
Submitted:
19 January 2024
Posted:
23 January 2024
You are already at the latest version
Abstract
Hepatitis D virus (HDV) infection represents the most severe form of chronic viral hepatitis. We have shown that the delivery of HDV replication-competent genomes to the hepatocytes using adenoassociated virus (AAV-HDV) as gene delivery vehicles offers a unique platform to investi-gate the molecular aspects of HDV and associated liver damage. Here, we generated HDV ge-nomes modified by site directed mutagenesis aimed to: i) prevent some post-translational modi-fications of HDV antigens (HDAgs) such as Large-HDAg (L-HDAg) isoprenylation or Short-HDAg (S-HDAg) phosphorylation ii) alter the localization of HDAgs within the subcellular compartments and iii) inhibit the right conformation of the delta ribozyme. First, the different HDV mutants were tested in vitro using plasmid-transfected Huh-7 cells and then in vivo in C57BL/6 mice using AAV vectors. We found that Ser 177 phosphorylation and the ribozymal ac-tivity are essential for HDV replication and HDAg expression. Mutation of the isoprenylation domain prevent the formation of infectious particles and increase cellular toxicity and liver dam-age. Finally, altering the intracellular localization od HDAg have a profound on viral replication in vivo but liver toxicity is maintained. Mutation in the nuclear export signal impair the for-mation of infectious viral particles. These findings contribute valuable insights into the intricate mechanisms of HDV biology and have implications for therapeutic considerations
Keywords:
HDV
; HDAg post-translational modification
; isoprenylation
; phosphorylation
; ribozyme
; intra-cellular localization: cell and mouse models
; liver damage
1. Introduction
Hepatitis Delta Virus (HDV) is the smallest known virus that infects humans. It belongs to the Kolmoviridae family and the Deltavirus genus [1]. It is a satellite virus that relies on the surface proteins of Hepatitis B Virus (HBsAg) for assembly, hepatocyte entry, and the release of infectious particles [2]. HDV virions consist of HBV surface antigens and host cell lipids surrounding a ribonucleoprotein (RNP) complex formed by a single-stranded circular RNA genome of about 1700 nucleotides, along with hepatitis delta antigens (HDAg) [2,3,4]. Upon hepatocyte infection, the HDV RNA genome translocates into the nucleus, where the host cell RNA polymerase initiates replication through a rolling-circle mechanism [5,6,7]. This replication strategy allows the production of linear concatemers of HDV genomes and antigenomes that are self-cleaved by the HDV ribozymes, intrinsic catalytic RNA domains present in each RNA strand [8,9]. The antigenomic RNA encompasses a single open reading frame that encodes the short hepatitis delta antigen (S-HDAg) [10]. Through editing by the host cell’s adenosine deaminase acting on RNA-1 (ADAR-1) at the UAG stop codon, an amber (W) site is formed. This modification results in an extended open reading frame (ORF) that gives rise to the large hepatitis delta antigen (L-HDAg) [10]. Despite sharing most of their amino acid sequence, both the short and the large antigens differ significantly in their functions. S-HDAg is essential for HDV replication, whilst L-HDAg blocks HDV replication and is essential for viral assembly [11,12]. Both HDAgs contain a nuclear localization signal (NLS) located between positions 66 and 75 (EGAPPARAR), being Glu-66 (E66), and Arg-75 (R75) indispensable for the nuclear import of HDV RNP [13,14,15]. The L-HDAg contains a Nuclear Export Signal (NES) and Viral Assembly Signal (VAS) domains [16] and is responsible for the translocation of HDV RNPs from the nucleus to the cytoplasm, which is essential for viral assembly. The NES is located within the aa 198-210 of the L-HDAg sequence. More specifically, Pro-205 is the critical residue for the correct functionality of NES [16].
The association between HBsAg and HDV RNP takes place in the ER via a lipid farnesyl-moiety present on the L-HDAg that binds to the cytoplasmic loop of the small HBV surface antigen (S-HBsAg) and is indispensable to promote viral spreading and to complete the HDV life cycle [17,18]. Furthermore, phosphorylation of some residues of S-HDAg plays an important role on viral replication [19]. More specifically, phosphorylation of Ser-177 promotes HDV replication by increasing the interaction of S-HDAg with RNA Pol II [19,20,21]. During the HDV replication, S-HDAg co-localizes with the host RNA pol II in the nucleoplasm, within the SC35 speckles sites that are highly active sites of transcription and RNA processing [7,23]. At earlier steps, S-HDAg is predominantly located in the nucleus, both in the nucleolus and the nucleoplasm. Later in the infection, when L-HDAg starts to be synthesized, the HDAgs can be found in non-SC-35 speckles sites, in the cytoplasm, and in the Golgi apparatus, where the post-translational modifications take place [24,25,26]. Thus correct localization of HDAg is an important aspect of HDV viral cycle.
HDV infection is recognized as the most severe form of viral hepatitis. HDV-infected patients have a higher risk of developing cirrhosis and hepatocellular carcinoma (HCC) as well as hepatic decompensation and increased mortality in comparison with HBV mono-infected patients [27]. Despite the severity of this disease, the underlying mechanism is still unknown and hence there is a lack of effective treatments to control HDV-induced liver damage [28]. One of the main reasons for the scarce knowledge of the molecular mechanism/s involved in the pathology of this disease is the absence of adequate animal models that resemble the main pathological features observed in HDV patients and amenable to experimentation [29]. Recently, we utilized adeno-associated vectors (AAVs) as delivery vehicles for HBV/HDV replication-competent viral genomes. The co-administration of recombinant AAV-HBV and AAV-HDV to WT mice resulted in the establishment of AAV-independent HDV replication and, more importantly, the animals developed a significant liver pathology characterized by transaminase elevation, lobular inflammation, cytoplasmic swelling, and sanded nuclei, that were not observed in AAV-HBV mice [30,31,32]. Thanks to the easy manipulation of the system, we were able to confirm in vivo what has been previously shown in cell culture HV model e.i. that S-HDAg is essential for HDV replication and cannot be replaced by L-HDAg or host cellular proteins, and that L-HDAg is essential to produce the HDV infectious particle and inhibits its replication [31]. Furthermore we found that the ratio L-HDAg/S-HDAg plays an important role on the magnitude of HDV-induced liver damage, the lowest it is the strongest is liver damage [31].
The aim of the present work was to identify the role of post-translational modifications and cellular location of HDV antigens on HDV viral cycle and in HDV-mediated liver damage. We found that Ser 177 phosphorylation and the ribozyme are essential for HDAg expression and HDV replication. Mutation in the NES and NLS signal clearly affect HDAg intracellular localization significantly affect HDV replication in vivo but not in vitro. More importantly, mutation of the isoprenylation domain prevent the formation of infectious particles and reduces the L-HDAg/S-HDAg and increase cellular toxicity both in vitro and in vivo.
2. Materials and Methods
2.1. Site-directed mutagenesis (SDM)
Site-directed mutagenesis (SDM) was handled using the TaKaRa In-Fusion Cloning Kit except for the HDV-NPrL-HDAg plasmid that was carried out using QuikChange II Site-Directed Mutagenesis. Briefly, using TaKaRa In-Fusion Cloning Kit, four oligonucleotides were designed for each HDV mutant, except for the HDV-∆HDAg mutant that required six oligonucleotides since it was necessary to introduce two mutations. Then, 10 ng of the HDV plasmid was used for the amplification of the new mutated inserts. In parallel, the plasmid was digested with the appropriate restriction enzymes. The empty vector obtained after the enzymatic digestion and the PCR products were purified and ligated for 15 minutes at 50 °C. Subsequently, Stellar competent cells were transformed by heat shock, and the obtained clones were sequenced to verify the presence of the desired mutations and the absence of unspecific mutations. When using QuikChange II Mutgenesis, two mutagenic oligonucleotide primers were designed to produce the HDV-NPrL-HDAg mutant as recommended in the QuikChange™ manual (Agilent, #200524). For the PCR amplification, 50 ng of the HDV plasmid, 125ng of primer Forward (Fw), 125ng of Primer Reverse (Rv), 1 μl of dNTP mix, 3 μl of QuikSolution and 1 μl of PfuUltra High Fidelity (HF) DNA polymerase (Agilent, #600380) were mixed at a final volume of 50 μl. And PCR amplified. Then, 10 μl of the PCR product was run in an electrophoresis gel to check the quality of the reaction. To remove the methylated DNA plasmid (in this case, the non-mutated HDV plasmid), 1 μl of the DpnI restriction enzyme was added to the reaction product and was incubated at 37 °C for 1 hour. Finally, the PCR product was electroporated on One Shot® Top10 Electrocomp™ E. coli (Thermo Fisher Scientific, #C404052). Plasmid DNA was purified using the NucleoSpin® Plasmid kit (Macherey-Nagel, #740588) and the positive colonies were identified by restriction enzyme digestion. Then, the DNA plasmids were sequenced to verify the introduction of the correct mutation and to dis-card unspecific mutations.
2.2. Cell lines
The human hepatoma cell line Huh-7 and 293T were acquired from the Glow Biologics (GBTC-099H) and ATCC (CRL-3216)., respectively. Huh-7 cells were used for the HDV plasmid transfection studies and 293-T cells for AAV vector production. Huh-7.5.1 stably expressing the human Na-taurocholate co-transporting polypeptide (hNTCP, Huh-7-hNTCP), which is essential for HBV and HDV cell entry [33,34] were kindly provided by Dr Urtzi Garaigorta and were employed for infectivity studies. Both cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 1% of L-glutamine, 1% of glucose, 100 U/ml of penicillin-streptomycin and no-essential amino acids, and incubated at 37 °C with 5% CO2 in humidified atmosphere. In the case of Huh-7-hNTCP DMEM was supplemented with 2.5 μg/ml of blasticidine to ensure the selection of hNTCP expressing cells.
2.3. DNA Transfection
For transfection, Huh-7 cells were used at a confluence of 80-90%. The day of transfection, the culture medium was replaced by Optimem. Briefly, Lipofectamine 3000 and Optimem were mixed, vortexed and incubated 5 minutes at room temperature (RT) (reaction mix A). In parallel, plasmid DNA, the P3000 reagent and Optimem were also mixed, vortexed and incubated 5 minutes at RT (reaction mix B). Then, mix A and B were mixed, without vortex, and incubated 15 minutes at RT. After that time, the transfection mix was added drop by drop. Six hours after adding the transfection reagents, fresh DMEM 10% FBS was added on the culture plates without removing the medium. Finally, 1 day after transfection, the culture medium was removed, and the cells were washed before adding fresh DMEM 10% FBS.
2.4. Generation of AAV vectors
HEK293T cells were used as packaging cells. The constructs containing the recombinant AAV genomes were transfected together with the helper plasmid pDP8.ape (Plasmid Factory, #PF478) that provides the genes requires for the replication and encapsidation of AAV serotype 8 (a liver tropic AAV serotype in mice). The transfection was carried out using poly-ethyleneimine (PEI, Sig-ma-Aldrich, #408727). Cells were incubated for 72 hours and during this time the viral particles were assembled inside the packaging cells. Cells were collected in lysis buffer (50 mM Tris, 150 mM NaCl, 2 mM MgCl2, 0.1% Triton X-1000) and various freeze-thaw cycles were performed to release the viral particles. Cell debris was eliminated by centrifugation. The supernatant of the cells was collected and incubated in 8% polyethylene glycol (PEG-800, Sigma-Aldrich, #P5416) for 48-72 hours to precipitate the particles. The supernatant was centrifuged, and the pellet was resuspended in lysis buffer. Cell lysate and precipitated supernatant were mixed and treated with 0.1mg/plate of DNaseI (Roche, #10104159001) and RNaseA (Roche, #10109169001) for 1 hour at 37ºC. The viral particles were purified by ultracentrifugation in iodioxanol gradient (OPTIPREP, Atom, #415468) gradient. Iodixanol was removed and the vector was concentrated using Amicon 100K columns (MERK Millipore, #UFC910008). Viral DNA was extracted using the High Pure Viral DNA Kit (Roche, #11858874001) following the manufacturer’s instructions. The vector titer in terms of viral genomes (vg) per mL was calculated by Real Time qPCR (RT-qPCR) (#1855196, BioRad Hercules, CA, USA) using specific primers for the AAV’s inverted terminal repeat (ITR) sequences.
2.5. Animal manipulation and procedures
WT C57BL/6 mice were purchased from Harlan Laboratories (Barcelona, Spain). Six- to eight-week-old male mice were used in all experiments. Mice were kept under controlled temperature, light, and pathogen-free conditions in a BSL3 animal facility.
The animals received the vectors intravenously diluted in saline solution (Braun, #651860) in a final volume of 100 μL/mouse under inhalatory anesthesia. All mice received 5x1010 vg of the corresponding AAV-HDV wt or mutant vector together with 5x1010 vg of AAV-HBV.
The experimental design was approved by the Ethics Committee for Animal Testing of the University of Navarra (R-132-19GN).
2.6. Protein extraction from cells and liver samples
RIPA Buffer (0.75M NaCl, 5% of Tris 1M pH 8, 0.1% SDS, 1% Triton X-100, 0.5% sodium deoxycholate diluted in water) was supplemented before each use with 1mM sodium orthovanadate, 1mM PMSF, 1mM sodium pyrophosphate and protease inhibitor cocktail. Cell pellets were resuspended and incubated for 30 minutes at 4ºC on a shaker with the RIPA lysis buffer and centrifuged at 13000 rpm for 20 minutes at 4ºC. Supernatants were collected in new tubes. Protein concentration was calculated using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, #23225).
2.7. Immunofluorescence (IF)
Cells were washed once with PBS, fixed with cold 4% formaldehyde (freshly pre-pared from methanol-free 16% formaldehyde, Thermo Scientific, #28908) for 15 minutes at RT and washed again for three times with PBS. For intranuclear staining, cells were permeabilized with 0.1% Triton X-100 in PBS for 15 minutes at RT and, after three washes with PBS, a blockade was performed with 5% BSA in PBS-0.1% tween for 30 minutes at 37ºC. Next, cells were incubated for 30 minutes at 37ºC with anti-HDV human serum provided by the BioBank of the Universidad de Navarra (CUN-28336) at dilution 1:2500. After washing three times, the secondary antibody (anti-human Alexa-Fluor 488,) was incubated for 30 minutes at 37ºC diluted 1:3000 in PBS and protected from light. Finally, coverslips were mounted in microscope slides using mounting medium with DAPI for fluorescent labelling (Vector Laboratories, #H-1200). Fluorescent samples were obtained with a confocal microscope (Zeiss LSM 880 NLO) at 40x, 60x and 100x of magnification.
2.8. Determination of HDV infectious viral particles
Huh-7-hNTCP cells seeded in BD FalconTM Culture Slides (8-well, BD Biosciences, US, #345118) or in 6- or 12-wells plate with a coverslip, were incubated 24 hours with HDV-containing cell supernatants. Supernatants were removed and cells were washed with fresh PBS. Then, cells were maintained with fresh medium that was changed every 2 days until 7 days post-infection. At this time point, cells were incubated 15 minutes at RT with 4% PFA, and HDAg expression was assessed by immunofluorescence, as described above.
2.9. Western Blot
20-30μg of extracted proteins were mixed with a SDS-PAGE loading buffer (70% Tris HCl pH 6.8, 30% glycerol, 0.35M SDS, 0.6M DTT, 0.18mM Bromophenol Blue) and boiled for 5 minutes at 95ºC. Samples were loaded on 12% SDS-polyacrylamide gels of 1.5mm and electrophoresis was performed for separating proteins. 5ul of Precision Plus Protein™ Dual Color Standard (BioRad, #161-0394), ranging from 10kDa to 250kDa, was used to determine protein size. Subsequently, the resolved proteins were transferred to a nitrocellulose membrane (Bio-Rad #162-0112) by wet electroelution at 80V for 1 hour and 15 minutes. Following, the membrane was stained with Ponceau Red dye to check the transference quality. After washing the dye with water, the membrane was blocked during 45 minutes with Tris-buffer saline (TBS)-tween 5% non-fat dry milk at RT on a shaker. Primary antibodies were added and incubated ON while shaking at 4ºC. Next, the membrane was washed 3 times with TBS-Tween 20 0.05% for 10 minutes and then, it was incubated with the appropriate horseradish peroxidase (HRP)-conjugated secondary antibody at RT for 1h. After 3 washes of 10 minutes, the peroxidase signal was developed using SuperSignalTM West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific, #34095). Odyssey CLx near-infrared Fluorescence Imaging System was used for image generation and images were analyzed with the Image Studio Lite software.
2.10. Histology and Immunohistochemistry (IHC)
Hematoxylin & Eosin (H&E): Liver sections were fixed with 4% paraformaldehyde (PFA), embedded in paraffin, sectioned (3 μm), and stained with hematoxylin and eosin. Sections were mounted and analysed by light microscopy for histologic evaluation.
Immunohistochemistry (IHC): the first steps were the same as for the H&E staining. Then, a step of antigen retrieval was performed that consisted of incubation for 30 min at 95 °C in 0,01 M Tris-1 mM EDTA pH 9. Subsequently, primary antibodies were incubated overnight at 4 °C. After rinsing in TBS-T, the sections were incubated with the corresponding secondary antibodies for 30 min at RT. Peroxidase activity was revealed using DAB+ and sections were lightly counterstained with Harris hematoxylin. Finally, slides were dehydrated in graded series of ethanol, cleared in xylene and mounted with Eukitt (Labolan, #28500, Navarra, Spain). Image acquisition was performed on an Aperio CS2 slide scanner using ScanScope Software (Leica Biosystems, Vista, CA, USA). The image analysis was performed using a plugin developed for Fiji, ImageJ (NIH, Bethesda, MD, USA). The antibodies employed were anti-HDV human serum sample (CUN-28336) and anti-F4/80 (BioLegend #123102).
2.11. RNA Extraction and RT-qPCR
Total RNA from liver samples was isolated using TRI Reagent®® (#T9424, Sigma-Aldrich,) according to the manufacturer’s instructions. Total RNA was pre-treated with DNAse I (#AM-1907, TURBO DNA-free ™ Kit, Applied Biosystems) and reverse-transcribed into complementary DNA (cDNA) using M-MLV reverse-transcriptase (Invitrogen, #28025013). Real-time quantitative polymerase chain reactions (RT-qPCR) were performed using iQ SYBR Green Supermix (#170-8884, BioRad) in a CFX96 Real-Time Detection System (#1855196, BioRad,) and primers as specified in supplementary materials. HDV strand-specificity was analyzed as described elsewhere [30]. GAPDH was used as a control housekeeping gene.
2.12. Statistical Analysis ç
Statistical analyses were performed using GraphPad Prism 8.0 software. The data are presented as individual values ± standard deviation. The statistical analysis performed in each experiment was specified in the legend of the figures. Significance *P<0.05, **P < 0.01, ***P < 0.001, ****P<0.0001).
3. Results
3.1. Analysis of HDVAg expression and HDV replication in Huh-7 cells transfected with HDV WT and mutants
We have constructed the mutants described in Figure 1A: an HDV mutant expressing a non-prenylated L-HDAg (HDV-NPrL-HDAg), where Cys-211 was replaced by a serine as described by Glenn et al. [17]; two mutants altering HDAg phosphorylation, one non-phosphorylated with Ser-177 substituted by alanine, and another with Ser-177 replaced by the phosphomimetic amino acid aspartate [19,20,21]; an HDV mutant expressing HDAgs lacking the nuclear localization signal (NLS), with Glu-66 and Arg-75 substituted by Ala (HDAg-∆NLS) [15]; an HDV mutant expressing L-HDAg without the Nuclear Export Signal (NES), where Pro-205 was replaced with Ala-205 [16]; and finally, we have introduced a mutation in helix 1 of the antigenomic HDV ribozyme (HDV-∆Ribozyme). More specifically, the nucleotide sequence CCGG located at position 691-694 was replaced by AGCC that results in a conformational change [9].
The Huh-7 cells were co-transfected with a plasmid carrying the HDV wild-type (WT) genome or the different mutants described, along with a plasmid carrying the HBV 1.3x genome [30]. After transfection, cells were harvested at days 1, 3, 7, 10 and 14 for the analyis of HDVAg expression by Western blot and at days 7 and 14 for RNA extraction to determine and quatify the presence of HDV genomes by quantitative PCR.
As shown in Figure 1B, in cells transfected with WT HDV at day 1, we detected S-HDAg, and by day 3, and more clearly at day 7, we detected both S-HDAg and L-HDAg, as previously described [30,31]. No protein expression was detected in cells transfected with the HDV mutants in which Ser-177 was mutated (HDV-Ser177Ala or HDV-Ser177Asp)or in the one we introduced a mutation that altered ribozyme conformation (Figure 1B). In cells transfected with HDV-NPrL-HDAg, S-HDAg was clearly detected from day 1 and its concentration increase with time (the decrease observe at day 10 was due to a protein loading problem). However, L-HDAg was only observed at day 14 at the expression levels were very low (Figure 2B). Cells transfected with HDV-∆NLS-HDAg or HDV-∆NES-HDAg mutants showed a very similar kinetic of HDAgs expression to the one shown by WT, but the levels of L-HDAg expression are slightly lower than in WT.
The analysis of the presence of HDV genomes showed no HDV replication activity in cells transfected with HDV mutants in which Ser-177 was mutated, nor in the ribozyme mutant. The levels of HDV replication was significanly lower in the cells transfected with the HDV-NPrL-HDAg and significarly higher with the HDV-∆NLS-HDAg or HDV--∆NES-HDAg mutants in comparison to WT at day 7, that might be explainned by the lower L-HDAg, however, at day 14 the differences among the groups dissapeared (Figure 1C).
3.2. Analysis of the Intracellular Localization of HDAgs and Assessment of the Formation of HDV Infectious Virions by HDV-NPrL-HDAg, HDV-∆NLS-HDAg and HDV-∆NES-HDAg Mutants
At 7 and 14 days post-transfection, Huh-7-cells were fixed, and HDAg expression was analyzed by immunofluorescence. As shown in Figure 2A, transfection with the plasmid carrying WT HDV resulted in mainly nuclear presence of HDAg at day 7, and by day 14, expression could be detected both in the nucleus and the cytoplasm. In cells transfected with NPrL-HDAg, both at day 7 and day 14, the antigen is predominantly detected in the nucleus, reflecting the lower levels of L-HDAg which is required for nuclear export. The mutant expressing ∆NLS-HDAg exhibited a distinct cytosolic presence at day 7, contrasting with the predominantly nuclear location observed in HDV WT. By day 14, the expression significantly diminished in both the nucleus and cytoplasm. On the contrary, cells transfected with HDV-∆NES-HDAg displayed a robust and nearly exclusive nuclear localization at both day 7 and day 14 (Figure 2A).
Interestingly, we observed that the morphology of the nucleus of HDV-NPrL-HDAg transfected cells was clearly altered. In fact, the analysis of the size of the transfected cells with the different HDV variants revealed a significant nuclear enlargement of the NPrL-HDAg with respect to the HDV WT and the other two mutants (Figure 2B).
Then, we analyzed the formation of HDV infectious particles in the supernatant of transfected cells. For this purpose, cell supernatant was harvested on days 7 and 14 and used to infect Huh-7 cells stably expressing hNTCP. A minimal number of infected cells were observed when cells were incubated with day 7 supernatants, but the count significantly increased when using day 14 HDV-WT supernatants. While infectious particles were detected in the supernatant from ∆NLS-HDAg transfected cells, none were observed in the supernatants of NPrL-HDAg or ∆NES-HDAg transfected cells (Figure 2C).
3.3. In vivo analysis of the HDV-NPrL-HDAg, HDV-∆NLS-HDAg, and HDV-∆NES-HDAg mutants
For in vivo evaluation, liver-tropic AAV serotype 8 vectors carrying HDV WT or the indicated mutants were generated. Subsequently, the AAV-HDV vectors were intravenously administered to C57BL/6 male mice along with AAV-HBV at a dose of 5x1010 vg/mouse each and the animals were sacrificed three weeks later (Figure 3A). Upon sacrifice, the liver was extracted, and the presence of HDAg was analyzed through western blot and immunohistochemistry, while HDV genomes were assessed via RT-qPCR.
Western blot analysis revealed the expression of both S-HDAg and L-HDAg in animals receiving HDV-WT and the different mutants (Figure 3B). However, significant differences were observed in the L-HDAg/S-HDAg ratio depending on the mutant. The ratio was lower in HDV-NPrL-HDAg and HDV-∆NES-HDAg, and higher in HDV-∆NLS-HDAg compared to HDV WT (Figure 3C). Immunohistochemistry analysis of HDAg in the liver of HDV WT revealed the expected nuclear-cytoplasmic distribution, with weak cytoplasmic staining. In the animals receiving the HDV-NPrL-HDAg and HDV-∆NES-HDAg mutants, a very similar pattern to HDV WT was observed, with the majority of hepatocytes showing cytoplasmic-nuclear staining, the cytoplasmic stainning was particular intense in HDV-NPrL-HDAg. However, a considerable number of cells exhibited almost only nuclear staining (Figure 3D). Moreover, animals receiving HDV-∆NLS-HDAg displayed a very intense cytoplasmic signal, correlating with higher L-HDAg/S-HDAg ratios (Figure 3D).
HDV genomes were significantly higher in the mice receiving HDV WT than in the animals receiving the different mutants, with more accumulation in the HDV-NPrL-HDAg and HDV-∆NLS-HDAg mutants (Figure 3E). This suggests deficiencies in HDV replication associated with the alteration of HDAg intracellular location or a mutant-related toxic effect.
3.4. Evaluation of liver damage
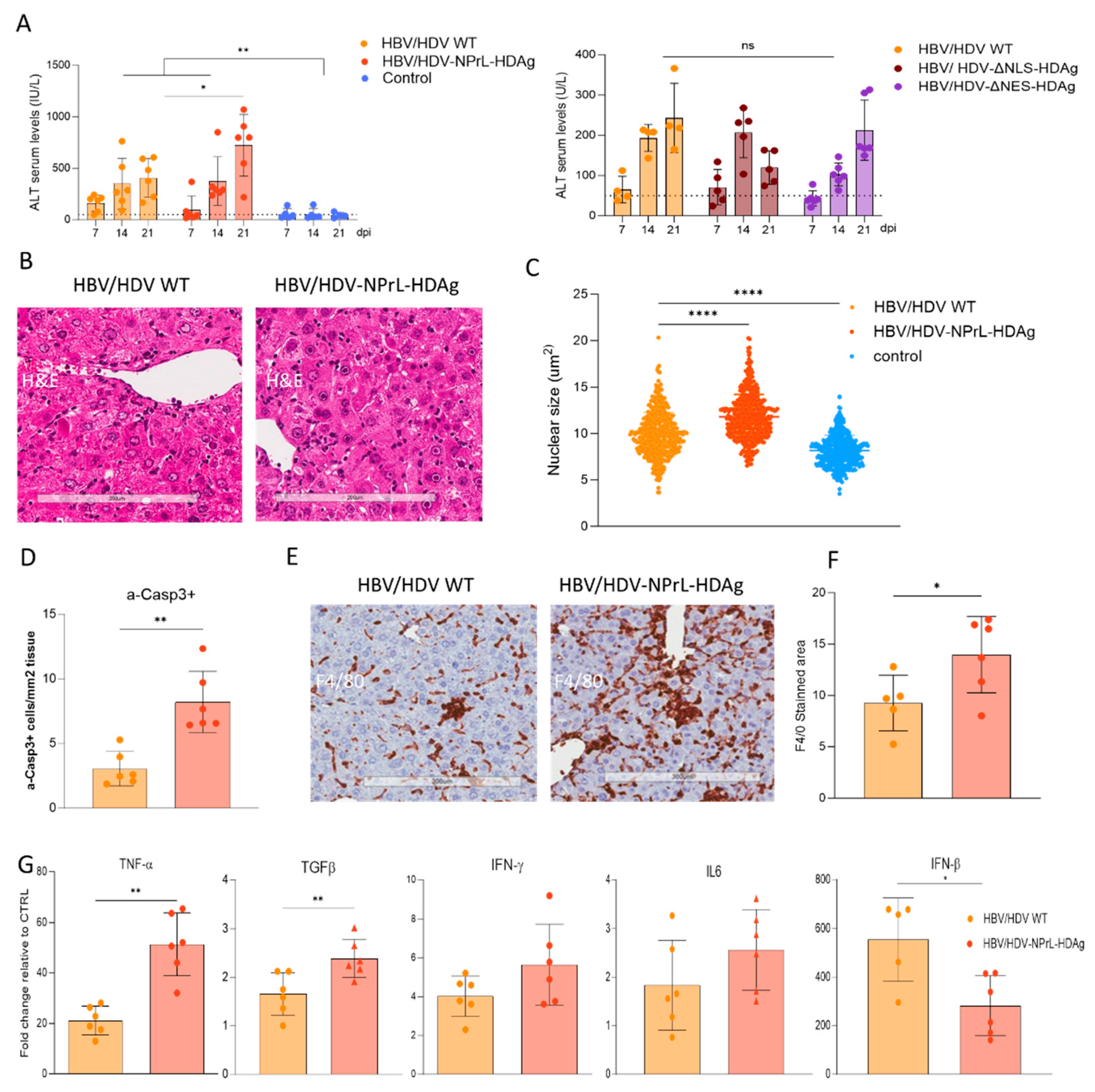
To evaluate liver damage we separate the study in two different experimental groups. The first group was composed by HBV HDV WT-, HBV-HDV-NPrl Ag-injected and untreated control animals and the second group was composed by HBV-HDV WT-, HDV-∆NLS-HDAg- and HDV-∆NES-HDAg-treated mice. Transaminase levels were analyzed at days 7, 14, and 21 after vector injection, as described in Figure 3A. As anticipated in both experiments, AAV-HDV WT mice exhibited ALT elevation above normal levels in both experiments. Transaminase levels were also elevated in the animals receiving the different mutants (Figure 4A). However, while in mice receiving HDV-∆NLS-HDAg or HDV-∆NES-HDAg, the levels were similar to that in HDV WT, in the animals receiving HDV-NPrL-HDAg, ALT levels they were significaly higher at day 21. The pattern of transaminase elevation was very similar in all cases, showing an increase from day 7 to day 21, except for HDV-∆NLS-HDAg, where the peak was achieved at day 14 (Figure 4A).
Due to the elevated transaminase levels in HDV-NPrL-HDAg, we conducted a more detailed analysis of the liver pathology in those mice. H&E staining revealed a more altered liver architecture in HDV-NPrL-HDAg than in HDV WT mice (Figure 4B). The analysis of the size of hepatocyte nuclei revealed, as previously observed, bigger nuclei in HDV WT animals compared to controls, which were even larger in HDV-NPrL-HDAg mice (Figure 4C). Previously, we demonstrated in this animal model that HDV-induced liver damage was associated with the induction of apoptosis, identified by the presence of activated Caspase 3 (a-Casp3), an increase in parenchymal macrophages, and TNF-α [30]. In fact, TNF-α inhibition can partially ameliorate HDV-induced liver damage [32]. Here, we observed that HDV-NPrL-HDAg increased the number of apoptotic hepatocytes compared to HDV-WT (Figure 4D), and we also observed a stronger inflammatory macrophage infiltrate (Figure 4E-F) and significantly higher TNF-α production (Figure 4H). The analysis of the expression of additional cytokines revealed a significant increase in TGF-β and a tendency to increase in IFN-γ and IL-6 (Figure 4H). Interestingly, IFN-β, which is strongly induced by HDV, is significantly lower in HDV-NPrL-HDAg (Figure 4H), probably as a consequence of lower genome levels (Figure 3E).
4. Discussion
In this study, we comprehensively assessed in vitro and in vivo the consequences of introducing various mutations into the HDV genome, specifically targeting HDAg intracellular localization, post-translational modification, and the functionality of the HDV ribozyme. Utilizing a surrogate system developed in our laboratory involving plasmids and a liver-tropic AAV vector carrying the HDV 1.2x sequence, developed by Dr. Taylor’s group [35], under the transcriptional control of a liver-specific promoter, we achieved transcription of the HDV anti-genome sequence. This sequence initiated HDV replication and the expression of the two HDAgs, both in vitro and, more importantly, in the livers of mice [30].
Our in vitro investigations were conducted in the human hepatic cell line Huh-7, known for sustaining HDV replication due to the absence of a type I response [31]. We observed that mutations in the Ser177 residue, either by replacing it with the neutral amino acid alanine or the phosphomimetic amino acid aspartic acid, resulted in the complete absence of protein expression and a substantial impact on HDV replication. Ser177 phosphorylation of S-HDAg is crucial for viral replication, as it is required for interaction with cellular RNA Pol II in the production of new genomes [19]. Unphosphorylated S-HDAg cannot interact with the host polymerase, preventing the replication of the HDV antigenome. Furthermore, the results obtained from transfection with the mutant, where the Ser residue was replaced by Asp, underscore the importance of maintaining a delicate equilibrium between unphosphorylated and phosphorylated S-HDAg at Ser-177 in the HDV life cycle. Indeed, it has been demonstrated that only non-phosphorylated S-HDAg is incorporated into HDV particles [22]. Our data confirm the importance of maintaining a proper balance between phosphorylated and unphosphorylated S-HDAg at Ser-177 for the correct establishment of the HDV life cycle.
Similar outcomes were observed following the transfection of the HDV mutant, where we introduced a conformational change in the ribozyme sequence of the antigenome. Drawing on previous studies involving the truncation of different regions of the HDV ribozyme, we engineered an HDV mutant with a mutation in helix 1 of the antigenomic HDV ribozyme. Transfection of this mutant into Huh-7 cells resulted in no HDAg production or HDV replication, confirming the anticipated importance of the correct folding of this sequence for establishing HDV replication [9]. Due to the lack of protein expression and HDV replication in vitro, Ser177 and ribozyme mutants were not evaluated in vivo.
Regarding the non-prenylated mutant, we observed an altered L-HDAg/S-HDAg ratio both in vitro and in vivo through Western Blot (WB) analysis. While normal levels of S-HDAg expression were detected, the expression of non-prenylated L-HDAg was significantly lower. These data suggest that the lack of isoprenylation might affect the stability of L-HDAg, resulting in a lower L-HDAg/S-HDAg ratio compared to HDV WT. Alternatively, the absence of prenylated L-HDAg might impact HDV genome editing, reducing the expression of this antigen. However, recent findings by Verrier et al. demonstrated that treating HepaRG cells with an isoprenylation inhibitor resulted in the accumulation of edited HDV genomes, reducing the likelihood of this hypothesis [36]. Furthermore, our in vitro experiments clearly indicated that isoprenylation plays a crucial role in the cellular localization of HDAgs, leading to a predominant nuclear localization. This observation was also noted in some mouse hepatocytes. The absence of isoprenylation likely disrupts the normal translocation of HDAgs to the cytoplasm and their association with cellular membranes. Similar observations were made in HDV mutants lacking L-HDAg [31] and in the HDV-∆NES-HDAg, highlighting the role of L-HDAg in the translocation of HDV RNPs from the nucleus to the cytoplasm.
We also found that the lack of isoprenylation leads to a delay in viral replication in vitro and a significant reduction in viral replication in vivo. This suggests that this translational modification might play a role in the regulation of HDV replication. Interestingly, according to a previous publication, an increase in viral replication would be expected since earlier findings attributed the inhibition of viral replication to prenylated L-HDAg, however, we observed the opposite situation [37]. One hypothesis to explain the decreased replicative capacity may be due to impaired HDAg localization and altered membrane association [38]. Isoprenylation facilitates the association of HDV ribonucleoproteins (RNPs) with cellular membranes. Without isoprenylation, this association may be compromised, affecting the interaction of HDAgs with cellular kinases [19,20,21,22]. As expected, this mutant was not capable of producing HDV infectious particles, as this motif is essential for the interaction with HBsAg. In summary, inhibiting the isoprenylation of L-HDAg has multifaceted effects on the composition, stability, cellular localization, and membrane association of HDAgs, ultimately impacting the efficiency of HDV replication.
However, our more important finding associated with this mutant is its association with exacerbated liver pathology. We observed both in vitro and in vivo a higher enlargement of the nuclei compared to HDV-WT. Nuclear enlargement is indicative of cellular stress and toxicity that might be associated to alterations in cellular processes, including damage to the DNA, disruption of normal cellular functions, or induction of cellular stress responses [39]. Even though the replicative capacity of this mutant was lower than the WT, it was able to induce stronger liver damage than HDV WT vector, as evidenced by significantly higher transaminase elevation and a higher number of apoptotic hepatocytes. This higher toxicity was accompanied by a stronger macrophage infiltrate and higher expression of proinflammatory cytokines like TNF-α or TGF-β. These results are in line with or previous that demonstrated a link between TNF-α and HDV-induced liver toxicity, as the administration of the TNF-α inhibitor etanercept resulted in the amelioration of liver injury. Interestingly, the expression of type-I IFN was lower than the one in the HDV WT, most likely an indicative of the lower replication capacity of the mutant, however, is in contradiction to observation in cell culture where treatment with isoprenylation inhibitors resulted in an increase in IFN-β production [40].
The higher toxicity of the non-prenylated mutant could be associated with the altered L-HDAg/S-HDAg 41]; we have previously shown that an HDV mutant lacking L-HDAg expression, only expressing S-HDAg, was more toxic than the HDV WT, and that this toxicity can be reduced by L-HDAg complementation. These data points toward S-HDAg as the antigen preferentially involved in HDV cytotoxicity [31]. However, it could not be discarded that the unprenylated L-HDAg interfered with host processes. As we know, the elimination of the isoprenylation site is detrimental for viral assembly, consequently, the HDV RNPs accumulate inside the cells. The intracellular accumulation of the HDAgs could impair biological functions and induce cell stress or hepatocyte death, as has been described for other viral proteins. One example is HCV, which induces ER stress and mitochondrial alterations through calcium signaling, leading to the production of ROS that are normally found in chronic HCV patients [42,43]. In the case of HBV, the HBx protein promotes the activation of the inflammasome, the production of mitochondrial ROS, and the induction of pyroptosis in the infected cell [44]. Additional studies are needed to clarify the role of the isoprenylated and non-isoprenylated L-HDAg within the HDV-induced pathology.
Next, we determined the role of the intracellular location of HDAgs in the HDV viral cycle. For that purpose, we altered the nuclear localization signal of HDAgs by the substitution of residues Glu-66 and Arg-75 by Ala, reported as essential for the correct functionality of the NLS domain [13,14,15]. The HDAgs expressed by the HDAg-ΔNLS mutant displayed a subcellular distribution pattern different from that of the HDV WT, with a predominant cytoplasmic localization of the HDAgs. However, this mutation doesn’t completely abrogate the presence of the HDAgs in the nucleus, as shown both in Huh-7 cells and mice, indicating that other sequences should be involved in the traffic of the antigens from the cytoplasm to the nucleus. The discrepancy between our results and those of Alves et al. could be due to differences in the HDV constructs since they deleted residues 66 and 75, while we substituted them with alanines [75]. Therefore, it should be considered to identify more residues implicated in the functionality of the NLS domain located between 66-75 aa. Furthermore, this mutant showed a decline in HDV replication that cannot be explained by the absence of HDAg in the nucleus but alternatively by an additional function of residue Arg-75, since it has been described to be essential for the proper recruitment of host RNA Pol by binding to chromatin-remodeling complexes [45,46].
Finally, we have generated a mutant in which we altered the capacity of the L-HDAg to exit the nucleus by mutating the NES signal. The NES domain is located exclusively in the L-HDAg between the residues 198-210 (ILFPADPPFSPQS). In particular, Pro-205 was demonstrated to be essential for the translocation of HDV RNP from the nuclear to the cytosol [16]. Based on those studies, we constructed an HDV mutant with a truncated NES by replacing Pro-205 with Ala-205. Despite the loss of nuclear export, HDAg-staining was detected in the cytoplasmic compartment in both transfected Huh-7 cells and C57BL/6 mice, so some amount of protein is retained after synthesis.
Although the replication capacity of this mutant was similar to or higher than that shown by HDV-WT in vitro, it is unable to form HDV infectious particles. These results indicate that once L-HDAg is inside the nucleus, if the NES domain is not functional, the RNP particles cannot be exported to the cytoplasm, and HDV infectious particles are not formed. These findings confirm previous data reported by Lee et al., demonstrating that this residue within the NES domain is indispensable for the nuclear export of HDV RNPs mediated by L-HDAg [16]. The trafficking of HDAgs from the nucleus to the cytoplasm is necessary for the interaction between L-HDAg and HBsAg in the ER and, consequently, is required for the production of infectious particles. Consequently, the HDVAg-ΔNES mutant was not able to form infectious progeny particles. In mice, we observed that the lack of NES altered the ratio of L-HDAg/S-HDAg and the replication capacity of HDV, which in both cases is lower than in HDV WT. The liver toxicity associated with the administration of this mutant is similar to the one of HDV-WT.
5. Conclusions
Taken together, our results highlight the relevance of HDAgs and their posttranslational modification in HDV biology and HDV-induced liver injury, demonstrated for the first time in an animal model. The increased toxicity of the mutant expressing a non-prenylated L-HDAg correlates with a higher macrophage infiltrate and TNF-α expression. Once again, our data showcase the versatility of the AAV-based HDV model in addressing fundamental questions in HDV biology in vivo.
Author Contributions
Conceptualization, R.A. and G.G.A.; methodology, S.M., A.V. and C.O.; formal analysis. S.M., C.U. and G.C.; investigation, S.M., C.U., N.G.E. and G.C.; writing—original draft preparation, S.M and C.U.; writing—review and editing, C.U., R.A. and G.G.A.; supervision, R.A. and G.G.A.; funding acquisition, G.G.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by RTI2018-101936-B-I00 financiado por FEDER, UE/Ministerio de Ciencia e Innovación—Agencia Estatal de Investigación. Carla Usai and Gracián Camps were supported by FPI fellowships from the Spanish Ministry of Economy and Competitiveness and Sheila Maestro was supported by FIMA’s AC fellowship.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Declaration of Helsinki, and approved by the Institutional Review Board. Patient serum was provided by the Biobank of the University of Navarra and processed following standard operating procedures approved by the Ethical and Scientific Committee (2019.217 CEI-CUN). The animal experimental design was approved by the Ethics Committee for Animal Testing of the University of Navarra (R-132-19GN).
Data Availability Statement
The original contributions presented in the study are included in the article material, further inquiries can be directed to the corresponding authors.
Acknowledgments
We particularly acknowledge the patients for their participation and the Biobank of the University of Navarra for its collaboration. We thank Professor John Taylor, Professor Frank Chisari and Dr Urtzi Garaigorta for providing us with essential reagents for our study. We are grateful to Elena Ciordia, Alberto Espinal, and CIFA staff for animal care and vivarium management and to Laura Guembe for technical assistance.
Conflicts of Interest
The authors declare no conflict of interest in relation with the work presented in this manuscript.
References
- https://www.ncbi.nlm.nih.gov/Taxonomy/Browser/.
- Bonino, F.; Heermann, K.H.; Rizzetto, M.; Gerlich, W.H. Hepatitis Delta Virus: Protein Composition of Delta Antigen and Its Hepatitis B Virus-Derived Envelope. J. Virol. 1986, 58, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Kos, A.; Dijkema, R.; Arnberg, A.C.; van der Meide, P.H.; Schellekens, H. The hepatitis delta (delta) virus possesses a circular RNA. Nature 1986, 323, 558–560. [Google Scholar] [CrossRef] [PubMed]
- Ryu, W.; Netter, H.J.; Bayer, M.; Taylor, J. Ribonucleoprotein Complexes of Hepatitis Delta Virus. J. Virol. 1993, 67, 3281–3287. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.J.; Kalpana, G.; Goldberg, J.; Mason, W.; Werner, B.; Gerin, J.; Taylor, J. Structure and replication of the genome of the hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1986, 83, 8774–8778. [Google Scholar] [CrossRef] [PubMed]
- Taylor, JM. Chapter 3. Replication of the hepatitis delta virus RNA genome. Adv Virus Res. 2009, 74, 103–121. [Google Scholar]
- Gudima, S.; Chang, J.; Moraleda, G.; Azvolinsky, A.; Taylor, J. Parameters of Human Hepatitis Delta Virus Genome Replication: The Quantity, Quality, and Intracellular Distribution of Viral Proteins and RNA. J. Virol. 2002, 76, 3709–3719. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.M. Hepatitis Delta Virus: Cis and Trans Functions Required for Replication. Cell 1990, 61, 371–373. [Google Scholar] [CrossRef] [PubMed]
- Jeng, K.S.; Daniel, A.; Lai, M.M. A pseudoknot ribozyme structure is active in vivo and required for hepatitis delta virus RNA replication. J. Virol. 1996, 70, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.S.; Choo, Q.L.; Weiner, A.J.; Ou, J.H.; Najarian, R.C.; Thayer, R.M.; Mullenbach, G.T.; Denniston, K.J.; Gerin, J.L.; Houghton, M. Structure, sequence and expression of the hepatitis delta (delta) viral genome. Nature 1986, 323, 508–514. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.Z.; Chen, P.J.; Chen, D.S. Large hepatitis delta antigen in packaging and replication inhibition: Role of the carboxyl-terminal 19 amino acids and amino-terminal sequences. J. Virol. 1995, 69, 5332–5336. [Google Scholar] [CrossRef]
- Chang, F.L.; Chen, P.J.; Tu, S.J.; Wang, C.J.; Chen, D.S. The large form of hepatitis delta antigen is crucial for assembly of hepatitis delta virus. Proc. Natl. Acad. Sci. USA 1991, 88, 8490–8494. [Google Scholar] [CrossRef]
- Chang, M.; Chang, S.C.; Chang, C.; Wu, K. Nuclear Localization Signals, but Not Putative Leucine Zipper Motifs, Are Essential for Nuclear Transport of Hepatitis Delta Antigen. J. Virol. 1992, 66, 6019–6027. [Google Scholar] [CrossRef]
- Xia, Y.; Yeh, C.; Ou, J.; Lai, M.M.C. Characterization of Nuclear Targeting Signal of Hepatitis Delta Antigen: Nuclear Transport as a Protein Complex. J. Virol. 1992, 66, 914–921. [Google Scholar] [CrossRef] [PubMed]
- Alves, C.; Freitas, N.; Cunha, C. Characterization of the nuclear localization signal of the hepatitis delta virus antigen. Virology 2008, 370, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Chang, S.C.; Wu, C.H.H.; Chang, M.F. A Novel Chromosome Region Maintenance 1-independent Nuclear Export Signal of the Large Form of Hepatitis Delta Antigen That Is Required for the Viral Assembly. J. Biol. Chem. 2001, 276, 8142–8148. [Google Scholar] [CrossRef] [PubMed]
- Glenn, J.S.; Watson, J.A.; Havel, C.M.; White, J.M. Identification of a Prenylation Site in Delta Virus Large Antigen. Science 1992, 256, 1331–1333. [Google Scholar] [CrossRef]
- Hwang, S.B.; Lai, M.M. Isoprenylation Mediates Direct Protein-Protein Interactions between Hepatitis Large Delta Antigen and Hepatitis B Virus Surface Antigen. J. Virol. 1993, 67, 7659–7662. [Google Scholar] [CrossRef]
- Mu, J.; Chen, D.; Irol, J.V. The Conserved Serine 177 in the Delta Antigen of Hepatitis Delta Virus Is One Putative Phosphorylation Site and Is Required for Efficient Viral RNA Replication. J. Virol. 2001, 75, 9087–9095. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Huang, W.; Hong, S.; Tsay, Y.; Chen, P. ERK1/2-Mediated Phosphorylation of Small Hepatitis Delta Antigen at Serine 177 Enhances Hepatitis Delta Virus Antigenomic RNA Replication. J. Virol. 2008, 82, 9345–9358. [Google Scholar] [CrossRef]
- Bichko, V.; Barik, S.; Taylor, J. Phosphorylation of the Hepatitis Delta Virus Antigens. J. Virol. 1997, 71, 512–518. [Google Scholar] [CrossRef]
- Yeh, T.S.; Lee, Y.H.W. Assembly of hepatitis delta virus particles: Package of multimeric hepatitis delta virus genomic RNA and role of phosphorylation. Virology 1998, 249, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Macnaughton, T.; Gao, L.; Lai, M.C. RNA-Templated Replication of Hepatitis Delta Virus: Genomic and Antigenomic RNAs Associate with Different Nuclear Bodies. J. Virol. 2006, 80, 6478–6486. [Google Scholar] [CrossRef] [PubMed]
- Bichko, V.V.; Taylor, J.M. Redistribution of the Delta Antigens in Cells Replicating the Genome of Hepatitis Delta Virus. J. Virol. 1996, 70, 8064–8070. [Google Scholar] [CrossRef]
- Chang, M.F.; Baker, S.C.; Soe, L.H.; Kamahora, T.; Keck, J.G.; Makino, S.; Govindarajan, S.; Lai, M.M. Human Hepatitis Delta Antigen Is a Nuclear Phosphoprotein with RNA Binding Activity. J. Virol. 1998, 62, 2403–2410. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Alves, C.; Gudima, S.; Taylor, J. Intracellular Localization of Hepatitis Delta Virus Proteins in the Presence and Absence of Viral RNA Accumulation. J. Virol. 2009, 83, 6457–6463. [Google Scholar] [CrossRef] [PubMed]
- Negro, F.; Lok, A.S. Hepatitis D: A Review. JAMA 2023, 330, 2376–2387. [Google Scholar] [CrossRef] [PubMed]
- Abdul Majeed, N.; Zehnder, B.; Koh, C.; Heller, T.; Urban, S. Hepatitis delta: Epidemiology to recent advances in therapeutic agents. Hepatology 2023, 78, 1306–1321. [Google Scholar] [CrossRef] [PubMed]
- Aldabe, R.; Suárez-Amarán, L.; Usai, C.; González-Aseguinolaza, G. Animal models of chronic hepatitis delta virus infection host-virus immunologic interactions. Pathogens 2015, 4, 46–65. [Google Scholar] [CrossRef]
- Suárez-Amarán, L.; Usai, C.; Di Scala, M.; Godoy, C.; Ni, Y.; Hommel, M.; Palomo, L.; Segura, V.; Olagüe, C.; Vales, A.; et al. A new HDV mouse model identifies mitochondrial antiviral signaling protein (MAVS) as a key player in IFN-β induction. J. Hepatol. 2017, 67, 669–679. [Google Scholar] [CrossRef]
- Maestro, S.; Gómez-Echarte, N.; Camps, G.; Usai, C.; Suárez-Amarán, L.; Vales, A.; Olagüe, C.; Aldabe, R.; González-Aseguinolaza, G. AAV-HDV: An Attractive Platform for the In Vivo Study of HDV Biology and the Mechanism of Disease Pathogenesis. Viruses 2021, 13, 788. [Google Scholar] [CrossRef]
- Usai, C.; Maestro, S.; Camps, G.; Olague, C.; Suárez-Amaran, L.; Vales, A.; Aragon, T.; Hommel, M.; Aldabe, R.; Gonzalez-Aseguinolaza, G. TNF-alpha inhibition ameliorates HDV-induced liver damage in a mouse model of acute severe infection. JHEP Rep. 2020, 2, 100098. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2013, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Fälth, M.; Stindt, J.; Königer, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef] [PubMed]
- Kuo, M.Y.; Chao, M.; Taylor, J. Initiation of replication of the human hepatitis delta virus genome from cloned DNA: Role of delta antigen. J. Virol. 1989, 63, 1945–1950. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Salvetti, A.; Pons, C.; Michelet, M.; Rivoire, M.; Baumert, T.F.; Durantel, D.; Lucifora, J. Loss of hepatitis D virus infectivity upon farnesyl transferase inhibitor treatment associates with increasing RNA editing rates revealed by a new RT-ddPCR method. Antiviral Res. 2022, 198, 105250. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.B.; Lai, M.M. Isoprenylation Masks a Conformational Epitope and Enhances trans-Dominant Inhibitory Function of the Large Hepatitis Delta Antigen. J. Virol. 1994, 68, 2958–2964. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Shih, K.; Lo, S.J. Ser-123 of the large antigen of hepatitis delta virus modulates its cellular localization to the nucleolus, SC-35 speckles or the cytoplasm. J. Gen. Virol. 2004, 85, 1685–1694. [Google Scholar] [CrossRef]
- Nakajima, T.; Nakashima, T.; Okada, Y.; Jo, M.; Nishikawa, T.; Mitsumoto, Y.; Katagishi, T.; Kimura, H.; Itoh, Y.; Kagawa, K.; Yoshikawa, T. Nuclear size measurement is a simple method for the assessment of hepatocellular aging in non-alcoholic fatty liver disease: Comparison with telomere-specific quantitative FISH and p21 immunohistochemistry. Pathol Int. 2010, 60, 175–183. [Google Scholar] [CrossRef]
- Lempp, F.A.; Schlund, F.; Rieble, L.; Nussbaum, L.; Link, C.; Zhang, Z.; Ni, Y.; Urban, S. Recapitulation of HDV infection in a fully permissive hepatoma cell line allows efficient drug evaluation. Nat Commun. 2019, 10, 2265. [Google Scholar] [CrossRef]
- Cole, S.M.; Gowans, E.J.; Macnaughton, T.B.; Hall, P.M.; Burrell, C.J. Direct Evidence for Cytotoxicity Associated with Expression of Hepatitis Delta Virus Antigen. Hepatology 1991, 13, 845–851. [Google Scholar] [CrossRef]
- Lee, C.; Chen, P.; Lai, M.M.C.; Chen, D. Isoprenylation of large hepatitis delta antigen is necessary but not sufficient for hepatitis delta virus assembly. J. Virol. 1994, 199, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus NS5A protein alters intracellular calcium levels, induces oxidative stress, and activates STAT-3 and NFκB. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [CrossRef] [PubMed]
- Tardif, K.D.; Mori, K.; Kaufman, R.J.; Siddiqui, A. Hepatitis C Virus Suppresses the IRE1-XBP1 Pathway of the Unfolded Protein Response. J. Biol. Chem. 2004, 279, 17158–17164. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.H.; Ding, J.; Xie, X.X.; Yang, X.H.; Wu, X.F.; Chen, Z.X.; Guo, Q.L.; Gao, W.Y.; Wang, X.Z.; Li, D. Hepatitis B virus X protein promotes liver cell pyroptosis under oxidative stress through NLRP3 inflammasome activation. Inflamm. Res. 2020, 69, 683–696. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Filipovska, J.; Yano, K.; Furuya, A.; Inukai, N.; Narita, T.; Wada, T.; Sugimoto, S.; Konarska, M.M.; Handa, H. Stimulation of RNA Polymerase II Elongation by Hepatitis Delta Antigen. Science 2001, 293, 124–127. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Mura, T.; Chanarat, S.; Okamoto, S.; Handa, H. Hepatitis delta antigen binds to the clamp of RNA polymerase II and affects transcriptional fidelity. Genes Cells 2007, 12, 863–875. [Google Scholar] [CrossRef]
Figure 1.
Analysis of HDAg expression and HDV genomes in Huh-7 cells transfected wit HDV-WT or the different mutants. (A). Schematic representation of the different mutants used in the study. (B). Huh-7 cells were transfected with HDV WT plasmid or the different mutans indicated in the figure together with a plasmid carrying HBV 1.3x genome. Cells were harvested at days 1, 3, 7, 10 and 14 post-transfection (dpt) and HDAg expression was analysed by Wester Blot in cell lysates. As control Huh-7 cells were co-tranfected with plasmids expressing S-HDAg or L-HDAg under the control of a liver specific promoterand collected 3 dpt [30]. (-) indicates non-transfected cells. (C) Cells were transfected as described in C and harvested at 7 and 14 dpt. Total RNA was extracted and HDV genome levels were quantified by RT-qPCR and normilized using RNA concentration. Statistical analysis was performed using two-way anova followed by Dunnett’s multiple comparison test (**P < 0.01, ***P < 0.001).
Figure 1.
Analysis of HDAg expression and HDV genomes in Huh-7 cells transfected wit HDV-WT or the different mutants. (A). Schematic representation of the different mutants used in the study. (B). Huh-7 cells were transfected with HDV WT plasmid or the different mutans indicated in the figure together with a plasmid carrying HBV 1.3x genome. Cells were harvested at days 1, 3, 7, 10 and 14 post-transfection (dpt) and HDAg expression was analysed by Wester Blot in cell lysates. As control Huh-7 cells were co-tranfected with plasmids expressing S-HDAg or L-HDAg under the control of a liver specific promoterand collected 3 dpt [30]. (-) indicates non-transfected cells. (C) Cells were transfected as described in C and harvested at 7 and 14 dpt. Total RNA was extracted and HDV genome levels were quantified by RT-qPCR and normilized using RNA concentration. Statistical analysis was performed using two-way anova followed by Dunnett’s multiple comparison test (**P < 0.01, ***P < 0.001).
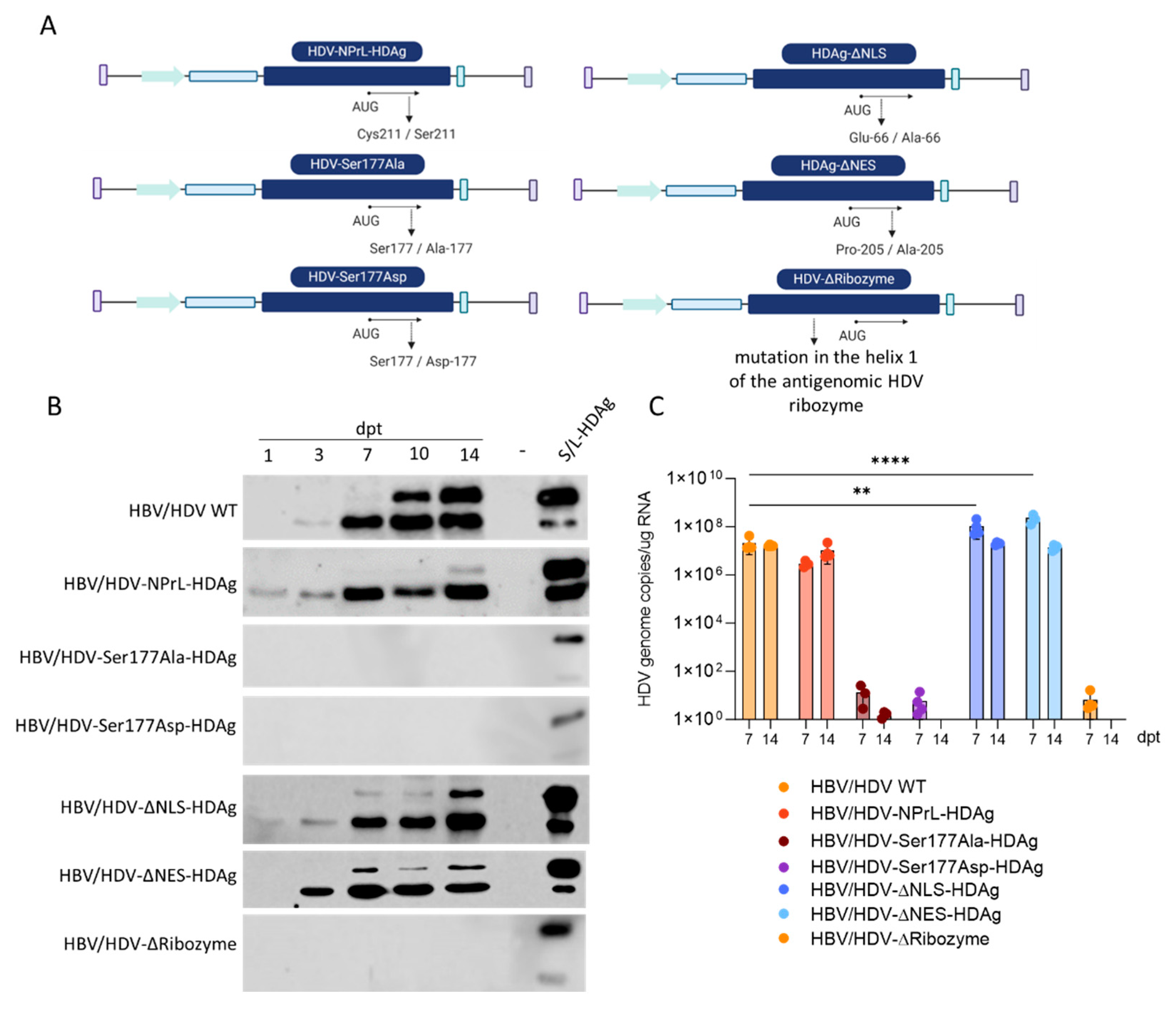
Figure 2.
Analysis of HDAg intracellular localization in Huh-7 cells transfected wit HDV-WT or the different mutants and evaluation of the capacitu to produce HDV infectious particles. (A). HDAg localization was examined by immunofluorescence (IF). (B) Nuclear diameter of HDAg positive hepatocytes were measured from IF images Aperio Image Scope v12.3 software with the ruler tool, at 20x magnification. Statistical analysis was performed using ordinary one-way Anova (****P < 0.0001) (C) Huh-7.5.1-hNTCP cells were infected with supernatants from co-transfected cells collected at 7- and 14-dpt. At 7 days post-infection, cells were fixed and immune stained with anti-HDV human serum for HDAg detection.
Figure 2.
Analysis of HDAg intracellular localization in Huh-7 cells transfected wit HDV-WT or the different mutants and evaluation of the capacitu to produce HDV infectious particles. (A). HDAg localization was examined by immunofluorescence (IF). (B) Nuclear diameter of HDAg positive hepatocytes were measured from IF images Aperio Image Scope v12.3 software with the ruler tool, at 20x magnification. Statistical analysis was performed using ordinary one-way Anova (****P < 0.0001) (C) Huh-7.5.1-hNTCP cells were infected with supernatants from co-transfected cells collected at 7- and 14-dpt. At 7 days post-infection, cells were fixed and immune stained with anti-HDV human serum for HDAg detection.
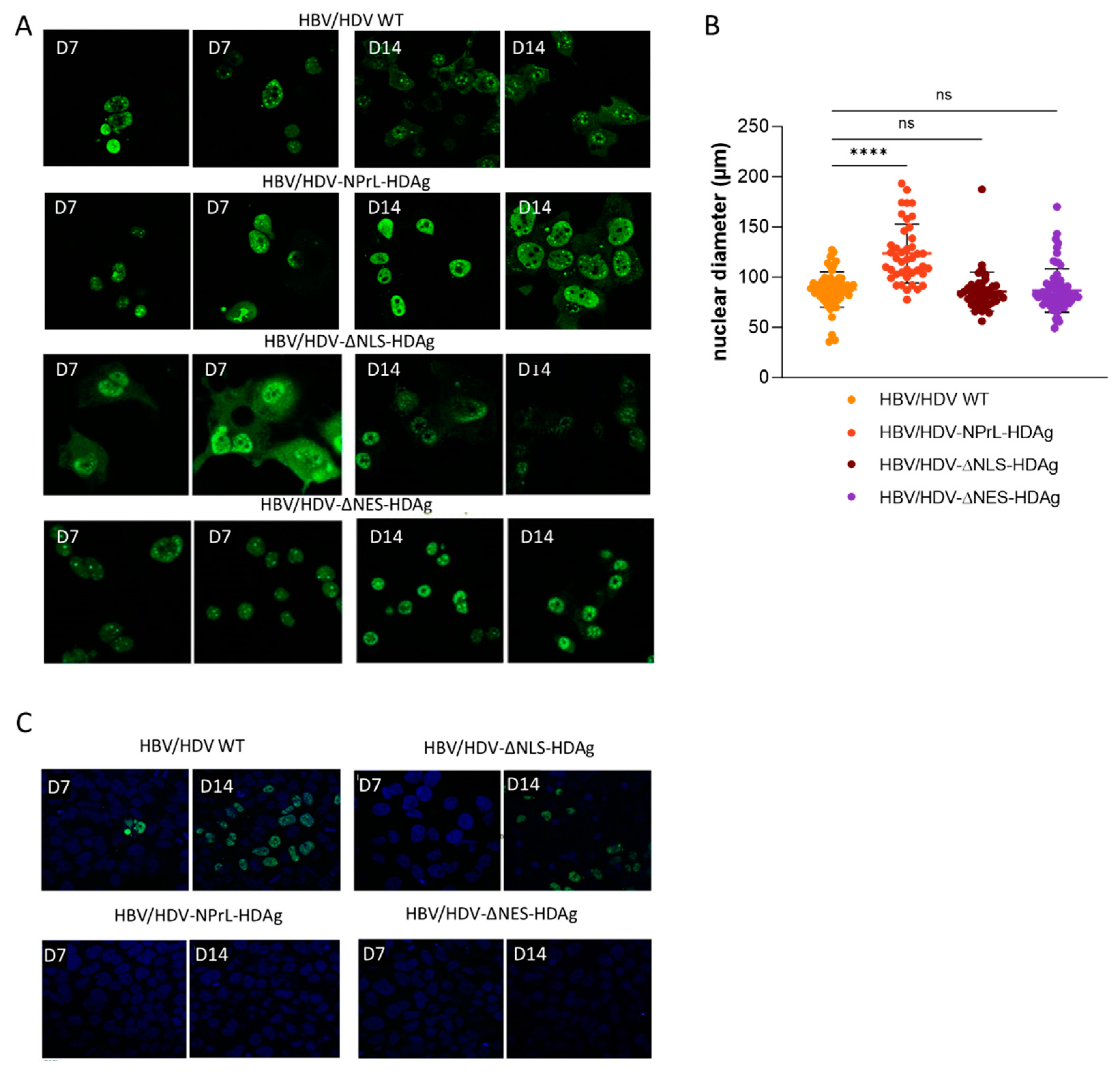
Figure 3.
In Vivo Evaluation of HDV Mutants. (A) Schematic representation of the experimental layout. C57BL/6 male mice received AAV HDV or AAV HDV mutants along with AAV-HBV at a dose of 5x1010 vg/mouse each intravenously. Serum samples were extracted at 7, 14, and 21 days post-injection (dpi), and animals were sacrificed at week 3, with the liver harvested. (B) Western blot analysis of liver lysates was performed to detect S-HDAg and L-HDAg. (C) HDAg expression levels were quantified, and the L-HDAg/S-HDAg ratio was calculated for each animal. (D) Immunostaining against HDAgs was conducted at 21 dpi in the liver sections of mice injected with AAV-HDV WT or the different mutants. Green arrows indicate hepatocytes where a clear nuclear signal is detected, while cytoplasmic staining was undetectable or very weak. Scale bar: 200 μm. (E) Total RNA was extracted from the livers, and HDV genome levels were assessed by RT-qPCR and normalized using GAPDH as a housekeeping gene. Statistical analysis was performed using ordinary one-way Anova followed by multiple comparisons (**p < 0.01, ***p < 0.001, ****p < 0.0001).
Figure 3.
In Vivo Evaluation of HDV Mutants. (A) Schematic representation of the experimental layout. C57BL/6 male mice received AAV HDV or AAV HDV mutants along with AAV-HBV at a dose of 5x1010 vg/mouse each intravenously. Serum samples were extracted at 7, 14, and 21 days post-injection (dpi), and animals were sacrificed at week 3, with the liver harvested. (B) Western blot analysis of liver lysates was performed to detect S-HDAg and L-HDAg. (C) HDAg expression levels were quantified, and the L-HDAg/S-HDAg ratio was calculated for each animal. (D) Immunostaining against HDAgs was conducted at 21 dpi in the liver sections of mice injected with AAV-HDV WT or the different mutants. Green arrows indicate hepatocytes where a clear nuclear signal is detected, while cytoplasmic staining was undetectable or very weak. Scale bar: 200 μm. (E) Total RNA was extracted from the livers, and HDV genome levels were assessed by RT-qPCR and normalized using GAPDH as a housekeeping gene. Statistical analysis was performed using ordinary one-way Anova followed by multiple comparisons (**p < 0.01, ***p < 0.001, ****p < 0.0001).
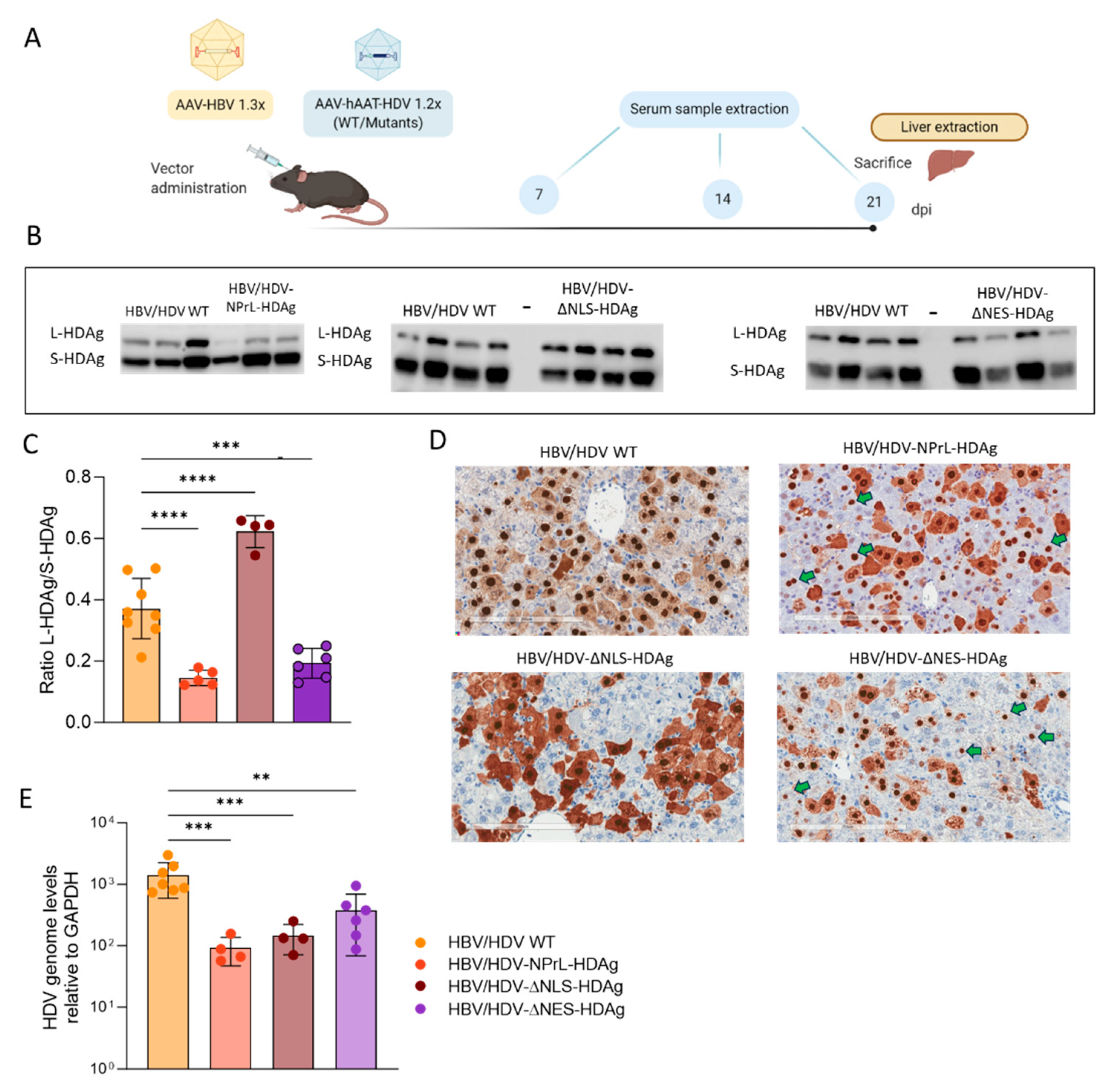
Figure 4.
Analysis of liver pathology. (A) Peripheral blood was collected every 7 days for 3 weeks to measure ALT concentration in serum (as described in Figure 3A). Individual data points and mean values ± standard deviation are plotted, and significant differences between groups at each time point were determined by a Mixed Effect analysis and multiple comparisons (*p < 0.05, **p < 0.01). (B) Liver sections from HBV/HDV WT- and HBV/HDV-NPrL-HDAg-injected mice obtained at 21 dpi were analyzed by H&E staining, revealing more abundant degenerated nuclei in the HDV-NPrL-HDAg mutant. Scale bar: 200 μm (C) The nuclear diameter was significantly bigger in the HDV-NPrL-HDAg group, as determined by an unpaired t-test. (D) Immunostaining against activated caspase 3 (a-casp3) revealed that the HDV-NPrL-HDAg mutant exacerbates hepatocyte death by apoptosis, with differences between the two groups revealed by a Mann–Whitney test. (D–F) Liver sections were stained with anti-F4/80 to determine the presence of macrophages, showing a significantly higher presence in the HDV-NPrL-HDAg group. Scale bar: 200 μm (G) The expression of different cytokines in the liver of HBV/HDV WT and HBV/HDV-NPrL-HDAg was analyzed by Real-time qPCR and compared to those in untreated control animals, revealing differences between the two groups (*p < 0.05, **p < 0.01). Mann-withney test was applied for the statistical comparison of the two groups.
Figure 4.
Analysis of liver pathology. (A) Peripheral blood was collected every 7 days for 3 weeks to measure ALT concentration in serum (as described in Figure 3A). Individual data points and mean values ± standard deviation are plotted, and significant differences between groups at each time point were determined by a Mixed Effect analysis and multiple comparisons (*p < 0.05, **p < 0.01). (B) Liver sections from HBV/HDV WT- and HBV/HDV-NPrL-HDAg-injected mice obtained at 21 dpi were analyzed by H&E staining, revealing more abundant degenerated nuclei in the HDV-NPrL-HDAg mutant. Scale bar: 200 μm (C) The nuclear diameter was significantly bigger in the HDV-NPrL-HDAg group, as determined by an unpaired t-test. (D) Immunostaining against activated caspase 3 (a-casp3) revealed that the HDV-NPrL-HDAg mutant exacerbates hepatocyte death by apoptosis, with differences between the two groups revealed by a Mann–Whitney test. (D–F) Liver sections were stained with anti-F4/80 to determine the presence of macrophages, showing a significantly higher presence in the HDV-NPrL-HDAg group. Scale bar: 200 μm (G) The expression of different cytokines in the liver of HBV/HDV WT and HBV/HDV-NPrL-HDAg was analyzed by Real-time qPCR and compared to those in untreated control animals, revealing differences between the two groups (*p < 0.05, **p < 0.01). Mann-withney test was applied for the statistical comparison of the two groups.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



