Submitted:
22 January 2024
Posted:
23 January 2024
You are already at the latest version
Abstract
Dystonia is a neurological disorder that results in atypical uncontrolled motions or postures caused by either continuous or intermittent contractions of the muscles. Subtypes of dystonia vary in their symptoms and severity; they can affect people of all ages and cause significant disability and poor quality of life. Identifying genes responsible for single or mixed dystonia variants has improved our understanding of its pathogenesis. The mechanisms underlying several of the most prevalent genetic dystonias, including idiopathic dystonia, TOR1A, THAP1, and KMT2B mutations, involve anomalies in transcriptional regulation, striatal dopaminergic signaling, synaptic plasticity, and a loss of inhibition at neuronal circuits. Dystonia is mostly diagnosed based on clinical indicators, and the diagnosis and etiology of this illness remain a problem. In this review, we tried to summarize novel updates about genetic causes and treatment of focal dystonia.
Keywords:
focal dystonia
; blepharospasm
; cervical dystonia
; oromandibular dystonia
; spasmodic dysphonia
1. Introduction
Dystonias are several disorders defined by abnormal repetitive motions or postures caused by excessive contractions of the muscles [1]. The most common forms of dystonia affect a single body region, such as the neck (cervical dystonia), the upper part of the face (blepharospasm), the larynx (spasmodic dystonia), or the top of a limb (focal hand dystonia) (Figure 1.). Although multiple parts of the body may be affected, the pathophysiology of focal dystonias is hypothesized to involve the brain's motor system [2,3].
The most common type of focal dystonia is cervical dystonia (CD). It is characterized by involuntary muscular contractions that result in aberrant head/neck/shoulder motions and postures, as well as tremor and pain [4]. Pain is an important cause of disability and social isolation in patients with cervical dystonia and is frequently the primary reason patients pursue treatment. Pain is regularly identified as a difficult and disabling component of dystonia symptoms in surveys of patients, with a considerable impact on daily activities and work. Mechanisms of pain in CD may be muscle-based and non-muscle-based. Accumulating evidence suggests that non-muscle-based mechanisms (such as dysfunction of descending pain inhibitory pathways as well as structural and network changes in the basal ganglia, cortex, and other areas) may also contribute to pain in CD alongside prolonged muscle contraction [5].
Blepharospasm (BSP) is a main adult-onset focal dystonia that includes involuntary eyelid closing and orbicularis oculi muscle spasms [6]. Henri Meige described ten patients with blepharospasm in 1910: these patients experienced involuntary eyelid closing in combination with jaw muscular contraction. Meige labeled this condition "Convulsions de la Face" in his study [7]. The most noticeable symptom is spasms of ocular closure. Another symptom is frequent blinking, which can be independent or coupled with spasms. A further feature is the inability to open the eyes spontaneously in the absence of orbicularis oculi spasm [8].
Oromandibular dystonia (OMD) represents a very infrequent type of focal idiopathic dystonia. OMD was clinically discovered at the turn of the twentieth century. However, some features of OMD remain unexplained, including the high risk of oral trauma, which is frequently associated with the development of motor symptoms [9]. OMD is a type of focal dystonia characterized by repeated, continuous spasms of the masticatory, lingual, and facial muscles, which results in uncontrolled jaw opening and closing, retractions, protrusion, or a combination of these movements. OMD can occur alone or in conjunction with limb or trunk dystonia caused by toxic, neurodegenerative, metabolic, infectious, postanoxic, post-traumatic, stroke, status epilepticus, primary faciobrachial, or generalized dystonia. The cause could not be determined in some patients [10].
Spasmodic dysphonia (SD) is a type of focal dystonia that involves excessive or incorrect laryngeal muscle contraction during speech. SD is characterized by either inadequate glottic closure (adductor type) or abrupt opening of the vocal folds (abductor type). The predominant symptom of adductor-type SD is a strained or strangled voice, whereas a breathy or absent voice characterizes abductor-type SD. Adductor-type SD accounts for 97% of all SD patients, with 70% exhibiting aberrant extra laryngeal muscle contractions [11].
Focal Hand Dystonia (FHD) is a movement disease that causes involuntary movements, cramps, and spasms. Pathological neuronal microcircuits in the cortical somatosensory system are linked to it. Invasive preclinical techniques allow researchers to explore individual cortical layer and column neuronal microcircuits [12].
A distinct subset of focal dystonia is referred to as occupational dystonia, which includes dystonic illnesses brought on by repeated motor activity and intimately linked to a particular vocation that the affected individual performs for a living. Thus, musicians are a group that is especially susceptible to this illness, which manifests itself when highly skilled motions are performed. It is currently unclear what pathophysiological factors lead to focal dystonia in musicians. Nonetheless, increasing evidence of abnormalities in sensory-motor unity, cortical and subcortical inhibition processes, and the handling of sensory information highlights this disease, thanks to the contributions of neurophysiological research and functional neuroimaging [13].
Focal task-specific dystonia (FTSD) of the hand in musicians is well-studied and is known as musicians' hand dystonia. However, musicians may also experience issues with their laryngeal muscles, lower limbs, and embouchure. When musicians experience dystonia in their perioral and facial muscles while playing embouchure instruments, it is referred to as embouchure dystonia (ED). Diagnosing ED is critical because if the musician keeps playing the instrument, the dystonia could become non-specific and persistent. The only musicians who have been documented to have task-specific dystonia of the lower limbs are drummers. The electromyogram (EMG) of the affected muscles during the task is the basis for the diagnosis. The term "singer's dystonia" (SD) describes a type of laryngeal dystonia that is exclusive to singing tasks [14].
Task-specific dystonia can be noticeable when performing fine motor control tasks like not only writing, and playing an instrument, but also other chores. It can also happen when playing specific sports and could even be harmful to the careers of professional athletes. Consequently, movement disorder neurologists and sports physicians must be knowledgeable about the phenomenology and presentation of sports-related dystonia (SRD) [15].
3. Classification and comorbidities in dystonia
The etiology of dystonia is incredibly varied. The deep grey matter structures called basal ganglia, which oversee initiating and regulating voluntary movement, are implicated in the final common pathway; nevertheless, the reasons for this failure span practically the whole area of neurology. According to Albanese et al., there are two subaxes along which to classify etiology: inherited vs acquired disease, and either the existence or absence of degenerative or structural lesions. The term "primary dystonia," which lacks a specific and widely accepted description, has been less and less common since the release of the more modern Albanese classification. Therefore, in the first instance, a different classification might only differentiate between acquired and hereditary causes of dystonia [16,17].
Dystonia can be acquired from any process that damages the basal ganglia. These processes include vascular (dystonic cerebral palsy is primarily caused by perinatal hypoxic-ischaemic encephalopathy, but stroke is also a significant cause of dystonia in all age groups), toxic (heavy metal poisoning, for example), iatrogenic (such as antipsychotic drugs), traumatic brain injury, or the existence of a space-occupying lesion [18].
Genetic dystonias encompass both situations where the primary symptom is dystonia (e.g., TOR1A variations, due to ANO3 mutations, etc.) and conditions where dystonia is a co-occurring feature of a more complex condition. There is some overlap in the neurological, neuropsychiatric, and systemic characteristics associated with almost all hereditary dystonias, making this distinction imprecise. Dystonia is one of many features of a wide range of genetic problems, such as neurotransmitter abnormalities, neurometabolic disorders, and neurodegenerative diseases. While many diseases have early onset, other significant ones, like Huntington's disease (HD), may not exhibit symptoms until late adulthood [19].
Parkinson's disease and dystonia are inextricably related. Flexion postures of the limbs and trunk are a main sign of Parkinson's disease that was notably prevalent prior to the use of levodopa. These occasionally resulted in severe abnormalities that were misdiagnosed as joint disorders such as rheumatoid arthritis [20]. Dystonia can be present in 30% or more of Parkinson's disease patients, and it is more common when the disease begins under the age of 40 [21]. Parkinsonism and dystonia co-occur in several disorders, and indeed parkinsonism is often associated with mutations in genes like TAF1, ATP1A3, PRKRA, including those involved in the dopamine synthesis pathway, e.g. GCH1, TH [22].
Both dystonia and LID (levodopa-induced dyskinesia) are hyperkinetic movement disorders. Dystonia usually develops on its own, but it can sometimes occur because of a stroke, an injury, or genetic factors. LID relates to Parkinson's disease (PD), arising due to chronic levodopa medication, and can be dystonic or choreiform. LID and dystonia share several phenomenological characteristics and processes. LID and dystonia are both caused by an integrated circuit that includes the cortex, basal ganglia, thalamus, and cerebellum. They also have problems in striatal synaptic plasticity and dysregulation of striatal cholinergic signaling. The extended duration of both LID and dystonia suggests that underlying epigenetic dysregulation could be a proximate cause [23].
4. Diagnostic of dystonia
Finding an underlying etiology for focal dystonia, including acquired, inherited, and idiopathic origins, requires accurate phenotyping of the condition. The last several years have seen a rise in interest in both motor symptoms and the related nonmotor symptoms, as well as their negative effects on quality of life. The growing number of recently identified genes linked to dystonia complicates the diagnosis process. The goal of recent initiatives has been to advance the development of algorithms and recommendations to help with diagnosis and with utilizing diagnostic instruments [24].
The term dystonia refers to a collection of illnesses in which dystonia is the primary clinical symptom, as well as a clinical description of an aberrant posture, sometimes with a twisting quality. There are various causes of dystonias, including idiopathic, hereditary, acquired, and others. While imaging and laboratory tests are usually normal in patients with dystonia, there is no good diagnostic test for idiopathic dystonia. In cases of genetic dystonia, however, genetic testing may provide a conclusive diagnosis. Effect by voluntary action, overflow/mirror dystonia, and a "null point" are among the clinical characteristics of dystonia. It is important to ask precise questions and evaluate sensory tricks through investigation. Clinical factors (onset age, body distribution, temporal pattern, and related clinical aspects) and origin (such as nervous system pathology or whether the disorder is inherited, acquired, or idiopathic) are used to categorize dystonia. Dystonia diagnosis is a difficult task. Patients with functional dystonia (also known as "psychogenic"), scoliosis, myoclonus, tics, Parkinson's disease, and headaches are frequently misdiagnosed [25,26].
Four different forms of dystonia can be distinguished based on their temporal pattern. The occurrence and intensity of dystonia follow a consistent pattern that is similar throughout the day. Dystonia that is specific to an action or task, such as typing, singing, performing an instrument, or writing, only manifests itself during that specific activity. Paroxysmal dystonia is characterized by abrupt, distinct dystonia episodes that end with a return to the neurologic baseline. In dopa-responsive dystonia, diurnal variation—mild symptoms upon arising that get worse as the day goes on—is typically observed. One can treat dystonia alone or in conjunction with another movement problem. Apart from tremor, which is typically phenomenologically dystonic in origin, dystonia is the sole clinical motor symptom associated with solitary dystonia. However, various movement disorders such as myoclonus, parkinsonism, ataxia, or chorea/dyskinesias are linked to combination dystonia. While neuropsychiatric symptoms are often linked to dystonia, cognitive impairment is often limited to degenerative/progressive dystonias. Differentiating dystonia can be based on how quickly symptoms manifest. At least initially, focal dystonias usually occur after a gradual or subacute deterioration of symptoms, which is then followed by a plateau. Although it usually plateaus, further expansion to segmental/multifocal dispersion is possible [27].
Not only can Parkinson's disease manifest with concurrent dystonia, but in some circumstances, parkinsonian characteristics might be mistaken for dystonia. Dystonia is a common presenting feature in patients with Parkinson's disease, which can complicate diagnosis. Of these, 30% generate off-state dystonia, and 10% to 15% may develop (usually lower) limb dystonia. Other possible complications include blepharospasm, apraxia of eyelid opening (primarily in atypical parkinsonism), as well as truncal/axial dystonia, which includes camptocormia and Pisa syndrome. Furthermore, dystonia can also imitate PD. For example, a dystonic limb's delayed movement might be mistaken for parkinsonian bradykinesia, dystonia's elevated tone can be mistaken for stiffness, and a dystonic tremor at rest can be mistaken for a parkinsonian tremor [28].
Misdiagnosis of Parkinson's disease can occur even in people with genetically dystonia syndromes, such as DYT-TOR1A and dopa-responsive dystonia.4 But aside from tremor, isolated dystonia lacks the development of diagnostic features of parkinsonism without true rigidity (the presence of "lead pipe" rigidity, with or without cogwheeling at the wrist), or bradykinesia (a necessary criterion of parkinsonism, which includes slowness with particular features of decrement/fatiguing accompanied by several iterations, or hesitations/interruptions). In mixed types of dystonia, especially in dystonia-parkinsonism, where actual parkinsonism does exist, it is more difficult to distinguish between dystonia and Parkinson's disease. Genetic dystonia parkinsonism causes typically manifest at a much younger age than conventional Parkinson's disease; nonetheless, there may be cases where there is no discernible clinical difference, in which case the medical professional may rely on pertinent symptoms, imaging, and family history to make a diagnosis [29].
5. The genetic background of dystonia
Pathogenic variations in multiple genes can cause isolated dystonia, and the number of identified dystonia genes is expanding [3], but the prevalence of recognized monogenic dystonias is limited. Notably, up to 30% of individuals with idiopathic dystonia report having first or second-degree family members with dystonia [30,31,32], implying an inherited/genetic role in these cases as well. Furthermore, the genetics of dystonia are characterized by low penetrance, possibly leading to an underestimate of hereditary causes [33]. Pathogenic variants in dystonia genes have been linked to a variety of pathways, including gene transcription during neurodevelopment, endoplasmic reticulum stress response, calcium homeostasis, striatal dopamine signaling, and autophagy [34].
- a)
- Endoplasmic reticulum stress response - TOR1A (torsin 1A)
TOR1A, the initially identified dystonia gene, was discovered about 25 years ago on human chromosome 9q34. A distinct 3-bp deletion resulted in the loss of one of a pair of glutamic-acid residues in a preserved area of torsinA [35].
A study with a cohort of Chinese patients revealed a novel missense variant in TOR1A. The first of three cases of previously known variant c.907_909del (p.Glu303del) in a 20-year-old male patient who began experiencing right-hand dysgraphia at the age of 15. Two years later, he acquired cervical dystonia accompanied head tremor. The mutation was passed on from his mother, although she experienced no symptoms [36].
The same variant originated from a 13-year-old male who began walking with a right foot drop at 9 and subsequently involved the neck, torso, and lower limbs. His mother had mobility difficulties and a moderate intellectual handicap; however, DNA samples were not accessible [36].
The last patient was a 17-year-old male who had dysgraphia in his right hand. Six years later, he still had limb dystonia [36].
The last 18-year-old male exhibited a new missense mutation c.765C>A (p.S255R) that caused aberrant trunk position as his first symptom, which proceeded to generalized dystonia within a year [36].
- b)
- Neurodevelopment - THAP1 (THAP domain-containing protein 1), KMT2B (lysine-specific methyltransferase 2B)
Most known mutations in the thanatos-associated protein 1 (THAP1) genes are missense or out-of-frame deletions, while intronic or splice site alterations are rare [37]. Many THAP1 variants, including truncating mutations, have been classified as possibly harmful since they have only been reported in individuals with no information on segregation. Although truncating mutations normally cause a loss of function, they are not always associated with disease [38].
A 54-year-old male with reported retrocollis and dysarthria, which began at 42 years old and progressed. He has no relatives experiencing neurological issues. A physical examination indicated spasmodic speech with perioral dystonia, severe retrocollis from the upper back to the neck, and bilateral upper limb dystonia. Various medications were used but had no effect. Toxin botulinum A cervical dystonia injection was similarly ineffective in treating him. A known pathogenic variation of THAP1 c.505C>T (p.Arg169*) was discovered during dystonia gene panel testing [39].
KMT2B-linked dystonia (DYT-KMT2B) is a dystonia syndrome that often begins in the lower limbs and progresses caudocranially to impact the upper limbs, with substantial craniocervical involvement.
The patient was of Asian heritage and appears to be the first case of DYT-KMT2B from Southeast Asia, providing more evidence for the pathogenic role of the KMT2B c.6210_6213delTGAG variation. At the age of 12, a patient of mixed ancestry (Indian father, Chinese mother) began having slurred speech, which worsened over time to include occasional choking and was linked with involuntary jaw opening and tongue motions. There were no other involuntary motions in the body [40].
- c)
- Striatal dopamine signaling – GNAL (guanine nucleotide-binding protein alpha-activating activity polypeptide)
Previous reports suggest that GNAL variations have been detected in less than 2% of dystonia cases of European origin and contribute to disease in late adulthood [41]. However, in the Hungarian cohort study, it was found a higher prevalence than previously reported. All three mutations identified there are thought to be pathogenic, and the clinical profile fits the characteristics already linked with GNAL variants.
The GNAL c.677G>T (p.Cys226Phe) variant was discovered in a male patient who has had left-sided neck pain, right-directed laterocollis, torticollis, and mild retrocollis since the age of 27 [42].
The symptoms of a female patient with another GNAL c.1315G>A (p.Val439Met) variation began when she was 30 years old. The patient complained that her head began to move slightly to the left and that it became more and more challenging for her to keep it in a neutral position [42].
The GNAL c.1288G>A (p.Ala430Thr) variant was discovered in a 19-year-old female patient with cervical dystonia, left-directed laterocollis, torticollis, and antecollis with platysmal contractions. She tried BoNT and deep cerebral stimulation but neither worked. The patient's concerns indicate that the disease progresses [42].
- d)
- Calcium homeostasis - ANO3 (anoctamin)
ANO3-linked dystonia (DYT-ANO3) has an autosomal-dominant inheritance pattern, yet only heterozygous variations have been identified so far. Most individuals experienced multifocal or segmental dystonia, with focal or generalized distribution being less prevalent. The most common onset was cervical dystonia, followed by upper limb dystonia. Lower limb onset has also been recorded in several cases and appears to relate to a younger age (under 20) [38].
The c.2497A>G (p.Ile833Val) sequence variation affects a highly conserved isoleucine in ANO3's fourth extracellular domain. The patient, a 56-year-old woman, had no family history of cervical dystonia and a dystonic head tremor that began at age 40 [43].
The ANO3 c.2917G>C (p.Gly973Arg) variation replaces glycine with arginine at position 973 in the terminal cytoplasmatic domain. From a clinical perspective, the 71-year-old female patient had sporadic segmental dystonia of the facial region that began at the age of 69 [43].
Since the age of 41, one male patient with the c.2276-6T>C ANO3 splice region mutation has developed focal dystonia, left-sided torticollis, right-sided laterocaput, and dysarthria, but it was a variant of uncertain significance. There were no family members available for testing [42].
- e)
- Autophagy - VPS16 (PS16 core subunit of corvet and HOPS complexes)
VPS16 variations were discovered in the context of early-onset global dystonia accompanied by lysosomal dysfunction [44]. Nevertheless, VPS16 variations have recently been discovered among patients suffering from focal dystonia [45]. Furthermore, these results imply that VPS16 variations should be evaluated in cases of focal dystonia. In the VPS16 gene, two uncommon variations of unknown relevance were discovered. A male cervical dystonia patient with retrocollis and dysarthria had the c.1370T>C (p.Leu457Pro) missense mutation, whereas a female with benign essential blepharospasm had the c.241-2A>C splice site variant [42].
It is also noteworthy that positive family history has been linked to the spread of dystonia from one body location to another [46]. This fact confirms that genes are highly associated with the onset of focal dystonias as well as their progression to the general form.
5. Treatment options for dystonia
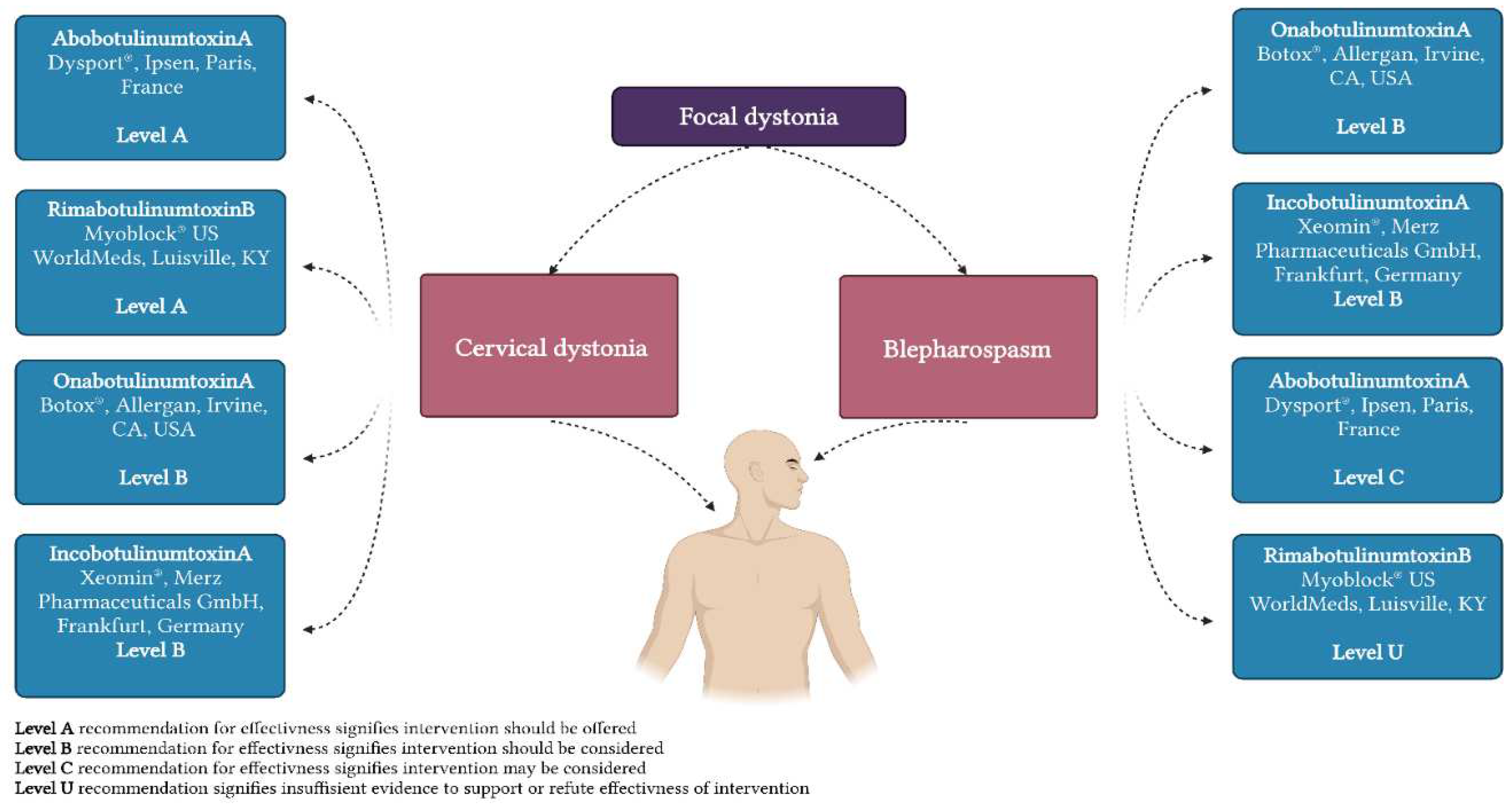
BoNT (botulinum neurotoxin) have been utilized since the 1980s and remains the primary treatment for focal dystonia. To prevent dystonic muscle contractions, BoNT's mechanism of action involves extracellular binding to glycoprotein structures on cholinergic nerve terminals, cleavage of the soluble N-ethylmaleimide-sensitive factor attachment protein (SNAP) receptor complex's components, and neuromuscular transmission through intracellular blockade of acetylcholine release. Notably, new research on various types of dystonia has shown that the main impact of BoNT therapy is the reduction of aberrant activity in the precuneus at the end of the injection cycle, the sensorimotor cortical and subcortical regions at the peak of injection benefits, and the prefrontal and cerebellar regions for years of treatment. [38,39] Its efficacy has been demonstrated numerous times in multiple research studies and trials, including long-term follow-ups also demonstrating continuing safety and efficacy after as much as two decades of therapy [49,50,51,52]. Botulinum toxin formulations are currently accessible in a variety of forms (Figure 2).
Nonetheless, a sizable fraction of patients receiving BoNT therapy fail to respond to treatment either initially or later [54]. There could be a variety of causes for this, some of which are yet unknown [55]. BoNT therapy is difficult; choosing the right muscles to inject and administering the neurotoxin precisely are essential steps toward a successful course of treatment. Certain dystonic symptoms, such as tremulous dystonia or complex multiaxial dystonic movements, are not well suited for treatment with selective chemodenervation. A further consideration is the issue of probable antibody development during long-term BoNT therapy because of its immunogenicity [56]. However, the actual role of immunerrorogenicity in the overall issue of treatment refractoriness can only be evaluated to a limited level, since a recent meta-analysis suggests that the predicted detection frequency of antibody neutralization in dystonic disorders is only approximately 1% [57]. Additionally, not all aspects of antibody formation are fully understood. However, some factors that are thought to play a role include those related to applications, like brief injection cycles or large doses per injecting, while additional variables have to do with the toxin itself, like its composition, production, and storage [58].
According to established dystonia treatment guidelines, deep brain stimulation (DBS) can be used as an alternative option if BoNT treatment fails completely [59,60]. BoNT therapy focuses on the effector organ muscle and is merely symptomatic, whereas DBS acts on the brain to correct the network dysfunction thought to be the cause of dystonic movement disorders [61]. However, there is currently no scientific evidence to support this therapy's superiority. Although DBS has been demonstrated to enhance symptom management in focal [62], segmental [63], and generalized [64] dystonia, there hasn't been much research done specifically on whether DBS can help patients whose BoNT therapy has failed. In this field, there are just two research with the strongest degree of evidence. Research studies were able to demonstrate a statistically significant improvement in dystonic symptoms with DBS can be reported despite this obvious limitation. Data on safety and tolerability seemed sufficient [65]. On the other hand, a large portion of the data is derived from retrospective data. More scientific research is required to fully understand the evidence basis in this field. Above all, additional double-blind, randomized, controlled trials are required; these may include a head-to-head comparison between DBS and BoNT [66].
As existing drugs are not suitable for many patients or become ineffective over time, attempts are being made to administer medicines previously developed for non-dystonia purposes, such as 5HT receptor agonists, various GABAergic pharmaceuticals (e.g., zolpidem or sodium oxybate), and glutamate regulating treatments, could be viable alternatives to traditional dystonia pharmacotherapeutics [67].
Zolpidem, one of the GABA agonists was reported as sufficient antidystonia medication. A man of 53 years old was given a PD diagnosis. He also went through a severe dystonia episode, characterized by a prolonged widening of his mouth and a movement of his head to the right. He was hardly able to move. Doctors gave him 10 mg of zolpidem at night to help with his insomnia. After around fifteen minutes, his dystonia subsided, and he could talk well. In addition, he was able to open his mouth, eat, and swallow. He could also get out of bed and walk with little assistance. Also, his dyskinesia got better. But the increase in motor function was quite temporary—it lasted for two hours. About 30 minutes was the length of both the antidystonia and anti-dyskinesia effects, which was longer than the improvement in motor function. This was not the case with other hypnotics (such as 2 mg of lorazepam) [68].
Dipraglurant, which affects the metabolic glutamate receptor type 5 (mGluR5), which contributes to the development of dystonia in DYT1 rat models, and sodium oxybate have been examined in patients with alcohol-responsive spasmodic dysphonia with promising results [69].
There are no reports of attempts being made to introduce gene therapies for diagnosed mutations in identified genes. The use of nanoparticles or adenoviruses as a vector could be a promising therapy in dystonia caused in most cases by deletions occurring in genes. Currently, the most developed field of gene therapies is attempting to introduce gene therapy using adenoviruses and nanoparticles in an anticancer approach [70].

Table 1.
Summary of potential benefits of treatment [71] based on the last guidelines for dystonia diagnosis and therapy [59].
| Primary focal dystonia |
Cervical dystonia |
Oromandibular dystonia | Blepharospasm |
Spasmodic dysphonia |
Focal hand dystonia |
| BoNT | + | + | + | + | + |
| DBS | + | +/- | +/- | - | + |
| Trihexyphenidyl | + | - | - | - | + |
| Tetrabenazine | + | - | - | - | - |
| Clonazepam | + | - | + | - | - |
| Baclofen | - | + | - | - | + |
| Levodopa | - | - | - | - | - |
| Amantadine | +/- | - | - | - | - |
| Haloperidol | - | - | - | - | - |
6. Conclusions and future perspectives
The mechanism behind dystonia is becoming clearer as genetically based types of the illness are discovered. The action of the protein products of numerous genes may explain the underlying mechanism of dystonias and provide a path to permanently alleviate patient symptoms. However, we still do not know all the mutations and genes involved in the development of focal dystonias, which forces the further development of the genomic background. There are symptomatic treatments available, including pharmaceutical therapy (anticholinergics), intramuscular botulinum toxin injection, and deep brain stimulation; nevertheless, future studies will hopefully lead to reliable biomarkers and better treatments. There is a significant therapeutic gap in current dystonia therapy options, as well as an obvious need for a reliable new treatment. Because most individuals with focal dystonia receive therapy by BoNT, testing any new medication is difficult. There are two key challenges to finding a unique therapeutic solution for dystonia sufferers. The first step is to recognize the current treatment gap. Second, moving some of our dystonia research goals to clinical trial preparation is critical if we are to test innovative treatment medicines. There are currently no reports of gene therapies regarding the introduction of correct genes, but the development of such a treatment modality may be considered in patients with primary dystonia.
Author Contributions
Each of the authors contributed to the data analysis and revision of this manuscript. J.K. and E.S. were responsible for the acquisition, analysis, and interpretation of the collected data and contributed to the concept and draft of the work. J.K., E.S. P.R., and O.K.S. were responsible for the preparation and creation of the published paper. J.K., A.K., and M.C. supervised the entire manuscript, provided critical revisions, and approved the final draft. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jinnah, H.A. The Dystonias. Contin. Lifelong Learn. Neurol. 2019, 25, 976–1000. [Google Scholar] [CrossRef]
- Jinnah, H.A.; Berardelli, A.; Comella, C.; DeFazio, G.; DeLong, M.R.; Factor, S.; Galpern, W.R.; Hallett, M.; Ludlow, C.L.; Perlmutter, J.S.; et al. The Focal Dystonias: Current Views and Challenges for Future Research. Mov. Disord. 2013, 28, 926–943. [Google Scholar] [CrossRef]
- Balint, B.; Mencacci, N.E.; Valente, E.M.; Pisani, A.; Rothwell, J.; Jankovic, J.; Vidailhet, M.; Bhatia, K.P. Dystonia. Nat. Rev. Dis. Primer 2018, 4, 25. [Google Scholar] [CrossRef]
- Tyślerowicz, M.; Kiedrzyńska, W.; Adamkiewicz, B.; Jost, W.H.; Sławek, J. Cervical Dystonia — Improving the Effectiveness of Botulinum Toxin Therapy. Neurol. Neurochir. Pol. 2020, 54, 232–242. [Google Scholar] [CrossRef]
- Rosales, R.L.; Cuffe, L.; Regnault, B.; Trosch, R.M. Pain in Cervical Dystonia: Mechanisms, Assessment and Treatment. Expert Rev. Neurother. 2021, 21, 1125–1134. [Google Scholar] [CrossRef]
- Ma, H.; Qu, J.; Ye, L.; Shu, Y.; Qu, Q. Blepharospasm, Oromandibular Dystonia, and Meige Syndrome: Clinical and Genetic Update. Front. Neurol. 2021, 12, 630221. [Google Scholar] [CrossRef]
- Titi-Lartey, O.A.; Patel, B.C. Benign Essential Blepharospasm. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2023. [Google Scholar]
- Aramideh, M.; Ongerboer De Visser, B.W.; Brans, J.W.; Koelman, J.H.; Speelman, J.D. Pretarsal Application of Botulinum Toxin for Treatment of Blepharospasm. J. Neurol. Neurosurg. Psychiatry 1995, 59, 309–311. [Google Scholar] [CrossRef]
- Manzo, N.; Ginatempo, F.; Belvisi, D.; Defazio, G.; Conte, A.; Deriu, F.; Berardelli, A. Pathophysiological Mechanisms of Oromandibular Dystonia. Clin. Neurophysiol. 2022, 134, 73–80. [Google Scholar] [CrossRef]
- Balasubramaniam, R.; Ram, S. Orofacial Movement Disorders. Oral Maxillofac. Surg. Clin. N. Am. 2008, 20, 273–285. [Google Scholar] [CrossRef]
- Lin, J.; Sadoughi, B. Spasmodic Dysphonia. In Advances in Oto-Rhino-Laryngology; S. Karger AG, 2020; Vol. 85, pp. 133–143. ISBN 978-3-318-06627-2. [Google Scholar]
- Huber, L.; Kassavetis, P.; Gulban, O.F.; Hallett, M.; Horovitz, S.G. Laminar VASO fMRI in Focal Hand Dystonia Patients. Dystonia 2023, 2, 10806. [Google Scholar] [CrossRef]
- Aránguiz, R.; Chana-Cuevas, P.; Alburquerque, D.; León, M. Distonía focales en los músicos. Neurología 2011, 26, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Ray, S.; Pal, P.K. Dystonia in Performing Artists: Beyond Focal Hand Dystonia. Can. J. Neurol. Sci. J. Can. Sci. Neurol. 2022, 49, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Lenka, A.; Jankovic, J. Sports-Related Dystonia. Tremor Hyperkinetic Mov. 2021, 11, 54. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Bhatia, K.; Bressman, S.B.; DeLong, M.R.; Fahn, S.; Fung, V.S.C.; Hallett, M.; Jankovic, J.; Jinnah, H.A.; Klein, C.; et al. Phenomenology and Classification of Dystonia: A Consensus Update. Mov. Disord. 2013, 28, 863–873. [Google Scholar] [CrossRef]
- Grütz, K.; Klein, C. Dystonia Updates: Definition, Nomenclature, Clinical Classification, and Etiology. J. Neural Transm. 2021, 128, 395–404. [Google Scholar] [CrossRef]
- Steel, D.; Reid, K.M.; Pisani, A.; Hess, E.J.; Fox, S.; Kurian, M.A. Advances in Targeting Neurotransmitter Systems in Dystonia. In International Review of Neurobiology; Elsevier, 2023; Vol. 169, pp. 217–258. ISBN 978-0-323-99026-4. [Google Scholar]
- Hernández, I.H.; Cabrera, J.R.; Santos-Galindo, M.; Sánchez-Martín, M.; Domínguez, V.; García-Escudero, R.; Pérez-Álvarez, M.J.; Pintado, B.; Lucas, J.J. Pathogenic SREK1 Decrease in Huntington’s Disease Lowers TAF1 Mimicking X-Linked Dystonia Parkinsonism. Brain 2020, 143, 2207–2219. [Google Scholar] [CrossRef]
- MacFarlane, D.G.; Dieppe, P.A. Pseudo-Rheumatoid Deformity in Elderly Osteoarthritic Hands. J. Rheumatol. 1983, 10, 489–490. [Google Scholar] [PubMed]
- Ishikawa, A.; Miyatake, T. A Family with Hereditary Juvenile Dystonia-parkinsonism. Mov. Disord. 1995, 10, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.S.; Bhatia, K.P.; Lang, A.E. Dystonia and Parkinson’s Disease: What Is the Relationship? Neurobiol. Dis. 2019, 132, 104462. [Google Scholar] [CrossRef]
- Calabresi, P.; Standaert, D.G. Dystonia and Levodopa-Induced Dyskinesias in Parkinson’s Disease: Is There a Connection? Neurobiol. Dis. 2019, 132, 104579. [Google Scholar] [CrossRef]
- Centen, L.M.; Van Egmond, M.E.; Tijssen, M.A.J. New Developments in Diagnostics and Treatment of Adult-Onset Focal Dystonia. Curr. Opin. Neurol. 2023, 36, 317–323. [Google Scholar] [CrossRef]
- Stephen, C.D. The Dystonias. Contin. Lifelong Learn. Neurol. 2022, 28, 1435–1475. [Google Scholar] [CrossRef]
- Albanese, A.; Lalli, S. Is This Dystonia? Mov. Disord. 2009, 24, 1725–1731. [Google Scholar] [CrossRef]
- Morales-Briceno, H.; Fung, V.S.C.; Bhatia, K.P.; Balint, B. Parkinsonism and Dystonia: Clinical Spectrum and Diagnostic Clues. J. Neurol. Sci. 2022, 433, 120016. [Google Scholar] [CrossRef]
- Lalli, S.; Albanese, A. The Diagnostic Challenge of Primary Dystonia: Evidence from Misdiagnosis. Mov. Disord. 2010, 25, 1619–1626. [Google Scholar] [CrossRef]
- Postuma, R.B.; Berg, D.; Stern, M.; Poewe, W.; Olanow, C.W.; Oertel, W.; Obeso, J.; Marek, K.; Litvan, I.; Lang, A.E.; et al. MDS Clinical Diagnostic Criteria for Parkinson’s Disease: MDS-PD Clinical Diagnostic Criteria. Mov. Disord. 2015, 30, 1591–1601. [Google Scholar] [CrossRef]
- Dhaenens, C.; Krystkowiak, P.; Douay, X.; Charpentier, P.; Bele, S.; Destée, A.; Sablonnière, B. Clinical and Genetic Evaluation in a French Population Presenting with Primary Focal Dystonia. Mov. Disord. 2005, 20, 822–825. [Google Scholar] [CrossRef]
- Leube, B.; Kessler, K.R.; Goecke, T.; Auburger, G.; Benecke, R. Frequency of Familial Inheritance among 488 Index Patients with Idiopathic Focal Dystonia and Clinical Variability in a Large Family. Mov. Disord. 1997, 12, 1000–1006. [Google Scholar] [CrossRef]
- Williams, L.; McGovern, E.; Kimmich, O.; Molloy, A.; Beiser, I.; Butler, J.S.; Molloy, F.; Logan, P.; Healy, D.G.; Lynch, T.; et al. Epidemiological, Clinical and Genetic Aspects of Adult Onset Isolated Focal Dystonia in Ireland. Eur. J. Neurol. 2017, 24, 73–81. [Google Scholar] [CrossRef]
- Loens, S.; Hamami, F.; Lohmann, K.; Odorfer, T.; Ip, C.W.; Zittel, S.; Zeuner, K.E.; Everding, J.; Becktepe, J.; Marth, K.; et al. Tremor Is Associated with Familial Clustering of Dystonia. Parkinsonism Relat. Disord. 2023, 110, 105400. [Google Scholar] [CrossRef]
- Thomsen, M.; Lange, L.M.; Zech, M.; Lohmann, K. Genetics and Pathogenesis of Dystonia. Annu. Rev. Pathol. Mech. Dis. 2024, 19, 321932111. [Google Scholar] [CrossRef]
- Ozelius, L.J.; Hewett, J.W.; Page, C.E.; Bressman, S.B.; Kramer, P.L.; Shalish, C.; De Leon, D.; Brin, M.F.; Raymond, D.; Corey, D.P.; et al. The Early-Onset Torsion Dystonia Gene (DYT1) Encodes an ATP-Binding Protein. Nat. Genet. 1997, 17, 40–48. [Google Scholar] [CrossRef]
- Li, L.; Liu, Y.; Huang, J.; Yang, Y.; Pan, Y.; Zhang, X.; Pan, L.; Jin, L. Genetic Spectrum and Clinical Features in a Cohort of Chinese Patients with Isolated Dystonia. Clin. Genet. 2023, 103, 459–465. [Google Scholar] [CrossRef]
- Holla, V.V.; Prasad, S.; Neeraja, K.; Kamble, N.; Yadav, R.; Pal, P.K. Late Adulthood Onset DYT-THAP1 Secondary to a Novel Splice Site Mutation-A Report from India. Parkinsonism Relat. Disord. 2020, 78, 36–37. [Google Scholar] [CrossRef]
- Lange, L.M.; Junker, J.; Loens, S.; Baumann, H.; Olschewski, L.; Schaake, S.; Madoev, H.; Petkovic, S.; Kuhnke, N.; Kasten, M.; et al. Genotype–Phenotype Relations for Isolated Dystonia Genes: MDSGene Systematic Review. Mov. Disord. 2021, 36, 1086–1103. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.J.; Woo, K.A.; Kim, H.-J.; Jeon, B. Late-Onset Dystonia With THAP1 Mutation (DYT6) in South Korea: A Case Report and Literature Review. J. Clin. Neurol. Seoul Korea 2023, 19, 198–200. [Google Scholar] [CrossRef]
- Closas, A.M.F.D.; Lohmann, K.; Tan, A.H.; Ibrahim, N.M.; Lim, J.L.; Tay, Y.W.; Muthusamy, K.A.; Ahmad-Annuar, A.B.; Klein, C.; Lim, S.-Y. A KMT2B Frameshift Variant Causing Focal Dystonia Restricted to the Oromandibular Region After Long-Term Follow-Up. J. Mov. Disord. 2023, 16, 91–94. [Google Scholar] [CrossRef]
- Erro, R.; Bhatia, K.P.; Hardy, J. GNAL Mutations and Dystonia. JAMA Neurol. 2014, 71, 1052. [Google Scholar] [CrossRef] [PubMed]
- Salamon, A.; Nagy, Z.F.; Pál, M.; Szabó, M.; Csősz, Á.; Szpisjak, L.; Gárdián, G.; Zádori, D.; Széll, M.; Klivényi, P. Genetic Screening of a Hungarian Cohort with Focal Dystonia Identified Several Novel Putative Pathogenic Gene Variants. Int. J. Mol. Sci. 2023, 24, 10745. [Google Scholar] [CrossRef]
- Zech, M.; Gross, N.; Jochim, A.; Castrop, F.; Kaffe, M.; Dresel, C.; Lichtner, P.; Peters, A.; Gieger, C.; Meitinger, T.; et al. Rare Sequence Variants in ANO3 and GNAL in a Primary Torsion Dystonia Series and Controls. Mov. Disord. 2014, 29, 143–147. [Google Scholar] [CrossRef]
- Steel, D.; Zech, M.; Zhao, C.; Barwick, K.E.S.; Burke, D.; Demailly, D.; Kumar, K.R.; Zorzi, G.; Nardocci, N.; Kaiyrzhanov, R.; et al. Loss-of-Function Variants in HOPS Complex Genes VPS16 and VPS41 Cause Early Onset Dystonia Associated with Lysosomal Abnormalities. Ann. Neurol. 2020, 88, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-X.; Jiang, L.-T.; Liu, Y.; Zhang, X.-L.; Pan, Y.-G.; Pan, L.-Z.; Nie, Z.-Y.; Wan, X.-H.; Jin, L.-J. Mutation Screening of VPS16 Gene in Patients with Isolated Dystonia. Parkinsonism Relat. Disord. 2021, 83, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Berman, B.D.; Groth, C.L.; Sillau, S.H.; Pirio Richardson, S.; Norris, S.A.; Junker, J.; Brüggemann, N.; Agarwal, P.; Barbano, R.L.; Espay, A.J.; et al. Risk of Spread in Adult-Onset Isolated Focal Dystonia: A Prospective International Cohort Study. J. Neurol. Neurosurg. Psychiatry 2020, 91, 314–320. [Google Scholar] [CrossRef] [PubMed]
- Hok, P.; Veverka, T.; Hluštík, P.; Nevrlý, M.; Kaňovský, P. The Central Effects of Botulinum Toxin in Dystonia and Spasticity. Toxins 2021, 13, 155. [Google Scholar] [CrossRef] [PubMed]
- O’Flynn, L.C.; Simonyan, K. Short- and Long-Term Central Action of Botulinum Neurotoxin Treatment in Laryngeal Dystonia. Neurology 2022, 99. [Google Scholar] [CrossRef]
- Zakin, E.; Simpson, D.M. Botulinum Toxin Therapy in Writer’s Cramp and Musician’s Dystonia. Toxins 2021, 13, 899. [Google Scholar] [CrossRef] [PubMed]
- Duarte, G.S.; Rodrigues, F.B.; Marques, R.E.; Castelão, M.; Ferreira, J.; Sampaio, C.; Moore, A.P.; Costa, J. Botulinum Toxin Type A Therapy for Blepharospasm. Cochrane Database Syst. Rev. 2020, 2020. [Google Scholar] [CrossRef]
- Ramirez-Castaneda, J.; Jankovic, J. Long-Term Efficacy, Safety, and Side Effect Profile of Botulinum Toxin in Dystonia: A 20-Year Follow-Up. Toxicon 2014, 90, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.B.; Duarte, G.S.; Marques, R.E.; Castelão, M.; Ferreira, J.; Sampaio, C.; Moore, A.P.; Costa, J. Botulinum Toxin Type A Therapy for Cervical Dystonia. Cochrane Database Syst. Rev. 2020, 2020. [Google Scholar] [CrossRef]
- Simpson, D.M.; Hallett, M.; Ashman, E.J.; Comella, C.L.; Green, M.W.; Gronseth, G.S.; Armstrong, M.J.; Gloss, D.; Potrebic, S.; Jankovic, J.; et al. Practice Guideline Update Summary: Botulinum Neurotoxin for the Treatment of Blepharospasm, Cervical Dystonia, Adult Spasticity, and Headache: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2016, 86, 1818–1826. [Google Scholar] [CrossRef]
- Erro, R.; Picillo, M.; Pellecchia, M.T.; Barone, P. Improving the Efficacy of Botulinum Toxin for Cervical Dystonia: A Scoping Review. Toxins 2023, 15, 391. [Google Scholar] [CrossRef] [PubMed]
- Jinnah, H.A.; Comella, C.L.; Perlmutter, J.; Lungu, C.; Hallett, M. Longitudinal Studies of Botulinum Toxin in Cervical Dystonia: Why Do Patients Discontinue Therapy? Toxicon 2018, 147, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kessler, K.R.; Skutta, M.; Benecke, R. Long-Term Treatment of Cervical Dystonia with Botulinum Toxin A: Efficacy, Safety, and Antibody Frequency. J. Neurol. 1999, 246, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Rahman, E.; Banerjee, P.S.; Asghar, A.; Gupta, N.K.; Mosahebi, A. Botulinum Toxin Type A Immunogenicity across Multiple Indications: An Overview Systematic Review. Plast. Reconstr. Surg. 2022, 149, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Bellows, S.; Jankovic, J. Immunogenicity Associated with Botulinum Toxin Treatment. Toxins 2019, 11, 491. [Google Scholar] [CrossRef] [PubMed]
- Albanese, A.; Asmus, F.; Bhatia, K.P.; Elia, A.E.; Elibol, B.; Filippini, G.; Gasser, T.; Krauss, J.K.; Nardocci, N.; Newton, A.; et al. EFNS Guidelines on Diagnosis and Treatment of Primary Dystonias. Eur. J. Neurol. 2011, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.; Zheng, Z.; Yin, Z.; Zhang, J.; Lu, G. Deep Brain Stimulation Treating Dystonia: A Systematic Review of Targets, Body Distributions and Etiology Classifications. Front. Hum. Neurosci. 2021, 15, 757579. [Google Scholar] [CrossRef] [PubMed]
- Filip, P.; Jech, R.; Fečíková, A.; Havránková, P.; Růžička, F.; Mueller, K.; Urgošík, D. Restoration of Functional Network State towards More Physiological Condition as the Correlate of Clinical Effects of Pallidal Deep Brain Stimulation in Dystonia. Brain Stimulat. 2022, 15, 1269–1278. [Google Scholar] [CrossRef] [PubMed]
- Volkmann, J.; Mueller, J.; Deuschl, G.; Kühn, A.A.; Krauss, J.K.; Poewe, W.; Timmermann, L.; Falk, D.; Kupsch, A.; Kivi, A.; et al. Pallidal Neurostimulation in Patients with Medication-Refractory Cervical Dystonia: A Randomised, Sham-Controlled Trial. Lancet Neurol. 2014, 13, 875–884. [Google Scholar] [CrossRef]
- Kupsch, A.; Benecke, R.; Müller, J.; Trottenberg, T.; Schneider, G.-H.; Poewe, W.; Eisner, W.; Wolters, A.; Müller, J.-U.; Deuschl, G.; et al. Pallidal Deep-Brain Stimulation in Primary Generalized or Segmental Dystonia. N. Engl. J. Med. 2006, 355, 1978–1990. [Google Scholar] [CrossRef]
- Vidailhet, M.; Vercueil, L.; Houeto, J.-L.; Krystkowiak, P.; Benabid, A.-L.; Cornu, P.; Lagrange, C.; Tézenas Du Montcel, S.; Dormont, D.; Grand, S.; et al. Bilateral Deep-Brain Stimulation of the Globus Pallidus in Primary Generalized Dystonia. N. Engl. J. Med. 2005, 352, 459–467. [Google Scholar] [CrossRef]
- Yin, F.; Zhao, M.; Yan, X.; Li, T.; Chen, H.; Li, J.; Cao, S.; Guo, H.; Liu, S. Bilateral Subthalamic Nucleus Deep Brain Stimulation for Refractory Isolated Cervical Dystonia. Sci. Rep. 2022, 12, 7678. [Google Scholar] [CrossRef]
- Odorfer, T.M.; Volkmann, J. Deep Brain Stimulation for Focal or Segmental Craniocervical Dystonia in Patients Who Have Failed Botulinum Neurotoxin Therapy—A Narrative Review of the Literature. Toxins 2023, 15, 606. [Google Scholar] [CrossRef]
- Pirio Richardson, S.; Jinnah, H.A. New Approaches to Discovering Drugs That Treat Dystonia. Expert Opin. Drug Discov. 2019, 14, 893–900. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Sy, H.-N.; Wu, S.-L. Zolpidem Improves Akinesia, Dystonia and Dyskinesia in Advanced Parkinson’s Disease. J. Clin. Neurosci. 2008, 15, 955–956. [Google Scholar] [CrossRef]
- Abu-hadid, O.; Jimenez-Shahed, J. An Overview of the Pharmacotherapeutics for Dystonia: Advances over the Past Decade. Expert Opin. Pharmacother. 2022, 23, 1927–1940. [Google Scholar] [CrossRef]
- Szewczyk-Roszczenko, O.; Barlev, N.A. The Role of P53 in Nanoparticle-Based Therapy for Cancer. Cells 2023, 12, 2803. [Google Scholar] [CrossRef] [PubMed]
- Batla, A.; Stamelou, M.; Bhatia, K.P. Treatment of Focal Dystonia. Curr. Treat. Options Neurol. 2012, 14, 213–229. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The most frequent locations of dystonia (Created with BioRender.com).
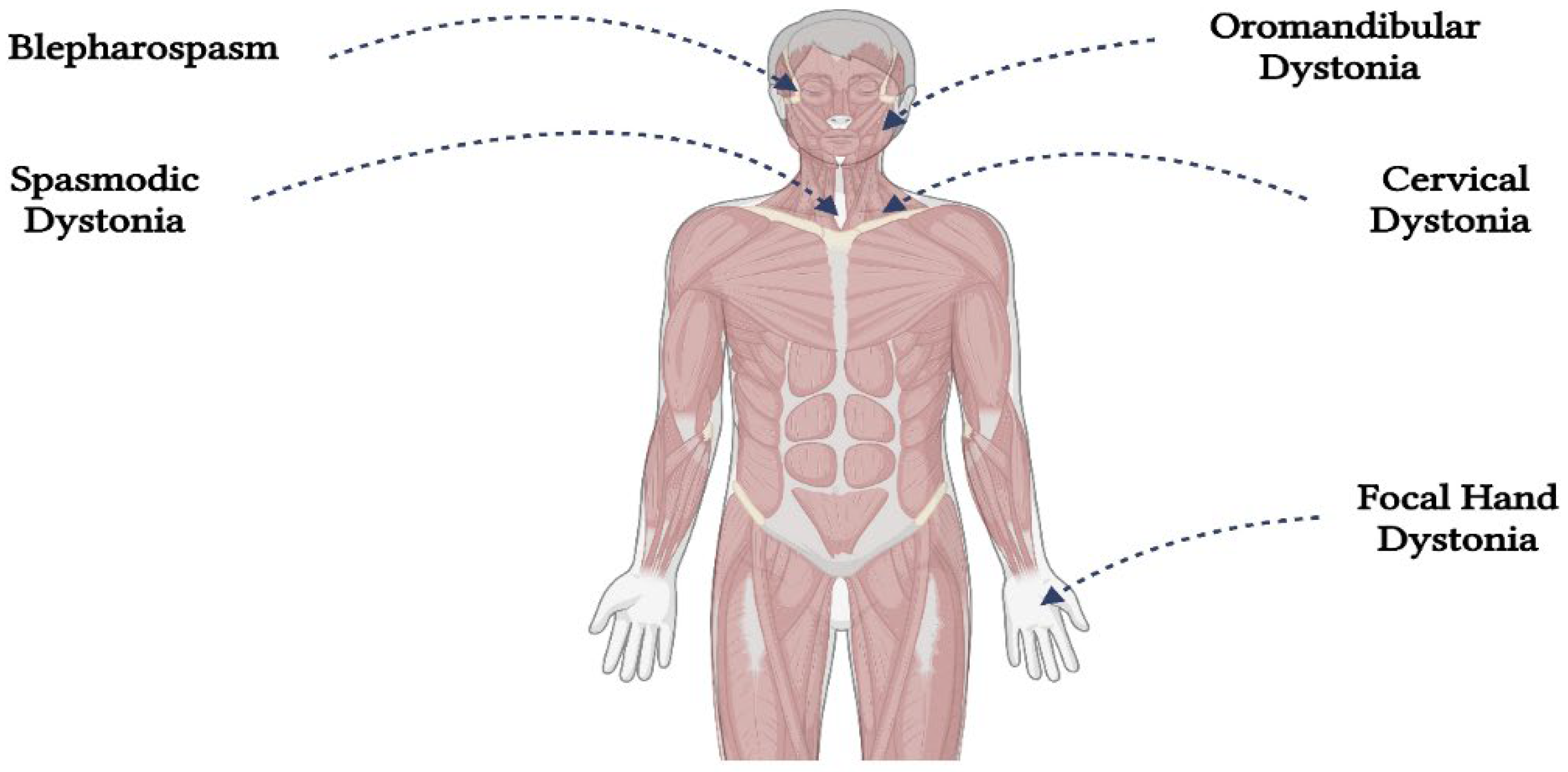
Figure 2.
Report of the Guideline Development Subcommittee of the American Academy of Neurology [44]. (Created with BioRender.com).
Figure 2.
Report of the Guideline Development Subcommittee of the American Academy of Neurology [44]. (Created with BioRender.com).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



