
Submitted:
22 January 2024
Posted:
23 January 2024
You are already at the latest version
Abstract
We investigated the potential genetic variants using whole exome sequencing (WES) and evalu-ated the disease course using T cell receptor (TCR) repertoire analysis in rheumatoid arthritis (RA). Fourteen patients with RA and five healthy controls (HCs) were enrolled. For patients with RA, only naïve patients were recruited before treatment, and data were collected at baseline and 6 and 12 months after the initiation of disease-modifying antirheumatic drug (DMARD) treatment. Laboratory data and disease parameters were also collected. Genetic variants were detected using WES, and the diversity of the TCR repertoire was assessed using Shannon-Weiner diversity. Some variants were detected by WES, but their clinical significance should be confirmed by further studies. The diversity of the TCR repertoire in patients with RA was lower than that in HCs; however, after DMARD treatment, it increased significantly. The diversity was negatively cor-related with laboratory findings and disease measures with statistical significance. Variants with a potential for RA pathogenesis were identified and the clinical significance of TCR repertoire was evaluated in Korean patients with RA. Further studies are required to confirm the findings of the present study.
Keywords:
whole exome sequencing
; T cell receptor repertoire
; diversity
; rheumatoid arthritis
1. Introduction
Rheumatoid arthritis (RA) is a major systemic autoimmune disease characterized by chronic inflammation of the synovial joints and bony destruction, resulting in a wide spectrum of symptoms such as polyarticular pain and swelling [1]. RA is associated with progressive damage, systemic complications, mortality, and morbidity, and imposes a significant socioeconomic burden [2]. Except for a few certain cases and populations, the prevalence of RA is generally constant at around 0.5 to 1.0% with tens of millions of patients worldwide [3].
RA has many complex etiologies involving genetic, immunological, and acquired factors [4]. Previous studies have reported that genetic background contributes to approximately 50% of patients with RA [5,6]. In addition, environmental factors, such as smoking, respiratory diseases, and changes in the gut microbiome, may increase the risk or cause epigenetic modifications in susceptible patients, which may lead to RA development [2,3]. As RA shows complex traits and rapid progression, identifying risk factors and preventing disease progression are important [1-4]. For these reasons, identifying genes for RA is very important because it can be used as basic data in medicine as well as in public healthcare, which can be applied to further understand the pathogenesis of RA and potential targets of RA therapy [7,8]. As part of the RA research, many studies have been conducted on human leukocyte antigen (HLA)-related alleles [1,2,9]. Many studies related to T cells have been conducted in RA. While existing studies have focused on the abnormal expression of T cells and the resulting autoantibody production, recent studies have focused on immunotolerance such as programmed cell death 1 (PD-1) [10,11,12]. Although many studies have been conducted on susceptibility to RA development using single nucleotide polymorphisms (SNPs) and have attempted to identify disease-specific genes or polymorphisms in RA, these SNPs alone cannot accurately explain the etiology because the RA-related genetic background is complex [13]. Genome-wide association studies (GWAS), suggested genetic loci for susceptibility to RA [14,15,16,17]. With the advent of next-generation sequencing (NGS), some potential genetic variants have been analyzed [18]. Abnormal and pathogenic T-cell responses that evade normal immune functions could be considered one of the mechanisms of RA development [2], and the T cell receptor (TCR) repertoire has been studied in patients with RA [19]. However, to date, there have been only a limited number of related studies, and there have been no published studies on Korean patients.
For these reasons, this study aimed to investigate the potential genetic variants for RA development in Korean patients using whole exome sequencing (WES) and the pathogenesis of disease course using T cell receptor (TCR) β & γ repertoire (TRB/TRG) analysis. This study aimed to provide basic data that could be applied to Korean patients by analyzing and evaluating results at the molecular genetic level.
2. Materials and Methods
2.1. Study design and data collection
From December 2020 to February 2022, study participants were recruited from patients who visited the outpatient department (OPD) of the Rheumatology of Internal Medicine at Wonju Severance Christian Hospital, a tertiary university-affiliated hospital located in Wonju, South Korea. The inclusion criterion of this study were naïve patients who were first diagnosed with RA in our hospital and those who have not yet started the disease-modifying antirheumatic drug (DMARD) treatment. A total of 17 patients were enrolled and scheduled to visit the OPD at various time points: the first time (before the initiation of DMARD treatment), and 6 and 12 months after treatment. However, three patients were lost to follow-up during the study period, and finally, 14 patients remained, and their blood samples were collected for up to 12 months and used for data analysis. Five healthy controls (HCs) without diagnosed RA were also enrolled for comparison, requiring only one visit. At each visit, blood samples were collected in two K2-ethylenediaminetetraacetic acid (EDTA) tubes, one for measuring the erythrocyte sedimentation rate (ESR) and the other for genomic testing, and one serum separation tube (SST) for measuring other laboratory data.
Baseline characteristics were collected as follows: age, sex, medical history of hypertension and diabetes mellitus, height, body weight, and the duration before the first visit for clinical information; tender joint count of 44/28 joints (TJC44/28), swollen joint count of 44/28 joints (SJC44/28), disease activity score in 28 joints (DAS28), health assessment questionnaire (HAQ), visual analogue scale (VAS) score, simplified disease activity (SDAI), clinical disease activity index (CDAI), fulfillment of the 1987 American College of Rheumatology (ACR) criteria (whether ≥4 out of 7 cases are met) [20] and the 2010 ACR/European Alliance of Associations for Rheumatology (EULAR) criteria (whether a score of ≥6 out of 10 was met) [21] for clinical classification or scoring system.
Laboratory data were collected on the following: anti-nuclear antibody (ANA) determined by the QUANTA Lite® ANA assay (Inova Diagnostics, Inc., San Diego, CA, USA), rheumatoid factor (RF) and C-reactive protein (CRP) were determined by the Cobas c 702 automated analyzer (Roche Diagnostics, Rotkreuz, Switzerland), anti-cyclic citrullinated peptide (ACCP) determined by the QUANTA® Flash CCP3 assay (Inova Diagnostics), and ESR determined by the TEST-1 analyzer (SIRE Analytical Systems, Udine, Italy).
This study was approved by the Institutional Review Board (IRB) of Wonju Severance Christian Hospital (IRB No. CR320086). All the participants voluntarily participated in the study and provided written informed consent.
2.2. Genomic analysis
Peripheral blood mononuclear cells (PBMCs) were isolated from EDTA-treated whole blood by Ficoll-Hypaque density-gradient centrifugation. The detailed protocols for WES and TRB/TRG were as follows.
For WES, genomic deoxyribonucleic acid (DNA) was extracted by using the ChemagicTM Magnetic Separation Module I (MSM I) method (PerkinElmer Chemagen, Baesweiler, Germany) with the DNA blood 200 μL kit. The MGIEasy Exome Capture V5 Probe Set (MGI Tech Co., Ltd., Shenzhen, China) was used for library preparation, and sequencing was performed on the MGI DNBSEQ-G400 platform (MGI Tech Co., Ltd.) to generate 2 × 100 bp paired-end reads. DNA sequence reads were aligned to the reference sequence based on the public human genome build GRCh37/UCSC hg19. Alignments were performed with BWA-mem (version 0.7.17), duplicate reads were marked with biobambam2 and base quality recalibration, variant calling was performed with the Genome Analysis Tool kit (GATK, version 4.1.8), and annotation was performed with VEP101 (Variant Effect Predictor 101) and dbNSFP v4.1 (Liu et al, 2020).
For the TCR/TRB analysis, genomic DNA was extracted using the ChemagicTM DNA Blood 200 kit (Chemagen, Baesweiler, Germany). For TCR repertoire sequencing, the Lymphotrack® TRB and TRG assay (Cat No. 72270009 and 72270019, respectively) (Invivoscribe Inc., San Diego, CA, USA) for NGS were used. These assays amplify the DNA between primers that target the conserved V and J regions of the T-cell receptor genes, TRB and TRG. Polymerase chain reaction (PCR) amplicons were purified and quantified using a Qubit fluorometer (Thermo Fisher Scientific). Equimolar amounts of libraries were pooled and loaded onto a flow cell using a MiSeq Reagent kit v2 (500 cycles) (Illumina Inc., San Diego, CA, USA) and sequenced on a MiSeq instrument (Illumina).
2.3. Statistical analysis
The results of the numerical data were checked for normality using the Kolmogorov–Smirnov test, which determined a p-value greater than 0.05 had normality. For parametric data, results are presented as means ± standard deviation (SDs), and the Student’s t-test was used for comparison. For non-parametric data, results are presented as median and interquartile range (IQR), and the Mann–Whitney U test was used for comparison. The results of categorical data were presented as frequencies and percentages (%), and the χ² test was used for comparison.
For patients with RA, the results at baseline (naïve) were compared to those at 6 and 12 months of follow-up. To compare DAS28, DAS28-ESR (=0.70 × In [ESR]) and DAS28-CRP (=0.36 × In [CRP + 1] + 0.96) [22] were calculated. All three VAS scores (pain VAS, global VAS, and physician VAS) were evaluated, but all showed almost the same value; therefore, only the pain VAS scores were used for comparison.
All statistical analyses were performed using Analyse-it version 6.15.4 (Analyse-it Software, Ltd., Leeds, UK), an add-on program to Microsoft Excel 2019 (Microsoft Corp., Redmond, WA, USA), and SPSS (version 25.0; IBM Corp., Armonk, NY, USA). Statistical significance was set at P-value < 0.05.
3. Results
3.1. Baseline characteristics
The baseline characteristics of the 14 patients with RA and 5 HCs are presented in Table 1. The median age of the patients with RA was 57 years (IQR, 45–69), and 12 (85.7%) of were female. There was no statistically significant difference in the proportions of patients with hypertension, diabetes mellitus, or obesity between patients with RA and HCs. When comparing RA criteria, 10 patients (71.4%) satisfied the 1987 ACR criteria, whereas all 14 patients (100%) satisfied the 2010 ACR/EULAR criteria. Laboratory findings showed that the proportions of people positive for ANA, RF, and ACCP were significantly higher in patients with RA than in HCs, and the median ESR and CRP values were also higher in patients with RA than in HCs.
3.2. Genomic variants
Using WES, disease-related variants that have been reported to be associated with RA among several small nucleotide polymorphisms (SNPs) were detected in four (28.6%) patients with RA. Table 2 lists the variants detected using WES that are likely to be related to RA pathogenesis. Most were variants of uncertain significance (VUS), but there was one case of a pathogenic variant (PV).
3.3. Changes in clinical features after DMARD treatment
Table 3 displays the results of comparing the clinical features of naïve RA patients before starting DMARD treatment with the clinical features at 6 and 12 months after the initiation of DMARD treatment. Compared to baseline (before treatment), laboratory findings after 12 months of treatment decreased significantly. There were also changes in the number of joints; TJC44, SJC44, TJC28, and SJC28 counts at 6 months decreased significantly compared to baseline. These facts are also reflected in the disease measures as they are, and DAS28, HQA, VAS, SDAI, and CDAI scores at 6 months decreased significantly at 6 months, and even at 12 months they remained significantly decreased compared to the baseline.
3.4. Changes in TCR diversity after DMARD treatment
TRB and TRG diversities were assessed using the Shannon–Weiner diversity index [23,24]. The results of the comparison of TCR diversity between HCs and patients with RA at baseline and at 6 and 12 months after starting DMARD treatment are illustrated in Figure 1. For TRB diversity, the Shannon–Weiner diversity indices in HCs and patients with RA at baseline, and 6 months and 12 months after treatment were 0.69 (IQR, 0.63–0.74), 0.55 (IQR, 0.41–0.58), 0.56 (IQR, 0.43–0.73), and 0.96 (IQR, 0.66–1.52), respectively (Figure 1(A)). For TRG diversity, the Shannon–Weiner diversity indices in HCs and patients with RA at baseline, and 6 months and 12 months after treatment were 0.42 (IQR, 0.37–0.48), 0.29 (IQR, 0.26–0.31), 0.32 (IQR, 0.24–0.43), and 0.51 (IQR, 0.35–0.76), respectively. Naϊve RA patients showed a relatively lower TCR diversity than HCs, but after the initiation of DMARD treatment, TCR diversity has risen significantly according to the duration of the treatment.
3.5. Relationship between TCR diversity and RA-related factors
The relationship between TCR diversity and disease-related factors in RA was analyzed using Spearman's rank correlation analysis. For laboratory findings, data from patients with RA and HCs were included and analyzed, and only ESR showed a significant correlation with both TRB and TRG. Disease measures showed significant correlations in all items evaluated, especially in DAS28-ESR, SDAI, and CDAI, with correlation coefficient exceeding 0.5 (Table 4).
Additionally, the relationship between TCR diversity (Shannon–Weiner diversity) and the value of each RA-related factor was obtained through linear regression. Figure 2 shows a linear regression diagram between TCR diversity and laboratory values, with the thick line representing the regression line and the dotted line representing the 95% confidence interval. In Table 5, the slope and 95% confidence interval of the linear regression equation between the Shannon–Weiner diversity and RA-related factors were analyzed and presented using data from patients with RA and HCs. Because the analytical measuring interval (AMI) of each test item varies for laboratory items and the scale of each scoring varies for disease measures, it is difficult to compare uniformly using only the slope and intercept. However, it can be seen that, except for ACCP, compared to TRG diversity, it shows a negative slope; therefore, it can be seen that the improvement of laboratory findings and disease measures (decreases in numeric values) is related to the increase in TCR diversity.
4. Discussion
RA is a systemic autoimmune disease that causes chronic inflammatory synovitis, joint destruction, and multi-organ manifestations, resulting in morbidity and mortality [1,4]. RA is an autoimmune disease and many studies have been conducted on autoantibodies; however, a consensus on the crucial autoantigens or genetic and environmental factors that trigger the autoimmune process has not yet been established [25]. The cause of RA is not yet clearly known because of the complex traits of various factors; however, genetic factors account for approximately 50% of RA cases [5,6]. However, it is complicated because it is known that the causes of RA are related to type 1 diabetes, autoimmune thyroid disease, infection, smoking, obesity, and hormones in addition to genetic factors [3]. However, similar to other diseases, many studies have been conducted to identify genetic mutations in RA and identify treatment targets. Owing to the development of GWAS, many genetic mutations related to RA have been discovered [26]. Genetic factors are important because they are related not only to preventive activities for individuals with a high probability of RA but also to the treatment process, response, severity, and prognosis [9]. Current conventional and biological treatments sometimes fail or show a partial response. If the genetic marker is well identified, it is expected that the response to treatment and prognosis can be improved while minimizing toxicity through targeted treatment [25].
For these reasons, this study aimed to find possible variations for the onset of RA in Korean naϊve patients and to evaluate the usefulness of TCR repertoire analysis as a factor related to the disease course and the response to DMARD treatment. Of the 14 patients with RA finally enrolled in this study, all satisfied the 2010 ACR/EULAR criteria, but only 10 patients (71.4%) met the criteria for diagnosing RA according to the 1987 ACR criteria, which is known to have lower sensitivity for early RA diagnosis than the 2010 ACR/EULAR [21].
Based on the WES results, variants expected to be associated with RA were detected in 4 of 14 patients with RA. In the Janus kinase 3 (JAK3) gene, the variant c.1333C>T, p.Arg445Ter was detected, which has been reported as a pathogenic variant in severe combined immunodeficiency [27] but has not yet been reported in RA. However, its association with RA may be considered because JAK3 inhibitors may be used for RA treatment [28]. and follow-up studies are needed. Peptidyl arginine deiminase type IV (PAID4) is a gene that converts arginine residues into citrulline residues in the presence of calcium ions, and a mutation in this gene is known to overproduce citrulline and cause a loss of tolerance, thereby increasing vulnerability to RA [29]. Tumor necrosis factor ligand superfamily member 18 (TNFSF18) is known to activate T cells and B cells in connection with cell signaling [30], and tumor necrosis factor receptor-associated factor (TRAF) is known to be associated with TNF-α and interleukin (IL)-1 and cause inflammation [31]. NF-κB is known to be activated in the synoviums of patients with RA to regulate genes contributing to inflammation, such as TNF-a, IL-6, and IL-8, and treatment therapy targeting this gene is under study [7]. However, it does not match the SNP found in the European [15] and Japanese studies [32]; the clinical significance of the SNP shown in this study is somewhat limited, and it is necessary to clarify the clinical significance of this SNP or to further discover other SNPs through follow-up studies on Korean patients. Of course, it will be helpful to develop a drug or treatment agent that targets gene mutations as they affect the activity of certain proteins, but more GWAS studies will be needed as human genetic patterns are complex, making it difficult to set and establish targets and produce effective therapeutic effects [33].
Genetic and specific environmental factors combine to trigger RA, which activates the immune reaction and causes loss of immune tolerance and increases the number and binding affinity of autoantibodies [1]. The disease progression of RA occurs through epigenetic remodeling [1]. In the immune response, T cells and B cells are activated, resulting in an increase in inflammatory cytokines, production of matrix metalloproteinases, and induction and activation of osteoclasts, resulting in bony destruction [2]. Because the aberrant expression of CD4+ T cells plays an important role in the pathogenesis of RA, the identification and quantitative comparison of CD4+ T cells are being studied in relation to the etiology of RA for early diagnosis and as a possible indicator of the disease course or response to treatment [23]. A previous study reported that the TCR repertoire helps in the early diagnosis of RA [19]. It is known that the TCR repertoire in patients with RA is known to be less diverse than in healthy individuals and related to the disease activity [23,34].
In this study, a TCR repertoire analysis was performed. Naïve patients without prior DMARD treatment were enrolled, and the data of their baseline (before treatment) and 6 and 12 months after the initiation of DMARD treatment were compared. The baseline laboratory findings of patients with RA were significantly different from those of the HCs. The TRB/TRG diversities of patients with RA were lower than those of HCs. However, TRB/TRG diversity in RA patients have increased after the initiation of DMARD treatment. Laboratory values, affected joint counts, and disease measures were significantly decreased. As symptoms and disease-related factors improved according to treatment response, TRB/TRG diversity increased. According to previous studies, as the progression or severity of the disease increases, clinical expansion of T cells occurs, and their diversity of T cells decreases [35]. The diversity of the TCR repertoire may change according to treatment, and monitoring changes in the TCR repertoire is expected to play an important role in monitoring the disease course, evaluating the response to treatment, confirming recurrence, and predicting the prognosis of patients with RA [36]. Correlations between TCR repertoire diversity and disease-related factors were also evaluated. The factors were negatively correlated with TRB/TRG diversity, and this is in line with the result of the previous RA cohort study [23]. The DAS, SDAI, and CDAI scores were more strongly correlated with TRB/TRG diversity than the laboratory findings. The relationships between TCR/TRG diversity and RA-related factors were obtained using linear regression. However, because the reference value and AMI of each item are different, and the scale of disease measures is different, the correlation should not be determined using only the slope values of the equations.
This study has several limitations. First, the number of enrolled patients with RA was small. This is because only naïve patients without prior treatment who were diagnosed with RA at our hospital were enrolled. Secondly, using WES, the variants detected in this study had no definite associations with previous reports. Third, this study focused only on TCR diversity according to DMARD treatment. Follow-up studies with a larger number of patients with RA will be able to clarify the clinical significance of the variants detected by WES in this study, detect other variants, and increase the diagnostic and predictive value of TRB/TRG diversity as a marker for the diagnosis, treatment, and prognosis of RA.
5. Conclusions
This study investigated potential genetic variants for RA development in Korean patients using WES and evaluated the clinical significance of the TCR repertoire by analyzing TCR/TRB diversity. Some potential variants were detected using WES; however, further studies are required to confirm their clinical significance. This study is meaningful in that Korean patients showed similar results and these factors and TCR diversities showed negative correlations and linear relationships because the correlation coefficient was high and statistically significant. This study provides basic data that can be applied to Korean patients by analyzing and evaluating results at the molecular genetic level.
Author Contributions
Conceptualization, J.C., J.-H.L., and H.S.L.; Methodology, J.C., J.-H.L., and Y.U.; Sample collection, H.S.L.; Measurement and analysis, J.S.S.; Statistical analysis, J.C., J. S. S., and J.K.; Writing-original draft, J.C.; Writing-review and editing, J.-H.L. and H.S.L.; Supervision, J.K. and Y.U. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean Government (NRF-2020R1G1A1011359).
Institutional Review Board Statement
This study was approved by the Institutional Review Board (IRB) of Wonju Severance Christian Hospital (IRB no. CR320086).
Informed Consent Statement
Informed consent was obtained from all participants involved in the study.
Data Availability Statement
Data available on request from the corresponding author.
Acknowledgments
We thank GC Genome (GC Labs, Yongin, Korea) for analyzing the WES and TRB/TRG assays.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Gravallese, E.M.; Firestein, G.S. Rheumatoid arthritis - common origins, divergent mechanisms. N Engl J Med 2023, 388, 529–542. [Google Scholar] [CrossRef]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N Engl J Med 2011, 365, 2205–2219. [Google Scholar] [CrossRef]
- Silman, A.J.; Pearson, J.E. Epidemiology and genetics of rheumatoid arthritis. Arthritis Res 2002, 4 Suppl 3, S265–272. [Google Scholar] [CrossRef]
- Klareskog, L.; Catrina, A.I.; Paget, S. Rheumatoid arthritis. Lancet 2009, 373, 659–672. [Google Scholar] [CrossRef]
- Seldin, M.F.; Amos, C.I.; Ward, R.; Gregersen, P.K. The genetics revolution and the assault on rheumatoid arthritis. Arthritis Rheum 1999, 42, 1071–1079. [Google Scholar] [CrossRef]
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum 2000, 43, 30–37. [Google Scholar] [CrossRef]
- Firestein, G.S. Evolving concepts of rheumatoid arthritis. Nature 2003, 423, 356–361. [Google Scholar] [CrossRef]
- Kurreeman, F.A.; Padyukov, L.; Marques, R.B.; Schrodi, S.J.; Seddighzadeh, M.; Stoeken-Rijsbergen, G.; van der Helm-van Mil, A.H.; Allaart, C.F.; Verduyn, W.; Houwing-Duistermaat, J.; et al. A candidate gene approach identifies the traf1/c5 region as a risk factor for rheumatoid arthritis. PLoS Med 2007, 4, e278. [Google Scholar] [CrossRef]
- Oliver, J.E.; Worthington, J.; Silman, A.J. Genetic epidemiology of rheumatoid arthritis. Curr Opin Rheumatol 2006, 18, 141–146. [Google Scholar] [CrossRef]
- Prokunina, L.; Padyukov, L.; Bennet, A.; de Faire, U.; Wiman, B.; Prince, J.; Alfredsson, L.; Klareskog, L.; Alarcón-Riquelme, M. Association of the pd-1.3a allele of the pdcd1 gene in patients with rheumatoid arthritis negative for rheumatoid factor and the shared epitope. Arthritis Rheum 2004, 50, 1770–1773. [Google Scholar] [CrossRef]
- Sandigursky, S.; Silverman, G.J.; Mor, A. Targeting the programmed cell death-1 pathway in rheumatoid arthritis. Autoimmun Rev 2017, 16, 767–773. [Google Scholar] [CrossRef]
- Johnson, D.B.; Frampton, G.M.; Rioth, M.J.; Yusko, E.; Xu, Y.; Guo, X.; Ennis, R.C.; Fabrizio, D.; Chalmers, Z.R.; Greenbowe, J.; et al. Targeted next generation sequencing identifies markers of response to pd-1 blockade. Cancer Immunol Res 2016, 4, 959–967. [Google Scholar] [CrossRef]
- Saad, M.N.; Mabrouk, M.S.; Eldeib, A.M.; Shaker, O.G. Genetic case-control study for eight polymorphisms associated with rheumatoid arthritis. PLoS One 2015, 10, e0131960. [Google Scholar] [CrossRef]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat Genet 2010, 42, 508–514. [Google Scholar] [CrossRef]
- Eyre, S.; Bowes, J.; Diogo, D.; Lee, A.; Barton, A.; Martin, P.; Zhernakova, A.; Stahl, E.; Viatte, S.; McAllister, K.; et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat Genet 2012, 44, 1336–1340. [Google Scholar] [CrossRef]
- Zhernakova, A.; Stahl, E.A.; Trynka, G.; Raychaudhuri, S.; Festen, E.A.; Franke, L.; Westra, H.J.; Fehrmann, R.S.; Kurreeman, F.A.; Thomson, B.; et al. Meta-analysis of genome-wide association studies in celiac disease and rheumatoid arthritis identifies fourteen non-hla shared loci. PLoS Genet 2011, 7, e1002004. [Google Scholar] [CrossRef]
- Kurreeman, F.A.; Stahl, E.A.; Okada, Y.; Liao, K.; Diogo, D.; Raychaudhuri, S.; Freudenberg, J.; Kochi, Y.; Patsopoulos, N.A.; Gupta, N.; et al. Use of a multiethnic approach to identify rheumatoid- arthritis-susceptibility loci, 1p36 and 17q12. Am J Hum Genet 2012, 90, 524–532. [Google Scholar] [CrossRef]
- Wiley, G.B.; Kelly, J.A.; Gaffney, P.M. Use of next-generation DNA sequencing to analyze genetic variants in rheumatic disease. Arthritis Res Ther 2014, 16, 490. [Google Scholar] [CrossRef]
- Turcinov, S.; Af Klint, E.; Van Schoubroeck, B.; Kouwenhoven, A.; Mia, S.; Chemin, K.; Wils, H.; Van Hove, C.; De Bondt, A.; Keustermans, K.; et al. Diversity and clonality of t cell receptor repertoire and antigen specificities in small joints of early rheumatoid arthritis. Arthritis Rheumatol 2023, 75, 673–684. [Google Scholar] [CrossRef]
- Arnett, F.C.; Edworthy, S.M.; Bloch, D.A.; McShane, D.J.; Fries, J.F.; Cooper, N.S.; Healey, L.A.; Kaplan, S.R.; Liang, M.H.; Luthra, H.S.; et al. The american rheumatism association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum 1988, 31, 315–324. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O., 3rd; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 rheumatoid arthritis classification criteria: An american college of rheumatology/european league against rheumatism collaborative initiative. Arthritis Rheum 2010, 62, 2569–2581. [Google Scholar] [CrossRef]
- Fleischmann, R.; van der Heijde, D.; Koenig, A.S.; Pedersen, R.; Szumski, A.; Marshall, L.; Bananis, E. How much does disease activity score in 28 joints esr and crp calculations underestimate disease activity compared with the simplified disease activity index? Ann Rheum Dis 2015, 74, 1132–1137. [Google Scholar] [CrossRef]
- Yang, P.; He, Y.; Qing, P.; Xu, W.; Xie, D.; Cazier, J.B.; Liu, X.; Varnai, C.; Zhou, Y.; Zhao, Y.; et al. Application of t-cell receptor repertoire as a novel monitor in dynamic tracking and assessment: A cohort-study based on ra patients. J Cell Mol Med 2022, 26, 6042–6055. [Google Scholar] [CrossRef]
- Viljakainen, J.; Raju, S.C.; Viljakainen, H.; Figueiredo, R.A.O.; Roos, E.; Weiderpass, E.; Rounge, T.B. Meal regularity plays a role in shaping the saliva microbiota. Front Microbiol 2020, 11, 757. [Google Scholar] [CrossRef]
- Imboden, J.B. The immunopathogenesis of rheumatoid arthritis. Annu Rev Pathol 2009, 4, 417–434. [Google Scholar] [CrossRef]
- Stahl, E.A.; Wegmann, D.; Trynka, G.; Gutierrez-Achury, J.; Do, R.; Voight, B.F.; Kraft, P.; Chen, R.; Kallberg, H.J.; Kurreeman, F.A.; et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat Genet 2012, 44, 483–489. [Google Scholar] [CrossRef]
- Schumacher, R.F.; Mella, P.; Badolato, R.; Fiorini, M.; Savoldi, G.; Giliani, S.; Villa, A.; Candotti, F.; Tampalini, A.; O'Shea, J.J.; et al. Complete genomic organization of the human jak3 gene and mutation analysis in severe combined immunodeficiency by single-strand conformation polymorphism. Hum Genet 2000, 106, 73–79. [Google Scholar] [CrossRef]
- Angelini, J.; Talotta, R.; Roncato, R.; Fornasier, G.; Barbiero, G.; Dal Cin, L.; Brancati, S.; Scaglione, F. Jak-inhibitors for the treatment of rheumatoid arthritis: A focus on the present and an outlook on the future. Biomolecules 2020, 10. [Google Scholar] [CrossRef]
- Tamiya, G.; Shinya, M.; Imanishi, T.; Ikuta, T.; Makino, S.; Okamoto, K.; Furugaki, K.; Matsumoto, T.; Mano, S.; Ando, S.; et al. Whole genome association study of rheumatoid arthritis using 27 039 microsatellites. Hum Mol Genet 2005, 14, 2305–2321. [Google Scholar] [CrossRef]
- Croft, M.; Siegel, R.M. Beyond tnf: Tnf superfamily cytokines as targets for the treatment of rheumatic diseases. Nat Rev Rheumatol 2017, 13, 217–233. [Google Scholar] [CrossRef]
- Raychaudhuri, S.; Remmers, E.F.; Lee, A.T.; Hackett, R.; Guiducci, C.; Burtt, N.P.; Gianniny, L.; Korman, B.D.; Padyukov, L.; Kurreeman, F.A.; et al. Common variants at cd40 and other loci confer risk of rheumatoid arthritis. Nat Genet 2008, 40, 1216–1223. [Google Scholar] [CrossRef]
- Okada, Y.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Kawaguchi, T.; Stahl, E.A.; Kurreeman, F.A.; Nishida, N.; et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the japanese population. Nat Genet 2012, 44, 511–516. [Google Scholar] [CrossRef]
- Plenge, R.M.; Scolnick, E.M.; Altshuler, D. Validating therapeutic targets through human genetics. Nat Rev Drug Discov 2013, 12, 581–594. [Google Scholar] [CrossRef]
- Jiang, X.; Wang, S.; Zhou, C.; Wu, J.; Jiao, Y.; Lin, L.; Lu, X.; Yang, B.; Zhang, W.; Xiao, X.; et al. Comprehensive tcr repertoire analysis of cd4(+) t-cell subsets in rheumatoid arthritis. J Autoimmun 2020, 109, 102432. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, W.; Zhao, M.; Fu, L.; Liu, L.; Wu, J.; Luo, S.; Wang, L.; Wang, Z.; Lin, L.; et al. T cell receptor β repertoires as novel diagnostic markers for systemic lupus erythematosus and rheumatoid arthritis. Ann Rheum Dis 2019, 78, 1070–1078. [Google Scholar] [CrossRef]
- Ria, F.; Penitente, R.; De Santis, M.; Nicolò, C.; Di Sante, G.; Orsini, M.; Arzani, D.; Fattorossi, A.; Battaglia, A.; Ferraccioli, G.F. Collagen-specific t-cell repertoire in blood and synovial fluid varies with disease activity in early rheumatoid arthritis. Arthritis Res Ther 2008, 10, R135. [Google Scholar] [CrossRef]
Figure 1.
Comparison of (a) TRB / (b) TRG diversity between healthy control and patients with RA at various time points after the initiation of DMARD treatment. TRB, T cell receptor β repertoires; TRG, T cell receptor γ repertoires; DMARD, disease-modifying antirheumatic drug. * p < 0.05 represents statistical significance.
Figure 1.
Comparison of (a) TRB / (b) TRG diversity between healthy control and patients with RA at various time points after the initiation of DMARD treatment. TRB, T cell receptor β repertoires; TRG, T cell receptor γ repertoires; DMARD, disease-modifying antirheumatic drug. * p < 0.05 represents statistical significance.
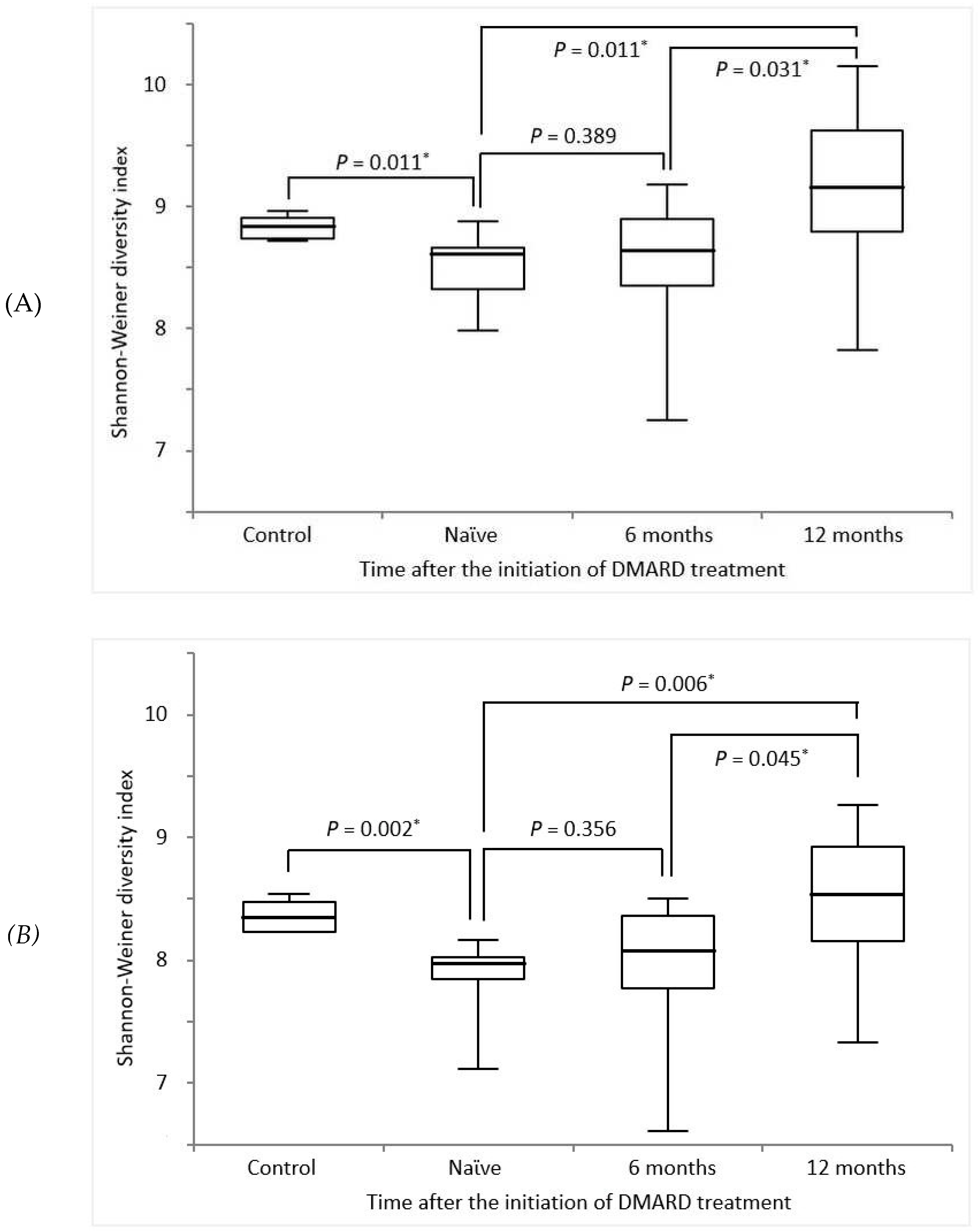
Figure 2.
Linear regression between (a) TRB / (b) TRG diversity and laboratory findings. TRB, T cell receptor β repertoires; TRG, T cell receptor γ repertoires; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein; RF, rheumatoid factor; DAS28, disease activity score in 28 joints; ACCP, anti-cyclic citrullinated peptide.
Figure 2.
Linear regression between (a) TRB / (b) TRG diversity and laboratory findings. TRB, T cell receptor β repertoires; TRG, T cell receptor γ repertoires; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein; RF, rheumatoid factor; DAS28, disease activity score in 28 joints; ACCP, anti-cyclic citrullinated peptide.
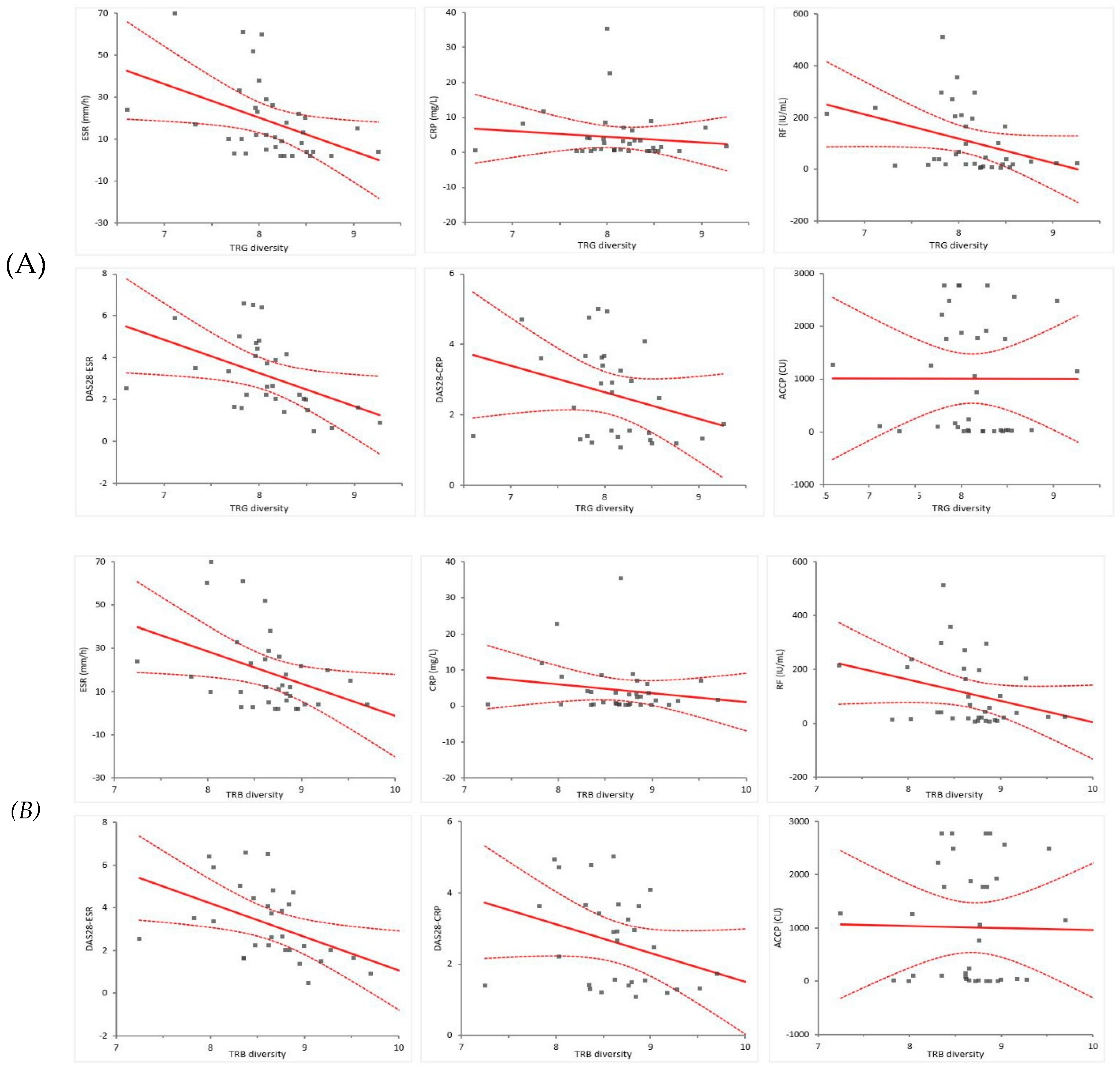

Table 1.
Baseline characteristics of the study participants.
| Baseline variables | Unit | Feature | Patients withRA (n=14) | HCs(n=5) | p-Value |
| Age | Years | Median (IQR) | 57 (45–69) | 38 (31–48) | 0.034* |
| Female sex | No. (%) | 12 (85.7) | 3 (60.0) | 0.046* | |
| Medical history of | |||||
| Hypertension | No. (%) | 3 (21.4) | 1 (20.0) | 0.946 | |
| Diabetes mellitus | No. (%) | 3 (21.4) | 0 (0.0) | 0.259 | |
| Obesity (BMI > 25 kg/m2) | No. (%) | 4 (28.6) | 1 (20.0) | 0.709 | |
| Duration before the first visit | Months | Median (IQR) | 8 (3-12) | - | - |
| RA criteria fulfillment | |||||
| ACR 1987 | No. (%) | 10 (71.4) | - | - | |
| ACR/EULAR 2010 | No. (%) | 14 (100.0) | - | - | |
| Disease-related variants | No. (%) | 4 (28.6) | - | - | |
| Laboratory findings | |||||
| ANA-positive | No. (%) | 12 (85.7) | 0 (0.0) | <0.001* | |
| RF-positive | No. (%) | 13 (92.9) | 0 (0.0) | <0.001* | |
| ACCP-positive | No. (%) | 13 (92.9) | 0 (0.0) | <0.001* | |
| ESR | mm/h | Median (IQR) | 29 (12–53) | 5 (2–8) | <0.001* |
| CRP | mg/L | Median (IQR) | 3.7 (2.2–8.2) | 1.3 (0.3–2.8) | 0.019* |
RA, rheumatoid arthritis; HC, healthy control; IQR, interquartile range; BMI, body mass index; ACR, American College of Rheumatology; EULAR, European Alliance of Associations for Rheumatology; ANA, antinuclear antibody; RF, rheumatoid factor; ACCP, anticyclic citrullinated peptide; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein. * p < 0.05 represents statistical significance.
Table 2.
Variants likely to be associated with rheumatoid arthritis detected using whole exome sequencing.
Table 2.
Variants likely to be associated with rheumatoid arthritis detected using whole exome sequencing.
| Patient | Gene | DNA change | AA change | Zygosity | Class |
| 1 | JAK3 | 1333C>T | Arg445Ter | Hetero | PV |
| PADI4 | 1861G>C | Glu621Gln | Hetero | VUS | |
| TNFSF18 | 167T>C | Met56Th | Hetero | VUS | |
| TRAF1 | 385C>T | Arg129Trp | Hetero | VUS | |
| 2 | NFKB1 | 2708A>G | His903Arg | Hetero | VUS |
| 3 | TNFSF18 | 93G>A | Met31Ile | Hetero | VUS |
| 4 | TNFSF18 | 94C>T | Pro32Ser | Hetero | VUS |
DNA, deoxyribonucleic acid; AA, amino acid; JAK3, Janus kinase 3; PV, pathogenic variant; PADI4, peptidyl arginine deiminase IV; VUS, variant of uncertain significance; TNFSF18, tumor necrosis factor ligand superfamily member 18; TRAF1, tumor necrosis factor receptor associated factor 1; NFKB1, nuclear factor κB subunit1.
Table 3.
Comparison of clinical features of patients with rheumatoid arthritis before and after DMARD treatment.
Table 3.
Comparison of clinical features of patients with rheumatoid arthritis before and after DMARD treatment.
| Variables | Unit | Feature | Baseline | Time after DMARD treatment | p-Value | ||
| 6 months | 12 months | Baseline vs6 months | Baseline vs12 months | ||||
| Laboratory findings | |||||||
| ESR | mm/h | Median (IQR) | 29 (12-53) | 11 (3-22) | 10 (5-20) | 0.006* | 0.005* |
| CRP | mg/L | Median (IQR) | 3.7 (2.2-8.2) | 1.6 (0.7-7.0) | 0.9 (0.4-3.5) | 0.148 | 0.029* |
| RF titer | IU/mL | Median (IQR) | 148.5 (55.7-239.8) | 42.0 (19.0-107.3) | 33.5 (22.8-99.1) | 0.051 | 0.033* |
| ACCP titer | CU | Median (IQR) | 1266.2 (37.8-1970.2) | 947.8 (36.6-1831.8) | 801.4 (150.6-1905.1) | 0.963 | 0.748 |
| Joint counts | |||||||
| TJC44 | no. | Median (IQR) | 4 (3-9) | 1 (0-3) | 1 (0-2) | 0.003* | <0.001* |
| SJC44 | no. | Median (IQR) | 3 (2-5) | 0 (0-1) | 0 (0-1) | <0.001* | <0.001* |
| TJC28 | no. | Median (IQR) | 3 (2-5) | 0 (0-1) | 0 (0-1) | <0.001* | <0.001* |
| SJC28 | no. | Median (IQR) | 2 (1-3) | 0 (0-1) | 0 (0-1) | <0.001* | <0.001* |
| Disease measures | |||||||
| DAS28-ESR | Mean ± SD | 4.75 ± 1.26 | 2.52 ± 0.89 | 2.16 ± 1.21 | <0.001* | <0.001* | |
| DAS28-CRP | Mean ± SD | 3.68 ± 0.89 | 2.03 ± 0.90 | 1.98 ± 0.85 | <0.001* | <0.001* | |
| HAQ score | Mean ± SD | 1.18 ± 0.65 | 0.64 ± 0.32 | 0.53 ± 0.35 | 0.013* | 0.008* | |
| Pain VAS score | Mean ± SD | 6.71 ± 1.98 | 2.14 ± 1.17 | 1.89 ± 1.18 | <0.001* | <0.001* | |
| SDAI | Mean ± SD | 16.54 ± 8.22 | 5.43 ± 3.62 | 3.68 ± 2.46 | <0.001* | <0.001* | |
| CDAI | Mean ± SD | 16.43 ± 8.20 | 5.23 ± 3.47 | 4.76 ± 4.25 | <0.001* | <0.001* | |
DMARD, disease-modifying antirheumatic drugs; ESR, erythrocyte sedimentation rate; IQR, interquartile range; CRP, C-reactive protein; RF, rheumatoid factor; ACCP, anti-cyclic citrullinated peptide; TJC44/28, tender joint counts of 44/28; SJC44/28, swollen joint counts of 44/28; DAS28, disease activity score in 28 joints; SD, standard deviation; KHAQ, health assessment questionnaire; VAS, visual analogue scale; SDAI, simplified disease activity index; CDAI, clinical disease activity index. * p < 0.05 represents statistical significance.
Table 4.
Correlations between TRB/TRG diversity and disease-related factors.
| TRB | TRG | ||||
| Spearman's r(95% CI) | p-Value | Spearman's r (95% CI) | p-Value | ||
| Laboratory findings | |||||
| ESR | -0.435 (-0.689 to -0.084) | 0.015* | -0.378 (-0.652 to -0.017) | 0.036* | |
| CRP | -0.163 (-0.489 to 0.213) | 0.380 | -0.063 (-0.417 to 0.308) | 0.737 | |
| RF titer | -0.267 (-0.575 to 0.107) | 0.146 | -0.277 (-0.583 to 0.096) | 0.131 | |
| ACCP titer | 0.076 (-0.488 to 0.226) | 0.684 | -0.019 (-0.380 to 0.347) | 0.919 | |
| Disease measures | |||||
| DAS28-ESR | -0.580 (-0.780 to -0.274) | <0.001* | -0.575 (-0.777 to -0.268) | <0.001* | |
| DAS28-CRP | -0.389 (-0.660 to -0.029) | 0.031* | -0.358 (-0.638 to -0.007) | 0.048* | |
| HAQ score | -0.382 (-0.655 to -0.020) | 0.034* | -0.337 (-0.624 to 0.031) | 0.064 | |
| Pain VAS score | -0.498 (-0.729 to -0.163) | 0.004* | -0.481 (-0.719 to -0.143) | 0.006* | |
| SDAI | -0.552 (-0.763 to -0.236) | 0.001* | -0.576 (-0.777 to -0.268) | <0.001* | |
| CDAI | -0.561 (-0.768 to -0.248) | 0.001* | -0.587 (-0.784 to -0.284) | <0.001* | |
TRB, T cell receptor β repertoires; TRG, T cell receptor γ repertoires; CI, confidence interval; ESR, erythrocyte sedimentation rate; IQR, interquartile range; CRP, C-reactive protein; RF, rheumatoid factor; ACCP, anti-cyclic citrullinated peptide; DAS28, disease activity score in 28 joints; KHAQ, health assessment questionnaire; VAS, visual analog scale; SDAI, simplified disease activity index; CDAI, clinical disease activity index. * p < 0.05 represents statistical significance.
Table 5.
Results of the linear regression between TRB/TRG diversity and disease-related factors.
| Slope | (95% CI) | Intercept | (95% CI) | ||
| TRB | Laboratory findings | ||||
| ESR (mm/h) | -14.89 | (-25.71 to -4.08) | 147.79 | (53.54 to 242.00) | |
| CRP (mg/L) | -2.52 | (-7.04 to 2.01) | 26.20 | (-13.22 to 65.62) | |
| RF (IU/mL) | -78.84 | (-156.60 to -1.04) | 793.42 | (115.55 to 1471.28) | |
| ACCP (CU) | -41.09 | (-756.07 to 673.89) | 1368.08 | (-4861.37 to 7597.43) | |
| Disease measures | |||||
| DAS28-ESR | -1.57 | (-2.59 to -0.55) | 16.78 | (7.89 to 25.66) | |
| DAS28-CRP | -0.81 | (-1.62 to 0.01) | 9.58 | (2.50 to 16.66) | |
| HAQ score | -0.64 | (-1.14 to -0.13) | 6.39 | (1.99 to 10.80) | |
| Pain VAS score | -1.99 | (-3.84 to -0.16) | 20.99 | (4.99 to 36.99) | |
| SDAI | -6.42 | (-11.85 to -0.98) | 64.68 | (17.54 to 111.93) | |
| CDAI | -6.40 | (-11.81 to -0.99) | 64.42 | (17.39 to 111.45) | |
| TRG | Laboratory findings | ||||
| ESR (mm/h) | -16.06 | (-27.64 to -4.48) | 148.62 | (54.42 to 242.82) | |
| CRP (mg/L) | -1.62 | (-6.52 to 3.29) | 17.43 | (-22.50 to 57.35) | |
| RF (IU/mL) | -94.32 | (-176.45 to -12.20) | 873.51 | (205.61 to 1541.40) | |
| ACCP (CU) | -2.10 | (-768.76 to 764.55) | 1027.77 | (-5207.48 to 7263.02) | |
| Disease measures | |||||
| DAS28-ESR | -1.60 | (-2.74 to -0.46) | 16.07 | (6.84 to 25.30) | |
| DAS28-CRP | -0.75 | (-1.66 to 0.15) | 8.67 | (1.34 to 16.00) | |
| HAQ score | -0.68 | (-1.23 to -0.12) | 0.64 | (1.85 to 10.85) | |
| Pain VAS score | -2.12 | (-4.14 to -0.10) | 20.79 | (4.45 to 37.14) | |
| SDAI | -6.15 | (-12.22 to -0.08) | 58.68 | (9.55 to 107.81) | |
| CDAI | -6.15 | (-12.19 to -0.11) | 58.54 | (9.65 to 107.43) | |
TRB, T cell receptor β repertoires; TRG, T cell receptor γ repertoires; CI, confidence interval; ESR, erythrocyte sedimentation rate; CRP, C-reactive protein; RF, rheumatoid factor; ACCP, anti-cyclic citrullinated peptide; DAS28, disease activity score in 28 joints; KHAQ, health assessment questionnaire; VAS, visual analog scale; SDAI, simplified disease activity index; CDAI, clinical disease activity index.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



