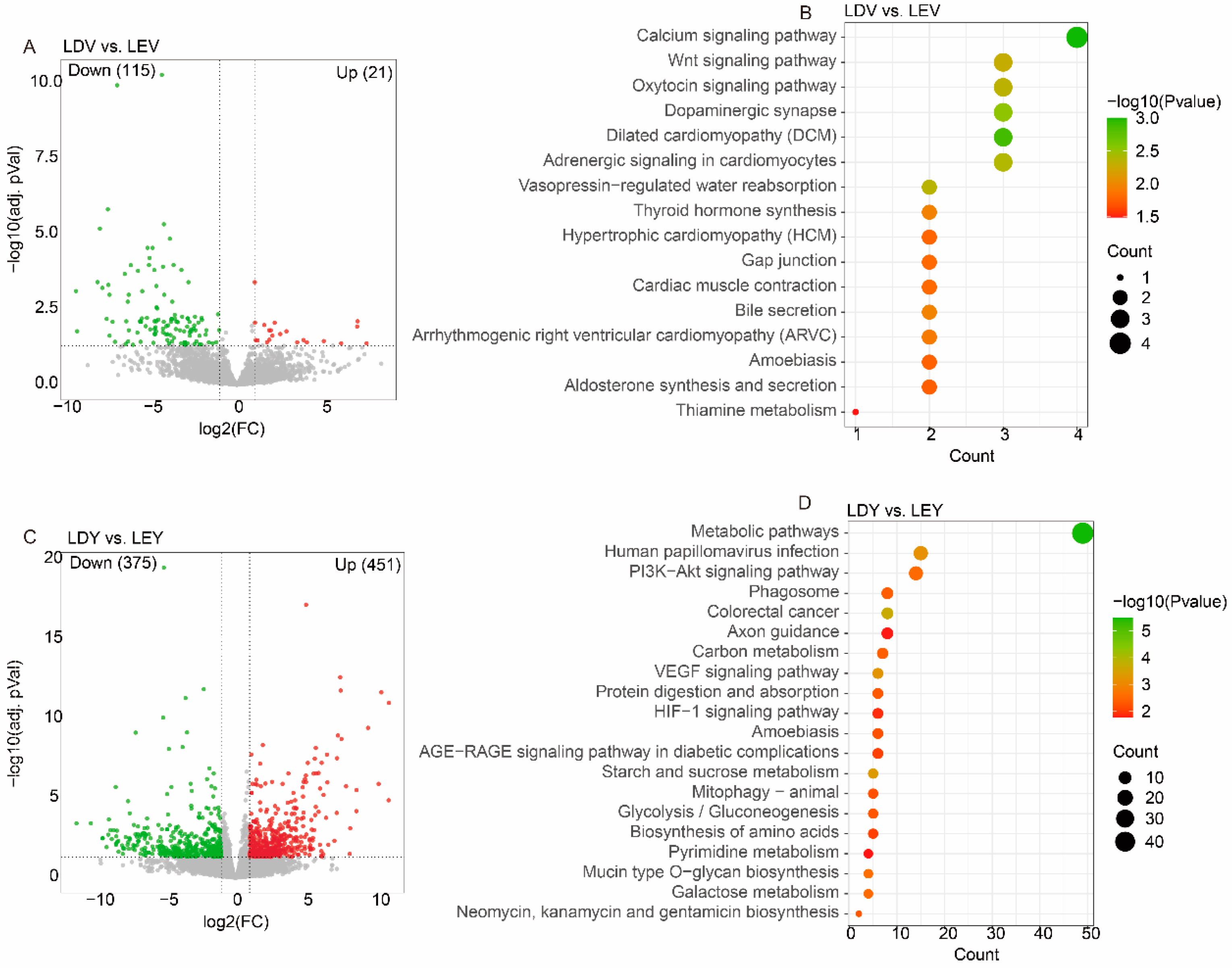
Figure 2.
Analysis of differentially expressed lincRNAs. The distribution of differentially expressed lincRNAs in four comparisons of the vulva (A). The distribution of differentially expressed lincRNAs in four comparisons of the vagina (B). Cis-acting target genes of lincRNA in the vulva (C), with red indicating lincRNA and green indicating target genes. Cis-acting target genes of lincRNA in the vulva (D).
Figure 2.
Analysis of differentially expressed lincRNAs. The distribution of differentially expressed lincRNAs in four comparisons of the vulva (A). The distribution of differentially expressed lincRNAs in four comparisons of the vagina (B). Cis-acting target genes of lincRNA in the vulva (C), with red indicating lincRNA and green indicating target genes. Cis-acting target genes of lincRNA in the vulva (D).
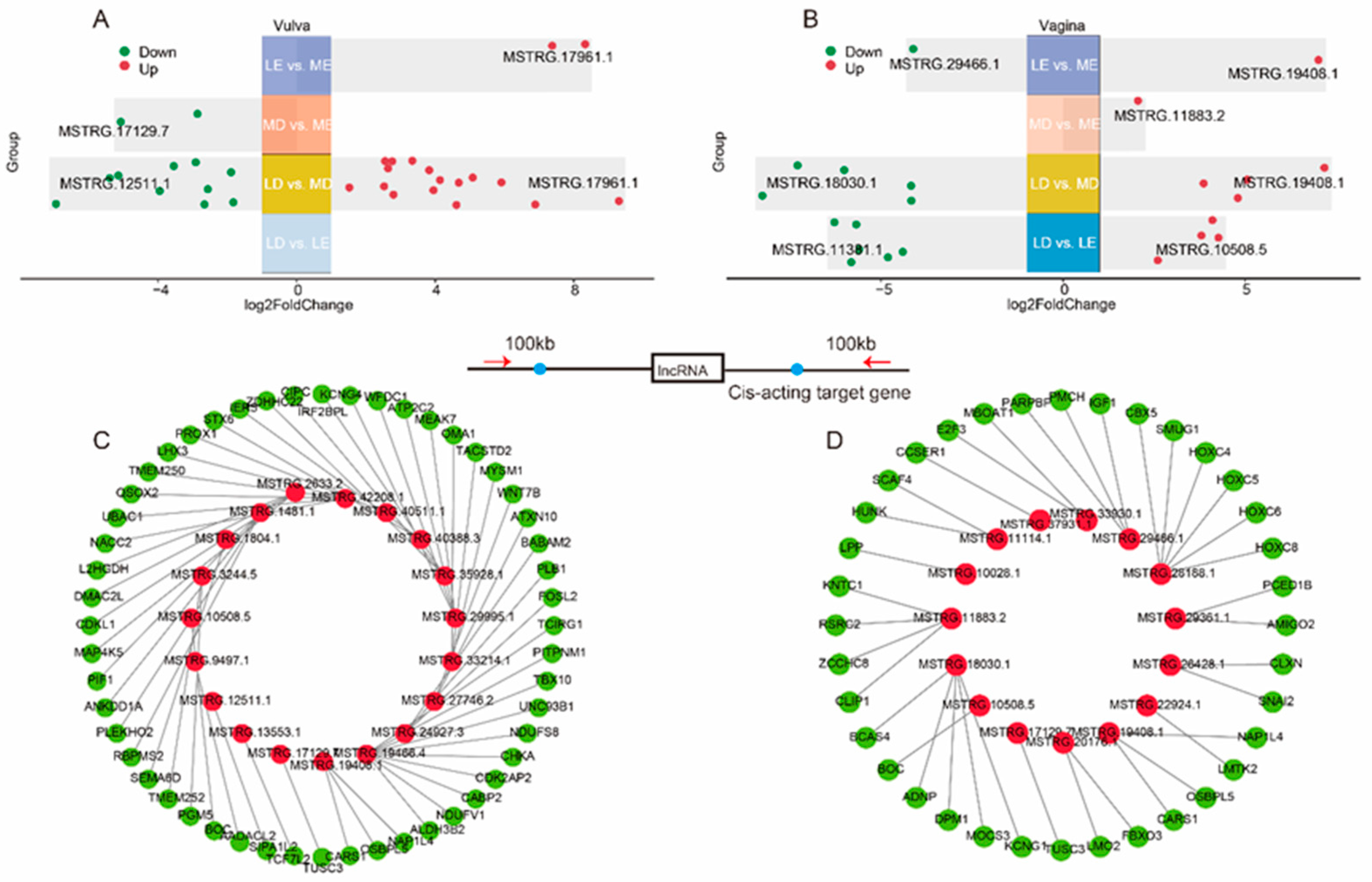
Figure 3.
Identification and analysis of APA sites in the estrus cycles of Large White gilts. The distribution of APA sites in the vulva (A) and vagina (E). PCA of transcripts undergoing APA vulva (B) and vagina (F). Distribution of shortened and lengthened 3'-UTRs vulva (C) and vagina (G). Functional enrichment analysis of transcripts undergoing APA vulva (D) and vagina (H). L: Large white gilts, M: Mi gilts, E: stage of estrus, D: stage of diestrus, Y: vagina, V: vulva. For instance, LEY: Samples were obtained from the vagina in Large White gilts during estrus.
Figure 3.
Identification and analysis of APA sites in the estrus cycles of Large White gilts. The distribution of APA sites in the vulva (A) and vagina (E). PCA of transcripts undergoing APA vulva (B) and vagina (F). Distribution of shortened and lengthened 3'-UTRs vulva (C) and vagina (G). Functional enrichment analysis of transcripts undergoing APA vulva (D) and vagina (H). L: Large white gilts, M: Mi gilts, E: stage of estrus, D: stage of diestrus, Y: vagina, V: vulva. For instance, LEY: Samples were obtained from the vagina in Large White gilts during estrus.
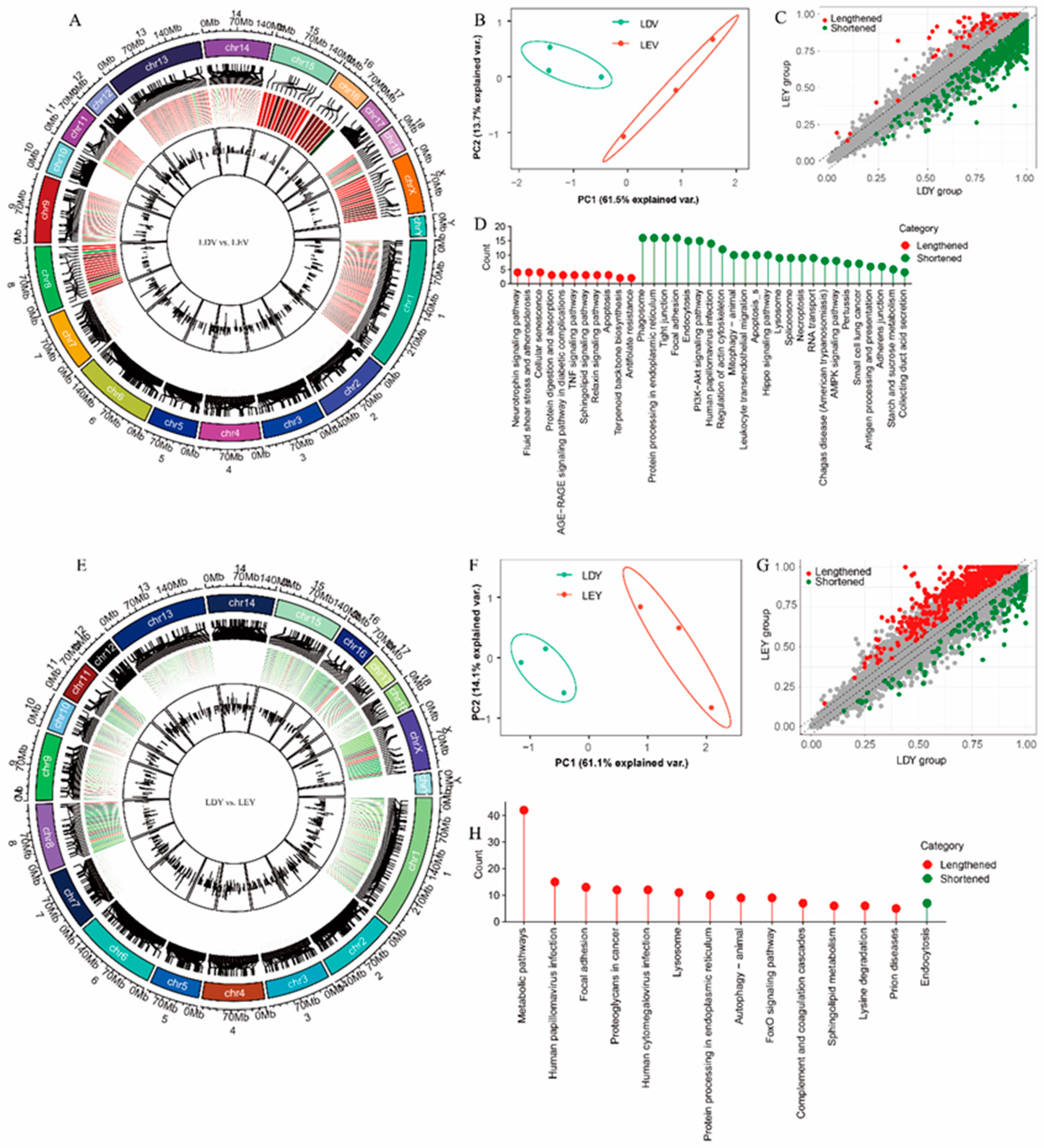
Figure 4.
Visualization and validation of 3'-UTR expression abundance of Large White gilts. The heatmap depicts the expression patterns of lengthened and shortened 3'-UTRs in the vulva (A) and vagina (B) during the estrus and diestrus stages. IGV to show 3'-UTRs length and abundance of TOMM40L, ABHD2 (C), POLA2, CEP162 (D) gene. RT-qPCR was employed to validate the expression levels of their lengthened 3'-UTRs.
Figure 4.
Visualization and validation of 3'-UTR expression abundance of Large White gilts. The heatmap depicts the expression patterns of lengthened and shortened 3'-UTRs in the vulva (A) and vagina (B) during the estrus and diestrus stages. IGV to show 3'-UTRs length and abundance of TOMM40L, ABHD2 (C), POLA2, CEP162 (D) gene. RT-qPCR was employed to validate the expression levels of their lengthened 3'-UTRs.
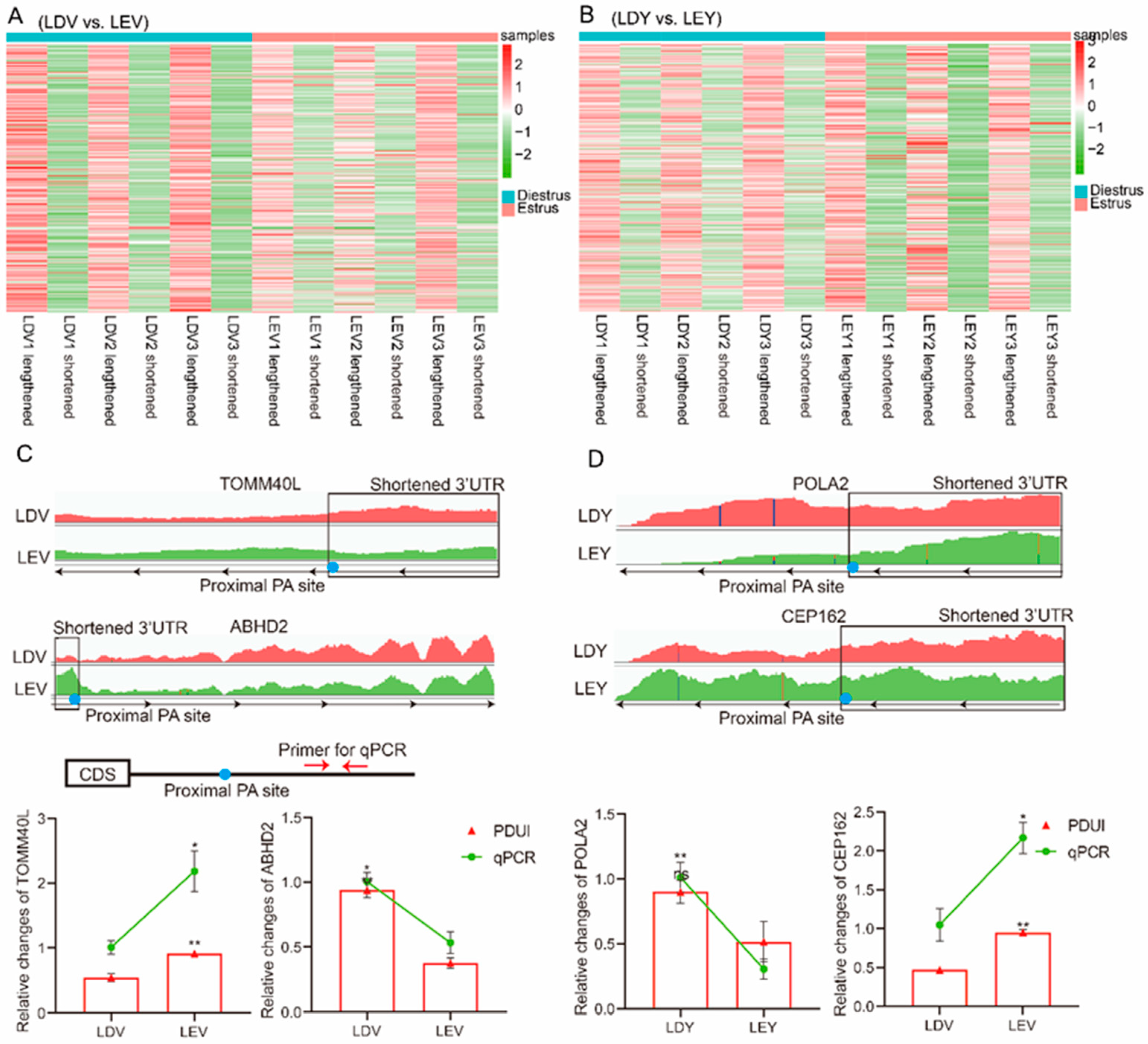
Figure 5.
Regulatory elements in lengthened 3'-UTRs of Large White gilts. The miRNA-gene interactome network of lengthened 3'-UTRs in the vulva (A) and vagina (B) during the estrus and diestrus stages. The green color represents miRNAs, while the red color represents genes with lengthened 3'-UTR. The CPEs on the lengthened 3'-UTRs in the vulva (C) and vagina (D) during the estrus and diestrus stages.
Figure 5.
Regulatory elements in lengthened 3'-UTRs of Large White gilts. The miRNA-gene interactome network of lengthened 3'-UTRs in the vulva (A) and vagina (B) during the estrus and diestrus stages. The green color represents miRNAs, while the red color represents genes with lengthened 3'-UTR. The CPEs on the lengthened 3'-UTRs in the vulva (C) and vagina (D) during the estrus and diestrus stages.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



