
Submitted:
22 January 2024
Posted:
23 January 2024
You are already at the latest version
Abstract
Genotypic testing is often recommended to improve the management of patients infected with human immunodeficiency virus (HIV). To help combat this major pandemic, next-generation sequencing (NGS) techniques are widely used to analyse resistance to antiretroviral drugs. In this study, we used a Vela Sentosa kit, which is usually used for the Ion Torrent personal genome machine (PGM) platform, to sequence HIV using the Illumina Miseq platform. After RNA extraction and reverse transcriptase-polymerase chain reaction (RT-PCR), minor modifications were applied to the Vela Sentosa kit to adapt it to Illumina Miseq platform. Analysis of the results showed the same mutations present in the samples using both sequencing platforms. The total number of reads varied from 185,069 to 752,343 and from 642,162 to 2,074,028 in the Ion Torrent PGM platform and the Illumina Miseq platform, respectively. The average depth was 21955 and 46856 for Ion Torrent PGM and Illumina Miseq platforms, respectively. The cost of sequencing a run of eight samples was closely similar between the two platforms (about 1 790 United States dollars [USD] for Illumina Miseq and about 1833 USD for Ion Torrent PGM platform). We have shown for the first time that it is possible to adapt and use the Vela Sentosa kit for the Illumina Miseq platform to obtain high quality results with a similar cost.
Keywords:
human immunodeficiency virus
; Vela Sentosa
; Miseq
; personal genome machine
1. Introduction
Human immunodeficiency virus (HIV) infection remains a major public health problem. Human immunodeficiency virus/acquired immunodeficiency syndrome (HIV/AIDS) has caused the deaths of 40.4 million people since the start of the epidemic in the 1980s [1]. According to the latest available data, in 2022, 630,000 people died of HIV/AIDS-related illnesses, and 1.3 million people contracted HIV worldwide [1]. In France, 6.5 million HIV serology tests were performed by medical diagnostic laboratories in 2022 [2]. According to the estimates made by the French Ministry of Health, between 4200 and 5700 persons were tested positive for HIV in 2022. This number has remained stable for several years, regardless of the mode of contamination or place of birth of those who were diagnosed to be HIV-positive. In 2022, 28% of HIV infections were discovered at an advanced stage of infection in France, and this proportion has remained stable over the past few years.
The World Health Organization (WHO) recommends two nucleoside reverse transcriptase inhibitors (NRTIs) plus a non-nucleoside reverse transcriptase inhibitor or an integrase inhibitor for the standard first-line antiretroviral (ARV) therapy in adults and adolescents [3]. In adults, the second-line ARV therapy consists of two NRTIs combined with a ritonavir-boosted protease inhibitor. ARV treatment works best when taken as prescribed by medical practitioners. Poor compliance can lead to drug resistance, enabling HIV to multiply and the disease to progress.
In addition to poor compliance, another major obstacle to a successful ARV therapy is the development and acquisition of drug resistance due to mutations in HIV strains. Genotypic tests for ARV resistance are widely used to improve the therapeutic management of patients [4,5]. They are recommended at the time of diagnosis, i.e. before the initiation of treatment, in order to search for possible transmission of resistance, which is detected in around 10% of new infections for non-NRTIs and 3% for anti-proteases [4]. Further genotypic tests are strongly recommended in the event of ARV treatment failure. Genotypic tests analyse the sequences of genes that code for viral reverse transcriptase, protease, and integrase, in comparison with those of a wild-type HIV reference strain, to identify the mutations associated with resistance [6,7,8,9].
At the Versailles Hospital Centre in Versailles, France, personal genomic machine (PGM) Ion Torrent high-throughput sequencing technology has been used, together with a commercial kit from Vela Diagnostics, since 2021 to diagnose HIV. Compared with the older diagnostic techniques, such as the Sanger dideoxy method, Next-Generation Sequencing (NGS) enables simultaneous reads of millions of copies of HIV genomes per run, enabling the detection of low-frequency mutations. In addition, NGS approaches enable the detection of admixtures, which is particularly important for patient follow-up [10]. In 2019, Vela Diagnostics NGS obtained the United States Food and Drug Administration (FDA) approval for HIV genotyping and resistance testing for in vitro diagnostic purposes [11].
A recent Italian study that evaluated the Vela Diagnostics system showed better results with this NGS technology, compared to Sanger sequencing, in terms of success rate, accuracy, repeatability, and reproducibility of results [12]. In the present study, for the first time, we used a Vela Sentosa kit to sequence HIV genomes using the Illumina Miseq platform. The aim of our study was to show that it is possible to adapt the Vela kit to Miseq and obtain better results at a lower cost than with the PGM Ion Torrent sequencer.
2. Materials and Methods
2.1. Test validation samples
The reference HIV samples for quality control (QC) and test validation were obtained from the French National Reference Centre for HIV (Centre National de Référence HIV; CNR) in Bichat hospital, Paris, France. These samples are produced from the supernatants of the reference HIV strains under in vitro cultivation. Each year, QC samples are distributed and tested in several laboratories in France and at the Pasteur Institute (Paris, France) to assess the quality of HIV tests being performed. In addition to the QCs, we added a positive HIV control (AcroMetrixTM Genotyping HIV RT/PR mutant control) and a negative control (HIV-free PCR).
2.2. RNA extraction
All samples were centrifuged at 17000 g for 2 h at 4°C. After centrifugation, 400 µl of the supernatant were used to extract the viral RNA using the EZ1 virus mini kit (Qiagen S.A.S., Courtaboeuf, France) as recommended by the manufacturer.
2.3. PCR amplification
Reverse transcriptase-polymerase chain reaction (RT-PCR) was performed using the Sentosa® SQ HIV drug resistance mutations (DRM) library kit (Vela Diagnostics, Hamburg, Germany) according to the manufacturer's recommendations. Four primer pools were used to amplify the protease (PR), reverse transcriptase (RT), and integrase (INT) genes. Each sample was tested in quadruplicate according to the primer pool used in a single PCR program.
2.4. Library preparation for Ion Torrent PGM platform
The library was prepared according to the Sentosa® SQ HIV DRM (Experienced User Card V1.0) manual protocol (Vela Diagnostics, Hamburg, Germany). After amplification, the PCR products were normalized and purified using the Sentosa® SQ virus solution prep kit. For each sample, 13 µl of the purified PCR product were used for enzymatic fragmentation using the Sentosa® SQ HIV DRM library kit. The ligation step was performed using 1 µl adapter and 1 µl barcode in the Ion Xpress Barcode Adapters kit (Thermo Fisher Scientific, Paris, France). The ligation program was 20°C for 15 min, 65°C for 5 min, and 85°C for 2 min. After ligation, each ligated PCR product was purified with magnetic beads and eluted with 25 µl elution buffer. For each sample, 10 µl were taken to constitute a pool of all the libraries. The pool was then diluted by 1/4 with the elution buffer, and PCR and emulsion enrichment were performed on the Ion Chef instrument prior to sequencing. The three viral genes, PR, RT, and INT, were sequenced on a Sentosa® SQ 316 chip (Ion Torrent PGM) using the dedicated Sentosa® reagent kits.
2.5. Library preparation for Illumina Miseq platform
After PCR, 20 µl of the purified product were used for enzymatic fragmentation using the same Sentosa® SQ HIV DRM manual protocol. Ligation was performed using 10 µl of TruSeq DNA UD Indexes v2 from the Illumina platform (Illumina Inc., San Diego, CA, USA) under the following conditions: 20°C for 20 min, 65°C for 5 min, and 85°C for 2 min. After ligation, each ligated PCR product was eluted with 18 µl elution buffer. For each sample, 5 µl were taken to form a pool of all the libraries. The concentration of the pooled library was set at 20 nM, and no dilution was performed. The pool was denatured with 0.05 N NaOH and diluted with HT1 to 30 pM. This diluted pool was incubated at 96°C for 4 min using a heat block, and 15% PhiX control was added. The library products were analysed by sequencing using the MiSeq v2 reagent kit (cycle 300) (Illumina Inc.) on the Illumina MiSeq platform in 2 × 151 cycles and a micro flow cell.
2.6. Sequence data analysis
The bioinformatic analyses were performed using the Advanced Sequencing Platform (ASP) v3.13.0 available from SmartGene (SmartGene, Lausanne, Switzerland). The SmartGene HIV-1 application is based on the proprietary IDNS (Integrated Database Network System) technology of SmartGene and is accessible via a secure web interface, which can handle base called sequencing files generated through different sequencing technologies. Related SmartGene patents are EP05700367 and EP07816282. *.bam or *.fastq files obtained through the Sentosa SQ platform (Vela) or through the MiSeq platform (Illumina) were uploaded to ASP. The application first performs the following steps in an automated manner:
(1) paired-end detection of read files, if uploaded in the same batch;
(2) technology-specific quality filtering to trim or remove low-quality reads; and
(3) establishment of a work list of the batch.
The user then selects the analysis pipeline to be used, in this case the HIV-1 PR+RT+INT targeted workflow, and selects the appropriate cut-off for ambiguous bases (background noise filtering, here 0.5%). The automated analysis pipeline performs the following actions:
- (1)
- quality filtering: R1 and R2 files are merged for Illumina data, then a sliding window (size of 25 nt) is applied to reads, enhancing the trimming of poor quality sections having a low Phred score (< 23 for the present study) and filtering short reads as well (< 20 nt in this study);
- (2)
- read mapping: reads passing the quality filters are mapped against HIV-1 profiles for PR, RT, and INT, generating a frame-aware nucleic acid alignment, using a proprietary alignment method of combined global and local alignments developed by SmartGene, followed by translation of the corrected alignments into amino acid sequences;
- (3)
- mapped reads are used for the creation of a frequency matrix;
- (4)
- mutations are determined and validated with respect to the reference (here HxB2, accession no. K03455) and their frequencies are calculated.
To generate the final report, the user selects the mutation cut-off (0.5–20%) above which mutations are assessed for resistance using one of the embedded HIV resistance algorithms, as well as a minimal read depth. In the present study, the algorithm developed by the French National Agency for AIDS Research (ANRS) was used [13], with a cut-off value of 20% and a minimum depth of 200. The drug resistance profile was then characterised, and the mutation frequencies were expressed as a percentage of the total number of HIV strains analysed. The final results were summarized in a list of all mutations per sample with their respective frequencies.
2.7. Statistical analysis
All statistical tests were carried out using Microsoft Excel version 16 (Microsoft Corp., Redmond, WA, USA). The cost per sample per run (eight samples) was calculated for the sequencing preparation workflow of Illumina Miseq and Ion Torrent PGM platforms. The prices of all kits used from PCR to sequencing were included, taking into account the total number of samples that can be processed per kit.
The average values of depths and the numbers of reads were compared using the two-tailed Student t-test. The significance level was set at P < 0.05.
Cohen's kappa coefficient was calculated to estimate the degree of agreement between the results of Ion torrent PGM platform and Miseq Illumina platform [14].
3. Results
A total of six QCs and the single positive control were correctly sequenced, and all mutations were found with both platforms. For the Ion torrent PGM platform, the total number of reads ranged from 185,069 to 752,343 (Figure 1). The average depth was 21,955, with an average mutation frequency of 99.3% (Figure 2). All HIV strains showed mutations that are associated with drug resistance. CQ022022, CQ032022, CQ012023, and CQ042023 strains had subtype B, while CQ022023 strain had subtype A, and CQ012022 strain had subtype F2 (Additional File S1).
For the Miseq Illumina platform, the total number of reads ranged from 642,162 to 2,074,028. The average depth was 46,856, with an average frequency of 99.5%. CQ022022, CQ032022, CQ012023, and CQ042023 strains were subtype B, while CQ022023 strain was subtype A1, and CQ012022 strain was subtype F2 (Additional File S2).
Concerning the number of mutations, those found in the INT gene were 100% concordant (Cohen’s kappa coefficient, 1; perfect agreement) for all samples. However, a few small, non-significant discrepancies (P > 0.05) were observed in the PR and RT genes (Table 1). These discordant nucleotides have not been reported to be associated with drug resistance.
The comparison of mean reads showed a statistically significant difference between Miseq (mean total 577,718) and PGM (mean total 445,178) (P = 0.0017). Although the average depth was higher with the Illumina Miseq platform than with the Ion torrent PGM platform, the difference did not reach statistical significance (P = 0.10).
Furthermore, the total duration of the preparation run was 26 h 43 min with the PGM and 26 h 28 min with Miseq. In all cases, the handling of the Illumina protocol was simpler and shorter (Figure 3). The price of a sequencing run on the Illumina Miseq platform was 1640 euros or about 1790 US dollars (exchange rate on 5 January 2024), and 1685 euros or about 1833 US dollars on the Ion Torrent PGM platform (Table 2).
4. Discussion
The NGS technology has become a powerful tool in the public health context of virus surveillance. The study of HIV drug resistance by sequencing is a key step in HIV/AIDS therapeutic management and strategy. Sequencing performed in the search for mutations in RT, PR, and INT genes is indicated in several clinical situations to optimise patient management [6,7,8,9]. Since the introduction of PGM Ion torrent sequencing technology at the Hospital Centre in Versailles in 2021, this technique has been used to study HIV resistance in over 677 patients. Ion Torrent sequencing technology is known for its simplicity, speed, and lower cost, due to its semiconductor chips [15].
The switch to a new sequencer, i.e. the Illumina Miseq platform, at our laboratory prompted us to make adjustments and adapt the Sentosa® kit supplied by Vela to the Miseq sequencer on the Illumina platform. Vela's Sentosa kit is usually used for the PGM; this is the first time that it has been adapted for use with the Miseq Illumina. The advantage of using the Vela kit is that a single PCR can amplify all three HIV genes of interest, namely RT, PR, and INT [16], compared with other techniques which require PCR amplifications of individual genes [17].
Overall, the results showed a perfect concordance (Cohen's kappa coefficient ranged from 0.97 to 1: perfect agreement) of mutations between the two techniques, validating the Miseq sequencing technique on the Illumina platform. Indeed, the latter assay performed with minor modifications met our predefined validation criteria based on recommended methods for validating an in-house genotyping assay for HIV drug resistance monitoring (Figure 3). Consequently, the test was deemed suitable for our needs and capable of replacing the Ion Torrent PGM test in our laboratory.
Interestingly, the total number of reads for each sample and the average depth were higher with the Miseq Illumina platform than with the Ion Torrent platform. This difference can be explained by the fact that the chip in Ion Torrent platform used for sequencing generates around 5 million reads, whereas Illumina platform's micro flow cell generates around 8 million reads. In addition, the preparation of the library was simpler and faster with Miseq Illumina platform, allowing time flexibility for laboratory technicians and rapid delivery of results. Moreover, the price of reagents was 1790 USD with the Miseq Illumina platform and 1833 USD with the Ion Torrent PGM platform. This small difference in price is due to the necessity of using several Thermo Fisher Scientific kits for library enrichment on the Ion Chef automated system prior to PGM sequencing. This enrichment step is specific to and necessary for the Ion Torrent platform, but not for the Miseq Illumina platform.
5. Conclusions
For the first time, we have shown that it is possible to use the Vela library to sequence HIV genome on the Miseq sequencer with Illumina platform. This platform is simpler and faster and provides high quality results similar to those of the Ion Torrent PGM platform.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Additional File S1: Reports of drug resistance results with the Ion torrent PGM platform; Additional File S2: Reports of drug resistance results with the Illumina Miseq platform; Additional File S3: Frequency of mutations with their depths found in all samples with both platforms.
Author Contributions
Conceptualization, S.M.J. and N.P.M.; methodology, N.P.M.; validation, S.M.J.; writing—original draft preparation, N.P.M.; writing—review and editing. C.F.L., S.D., and O.Z. carried out the RNA extractions and RT-PCR. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Hospital Centre of Versailles, grant number O2024/04.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
We thank the personnel of the laboratory of Hospital Centre of Versaille, especially Benjamin Maneglier, Franck Mausoleo, and Alexandra Leocadie. A Big thank to Leonardo Basco for reading and correcting this article. We thank also the SmartGene team, especially Jean Ruelle. We thank Leonardo Basco.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
AIDS, acquired immunodeficiency syndrome; ANRS, French National Agency for AIDS Research; ARV, antiretroviral; ASP, Advanced Sequencing Platform; CNR, Centre National de Référence (French National Reference Centre for HIV); DRM, drug resistance mutations; FDA, Food and Drug Administration; HIV, human immunodeficiency virus; IDNS, Integrated Database Network System; INT, integrase; NGS, next-generation sequencing; NRTIs, nucleoside reverse transcriptase inhibitors; PGM, personal genomic machine; PR, protease; QC, quality control; RT, reverse transcriptase; RT-PCR, reverse transcriptase-polymerase chain reaction; USD, United States dollars; WHO, World Health Organization.
References
- Joint United Nations Programme on HIV/AIDS (UNAIDS). The path that ends AIDS: UNAIDS Global AIDS Update 2023. Geneva, Switzerland: Joint United Nations Programme on HIV/AIDS, 2023. Available online: https://thepath.unaids.org/wp-content/themes/unaids2023/assets/files/2023_report.pdf (accessed on 5 January 2024).
- Santé Publique France. Infection par le VIH. Bulletin de santé publique VIH-IST. November 2023. Available online: https://www.santepubliquefrance.fr/ (accessed on 5 January 2024).
- UNAIDS. HIV treatment. Geneva, Switzerland: Joint United Nations Programme on HIV/AIDS, 2024. 2024. Available online: https://www.unaids.org/en/topic/treatment (accessed on 5 January 2024).
- Assoumou, L.; Bocket, L.; Pallier, C.; Grude, M.; Ait-Namane, R.; Izopet, J.; Raymond, S.; Charpentier, C.; Visseaux, B.; Wirden, M.; et al. Stable prevalence of transmitted drug resistance mutations and increased circulation of non-B subtypes in antiretroviral-naive chronically HIV-infected patients in 2015/2016 in France. J. Antimicrob. Chemother. 2019, 74, 1417–1424. [Google Scholar] [CrossRef]
- Aulicino, P.C.; Zapiola, I.; Kademian, S.; Valle, M.M.; Giuliano, S.F.; Toro, R.; Barbas, G.; Cañizal, A.M.; Mayon, P.; Golemba, M.D.; et al. Pre-treatment drug resistance and HIV-1 subtypes in infants from Argentina with and without exposure to antiretroviral drugs for prevention of mother-to-child transmission. J. Antimicrob. Chemother. 2019, 74, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Hertogs, K.; de Béthune, M.-P.; Miller, V.; Ivens, T.; Schel, P.; Van Cauwenberge, A.; Eynde, C.V.D.; van Gerwen, V.; Azijn, H.; van Houtte, M.; et al. A Rapid Method for Simultaneous Detection of Phenotypic Resistance to Inhibitors of Protease and Reverse Transcriptase in Recombinant Human Immunodeficiency Virus Type 1 Isolates from Patients Treated with Antiretroviral Drugs. Antimicrob. Agents Chemother. 1998, 42, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Johnston, E.; Dupnik, K.M.; Gonzales, M.J.; A Winters, M.; Rhee, S.-Y.; Imamichi, T.; Shafer, R.W. Panel of prototypical infectious molecular HIV-1 clones containing multiple nucleoside reverse transcriptase inhibitor resistance mutations. AIDS 2005, 19, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.; Vazquez, A.C.; Winner, D.; Rose, J.D.; Wylie, D.; Rhea, A.M.; Henry, K.; Pappas, J.; Wright, A.; Mohamed, N.; et al. Novel Method for Simultaneous Quantification of Phenotypic Resistance to Maturation, Protease, Reverse Transcriptase, and Integrase HIV Inhibitors Based on 3′Gag(p2/p7/p1/p6)/PR/RT/INT-Recombinant Viruses: a Useful Tool in the Multitarget Era of Antiretroviral Therapy. Antimicrob. Agents Chemother. 2011, 55, 3729–3742. [Google Scholar] [CrossRef]
- Saladini, F.; Giannini, A.; Boccuto, A.; Vicenti, I.; Zazzi, M. Agreement between an in-house replication competent and a reference replication defective recombinant virus assay for measuring phenotypic resistance to HIV-1 protease, reverse transcriptase, and integrase inhibitors. J. Clin. Lab. Anal. 2018, 32. [Google Scholar] [CrossRef] [PubMed]
- Stella-Ascariz, N.; Arribas, J.R.; Paredes, R.; Li, J.Z. The Role of HIV-1 Drug-Resistant Minority Variants in Treatment Failure. J. Infect. Dis. 2017, 216 (Suppl. S9), S847–S850. [Google Scholar] [CrossRef]
- May, S.; Adamska, E.; Tang, J. Evaluation of Vela Diagnostics HIV-1 genotyping assay on an automated next generation sequencing platform. J. Clin. Virol. 2020, 127, 104376. [Google Scholar] [CrossRef] [PubMed]
- Bonifacio, M.A.; Genchi, C.; Lagioia, A.; Talamo, V.; Volpe, A.; Mariggiò, M.A. Analytical Assessment of the Vela Diagnostics NGS Assay for HIV Genotyping and Resistance Testing: The Apulian Experience. Int. J. Mol. Sci. 2022, 23, 2727. [Google Scholar] [CrossRef] [PubMed]
- Descamps D, Calvez V, Chaix ML, Charpentier C, Delaugerre C, Flandre P, Izopet J, Marcellin AG, Morand-Joubert L, Peytavin G, Plantier JC, Yerly S. HIV-1 genotypic drug resistance interpretation’s algorithms. Paris, France: HIV French resistance and French ANRS-MIE (National Agency for AIDS Research – Emergent Infectious Diseases) AC43 Resistance group, 2023. Available online: https://hivfrenchresistance.org (accessed on 17 January 2024).
- Landis, J.R.; Koch, G.G. The measurement of observer agreement for categorical data. Biometrics 1977, 33, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Thermo Fisher Scientific. Ion Torrent. Available online: https://www.thermofisher.com/fr/fr/home/brands/ion-torrent.html (accessed on 5 January 2024).
- Dessilly, G.; Goeminne, L.; Vandenbroucke, A.-T.; Dufrasne, F.E.; Martin, A.; Kabamba-Mukabi, B. First evaluation of the Next-Generation Sequencing platform for the detection of HIV-1 drug resistance mutations in Belgium. PLOS ONE 2018, 13, e0209561. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.T.; Simmon, K.E.; Mallory, M.A.; Hymas, W.C.; Stevenson, J.; Barker, A.P.; Hillyard, D.R. HIV-1 Drug Resistance Assay Using Ion Torrent Next Generation Sequencing and On-Instrument End-to-End Analysis Software. J. Clin. Microbiol. 2022, 60, e0025322. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Comparison of the total number of reads with Ion torrent PGM and Illumina Miseq platforms.
Figure 1.
Comparison of the total number of reads with Ion torrent PGM and Illumina Miseq platforms.
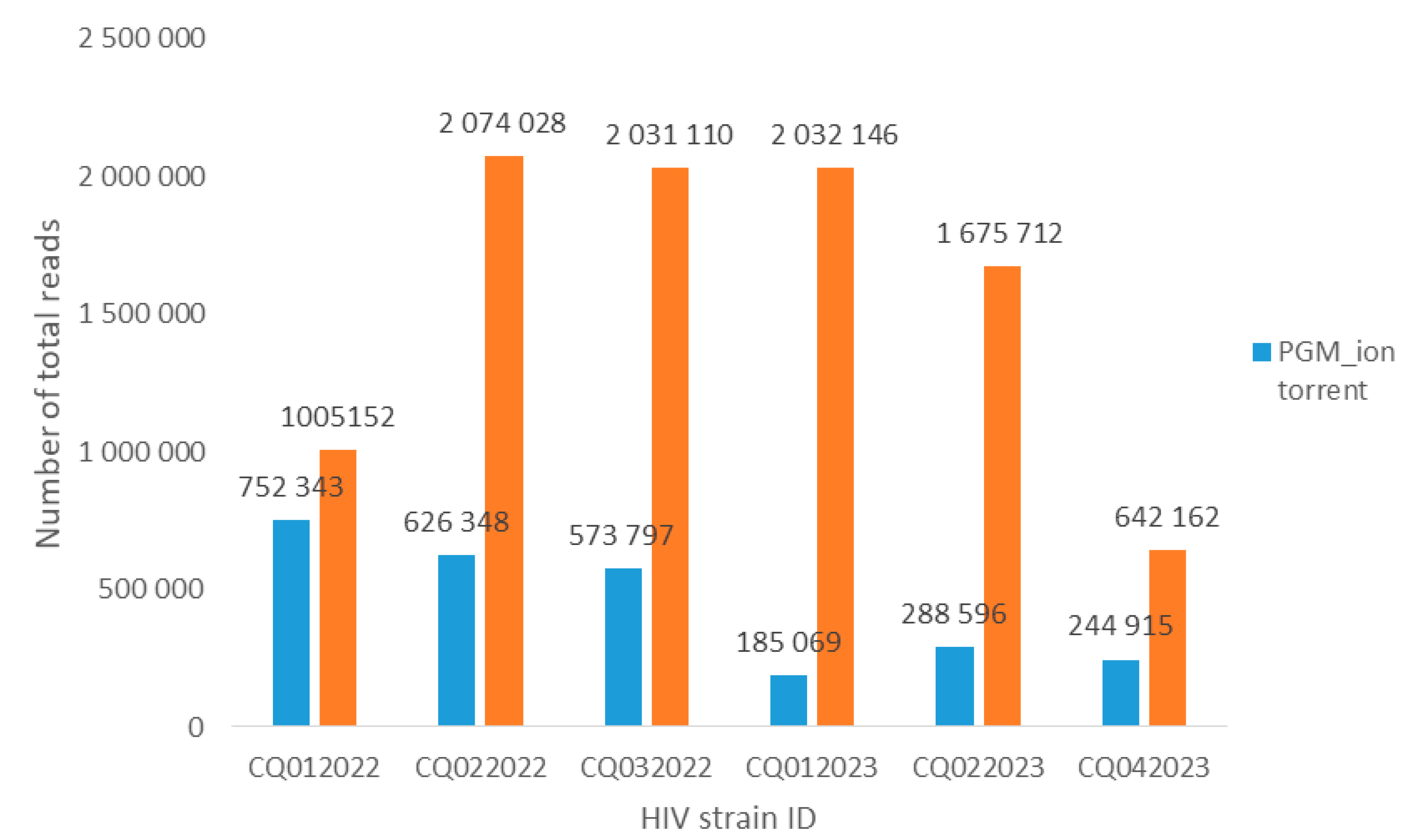
Figure 2.
Comparison of depth between Ion torrent PGM and Illumina Miseq platforms. The figure presents the average coverage for each HIV gene with PGM Ion torrent (blue squares) and Miseq Illumina (brown squares). PR, protease gene; RT, reverse transcriptase gene; INT, integrase gene.
Figure 2.
Comparison of depth between Ion torrent PGM and Illumina Miseq platforms. The figure presents the average coverage for each HIV gene with PGM Ion torrent (blue squares) and Miseq Illumina (brown squares). PR, protease gene; RT, reverse transcriptase gene; INT, integrase gene.
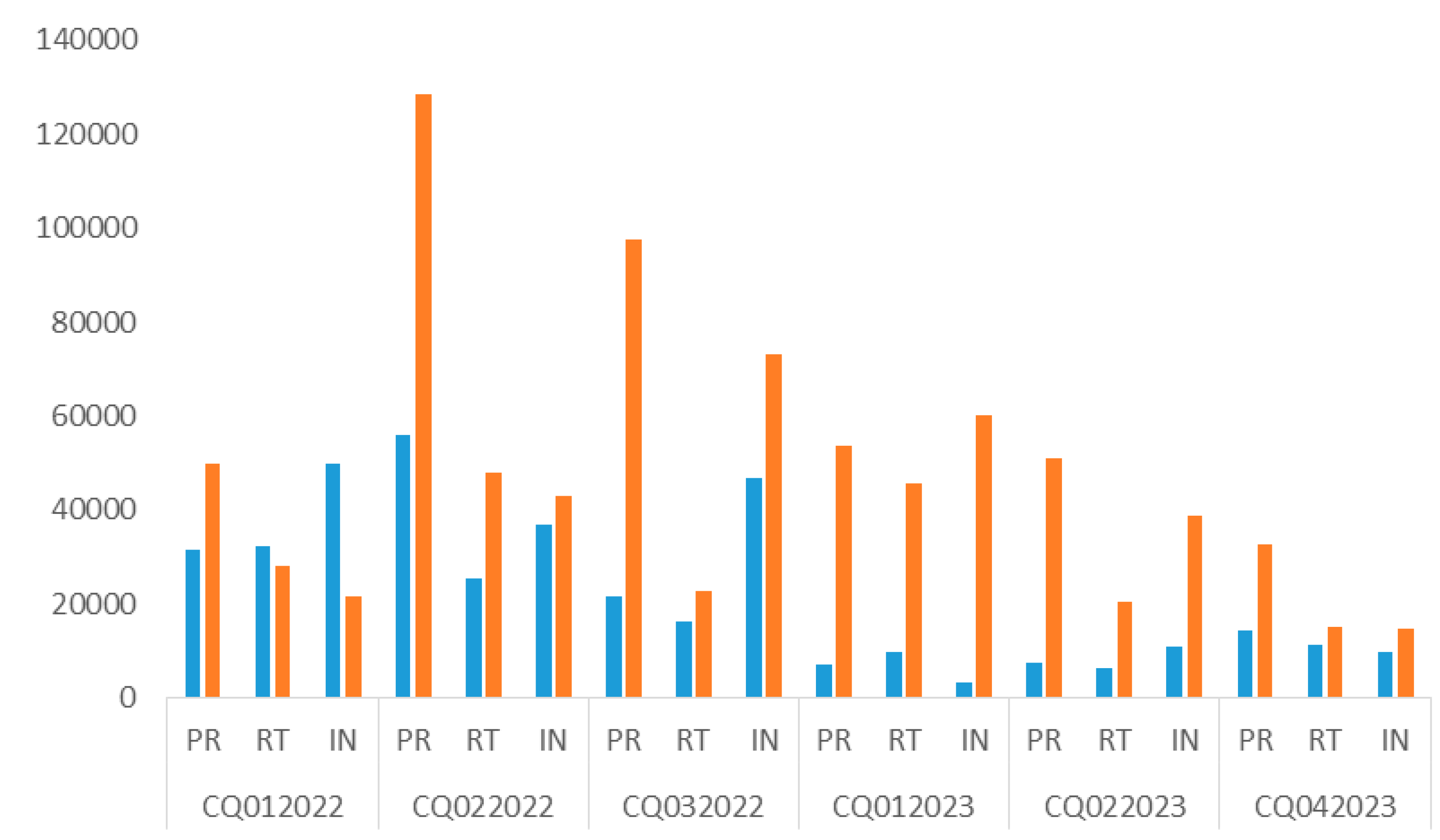
Figure 3.
Sequencing times with Illumina Miseq and Ion Torrent PGM.
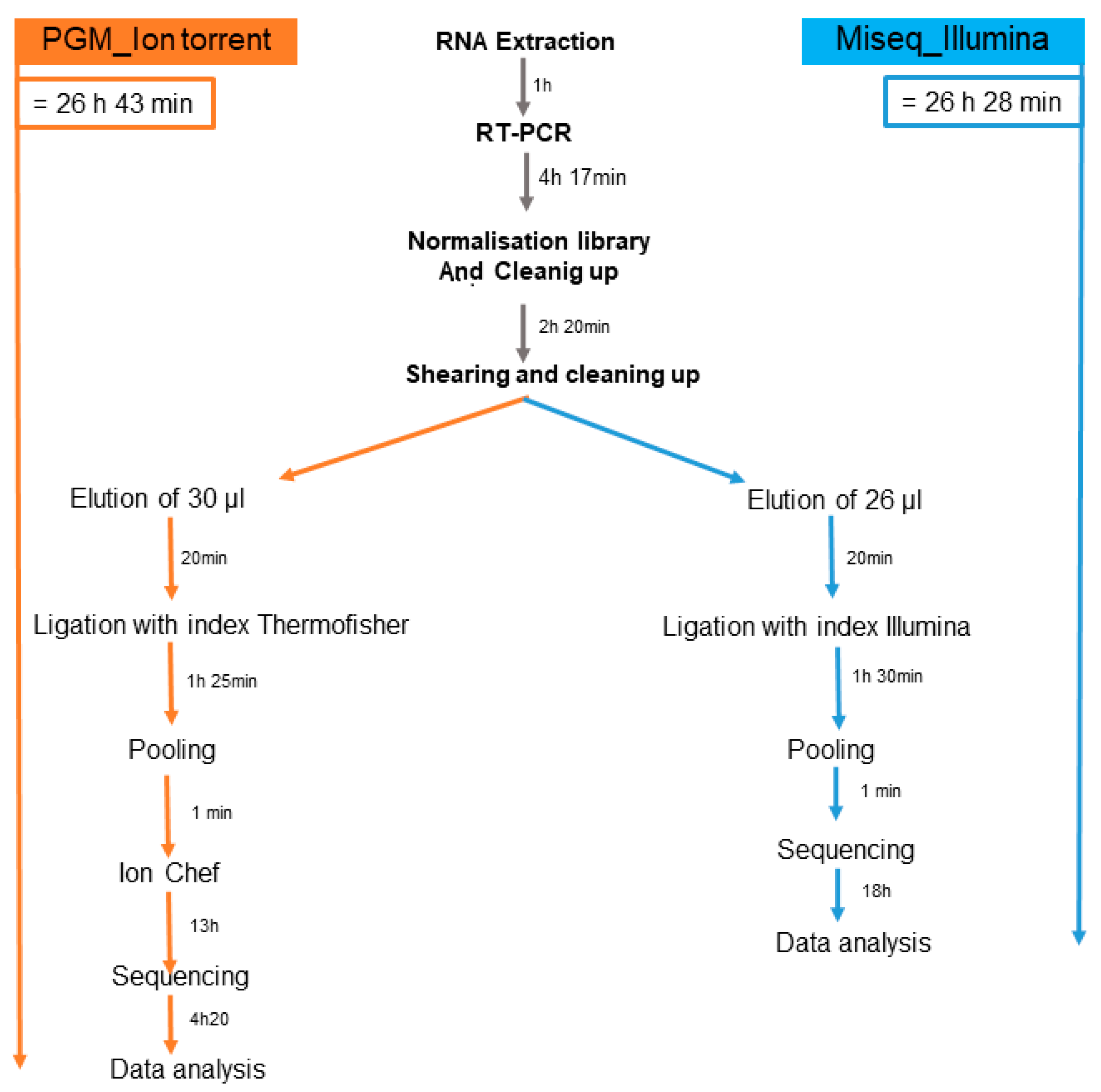

Table 1.
Agreement and discordance in the number of mutations (> 20%).
| Gene | ID HIV strain number | Mutation number PGM >20% | Mutation number Miseq >20% | Number of discordance | Resistance yes or no | Concordance(%) |
|---|---|---|---|---|---|---|
| Protease | CQ012022 | 24 | 23 | 1 | no | 99 |
| CQ022022 | 18 | 18 | 0 | yes | 100 | |
| CQ032022 | 19 | 20 | 1 | no | 99 | |
| CQ012023 | 24 | 24 | 0 | yes | 100 | |
| CQ022023 | 11 | 11 | 0 | yes | 100 | |
| CQ024023 | 14 | 14 | 0 | yes | 100 | |
|
Reverse Transcriptase |
CQ012022 | 43 | 43 | 0 | yes | 100 |
| CQ022022 | 38 | 37 | 1 | no | 99.5 | |
| CQ032022 | 22 | 23 | 1 | yes | 99.5 | |
| CQ012023 | 31 | 31 | 0 | yes | 100 | |
| CQ022023 | 34 | 34 | 0 | yes | 100 | |
| CQ024023 | 26 | 25 | 1 | no | 99.5 | |
| Integrase | CQ012022 | 20 | 20 | 0 | yes | 100 |
| CQ022022 | 10 | 10 | 0 | yes | 100 | |
| CQ032022 | 18 | 18 | 0 | yes | 100 | |
| CQ012023 | 22 | 22 | 0 | yes | 100 | |
| CQ022023 | 21 | 21 | 0 | yes | 100 | |
| CQ024023 | 15 | 15 | 0 | yes | 100 |
Table 2.
Price comparison between Ion Torrent PGM and Illumina Miseq platform.
| Library reagent | Manufacturer | Price (€) | Price, 1 run (€) | Total price | |
|---|---|---|---|---|---|
| PGM_ion torrent | Miseq_Illumina | ||||
| Sentosa SQ HIV DRM Library Kit (96 samples) | Vela | 2644 | 220.33 | 1685.05 | 1640.24 |
| Sentosa SQ Virus Solution Prep Kit (96 samples) | Vela | 974.05 | 81.17 | ||
| MiseqR Reagent kit v2 (300 cycles) | Illumina | 1273 | 1273 | ||
| IDT for Illumina – TruSeq DNA UD Indexes v2 (96) | Illumina | 788,88 | 65.74 | ||
| Ion Xpress Barcode Adapters Kit (96 samples) | ThermoFisher | 1757,97 | 146.49 | ||
| Ion PGMTM Hi-QTM View Chef Reagent (4 reactions) | ThermoFisher | 3171.92 | 792.98 | ||
| Ion PGMTM Hi-QTM View Chef Supplies (4 reactions) | ThermoFisher | ||||
| Ion PGMTM Hi-QTM View Chef Solutions (4 reactions) | ThermoFisher | ||||
| Ion PGMTM Hi-QTM View Sequencing Supplies (4 reactions) | ThermoFisher | ||||
| Ion PGM Hi-Q View Sequencing Solutions (4 reactions) | ThermoFisher | ||||
| Ion PGM Hi-Q View Sequencing dNTPs (4 reactions) | ThermoFisher | ||||
| Sentosa SQ 318 Chip Kit (1 reaction) | Vela | 3552.6 | 444.07 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



