
Submitted:
20 January 2024
Posted:
22 January 2024
You are already at the latest version
Abstract
The incidence of hepatocellular carcinoma (HCC) is increasing and 40% of patients are diagnosed at advanced stages. Over the past 5 years, the number of clinical available treatments have dramatically increased for HCC, making patient management particularly complex. Immune checkpoint inhibitors (ICIs) have improved the overall survival of patients showing a durable treatment benefit over time, and a different response pattern with respect to tyrosine kinase inhibitors (TKIs). Although improved survival in responder cases, a sizeable group of patients are primary progressors or are ineligible for immunotherapy. Indeed, patients with non-viral etiologies, such non-alcoholic steatohepatitis (NASH), and alterations in specific driver genes might be less responsive to immunotherapy. Therefore, improving the comprehension of mechanisms of drug resistance and identifying biomarkers informative of the best treatment approach are required actions to improve patient survival. Numerous evidence indicated non-coding RNAs (ncRNAs) as pivotal players in cancer. Molecular mechanisms through which ncRNAs exert their effects in cancer progression and drug resistance have been widely investigated. Nevertheless, there are no studies summarizing the synergistic effect between ncRNA-based strategies and TKIs or ICIs in the preclinical setting. This review aims to provide up-to-date information regarding the possible use of ncRNAs as therapeutic targets in association with molecular targeted agents and immunotherapies and as predictive tools for the selection of optimized treatment options in advanced HCCs.
Keywords:
HCC
; non-coding RNA
; microRNA
; TKI
; ICI
; biomarkers
1. Introduction
The incidence of hepatocellular carcinoma (HCC) is increasing globally representing the sixth more common cancer and the third leading cause of cancer related death in 2020 [1]. Over the past decade, there has been a remarkable breakthrough in the availability of systemic treatment options for advanced HCC. Despite the robust efforts in clinical trials testing several molecular-targeted compounds, after its approval in 2007 [2], sorafenib remained the only systemic treatment for patients at advanced stages until 2017 when regorafenib granted approval in the second-line setting [3]. Subsequently, other positive phase III studies for tyrosine kinase inhibitors (TKIs) led to the approval of lenvatinib in the first line [4] and cabozantinib and ramucirumab in the second-line after sorafenib progression [5,6]. Remarkably, the advent of immune checkpoint inhibitors (ICIs) in the oncologic field further revolutionized the management of HCC patients with the IMbrave150 phase III trial demonstrating the superior efficacy of atezolizumab–bevacizumab combination in terms of overall survival (OS) with respect to sorafenib, becoming the new front-line standard of care for HCC. More recently, a second immunotherapy-based combination, durvalumab plus tremelimumab, has been qualified as a further front-line regimen [7]. Since the advent of immunotherapy, the overall survival has gradually increased over time showing benefits for patients with sensitive tumors and preserved liver function, the latter being a clinical requirement for treatment eligibility. On the contrary, vascular disorders and arterial hypertension may prevent the use of ICIs. Sorafenib and lenvatinb represent the first line treatments of choice for patients not eligible to immunotherapy [8]. As no head-to-head comparisons are available for all the first-line treatments, the recommendation for the most appropriate choice and sequence relies only on the analysis of clinical, radiological, and biochemical profiles of the patient. In this scenario, the identification of biomarkers for patient stratification, for the delivery of sequential treatment lines and for the optimization of clinical outcomes remains an urgent matter to be addressed in HCC research. Indeed, except for ramucirumab, for which elevated AFP levels (>400 ng/ml) are used to select patients, no biomarkers identify responder cases.
Despite substantial improvements in survival outcomes relative to sorafenib, most advanced patients do not derive durable benefit from immunotherapy regimens. Remarkably, the etiology of HCC seems to affect the immune system response impairing the efficacy of immunotherapy. In particular, non-alcoholic steatohepatitis (NASH)-derived HCCs resulted to be less responsive to ICIs, probably due to aberrant activation of CD8+ T cells causing tissue damage and impaired immune surveillance [9]. Interestingly, driver mutations hindered the response to both TKIs and ICIs. PI3K–mTOR pathway alterations and aberrant activation of WNT/β-catenin signaling were associated with shorter OS in patients treated with sorafenib and immunotherapy, respectively [10]. Although several studies gave a better understanding of molecular mechanisms involved in the onset of drug resistance in HCC with a particular focus on non-coding RNAs (ncRNAs), no drugs targeting tumor-associated ncRNAs have entered the clinical practice so far [11,12].
Here, we will describe the most recent preclinical studies, employing at least one animal model, testing the synergistic effect between ncRNAs (microRNAs, long non-coding RNAs and circular RNAs) and TKIs or ICIs, providing the rational for unconventional combination strategies. As the lack of predictive biomarkers for successful patient stratification remains an open issue, the last part of this review will focus on ncRNAs, and especially microRNAs, as possible circulating candidates of treatment response and tumor escape in HCC.
2. Role of non-coding RNAs in hepatocarcinogenesis
Despite the most studied sequences are those of protein-coding genes, they account for only 1-2% of the human genome. Indeed, the vast majority of the human genome encodes for ncRNAs [13]. Non-coding sequences can be transcribed into structural RNAs (rRNA, tRNA) and regulatory RNAs with essential roles in the fine tuning of gene expression and genome organization, comprehending small, medium and long non-coding RNAs. Since their discovery, it has become increasingly evident the central role of these “supposedly inert sequences”, transcribed into a plethora of different molecules, in both physiologic and pathologic conditions, resulting involved in most of the human diseases [14]. The deregulation of ncRNAs is associated with genome structural modifications or copy number variations, as well as with epigenetic or transcription factor alterations. The study by Calin et al. reported the localization of most of aberrantly expressed microRNAs (miRNAs) within fragile sites of the genome associated with cancer. In the last two decades, the myriad of studies on miRNA activities in human diseases clearly proved their deregulation as a common cancer hallmark [15]. Recently, also long non-coding RNAs (lncRNAs) and circular RNAs (circRNAs) are gaining attention in cancer research. Below are reported few recent studies on the biologic effects of miRNAs, lncRNAs and circRNAs in hepatocarcinogenesis.
Long noncoding RNAs are > 200 nucleotides long molecules, and regulate several cellular processes such as proliferation, differentiation, and development. They can positively or negatively regulate gene expression by acting as signals, decoy, guide, or scaffold. They are also referred to competing endogenous RNAs (ceRNA), modulating protein translation and other ncRNAs, acting as miRNA sponges, or regulating small nucleolar RNAs (snoRNAs). Recent studies have also identified lncRNAs bearing ORFs, thus they are able to codify for proteins [16]. Qualitative or quantitative alterations of lncRNAs have been described in HCC contributing to cancerous phenotype (e.g., metabolic reprogramming, persistent proliferation, metastasis, migration, accelerated angiogenesis, epithelial to mesenchymal transition, and apoptotic cell death evasion). A deeper understanding of molecular mechanism underlying the deregulation of lncRNAs would provide new insights into cancer progression and is crucial to the discovery of new therapeutic agents in HCC [17]. To give a couple of examples, the lncRNA CEBPA-DT is upregulated in HCC with distant metastasis, associating with poor prognosis. Cai et al. demonstrated that CEBPA-DT induced epithelial-mesenchymal transition (EMT) through upregulation of Snail1 by promoting nuclear translocation of β-catenin [18]. Similarly, FTO-IT1 is overexpressed in HCC and emerged as a glycolysis-associated lncRNA influencing metabolic shift of cancer cells, by increasing their glycolytic capacity and proliferation rate. Its effect is mediated by FTO stabilization, which in turn increases the expression of GLUT1 and PKM2 glycolytic genes in HCC cells. FTO-IT1 silencing in vivo with lentiviral vectors gave rise to smaller tumor masses and reduced FTO, GLUT1 and PKM2 expression [19]. These and other studies demonstrated that cancer-associated lncRNAs may be used as a potential therapeutic targets for HCC treatment.
Circular RNAs represent another class of ncRNAs which are generated by back-splicing of linear transcripts, resulting in a circular structure that confers resistance to exonuclease activity. CircRNAs activate or repress gene expression, act as miRNA or protein sponges, enhance protein activity by forming circRNA-protein complexes acting as scaffold, or sequester proteins in specific cellular compartments [20]. Regarding HCC, hsa_circRNA_104348 is upregulated in liver tumors, particularly at advanced stages, and correlates with poor prognosis. Mechanistically, hsa_circRNA_104348 exerts its biological function by sponging miR-187-3p, which in turn regulates the Rho effector RTKN2, promoting proliferation, migration, and invasion through Wnt/β-catenin signaling pathway activation. As a proof of concept, knockdown of hsa_circRNA_104348 inhibits liver tumorigenesis and lung metastasis in xenograft mice [21]. CircRHOT1 is upregulated in HCC and associates with decreased OS and disease-free survival (DFS). Specifically, circRHOT1 promotes HCC cell growth, invasion and tumor formation via recruiting TIP60 (part of NuA4 chromatin remodeling complex) to NR2F6 promoter, thus triggering its transcription. In turn, NR2F6 modulates gene expression by recognizing DNA response elements linked to the regulation of adaptive immunity [22]. Recently, exosome-mediated transfer of circRNAs is emerging as a novel mechanism in cancer progression. As an example, exosomal circRNA-100338 exerts a pro-invasive role in HCC by increasing angiogenesis, as demonstrated in preclinical models where exosomal circRNA-100338 promotes HUVEC cells proliferation, permeability, and tube formation, while enhancing angiogenesis and tumor metastasis in vivo. High circRNA-100338 serum levels post-hepatectomy may predict pulmonary metastasis and poor survival in HCC [23], confirming the role of circRNAs in cancer development and aggressiveness.
MicroRNAs are short non-coding RNAs 22-25 nucleotides long, first identified in C. elegans 30 years ago. They play key roles in the main biological processes such as cell proliferation, differentiation, and embryonic development, having also tissue-specific functions. As concerns miRNAs mechanism of action, they bind to the 3’-UTR regions of their target mRNAs, repressing their translation or triggering their degradation [24]. They can also act as intercellular communication molecules, when secreted in extracellular vesicles [25]. For instance, HCC-derived exosomal miR-21 contributes to tumor progression by converting hepatic stellate cells to cancer-associated fibroblasts through PTEN downregulation, favoring tumor progression by enhancing neoangiogenesis [26]. In tumors, miRNAs can act as tumor suppressors (TS) or oncogenes based on tumor type, stadium, and tumor microenvironment [27]. Moreover, depending on the basal expression levels of targets core or mutational background they can act as either TS or oncogenes not only in different tumor types but also within the same tumor, as we previously reported for miR-221 and miR-30e-3p in HCC [28,29].
MiRNA signatures of human tumors associated with diagnosis, staging, progression, prognosis and response to treatment [30]. Others and our group firstly reported miRNA signatures in HCC by means of genome-wide microarray profiling, identifying HCC-specific miRNAs associated with risk factors, metastasis and oncogene/TS alterations [31,32,33]. Remarkably, the hepato-specific miR-122 was demonstrated to promote HCV replication [34] and represented the first miRNA to be silenced in vivo by chemically-modified oligonucleotides in rodents and non-human primates [35,36]. MiRNAs can also modulate the metabolic reprogramming of HCC cells, as in the case of miR-342-3p whose expression is high in regressing tumors. Mechanistically, miR-342-3p targets the lactate transporter MCT1 in HCC cells, thus affecting their lactate intake. In vivo, miR-342-3p delivery improved animal survival highlighting its promising therapeutic potential [37]. Other studies confirmed miRNAs modulation in vivo as effective strategies to slow down HCC progression or prevent tumor development [38,39], opening the path towards the use of miRNAs as promising therapeutic candidates. Given their potential clinical applications, miRNAs have been the focus of cancer research in the last 20 years. Despite the early termination of the first miRNA-based clinical trial in the oncologic field [40], currently, several clinical trials using miRNAs as diagnostic biomarkers or therapeutic targets are underway in cancer [41].
In this scenario, a deeper understanding of ncRNAs as actionable targets in the preclinical setting will allow to pinpoint mechanisms of drug resistance and synergies among treatments and to identify biomarkers for patient stratification and therapeutic sequences in HCC.
3. Combination of non-coding RNAs-based strategies with TKIs in HCC
Sorafenib and lenvatinib represent the two first-line treatments for patient not eligible to immunotherapy [42]. Due to sorafenib being the only systemic drug for advanced patients for almost a decade [2], a considerable number of literature articles relies on molecular mechanisms underneath deregulated ncRNAs contributing to sorafenib resistance. On the other hand, lenvatinib entry into clinical practice just preceded the accelerated approval of ‘Atezo/Beva’ combination therapy for the treatment of advanced HCC, directing the interest of the scientific community toward immunotherapy with few studies analyzing the effect of ncRNAs following lenvatinib administration. In the next two chapters, we will address the role of ncRNAs on the onset of TKIs resistance focusing on preclinical studies reporting the evaluation of combined strategies in at least one animal model.
3.1. Non-coding RNAs and sorafenib combination improves the therapeutic response
Non-coding RNAs are often deregulated in HCC and are extensively involved in the modulation of molecular mechanisms leading to sorafenib resistance, such as hypoxia, autophagy, metabolic reprogramming, and activation of oncogenic pathways [12]. Sorafenib is an oral TKI that blocks tumor cell proliferation by targeting Raf/MEK/ERK signaling at the level of Raf kinase, and exerts an antiangiogenic effect by targeting vascular endothelial growth factor receptor-2/-3 (VEGFR-2/-3), and platelet derived growth factor receptor beta (PDGFR-β) tyrosine kinases [43].
Due to the antiangiogenic properties of sorafenib, blocking factors having a mitogenic effect on endothelial cells and interfering with HIF-1A signaling represent crucial actions to potentiate sorafenib efficacy and to prevent drug resistance. MicroRNA-494 is an oncogenic HCC-associated miRNA which is upregulated in 30% of cases and associates with stem cell-like characteristics and poor prognosis [44,45]. Regarding its involvement in sorafenib resistance, we previously demonstrated that miR-494 activates the AKT/mTOR pathway by targeting PTEN and reported a stronger antitumor effect of antagomiR-494 plus sorafenib treatment with respect to sorafenib alone in the DEN-HCC rat model [46]. Notably, the Golgi phosphoprotein GOLPH3 is involved in sorafenib resistance in vivo by increasing the microvascular density of xenograft tumors. Gao et al. showed that exosomes released by GOLPH3 overexpressing HCC cells are enriched in miR-494 content enhancing tube formation and migration of the umbilical endothelial HUVEC cell line. MiR-494-loaded extracellular vesicles increased sorafenib resistance of HCC cells, highlighting both autologous and heterologous mechanisms of action for this miRNA [47]. These data proved the biologic activity of exosome-associated miR-494 in cell-to-cell crosstalk and confirmed that miR-494 as an important tumor-derived autocrine and paracrine signal promoting angiogenesis, HIF-1A activation, and tumor growth under hypoxic condition in different cancer types [48]. Similarly, the polypeptide 14-3-3η is a growth-promoting factor highly expressed in tumor and vascular endothelial cells contributing to poor survival of HCC patients [49]. The study by Shen and coworkers described the overexpression of 14-3-3η in sorafenib-resistant (SR) Huh-7 cells and demonstrated that its silencing restores drug sensitivity and reduces cancer stem cell (CSC) properties. Interestingly, 14-3-3η polypeptide post-transcriptionally activates HIF-1A via inhibition of the proteasome machinery. MiR-16 was identified as the epigenetic regulator of this polypeptide showing an inverse correlation in HCC patients treated with combined transartherial chemoembolization (TACE) and sorafenib. In line, low miR-16/high 14-3-3η HCC subgroup showed the worst OS after combined treatment. MiR-16 overexpression or 14-3-3η silencing in combination with sorafenib determined a higher anti-tumor effect in xenograft mice with respect to sorafenib alone, highlighting miR-16 restoration as a promising strategy to improve sorafenib efficacy in HCC [50]. A genome-wide CRISPR/Cas9 library screening identified the deficiency of miR-15a (belonging to the tumor suppressor miR-15a/16-1 miRNA cluster) and miR-20b (belonging to the oncogenic miR-19~92 miRNA cluster) to contribute to sorafenib resistance in HCCLM3 cell line. In agreement with the opposite role of these two miRNA clusters in tumors, miR-15a overexpressing cells decreased in vivo tumorigenesis while miR-20b overexpression slightly increased tumor size; nevertheless, the overexpression of both miRNAs led to inhibition of tumorigenesis in the xenograft model subjected to sorafenib treatment, confirming their role in drug sensitization. Target prediction algorithms identified the cochaperone CDC37L1 as the only common target of these two miRNAs. Functional analysis and luciferase assays proved its inhibition by both miR-15a and miR-20b. Mechanistically, CDC37L1 binds to the heat shock protein HSP90 to activate the peptidyl-prolyl cis-trans isomerase A (PPIA) that accelerates protein folding; higher mRNA levels associated with poorer OS and DFS in sorafenib-treated patients [51]. Another study employed a CRISPR-based screening method in vivo by using a sorafenib-treated xenograft model to improve the translational value of preclinical data with respect to in vitro tools. The Authors identified miR-3689a-3p as the most overexpressed miRNA in sorafenib-sensitive tumors and reported the targeting of the copper chaperone for superoxide dismutase (CCS) which, by reducing SOD1 ability to scavenge mitochondrial ROS, increased cellular oxidative stress that eventually mediated the antitumor effect of sorafenib. Orthotopic mouse models showed that miR-3689a-3p downregulation decreased sorafenib efficacy. Since lower miR-3689a-3p levels were detected in tumor specimens from HCC patient cohorts, this study paves the way towards a combined miRNA mimics and sorafenib strategy to boost sorafenib anticancer efficacy [52]. Notably, this high throughput screening procedure is particularly suitable for the discovery of driver genes and therapeutic targets that modulate drug efficacy. An example is represented by the metabolic gene phosphoglycerate dehydrogenase (PHGDH), regulating the serine synthesis pathway, whose specific inhibition by NCT-503 acted synergistically with sorafenib to abolish in vivo tumorigenesis [53]. Similarly, the downregulation of Kelch-like ECH-associated protein 1 (KEAP1) in response to sorafenib administration increased the activity of Nrf2, a key transcription factor controlling antioxidant responses, which contributed to enhance drug resistance to sorafenib, lenvatinib, and regorafenib in HCC [54].
Metabolic reprogramming from oxidative phosphorylation to aerobic glycolysis, also known as ‘Warburg effect’, is a core hallmark of cancer cells [55] influencing the response to sorafenib. Even though ATP production during aerobic glycolysis is much lower, the Warburg effect confers advantages to cancer cells growth by providing the carbon sources required for rapid cell proliferation and, in the meantime, by minimizing the production of toxic ROS [56]. Several HCC-specific miRNAs are involved in this glycolytic shift, as in the case of miR-3662, which is downregulated in liver tumors. Its reinforced expression in HCC cell lines associated with a decrease in glucose and oxygen consumption, ATP and lactate production, and in vivo tumorigenesis. Interestingly, both HIF-1A and esokinase 2 (HK2) are direct targets of this miRNA and their overexpression mitigates the above-mentioned effects confirming their key activity in mediating miR-3662 biologic processes [57,58]. We recently reported that the oncomiR-494 can rewire tumor metabolism of HCC cells by targeting the catalytic subunit of Glucose-6 phosphatase (G6pc) which is a multi-subunit complex catalyzing the de-phosphorylation of G6P to free glucose playing a central role in glucose homeostasis. A negative correlation was displayed between miR-494 and G6pc in HCC patient cohorts where lower G6pc levels associated with high tumor grade, microvascular invasion (MVI) and larger tumor size. We demonstrated that miR-494/G6pc axis contributes to metabolic plasticity of cancer cells favoring the accumulation of glycogen and lipid droplets that are exploited in case of critical metabolic conditions (e.g., glucose deprivation) giving an advantage to uncontrolled proliferation of malignant cells. We also showed that miR-494/G6pc axis promotes sorafenib resistance and proposed combined antagomiR-based treatments with sorafenib or 2-deoxyglucose (2-DG) for HCC patients who may develop sorafenib resistance and that are ineligible for immunotherapy [59]. An interesting study by Zhang et al. [60] demonstrated the pivotal role of miR-30a-5p/CLCF1 axis in modulating the metabolic shift toward aerobic glycolysis of SR HepG2 cells and xenograft tumors. Specifically, they found a time-dependent decrease of miR-30a-5p in sorafenib treated cells and demonstrated by functional analysis the direct targeting of the pro-inflammatory cytokine CLCF1 that activates the downstream PI3K/AKT pathway controlling proliferation and metabolic reprogramming of cancer cells. Indeed, the treatment with the AKT inhibitor MK-2206 reverted the glycolytic phenotype of SR HepG2 cells decreasing ATP and lactate production as well as mRNA expression of metabolic genes GLUT3, HK2, and PDK1. Strikingly, the Authors proved the therapeutic efficacy of a lipid formulation containing a chemically modified oligonucleotide (2′-O-methyl-modified miRNA conjugated with cholesterol) mimicking miR-30a-5p, which was injected into the tail vein of immunocompromised mice (once a week for five weeks). This miRNA formulation effectively inhibited the tumor growth of SR HepG2 cells, proving the feasibility and safety of miRNA delivery in vivo and its efficacy against sorafenib resistant tumors. An inverse correlation between miR-30a-5p and CLCF1 was found in HCC patients confirming the importance of this signaling axis in human tumors and suggesting the combination of agomiR-30a-5p and sorafenib as a promising strategy to improve TKIs efficacy and overcome acquired resistance.
In a treatment perspective, small extracellular vesicles (EV), which are loaded with miRNAs, proteins, and mRNAs protecting them from degradation, represent promising drug delivery vehicles and ideal miRNAs carriers to cancer cells [61]. Mesenchymal stem cells are a precious source of EVs retaining the characteristics of their parental cells and showing low immunogenicity and tumor-delivery properties [62]. An elegant study by Sun et al. described the engineering of EVs with miR-654-5p by in vitro electroporation (m654-sEV) and reported their effectiveness in sorafenib sensitization in preclinical models derived from SR resistant HCC cells through the direct targeting of the ferroptosis inhibitor HSPB1 [63]. In vivo findings proved that the combination of sorafenib and m654-sEV strongly suppressed tumor growth in comparison to sorafenib treatment alone by modulating ferroptosis-associated markers. In particular, the combined treatment effectively inhibited HSPB1 expression, increased levels of transferrin receptor (TFRC), COX2, Fe2+, and ROS, together with a decrease in glutathione (GSH) levels, suggesting this strategy as a reliable one to overcome sorafenib resistance in HCC. Another miRNA involved in sorafenib resistance via impairment of iron-associated programmed cell death, ferroptosis, is miR-23a-3p which is overexpressed in sorafenib-resistant patients and associated with tumor recurrence. A sorafenib resistant xenograft model obtained by inoculation of MHCC97L cells showed that miR-23a-3p is overexpressed in tumors acquiring resistance after long drug exposure. Tumor-derived resistant cell lines displayed a transcriptional activation of pri-miR-23a mediated by ETS1 transcription factor. A consistent reduction in cell growth was obtained in an orthotopic model when miR-23a-3p knockout HCC cells where injected in the presence of sorafenib treatment. A functional analysis assessed the targeting of Acyl-CoA synthetase long-chain family member 4 (ACSL4), a necessary enzyme for catalyzing lipid peroxidation during ferroptosis, suggesting the silencing of miR-23a-3p as a promising option in sorafenib resistant HCC patients [64].
The reactivation of oncogenic pathways is a common mechanism of drug resistance to TKIs in HCC [65] and PI3K/AKT alterations might predict sorafenib resistance [10]. We described the dual role of miR-30e-3p on tumorigenesis and sorafenib resistance based on TP53 status [29]. We showed that miR-30e-3p behaves as a TS miRNA in p53 wild type cells establishing a feedforward loop with TP53/MDM2 axis while it behaves as an oncogene in p53 mutated backgrounds targeting PTEN/AKT pathway and driving drug resistance. In the DEN-HCC rat model treated with sorafenib, which highly mirrors the human disease [66], a lower miR-30e-3p expression was detected in non-responder tumors displaying a negative correlation between miR-30e-3p and tumor size and a positive correlation with apoptotic markers, demonstrating the involvement of this miRNA in sorafenib sensitization. Another study reported the downregulation of miR-124-3p.1 in liver tumors and described its role in sorafenib response by targeting SIRT1 and AKT2 preventing nuclear translocation of FOXO3a transcription factor. Treatment combination between miR-124-3p.1 mimics and sorafenib improved its antitumor effect in a nude mouse model [67]. Several evidence reported the upregulation of the IGF/FGF pathways during acquired resistance to sorafenib [68,69]. Lin and colleagues investigated the mechanisms that lead to miRNA deregulation in SR cells. They identified the downregulation of exportin 5 (XPO5) via DNA promoter methylation to be responsible for impaired miR-378a maturation driving IGF1R signaling activation [70]. The anti-cancer strategy pursued by these Authors took advantage of GW3965, an agonist molecule of the transcription factor LXRα which mediates miR-378a transcription. Sorafenib plus GW3965 therapy demonstrated a consistent inhibition of tumor growth compared with sorafenib alone in both orthotopic and patient-derived xenograft (PDX) mouse models demonstrating the regulation of miRNA biogenesis as a promising option to improve sorafenib effectiveness in HCC. FGFR4 and EGFR oncogenes are upregulated in SR resistant cell lines and are direct targets of miR-486-3p which is downregulated in liver tumors and correlates with poor survival [71]. Intratumor injection of lentiviral particles carrying miR-486-3p in sorafenib resistant SK-Hep1-derived orthotopic mice synergistically improved sorafenib efficacy. Similarly, circular RNA named circRNA-SORE resulted upregulated in SR HCC cells, xenograft and PDX models due to increased N6-methyladenosine (m6A) levels that positively influenced its mRNA stability. Lower circRNA-SORE associated with better OS and recurrence-free survival in sorafenib-treated patients. It acted as a ceRNA by sequestering miR-103a-2-5p and miR-660-3p, promoting Wnt/β-catenin pathway activation that triggers and maintains a drug resistant phenotype. Orthotopic models with sorafenib resistant SK-Hep1 cells silenced for circRNA-SORE displayed a higher sensitization to sorafenib treatment. In agreement, intratumor injection of short hairpin (sh-circRNA-SORE) lentiviral particles in sorafenib resistant HCCLM3-derived xenograft mice potentiated the antitumor effect of sorafenib, suggesting the clinical potential of ncRNAs-based combined strategies [72]. The only concern relative to the last two studies regards the use of the SK-Hep1 cell line for the establishment of orthotopic animal models. Indeed, SK-Hep1 cells originate from liver endothelial cells and not from parenchymal tumor hepatocytes. The use of inappropriate animal models may be one of the causes affecting preclinical data translation into the clinical practice, therefore particular attention should be paid when choosing preclinical tools. Circular RNA cDCBLD2 is upregulated in SR cell lines where it sponged miR-345-5p increasing type IIA topoisomerase expression (TOP2A) that reduced the sorafenib-mediated apoptotic effect. Higher TOP2A expression associated with recurrence, and metastasis of HCC patients treated with sorafenib and with worse OS and recurrence-free survival. Local injection of cholesterol-conjugated small interfering RNA molecules (si-cDCBLD2) in a sorafenib-resistant PDX model increased drug sensitivity, supporting the clinical potential of cDCBLD2 silencing to enhance sorafenib efficacy in resistant patients [73].
Regarding the role played by autophagy on sorafenib resistance, Li and coworkers reported the nuclear activation of the lncRNA SNHG1 by miR-21 in SR HCC cells and described the activation of the AKT pathway via SLC3A2 upregulation [74]. Interestingly, in vitro inhibition of this lncRNA by an anti-SNHG1 siRNA strategy induced sorafenib sensitization through the activation of autophagy and apoptotic cascade; moreover, it showed tumor inhibition in vivo exerting a synergistic effect with sorafenib co-administration. On the contrary, miR-541 sensitized HCC cells to sorafenib treatment by inhibiting the expression of two autophagy-related genes, ATG2A and RAB1B, highlighting the opposite role attributed to autophagy on drug sensitization [75]. MiR-541 is downregulated in HCC and its low expression correlates with shorter OS and high recurrence rate and predicts sorafenib resistance. Notably, intratumor injection of Ad-miR-541 potentiated the effects of sorafenib in xenograft mice resulting in maximal tumor growth inhibition [76].
Considering the central activity of liver cytochrome P450 family in drug metabolism, He et al. investigated the effect of CYP3A4 on sorafenib metabolism and clearance and displayed that a higher expression associates with poor survival in sorafenib-treated patients [77]. The Authors demonstrated CYP3A4 targeting by miR-4277 and reported an addictive antitumor effect of miRNA mimics plus sorafenib in immunocompromised mice. To this regard, the study by Li and colleagues dissected the mechanisms downstream the decreased expression of miR-138-1-3p in HCC and found the serine/threonine kinase PAK5 among its targets. PAK5 upregulation triggered β-catenin phosphorylation causing its nuclear translocation which, in turn, activated the transcription of the multidrug resistant transporter ABCB1 responsible for sorafenib efflux and decreased effect. Notably, combined treatments with lentiviral vectors for miR-138-1-3p or PAK5 shRNA together with sorafenib had an enhanced anticancer effect with respect to sorafenib monotherapy in SR HepG2-derived xenograft mice [78].
In conclusion, ncRNAs regulate sorafenib resistance through a variety of molecular mechanisms (Figure 1). Preclinical studies demonstrated that combined strategies designed to restore the deregulated expression of ncRNAs enhance sorafenib sensitization in HCC, opening the path towards the design of focused clinical trials to improve treatment efficacy and patient survival.
3.2. Non-coding RNAs and lenvatinib combination improves the therapeutic response
Lenvatinib is an oral TKI targeting VEGFR, FGFR, PDGFRa, RET, KIT [79]. A randomized phase III clinical trial demonstrated that lenvatinib is non-inferior to sorafenib, showing an OS of 13.6 months; therefore, it granted approval as first line treatment in 2018 [4]. However, only a low percentage of advanced HCC patients benefit from lenvatinib, with the great majority being non-responder or developing drug resistance before or during treatment [80]. For this reason, the knowledge of the underlying molecular mechanisms and the discovery of new target genes for combination strategies are urgent clinical needs aimed at improving lenvatinib efficacy.
Wei et al. showed that miR-3154 influence lenvatinib response, being upregulated in lenvatinib-resistant (LR) HCC cells. To validate this hypothesis, miR-3154 was silenced in HCC cells treated with lenvatinib, resulting in reduced cancer stem cell markers, colony formation and increased apoptosis. These effects were confirmed in PDX mouse models, where tumor volume was reduced upon lenvatinib treatment in low miR-3154 tumors only. Mechanistically, miR-3154 target the transcription factor hepatocyte nuclear factor 4 alpha (HNF4α) which is indispensable for hepatocyte differentiation and critical for maintaining liver health, preventing its nuclear translocation. Moreover, in a cohort of HCCs receiving lenvatinib after surgical resection, patients with low miR-3154 levels had a better survival compared to those bearing high levels. Considering that low miRNA levels correlate with a better response, the preliminary evaluation of miR-3154 in HCC tissue could help identify in advance patients who may benefit from lenvatinib before treatment start. On the other hand, if confirmed by other studies, miR-3154 could represent a therapeutic target improving lenvatinib sensitivity in HCC [81]. MiR-183-5p.1 promotes the expansion of liver tumor-initiating cells (T-ICs) by regulating the expression of MUC15, a membrane-associated mucin whose downregulation was previously associated with advanced stages, poorly differentiated and metastatic liver cancers [82]. Han et al. showed that downregulation of MUC15 elevated the expression of T-IC associated markers, promoting malignant transformation of hepatocytes and spheroid formation in vitro. Consistently, downregulation or deletion of MUC15 in murine models dramatically increased tumor number, size, and liver-to-body weight ratio. These effects were mediated by increased levels of c-MET, PI3K and p-AKT, revealing the existence of a miR-183-5p.1/MUC15/c-MET/PI3K/AKT/SOX2 regulatory circuit in liver T-ICs. In line with these results, miR-183-5p was upregulated in HCCs compared with normal tissues. On the contrary, HCC patients with high MUC15 expression displayed a prolonged survival following lenvatinib treatment, suggesting its evaluation as a predictor of lenvatinib response. In agreement, patient-derived organoid (PDO) and PDX models expressing low MUC15 levels were resistant to lenvatinib treatment. Given these evidence, the administration of anti-miR-183-5p.1 could be a possible strategy to increase MUC15 levels in HCC patients treated with lenvatinib [83]. Another miRNA involved in the regulation of oncogenic pathways is miR-128-3 that mediates lenvatinib resistance in HCC cells by downregulating c-Met [84]. In LR HCC cells, miR-128-3p mimics strengthened the anti-proliferative effects of lenvatinib by directly targeting c-Met resulting in the downregulation of the ERK/cyclin D1 pathway, which is involved in cell cycle progression. In addition, miR-128-3p mimics enhanced lenvatinib-induced apoptosis in LR-HCC cells through the downregulation of p-Akt and p-GSK-3β, and the increase of caspase-9 and -3 cleavage. In xenograft mice injected with LR HCC cells, both lenvatinib treatment and miR-128-3p mimics resulted in significantly smaller tumors compared to controls. Notably, the combination therapy led to even smaller tumors than each monotherapy, showing higher apoptosis and lower proliferation indexes together with reduced p-Akt and p-GSK-3β expression. These evidence suggest that the combinatorial therapy of lenvatinib plus miR-128-3p mimics could be explored in clinical trials to further increase the efficacy of lenvatinib and possibly overcome the development of resistance.
Liu et al. revealed that low basal circKCNN2 levels associate with worse prognosis and tumor recurrence in HCC patients but, on the other side, predispose to a stronger anti-tumor effect of lenvatinib via miR-520c-3p/MBD2 axis [85]. Mechanistically, by sponging miR-520c-3p, circKCNN2 avoids its binding to methyl-DNA-binding domain protein 2 (MBD2), resulting in reduced proliferation, migration, colony formation and cell cycle progression in HCC cells, and lower tumor burden in vivo. Moreover, cells and PDOs with lower intrinsic circKCNN2 levels were more sensitive to lenvatinib treatment but have a higher risk of tumor recurrence. On the contrary, ectopic expression of circKCNN2 together with lenvatinib treatment have synergistic effects, possibly because they both downregulate the FGF19/FGFR4/FRS2 pathway. Indeed, circKCNN2 represses FGFR4 through miR-520c-3p/MBD2 axis. In turn, intrinsic high levels of circKCNN2 may reduce the effectiveness of lenvatinib because the FGF19/FGFR4/FRS2 pathway is already inhibited. Conclusively, this work revealed that circKCNN2 may be a promising predictive biomarker of HCC recurrence and treatment sensitivity, as well as a therapeutic agent in low-expressing patients in combination with lenvatinib, even though caution should be paid due to its dual role on drug sensitivity and tumor recurrence.
MT1JP is a lncRNA acting as a ceRNA for miR-24-3p. Yu et al. found the upregulation of MT1JP in LR HCC cells and showed that lenvatinib itself promotes MT1JP expression in vitro. Conversely, viability and apoptosis assays showed that the overexpression of miR-24-3p sensitizes HCC cells to lenvatinib. To better understand the molecular mechanisms governing MT1JP/miR-24-3p-mediated lenvatinib response, the authors demonstrated that the antiapoptotic factor BCL2L2 is a miR-24-3p target gene, and its expression confers a survival advantage to lenvatinib-treated cells. In PDXs treated with lenvatinib, responder tumors had low MT1JP and high miR-24-3p levels, together with increased apoptotic markers, compared to non-responders. Moreover, injection of MT1JP-overexpressing SMMC-7721 cells gave rise to bigger tumors in lenvatinib-treated xenograft mice. These data suggested that MTJ1P silencing or miR-24-3p mimics could be used as co-treatments to increase lenvatinib efficacy in HCC [86]. Another lncRNA upregulated in LR HCC cells, PDOs and patients is LINC01607. Notably, a significant reduction of ROS production was found in an orthotopic HCC model following LINC01607 overexpression being responsible for in vivo lenvatinib resistance. Mitophagy was activated following lenvatinib treatment, suggesting its contribution to the enhanced antioxidant capacity of LR HCC cells helping them to maintain low oxidative stress levels. To explain molecular mechanisms underneath LINC01607 deregulation, it emerged that it acts as a ceRNA for miR-892b increasing p62-associated mitophagy. P62 also regulates Nrf2 expression, which in turn protects cancer cells from oxidative stress regulating the expression of several antioxidant genes. Finally, a xenograft model with LR Hep3B cells silenced for LINC01607 demonstrated its synergistic effect with lenvatinib treatment and the same was confirmed in the PDO model. Taken together, these results indicated that LINC01607 promotes antioxidant capacity of LR HCC cells through miR-892b/p62/Nrf2 axis [87], suggesting non-coding RNAs as promising therapeutic targets to overcome lenvatinib resistance in HCC.
Given the central role of miRNAs in regulating HCC progression and response to treatment, they were also explored as therapies on their own. This is the case of miR-22, whose reduced expression was linked to poor survival outcome in patients with HCC. Hu et al. proved that miR-22 gene therapy is an effective treatment in two orthotopic HCC mouse models, ensuring prolonged survival compared to lenvatinib, without causing detectable toxicity [88]. The anti-HCC effects of miR-22 were mediated by its immunomodulatory functions on T cells. Indeed, miR-22 silenced HIF-1A and increased retinoic acid (RA) signaling in both hepatocytes and T cells, therefore repressing IL-17 pro-inflammatory signaling and inhibiting Th17 and Treg cells expansion, while enhancing cytotoxic CD8+ T cells recruitment, activation, and survival. Additionally, miR-22 treatment improved metabolism, inhibited inflammation, and reduced hypoxia signaling. These data suggested miR-22 gene therapy as a novel effective option for HCC treatment that may also empower the effect of immunotherapy by favoring a cytotoxic immune response against HCC. There is evidence of using lenvatinib as second line therapy for HCC patients undergoing sorafenib resistance. In this context, Shi et al. underlined the importance of considering lenvatinib influence on miRNAs expression profile in SR Huh-7 cells, thus identifying possible targets influencing HCC cells sensitivity to TKIs. For instance, lenvatinib treatment reduced the expression of two HCC-associated miRNAs (miR-130b and miR-106b) whose high levels associated with reduced OS in HCC patients. For this reason, studying this aspect and its molecular implications in depth could be of great significance to improve HCC management in sorafenib-resistant patients [89].
In summary, despite the low number of studies reporting mechanisms underneath lenvatinib resistance, here we showed that a variety of ncRNAs modulate lenvatinib response in HCC preclinical models (Figure 2) deserving attention as promising candidates for combined strategies.
4. Combination of ncRNAs-based strategies and ICIs improves therapeutic efficacy in HCC preclinical models
Immune checkpoint inhibitors greatly improved the survival of advanced HCCs. However, certain etiologies (e.g. NAFLD) or genetic backgrounds (e.g. β-catenin mutations) may affect ICIs therapeutic response in a substantial proportion of patients [9,10]. Moreover, therapeutic efficacy could be further improved in responders. An entangled network of interactions exists between tumor cells and cellular subpopulations belonging to the tumor microenvironment (TME). HCC cells can reprogram the expression pattern of several ncRNAs to: (I) alter cancer cell immunogenicity by decreasing the exposure of cancer-associated antigens, (II) promote the immune exhaustion of CD8+ T cells by regulating the expression of immune checkpoint inhibitors and (III) decrease the tumor infiltration of immune system cells such as CD8 + T cells, natural killer (NK) cells, dendritic cells (DCs), and macrophages by modulating the release of pro-inflammatory and anti-inflammatory cytokines. The specific deregulation of ncRNAs in immune cells also contributes to impaired antitumor immunity. The peculiar ability of miRNAs to fine-tune gene expression makes them ideal candidates to precisely modulate immune system components. This is the case of miR-206 whose expression is reduced not only in neoplastic hepatocytes but also in Kupffer cells (KCs) driving M2 polarization and depletion of cytotoxic T cells (CTLs) in AKT/Ras mice leading to early tumor lethality. Specific overexpression of miR-206 in KCs promoted M1 polarization by targeting kruppel like factor 4 (Klf4) and, thereby, enhancing the production of M1 markers such as C-C motif chemokine ligand 2 (CCL2) that favored hepatic recruitment of CTLs. MiR-206 prevented HCC onset in mice with AKT/Ras genetic background, suggesting its potential use as an immunotherapeutic target [90]. Similarly, hydrodynamic injection of microRNA-15a/16-1 prevented HCC in 100% of AKT/Ras and c-Myc mice and, when used in therapeutic settings, promoted tumor regression in both animal models. Mechanistically, microRNA-15a/16-1 inhibited the transcriptional activation of the chemokine CCL22 by nuclear factor-kB (Nf-kB) targeting in KCs, therefore preventing Tregs recruitment and dysregulation of CD8+ T cells. This miRNA cluster represents a potential candidate for immunotherapy against HCC alone or in combination with current clinical agents [91].
Interestingly, the metabolic reprogramming occurring in cancer cells can induce nutrient (e.g., glucose) competition with immune system cells or can lead to the accumulation of immunosuppressive metabolites that hamper the antitumor immunity. Clinical trials evaluating the combined effect of metabolic modulators and immune checkpoint inhibitors are underway in many cancer types [92]. Cai and coworkers discovered a novel circular RNA upregulated in HCC, named circRHBDD1, which correlates with tumor number and size, MVI and AFP levels and predicts poor prognosis. Experimental models showed that circRHBDD1 triggers aerobic glycolysis by regulating the expression of metabolic genes HK2 and GLUT1 leading to a decrease in the oxygen consumption rate while increasing lactate and ATP production and promoting in vivo tumorigenesis in a PDX mouse model. Mechanistically, circRHBDD1 interacts with the m6A-binding protein YTHDF1 favoring PIK3R1 translation and activating the PI3K/AKT pathway that contributes to the metabolic shift of cancer cells. Notably, higher levels of circRHBDD1 were detected in HCC patients with disease progression after receiving an anti-PD1 therapy. An immunocompetent xenograft model inoculated with circRHBDD1 silenced Hepa1-6 cells showed a stronger antitumor response and an improved survival in the presence of anti-PD1 treatment with respect to control cells, with a higher staining for CD8+ T cell infiltrates. These findings suggest that targeting cancer metabolism might synergistically enhance immunotherapy via metabolic reprogramming not only of cancer cells but also by reshaping the TME [93]. In line, dysregulation of genes belonging to fatty acid (FA) metabolism is considered an emerging cause of tumor aggressiveness and poor prognosis in several cancer types [94]. An in-silico study identified a FA metabolism-related lncRNAs signature which categorized patients from two online datasets on the basis of high or low FA metabolic score. Notably, patients with a lower FA metabolic score presented a higher immune cell infiltration score, an upregulation of critical immune checkpoint inhibitor genes and a higher tumor immune dysfunction and exclusion (TIDE) score, suggesting that this patient subgroup may experience immune escape and may have a lower probability of benefit from immunotherapy [95]. Similarly, a signature based on the expression of the lncRNA SNHG1 and on its target genes FANCD2 and G6PD, which expression is increased due to miR-199a sponging activity, displayed an association with poorer OS in patients within the high-risk group. These patients also showed the upregulation of multiple checkpoint molecules and a higher TIDE score suggesting that this signature might predict an immune suppressive tumor milieu, representing a potential marker for the decision-making of tailored therapeutic strategies in HCC [96].
An interesting study by Huang et al. identified the overexpression of circular RNA circMET in human HCCs and highlighted its correlation with tumor progression and shorter OS, as well as with EMT in HCC cells. Mechanistically, the ceRNA activity of circMET with respect to miR-30-5p family members drove the upregulation of the downstream target Snail that, in turn, transcriptionally activated dipeptidyl peptidase 4 (DPP4) whose expression deeply impact on insulin and glucose metabolism and immune cells regulation. Regarding this last aspect, immunocompetent animal models implanted with circMET Hepa1-6 cells showed lower CXCL10 serum levels and CD8+ T cells tumor infiltration. Strikingly, the combination between anti-PD1 inhibitor and anti-DPP4 inhibitor, sitagliptin, in the xenograft model induced a complete tumor regression, emphasizing the improved therapeutic efficacy of ICIs when combined with inhibitors of genes targeted by HCC-specific ncRNAs [97]. Regarding the modulation of DPP4 gene, Zhang et al. identified an oncogenic lncRNA LINC01132 which is overexpressed in human HCCs due to copy number amplification. Specifically, LINC01132 binds to NRF1 transcription factor that activates DPP4 promoter mediating its upregulation in HCC cells. LINC01132 silencing determined tumor growth inhibition in vivo in two HCC animal models (xenograft and PDX). Due to the well-known immune suppressive activity of DPP4 [98], and its association with decreased lymphocyte trafficking, a combination therapy of LINC01132 shRNAs and PD-L1 inhibitor was tested in the Hepa1–6 xenograft model, showing a clear tumor regression with respect to LINC01132 silencing alone and the highest positivity for CD8+ infiltrates, proving the efficacy of LINC01132 knockdown together with PD-L1 blockade [99].
An outstanding study by Fu et al. demonstrated the relevance of the myeloid-associated miRNA, miR-223, in cell-to-cell crosstalk modulating tumor hypoxia, angiogenesis and inflammatory tumor microenvironment that control HCC progression. The Authors proposed an intriguing model for chronic inflammation-associated HCCs in which myeloid cells represent the source for miR-223 transfer to cancer cells where it inhibits HIF-1α expression and indirectly influence the composition of the TME by suppressing HIF-1α-driven CD39/CD73-adenosine pathway that contributes to PD-1 and PD-L1 upregulation in immune cells. Adenovirus-mediated gene delivery of miR-223 in two inflammatory-associated models of HCC hindered tumor development and progression by inhibiting angiogenesis and hypoxia-mediated PD1/PD-L1 activation in T cells and macrophages, proving the therapeutic potential of miR-223 in blocking the immunosuppressive tumor microenvironment in HCC [100]. Notably, “RNA–RNA” crosstalk relies not only on non-coding RNAs functioning as “microRNA sponges”, but also on coding mRNAs which can relieve the inhibitory effect of miRNAs on their target genes by ceRNA activity. An example is represented by HMGB1 mRNA whose overexpression in HBV+ early-stage HCCs acts as a miRNA sponge to competitively bind the miR-200 family (miR-200a/200b/429) leading to RICTOR mRNA upregulation which, in turn, activates the AKT/mTORC1 pathway. This epigenetic crosstalk led to increased glutamine metabolism and release of PD-L1+ exosomes that affect immunotherapy response. In this context, HMGB1 is a new therapeutic target and biomarker of anti-PD-L1 efficacy in early HCC patients [101]. Other studies reported the incorporation of circRNAs into HCC-derived exosomes to be delivered to immune cell subpopulations and promoting their dysfunction. Hu and colleagues found the upregulation of circCCAR1 in tumor tissues and exosomal fraction from HCC which correlated with poor prognosis. In vivo experiments revealed an increase of tumor growth and metastasis of circCCAR1-OE HCCLM3 cells. Notably, by sponging miR-127-5p this circRNA increased the expression of Wilms tumor 1-associated protein (WTAP) that, in turn, mediated m6A modification and enhanced the stability of circCCAR1 itself. The extracellular secretion of circCCAR1 into exosomes determined CD8+ T cells dysfunction due to direct PD1 stabilization. Experiments in humanized NOD/SCID gamma (HuNSG) mice confirmed the decrease of CD8+ T cells infiltrate in tumors from circCCAR1-OE HCCLM3 cells and showed immune resistance to anti-PD1 therapy (Opdivo) with a decreased survival. In agreement, tissue and exosomal circCCAR1 levels were negatively related to CD8 + T-cells in HCC patients [102]. Similarly, a higher expression of circUHRF1 was reported in HCC tissues associating with poor prognosis and NK cell dysfunction. Indeed, exosomal embedding and extrusion of circUHRF1 inhibited NK population decreasing the secretion of pro-inflammatory cytokines IFN-γ and TNF-α. Mechanistically, circUHRF1 sponged miR-449c-5p determining the upregulating of TIM-3 expression which promoted NK cells exhaustion. A xenograft model with circUHRF1-knockdown HCCLM3 cells was established and NK cells were injected after tumor growth. Anti-PD1 sensitization and improved OS were observed in circUHRF1-knockdown mice, highlighting that circUHRF1 inhibition might be a promising strategy to ameliorate anti-PD1 efficacy in HCC [103].
Dong and coworkers demonstrated that oncogenic miR-93-5p is early deregulated during tumorigenesis. Its overexpression caused changes towards mesenchymal phenotype and malignant transformation of liver progenitor cells (LPCs) giving rise to 100% of tumors and metastasis when inoculated in animal models. A proteomic analysis revealed GAL-9 upregulation in miR-93-OE LPCs suggesting its inhibition in a therapeutic perspective. Indeed, it functions as a negative regulator of the innate response favoring antitumor immunity evasion of cancer cells. Despite anti-PD1 monoclonal antibodies had no effect against miR-93-OE-derived xenografts, tumor shrinkage was observed when combining anti-GAL-9 and anti-PD1 treatments, providing evidence for promising GAL-9 targeting in combined strategies for the treatment of LPC-like HCC subtypes [104].
In summary, the deregulation of several non-coding RNAs influence the relationship between tumor and immune cells often promoting the adoption of elusive mechanisms to escape from innate and adaptative immune response allowing cancer cells to expand and invade distant sites. In Table 1 are listed the therapeutic strategies adopted by different preclinical studies to prove the efficacy of novel candidates as immunotherapeutic agents alone or in combination with ICIs for the treatment of HCC.
5. Non-coding RNAs as biomarkers of treatment response in HCC
Non-coding RNAs are dysregulated in many cancer types including HCC and showed promise as treatment response biomarkers at tissue level [105]. In 2008, an outstanding study demonstrated for the first time the presence of miRNAs in body fluids such as serum and plasma [106]. MiRNAs can be secreted outside the cell through active or passive extrusion mechanisms, being incorporated into microvesicles or exosomes that protect them from RNase activity. Moreover, the intrinsic nature of miRNAs made them robust candidate in the case of repeated freeze-thaw cycles of biologic specimens. The ease of miRNA detection methods (e.g. Real Time PCR and digital PCR) [107] allows their analysis in liquid biopsy, a non-invasive procedure that favors the evaluation of repeated blood withdrawals to monitor patients during follow up assessments. Several studies attempted to identify tissue and blood biomarkers predictive to immunotherapy, but no robust results were obtained so far [108]. Despite the tissue may results a preferred source, biomarkers discovery could take advantage of liquid biopsy to overcome the matter of tumor heterogeneity and to avoid the hazard of liver biopsy at advanced stages.
Since sorafenib has been the only first-line therapeutic option for nearly a decade [2], showing modest survival benefit due to the onset of innate or early-acquired resistance [109], great efforts have been done to search for circulating candidates for an early switch to second-line agents. In this setting, our group reported sorafenib-mediated extrusion of oncomiR-221 in preclinical models showing an inverse correlation with tissue levels in treated animals (DEN-HCC rats and xenograft mice). To test whether miR-221 could be a predictive biomarker of sorafenib response, miR-221 levels were analyzed in the sera of two cohorts of HCC patients both before treatment start and during treatment (two-month follow up). In line with its oncogenic properties, lower miR-221 levels were detected in responder patients at basal level. On the contrary, comparisons between different time points showed an increase of serum miR-221 levels in responder patients when evaluated on treatment with respect to pre-treatment values. We suggested that, if confirmed in future studies, monitoring miR-221 circulating levels overtime in sorafenib-treated patients might represent a promising strategy to discriminate patients with prolonged response from those with early tumor escape, increasing the treatment window with second-line options for the last ones. Moreover, we demonstrated that high miR-221 tumor levels influence sorafenib resistance due to caspase-3 inhibition leading to decreased apoptotic cell death [110]. De la Cruz-Ojeda investigated the relationship between three sorafenib-deregulated miRNAs (miR-200c-3p, miR-222-5p and miR-512-3p) and sorafenib response in advanced HCCs. Notably, hazard ratio analysis showed an association between miR-200c-3p baseline levels and increased survival, while miR-222-5p and miR-512-3p analyzed at one month after sorafenib treatment associated with a poorer prognosis [111]. Since miR-221 and miR-222 belong to the same bicistronic cluster, findings between this and our study seem discordant, pointing out that further efforts need to be put in place to obtain robust data to translate into the laboratory routine, in terms of methodology and evaluation of different patient cohorts. Regarding the identification of early escape biomarkers, Gramantieri et al. demonstrated that sorafenib modulates exosome-mediated miR-30e-3p extrusion in a TP53-dependent manner promoting the increase of its circulating fraction. This finding was supported by the inverse correlation between tissue and serum miR-30e-3p levels observed in DEN-HCC rats and xenograft mice subjected to sorafenib treatment. In a preliminary cohort of sorafenib-treated HCC patients, higher circulating miR-30e-3p levels were found in the sorafenib-resistant group when evaluating samples collected after 2 months of treatment, suggesting miR-30e-3p as a possible candidate predicting the development of sorafenib resistance [29].
The recent approval of novel molecular-targeted drugs and immunotherapy-based therapeutic strategies has revolutionized the management of advanced HCC patients; notwithstanding, no biomarker entered the clinical practice to support clinicians in stratifying patients not eligible for ICIs. In sorafenib-treated patients, Fernández-Tussy and collaborators reported the relationship between higher miR-518d-5p circulating levels and shorter treatment duration and OS in BCLC-C patient subgroup only. Mechanistically, this oncogenic miRNA belonging to the C19MC targets PUMA and confers a survival advantage to cancer cells by enhancing their buffering capacity against ROS, maintaining membrane integrity and avoiding apoptosis during sorafenib treatment [112]. Nishida et al. performed a miRNA screening in the serum of 16 HCC patients treated with sorafenib, identifying miR-181a-5p and miR-339-5p as associated with disease response at 1 month of follow up. In a validation cohort (53 patients), miRNA levels decreased progressively among patient groups, showing high levels in partial responders, intermediate levels in patients with stable disease, and low levels in those with progressive disease. Again, when BCLC-C cases were considered, multivariate analysis at the 3-month follow up revealed miR-181a-5p as an independent factor for predicting disease control and OS. These last two studies underline the high heterogeneity of advanced HCCs and, as expected, report a better predictive performance of circulating miRNAs within more homogeneous patient subgroups [113]. The study by Shao et al. investigated miR-10b-3p role in regulating tumor response to sorafenib treatment. In preclinical models, sorafenib induced miR-10b-3p extrusion and higher miR-10b-3p levels associated with sorafenib sensitization, hypothesizing its possible use as biomarker for patients treated with sorafenib. In a patient cohort (N=45), higher miR-10b-3p serum levels predicted a better OS but not PFS when analyzed before treatment [114]. Interestingly, miR-10b is overexpressed in several tumors including HCC and its expression in the exosomal fraction from early-HCC patients is closely associated with tumor size and recurrence representing an independent prognostic factor for poor survival [115]. Since two miR-10b inhibitors has been developed based on straightforward advances in nanotechnology (TTX-MC138 and RGLS5579) demonstrating an effective anticancer activity in preclinical models [41], this miRNA investigation holds promise both as therapeutic target and predictive biomarker. Finally, a study by our research group reported the association between high miR-494 serum levels and sorafenib resistance in HCC patients at baseline [59]. In addition, we underlined the relationship between miR-494 serum levels and genes involved in metabolic reprogramming pointing out the possibility that circulating levels of this miRNA might not only predict sorafenib response but also identify tumors with a deregulated metabolism that could benefit from combined TKIs and antagomiRs or metabolic interference strategies. This preliminary study needs to be confirmed in larger patient cohorts to prove the predictive value of miR-494 for patient stratification to tailored treatments.
As for lenvatinib, one study assessed the correlations between serum biomarkers and efficacy outcomes from the REFLECT clinical trial. Remarkably, only serum proteins were tested by ELISA, whereas no data on circulating ncRNAs are reported. Briefly, higher baseline VEGF, ANG2, and FGF21 correlated with shorter OS with both sorafenib and lenvatinib treatments while a longer OS correlated with higher baseline FGF21 in the lenvatinb group compared with sorafenib, needing further confirmation [116].
Regarding second line agents, regorafenib is a multiple TKI blocking the activity of protein kinases regulating angiogenesis, proliferation, tumor microenvironment, and metastasis, including vascular endothelial growth factor receptors (VEGFR1-3), TIE2, KIT, RET, RAF-1, BRAF, platelet-derived growth factor receptor (PDGFR), and fibroblast growth factor receptor (FGFR). There is only one retrospective study performed in the RESORCE trial reporting the association between circulating miRNAs and prolonged OS in regorafenib-treated patients [117]. Plasma specimens from 343 HCC patients (234 regorafenib and 109 placebo) were assessed for the expression of 750 miRNAs. Nine circulating miRNAs (increased: MIR30A, MIR122, MIR125B, MIR200A, MIR374B; decreased: MIR15B, MIR107, MIR320; and absent: MIR645) were predictive of survival benefit with regorafenib. Top gene sets related to these miRNAs included liver cancer progression and metabolic pathways such as lipids, amino acids, bile acids, and xenobiotics metabolism and glucuronidation. Bioinformatics analysis revealed that patients with improved regorafenib response overlap with the well-differentiated S3 subtype of the Hoshida classification [118], which is characterized by a hepatocyte-like phenotype, well-differentiated, and smaller tumors. Notably, AFP and c-MET plasma levels were associated with decreased overall survival independently of regorafenib treatment while only five plasma proteins associated (angiopoietin 1 [ANG-1], cystatin B, the latency-associated peptide of transforming growth factor beta 1 [LAP TGF-b1], oxidized low-density lipoprotein receptor 1 [LOX-1], C-C motif chemokine ligand 3 [MIP-1a]) with treatment benefit.
Despite the measurement of tumor derived miRNAs in body fluids might represent an easy and promising approach for the blood-based detection of treatment response or early tumor escape in HCC (Table 2), further studies in larger patient cohorts or well-defined patient subgroups are needed to translate these findings into routine lab tests. Studies designed to validate biomarkers and to identify new ones are critical for the improvement of tailored treatments in HCC.
6. Conclusions
Preclinical studies unraveled molecular mechanisms underneath deregulated non-coding RNAs associated with drug resistance phenotype in HCC and demonstrated their potential in combined therapeutic strategies to be evaluated in ‘ad hoc’ clinical trials with well-defined nanoparticle delivery systems. Because of the numerous non-coding RNAs with a role in treatment response, it remains challenging to select the most promising candidates for combined therapeutic interventions. Notably, some studies reported the feasibility and efficacy of blocking downstream targets of HCC-associated ncRNAs by using inhibitors or monoclonal antibodies, increasing the range of possible therapeutic approaches for overcoming HCC drug resistance, contributing to a better outcome in advanced patients. Since liver biopsy is often not feasible in patients with advanced diseases, the use of preclinical models can provide useful information about the relationship between circulating and tissue ncRNAs which may help selecting subgroups of patients eligible for combined treatments based on extracellular levels. Notably, the first phase 1 clinical trial testing a miRNA mimic formulation in various metastatic cancer types, including HCC, was terminated owing to serious immune mediated adverse events [40]. The tested drug was MRX34 which is a synthetic double-stranded miR-34a mimic encapsulated in liposomal nanoparticles (ClinicalTrials.gov identifier: NCT01829971). The lesson from this study was that improvements in synthetic miRNA mimics and delivery systems are necessary. Despite this first failure, other clinical trials testing miRNA formulations in cancer are ongoing [41], pointing out the potential of miRNA therapy as a next-generation strategy.
Regarding blood biomarkers, the translation of preclinical findings is still far from being put into practice, and several causes may be responsible for this issue. First, there are too many variables that prevent a complete comparison between the different studies, to name a few: different samples (plasma or serum or exosomes), different analytical methodologies (qPCR, ddPCR, RNAseq), different etiologies among the human cohorts, and different time points (at baseline or on treatment). In addition, because of the heterogeneity of advanced HCCs it could be necessary to divide patients in more homogeneous subgroups (e.g. BCLC-C, etiology, driver gene mutations, etc.) where the predictive value of biomarkers might perform better. Additional clinical needs concern patients not eligible for immunotherapy for whom remain the choice among sorafenib and lenvatinib, and patients undergoing tumor escape to first-line treatments to improve the likelihood of success to second-line agents. Finally, the identification of multi-parameter signatures combining one or more noncoding RNAs and clinicopathological variables (e.g., serum AFP, albumin, bilirubin, etc.) might increase the predictive score in terms of treatment response.
In conclusion, giant strides have been made in preclinical studies regarding the possible use of non-coding RNAs-based strategies to improve the efficacy of current therapies. Results from ongoing clinical trials in cancer patients will allow a step forward in the near future, when combined approaches to overcome the onset of drug resistance in HCC patients can be tested.
Author Contributions
F.F. planned the manuscript’s structure and content; C.V., E.M., I.L., G.G., and F.F. performed accurate literature review and wrote the paper; C.V., E.M., I.L., and G.G. drew the figures; L.G., C.G., F.P., C.S., and F.F. performed manuscript supervision and revision. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by: AIRC under IG 2020 - ID. 25187 – P.I. Fornari Francesca: “Identification of circulating biomarkers for patient allocation to the best treatment in hepatocellular carcinoma”. Bando “Alma Idea 2022” University of Bologna – P.I. Fornari Francesca: “Development of 3D models, preclinical tools preclinical and circulating biomarkers for the optimization of personalized treatment approaches in hepatocellular carcinoma (SIMPLEMEDICINE)”. The research leading to these results has received funding from the European Union - NextGenerationEU through the Italian Ministry of University and Research under PNRR - M4C2-I1.3 Project PE_00000019 "HEAL ITALIA", CUP of Institution: I53C22001440006. The views and opinions expressed are those of the authors only and do not necessarily reflect those of the European Union or the European Commission. Neither the European Union nor the European Commission can be held responsible for them.
Acknowledgments
The authors wish to acknowledge Dr. Marco Malaguti (University of Bologna, Italy) for the license to use BioRender.com for creating the images.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.-F.; de Oliveira, A.C.; Santoro, A.; Raoul, J.-L.; Forner, A.; et al. Sorafenib in Advanced Hepatocellular Carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef]
- Bruix, J.; Qin, S.; Merle, P.; Granito, A.; Huang, Y.-H.; Bodoky, G.; Pracht, M.; Yokosuka, O.; Rosmorduc, O.; Breder, V.; et al. Regorafenib for Patients with Hepatocellular Carcinoma Who Progressed on Sorafenib Treatment (RESORCE): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet 2017, 389, 56–66. [Google Scholar] [CrossRef]
- Kudo, M.; Finn, R.S.; Qin, S.; Han, K.-H.; Ikeda, K.; Piscaglia, F.; Baron, A.; Park, J.-W.; Han, G.; Jassem, J.; et al. Lenvatinib versus Sorafenib in First-Line Treatment of Patients with Unresectable Hepatocellular Carcinoma: A Randomised Phase 3 Non-Inferiority Trial. Lancet 2018, 391, 1163–1173. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Meyer, T.; Cheng, A.-L.; El-Khoueiry, A.B.; Rimassa, L.; Ryoo, B.-Y.; Cicin, I.; Merle, P.; Chen, Y.; Park, J.-W.; et al. Cabozantinib in Patients with Advanced and Progressing Hepatocellular Carcinoma. N. Engl. J. Med. 2018, 379, 54–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.X.; Kang, Y.-K.; Yen, C.-J.; Finn, R.S.; Galle, P.R.; Llovet, J.M.; Assenat, E.; Brandi, G.; Pracht, M.; Lim, H.Y.; et al. Ramucirumab after Sorafenib in Patients with Advanced Hepatocellular Carcinoma and Increased α-Fetoprotein Concentrations (REACH-2): A Randomised, Double-Blind, Placebo-Controlled, Phase 3 Trial. Lancet Oncol. 2019, 20, 282–296. [Google Scholar] [CrossRef]
- Abou-Alfa, G.K.; Chan, S.L.; Kudo, M.; Lau, G.; Kelley, R.K.; Furuse, J.; Sukeepaisarnjaroen, W.; Kang, Y.-K.; Dao, T.V.; De Toni, E.N.; et al. Phase 3 Randomized, Open-Label, Multicenter Study of Tremelimumab (T) and Durvalumab (D) as First-Line Therapy in Patients (Pts) with Unresectable Hepatocellular Carcinoma (uHCC): HIMALAYA. J. Clin. Oncol. 2022, 40, 379–379. [Google Scholar] [CrossRef]
- Reig, M.; Forner, A.; Rimola, J.; Ferrer-Fàbrega, J.; Burrel, M.; Garcia-Criado, Á.; Kelley, R.K.; Galle, P.R.; Mazzaferro, V.; Salem, R.; et al. BCLC Strategy for Prognosis Prediction and Treatment Recommendation: The 2022 Update. J. Hepatol. 2022, 76, 681–693. [Google Scholar] [CrossRef]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH Limits Anti-Tumour Surveillance in Immunotherapy-Treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Harding, J.J.; Nandakumar, S.; Armenia, J.; Khalil, D.N.; Albano, M.; Ly, M.; Shia, J.; Hechtman, J.F.; Kundra, R.; El Dika, I.; et al. Prospective Genotyping of Hepatocellular Carcinoma: Clinical Implications of Next-Generation Sequencing for Matching Patients to Targeted and Immune Therapies. Clin. Cancer Res. 2019, 25, 2116–2126. [Google Scholar] [CrossRef]
- Afra, F.; Mahboobipour, A.A.; Salehi Farid, A.; Ala, M. Recent Progress in the Immunotherapy of Hepatocellular Carcinoma: Non-Coding RNA-Based Immunotherapy May Improve the Outcome. Biomed. Pharmacother. 2023, 165, 115104. [Google Scholar] [CrossRef]
- Fornari, F.; Giovannini, C.; Piscaglia, F.; Gramantieri, L. Elucidating the Molecular Basis of Sorafenib Resistance in HCC: Current Findings and Future Directions. J. Hepatocell. Carcinoma 2021, 8, 741–757. [Google Scholar] [CrossRef]
- Mattick, J.S.; Makunin, I.V. Non-Coding RNA. Hum. Mol. Genet. 2006, 15 Spec No 1, R17-29. [Google Scholar] [CrossRef]
- Esteller, M. Non-Coding RNAs in Human Disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef]
- Nemeth, K.; Bayraktar, R.; Ferracin, M.; Calin, G.A. Non-Coding RNAs in Disease: From Mechanisms to Therapeutics. Nat. Rev. Genet. 2023. [Google Scholar] [CrossRef]
- Kopp, F.; Mendell, J.T. Functional Classification and Experimental Dissection of Long Noncoding RNAs. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef]
- Huang, Z.; Zhou, J.-K.; Peng, Y.; He, W.; Huang, C. The Role of Long Noncoding RNAs in Hepatocellular Carcinoma. Mol. Cancer 2020, 19, 77. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Lyu, T.; Li, H.; Liu, C.; Xie, K.; Xu, L.; Li, W.; Liu, H.; Zhu, J.; Lyu, Y.; et al. LncRNA CEBPA-DT Promotes Liver Cancer Metastasis through DDR2/β-Catenin Activation via Interacting with hnRNPC. J. Exp. Clin. Cancer Res. 2022, 41, 335. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Hu, Y.; Wang, H.; Hu, P.; Xiong, H.; Zeng, Z.; Han, S.; Wang, D.; Wang, J.; Zhao, Y.; et al. LncRNA FTO-IT1 Promotes Glycolysis and Progression of Hepatocellular Carcinoma through Modulating FTO-Mediated N6-Methyladenosine Modification on GLUT1 and PKM2. J. Exp. Clin. Cancer Res. 2023, 42, 267. [Google Scholar] [CrossRef]
- Zhou, W.-Y.; Cai, Z.-R.; Liu, J.; Wang, D.-S.; Ju, H.-Q.; Xu, R.-H. Circular RNA: Metabolism, Functions and Interactions with Proteins. Mol. Cancer 2020, 19, 172. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Liang, M.; Liu, H.; Huang, J.; Li, P.; Wang, C.; Zhang, Y.; Lin, Y.; Jiang, X. CircRNA hsa_circRNA_104348 Promotes Hepatocellular Carcinoma Progression through Modulating miR-187-3p/RTKN2 Axis and Activating Wnt/β-Catenin Pathway. Cell Death Dis. 2020, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Long, H.; Zheng, Q.; Bo, X.; Xiao, X.; Li, B. Circular RNA circRHOT1 Promotes Hepatocellular Carcinoma Progression by Initiation of NR2F6 Expression. Mol. Cancer 2019, 18, 119. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-Y.; Huang, Z.-L.; Huang, J.; Xu, B.; Huang, X.-Y.; Xu, Y.-H.; Zhou, J.; Tang, Z.-Y. Exosomal circRNA-100338 Promotes Hepatocellular Carcinoma Metastasis via Enhancing Invasiveness and Angiogenesis. J. Exp. Clin. Cancer Res. 2020, 39, 20. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many Roads to Maturity: microRNA Biogenesis Pathways and Their Regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef]
- Hu, W.; Liu, C.; Bi, Z.-Y.; Zhou, Q.; Zhang, H.; Li, L.-L.; Zhang, J.; Zhu, W.; Song, Y.-Y.-Y.; Zhang, F.; et al. Comprehensive Landscape of Extracellular Vesicle-Derived RNAs in Cancer Initiation, Progression, Metastasis and Cancer Immunology. Mol. Cancer 2020, 19, 102. [Google Scholar] [CrossRef]
- Zhou, Y.; Ren, H.; Dai, B.; Li, J.; Shang, L.; Huang, J.; Shi, X. Hepatocellular Carcinoma-Derived Exosomal miRNA-21 Contributes to Tumor Progression by Converting Hepatocyte Stellate Cells to Cancer-Associated Fibroblasts. J. Exp. Clin. Cancer Res. 2018, 37, 324. [Google Scholar] [CrossRef]
- Shenouda, S.K.; Alahari, S.K. MicroRNA Function in Cancer: Oncogene or a Tumor Suppressor? Cancer Metastasis Rev. 2009, 28, 369–378. [Google Scholar] [CrossRef]
- Fornari, F.; Milazzo, M.; Galassi, M.; Callegari, E.; Veronese, A.; Miyaaki, H.; Sabbioni, S.; Mantovani, V.; Marasco, E.; Chieco, P.; et al. P53/Mdm2 Feedback Loop Sustains miR-221 Expression and Dictates the Response to Anticancer Treatments in Hepatocellular Carcinoma. Mol. Cancer Res. 2014, 12, 203–216. [Google Scholar] [CrossRef]
- Gramantieri, L.; Pollutri, D.; Gagliardi, M.; Giovannini, C.; Quarta, S.; Ferracin, M.; Casadei-Gardini, A.; Callegari, E.; De Carolis, S.; Marinelli, S.; et al. MiR-30e-3p Influences Tumor Phenotype through MDM2/TP53 Axis and Predicts Sorafenib Resistance in Hepatocellular Carcinoma. Cancer Res. 2020, 80, 1720–1734. [Google Scholar] [CrossRef]
- Calin, G.A.; Croce, C.M. MicroRNA Signatures in Human Cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef]
- Gramantieri, L.; Ferracin, M.; Fornari, F.; Veronese, A.; Sabbioni, S.; Liu, C.-G.; Calin, G.A.; Giovannini, C.; Ferrazzi, E.; Grazi, G.L.; et al. Cyclin G1 Is a Target of miR-122a, a microRNA Frequently down-Regulated in Human Hepatocellular Carcinoma. Cancer Res. 2007, 67, 6092–6099. [Google Scholar] [CrossRef]
- Ladeiro, Y.; Couchy, G.; Balabaud, C.; Bioulac-Sage, P.; Pelletier, L.; Rebouissou, S.; Zucman-Rossi, J. MicroRNA Profiling in Hepatocellular Tumors Is Associated with Clinical Features and Oncogene/Tumor Suppressor Gene Mutations. Hepatology 2008, 47, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Budhu, A.; Jia, H.-L.; Forgues, M.; Liu, C.-G.; Goldstein, D.; Lam, A.; Zanetti, K.A.; Ye, Q.-H.; Qin, L.-X.; Croce, C.M.; et al. Identification of Metastasis-Related microRNAs in Hepatocellular Carcinoma. Hepatology 2008, 47, 897–907. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef] [PubMed]
- Elmén, J.; Lindow, M.; Schütz, S.; Lawrence, M.; Petri, A.; Obad, S.; Lindholm, M.; Hedtjärn, M.; Hansen, H.F.; Berger, U.; et al. LNA-Mediated microRNA Silencing in Non-Human Primates. Nature 2008, 452, 896–899. [Google Scholar] [CrossRef]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in Vivo with “Antagomirs”. Nature 2005, 438, 685–689. [Google Scholar] [CrossRef]
- Komoll, R.-M.; Hu, Q.; Olarewaju, O.; von Döhlen, L.; Yuan, Q.; Xie, Y.; Tsay, H.-C.; Daon, J.; Qin, R.; Manns, M.P.; et al. MicroRNA-342-3p Is a Potent Tumour Suppressor in Hepatocellular Carcinoma. J. Hepatol. 2021, 74, 122–134. [Google Scholar] [CrossRef]
- Kota, J.; Chivukula, R.R.; O’Donnell, K.A.; Wentzel, E.A.; Montgomery, C.L.; Hwang, H.-W.; Chang, T.-C.; Vivekanandan, P.; Torbenson, M.; Clark, K.R.; et al. Therapeutic microRNA Delivery Suppresses Tumorigenesis in a Murine Liver Cancer Model. Cell 2009, 137, 1005–1017. [Google Scholar] [CrossRef] [PubMed]
- Callegari, E.; Domenicali, M.; Shankaraiah, R.C.; D’Abundo, L.; Guerriero, P.; Giannone, F.; Baldassarre, M.; Bassi, C.; Elamin, B.K.; Zagatti, B.; et al. MicroRNA-Based Prophylaxis in a Mouse Model of Cirrhosis and Liver Cancer. Mol. Ther. Nucleic Acids 2019, 14, 239–250. [Google Scholar] [CrossRef]
- Hong, D.S.; Kang, Y.-K.; Borad, M.; Sachdev, J.; Ejadi, S.; Lim, H.Y.; Brenner, A.J.; Park, K.; Lee, J.-L.; Kim, T.-Y.; et al. Phase 1 Study of MRX34, a Liposomal miR-34a Mimic, in Patients with Advanced Solid Tumours. Br. J. Cancer 2020, 122, 1630–1637. [Google Scholar] [CrossRef]
- Kim, T.; Croce, C.M. MicroRNA: Trends in Clinical Trials of Cancer Diagnosis and Therapy Strategies. Exp. Mol. Med. 2023, 55, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Zanuso, V.; Rimassa, L.; Braconi, C. The Rapidly Evolving Landscape of HCC: Selecting the Optimal Systemic Therapy. Hepatology 2023. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 Exhibits Broad Spectrum Oral Antitumor Activity and Targets the RAF/MEK/ERK Pathway and Receptor Tyrosine Kinases Involved in Tumor Progression and Angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef]
- Luk, J.M.; Burchard, J.; Zhang, C.; Liu, A.M.; Wong, K.F.; Shek, F.H.; Lee, N.P.; Fan, S.T.; Poon, R.T.; Ivanovska, I.; et al. DLK1-DIO3 Genomic Imprinted microRNA Cluster at 14q32.2 Defines a Stemlike Subtype of Hepatocellular Carcinoma Associated with Poor Survival. J. Biol. Chem. 2011, 286, 30706–30713. [Google Scholar] [CrossRef]
- Lim, L.; Balakrishnan, A.; Huskey, N.; Jones, K.D.; Jodari, M.; Ng, R.; Song, G.; Riordan, J.; Anderton, B.; Cheung, S.-T.; et al. MicroRNA-494 within an Oncogenic microRNA Megacluster Regulates G1/S Transition in Liver Tumorigenesis through Suppression of Mutated in Colorectal Cancer. Hepatology 2014, 59, 202–215. [Google Scholar] [CrossRef]
- Pollutri, D.; Patrizi, C.; Marinelli, S.; Giovannini, C.; Trombetta, E.; Giannone, F.A.; Baldassarre, M.; Quarta, S.; Vandewynckel, Y.P.; Vandierendonck, A.; et al. The Epigenetically Regulated miR-494 Associates with Stem-Cell Phenotype and Induces Sorafenib Resistance in Hepatocellular Carcinoma. Cell Death Dis. 2018, 9, 4. [Google Scholar] [CrossRef]
- Gao, Y.; Yin, Z.; Qi, Y.; Peng, H.; Ma, W.; Wang, R.; Li, W. Golgi Phosphoprotein 3 Promotes Angiogenesis and Sorafenib Resistance in Hepatocellular Carcinoma via Upregulating Exosomal miR-494-3p. Cancer Cell Int. 2022, 22, 35. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Liu, Y.; Fang, X.; Liu, Y.; Fang, L.; Lin, L.; Liu, X.; Wang, N. Tumor-Derived microRNA-494 Promotes Angiogenesis in Non-Small Cell Lung Cancer. Angiogenesis 2015, 18, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Jiang, F.; Yang, Y.; Huang, G.; Pu, F.; Liu, Q.; Chen, L.; Ju, L.; Lu, M.; Zhou, F.; et al. 14-3-3η Is a Novel Growth-Promoting and Angiogenic Factor in Hepatocellular Carcinoma. J. Hepatol. 2016, 65, 953–962. [Google Scholar] [CrossRef]
- Qiu, Y.; Shan, W.; Yang, Y.; Jin, M.; Dai, Y.; Yang, H.; Jiao, R.; Xia, Y.; Liu, Q.; Ju, L.; et al. Reversal of Sorafenib Resistance in Hepatocellular Carcinoma: Epigenetically Regulated Disruption of 14-3-3η/Hypoxia-Inducible Factor-1α. Cell Death Discov. 2019, 5, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Yu, S.; Chen, J.; Quan, M.; Gao, Y.; Li, Y. miR-15a and miR-20b Sensitize Hepatocellular Carcinoma Cells to Sorafenib through Repressing CDC37L1 and Consequent PPIA Downregulation. Cell Death Discov. 2022, 8, 1–11. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.-T.; Wu, J.; Feng, Z.; Yuan, H.; Li, Q.; Xing, T.; Xu, L.; Zhang, C.; Tan, H.-Y.; et al. CRISPR/Cas9 Screens Unravel miR-3689a-3p Regulating Sorafenib Resistance in Hepatocellular Carcinoma via Suppressing CCS/SOD1-Dependent Mitochondrial Oxidative Stress. Drug Resist. Updat. 2023, 71, 101015. [Google Scholar] [CrossRef]
- Wei, L.; Lee, D.; Law, C.-T.; Zhang, M.S.; Shen, J.; Chin, D.W.-C.; Zhang, A.; Tsang, F.H.-C.; Wong, C.L.-S.; Ng, I.O.-L.; et al. Genome-Wide CRISPR/Cas9 Library Screening Identified PHGDH as a Critical Driver for Sorafenib Resistance in HCC. Nat. Commun. 2019, 10, 4681. [Google Scholar] [CrossRef]
- Zheng, A.; Chevalier, N.; Calderoni, M.; Dubuis, G.; Dormond, O.; Ziros, P.G.; Sykiotis, G.P.; Widmann, C. CRISPR/Cas9 Genome-Wide Screening Identifies KEAP1 as a Sorafenib, Lenvatinib, and Regorafenib Sensitivity Gene in Hepatocellular Carcinoma. Oncotarget 2019, 10, 7058–7070. [Google Scholar] [CrossRef]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg Effect: The Metabolic Requirements of Cell Proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Chen, Z.; Zuo, X.; Zhang, Y.; Han, G.; Zhang, L.; Wu, J.; Wang, X. MiR-3662 Suppresses Hepatocellular Carcinoma Growth through Inhibition of HIF-1α-Mediated Warburg Effect. Cell Death Dis. 2018, 9, 549. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Xiao, X.; Han, Y.; Fan, D.; Zhu, Y.; Yang, L. MiR-3662 Suppresses Cell Growth, Invasion and Glucose Metabolism by Targeting HK2 in Hepatocellular Carcinoma Cells. Neoplasma 2020, 67, 773–781. [Google Scholar] [CrossRef] [PubMed]
- Bergamini, C.; Leoni, I.; Rizzardi, N.; Melli, M.; Galvani, G.; Coada, C.A.; Giovannini, C.; Monti, E.; Liparulo, I.; Valenti, F.; et al. MiR-494 Induces Metabolic Changes through G6pc Targeting and Modulates Sorafenib Response in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2023, 42, 145. [Google Scholar] [CrossRef]
- Zhang, Z.; Tan, X.; Luo, J.; Yao, H.; Si, Z.; Tong, J.-S. The miR-30a-5p/CLCF1 Axis Regulates Sorafenib Resistance and Aerobic Glycolysis in Hepatocellular Carcinoma. Cell Death Dis. 2020, 11, 902. [Google Scholar] [CrossRef]
- Li, J.; Zhang, Y.; Dong, P.-Y.; Yang, G.-M.; Gurunathan, S. A Comprehensive Review on the Composition, Biogenesis, Purification, and Multifunctional Role of Exosome as Delivery Vehicles for Cancer Therapy. Biomed. Pharmacother. 2023, 165, 115087. [Google Scholar] [CrossRef] [PubMed]
- Nicodemou, A.; Bernátová, S.; Čeháková, M.; Danišovič, Ľ. Emerging Roles of Mesenchymal Stem/Stromal-Cell-Derived Extracellular Vesicles in Cancer Therapy. Pharmaceutics 2023, 15, 1453. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Liu, Q.; Jiang, Y.; Cai, Z.; Liu, H.; Zuo, H. Engineered Small Extracellular Vesicles Loaded with miR-654-5p Promote Ferroptosis by Targeting HSPB1 to Alleviate Sorafenib Resistance in Hepatocellular Carcinoma. Cell Death Discov. 2023, 9, 362. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Chan, Y.-T.; Tan, H.-Y.; Zhang, C.; Guo, W.; Xu, Y.; Sharma, R.; Chen, Z.-S.; Zheng, Y.-C.; Wang, N.; et al. Epigenetic Regulation of Ferroptosis via ETS1/miR-23a-3p/ACSL4 Axis Mediates Sorafenib Resistance in Human Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 3. [Google Scholar] [CrossRef] [PubMed]
- Eun, J.W.; Yoon, J.H.; Ahn, H.R.; Kim, S.; Kim, Y.B.; Lim, S.B.; Park, W.; Kang, T.W.; Baek, G.O.; Yoon, M.G.; et al. Cancer-Associated Fibroblast-Derived Secreted Phosphoprotein 1 Contributes to Resistance of Hepatocellular Carcinoma to Sorafenib and Lenvatinib. Cancer Commun. 2023, 43, 455–479. [Google Scholar] [CrossRef]
- Fornari, F.; Gramantieri, L.; Callegari, E.; Shankaraiah, R.C.; Piscaglia, F.; Negrini, M.; Giovannini, C. MicroRNAs in Animal Models of HCC. Cancers 2019, 11, 1906. [Google Scholar] [CrossRef]
- Dong, Z.-B.; Wu, H.-M.; He, Y.-C.; Huang, Z.-T.; Weng, Y.-H.; Li, H.; Liang, C.; Yu, W.-M.; Chen, W. MiRNA-124-3p.1 Sensitizes Hepatocellular Carcinoma Cells to Sorafenib by Regulating FOXO3a by Targeting AKT2 and SIRT1. Cell Death Dis. 2022, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Tovar, V.; Cornella, H.; Moeini, A.; Vidal, S.; Hoshida, Y.; Sia, D.; Peix, J.; Cabellos, L.; Alsinet, C.; Torrecilla, S.; et al. Tumour Initiating Cells and IGF/FGF Signalling Contribute to Sorafenib Resistance in Hepatocellular Carcinoma. Gut 2017, 66, 530–540. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, J.; Ma, L.; Shan, J.; Shen, J.; Yang, Z.; Liu, L.; Luo, Y.; Yao, C.; Qian, C. MicroRNA-122 Confers Sorafenib Resistance to Hepatocellular Carcinoma Cells by Targeting IGF-1R to Regulate RAS/RAF/ERK Signaling Pathways. Cancer Lett. 2016, 371, 171–181. [Google Scholar] [CrossRef]
- Lin, Z.; Xia, S.; Liang, Y.; Ji, L.; Pan, Y.; Jiang, S.; Wan, Z.; Tao, L.; Chen, J.; Lin, C.; et al. LXR Activation Potentiates Sorafenib Sensitivity in HCC by Activating microRNA-378a Transcription. Theranostics 2020, 10, 8834–8850. [Google Scholar] [CrossRef]
- Ji, L.; Lin, Z.; Wan, Z.; Xia, S.; Jiang, S.; Cen, D.; Cai, L.; Xu, J.; Cai, X. miR-486-3p Mediates Hepatocellular Carcinoma Sorafenib Resistance by Targeting FGFR4 and EGFR. Cell Death Dis. 2020, 11, 250. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wan, Z.; Tang, M.; Lin, Z.; Jiang, S.; Ji, L.; Gorshkov, K.; Mao, Q.; Xia, S.; Cen, D.; et al. N6-Methyladenosine-Modified CircRNA-SORE Sustains Sorafenib Resistance in Hepatocellular Carcinoma by Regulating β-Catenin Signaling. Mol. Cancer 2020, 19, 163. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.; Chen, T.; Zheng, L.; Cai, J.; Zhao, H.; Wang, Y.; Tao, L.; Xu, J.; Ji, L.; Cai, X. cDCBLD2 Mediates Sorafenib Resistance in Hepatocellular Carcinoma by Sponging miR-345-5p Binding to the TOP2A Coding Sequence. Int. J. Biol. Sci. 2023, 19, 4608–4626. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Dong, X.; He, C.; Tan, G.; Li, Z.; Zhai, B.; Feng, J.; Jiang, X.; Liu, C.; Jiang, H.; et al. LncRNA SNHG1 Contributes to Sorafenib Resistance by Activating the Akt Pathway and Is Positively Regulated by miR-21 in Hepatocellular Carcinoma Cells. J. Exp. Clin. Cancer Res. 2019, 38, 183. [Google Scholar] [CrossRef] [PubMed]
- Zhai, B.; Hu, F.; Jiang, X.; Xu, J.; Zhao, D.; Liu, B.; Pan, S.; Dong, X.; Tan, G.; Wei, Z.; et al. Inhibition of Akt Reverses the Acquired Resistance to Sorafenib by Switching Protective Autophagy to Autophagic Cell Death in Hepatocellular Carcinoma. Mol. Cancer Ther. 2014, 13, 1589–1598. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.-P.; Liu, J.-P.; Feng, J.-F.; Zhu, C.-P.; Yang, Y.; Zhou, W.-P.; Ding, J.; Huang, C.-K.; Cui, Y.-L.; Ding, C.-H.; et al. miR-541 Potentiates the Response of Human Hepatocellular Carcinoma to Sorafenib Treatment by Inhibiting Autophagy. Gut 2020, 69, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Sun, H.; Jiang, Q.; Chai, Y.; Li, X.; Wang, Z.; Zhu, B.; You, S.; Li, B.; Hao, J.; et al. Hsa-miR-4277 Decelerates the Metabolism or Clearance of Sorafenib in HCC Cells and Enhances the Sensitivity of HCC Cells to Sorafenib by Targeting Cyp3a4. Front. Oncol. 2021, 11, 735447. [Google Scholar] [CrossRef]
- Li, T.-T.; Mou, J.; Pan, Y.-J.; Huo, F.-C.; Du, W.-Q.; Liang, J.; Wang, Y.; Zhang, L.-S.; Pei, D.-S. MicroRNA-138-1-3p Sensitizes Sorafenib to Hepatocellular Carcinoma by Targeting PAK5 Mediated β-Catenin/ABCB1 Signaling Pathway. J. Biomed. Sci. 2021, 28, 56. [Google Scholar] [CrossRef]
- Spallanzani, A.; Orsi, G.; Andrikou, K.; Gelsomino, F.; Rimini, M.; Riggi, L.; Cascinu, S. Lenvatinib as a Therapy for Unresectable Hepatocellular Carcinoma. Expert Rev. Anticancer Ther. 2018, 18, 1069–1076. [Google Scholar] [CrossRef]
- Ladd, A.D.; Duarte, S.; Sahin, I.; Zarrinpar, A. Mechanisms of Drug Resistance in HCC. Hepatology 2023. [Google Scholar] [CrossRef]
- Wei, Y.; Wei, L.; Han, T.; Ding, S. miR-3154 Promotes Hepatocellular Carcinoma Progression via Suppressing HNF4α. Carcinogenesis 2022, 43, 1002–1014. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.-Y.; Chen, L.; Chen, H.-Y.; Hu, L.; Li, L.; Sun, H.-Y.; Jiang, F.; Zhao, J.; Liu, G.-M.-Y.; Tang, J.; et al. MUC15 Inhibits Dimerization of EGFR and PI3K-AKT Signaling and Is Associated with Aggressive Hepatocellular Carcinomas in Patients. Gastroenterology 2013, 145, 1436–1448.e1-12. [Google Scholar] [CrossRef] [PubMed]
- Han, T.; Zheng, H.; Zhang, J.; Yang, P.; Li, H.; Cheng, Z.; Xiang, D.; Wang, R. Downregulation of MUC15 by miR-183-5p.1 Promotes Liver Tumor-Initiating Cells Properties and Tumorigenesis via Regulating c-MET/PI3K/AKT/SOX2 Axis. Cell Death Dis. 2022, 13, 200. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Jiang, W.; Han, P.; Zhang, J.; Tong, L.; Sun, X. MicroRNA-128-3p Mediates Lenvatinib Resistance of Hepatocellular Carcinoma Cells by Downregulating c-Met. J. Hepatocell. Carcinoma 2022, 9, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liu, W.; Chen, X.; Yin, J.; Ma, L.; Liu, M.; Zhou, X.; Xian, L.; Li, P.; Tan, X.; et al. circKCNN2 Suppresses the Recurrence of Hepatocellular Carcinoma at Least Partially via Regulating miR-520c-3p/Methyl-DNA-Binding Domain Protein 2 Axis. Clin. Transl. Med. 2022, 12, e662. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Yu, J.; Lu, L.; Zhang, Y.; Zhou, Y.; Zhou, Y.; Huang, F.; Sun, L.; Guo, Z.; Hou, G.; et al. MT1JP-Mediated miR-24-3p/BCL2L2 Axis Promotes Lenvatinib Resistance in Hepatocellular Carcinoma Cells by Inhibiting Apoptosis. Cell. Oncol. 2021, 44, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Tao, H.; Zhu, J.; Lu, Y.; Cheng, F.; Xiong, Y.; Liu, J.; Cai, G.; Zhang, Z.; et al. Targeting LINC01607 Sensitizes Hepatocellular Carcinoma to Lenvatinib via Suppressing Mitophagy. Cancer Lett. 2023, 576, 216405. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Setayesh, T.; Vaziri, F.; Wu, X.; Hwang, S.T.; Chen, X.; Yvonne Wan, Y.-J. miR-22 Gene Therapy Treats HCC by Promoting Anti-Tumor Immunity and Enhancing Metabolism. Mol. Ther. 2023, 31, 1829–1845. [Google Scholar] [CrossRef]
- Shi, T.; Iwama, H.; Fujita, K.; Kobara, H.; Nishiyama, N.; Fujihara, S.; Goda, Y.; Yoneyama, H.; Morishita, A.; Tani, J.; et al. Evaluating the Effect of Lenvatinib on Sorafenib-Resistant Hepatocellular Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 13071. [Google Scholar] [CrossRef]
- Liu, N.; Wang, X.; Steer, C.J.; Song, G. MicroRNA-206 Promotes the Recruitment of CD8+ T Cells by Driving M1 Polarisation of Kupffer Cells. Gut 2022, 71, 1642–1655. [Google Scholar] [CrossRef]
- Liu, N.; Chang, C.W.; Steer, C.J.; Wang, X.W.; Song, G. MicroRNA-15a/16-1 Prevents Hepatocellular Carcinoma by Disrupting the Communication Between Kupffer Cells and Regulatory T Cells. Gastroenterology 2022, 162, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wenes, M.; Romero, P.; Huang, S.C.-C.; Fendt, S.-M.; Ho, P.-C. Navigating Metabolic Pathways to Enhance Antitumour Immunity and Immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 425–441. [Google Scholar] [CrossRef]
- Cai, J.; Chen, Z.; Zhang, Y.; Wang, J.; Zhang, Z.; Wu, J.; Mao, J.; Zuo, X. CircRHBDD1 Augments Metabolic Rewiring and Restricts Immunotherapy Efficacy via m6A Modification in Hepatocellular Carcinoma. Mol. Ther. Oncolytics 2022, 24, 755–771. [Google Scholar] [CrossRef]
- Xu, K.; Xia, P.; Chen, X.; Ma, W.; Yuan, Y. ncRNA-Mediated Fatty Acid Metabolism Reprogramming in HCC. Trends Endocrinol. Metab. 2023, 34, 278–291. [Google Scholar] [CrossRef]
- Chen, E.; Yi, J.; Jiang, J.; Zou, Z.; Mo, Y.; Ren, Q.; Lin, Z.; Lu, Y.; Zhang, J.; Liu, J. Identification and Validation of a Fatty Acid Metabolism-Related lncRNA Signature as a Predictor for Prognosis and Immunotherapy in Patients with Liver Cancer. BMC Cancer 2022, 22, 1037. [Google Scholar] [CrossRef]
- Zhou, L.; Zhang, Q.; Cheng, J.; Shen, X.; Li, J.; Chen, M.; Zhou, C.; Zhou, J. LncRNA SNHG1 Upregulates FANCD2 and G6PD to Suppress Ferroptosis by Sponging miR-199a-5p/3p in Hepatocellular Carcinoma. Drug Discov. Ther. 2023, 17, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.-Y.; Zhang, P.-F.; Wei, C.-Y.; Peng, R.; Lu, J.-C.; Gao, C.; Cai, J.-B.; Yang, X.; Fan, J.; Ke, A.-W.; et al. Circular RNA circMET Drives Immunosuppression and Anti-PD1 Therapy Resistance in Hepatocellular Carcinoma via the miR-30-5p/Snail/DPP4 Axis. Mol. Cancer 2020, 19, 92. [Google Scholar] [CrossRef] [PubMed]
- Hollande, C.; Boussier, J.; Ziai, J.; Nozawa, T.; Bondet, V.; Phung, W.; Lu, B.; Duffy, D.; Paradis, V.; Mallet, V.; et al. Inhibition of the Dipeptidyl Peptidase DPP4 (CD26) Reveals IL-33-Dependent Eosinophil-Mediated Control of Tumor Growth. Nat. Immunol. 2019, 20, 257–264. [Google Scholar] [CrossRef]
- Zhang, J.; Pan, T.; Zhou, W.; Zhang, Y.; Xu, G.; Xu, Q.; Li, S.; Gao, Y.; Wang, Z.; Xu, J.; et al. Long Noncoding RNA LINC01132 Enhances Immunosuppression and Therapy Resistance via NRF1/DPP4 Axis in Hepatocellular Carcinoma. J. Exp. Clin. Cancer Res. 2022, 41, 270. [Google Scholar] [CrossRef]
- Fu, Y.; Mackowiak, B.; Feng, D.; Lu, H.; Guan, Y.; Lehner, T.; Pan, H.; Wang, X.W.; He, Y.; Gao, B. MicroRNA-223 Attenuates Hepatocarcinogenesis by Blocking Hypoxia-Driven Angiogenesis and Immunosuppression. Gut 2023, 72, 1942–1958. [Google Scholar] [CrossRef]
- Wei, Y.; Tang, X.; Ren, Y.; Yang, Y.; Song, F.; Fu, J.; Liu, S.; Yu, M.; Chen, J.; Wang, S.; et al. An RNA-RNA Crosstalk Network Involving HMGB1 and RICTOR Facilitates Hepatocellular Carcinoma Tumorigenesis by Promoting Glutamine Metabolism and Impedes Immunotherapy by PD-L1+ Exosomes Activity. Signal Transduct. Target. Ther. 2021, 6, 421. [Google Scholar] [CrossRef]
- Hu, Z.; Chen, G.; Zhao, Y.; Gao, H.; Li, L.; Yin, Y.; Jiang, J.; Wang, L.; Mang, Y.; Gao, Y.; et al. Exosome-Derived circCCAR1 Promotes CD8 + T-Cell Dysfunction and Anti-PD1 Resistance in Hepatocellular Carcinoma. Mol. Cancer 2023, 22, 55. [Google Scholar] [CrossRef]
- Zhang, P.-F.; Gao, C.; Huang, X.-Y.; Lu, J.-C.; Guo, X.-J.; Shi, G.-M.; Cai, J.-B.; Ke, A.-W. Cancer Cell-Derived Exosomal circUHRF1 Induces Natural Killer Cell Exhaustion and May Cause Resistance to Anti-PD1 Therapy in Hepatocellular Carcinoma. Mol. Cancer 2020, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Dong, Z.-R.; Cai, J.-B.; Shi, G.-M.; Yang, Y.-F.; Huang, X.-Y.; Zhang, C.; Dong, R.-Z.; Wei, C.-Y.; Li, T.; Ke, A.-W.; et al. Oncogenic miR-93-5p/Gal-9 Axis Drives CD8 (+) T-Cell Inactivation and Is a Therapeutic Target for Hepatocellular Carcinoma Immunotherapy. Cancer Lett. 2023, 564, 216186. [Google Scholar] [CrossRef]
- Wei, L.; Wang, X.; Lv, L.; Liu, J.; Xing, H.; Song, Y.; Xie, M.; Lei, T.; Zhang, N.; Yang, M. The Emerging Role of microRNAs and Long Noncoding RNAs in Drug Resistance of Hepatocellular Carcinoma. Mol. Cancer 2019, 18, 147. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as Stable Blood-Based Markers for Cancer Detection. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Miotto, E.; Saccenti, E.; Lupini, L.; Callegari, E.; Negrini, M.; Ferracin, M. Quantification of Circulating miRNAs by Droplet Digital PCR: Comparison of EvaGreen- and TaqMan-Based Chemistries. Cancer Epidemiol. Biomarkers Prev. 2014, 23, 2638–2642. [Google Scholar] [CrossRef] [PubMed]
- Greten, T.F.; Villanueva, A.; Korangy, F.; Ruf, B.; Yarchoan, M.; Ma, L.; Ruppin, E.; Wang, X.W. Biomarkers for Immunotherapy of Hepatocellular Carcinoma. Nat. Rev. Clin. Oncol. 2023, 20, 780–798. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Chen, Z.; Zhang, W.; Cheng, Y.; Zhang, B.; Wu, F.; Wang, Q.; Wang, S.; Rong, D.; Reiter, F.P.; et al. The Mechanisms of Sorafenib Resistance in Hepatocellular Carcinoma: Theoretical Basis and Therapeutic Aspects. Signal Transduct. Target. Ther. 2020, 5, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fornari, F.; Pollutri, D.; Patrizi, C.; La Bella, T.; Marinelli, S.; Casadei Gardini, A.; Marisi, G.; Baron Toaldo, M.; Baglioni, M.; Salvatore, V.; et al. In Hepatocellular Carcinoma miR-221 Modulates Sorafenib Resistance through Inhibition of Caspase-3-Mediated Apoptosis. Clin. Cancer Res. 2017, 23, 3953–3965. [Google Scholar] [CrossRef]
- de la Cruz-Ojeda, P.; Schmid, T.; Boix, L.; Moreno, M.; Sapena, V.; Praena-Fernández, J.M.; Castell, F.J.; Falcón-Pérez, J.M.; Reig, M.; Brüne, B.; et al. miR-200c-3p, miR-222-5p, and miR-512-3p Constitute a Biomarker Signature of Sorafenib Effectiveness in Advanced Hepatocellular Carcinoma. Cells 2022, 11, 2673. [Google Scholar] [CrossRef]
- Fernández-Tussy, P.; Rodríguez-Agudo, R.; Fernández-Ramos, D.; Barbier-Torres, L.; Zubiete-Franco, I.; Davalillo, S.L. de; Herraez, E.; Goikoetxea-Usandizaga, N.; Lachiondo-Ortega, S.; Simón, J.; et al. Anti-miR-518d-5p Overcomes Liver Tumor Cell Death Resistance through Mitochondrial Activity. Cell Death Dis. 2021, 12, 555. [Google Scholar] [CrossRef] [PubMed]
- Nishida, N.; Arizumi, T.; Hagiwara, S.; Ida, H.; Sakurai, T.; Kudo, M. MicroRNAs for the Prediction of Early Response to Sorafenib Treatment in Human Hepatocellular Carcinoma. Liver Cancer 2017, 6, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Shao, Y.-Y.; Chen, P.-S.; Lin, L.-I.; Lee, B.-S.; Ling, A.; Cheng, A.-L.; Hsu, C.; Ou, D.-L. Low miR-10b-3p Associated with Sorafenib Resistance in Hepatocellular Carcinoma. Br. J. Cancer 2022, 126, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Tian, X.-P.; Wang, C.-Y.; Jin, X.-H.; Li, M.; Wang, F.-W.; Huang, W.-J.; Yun, J.-P.; Xu, R.-H.; Cai, Q.-Q.; Xie, D. Acidic Microenvironment Up-Regulates Exosomal miR-21 and miR-10b in Early-Stage Hepatocellular Carcinoma to Promote Cancer Cell Proliferation and Metastasis. Theranostics 2019, 9, 1965–1979. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Kudo, M.; Cheng, A.-L.; Wyrwicz, L.; Ngan, R.K.C.; Blanc, J.-F.; Baron, A.D.; Vogel, A.; Ikeda, M.; Piscaglia, F.; et al. Pharmacodynamic Biomarkers Predictive of Survival Benefit with Lenvatinib in Unresectable Hepatocellular Carcinoma: From the Phase III REFLECT Study. Clin. Cancer Res. 2021, 27, 4848–4858. [Google Scholar] [CrossRef]
- Teufel, M.; Seidel, H.; Köchert, K.; Meinhardt, G.; Finn, R.S.; Llovet, J.M.; Bruix, J. Biomarkers Associated With Response to Regorafenib in Patients With Hepatocellular Carcinoma. Gastroenterology 2019, 156, 1731–1741. [Google Scholar] [CrossRef]
- Hoshida, Y.; Toffanin, S.; Lachenmayer, A.; Villanueva, A.; Minguez, B.; Llovet, J.M. Molecular Classification and Novel Targets in Hepatocellular Carcinoma: Recent Advancements. Semin. Liver Dis. 2010, 30, 35–51. [Google Scholar] [CrossRef]
Figure 1.
Molecular mechanisms underneath deregulated non-coding RNAs influencing sorafenib resistance in HCC. Schematic representation of molecular pathways playing a role in sorafenib-resistant cells following aberrant expression of HCC-specific ncRNAs.
Figure 1.
Molecular mechanisms underneath deregulated non-coding RNAs influencing sorafenib resistance in HCC. Schematic representation of molecular pathways playing a role in sorafenib-resistant cells following aberrant expression of HCC-specific ncRNAs.
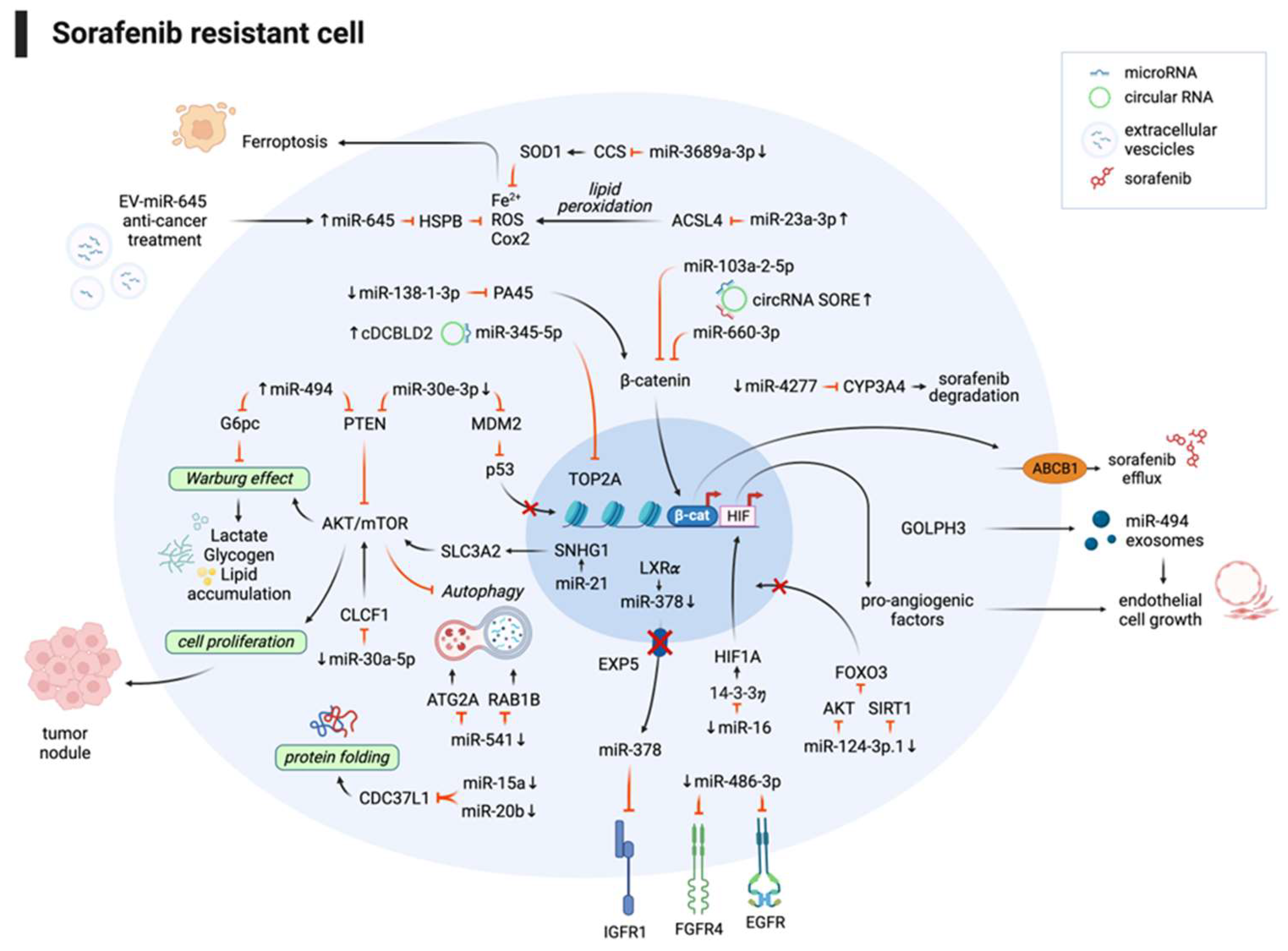
Figure 2.
Molecular mechanisms underneath deregulated non-coding RNAs influencing lenvatinb resistance in HCC. Schematic representation of molecular pathways playing a role in lenvatinib-resistant cells following aberrant expression of HCC-specific ncRNAs.
Figure 2.
Molecular mechanisms underneath deregulated non-coding RNAs influencing lenvatinb resistance in HCC. Schematic representation of molecular pathways playing a role in lenvatinib-resistant cells following aberrant expression of HCC-specific ncRNAs.
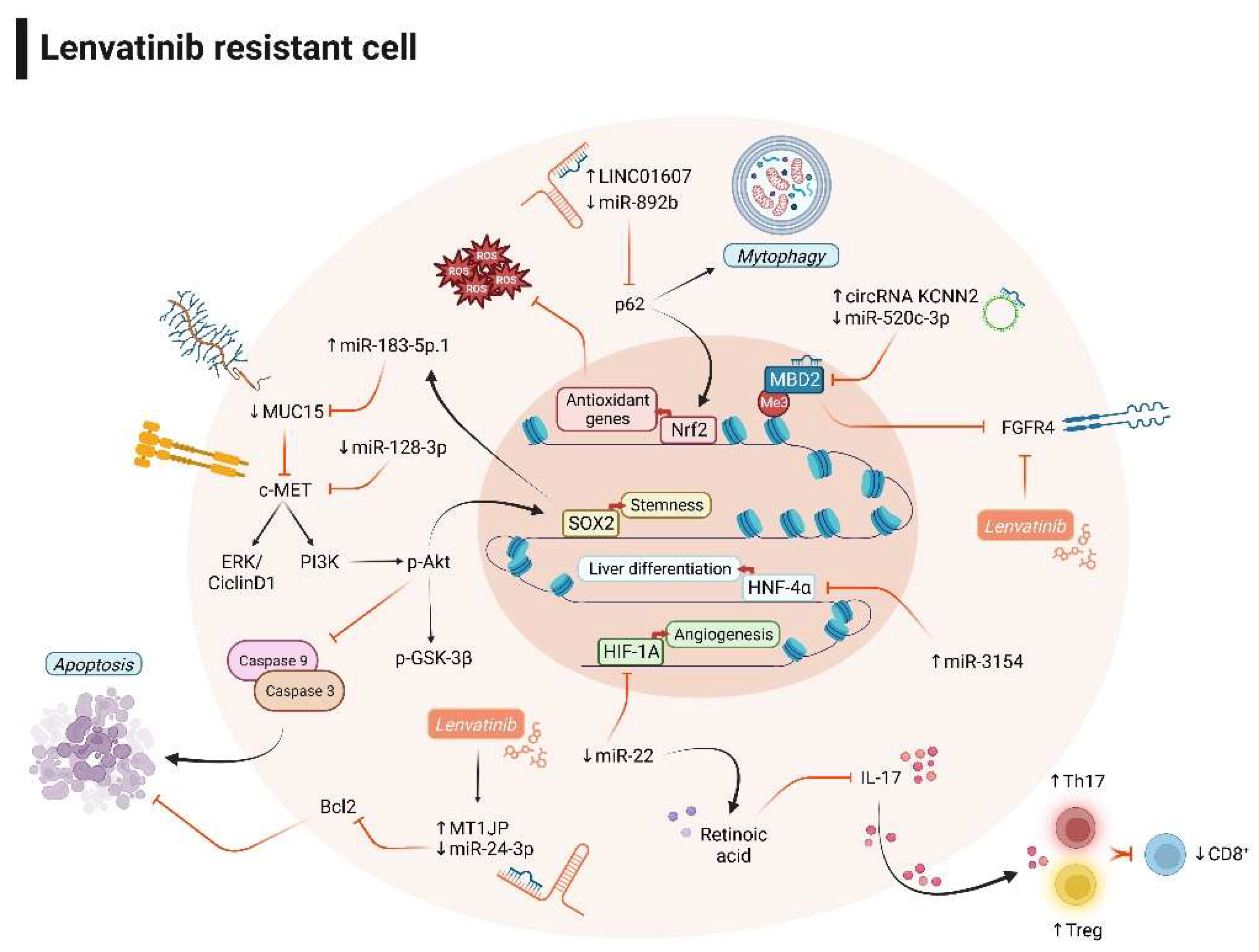

Table 1.
Experimental models for evaluating non-coding RNAs as immunotherapy targets.
| Non-coding RNA | Target gene/sponged miRNA/other targets | Experimental in vivo models | Therapeutic/experimental strategy | Effect on immune cells | Treatment combination | Ref. N. |
|---|---|---|---|---|---|---|
| miR-206 | Klf4/CCL2 | AKT/Ras and Sleeping Beauty transposon hydrodynamic injection in FVB/NJ mice | Mini-circle and Sleeping Beauty hydrodynamic injection for miR-206 overexpression | Decreased Treg recruitment | None | 90 |
| miR-15a/16-1 | Nf-kB/CCL22 | AKT/Ras, Myc hydrodynamic injection in FVB/NJ mice | Hydrodynamic injection for miRNA overexpression | M1 macrophage polarization | None | 91 |
| circRHBDD1 | YTHDF1/PIK3R1 | PDX NOD/SCID, BALBc mice; Hepa1-6 cells in xenograft C57BL/6 mice | circRHBDD1 interference vector | N/A | Anti-PD1 | 93 |
| circMET | miR-30-5p/SNAI1/DPP4/CXCL10 | Hepa1-6 cells in xenograft C57BL/6 mice | Sitagliptin (DPP4 inhibitor) | Increased CD8+ T cells recruitment | Anti-PD1 | 97 |
| LINC01132 | NRF1/DPP4 | PDX nude mice; Hepa1-6 cells in C57BL/6 xenograft mice | LINC01132 adenovirus interference vector | Increased CD8+ T cells recruitment | Anti-PD-L1 | 99 |
| miR-223 | HIF1/CD39/CD73 | miR-223 KO mice + DEN or CCL4; C57BL/4J mice + DEN+CCl4 | miR-223 adenovirus vector | Decreased PD1/PD-L1 expression | None | 100 |
| CircCCAR1 | miR-127-5p/WTAP | HCCLM3 cells in BALBc, HuNSG xenograft mice | circCCAR1 overexpression vector | CD8+ T cells dysfunction | Anti-PD1 | 102 |
| circUHRF1 | miR-449c-5p/TIM3 | HCCLM3 cells in NOD/SCID xenograft mice | circUHRF1 interference vector | Increased NK activity | Anti-PD1 | 103 |
Table 2.
microRNAs as biomarkers in advanced HCCs.
| miRNA name | Blood specimen | Timepoint of analysis | Circulating levels in responders | Treatment | Ref. N. |
|---|---|---|---|---|---|
| miR-221 | Serum | Basal On treatment (2 m) |
Low High |
Sorafenib | 110 |
|
miR-200c-3p miR-222-5p miR-512-3p |
Plasma | Basal On treatment (1 m) On treatment (1 m) |
High Low Low |
Sorafenib | 111 |
| miR-30e-3p | Serum | On treatment (2 m) | Low | Sorafenib | 29 |
| miR-518d-5p | Serum | Basal | Low | Sorafenib | 112 |
| miR-181a-5p | Serum | Basal | High | Sorafenib | 113 |
| miR-10b-3p | Serum | Basal | High | Sorafenib | 114 |
| miR-494 | Serum | Basal | Low | Sorafenib | 59 |
|
miR-30a, miR-122, miR-125b, miR-200a, miR-347b; miR-15b, miR-107, miR-320; miR-645 |
Plasma | Basal | High Low Absent |
Regorafenib | 117 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



