
Submitted:
23 January 2024
Posted:
24 January 2024
You are already at the latest version
Abstract
Long-acting (LA) drug-delivery system (DDS) successes are linked to their abilities to harness biocompatible polymers and materials for sustained and predictable release of therapeutic agents over an extended period of time. For human immune deficiency virus type one (HIV-1) infections, LA DDSs hold great promise in closing the gaps of daily oral therapy for treatment and prevention. Examples are Cabenuva, Apretude and Sunlenca, which have been shown to be safe and effective. Alternative promising drug delivery technologies such as implants, prodrugs, vaginal rings, and microarray patches are being explored to further meet patients’ needs. We posit that improved physicochemical properties of the formulation payloads and material chemistry design with optimal drug release kinetics holds the potential to optimize release profiles. Furthermore, for the treatment and prevention of HIV-1 infection, the landscape of LA medicines now in clinical or preclinical development offers hope that the strategic design of polymers will further improve upon controlled delivery of drugs to simplify dosing schedules.
Keywords:
long-acting formulations
; polymer
; antiretroviral therapy
; HIV
; chronic infectious diseases
; implants
; vaginal rings
; prodrug nanoformulations
; microarray patches
1. Introduction
Long-acting (LA) drug delivery systems (DDSs) harness polymer properties to achieve spatiotemporal control over release and drug biodistribution. This allows the LA dosage regimens to extend from days to months. Such DDSs significantly reduce the burden of chronic disease. Treatment success parallel medication adherence. The development of LA medicines are traced back to the 1930s with the discovery of hydrophobic drug release from implants [8]. However, the discovery of low molecular weight (MW) dye diffusion through silicone tubing laid the groundwork for rate-controlling LA DDS polymers [9]. Biopharmaceutical research activities directed their efforts to convert biocompatible silicone tubing into versatile drug-delivery materials. These efforts included improved uses of atropine, histamines, anesthetics, steroids, antimalarial, and antischistosomal agents [9]. Since the 1960s, the work of Drs. Folkman, Langer, Higuchi, Roseman, Peppas, Heller, Ringsdorf, and Speiser pioneered the LA DDSs advancements seen today. Widespread research resulted in the development of LA DDS which extended the dosage intervals for a broad range of drug regimens. This resulted in improved treatment outcomes for contraception and for treatments of psychosis, diabetes, osteoporosis, and ocular diseases.
A key example of a treatable chronic disease that would benefit from LA DDSs is human immune deficiency virus type one (HIV-1) infection. Here, prior studies demonstrated that non-adherence to therapeutic regimens was a significant limiting factor in achieving successful treatment outcomes. One notable is the development of viral resistance. Another is simply remembering to take the medicines on daily prescribed requirements. Indeed, patients have repeatedly expressed treatment preferences for infrequent dosing regimens as without a viable cure or preventive vaccine, daily oral antiretroviral therapy (ART) is a principal means for HIV-1 treatment and prevention. Over the past two decades, considerable efforts have been focused on the development of LA ARTs that includes a specific focus on controlled-release LA formulations. Such formulations include solid implants, vaginal rings (VRs), and surfactant-stabilized aqueous nanocrystal suspensions. In these depot-forming DDSs, the drug is either encapsulated in biodegradable polymers for implantation in subcutaneous (SC) spaces, in reproductive organs, or formulated as drug nanocrystals for either intramuscular (IM) or SC injections. Extensive tests have culminated in the approval of surfactant-stabilized aqueous nanocrystal LA cabotegravir and rilpivirine (CAB and RPV LA) suspensions by the US Food and Drug Administration (FDA). CAB and RPV LA offers the convenience of monthly or bimonthly dosing for HIV-1 treatment [11,12,13].
Multiple randomized clinical trials have demonstrated comparable efficacy between CAB and RPV LA and a standard daily oral regimen [12,14,15]. Based on the data that 91% of prior clinical trial participants preferred LA ART over daily oral medicines, CAB LA proved more effective in preventing HIV-1 infection when compared to daily oral Truvada [16]. This propelled its US FDA approval in December 2021 [13]. The remarkable efficacy of CAB LA was linked to improved treatment satisfaction and therapeutic adherence. However, the limitations associated with CAB LA include the need for frequent clinical visits, injection site reactions, variable pharmacokinetics (PK) profiles, prolonged terminal phase tailings, high costs, and co-morbid conditions.
Alternative approaches that include in situ forming implants (ISFIs), solid implants (SIs), and prodrug nanocrystals are being developed to further improve the PK profiles and other listed limitations associated with CAB LA. Notably, surfactant-coated stearoylated CAB prodrug nanocrystals achieved sustained therapeutic drug levels for up to a year in preclinical models [17]. Other studies demonstrated that poly(lactic acid-co-glycolic acid) (PLGA) based ISFI could potentially extend CAB dosing intervals to every 6-months [18]. Other studies have also shown that solid implants containing highly potent ARVs, such as tenofovir alafenamide (TAF) and islatravir (ISL) can simplify ART dosages [2,19-21]. However, in November 2021, Merck announced that participants who received once-weekly ISL plus MK-8507 experienced a decline in CD4 T cells. Further review found that CD4 counts also fell in people taking once-daily ISL plus doravirine for HIV-1 treatment, while those taking ISL alone for pre-exposure prophylaxis (PrEP) experienced a decline in total lymphocyte counts. As a result, studies of ISL for PrEP have been discontinued. These findings underscore the importance of establishing safety profiles of new chemical entities prior to combining them with other therapies. Polymeric TAF implant technologies have demonstrated a zero-order release kinetics of the pharmacologically active tenofovir diphosphate (TFV-DP) metabolite in preclinical models of PK studies [3]. These include non-degradable materials that provide sustained drug delivery. The use of self-administered microbicides can significantly reduce the risk of HIV-1 infection. Vaginal rings (VRs) that consist of non-biodegradable elastomeric polymers are being developed as self-administrable PrEP treatment targeting at-risk populations [22]. Several VRs of TAF and dapivirine (DPV), a non-nucleoside reverse transcriptase inhibitor (NNRTI), have demonstrated that such formulations could extend ART dosing intervals [22,23,24]. In two separate phase 3 clinical trials, known as the RING and the ASPIRE studies, DPV VRs demonstrated their effectiveness in reducing HIV-1 transmission. Compared to a placebo ring, the DPV ring reduced transmission rates by 31% in the RING study and 27% in the ASPIRE study [25,26]. These results suggest that DPV ring could supplement existing HIV prevention practices. Microneedles (MNs), also referred to as microarray patches (MAPs), are another class of LA DDSs that use drug-diffusible, rate-controlling, non-biodegradable, or slowly biodegradable polymer membranes to extend the apparent half-lives of drugs [10]. Due to the non-invasive and self-administrable nature, the goal of MNs is to expand LA ART options and to appeal to a wide variety of users that include pediatric populations. Examples include CAB and RPV-loaded MNs that are currently being explored as alternative treatment options to daily oral therapy as well as injectables [4,27]. However, further work is required to demonstrate improved or comparable PK and efficacy profiles to the existing CAB and RPV LA injectable formulations.
This review summarizes the physiochemical and biological parameters of the commonly used polymers used for LA DDSs in treatment and prevention regimens for HIV-1 infection. LA ART formulations either in pre-clinical or clinical development are discussed. For each of these delivery systems, polymer compositions and drug release kinetics are discussed. Additionally, an expert opinion section highlights LA ART DDS design considerations which could potentially encourage widespread acceptance and utilization among the key target populations.
2. LA DDSs
The emergence of polymer-based DDSs can be traced back to the 1930s when the implantation of pellets with hydrophobic substances were identified for their ability to facilitate sustained drug release [28]. Examples include pellets with estradiol for prostate cancer treatment and testosterone pellets for the treatment of hypogonadism [29]. Later, clinical use of depot-forming formulations loaded with hydrophobic drugs in either water-based or oily mediums, such as procaine penicillin G in water and fluphenazine decanoate in sesame oil, became popular [30,31]. Since then, more drug delivery approaches and models have been developed to improve our understanding of the materials and drug release mechanisms. In the 1960s, T. Higuchi presented his renowned "Higuchi model" to describe the drug release kinetics from various sustained release matrix systems [32]. The model suggested that the release of the solid drugs dispersed in a matrix varies with the square root of time. At the same time, Folkman discovered that silicone rubber could act as a drug reservoir, allowing for constant drug release after implantation. This breakthrough led to the conceptualization of the rate-controlling membranes or reservoir-based implants [33]. Notable examples of this concept in action are Ocuserts®, which are designed to deliver ocular drugs at predictable and controlled rates [34].
The challenges associated with non-biodegradable implants, such as the necessity of surgical removal after product life ends and adverse implant site reactions, spurred the development of biodegradable implants. The development and clinical utilization of biodegradable polymers like polylactic acid (PLA), polyglycolide (PGA), and PLGA dates back to the 1970s [35]. At the early stage of development, clinical applications of these biodegradable polymers were restricted to surgical sutures [35]. Later on, PLA and PLGA microparticles and pellet depot systems for delivering contraceptive drugs and luteinizing hormone-releasing hormone (LHRH) analogs were developed [36]. Other notable examples of commercial products where the biodegradable copolymers were utilized include Lupron Depot and Zoladex® implants for treatment of prostate cancer, breast cancer, and endometriosis-like disease [36]. Furthermore, a polymer based VR was patented by Upjohn Company for sustained drug release [37]. These developments marked significant milestones in the evolution of polymer-based LA DDSs. Additionally, extensive research was undertaken to develop silicone-based VRs for contraception. In-depth research, primarily sponsored by the World Health Organization (WHO), paved the way for the clinical approval of multiple contraceptive implants in the early 1990s.
The development of LA ARTs began in the early 2000s, with an initial focus on VRs and implants. Later, attention shifted towards nanocrystal formulations[38]. Clinical trials for the nanocrystal aqueous suspensions started around 2008 (Figure 1), including phase II trials for HIV-1 prevention (PrEP) with LA RPV [39]. However, the development of this formulation as a single agent for PrEP faced challenges linked to the high prevalence of NNRTI mutations [39,40]. After a decade of research and several clinical trials, the first LA complete therapy for HIV-1 treatment was approved in 2020 [41].
3. Drug Release Kinetics from LA DDSs
In polymer-based DDSs, drug release refers to a transfer process in which drug molecules are released from the inner core or matrix to the outer surface of the delivery system and eventually into the surrounding environment or tissue [42]. The rate of drug release from polymer-based DDSs can be modulated by choosing an appropriate polymer with a suitable DDS design. The terminology ‘long-acting’ is the ability to extend the duration of action of a therapeutic agent for a longer period of time. Most often, the terms long-acting, controlled-release, sustained-release, and extended-release are used synonymously [43]. According to the United States Pharmacopeia (USP), the term ‘extended-release’ or ‘long-acting’ or ‘sustained-release’ is defined as 'a deliberate modification to protract the release rate of an active pharmaceutical ingredient (API) in comparison to an immediate release dosage form' [44]. In this review, the terminology ‘long-acting’ ‘sustained-’ or ’controlled- release’ are used to designate formulations that can extend the apparent half-lives of drugs. The release of drugs from a delivery system can follow one or more mechanisms, which can be correlated with a number of existing release kinetics models. The commonly used models are zero-order kinetic, first-order-order kinetic, Higuchi, Korsmeyer-Peppas, Peppas-Sahlin, and Hixson-Crowell. The zero-order kinetic model considers that there is no drug concentration influence on drug release rates. The zero-order kinetic models are reflected by the mathematical equation -1, where C0 and Ct represent the amount of drug at the start and the amount released at time ‘t’.
For first-order kinetics, the rate of drug release is proportional to the concentration of the remaining drug in the delivery system and represented by Eq-2, where dc/dt is the rate of drug release, k1 is the first-order rate constant, and ‘C’ is the concentration of the remaining drug at time, t.
The Korsmeyer-Peppas model (Eq-3) reflects dissolution-mediated drug release. ‘Mt’ and ‘M∞’ is the amount of eluted drug, ‘t’ is the recorded time ‘∞’. The ratio of ‘Mt/M∞’ denoted the fraction of drug release at the time, ‘t’. Kk is the Korsmeyer rate constant.
The Peppas–Sahlin equation (Eq-4) links too the diffusion and relaxation-mediated drug release. The diffusion exponent is represented by ‘m’, and kinetic constants are represented by K1 and K2.
The Higuchi model (Eq-5) is based on diffusion-mediated drug release. Here ‘Q’ indicates the amount of drug released per unit area at time t and KH is the Higuchi constant.
The Hixson-Crowell model (Eq-6) applies to uniform-size drug particles, where the rate of drug release is controlled by drug dissolution. This is governed by the surface area or diameter of the drug-encased particles. The rate of drug dissolution is based on the cube root of the drug mass. The M0 and Mt are the mass of the drug at the initial and at the recorded time ‘t’. KH-C symbolizes the Hixson-Crowell release constant.
An ideal LA DD should exhibit a zero-order drug release kinetics where a constant amount of drug is released per unit time. However, maintaining zero-order kinetics is very challenging, and it majorly depends on the physicochemical properties of the drug and excipients [45,46]. Diffusion, osmotic pumping, swelling, degradation or erosion-induced release are the commonly reported mechanisms for polymer-based DDSs.
Non-degradable polymer-based DDSs can be either reservoir or matrix form. In the reservoir form of DDSs, the release rate is governed by the thickness of the polymer membrane and permeability of the drug through the polymer membrane. Whereas, in matrix form of DDSs, Fickian diffusion remains the underlying release mechanism [47]. Diffusion refers to the random movement of the drug molecules from the higher to the lower concentration region in the matrix. The rate of diffusion in LA DDSs can be described by Fick’s law [47]. According to this law, the drug release rate depends on the concentration gradient () and diffusibility (D) of the drug through the polymer matrix (Equation-7). Particularly for slab-like matrix, Equation-7 can be simply transformed to Equation-8.
……… ….Eq-7;
D is the diffusion coefficient or diffusivity, C is the concentration.
… …………Eq-8;
Mt is the sum of drug released at time t, M0 is the total of the drug-loaded mass, D is the diffusion coefficient, and h is the thickness of the slab-like matrix.
According to Eq-8, the release rate is directly proportional to the drug's diffusibility (D) through the slab and inversely proportional to the thickness (h) of the polymer [47]. Although, Eq-8 can be transformed to other various forms based on the geometry of the DDSs, the parameters, such as the drug’s diffusibility and slab thickness, play their role in a similar way. For a particular DDS, the release kinetics can be zero-order when ‘h’ and ‘D’ remain constant over time (Eq-8) [47]. The diffusibility is influenced by the size of the drug molecule relative to the pore size of the matrix. Furthermore, matrix pore size and density have been governed by the properties of the polymer used to fabricate the matrix, such as nature of the monomers and the molar composition used to synthesize the polymer [48]. Matrix systems lack a rate-controlling membrane, so the diffusion rate is affected by non-constant drug concentration gradient and diffusion distance. Additionally, the diffusion distance is reliant on the polymer's swelling [48].
In degradable polymer-based DDSs, drug release is majorly controlled by the rate of polymer degradation or erosion and osmotic pumping of the drugs. The chemical degradation of the polymer is influenced by its hydrophilicity. Hydrophilic polymers can absorb water, resulting in an increase in their pore size. This allows them to initiate drug release [49]. Over time, the polymer undergoes degradation, which increases the number and size of pores, eventually leading to continuous release of drug. Unlike degradation, erosion is an alternative method of the drug release process where polymeric chain segments are dissolved by keeping their chemical structure intact [48]. Erosion processes can happen either on the surface or bulk, or a combination of both places on the DDS. Surface erosion gradually reduces the size of DDS from the outward to inwards [50,51]. In bulk erosion, water permeates the entire bulk of the polymer matrix, leading to a uniform degradation with no significant change in their initial size [51]. In addition, bulk erosion may produce faster and unpredictable drug release kinetics, making it less favorable for LA DDSs [52]. Osmotic pumping is another method of drug release, where osmotic pressure drives the influx of water into the non-swelling segment of DDS, resulting in drug release [53].
Interestingly, the prevalent drug release pattern from LA DDSs is the triphasic rather than monophasic drug release pattern [45]. The first phase of drug release from an LA DDS demonstrates an initial burst release due to the rapid release of drug molecules located near the surface of the DDS or the surrounding tissue [54]. The extent of this burst release is influenced by factors such as the design and morphology of the DDS, polymer properties, fabrication process, storage conditions, and homogeneity of the drug-matrix concentration [54]. While an initial burst release may be desired for rapid drug action, it can significantly reduce the drug depot concentration, consequently impacting the DDS longevity. Strategies such as hydrophobic polymer coating on the outer surface of DDS can curtail this challenge [54]. The second phase of drug release involves a slow-release period, where the drug diffuses through the polymer matrix. This phase concurrently occurs with polymer degradation in biodegradable systems [45]. The third phase may exhibit faster release as bulk erosion of the matrix starts. Drug release can also follow a biphasic trend, transitioning from the initial burst release to the zero-order kinetics [55]. Overall, the kinetics of drug release are complex and influenced by multiple factors. Understanding and optimizing the release profile of DDSs are crucial for achieving desired therapeutic outcomes and ensuring the efficacy of DDSs.
Molecular weight (MW) and the molar ratio of the monomeric units of a polymer play a crucial role in determining its physicochemical properties, including solubility, crystallinity, glass transition temperature (Tg), and mechanical strength. Polymers with less elasticity result in non-deformable matrices and smaller pore formation, leading to a slower drug release [56]. Moreover, the degradation of the copolymer is influenced by the nature and molar ratio of monomeric units [57]. For example, a higher molar ratio of hydrophilic monomer, glycolic acid (GA) in PLGA composition leads to its faster biodegradation and subsequent drug release [58].
Polymer crystallinity refers to the proportion of crystalline and amorphorus region within the polymer structure [59]. The ratio of different monomeric units in a copolymer affects the polymer's crystallinity and Tg. Most polymers are semi-crystalline in nature, in which the amorphous domains separate the crystalline domains. The water permeability of the polymer is primarily controlled by the percentage of crystallinity. An increased level of polymer crystallinity decreases the overall water permeability, resulting in a slower rate of polymer biodegradation and a decrease in the drug release rate [60]. In high MW polymers, the influence of crystallinity on drug release depends on the presence of monomer crystallinity [61]. A notable example is the biodegradability of PLGA, where the ratio of its monomers, lactic acid and GA, determines the degree of crystallinity, subsequent biodegradation and drug release profile. PLLA, composed of L-lactic acid, is highly crystalline, while PDLA, composed of D-lactic acid, is completely amorphous. Like PLLA, PGA is also a highly crystalline polymer. The degree of crystallinity and biodegradability of PLGA copolymer depends on the molar composition of lactic acid and GA comonomers in the copolymer [62]. Furthermore, the ratio of the amorphous and crystalline regions also influences the polymer's Tg, which in turn affects its mechanical strength and water permeability. For example, a polymer with Tg equal or close to physiological temperature can transform into a rubbery state, facilitating water diffusion and promoting drug release [48].
4. Common Polymers in LA DDSs
Advancements in synthetic polymer chemistry have led to the development of numerous biocompatible and biodegradable polymers. Synthetic polymers are derivatized from chemically synthesized monomers and are typically biologically inert, causing minimal adverse immune reactions. They also exhibit more predictable physical and chemical properties compared to natural polymers [63]. The ability to adjust the physicochemical properties of synthetic polymers allows for tailored drug release kinetics, making them more favorable than natural polymers [64]. Polymers used in the fabrication of LA DDSs can be categorized as either biodegradable or non-biodegradable [65]. The injection site reactions and requirement for removal of DDSs after the lifespan spurred the development of biodegradable polymers. The use of biocompatible and biodegradable synthetic polymers in LA DDSs eliminates the need for removal after its lifespan. In the following discussion, we will explore the polymers commonly employed in LA DDSs, along with their physiochemical characteristics and mechanisms of drug release.
4.1. Polyethylene Glycol (PEG)
PEG, also known as macrogol, is synthesized by ring-opening polymerization of ethylene oxide. Its outstanding biocompatibility, hydrophilicity, non-immunogenicity, and antifouling properties towards serum protein contribute to its extensive use in DDSs [66]. Chemical modification with PEG, commonly referred to as PEGylation, enables the conjugation of biomolecules to the surface of the micro- or nanoparticles. Surface PEGylation of DDSs substantially reduced its opsonization by decreasing the chances of protein corona formation, which eventually increases the plasma apparent half-life and reduces the immunogenicity in vivo [67,68]. Based on structure, PEG can be classified as linear, branched, and star-shaped [69]. The end hydroxyl group of the PEG can be transformed into various active functional groups, such as amino, maleic amide, and carboxyl moieties, suitable for functionalization with various drugs and other pharmacologically active biomolecules [70,71]. However, the minimal availability of active functional groups in linear PEG offers limited PEG-drug covalent conjugates, resulting in low drug-loading content. Therefore, employing a four- or eight-arm PEG structure can increase the number of active end functional groups, resulting in improved drug-loading in DDSs [72,73]. PEG cannot undergo biodegradation; hence, a low MW PEG has been recommended for use in DDSs so that it can easily be eliminated from the biological system. In addition, PEG can be copolymerized with biodegradable polyesters, such as PLA or PLGA, to improve the overall biodegradability of the copolymer [74].
4.2. Poloxamers
Poloxamers are proprietary polymers of BASF, also known as Pluronics®. These are triblock copolymers and have been synthesized by sequential polymerization of ethylene oxide (EO) and propylene oxide (PO). The arrangement of blocks in the copolymer can either be PEO-b-PPO-b-PEO or PPO-b-PEO-b-PPO [75]. Poloxamers are amphiphilic in nature because of the hydrophilic and hydrophobic properties of PEO and PPO, respectively. Amphiphilic poloxamers self-assemble to a spherical shape micelle at the concentration above their critical micelle concentration (CMC) at a specified temperature. These micelles adopt a core-shell morphology with PPO block in the core and PEO block on the shell. Some poloxamers can transform their morphology from micellar structures to solid micellar cubes, producing thermo-responsive clear and rigid hydrogel at elevated temperatures and concentrations [76]. For example, Poloxamer 407 can form hydrogels in 20% w/w solution [77]. The rate of drug release from the poloxamer-based DDSs primarily depends on the drug’s hydrophilicity and relative block lengths of PPO and PEO [78,79]. It has been seen that hydrophilic drugs typically followed diffusion-induced release, whereas hydrophobic drugs followed erosion-induced release [80]. In addition, factors such as poloxamer concentration, lipophilicity, physicochemical properties of the used solute, and the amount of the aqueous channel network in poloxamer gel significantly influence drug release. In an abundant aqueous environment, the structure of poloxamer micelles breaks down quickly, leading to gel matrix degradation [81].
4.3. Ethylene-Vinyl Acetate (EVA)
The random copolymer of ethylene (E) and vinyl acetate (VA) is often referred to as ethylene-vinyl acetate (EVA). The copolymer has been synthesized by radical copolymerization of ethylene and vinyl acetate with various feed molar ratios of monomers depending on its applicability [82]. EVA has been extensively used in clinical settings, especially for ocular implants, contraceptives, and hormone replacement therapy. NuvaRing®, Progestasert®, and Implanon® are the three most widely used EVA-based contraceptive products, where EVA acts as a release rate-controlling membrane [83]. EVA has also been used in the delivery of buprenorphine/hydromorphone for treatment of opioid withdrawal.
As like other polymers, EVA’s release kinetics is influenced by its physicochemical properties, such as hydrophobicity, crystallinity, etc. Furthermore, the EVA’s physicochemical properties have been governed by wt% of VA. The commercially available EVA contains 1 to 40 wt% VA, randomly distributed throughout the EVA backbone [84]. The acetate side chain of VA creates steric hindrance that restricts the alignment of the polymeric chain. As a result, an increasing percentage of VA decreases the crystallinity [85]. Eventually, EVA becomes nearly amorphous when the VA content increases to 40 wt% [86]. EVA exhibits a complex combination of amorphous and crystalline phase regions, characterized by at least two Tg (between -35 to -25°C), which is barely influenced by the overall VA content [87,88]. In addition, the MW distribution, branching, and morphology also have a prominent impact on the physicochemical properties of EVA. In hydrogel reservoir-based delivery systems, EVA is commonly employed as a membrane for controlling the release rate. Additionally, EVA finds extensive application as an excipient in the development of nano- and microparticles as it is compatible with a wide range of excipients that can be used in complex multilayer polymer blends to further tune the release rate [83,89].
Figure 2.
Chemical structures of the polymers used in LA delivery systems.
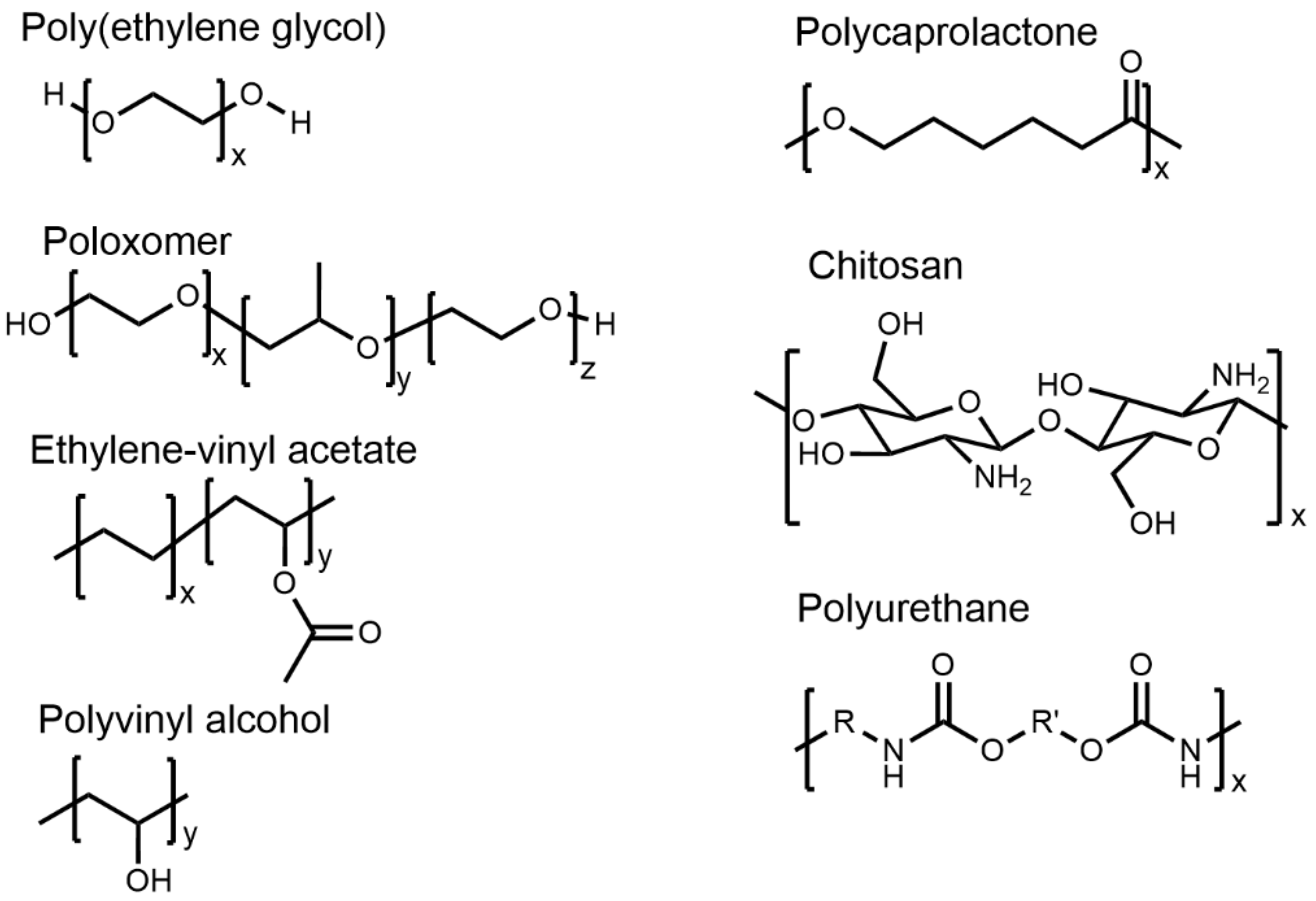
4.4. Polyvinyl Alcohol (PVOH)
Polyvinyl alcohol (PVOH) is a highly biocompatible and biodegradable semicrystalline polymer used in LA DDSs [90]. PVOH is synthesized by sodium hydroxide-mediated hydrolysis of polyvinyl acetate and can be classified as fully or partially hydrolyzed PVOH [91,92]. PVOH is highly water soluble because of its pendent hydroxyl groups. The solubility, mechanical, and adhesion properties of PVOH depend on its MW and percentage of hydrolysis. Partially hydrolyzed PVOH (87–89%) shows improved solubility, flexibility, and adhesion to hydrophobic surfaces, while highly hydrolyzed PVOH (91–99%) exhibits enhanced water stability, increased tensile strength and adhesion to hydrophilic surfaces [93]. The hydroxyl groups in PVOH can form inter and intramolecular hydrogen bonding, which influences its rheological, viscoelastic, and solution properties. PVOH's characteristics, including high surface stabilization and chelation, make it an excellent candidate for drug delivery [94,95]. PVOH has been used in many clinically approved products over the last few decades. For example, Iluvien® implant has been fabricated by using PVOH and silicone, and used for macular edema [96]. In addition, PVOH has been found to be extensively applied in preclinical research for developing hydrogels, microspheres, and nanoparticles for various therapeutic purposes [90]. PVOH hydrogels can be physically or chemically cross-linked, holding drug molecules into their 3D crosslinked structure [97,98]. These hydrogels have gained popularity as the drug release kinetics from these hydrogels can be predicted by observing water uptake and swelling index [97,99].
PVOH has demonstrated remarkable biocompatibility in various studies conducted since the late 1980s, as evidenced by its successful use in biomedical applications [100,101]. Examples include its inclusion in ophthalmic solutions and tear replacement solutions without causing any discomfort to the eyes [102]. In recent years, PVOH hydrogels have found extensive use in cartilage replacement, tissue sealants for emphysema treatment, wound dressings with healing agents, and as a barrier against secondary infections [103,104,105]. The toxicity profile of PVOH was extensively evaluated after oral and intravenous injections of PVOH in animal models [106,107]. Oral administration of PVOH to rats demonstrated no reproductive, neurological, or systemic toxicity [108]. Acute oral toxicity of PVOH was found to be very low with poor gastrointestinal absorption and no mutagenic or clastogenic effects [106].The highest oral dose with no observed adverse effect level (NOAEL) was determined to be 5,000 mg/kg/day [106]. After oral administration, PVOH was primarily excreted in feces and minimally in urine [106]. PVOH is usually slowly eliminated from plasma, and the liver. It was identified as the main organ of PVOH deposition [109]. Furthermore, genotoxicity tests indicated no evidence of mutagenicity or carcinogenic activity [110].
4.5. Polyurethanes (PUs)
PUs are condensation polymers, synthesized by step-growth polymerization of isocyanates and polyols. Chain extenders are often added during the synthesis process of PUs to enhance their mechanical strength. The stiffer hard segments in PUs are made up of chain extenders and isocyanates, and flexible soft segments of PUs are made up of polyols. The interplay between these segments endows polyurethanes with a balance between rigidity and elasticity [111]. Both the aliphatic or aromatic diisocyanates are employed for the synthesis of PU. PUs derived from aliphatic diisocyanates tend to be more biocompatible and show enhanced resistance to biodegradation than those produced from aromatic diisocyanates [112,113]. Polyether and polycarbonate are commonly employed as polyols, with MW typically ranging between 1 to 5 kDa. Notable advancements were made in developing biodegradable polyester-based mechanically strong PUs, widely used in manufacturing VR-like implants [114]. For instance, a biodegradable intravaginal ring was developed using a poly(ester urethane) that released antiretroviral DPV for more than a month [115]. PUs derived from polyethers display enhanced hydrolytic stability and remain elastic at low temperatures. However, they are susceptible to both oxidation and thermal degradation. The addition of antioxidants has further increased their stability. For example, PEG, a widely used hydrophilic polyol, significantly influences the drug release rate from PU-based DDSs [116,117].
The drug release from the PU-based DDSs follows three primary mechanisms. These include solute diffusion, polymer swelling, and polymer degradation [118]. PU-based DDSs show an initial burst release which is mostly because of solute diffusion through PUs. In non-degradable PU-based DDSs, the drug release rate is determined by the thickness, swelling capacity, and permeability as well as the drug concentration gradient and polymer-drug compatibility [47]. In biodegradable PU-based DDSs, the release rate is determined by its own swelling capacity and rate of degradation or erosion. The PU can be hydrophilic or hydrophobic depending on the nature of the diol/polyol and chain extender employed during synthesis. Unlike hydrophobic PUs, hydrophilic PUs can easily swell and demonstrate a faster rate of drug release. For example, the rate of DPV release from swellable PU-based DDSs is significantly higher than its non-swellable counterpart [119]. Hydrophobic PUs tend to experience surface erosion, whereas hydrophilic PUs with less reactive ester linkages typically undergo bulk erosion [120,121]. The type of diol/polyol used in PU-based DDSs determine its swelling capacity which in turn control its rate of degradation/erosion. The degradation of PUs entirely depends on the hydrophilicity and biodegradability of diol/polyol together with the chain extender [122,123]. PUs with PEG as diol can easily get hydrated and accelerate its rate of degradation. Moreover, PUs can also undergo chemical degradation or enzymatic degradation [124].
4.6. Polyesters
Biodegradability of polyesters, such as PGA, PLA, and PLGA, make them the most extensively used polymers in LA DDSs. These polymers serve as vehicles for delivering various pharmaceutically active biomolecules like proteins, nucleic acid, and small drug molecules, etc. PLA is synthesized through the lactide ring-opening polymerization. The physiochemical properties and release profile of PLA can be tuned by tailoring the degree of racemization of lactic acid. PLLA, derived from L-lactide, exhibits a semicrystalline structure characterized by its hardness and transparency, with a Tg of 53 °C. In contrast, PDLA, derived from D-lactide, is amorphous in nature, displaying lower mechanical strength and a Tg of 55°C [125]. Due to the high thermal stability of PLA, it can be fabricated using various thermal processing methods, such as injection molding, extrusion, etc. PLA degrades through the hydrolysis of its ester bond and form lactic acid as an intermediate degradation product which further converted to carbon dioxide and water, a biocompatible and non-toxic end product [126]. Moreover, the intermediate lactic acid can accelerate the degradation by catalyzing the degradation process. Biodegradation of PLA is very slow due to its semicrystalline nature. Furthermore, side chain methyl group brings hydrophobicity in PLA which further lower its rate of degradation. Therefore, depending on the implant size and MW used, PLA can take as high as 2 years to degrade inside the body [125]. The slow biodegradation rate of PLA has been utilized in the development of Brimo DDS®, an intravitreal implant that incorporates PLA for the sustained release of brimonidine to treat the geographic atrophy [127].
PGA is another polyester. PGA is synthesized by the ring-opening polymerization of glycolide [50]. In comparison with PLA, PGA is highly crystalline providing it with notable mechanical properties that includes enhanced strength, rigidity, and thermal resilience [128]. However, PGA has limited potential as a standalone polymer for several reasons. First, the hydrolysis of the ester in PGA is faster than PLA due to its high hydrophilicity. Additionally, the degraded byproduct, glycolic acid, can potentially cause inflammation [129]. Second, PGA possesses inherent brittleness and limited solubility in commonly used organic solvents which further restricts its processibility to formulate a desired DDS. Therefore, PGA is frequently employed in combination with other polymers to overcome any usage limitations.
PLGA, one of the extensively utilized polymers in LA formulations, comprises a substantial 46% of the approved LA injectable medications made from polymers [130]. PLGA is synthesized by combination of ring opening and condensation polymerization reaction. PLGA can be synthesized in various MWs and lactide-to-glycolide (L/G) ratios, where an increase in the L/G ratio corresponds to a slower rate of polymer degradation. For instance, in aqueous conditions, PLGA polymers with L/G ratios of 50:50, 75:25, and 85:15 have degradation times of approximately 1-2, 4-5, and 5-6 months, respectively [125].
Drug release from PLGA or PLA-like polyester mainly followed two mechanisms: diffusion and bulk erosion or degradation [85]. At the early stage of release, diffusion is the main release mechanism which depends on the concentration gradient and the shape of the DDS [45]. Over the time, PLGA erosion becomes the dominating mechanism. The drug release process form PLGA-based DDSs starts with slow water infiltration followed by swelling of polymer. The swelling of the polymer core increases the pore's size and makes way for the drug molecule to diffuse through the polymer matrix or membrane [131]. Soon after, PLGA undergoes degradation, eventually formation of lactic acid and GA as byproducts. These acids act as auto-catalyst and further speed up the degradation process [45]. Collectively, the MW, PLA:PGA ratio and geometry of the PLGA-based DDS influenced the water absorption and, eventually, its degradation. Moreover, PLGA-drug interaction, drug solubility, pH of the surrounding environment, osmolarity, and porosity of the DDS have a considerable impact on the release rate. PLGA-based DDSs mostly followed the triphasic release kinetics [45].
Polycaprolactone (PCL) is a semicrystalline and hydrophobic polyester which is typically synthesized by the ring-opening polymerization of ε-caprolactone. Compared to other hydrophilic polyesters, PCL shows a very slow rate of biodegradation [132]. Microorganisms can slowly degrade the PCL by using lipase, an ester hydrolysis enzyme. However, the hydrophobicity limits the interaction between PLA and lipase, therefore chemical degradation remains the primary degradation mechanism [133,134]. PCL-based DDSs can maintain a sustained drug release over several months. PCL is also known for its mechanical strength, which has made it a popular choice in applications like surgical dressing, wound healing, and contraceptive implants [135]. By taking advantage of the compatibility between PCL and various hydrophobic drugs, the drugs can be uniformly dispersed in the PCL matrix [136]. Additionally, it can be blended with an array of both natural and synthetic polymers, including starch, hydroxyapatite, chitosan, PEG, PU, oxazolines, PEO, and PVA. The blending allows fine-tuning of PCL’s mechanical strength, crystallinity and degradation behavior, and accomplishes an ideal DDS [137].
For PCL-based DDS, the hydrophobicity and crystallinity are two important factors that dictate the degradation and drug release kinetics. The chemical degradation of PCL-based DDS happens in two steps. The first step is the rate-limiting step, where water infiltrates and hydrolyzes the amorphous region, which causes mass loss and is followed by an increase of porosity in the DDS. In the second step, PCL degradation product, 6-Hydroxyhexanoic acid further catalyze the hydrolysis process which eventually propagated to the crystalline region of the PCL [136]. Interestingly, the degraded form of PCL can be phagocytosed by cells [135]. PCL-based DDS usually follows the biphasic release kinetics, where there is an initial burst release of the drugs followed by a sustained release phase. It has been evidently seen that the initial burst release is much higher for hydrophilic drugs than the hydrophobic drugs [113,138,139].
4.7. Chitosan and Hyaluronic Acid (HA)
Chitosan, a natural biopolymer has widely been used in tissue engineering, bio-scaffold creation, and drug delivery application. Chitosan has been derived from chitin (a long-chain natural polysaccharide) by adopting the alkaline or enzymatic deacetylation process [140,141]. Chitosan is a copolymer of D-glucosamine and N-acetyl-D-glucosamine linked through the β-(1 → 4)-glycosidic bonds, giving it a rigid crystalline form. The chitosan backbone contains ‘–NH2’ and ‘–OH’ like active functional groups that can be easily functionalized with active biomolecules, making it so popular for nano or microparticle-based DDS. Chitosan, a biodegradable polysaccharide, is particularly notable for its versatility in sustained drug delivery [142]. The physicochemical characteristics of chitosan depends on its MW and degree of acetylation. Chitosan exhibits low water solubility, enabling it to remain in the body for extended periods. This property has made it particularly useful for sustained-release in oral drug delivery applications [143]. Moreover, inspired by anti-inflammatory, anti-bacterial, and mucoadhesive properties, chitosan has been widely used in wound dressing, healing, and pulmonary DDSs.
HA is a naturally occurring nonimmunogenic and hydrophilic polysaccharide. It is commonly used in the development of DDS. The HA contains two basic sugar units namely, D-glucuronic acid and N-acetyl-D-glucosamine, conjugated through β(1–4) and β(1–3) glycoside bonds [144]. HA is known to interact with CD44 cell surface receptors, and exogenous HA can reduce the HIV-1 infection to CD4+ T-cells in a CD44-dependent manner [144]. However, endogenous HA failed to do so as they undergo a rapid degradation at the cell surface by the enzyme called hyaluronidase. Moreover, HA is one of the major components of mucosa, which results in low mucoadhesive properties of exogenous HA. The thiol group (SH) modification on the backbone of HA can further increase its mucoadhesive property. In this context, Agrahari et al. formulated TFV-encapsulated nanofibers (NFs) by using thiolate hyaluronic acid to stop vaginal transmission of HIV [145]. The NFs were demonstrated to be safe toward the genital tract and other major organs of 57BL/6 mice. The NFs show hyaluronidase responsive TFV release, resulting in the prevention of HIV vaginal transmission.
4.8. Polymers and Polymer Blends
Over the past few decades, research into LA DDSs has shown significant strides forward. The introduction of novel polymers and polymer blends has improved the efficiency of the existing drug delivery systems. This includes better biocompatibility, reductions in local and systemic toxicities, and optimal release profiles. FDA-approved ISFI is one of them, which employed dimethyl sulfoxide (DMSO) and N-methyl-2-pyrrolidone (NMP) like organic solvents to lower the solution viscosity of PLGA and induce phase conversion. Compared with DMSO, NMP has more carcinogenic, teratogenic, and mutagenic toxicities [93,146,147]. Therefore, to avoid NMP and minimize the use of PLGA, poly(3-hydroxybutyrate-co-3-hydroxyvalerate (PHBV), and poly (ethylene carbonate) (PEC) were developed.
PHBV is a hydrophobic, thermoplastic, semicrystalline, biocompatible, and biodegradable linear aliphatic polyester that has been synthesized by incorporating the 3-hydroxy valerate units into the poly(3-hydroxybutyrate) (PHB) [148]. PHBV is very brittle and has a melting temperature lower than PHB [149]. The thermal properties of PHBV can be improved through blending with PCL or TiO2 nanoparticles [150]. The PHBV was used to formulate the aripiprazole (an antipsychotic drug) encapsulated nanoporous ISFI [151]. Rheological analyses demonstrated the existence of a highly crosslinked network structure in the PHBV ISFI. The spectroscopic analysis revealed the intactness of aripiprazole with the network structure. The ISFI demonstrated a burst release followed by a sustained release of aripiprazole over 18 days.
PEC is a biodegradable polymer, synthesized by co-polymerization of CO2 with ethylene oxide [152]. Generally, PEC is characterized by high ethylene carbonate content with low polydispersity and high glass transition temperature. PEC undergoes biodegradation through surface erosion, followed by the formation of ethylene glycol as a degradation product [153]. The in vivo biodegradability of PECs is influenced by the presence of reactive oxygen species, which are associated with inflammatory cells. Moreover, the molecular weight and percent of ether contained in PEC also have an immense effect on its degradation rate [154]. It was successfully demonstrated an LA ISFI using PEC as a rate-controlling polymer and bovine serum albumin as a model drug. To compare the effects of DMSO and NMP on the microstructure and drug release of PEC-containing ISFIs, bovine serum albumin-encased ISFI containing either PEC/DMSO or PEC/NMP were developed [155]. In comparison with PEC/NMP ISFI, PEC/DMSO ISFI demonstrated a quick escape of DMSO followed by the formation of a depot with low porosity, which minimizes burst drug release.
The emergence of stimuli-responsive polymeric biomaterials demonstrates improved spatio-temporal drug release. These smart biopolymers are responsive to various internal and external stimuli, including pH, redox potential, temperature, and enzyme activity [156,157,158]. Phenylboronic acid (PBA)-containing polymers are an example of such smart biomaterials which are sensitive to pH, sugars, and reactive oxygen species) [159]. PBA-conjugated polymers have proven to be effective in pH-responsive delivery of anticancer and antiretroviral drugs. For instance, doxorubicin-encapsulated, PBA-modified poly(maleic anhydride)-F127 micelles show a pH-responsive release [160]. The use of PBA-conjugated polymers in LA ARV formulations could offer enhanced pharmacologic benefits. In this circumstance, a PBA-based mucin-like polymer system that blocks HIV-1 migration was developed within the genitourinary system [161,162].
5. LA ARV Delivery
LA DDSs, such as implants, VRs, MNs, and deport-forming nanoformulations, offer a promising avenue for extended release of ARVs beyond two months (Figure 3). In HIV-1 infected patients, the viral load is always persistent in tissue along with systemic circulation. This necessitates achieving continuous effective drug concentration both at the target tissue and plasma without reducing the longevity of the drug depot. Notably, for sexually transmitted diseases like HIV-1, precise drug concentrations within reproductive organs are pivotal for effective disease prevention. While local administration methods, such as VRs, offer a potential solution, they also face the challenges of maintaining the local therapeutic drug concentration without causing any toxicity to the surrounding tissues. Furthermore, the drug can be systemically absorbed, potentially diminishing local drug concentrations. In this context, implants or depot-forming injectables emerge as an alternative strategy as they can deliver the drug to reproductive organs through systemic drug exposure. Yet, achieving optimal drug concentrations without inducing local adverse effects in the implanted or injected site presents a multifaceted obstacle. The intricacies of polymer-drug interactions and device design play a pivotal role in dictating drug exposure and its release kinetics. Here, we will discuss recent advancements in LA ARV formulations, specifically focusing on the interplay between drug and polymer interaction and factors that influence the drug release kinetics. By deciphering the complex dynamics underlying polymer-mediated drug delivery, this exploration aims to pave the way to achieve an efficacious long-acting ARV formulation by fine-tuning their controllable parameters.
5.1. Implants
In 1938, Deansby and Parkes pioneered the implantable drug systems by observing the effects of estrone pellet implants on male chickens [163]. Folkman and Long later refined this delivery system with a silicone rubber capsule for continuous drug release. This led to major developments in implantable drug delivery, especially for contraception and ocular treatments [163]. The approval of various contraceptive and ocular implants boosted their acceptance and uptake by patients. This progress influenced implants' development for HIV-1 treatment and PrEP. In the early 2000s, research focused on implants for the sustained release of ARVs. TAF's potency and suitable physiochemical properties, such as hydrophobicity, MW, and solubility in polymer matrices, make it an ideal candidate for deliverying through implants. Other drugs like ISL, CAB, DTG, and entecavir are also being explored for implantable-based delivery [3,164,165].
The formulations and drug release mechanisms of implantable devices are multifaceted and varied, making their categorization challenging. Nevertheless, those designed for HIV treatment and prevention can be broadly divided into two types. The first includes preformed solid implants, which are made of either biodegradable or non-biodegradable polymers and require surgical or invasive procedures for subdermal placement [3,166]. The second type involves injectable in-situ forming implants (ISFIs). ISFIs can be injected in the form of suspension or solution in subdermal spaces where solvent induced phase inversion produces a drug loaded implant [18,167]. Even though most ARVs and the polymers used in the fabrication of implants are FDA-approved with established systemic safety profiles, local toxicity concerns that are either drug specific or associated with the nature of implant materials remain. Improved understanding of how ARVs and their metabolites interact with and get eliminated from the surrounding tissues and extracellular environments is of significance. For instance, high concentrations of TFV and TAF have shown significant implant site toxicities in some preclinical studies and significantly affected rapid development and translation of these devices [168]. Nonetheless, advancements are being made in implant design to regulate TAF release and stability within the device.
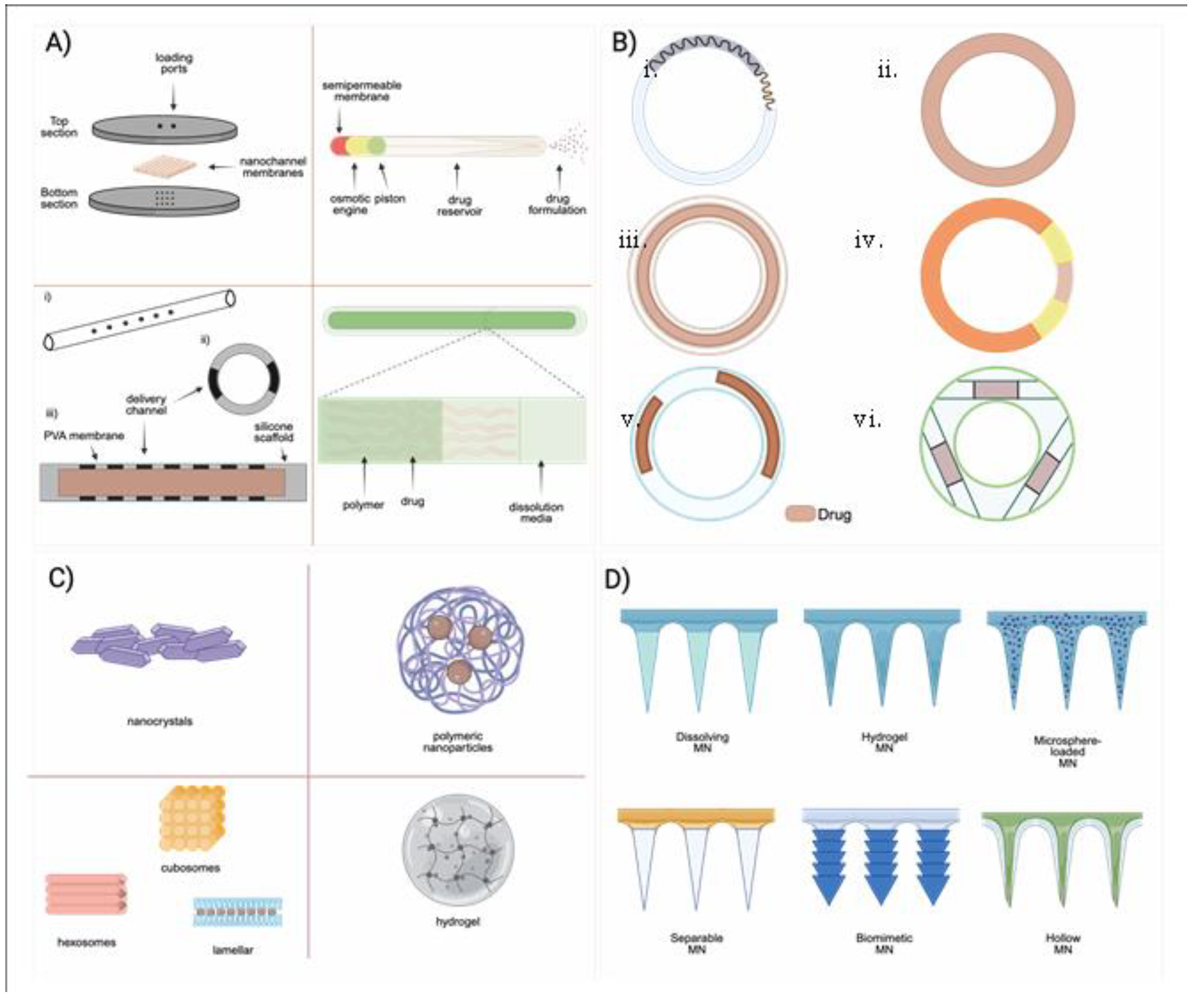
Figure 3.
Different types of LA DDSs that are in either clinical or preclinical development. A) Diffeent types of solid implants. Reproduce with permission from Ref [5], . Copyright 2018 Antimicrobial Agents and Chemotherapy. B) Different types of Vaginal rings- i) over-moldeed metal spring desing ii) martix-type VR iii) Full length reservoir-type VR iv) sandwich-shape VR v) Partial reservoir-type VR vi) Insertable reservoir-type VR. Images were adapted under the terms of Creative Commons Attrobution- Non commercial from ref. [7] .C) Nanocrystal formulations, polymeric nanoformulations, liposomes, and hydrogels. D) Different types of microarray patches. Reproduced with permission [10].
Figure 3.
Different types of LA DDSs that are in either clinical or preclinical development. A) Diffeent types of solid implants. Reproduce with permission from Ref [5], . Copyright 2018 Antimicrobial Agents and Chemotherapy. B) Different types of Vaginal rings- i) over-moldeed metal spring desing ii) martix-type VR iii) Full length reservoir-type VR iv) sandwich-shape VR v) Partial reservoir-type VR vi) Insertable reservoir-type VR. Images were adapted under the terms of Creative Commons Attrobution- Non commercial from ref. [7] .C) Nanocrystal formulations, polymeric nanoformulations, liposomes, and hydrogels. D) Different types of microarray patches. Reproduced with permission [10].
A wide range of non-biodegradable (such as silicone and PUs) and biodegradable polymers (PCL or PLA) have been used for ARV implant generation (Figure 4). Biomedical-grade silicones have been used for implant development for various diseases over six decades and are also the preferred materials for the development of ARVs, such as TAF implants. Irrespective of the extensive use of silicone based TAF implants, TAF permeability through the silicone implant is low. To improve TAF permeability, the implants have been fabricated with nano- or microchannels that aid in improved TAF release kinetics. To further control drug release rates, the implants were either coated with a rate-controlling membrane or through readjustment of the diameter and number of drug release channels [3]. Interestingly, TAF release rate from this implant was independent of initial TAF loading. PK evaluation of this device in beagle dogs demonstrated a zero order release kinetics of TAF (1.07 ± 0.02 mg day−1) that sustained therapeutic TFV-DP levels in PBMCs for 40 days. The implant is currently in an early phase-1 clinical trial to assess its safety and tolerability [169]. The ability to remove the implant can be advantageous in the case of adverse reactions. However, removal of this device requires invasive surgical procedures that may limit broader use. In such cases, transcutaneously refillable silicone-coated nanochannels may offers enhanced pateint convinence and acceptability over conventional solid impant by offering refillability with explantation [19]. This design is primarily comprised of a transcutaneously refillable medical-grade titanium drug reservoir and a uniquely crafted silicone membrane housing TaN-coated slit nanochannels [19]. These nanochannels are arranged in square arrays and connected to the inlet and outlet of the membrane through a network of microchannels. Drug release rate from the implant is dependent on interactions with the interior surface of the membrane and can be further controlled by adjusting the number of nanochannels.
Hydrophobic poly (tetramethylene oxide) subunit-containing PUs have also been used to create TAF implants [166]. As hydrophobic PU demonstrates minimal swelling, the drug inside the hydrophobic PU tube forms a suspension encircled by an outer layer of saturated drug. The difference in concentration of these two layers drives the drug release rate, which follows a zero-order kinetics. This release rate can be fine-tuned by modifying the membrane wall thickness or composition of PU. PU-based pellets-in-a-tube types TAF implant where a mixture of TAF, NaCl, and magnesium stearate were converted into a pellet and impulse sealed into PU tubes were created [166]. In this report, the release kinetics of TAF from the implant did not adhere to a zero-order. Interestingly, incorporating a hydrophilic excipient like PEG into the TAF pellet or increasing the crystallinity of the PU membrane had been found to enhance the drug's release from the implant [20]. However, preclinical in vivo, safety, and tolerability studies in different animal models demonstrated that the PU-based TAF implant results in local inflammation and toxicities. The local toxicities observed in those implants may be the results of high TFV and TAF concentrations in the implant site, as observed in those studies. A similar implant design has also been employed to develop CAB implants [170]. In a comparative study, the release rate of CAB from various elastomeric PUs membranes, including poly(dimethylsiloxane), poly(ethylene-co-vinyl acetate), and hydrophilic poly(ether) urethanes containing implant was tested. Interestingly, only the segmented hydrophilic poly(ether) urethanes demonstrated sufficient CAB release rate to reach a pharmacologically relevant concentration. Furthermore, using polyvinylpyrrolidone (PVP), instead of PEG as an excipient caused less mass swelling and higher implant drug release [170].
Biodegradable polymers are widely used for the development of reservoir-type TAF implants. PCL are TAF compatible. When TAF-containing PCL implant is inserted subcutaneously, the biological fluid from the surroundings slowly penetrates the PCL implant, creating water-filled channels and dissolving TAF to facilitate its release. As more water enters the implant, it induces the bulk erosion of PCL. However, PCL’s bulk erosion rate is so slow that it can persist for up to a year [171]. A biodegradable PCL-containing solid TAF implant was recently developed [2]. This implant was 2.5 mm in diameter but of varying thicknesses and produced by hot-melt extrusion process. The PCL tube was filled with a formulation containing TAF and castor oil and sealed at both ends through heat or injection sealing. The TAF release rate from PCL implant depended on factors like the implant's surface area, PCL's MW, membrane thickness, and TAF solubility. TAF release studies were found to correlate with the implant's surface area and inversely with PCL wall thickness [2]. Interestingly, thinner PCL wall-containing implants demonstrated a rapid initial TAF release, while high MW PCL implants released TAF more quickly. The effect of PCL's MW on release rates was common in thinner walls rather than thicker ones. This is possibly due to differences in crystallinity between PCL types [2]. Incorporation of hydrophilic excipients in a PCL Implant can increase the TAF release rate. For example, a PCL implant where TAF was formulated with PEG3350 with various weight ratios [164]. The PEG3350 elevates the TAF dissolution in the implant. This eventually increases the drug release rate. It has been reported that the rate of TAF release is proportional to the mass of the PEG3350 used and the surface area of the implant.
ISL, a novel nucleoside reverse-transcriptase translocation inhibitor (NRTTI) has gained attention in implant generation due to the potency and extended intracellular apparent half-life of its triphosphate metabolite [21,172]. ISL was incorporated in both biodegradable and non-biodegradable polymer-containing implants for extended-release [5]. Two types monolithic matrix-type implant were developed by using either biodegradable PLA and PCL or non-degradable EVA polymer for SC delivery of ISL [5]. The uniform dispersion of crystalline ISL in the polymer matrix resulted in a minimal implant surface porosity, which limited the water infiltration to the implant. Therefore, the implant showed a small transient initial burst release followed by a slow first-order release of ISL. The initial small transient burst release was presumably because of the rapid dissolution of ISL, persisting on the implant surface. Furthermore, the rate of ISL release showed an increasing trend with its loading content. Several clinical trials involving ISL implants and oral regimens have been halted as the reduction of the participant's body's CD4+ T-cells and lymphocyte counts have been reported [169].
Given the instability and safety concerns associated with TAF and ISF-based implants, alternative approaches such as the development of stable TFV prodrugs and salts, new ISF analogs, ISFIs and other potent drugs from different classes such as CAB and DTG (INSTIs) have gained considerable attention. In-situ forming implants (ISFIs) are injectable drug formulations that turn into solid/semisolid depots after administration [173]. A combination of the drug, excipient or release rate limiting agents, and water-miscible organic solvents have been used in the formation of ISFIs. N-methylpyrrolidone (NMP), and dimethyl sulfoxide (DMSO) are the commonly used water-miscible organic solvents used in ISFIs. After SC dministration of ISFIs, the water-miscible solvent starts diffusing out from the implant, and surrounding water starts to infiltrate. Subsequently, there is an immediate phase separation of solvents, solidification of formulations, generation of microchannels for water influx, and eventually formation of ISFI [174]. The drug release rate largely depends on the phase-inversion kinetics of the implant, MW and hydrophobicity of the used drug molecules, and biodegradability of the excipient [175,176,177].
Biopolymers such as hydroxypropyl methylcellulose, PLGA, PEG, poloxamers, PCL, and poly(N-isopropyl acrylamide) have been employed to develop ISFIs for various indications [178]. The selection of polymer depends significantly on the drug's physicochemical properties and the desired release rate. Generally, a polymer's solubility should be compatible with the drug's solubility characteristics for optimal drug release kinetics [179,180,181]. PLGA was used as an excipient and NMP/DMSO as water-miscible solvents to formulate DTG and CAB ISFIs [1,18]. The authors investigated the impact of physiochemical properties of the polymer, used solvents, drug-polymer miscibility into the solvent, and polymer-to-solvent ratio on the drug release kinetic.
The rate of drug release from the ISFIs is linked to the microstructure of ISFIs at the adminsitration site. For example, scanning electron microscopy (SEM) imaging of DTG ISFIs demonstrated a central pore surrounded by a highly interconnected porous outer PLGA shell (Figure 5). Over time, the thickness of the outer PLGA shell decreased due to PLGA erotion, leading to an increase in the central pore size. The implant with a thinner shell and larger central pore size facilitated a high rate of drug release from ISFIs. The multispecies tolerability studies of ISFIs support their safety. The acceptability of ISFIs stems from their easy manufacturing and patient-comfortable administration process. Moreover, the clinical availability of leuprolide acetate ISFI (Atrix Laboratories Inc.) and Sanofi Aventis (Eligard®) supports the safety of this delivery system [182]. The overall limitations of ISFIs are the injection site reaction, injection intervention, minimal control of the size and shape of the implant, and heterogeneous drug release [183,184].
5.2. Vaginal Rings (VRs)
Building on the VR’s clinical progress for contraception and hormone therapy, rings containing ARVs are developed to prevent HIV-1 infection. In addition, VRs are gaining preference over other vaginal products that include gels or creams based on their efficacy in delivering ARVs to the vaginal epithelium over extended time periods [37,185,186]. VRs are also widely accepted among women compared to other LA DDSs due to their user-friendly and discreet method of administration and easy reversibility of treatment when needed.
The traditional approach to VRs design involves uniform dispersion of drugs within polymer matrices. This is accomplished by molding a drug-polymer matrix into a ring-shaped device. Conventional VRs are classified into a sandwich (or matrix)-type VR and core (or reservoir)-type VR [115]. In the sandwich design, a drug-dispersed polymer matrix is placed between two non-medicated rate-controlling polymer layers [7]. The core-type VR involves creating a small pellet containing a mixture of drugs and polymers. Several cores are combined into a single ring. The ring is covered by a drug-free secondary polymer membrane [7]. The drug release rate in VRs is typically governed by its drug diffusion through the polymer membrane [7]. When the VR is placed vaginally, water enters its wall, leading to polymer swelling and the dissolution of drugs [7]. The dissolved drugs near the matrix's surface are diffused first. This action also forms a drug-depleted layer near the matrix surfaces. Subsequently, the remaining undissolved drugs undergo dissolution and diffuse through the polymer matrix. Factors such as the drug's solubility in the polymer, the amount of drug loaded into the VR, and the surface area and thickness of the VR membrane influence this diffusion process. DPV, maraviroc, TAF, TDF, UC781, and MC1220 are being utilized in VR fabrication. Among these, DPV, TDF, and TAF, due to their ideal properties, have been predominantly studied for VR development. However, only the DPV VR has progressed to phase III clinical trials and received approval in several African nations for the prevention of HIV-1 [187].
Silicone, EVA, and PUs, have been extensively used to develop DPV and TDF VRs [187,188]. They are clinically proven safe polymers for VR development. For instance, five of seven clinically approved VRs are made of silicone [115,189,190]. NuvaRing is the only VR that solely uses EVA, whereas another VR, Ornibel, uses PU as either a drug reservoir or a coating agent to control the drug release [187]. However, these polymers come with drawbacks. For instance, condensation-type silicone elastomers can produce a propanol by-product. This can increase the drug solubility, prompting more solubilized drugs to move to the polymer surface. This then would lead to an increased initial drug burst release [191]. Furthermore, reports have emerged of biofilm formation due to enhanced bacterial adherence on EVA-based VRs [192,193,194]. However, biofilm generation can be significantly reduced by surface modifications such as in biocompatible surfactant, argon plasma treatment, or perfluoro-alkyl siloxane grafting use present on the elastomer [195,196,197,198].
The exploration of DPV VR development began nearly 25 years ago. The VRs were then developed using a condensation-cure silicone elastomer system [199,200]. To produce the core or reservoir type DPV VRs, DPV was blended with silicone elastomer and reaction injection molded at 50°C to form an active core. This core was then segmented and enclosed within a non-medicated outer silicone membrane through another reaction injection molding process. The resultant VRs had a 5.50 mm and 9.00 mm outer cross-sectional diameter, while the total diameter of the VR was 55.00 mm. DPV release rate from the reservoir VRs depends on the solubility of DPV in the silicone matrix, and the diffusivity of the silicone solubilize DPV through the non-medicated silicone sheath layer. In contrast, the matrix-type DPV VR was developed through a curing process involving DPV and silicone elastomer with a normal propyl orthosilicate cross-linker mixture. The implants didn’t show any significant local site reactions and proved to be well tolerated in the phase I clinical trial [199,200]. The PK and release kinetics of these two VR types showed notable differences. Matrix-type VRs exhibited a substantial initial burst release of DPV, reaching peak plasma concentration within 8 hours. On the other hand, reservoir-type VRs took 10 days to reach the peak plasma concentration. Moreover, the average plasma concentration in matrix-type VRs was almost 50 times higher than in reservoir-type VRs. Interestingly, the matrix-type VRs demonstrated a rapid decline in plasma concentration over time, while the drop in plasma concentration from reservoir-type VRs was relatively slow [200,201]. From the clinical trial findings to date, the matrix-type silicone DPV VR stands out as the most effective one. There are several phase III clinical trials conducted with DPV matrix-type silicone-based VR, and DPV VR has been reported to reduce the risk of HIV-1 acquisition by 27% to 37% in African women [202,203].
Apart from DPV, TFV was the only other vaginal microbicide that demonstrated potential as a vaginal PrEP agent during clinical trials. As per the trial result, gel containing 1% TFV exhibited a 39% protection rate against HIV-1 infections in women [204]. Inspired by these findings, considerable research efforts were undertaken towards developing TFV or TFV prodrug-based VRs. While various polymers, including silicone, EVA, and PU, were investigated for creating TFV VRs, it was only the PU-based TFV or TFV prodrugs-VRs that demonstrated promising results in preclinical studies. For instance, Mesquita et al. developed silicone, EVA, and PU-based TDF-VRs and compared the TDF release rate from these VRs [205]. The in vitro release study revealed that the release rate of TDF from PU-based VR was 5 to 15 times higher, in comparison to those created from EVA and silicone [205]. The differences in TDF release rate were attributed to the varying solubility of TDF in employed polymers. Specifically, TDF showed lower solubility in silicone and EVA as opposed to PU, resulting in a decreased diffusion of TDF through the polymer membrane [205].
The higher release rate of TDF from hydrophilic PU-based TDF VR has also been investigated [205]. Additionally, the authors compared both matrix and reservoir types of TDF VRs, made with hydrophilic polyether urethane (PEU). To prepare the matrix-type TDF VR, TDF-loaded hydrophilic PEU was extruded first using a twin-screw extruder and then, cut into small pellets. Then, the pelletized segments were re-extruded, cut into 15.5 cm segments, and end-joined using an induction welding [205]. For preparing reservoir type TDF loaded VR, the hydrophilic PEU was extruded into tubes, and the empty tubes were filled with either TDF only or a formulation of TDF and NaCl (86:14 wt% ratio) [206]. The matrix type facilitated an immediate drug release, which then decreased by a factor of at least 20-fold from day 1 to day 28. In contrast, reservoir-type VRs showed a prolonged lag time to start the drug release. Interestingly, when an osmotic agent like NaCl was added to the reservoir-type VR, there was an increased influx of vaginal fluid into the VR, prompting a quicker TDF release. To establish human safety and tolerability, a phase I clinical trial was conducted using reservoir type HPEU -TDF VR. However, the trial was discontinued due to the observed genital ulcers among the participants. However, no observed ulceration was observed among the participants who received placebo-containing VRs manufactured using the same technique, indicating the observed local adverse effect is not due to the polymer [23].
Hydrophilic PU (HPU) membrane-based reservoir type for TFV VRs was also reported [204]. The VR was produced by making an HPU tube by hot-melt extrusion and then, filling the tube with either TFV or TFV/glycerol/water-containing semisolid formulation. The end of the tube was sealed with induction welder. Only TFV-containing VR showed a slow but steadily increasing TFV release rate in the in vitro release study. However, adding HPU permeable substance, such as glycerol with TFV in the polymer matrix increased its solubility in HPU and release rate [204].
PU-based VRs were also used for the vaginal delivery of IQP-0528 and MIV-150. IQO-0528 is an NNRTI that has physiochemical properties similar to DPV. A matrix-type PU-based VR was developed to deliver the IQP-0528 [207]. The VR was prepared by injection of a molding technique after mixing the drug and Tecoflex EG85A PU. In vitro studies with this VR indicated a linear relationship among the drug release rate, VR surface area, and drug loading to the VR. However, in vivo findings indicated that drug release was not solely determined by drug loading or VR surface area. Instead, it was primarily driven by diffusion, contingent on both the VR drug concentration and drug presence in the surrounding media [207].
5.3. Microneedles (MNs)
Microneedles (MNs), also known as microneedle arrays or patches, are transdermal drug delivery systems featuring tiny projections around 200 µm tall. While initially developed for immediate-release transdermal drug delivery, MNs have now evolved as innovative LA DDSs due to the advancement in polymer chemistry. MNs penetrate through the skin up to the dermis, an area rich in blood vessels, ensuring efficient drug absorption while reducing systemic exposure and bypassing first-pass metabolism [208,209,210]. Their small size and length cause less pain, as pain receptors reside deeper in the skin. The design and composition of MNs, including tip length, spacing, diameter, and overall geometry, can be tuned to optimize drug delivery. Further, MNs can be combined with other drug delivery technologies like nano- or microparticles or hydrogels for extended drug delivery [211,212]. Their benefits have spurred a significant increase in research in recent decades. Particularly in ARV delivery, their discreet, easy, and minimally invasive nature is recognized and widely accepted [213]. For instance, a survey study was conducted to gather opinions from patients and healthcare professionals regarding the use of MNs for ARV delivery. The results showed a positive attitude towards MNs technology, with both groups acknowledging the benefits of discreet and self-administrable MNs for administering ARV drugs [214]. Given the constraints related to permeability through the skin's stratum corneum, MN technology proves particularly effective for potent, lipophilic, and low MW drugs. ARVs such as RPV, CAB, and BIC have demonstrated significant potential when employed with MN technology.
There are several types of MNs based on their design and drug-releasing mechanism. Each of the MN's drug release kinetics varies based on the MN’s geometry, surface area, number of MNs per patch, polymer used, nature of the drugs, and its loading content [215]. In general, MNs are classified into solid MNs, drug-coated MNs, dissolving MNs, and hollow MNs. Solid MNs are usually used to pretreat the skin to create microchannels to facilitate drug absorption before applying any transdermal topical gel or formulation [215]. Polymers with good mechanical strength, such as silicone, PMMA, PCL, PLGA, PLA, and PGA, and metals such as stainless steel, titanium, and ceramics have been used to prepare the solid MNs [216]. In drug coated MNs, the outer side of solid inert MN tips are coated with drug formulation. Upon insertion of the drug coated MNs into the skin, the drug formulation is dissolved in the surrounding medium to release the drugs from MNs. These MNs are less utilized for LA DDSs because of their inability to form a drug depot. However, for LA drug delivery, dissolving MNs containing nanocrystal formulation of ARVs have utility.
Dissolving MNs are made of biocompatible polymers that can encapsulate drug-containing matrices and, upon administration, dissolve in the biological fluid and release the drug content [217]. Nanocrystal suspension of ARVs, such as CAB, RPV, and bictegravir (BIC), when delivered through dissolving MNs, can easily form dissolution-controlled drug depots (Figure 6). This strategy can allow a less painful way of self-administration of ARVs. The tips of dissolving MNs can be made of biodegradable or non-biodegradable polymer or sugar, and the rate of polymer or sugar degradation determines the MNs' release duration [217,218,219,220,221,222]. The fast-dissolving MNs use hydrophilic polymers like dextrose, maltose, and chitosan, while slow-dissolving ones employ hydrophobic polymers like PLGA, PVA, and PLA, due to their biocompatibility, mechanical strength, and slow erosion [223,224,225]. PLGA MNs are mainly loaded with hydrophobic drugs and can be designed to separate from their baseplates upon insertion [224,226,227]. PCL is also used in MNs production which allows controlled drug release using near-infrared light as an external triggers [228].
To creat a bilayer dissolving MNs for transdermal delivery of RPV , a first layer or MNs tips were developed using varying compositions of PVP and RPV nanocrystal suspension [27]. The drug-free baseplate of the MNs was made using an aqueous solution of PVA and glycerol. Those fast-dissolving MNs dissolved within few minutes after the insertion into the skin and formed an RPV depot in the application site. However, the dissolution testing of MNs in the excised skin demonstrated a slower dissolution profile of RPV suspension, which may be due to the hydrophobicity of RPV in the MN tips. Also, the concentration of RPV in the skin tissue was proportional to the MNs application time. Although the varying composition of RPV nanocrystal suspension and PVP was used to manufacture MNs, the MN with 70% RPV, 15% PVP, and 15% water demonstrated better plasma RPV levels compared to other MNs. Due to the discreet nature of the MNs and their ability to localized delivery of the drugs, the same type of RPV MNs also generated by the same group for vaginal and CNS delivery of the RPV [229,230].
A similar type of bilayer MNs has also been developed by the same group for the transdermal delivery of BIC and CAB nanocrystal formulation (Figure 6). For CAB MNs generation, bilayer MNs were developed, where the MNs tips were generated by using a hydrogel composed of 20:20:60 of PVA:PVP:CAB LA nanoformulation [4]. The baseplate was made using PVA and PVP. The two polymer blends formed a sufficiently strong MAP tip, which could be attributed to the hydrogen bond formed between the carbonyl group of PVP and the hydroxyl group of PVA. This hydrogen bonding also improved the crystallinity and cohesiveness of the polymer. In vitro MNs tip dissolution study showed that CAB MNs tips dissolved almost 100% within the first 60 mins after the administrations. Importantly, after a single MNs administration in rats, these MNs maintained the CAB therapeutic levels for up to a month [4]. For developing the BIC-containing MNs, first, PVP and PVOH-containing BIC nanosuspension was prepared and lyophilized. Then, the lyophilized powder was reconstituted in a very small amount of deionized water, and MNs tips were generated from the resultant suspension. The baseplate of the MNs was made using PVP and glycerol. Similar to CAB MNs, BIC MNs also maintained plasma BIC levels equal to the human therapeutic level for up to a month [231].
Newer types of MNs, such nano- or microparticle-loaded MNs, hydrogel-forming MNs (HFMNs), and separable MNs, are also becoming very popular and useful in the drug delivery [224,232]. HFMNs are particularly important for LA drug delivery and are made from swellable cross-linking polymers. At the dry state, HFMNs are sturdy enough to penetrate the skin. Upon insertion, HFMNs forms hydrogels by soaking up water from the interstitial fluid, causing the formation of water-filled channels in the MN tips [233,234]. The drug's release rate from HFMNs is influenced by the dimensions and number of channels, as well as the size of the drug particles [235]. Unlike dissolving MNS, HFMNs have a higher drug-loading capacity [236]. However, this capacity varies based on the polymer type and cross-linker used. Common polymers for HFMNs include PMVE/MA, PVA, EVA, chitosan, and silk fibroin, often crosslinked with hydrophilic polymers or compounds like PEG, dextran, and CMC [232,236]. The drug release rate can be tailored by adjusting the percent of cross-linker and swelling ratio.
5.4. Polymeric Micro- and Nanoparticles
Polymeric microparticles are microscopic particles with sizes between 1 to 1,000 μm, where a core substance is encapsulated by one or more layers of the polymer membrane. Microparticles can further be divided into microspheres and microcapsules. While microspheres consist of a uniform matrix with the API dispersed throughout, microcapsules feature a distinct core, which can be liquid, solid, or semisolid, enveloped by a continuous polymer coating [237]. Nanoparticles have a similar feature, but the size between 1 to 1,000 nm. Micro- and nanoparticles can be prepared through a process called micro- or nanoencapsulation, respectively. This process entails encapsulating solid or liquid drug particles either dispersed or dissolved within a polymeric matrix or as a core surrounded by a polymeric shell. The polymer used in these particulate systems is alginate, chitosan, PLA, and PLGA. However, PLGA is the most widely used polymer for micro- or nanoencapsulation, which is used in almost more than 50% of clinically approved LA injectables [238]. The proven clinical safety and tunable drug release kinetics make PLGA a widely used polymer in particulate drug delivery systems. The encapsulation of drugs in micro- or nanoparticulate systems can be achieved through either physicochemical or mechanical processes.
The physiochemical process of drug entrapment involves coacervation or phase separation and inotropic gelation techniques [239,240]. There are three steps of microencapsulation by the coacervation-phase separation technique, which is carried out under continuous agitation. The first step involves the dispersion of the drugs in the polymer solution or suspension, then the coacervation process starts with a stimulus such as an addition of acid or salt (pH change), an input of non-solvent to the dispersion, or a temperature change of the emulsion which causes the precipitation of the polymer and continuous coating of the polymer to the core drug surface. The last step is the hardening or solidifying of the coated polymer materials to the core drugs surface [241,242,243]. The mechanical process of drug entrapment in micro- or nanoparticulate systems includes fluid bed coating and solvent evaporation technique [239]. In fluid bed coating, the drug particles are initially suspended in the airflow and then coated with a polymer solution through spraying. Subsequently, the solvent is evaporated to solidify the polymer coating around the drug particles [242,244]. However, the most widely used method for polymeric drug entrapment is the solvent evaporation or extraction method [245,246]. This method involves the emulsification of the polymer and drug solution, followed by the evaporation of the solvent. During the evaporation process, the polymer materials shrink around the core drug particles and produce micro- or nanosized particles [241,242]. The emulsification process of the polymer and drug solution depends on the physiochemical properties of the drug, which can be either a single emulsification process (o/w) for hydrophobic drugs or a double emulsification process (w/o/w) process for hydrophilic drugs [242,247,248]. However, challenges still exist in using the above-mentioned processes for nano- or microparticle formulation as controlling size, size distribution, shape, and batch-to-batch variation remains unmanageable. Moreover, encapsulating biologics using these techniques is considerably more challenging due to the inherent nature of proteins and biologics to interact with the polymer surface, as well as concerns about the stability of the biologics [249,250].
5.4.1. PLGA Based Micro- and Nanoparticles
PLGA has ideal physiochemical properties as a drug carrier, such as biocompatibility and both gel and capsule-forming ability [55,251]. The superior suitability of PLGA has been acknowledged in clinical settings, and the introduction of PLGA-based depot-forming injectables began with the US FDA's approval of PLGA-based Lupron Depot in 1989. Since then, PLGA microparticles have been used for both delivering small molecules and protein, such as Nutropin Depot.
A lot of preclinical efforts were also employed to develop ARV-loaded PLGA micro- and nanoparticles, as the PLGA particle can encapsulate multiple ARVs in the same formulation. Those particles have shown promise to prolong dosage regimens for both treatment and pre-exposure prophylaxis (PrEP) as they improved the PK and drug release properties of the ARV. Tahir Khuroo et al. developed a PLGA microparticle encapsulating a prodrug of DTG by emulsification-evaporation method. This PLGA microparticle increased the plasma apparent half-life of the DTG by almost 8.6-fold. However, the PLGA microparticle showed an initial burst release of DTG at day 1. However, the plasma DTG level has been gradually decreased by 4-fold over a 60-day period [252]. Initial burst release is a common phenomenon for PLGA-based formulations. Several studies on the morphology and drug distribution of PLGA microparticle indicated that PLGA microparticle is very porous and the drugs in the PLGA microparticle are distributed on the surface or the orifice of the large, interconnected pores [253,254]. Initially, water is absorbed through the pores, and then, fast dissolution of the neighboring drugs may cause the initial burst release of the drugs. In an alternative scenario, water uptake into PLGA microspheres with a seemingly smooth, thin surface layer can directly initiate drug release through various processes [254]. These processes may involve polymer swelling, leading to the formation of surface pores or the creation of new pore networks. However, after the initial burst release, the PLGA microparticle often undergoes pore rearrangement or a process called pore healing, causing the new pore formation and pore closing [254,255]. This may also explain the steady drug release kinetics after the initial burst release from PLGA microparticles. The drug release kinetics from the PLGA-based micro- and nanoparticle can be controlled by controlling the MW of PLGA, the glycolic and lactic acid ratio of the PLGA, size, and surface porosity of the particle [256,257,258].
PLGA nanoparticle is widely employed by Mandal et al. and colleagues for delivering combination ARV (cARV), such as (EVG + TAF + FTC) or (TFV + EVG) or (TAF + FTC) or (BIC + TAF) [259,260,261,262]. These nanoparticles were developed by w/o/w emulsion-solvent evaporation method, where the aqueous solution of the drugs was first emulsified with organic solution of PLGA (75:25 lactide:glycolide ratio) (MW 4,000–15,000 Da) and poloxamer, then the resultant emulsion is again emulsified in 1% PVA solution to generate a nanoparticle with a size of 250nm. The nanoparticle improved the biodistribution of all three encapsulated drugs. Four subsequent injections of these nanoparticles over a 6-week period protect the humanized mice from HIV-1 infection for 19 weeks [262]. A similar nanoparticle encapsulating either TAF+FTC or TAF+BIC, or TAF + EVG also improved the drug PK and biodistribution of those drugs [259,261,263].
Despite numerous clinically approved depot-forming PLGA-based implants and a significant increase in PLGA-based drug delivery development efforts over the last few decades, there are still notable limitations in PLGA-based drug delivery systems. Such limitations include the degradation of PLGA suspension or solution over the period during storage due to its hydrolytic properties. To improve the efficiency and effectiveness of PLGA-based drug delivery, it is crucial to thoroughly understand the factors that influence the degradation rate and drug release kinetics of PLGA-based polymers [264]. In addition to that, injection site reaction, accumulation of immune cells, and deposition of micro- and nanoparticle in the spleen and lung can pose significant challenges for developing polymeric nano- and microparticles [265].
5.4.2. Nanocrystal Formulations
Nanocrystal technology has revolutionized HIV-1 treatment and prevention. This technology facilitates formulation of water insoluble molecules into LA surfactant stabilized drug nanosuspensions in aqueos buffers. Using either top-down or bottom-up technology, such as high-pressure homogenization, wet bead milling, microfluidization and emulsion template freeze-drying technique, solid drug microparticles are suspended in an aqueous buffer with stabilizing surfactants or water soluble polymers and nanoformulated into stable solid drug nanoparticles. Compared to other formulation approaches, nanocrystals exhibit superior properties that include high drug loading to affect administration volumes, improved drug dissolution through reduced particle size, protect drug molecules from rapid metabolism to extend their apparent half-lives, and enhance drug stability by keeping it in solid form within the formulation. The use of aqueous buffers to produce drug nanocrystals also minimizes solvent related toxicities in the final product. The success of this technology in HIV therapy is highlighted by the LA injcetable Cabenuva (CAB and RPV LA) and Apretude (CAB LA) which are currently approved for treatment and prevention, respectively. LA CAB consists of pure CAB nanocrystals with an average particle size of 200 nm dispersed in a stabilizing aquoeus buffer solution of Tween 20, PEG3350, and mannitol [266]. For LA RPV nanocrystals, Poloxamer 338 (P338) is used as the stabilizing surfactant [39].
Upon IM injections, these nanocrystals form a drug depot at the injection site that slowly dissociates to release the drug for months. Drug release and absorption in biological matrices is dependent on several factors such as nanoparticle agglomeration or dispersion at the site of injection site, particle size and surface properties that affect host protein adsorption to the particle surface to facilitate absorption [266]. The longevity of the depot is heavily influenced by the hydrophobicity of the formulated compounds. However, to ensure sufficient dissolution of the nanocrystals to the surrounding medium and eventually demonstrate the effective plasma and tissue drug levels, the hydrophobicity of the formulated compound needs to be optimal. The particle size, in addition to the hydrophobicity, also influences the dissolution properties. With a higher surface area, the nano-sized crystal improves the dissolution properties of the drugs, helping maintain a sufficient concentration for efficacy. The benefit of smaller particle size is also evident in the improved syringeability of the formulation [169,266].

Table 1.
A comprehensive summary of CAB and RPV based LA delivery system for the treatment and prevention of HIV-1 infection.
Table 1.
A comprehensive summary of CAB and RPV based LA delivery system for the treatment and prevention of HIV-1 infection.
| Drugs | (CAB + RPV LA) | CAB LA |
| Brand name | Cabenuva | Apretude or Vocabria |
| Formulation characteristics | CAB nanocrystals are produced in an aqueous solution of polysorbate 20 (Tween 20), PEG3350, and mannitol with particle size of 200 nm.RPV nanocrystals are produced in an aqueous solution of Poloxamer 338 with an average particle size of 200 nm | Same as Cabenuva. |
| Indication | Treatment of HIV-1 infection | Prevention of HIV-1 infection |
| Population | Virologically suppressed PLWH | Adults and adolescents weighing at least 35 kg (77 lbs) who are at risk of sexually acquiring HIV |
| Dosage regimen | Two initial injection of CAB 600 mg/ 3 mL and RPV 900 mg/ 3 mL given 1 month apart for two consecutive months and then given every two months (CAB 600 mg/ 3 mL and 900 mg/ 3 mL) thereafter. Injection is given in separate gluteal muscles. | Two initial injections (600 mg; 3 mL) given one month apart for two consecutive months, followed by maintenance doses (600 mg; 3 mL) given every 2 months thereafter. An OLI of CAB tablets may, or may not, be given for one month prior to starting CAB to assess tolerability. |
| Approval | First approved by Health Canada in March 2020, followed by the EMA in October of 2020, and the US FDA in January of 2021. | First approved by the US FDA in December of 2021 and the EMA in October of 2022. |
| Efficacy in the clinical trials | Several phase IIb and phase III clinical trials (LATTE-2, FLAIR, ATLAS, ATLAS-2M) proved non-inferiority for maintaining viral suppression compared to standard daily oral therapy [11,14,16,267]. | Proved superior than approved oral PrEP agent TDF/FTC in 2 phase III clinical trials (HPTN-083 and 084) [268,269]. |
| Side effects | Mild ISRs were the most reported AEs during clinical trials, but none of them caused treatment withdrawal. | Same as Cabenuva. |
| PLWH – people living with HIV. EMA – European Medicines Agency. US FDA – US Food and Drug Administration. OLI – oral lead in. AEs – adverse events. ISRs – injection site reactions. | ||
To further extend ARVs dosage regimen, a prodrug strategy that is coined ‘Long-Acting Slow Effective Anti-retroviral Therapy (LASER ART),” was developed. The strategy relies on controlled drug release from a tissue macrophage depot. LASER ART was applied to all classes of ARVs with the goal of improving their physicochemical properties to facilitate their formulation into scalable LA agents with improved apparent half-lives. Some are currently in advanced preclinical development with the potential to improve the HIV treatment dosage regimen by up to 6 months or longer [6].
Prodrug nanocrystals of a stearoylated CAB were shown to sustain CAB concentrations for up to one year in rodent and non-human primate (NHP) models [17]. A similar modification of DTG significantly improved the PK profile of the parent drug [6]. However, esterification of CAB and DTG with either shorter or longer than 18-carbon fatty acid failed to maintain plasma CAB and DTG levels beyond two months [6,270,271]. The prodrug nanocrystal strategy was extended to NRTIs where FTC, 3TC, and TFV were converted into hydrophobic ProTide prodrugs with improved PK profiles [272,273,274]. Notably, hydrophobic stable TFV ProTide nanocrystals demonstrated effective TFV-diphosphate levels in a two months PK study in rats after a single IM injection [272]. This formulation demonstrated anti-HBV activity in both HBV-infected transgenic and human hepatocyte-transplanted TK-NOG mice, for at least three months [275,276].
Research across a broad spectrum of hydrophobic ARV prodrugs sheds light on the correlation between prodrug hydrophobicity and drug release. The prolonged PK profile observed in the INSTI-esterified prodrugs is influenced by multiple factors that contribute to the observed extended apparent half-lives. These influencing factors encompass i) the physiochemical attributes of the prodrug, ii) the chemical and enzymatic conversion of the prodrug to the parent drug, and iii) the absorption of the prodrug and parent drug by the lymphatic systems and tissues (Figure 7). Yet, there's a noticeable gap in research dedicated to understanding the role of polymer/surfactants in drug release and how they interact with cells that undergo mononuclear phagocytosis. Many of these formulations require hydrophilic low MW polymers like poloxamer or a combination of PEG and polysorbate. This ensures that the particles remain stable in aqueous suspensions. One crucial facet of nanocrystal formulations is their uptake by phagocytic cells, like macrophages. The nature of the polymer in the formulation affects how macrophages interact with the nanoparticle and how stable the nanoparticle remains within phagocytosed cells. Numerous studies highlight the stability of these nanoparticles within macrophages, pointing to the creation of a depot within these cells.. Comprehensive research focusing on prodrug nanoparticle size, charge, polymer/surfactant choices, and their effects on macrophage interactions is crucial. Deodhar et al. demonstrate improved stability of surfactant-coated nanoformulated DTG prodrugs in various environments like acidic pH, macrophages, plasma, and various tissues, compared to its counterpart in a free prodrug solution [6]. Nevertheless, the foundational mechanism promoting the heightened stability of the nanoparticle calls for further exploration. Furthermore, various challenges are associated with this technology when the native parent is used in the formulation, including an initial burst release, a prolonged PK tail, and population-level PK variability [266,277]. This variability in CAB LA’s PK is also evident between genders. Some females exhibit a smoothed concentration-time curve, presenting a lower peak drug concentration (Cmax) and a prolonged period to reach this Cmax compared to males. However, both genders show comparable overall drug exposure (AUC[0-∞]) following a single LA dose of CAB. This suggests that the absorption rate might differ between genders, rather than the total absorption [278]. Additionally, the terminal phase PK tail for CAB LA shows variations between male and female patients.
6. Conclusion and Future Perspective of la Drug Delivery Systems
The prospects for polymer-based LA formulations are bright, given our rapidly expanding knowledge of polymer properties and drug release mechanisms. While current LA drugs for HIV offer improvement over traditional oral HIV medications, there are user and treatment related gaps that could be addressed by the emerging LA drug delivery technologies such as prodrug approaches and implants. Ideally, the future LA ART medicines should possess three main characteristics.
First, they should be easy to manufacture, scalable, and user-friendly, with a dosing schedule extended at least beyond two months to synchronize dosing with the existing routine patient laboratory test clinic visits. A desirable target product profile is therefore a six-month ART dosage regimen. However, achieving a six-month dosage regimen with the exixting parent drugs is challenging due to their inherent features that include rapid drug metabolism upon absorption from the implant/injection site depot. The constraints on the allowable volume for injections or the size of implantable products, especially for SC or IM delivery, further intensify this challenge. Sometimes, the required drug amount surpasses these permissible limits. A potential solution lies in choosing drugs that are highly potent and have a high resistance to mutations. Prodrug approaches could also be used to achieve this goal by improving intracellular drug delivery and absorption, distribution, metabolism, and excretion (ADME) properties of parent drugs to extend their half-lives. When identifying the optimal drug for LA formulation development, the co-existing medical conditions in patients should also be considered. Given that a significant number of HIV patients (~ 7.5%) are co-infected with hepatitis B or tuberculosis, LA drugs that can concurrently target multiple viruses are especially valuable [279].
The second desired feature of LA formulations is to minimize post-market complications. This includes curtailing prolonged PK tail in the terminal phase of drug release, inhibiting the emergence of drug-resistant mutations, and ensuring that the treatment can be reversed, especially for newer drugs with no established safety profiles, if adverse reactions occur. For example, the FDA-approved CAB LA exhibits a long PK tail after the last injection [280]. The CDC recommends that those at risk of HIV continue with oral CAB or PrEP for at least a year after discontinuing CAB LA. Yet, making the shift from LA drugs to oral PrEP isn't always straightforward, as many prefer LA treatments over daily oral ART. There are also concerns about how easily LA treatments can be reversed in the event of complications. These have been circumvented through oral lead-ins prior to administering LA injectable agents. Solid LA implantable devices might offer an alterantive reversal solution, but the need for surgical removal post-lifespan dilutes this advantage.
The third desired feature of LA formulations is their widespread adoption and utilization by the end users. Factors like cost, storage stability, transportation requirements, and specific storage conditions can significantly influence the acceptance and use of LA medicines. Given that HIV is most common in resource-limited settings with limited financial capacity for medication, inadequate cold storage infrastructure, and a scarcity of medical professionals, an LA medicine that is affordable, stable at room temperature, and self-administrable would greatly boost its adoption and application.
Currently, no LA technology, whether in clinical or preclinical development, embodies all three of these ideal features, suggesting that a "one size fits all" approach to LA therapy is inappropriate. Given this, there's a push to tailor LA medicines to the unique needs of specific sub-groups. Understanding the interactions between the polymers, drugs, and their release mechanisms will enable more precise tuning of drug release rates from these formulations. Additionally, to ensure the clinical success of LA ARVs in such resource-limited settings, it is imperative to involve all stakeholders in the early phase of formulation development. These stakeholders include end-users, healthcare professionals, business developers, and policymakers. Collaboration, careful planning, and implementation consideration are essential to prioritize and design the most promising ARVs and LA DDSs that facilitate effective implementation in the most HIV-burdened settings. Moreover, the development of LA ARVs may present regulatory challenges, such as establishing optimal dosing regimens to address the need for combination antiretroviral therapy, and managing missed doses and potential drug-drug interactions in patients with co-morbidities. Some of these challenges are particularly important for PrEP dosing schedules, as there is no precise biomarker to gauge outcomes. In such situations, conducting thorough in-vivo PK studies and evaluating PrEP effectiveness in preclinical animal models becomes imperative. Nonetheless, selecting an appropriate animal model to determine PrEP efficacy remains a significant challenge in preclinical evaluation, given that none of the existing models can fully replicate the human transmission and immunogenicity of HIV infection. Additionally, since LA ARVs can persist in the body for extended periods, ensuring the long-term safety of both the active ingredients and excipients used in LA ARVs in preclinical in-vivo models is of significance. While the excipients used in LA ARVs are generally recognized as safe (GRAS), extensive GLP toxicology studies in relevant models are needed to broaden the usage in special populations such as pediatric patients. The requirement for extensive, extended safety study timelines introduces further challenges, including increased overall product development costs and time. In this context, the use of physiologically based pharmacokinetic (PBPK) modeling can be a valuable tool for predicting drug PK. This approach can also streamline formulation screening and expedite the development of LA products.
Overall, advancements in polymer chemistry and insights into their drug release mechanisms have paved the way for promising strategies to extend HIV-1 treatment dosage regimens beyond two months. Nevertheless, there are developmental and translational hurdles that must be overcome for these products to successfully transition to clinical application.
Author Contributions
Conceptualization, B.E and H.E.G; literature collection and analysis M.U.N, S.P, A.S, L.A, L.K.V., and B.S..; figure preparation, M.U.N and S.P.; writing—original draft preparation, M.U.N, S.P, A.S, L.A, L.K.V. and B.S.; writing—review and editing, B.E and H.E.G.; funding acquisition, B.E. and H.E.G. All authors have read and agreed to the published version of the manuscript.
Funding
The work is supported by National Institutes of Health grants R01 NS034239; R01 NS036126; R01 MH115860; R01 NS126089 (HEG); R01 AI145542 (BE and HEG); R01 AI158160 (HEG and BE).”.
References
- Benhabbour, S.R.; Kovarova, M.; Jones, C.; Copeland, D.J.; Shrivastava, R.; Swanson, M.D.; Sykes, C.; Ho, P.T.; Cottrell, M.L.; Sridharan, A. Ultra-long-acting tunable biodegradable and removable controlled release implants for drug delivery. Nature communications 2019, 10, 4324. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.M.; Krovi, S.A.; Li, L.; Girouard, N.; Demkovich, Z.R.; Myers, D.; Creelman, B.; van der Straten, A. Characterization of a Reservoir-Style Implant for Sustained Release of Tenofovir Alafenamide (TAF) for HIV Pre-Exposure Prophylaxis (PrEP). Pharmaceutics 2019, 11, 315. [Google Scholar] [CrossRef] [PubMed]
- Gunawardana, M.; Remedios-Chan, M.; Miller, C.S.; Fanter, R.; Yang, F.; Marzinke, M.A.; Hendrix, C.W.; Beliveau, M.; Moss, J.A.; Smith, T.J. Pharmacokinetics of long-acting tenofovir alafenamide (GS-7340) subdermal implant for HIV prophylaxis. Antimicrobial agents and chemotherapy 2015, 59, 3913–3919. [Google Scholar] [CrossRef] [PubMed]
- Tekko, I.A.; Vora, L.K.; Volpe-Zanutto, F.; Moffatt, K.; Jarrahian, C.; McCarthy, H.O.; Donnelly, R.F. Novel Bilayer Microarray Patch-Assisted Long-Acting Micro-Depot Cabotegravir Intradermal Delivery for HIV Pre-Exposure Prophylaxis. Advanced Functional Materials 2022, 32, 2106999. [Google Scholar] [CrossRef]
- Barrett, S.E.; Teller, R.S.; Forster, S.P.; Li, L.; Mackey, M.A.; Skomski, D.; Yang, Z.; Fillgrove, K.L.; Doto, G.J.; Wood, S.L.; et al. Extended-Duration MK-8591-Eluting Implant as a Candidate for HIV Treatment and Prevention. Antimicrobial Agents and Chemotherapy 2018, 62. [Google Scholar] [CrossRef]
- Deodhar, S.; Sillman, B.; Bade, A.N.; Avedissian, S.N.; Podany, A.T.; McMillan, J.M.; Gautam, N.; Hanson, B.; Dyavar Shetty, B.L.; Szlachetka, A. Transformation of dolutegravir into an ultra-long-acting parenteral prodrug formulation. Nature communications 2022, 13, 3226. [Google Scholar] [CrossRef]
- Malcolm, R.K.; Fetherston, S.M.; McCoy, C.F.; Boyd, P.; Major, I. Vaginal rings for delivery of HIV microbicides. Int J Womens Health 2012, 4, 595–605. [Google Scholar] [CrossRef] [PubMed]
- Deanesly, R.; Parkes, A.S. Testosterone. British Medical Journal 1936, 1, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Paolini, M.S.; Fenton, O.S.; Bhattacharya, C.; Andresen, J.L.; Langer, R. Polymers for extended-release administration. Biomedical Microdevices 2019, 21, 45. [Google Scholar] [CrossRef]
- Vora, L.K.; Moffatt, K.; Tekko, I.A.; Paredes, A.J.; Volpe-Zanutto, F.; Mishra, D.; Peng, K.; Raj Singh Thakur, R.; Donnelly, R.F. Microneedle array systems for long-acting drug delivery. European Journal of Pharmaceutics and Biopharmaceutics 2021, 159, 44–76. [Google Scholar] [CrossRef]
- Swindells, S.; Andrade-Villanueva, J.-F.; Richmond, G.J.; Rizzardini, G.; Baumgarten, A.; Masiá, M.; Latiff, G.; Pokrovsky, V.; Bredeek, F.; Smith, G. Long-acting cabotegravir and rilpivirine for maintenance of HIV-1 suppression. New England Journal of Medicine 2020, 382, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.H.R.; Henry, W.K.; Podzamczer, D.; Masiá, M.D.M.; Bettacchi, C.J.; Arasteh, K.; Jaeger, H.; Khuong-Josses, M.A.; Montes-Ramírez, M.L.; Stellbrink, H.J.; et al. Efficacy, Safety, and Durability of Long-Acting Cabotegravir and Rilpivirine in Adults With Human Immunodeficiency Virus Type 1 Infection: 5-Year Results From the LATTE-2 Study. Open Forum Infect Dis 2021, 8, ofab439. [Google Scholar] [CrossRef] [PubMed]
- Parikh, U.M.; Koss, C.A.; Mellors, J.W. Long-Acting Injectable Cabotegravir for HIV Prevention: What Do We Know and Need to Know about the Risks and Consequences of Cabotegravir Resistance? Curr HIV/AIDS Rep 2022, 19, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Margolis, D.A.; Gonzalez-Garcia, J.; Stellbrink, H.-J.; Eron, J.J.; Yazdanpanah, Y.; Podzamczer, D.; Lutz, T.; Angel, J.B.; Richmond, G.J.; Clotet, B. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. The Lancet 2017, 390, 1499–1510. [Google Scholar] [CrossRef] [PubMed]
- Orkin, C.; Oka, S.; Philibert, P.; Brinson, C.; Bassa, A.; Gusev, D.; Degen, O.; García, J.G.; Morell, E.B.; Tan, D.H.S.; et al. Long-acting cabotegravir plus rilpivirine for treatment in adults with HIV-1 infection: 96-week results of the randomised, open-label, phase 3 FLAIR study. Lancet HIV 2021, 8, e185–e196. [Google Scholar] [CrossRef] [PubMed]
- Orkin, C.; Arasteh, K.; Górgolas Hernández-Mora, M.; Pokrovsky, V.; Overton, E.T.; Girard, P.-M.; Oka, S.; Walmsley, S.; Bettacchi, C.; Brinson, C. Long-acting cabotegravir and rilpivirine after oral induction for HIV-1 infection. New England Journal of Medicine 2020, 382, 1124–1135. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, T.A.; Bade, A.N.; Sillman, B.; Shetty, B.L.D.; Wojtkiewicz, M.S.; Gautam, N.; Hilaire, J.R.; Sravanam, S.; Szlachetka, A.; Lamberty, B.G. A year-long extended release nanoformulated cabotegravir prodrug. Nature materials 2020, 19, 910–920. [Google Scholar] [CrossRef] [PubMed]
- Young, I.C.; Massud, I.; Cottrell, M.L.; Shrivastava, R.; Maturavongsadit, P.; Prasher, A.; Wong-Sam, A.; Dinh, C.; Edwards, T.; Mrotz, V.; et al. Ultra-long-acting in-situ forming implants with cabotegravir protect female macaques against rectal SHIV infection. Nature Communications 2023, 14, 708. [Google Scholar] [CrossRef] [PubMed]
- Chua, C.Y.X.; Jain, P.; Ballerini, A.; Bruno, G.; Hood, R.L.; Gupte, M.; Gao, S.; Di Trani, N.; Susnjar, A.; Shelton, K.; et al. Transcutaneously refillable nanofluidic implant achieves sustained level of tenofovir diphosphate for HIV pre-exposure prophylaxis. J Control Release 2018, 286, 315–325. [Google Scholar] [CrossRef]
- Simpson, S.M.; Widanapathirana, L.; Su, J.T.; Sung, S.; Watrous, D.; Qiu, J.; Pearson, E.; Evanoff, A.; Karunakaran, D.; Chacon, J.E.; et al. Design of a Drug-Eluting Subcutaneous Implant of the Antiretroviral Tenofovir Alafenamide Fumarate. Pharmaceutical Research 2020, 37, 83. [Google Scholar] [CrossRef]
- Markowitz, M.; Grobler, J.A. Islatravir for the treatment and prevention of infection with the human immunodeficiency virus type 1. Current Opinion in HIV and AIDS 2020, 15, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Baeten, J.M.; Palanee-Phillips, T.; Mgodi, N.M.; Mayo, A.J.; Szydlo, D.W.; Ramjee, G.; Gati Mirembe, B.; Mhlanga, F.; Hunidzarira, P.; Mansoor, L.E.; et al. Safety, uptake, and use of a dapivirine vaginal ring for HIV-1 prevention in African women (HOPE): an open-label, extension study. Lancet HIV 2021, 8, e87–e95. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.J.; Wood, L.; Billingsley, J.M.; Ray, L.L.; Goymer, J.; Sinclair, S.; McGinn, A.P.; Marzinke, M.A.; Frank, B.; Srinivasan, S.; et al. Tenofovir disoproxil fumarate intravaginal ring for HIV pre-exposure prophylaxis in sexually active women: a phase 1, single-blind, randomised, controlled trial. Lancet HIV 2019, 6, e498–e508. [Google Scholar] [CrossRef] [PubMed]
- Boyd, P.; Variano, B.; Spence, P.; McCoy, C.F.; Murphy, D.J.; Dallal Bashi, Y.H.; Malcolm, R.K. In vitro release testing methods for drug-releasing vaginal rings. Journal of Controlled Release 2019, 313, 54–69. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, E.T.; van der Straten, A.; Chitukuta, M.; Reddy, K.; Woeber, K.; Atujuna, M.; Bekker, L.G.; Etima, J.; Nakyanzi, T.; Mayo, A.J.; et al. Acceptability and use of a dapivirine vaginal ring in a phase III trial. Aids 2017, 31, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.; van Niekerk, N.; Kapiga, S.; Bekker, L.-G.; Gama, C.; Gill, K.; Kamali, A.; Kotze, P.; Louw, C.; Mabude, Z.; et al. Safety and Efficacy of a Dapivirine Vaginal Ring for HIV Prevention in Women. New England Journal of Medicine 2016, 375, 2133–2143. [Google Scholar] [CrossRef] [PubMed]
- Mc Crudden, M.T.C.; Larrañeta, E.; Clark, A.; Jarrahian, C.; Rein-Weston, A.; Lachau-Durand, S.; Niemeijer, N.; Williams, P.; Haeck, C.; McCarthy, H.O.; et al. Design, formulation and evaluation of novel dissolving microarray patches containing a long-acting rilpivirine nanosuspension. Journal of Controlled Release 2018, 292, 119–129. [Google Scholar] [CrossRef] [PubMed]
- Deanesly, R.; Parkes, A.S. Biological properties of some new derivatives of testosterone. Biochemical Journal 1937, 31, 1161. [Google Scholar] [CrossRef] [PubMed]
- Chien, Y.W. Novel drug delivery systems. Drugs and the pharmaceutical sciences 1992, 50. [Google Scholar]
- Dreyfuss, J.; Ross Jr, J.J.; Shaw, J.M.; Miller, I.; Schreiber, E.C. Release and elimination of 14C-fluphenazine enanthate and decanoate esters administered in sesame oil to dogs. Journal of pharmaceutical sciences 1976, 65, 502–507. [Google Scholar] [CrossRef]
- Wright, J.C.; Hoffman, A.S. Historical overview of long acting injections and implants. In Long acting injections and implants; Springer, 2011; pp. 11–24. [Google Scholar]
- Higuchi, T. Mechanism of sustained-action medication. Theoretical analysis of rate of release of solid drugs dispersed in solid matrices. Journal of pharmaceutical sciences 1963, 52, 1145–1149. [Google Scholar] [CrossRef]
- Folkman, J.; Long Jr, D.M.; Rosenbaum, R. Silicone rubber: a new diffusion property useful for general anesthesia. Science 1966, 154, 148–149. [Google Scholar] [CrossRef] [PubMed]
- Heilmann, K. Therapeutic systems: rate-controlled drug delivery: concept and development; 1984.
- Frazza, E.; Schmitt, E. A new absorbable suture. Journal of biomedical materials research 1971, 5, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Kent, J.S.; Lewis, D.H.; Sanders, L.M.; Tice, T.R. Microencapsulation of water soluble active polypeptides. 1987.
- Hoffman, A.S. The origins and evolution of “controlled” drug delivery systems. Journal of controlled release 2008, 132, 153–163. [Google Scholar] [CrossRef]
- Nowacek, A.S.; Miller, R.L.; McMillan, J.; Kanmogne, G.; Kanmogne, M.; Mosley, R.L.; Ma, Z.; Graham, S.; Chaubal, M.; Werling, J.; et al. NanoART synthesis, characterization, uptake, release and toxicology for human monocyte-macrophage drug delivery. Nanomedicine (Lond) 2009, 4, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Baert, L.; van‘t Klooster, G.; Dries, W.; François, M.; Wouters, A.; Basstanie, E.; Iterbeke, K.; Stappers, F.; Stevens, P.; Schueller, L. Development of a long-acting injectable formulation with nanoparticles of rilpivirine (TMC278) for HIV treatment. European Journal of Pharmaceutics and Biopharmaceutics 2009, 72, 502–508. [Google Scholar] [CrossRef]
- Penrose, K.J.; Parikh, U.M.; Hamanishi, K.A.; Else, L.; Back, D.; Boffito, M.; Jackson, A.; Mellors, J.W. Selection of rilpivirine-resistant HIV-1 in a seroconverter from the SSAT 040 trial who received the 300-mg dose of long-acting rilpivirine (TMC278LA). The Journal of infectious diseases 2016, 213, 1013–1017. [Google Scholar] [CrossRef]
- Venkatesan, P. Long-acting injectable ART for HIV: a (cautious) step forward. Lancet Microbe 2022, 3, e94. [Google Scholar] [CrossRef]
- Langer, R. New methods of drug delivery. Science 1990, 249, 1527–1533. [Google Scholar] [CrossRef]
- Rahnfeld, L.; Luciani, P. Injectable lipid-based depot formulations: where do we stand? Pharmaceutics 2020, 12, 567. [Google Scholar] [CrossRef]
- U.S. Pharmacopeia, N.F.; Effective date, pp. 1–36.
- Fredenberg, S.; Wahlgren, M.; Reslow, M.; Axelsson, A. The mechanisms of drug release in poly (lactic-co-glycolic acid)-based drug delivery systems—A review. International journal of pharmaceutics 2011, 415, 34–52. [Google Scholar] [CrossRef] [PubMed]
- Berchane, N.; Carson, K.; Rice-Ficht, A.; Andrews, M. Effect of mean diameter and polydispersity of PLG microspheres on drug release: Experiment and theory. International Journal of Pharmaceutics 2007, 337, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Kao, W.J. Drug release kinetics and transport mechanisms of non-degradable and degradable polymeric delivery systems. Expert opinion on drug delivery 2010, 7, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Liechty, W.B.; Kryscio, D.R.; Slaughter, B.V.; Peppas, N.A. Polymers for drug delivery systems. Annual review of chemical and biomolecular engineering 2010, 1, 149–173. [Google Scholar] [CrossRef] [PubMed]
- Webber, W.L.; Lago, F.; Thanos, C.; Mathiowitz, E. Characterization of soluble, salt-loaded, degradable PLGA films and their release of tetracycline. Journal of Biomedical Materials Research: An Official Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and the Australian Society for Biomaterials 1998, 41, 18–29. [Google Scholar] [CrossRef]
- Marin, E.; Briceño, M.I.; Caballero-George, C. Critical evaluation of biodegradable polymers used in nanodrugs. International journal of nanomedicine 2013, 3071–3091. [Google Scholar]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric systems for controlled drug release. Chemical reviews 1999, 99, 3181–3198. [Google Scholar] [CrossRef] [PubMed]
- Maroni, A.; Zema, L.; Cerea, M.; Foppoli, A.; Palugan, L.; Gazzaniga, A. Erodible drug delivery systems for time-controlled release into the gastrointestinal tract. Journal of Drug Delivery Science and Technology 2016, 32, 229–235. [Google Scholar] [CrossRef]
- Keraliya, R.A.; Patel, C.; Patel, P.; Keraliya, V.; Soni, T.G.; Patel, R.C.; Patel, M. Osmotic drug delivery system as a part of modified release dosage form. International Scholarly Research Notices 2012, 2012. [Google Scholar] [CrossRef]
- Huang, X.; Brazel, C.S. On the importance and mechanisms of burst release in matrix-controlled drug delivery systems. Journal of controlled release 2001, 73, 121–136. [Google Scholar] [CrossRef]
- Makadia, H.K.; Siegel, S.J. Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 2011, 3, 1377–1397. [Google Scholar] [CrossRef]
- Saltzman, W.M. Drug delivery: engineering principles for drug therapy. Oxford University Press, 2001. [Google Scholar]
- Kim, J.M.; Seo, K.S.; Jeong, Y.K.; Lee, H.B.; Kim, Y.S.; Khang, G. Co-effect of aqueous solubility of drugs and glycolide monomer on in vitro release rates from poly (D, L-lactide-co-glycolide) discs and polymer degradation. Journal of Biomaterials Science, Polymer Edition 2005, 16, 991–1007. [Google Scholar] [CrossRef]
- Lu, L.; Peter, S.J.; Lyman, M.D.; Lai, H.-L.; Leite, S.M.; Tamada, J.A.; Uyama, S.; Vacanti, J.P.; Langer, R.; Mikos, A.G. In vitro and in vivo degradation of porous poly (DL-lactic-co-glycolic acid) foams. Biomaterials 2000, 21, 1837–1845. [Google Scholar] [CrossRef]
- Ehrenstein, G.W. Polymeric materials: structure, properties, applications. Carl Hanser Verlag GmbH Co KG, 2012. [Google Scholar]
- Odian, G. Principles of polymerization. John Wiley & Sons, 2004. [Google Scholar]
- Allcock, H.R.; Lampe, F.W.; Mark, J.E.; Allcock, H. Contemporary polymer chemistry. Prentice-Hall Englewood Cliffs: NJ, USA, 1981. [Google Scholar]
- Muller, W.E.; Wang, X.; Schroder, H. Biomedical Inorganic Polymers; Springer, 2016. [Google Scholar]
- Ginjupalli, K.; Shavi, G.V.; Averineni, R.K.; Bhat, M.; Udupa, N.; Upadhya, P.N. Poly (α-hydroxy acid) based polymers: A review on material and degradation aspects. Polymer Degradation and Stability 2017, 144, 520–535. [Google Scholar] [CrossRef]
- Nair, L.S.; Laurencin, C.T. Biodegradable polymers as biomaterials. Progress in polymer science 2007, 32, 762–798. [Google Scholar] [CrossRef]
- Dash, A.; Cudworth II, G. Therapeutic applications of implantable drug delivery systems. Journal of pharmacological and toxicological methods 1998, 40, 1–12. [Google Scholar] [CrossRef]
- Gan, M.; Zhou, Q.; Ge, J.; Zhao, J.; Wang, Y.; Yan, Q.; Wu, C.; Yu, H.; Xiao, Q.; Wang, W. Precise in-situ release of microRNA from an injectable hydrogel induces bone regeneration. Acta Biomaterialia 2021, 135, 289–303. [Google Scholar] [CrossRef]
- Rahme, K.; Dagher, N. Chemistry Routes for Copolymer Synthesis Containing PEG for Targeting, Imaging, and Drug Delivery Purposes. Pharmaceutics 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Cano, C.; Carril, M. Recent Developments in the Design of Non-Biofouling Coatings for Nanoparticles and Surfaces. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Dirisala, A.; Uchida, S.; Toh, K.; Li, J.; Osawa, S.; Tockary, T.A.; Liu, X.; Abbasi, S.; Hayashi, K.; Mochida, Y. Transient stealth coating of liver sinusoidal wall by anchoring two-armed PEG for retargeting nanomedicines. Science Advances 2020, 6, eabb8133. [Google Scholar] [CrossRef]
- Zhang, D.; Zheng, H.; Geng, K.; Shen, J.; Feng, X.; Xu, P.; Duan, Y.; Li, Y.; Wu, R.; Gou, Z. Large fuzzy biodegradable polyester microspheres with dopamine deposition enhance cell adhesion and bone regeneration in vivo. Biomaterials 2021, 272, 120783. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, K.; Hussain, I.; Oderinde, O.; Yao, F.; Zhang, J.; Fu, G. A Conductive Self-Healing Double Network Hydrogel with Toughness and Force Sensitivity. Chemistry–A European Journal 2018, 24, 6632–6638. [Google Scholar] [CrossRef] [PubMed]
- Apostolides, D.E.; Patrickios, C.S.; Sakai, T.; Guerre, M.; Lopez, G.r.; Améduri, B.; Ladmiral, V.; Simon, M.; Gradzielski, M.; Clemens, D. Near-model amphiphilic polymer conetworks based on four-arm stars of poly (vinylidene fluoride) and poly (ethylene glycol): Synthesis and characterization. Macromolecules 2018, 51, 2476–2488. [Google Scholar] [CrossRef]
- Xue, S.; Li, X.; Li, S.; Chen, N.; Zhan, Q.; Long, L.; Zhao, J.; Hou, X.; Yuan, X. Bone fracture microenvironment responsive hydrogel for timing sequential release of cargoes. Colloids and Surfaces A: Physicochemical and Engineering Aspects 2021, 629, 127413. [Google Scholar] [CrossRef]
- Le Fer, G.; Dilla, R.A.; Wang, Z.; King, J.; Chuang, S.S.; Becker, M.L. Clustering and Hierarchical Organization of 3D Printed Poly (propylene fumarate)-block-PEG-block-poly (propylene fumarate) ABA Triblock Copolymer Hydrogels. Macromolecules 2021, 54, 3458–3468. [Google Scholar] [CrossRef]
- Roy, D.; Cambre, J.N.; Sumerlin, B.S. Future perspectives and recent advances in stimuli-responsive materials. Progress in Polymer Science 2010, 35, 278–301. [Google Scholar] [CrossRef]
- Alexandridis, P.; Zhou, D.; Khan, A. Lyotropic liquid crystallinity in amphiphilic block copolymers: temperature effects on phase behavior and structure for poly (ethylene oxide)-b-poly (propylene oxide)-b-poly (ethylene oxide) copolymers of different composition. Langmuir 1996, 12, 2690–2700. [Google Scholar] [CrossRef]
- Pozzo, D.C.; Hollabaugh, K.R.; Walker, L.M. Rheology and phase behavior of copolymer-templated nanocomposite materials. Journal of Rheology 2005, 49, 759–782. [Google Scholar] [CrossRef]
- Bodratti, A.M.; Alexandridis, P. Formulation of poloxamers for drug delivery. Journal of functional biomaterials 2018, 9, 11. [Google Scholar] [CrossRef]
- Zhang, K.; Shi, X.; Lin, X.; Yao, C.; Shen, L.; Feng, Y. Poloxamer-based in situ hydrogels for controlled delivery of hydrophilic macromolecules after intramuscular injection in rats. Drug delivery 2015, 22, 375–382. [Google Scholar] [CrossRef]
- Yang, L.; Alexandridis, P. Controlled Drug Delivery. 2000.
- Van Hemelryck, S.; Dewulf, J.; Niekus, H.; van Heerden, M.; Ingelse, B.; Holm, R.; Mannaert, E.; Langguth, P. In vitro evaluation of poloxamer in situ forming gels for bedaquiline fumarate salt and pharmacokinetics following intramuscular injection in rats. International journal of pharmaceutics: X 2019, 1, 100016. [Google Scholar] [CrossRef] [PubMed]
- Demarteau, J.; Kermagoret, A.; Jérôme, C.; Detrembleur, C.; Debuigne, A. Controlled Synthesis of Ethylene-Vinyl Acetate Based Copolymers by Organometallic Mediated Radical Polymerization. In Controlled Radical Polymerization: Materials; ACS Symposium Series; American Chemical Society: 2015; Volume 1188, pp. 47–61.
- Schneider, C.; Langer, R.; Loveday, D.; Hair, D. Applications of ethylene vinyl acetate copolymers (EVA) in drug delivery systems. Journal of Controlled Release 2017, 262, 284–295. [Google Scholar] [CrossRef]
- Arsac, A.; Carrot, C.; Guillet, J. Determination of primary relaxation temperatures and melting points of ethylene vinyl acetate copolymers. Journal of Thermal Analysis and Calorimetry 2000, 61, 681–685. [Google Scholar] [CrossRef]
- Wang, X.; Venkatraman, S.S.; Boey, F.Y.; Loo, J.S.; Tan, L.P. Controlled release of sirolimus from a multilayered PLGA stent matrix. Biomaterials 2006, 27, 5588–5595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lin, W.; Yang, G.; Chen, Q. Studies on the phase structure of ethylene-vinyl acetate copolymers by solid-state 1H and 13C NMR. Journal of Polymer Science Part B: Polymer Physics 2002, 40, 2199–2207. [Google Scholar] [CrossRef]
- Wang, L.; Fang, P.; Ye, C.; Feng, J. Solid-state NMR characterizations on phase structures and molecular dynamics of poly(ethylene-co-vinyl acetate). Journal of Polymer Science Part B: Polymer Physics 2006, 44, 2864–2879. [Google Scholar] [CrossRef]
- Almeida, A.; Possemiers, S.; Boone, M.N.; De Beer, T.; Quinten, T.; Van Hoorebeke, L.; Remon, J.P.; Vervaet, C. Ethylene vinyl acetate as matrix for oral sustained release dosage forms produced via hot-melt extrusion. European Journal of Pharmaceutics and Biopharmaceutics 2011, 77, 297–305. [Google Scholar] [CrossRef]
- McKeen, L.W. The effect of sterilization on plastics and elastomers; William Andrew, 2018. [Google Scholar]
- Gaaz, T.S.; Sulong, A.B.; Akhtar, M.N.; Kadhum, A.A.; Mohamad, A.B.; Al-Amiery, A.A. Properties and Applications of Polyvinyl Alcohol, Halloysite Nanotubes and Their Nanocomposites. Molecules 2015, 20, 22833–22847. [Google Scholar] [CrossRef]
- Song, S.I.; Kim, B.C. Characteristic rheological features of PVA solutions in water-containing solvents with different hydration states. Polymer 2004, 45, 2381–2386. [Google Scholar] [CrossRef]
- Marin, E.; Rojas, J.; Ciro, Y. A review of polyvinyl alcohol derivatives: promising materials for pharmaceutical and biomedical applications. African J Pharm Pharmacol 2014, 8, 674–684. [Google Scholar]
- Rowe, R.C.; Sheskey, P.; Quinn, M. Handbook of pharmaceutical excipients; Libros Digitales-Pharmaceutical Press: 2009.
- Dediu, V.; Busila, M.; Tucureanu, V.; Bucur, F.I.; Iliescu, F.S.; Brincoveanu, O.; Iliescu, C. Synthesis of ZnO/Au nanocomposite for antibacterial applications. Nanomaterials 2022, 12, 3832. [Google Scholar] [CrossRef] [PubMed]
- Fatema, U.K.; Rahman, M.M.; Islam, M.R.; Mollah, M.Y.A.; Susan, M.A.B.H. Silver/poly (vinyl alcohol) nanocomposite film prepared using water in oil microemulsion for antibacterial applications. Journal of colloid and interface science 2018, 514, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Meira, J.; Madeira, C.; Falcão-Reis, F.; Figueira, L. Sustained control from recurring non-infectious uveitic macular edema with 0.19 mg fluocinolone acetonide intravitreal implant–a case report. Ophthalmology and Therapy 2019, 8, 635–641. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Highley, C.B.; Yeh, Y.C.; Galarraga, J.H.; Uman, S.; Burdick, J.A. Three-dimensional extrusion bioprinting of single-and double-network hydrogels containing dynamic covalent crosslinks. Journal of Biomedical Materials Research Part A 2018, 106, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, J.; Liu, J.; Long, Y.; Fang, L.; Wang, Q.; Liu, T. Highly compressible and superior low temperature tolerant supercapacitors based on dual chemically crosslinked PVA hydrogel electrolytes. Journal of Materials Chemistry A 2020, 8, 6219–6228. [Google Scholar] [CrossRef]
- Chao, Y.; Chen, Q.; Liu, Z. Smart injectable hydrogels for cancer immunotherapy. Advanced Functional Materials 2020, 30, 1902785. [Google Scholar] [CrossRef]
- Lanman, T.; Martin, N.; Vinters, H. The pathology of encephalic arteriovenous malformations treated by prior embolotherapy. Neuroradiology 1988, 30, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hulman, G.; Kirkham, J. Ivalon Sponge Presenting as an Extrarectal Mass. In Proceedings of the Journal of Pathology; 1990; p. 171. [Google Scholar]
- Fassi, A.; Naidoo, N. Irritation associated with tear-replacement ophthalmic drops-a pharmaceutical and subjective investigation. South African Medical Journal 1989, 75, 233–235. [Google Scholar]
- Singh, B.; Varshney, L.; Francis, S. Designing tragacanth gum based sterile hydrogel by radiation method for use in drug delivery and wound dressing applications. International journal of biological macromolecules 2016, 88, 586–602. [Google Scholar] [CrossRef]
- Nasef, S.M.; Khozemy, E.E.; Kamoun, E.A.; El-Gendi, H. Gamma radiation-induced crosslinked composite membranes based on polyvinyl alcohol/chitosan/AgNO3/vitamin E for biomedical applications. International journal of biological macromolecules 2019, 137, 878–885. [Google Scholar] [CrossRef]
- Yuan, H.J.; Wei, Z.J.; Yu, X.Z. Study on Polyvinyl Alcohol Hydrogel Materials for Improving the Performance of Artificial Articular Cartilage. Advanced Materials Research 2013, 703, 29–32. [Google Scholar] [CrossRef]
- DeMerlis, C.; Schoneker, D. Review of the oral toxicity of polyvinyl alcohol (PVA). Food and chemical Toxicology 2003, 41, 319–326. [Google Scholar] [CrossRef]
- Rodwell, D.; Kelly, C.; DeMerlis, C.; Schoneker, D.; Borzelleca, J. Effects of polyvinyl alcohol administered in the diet to rats on fertility, early embryonic development, growth and development. Food and chemical toxicology 2003, 41, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.; DeMerlis, C.; Schoneker, D.; Borzelleca, J. Subchronic toxicity study in rats and genotoxicity tests with polyvinyl alcohol. Food and chemical toxicology 2003, 41, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Sun, W.; Tian, J.; Chen, L.; He, S.; Wang, J. Improvement of biodegradability of PVA-containing wastewater by ionizing radiation pretreatment. Environmental Science and Pollution Research 2012, 19, 3178–3184. [Google Scholar] [CrossRef]
- Chou, W.-L.; Wang, C.-T.; Huang, K.-Y. Investigation of process parameters for the removal of polyvinyl alcohol from aqueous solution by iron electrocoagulation. Desalination 2010, 251, 12–19. [Google Scholar] [CrossRef]
- Jabbari, E.; Khakpour, M. Morphology of and release behavior from porous polyurethane microspheres. Biomaterials 2000, 21, 2073–2079. [Google Scholar] [CrossRef]
- Kääriä, K.; Hirvonen, A.; Norppa, H.; Piirilä, P.; Vainio, H.; Rosenberg, C. Exposure to 4, 4′-methylenediphenyl diisocyanate (MDI) during moulding of rigid polyurethane foam: determination of airborne MDI and urinary 4, 4′-methylenedianiline (MDA). Analyst 2001, 126, 476–479. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Wei, K.; Zhao, N.; Zhang, S.; Chen, J. Drug distribution within poly (ɛ-caprolactone) microspheres and in vitro release. Journal of Materials Processing Technology 2009, 209, 348–354. [Google Scholar] [CrossRef]
- Thompson, D.G.; Osborn, J.C.; Kober, E.M.; Schoonover, J.R. Effects of hydrolysis-induced molecular weight changes on the phase separation of a polyester polyurethane. Polymer degradation and stability 2006, 91, 3360–3370. [Google Scholar] [CrossRef]
- Kaur, M.; Gupta, K.M.; Poursaid, A.E.; Karra, P.; Mahalingam, A.; Aliyar, H.A.; Kiser, P.F. Engineering a degradable polyurethane intravaginal ring for sustained delivery of dapivirine. Drug delivery and translational research 2011, 1, 223–237. [Google Scholar] [CrossRef] [PubMed]
- Christenson, E.M.; Dadsetan, M.; Wiggins, M.; Anderson, J.M.; Hiltner, A. Poly (carbonate urethane) and poly (ether urethane) biodegradation: in vivo studies. Journal of Biomedical Materials Research Part A: An Official Journal of The Society for Biomaterials, The Japanese Society for Biomaterials, and The Australian Society for Biomaterials and the Korean Society for Biomaterials 2004, 69, 407–416. [Google Scholar] [CrossRef] [PubMed]
- Rychlý, J.; Lattuati-Derieux, A.; Lavédrine, B.; Matisová-Rychlá, L.; Malíková, M.; Csomorová, K.; Janigová, I. Assessing the progress of degradation in polyurethanes by chemiluminescence and thermal analysis. II. Flexible polyether-and polyester-type polyurethane foams. Polymer degradation and stability 2011, 96, 462–469. [Google Scholar] [CrossRef]
- Arifin, D.Y.; Lee, L.Y.; Wang, C.-H. Mathematical modeling and simulation of drug release from microspheres: Implications to drug delivery systems. Advanced drug delivery reviews 2006, 58, 1274–1325. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.J.; Gupta, K.M.; Fabian, J.; Albright, T.H.; Kiser, P.F. Segmented polyurethane intravaginal rings for the sustained combined delivery of antiretroviral agents dapivirine and tenofovir. European Journal of Pharmaceutical Sciences 2010, 39, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Ritger, P.L.; Peppas, N.A. A simple equation for description of solute release II. Fickian and anomalous release from swellable devices. Journal of controlled release 1987, 5, 37–42. [Google Scholar] [CrossRef]
- Siepmann, J.; Siepmann, F. Mathematical modeling of drug delivery. International journal of pharmaceutics 2008, 364, 328–343. [Google Scholar] [CrossRef]
- Storey, R.F.; Hickey, T.P. Degradable polyurethane networks based on d, l-lactide, glycolide, ε-caprolactone, and trimethylene carbonate homopolyester and copolyester triols. Polymer 1994, 35, 830–838. [Google Scholar] [CrossRef]
- Guelcher, S.; Srinivasan, A.; Hafeman, A.; Gallagher, K.; Doctor, J.; Khetan, S.; McBride, S.; Hollinger, J. Synthesis, in vitro biocompatibility and biodegradation, and mechanical properties of two-component polyurethane scaffolds: effects of water and polyol composition. Tissue Eng 2007, 13, 2321–2333. [Google Scholar] [CrossRef]
- Labow, R.S.; Erfle, D.J.; Santerre, J.P. Elastase-induced hydrolysis of synthetic solid substrates: poly (ester-urea-urethane) and poly (ether-urea-urethane). Biomaterials 1996, 17, 2381–2388. [Google Scholar] [CrossRef]
- Ulery, B.D.; Nair, L.S.; Laurencin, C.T. Biomedical Applications of Biodegradable Polymers. J Polym Sci B Polym Phys 2011, 49, 832–864. [Google Scholar] [CrossRef]
- Feng, P.; Jia, J.; Liu, M.; Peng, S.; Zhao, Z.; Shuai, C. Degradation mechanisms and acceleration strategies of poly (lactic acid) scaffold for bone regeneration. Materials & Design 2021, 210, 110066. [Google Scholar] [CrossRef]
- Kuppermann, B.D.; Patel, S.S.; Boyer, D.S.; Augustin, A.J.; Freeman, W.R.; Kerr, K.J.; Guo, Q.; Schneider, S.; López, F.J. Phase 2 Stusy of the Safety and Efficacy of Brimonidine Drug Delivery System (BRIMO DDS) Generation 1 in Patients with Geographic Atrophy Secondary to Age-related Macular Degeneration. Retina 2021, 41, 144–155. [Google Scholar] [CrossRef]
- Pişkin, E. Biodegradable polymers as biomaterials. J Biomater Sci Polym Ed 1995, 6, 775–795. [Google Scholar] [CrossRef] [PubMed]
- Pisecky, L.; Luger, M.; Klasan, A.; Gotterbarm, T.; Klotz, M.C.; Hochgatterer, R. Bioabsorbable implants in forefoot surgery: a review of materials, possibilities and disadvantages. EFORT Open Rev 2021, 6, 1132–1139. [Google Scholar] [CrossRef]
- Wan, B.; Bao, Q.; Burgess, D. Long-acting PLGA microspheres: Advances in excipient and product analysis toward improved product understanding. Advanced Drug Delivery Reviews 2023, 198, 114857. [Google Scholar] [CrossRef] [PubMed]
- Blasi, P.; D'Souza, S.S.; Selmin, F.; DeLuca, P.P. Plasticizing effect of water on poly (lactide-co-glycolide). Journal of Controlled Release 2005, 108, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Chandra, R.; Rustgi, R. Biodegradable polymers. Progress in polymer science 1998, 23, 1273–1335. [Google Scholar] [CrossRef]
- Cayuela, J.; Bounor-Legaré, V.; Cassagnau, P.; Michel, A. Ring-opening polymerization of ε-caprolactone initiated with titanium n-propoxide or titanium phenoxide. Macromolecules 2006, 39, 1338–1346. [Google Scholar] [CrossRef]
- Natta, F.J.v.; Hill, J.W.; Carothers, W.H. Studies of polymerization and ring formation. XXIII. 1 ε-Caprolactone and its polymers. Journal of the American Chemical Society 1934, 56, 455–457. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Progress in polymer science 2010, 35, 1217–1256. [Google Scholar] [CrossRef]
- Sinha, V.; Bansal, K.; Kaushik, R.; Kumria, R.; Trehan, A. Poly-ϵ-caprolactone microspheres and nanospheres: an overview. International journal of pharmaceutics 2004, 278, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Lu, X.; Dalai, S.; Zhang, J. Thermophysical properties of polycaprolactone/chitosan blend membranes. Thermochimica Acta 2009, 487, 33–38. [Google Scholar] [CrossRef]
- Perez, M.H.; Zinutti, C.; Lamprecht, A.; Ubrich, N.; Astier, A.; Hoffman, M.; Bodmeier, R.; Maincent, P. The preparation and evaluation of poly (ϵ-caprolactone) microparticles containing both a lipophilic and a hydrophilic drug. Journal of controlled release 2000, 65, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Gorna, K.; Gogolewski, S. In vitro degradation of novel medical biodegradable aliphatic polyurethanes based on ϵ-caprolactone and Pluronics® with various hydrophilicities. Polymer Degradation and Stability 2002, 75, 113–122. [Google Scholar] [CrossRef]
- Ren, D.; Yi, H.; Wang, W.; Ma, X. The enzymatic degradation and swelling properties of chitosan matrices with different degrees of N-acetylation. Carbohydrate Research 2005, 340, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Ravi Kumar, M.N.V. A review of chitin and chitosan applications. Reactive and Functional Polymers 2000, 46, 1–27. [Google Scholar] [CrossRef]
- Mohammed, M.A.; Syeda, J.T.M.; Wasan, K.M.; Wasan, E.K. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Fonte, P.; Araújo, F.; Silva, C.; Pereira, C.; Reis, S.; Santos, H.A.; Sarmento, B. Polymer-based nanoparticles for oral insulin delivery: Revisited approaches. Biotechnology advances 2015, 33, 1342–1354. [Google Scholar] [CrossRef]
- Li, P.; Fujimoto, K.; Bourguingnon, L.; Yukl, S.; Deeks, S.; Wong, J.K. Exogenous and endogenous hyaluronic acid reduces HIV infection of CD4(+) T cells. Immunol Cell Biol 2014, 92, 770–780. [Google Scholar] [CrossRef]
- Agrahari, V.; Meng, J.; Ezoulin, M.J.; Youm, I.; Dim, D.C.; Molteni, A.; Hung, W.T.; Christenson, L.K.; Youan, B.C. Stimuli-sensitive thiolated hyaluronic acid based nanofibers: synthesis, preclinical safety and in vitro anti-HIV activity. Nanomedicine (Lond) 2016, 11, 2935–2958. [Google Scholar] [CrossRef]
- Jouyban, A.; Fakhree, M.A.A.; Shayanfar, A. Review of pharmaceutical applications of N-methyl-2-pyrrolidone. Journal of Pharmacy & Pharmaceutical Sciences 2010, 13, 524–535. [Google Scholar]
- Guide, M. Highlights of Prescribing Information.
- Rivera-Briso, A.L.; Serrano-Aroca, Á. Poly(3-Hydroxybutyrate-co-3-Hydroxyvalerate): Enhancement Strategies for Advanced Applications. Polymers (Basel) 2018, 10. [Google Scholar] [CrossRef]
- Avella, M.; Martuscelli, E.; Raimo, M. Review Properties of blends and composites based on poly (3-hydroxy) butyrate (PHB) and poly (3-hydroxybutyrate-hydroxyvalerate)(PHBV) copolymers. Journal of materials science 2000, 35, 523–545. [Google Scholar] [CrossRef]
- Mofokeng, J.; Luyt, A. Morphology and thermal degradation studies of melt-mixed poly (hydroxybutyrate-co-valerate)(PHBV)/poly (ε-caprolactone)(PCL) biodegradable polymer blend nanocomposites with TiO 2 as filler. Journal of Materials Science 2015, 50, 3812–3824. [Google Scholar] [CrossRef]
- Hedayati, A.; Yazdi, S.G.; Dehghankelishadi, P.; Javan, N.B.; Akbari, H.; Dorkoosh, F.A. Preparation, Optimization and Physicochemical Characterization of Aripiprazole Loaded Nano-porous in situ Forming Implant. Pharm Nanotechnol 2017, 5, 138–147. [Google Scholar] [CrossRef]
- Acemoglu, M.; Nimmerfall, F.; Bantle, S.; Stoll, G.H. Poly(ethylene carbonate)s, part I: Syntheses and structural effects on biodegradation. Journal of Controlled Release 1997, 49, 263–276. [Google Scholar] [CrossRef]
- Sasanuma, Y.; Takahashi, Y. Structure–Property Relationships of Poly(ethylene carbonate) and Poly(propylene carbonate). ACS Omega 2017, 2, 4808–4819. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.H.; Nimmerfall, F.; Acemoglu, M.; Bodmer, D.; Bantle, S.; Müller, I.; Mahl, A.; Kolopp, M.; Tullberg, K. Poly(ethylene carbonate)s, part II: degradation mechanisms and parenteral delivery of bioactive agents. J Control Release 2001, 76, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Kemmer, A.; Keim, K.; Curdy, C.; Petersen, H.; Kissel, T. Poly(ethylene carbonate) as a surface-eroding biomaterial for in situ forming parenteral drug delivery systems: A feasibility study. European Journal of Pharmaceutics and Biopharmaceutics 2010, 76, 222–229. [Google Scholar] [CrossRef]
- Panja, S.; Maji, S.; Maiti, T.K.; Chattopadhyay, S. A Smart Magnetically Active Nanovehicle for on-Demand Targeted Drug Delivery: Where van der Waals Force Balances the Magnetic Interaction. ACS Applied Materials & Interfaces 2015, 7, 24229–24241. [Google Scholar] [CrossRef]
- Panja, S.; Dey, G.; Bharti, R.; Mandal, P.; Mandal, M.; Chattopadhyay, S. Metal Ion Ornamented Ultrafast Light-Sensitive Nanogel for Potential in Vivo Cancer Therapy. Chemistry of Materials 2016, 28, 8598–8610. [Google Scholar] [CrossRef]
- Panja, S.; Dey, G.; Bharti, R.; Kumari, K.; Maiti, T.K.; Mandal, M.; Chattopadhyay, S. Tailor-Made Temperature-Sensitive Micelle for Targeted and On-Demand Release of Anticancer Drugs. ACS Applied Materials & Interfaces 2016, 8, 12063–12074. [Google Scholar] [CrossRef]
- Lan, T.; Guo, Q. Phenylboronic acid-decorated polymeric nanomaterials for advanced bio-application. Nanotechnology Reviews 2019, 8, 548–561. [Google Scholar] [CrossRef]
- Feng, R.; Zhu, L.; Teng, F.; Wang, M.; Chen, S.; Song, Z.; Li, H. Phenylboronic acid-modified polymaleic anhydride-F127 micelles for pH-activated targeting delivery of doxorubicin. Colloids and Surfaces B: Biointerfaces 2022, 216, 112559. [Google Scholar] [CrossRef]
- Mahalingam, A.; Jay, J.I.; Langheinrich, K.; Shukair, S.; McRaven, M.D.; Rohan, L.C.; Herold, B.C.; Hope, T.J.; Kiser, P.F. Inhibition of the transport of HIV in vitro using a pH-responsive synthetic mucin-like polymer system. Biomaterials 2011, 32, 8343–8355. [Google Scholar] [CrossRef]
- Jay, J.I.; Shukair, S.; Langheinrich, K.; Hanson, M.C.; Cianci, G.C.; Johnson, T.J.; Clark, M.R.; Hope, T.J.; Kiser, P.F. Modulation of Viscoelasticity and HIV Transport as a Function of pH in a Reversibly Crosslinked Hydrogel. Adv Funct Mater 2009, 19, 2969–2977. [Google Scholar] [CrossRef]
- Folkman, J.; Long, D.M. The use of silicone rubber as a carrier for prolonged drug therapy. Journal of Surgical Research 1964, 4, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, E.; Johengen, D.; Luecke, E.; Rothrock, G.; McGowan, I.; van der Straten, A.; Desai, T. A tunable, biodegradable, thin-film polymer device as a long-acting implant delivering tenofovir alafenamide fumarate for HIV pre-exposure prophylaxis. Pharmaceutical research 2016, 33, 1649–1656. [Google Scholar] [CrossRef]
- Li, L.; Johnson, L.M.; Krovi, S.A.; Demkovich, Z.R.; van der Straten, A. Performance and stability of tenofovir alafenamide formulations within subcutaneous biodegradable implants for HIV pre-exposure prophylaxis (PrEP). Pharmaceutics 2020, 12, 1057. [Google Scholar] [CrossRef]
- Su, J.T.; Simpson, S.M.; Sung, S.; Tfaily, E.B.; Veazey, R.; Marzinke, M.; Qiu, J.; Watrous, D.; Widanapathirana, L.; Pearson, E. A subcutaneous implant of tenofovir alafenamide fumarate causes local inflammation and tissue necrosis in rabbits and macaques. Antimicrobial agents and chemotherapy 2020, 64, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Joiner, J.B.; Prasher, A.; Young, I.C.; Kim, J.; Shrivastava, R.; Maturavongsadit, P.; Benhabbour, S.R. Effects of Drug Physicochemical Properties on In-Situ Forming Implant Polymer Degradation and Drug Release Kinetics. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Romano, J.W.; Baum, M.M.; Demkovich, Z.R.; Diana, F.; Dobard, C.; Feldman, P.L.; Garcia-Lerma, J.G.; Grattoni, A.; Gunawardana, M.; Ho, D.K.; et al. Tenofovir Alafenamide for HIV Prevention: Review of the Proceedings from the Gates Foundation Long-Acting TAF Product Development Meeting. AIDS Res Hum Retroviruses 2021, 37, 409–420. [Google Scholar] [CrossRef] [PubMed]
- Ullah Nayan, M.; Sillman, B.; Hasan, M.; Deodhar, S.; Das, S.; Sultana, A.; Thai Hoang Le, N.; Soriano, V.; Edagwa, B.; Gendelman, H.E. Advances in long-acting slow effective release antiretroviral therapies for treatment and prevention of HIV infection. Advanced Drug Delivery Reviews 2023, 200, 115009. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, D.; Simpson, S.M.; Su, J.T.; Bryndza-Tfaily, E.; Hope, T.J.; Veazey, R.; Dobek, G.; Qiu, J.; Watrous, D.; Sung, S.; et al. Design and Testing of a Cabotegravir Implant for HIV Prevention. J Control Release 2021, 330, 658–668. [Google Scholar] [CrossRef]
- Nevoralová, M.; Koutný, M.; Ujčić, A.; Starý, Z.; Šerá, J.; Vlková, H.; Šlouf, M.; Fortelný, I.; Kruliš, Z. Structure Characterization and Biodegradation Rate of Poly(ε-caprolactone)/Starch Blends. Frontiers in Materials 2020, 7. [Google Scholar] [CrossRef]
- Markowitz, M.; Sarafianos, S.G. EFdA (4′-ethynyl-2-fluoro-2′-deoxyadenosine, MK-8591): a novel HIV-1 reverse transcriptase translocation inhibitor. Current opinion in HIV and AIDS 2018, 13, 294. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Burgess, D.J. Drug release from in situ forming implants and advances in release testing. Adv Drug Deliv Rev 2021, 178, 113912. [Google Scholar] [CrossRef]
- Phaechamud, T.; Senarat, S.; Puyathorn, N.; Praphanwittaya, P. Solvent exchange and drug release characteristics of doxycycline hyclate-loaded bleached shellac in situ-forming gel and-microparticle. International journal of biological macromolecules 2019, 135, 1261–1272. [Google Scholar] [CrossRef]
- Amini-Fazl, M.S. Biodegradation study of PLGA as an injectable in situ depot-forming implant for controlled release of paclitaxel. Polymer Bulletin 2021, 1–14. [Google Scholar] [CrossRef]
- Kempe, S.; Mäder, K. In situ forming implants—an attractive formulation principle for parenteral depot formulations. Journal of controlled release 2012, 161, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, N.; Sinha, V.R. In situ forming depot as sustained-release drug delivery systems. Critical Reviews™ in Therapeutic Drug Carrier Systems 2019, 36. [Google Scholar] [CrossRef]
- Yu, L.; Ding, J. Injectable hydrogels as unique biomedical materials. Chemical Society Reviews 2008, 37, 1473–1481. [Google Scholar] [CrossRef]
- Ibrahim, T.M.; Ayoub, M.M.; El-Bassossy, H.M.; El-Nahas, H.M.; Gomaa, E. Investigation of Alogliptin-Loaded In Situ Gel Implants by 23 Factorial Design with Glycemic Assessment in Rats. Pharmaceutics 2022, 14, 1867. [Google Scholar] [CrossRef]
- Wex, J.; Sidhu, M.; Odeyemi, I.; Abou-Setta, A.M.; Retsa, P.; Tombal, B. Leuprolide acetate 1-, 3-and 6-monthly depot formulations in androgen deprivation therapy for prostate cancer in nine European countries: evidence review and economic evaluation. ClinicoEconomics and Outcomes Research 2013, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Johnson, C.E.; Schmalstig, A.A.; Annis, A.; Wessel, S.E.; Van Horn, B.; Schauer, A.; Exner, A.A.; Stout, J.E.; Wahl, A. A long-acting formulation of rifabutin is effective for prevention and treatment of Mycobacterium tuberculosis. Nature communications 2022, 13, 4455. [Google Scholar] [CrossRef]
- Cox, M.C.; Scripture, C.D.; Figg, W.D. Leuprolide acetate given by a subcutaneous extended-release injection: less of a pain? Expert review of anticancer therapy 2005, 5, 605–611. [Google Scholar] [CrossRef]
- Agarwal, P.; Rupenthal, I.D. Injectable implants for the sustained release of protein and peptide drugs. Drug Discov Today 2013, 18, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Packhaeuser, C.; Schnieders, J.; Oster, C.; Kissel, T. In situ forming parenteral drug delivery systems: an overview. European Journal of Pharmaceutics and Biopharmaceutics 2004, 58, 445–455. [Google Scholar] [CrossRef]
- Griffin, J.B.; Ridgeway, K.; Montgomery, E.; Torjesen, K.; Clark, R.; Peterson, J.; Baggaley, R.; van der Straten, A. Vaginal ring acceptability and related preferences among women in low-and middle-income countries: a systematic review and narrative synthesis. PloS one 2019, 14, e0224898. [Google Scholar] [CrossRef]
- Macht, D.I. On the absorption of drugs and poisons through the vagina. Journal of Pharmacology and Experimental Therapeutics 1918, 10, 509–522. [Google Scholar]
- Malcolm, R.K.; Boyd, P.J.; McCoy, C.F.; Murphy, D.J. Microbicide vaginal rings: Technological challenges and clinical development. Advanced drug delivery reviews 2016, 103, 33–56. [Google Scholar] [CrossRef]
- McConville, C.; Andrews, G.P.; Laverty, T.P.; Woolfson, A.D.; Malcolm, R.K. Rheological evaluation of the isothermal cure characteristics of medical grade silicone elastomers. Journal of applied polymer science 2010, 116, 2320–2327. [Google Scholar] [CrossRef]
- Monteiro, I.; Guazzelli, C.F.; Bahamondes, L. Advances in contraceptive vaginal rings: What does the future hold? Expert Opinion on Pharmacotherapy 2018, 19, 1685–1691. [Google Scholar] [CrossRef]
- Speroff, L.; Group, U.S.V.I. Efficacy and tolerability of a novel estradiol vaginal ring for relief of menopausal symptoms. Obstetrics & Gynecology 2003, 102, 823–834. [Google Scholar]
- Woolfson, A.; Malcolm, R.; Gallagher, R. Design of a silicone reservoir intravaginal ring for the delivery of oxybutynin. Journal of controlled release 2003, 91, 465–476. [Google Scholar] [CrossRef]
- Leonhard, M.; Moser, D.; Reumueller, A.; Mancusi, G.; Bigenzahn, W.; Schneider-Stickler, B. Comparison of biofilm formation on new phonax and provox 2 voice prostheses—a pilot study. Head & neck 2010, 32, 886–895. [Google Scholar]
- MacCallum, N.; Howell, C.; Kim, P.; Sun, D.; Friedlander, R.; Ranisau, J.; Ahanotu, O.; Lin, J.J.; Vena, A.; Hatton, B. Liquid-infused silicone as a biofouling-free medical material. ACS Biomaterials Science & Engineering 2015, 1, 43–51. [Google Scholar]
- Fundeanu, I.; Klee, D.; Schouten, A.J.; Busscher, H.J.; Van der Mei, H.C. Solvent-free functionalization of silicone rubber and efficacy of PAAm brushes grafted from an amino-PPX layer against bacterial adhesion. Acta biomaterialia 2010, 6, 4271–4276. [Google Scholar] [CrossRef]
- Rodrigues, L.; Van der Mei, H.; Teixeira, J.A.; Oliveira, R. Biosurfactant from Lactococcus lactis 53 inhibits microbial adhesion on silicone rubber. Applied microbiology and biotechnology 2004, 66, 306–311. [Google Scholar] [CrossRef]
- Everaert, E.; Belt-Gritter, B.V.D.; Van der Mei, H.; Busscher, H.; Verkerke, G.; Dijk, F.; Mahieu, H.; Reitsma, A. In vitro and in vivo microbial adhesion and growth on argon plasma-treated silicone rubber voice prostheses. Journal of Materials Science: Materials in Medicine 1998, 9, 147–157. [Google Scholar]
- Everaert, E.P.; Mahieu, H.F.; van de Belt-Gritter, B.; Peeters, A.J.G.; Verkerke, G.J.; van der Mei, H.C.; Busscher, H.J. Biofilm formation in vivo on perfluoro-alkylsiloxane–modified voice prostheses. Archives of Otolaryngology–Head & Neck Surgery 1999, 125, 1329–1332. [Google Scholar]
- Traore, Y.L.; Chen, Y.; Bernier, A.-M.; Ho, E.A. Impact of hydroxychloroquine-loaded polyurethane intravaginal rings on lactobacilli. Antimicrobial Agents and Chemotherapy 2015, 59, 7680–7686. [Google Scholar] [CrossRef] [PubMed]
- Roddy, R.E.; Zekeng, L.; Ryan, K.A.; Tamoufe, U.; Weir, S.S.; Wong, E.L. A controlled trial of nonoxynol 9 film to reduce male-to-female transmission of sexually transmitted diseases. New England Journal of Medicine 1998, 339, 504–510. [Google Scholar] [CrossRef]
- Malcolm, R.K.; Woolfson, A.D.; Toner, C.F.; Morrow, R.J.; McCullagh, S.D. Long-term, controlled release of the HIV microbicide TMC120 from silicone elastomer vaginal rings. Journal of Antimicrobial Chemotherapy 2005, 56, 954–956. [Google Scholar] [CrossRef] [PubMed]
- Nel, A.; Smythe, S.; Young, K.; Malcolm, K.; McCoy, C.; Rosenberg, Z.; Romano, J. Safety and Pharmacokinetics of Dapivirine Delivery From Matrix and Reservoir Intravaginal Rings to HIV-Negative Women. JAIDS Journal of Acquired Immune Deficiency Syndromes 2009, 51, 416–423. [Google Scholar] [CrossRef]
- Nel, A.; Bekker, L.-G.; Bukusi, E.; Hellstrӧm, E.; Kotze, P.; Louw, C.; Martinson, F.; Masenga, G.; Montgomery, E.; Ndaba, N. Safety, acceptability and adherence of dapivirine vaginal ring in a microbicide clinical trial conducted in multiple countries in Sub-Saharan Africa. PloS one 2016, 11, e0147743. [Google Scholar] [CrossRef] [PubMed]
- Baeten, J.M.; Palanee-Phillips, T.; Brown, E.R.; Schwartz, K.; Soto-Torres, L.E.; Govender, V.; Mgodi, N.M.; Matovu Kiweewa, F.; Nair, G.; Mhlanga, F. Use of a vaginal ring containing dapivirine for HIV-1 prevention in women. New England Journal of Medicine 2016, 375, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Johnson, T.J.; Clark, M.R.; Albright, T.H.; Nebeker, J.S.; Tuitupou, A.L.; Clark, J.T.; Fabian, J.; McCabe, R.T.; Chandra, N.; Doncel, G.F.; et al. A 90-day tenofovir reservoir intravaginal ring for mucosal HIV prophylaxis. Antimicrob Agents Chemother 2012, 56, 6272–6283. [Google Scholar] [CrossRef]
- Mesquita, P.M.; Rastogi, R.; Segarra, T.J.; Teller, R.S.; Torres, N.M.; Huber, A.M.; Kiser, P.F.; Herold, B.C. Intravaginal ring delivery of tenofovir disoproxil fumarate for prevention of HIV and herpes simplex virus infection. Journal of antimicrobial chemotherapy 2012, 67, 1730–1738. [Google Scholar] [CrossRef]
- Smith, J.M.; Rastogi, R.; Teller, R.S.; Srinivasan, P.; Mesquita, P.M.; Nagaraja, U.; McNicholl, J.M.; Hendry, R.M.; Dinh, C.T.; Martin, A.; et al. Intravaginal ring eluting tenofovir disoproxil fumarate completely protects macaques from multiple vaginal simian-HIV challenges. Proc Natl Acad Sci U S A 2013, 110, 16145–16150. [Google Scholar] [CrossRef]
- Su, J.T.; Teller, R.S.; Srinivasan, P.; Zhang, J.; Martin, A.; Sung, S.; Smith, J.M.; Kiser, P.F. A Dose Ranging Pharmacokinetic Evaluation of IQP-0528 Released from Intravaginal Rings in Non-Human Primates. Pharm Res 2017, 34, 2163–2171. [Google Scholar] [CrossRef] [PubMed]
- Quinn, H.L.; Kearney, M.-C.; Courtenay, A.J.; McCrudden, M.T.; Donnelly, R.F. The role of microneedles for drug and vaccine delivery. Expert opinion on drug delivery 2014, 11, 1769–1780. [Google Scholar] [CrossRef]
- Permana, A.D.; McCrudden, M.T.; Donnelly, R.F. Enhanced intradermal delivery of nanosuspensions of antifilariasis drugs using dissolving microneedles: A proof of concept study. Pharmaceutics 2019, 11, 346. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.J.; Kaestner, S.A.; Sutter, D.E.; Harvey, N.G.; Mikszta, J.A.; Pettis, R.J. Microneedle-based intradermal delivery enables rapid lymphatic uptake and distribution of protein drugs. Pharmaceutical research 2011, 28, 107–116. [Google Scholar] [CrossRef]
- Moga, K.A.; Bickford, L.R.; Geil, R.D.; Dunn, S.S.; Pandya, A.A.; Wang, Y.; Fain, J.H.; Archuleta, C.F.; O'Neill, A.T.; DeSimone, J.M. Rapidly–dissolvable microneedle patches via a highly scalable and reproducible soft lithography approach. Advanced Materials 2013, 25, 5060–5066. [Google Scholar] [CrossRef]
- Römgens, A.; Bader, D.; Bouwstra, J.; Baaijens, F.; Oomens, C. Monitoring the penetration process of single microneedles with varying tip diameters. journal of the mechanical behavior of biomedical materials 2014, 40, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Gill, H.; Prausnitz, M. Effect of microneedle design on pain in human subjects. clin. J. Pain 2008, 24, 585–594. [Google Scholar]
- Moffatt, K.; Quinn, C.; McCague, P.J.; Donnelly, R.F. Exploration into the opinions of patients with HIV, healthcare professionals and the lay public of the use of microneedles in clinical practice: highlighting the translational potential for their role in HIV infection. Drug Delivery and Translational Research 2021, 11, 1199–1217. [Google Scholar] [CrossRef]
- Chen, M.; Quan, G.; Sun, Y.; Yang, D.; Pan, X.; Wu, C. Nanoparticles-encapsulated polymeric microneedles for transdermal drug delivery. Journal of Controlled Release 2020, 325, 163–175. [Google Scholar] [CrossRef]
- Xie, L.; Zeng, H.; Sun, J.; Qian, W. Engineering microneedles for therapy and diagnosis: A survey. Micromachines 2020, 11, 271. [Google Scholar] [CrossRef]
- Lee, J.W.; Park, J.-H.; Prausnitz, M.R. Dissolving microneedles for transdermal drug delivery. Biomaterials 2008, 29, 2113–2124. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Hirono, M.; Fukushima, K.; Sugioka, N.; Takada, K. Two-layered dissolving microneedles formulated with intermediate-acting insulin. International journal of pharmaceutics 2012, 436, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.Y.; Choi, S.-O.; Prausnitz, M.R. Fabrication of dissolving polymer microneedles for controlled drug encapsulation and delivery: bubble and pedestal microneedle designs. Journal of pharmaceutical sciences 2010, 99, 4228–4238. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, C.; Yan, L.; Huang, L.; Zhu, X.; Chen, B.; Sant, H.J.; Niu, X.; Zhu, G.; Yu, K. Improved polyvinylpyrrolidone microneedle arrays with non-stoichiometric cyclodextrin. Journal of Materials Chemistry B 2014, 2, 1699–1705. [Google Scholar] [CrossRef] [PubMed]
- Vora, L.K.; Vavia, P.R.; Larrañeta, E.; Bell, S.E.; Donnelly, R.F. Novel nanosuspension-based dissolving microneedle arrays for transdermal delivery of a hydrophobic drug. Journal of interdisciplinary nanomedicine 2018, 3, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Park, J.-H.; Allen, M.G.; Prausnitz, M.R. Polymer microneedles for controlled-release drug delivery. Pharmaceutical research 2006, 23, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Abdelghany, S.; Tekko, I.A.; Vora, L.; Larrañeta, E.; Permana, A.D.; Donnelly, R.F. Nanosuspension-based dissolving microneedle arrays for intradermal delivery of curcumin. Pharmaceutics 2019, 11, 308. [Google Scholar] [CrossRef]
- Li, W.; Terry, R.N.; Tang, J.; Feng, M.R.; Schwendeman, S.P.; Prausnitz, M.R. Rapidly separable microneedle patch for the sustained release of a contraceptive. Nature Biomedical Engineering 2019, 3, 220–229. [Google Scholar] [CrossRef]
- Zhao, X.; Zhang, S.; Yang, G.; Zhou, Z.; Gao, Y. Exploring Trehalose on the release of levonorgestrel from implantable PLGA microneedles. Polymers 2020, 12, 59. [Google Scholar] [CrossRef]
- He, M.; Yang, G.; Zhao, X.; Zhang, S.; Gao, Y. Intradermal implantable PLGA microneedles for etonogestrel sustained release. Journal of Pharmaceutical Sciences 2020, 109, 1958–1966. [Google Scholar] [CrossRef] [PubMed]
- Than, A.; Liang, K.; Xu, S.; Sun, L.; Duan, H.; Xi, F.; Xu, C.; Chen, P. Transdermal delivery of anti-obesity compounds to subcutaneous adipose tissue with polymeric microneedle patches. Small Methods 2017, 1, 1700269. [Google Scholar] [CrossRef]
- Chen, M.-C.; Wang, K.-W.; Chen, D.-H.; Ling, M.-H.; Liu, C.-Y. Remotely triggered release of small molecules from LaB6@ SiO2-loaded polycaprolactone microneedles. Acta biomaterialia 2015, 13, 344–353. [Google Scholar] [CrossRef] [PubMed]
- Mc Crudden, M.T.C.; Larrañeta, E.; Clark, A.; Jarrahian, C.; Rein-Weston, A.; Creelman, B.; Moyo, Y.; Lachau-Durand, S.; Niemeijer, N.; Williams, P.; et al. Design, Formulation, and Evaluation of Novel Dissolving Microarray Patches Containing Rilpivirine for Intravaginal Delivery. Advanced Healthcare Materials 2019, 8, 1801510. [Google Scholar] [CrossRef] [PubMed]
- Abbate, M.T.A.; Ramöller, I.K.; Sabri, A.H.; Paredes, A.J.; Hutton, A.J.; McKenna, P.E.; Peng, K.; Hollett, J.A.; McCarthy, H.O.; Donnelly, R.F. Formulation of antiretroviral nanocrystals and development into a microneedle delivery system for potential treatment of HIV-associated neurocognitive disorder (HAND). International Journal of Pharmaceutics 2023, 640, 123005. [Google Scholar] [CrossRef]
- Zhang, C.; Vora, L.K.; Tekko, I.A.; Volpe-Zanutto, F.; Peng, K.; Paredes, A.J.; McCarthy, H.O.; Donnelly, R.F. Development of dissolving microneedles for intradermal delivery of the long-acting antiretroviral drug bictegravir. Int J Pharm 2023, 642, 123108. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, R.F.; McCrudden, M.T.; Zaid Alkilani, A.; Larrañeta, E.; McAlister, E.; Courtenay, A.J.; Kearney, M.-C.; Singh, T.R.R.; McCarthy, H.O.; Kett, V.L. Hydrogel-forming microneedles prepared from “super swelling” polymers combined with lyophilised wafers for transdermal drug delivery. PloS one 2014, 9, e111547. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Hu, L.; Xu, C. Recent advances in the design of polymeric microneedles for transdermal drug delivery and biosensing. Lab on a Chip 2017, 17, 1373–1387. [Google Scholar] [CrossRef]
- Chai, Q.; Jiao, Y.; Yu, X. Hydrogels for biomedical applications: their characteristics and the mechanisms behind them. Gels 2017, 3, 6. [Google Scholar] [CrossRef]
- Lin, C.-C.; Metters, A.T. Hydrogels in controlled release formulations: network design and mathematical modeling. Advanced drug delivery reviews 2006, 58, 1379–1408. [Google Scholar] [CrossRef]
- Donnelly, R.F.; Singh, T.R.R.; Garland, M.J.; Migalska, K.; Majithiya, R.; McCrudden, C.M.; Kole, P.L.; Mahmood, T.M.T.; McCarthy, H.O.; Woolfson, A.D. Hydrogel-forming microneedle arrays for enhanced transdermal drug delivery. Advanced functional materials 2012, 22, 4879–4890. [Google Scholar] [CrossRef] [PubMed]
- Blasi, P. Poly (lactic acid)/poly (lactic-co-glycolic acid)-based microparticles: An overview. Journal of Pharmaceutical Investigation 2019, 49, 337–346. [Google Scholar] [CrossRef]
- Abdelkader, H.; Fathalla, Z.; Seyfoddin, A.; Farahani, M.; Thrimawithana, T.; Allahham, A.; Alani, A.W.G.; Al-Kinani, A.A.; Alany, R.G. Polymeric long-acting drug delivery systems (LADDS) for treatment of chronic diseases: Inserts, patches, wafers, and implants. Advanced Drug Delivery Reviews 2021, 177, 113957. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K. Functional coatings and microencapsulation: a general perspective. Functional Coatings: by polymer microencapsulation, 2006; 1–28. [Google Scholar]
- Yeo, Y.; Baek, N.; Park, K. Microencapsulation methods for delivery of protein drugs. Biotechnology and Bioprocess Engineering 2001, 6, 213–230. [Google Scholar] [CrossRef]
- Tomaro-Duchesneau, C.; Saha, S.; Malhotra, M.; Kahouli, I.; Prakash, S. Microencapsulation for the therapeutic delivery of drugs, live mammalian and bacterial cells, and other biopharmaceutics: current status and future directions. Journal of pharmaceutics 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Jyothi, N.V.N.; Prasanna, P.M.; Sakarkar, S.N.; Prabha, K.S.; Ramaiah, P.S.; Srawan, G. Microencapsulation techniques, factors influencing encapsulation efficiency. Journal of microencapsulation 2010, 27, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Bansode, S.; Banarjee, S.; Gaikwad, D.; Jadhav, S.; Thorat, R. Microencapsulation: a review. International journal of pharmaceutical sciences review and research 2010, 1, 38–43. [Google Scholar]
- Dubey, R. Microencapsulation technology and applications. Defence Science Journal 2009, 59, 82. [Google Scholar]
- Sharifi, F.; Otte, A.; Yoon, G.; Park, K. Continuous in-line homogenization process for scale-up production of naltrexone-loaded PLGA microparticles. Journal of Controlled Release 2020, 325, 347–358. [Google Scholar] [CrossRef]
- Chereddy, K.K.; Vandermeulen, G.; Préat, V. PLGA based drug delivery systems: Promising carriers for wound healing activity. Wound Repair and Regeneration 2016, 24, 223–236. [Google Scholar] [CrossRef]
- Ramazani, F.; Chen, W.; van Nostrum, C.F.; Storm, G.; Kiessling, F.; Lammers, T.; Hennink, W.E.; Kok, R.J. Strategies for encapsulation of small hydrophilic and amphiphilic drugs in PLGA microspheres: state-of-the-art and challenges. International journal of pharmaceutics 2016, 499, 358–367. [Google Scholar] [CrossRef]
- Lee, J.; Kwon, H.J.; Ji, H.; Cho, S.H.; Cho, E.-H.; Han, H.D.; Shin, B.C. Marbofloxacin-encapsulated microparticles provide sustained drug release for treatment of veterinary diseases. Materials Science and Engineering: C 2016, 60, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Shahjin, F.; Patel, M.; Machhi, J.; Cohen, J.D.; Nayan, M.U.; Yeapuri, P.; Zhang, C.; Waight, E.; Hasan, M.; Abdelmoaty, M.M.; et al. Multipolymer microsphere delivery of SARS-CoV-2 antigens. Acta Biomaterialia 2023, 158, 493–509. [Google Scholar] [CrossRef]
- van de Weert, M.; Hennink, W.E.; Jiskoot, W. Protein instability in poly (lactic-co-glycolic acid) microparticles. Pharmaceutical research 2000, 17, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Cabezas, L.I.; Gracia, I.; de Lucas, A.; Rodríguez, J.F. Novel model for the description of the controlled release of 5-fluorouracil from PLGA and PLA foamed scaffolds impregnated in supercritical CO2. Industrial & Engineering Chemistry Research 2014, 53, 15374–15382. [Google Scholar]
- Khuroo, T.; Dharani, S.; Mohamed, E.M.; Immadi, S.; Wu, Z.; Khan, M.A.; Lu, D.; Nehete, P.; Rahman, Z. Ultra-long acting prodrug of dolutegravir and delivery system – Physicochemical, pharmacokinetic and formulation characterizations. International Journal of Pharmaceutics 2021, 607, 120889. [Google Scholar] [CrossRef]
- Collett, B.M. Scanning electron microscopy: A review and report of research in wood science. Wood and Fiber Science 1970, 113–133. [Google Scholar]
- Doty, A.C.; Weinstein, D.G.; Hirota, K.; Olsen, K.F.; Ackermann, R.; Wang, Y.; Choi, S.; Schwendeman, S.P. Mechanisms of in vivo release of triamcinolone acetonide from PLGA microspheres. Journal of Controlled Release 2017, 256, 19–25. [Google Scholar] [CrossRef]
- Shah, R.B.; Schwendeman, S.P. A biomimetic approach to active self-microencapsulation of proteins in PLGA. Journal of Controlled Release 2014, 196, 60–70. [Google Scholar] [CrossRef]
- Srikar, G.; Rani, A.P. Study on influence of polymer and surfactant on in vitro performance of biodegradable aqueous-core nanocapsules of tenofovirdisoproxil fumarate by response surface methodology. Brazilian Journal of Pharmaceutical Sciences 2019, 55. [Google Scholar] [CrossRef]
- Kazazi-Hyseni, F.; Landin, M.; Lathuile, A.; Veldhuis, G.J.; Rahimian, S.; Hennink, W.E.; Kok, R.J.; van Nostrum, C.F. Computer modeling assisted design of monodisperse PLGA microspheres with controlled porosity affords zero order release of an encapsulated macromolecule for 3 months. Pharmaceutical research 2014, 31, 2844–2856. [Google Scholar] [CrossRef] [PubMed]
- Samadi, N.; Abbadessa, A.; Di Stefano, A.; Van Nostrum, C.; Vermonden, T.; Rahimian, S.; Teunissen, E.; Van Steenbergen, M.; Amidi, M.; Hennink, W. The effect of lauryl capping group on protein release and degradation of poly (d, l-lactic-co-glycolic acid) particles. Journal of Controlled Release 2013, 172, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Kang, G.; Prathipati, P.K.; Zhou, Y.; Fan, W.; Li, Q.; Destache, C.J. Nanoencapsulation introduces long-acting phenomenon to tenofovir alafenamide and emtricitabine drug combination: A comparative pre-exposure prophylaxis efficacy study against HIV-1 vaginal transmission. Journal of controlled release 2019, 294, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Prathipati, P.K.; Kang, G.; Zhao, Y.; Yuan, Z.; Fan, W.; Li, Q.; Destache, C.J. Tenofovir alafenamide and elvitegravir loaded nanoparticles for long-acting prevention of HIV-1 vaginal transmission. AIDS (London, England) 2017, 31, 469. [Google Scholar] [CrossRef] [PubMed]
- Mandal, S.; Prathipati, P.K.; Sunagawa, S.W.; Destache, C.J. A concept evaluation study of a new combination bictegravir plus tenofovir alafenamide nanoformulation with prolonged sustained-drug-release potency for HIV-1 preexposure prophylaxis. Antimicrobial Agents and Chemotherapy 2021, 65, 10–1128. [Google Scholar] [CrossRef]
- Mandal, S.; Kang, G.; Prathipati, P.K.; Fan, W.; Li, Q.; Destache, C.J. Long-acting parenteral combination antiretroviral loaded nano-drug delivery system to treat chronic HIV-1 infection: A humanized mouse model study. Antiviral research 2018, 156, 85–91. [Google Scholar] [CrossRef]
- Prathipati, P.K.; Mandal, S.; Pon, G.; Vivekanandan, R.; Destache, C.J. Pharmacokinetic and Tissue Distribution Profile of Long Acting Tenofovir Alafenamide and Elvitegravir Loaded Nanoparticles in Humanized Mice Model. Pharmaceutical Research 2017, 34, 2749–2755. [Google Scholar] [CrossRef] [PubMed]
- DeYoung, M.B.; MacConell, L.; Sarin, V.; Trautmann, M.; Herbert, P. Encapsulation of exenatide in poly-(D, L-lactide-co-glycolide) microspheres produced an investigational long-acting once-weekly formulation for type 2 diabetes. Diabetes technology & therapeutics 2011, 13, 1145–1154. [Google Scholar]
- Jindal, A.B.; Bhide, A.R.; Salave, S.; Rana, D.; Benival, D. Long-acting parenteral drug delivery systems for the treatment of chronic diseases. Advanced Drug Delivery Reviews 2023, 198, 114862. [Google Scholar] [CrossRef]
- Trezza, C.; Ford, S.L.; Spreen, W.; Pan, R.; Piscitelli, S. Formulation and pharmacology of long-acting cabotegravir. Current Opinion in HIV and AIDS 2015, 10, 239. [Google Scholar] [CrossRef]
- Overton, E.T.; Richmond, G.; Rizzardini, G.; Jaeger, H.; Orrell, C.; Nagimova, F.; Bredeek, F.; García Deltoro, M.; Swindells, S.; Andrade-Villanueva, J.F.; et al. Long-acting cabotegravir and rilpivirine dosed every 2 months in adults with HIV-1 infection (ATLAS-2M), 48-week results: a randomised, multicentre, open-label, phase 3b, non-inferiority study. Lancet 2021, 396, 1994–2005. [Google Scholar] [CrossRef] [PubMed]
- Delany-Moretlwe, S.; Hughes, J.P.; Bock, P.; Ouma, S.G.; Hunidzarira, P.; Kalonji, D.; Kayange, N.; Makhema, J.; Mandima, P.; Mathew, C. Cabotegravir for the prevention of HIV-1 in women: results from HPTN 084, a phase 3, randomised clinical trial. The Lancet 2022, 399, 1779–1789. [Google Scholar] [CrossRef]
- Landovitz, R.J.; Donnell, D.; Clement, M.E.; Hanscom, B.; Cottle, L.; Coelho, L.; Cabello, R.; Chariyalertsak, S.; Dunne, E.F.; Frank, I. Cabotegravir for HIV prevention in cisgender men and transgender women. New England Journal of Medicine 2021, 385, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Sillman, B.; Bade, A.N.; Dash, P.K.; Bhargavan, B.; Kocher, T.; Mathews, S.; Su, H.; Kanmogne, G.D.; Poluektova, L.Y.; Gorantla, S. Creation of a long-acting nanoformulated dolutegravir. Nature communications 2018, 9, 443. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.; Su, H.; Dash, P.; Lin, Z.; Shetty, B.L.D.; Kocher, T.; Szlachetka, A.; Lamberty, B.; Fox, H.S.; Poluektova, L. Creation of a nanoformulated cabotegravir prodrug with improved antiretroviral profiles. Biomaterials 2018, 151, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Cobb, D.A.; Smith, N.; Deodhar, S.; Bade, A.N.; Gautam, N.; Shetty, B.L.D.; McMillan, J.; Alnouti, Y.; Cohen, S.M.; Gendelman, H.E. Transformation of tenofovir into stable ProTide nanocrystals with long-acting pharmacokinetic profiles. Nature communications 2021, 12, 5458. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Zhou, T.; Araínga, M.; Palandri, D.; Gautam, N.; Bronich, T.; Alnouti, Y.; McMillan, J.; Edagwa, B.; Gendelman, H.E. Creation of a long-acting nanoformulated 2′, 3′-dideoxy-3′-thiacytidine. Journal of Acquired Immune Deficiency Syndromes (1999) 2017, 74, e75. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, I.M.; Bade, A.N.; Lin, Z.; Soni, D.; Wojtkiewicz, M.; Dyavar Shetty, B.L.; Gautam, N.; McMillan, J.M.; Alnouti, Y.; Edagwa, B.J. Synthesis and characterization of a long-acting emtricitabine prodrug nanoformulation. International journal of nanomedicine 2019, 6231–6247. [Google Scholar] [CrossRef]
- Ogunnaike, M.; Das, S.; Raut, S.S.; Sultana, A.; Nayan, M.U.; Ganesan, M.; Edagwa, B.J.; Osna, N.A.; Poluektova, L.Y. Chronic Hepatitis B Infection: New Approaches towards Cure. Biomolecules 2023, 13, 1208. [Google Scholar] [CrossRef]
- Das, S.; Wang, W.; Ganesan, M.; Fonseca-Lanza, F.; Cobb, D.A.; Bybee, G.; Sun, Y.; Guo, L.; Hanson, B.; Cohen, S.M. An ultralong-acting tenofovir ProTide nanoformulation achieves monthslong HBV suppression. Science Advances 2022, 8, eade9582. [Google Scholar] [CrossRef]
- Spreen, W.; Ford, S.L.; Chen, S.; Wilfret, D.; Margolis, D.; Gould, E.; Piscitelli, S. GSK1265744 Pharmacokinetics in Plasma and Tissue After Single-Dose Long-Acting Injectable Administration in Healthy Subjects. JAIDS Journal of Acquired Immune Deficiency Syndromes 2014, 67, 481–486. [Google Scholar] [CrossRef] [PubMed]
- Ford, S.; Chiu, J.; Lovern, M.; Spreen, W.; Kim, J. Population PK approach to predict cabotegravir (CAB, GSK1265744) long-acting injectable doses for phase 2b. In Proceedings of the Proceedings of the 54th Interscience Conference on Antimicrobial Agents and Chemotherapy, 2014; pp. 5–9.
- Soriano, V.; Barreiro, P.; Benitez, L.; Peña, J.M.; de Mendoza, C. New antivirals for the treatment of chronic hepatitis B. Expert opinion on investigational drugs 2017, 26, 843–851. [Google Scholar] [CrossRef] [PubMed]
- Scarsi, K.K. Chasing the cabotegravir tail: implications for prevention. The Lancet HIV 2020, 7, e451–e453. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Timeline for LA drug delivery formulations for the treatment and prevention of HIV-1.
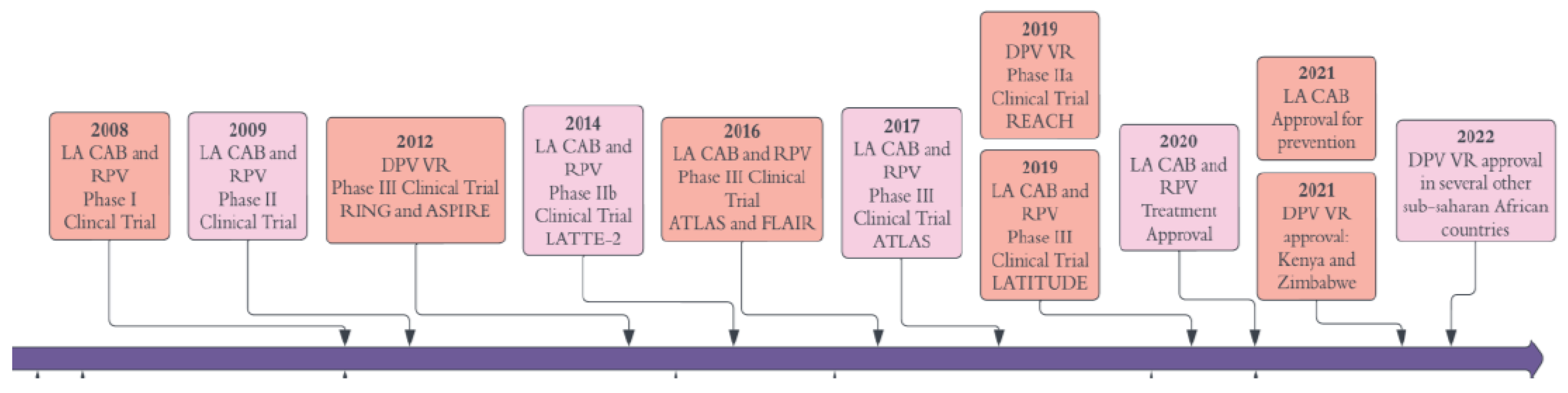
Figure 4.
Different types of non-erodible implants are illustrated. A-C) TAF implants contain silicone and PVA. Reproduced with permission from [3]. D) PCL reservoir-style implant for TAF delivery, which comprises a formulated drug core (Reproduced under the Cretive Commons CC-BY licence from [2]. E) and F) Cross-sectional depiction of nanochannel containing implant showing drug refill needles through the loading ports with resealable silicon plugs. Two different size and shape implants of TAF and FTC. Reprinted with permission [3].
Figure 4.
Different types of non-erodible implants are illustrated. A-C) TAF implants contain silicone and PVA. Reproduced with permission from [3]. D) PCL reservoir-style implant for TAF delivery, which comprises a formulated drug core (Reproduced under the Cretive Commons CC-BY licence from [2]. E) and F) Cross-sectional depiction of nanochannel containing implant showing drug refill needles through the loading ports with resealable silicon plugs. Two different size and shape implants of TAF and FTC. Reprinted with permission [3].
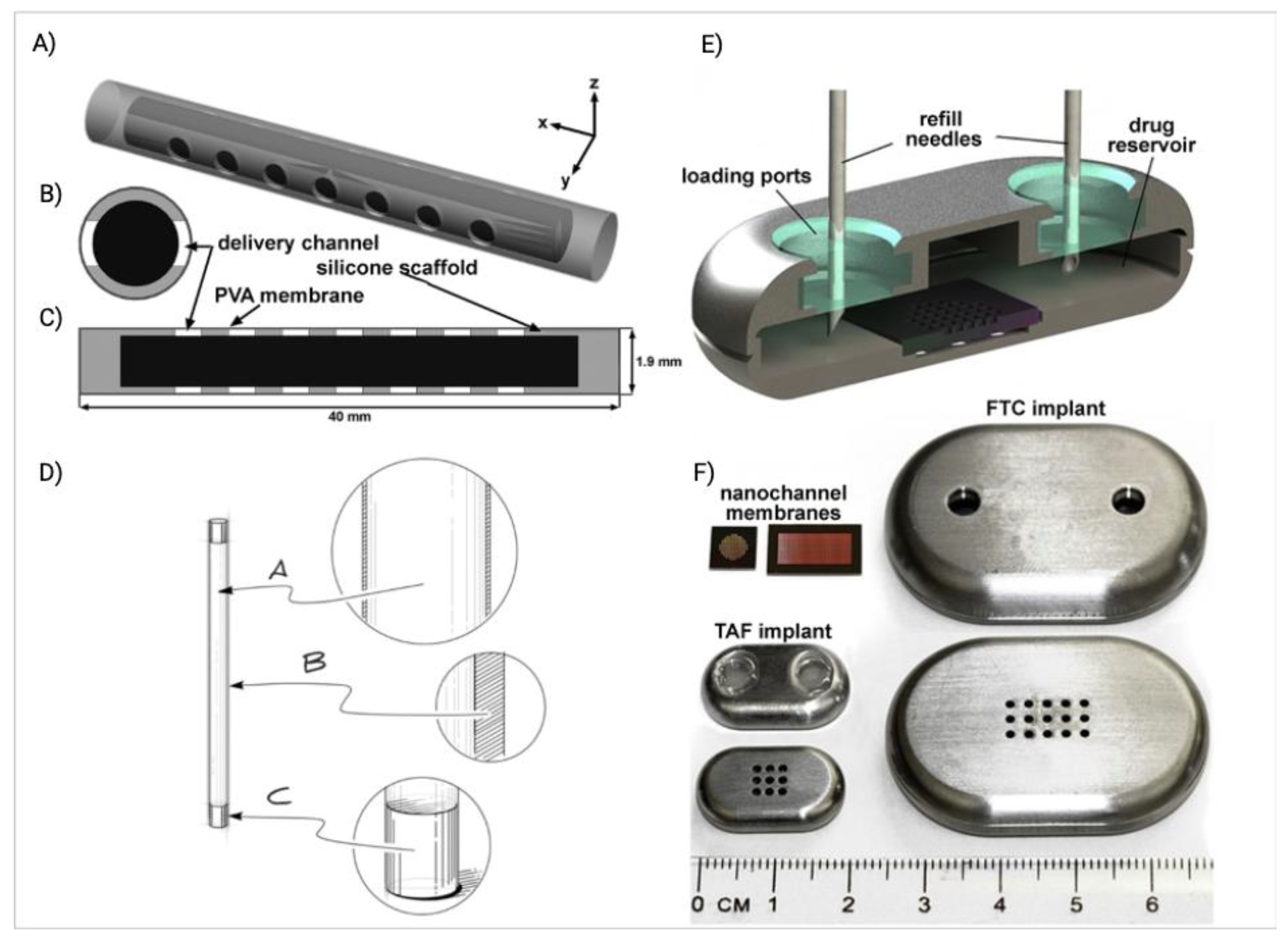
Figure 5.
ISFIs microstaructure in PLGA BASED ISFI’s: A) Cumulative release of CAB ISFIs PLGA formulations. B) Effect of drug loading on CAB release. C) PLGA molecular weight effects CAB release. D-L) Effect of PLGA degradation on implant microstructure. SEM cross-section images of placebo-ISFIs (1:2 w/w PLGA/NMP, PLGA MW 27 kDa) over a 30-day period. D-F) Low magnification image (100×) of the entire implant (scale bar = 100 µm). G-I) Higher magnification (200×) of the implant shell (shell thickness was measured using SEM scale; scale bar = 50 µm). J-L) Higher (500×) magnification of the center of the implant (scale bar = 20 µm). D/G/J) Implants imaged at day 3 post incubation in 0.01 M PBS pH 7.4 at 37 °C. E/H/K) Implants imaged at day 14 post incubation. F/I/L) Implants imaged at day 30 post incubation. Images were reproduced under the terms of the Creative Commons CC-BY license Error! Bookmark not defined..
Figure 5.
ISFIs microstaructure in PLGA BASED ISFI’s: A) Cumulative release of CAB ISFIs PLGA formulations. B) Effect of drug loading on CAB release. C) PLGA molecular weight effects CAB release. D-L) Effect of PLGA degradation on implant microstructure. SEM cross-section images of placebo-ISFIs (1:2 w/w PLGA/NMP, PLGA MW 27 kDa) over a 30-day period. D-F) Low magnification image (100×) of the entire implant (scale bar = 100 µm). G-I) Higher magnification (200×) of the implant shell (shell thickness was measured using SEM scale; scale bar = 50 µm). J-L) Higher (500×) magnification of the center of the implant (scale bar = 20 µm). D/G/J) Implants imaged at day 3 post incubation in 0.01 M PBS pH 7.4 at 37 °C. E/H/K) Implants imaged at day 14 post incubation. F/I/L) Implants imaged at day 30 post incubation. Images were reproduced under the terms of the Creative Commons CC-BY license Error! Bookmark not defined..
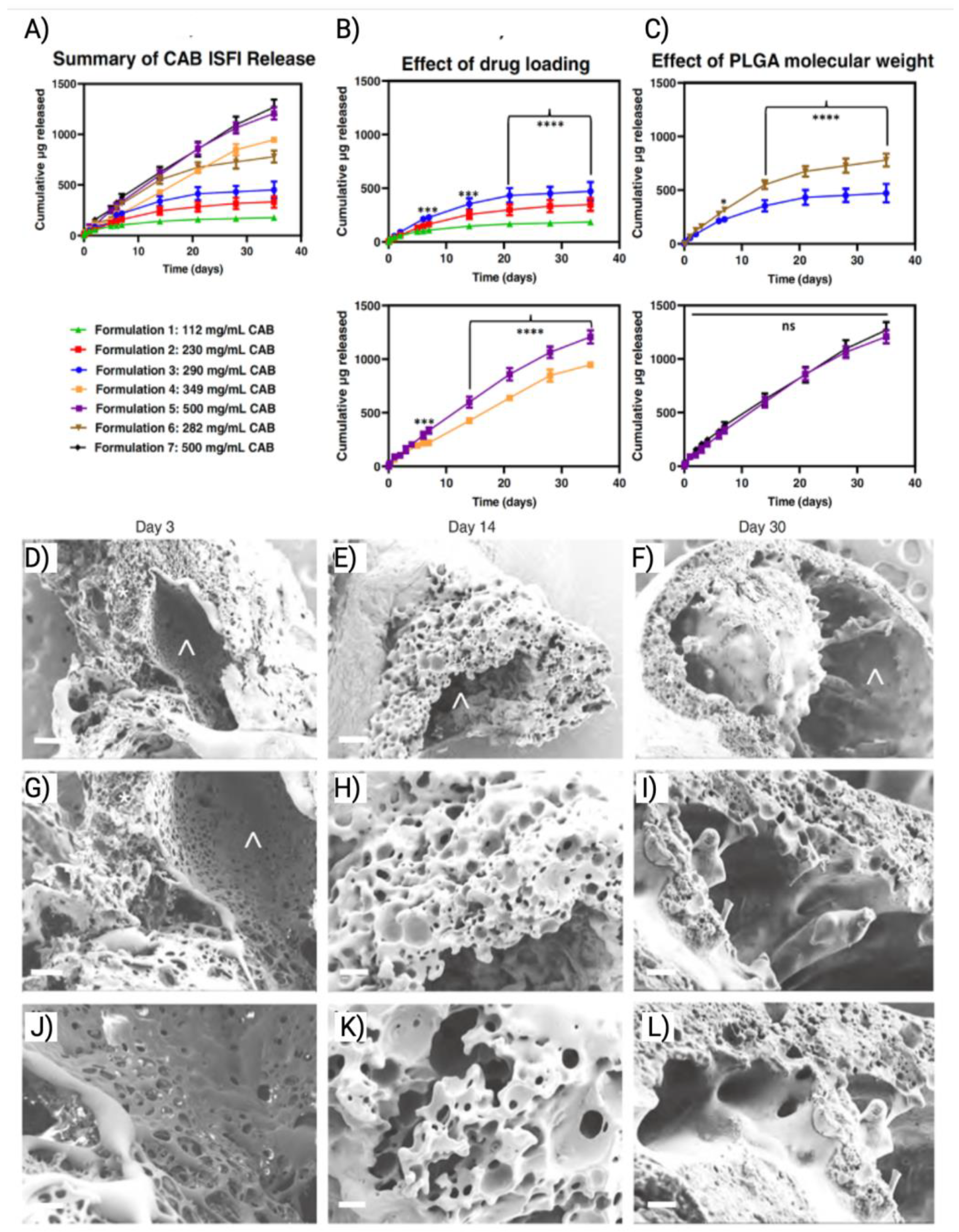
Figure 6.
Fast dissolving MNs have been made for ARV delivery. A) Schematic depiction of programmed-dissolving bilayer MAP with a high drug loading of LA micro-depots for potential monthly human application. B) Schematic presentation of the MN preparation process. The figure was reproduced under the terms of the Creative Commons CC-BY license Error! Bookmark not defined..
Figure 6.
Fast dissolving MNs have been made for ARV delivery. A) Schematic depiction of programmed-dissolving bilayer MAP with a high drug loading of LA micro-depots for potential monthly human application. B) Schematic presentation of the MN preparation process. The figure was reproduced under the terms of the Creative Commons CC-BY license Error! Bookmark not defined..
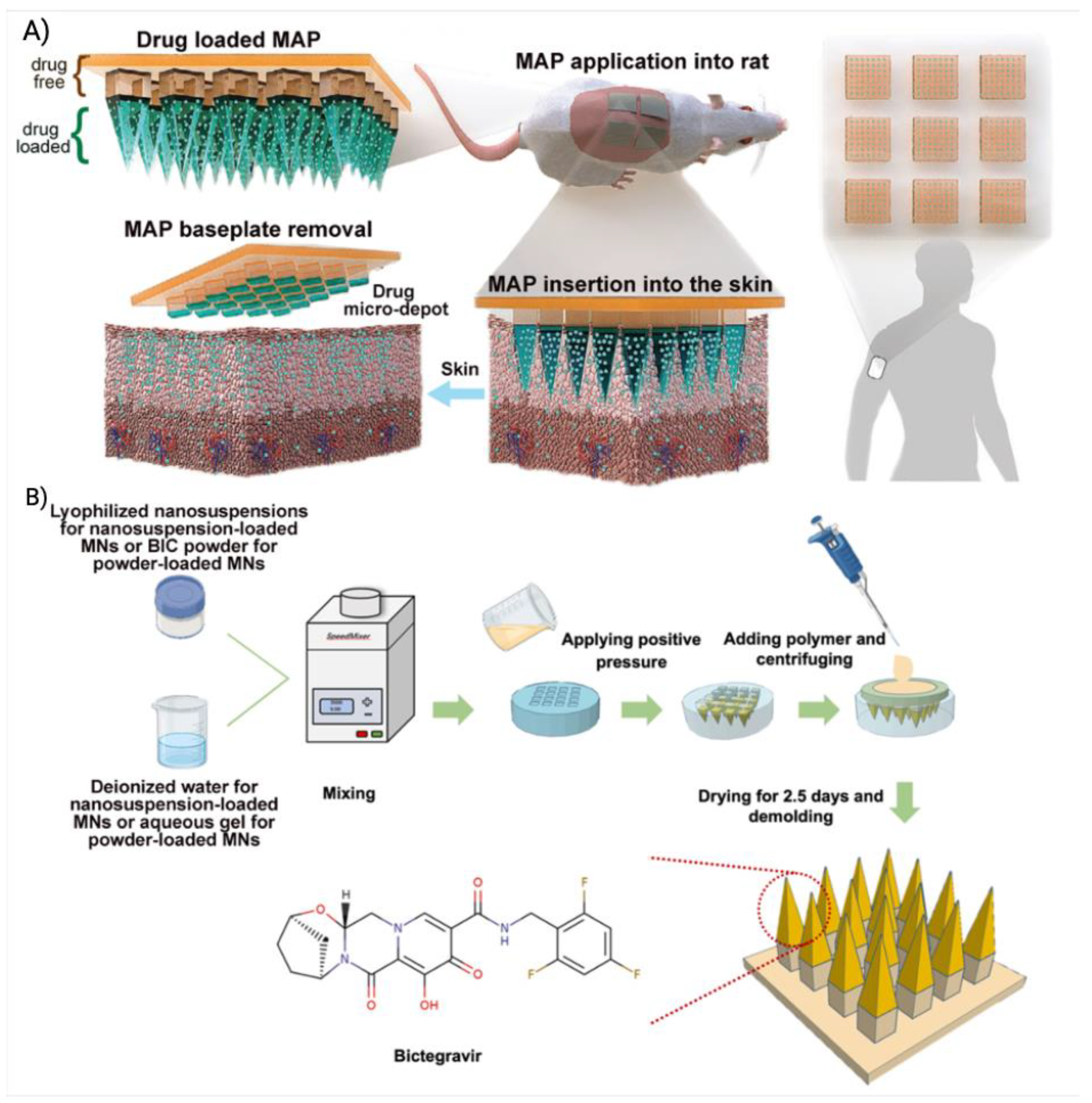
Figure 7.
The illustration shows the in-vivo fate of LASER ART prodrug nanoformulation. A) Chemical synthesis of prodrug from parent ARV. B) IM injection of LASER ART nanoformulation illustrates the muscle as the primary drug depot. C-D) Macrophages phagocytosized and store the nanocrystal from the site of injection. E) Biodistribution pf prodrug and active drug in tissues including HIV-1 reservoir site. F) Slow prodrug release in the low pH microenvironment in macrophages and then hydrolysis to release active drug. G-H) Slow dissolution and hydrolysis of prodrug in the blood and tissues. The figure was reproduced under the terms of the Creative Commons CC-BY license Error! Bookmark not defined..
Figure 7.
The illustration shows the in-vivo fate of LASER ART prodrug nanoformulation. A) Chemical synthesis of prodrug from parent ARV. B) IM injection of LASER ART nanoformulation illustrates the muscle as the primary drug depot. C-D) Macrophages phagocytosized and store the nanocrystal from the site of injection. E) Biodistribution pf prodrug and active drug in tissues including HIV-1 reservoir site. F) Slow prodrug release in the low pH microenvironment in macrophages and then hydrolysis to release active drug. G-H) Slow dissolution and hydrolysis of prodrug in the blood and tissues. The figure was reproduced under the terms of the Creative Commons CC-BY license Error! Bookmark not defined..
Table 2.
LA ARV formulations that are either in clinical or preclinical development.
| Delivery system | ARV drug | Polymer | Formulation details | Predicted dosage regimen (month) | Clinical trial | Ref. |
|---|---|---|---|---|---|---|
| Implant | TAF | Silicone and PVA | TAF powder is encapsulated in a silicone tube, which is curated with several drug delivery channel coated with PVA. Drug release rate can be controlled by controlling the number of channel or thickness of PVA membrane | 6 | Phase I | [3] |
| TAF | Silicone | Transcutaneous refillable nanochannel delivery implant, where drug reservoir is made of titanium and drug is released through TaN coated slit nanochannel. | 6 | Preclinical | [19] | |
| TAF |
PUs |
TAF formulation pellet is encased in PU-based drug reservoir and the drug is released through diffusion through the PU membrane. | 3 | [166] | ||
| CAB | CAB formulation is converted into pellets and then encased in PUs membrane containing drug reservoir. PUs membrane act as RCM for controlling drug release through diffusion. | 3 | [170] | |||
| TAF | PCL | TAF formulation containing TAF and castor oil was loaded into PCL tubes and the drug was released through the diffusion and bulk erosion of PCL. Drug release rate can be controlled through controlling MW of PCL. | 3 | [164] | ||
| ISL | EVA | Crystalline ISL is uniformly dispersed in EVA and the implant is prepared through hot melt extrusion. | 12 | On hold | [5] | |
| ISFIs | CAB |
PLGA |
CAB and PLGA were solubilized in NMP:DMSO and injected subcutaneously. Upon injection, the solution went through phase inversion and formed solid implant. | 6 | Preclinical | [55] |
| DTG | Same as CAB PLGA ISFI | 6 | [1] | |||
| VRs | TDF | PUs | TDF formulation was loaded in hydrophilic PUs tubes and the PUs tube was end-sealed using induction welding. | 1 | [204] | |
| TDF | PUs | TDF and NaCl formulation was loaded onto PUs extruded tube and end sealed to prepare the VRs. | 1 | [23] | ||
| MNs | CAB |
PVP and PVA |
MNs tips were generated by using a hydrogel composed of 20:20:60 of PVA:PVP:CAB and the baseplate of MNs was made using PVP and PVA. The MNs tips dissolved quick to release the CAB and the CAB formed drug depot at the administration site. | 1 | Preclinical | [4] |
| BIC | MNs tips were made of PVP, PVA and BIC. The baseplate was made of PVP and glycerol. | 1 | [231] | |||
| RPV | MNs tips were made of PVP and RPV. The baseplate was made of PVA and glycerol. | 1 | [27] | |||
| Microparticle | DTG | PLGA | DTG was transformed into a hydrophobic prodrug and encapsulated into PLGA-based microparticles by organic and aqueous solvents emulsification-evaporation. | 3 | [252] | |
| Prodrug nanocrystal | TFV |
Poloxamer, Polysorbate and PEG |
TFV was converted into lipophilic ProTideS and then formulated as aqueous nanocrystals | ≥3 | [272] | |
| CAB | Lipohilic ester prodrugs of cabotegravir were synthesized and formulated as aqueous nanocrystals | 12 | [17] | |||
| DTG | Fatty acid ester prodrugs of DTG were syntheiszed and nanoformulated as aqueos nanocrystals | ≥6 | [6] | |||
| ISL – islatravir. TFV – tenofovir. TDF – tenofovir disoproxil fumarate. TAF – tenofovir alafenamide fumarate, CAB – cabotegravir, BIC – bictegravir, DTG – dolutegravir, RPV – rilpivirine. | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



