
Submitted:
18 January 2024
Posted:
19 January 2024
You are already at the latest version
Abstract
This study examined the efficiency of a combined diagnostic approach in which bioamplification of Arizona 861 S1 citron was integrated with RT‒PCR to detect Citrus tristeza virus (CTV) and Hop Stunt Viroid (HSVd) in Citrus plants. The capacity of the combined method to diagnose mixed infections of CTV and HSVd, along with early single infections of both pathogens, was assessed by evaluating the incubation periods and differential detection capabilities between the combined and direct methods. The bioamplification method exhibited high sensitivity (100%) and specificity (100%) for CTV, enabling early detection even in patients with low-titer infections compared to direct RT‒PCR. However, for HSVd, the combined method achieved 50% sensitivity in early infections, highlighting the need for additional replicates to enhance diagnostic reliability. The relative concentrations of pathogens in mixed and simple infections were evaluated; the analysis revealed no significant differences, suggesting minimal interference between CTV and HSVd. This approach allows early detection of CTV but requires further refinement for HSVd diagnosis. These findings underscore the complementary roles of the cost-effectiveness of direct RT‒PCR and the sensitivity of the combined method in implementing effective disease control strategies in citrus crops.
Keywords:
Virus-viroid detection
; Mixed infections
; Comparison of diagnostic methods
; CTV
; HSVd
; Bioamplification
1. Introduction
Citrus fruits are an essential industry worldwide; their fruits are widely demanded for their organoleptic and nutritional properties and health benefits [1]. These plants represent approximately 17% of global fruit production [2,3]. In the case of Chile, citrus exports in 2020 were approximately 365 thousand tons, and constant growth is forecast for the coming years [4]. Citrus graft-transmissible disease (CGTD) is one of the main threats to this industry, causing estimated losses of 15% to 25% within infected orchards [5].
These diseases are highly relevant internationally. Diseases such as citrus tristeza, caused by Citrus Tristeza Virus (CTV), and citrus cachexia caused by Hop stunt viroid (HSVd) are distributed worldwide, resulting in yield losses and reduced tree longevity [6]. Yield losses due to HSVd have been reported to range from 34% to 76%, depending on the combination of varieties and rootstocks used. In clementines, HSVd caused a 34% decrease in yield; however, in Maltese orange plants grafted onto Citrus macrophylla, yield losses reached 76% [7,8,9]. Citrus tristeza virus is responsible for one of the most significant diseases in the history of these crops, causing epidemics that devastated the citrus industry during the 20th century. Sour orange (Citrus ariantum), one of the most widely used rootstocks at one time, is extremely susceptible to certain CTV variants, causing the syndrome known as quick decline, leading to plant death propagated on this rootstock. This resulted in the death of more than 100 million trees in affected areas during the 20th century, prompting major varietal replacements with rootstocks tolerant of this disease in all regions where tristeza and sour orange coexisted [10].
In Chile, the first reports of the presence of important CGTD throughout the main citrus-growing area of the country were made by Weathers et al. [11]. Later, Besoain [12] and Besoain et al. [13] confirmed the presence of citrus tristeza, which is associated with aggressive CTV isolates in northern Chile and affects grapefruit and Mexican lime trees through the presence of stem pitting on the affected plants. Additionally, attenuated isolates of CTV in sweet orange and lemon trees were detected in central Chile. In other investigations, cachexia, exocortis and psoriasis were reported in the central area of Chile [14,15]. In a study carried out by Pizarro [16], at the Mallarauco locality close, it was determined that the cachexia incidence in this area varied between 10.3 and 36.7%; this area of the country was considered the most affected by this disease [15]. Although the reports by Weathers and collaborators were based on the symptoms observed in the field and with Mexican lima indicator plants, most modern reports have also used serological and/or molecular methods to diagnose the presence of these pathogens, especially CTV and cachexia isolates [12,13,16,17].
The primary means of spreading these diseases is through grafting, although tristeza is also spread by aphids. In Chile, a study carried out by Camps et al. [17] demonstrated that Aphis gossypii was capable of transmitting the virus from infected plants to healthy plants, producing a change in the characteristics of the virus populations, where the attenuated populations acquired by aphids after being transmitted to a new host (from Mexican lime to sweet orange) became populations where an aggressive variant predominated. On the other hand, the causal agent of cachexia, Hop stunt viroid (HSVd), can be transmitted highly efficiently through contaminated cutting tools. Therefore, the risk of spreading these diseases at the noncertified nursery level and in the field is high.
Consequently, although citrus farming is a growing industry, CGTDs are a threat to citrus plants and have caused significant losses in Chilean and global citrus farming. To confront these threats, countries where citrus farming is important have developed protection programs against these diseases to prevent their spread. In Chile, the Servicio Agrícola y Ganadero (SAG) issued an exempt resolution, N° 8911 [18], that regulates the propagation of citrus plant material in the country. This resolution requires that all plants used as a source of propagation material (mother plants) must be free of CTV, CPsV and HSVd. To comply with this resolution, subjecting maternal plants to periodic analyses is necessary to diagnose the presence of these pathogens. Therefore, reliable, efficient and reproducible diagnostic methods are important.
In general, in the field, this type of disease usually has a relatively long incubation period, and its diagnosis through observation of symptoms in the field is usually confusing. Therefore, among the diagnostic methods most commonly used to determine the presence of these pathogens, there are biological, serological and molecular methods. Specifically, techniques such as biological indexing (BI) are used; this method is based on the expression of specific symptoms in indicator plants that are very sensitive to a specific pathogen. In addition, laboratory analyses, such as ELISA and its variants, and molecular techniques, such as RT‒PCR and qPCR, are used. Although these techniques are highly reliable, they have several limitations. BI is highly expensive in terms of time, equipment, infrastructure and personnel. The most significant limitations in laboratory techniques are related to sampling since poor sample handling before analysis or possible cross-contamination can lead to incorrect diagnoses [19]. Climatic conditions, time of year, maturity of infection, and uneven distribution of the pathogen in the plant can also result in false-negatives.
To overcome the limitations mentioned above, techniques such as bioamplification, which seek to multiply the virus or viroid naturally by inoculating it into a living plant under ideal climatic conditions for pathogen development, are used. Duran-Vila and colleagues [20] proposed an improved method of biological indexing to detect cachexia using Arizona 861-S1 citron combined with the molecular technique sPAGE. They successfully diagnosed the disease 3 months postinoculation, suggesting that this variety allows for a rapid increase in the viroid titer detectable by the molecular technique. Later, Camps et al. [21] successfully utilized the bioamplification technique in Arizona 861-S1 citrus combined with RT‒PCR to diagnose the presence of CTV, HSVd, and CEVd in both single and mixed infections. These infections were detected after an incubation period of 8 months. While single infections could be diagnosed at 4 months, multiple infections were not detectable until 8 months. This finding suggested possible interference between pathogens in the case of multiple infections. There is diverse evidence indicating that mixed infections in citrus plants are common and that there may be interactions between different pathogens, as observed for CTV and citrus dwarfing viroid (CDVd) [22] or between HSVd and CEVd [23], where mixed infection promotes an increase in the titer of one or both pathogens [22,23]. The nature of the interaction between viruses and viroids has been poorly studied and depends on factors such as the host, the variants involved, and competition for resources within the cell [24].
Therefore, the main objective of this research was to determine whether the bioamplification technique in Arizona 861-S-1 citron combined with RT‒PCR is effective at detecting incipient and mixed infections of CTV and HSVd in comparison to direct RT‒PCR.
2. Results
2.1. Analysis of mixed infections
The analysis of mixed infections showed that it is possible to detect both pathogens via the bioamplification method combined with RT‒PCR for both mixed infections and simple infections. The Kruskall Wallis test for relative band intensity using densitometry with ImageJ showed no significant differences in the concentration of the pathogens between simple infections and mixed infections for the different treatments (p value = 0.8) (Figure 1).
2.2. Comparison of combined bioamplification and RT‒PCR methods with direct RT‒PCR for the detection of CTV and HSV
Citrus tristeza virus was detected using direct detection to determine the presence of the virus during infections from 3 months onward for all treatments and repetitions. On the other hand, through the analysis carried out with the mixed method, disease could be diagnosed from 1 month onwards. In this context, it was possible to identify the presence of the virus in 50% of the inoculated plants in the first month of infection and 100% of the treated plants for 3 and 6 months of infection, as illustrated in Figure 2.
In the case of HSVd, the direct detection method identified the presence of the viroid in infections from 3 months onward for all treatments and repetitions. On the other hand, through the analysis carried out with the mixed method, the disease could be diagnosed from 3-month infections. In this context, viroids were detected in 100% of the treatments at 3 and 6 months after infection using the direct method and in 50% of the infected plants in each treatment using the bioamplification method, as presented in Figure 3.
2.2. Validation parameters for the combined bioamplification and RT‒PCR detection method
The validation parameters for the combined test for the detection of CTV are presented in Table 1. The Arizona citron bioamplification test for CTV showed high sensitivity (100%), which indicated that a negative result ruled out the presence of CTV. Likewise, the test exhibited 100% specificity, confirming the diagnosis if the test was positive. Cohen’s kappa index for CTV was 0.75, with a p value = 0.028. These results indicate that the agreement between the two methods for diagnosing CTV was “good”.
The validation parameters for the detection of HSVd are shown in Table 2. For HSVd, the bioamplification test has a low sensitivity of 50%. This result indicates that a negative test result does not ensure the absence of the pathogen since 50% of the results were false-negatives. On the other hand, a specificity of 100% indicates that a positive test result ensures the diagnosis of the pathogen. Cohen’s kappa index for the analysis was 0.5, and the p value was 0.102. This result indicates moderate agreement between the two methods, although this agreement is not significant.
3. Discussion
This study compared the combined diagnostic method of bioamplification in Arizona citron with RT‒PCR and direct RT‒PCR diagnostic techniques on samples obtained under favorable sampling conditions. This comparison was carried out by evaluating the validation parameters of diagnostic methods [25,26,27]. Additionally, the ability of the combined method to detect mixed infections of CTV and HSVd was examined, along with potential evidence of interference in pathogen concentrations in mixed infections through the comparison of the relative intensity of bands in agarose gel electrophoresis of PCR products from the analyzed samples.
In this study, it was observed that the combined method allowed early detection of CTV in samples from plants after one month of incubation, i.e., in plants with a low virus titer, unlike direct detection, which failed to diagnose the pathogen’s presence in plants after the same incubation time. The main explanation for this phenomenon lies in the ability of the combined method to promote virus multiplication in plant tissues naturally by maintaining plants at temperature conditions conducive to pathogen development. Kokane et al. [28] noted that the concentration of CTV viral particles in leaf midribs, measured through RT–qPCR, decreased by one to two orders of magnitude when the average temperature increased from 29 to 37 °C. Furthermore, an increase in temperature causes a shift in the distribution of viral particles in the plant, a decrease in the concentration of the virus in leaves and an increase in the concentration in roots [28].
The validation parameters for the CTV bioamplification test and the sensitivity and diagnostic specificity values were both equal to 1 in both cases. These findings align with those of Vidal et al. [25], who calculated these parameters for CTV diagnosis using real-time RT‒PCR and compared them with those of immunodetection-ELISA, obtaining a sensitivity of 0.9820 and a specificity of 0.8519. Similarly, Simeone et al. [26] used BI with RT‒qPCR for psorosis diagnosis, achieving a sensitivity and specificity equal to 1. Values close to 1 were obtained by Massar et al. [19] and Massar et al. [29], who evaluated methodologies for RT‒PCR detection of viruses in stone fruits and apples in virus-free material certification programs. These authors concluded that tests with these sensitivity and specificity values can be used in virus-free material certification programs due to their low false diagnosis rates, reducing the risk of spreading GTDs by releasing incorrectly diagnosed contaminated material.
When assessing the agreement between the two methods, a kappa value of 0.75 (p value = 0.028) was obtained. These findings coincide with those of Massar et al. [19] who evaluated interlaboratory agreement using an identical RT‒PCR protocol for the detection of Prune dwarf virus and Prunus necrotic ringspot virus. Massar and colleagues concluded that there was strong consensus among laboratories utilizing the same methodology, indicating the high reproducibility of this test. In other words, the outcomes of these procedures remain consistent irrespective of changes in the operator or the laboratory conducting the technique [29].
On the other hand, both diagnostic methods for HSVd detected the pathogen after 3 months of incubation. However, the combined amplification and RT‒PCR method detected HSVd in 50% of the samples during this specific incubation period, unlike the direct method, which detected 100% of the pathogens. This observation could highlight one of the limitations of the combined technique, as inoculation may not result in successful infection due to factors such as the uneven distribution of the viroid in the plant, as indicated by Garnsey et al. [30], who found that the HSVd viroid accumulates differentially in various plant tissues. Additionally, viroid RNA silencing signals translocated via the phloem compete with the systemic translocation of viroid particles, generating tissues with infected and healthy zones and resulting in a nonuniform viroid distribution in the plant [31]. This approach necessitates the use of a certain number of replicates of bioamplifying plants for each test, similar to biological indexing, where if at least one of the test indicator plants shows symptoms, the test is considered positive [32].
The combined test of bioamplification in Arizona citron 861 S1 and RT‒PCR in the case of HSVd achieved a sensitivity of 0.5 and specificity of 1. According to Bravo-Grau and Cruz [33], this approach constitutes a low level of sensitivity, indicating a 50% probability of diagnosing a false-negative if only one plant is used per test. On the other hand, Cohen’s kappa coefficient was 0.5 in this case, indicating moderate agreement between the results of both diagnostic tests [19]. Once again, the results indicate that the bioamplification test for HSVd needs to be performed with a certain number of replicates to increase its diagnostic sensitivity [32]. However, the obtained p value for Cohen’s kappa index indicates that this agreement could be attributable to chance. Therefore, it is advisable to conduct a comprehensive analysis to assess the minimum number of replicates needed to ensure a reliable diagnosis of HSVd in incipient infections using the Arizona citron bioamplification method.
In studies performed by Bertolini et al. [34], the analytical sensitivity of RT‒PCR was compared with that of real-time RT‒PCR (RT‒qPCR) for CTV detection. In that case, they found that the RT‒qPCR test is 1000 times more sensitive than the RT‒PCR test and can detect low-titer viral infections, as observed in incipient infections. The same trend occurred for HSVd, for which the RT‒qPCR protocol demonstrated an analytical sensitivity 1000 times greater than that of conventional RT‒PCR. Furthermore, it presented a diagnostic sensitivity of 100% compared to traditional RT‒PCR, which showed a diagnostic sensitivity of 94% [35]. This finding suggested that real-time RT‒PCR is more suitable for routine use in incipient infections.
Mixed infections of CTV and HSVd were successfully diagnosed after four months of incubation on all the inoculated plants; however, symptoms of the disease could not be observed during this period. Using the same technique, Camps et al. [21] diagnosed both pathogens for four months by observing specific symptoms of each disease expressed in the same plant. Nonetheless, detecting both pathogens via RT‒PCR was only possible after eight months of incubation. In the case of CTV, although CTV isolates naturally correspond to mixed populations of the virus, according to Camps et al. [21], they used a VT-type isolate, classified as aggressive because it reacts to the MCA13 antibody [17], and were able to observe vein clearing in the leaves of Arizona citron 861-S1-inoculated plants. In this study, 334 isolates from Mexican lime were obtained initially from a sweet orange in the Pica locality and were classified as VT-type or MCA13-positive [13]. After being inoculated in Mexican lime, these isolates were classified as corresponding to a mixed isolate of the VT+T30K17 type and did not react to the monoclonal antibody MCA13 [17]. The MCA13 was developed by Permar et al. [36] to recognize aggressive CTV isolates. This change could explain why the inoculated cider plants with the 344 CTV isolate did not develop vein-clearing symptoms during the four-month inoculation period.
Densitometric analysis did not reveal significant differences in the relative concentrations of pathogens in plant tissues between simple and mixed infections. This result indicates no evidence of interference between the two pathogens. Densitometry has previously been used to assess HSVd concentrations in Citrus tissues, but it has been used on autoradiograms resulting from the Northern blot technique, according to previous studies [37]. With this technique, obtaining a relative quantification of the viroid concentration is possible, allowing the determination of the method’s analytical sensitivity for detection. To our understanding, our study is the first to report the quantification of HSVd and CTV cDNA via densitometry. Other techniques that allow more concrete quantification, such as RT‒qPCR, have been used to evaluate the interaction and distribution of HSVd and CEVd in plant tissues [23]. Therefore, quantitative techniques such as RT‒qPCR are suggested for improving the understanding of how HSVd and CTV titers behave in mixed infections. These findings will aid in the development of better techniques, sampling guidelines, and diagnostics for application in quarantine or plant health certification programs for all types of infections, be they simple, mixed, or recent, and associated with low pathogen titers.
Although the differences in CTV and HSVd titers between simple and mixed infections were nonsignificant, the results suggested that the HSVd titer was lower in patients coinfected with CTV. This result differs from the mixed infection of CTV and Citrus Dwarfing Viroid (CDVd) observed by Serra et al. [22], who determined that the mixed infection of CTV and CDVd promotes an increase in the viroid concentration in the tissues of coinfected plants. These authors speculate that the proteins P23 and P25, encoded by CTV, possess the suppressor activity of the viroid RNA silencing system [38], resulting in the promotion of viroid infection. On the other hand, HSVd replication occurs in the plant cell nucleus with the accumulation of mature viroids in the nucleolus [31], in addition to HSVd being found in different tissues, such as phloem, palisade parenchyma of leaves, or cortical cells near the cambium [23]. Unlike CTV, whose viral RNA replication occurs in vesicle complexes, the accumulation of mature virions occurs mainly in the phloem cell cytoplasm [10] may influence the ability of both pathogens to coexist in the host plant.
Lin et al. [23], when studying mixed infections of HSVd and CEVd, observed that coinfection promoted an increase in the titer of both pathogens compared to single infections. On the other hand, Serra et al. [22] reported that mixed infections of CTV and citrus dwarfing viroid (CDVd) promoted the accumulation of the latter pathogen in plant cells. These authors indicate that this phenomenon depends on the host, as the CDVd titer is promoted in Mexican lime but does not increase when the host is a citron plant. This result aligns with what was observed in this study, specifically the results of mixed infection RT‒PCR bioamplification in Arizona citron.
4. Materials and Methods
4.1.___
The study was carried out in the PUCV Citrus Clonal Propagation Program, specifically in greenhouses designated for the diagnosis of citrus viruses and viroids of the PUCV Phytopathology Laboratory, located in La Palma, Quillota (32°53’45.7”S 71°12’34.1”W).
The plants used in the experiments were Cidro Arizona 861-S1 (CA) (Citrus medica Linnaeus), Frost Eureka (FE) lemon (Citrus limon (L.) N. Burman) and Cara Cara (CC) sweet orange (Citrus sinensis (L.) Osbeck) plants, all of which were grafted on rough lemon (Citrus jambhiri Lushington). These plants were placed in 3-liter containers with a substrate mixture composed of peat and perlite at a 1:1 ratio, and 6 g of slow-release granular fertilizer (Ferpac cote 6 M, FERPAC) was applied per plant. At the time of inoculation, the plants were approximately 12 months old. Inoculations were carried out using patches of green bark taken from only one donor branch of each plant infected with CTV and HSVd. Once the inoculations were carried out, the plants were kept in greenhouses free of pests and other diseases, and the temperature was controlled according to the specific regimes of each trial.
The CTV and HSVd strains used in the study were obtained from the Virus and Viroid Strain Bank of the Phytopathology Laboratory of the PUCV. These strains correspond to isolate 334 for CTV [12] from a Mexican lime plant and isolate 619-21 for HSVd, which is conserved in the Cidro Arizona plant. The plants selected as donors of each pathogen were previously checked to verify the presence of CTV and HSVd using RT‒PCR following the methodologies of Hilf et al. [39] and Yang et al. [40], respectively.
4.2. Analysis of mixed infections:
In this trial, bark patches obtained from each donor plant were used to inoculate plants of the Arizona 861-S1 variety cider grafted on rough lemon (CA-RL) with CTV and HSVd in individual and mixed infections for each pathogen. Four patch grafts of bark infected with each pathogen were made for single infections. For mixed infections, grafts of two patches of each donor plant were generated for each replicate of each treatment (Figure S1). The negative controls were grafted with four patches of healthy Eureka Frost lemon (HSVd) or Cara-Cara sweet orange (CTV) plants. Four treatments were established with four repetitions: simple infection with CTV, simple infection with HSVd, mixed infection and healthy control. The inoculated plants were kept in a temperature-controlled environment for four months: between 25 and 28 °C for the first two months and between 28 and 31 °C for the next two months. At the end of the incubation period, four twigs were collected from each inoculated plant for RNA extraction and subsequent analysis via RT‒PCR according to the methodology used for each pathogen, which is described later.
4.3. Differential periods of incubation for CTV and HSVd:
To achieve variable levels of infectious particles in the plants, two bark patches obtained from the donor plants were used to inoculate plants of sweet orange CC-RL with CTV and plants of lemon EF-RL with HSVd. Inoculations consisting of three different incubation periods (1, 3, and 6 months) plus 0 months as the healthy control treatment were carried out for each pathogen (Figure S2). All inoculations were performed according to the specific incubation intervals required for each treatment. All the inoculations were carried out on the same date. The inoculated plants were maintained at 25-28 °C until the corresponding incubation time was reached. At the end of the incubation period, four twigs were taken from each inoculated plant for analysis by RT‒PCR.
4.4. Bioamplification in Arizona 861-S1 citron Plus RT‒PCR:
In this assay, two bark patches taken from the inoculated plants of each treatment in the direct detection assay were used to inoculate plants of the AC‒RL variety in the CTV and HSVd treatments. Four treatments were carried out, which consisted of inoculation with tissue from plants with three different disease incubation intervals, and a healthy control treatment was included for each pathogen (Figure S3). The inoculated plants were kept in a temperature-controlled environment for four months: between 25 and 28 °C for the first two months and between 28 and 31 °C for the next two months. At the end of the incubation period, four twigs were collected from each inoculated plant for RT‒PCR analysis.
4.5. Total RNA Extraction:
Briefly, 100 mg of bark and petiole tissue was taken for RNA extraction, pulverized and homogenized in liquid nitrogen. An RNeasy® Plant Mini Kit (Qiagen Hilden, Germany) was used following the manufacturer’s instructions to extract the RNA.
4.6. RT‒PCR analysis:
The protocols described by Hilf et al.[39] and Yang et al.[40] were used for RT‒PCR analysis of the samples for CTV and HSVd, respectively; some modifications were made to both protocols. In the case of CTV, RT‒PCR was carried out as follows: for the RT‒PCR, 3 μL of RNA and 1 μL of primer F were utilized. The mixture was incubated at 70 °C for 10 minutes, followed by rapid transfer to an ice bath for 5 minutes. Then, 2 μL of Buffer M-MuLV, 0.25 μL of M-MuLV enzyme, 0.25 μL of RNase inhibitor, 1 μL of dNTPs, and 1.5 μL of H2O were added to the mixture, resulting in a total reaction volume of 9 μL, which was incubated at 42 °C for 60 minutes. PCR was performed using 1 μL of cDNA. The reaction mixture consisted of 12.5 μL of SapphireAmp Fast PCR Master Mix™ (Takara), 1.0 μL of Pf (10 μM), 1.0 μL of Pr (10 μM), and 7.5 μL of H2O, resulting in a final volume of 22 μL. The primers used corresponded to the sequence of the coat protein of isolate T36,T36 CP (F) 5’ATGGACGACGAAACAAAGAAATTG3’; (R) 5’TCAACGTGTGTTGAATTTCCCA3’ (672 bp). The thermocycling cycle consisted of a predenaturation step at 42 °C for 45 minutes; a denaturation step at 94 °C for 2 minutes; 30 cycles at 94 °C for 30 seconds, 55 °C for 30 seconds, and 72 °C for 45 seconds; and a final extension at 72 °C for 5 minutes.
In the case of HSVd, 4 µL of RNA, 1 µL of reverse primer and 3 µL of water were used. The mixture was incubated at 100 °C for 5 minutes, cooled on ice for 2 minutes, and then kept at room temperature for 45 minutes. The mixture contained 1 µL of M-MLV buffer (New England BioLabs, United Kingdom), 1 µL of 1.25 mM dNTPs, 40 U of RNase inhibitor (New England BioLabs, United Kingdom) and 400 U of MLV M-Reverse Transcriptase (New England BioLabs, United Kingdom). The volume was adjusted to 50 µL with sterile water, and the mixture was incubated at 42 °C for 1 hour. PCR included the addition of a 4 µL aliquot of the above reaction mixture to 5.5 μL of SapphireAmp Fast PCR Master Mix™ (Takara), 1.0 μL of Pf (10 μM), 1.0 μL of Pr (10 μM), and 13.5 μL of H2O, resulting in a final volume of 25 μL. Amplification consisted of a denaturation step at 94 °C for 3 minutes; 40 cycles at 94 °C for 1 minute, 60 °C for 1 minute, and 72 °C for 1 minute; and a final extension at 72 °C for 7 minutes. Finally, electrophoresis was performed on a 1.5% agarose gel. The resulting bands were observed in a UV transillumination chamber.
4.7. Densitometric analysis:
Photographs were taken of the electrophoresis gels of the RT‒PCR analyses carried out in the mixed infections assay; these were used to measure the relative intensity of each amplified band; for this purpose, ImageJ software was used. These measurements were compared with a calibration curve generated by serial dilution of a sample of known DNA at a concentration of 30 µg/band to determine the relative concentration of the amplified band.
4.8. Statistical analysis:
The diagnostic sensitivity (SE) and specificity (SP) were calculated according to the methods of Altman and Bland [41]. The sensitivity is the proportion of true positives diagnosed by the evaluated test over the total number of positives according to the standard test. Specificity is the proportion of true negatives diagnosed by the test evaluated over the total negatives according to the standard test. In this case, the standard test was direct RT‒PCR, and the test evaluated was the combined bioamplification and RT‒PCR method. Cohen’s kappa coefficient was calculated according to Cohen [42] considering the criteria of Landis and Koch [43], where weak agreement was considered if kappa was between 0.21 and 0.40, moderate agreement was between 0.41 and 0.60, strong agreement was between 0.61 and 0.80, and almost perfect agreement was between 0.81 and 1.00. The probability that the degree of agreement indicated by Cohen’s kappa index was due to chance was analyzed with the chi-square test, with a p value = 0.05. Differences between treatments for measurement percentages for each test were calculated using Fisher’s exact test. Differences between mixed and single infection treatments were calculated using the chi-square test, and separation of means was performed using Tukey’s test.
5. Conclusions
The analysis of the results from this study suggested that the detection of CTV through the bioamplification method in Arizona 861 S1 citron and RT‒PCR can be employed as diagnostic tools for CTV disease within a virus and viroid detection program in plant material intended for propagation. In this context, this methodology can be used to identify early infections with low viral titers, providing an advantage compared to the direct analysis of samples collected in the field. Moreover, this combined technique detects mixed infections of CTV and HSVd. However, concerning the latter pathogen, further investigations are necessary to evaluate the utility of using only the combined method of bioamplification in Arizona 861 S1 citron plus RT‒PCR for diagnosing single or mixed infections. Specifically, it would be advisable to determine the minimum number of bioamplifying plant replicas needed for a reliable diagnosis of HSVd in early infections.
On the other hand, the RT‒PCR technique applied directly would be more cost-effective and faster for virus and viroid detection. However, incipient infections with low titers may go undetected. In this regard, it would be essential to compare RT‒PCR with qPCR to detect early HSVd infections. An alternative to the latter is the use of the bioamplification method as a second confirmatory test for the phytosanitary status of the plants analyzed via direct RT‒PCR. Finally, these results provide valuable information for decision-making in detecting CTV and HSVd in mother plants to prevent the spread of graft-transmissible diseases in citrus crops.
Supplementary Materials
Not applicable now.
Author Contributions
Investigation and writing, C.H.; original draft preparation, C.H. and X.B.; formal analysis, C.H., N.R.; investigation, C.H., N.R. and X.B.; statistical analysis: C.H.; writing—review and editing, C.H., A.L. and X.B.; supervision and writing—review and editing, X.B. All the authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fundación para la Innovación Agraria de Chile (FIA PYT-2020 0219) and Comité de Cítricos de Chile.
Data Availability Statement
Not applicable now.
Acknowledgments
The authors thank Iván Cortés for his collaboration in the field maintenance trial. The authors thank Professors Roberto Bastías and Ricardo Cautín for their technical advice.
Conflicts of Interest
The authors declare that there are no conflicts of interest.
References
- Liu, Y.Q.; Heying, E.; Tanumihardjo, S.A. History, global distribution, and nutritional importance of citrus fruits. Comprehensive Reviews in Food Science and Food Safety 2012, 11, 530–545. [Google Scholar] [CrossRef]
- Knoema. (2020). Citrus fruit production quantity. Recuperado de: https://knoema.com/atlas/topics/Agriculture/Crops-Production-Quantity-tonnes/Citrus-fruit-production.
- World Citrus Organization. (2020). Global Citrus Outlook. Recuperado de: https://worldcitrusorganisation.org/wp-content/uploads/2020/01/Citrus-Market-Trends-2019.pdf.
- Comité de Cítricos Chile. (2021). Comité de Cítricos Asoex: Estiman crecimiento de un 6% en las exportaciones de cítricos para temporada 2021. Recuperado de https://www.comitedecitricos.cl/noticias-y-actividades/noticias/1092-comite-de-citricos-asoex-estiman-crecimiento-de-un-6-en-las-exportaciones-de-citricos-para-temporada-2021 [16-04-21].
- Moreno, P.; Guerri, J.; García, M.L. The Psorosis disease of citrus: A pale light at the end of the tunnel. Journal of Citrus Pathology 2015, 2. [Google Scholar] [CrossRef]
- Moreno, P.; Ambrós, S.; Albiach-Martí, M.R.; Guerri, J.; Peña, L. Citrus tristeza virus: a pathogen that changed the course of the citrus industry. Molecular Plant Pathology 2008, 9, 251–268. [Google Scholar] [CrossRef] [PubMed]
- Belabess, Z.; Radouane, N.; Sagouti, T.; Tahiri, A.; Lahlali, R. (2021). A Current Overview of Two Viroids Prevailing in Citrus Orchards: Citrus Exocortis Viroid and Hop Stunt Viroid. IntechOpen. [CrossRef]
- Vernière, C.; Perrier, X.; Dubois, C.; Dubois, A.; Botella, L.; Chabrier, C.; Bové, J.M.; Duran Vila, N. Citrus viroids: Symptom expression and effect on vegetative growth and yield of clementine trees grafted on trifoliate orange. Plant Disease 2004, 88, 1189–1197. [Google Scholar] [CrossRef] [PubMed]
- Najar, A.; Hamdi, I.; Varsani, A.; Duran-Vila, N. Citrus viroids in Tunisia: Prevalence and molecular characterization. Journal of Plant Pathology 2017, 99, 787–792. [Google Scholar] [CrossRef]
- Folimonova, S.Y.; Sun, Y.-D. Citrus Tristeza Virus: From Pathogen to Panacea. Annual Review of Virology 2022, 9, 417–435. [Google Scholar] [CrossRef] [PubMed]
- Weathers, L.; Sánchez, L.; Platt, G. Naturaleza y distribución de las enfermedades virosas de cítricos en Chile. Agricultura Técnica 1969, 29, 166–170. [Google Scholar]
- Besoain, X. (2008). Incidencia, caracterización y epidemiología del virus de la tristeza de los cítricos en Chile. Tesis Doctoral. Universidad Politécnica de Valencia. Escuela Técnica Superior de Ingenieros Agrónomos. Valencia, España. 129 pp.
- Besoain, X.; Bertolini, E.; Camps, R.; Ramella, F.; Gorris, M.T.; Torres, M.; Cambra, M. Aggressive Citrus tristeza virus isolates in Chile are MCA13-positive and VT type, while mild isolates are MCA13-negative and T30 type. Ciencia e Investigación Agraria 2015, 42, 251–262. [Google Scholar] [CrossRef]
- Besoain, X.; Valenzuela, M.; Castro, M.; Ballester Olmos, J.F. Situación actual de las enfermedades tipo virus que afectan a los cítricos en Chile. Fitopatología 2000, 35, 98–104. [Google Scholar]
- Valenzuela, Miryam & Besoain, Ximena & Castro, Mónica & Pizarro, Carmen & Ballester-Olmos, José. El Problema de la cachexia en Chile. Fitopatologia 2000, 35, 105–110. [Google Scholar]
- Pizarro Zamorano, C.G. (1999). Detección del viroide Cachexia y Exocortis mediante método combinado de planta indicadora y electroforesis reversa en geles de poliacrilamida. Tesis de ingeniero agrónomo, Universidad Católica de Valparaíso. Quillota, Chile.
- Camps, R.; Fiore, N.; Riquelme, N.; Barros-Parada, W.; Besoain, X. Genotype variation of citrus tristeza virus after passage on different hosts, and changes in the virus genotype populations by the vector Aphis gossypii. Phytopathologia Mediterranea 2022, 61, 55–63. [Google Scholar] [CrossRef]
- Servicio Agrícola y Ganadero (SAG). (2020). Resolución Nº 8.911/2020 Establece lista de plagas no cuarentenarias reglamentadas para material de propagación de especies de cítricos y las medidas fitosanitarias para su supresión.
- Massart, S.; Brostaux, Y.; Barbarossa, L.; César, V.; Cieslinska, M.; Dutrecq, O.; Fonseca, F.; Guillem, R.; Laviña, A.; Olmos, A.; Steyer, S.; Wetzel, T.; Kummert, J.; Jijakli, M.H. Inter-laboratory evaluation of a duplex RT-PCR method using crude extracts for the simultaneous detection of Prune dwarf virus and Prunus necrotic ringspot virus. European Journal of Plant Pathology 2008, 122, 539–547. [Google Scholar] [CrossRef]
- Duran-Vila, N.; Pina, J.A.; Molins, M.I.; Navarro, L. (1991). A new indexing method for cachexia. In: Proc. llth Conf. IOCV. IOCV, Riverside, 224-229.
- Camps, R.; Castro, M.; Besoain, X. Simultaneous detection of CTV, CEVd and HSVd using Arizona 861 S1 Citron and RT-PCR. Ciencia e Investigación Agraria 2014, 41, 23–24. [Google Scholar] [CrossRef]
- Serra, P.; Bani Hashemian, S.M.; Fagoaga, C.; Romero, J.; Ruiz-Ruiz, S.; Gorris, M.T.; Bertolini, E.; Duran-Vila, N. Virus-viroid interactions: Citrus Tristeza Virus enhances the accumulation of Citrus Dwarfing Viroid in Mexican lime via virus-encoded silencing suppressors. Journal of Virology 2014, 88, 1394–1397. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-Y.; Wu, M.-L.; Shen, T.-L.; Hung, T.-H. A mutual titer-enhancing relationship and similar localization patterns between Citrus exocortis viroid and Hop stunt viroid co-infecting two citrus cultivars. Virology Journal 2015, 12, 142. [Google Scholar] [CrossRef] [PubMed]
- Syller, J. Facilitative and antagonistic interactions between plant viruses in mixed infections. Molecular Plant Pathology 2012, 13, 204–216. [Google Scholar] [CrossRef] [PubMed]
- Vidal, E.; Yokomi, R.K.; Moreno, A.; Bertolini, E.; Cambra, M. Calculation of Diagnostic Parameters of Advanced Serological and Molecular Tissue-Print Methods for Detection of Citrus tristeza virus: A Model for Other Plant Pathogens. Phytopathology 2012, 102, 114–121. [Google Scholar] [CrossRef]
- Simeone, M.; Gómez, C.; Bertalmío, A.; Ruiz, E.; Hauteville, C.; Suarez, L.G.; Tito, B.; García, M.L. Detection of citrus psorosis virus by RT-qPCR validated by diagnostic parameters. Plant Pathology 2021, 70, 980–986. [Google Scholar] [CrossRef]
- Massart, S.; Lebas, B.; Chabirand, A.; Chappé, A.-M.; Dreo, T.; Faggioli, F.; et al. Guidelines for improving statistical analyses of validation datasets for plant pest diagnostic tests. EPPO Bulletin 2022, 52, 419–433. [Google Scholar] [CrossRef]
- Kokane, S.B.; Misra, P.; Kokane, A.D.; Gubyad, M.G.; Warghane, A.J.; Surwase, D.; Reddy, M.K.; Ghosh, D.K. Development of a real-time RT-PCR method for the detection of Citrus tristeza virus (CTV) and its implication in studying virus distribution in planta. 3 Biotech 2021, 11, 431. [Google Scholar] [CrossRef]
- Massart, S.; Brostaux, Y.; Barbarossa, L.; Batlle, A.; Cesar, V.; Dutrecq, O.; Fonseca, F.; Guillem, R.; Komorowska, B.; Olmos, A.; Steyer, S.; Wetzel, T.; Kummert, J.; Jijakli, M.H. Interlaboratory evaluation of two Reverse-transcriptase Polymeric Chain Reaction-based methods for detection of four fruit tree viruses. Annals of Applied Biology 2009, 154, 133–141. [Google Scholar] [CrossRef]
- Garnsey, S.M.; Zies, D.L.; Irey, M.; Sieburth, P.J.; Semancik, J.S.; Levy, L.; Hilf, M.E. (2002). Practical field detection of citrus viroids in Florida by RT-PCR. In International Organization of Citrus Virologists Conference Proceedings (1957-2010) (Vol. 15, No. 15).
- Márquez-Molins, J.; Gómez, G.; Pallás, V. Hop stunt viroid: A polyphagous pathogenic RNA that has shed light on viroid–host interactions. Molecular Plant Pathology 2020, 00, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vidalakis, G.; Garnsey, S.M.; Bash, J.A.; Greer, G.D.; Gumpf, D.J. Efficacy of bioindexing for graft-transmissible citrus pathogens in mixed infections. Plant Disease 2004, 88, 1328–1334. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Grau, S.; Cruz, Q., J. P. Estudios de exactitud diagnóstica: Herramientas para su Interpretación. Revista chilena de radiología 2015, 21, 158–164. [Google Scholar] [CrossRef]
- Bertolini, E.; Moreno, A.; Capote, N.; Olmos, A.; de Luis, A.; Vidal, E. . & Cambra, M. Quantitative detection of Citrus tristeza virus in plant tissues and single aphids by real-time RT-PCR. European Journal of Plant Pathology 2008, 120, 177–188. [Google Scholar]
- Papayiannis, L.C. Diagnostic real-time RT-PCR for the simultaneous detection of Citrus exocortis viroid and Hop stunt viroid. Journal of Virological Methods 2014, 196, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Permar, T.A.; Garnsey, S.M. (1991). Comparison of biological indexing and immunological assays for identifying severe Florida isolates of citrus tristeza virus. In International Organization of Citrus Virologists Conference Proceedings (1957-2010) (Vol. 11, No. 11).
- Umaña Castro, R.; Pritsch, C.; Molina Bravo, R.; Pagliano, G. Diagnostic parameters of Northern blot hybridization technique for detection of citrus viroids in field-grown plants. Asian Journal of Plant Pathology 2017, 11, 71–80. [Google Scholar] [CrossRef]
- Saponari, M.; Zicca, S.; Loconsole, G.; Navarro, B.; Di Serio, F. Detection of Citrus tristeza virus and Coinfecting Viroids. Citrus Tristeza Virus: Methods and Protocols, 67-78.
- Hilf, M.; Mavrodieva, V.; and Garnsey, S. Genetic marker analysis of a global collection of a Citrus tristeza virus: Characterization and distribution of CTV genotypes and association with symptoms. Phytopathology 2005, 95, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Hadidi, A.; Garnsey, S.M. Enzymatic cDNA amplification of citrus exocortis and cachexia viroids from infected citrus hosts. Phytopathology 1992, 82, 279–285. [Google Scholar] [CrossRef]
- Altman, D.G.; Bland, J.M. Statistics Notes: Diagnostic tests 1: sensitivity and specificity. British Medical Journal 1994, 308, 1552. [Google Scholar] [CrossRef]
- Cohen, J. A coefficient of agreement for nominal scales. Educational and Psychological Measurement 1960, 20, 37–46. [Google Scholar] [CrossRef]
- Landis, R.; Koch, G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Roistacher, C.N. Psorosis complex: Psorosis-A, psorosis-B and ringspot. In Graft-Transmissible Diseases of Citrus (Handbook for Detection and Diagnosis); FAO: Rome, Italy, 1991; pp. 115–126. [Google Scholar]
- Bester, R.; Steyn, C.; Breytenbach, J.H.J.; de Bruyn, R.; Cook, G.; Maree, H.J. Reproducibility and Sensitivity of High-Throughput Sequencing (HTS)-Based Detection of Citrus Tristeza Virus and Three Citrus Viroids. Plants 2022, 11, 1939. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Average cDNA concentration per amplified band. CTV: cDNA concentration of simple CTV infection. CTV MI: CTV cDNA concentration in mixed infection. HSVd: HSVd cDNA concentration in simple infection. HSVd MI: concentration of HSVd cDNA in mixed infection.
Figure 1.
Average cDNA concentration per amplified band. CTV: cDNA concentration of simple CTV infection. CTV MI: CTV cDNA concentration in mixed infection. HSVd: HSVd cDNA concentration in simple infection. HSVd MI: concentration of HSVd cDNA in mixed infection.
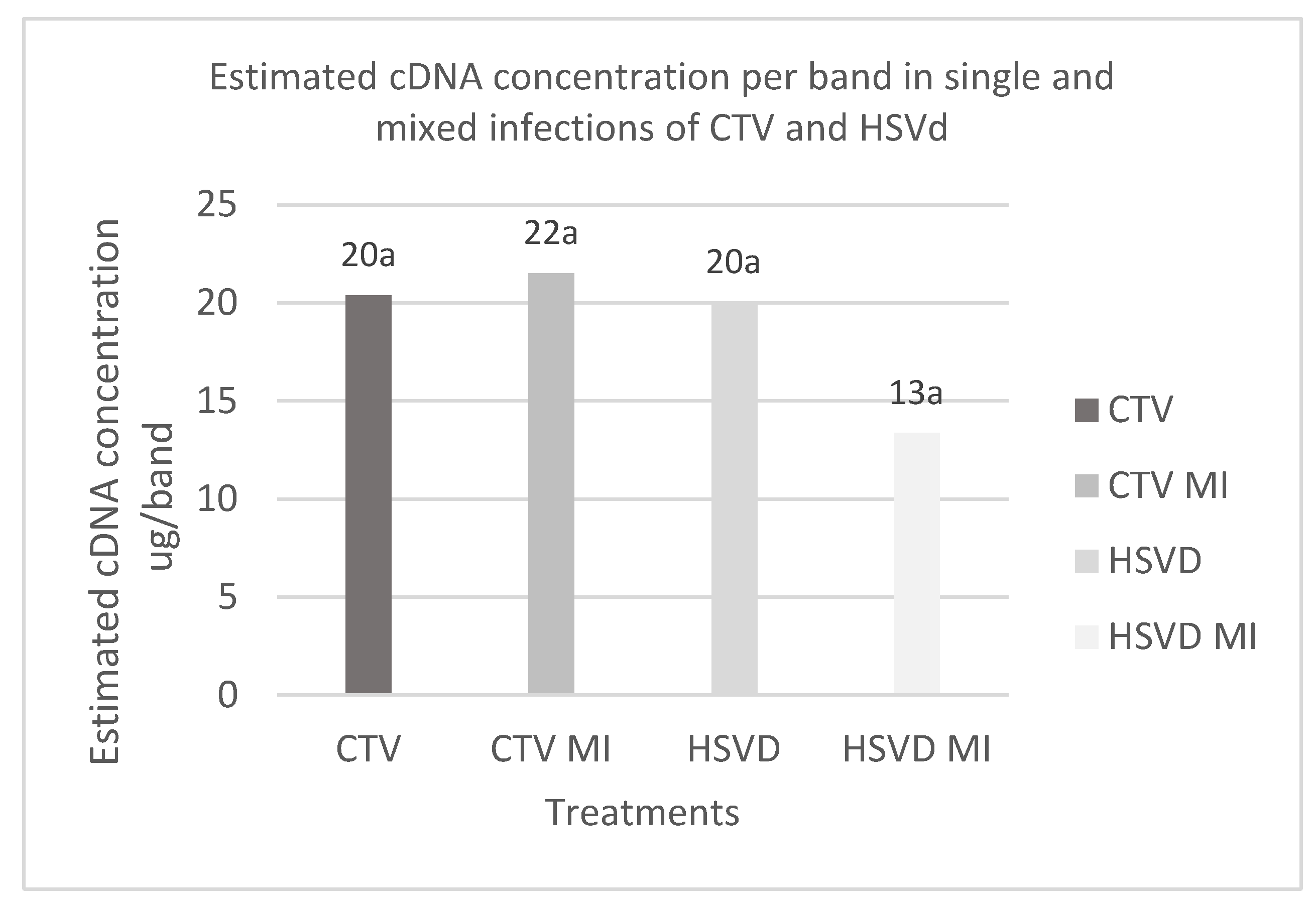
Figure 2.
Comparison of CTV detection percentages according to diagnostic method and infection time. T1: Direct method. T2: Bioamplification. Infections of 6, 3, 1 and 0 months.
Figure 2.
Comparison of CTV detection percentages according to diagnostic method and infection time. T1: Direct method. T2: Bioamplification. Infections of 6, 3, 1 and 0 months.
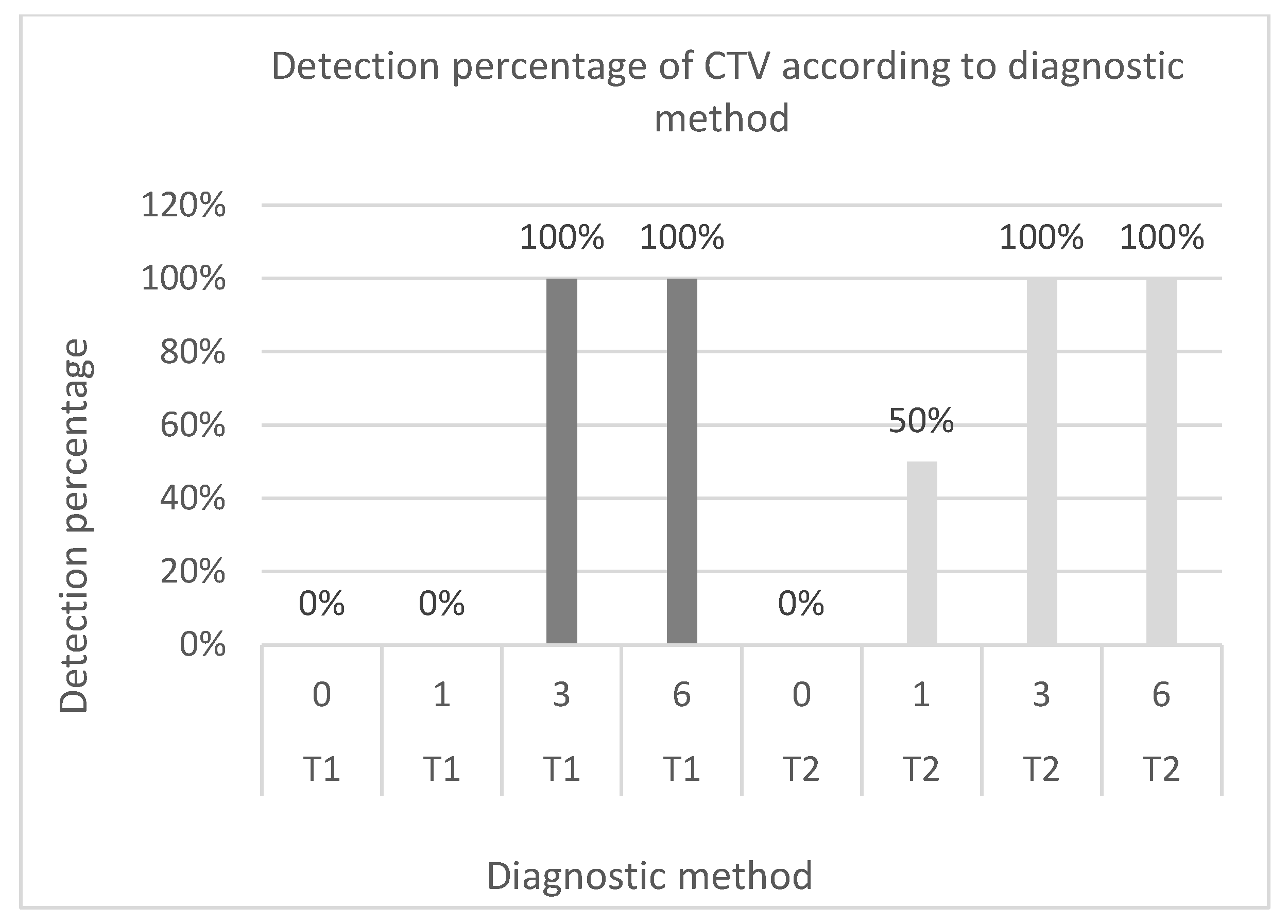
Figure 3.
Comparison of HSVd detection percentages according to diagnostic method and infection time. T1: Direct method. T2: Bioamplification. Infections of 6, 3, 1 and 0 months.
Figure 3.
Comparison of HSVd detection percentages according to diagnostic method and infection time. T1: Direct method. T2: Bioamplification. Infections of 6, 3, 1 and 0 months.
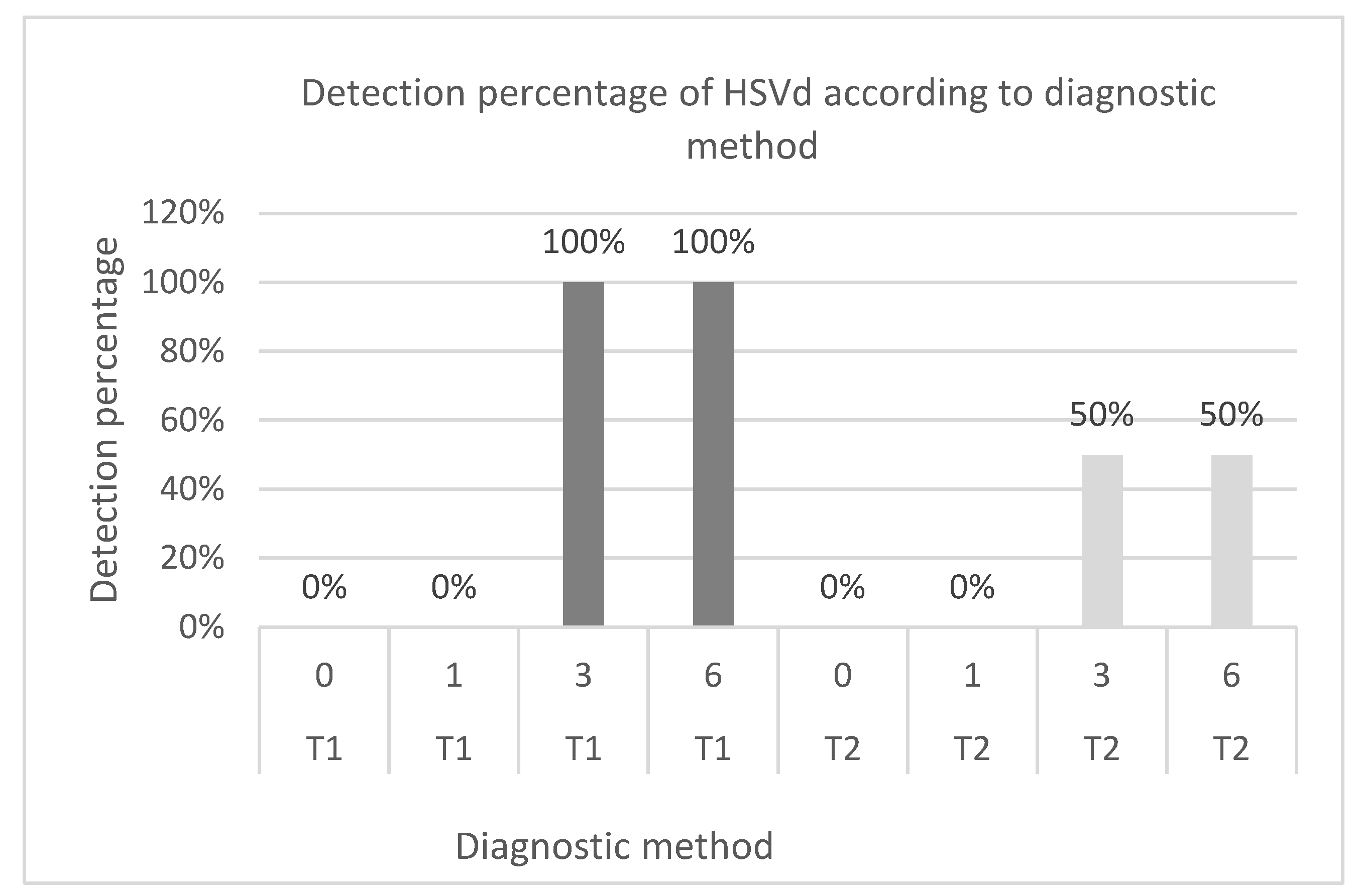

Table 1.
Comparison of the results of direct analysis and bioamplification for CTV detection Contingency table with coincidences and differences between the results of both methods. Validation parameters for the bioamplification method for CTV.
Table 1.
Comparison of the results of direct analysis and bioamplification for CTV detection Contingency table with coincidences and differences between the results of both methods. Validation parameters for the bioamplification method for CTV.
| Direct method | Total | |||
|---|---|---|---|---|
| Bioamplification | + | - | ||
| + | 5 | 0 | ||
| - | 0 | 3 | ||
| Total | 8 | |||
| Validation parameters | ||||
| Sensitivity | 1.0 | |||
| Specificity | 1.0 | |||
| Cohen’s Kappa index (p value) | 0.75(0.028) | |||
Table 2.
Comparison of the results of direct analysis and bioamplification for detecting HSVd. Contingency table with coincidences and differences between the results of both methods. Validation parameters for the bioamplification method for HSVd.
Table 2.
Comparison of the results of direct analysis and bioamplification for detecting HSVd. Contingency table with coincidences and differences between the results of both methods. Validation parameters for the bioamplification method for HSVd.
| Direct method | Total | |||
|---|---|---|---|---|
| Bioamplification | + | - | ||
| + | 2 | 0 | ||
| - | 2 | 4 | ||
| Total | 8 | |||
| Validation parameters | ||||
| Sensitivity | 0.5 | |||
| Specificity | 1.0 | |||
| Cohen’s Kappa index(p value) | 0.5(0.102) | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



