
Submitted:
23 January 2024
Posted:
23 January 2024
You are already at the latest version
Abstract
Cellular Integrated Stress Response (ISR), the mitochondrial Unfolded Protein Response (UP-Rmt) and the IFN signaling are associated with viral infections. The Activating transcription fac-tor 4 (ATF4) plays a pivotal role in these pathways and controls the expression of many genes involved in redox processes, amino acid metabolism, protein misfolding, autophagy and apop-tosis. The precise role of ATF4 during viral infection is unclear and depends on cell hosts, viral agents and models. Furthermore, ATF4 signaling can be hijacked by pathogens to favor viral in-fection and replication. In this review, we summarize the ATF4-mediated signaling pathways in response to viral infections, focusing on human immunodeficiency virus (HIV). We examine the consequences of ATF4 activation for HIV replication and reactivation. The role of ATF4 in au-tophagy and apoptosis is explored as in the context of HIV infection programmed cell deaths contribute to the depletion of CD4 T cells. Furthermore, ATF4 can also participate in the estab-lishment of innate and adaptive immunity that are essential for the host to control viral infec-tions. We finally discuss the putative role of the ATF4 paralogue, named ATF5, in HIV infection. This review underlines the role of ATF4 at the crossroads of multiple processes reflecting host-pathogen interactions.
Keywords:
ISR
; AIDS
; immunity
; mitochondria
; ER stress
; UPR
1. Introduction
One of the main regulators of cellular homeostasis is the Activating Transcription Factor 4 (ATF4). This bZIP domain transcription factor plays a crucial role in both the integrated stress response (ISR) and the mitochondrial stress response (MSR) [1,2,3]. In reaction to oxidative stress, misfolded protein accumulation, or nutrient deprivation, ATF4 triggers the expression of genes involved in cellular processes participating in the control of autophagy or cell death. Furthermore, the role of ATF4 in cellular responses to stress is intimately linked to its dimerization partners such as the transcription factor ATF5, an ATF4 paralogue found in mammals, which belongs to the ATF4 bZIP-domain transcription factor family [4,5]. ATF5 is also strongly involved in stress responses and can be a target of ATF4. Thus, ATF4 and ATF5 are key cellular factors that integrate various stress signals.
During viral infections, these cellular processes can be modulated by virus machineries. In this context, it has been shown that numerous viral infections modulate ATF4 activity, which contributes to or represses viral replication, depending on the virus (Table 1) and the targeted host cell. Whereas ATF4 levels have been shown to be increased by human immunodeficiency virus-1 (HIV-1) infection and to promote viral replication [6,7,8,9,10,11,12], data concerning the implementation of this regulation remain scarce. Thus, apart from its transcription activity at the HIV-1 promoter, our understanding of the impact of its various cellular functions on the HIV-1 viral cycle remains patchy. Understanding the mechanisms of ATF4 activation in the context of HIV-1 infection also merits to be further explored as ATF4 is envisaged as a latency-reversing agent (LRA), that could be capable to reactivate HIV-1, thereby giving a chance to eliminate viral reservoirs that are a major obstacle to the eradication of the virus from the infected organism [10].
This article aims to provide an overview of the role of ATF4 in HIV infection. We summarize models in which ATF4 activation has been reported during HIV-1 infection and focus on how HIV-1 can induce ATF4 activation. We then explore the consequences of ATF4 activation on host cells and viral cycle. Finally, we also consider that ATF5, the paralogue of ATF4 in mammals, could be a potent actor during viral infections.
2. HIV infection regulates ATF4
2.1. ATF4 is up-regulated during HIV and SIV infections
HIV-1 infection leads to the development of acquired immunodeficiency syndrome (AIDS) associated with the depletion of CD4+ T cells, mainly by apoptosis [66,67,68] which predicts further pathogenicity [69,70]. Of interest, the induction of ATF4 following HIV-1 infection has been observed in several in vitro models. It was first described in Jurkat T cells, in which ATF4 was up-regulated at both the transcript and protein levels 8 hours post-infection and remained elevated 48 hours later [6]. This increase in ATF4 transcript levels was further observed in primary human CD4+ T cells at day 5 post-infection [8]. In a model of HIV-1 latency, ATF4 transcript and protein levels are weakly detectable [10] but increased with viral reactivation [6,8,10]. In vivo, in monkeys infected with simian immunodeficiency virus (SIV), ATF4 transcripts are also up-regulated in the gut mucosa. This increase occurred during the acute phase of SIV infection, i.e. 1 to 2 weeks post-infection, but are not observed during the chronic phase [8]. Viral proteins released in the microenvironment of infected cells such as Tat has been proposed to be sufficient for inducing ATF4 gene expression through ER stress in non-infected cells [11,12]. Altogether, these observations show that ATF4 expression is differentially regulated during both the early and chronic phases of viral replication, in infected but also non-infected cells, which are close to the former, suggesting that ATF4 induction is related to viral stress and plays a potential role in the establishment of latency.
2.2. How can HIV-1 regulate ATF4?
2.2.1. HIV-1 induced ISR/ATF4 signaling
The ISR leads to global translation blockade through the phosphorylation of eIF2α, which increases ATF4 translation [3]. Several kinases are responsible for eIF2α phosphorylation: double-stranded RNA-dependent protein kinase (PKR), PKR-like ER kinase (PERK), general control non-derepressible 2 (GCN2), heme-regulated eIF2α kinase (HRI), microtubule affinity regulating kinase 2 (MARK2) and family with sequence similarity 69 member C (FAM69C) [3,71,72] (Figure 1). The role of eIF2α and its kinases during viral infection has been recently reviewed by Liu et al. [73]. We focus on HIV studies providing evidence that eIF2α phosphorylation by ISR kinases can be associated with ATF4 induction, and that a direct control of these kinases by HIV-1 proteins may affect ATF4 activity.
Among the ISR kinases, PKR is the best characterized viral nucleic acid sensor [74,75]. It is activated mainly by double-stranded RNAs (dsRNA) [76,77,78,79]. PKR regulates HIV infection as it can bind the hairpin structure within the transactivation-response region of the HIV-1 genome [80]. Whereas PKR contributes to ATF4 translation in response to endoplasmic reticulum (ER) stress [81], paradoxically, to our knowledge, no study reports the induction of ATF4 in the context of HIV-1 infection through an endogenous PKR/eIF2α pathway. A reason that may explain this lack of evidence is the rapid inhibition of PKR by multiple mechanisms during HIV-1 replication [80] (Figure 1): PKR activation is inhibited by high levels of dsRNA and by the direct binding of cellular proteins, including TAR RNA binding protein (TRBP) and adenosine deaminase acting on RNA (ADAR1) [82,83,84,85,86,87,88]. Moreover, the viral Tat protein may counteract PKR activation by preventing PKR autophosphorylation. Tat also serves as a competitive inhibitor limiting PKR binding to eIF2α thanks to a sequence homology between one of its motifs and eIF2α [89,90,91].
Cells experiencing ER stress due to nutrient deprivation or viral replication may lead to the activation of PERK. Like many other viruses, HIV is known to induce protein misfolding and ER stress [92]. Campestrini et al. have previously reported induction of ER Unfolded Protein Response (UPR) genes such as PERK and ATF4, following the treatment of Jurkat cells with the HIV-1 Tat protein [11]. Additionally, PERK activation has been observed in Tat-treated human brain microvascular endothelial cells (HBMECs) and in HIV-infected CD4+ T cell lines [12,93]. Whereas PERK is one of the sensors of the UPR with IRE-1 and ATF6, the functional role of PERK in ATF4 induction in response to HIV infection is not fully demonstrated.
GCN2 is activated in response to amino acid deprival and metabolic dysfunction (Figure 1). These alterations are reported in plasma of HIV-infected patients and in the gut of SIV-infected monkeys [8,94,95,96,97]. Moreover, GCN2 is activated in vitro by HIV or SIV infection [8,98] suggesting that metabolic disorders observed in HIV-infected patients could also lead to GCN2 activation. GCN2 phosphorylates the HIV 1 integrase, which reduces viral integration [99]. This property is not limited to HIV-1 since various integrases from other retroviruses are also recognized as substrates by GCN2 [99]. The anti-viral activity of GCN2 can be overcame by the HIV-1 protease that can cleave GCN2 [100]. Jiang et al. have reported that HIV-induced ATF4 transcription would result from the activation of the GCN2/ATF4 pathway [8]. Serum deprivation triggers HIV-1 replication of CD4+ T cells, correlating with the GCN2-mediated activation of ATF4, which is recruited to the HIV-1 long terminal repeat (LTR) to facilitate viral transcription.
Both heme/iron depletion and arsenite-induced oxidative stress activate HRI [3]. Arsenite-induced HRI activation increases viral protein production and infection rate of reovirus[101]. It can also regulate the level of the viral factor Zta that plays a role in the lytic cycle of EBV [102,103]. HRI has been shown to be able to activate ATF4, triggering the expression of genes involved in autophagy [104,105,106] and oxidative-stress response including the antioxidant heme oxygenase-1 (HO-1) gene [106]. Several groups have reported a role of HO-1 in regulating viral replication [107,108] including HIV’s [109]. HO-1 dysfunction has been associated with neuronal diseases in people living with HIV [110]. For instance, despite a link between HO-1 and viral replication, to date no study has addressed the role of an HRI pathway regulating HIV infection and replication.
Recently, two other kinases able to phosphorylate eIF2α have been characterized, namely MARK2 and FAM69C [71,72]. Both kinases are induced by proteotoxic stress. FAM69C KO mice-derived microglia displayed an indirect increase in inflammation, and HIV-1 binds MARK2 to control motor adaptor function on viral cores [72,111]. However, no HIV-induced ATF4 activation depending on these kinases has been reported yet.
To conclude, PERK and GCN2 may lead to ATF4 activation after HIV-1 infection, and the direct interaction of HIV-1 components with GCN2 and MARK2 may in turn control their ability to activate ATF4. Thus, the control of ATF4 by these additional eIF2α kinases in this context is highly probable.
2.2.2. Mitochondrial Stress response, ATF4 and HIV.
The contribution of HIV-1 infection to the induction of mitochondrial dysfunctions has been described earlier [112,113,114]. During HIV-1 infection, mitochondrial functions are compromised resulting in reduced oxidative phosphorylation (OXPHOS), ATP synthesis, gluconeogenesis, and β-oxidation. In addition to membrane depolarization and release of cytochrome C [115,116], HIV-1-induced alterations may also disrupt cellular homeostasis, increase oxidative stress, affect mitochondrial dynamics, and lead to the loss in mitochondrial DNA [115,117,118]. Mitochondrial dysfunctions are not only observed in CD4+ T cells, but also in myeloid cells such as neutrophils [119,120], monocytes [121] and CD8+ T cells [122], which are non-infected T cells. Therefore, indirect mechanisms may contribute to the alteration of mitochondrial functions during HIV and SIV infections.
ATF4 is induced by several mitochondrial stress-like alterations affecting proteostasis, respiration, and mitochondrial membrane potential (MMP) loss [123,124,125]. A multi-omics study has suggested that ATF4 coordinates the mitochondrial stress response [125]. ATF4 induction was shown to depend on HRI in response to the respiratory chain and ATP synthesis disruptions [126,127]. Individual knockdown of each of the four original ISR kinases (i.e. HRI, PERK, PKR and GCN2) did not abolish the induction of ATF4 and its target genes. These results suggest the role of either an unknown or newly identified ISR kinases (as indicated above), or some redundancy.
In response to mitochondrial misfolded protein accumulation, ATF4 plays a significant role in cell homeostasis as a transcription factor of the canonical mitochondrial UPR (UPRmt) [124]. ATF4 is also associated with the transcription of genes encoding mitochondrial chaperonin and proteases [124]. For instance, Li et al. suggest that unfolded mitochondrial proteins would be degraded by lysosomes, leading to the increase in amino acids that would activate mTORC1 via the lysosomal v-ATPase through a still unknown mechanism during the UPRmt [128,129,130,131]. mTORC1 would then phosphorylate and activate ATF4, thereby triggering transcription of chaperone-encoding genes, and increasing the mitochondrial folding capacity [129].
ATF4 is also activated in response to alterations in mitochondrial dynamics. The deletion of Optic atrophy protein 1 (OPA1), which is crucial for the fusion of inner membranes during mitochondrial fusion, and contributes to the release of apoptogenic factors [132,133], generates mitochondria-derived reactive oxygen species (ROS) and thus causes increased oxidative stress and death [117]. This process triggers ER stress and a PERK-dependent UPR, resulting in the transcriptional activation of ATF4 and other genes. This response initiates a catabolic program contributing to muscle loss and systemic aging [134]. The knockdown of Drp1, a major effector of mitochondria fission [135], leads to eIF2α phosphorylation and ATF4 activation in the liver [136]. Although modulation of OPA1 was not directly associated with ATF4 activation, it has been show that the OMA1 protease, which cleaves the mitochondrial protein DELE1, but also OPA1 [137,138], leads to the release in the cytosol of the DELE1’s carboxy-terminal domain that oligomerizes with HRI (Figure 1) [126,127,139]. The authors propose that in turn, HRI increases ATF4 protein level [127]. Therefore, affecting mitochondrial fission or fusion can be associated with an ATF4-mediated stress response. To date, ATF4 activation in the context of HIV infection has not been reported to originate from mitochondrial stress. Nevertheless, this lack of data certainly stems from the fact that ATF4 is also involved in the response to other HIV-1-induced stresses that-trigger ISR, like ER stress and amino acid depletion. It is thus challenging to determine the respective contribution of HIV-1-induced stress sources to ATF4 activation.
2.2.3. The viral Vpu protein stabilizes the ATF4 protein
The HIV viral protein U (Vpu) is an HIV-1 accessory protein that down-modulates CD4 and BST-2/tetherin. A cellular Skp, Cullin, F-Box (SCF) E3 ubiquitin ligase complex is recruited by Vpu to target CD4 for ubiquitination and proteasomal degradation [140]. This process involves the recruitment of the F-box β-transducin repeat-containing protein (βTrCP) [141,142,143,144]. Additionally, Vpu reduces BST-2/tetherin from the cell surface by preventing the trafficking of BST-2/tetherin to the plasma membrane from the trans Golgi network and/or the recycling endosome [145,146,147]. Furthermore, Vpu targets BST-2/tetherin for degradation, thus promoting viral progeny release and inhibiting NF-κB signaling [143,148].
The SCF-βTrCP E3 ubiquitin ligase complex has been reported to contribute in the degradation of ATF4 (Figure 1) [149]. Unlike its effect on CD4 and BST-2/tetherin, Vpu inhibits ATF4 βTrCP-mediated proteasomal degradation [150]. This apparent discrepancy in the effect of Vpu on βTrCP-dependent proteasomal degradation may be explained by the existence of two distinct paralogs of β-TrCP, βTrCP1/BTRC and βTrCP2/FBXW11 [151]. Recent work by Pickering et al. demonstrates that Vpu has contrasting effects on βTrCP1 and βTrCP2 and suggests that Vpu would induce proteasomal degradation mediated by βTrCP2 and inhibit βTrCP1-dependent protein degradation [152]. Thus, the contribution of viral proteins encoded by HIV merits further investigation regarding the role of ATF4.
3. ATF4 role during HIV-1 replication
3.1. ATF4 positively regulates HIV-1 cycle
The induction of ATF4 during HIV-1 infection raises the possibility that ATF4 may play a role in viral replication (Table 2). Thus, it has been shown that the overexpression of ATF4 increases viral replication whereas its silencing decreases HIV-1 replication [6,7]. Using compounds that inhibit GCN2 or PERK or that lead to ATF4 levels increase, it has been shown that the induction of the ISR/ATF4 pathway reactivates HIV-1 in models of HIV-1 latency [8,9,10]. Altogether, these data strongly suggest a role for ATF4 in regulating HIV-1 replication both during the acute infection and the exit from latency. However, given the nature of infected cells in particular because memory CD4 T cells and T follicular helper cells (Tfh) [153,154,155,156,157], which represent the main reservoirs in visceral tissues, further analyses should be performed to elucidate the role of ATF4 in primary T cell subsets.
3.2. How ATF4 favorizes HIV-1 replication
3.2.1. ATF4 binds to the HIV-1 LTR and promotes viral gene transcription
ATF4 can be a direct regulator of HIV-1 transcription. During HIV-1 infection, various cellular transcription factors including some bZIP domain proteins have been shown to bind the 5’ long terminal repeat (LTR) -that contains the transcriptional promoter of the viral genome of HIV-1- and regulates transcription of HIV-1 genes [158] (Figure 2).
ATF4 induces HIV’s gene transcription and regulates the Human T-cell Leukemia Virus (HTLV) promoter [159]. As an ATF/CREB family member, ATF4 binds to the C/EBP-ATF consensus sequence (TGACGT (C/A) (G/A)). Two C/EBP-ATF binding sites were first identified in the LTR of HIV-1, but super-electrophoretic mobility shift assay (EMSAs) failed to show the binding of ATF4 to these regions [160,161]. A bioinformatics approach led by Jiang et al. identified additional potential C/EBP-ATF binding sites in the LTR [8]. Caselli et al. reported that co-expression of Tat and ATF4 induced higher LTR transcription than the sole expression of Tat, suggesting a synergistic interaction between the two proteins [6]. ChIP experiments showed that ATF4 binds to the LTR sequence. This interaction is positively regulated by Forkhead box O 1 (FOXO1) inhibition and amino acids deprival [8,9]. Further works suggest that Tat may be unnecessary for ATF4-mediated HIV-1 promoter activation [6,7]. However, this effect has only been observed in HeLa cells, but not in Jurkat and 293T cells [6].
ATF4 can also bind to sequences that differ from the C/EBP-ATF consensus sequence, depending on its dimerization partners (Figure 3) [1,162,163]. ATF4 can dimerize with Jun and Fos [164] that heterodimerize to form the AP-1 transcription factor [164]. The fact that the HIV-1 promoter presents an AP-1 binding site could suggest that a heterodimer of ATF4 with Jun or Fos can be formed to activate the LTR activity. However, the partners able to dimerize with ATF4 to control HIV-1 transcription are still unknown.
Five to twenty percent of HIV-infected patients are co-infected by the Hepatitis B virus (HBV). The X protein of HBV has been shown to regulate HIV-1 transcription by stimulating the binding of ATF4 to the LTR of HIV-1 [165]. ATF4 can also directly interact with the HTLV-1 transcriptional activator Tax protein and act as an adapter between Tax and the HTLV-1 promoter [159,166]. Therefore, co-infections may provide a permissive environment for ATF4 to activate the HIV-1 promoter.
3.2.2. ATF4, HIV and apoptosis
It is well known that HIV pathogenicity is associated with the depletion of CD4+ T cells leading to the development of AIDS. In this context, it has been shown that a form of programmed cell death, namely apoptosis, may contribute to the depletion of CD4+ T cells and be associated with pathogenicity [66,167,168]. In vitro and in vivo, HIV and SIV infections mediate T cell death [68,169] through the intrinsic apoptotic pathway that is characterized by the expression of the pro-apoptotic proteins Bax, Bak and Bim [170], leading to the release of apoptogenic mitochondrial factors and consequently to caspase activation. CD4+ T cell apoptosis also involves the extrinsic pathway in which Fas (CD95) is critical [171,172,173,174]. Thus, caspase inhibition prevents in vivo the depletion of CD4+ T cells and delays the progression to AIDS [67].
It has been shown that Tat up-regulates UPR mediators with an increase in the transcript levels of ATF4, CHOP and the pro-apoptotic BH3-only BIM (BCL2L11), thus leading to apoptosis [11]. Of interest, ATF4 mediates cell death through the activation of the bZIP transcription factor CHOP [3], which also induces the transcription of BIM and of the pro-apoptotic BH3-only PUMA (also known as BBC3) [175,176]. Additionally, the CHOP-ATF4 dimer up-regulates ATF5, NOXA (also known as PMAIP1), APAF-1, and TXNIP, which in turn amplify cell death [177,178,179]. ATF4 also promotes degradation of the caspase inhibitor X chromosome-linked inhibitor of apoptosis (XIAP) protein through the ubiquitin-proteasome system, which allows the activation of caspases [180]. Neill and Masson recently reviewed ATF4 target genes that may take part in various potent routes through which ATF4 may promote apoptosis [162] (Table 3). In a model of HIV latency, inducing ISR/ATF4 by a prolonged pharmacological treatment, which is significantly associated with cell death [10], increases ATF4 and CHOP protein levels. Thus, several apoptotic genes have been found induced by ATF4 in CD4+ T cells during HIV and SIV infections. However, the role of ATF4 in regulating the death of CD4+ T cells in the context of HIV-infection has been poorly explored.
Other genes have been identified [162] that could contribute to ATF4-mediated death of CD4+ T cells such as Tumorous imaginal disc 1 (TID1), Transmembrane Bax inhibitor-1 motif-containing protein 5 (TMBIM5), G0/G1 switch gene 2 (G0S2) and p53-binding protein 2 (TP53BP2).
TID1, also known as DnaJ homolog subfamily A member 3 (DNAJA3), which belongs to the DnaJ family of proteins is involved both in apoptosis and autophagy. It has been proposed that TID1 helps the oncosuppressor protein p53 to translocate to the mitochondria under hypoxic conditions, thus leading to a transcription-independent mitochondrial apoptotic pathway [205]. Given the role of p53-mediated death in productive HIV-infected cells [206,207], TID1 regulation of p53 merits to be further explored. Interestingly, TID1 transcript levels are increased in T cells following HIV-1 infection, and a recent study shows that TID1 positively regulates HIV-1 replication [186,208].
TP53BP2, also known as apoptosis-stimulating of p53 protein 2 (ASPP2), can interact with p53, thereby enhancing its DNA binding and transactivation functions on the promoters of pro-apoptotic genes [209]. ATF4 regulates the TP53BP2 gene in liver ischemia-reperfusion (I/R) injury and contributes to hepatocyte apoptosis [210]. In cells exposed to the HIV glycoprotein gp120, the role of TP53BP2 would depend on stress intensity as exemplified by the use of varying doses of gp120 and could control both autophagy and apoptosis [203,211].
Another interesting but poorly described target of ATF4 is the TMBIM5 protein, also known as Growth hormone inducible transmembrane protein (GHITM, or MICS1). TMBIM5 localizes at the mitochondrial inner-membrane and is essential to maintain both mitochondrial structure and function [212,213]. This protein seems to ensure cell survival by allowing Ca2+ efflux from mitochondria and limiting mitochondrial hyperpolarization [214,215,216]. TMBIM5-knockout cells are more sensitive to apoptosis elicited by staurosporine or BH3 mimetic inhibitors of Bcl-2 and Bcl-xL [212,213]. Interestingly, global gene expression data show that TMBIM5 transcript levels are down-regulated in infected monocytes and increased in astrocytes of HIV HAND patients [201,202]. Given the limited data available, further investigations are needed to determine whether TMBIM5 plays a role in the induction of apoptosis downstream of ATF4 in response to HIV-1 infection.
Lastly, the transcription of G0/G1 switch gene 2 (G0S2) that regulate lipid production [217] has also been found to be a target of ATF4. Initially identified in cultured mononuclear cells in response to drug-induced cell cycle transition from the G0 to G1 phase [218,219,220], G0S2 displays opposite roles toward apoptosis. It prevents cells from ATP depletion, induces a cellular tolerance for hypoxic stress [221], and acts as a survival molecule in endothelial cells, protecting them from serum starvation- and hydrogen peroxide (H2O2)-induced apoptosis [222]. Interestingly, G0S2 is down-regulated in macrophages but induced in dendritic cells upon HIV-1 infection [187,188]. The biological significance of these observations has yet to be established.
Altogether, the identified ATF4 targets represent multiple new mechanisms to be explored by which ATF4 could regulate cell death or survival of HIV-infected cells.
3.2.3. ATF4, HIV and autophagy
Autophagy is a pro-survival intracellular catabolic process that targets protein aggregates and damaged organelles in response to stress. ATF4 activates the transcription of genes involved in the initiation (BECN1), in the elongation of the phagophore and the maturation of the autophagosome (WIPI1, ATG12, ATG5, ATG10, ATG16L1, ATG3, ATG7, MAP1lc3B, GABARAPL2), and in the selective clearance of cargo (p62/SQSTM1 and NBR1) [162,223]. Interestingly, ATF4 triggers the transcriptional activation of the autophagy genes directly as a homo or heterodimer with CHOP or indirectly through CHOP induction (Figure 3) [223]. The ratio between ATF4 and CHOP is decisive for the regulation of these genes, underpinning a fine-tuned control of the different stages of autophagy, the subtleties of which remain to be elucidated. ATF4 can also act upstream of autophagy to induce this process; ATF4 can increase the expression of the gene encoding Regulated in development and DNA damage response 1 (REDD1; also known as Ddit4), which suppresses mTOR complex 1 (mTORC1) activity, allowing autophagy induction upon ER stress and starvation [224,225,226].
HIV-1 modulates autophagy in a cell-type-dependent manner [227,229]. The Env protein found at the cell surface of infected cells induces an autophagy-dependent cell death of bystander CD4+ T cells [231]. Thus, ARF6, which promotes autophagosome biogenesis by facilitating endocytic uptake of the plasma membrane into autophagosome precursors, has been proposed to promote the fusion of HIV-1 envelope with the plasma membrane, therefore permitting the entry of HIV-1 in CD4+ T lymphocytes [232,233]. Other groups have reported that HIV inhibits autophagy [234,235]. After virus entry, the Vpr protein decreases the amount of proteins like MAP1LC3 and BECN1 [236]. Furthermore, although autophagy is initiated in primary CD4+ T cells infected with HIV, this process aborts due to a lysosome destabilization by DRAM, a p53-inducible gene. This permeabilization of lysosomes and resulting cell death has been proposed has an altruistic self-defense mechanism limiting viral dissemination by the elimination of infected cells [207]. Despite the obvious link between ATF4 and autophagy, the role of ATF4 in autophagy in the context of HIV infection remains poorly documented.
3.2.4. Immune response and ATF4 activation during HIV infection
ATF4, innate immunity and HIV
Pattern recognition receptors (PRRs) are fundamental molecules for innate immune response recognizing pathogen-associated molecular patterns (PAMPs). PRRs bind to PAMPs and trigger cell signaling cascades. PRRs include Toll-like receptors (TLRs), Nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs), RIG-I-like receptors (RLRs) and C-type lectin receptors (CLRs). For example, TLR7 and TLR8 can recognize single-stranded RNAs, which are present in the HIV genome [237], leading to Interferon regulatory factor 3 and 7 (IRF3 and IRF7) activations and inducing robust expression of type I interferon (IFN) genes [238]. ATF4 inhibits IRF7 transcription but also its activation by inhibiting TANK-binding kinase 1 (TBK1) and IκB kinase (IKK)ε-mediated IRF7 phosphorylation [58]. To be activated, IRF7 also requires its ubiquitination by Tumor necrosis factor receptor-associated factor 6 (TRAF6) (Figure 3). GADD34 that binds to and inhibits TRAF6 activity is an ATF4 target [239]. Its transcript levels have been shown to be increased in HIV-infected cells in a p53-dependent manner [240]. Inhibition of TRAF6 enhanced HIV-1 replication in macrophages [241] whereas GADD34 reduced viral protein expression [242]. Therefore, the role of ATF4 and its targets merits to be further explored in innate immunity.
HIV-1 also up-regulates TLR2 levels and signaling in vivo and in vitro [243]. This is of particular significance since HIV-1 provirus integration is increased in TLR2-bearing cells compared to cells that do not express TLR2 [244]. Although a role of ATF4 in this regulation of TLR2 has not been so far demonstrated, ER and oxidative stresses up-regulate TLR2 mRNA levels in an ATF4-dependent manner [56,245]. A ChIP assay revealed a binding site for ATF4 in an intronic region of the TLR2 gene [56]. Therefore, it can be hypothesized that ER stress-induced by HIV infections could activate ATF4 and regulate TLR2 gene expression in infected cells. ATF4 also mediates TLR4-triggered cytokine production [246], and it has been shown that the level of TLR4 is increased upon HIV-1 infection [243]. Given the role of ATF4 in the regulation of TLRs, and that microbial antigens may activate innate cells, ATF4 activation could explain the inflammation that characterizes people living with HIV and therefore merits further clarification.
ATF4, cellular immunity and HIV
Viruses have developed a diverse array of strategies to manipulate host cell metabolism and reorientate metabolic resources toward their benefit [118]. Current knowledge on T-cell metabolism in HIV infection suggests that HIV-1 takes advantage of the glycolytic process in CD4+ T cells to infect them and to boost viral replication [247,248,249]. It has been shown that IL-7-mediated thymocytes survival through the increase of Glut1 protein level at the cell surface, leading to glucose uptake and favoring viral infection [250]. Interestingly, ATF4 can influence a network of genes responsible for metabolic flux when activated by an extracellular oxidizing environment [251]. Thus, ATF4 up-regulates genes involved in glycolysis and promotes the anaplerotic flux through enhanced glutaminolysis, which plays a role in T cell growth in oxygen- or amino acid-deprived environments [251]. Glutaminolysis fuels the tricarboxylic acid (TCA) cycle and oxidative phosphorylation (OXPHOS) in T-cell receptor-stimulated naïve CD4 subsets, as well as memory CD4 subsets [252]. This balance in the bioenergetics pathways is essential for T cell polarization [253]. ATF4 has also been implicated in regulating the differentiation of naïve T cells in T helper 17 cells (Th17). T cells from ATF4-deficient mice are characterized by a diminished Th1 differentiation and an elevation in factors promoting Th17 commutation, such as Interleukin-17 (IL-17) [251]. This association is further supported by the effect of the small molecule halofuginone (HF), which inhibits Th17 differentiation in both mice and humans and increasesATF4 protein levels [254]. Notably, adding amino acids in excess counteracts the inhibition of Th17 differentiation by HF, suggesting the involvement of the amino acid starvation response mediated by ATF4 in targeting Th17 differentiation. Interestingly, Th17 cells are highly vulnerable to HIV-1 infection [255] and this population represents a major viral reservoir under ART [256]. The loss of Th17 homeostasis contributes to disease progression in SIV-infected rhesus macaques associated with the loss of intestinal epithelial integrity, leading to microbial translocation [257,258,259,260]. This alteration in the balance of T cell subsets persists despite ART [261]. Given the role of ATF4 in regulating mitochondrial and reticulum stress-associated apoptosis, a contribution of ATF4 dysregulation in the alteration of the Th17 balance at the mucosal barrier cannot be excluded. Taken together, these results suggest that the induction of ATF4 observed in CD4+ T lymphocytes during HIV-1 infection [10] may contribute to the increased production of energy and amino acids required for viral replication.
4. ATF5 the paralog of ATF4
ATF5 is a member of the bZIP transcription factor family, like CREB, Fos and NRF2 [262]. Based on its bZIP domain sequence, ATF5 has been classified into the ATF4 subgroup family (Figure 4). ATF5 also shares many functional features with ATF4, especially in stress cellular responses [263,264]. Interest in ATF5 has risen sharply in recent years, and its role in regulating cellular stress, cell survival, immunity, and cancer has recently been reviewed [265,266]. ATF5 is also involved in cell proliferation and differentiation of various cell types [262]. Thus, a growing number of studies implicate ATF4 and ATF5 in the regulation of the immune response and reveal how the dialogue between stress responses and immune pathways can be fundamental for the regulation of tissue homeostasis by the cell in response to infection [267]. Of particular interest, how ATF4/ATF5 and NF-κB coordinate their activity and their effects on viral replication, latency, and cell survival need further investigation.
Data concerning the involvement of ATF5 in the viral stress response and its possible role during infections remain sparse. During Epstein-Barr virus (EBV) infection, the viral protein Latent membrane protein 1 (LMP-1) positively regulates the ATF5 protein level via the NF-κB pathway [268]. In this context, ATF5 transcriptionally inhibits the Signaling lymphocyte activation molecule-associated protein (SAP) gene, promoting a Th1-like cytokine profile in T lymphocytes. Intriguingly, LMP-1 not only activates ATF5 but also positively controls PERK to induce phosphorylation of eIF2α, which up-regulates ATF4 expression in B cells [44]. ATF4, in turn, transactivates LMP-1’s promoter in a positive feedback loop to enhance LMP-1 protein levels and activity. Thus, LMP-1 activates both ATF4 and ATF5, which seem to have different functions in the viral cycle.
ATF5 has also been shown to play a role in the replication of Herpes simplex virus type 1 (HSV-1). It promotes the replication of the virus by activating the transcription of viral genes, but this activity is counteracted by the cellular HSV-1 infection response repressive protein (HIRRP), through physical interaction [269]. Interestingly, an elevation in the eIF2α/ATF4 signaling is observed towards the end of HSV-1 replication, which leads to the completion of virion production and release [51]. Therefore, as in EBV infection, ATF4 and ATF5 act at different steps of the HSV-1 infection.
In glioma cells that are highly permissive to Human cytomegalovirus (HCMV) infection, the viral immediate-early protein of 86-kDa (IE86) interacts with and acetylates ATF5, thereby promoting cell survival [270]. Furthermore, the HCMV protein pUL38 is required to maintain the viability of infected fibroblasts by increasing phosphorylation of both PERK and eIF-2α and inducing robust accumulation of ATF4 protein [46]. Thus, both ATF4 and ATF5 contribute to the survival of infected cells.
Microarray and RNAseq analyses have shown that the levels of ATF5 transcripts increase in cells infected with viruses such as ZIKV or dengue [201]. ZIKV infection also increases ATF4 transcript and protein levels [39,40,271]. In the context of HIV-1 infection, ATF5 transcript levels are up-regulated in Th1Th17 vs Th1 cells [272]. However, the roles of ATF4 and ATF5 are still poorly understood in these models. ATF5, like ATF4, can be activated by various stresses and involved in the ISR and UPRmt pathways. Therefore, whether ATF5 is activated in response to HIV-1 infection and regulated by HIV proteins have to be determined.
5. Conclusions
In this review, we provide an overview of ATF4 functions during HIV-1 infection and explore the role of some of the poorly characterized ATF4 transcriptional targets that are involved in cell death, autophagy, metabolism or in the immune response. This review thus proposes some molecular mechanisms that should be investigated for understanding the role of ATF4 in the context of HIV-1 but also by other viruses. Although most of the research on ATF4 is generally associated with the demonstration of protein and transcript levels modulation, it is also of interest to determine proteins relocation and ATF4 binding partners at promoter levels. It is also becoming well-established that post-translational modifications regulate ATF4 activity. This last point is less understood, and advances in our knowledge of the impact of post-translational modifications, particularly on the choice of ATF4 heterodimerization partners, should be decisive in understanding the control of ATF4 activity in the context of HIV infection. Given its similarity with ATF5, it is crucial to decipher the roles of ATF5, depending on cell types and its fine regulations. Understanding the role of theses transcriptional factors may pave the way to identifying new drug therapies.
Author Contributions
JMC was responsible for the ideation. AC, FA, JE and JMC performed the literature search. AC, JE, SG and JMC drafted the work. AC, FA, JE, and SG revised the work. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by grants from the Canadian Institutes of Health Research (CIHR) (HBF-123682, HBF-126786, and MOP-133476), and by the Canadian HIV Cure Enterprise Grant HIG-133050 from the CIHR partnership with CANFAR and IAS to JE. JE was also supported by a grant from ANRS-MIE. JE thanks the Canada Research Chair program for financial assistance.
Data Availability Statement
Data sharing not applicable.
Acknowledgments
The authors thank Bernard Mignotte for his careful reading and relevant comments that helped improve this article.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
References
- Ameri, K.; Harris, A.L. Activating Transcription Factor 4. Int J Biochem Cell Biol 2008, 40, 14–21. [Google Scholar] [CrossRef]
- Kasai, S.; Yamazaki, H.; Tanji, K.; Engler, M.J.; Matsumiya, T.; Itoh, K. Role of the ISR-ATF4 Pathway and Its Cross Talk with Nrf2 in Mitochondrial Quality Control. J Clin Biochem Nutr 2019, 64, 1–12. [Google Scholar] [CrossRef]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The Integrated Stress Response. EMBO Rep 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed]
- Angelastro, J.M. Targeting ATF5 in Cancer. Trends Cancer 2017, 3, 471–474. [Google Scholar] [CrossRef]
- Greene, L.A.; Lee, H.Y.; Angelastro, J.M. The Transcription Factor ATF5: Role in Neurodevelopment and Neural Tumors. J Neurochem 2009, 108, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Benedetti, S.; Gentili, V.; Grigolato, J.; Di Luca, D. Short Communication: Activating Transcription Factor 4 (ATF4) Promotes HIV Type 1 Activation. AIDS Res Hum Retroviruses 2012, 28, 907–912. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-D.; Yu, K.-L.; Park, S.-H.; Jung, Y.-M.; Kim, M.-J.; You, J.-C. Understanding of the Functional Role(s) of the Activating Transcription Factor 4(ATF4) in HIV Regulation and Production. BMB Rep 2018, 51, 388–393. [Google Scholar] [CrossRef]
- Jiang, G.; Santos Rocha, C.; Hirao, L.A.; Mendes, E.A.; Tang, Y.; Thompson, G.R.; Wong, J.K.; Dandekar, S. HIV Exploits Antiviral Host Innate GCN2-ATF4 Signaling for Establishing Viral Replication Early in Infection. mBio 2017, 8, e01518–16. [Google Scholar] [CrossRef]
- Vallejo-Gracia, A.; Chen, I.P.; Perrone, R.; Besnard, E.; Boehm, D.; Battivelli, E.; Tezil, T.; Krey, K.; Raymond, K.A.; Hull, P.A.; et al. FOXO1 Promotes HIV Latency by Suppressing ER Stress in T Cells. Nat Microbiol 2020, 5, 1144–1157. [Google Scholar] [CrossRef]
- Li, D.; Wong, L.M.; Tang, Y.; Allard, B.; James, K.S.; Thompson, G.R.; Dandekar, S.; Browne, E.P.; Li, Q.; Simon, J.M.; et al. Depletion of HIV Reservoir by Activation of ISR Signaling in Resting CD4+T Cells. iScience 2023, 26, 105743. [Google Scholar] [CrossRef]
- Campestrini, J.; Silveira, D.B.; Pinto, A.R. HIV-1 Tat-Induced Bystander Apoptosis in Jurkat Cells Involves Unfolded Protein Responses. Cell Biochem Funct 2018, 36, 377–386. [Google Scholar] [CrossRef]
- Ma, R.; Yang, L.; Niu, F.; Buch, S. HIV Tat-Mediated Induction of Human Brain Microvascular Endothelial Cell Apoptosis Involves Endoplasmic Reticulum Stress and Mitochondrial Dysfunction. Mol Neurobiol 2016, 53, 132–142. [Google Scholar] [CrossRef]
- Granberg, F.; Svensson, C.; Pettersson, U.; Zhao, H. Adenovirus-Induced Alterations in Host Cell Gene Expression Prior to the Onset of Viral Gene Expression. Virology 2006, 353, 1–5. [Google Scholar] [CrossRef]
- Gao, P.; Chai, Y.; Song, J.; Liu, T.; Chen, P.; Zhou, L.; Ge, X.; Guo, X.; Han, J.; Yang, H. Reprogramming the Unfolded Protein Response for Replication by Porcine Reproductive and Respiratory Syndrome Virus. PLoS Pathog 2019, 15, e1008169. [Google Scholar] [CrossRef]
- Zhang, F.; Moon, A.; Childs, K.; Goodbourn, S.; Dixon, L.K. The African Swine Fever Virus DP71L Protein Recruits the Protein Phosphatase 1 Catalytic Subunit to Dephosphorylate eIF2alpha and Inhibits CHOP Induction but Is Dispensable for These Activities during Virus Infection. J Virol 2010, 84, 10681–10689. [Google Scholar] [CrossRef]
- Williams, B.L.; Lipkin, W.I. Endoplasmic Reticulum Stress and Neurodegeneration in Rats Neonatally Infected with Borna Disease Virus. J Virol 2006, 80, 8613–8626. [Google Scholar] [CrossRef]
- Tuñón, M.J.; San-Miguel, B.; Crespo, I.; Laliena, A.; Vallejo, D.; Álvarez, M.; Prieto, J.; González-Gallego, J. Melatonin Treatment Reduces Endoplasmic Reticulum Stress and Modulates the Unfolded Protein Response in Rabbits with Lethal Fulminant Hepatitis of Viral Origin. J Pineal Res 2013, 55, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Qi, B.; Gu, Y.; Xu, F.; Du, H.; Li, X.; Fang, W. Porcine Circovirus 2 Deploys PERK Pathway and GRP78 for Its Enhanced Replication in PK-15 Cells. Viruses 2016, 8, 56. [Google Scholar] [CrossRef]
- Zhou, Y.-S.; Gu, Y.-X.; Qi, B.-Z.; Zhang, Y.-K.; Li, X.-L.; Fang, W.-H. Porcine Circovirus Type 2 Capsid Protein Induces Unfolded Protein Response with Subsequent Activation of Apoptosis. J Zhejiang Univ Sci B 2017, 18, 316–323. [Google Scholar] [CrossRef]
- Lv, J.; Jiang, Y.; Feng, Q.; Fan, Z.; Sun, Y.; Xu, P.; Hou, Y.; Zhang, X.; Fan, Y.; Xu, X.; et al. Porcine Circovirus Type 2 ORF5 Protein Induces Autophagy to Promote Viral Replication via the PERK-eIF2α-ATF4 and mTOR-ERK1/2-AMPK Signaling Pathways in PK-15 Cells. Front Microbiol 2020, 11, 320. [Google Scholar] [CrossRef]
- Liao, Y.; Fung, T.S.; Huang, M.; Fang, S.G.; Zhong, Y.; Liu, D.X. Upregulation of CHOP/GADD153 during Coronavirus Infectious Bronchitis Virus Infection Modulates Apoptosis by Restricting Activation of the Extracellular Signal-Regulated Kinase Pathway. J Virol 2013, 87, 8124–8134. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Huang, C.; Shi, Y.; Li, N.; Wang, E.; Hu, R.; Li, G.; Yang, F.; Zhuang, Y.; Liu, P.; et al. Investigation of the Crosstalk between GRP78/PERK/ATF-4 Signaling Pathway and Renal Apoptosis Induced by Nephropathogenic Infectious Bronchitis Virus Infection. J Virol 2022, 96, e0142921. [Google Scholar] [CrossRef] [PubMed]
- Fang, P.; Tian, L.; Zhang, H.; Xia, S.; Ding, T.; Zhu, X.; Zhang, J.; Ren, J.; Fang, L.; Xiao, S. Induction and Modulation of the Unfolded Protein Response during Porcine Deltacoronavirus Infection. Vet Microbiol 2022, 271, 109494. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-M.; Gabler, N.K.; Burrough, E.R. Porcine Epidemic Diarrhea Virus Infection Induces Endoplasmic Reticulum Stress and Unfolded Protein Response in Jejunal Epithelial Cells of Weaned Pigs. Vet Pathol 2022, 59, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Christ, W.; Klingström, J.; Tynell, J. SARS-CoV-2 Variant-Specific Differences in Inhibiting the Effects of the PKR-Activated Integrated Stress Response. Virus Res 2024, 339, 199271. [Google Scholar] [CrossRef]
- Wang, J.; Chen, K.-Y.; Wang, S.-H.; Liu, Y.; Zhao, Y.-Q.; Yang, L.; Yang, G.-H.; Wang, X.-J.; Zhu, Y.-H.; Yin, J.-H.; et al. Effects of Spatial Expression of Activating Transcription Factor 4 on the Pathogenicity of Two Phenotypes of Bovine Viral Diarrhea Virus by Regulating the Endoplasmic Reticulum-Mediated Autophagy Process. Microbiol Spectr 2023, 11, e0422522. [Google Scholar] [CrossRef]
- Fraser, J.E.; Wang, C.; Chan, K.W.K.; Vasudevan, S.G.; Jans, D.A. Novel Dengue Virus Inhibitor 4-HPR Activates ATF4 Independent of Protein Kinase R-like Endoplasmic Reticulum Kinase and Elevates Levels of eIF2α Phosphorylation in Virus Infected Cells. Antiviral Res 2016, 130, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Diosa-Toro, M.; Troost, B.; van de Pol, D.; Heberle, A.M.; Urcuqui-Inchima, S.; Thedieck, K.; Smit, J.M. Tomatidine, a Novel Antiviral Compound towards Dengue Virus. Antiviral Res 2019, 161, 90–99. [Google Scholar] [CrossRef]
- Ciccaglione, A.R.; Marcantonio, C.; Tritarelli, E.; Equestre, M.; Vendittelli, F.; Costantino, A.; Geraci, A.; Rapicetta, M. Activation of the ER Stress Gene Gadd153 by Hepatitis C Virus Sensitizes Cells to Oxidant Injury. Virus Res 2007, 126, 128–138. [Google Scholar] [CrossRef]
- Merquiol, E.; Uzi, D.; Mueller, T.; Goldenberg, D.; Nahmias, Y.; Xavier, R.J.; Tirosh, B.; Shibolet, O. HCV Causes Chronic Endoplasmic Reticulum Stress Leading to Adaptation and Interference with the Unfolded Protein Response. PLoS One 2011, 6, e24660. [Google Scholar] [CrossRef]
- Wang, J.; Kang, R.; Huang, H.; Xi, X.; Wang, B.; Wang, J.; Zhao, Z. Hepatitis C Virus Core Protein Activates Autophagy through EIF2AK3 and ATF6 UPR Pathway-Mediated MAP1LC3B and ATG12 Expression. Autophagy 2014, 10, 766–784. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-C.; Zhang, M.-Q.; Zhang, J.-P. Opposite Effects of Two Human ATG10 Isoforms on Replication of a HCV Sub-Genomic Replicon Are Mediated via Regulating Autophagy Flux in Zebrafish. Front Cell Infect Microbiol 2018, 8, 109. [Google Scholar] [CrossRef] [PubMed]
- Ríos-Ocampo, W.A.; Navas, M.-C.; Buist-Homan, M.; Faber, K.N.; Daemen, T.; Moshage, H. Hepatitis C Virus Proteins Core and NS5A Are Highly Sensitive to Oxidative Stress-Induced Degradation after eIF2α/ATF4 Pathway Activation. Viruses 2020, 12, 425. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Xu, A.; Wu, X.; Zhang, Y.; Guo, Y.; Guo, F.; Pan, Z.; Kong, L. Japanese Encephalitis Virus Induces Apoptosis by the IRE1/JNK Pathway of ER Stress Response in BHK-21 Cells. Arch Virol 2016, 161, 699–703. [Google Scholar] [CrossRef] [PubMed]
- Carr, M.; Gonzalez, G.; Martinelli, A.; Wastika, C.E.; Ito, K.; Orba, Y.; Sasaki, M.; Hall, W.W.; Sawa, H. Upregulated Expression of the Antioxidant Sestrin 2 Identified by Transcriptomic Analysis of Japanese Encephalitis Virus-Infected SH-SY5Y Neuroblastoma Cells. Virus Genes 2019, 55, 630–642. [Google Scholar] [CrossRef]
- Wang, Q.; Xin, X.; Wang, T.; Wan, J.; Ou, Y.; Yang, Z.; Yu, Q.; Zhu, L.; Guo, Y.; Wu, Y.; et al. Japanese Encephalitis Virus Induces Apoptosis and Encephalitis by Activating the PERK Pathway. Journal of Virology 2019, 93, 10–1128. [Google Scholar] [CrossRef]
- Zhao, D.; Yang, J.; Han, K.; Liu, Q.; Wang, H.; Liu, Y.; Huang, X.; Zhang, L.; Li, Y. The Unfolded Protein Response Induced by Tembusu Virus Infection. BMC Vet Res 2019, 15, 34. [Google Scholar] [CrossRef] [PubMed]
- Basu, M.; Courtney, S.C.; Brinton, M.A. Arsenite-Induced Stress Granule Formation Is Inhibited by Elevated Levels of Reduced Glutathione in West Nile Virus-Infected Cells. PLoS Pathog 2017, 13, e1006240. [Google Scholar] [CrossRef]
- Tan, Z.; Zhang, W.; Sun, J.; Fu, Z.; Ke, X.; Zheng, C.; Zhang, Y.; Li, P.; Liu, Y.; Hu, Q.; et al. ZIKV Infection Activates the IRE1-XBP1 and ATF6 Pathways of Unfolded Protein Response in Neural Cells. J Neuroinflammation 2018, 15, 275. [Google Scholar] [CrossRef]
- Mufrrih, M.; Chen, B.; Chan, S.-W. Zika Virus Induces an Atypical Tripartite Unfolded Protein Response with Sustained Sensor and Transient Effector Activation and a Blunted BiP Response. mSphere 2021, 6, e0036121. [Google Scholar] [CrossRef]
- Cho, H.K.; Cheong, K.J.; Kim, H.Y.; Cheong, J. Endoplasmic Reticulum Stress Induced by Hepatitis B Virus X Protein Enhances Cyclo-Oxygenase 2 Expression via Activating Transcription Factor 4. Biochem J 2011, 435, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; He, J.; Fu, Y.; Hu, X.; Sun, L.-Q.; Huang, Y.; Fan, X. Hepatitis B Virus X Protein Inhibits Apoptosis by Modulating Endoplasmic Reticulum Stress Response. Oncotarget 2017, 8, 96027–96034. [Google Scholar] [CrossRef]
- Choi, Y.-M.; Kim, D.H.; Jang, J.; Choe, W.H.; Kim, B.-J. rt269L-Type Hepatitis B Virus (HBV) in Genotype C Infection Leads to Improved Mitochondrial Dynamics via the PERK-eIF2α-ATF4 Axis in an HBx Protein-Dependent Manner. Cell Mol Biol Lett 2023, 28, 26. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.Y.; Sugden, B. The LMP1 Oncogene of EBV Activates PERK and the Unfolded Protein Response to Drive Its Own Synthesis. Blood 2008, 111, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Isler, J.A.; Skalet, A.H.; Alwine, J.C. Human Cytomegalovirus Infection Activates and Regulates the Unfolded Protein Response. J Virol 2005, 79, 6890–6899. [Google Scholar] [CrossRef] [PubMed]
- Xuan, B.; Qian, Z.; Torigoi, E.; Yu, D. Human Cytomegalovirus Protein pUL38 Induces ATF4 Expression, Inhibits Persistent JNK Phosphorylation, and Suppresses Endoplasmic Reticulum Stress-Induced Cell Death. J Virol 2009, 83, 3463–3474. [Google Scholar] [CrossRef]
- Siddiquey, M.N.A.; Zhang, H.; Nguyen, C.C.; Domma, A.J.; Kamil, J.P. The Human Cytomegalovirus Endoplasmic Reticulum-Resident Glycoprotein UL148 Activates the Unfolded Protein Response. J Virol 2018, 92, e00896–18. [Google Scholar] [CrossRef] [PubMed]
- Sharon, E.; Frenkel, N. Human Herpesvirus 6A Exhibits Restrictive Propagation with Limited Activation of the Protein Kinase R-eIF2α Stress Pathway. J Virol 2017, 91, e02120–16. [Google Scholar] [CrossRef] [PubMed]
- Caselli, E.; Benedetti, S.; Grigolato, J.; Caruso, A.; Di Luca, D. Activating Transcription Factor 4 (ATF4) Is Upregulated by Human Herpesvirus 8 Infection, Increases Virus Replication and Promotes Proangiogenic Properties. Arch Virol 2012, 157, 63–74. [Google Scholar] [CrossRef]
- Sun, Q.; Wang, F.; Chen, Q.; Sarid, R.; Li, X.; Kuang, E. Upregulation of ATF4-LAMP3 Axis by ORF45 Facilitates Lytic Replication of Kaposi’s Sarcoma-Associated Herpesvirus. J Virol 2022, 96, e0145622. [Google Scholar] [CrossRef]
- Burnett, H.F.; Audas, T.E.; Liang, G.; Lu, R.R. Herpes Simplex Virus-1 Disarms the Unfolded Protein Response in the Early Stages of Infection. Cell Stress Chaperones 2012, 17, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Qian, Z.; Xuan, B.; Chapa, T.J.; Gualberto, N.; Yu, D. Murine Cytomegalovirus Targets Transcription Factor ATF4 to Exploit the Unfolded-Protein Response. J Virol 2012, 86, 6712–6723. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.-C.; Dong, S.-H.; Liu, Z.-S.; Liu, S.; Zhang, C.-C.; Liang, X.-Z. Regulation of Gammaherpesvirus Lytic Replication by Endoplasmic Reticulum Stress-Induced Transcription Factors ATF4 and CHOP. J Biol Chem 2018, 293, 2801–2814. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhu, J.; Zhou, X.; Wang, H.; Li, X.; Zhao, A. Induction of the Unfolded Protein Response (UPR) during Pseudorabies Virus Infection. Vet Microbiol 2019, 239, 108485. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Ni, M.; Ahmed, W.; Xu, Y.; Bao, X.; Zhuang, T.; Feng, L.; Guo, M. Pseudorabies Virus Infection Induces Endoplasmic Reticulum Stress and Unfolded Protein Response in Suspension-Cultured BHK-21 Cells. J Gen Virol 2022, 103. [Google Scholar] [CrossRef]
- Liao, Y.; Gu, F.; Mao, X.; Niu, Q.; Wang, H.; Sun, Y.; Song, C.; Qiu, X.; Tan, L.; Ding, C. Regulation of de Novo Translation of Host Cells by Manipulation of PERK/PKR and GADD34-PP1 Activity during Newcastle Disease Virus Infection. J Gen Virol 2016, 97, 867–879. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, R.; Li, Y.; Sun, Y.; Song, C.; Zhan, Y.; Tan, L.; Liao, Y.; Meng, C.; Qiu, X.; et al. Newcastle Disease Virus Induces G0/G1 Cell Cycle Arrest in Asynchronously Growing Cells. Virology 2018, 520, 67–74. [Google Scholar] [CrossRef]
- Liang, Q.; Deng, H.; Sun, C.-W.; Townes, T.M.; Zhu, F. Negative Regulation of IRF7 Activation by Activating Transcription Factor 4 Suggests a Cross-Regulation between the IFN Responses and the Cellular Integrated Stress Responses. J Immunol 2011, 186, 1001–1010. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Xue, M.; Chen, J.; Shi, H.; Zhang, X.; Shi, D.; Liu, J.; Huang, L.; Wei, Y.; Liu, C.; et al. Porcine Parvovirus Replication Is Suppressed by Activation of the PERK Signaling Pathway and Endoplasmic Reticulum Stress-Mediated Apoptosis. Virology 2020, 539, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Zhang, S.; Qin, X.; Chang, X.; Cui, X.; Li, H.; Zhang, S.; Gao, H.; Wang, P.; Zhang, Z.; et al. Foot-and-Mouth Disease Virus Capsid Protein VP2 Activates the Cellular EIF2S1-ATF4 Pathway and Induces Autophagy via HSPB1. Autophagy 2018, 14, 336–346. [Google Scholar] [CrossRef]
- Colli, M.L.; Paula, F.M.; Marselli, L.; Marchetti, P.; Roivainen, M.; Eizirik, D.L.; Op de Beeck, A. Coxsackievirus B Tailors the Unfolded Protein Response to Favour Viral Amplification in Pancreatic β Cells. J Innate Immun 2019, 11, 375–390. [Google Scholar] [CrossRef]
- Dunlap, K.M.; Bartee, M.Y.; Bartee, E. Myxoma Virus Attenuates Expression of Activating Transcription Factor 4 (ATF4) Which Has Implications for the Treatment of Proteasome Inhibitor-Resistant Multiple Myeloma. Oncolytic Virother 2015, 4, 1–11. [Google Scholar] [CrossRef]
- Smith, J.A.; Schmechel, S.C.; Raghavan, A.; Abelson, M.; Reilly, C.; Katze, M.G.; Kaufman, R.J.; Bohjanen, P.R.; Schiff, L.A. Reovirus Induces and Benefits from an Integrated Cellular Stress Response. J Virol 2006, 80, 2019–2033. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Major, L.D.; Scholte, F.E.M.; Gardner, J.; van Hemert, M.J.; Suhrbier, A.; Pijlman, G.P. Chikungunya Virus Non-Structural Protein 2-Mediated Host Shut-off Disables the Unfolded Protein Response. J Gen Virol 2015, 96, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Baer, A.; Lundberg, L.; Swales, D.; Waybright, N.; Pinkham, C.; Dinman, J.D.; Jacobs, J.L.; Kehn-Hall, K. Venezuelan Equine Encephalitis Virus Induces Apoptosis through the Unfolded Protein Response Activation of EGR1. J Virol 2016, 90, 3558–3572. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Idziorek, T.; de Bels, F.; Barré-Sinoussi, F.; Hurtrel, B.; Aubertin, A.M.; Venet, A.; Mehtali, M.; Muchmore, E.; Michel, P.; et al. Programmed Cell Death and AIDS: Significance of T-Cell Apoptosis in Pathogenic and Nonpathogenic Primate Lentiviral Infections. Proc Natl Acad Sci U S A 1994, 91, 9431–9435. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Silvestre, R.; Rodrigues, V.; Garibal, J.; Campillo-Gimenez, L.; Mouhamad, S.; Monceaux, V.; Cumont, M.-C.; Rabezanahary, H.; Pruvost, A.; et al. The Anti-Caspase Inhibitor Q-VD-OPH Prevents AIDS Disease Progression in SIV-Infected Rhesus Macaques. J Clin Invest 2018, 128, 1627–1640. [Google Scholar] [CrossRef]
- Monceaux, V.; Estaquier, J.; Février, M.; Cumont, M.-C.; Rivière, Y.; Aubertin, A.-M.; Ameisen, J.C.; Hurtrel, B. Extensive Apoptosis in Lymphoid Organs during Primary SIV Infection Predicts Rapid Progression towards AIDS. AIDS 2003, 17, 1585–1596. [Google Scholar] [CrossRef] [PubMed]
- Cumont, M.-C.; Diop, O.; Vaslin, B.; Elbim, C.; Viollet, L.; Monceaux, V.; Lay, S.; Silvestri, G.; Le Grand, R.; Müller-Trutwin, M.; et al. Early Divergence in Lymphoid Tissue Apoptosis between Pathogenic and Nonpathogenic Simian Immunodeficiency Virus Infections of Nonhuman Primates. J Virol 2008, 82, 1175–1184. [Google Scholar] [CrossRef]
- Viollet, L.; Monceaux, V.; Petit, F.; Ho Tsong Fang, R.; Cumont, M.-C.; Hurtrel, B.; Estaquier, J. Death of CD4+ T Cells from Lymph Nodes during Primary SIVmac251 Infection Predicts the Rate of AIDS Progression. J Immunol 2006, 177, 6685–6694. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.-N.; Kavianpour, S.; Zhang, T.; Zhang, X.; Nguyen, D.; Thombre, R.; He, L.; Wang, J. MARK2 Phosphorylates eIF2α in Response to Proteotoxic Stress. PLoS Biol 2021, 19, e3001096. [Google Scholar] [CrossRef]
- Wu, Z.; Mei, F.; Gan, Y.; Liu, A.; Hu, J.; Jin, Y.; Yin, Y. FAM69C Functions as a Kinase for eIF2α and Promotes Stress Granule Assembly. EMBO Rep 2023, 24, e55641. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, M.; Cheng, A.; Yang, Q.; Wu, Y.; Jia, R.; Liu, M.; Zhu, D.; Chen, S.; Zhang, S.; et al. The Role of Host eIF2α in Viral Infection. Virol J 2020, 17, 112. [Google Scholar] [CrossRef]
- Galabru, J.; Hovanessian, A. Autophosphorylation of the Protein Kinase Dependent on Double-Stranded RNA. J Biol Chem 1987, 262, 15538–15544. [Google Scholar] [CrossRef]
- García, M.A.; Meurs, E.F.; Esteban, M. The dsRNA Protein Kinase PKR: Virus and Cell Control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef]
- Carpick, B.W.; Graziano, V.; Schneider, D.; Maitra, R.K.; Lee, X.; Williams, B.R. Characterization of the Solution Complex between the Interferon-Induced, Double-Stranded RNA-Activated Protein Kinase and HIV-I Trans-Activating Region RNA. J Biol Chem 1997, 272, 9510–9516. [Google Scholar] [CrossRef]
- Kim, I.; Liu, C.W.; Puglisi, J.D. Specific Recognition of HIV TAR RNA by the dsRNA Binding Domains (dsRBD1-dsRBD2) of PKR. J Mol Biol 2006, 358, 430–442. [Google Scholar] [CrossRef] [PubMed]
- Maitra, R.K.; McMillan, N.A.; Desai, S.; McSwiggen, J.; Hovanessian, A.G.; Sen, G.; Williams, B.R.; Silverman, R.H. HIV-1 TAR RNA Has an Intrinsic Ability to Activate Interferon-Inducible Enzymes. Virology 1994, 204, 823–827. [Google Scholar] [CrossRef] [PubMed]
- Spanggord, R.J.; Vuyisich, M.; Beal, P.A. Identification of Binding Sites for Both dsRBMs of PKR on Kinase-Activating and Kinase-Inhibiting RNA Ligands. Biochemistry 2002, 41, 4511–4520. [Google Scholar] [CrossRef]
- Clerzius, G.; Gélinas, J.-F.; Gatignol, A. Multiple Levels of PKR Inhibition during HIV-1 Replication. Rev Med Virol 2011, 21, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.-S.; Yoon, C.-H.; Kim, Y.-S.; Bae, Y.-S. The Double-Strand RNA-Dependent Protein Kinase PKR Plays a Significant Role in a Sustained ER Stress-Induced Apoptosis. FEBS Lett 2007, 581, 4325–4332. [Google Scholar] [CrossRef]
- Benkirane, M.; Neuveut, C.; Chun, R.F.; Smith, S.M.; Samuel, C.E.; Gatignol, A.; Jeang, K.T. Oncogenic Potential of TAR RNA Binding Protein TRBP and Its Regulatory Interaction with RNA-Dependent Protein Kinase PKR. EMBO J 1997, 16, 611–624. [Google Scholar] [CrossRef]
- Blair, E.D.; Roberts, C.M.; Snowden, B.W.; Gatignol, A.; Benkirane, M.; Jeang, K.-T. Expression of TAR RNA-Binding Protein in Baculovirus and Co-Immunoprecipitation with Insect Cell Protein Kinase. J Biomed Sci 1995, 2, 322–329. [Google Scholar] [CrossRef]
- Clerzius, G.; Gélinas, J.-F.; Daher, A.; Bonnet, M.; Meurs, E.F.; Gatignol, A. ADAR1 Interacts with PKR during Human Immunodeficiency Virus Infection of Lymphocytes and Contributes to Viral Replication. J Virol 2009, 83, 10119–10128. [Google Scholar] [CrossRef]
- Cole, J.L. Activation of PKR: An Open and Shut Case? Trends Biochem Sci 2007, 32, 57–62. [Google Scholar] [CrossRef]
- Daher, A.; Longuet, M.; Dorin, D.; Bois, F.; Segeral, E.; Bannwarth, S.; Battisti, P.L.; Purcell, D.F.; Benarous, R.; Vaquero, C.; et al. Two Dimerization Domains in the Trans-Activation Response RNA-Binding Protein (TRBP) Individually Reverse the Protein Kinase R Inhibition of HIV-1 Long Terminal Repeat Expression. J Biol Chem 2001, 276, 33899–33905. [Google Scholar] [CrossRef]
- Duarte, M.; Graham, K.; Daher, A.; Battisti, P.L.; Bannwarth, S.; Segeral, E.; Jeang, K.T.; Gatignol, A. Characterization of TRBP1 and TRBP2. Stable Stem-Loop Structure at the 5’ End of TRBP2 mRNA Resembles HIV-1 TAR and Is Not Found in Its Processed Pseudogene. J Biomed Sci 2000, 7, 494–506. [Google Scholar] [CrossRef]
- Lemaire, P.A.; Anderson, E.; Lary, J.; Cole, J.L. Mechanism of PKR Activation by dsRNA. J Mol Biol 2008, 381, 351–360. [Google Scholar] [CrossRef]
- Brand, S.R.; Kobayashi, R.; Mathews, M.B. The Tat Protein of Human Immunodeficiency Virus Type 1 Is a Substrate and Inhibitor of the Interferon-Induced, Virally Activated Protein Kinase, PKR. J Biol Chem 1997, 272, 8388–8395. [Google Scholar] [CrossRef]
- Cai, R.; Carpick, B.; Chun, R.F.; Jeang, K.T.; Williams, B.R. HIV-I TAT Inhibits PKR Activity by Both RNA-Dependent and RNA-Independent Mechanisms. Arch Biochem Biophys 2000, 373, 361–367. [Google Scholar] [CrossRef]
- McMillan, N.A.; Chun, R.F.; Siderovski, D.P.; Galabru, J.; Toone, W.M.; Samuel, C.E.; Mak, T.W.; Hovanessian, A.G.; Jeang, K.T.; Williams, B.R. HIV-1 Tat Directly Interacts with the Interferon-Induced, Double-Stranded RNA-Dependent Kinase, PKR. Virology 1995, 213, 413–424. [Google Scholar] [CrossRef]
- Mehrbod, P.; Ande, S.R.; Alizadeh, J.; Rahimizadeh, S.; Shariati, A.; Malek, H.; Hashemi, M.; Glover, K.K.M.; Sher, A.A.; Coombs, K.M.; et al. The Roles of Apoptosis, Autophagy and Unfolded Protein Response in Arbovirus, Influenza Virus, and HIV Infections. Virulence 2019, 10, 376–413. [Google Scholar] [CrossRef]
- Tripathi, A.; Iyer, K.; Mitra, D. HIV-1 Replication Requires Optimal Activation of the Unfolded Protein Response. FEBS Lett 2023, 597, 2908–2930. [Google Scholar] [CrossRef]
- Hortin, G.L.; Landt, M.; Powderly, W.G. Changes in Plasma Amino Acid Concentrations in Response to HIV-1 Infection. Clin Chem 1994, 40, 785–789. [Google Scholar] [CrossRef]
- Dröge, W.; Murthy, K.K.; Stahl-Hennig, C.; Hartung, S.; Plesker, R.; Rouse, S.; Peterhans, E.; Kinscherf, R.; Fischbach, T.; Eck, H.P. Plasma Amino Acid Dysregulation after Lentiviral Infection. AIDS Res Hum Retroviruses 1993, 9, 807–809. [Google Scholar] [CrossRef] [PubMed]
- Mody, A.; Bartz, S.; Hornik, C.P.; Kiyimba, T.; Bain, J.; Muehlbauer, M.; Kiboneka, E.; Stevens, R.; St Peter, J.V.; Newgard, C.B.; et al. Effects of HIV Infection on the Metabolic and Hormonal Status of Children with Severe Acute Malnutrition. PLoS One 2014, 9, e102233. [Google Scholar] [CrossRef]
- Stone, J.D.; Heise, C.C.; Miller, C.J.; Halsted, C.H.; Dandekar, S. Development of Malabsorption and Nutritional Complications in Simian Immunodeficiency Virus-Infected Rhesus Macaques. AIDS 1994, 8, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Cosnefroy, O.; Jaspart, A.; Calmels, C.; Parissi, V.; Fleury, H.; Ventura, M.; Reigadas, S.; Andréola, M.-L. Activation of GCN2 upon HIV-1 Infection and Inhibition of Translation. Cell Mol Life Sci 2013, 70, 2411–2421. [Google Scholar] [CrossRef]
- Jaspart, A.; Calmels, C.; Cosnefroy, O.; Bellecave, P.; Pinson, P.; Claverol, S.; Guyonnet-Dupérat, V.; Dartigues, B.; Benleulmi, M.S.; Mauro, E.; et al. GCN2 Phosphorylates HIV-1 Integrase and Decreases HIV-1 Replication by Limiting Viral Integration. Sci Rep 2017, 7, 2283. [Google Scholar] [CrossRef] [PubMed]
- del Pino, J.; Jiménez, J.L.; Ventoso, I.; Castelló, A.; Muñoz-Fernández, M.Á.; de Haro, C.; Berlanga, J.J. GCN2 Has Inhibitory Effect on Human Immunodeficiency Virus-1 Protein Synthesis and Is Cleaved upon Viral Infection. PLoS One 2012, 7, e47272. [Google Scholar] [CrossRef]
- Lutz, M.M.; Worth, M.P.; Hinchman, M.M.; Parker, J.S.L.; Ledgerwood, E.D. Mammalian Orthoreovirus Infection Is Enhanced in Cells Pre-Treated with Sodium Arsenite. Viruses 2019, 11, 563. [Google Scholar] [CrossRef]
- Lee, J.; Stone, J.; Desai, P.; Kosowicz, J.G.; Liu, J.O.; Ambinder, R.F. Arsenicals, the Integrated Stress Response, and Epstein-Barr Virus Lytic Gene Expression. Viruses 2021, 13, 812. [Google Scholar] [CrossRef]
- McEwen, E.; Kedersha, N.; Song, B.; Scheuner, D.; Gilks, N.; Han, A.; Chen, J.-J.; Anderson, P.; Kaufman, R.J. Heme-Regulated Inhibitor Kinase-Mediated Phosphorylation of Eukaryotic Translation Initiation Factor 2 Inhibits Translation, Induces Stress Granule Formation, and Mediates Survival upon Arsenite Exposure. J Biol Chem 2005, 280, 16925–16933. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Ramaglia, V.; Abdel-Nour, M.; Bianchi, A.A.; Tsalikis, J.; Chau, H.N.; Kalia, S.K.; Kalia, L.V.; Chen, J.-J.; Arnoult, D.; et al. The eIF2α Kinase HRI Triggers the Autophagic Clearance of Cytosolic Protein Aggregates. J Biol Chem 2021, 296, 100050. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.; Peslak, S.A.; Lan, X.; Khandros, E.; Yano, J.A.; Sharma, M.; Keller, C.A.; Giardine, B.; Qin, K.; Abdulmalik, O.; et al. The HRI-Regulated Transcription Factor ATF4 Activates BCL11A Transcription to Silence Fetal Hemoglobin Expression. Blood 2020, 135, 2121–2132. [Google Scholar] [CrossRef] [PubMed]
- Suragani, R.N.V.S.; Zachariah, R.S.; Velazquez, J.G.; Liu, S.; Sun, C.-W.; Townes, T.M.; Chen, J.-J. Heme-Regulated eIF2α Kinase Activated Atf4 Signaling Pathway in Oxidative Stress and Erythropoiesis. Blood 2012, 119, 5276–5284. [Google Scholar] [CrossRef] [PubMed]
- Protzer, U.; Seyfried, S.; Quasdorff, M.; Sass, G.; Svorcova, M.; Webb, D.; Bohne, F.; Hösel, M.; Schirmacher, P.; Tiegs, G. Antiviral Activity and Hepatoprotection by Heme Oxygenase-1 in Hepatitis B Virus Infection. Gastroenterology 2007, 133, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Wilson, A.T.; Mathahs, M.M.; Wen, F.; Brown, K.E.; Luxon, B.A.; Schmidt, W.N. Heme Oxygenase-1 Suppresses Hepatitis C Virus Replication and Increases Resistance of Hepatocytes to Oxidant Injury. Hepatology 2008, 48, 1430–1439. [Google Scholar] [CrossRef]
- Devadas, K.; Dhawan, S. Hemin Activation Ameliorates HIV-1 Infection via Heme Oxygenase-1 Induction. J Immunol 2006, 176, 4252–4257. [Google Scholar] [CrossRef]
- Ambegaokar, S.S.; Kolson, D.L. Heme Oxygenase-1 Dysregulation in the Brain: Implications for HIV-Associated Neurocognitive Disorders. Curr HIV Res 2014, 12, 174–188. [Google Scholar] [CrossRef]
- Malikov, V.; Naghavi, M.H. Localized Phosphorylation of a Kinesin-1 Adaptor by a Capsid-Associated Kinase Regulates HIV-1 Motility and Uncoating. Cell Reports 2017, 20, 2792–2799. [Google Scholar] [CrossRef] [PubMed]
- Cossarizza, A.; Mussini, C.; Mongiardo, N.; Borghi, V.; Sabbatini, A.; De Rienzo, B.; Franceschi, C. Mitochondria Alterations and Dramatic Tendency to Undergo Apoptosis in Peripheral Blood Lymphocytes during Acute HIV Syndrome. AIDS 1997, 11, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Macho, A.; Castedo, M.; Marchetti, P.; Aguilar, J.J.; Decaudin, D.; Zamzami, N.; Girard, P.M.; Uriel, J.; Kroemer, G. Mitochondrial Dysfunctions in Circulating T Lymphocytes from Human Immunodeficiency Virus-1 Carriers. Blood 1995, 86, 2481–2487. [Google Scholar] [CrossRef] [PubMed]
- Petit, F.; Arnoult, D.; Lelièvre, J.-D.; Moutouh-de Parseval, L.; Hance, A.J.; Schneider, P.; Corbeil, J.; Ameisen, J.C.; Estaquier, J. Productive HIV-1 Infection of Primary CD4+ T Cells Induces Mitochondrial Membrane Permeabilization Leading to a Caspase-Independent Cell Death. J Biol Chem 2002, 277, 1477–1487. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Petit, F.; Lelièvre, J.-D.; Estaquier, J. Mitochondria in HIV-1-Induced Apoptosis. Biochem Biophys Res Commun 2003, 304, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Arnoult, D.; Petit, F.; Lelièvre, J.D.; Lecossier, D.; Hance, A.; Monceaux, V.; Hurtrel, B.; Ho Tsong Fang, R.; Ameisen, J.C.; Estaquier, J. Caspase-Dependent and -Independent T-Cell Death Pathways in Pathogenic Simian Immunodeficiency Virus Infection: Relationship to Disease Progression. Cell Death Differ 2003, 10, 1240–1252. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Vallette, F.; Vayssiere, J.-L.; Mignotte, B. The Mitochondrial Pathways of Apoptosis. Adv Exp Med Biol 2012, 942, 157–183. [Google Scholar] [CrossRef] [PubMed]
- Mesquita, I.; Estaquier, J. Viral Manipulation of the Host Metabolic Network. Exp Suppl 2018, 109, 377–401. [Google Scholar] [CrossRef]
- Elbim, C.; Monceaux, V.; François, S.; Hurtrel, B.; Gougerot-Pocidalo, M.-A.; Estaquier, J. Increased Neutrophil Apoptosis in Chronically SIV-Infected Macaques. Retrovirology 2009, 6, 29. [Google Scholar] [CrossRef]
- Elbim, C.; Monceaux, V.; Mueller, Y.M.; Lewis, M.G.; François, S.; Diop, O.; Akarid, K.; Hurtrel, B.; Gougerot-Pocidalo, M.-A.; Lévy, Y.; et al. Early Divergence in Neutrophil Apoptosis between Pathogenic and Nonpathogenic Simian Immunodeficiency Virus Infections of Nonhuman Primates. J Immunol 2008, 181, 8613–8623. [Google Scholar] [CrossRef]
- Elbim, C.; Pillet, S.; Prevost, M.H.; Preira, A.; Girard, P.M.; Rogine, N.; Matusani, H.; Hakim, J.; Israel, N.; Gougerot-Pocidalo, M.A. Redox and Activation Status of Monocytes from Human Immunodeficiency Virus-Infected Patients: Relationship with Viral Load. J Virol 1999, 73, 4561–4566. [Google Scholar] [CrossRef] [PubMed]
- Cumont, M.C.; Monceaux, V.; Viollet, L.; Lay, S.; Parker, R.; Hurtrel, B.; Estaquier, J. TGF-Beta in Intestinal Lymphoid Organs Contributes to the Death of Armed Effector CD8 T Cells and Is Associated with the Absence of Virus Containment in Rhesus Macaques Infected with the Simian Immunodeficiency Virus. Cell Death Differ 2007, 14, 1747–1758. [Google Scholar] [CrossRef] [PubMed]
- Kasai, S.; Yasumoto, K.-I.; Sogawa, K. Attenuation of Inhibitory PAS Domain Protein-Induced Cell Death by Synthetic Peptides Derived from Mcl-1 Transmenbrane Domain. Cell Death Discov 2021, 7, 92. [Google Scholar] [CrossRef]
- Münch, C. The Different Axes of the Mammalian Mitochondrial Unfolded Protein Response. BMC Biol 2018, 16, 81. [Google Scholar] [CrossRef]
- Quirós, P.M.; Prado, M.A.; Zamboni, N.; D’Amico, D.; Williams, R.W.; Finley, D.; Gygi, S.P.; Auwerx, J. Multi-Omics Analysis Identifies ATF4 as a Key Regulator of the Mitochondrial Stress Response in Mammals. J Cell Biol 2017, 216, 2027–2045. [Google Scholar] [CrossRef]
- Fessler, E.; Eckl, E.-M.; Schmitt, S.; Mancilla, I.A.; Meyer-Bender, M.F.; Hanf, M.; Philippou-Massier, J.; Krebs, S.; Zischka, H.; Jae, L.T. A Pathway Coordinated by DELE1 Relays Mitochondrial Stress to the Cytosol. Nature 2020, 579, 433–437. [Google Scholar] [CrossRef]
- Guo, X.; Aviles, G.; Liu, Y.; Tian, R.; Unger, B.A.; Lin, Y.-H.T.; Wiita, A.P.; Xu, K.; Correia, M.A.; Kampmann, M. Mitochondrial Stress Is Relayed to the Cytosol by an OMA1-DELE1-HRI Pathway. Nature 2020, 579, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, R.E.; Zoncu, R. The Lysosome as a Cellular Centre for Signalling, Metabolism and Quality Control. Nat Cell Biol 2019, 21, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Li, T.Y.; Wang, Q.; Gao, A.W.; Li, X.; Sun, Y.; Mottis, A.; Shong, M.; Auwerx, J. Lysosomes Mediate the Mitochondrial UPR via mTORC1-Dependent ATF4 Phosphorylation. Cell Discov 2023, 9, 92. [Google Scholar] [CrossRef] [PubMed]
- Wolfson, R.L.; Sabatini, D.M. The Dawn of the Age of Amino Acid Sensors for the mTORC1 Pathway. Cell Metab 2017, 26, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses Lysosomal Amino Acids through an Inside-out Mechanism That Requires the Vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef]
- Arnoult, D.; Grodet, A.; Lee, Y.-J.; Estaquier, J.; Blackstone, C. Release of OPA1 during Apoptosis Participates in the Rapid and Complete Release of Cytochrome c and Subsequent Mitochondrial Fragmentation. J Biol Chem 2005, 280, 35742–35750. [Google Scholar] [CrossRef]
- Arnoult, D.; Rismanchi, N.; Grodet, A.; Roberts, R.G.; Seeburg, D.P.; Estaquier, J.; Sheng, M.; Blackstone, C. Bax/Bak-Dependent Release of DDP/TIMM8a Promotes Drp1-Mediated Mitochondrial Fission and Mitoptosis during Programmed Cell Death. Curr Biol 2005, 15, 2112–2118. [Google Scholar] [CrossRef] [PubMed]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab 2017, 25, 1374–1389. [Google Scholar] [CrossRef] [PubMed]
- Estaquier, J.; Arnoult, D. Inhibiting Drp1-Mediated Mitochondrial Fission Selectively Prevents the Release of Cytochrome c during Apoptosis. Cell Death Differ 2007, 14, 1086–1094. [Google Scholar] [CrossRef]
- Steffen, J.; Ngo, J.; Wang, S.-P.; Williams, K.; Kramer, H.F.; Ho, G.; Rodriguez, C.; Yekkala, K.; Amuzie, C.; Bialecki, R.; et al. The Mitochondrial Fission Protein Drp1 in Liver Is Required to Mitigate NASH and Prevents the Activation of the Mitochondrial ISR. Mol Metab 2022, 64, 101566. [Google Scholar] [CrossRef] [PubMed]
- Ehses, S.; Raschke, I.; Mancuso, G.; Bernacchia, A.; Geimer, S.; Tondera, D.; Martinou, J.-C.; Westermann, B.; Rugarli, E.I.; Langer, T. Regulation of OPA1 Processing and Mitochondrial Fusion by M-AAA Protease Isoenzymes and OMA1. J Cell Biol 2009, 187, 1023–1036. [Google Scholar] [CrossRef]
- Head, B.; Griparic, L.; Amiri, M.; Gandre-Babbe, S.; van der Bliek, A.M. Inducible Proteolytic Inactivation of OPA1 Mediated by the OMA1 Protease in Mammalian Cells. J Cell Biol 2009, 187, 959–966. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Baron, K.R.; Pride, D.E.; Schneemann, A.; Guo, X.; Chen, W.; Song, A.S.; Aviles, G.; Kampmann, M.; Luke Wiseman, R.; et al. DELE1 Oligomerization Promotes Integrated Stress Response Activation. Nat Struct Mol Biol 2023, 30, 1295–1302. [Google Scholar] [CrossRef] [PubMed]
- Schubert, U.; Antón, L.C.; Bacík, I.; Cox, J.H.; Bour, S.; Bennink, J.R.; Orlowski, M.; Strebel, K.; Yewdell, J.W. CD4 Glycoprotein Degradation Induced by Human Immunodeficiency Virus Type 1 Vpu Protein Requires the Function of Proteasomes and the Ubiquitin-Conjugating Pathway. J Virol 1998, 72, 2280–2288. [Google Scholar] [CrossRef]
- Butticaz, C.; Michielin, O.; Wyniger, J.; Telenti, A.; Rothenberger, S. Silencing of Both Beta-TrCP1 and HOS (Beta-TrCP2) Is Required to Suppress Human Immunodeficiency Virus Type 1 Vpu-Mediated CD4 down-Modulation. J Virol 2007, 81, 1502–1505. [Google Scholar] [CrossRef]
- Douglas, J.L.; Viswanathan, K.; McCarroll, M.N.; Gustin, J.K.; Früh, K.; Moses, A.V. Vpu Directs the Degradation of the Human Immunodeficiency Virus Restriction Factor BST-2/Tetherin via a {beta}TrCP-Dependent Mechanism. J Virol 2009, 83, 7931–7947. [Google Scholar] [CrossRef] [PubMed]
- Mangeat, B.; Gers-Huber, G.; Lehmann, M.; Zufferey, M.; Luban, J.; Piguet, V. HIV-1 Vpu Neutralizes the Antiviral Factor Tetherin/BST-2 by Binding It and Directing Its Beta-TrCP2-Dependent Degradation. PLoS Pathog 2009, 5, e1000574. [Google Scholar] [CrossRef] [PubMed]
- Margottin, F.; Bour, S.P.; Durand, H.; Selig, L.; Benichou, S.; Richard, V.; Thomas, D.; Strebel, K.; Benarous, R. A Novel Human WD Protein, h-Beta TrCp, That Interacts with HIV-1 Vpu Connects CD4 to the ER Degradation Pathway through an F-Box Motif. Mol Cell 1998, 1, 565–574. [Google Scholar] [CrossRef]
- Iwabu, Y.; Fujita, H.; Kinomoto, M.; Kaneko, K.; Ishizaka, Y.; Tanaka, Y.; Sata, T.; Tokunaga, K. HIV-1 Accessory Protein Vpu Internalizes Cell-Surface BST-2/Tetherin through Transmembrane Interactions Leading to Lysosomes. J Biol Chem 2009, 284, 35060–35072. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, R.S.; Katsura, C.; Skasko, M.A.; Fitzpatrick, K.; Lau, D.; Ruiz, A.; Stephens, E.B.; Margottin-Goguet, F.; Benarous, R.; Guatelli, J.C. Vpu Antagonizes BST-2-Mediated Restriction of HIV-1 Release via Beta-TrCP and Endo-Lysosomal Trafficking. PLoS Pathog 2009, 5, e1000450. [Google Scholar] [CrossRef] [PubMed]
- Stoneham, C.A.; Singh, R.; Jia, X.; Xiong, Y.; Guatelli, J. Endocytic Activity of HIV-1 Vpu: Phosphoserine-Dependent Interactions with Clathrin Adaptors. Traffic 2017, 18, 545–561. [Google Scholar] [CrossRef] [PubMed]
- Goffinet, C.; Allespach, I.; Homann, S.; Tervo, H.-M.; Habermann, A.; Rupp, D.; Oberbremer, L.; Kern, C.; Tibroni, N.; Welsch, S.; et al. HIV-1 Antagonism of CD317 Is Species Specific and Involves Vpu-Mediated Proteasomal Degradation of the Restriction Factor. Cell Host Microbe 2009, 5, 285–297. [Google Scholar] [CrossRef]
- Lassot, I.; Ségéral, E.; Berlioz-Torrent, C.; Durand, H.; Groussin, L.; Hai, T.; Benarous, R.; Margottin-Goguet, F. ATF4 Degradation Relies on a Phosphorylation-Dependent Interaction with the SCF(betaTrCP) Ubiquitin Ligase. Mol Cell Biol 2001, 21, 2192–2202. [Google Scholar] [CrossRef]
- Besnard-Guerin, C.; Belaïdouni, N.; Lassot, I.; Segeral, E.; Jobart, A.; Marchal, C.; Benarous, R. HIV-1 Vpu Sequesters Beta-Transducin Repeat-Containing Protein (betaTrCP) in the Cytoplasm and Provokes the Accumulation of Beta-Catenin and Other SCFbetaTrCP Substrates. J Biol Chem 2004, 279, 788–795. [Google Scholar] [CrossRef]
- Putters, J.; Slotman, J.A.; Gerlach, J.P.; Strous, G.J. Specificity, Location and Function of βTrCP Isoforms and Their Splice Variants. Cell Signal 2011, 23, 641–647. [Google Scholar] [CrossRef]
- Pickering, S.; Sumner, J.; Kerridge, C.; Perera, M.; Neil, S. Differential Dysregulation of β-TrCP1 and -2 by HIV-1 Vpu Leads to Inhibition of Canonical and Non-Canonical NF-κB Pathways in Infected Cells. mBio 2023, 14, e0329322. [Google Scholar] [CrossRef]
- Xu, Y.; Weatherall, C.; Bailey, M.; Alcantara, S.; De Rose, R.; Estaquier, J.; Wilson, K.; Suzuki, K.; Corbeil, J.; Cooper, D.A.; et al. Simian Immunodeficiency Virus Infects Follicular Helper CD4 T Cells in Lymphoid Tissues during Pathogenic Infection of Pigtail Macaques. J Virol 2013, 87, 3760–3773. [Google Scholar] [CrossRef]
- Moukambi, F.; Rabezanahary, H.; Rodrigues, V.; Racine, G.; Robitaille, L.; Krust, B.; Andreani, G.; Soundaramourty, C.; Silvestre, R.; Laforge, M.; et al. Early Loss of Splenic Tfh Cells in SIV-Infected Rhesus Macaques. PLoS Pathog 2015, 11, e1005287. [Google Scholar] [CrossRef]
- Rabezanahary, H.; Moukambi, F.; Palesch, D.; Clain, J.; Racine, G.; Andreani, G.; Benmadid-Laktout, G.; Zghidi-Abouzid, O.; Soundaramourty, C.; Tremblay, C.; et al. Despite Early Antiretroviral Therapy Effector Memory and Follicular Helper CD4 T Cells Are Major Reservoirs in Visceral Lymphoid Tissues of SIV-Infected Macaques. Mucosal Immunol 2020, 13, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Banga, R.; Procopio, F.A.; Noto, A.; Pollakis, G.; Cavassini, M.; Ohmiti, K.; Corpataux, J.-M.; de Leval, L.; Pantaleo, G.; Perreau, M. PD-1(+) and Follicular Helper T Cells Are Responsible for Persistent HIV-1 Transcription in Treated Aviremic Individuals. Nat Med 2016, 22, 754–761. [Google Scholar] [CrossRef]
- Perreau, M.; Savoye, A.-L.; De Crignis, E.; Corpataux, J.-M.; Cubas, R.; Haddad, E.K.; De Leval, L.; Graziosi, C.; Pantaleo, G. Follicular Helper T Cells Serve as the Major CD4 T Cell Compartment for HIV-1 Infection, Replication, and Production. J Exp Med 2013, 210, 143–156. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.; Jaworski, J.P.; Cortés-Rubio, C.N. Balance between Retroviral Latency and Transcription: Based on HIV Model. Pathogens 2020, 10, 16. [Google Scholar] [CrossRef] [PubMed]
- Reddy, T.R.; Tang, H.; Li, X.; Wong-Staal, F. Functional Interaction of the HTLV-1 Transactivator Tax with Activating Transcription Factor-4 (ATF4). Oncogene 1997, 14, 2785–2792. [Google Scholar] [CrossRef]
- Pereira, L.A.; Bentley, K.; Peeters, A.; Churchill, M.J.; Deacon, N.J. A Compilation of Cellular Transcription Factor Interactions with the HIV-1 LTR Promoter. Nucleic Acids Res 2000, 28, 663–668. [Google Scholar] [CrossRef]
- Rabbi, M.F.; Saifuddin, M.; Gu, D.S.; Kagnoff, M.F.; Roebuck, K.A. U5 Region of the Human Immunodeficiency Virus Type 1 Long Terminal Repeat Contains TRE-like cAMP-Responsive Elements That Bind Both AP-1 and CREB/ATF Proteins. Virology 1997, 233, 235–245. [Google Scholar] [CrossRef]
- Neill, G.; Masson, G.R. A Stay of Execution: ATF4 Regulation and Potential Outcomes for the Integrated Stress Response. Front Mol Neurosci 2023, 16, 1112253. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Martínez, J.A.; Reinke, A.W.; Bhimsaria, D.; Keating, A.E.; Ansari, A.Z. Combinatorial bZIP Dimers Display Complex DNA-Binding Specificity Landscapes. Elife 2017, 6, e19272. [Google Scholar] [CrossRef] [PubMed]
- Hai, T.; Curran, T. Cross-Family Dimerization of Transcription Factors Fos/Jun and ATF/CREB Alters DNA Binding Specificity. Proc Natl Acad Sci U S A 1991, 88, 3720–3724. [Google Scholar] [CrossRef] [PubMed]
- Mu, Y.; Yu, Y.; Yue, X.; Musarat, I.; Gong, R.; Zhu, C.; Liu, Y.; Liu, F.; Zhu, Y.; Wu, J. The X Protein of HBV Induces HIV-1 Long Terminal Repeat Transcription by Enhancing the Binding of C/EBPβ and CREB1/2 Regulatory Proteins to the Long Terminal Repeat of HIV-1. Virus Res 2011, 156, 81–90. [Google Scholar] [CrossRef]
- Gachon, F.; Thebault, S.; Peleraux, A.; Devaux, C.; Mesnard, J.M. Molecular Interactions Involved in the Transactivation of the Human T-Cell Leukemia Virus Type 1 Promoter Mediated by Tax and CREB-2 (ATF-4). Mol Cell Biol 2000, 20, 3470–3481. [Google Scholar] [CrossRef]
- Ameisen, J.C.; Estaquier, J.; Idziorek, T. From AIDS to Parasite Infection: Pathogen-Mediated Subversion of Programmed Cell Death as a Mechanism for Immune Dysregulation. Immunol Rev 1994, 142, 9–51. [Google Scholar] [CrossRef]
- Clerici, M.; Sarin, A.; Coffman, R.L.; Wynn, T.A.; Blatt, S.P.; Hendrix, C.W.; Wolf, S.F.; Shearer, G.M.; Henkart, P.A. Type 1/Type 2 Cytokine Modulation of T-Cell Programmed Cell Death as a Model for Human Immunodeficiency Virus Pathogenesis. Proc Natl Acad Sci U S A 1994, 91, 11811–11815. [Google Scholar] [CrossRef]
- Finkel, T.H.; Tudor-Williams, G.; Banda, N.K.; Cotton, M.F.; Curiel, T.; Monks, C.; Baba, T.W.; Ruprecht, R.M.; Kupfer, A. Apoptosis Occurs Predominantly in Bystander Cells and Not in Productively Infected Cells of HIV- and SIV-Infected Lymph Nodes. Nat Med 1995, 1, 129–134. [Google Scholar] [CrossRef]
- Arnoult, D.; Viollet, L.; Petit, F.; Lelièvre, J.-D.; Estaquier, J. HIV-1 Triggers Mitochondrion Death. Mitochondrion 2004, 4, 255–269. [Google Scholar] [CrossRef]
- Estaquier, J.; Tanaka, M.; Suda, T.; Nagata, S.; Golstein, P.; Ameisen, J.C. Fas-Mediated Apoptosis of CD4+ and CD8+ T Cells from Human Immunodeficiency Virus-Infected Persons: Differential in Vitro Preventive Effect of Cytokines and Protease Antagonists. Blood 1996, 87, 4959–4966. [Google Scholar] [CrossRef]
- Estaquier, J.; Idziorek, T.; Zou, W.; Emilie, D.; Farber, C.M.; Bourez, J.M.; Ameisen, J.C. T Helper Type 1/T Helper Type 2 Cytokines and T Cell Death: Preventive Effect of Interleukin 12 on Activation-Induced and CD95 (FAS/APO-1)-Mediated Apoptosis of CD4+ T Cells from Human Immunodeficiency Virus-Infected Persons. J Exp Med 1995, 182, 1759–1767. [Google Scholar] [CrossRef]
- Katsikis, P.D.; Wunderlich, E.S.; Smith, C.A.; Herzenberg, L.A.; Herzenberg, L.A. Fas Antigen Stimulation Induces Marked Apoptosis of T Lymphocytes in Human Immunodeficiency Virus-Infected Individuals. J Exp Med 1995, 181, 2029–2036. [Google Scholar] [CrossRef]
- Sloand, E.M.; Young, N.S.; Kumar, P.; Weichold, F.F.; Sato, T.; Maciejewski, J.P. Role of Fas Ligand and Receptor in the Mechanism of T-Cell Depletion in Acquired Immunodeficiency Syndrome: Effect on CD4+ Lymphocyte Depletion and Human Immunodeficiency Virus Replication. Blood 1997, 89, 1357–1363. [Google Scholar] [CrossRef]
- Galehdar, Z.; Swan, P.; Fuerth, B.; Callaghan, S.M.; Park, D.S.; Cregan, S.P. Neuronal Apoptosis Induced by Endoplasmic Reticulum Stress Is Regulated by ATF4-CHOP-Mediated Induction of the Bcl-2 Homology 3-Only Member PUMA. J Neurosci 2010, 30, 16938–16948. [Google Scholar] [CrossRef]
- Puthalakath, H.; O’Reilly, L.A.; Gunn, P.; Lee, L.; Kelly, P.N.; Huntington, N.D.; Hughes, P.D.; Michalak, E.M.; McKimm-Breschkin, J.; Motoyama, N.; et al. ER Stress Triggers Apoptosis by Activating BH3-Only Protein Bim. Cell 2007, 129, 1337–1349. [Google Scholar] [CrossRef] [PubMed]
- Bagheri-Yarmand, R.; Sinha, K.M.; Gururaj, A.E.; Ahmed, Z.; Rizvi, Y.Q.; Huang, S.-C.; Ladbury, J.E.; Bogler, O.; Williams, M.D.; Cote, G.J.; et al. A Novel Dual Kinase Function of the RET Proto-Oncogene Negatively Regulates Activating Transcription Factor 4-Mediated Apoptosis. J Biol Chem 2015, 290, 11749–11761. [Google Scholar] [CrossRef]
- Teske, B.F.; Fusakio, M.E.; Zhou, D.; Shan, J.; McClintick, J.N.; Kilberg, M.S.; Wek, R.C. CHOP Induces Activating Transcription Factor 5 (ATF5) to Trigger Apoptosis in Response to Perturbations in Protein Homeostasis. Mol Biol Cell 2013, 24, 2477–2490. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Mora-Jensen, H.; Weniger, M.A.; Perez-Galan, P.; Wolford, C.; Hai, T.; Ron, D.; Chen, W.; Trenkle, W.; Wiestner, A.; et al. ERAD Inhibitors Integrate ER Stress with an Epigenetic Mechanism to Activate BH3-Only Protein NOXA in Cancer Cells. Proc Natl Acad Sci U S A 2009, 106, 2200–2205. [Google Scholar] [CrossRef]
- Hiramatsu, N.; Messah, C.; Han, J.; LaVail, M.M.; Kaufman, R.J.; Lin, J.H. Translational and Posttranslational Regulation of XIAP by eIF2α and ATF4 Promotes ER Stress-Induced Cell Death during the Unfolded Protein Response. Mol Biol Cell 2014, 25, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Wang, M.; Zhou, S.; Zhou, Q. HIV-1 Tat Targets Microtubules to Induce Apoptosis, a Process Promoted by the pro-Apoptotic Bcl-2 Relative Bim. EMBO J 2002, 21, 6801–6810. [Google Scholar] [CrossRef]
- Castellano, P.; Prevedel, L.; Eugenin, E.A. HIV-Infected Macrophages and Microglia That Survive Acute Infection Become Viral Reservoirs by a Mechanism Involving Bim. Sci Rep 2017, 7, 12866. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Ye, X.; Zhong, L.; Xu, J.; Qiu, J.; Wang, J.; Shao, Y.; Xing, H. Role of FOXO3 Activated by HIV-1 Tat in HIV-Associated Neurocognitive Disorder Neuronal Apoptosis. Front Neurosci 2019, 13, 44. [Google Scholar] [CrossRef] [PubMed]
- Galvão-Lima, L.J.; Zambuzi, F.A.; Soares, L.S.; Fontanari, C.; Meireles, A.F.G.; Brauer, V.S.; Faccioli, L.H.; Gama, L.; Figueiredo, L.T.M.; Bou-Habib, D.C.; et al. HIV-1 Gag and Vpr Impair the Inflammasome Activation and Contribute to the Establishment of Chronic Infection in Human Primary Macrophages. Mol Immunol 2022, 148, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Chand, K.; Iyer, K.; Mitra, D. Comparative Analysis of Differential Gene Expression of HSP40 and HSP70 Family Isoforms during Heat Stress and HIV-1 Infection in T-Cells. Cell Stress Chaperones 2021, 26, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Chand, K.; Barman, M.K.; Ghosh, P.; Mitra, D. DNAJB8 Facilitates Autophagic-Lysosomal Degradation of Viral Vif Protein and Restricts HIV-1 Virion Infectivity by Rescuing APOBEC3G Expression in Host Cells. FASEB J 2023, 37, e22793. [Google Scholar] [CrossRef] [PubMed]
- Buonaguro, L.; Monaco, A.; Aricò, E.; Wang, E.; Tornesello, M.L.; Lewis, G.K.; Marincola, F.M.; Buonaguro, F.M. Gene Expression Profile of Peripheral Blood Mononuclear Cells in Response to HIV-VLPs Stimulation. BMC Bioinformatics 2008, 9 Suppl 2, S5. [Google Scholar] [CrossRef]
- Lim, A.L.; Moos, P.; Pond, C.D.; Larson, E.C.; Martins, L.J.; Szaniawski, M.A.; Planelles, V.; Barrows, L.R. HIV-1 Provirus Transcription and Translation in Macrophages Differs from Pre-Integrated cDNA Complexes and Requires E2F Transcriptional Programs. Virulence 2022, 13, 386–413. [Google Scholar] [CrossRef]
- Grelli, S.; Balestrieri, E.; Matteucci, C.; Minutolo, A.; D’Ettorre, G.; Lauria, F.; Montella, F.; Vullo, V.; Vella, S.; Favalli, C.; et al. Apoptotic Cell Signaling in Lymphocytes from HIV+ Patients during Successful Therapy. Ann N Y Acad Sci 2006, 1090, 130–137. [Google Scholar] [CrossRef]
- Swingler, S.; Mann, A.M.; Zhou, J.; Swingler, C.; Stevenson, M. Apoptotic Killing of HIV-1-Infected Macrophages Is Subverted by the Viral Envelope Glycoprotein. PLoS Pathog 2007, 3, 1281–1290. [Google Scholar] [CrossRef]
- Balestrieri, E.; Grelli, S.; Matteucci, C.; Minutolo, A.; d’Ettorre, G.; Di Sora, F.; Montella, F.; Vullo, V.; Vella, S.; Favalli, C.; et al. Apoptosis-Associated Gene Expression in HIV-Infected Patients in Response to Successful Antiretroviral Therapy. J Med Virol 2007, 79, 111–117. [Google Scholar] [CrossRef]
- Laforge, M.; Campillo-Gimenez, L.; Monceaux, V.; Cumont, M.-C.; Hurtrel, B.; Corbeil, J.; Zaunders, J.; Elbim, C.; Estaquier, J. HIV/SIV Infection Primes Monocytes and Dendritic Cells for Apoptosis. PLoS Pathog 2011, 7, e1002087. [Google Scholar] [CrossRef] [PubMed]
- Busca, A.; Saxena, M.; Kumar, A. Critical Role for Antiapoptotic Bcl-xL and Mcl-1 in Human Macrophage Survival and Cellular IAP1/2 (cIAP1/2) in Resistance to HIV-Vpr-Induced Apoptosis. J Biol Chem 2012, 287, 15118–15133. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Fu, C.; Cong, Z.; Peng, L.; Peng, Z.; Chen, T.; Wang, W.; Jiang, H.; Wei, Q.; Qin, C. Galectin-3 Promotes Caspase-Independent Cell Death of HIV-1-Infected Macrophages. FEBS J 2017, 284, 97–113. [Google Scholar] [CrossRef] [PubMed]
- Serrano, A.; El Haddad, S.; Moal, F.; Prazuck, T.; Legac, E.; Robin, C.; Brulé, F.; Charpentier, S.; Normand, T.; Legrand, A.; et al. Dysregulation of Apoptosis and Autophagy Gene Expression in Peripheral Blood Mononuclear Cells of Efficiently Treated HIV-Infected Patients. AIDS 2018, 32, 1579–1587. [Google Scholar] [CrossRef] [PubMed]
- Muraduzzaman, A.K.M.; Islam, N.M.; Tabassum, S.; Munshi, S.U. Intrinsic Apoptotic Pathway Genes of Circulating Blood Neutrophils Triggered during HIV Infection and Remained Stimulated in ART Patients. Curr HIV Res 2023, 21, 122–127. [Google Scholar] [CrossRef]
- Dabrowska, A.; Kim, N.; Aldovini, A. Tat-Induced FOXO3a Is a Key Mediator of Apoptosis in HIV-1-Infected Human CD4+ T Lymphocytes. J Immunol 2008, 181, 8460–8477. [Google Scholar] [CrossRef] [PubMed]
- Perfettini, J.-L.; Roumier, T.; Castedo, M.; Larochette, N.; Boya, P.; Raynal, B.; Lazar, V.; Ciccosanti, F.; Nardacci, R.; Penninger, J.; et al. NF-kappaB and P53 Are the Dominant Apoptosis-Inducing Transcription Factors Elicited by the HIV-1 Envelope. J Exp Med 2004, 199, 629–640. [Google Scholar] [CrossRef]
- Nardacci, R.; Antinori, A.; Larocca, L.M.; Arena, V.; Amendola, A.; Perfettini, J.-L.; Kroemer, G.; Piacentini, M. Characterization of Cell Death Pathways in Human Immunodeficiency Virus-Associated Encephalitis. Am J Pathol 2005, 167, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, S.; Khan, M.Z.; Hippensteel, R.L.; Parkar, A.; Raghupathi, R.; Meucci, O. Role of the Transcription Factor E2F1 in CXCR4-Mediated Neurotoxicity and HIV Neuropathology. Neurobiol Dis 2007, 25, 17–26. [Google Scholar] [CrossRef]
- Moni, M.A.; Liò, P. Network-Based Analysis of Comorbidities Risk during an Infection: SARS and HIV Case Studies. BMC Bioinformatics 2014, 15, 333. [Google Scholar] [CrossRef]
- Petralia, M.C.; Nicoletti, F.; Tancheva, L.; Kalfin, R.; Fagone, P.; Mangano, K. Gene Co-Expression Network Modular Analysis Reveals Altered Immune Mechanisms in HIV-HAND. Brain Sci 2022, 12, 1378. [Google Scholar] [CrossRef]
- Liu, Z.; Zang, Y.; Qiao, L.; Liu, K.; Ouyang, Y.; Zhang, Y.; Chen, D. ASPP2 Involvement in P53-Mediated HIV-1 Envelope Glycoprotein Gp120 Neurotoxicity in Mice Cerebrocortical Neurons. Sci Rep 2016, 6, 33378. [Google Scholar] [CrossRef]
- Liu, Z.; Xiao, Y.; Torresilla, C.; Rassart, É.; Barbeau, B. Implication of Different HIV-1 Genes in the Modulation of Autophagy. Viruses 2017, 9, 389. [Google Scholar] [CrossRef]
- Ahn, B.Y.; Trinh, D.L.N.; Zajchowski, L.D.; Lee, B.; Elwi, A.N.; Kim, S.-W. Tid1 Is a New Regulator of P53 Mitochondrial Translocation and Apoptosis in Cancer. Oncogene 2010, 29, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Genini, D.; Sheeter, D.; Rought, S.; Zaunders, J.J.; Susin, S.A.; Kroemer, G.; Richman, D.D.; Carson, D.A.; Corbeil, J.; Leoni, L.M. HIV Induces Lymphocyte Apoptosis by a P53-Initiated, Mitochondrial-Mediated Mechanism. FASEB J 2001, 15, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Laforge, M.; Limou, S.; Harper, F.; Casartelli, N.; Rodrigues, V.; Silvestre, R.; Haloui, H.; Zagury, J.-F.; Senik, A.; Estaquier, J. DRAM Triggers Lysosomal Membrane Permeabilization and Cell Death in CD4(+) T Cells Infected with HIV. PLoS Pathog 2013, 9, e1003328. [Google Scholar] [CrossRef] [PubMed]
- Niu, G.; Zhang, H.; Liu, D.; Chen, L.; Belani, C.; Wang, H.-G.; Cheng, H. Tid1, the Mammalian Homologue of Drosophila Tumor Suppressor Tid56, Mediates Macroautophagy by Interacting with Beclin1-Containing Autophagy Protein Complex. J Biol Chem 2015, 290, 18102–18110. [Google Scholar] [CrossRef]
- Samuels-Lev, Y.; O’Connor, D.J.; Bergamaschi, D.; Trigiante, G.; Hsieh, J.K.; Zhong, S.; Campargue, I.; Naumovski, L.; Crook, T.; Lu, X. ASPP Proteins Specifically Stimulate the Apoptotic Function of P53. Mol Cell 2001, 8, 781–794. [Google Scholar] [CrossRef]
- Zhou, L.; Yang, X.; Shu, S.; Wang, S.; Guo, F.; Yin, Y.; Zhou, W.; Han, H.; Chai, X. Sufentanil Protects the Liver from Ischemia/Reperfusion-Induced Inflammation and Apoptosis by Inhibiting ATF4-Induced TP53BP2 Expression. Inflammation 2021, 44, 1160–1174. [Google Scholar] [CrossRef]
- Liu, Z.; Qiao, L.; Zhang, Y.; Zang, Y.; Shi, Y.; Liu, K.; Zhang, X.; Lu, X.; Yuan, L.; Su, B.; et al. ASPP2 Plays a Dual Role in Gp120-Induced Autophagy and Apoptosis of Neuroblastoma Cells. Front Neurosci 2017, 11, 150. [Google Scholar] [CrossRef] [PubMed]
- Oka, T.; Sayano, T.; Tamai, S.; Yokota, S.; Kato, H.; Fujii, G.; Mihara, K. Identification of a Novel Protein MICS1 That Is Involved in Maintenance of Mitochondrial Morphology and Apoptotic Release of Cytochrome c. Mol Biol Cell 2008, 19, 2597–2608. [Google Scholar] [CrossRef] [PubMed]
- Seitaj, B.; Maull, F.; Zhang, L.; Wüllner, V.; Wolf, C.; Schippers, P.; La Rovere, R.; Distler, U.; Tenzer, S.; Parys, J.B.; et al. Transmembrane BAX Inhibitor-1 Motif Containing Protein 5 (TMBIM5) Sustains Mitochondrial Structure, Shape, and Function by Impacting the Mitochondrial Protein Synthesis Machinery. Cells 2020, 9, 2147. [Google Scholar] [CrossRef]
- Austin, S.; Mekis, R.; Mohammed, S.E.M.; Scalise, M.; Wang, W.-A.; Galluccio, M.; Pfeiffer, C.; Borovec, T.; Parapatics, K.; Vitko, D.; et al. TMBIM5 Is the Ca2+ /H+ Antiporter of Mammalian Mitochondria. EMBO Rep 2022, 23, e54978. [Google Scholar] [CrossRef]
- Cui, E.; Zhang, L.; Pan, X.; Zhang, Q.; Zhang, L.; Wu, F.; Chen, N.; Lv, L.; Chen, W.; Chen, H.; et al. RNA-Sequencing Approach for Exploring the Therapeutic Effect of Umbilical Cord Mesenchymal Stem/Stromal Cells on Lipopolysaccharide-Induced Acute Lung Injury. Front Immunol 2022, 13, 1021102. [Google Scholar] [CrossRef]
- Zhang, L.; Dietsche, F.; Seitaj, B.; Rojas-Charry, L.; Latchman, N.; Tomar, D.; Wüst, R.C.; Nickel, A.; Frauenknecht, K.B.; Schoser, B.; et al. TMBIM5 Loss of Function Alters Mitochondrial Matrix Ion Homeostasis and Causes a Skeletal Myopathy. Life Sci Alliance 2022, 5, e202201478. [Google Scholar] [CrossRef]
- Ma, Y.; Zhang, M.; Yu, H.; Lu, J.; Cheng, K.K.Y.; Zhou, J.; Chen, H.; Jia, W. Activation of G0/G1 Switch Gene 2 by Endoplasmic Reticulum Stress Enhances Hepatic Steatosis. Metabolism 2019, 99, 32–44. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.; Forsdyke, D.R. A Human Putative Lymphocyte G0/G1 Switch Gene Containing a CpG-Rich Island Encodes a Small Basic Protein with the Potential to Be Phosphorylated. DNA Cell Biol 1991, 10, 581–591. [Google Scholar] [CrossRef]
- Siderovski, D.P.; Blum, S.; Forsdyke, R.E.; Forsdyke, D.R. A Set of Human Putative Lymphocyte G0/G1 Switch Genes Includes Genes Homologous to Rodent Cytokine and Zinc Finger Protein-Encoding Genes. DNA Cell Biol 1990, 9, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Heckmann, B.L.; Campbell, L.E.; Liu, J. G0S2: A Small Giant Controller of Lipolysis and Adipose-Liver Fatty Acid Flux. Biochim Biophys Acta Mol Cell Biol Lipids 2017, 1862, 1146–1154. [Google Scholar] [CrossRef]
- Kioka, H.; Kato, H.; Fujikawa, M.; Tsukamoto, O.; Suzuki, T.; Imamura, H.; Nakano, A.; Higo, S.; Yamazaki, S.; Matsuzaki, T.; et al. Evaluation of Intramitochondrial ATP Levels Identifies G0/G1 Switch Gene 2 as a Positive Regulator of Oxidative Phosphorylation. Proc Natl Acad Sci U S A 2014, 111, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, Y.; Zhu, Y.; Zhang, P. Lipolytic Inhibitor G0/G1 Switch Gene 2 Inhibits Reactive Oxygen Species Production and Apoptosis in Endothelial Cells. Am J Physiol Cell Physiol 2015, 308, C496–504. [Google Scholar] [CrossRef]
- B’chir, W.; Maurin, A.-C.; Carraro, V.; Averous, J.; Jousse, C.; Muranishi, Y.; Parry, L.; Stepien, G.; Fafournoux, P.; Bruhat, A. The eIF2α/ATF4 Pathway Is Essential for Stress-Induced Autophagy Gene Expression. Nucleic Acids Res 2013, 41, 7683–7699. [Google Scholar] [CrossRef] [PubMed]
- Whitney, M.L.; Jefferson, L.S.; Kimball, S.R. ATF4 Is Necessary and Sufficient for ER Stress-Induced Upregulation of REDD1 Expression. Biochem Biophys Res Commun 2009, 379, 451–455. [Google Scholar] [CrossRef]
- Wolff, N.C.; Vega-Rubin-de-Celis, S.; Xie, X.-J.; Castrillon, D.H.; Kabbani, W.; Brugarolas, J. Cell-Type-Dependent Regulation of mTORC1 by REDD1 and the Tumor Suppressors TSC1/TSC2 and LKB1 in Response to Hypoxia. Mol Cell Biol 2011, 31, 1870–1884. [Google Scholar] [CrossRef]
- Xu, D.; Dai, W.; Kutzler, L.; Lacko, H.A.; Jefferson, L.S.; Dennis, M.D.; Kimball, S.R. ATF4-Mediated Upregulation of REDD1 and Sestrin2 Suppresses mTORC1 Activity during Prolonged Leucine Deprivation. J Nutr 2020, 150, 1022–1030. [Google Scholar] [CrossRef]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Lorenzo-Sánchez, I.; Trujillo-González, R.; Estévez-Herrera, J.; García-Luis, J.; Valenzuela-Fernández, A. HIV Infection: Shaping the Complex, Dynamic, and Interconnected Network of the Cytoskeleton. Int J Mol Sci 2023, 24, 13104. [Google Scholar] [CrossRef] [PubMed]
- Cabrera-Rodríguez, R.; Pérez-Yanes, S.; Estévez-Herrera, J.; Márquez-Arce, D.; Cabrera, C.; Espert, L.; Blanco, J.; Valenzuela-Fernández, A. The Interplay of HIV and Autophagy in Early Infection. Front Microbiol 2021, 12, 661446. [Google Scholar] [CrossRef] [PubMed]
- Dinkins, C.; Pilli, M.; Kehrl, J.H. Roles of Autophagy in HIV Infection. Immunol Cell Biol 2015, 93, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Nardacci, R.; Ciccosanti, F.; Marsella, C.; Ippolito, G.; Piacentini, M.; Fimia, G.M. Role of Autophagy in HIV Infection and Pathogenesis. J Intern Med 2017, 281, 422–432. [Google Scholar] [CrossRef]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy Is Involved in T Cell Death after Binding of HIV-1 Envelope Proteins to CXCR4. J Clin Invest 2006, 116, 2161–2172. [Google Scholar] [CrossRef]
- García-Expósito, L.; Barroso-González, J.; Puigdomènech, I.; Machado, J.-D.; Blanco, J.; Valenzuela-Fernández, A. HIV-1 Requires Arf6-Mediated Membrane Dynamics to Efficiently Enter and Infect T Lymphocytes. Mol Biol Cell 2011, 22, 1148–1166. [Google Scholar] [CrossRef]
- Moreau, K.; Ravikumar, B.; Puri, C.; Rubinsztein, D.C. Arf6 Promotes Autophagosome Formation via Effects on Phosphatidylinositol 4,5-Bisphosphate and Phospholipase D. J Cell Biol 2012, 196, 483–496. [Google Scholar] [CrossRef]
- Van Grol, J.; Subauste, C.; Andrade, R.M.; Fujinaga, K.; Nelson, J.; Subauste, C.S. HIV-1 Inhibits Autophagy in Bystander Macrophage/Monocytic Cells through Src-Akt and STAT3. PLoS One 2010, 5, e11733. [Google Scholar] [CrossRef]
- Zhou, D.; Spector, S.A. Human Immunodeficiency Virus Type-1 Infection Inhibits Autophagy. AIDS 2008, 22, 695–699. [Google Scholar] [CrossRef]
- Alfaisal, J.; Machado, A.; Galais, M.; Robert-Hebmann, V.; Arnauné-Pelloquin, L.; Espert, L.; Biard-Piechaczyk, M. HIV-1 Vpr Inhibits Autophagy during the Early Steps of Infection of CD4 T Cells. Biol Cell 2019, 111, 308–318. [Google Scholar] [CrossRef]
- Rozman, M.; Zidovec-Lepej, S.; Jambrosic, K.; Babić, M.; Drmić Hofman, I. Role of TLRs in HIV-1 Infection and Potential of TLR Agonists in HIV-1 Vaccine Development and Treatment Strategies. Pathogens 2023, 12, 92. [Google Scholar] [CrossRef] [PubMed]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, Regulation, Modification and Function. Genes Immun 2011, 12, 399–414. [Google Scholar] [CrossRef] [PubMed]
- Farook, J.M.; Shields, J.; Tawfik, A.; Markand, S.; Sen, T.; Smith, S.B.; Brann, D.; Dhandapani, K.M.; Sen, N. GADD34 Induces Cell Death through Inactivation of Akt Following Traumatic Brain Injury. Cell Death Dis 2013, 4, e754. [Google Scholar] [CrossRef] [PubMed]
- Imbeault, M.; Ouellet, M.; Tremblay, M.J. Microarray Study Reveals That HIV-1 Induces Rapid Type-I Interferon-Dependent P53 mRNA up-Regulation in Human Primary CD4+ T Cells. Retrovirology 2009, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Sirois, M.; Robitaille, L.; Allary, R.; Shah, M.; Woelk, C.H.; Estaquier, J.; Corbeil, J. TRAF6 and IRF7 Control HIV Replication in Macrophages. PLoS One 2011, 6, e28125. [Google Scholar] [CrossRef] [PubMed]
- Ishaq, M.; Marshall, H.; Natarajan, V. GADD34 Attenuates HIV-1 Replication by Viral 5’-UTR TAR RNA-Mediated Translational Inhibition. Virology 2020, 540, 119–131. [Google Scholar] [CrossRef]
- Hernández, J.C.; Stevenson, M.; Latz, E.; Urcuqui-Inchima, S. HIV Type 1 Infection Up-Regulates TLR2 and TLR4 Expression and Function in Vivo and in Vitro. AIDS Res Hum Retroviruses 2012, 28, 1313–1328. [Google Scholar] [CrossRef]
- Henrick, B.M.; Yao, X.-D.; Rosenthal, K.L. HIV-1 Structural Proteins Serve as PAMPs for TLR2 Heterodimers Significantly Increasing Infection and Innate Immune Activation. Front Immunol 2015, 6, 426. [Google Scholar] [CrossRef] [PubMed]
- Shimasaki, S.; Koga, T.; Shuto, T.; Suico, M.A.; Sato, T.; Watanabe, K.; Morino-Koga, S.; Taura, M.; Okada, S.; Mori, K.; et al. Endoplasmic Reticulum Stress Increases the Expression and Function of Toll-like Receptor-2 in Epithelial Cells. Biochem Biophys Res Commun 2010, 402, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Bai, N.; Chang, A.; Zhang, Z.; Yin, J.; Shen, W.; Tian, Y.; Xiang, R.; Liu, C. ATF4 Is Directly Recruited by TLR4 Signaling and Positively Regulates TLR4-Trigged Cytokine Production in Human Monocytes. Cell Mol Immunol 2013, 10, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Wang, Q.; Ghneim, K.; Wang, L.; Rampanelli, E.; Holley-Guthrie, E.; Cheng, L.; Garrido, C.; Margolis, D.M.; Eller, L.A.; et al. Multi-Omics Analyses Reveal That HIV-1 Alters CD4+ T Cell Immunometabolism to Fuel Virus Replication. Nat Immunol 2021, 22, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Ostrowski, M.; Gouillou, M.; Tsai, L.; Yu, D.; Zhou, J.; Henstridge, D.C.; Maisa, A.; Hearps, A.C.; Lewin, S.R.; et al. Increased Glucose Metabolic Activity Is Associated with CD4+ T-Cell Activation and Depletion during Chronic HIV Infection. AIDS 2014, 28, 297–309. [Google Scholar] [CrossRef] [PubMed]
- Valle-Casuso, J.C.; Angin, M.; Volant, S.; Passaes, C.; Monceaux, V.; Mikhailova, A.; Bourdic, K.; Avettand-Fenoel, V.; Boufassa, F.; Sitbon, M.; et al. Cellular Metabolism Is a Major Determinant of HIV-1 Reservoir Seeding in CD4+ T Cells and Offers an Opportunity to Tackle Infection. Cell Metab 2019, 29, 611–626. [Google Scholar] [CrossRef] [PubMed]
- Loisel-Meyer, S.; Swainson, L.; Craveiro, M.; Oburoglu, L.; Mongellaz, C.; Costa, C.; Martinez, M.; Cosset, F.-L.; Battini, J.-L.; Herzenberg, L.A.; et al. Glut1-Mediated Glucose Transport Regulates HIV Infection. Proc Natl Acad Sci U S A 2012, 109, 2549–2554. [Google Scholar] [CrossRef]
- Yang, X.; Xia, R.; Yue, C.; Zhai, W.; Du, W.; Yang, Q.; Cao, H.; Chen, X.; Obando, D.; Zhu, Y.; et al. ATF4 Regulates CD4+ T Cell Immune Responses through Metabolic Reprogramming. Cell Rep 2018, 23, 1754–1766. [Google Scholar] [CrossRef] [PubMed]
- Clerc, I.; Moussa, D.A.; Vahlas, Z.; Tardito, S.; Oburoglu, L.; Hope, T.J.; Sitbon, M.; Dardalhon, V.; Mongellaz, C.; Taylor, N. Entry of Glucose- and Glutamine-Derived Carbons into the Citric Acid Cycle Supports Early Steps of HIV-1 Infection in CD4 T Cells. Nat Metab 2019, 1, 717–730. [Google Scholar] [CrossRef] [PubMed]
- Yero, A.; Bouassa, R.-S.M.; Ancuta, P.; Estaquier, J.; Jenabian, M.-A. Immuno-Metabolic Control of the Balance between Th17-Polarized and Regulatory T-Cells during HIV Infection. Cytokine Growth Factor Rev 2023, 69, 1–13. [Google Scholar] [CrossRef]
- Sundrud, M.S.; Koralov, S.B.; Feuerer, M.; Calado, D.P.; Kozhaya, A.E.; Rhule-Smith, A.; Lefebvre, R.E.; Unutmaz, D.; Mazitschek, R.; Waldner, H.; et al. Halofuginone Inhibits TH17 Cell Differentiation by Activating the Amino Acid Starvation Response. Science 2009, 324, 1334–1338. [Google Scholar] [CrossRef]
- Gosselin, A.; Monteiro, P.; Chomont, N.; Diaz-Griffero, F.; Said, E.A.; Fonseca, S.; Wacleche, V.; El-Far, M.; Boulassel, M.-R.; Routy, J.-P.; et al. Peripheral Blood CCR4+CCR6+ and CXCR3+CCR6+CD4+ T Cells Are Highly Permissive to HIV-1 Infection. J Immunol 2010, 184, 1604–1616. [Google Scholar] [CrossRef]
- Gosselin, A.; Wiche Salinas, T.R.; Planas, D.; Wacleche, V.S.; Zhang, Y.; Fromentin, R.; Chomont, N.; Cohen, É.A.; Shacklett, B.; Mehraj, V.; et al. HIV Persists in CCR6+CD4+ T Cells from Colon and Blood during Antiretroviral Therapy. AIDS 2017, 31, 35–48. [Google Scholar] [CrossRef]
- Estes, J.D.; Harris, L.D.; Klatt, N.R.; Tabb, B.; Pittaluga, S.; Paiardini, M.; Barclay, G.R.; Smedley, J.; Pung, R.; Oliveira, K.M.; et al. Damaged Intestinal Epithelial Integrity Linked to Microbial Translocation in Pathogenic Simian Immunodeficiency Virus Infections. PLoS Pathog 2010, 6, e1001052. [Google Scholar] [CrossRef] [PubMed]
- Favre, D.; Lederer, S.; Kanwar, B.; Ma, Z.-M.; Proll, S.; Kasakow, Z.; Mold, J.; Swainson, L.; Barbour, J.D.; Baskin, C.R.; et al. Critical Loss of the Balance between Th17 and T Regulatory Cell Populations in Pathogenic SIV Infection. PLoS Pathog 2009, 5, e1000295. [Google Scholar] [CrossRef] [PubMed]
- McGary, C.S.; Alvarez, X.; Harrington, S.; Cervasi, B.; Ryan, E.S.; Iriele, R.I.; Paganini, S.; Harper, J.L.; Easley, K.; Silvestri, G.; et al. The Loss of CCR6+ and CD161+ CD4+ T-Cell Homeostasis Contributes to Disease Progression in SIV-Infected Rhesus Macaques. Mucosal Immunol 2017, 10, 1082–1096. [Google Scholar] [CrossRef] [PubMed]
- Raffatellu, M.; Santos, R.L.; Verhoeven, D.E.; George, M.D.; Wilson, R.P.; Winter, S.E.; Godinez, I.; Sankaran, S.; Paixao, T.A.; Gordon, M.A.; et al. Simian Immunodeficiency Virus-Induced Mucosal Interleukin-17 Deficiency Promotes Salmonella Dissemination from the Gut. Nat Med 2008, 14, 421–428. [Google Scholar] [CrossRef]
- Yero, A.; Farnos, O.; Rabezanahary, H.; Racine, G.; Estaquier, J.; Jenabian, M.-A. Differential Dynamics of Regulatory T-Cell and Th17 Cell Balance in Mesenteric Lymph Nodes and Blood Following Early Antiretroviral Initiation during Acute Simian Immunodeficiency Virus Infection. J Virol 2019, 93, e00371–19. [Google Scholar] [CrossRef]
- Sears, T.K.; Angelastro, J.M. The Transcription Factor ATF5: Role in Cellular Differentiation, Stress Responses, and Cancer. Oncotarget 2017, 8, 84595–84609. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.B.; Mitchelmore, C.; Kjaerulff, K.M.; Rasmussen, T.E.; Pedersen, K.M.; Jensen, N.A. Mouse Atf5: Molecular Cloning of Two Novel mRNAs, Genomic Organization, and Odorant Sensory Neuron Localization. Genomics 2002, 80, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Vinson, C.; Myakishev, M.; Acharya, A.; Mir, A.A.; Moll, J.R.; Bonovich, M. Classification of Human B-ZIP Proteins Based on Dimerization Properties. Mol Cell Biol 2002, 22, 6321–6335. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Liu, Y.; Yang, Y.; Qiu, Y.; Wang, Z.; Li, X.; Zhang, W. Emerging Roles of Activating Transcription Factor (ATF) Family Members in Tumourigenesis and Immunity: Implications in Cancer Immunotherapy. Genes Dis 2022, 9, 981–999. [Google Scholar] [CrossRef] [PubMed]
- Paerhati, P.; Liu, J.; Jin, Z.; Jakoš, T.; Zhu, S.; Qian, L.; Zhu, J.; Yuan, Y. Advancements in Activating Transcription Factor 5 Function in Regulating Cell Stress and Survival. Int J Mol Sci 2022, 23, 7129. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, M.L.; Shaban, M.S.; Albert, B.V.; Gökçen, A.; Kracht, M. The Crosstalk of Endoplasmic Reticulum (ER) Stress Pathways with NF-κB: Complex Mechanisms Relevant for Cancer, Inflammation and Infection. Biomedicines 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.-C.; Wang, J.-M.; Hsieh, W.-C.; Chang, Y.; Su, I.-J. Up-Regulation of Activating Transcription Factor-5 Suppresses SAP Expression to Activate T Cells in Hemophagocytic Syndrome Associated with Epstein-Barr Virus Infection and Immune Disorders. Am J Pathol 2008, 173, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Zhang, X.; Che, Y.; Zhang, Y.; Tang, S.; Liao, Y.; Na, R.; Xiong, X.; Liu, L.; Li, Q. A Cellular Response Protein Induced during HSV-1 Infection Inhibits Viral Replication by Interacting with ATF5. Sci China Life Sci 2013, 56, 1124–1133. [Google Scholar] [CrossRef]
- Hu, M.; Wang, B.; Qian, D.; Wang, M.; Huang, R.; Wei, L.; Li, L.; Zhang, L.; Liu, D.X. Human Cytomegalovirus Immediate-Early Protein Promotes Survival of Glioma Cells through Interacting and Acetylating ATF5. Oncotarget 2017, 8, 32157–32170. [Google Scholar] [CrossRef]
- Badu, P.; Pager, C.T. Activation of ATF3 via the Integrated Stress Response Pathway Regulates Innate Immune and Autophagy Processes to Restrict Zika Virus. bioRxiv 2023, 2023.07.26.550716. [Google Scholar] [CrossRef]
- Bernier, A.; Cleret-Buhot, A.; Zhang, Y.; Goulet, J.-P.; Monteiro, P.; Gosselin, A.; DaFonseca, S.; Wacleche, V.S.; Jenabian, M.-A.; Routy, J.-P.; et al. Transcriptional Profiling Reveals Molecular Signatures Associated with HIV Permissiveness in Th1Th17 Cells and Identifies Peroxisome Proliferator-Activated Receptor Gamma as an Intrinsic Negative Regulator of Viral Replication. Retrovirology 2013, 10, 160. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A model of regulation of ATF4 and integrated stress kinases during HIV infection. Blue arrows correspond to interactions demonstrated by the literature in the context of HIV infection. Yellow arrows correspond to reports in a different context. Besides ATF4, cellular proteins are indicated in green and HIV proteins in red. Activation of ISR kinases by either ER stress (PERK and PKR), HIV RNA (PKR), heme depletion and cleavage of DELE1 by OMA1 after mitochondrial stress (HRI), amino acid deprival (GCN2), oxidative stress (FAM69C) and proteotoxic stress (MARK2). Several cellular and viral proteins modulate ISR kinases during HIV infection, as TRBP and ADAR1 and the HIV-1 Tat protein preventing the phosphorylation of eIF2α and increasing ATF4 synthesis. The viral Vpu protein stabilizes ATF4 by opposing its ubiquitination by the SCF-βTrCP complex. Created with BioRender.com.
Figure 1.
A model of regulation of ATF4 and integrated stress kinases during HIV infection. Blue arrows correspond to interactions demonstrated by the literature in the context of HIV infection. Yellow arrows correspond to reports in a different context. Besides ATF4, cellular proteins are indicated in green and HIV proteins in red. Activation of ISR kinases by either ER stress (PERK and PKR), HIV RNA (PKR), heme depletion and cleavage of DELE1 by OMA1 after mitochondrial stress (HRI), amino acid deprival (GCN2), oxidative stress (FAM69C) and proteotoxic stress (MARK2). Several cellular and viral proteins modulate ISR kinases during HIV infection, as TRBP and ADAR1 and the HIV-1 Tat protein preventing the phosphorylation of eIF2α and increasing ATF4 synthesis. The viral Vpu protein stabilizes ATF4 by opposing its ubiquitination by the SCF-βTrCP complex. Created with BioRender.com.
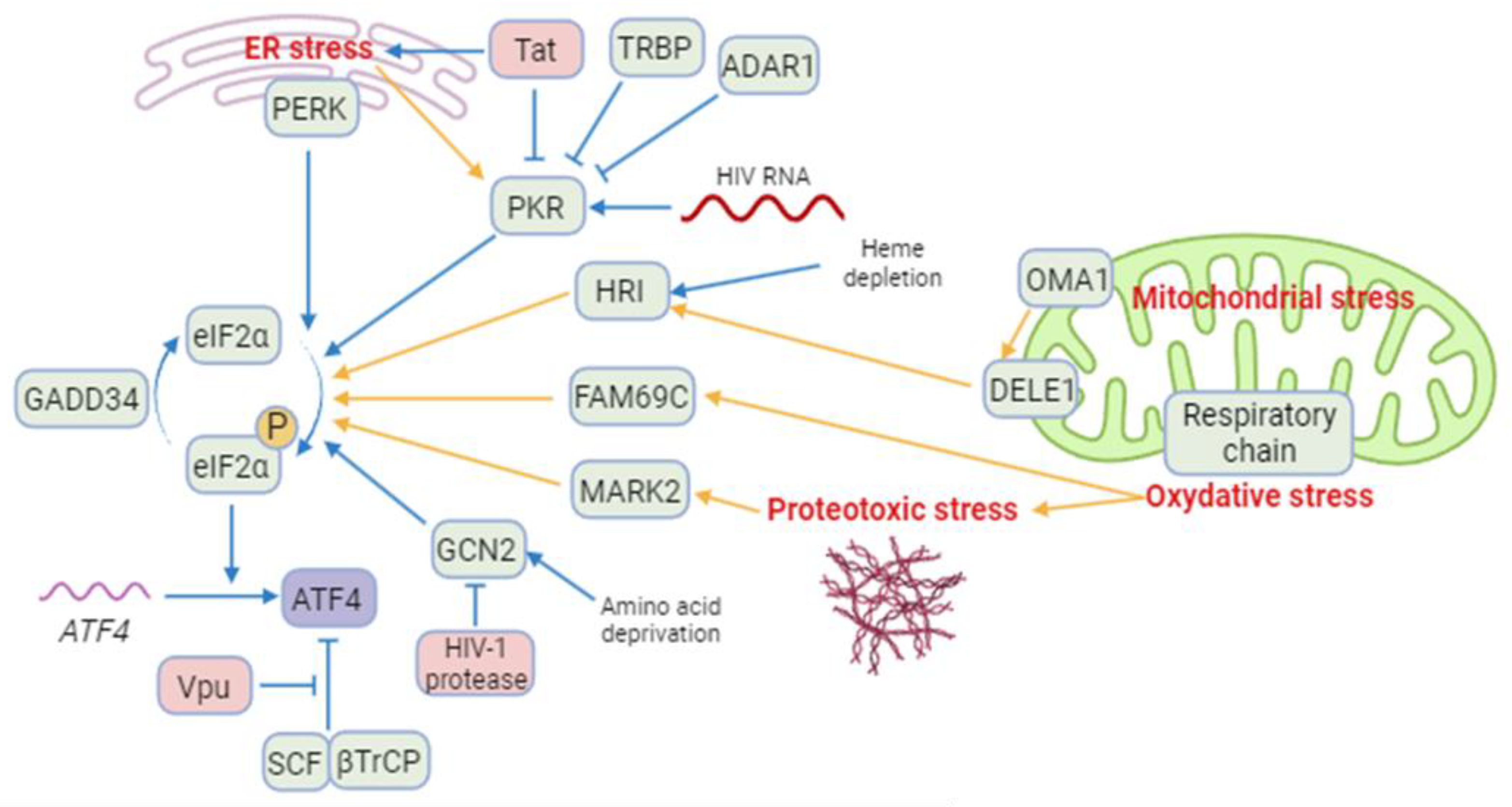
Figure 2.
ATF4 in the HIV cycle. The HIV cycle begins with the attachment of the virus to the cell surface thanks to the CD4 and CCR5 receptors (1). This allows the fusion between the viral envelope and the host cell membrane (2). During this phase, the viral nucleocapsid enters the cell cytoplasm. Single-stranded RNA molecules are then retro-transcribed in the nucleus (3). The viral DNA then integrates into the host cell genome (4), where it can remain latent until reactivation. The exit from the latency is stimulated by ATF4, which, in cooperation with the viral protein Tat, activates the HIV-1 promoter located in the LTR region of the viral DNA leading to viral RNA (5). These HIV messenger RNAs are exported from the nucleus to be translated (6) into proteins that assemble to form the internal structure of viral particles (7). Budding from the cell membrane (8) culminates with the release and maturation of viral particles in the extracellular environment (9). ATF4 is also capable of inducing the activation of target genes involved in cellular processes such as autophagy, inflammation and apoptosis. Adapted from “Disease Mechanism - Infectious Diseases, HIV Replication Cycle”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates.
Figure 2.
ATF4 in the HIV cycle. The HIV cycle begins with the attachment of the virus to the cell surface thanks to the CD4 and CCR5 receptors (1). This allows the fusion between the viral envelope and the host cell membrane (2). During this phase, the viral nucleocapsid enters the cell cytoplasm. Single-stranded RNA molecules are then retro-transcribed in the nucleus (3). The viral DNA then integrates into the host cell genome (4), where it can remain latent until reactivation. The exit from the latency is stimulated by ATF4, which, in cooperation with the viral protein Tat, activates the HIV-1 promoter located in the LTR region of the viral DNA leading to viral RNA (5). These HIV messenger RNAs are exported from the nucleus to be translated (6) into proteins that assemble to form the internal structure of viral particles (7). Budding from the cell membrane (8) culminates with the release and maturation of viral particles in the extracellular environment (9). ATF4 is also capable of inducing the activation of target genes involved in cellular processes such as autophagy, inflammation and apoptosis. Adapted from “Disease Mechanism - Infectious Diseases, HIV Replication Cycle”, by BioRender.com (2023). Retrieved from https://app.biorender.com/biorender-templates.
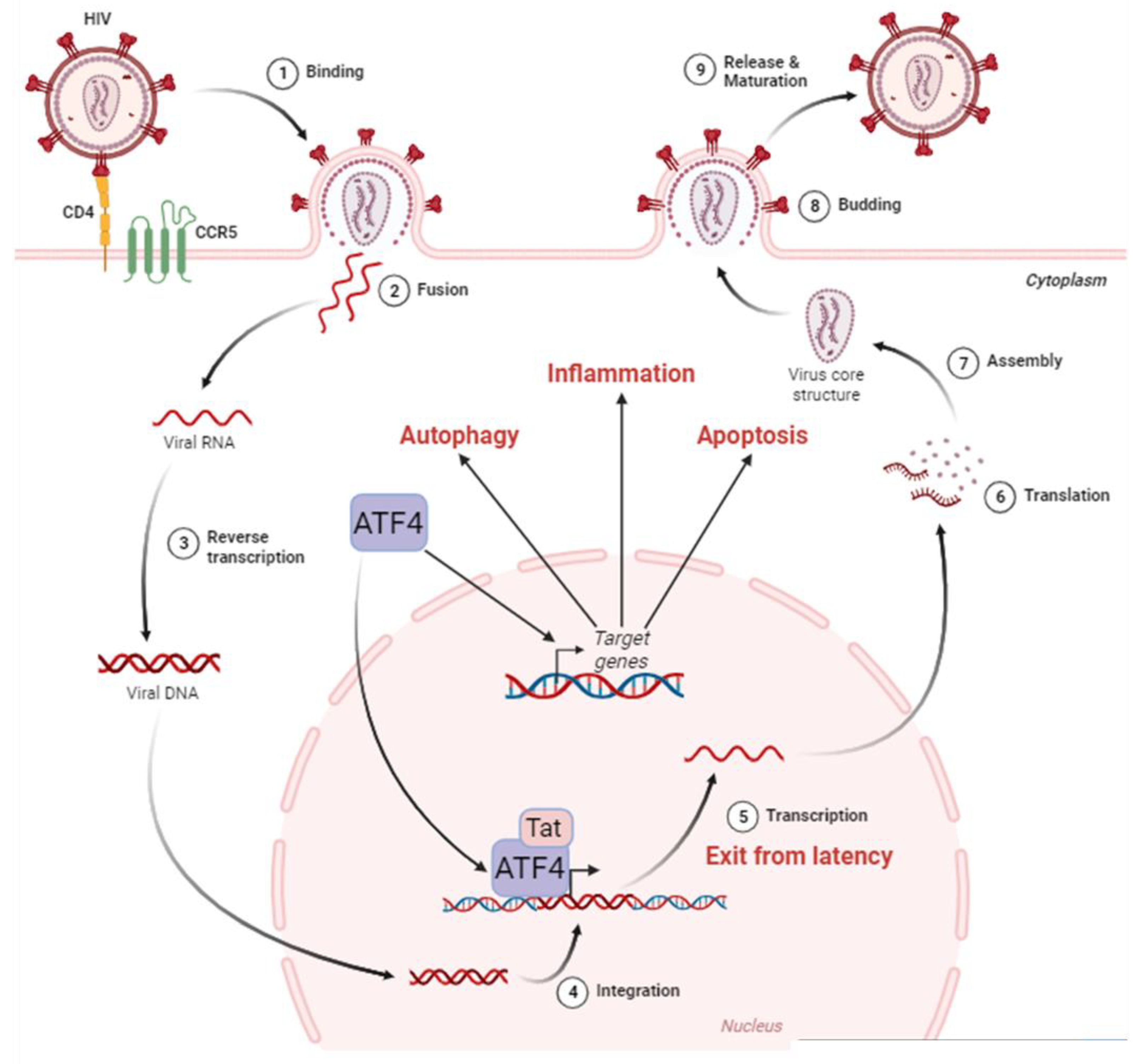
Figure 3.
ATF4 signaling pathways during HIV infection. Blue arrows correspond to interactions demonstrated in HIV literature. Yellow arrows correspond to other contexts. Besides ATF4, cellular proteins appear in green and HIV proteins in red. The genes in red are some of the pro-apoptotic genes induced by ATF4/ATF3 or ATF4/CHOP dimers and ATF4 dimerizing with a partner that remains to be determined. ATF5 also regulates the immune response at least by controlling immune cell differentiation. ATF4 also controls the expression of several pro-apoptotic genes including TID1/DNAJA3, G0S2 and TP53BP2. The genes in green are involved in ATF4-mediated survival as TMBIM5. Genes in purple are implicated in ATF4-induced autophagy like WIPI1, ATG12, ATG10, ATG16L1, ATG7, MAP1lc3B, GABARAPl2, p62/SQSTM1, NBR1 and REDD1. Finally, genes in pink are genes related to inflammation that are induced by ATF4 in a dimer with phosphorylated c-Jun or an undefined partner. Other genes activated by the phosphorylated ATF4/c-Jun dimer include RANTES and sICAM-1. Not shown in this diagram is IRF7 activation, which induces the production of type I and II interferons, and then activate the PKR/eIF2α/ATF4 pathway. Created with BioRender.com.
Figure 3.
ATF4 signaling pathways during HIV infection. Blue arrows correspond to interactions demonstrated in HIV literature. Yellow arrows correspond to other contexts. Besides ATF4, cellular proteins appear in green and HIV proteins in red. The genes in red are some of the pro-apoptotic genes induced by ATF4/ATF3 or ATF4/CHOP dimers and ATF4 dimerizing with a partner that remains to be determined. ATF5 also regulates the immune response at least by controlling immune cell differentiation. ATF4 also controls the expression of several pro-apoptotic genes including TID1/DNAJA3, G0S2 and TP53BP2. The genes in green are involved in ATF4-mediated survival as TMBIM5. Genes in purple are implicated in ATF4-induced autophagy like WIPI1, ATG12, ATG10, ATG16L1, ATG7, MAP1lc3B, GABARAPl2, p62/SQSTM1, NBR1 and REDD1. Finally, genes in pink are genes related to inflammation that are induced by ATF4 in a dimer with phosphorylated c-Jun or an undefined partner. Other genes activated by the phosphorylated ATF4/c-Jun dimer include RANTES and sICAM-1. Not shown in this diagram is IRF7 activation, which induces the production of type I and II interferons, and then activate the PKR/eIF2α/ATF4 pathway. Created with BioRender.com.
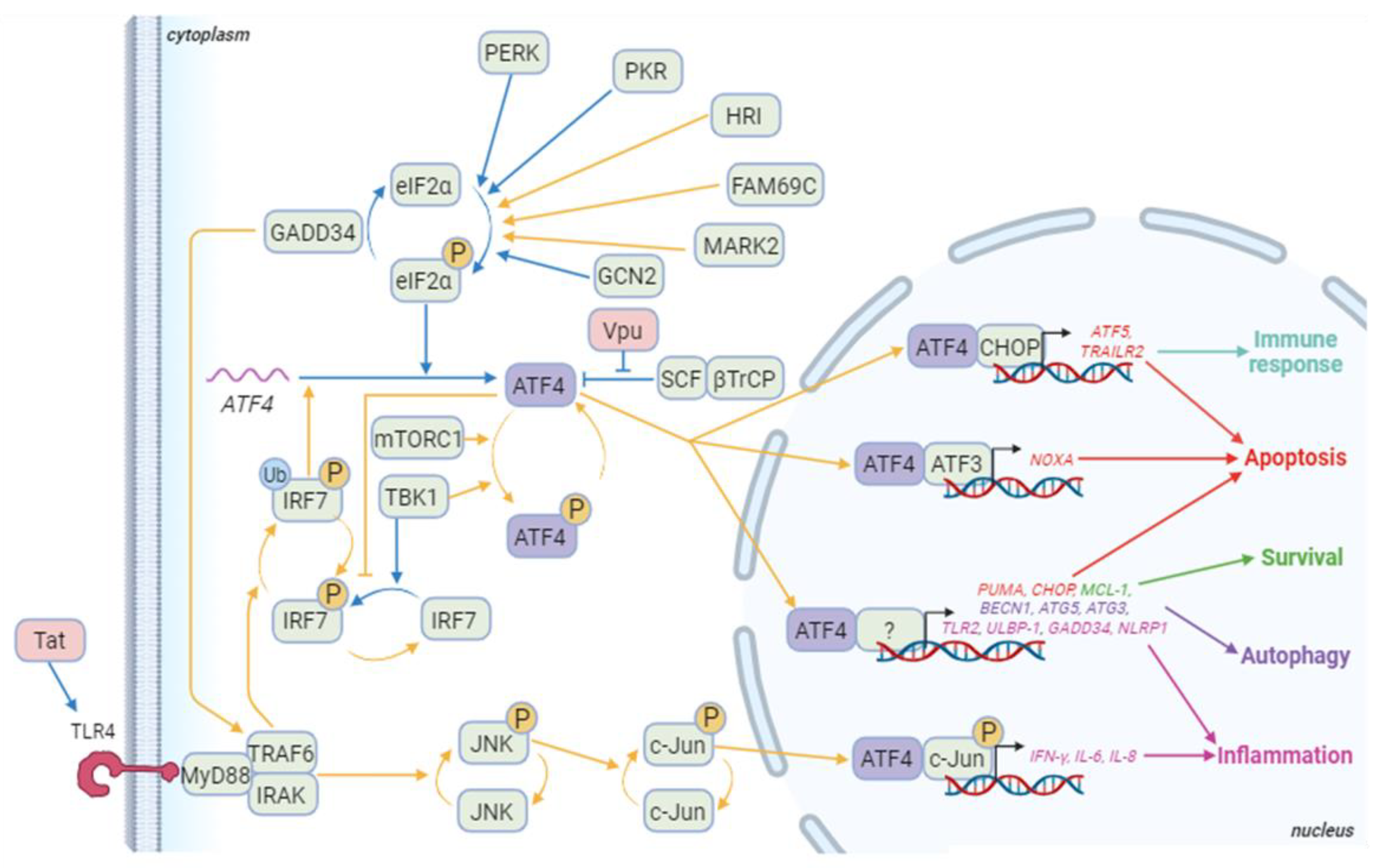
Figure 4.
Comparison of ATF4 and ATF5 proteins. Percentages of identity and similarity between amino acid sequences of ATF4 and ATF5 are shown. Both proteins contain a bZIP domain comprising a basic region for DNA binding and a leucine zipper motif for dimerization. ATF4 interacts with the p300 protein thanks to its N-terminal region (1-85), allowing its acetylation by p300 on lysine 311. Hydroxylation on prolines 60 and 235 results in a decrease in ATF4 transcriptional activity. ATF4 has a DSGICMS motif recognized by the β-TrCP factor to induce its degradation by the proteasome, following its phosphorylation by β-TrCP on its serine 219. Phosphorylation of ATF4 by β-TrCP on serine 224 also participates in this regulation. ATF5 can be acetylated on its lysine 29 by p300. The first 21 amino acids of ATF5 are involved in stabilizing the protein in response to interleukin-1β. The post-translational modifications of ATF4 and ATF5 are shown in the following color code: acetylation in orange, hydroxylation in blue, phosphorylation in green, ubiquitination in yellow, and SUMOylation in purple. Created with BioRender.com.
Figure 4.
Comparison of ATF4 and ATF5 proteins. Percentages of identity and similarity between amino acid sequences of ATF4 and ATF5 are shown. Both proteins contain a bZIP domain comprising a basic region for DNA binding and a leucine zipper motif for dimerization. ATF4 interacts with the p300 protein thanks to its N-terminal region (1-85), allowing its acetylation by p300 on lysine 311. Hydroxylation on prolines 60 and 235 results in a decrease in ATF4 transcriptional activity. ATF4 has a DSGICMS motif recognized by the β-TrCP factor to induce its degradation by the proteasome, following its phosphorylation by β-TrCP on its serine 219. Phosphorylation of ATF4 by β-TrCP on serine 224 also participates in this regulation. ATF5 can be acetylated on its lysine 29 by p300. The first 21 amino acids of ATF5 are involved in stabilizing the protein in response to interleukin-1β. The post-translational modifications of ATF4 and ATF5 are shown in the following color code: acetylation in orange, hydroxylation in blue, phosphorylation in green, ubiquitination in yellow, and SUMOylation in purple. Created with BioRender.com.
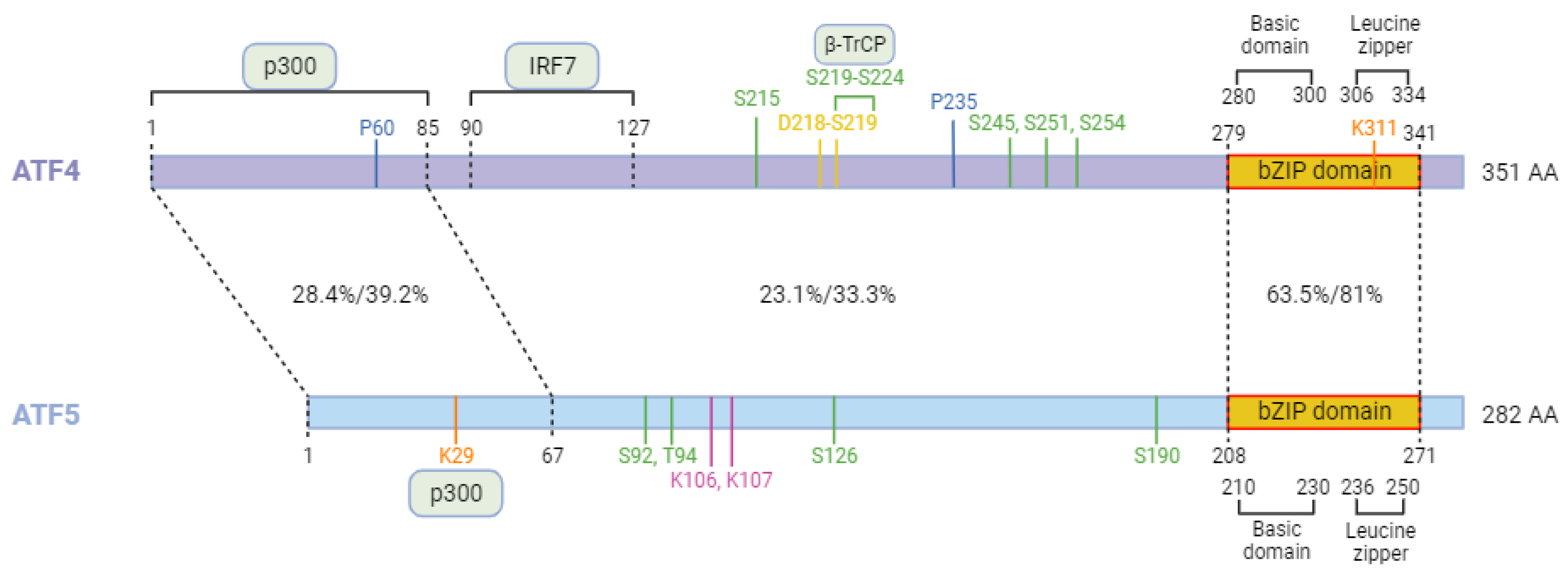

Table 1.
ATF4 regulation by viral infection and ATF4 role in viral replication.
| Virus family | Virus | Regulation of ATF4 (*) |
Effect of ATF4 regulation on viral replication (**) | Other major findings related to ATF4 and viral infection. |
Ref. |
|---|---|---|---|---|---|
| Adenoviridae | Adenovirus type 2 (AdV-2) | + (t) | ND | ATF4 transcript is transiently increased before being down-regulated after the onset of the adenovirus early gene expression. | [13] |
| Arteriviridae | Porcine reproductive and respiratory syndrome virus (PRRSV) | + (p) | [+] | ATF4 localizes to cytoplasmic viral replication complexes by the viral non-structural proteins nsp2/3. | [14] |
| Asfaviridae | African swine fever virus (ASFV) | - (p) | [+] | The viral protein DP71L inhibits the induction of ATF4 and its downstream target, CHOP, by promoting eIF2α dephosphorylation. | [15] |
| Bornaviridae | Borna disease virus (BDV) |
+ (p, n) | ND | ATF4 nuclear localization increases in cerebellar cells but not in the hippocampus of infected animals. | [16] |
| Caliciviridae | Rabbit hemorrhagic disease virus (RHDV) | + (t) | ND | ATF4 and CHOP mRNA levels increase are associated with apoptosis induction. | [17] |
| Circoviridae | Porcine circovirus type 2 (PCV2) | + (p) | [+] | The infection activates the PERK/eIF2α/ATF4/CHOP axis. | [18] |
| + (p) | ND | The viral proteins Replicase and Capsid induce the PERK/eIF2α/ATF4/CHOP axis. | [19] | ||
| + (t, p) | [+] | The viral protein ORF5 induces autophagy via the PERK/eIF2α/ATF4 and mTOR/ERK1/2/AMPK signaling pathways. | [20] | ||
| Coronaviridae | Coronavirus infectious bronchitis virus (IBV) |
+ (p) | [+] | ATF4 is up-regulated through PERK- and PKR-mediated eIF2α phosphorylation. | [21] |
| Nephropathogenic infectious bronchitis virus (NIBV) | + (t, p) | ND | Upon infection, the BiP/PERK/ATF4 signaling pathway is activated and induction of renal apoptosis is observed. | [22] | |
| Porcine deltacoronavirus (PDCoV) | + (t) | [-] | The infection activates the PERK/eIF2α/ATF4 axis and induces host translation attenuation. | [23] | |
| Porcine epidemic diarrhea virus (PEDV) | + (t, p, n) | ND | The ATF4 protein is present in apoptotic cells. | [24] | |
| Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) | - (p) | ND | Despite ISR activation and translational arrest, ATF4 and CHOP protein levels are not increased in infected cells. | [25] | |
| Flaviviridae | Bovine viral diarrhea virus (BVDV) |
+ (p) +/- (n) | [+] | Cytopathic BVDV induces ATF4 nuclear translocation and activates autophagy. Non-cytopathic BVDV induces ATF4 perinuclear localization but no autophagy. | [26] |
| Dengue virus (DENV) | + (n) | ND | None. | [27] | |
| + (p) | [+] |
None. | [28] | ||
| Hepatitis C virus (HCV) | + (p) | ND | ATF4 and the ATF6 pathways, contribute to the induction of CHOP in HCV replicon cells that showed an increased vulnerability to oxidant injury. | [29] | |
| + (t) | ND | HCV induces chronic ER stress. | [30] | ||
| + (p) | ND | The viral core protein induces the PERK/ATF4 branch of the UPR which up-regulates the autophagy gene ATG12. | [31] | ||
| + (t) | ND | ATF4 may contribute to autophagy regulation during infection. | [32] | ||
| + (t, p) | ND | Cells expressing HCV proteins and exposed to oxidative stress adapt to cellular stress through eIF2α/ATF4 activation. | [33] | ||
| Japanese encephalitis virus (JEV) |
+ (t) | ND | None. | [34] | |
| + (t) | ND | None. | [35] | ||
| + (p) | [-] | The viral protein NS4B activates PERK, which induces apoptosis via the PERK/ATF4/CHOP pathway. | [36] | ||
| Tembusu virus (TMUV) |
+ (t, p) | ND | CHOP induction leads to caspase-3 activation. | [37] | |
| West Nile virus (WNV) |
+ (p, n) | [+] | ATF4 is involved in the up-regulation of GSH levels and the inhibition of stress granule formation induced by -infection. | [38] | |
| Zika virus (ZIKV) | + (t) | ND | Upon infection, ATF4 transcript level is weakly increased. | [39] | |
| - (t) | ND | None. | [35] | ||
| + (p) | ND | The infection transiently activates ATF4 but phosphorylation of PERK and eIF2α is sustained. | [40] | ||
| Hepadnaviridae | Hepatitis B virus (HBV) | + (p) | ND | The reduction of intracellular ATP levels by the viral protein HBx induces ATF4 binding to the promoter of the COX2 gene and its transcription. | [41] |
| - (p) | ND | The viral HBx protein localizes in the ER lumen and directly interacts with BiP. This interaction results in suppression of eIF2α phosphorylation, which decreases the levels of ATF4/CHOP/Bcl-2. | [42] | ||
| + (t, n) | ND | HBV, with viral polymerase carrying the rt269L polymorphism, improves mitochondrial dynamics and enhances the autophagic flux, mainly thanks to the activation of the PERK/eIF2α/ATF4 signaling. | [43] | ||
| Herpesviridae | Epstein-Barr virus (EBV) | + (p) | ND | LMP1 increases the ATF4 protein level through PERK/eIF2α phosphorylation. ATF4 transactivates LMP1. | [44] |
| Human cytomegalovirus (HCMV) | + (t,p) | ND | The infection activates PERK, but the amount of phosphorylated eIF2α is limited and no translation attenuation is detected. | [45] | |
| + (p) | ND | The viral protein pUL38 induces phosphorylation of PERK and eIF2α, resulting in the accumulation of the ATF4 protein and cell protection against ER stress. | [46] | ||
| + (p) | ND | The viral protein UL148 activates ATF4 mainly through the PERK/eIF2α pathway. | [47] | ||
| Human herpes virus 6A (HHV-6A) |
+ (p) | ND | Induction of the PKR/eIF2α pathway results in a moderate increase of the ATF4 protein level, which peaks at the final stages of infection. | [48] | |
| Human herpes virus-8 (HHV-8) |
+ (t, p) | [+] | ATF4 induces MCP-1 production and pro-angiogenic properties in endothelial cells. | [49] | |
| + (p) | [+] | The viral protein ORF45 increases eIF2α phosphorylation and ATF4 translation, which in turn up-regulates the expression of lysosome-associated membrane protein 3 (LAMP3). | [50] | ||
| Herpes simplex virus-1 (HSV-1) |
+ (t, p) | ND | HSV-1 disarms the ER UPR in the early stages of viral infection. The activity of the eIF2α/ATF4 signaling increases at the final stage of HSV-1 replication. | [51] | |
| Murine cytomegalovirus (MCMV) | + (p) | [+] | MCMV activates the PERK/ATF4 pathway but only induces a subset of ATF4 targets. ATF4 is required for efficient viral DNA synthesis and late gene expression during a low-multiplicity infection. | [52] | |
| Murine gamma herpes virus 68 (MHV68) |
+ (p) | [-] | In response to ER stress, ATF4 inhibits B-cell receptor (BCR)-mediated MHV68 lytic gene expression by directly inhibiting the transcription of RTA, the MHV68 lytic switch transactivator. In a negative feedback loop, UPR-induced CHOP is required for and promotes BCR-mediated MHV68 lytic replication by decreasing upstream BiP and ATF4 protein levels. | [53] | |
| Pseudorabies virus (PRV) | + (t) | [+] | The eIF2α/ATF4 pathway is activated during infection. PRV-induces apoptosis in later stages of infection through the CHOP/Bcl-2 axis. Overexpression of BiP or ER stress-inducing treatment can enhance PRV production. | [54] | |
| + (t, p) | [+] | Infection-induced ER stress leads to PERK activation and up-regulation of ATF4, CHOP, and GADD34. | [55] | ||
| Paramyxoviridae | Newcastle disease virus (NDV) | + (p, n) | [+] | The PKR/eIF2α/ATF4 pathway leads to an increase in GADD34 protein level. GADD34, in conjunction with PP1, dephosphorylates eIF2α and restores global protein translation, benefiting virus protein synthesis. | [56] |
| + (p) | [+] | Induction of the PERK/eIF-2α/ATF4/CHOP signaling pathway is involved in the cyclin D1 dependent G0/G1 phase cell cycle arrest. | [57] | ||
| Sendai Virus (SV) | + (p) | ND | IRF7 up-regulates ATF4 activity and protein level, whereas ATF4 in return inhibits IRF7 activation. | [58] | |
| Parvoviridae | Porcine parvovirus (PPV) |
+ (t) | [-] | CHOP inhibits PPV replication by promoting apoptosis. ATF4 knockdown promotes PPV replication. | [59] |
| Picornaviridae | Foot-and-mouth disease virus (FMDV) |
+ (p) | [+] | The capsid protein VP2 induces autophagy through the eIF2α/ATF4/AKT/mTOR cascade, and interacts with HSPB1. | [60] |
| Group B coxsackievirus (CVB) | - (p) | [+] | PERK is activated and eIF2α is phosphorylated, but ATF4 protein levels do not increase. The ATF4/CHOP branch is blunted, thus inhibiting cell death. | [61] | |
| Poxviridae | Myxoma virus (MYXV) |
+ (t) - (p) | ND | PERK is activated and eIF2α is phosphorylated, but ATF4 translation is inhibited, which prevents MCL1 and CHOP transactivation. | [62] |
| Reoviridae | Reovirus | + (p) | [+] | The relative impact of ATF4 on viral replication depends on the infecting viral strain. | [63] |
| Rhabdoviridae | Vesicular stomatitis virus (VSV) |
ND | [+] | None. | [58] |
| Togaviridae | Chikungunya virus (CHIKV) | - (t) | ND | ER UPR induction is primed since the phosphorylation of eIF2α and partial splicing of the XBP1 mRNA are detected, but the viral protein nsP2 inhibits the transcription of a reporter gene under the control of the ATF4 promoter. | [64] |
| Venezuelan equine encephalitis virus (VEEV) |
+ (p) | ND | None. | [65] |
(*) ATF4 activation induced by viral infection is assessed by the increase (+) or not (-) of ATF4 protein (p) or transcript (t) levels or by ATF4 nuclear localization (n). (**) The effect of ATF4 on viral replication is positive [+], negative [-] or not determined (ND).
Table 2.
ATF4 promotes HIV-1 replication and reactivation.
| Models | Major findings | Ref. | |
|---|---|---|---|
| Replication | HIV infected CD4+ Jurkat T cells. | Cell transfection with an ATF4-encoding plasmid up-regulates the HIV-1 proviral genome levels (qPCR of gag gene) and increases viral release (ELISA of p24). | [6] |
| 293T cells transiently transfected with a plasmid encoding the HIV-1 genome and GFP gene. | siRNA directed against ATF4 transcripts decreases the viral titer (ELISA of p24) and Gag protein level (WB). | [7] | |
| Reactivation | U1 cells * | Cell nucleofection with an ATF4-encoding plasmid increases the viral load in the cell culture supernatant (qPCR of gag gene and p24 levels by WB). | [6] |
| J-Lat A1** and U1 cells* treated by a GCN2 inhibitor or supplemented with amino acids. | Inhibition of GCN2/ATF4 signaling represses the transcription of HIV-1 (real time qPCR with LTR primers). |
[8] | |
| J-Lat cells and CD4+ T cells from HIV-1 infected individuals | FOXO1 inhibitor-induced reactivation of HIV-1 is reduced by pharmacological inhibition of PERK/ATF4 (GFP reporter gene or dddPCR with LTR primers). | [9] | |
| J-Lat A1, 2D10*** cells and primary CD4+ T cells | Induction of the ISR/ATF4 signaling with a specific agonist of BiP, induces HIV-1 transcriptional activity (real time qPCR with LTR primers). | [10] | |
*U1 cell lines: subclone of the human U937 monocytic cell line that is infected with HIV-1. **J-Lat A1 cell lines: Jurkat T cells containing one integrated copy of HIV-1 LTR that controls expression of the EGFP reporter gene. ***2D10 cells: Jurkat T cells carrying a lentiviral vector that expresses a mutated Tat carrying the H13L mutation, which enhances viral gene silencing, Rev and a destabilized green fluorescent protein-encoding gene, which has a reduced half-life, in place of the Nef gene.
Table 3.
ATF4 target genes involved in apoptosis and HIV-1 infection.
| ATF4 target genes |
Model related to HIV infection | Major findings | Ref. |
|---|---|---|---|
| BIM/BCL2L11 | T cells derived from BIM−/− knockout mice treated with Tat. | BIM facilitates Tat-induced apoptosis. | [181] |
| CD4+ T cells from pathogenic SIVmac251-infected rhesus macaques. | Infection by SIV up-regulates death ligand CD95L and proapoptotic BIM and BAK but not BAX protein levels. | [116] | |
| Latently HIV-1-infected macrophages and lymph nodes, and brain of HIV-infected individuals without detectable viral replication. | BIM is up-regulated and recruited into mitochondria both in vitro and in vivo in latently infected cells that are protected from apoptosis. | [182] | |
| SH-SY5Y cells treated with Tat. |
FOXO3 down-regulates BCL2 transcript and protein levels and up-regulates BIM transcript and protein levels after entering the nucleus, eventually causing cellular apoptosis. | [183] | |
| Monocytes-derived macrophages purified from PBMCs*. | Immunofluorescence analysis shows structural alterations in the mitochondrial architecture and an increase of BIM protein levels in the cytoplasm of infected cells. | [184] | |
|
TID1/ DNAJA3 |
CEM-GFP cells transfected with a plasmid encoding the HIV-1 genome and GFP gene. | HIV-1 infection increases TID1 transcript levels. | [185] |
| HEK-293T cells transfected with either a Luciferase-encoding reporter vector or a plasmid encoding the HIV-1 genome and GFP gene. | TID1 increases HIV-1 LTR-driven gene expression and the viral p24 antigen release. | [186] | |
| G0S2 | PBMCs and MDDCs treated with virus-like particles containing the HIV-1 Pr55gag precursor protein and gp120 molecule anchored through the trans-membrane portion of the Epstein-Barr virus gp220/350. | G0S2 transcript levels are increased in dendritic cells. | [187] |
| THP-1 cells infected with a replication-deficient HIV-1 encoding the envelope glycoproteins from the vesicular stomatitis virus (VSV-G). | G0S2 transcript levels are down-regulated in cells containing an integrated provirus, compared to bystander uninfected cells or cells harboring pre-integration viral complexes. | [188] | |
| MCL1 | PBMCs of HIV-1-infected individuals. | Apoptosis and viral load are inversely correlated with MCL1 mRNA levels. | [189] |
| Monocyte-derived macrophages purified from PBMCs. | The expression of the MCL1 gene is up-regulated in macrophages infected with wild-type HIV-1 and in mock-infected macrophages that had been stimulated with M-CSF. However, MCL1 is not up-regulated in macrophages infected with a Δenv HIV-1. | [190] | |
| PBMCs of HIV-infected patients before and during successful ART. | After 12 months of therapy, the expression of MCL1 appears significantly up-regulated. | [191] | |
| Monocyte-derived macrophages or monocyte-derived dendritic cells incubated with R5 HIV-1Bal. | HIV-1 infection decreases the Mcl-1 protein level but increases Bax and Bak. | [192] | |
| Vpr-treated monocyte-derived macrophages. | Resistance to Vpr-induced apoptosis is specifically mediated by cIAP1/2 genes independently from Bcl-xL and Mcl-1, which play a key role in maintaining cell viability independently of the viral protein. | [193] | |
| HIV-infected macrophages and microglia. | Cells become viral reservoirs in response to acute infection through a BIM-dependent mechanism. | [182] | |
| THP-1-derived macrophages. | HIV-1 infection increases expression of the anti-apoptotic genes MCL1, BCL2 and BCL2L1 that encodes Bcl-xL. | [194] | |
| PBMCs of uninfected donors and HIV-positive patients treated by cART*. | Overexpression of MCL1 is detected in PBMCs of cART-treated patients. | [195] | |
| Neutrophils from either healthy individuals, or HIV patients whether asymptomatic, symptomatic, or ART receivers. | HIV-1 infection increases MCL1 transcript levels in vivo, and ART partially reduces this increase. | [196] | |
|
NOXA/ PMAIP1 |
Human CD4+ T cells infected with HIV-1 viruses lacking Env, Vpr, or Nef. Human PBMCs infected with wild-type HIV-1 viruses of different tropisms. | HIV-1 infection increases NOXA transcript levels, which is associated with cell death. | [197] |
| PUMA/BBC3 | Circulating CD4+ lymphocytes from untreated HIV-1 infected donors. | HIV-1 infection increases Puma protein levels, which drop upon ART. | [198] |
| HIV-associated encephalitis brain sections. | HIV infection increases the Puma protein level in dying syncytia and neurons. | [199] | |
| Murine cortical neuron culture treated with gp120 III. | Gp120 III is sufficient to increase Puma protein levels and induce cell death. | [200] | |
| CD4+ primary T cells infected with HIV-1 lacking Env, Vpr, or Nef genes. | The Env, Vpr and Nef are not necessary for HIV-1-induced PUMA transcript levels increase and HIV-mediated cell death. | [197] | |
| TMBIM5/GHITM | Monocytes from control and HIV patients. | TMBIM5 transcript levels are decreased in HIV-infected monocytes. | [201] |
| Brain from HIV-HAND* patients. | TMBIM5 transcript levels are increased mainly in HIV-HAND patient astrocytes. | [202] | |
|
TP53BP2/ ASPP2 |
Primary cortical neuron cultures treated with gp120 protein. | A high dose of gp120 stimulates the interaction of TP53BP2 with p53, which induces BAX transcription and contributes to caspase-3 cleavage. | [203] |
| SH-SY5Y neuroblastoma cells treated with gp120 protein. | TP53BP2 regulates autophagy and apoptosis differently depending on the dose of gp120. | [204] |
*PBMC: Peripheral blood mononuclear cell; cART: combined antiretroviral therapy; VLDL: Very low density lipoprotein; HAND: HIV-associated neurocognitive disorder.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



