
Submitted:
20 January 2024
Posted:
22 January 2024
You are already at the latest version
Abstract
Streptomyces sp. HNS054, which was previously isolated from a marine sponge, demonstrates vigorous growth and facile genetic modification. This study explores its potential as a marine ac-tinomycete chassis through complete genome sequencing and genetic engineering. With a 7.5 Mb genome containing 6678 predicted genes, 21 secondary metabolic biosynthetic gene clusters (BGCs), and 8 common site-specific recombination (SSR) system attB loci, HNS054 shows a close phylogenetic relation to S. griseoincarnatus strain RB7AG. Utilizing multiplexed site-specific ge-nome engineering (MSGE) and the CRISPR/Cas9 method, engineered strains derived from HNS054 (with three copies of the target BGCs) produce higher amounts of aborycin and ac-tinorhodin than S. coelicolor M1346 (containing three copies of the same BGCs). HNS054 stands out for its remarkable characteristics, including salt tolerance, rapid growth, and compatibility with synthetic biology tools, and positions it as a promising candidate for developing marine ac-tinomycete hosts to enhance secondary metabolite production.
Keywords:
marine actinomycete
; chassis
; natural product
; biosynthetic gene cluster
; rapid growth
; MSGE
1. Introduction
Recent advances in genome sequencing, especially marine microbial metagenomics, have amassed a rich wealth of marine biosynthetic gene clusters (BGCs) [1]. These BGCs represent a valuable resource for drug discovery and development, but they need to be expressed in suitable host cells. Host cells should be able to perform complex biosynthetic pathways and tolerate possible toxic effects of heterologous products. Actinomycetes serve as excellent hosts for expressing a wide range of bioactive compounds, including antibiotics, antitumor agents, immunosuppressants, and pigments. Their versatility makes them ideal for overexpressing complex and biotoxic natural products, finding applications across diverse sectors such as medicine, agriculture, food, and industry [2,3].
Noteworthy examples such as S. coelicolor [4], S. lividans [5,6], S. albus [7], and S. avermitilis [8], which originate from terrestrial environments, serve as mature platforms for the heterologous expression of various complex natural products. However, marine natural products may have different structures and activities than terrestrial ones, and marine actinomycetes may be more compatible with them. Nevertheless, progress in the development of marine actinomycete chassis has been relatively constrained. To date, only two instances stand out: the establishment of Salinispora tropica CNB-4401 [9] and MGCEP 1.0 [10], derived from the marine actinomycetes S. tropica CNB-440 and S. atratus SCSIO ZH16, respectively.
Actinomycete chassis cells undergo diverse genetic enhancements, involving genome simplification [11], removal of native secondary metabolite BGCs [12], insertion of site-specific recombination (SSR) system attB loci to integrate multiple foreign BGCs [13], and removal of negative regulators of BGC expression [14,15]. These modifications aim to enhance the heterologous expression of foreign BGCs. Consequently, potential hosts earmarked as chassis should be compatible with genetic engineering tools.
In our prior investigations, Streptomyces sp. HNS054, isolated from a marine sponge Mycale sp. [16], underwent extensive examination for its proficiency in novel natural product synthesis [17]. HNS054 exhibits robust growth and is amenable to genetic modification. In this study, the complete genome of HNS054 was sequenced, and genetic engineering methods were applied to evaluate its potential as a host for foreign BGC expression. The aborycin and actinorhodin BGCs served as test cases. Salinity tolerance, growth rates, biomass accumulation, and antibiotic sensitivity were also scrutinized. The results indicate that hosts derived from HNS054 have the potential to be developed as marine actinomycete chassis.
2. Results
2.1. Genomic Features and Annotation of HNS054
The complete genome of HNS054 (GenBank: CP139576) consists of the linear chromosome approximately 7.5 Mb in size, with a GC content of 72.3%. It encompasses 6678 putative protein-coding genes, 72 transfer RNA (tRNA) genes, and 18 ribosomal RNA (rRNA) genes. A summary of the genome information for HNS054 is presented in Figure 1 and Table S1. Although no CRISPR/Cas systems were detected by CRISPRCasFinder, 24 CRISPR arrays were identified (Table S2).
The COG, GO and KEGG database annotations assigned functional categories to 5121, 4766, and 2380 genes in the genome of HNS054, respectively (Figure S1, Figure S2, Figure S3). Among them, 129 genes were associated with secondary metabolite biosynthesis, transport, and metabolism, indicating the strain's potential to produce various secondary metabolites. Notably, 1194 genes with unknown functions were identified, warranting further exploration.
2.2. Comparative Genome and Pan-Genome Analysis
The genomic study of Streptomyces reveals that essential genes are distributed in the core chromosomal region centered around the replication origin (oriC). The subtelomeric regions at both ends of the linear chromosome, containing genes with lower conservation, are identified as non-essential gene regions [18]. Therefore, the delineation of these genomic regions is crucial for guiding the construction of a genome-minimized chassis. The location of the OriC of HNS054 was identified to be at positions 3740805-3741749 bp, approximately 19.7 Kb away from the chromosomal center at 3761515 bp (Figure S4). The OriC has 22 DnaA boxes and an AT-rich region: ACAAAAAA. Multi-genome alignment analysis revealed that the genome of HNS054 is symmetrical, consisting of a core region spanning about 5.73 Mb and two subtelomeric regions housing the accessory genome (Figure S5). The core region features highly conserved and homologous genes, while the accessory genome encompasses dispensable elements, including 18 genomic islands (GIs) predicted by IslandViewer4 (Figure S6). These GIs are primarily concentrated in the subtelomeric regions, potentially contributing to the strain’s adaptation and diversity.
Core genome phylogeny revealed a close association between strain HNS054 and S. griseoincarnatus RB7AG (Figure 2A). The most recent 16S rRNA data (Figure S7) positioned strain HNS054 within the same cluster as S. griseoincarnatus LMG 19316T. Average Nucleotide Identity (ANI), reflecting the average nucleotide identity of all orthologous genes shared between any two genomes, provided a robust distinction between strains of the same or closely related species, typically showing an 80-100% ANI range. Digital DNA-DNA hybridization (dDDH) values, offering a direct measure of shared gene content weighted by shared sequence similarity in silico, tended to be higher for closer related species. Both ANI and dDDH data consistently indicated that strain HNS054 was the closest relative to S. griseoincarnatus (Table S3). Therefore, HNS054 was confidently classified as S. griseoincarnatus.
The core genome phylogeny also shows that HNS054 is closely related to other well-studied Streptomyces chassis strains, such as S. coelicolor A3(2) and S. lividans TK24 (Figure 2A). This suggests shared evolutionary and metabolic characteristics among these strains. Through multiple genome-wide alignments and comparative genomics analyses, a total of 5179 protein clusters were annotated in HNS054, encompassing 238 highly conserved orthologous gene clusters and 48 clusters unique to this strain (Figure 2B). The functionally enriched proteins in the unique clusters are related to biosynthetic processes, including polysaccharide biosynthesis, ATP binding biosynthesis, DNA-mediated transposition, and plasma membrane biosynthetic processes. These proteins may confer HNS054 strong cell division abilities, leading to rapid growth and high biomass accumulation. The unique clusters also suggest distinctive metabolic capabilities, indicating potential applications in synthetic biology.
2.3. Analysis of Secondary Metabolite BGCs
The antiSMASH software predicted 21 BGCs in the genome of HNS054 (Table S4). These BGCs are involved in the synthesis of various types of secondary metabolites, including ribosomally synthesized and post-translationally modified peptides (RiPPs), non-ribosomal peptide synthetases (NRPSs), polyketide synthases (PKSs), NRPS-PKS hybrids, terpenes, ectoines, siderophores, and others. The chromosomal distribution of these BGCs is illustrated in Figure S8. Some of these BGCs are novel, with low similarity to known ones, indicating that HNS054 may produce new secondary metabolites during fermentation.
2.4. Analysis of SSR Systems in Strain HNS054
The genome of HNS054 possesses 10 common-type attB sites suitable for 8 SSR systems (φC31, φBT1, SV1, TG1, VWB, φK38-1, CBG73463, and φJoe), as depicted in Figure S9 and detailed in Table S5. These attB sites exhibit highly conserved core regions, with sequence identities ranging from 89% to 100%. Their distribution spans throughout the genome (Figure S10). SSR systems are powerful tools for Streptomyces’ genetic engineering, facilitating targeted and stable integration of the foreign DNA into the chromosome. HNS054 has multiple attB sites, which allow the use of the “multiple integrases multiple attB sites” method, for simultaneous multi-site insertion of genes or gene clusters in one step [19]. They also provide a rich resource for various applications, including genome editing, gene expression, plasmid construction, strain engineering, and natural product discovery.
2.5. Growth, Salt Tolerance, and Antibiotic Susceptibility of Strain HNS054
HNS054 grew faster and had higher biomass compared to S. coelicolor M1146 under various salinity conditions (Figure 3A). At 30‰ salinity, the growth rate of HNS054 was 1.8-fold that of M1146. Additionally, the biomass of HNS054 was 1.6-fold that of S. coelicolor M1146 during the plateau phase. This suggests that HNS054 possesses a stronger cell division ability than S. coelicolor M1146.
HNS054 has a maximum salinity tolerance of 60‰, while S. coelicolor M1146 has a maximum salinity tolerance of 45‰ (Figure 3B). The notable high salinity tolerance of HNS054 positions it as an advantageous marine Streptomyces, capable of thriving and producing secondary metabolites in seawater or saline mediums.
The antibiotic sensitivity of HNS054 was tested to find suitable screening markers for genetic engineering. HNS054 was resistant to ampicillin, chloramphenicol, thiostrepton, and nalidixic acid, while displaying sensitivity to kanamycin, apramycin, and tetracycline (Table S6). The minimum inhibitory concentrations (MICs) for kanamycin and apramycin were determined to be 25 μg/mL, and for tetracycline, it was 12.5 μg/mL. Consequently, kanamycin, apramycin, or tetracycline could serve as effective screening markers. For spectinomycin or hygromycin antibiotics, higher concentrations of 200 μg/mL spectinomycin and 100 μg/mL hygromycin were required for screening.
2.6. Development of MSGE Hosts from Strain HNS054 Using CRISPR/Cas9 Methodology
Employing the CRISPR/Cas9 methodology, a diverse set of enhanced heterologous hosts was derived from the wild-type HNS054. The initial knockout operation focused on eliminating aborycin production capacity in the wild strain by targeting BGC11. This manipulation resulted in the creation of the HNS1151 host, featuring a single native φC31 attB site. Subsequently, strain HNS1251 emerged through the integration of an artificial φC31 attB site into the location formerly occupied by BGC11. Expanding on this groundwork, strains HNS1351 to HNS1551 were generated by substituting BGC14, BGC17, and BGC2 with additional artificial φC31 attB sites, respectively. Each resulting strain, rigorously validated through PCR and sequencing (Figure S11), had one to five φC31 attB sites. A growth analysis showed that strains HNS1151-1551 had similar growth performance to the parent HNS054 (Figure S12).
2.7. Improvement of Aborycin Production
The aborycin BGC was integrated into the chromosome of strains HNS1151-1551 using the pSET152::gul plasmid, resulting in strain HNS1151::gul, HNS1251::2gul, HNS1351::3gul, HNS1451::4gul, and HNS1551::5gul (Figure S13). Aborycin production was analyzed through high-resolution HPLC-MS (Figure S14), and a standard curve for aborycin (Figure S15) was utilized to calibrate the aborycin yield based on HPLC peak areas.
The fermentation broth of engineered strains was compared for aborycin production (Figure 4A). Both supernatant and mycelial analyses demonstrated that strains HNS054 and HNS054::pSET152 produced approximately 7.1 ± 0.1 mg/L of aborycin (Figure 4B). In contrast, strains with two or three copies of the aborycin BGC produced 38.4 ± 0.9 mg/L (HNS1251::2gul) or 45.6 ± 2.1 mg/L (HNS1351::3gul), respectively. Interestingly, strains with four or five copies of the aborycin BGC did not exhibit further production enhancement; instead, a decrease was observed. This suggested that the overexpression of the aborycin BGC genes was no longer the limiting factor for aborycin production. To achieve further improvements, additional metabolic engineering strategies, such as enhancing precursor availability and antibiotic tolerance, need to be considered.
The enhancement of aborycin production in strain HNS1351::3gul was significant, representing a 6.4-fold increase compared to native HNS054. In comparison, the three-copy integration counterpart in S. coelicolor, strain M1346::3gul, produced aborycin at 10.2 ± 0.7 mg/L. Consequently, the aborycin yield of strain HNS1351::3gul surpassed that of strain M1346::3gul by 4.5-fold. These results highlight the effectiveness of the MSGE strategy in HNS054-based hosts, leading to a remarkable improvement in aborycin production.
2.8. Improvement of Actinorhodin Production
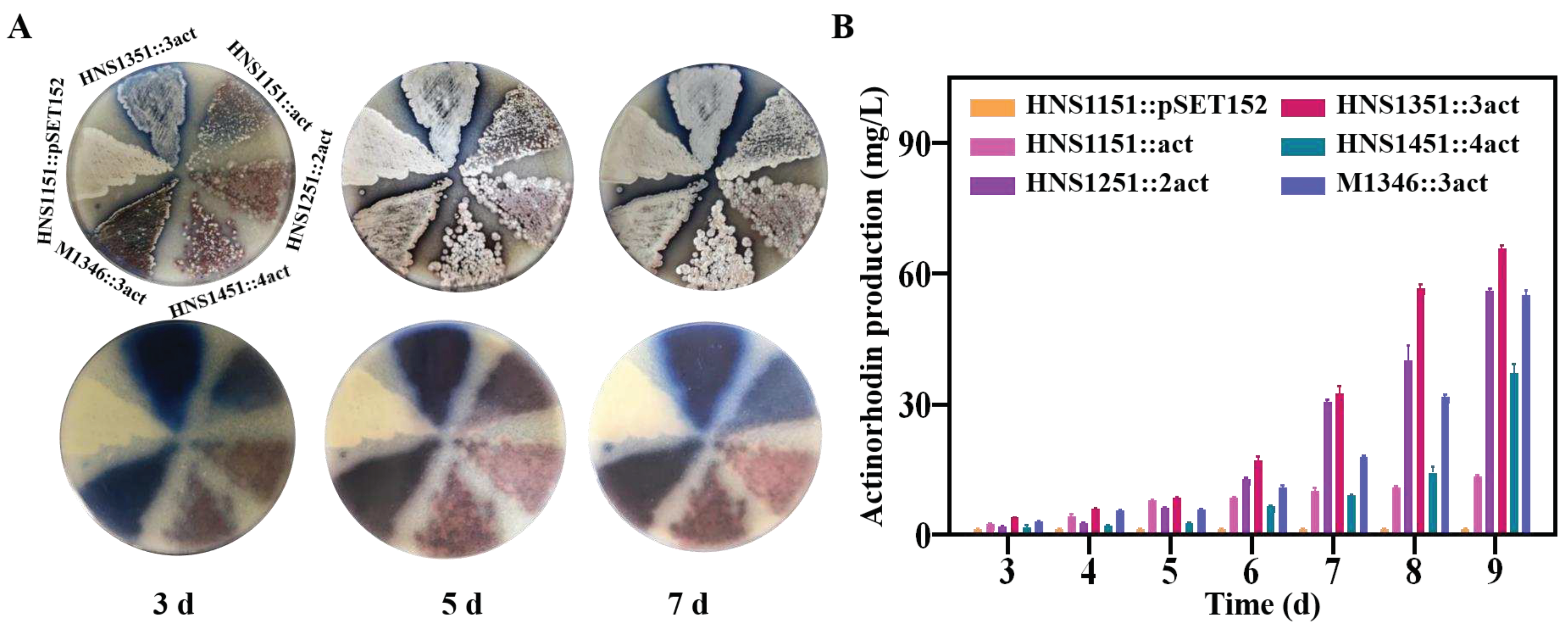
The actinorhodin BGC was incorporated into the chromosomes of strains HNS1151-1551 using the plasmid pSET152::act, generating strains HNS1151::act, HNS1251::2act, HNS1351::3act, and HNS1451::4act, respectively (Figure S16). Unfortunately, strain HNS1551::5act was not obtained, possibly due to limitations in integration capacity (Figure S17). The actinomycin yield gradually increased with the extension of culture time (Figure 5). Strain HNS1351::3act, which harbored three copies of the actinorhodin BGC, exhibited more intense pigmentation on MS agar media relative to other strains HNS054-based hosts and M1346::3act, indicating higher actinorhodin production (Figure 5A). On day 9, strain HNS1351::3act produced actinorhodin at a 4.9-fold, 1.2-fold, and 1.2-fold higher rate than strain HNS1151::act, HNS1251::2act, and M1346::3act, respectively (Figure 5B). However, actinorhodin production decreased by 43% in strain HNS1451::4act, which had four copies of the actinorhodin BGC. The expression of the empty plasmid pSET152 in strain HNS1151 had no effect on actinorhodin production. These results highlight the superiority of HNS054-based hosts in heterogeneously expressing foreign BGCs.
3. Discussion
The genomic sequencing of HNS054 has revealed intricate insights into its biological information, revealing a clear genome structure and rich functional potential. HNS054 harbors 6678 putative protein-coding genes, showcasing a relatively high gene density. Functional annotation using COG, GO, and KEGG databases classified the genes into various categories and pathways, reflecting the diverse metabolic capabilities of this strain. Among the putative genes, 1194 have unknown functions, implying the possibility of novel secondary metabolite biosynthesis. Through the antiSMASH software, 21 BGCs were predicted, spanning various secondary metabolic pathways like RiPPs, NRPSs, PKSs, and NRPS-PKS hybrids, suggesting HNS054 offered a rich resource in secondary metabolites. The genome size of HNS054 is 7.5 Mb, falling within the typical range of conventional Streptomyces genomes (6-12 Mb). Notably, it is comparable to the genome sizes of well-known Streptomyces chassis strains, such as S. coelicolor A3(2) (8.7 Mb), S. griseus NBRC 13350 (8.5 Mb), S. albus J1074 (6.8 Mb), and S. atratus SCSIO ZH16 (9.6 Mb) [20,21]. In comparison, the smallest reported Streptomyces genome is that of S. xiamenensis 318, with a size of 5.96 Mb and 21 BGCs [22]. The relatively moderate genome size of HNS054 positions it favorably for genome-scale modeling and engineering, facilitating the optimization of its metabolic performance and productivity. Furthermore, HNS054 exhibits fewer BGCs compared to the aforementioned chassis strains, which have 30, 38, 22, and 26 BGCs, respectively. This suggests that HNS054 boasts a cleaner metabolic background, reducing interference and competition from endogenous secondary metabolites with heterologous ones.
Despite exhibiting rapid growth compared to S. coelicolor M1146, unraveling the underlying mechanisms through genome comparison alone proves challenging. Previous reports suggest a positive correlation between rRNA operon and tRNA gene count with bacterial growth rate and metabolic activity [23]. While HNS054 possesses more ribosomal operons (6) and tRNA genes (72) than S. xiamenensis 318 (5 and 55, respectively), these values fall within the range observed in common chassis strains (6-7 rRNA operons, 63-69 tRNA genes). HNS054 also shares a characteristic symmetrical distribution of conserved genes around the oriC with these chassis strains. This arrangement potentially facilitates early expression of essential genes and discourages subtelomeric rearrangements, enhancing strain stability and adaptability [20,21,22]. Elucidating the precise genomic basis of HNS054's rapid growth requires the application of contemporary functional genomics approaches.
It has been reported that salt-resistant Halomonas spp. are preferred hosts due to their ability to conduct contamination-free fermentation without the need for strict sterilization under high salt conditions [24]. While HNS054 may not exhibit resistance levels as high as Halomonas spp. (moderate strains can tolerate 30-150‰ NaCl (w/v), and extreme strains can tolerate > 200‰ NaCl (w/v)), HNS054 demonstrated faster growth and higher bioaccumulation than S. coelicolor M1146 in the salinity range of 0-45‰ NaCl (w/v) (Figure 3). Furthermore, HNS054-derived strains show a lower susceptibility to contamination than M1146-derived strains during fermentation, genetic engineering, and sporing under 35‰ salinity. It can be deduced that the large-scale fermentation performance of the engineered HNS054 would reduce the risk of bacterial contamination under optimized salinity conditions.
HNS054 shines not only in growth and salt tolerance but also in its inherent potential for genetic manipulation. Compared to other Streptomyces chassis, its enriched repertoire of natural attB sites synergizes with its conventional SSR system (Table S5) and facilitates advanced multiplex site-specific genome editing [19]. Furthermore, improved HNS054-derived strains were constructed via CRISPR/Cas9-mediated precise deletion of specific BGCs and insertion of artificial φC31 attB sites. Notably, removing S. griseoincarnatus HNS054's native secondary metabolite BGCs and introducing attB sites maintained its growth and morphology (Figure S12), indicating a simplified background, potentially leading to increased precursor availability and secondary metabolite production. The presence of multiple homologous attB sites enables these HNS054-based hosts to leverage the "one integrase-multiple attB sites" concept for the MSGE strategy. This approach has proven to enhance secondary metabolite production in various Streptomyces strains [5,7,15,25,26]. Thus, HNS054's genomic features and amenability to MSGE make it a promising platform for overproducing valuable secondary metabolites.
However, the results indicate that the MSGE method has limitations in two key aspects. Firstly, as the number of attB sites of the same type increased from one to five, the conjugation efficiency of HNS054-based hosts and S. coelicolor M1146-based hosts declined from thousands to single conjugon (Figure S17). This observation suggests that introducing foreign gene clusters using the MSGE method becomes more challenging with an increasing number of attB sites of the same type in a strain. Secondly, in both case studies, an increase in the BGC copy number led to a declining trend in yield (Figure 4B and 5B), a phenomenon consistent with similar reports. For instance, strain S. albus B4, which harbors four copies of pyridinopyrone A, aloesaponarin II, and didemethoxyaranciamycinone BGCs, respectively, exhibited lower production than strain S. albus B2P1, which has three copies of the corresponding BGCs [7]. The mechanism behind this phenomenon is unclear, but it may involve the strain's self-protection mechanism. These limitations underscore the necessity of employing additional metabolic engineering strategies to overcome diminishing returns associated with higher BGC copy numbers.
Compared to the well-established model strain, S. coelicolor M145, and its derived strains, marine actinomycetes possess a unique metabolic system that facilitates the discovery and development of novel drug lead compounds from the marine environment. Zhang et al. [9] eliminated salinosporamide synthesis in the marine strain S. tropica CNB-440 and introduced the phage φC31 attB site of S. coelicolor, generating strain S. tropica CNB-4401. The thiolactinomycin BGC from S. pacifica was heterologously expressed, and the thiolactomycin production in S. tropica CNB-4401 was approximately 3-fold that of S. coelicolor M1152. Yang et al. [10] disrupted the synthetic pathway of two main metabolites (ilamycins and atratumycin) in S. atratus SCSIO ZH16 and used the natural specific integration site of this strain, resulting in the marine Streptomyces expression platform MGCEP 1.0. Alkaloids, aminonucleosides, nonribosomal peptides, and polyketides BGCs were heterologously expressed in MGCEP 1.0, leading to the identification of 19 compounds.
Being a marine actinomycete, HNS054 boasts faster growth and high salinity tolerance. The manipulation of HNS054 and its derivative strains offers notable advantages, including ease of operation, reduced susceptibility to contamination, and expedited completion of conjugation experiments. These inherent traits of the HNS054 series not only facilitate genetic engineering manipulations but also position them as promising candidates for future large-scale fermentation processes.
4. Materials and Methods
4.1. Strains, Plasmids, Primers, and Culture Conditions
The primers, plasmids, and strains utilized in this study are outlined in Tables S7 and S8. Streptomyces were cultured either on mannitol soya flour agar medium (MS, 20 g/L mannitol, 20 g/L soy flour, 15 g/L sea salt, 20 g/L agar) or in R5 liquid medium (10 g/L glucose, 0.1 g/L casamino acids, 5 g/L yeast extract, 5.73 g/L [Tris(hydroxymethyl)metyl]-2-aminopropanesulfonic acid (TES), 10 mL of 0.5% KH2PO4, 4 mL of 5 M CaCl2.2H2O, 15 mL of 20% L-proline, and 7 mL of 1 M NaOH, 2 mL trace element solution) with suitable antibiotics. Escherichia coli were cultured at 37 °C in Luria-Bertani (LB) medium supplemented with appropriate antibiotics with shaking at 300 rpm for 16 h. Streptomyces was cultivated on MS salt agar at 28 °C for 7 d for sporulation. Liquid cultures were involved ISP2 medium (10 g/L yeast extract, 10 g/L malt extract, 4 g/L glucose, 30 g/L sea salt), YEME medium (3 g/L yeast extract, 5 g/L peptone, 3 g/L malt extract, 10 g/L glucose, 340 g/L sucrose, 30 g/L sea salt), Gauze's Medium No.1 (1 g/L KNO3, 0.5 g/L K2HPO4, 0.5 g/L MgSO4.7H2O, 0-100 g/L NaCl, 0.01 g/L FeSO4.7H2O, 20 g/L soluble starch) or R5 medium and incubated at 28 °C with shaking at 200 rpm for 5-7 d.
4.2. Genome Sequencing, Assembly and Annotation
The genome sequencing of Streptomyces sp. HNS054 was conducted by Biomarker Technologies (Beijing, China) using Pacbio Sequel II and Illumina NovaSeq 6000 platform. The assembly of long reads was executed using Hifiasm v0.12 software [27], resulting in a single contig spanning 7.5 Mb. Quality control for short reads was conducted through FastQC (https://sourceforge.net/projects/fastuniq/), followed by trimming with Adapter Removal [28]. To rectify errors in long reads, SOAPdenovo2 (https://github.com/aquaskyline/SOAPdenovo2) with a k-mer setting of 17 was applied to the trimmed reads. The corrected long reads underwent further refinement with Pilon v1.22 software (https://github.com/broadinstitute/pilon) and circularization with Circlator v1.5.5 software (https://github.com/sanger-pathogens/circlator), culminating in the acquisition of the complete genome sequence of HNS054. The complete genome sequence has been submitted to the NCBI GenBank database under the accession number CP139576.1 (https://www.ncbi.nlm.nih.gov/search/all/?term=CP139576.1). The 16S rRNA gene sequence has also been submitted to the NCBI Nucleotide database under the accession number OR898379.1 (https://www.ncbi.nlm.nih.gov/search/all/?term=OR898379).
Coding genes were identified using Prodigal v2.6.3 software (https://github.com/hyattpd/Prodigal). Infernal v1.1.3 software (http://eddylab.org/infernal/) was employed for the prediction of rRNA genes, while tRNAscan-SE v2.0 software (https://github.com/UCSC-LoweLab/tRNAscan-SE) predicted tRNA genes. Additionally, Infernal v1.1.3 software was utilized for the prediction of other non-coding RNAs. CRISPRs and Cas genes were discerned using CRISPRCasFinder (https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index). For functional annotation, the predicted protein underwent a blast analysis against diverse databases, including Nr (Non-Redundant Protein Sequence Database), Swiss-Prot, Pfam, TrEMBL (Translation of EMBL), KEGG (Kyoto Encyclopedia of Genes and Genomes), and eggNOG (evolutionary genealogy of genes: Non-supervised Orthologous Groups). GO (Gene Ontology) annotation was carried out using Blast2go (https://www.blast2go.com/). Orthologous proteins were predicted and clustered across the genomes using the OrthoVenn software (http://www.bioinfogenome.net/OrthoVenn/start.php), employing the OrthoMCL algorithm. AntiSMASH 7.0 (https://antismash.secondarymetabolites.org/) was employed for predicting secondary metabolite BGCs and forecasting the synthesis of metabolic products in HNS054.
RepeatMasker (https://www.repeatmasker.org/RepeatMasker/) was employed for predicting repetitive sequences. IslandViewer 4 (http://www.pathogenomics.sfu.ca/islandviewer/) was used to identify GIs. The OriC was determined through the Ori-Finder web server (http://tubic.tju.edu.cn/Ori-Finder/). SSR system attB loci were identified by employing local blast to search for the core regions of nine prevalent types of SSR system attB nucleotide sequences within the HNS054 genome.
4.3. Comparative Genomic Analysis
The Mauve 2.3.1 software (http://darlinglab.org/mauve/mauve.html) was utilized for the creation and visualization of multiple genome alignments. The phylogenetic tree based on the 16S rRNA gene was constructed using a neighbor-joining approach in MEGA 11, with 1000 bootstrap replicates. dDDH estimates were determined using the Genome-to-Genome Distance Calculator (GGDC) web server (https://ggdc.dsmz.de/). ANI values were calculated by the JSpeciesWS online service (https://jspecies.ribohost.com/jspeciesws/#analyse). To conduct pan-genome analysis for strain HNS054 and other Streptomyces species, the Bacterial Pan Genome Analysis Pipeline (BPGA, http://www.iicb.res.in/bpga/index.html) software was employed and constructed a whole-genome-based phylogenetic tree. The resulting phylogenetic tree was visualized using the online software iTOL (Interaction Tree of Life, https://itol.embl.de/). Detailed information regarding Streptomyces species utilized for comparative genomic analysis, along with their GenBank accession numbers, is available in Table S9.
4.4. Assessment of Growth, Salt Tolerance, and Antibiotic Sensitivity
The biomass of HNS054 and S. coelicolor M1146 in the fermentation broth was determined utilizing a simplified diphenylamine colorimetric method [29].
Salt tolerance testing of the strains involved the preparation of Gauze's Medium No.1 with varying NaCl concentrations (0‰, 15‰, 30‰, 45‰, 60‰, 80‰, and 100‰). The tested strains were then inoculated onto the respective media and cultured at 28 °C for 5-7 d. The growth of the strains was monitored, and the results were documented.
The antibiotic sensitivity of HNS054 was tested on MS salt agar plates with various concentrations of antibiotics. Kanamycin, ampicillin, chloramphenicol, apramycin, nalidixic acid, thiostrepton, and tetracycline were assessed with concentrations ranging from 0-100 μg/mL. Additionally, hygromycin and spectinomycin were examined with concentrations ranging from 50-300 μg/mL. The spore suspension of HNS054 was inoculated on the plates at 28 °C for 5-9 d. The growth of the strain on the plates was observed to determine its antibiotic sensitivity.
4.5. Construction of the HNS054-derived Hosts
The construction of HNS054-derived hosts involved the CRISPR/Cas9 genome editing method to generate a series of strains (HNS1151-HNS1551). The strain HNS1151 was generated by knocking out BGC11 to eliminate aborycin production. Subsequently, in strains HNS1251-HNS1551, artificial φC31 attB sites were incorporated at the locations of deleted target BGCs (e.g., BGC11, BGC14, BGC17, BGC2). The general construction process is outlined below:
- Amplification of Upstream and Downstream Regions: PCR amplification of the upstream and downstream regions of the targeted BGCs (e.g., BGC11, BGC14, BGC17, BGC2) using specific primer pairs (e.g., Del-BGC11-up-B-fwd/rev, Del-BGC11-down-B-fwd/rev).
- Amplification of sgRNA Expression Cassette: PCR amplification of the sgRNA expression cassette using a plasmid template (e.g., pKCcas9dO) and specific primers (e.g., BGC11-sgRNA-B-fwd, sgRNA-rev).
- Gibson Assembly: assembly of the three DNA fragments (upstream region, downstream region, sgRNA expression cassette) using the Gibson assembly method to generate plasmids (e.g., pKY01dB11::attB).
- Plasmid Introduction: introduction of the constructed plasmids into the HNS054-derived hosts through conjugal transfer on MS solid media.
- Verification of Double Crossovers: verification of correct double crossovers in the transformed strains by PCR using specific primers (e.g., ID-BGC11-B-fwd/rev).
This general strategy was repeated to construct strains HNS1251, HNS1351, HNS1451, and HNS1551, each with an increasing number of artificial φC31 attB sites in the locations of the deleted BGCs. The resulting strains serve as engineered hosts with modified genomic features for further studies or applications.
4.6. Production of Aborycin
The plasmid pSET152::gul, harboring the aborycin BGC, was constructed using a one-step cloning method. Firstly, the pSET152 plasmid was linearized by PCR using the primers pSET152-fwd/pSET152-rev. Subsequently, the aborycin BGC was amplified from the genome DNA of S. griseoincarnatus HNS054 using the primers 054 gul-fwd/054 gul-rev. Finally, the aborycin BGC and the linearized pSET152 were assembled into the circular plasmid pSET152::gul using a one-step cloning kit.
The plasmid pSET152::gul was transferred into strains HNS1151-1551 by intergeneric conjugation with E. coli ET12567/pUZ8002 as the donor. Exconjugants were acquired by selecting for resistance to apramycin and were subsequently verified through PCR analysis using the primers ID-oriT-fwd/Native B-rev, ID-oriT-fwd/BGC11 B-rev, ID-oriT-fwd/BGC14 B-rev, ID-oriT-fwd/BGC17 B-rev, and ID-BGC2-fwd/BGC2 B-rev to confirm attP-attB recombination, respectively. In parallel, the plasmid pSET152 was introduced into strain HNS054 as a control.
Aborycin production in the engineered strains and S. coelicolor M1346::3gul were assessed using the same methodologies [15].
4.7. Production of Actinorhodin
The plasmid pSET152::act, harboring the actinorhodin BGC [30], was introduced into strains HNS1151-1551 through conjugal transfer. Exconjugants were obtained by selecting for apramycin resistance and confirmed via PCR analysis using the aforementioned primer pairs to verify attP-attB recombination. Plasmid pSET152 was also transferred into strain HNS1151 as a control. To assess whether the actinorhodin production ability of HNS054-based hosts is comparable to that of S. coelicolor M1346, the plasmid pSET152::act was additionally transferred into S. coelicolor M1346, generating the strain S. coelicolor M1346::3act.
The quantification of actinorhodin production in the engineered strains and S. coelicolor M1346::3act was conducted using the following method. The presence of the antibiotic was indicated by the blue coloration of actinorhodin on MS agar media. For actinorhodin extraction, 1 mL of culture samples was mixed with 250 μL of 5 M KOH and then centrifuged at 12000 rpm for 5 min. The concentration of actinorhodin in the supernatant was determined by measuring the absorbance at 640 nm, employing an extinction coefficient of 2.53 × 104 L/(mol·cm) [31].
4.8. Statistical Analysis
All of the results were expressed as the mean ± standard deviation (SD), which resulted from at least three independent experiments. The statistical analysis was performed using one-way ANOVA. The significance level was set at p < 0.05. The software used for the analysis was GraphPad Prism 8.3.
5. Conclusions
This study presents the comprehensive genomic analysis and annotation of the marine actinomycete S. griseoincarnatus HNS054. The genome of HNS054 revealed its phylogenetic position, diverse repertoire of secondary metabolites, and multiple site-specific recombination system attB sites. The strain exhibits high salinity tolerance, a fast growth rate, high biomass production, and sensitivity to antibiotics, which are desirable traits for industrial applications. Furthermore, a series of HNS054-based hosts were constructed by deleting BGCs and introducing the φC31 attB sites. Amplification of the aborycin and the actinorhodin BGCs in engineered strains significantly improved the production of these antibiotics. This study identifies HNS054 as a promising foundation for further developing industrial chassis adept at overproducing marine natural products.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: COG annotation of the HNS054 genome; Figure S2: GO annotation of the HNS054 genome; Figure S3: KEGG annotation of the HNS054 genome; Figure S4: Characterization of the replication origin site (OriC) on the HNS054 chromosome; Figure S5: Multigenome comparison of HNS054 and other Streptomyces chromosomes; Figure S6: Genomic island (GI) analysis of the HNS054 genome; Figure S7: Neighbor-joining phylogenetic tree based on 16S rRNA gene sequences of HNS054; Figure S8: Distribution of BGCs in the HNS054 genome; Figure S9: Nucleotide sequence alignment at attB sites of HNS054; Figure S10: Distribution of ten natural attB sites in the HNS054 chromosome; Figure S11: PCR verification of strains featuring BGC knockout and φC31 attB site introduction; Figure S12: Growth curve of strains; Figure S13: Identification of the plasmid pSET152⸬gul introduced into the strains HNS1151-1551; Figure S14: Positive ion peak in the mass spectra of aborycin; Figure S15: The standard curve depicting the relationship between aborycin concentrations and the HPLC peak areas; Figure S16: Identification of the plasmid pSET152⸬act introduced into the strains HNS1151-1451; Figure S17: The conjugation effect of attB numbers on strains; Table S1: Basic genome sequencing features of HNS054; Table S2: Endogenous CRISPR sequences in HNS054 genome; Table S3: ANI and dDDH analysis of HNS054 to related species; Table S4: AntiSMASH predicted BGCs in the HNS054 genome; Table S5: Identification of natural attB sites in strains; Table S6: The antibiotic sensitivity of HNS054; Table S7: Primers used in this study; Table S8: Plasmids and strains used in this study; Table S9: Basic information of Streptomyces genomes.
Author Contributions
Conceptualization, Z.H. and J.C.; Data curation, Q.W. and J.C.; Formal analysis, Z.H. and J.C.; Funding acquisition, J.Z. and J.C.; Investigation, J.Z. and J.C.; Methodology, Q.W., J.Z. and Z.L.; Project administration, J.C.; Resources, J.Z., S.D., Z.H. and J.C.; Software, Q.W., Z.L. and S.D.; Supervision, J.Z. and J.C.; Validation, Z.L. and S.D.; Visualization, Q.W.; Writing—original draft, Q.W.; Writing—review & editing, Q.W., J.Z., S.D. and J.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the General Program from the National Natural Science Foundation of China (Grant No. 41977200) and the Xiamen Science and Technology Program (Grant No. 3502Z20226031).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original data are included in the paper and can be obtained from the corresponding author upon reasonable request.
Acknowledgments
The authors express their sincere gratitude to Prof. Yinhua Lu from Shanghai Normal University, Shanghai, China, for generously providing the S. coelicolor M1146-M1546 strain. Special appreciation is extended to Prof. Guoqing Niu from Southwest University, Chongqing, China, for supplying the pSET152::act plasmid. The technical support provided by Yuanhuan Liu and Rong Ding, the esteemed technical managers of the College of Ocean and Earth Sciences and the School of Pharmaceutical Sciences at Xiamen University, Xiamen, China, is gratefully acknowledged. Their invaluable assistance with the instruments employed in this study has significantly contributed to the successful completion of our research.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Paoli, L.; Ruscheweyh, H.-J.; Forneris, C. C.; Hubrich, F.; Kautsar, S.; Bhushan, A.; Lotti, A.; Clayssen, Q.; Salazar, G.; Milanese, A. Biosynthetic potential of the global ocean microbiome. Nature 2022, 607, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Jose, P. A.; Maharshi, A.; Jha, B. Actinobacteria in natural products research: Progress and prospects. Microbiol. Res. 2021, 246, 126708. [Google Scholar] [CrossRef] [PubMed]
- Harir, M.; Bendif, H.; Bellahcene, M.; Fortas, Z.; Pogni, R. Streptomyces Secondary Metabolites. In Basic Biology and Applications of Actinobacteria; Enany, S., Ed.; IntechOpen: London, UK, 2018; pp. 99–122. [Google Scholar]
- Gomez-Escribano, J. P.; Bibb, M. J. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 2011, 4, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Y.; Rebets, Y.; Estévez, M. R.; Zapp, J.; Myronovskyi, M.; Luzhetskyy, A. Engineering of Streptomyces lividans for heterologous expression of secondary metabolite gene clusters. Microb. Cell. Fact. 2020, 19, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Peng, Q.; Gao, G.; Lu, J.; Long, Q.; Chen, X.; Zhang, F.; Xu, M.; Liu, K.; Wang, Y.; Deng, Z.; et al. Engineered Streptomyces lividans strains for optimal identification and expression of cryptic biosynthetic gene clusters. Front. Microbiol. 2018, 9, 3042. [Google Scholar] [CrossRef] [PubMed]
- Myronovskyi, M.; Rosenkränzer, B.; Nadmid, S.; Pujic, P.; Normand, P.; Luzhetskyy, A. Generation of a cluster-free Streptomyces albus chassis strains for improved heterologous expression of secondary metabolite clusters. Metab. Eng. 2018; 49, 316–324. [Google Scholar]
- Komatsu, M.; Komatsu, K.; Koiwai, H.; Yamada, Y.; Kozone, I.; Izumikawa, M.; Hashimoto, J.; Takagi, M.; Omura, S.; Shin-ya, K.; et al. Engineered Streptomyces avermitilis host for heterologous expression of biosynthetic gene cluster for secondary metabolites. ACS Synth. Biol. 2013, 2, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. J.; Moore, B. S.; Tang, X. Engineering Salinispora tropica for heterologous expression of natural product biosynthetic gene clusters. Appl. Microbiol. Biotechnol. 2018, 102, 8437–8446. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Liu, C.; Wang, Y.; Chen, Y.; Li, Q.; Zhang, Y.; Chen, Q.; Ju, J.; Ma, J. MGCEP 1.0: A genetic-engineered marine-derived chassis cell for a scaled heterologous expression platform of microbial bioactive metabolites. ACS Synth. Biol. 2022, 11, 3772–3784. [Google Scholar] [CrossRef] [PubMed]
- Komatsua, M.; Uchiyama, T.; Ōmura, S.; Cane, D. E.; Ikeda, H. Genome-minimized Streptomyces host for the heterologous expression of secondary metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 2646–2651. [Google Scholar] [CrossRef]
- Hwang, S.; Lee, Y.; Kim, J. H.; Kim, G.; Kim, H.; Kim, W.; Cho, S.; Palsson, B. O.; Cho, B.-K. Streptomyces as microbial chassis for heterologous protein expression. Front. Bioeng. Biotechnol. 2021, 9, 1–23. [Google Scholar] [CrossRef]
- Myronovskyi, M.; Luzhetskyy, A. Genome engineering in actinomycetes using site-specific recombinases. Appl. Microbiol. Biotechnol. 2013, 97, 4701–4712. [Google Scholar] [CrossRef] [PubMed]
- Nah, H.-J.; Park, J.; Choi, S.; Kim, E.-S. WblA, a global regulator of antibiotic biosynthesis in Streptomyces. J. Ind. Microbiol. Biotechnol. 2021, 48, kuab007. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liang, J.; Liu, Z.; Yi, Y.; Zhao, J.; Huang, Z.; Chen, J. Multicopy chromosome integration and deletion of negative global regulators significantly increased the heterologous production of aborycin in Streptomyces coelicolor. Mar. Drugs 2023, 21, 534. [Google Scholar] [CrossRef]
- Su, P.; Wang, D.; Ding, S.; Zhao, J. Isolation and diversity of natural product biosynthetic genes of cultivable bacteria associated with marine sponge Mycale sp. from the coast of Fujian, China. Can. J. Microbiol. 2014; 60, 217–225. [Google Scholar]
- Liu, T.; Huang, Z.; Gui, X.; Xiang, W.; Jin, Y.; Chen, J.; Zhao, J. Multi-omics comparative analysis of Streptomyces mutants obtained by iterative atmosphere and room-temperature plasma mutagenesis. Front. Microbiol. 2021, 11, 630309. [Google Scholar] [CrossRef] [PubMed]
- Bentley, S. D.; Chater, K. F.; Cerdeño-Tárraga, A. M.; Challis, G. L.; Thomson, N. R.; James, K. D.; Harris, D. E.; Quail, M. A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Wei, K.; Liu, X.; Wu, Y.; Zheng, G.; Chen, S.; Jiang, W.; Lu, Y. aMSGE: advanced multiplex site-specific genome engineering with orthogonal modular recombinases in actinomycetes. Metab. Eng. 2019, 52, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Bu, Q.; Li, Y.; Xie, H.; Wang, J.; Li, Z.; Chen, X.; Mao, X.; Li, Y. Comprehensive dissection of dispensable genomic regions in Streptomyces based on comparative analysis approach. Microb. Cell. Fact. 2020, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, C.; Liu, C.; Ju, J.; Ma, J. Genome sequencing of Streptomyces atratus SCSIOZH16 and activation production of nocardamine via metabolic engineering. Front. Microbiol. 2018, 9, 1269. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Wang, J.; Bu, X.; Yu, H.; Li, P.; Ou, H.; He, Y.; Xu, F.; Hu, X.; Zhu, X.; et al. Deciphering the streamlined genome of Streptomyces xiamenensis 318 as the producer of the anti-fibrotic drug candidate xiamenmycin. Sci. Rep. 2016, 6, 18977. [Google Scholar] [CrossRef]
- Yano, K.; Wada, T.; Suzuki, S.; Tagami, K.; Matsumoto, T.; Shiwa, Y.; Ishige, T.; Kawaguchi, Y.; Masuda, K.; Akanuma, G.; et al. Multiple rRNA operons are essential for efficient cell growth and sporulation as well as outgrowth in Bacillus subtilis. Microbiology 2013, 159, 2225–2236. [Google Scholar] [CrossRef]
- Ye, J.; Chen, G. Halomonas as a chassis. Essays Biochem. 2021, 65, 393–403. [Google Scholar] [PubMed]
- Pikl, Š.; Carrillo Rincón, A. F.; Slemc, L.; Goranovič, D.; Avbelj, M.; Gjuračić, K.; Sucipto, H.; Stare, K.; Baebler, Š.; Šala, M.; et al. Multiple copies of the oxytetracycline gene cluster in selected Streptomyces rimosus strains can provide significantly increased titers. Microb. Cell. Fact. 2021, 20, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zheng, G.; Chen, J.; Ge, M.; Jiang, W.; Lu, Y. Multiplexed site-specific genome engineering for overproducing bioactive secondary metabolites in actinomycetes. Metab. Eng. 2017, 40, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G. T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef] [PubMed]
- Lindgreen, S. AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res. Notes 2012, 5, 337. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Xiang, S.; Dai, X.; Yang, K. A simplified diphenylamine colorimetric method for growth quantification. Appl. Microbiol. Biotechnol. 2013, 97, 5069–5077. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Fu, Y.; Wang, M.; Niu, G. Synthetic cellobiose-inducible regulatory systems allow tight and dynamic controls of gene expression in Streptomyces. ACS Synth. Biol. 2021, 10, 1956–1965. [Google Scholar] [CrossRef]
- Demir, Z.; Bayraktar, A.; Tunca, S. One extra copy of ion gene causes a dramatic increase in actinorhodin production by Streptomyces coelicolor A3(2). Curr. Microbiol. 2019, 76, 1045–1054. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of the linear chromosome of HNS054. The outermost circle shows the genome size. Circles 2 and 3 depict genes on the positive and negative strands, respectively. Different colors denote various functional classifications based on COG categories. Circle 4 displays repeating sequences. Circle 5 illustrates the BGCs. Circle 6 marks tRNA genes. Circle 7 labels rRNA genes. Circle 8 reflects the GC content. Light yellow sections indicate regions with GC content higher than the average genomic GC content, while blue sections indicate regions with GC content lower than the average genomic GC content. Circle 9 reveals GC-skew ([G − C/G + C]). Dark gray areas indicate regions with higher G content than C, and red areas represent regions with higher C content than G.
Figure 1.
Schematic representation of the linear chromosome of HNS054. The outermost circle shows the genome size. Circles 2 and 3 depict genes on the positive and negative strands, respectively. Different colors denote various functional classifications based on COG categories. Circle 4 displays repeating sequences. Circle 5 illustrates the BGCs. Circle 6 marks tRNA genes. Circle 7 labels rRNA genes. Circle 8 reflects the GC content. Light yellow sections indicate regions with GC content higher than the average genomic GC content, while blue sections indicate regions with GC content lower than the average genomic GC content. Circle 9 reveals GC-skew ([G − C/G + C]). Dark gray areas indicate regions with higher G content than C, and red areas represent regions with higher C content than G.
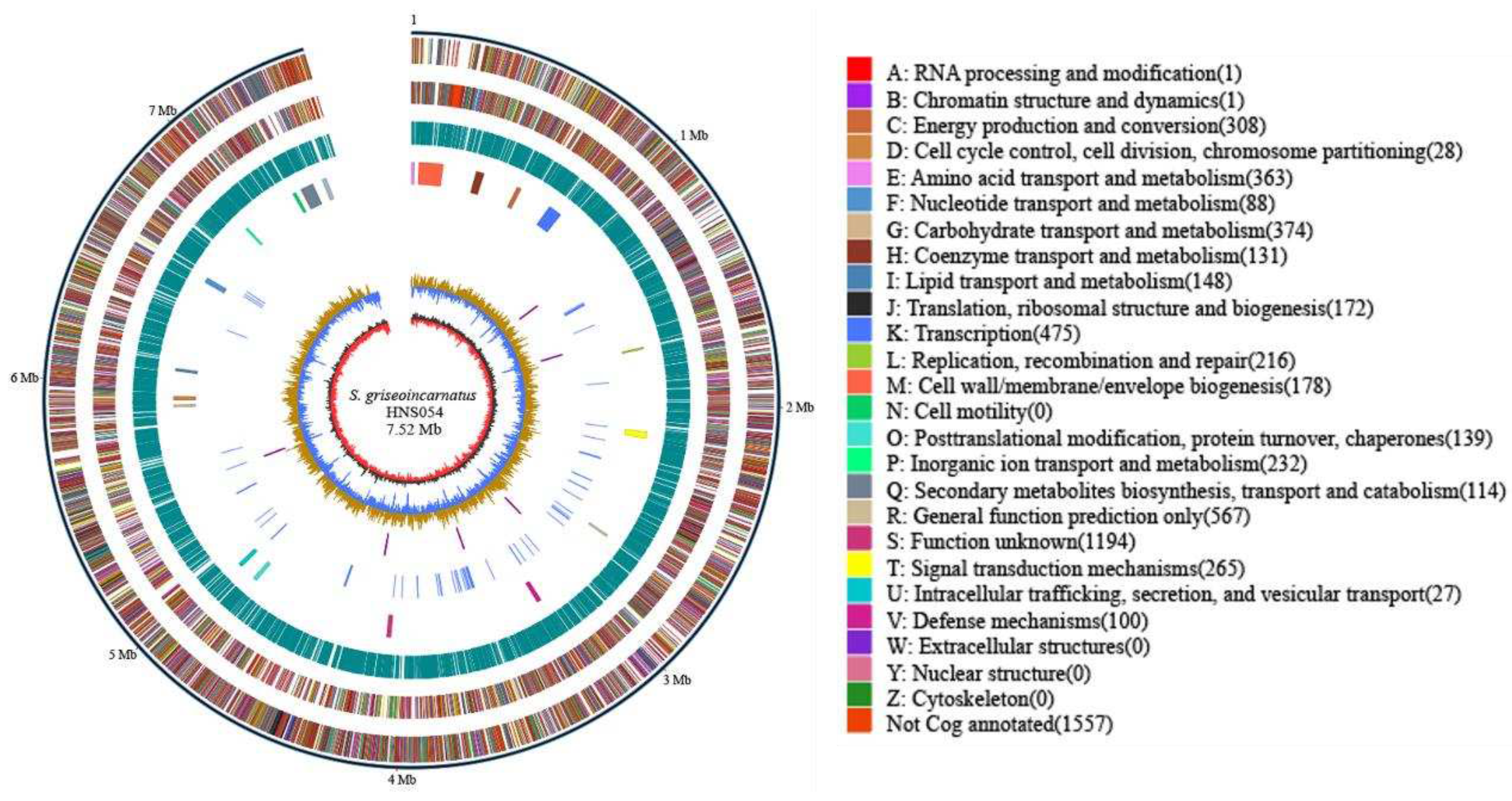
Figure 2.
Comparative genomics analysis of strain HNS054. (A) Whole-genome-based phylogenetic tree of HNS054 and other Streptomyces. Different strain sources and types of BGC are color-coded. (B) Comparison and annotation of orthologous gene clusters among HNS054 (GenBank Accession: CP139576), S. coelicolor A3(2) (GenBank Accession: CP042324), S. albus DSM 41398 (GenBank Accession: CP010519), S. avermitilis MA-4680 (GenBank Accession: NC_003155), and S. venezuelae ATCC 21782 (GenBank Accession: CP029190).
Figure 2.
Comparative genomics analysis of strain HNS054. (A) Whole-genome-based phylogenetic tree of HNS054 and other Streptomyces. Different strain sources and types of BGC are color-coded. (B) Comparison and annotation of orthologous gene clusters among HNS054 (GenBank Accession: CP139576), S. coelicolor A3(2) (GenBank Accession: CP042324), S. albus DSM 41398 (GenBank Accession: CP010519), S. avermitilis MA-4680 (GenBank Accession: NC_003155), and S. venezuelae ATCC 21782 (GenBank Accession: CP029190).
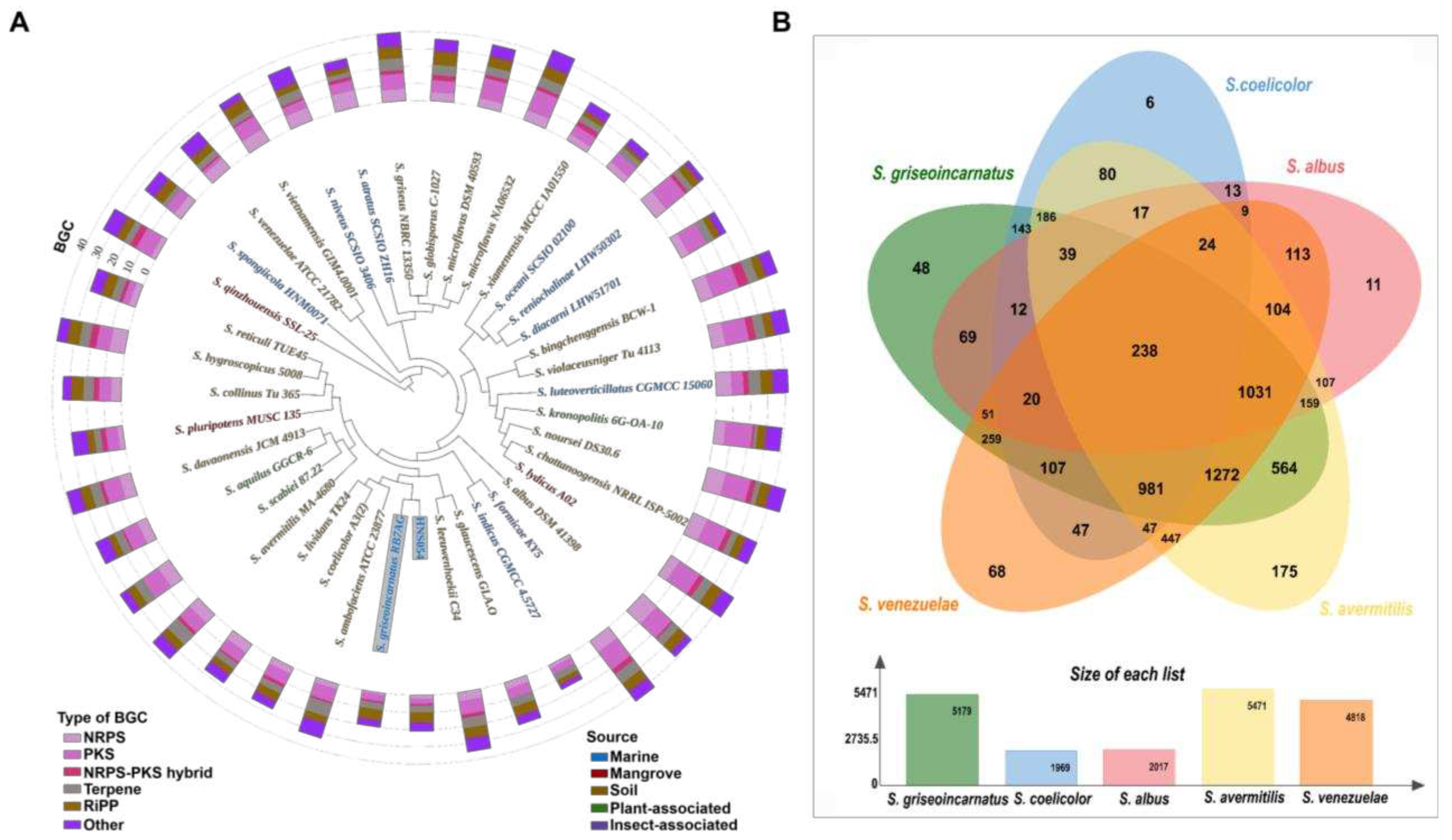
Figure 3.
(A) Growth curves of HNS054 and S. coelicolor M1146 under 30‰ salinity conditions. (B) Salt tolerance of HNS054 and S. coelicolor M1146.
Figure 3.
(A) Growth curves of HNS054 and S. coelicolor M1146 under 30‰ salinity conditions. (B) Salt tolerance of HNS054 and S. coelicolor M1146.
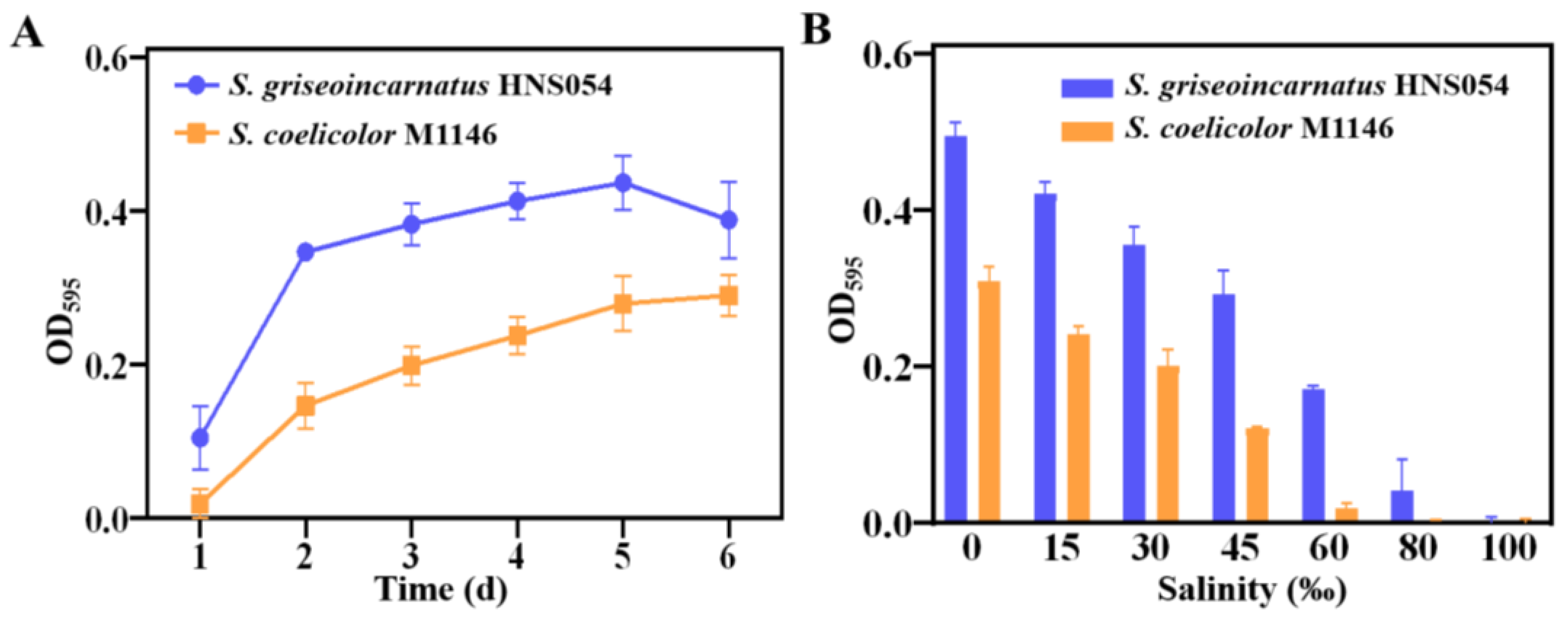
Figure 4.
Improvement of aborycin production. (A) Comparative HPLC analysis of aborycin in the culture extracts of corresponding strains. (B) Titers of different strains. ****: p < 0.0001; **: p < 0.01.
Figure 4.
Improvement of aborycin production. (A) Comparative HPLC analysis of aborycin in the culture extracts of corresponding strains. (B) Titers of different strains. ****: p < 0.0001; **: p < 0.01.
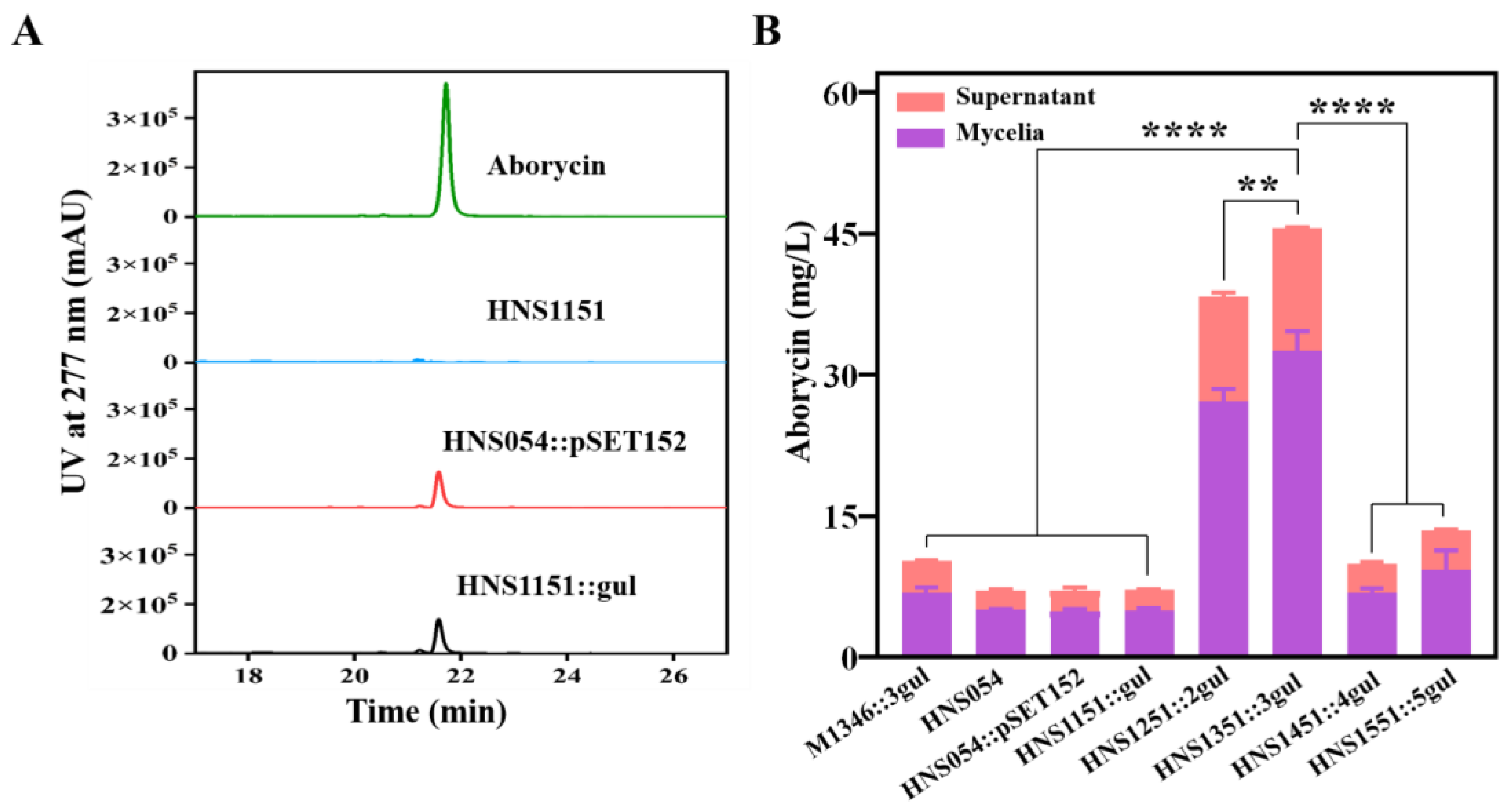
Figure 5.
Detection and comparison of actinorhodin concentrations in strains. (A) Visual observation of actinorhodin production. (B) Quantification of actinorhodin concentrations.
Figure 5.
Detection and comparison of actinorhodin concentrations in strains. (A) Visual observation of actinorhodin production. (B) Quantification of actinorhodin concentrations.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



