Submitted:
23 January 2024
Posted:
24 January 2024
You are already at the latest version
Abstract
Cancer is a devastating disease with female breast cancer becoming the leading cause of global cancer morbidity. Herein we report the design and synthesis of a sorafenib analog 1A and its base-promoted arranged compound 1A2. Their in vitro cytotoxicity and potencies in EFGR inhibition study revealed that compound 1A displayed good antiproliferative activity in MDA-MB-231 (breast) cells with an IC50 value of 16.18±1.42 μM. Crystallization 1A2 affored 1A2a as a solvate with dichloromethane which was explicitly confirmed by the single crystal X-ray diffraction analysis.
Keywords:
Sorafenib analogue
; crystal structure
; rearranged compound
; anticancer activity
1. Introduction
Cancer is a devastating disease, ranking as a leading cause of premature death worldwide. According to the International Agency for Research on Cancer, 19.3 million new cancer cases were diagnosed and almost 10.0 million cancer deaths were occurred worldwide in 2020, with female breast cancer occupying 11.7% of all cancer cases as the leading cause of global cancer morbidity[1]. An estimated 34 million new cancers are projected to occur in 2070, doubling the number relative to 2020, highlighting the desire need for potent anticancer agents[2].
Sorafenib, an orally active anticancer drug marketed as Nexavar by the companies Bayer (Leverkusen, Germany) and Onyx Pharmaceuticals (San Francisco, California)[3], was approved for the standard treatment of advanced renal cell carcinoma and advanced HCC[4]. It exhibited activity against a wide spectrum of tumor types, including renal cell, hepatocellular, breast, and colorectal carcinomas in the preclinical setting. Mechanism of actions can be ascribed to its multiple known protein kinase targets[5]. It is likely to modulate two biological pathways that are implicated in cancer by binding to and shutting down key receptor tyrosine-kinase enzymes[3].
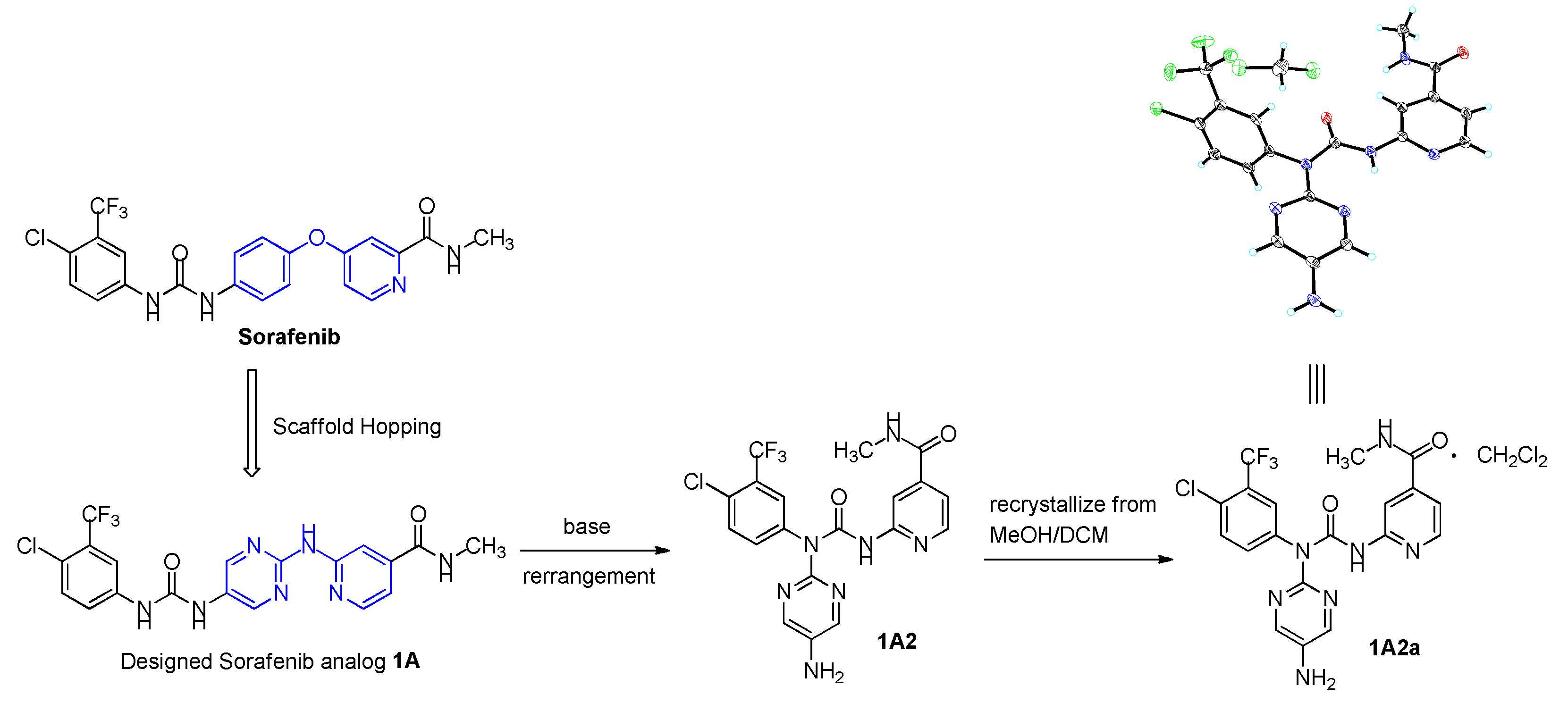
Design and synthesis sorafenib analogs can be an efficient way to discover potent cancer therapeutic agents. The epidermal growth factor receptor (EGFR) is a receptor tyrosine kinase, functions an important role in cell proliferation, survival, migration, adhesion, and differentiation, and is a precious target for cancer drug development. Curiously, many third-generation of EGFR tyrosine kinase inhibitors contain a 2-amino-pyrimidine scaffold (Figure 1)[6]. Inspired by these structural property, in this study, we design to keep the left wing of Sorafenib unchanged, and the central 4-phenoxy-pyridine was changed to 2-(pyrimidin-2-ylamino)pyridine, and identified a novel Sorafenib analogue 1A, capitalizing upon the scaffold hopping strategy (Figure 2). Captivating, 1A could be rearranged to 1A2 under an efficient base condition. Crystallization 1A2 from mixed solvent of methanol and dichloromethane identified 1A2a as a dichloromethane solvate (Figure 3). Then, the new compounds of 1A and 1A2 were biologically evaluated for their in vitro cytotoxicity and potencies in EFGR inhibition (Table 3).
2. Results
2.1. Chemistry
A five-step sequence was developed to forge 1A (Scheme 1). SN 2 reaction of methyl 2-aminopyridine-4-carboxylate with 2-chloro-5-nitropyrimidine, followed by nitro reduction (H2, Raney Ni), afforded intermediate 3, that underwent addition to 1-chloro-4-isocyanato-2-(trifluoromethyl)benzene, accomplishing the formation of intermediate 4. Then hydrolysis of methyl ester of 4, afforded carboxylic acid 5 that was transformed to carboxamide 1A under coupling reagents condition (HATU, DIEA). Finally, base-mediated (NaH, DMF) rearrangement of 1A delivered 1A2 (Figure 3) in high yield. Crystallization 1A2 from mixed solvent of methanol and dichloromethane obtained 1A2a as a solvate with dichloromethane.
2.2. Crystal structure of compound 1A2a
Single-crystal X-ray structure analysis reveals that compound 1A2a is a dichloromethane solvate at the stoichiometric ratio 1:1 and crystallizes in the triclinic system space group P1 with lattice parameters of a = 0.98517(7) nm, b = 1.02973(7) nm and c = 1.14712(8) nm). The crystal size is 0.340×0.270×0.080 mm3 (Table 1). The crystal structure analysis of interatomic distances in crystal 1A2a indicates the presence of a set of intramolecular C-F7…H51-C, C-O10…H28-C, C-Cl1…C42-F, C-O10…C41 interaction, one N17…H12-N hydrogen bond and intermolecular C-O10…H48-C interaction formed between the carbonyl group and the solvate molecule (Table 2). By study and comparison their geometrical parameters, it is possible to suggest that all of these interactions are weak, except for N17…H12-N hydrogen bond (Figure 3). View of the pack drawing of 1A2a is shown in Figure 4.
2.3. Biology
The cytotoxicity of compound 1A, 1A2 on five human cancer cell lines, including A549 (lung), HepG-2 (hepatocellular carcinoma, HCC), HCT116 (colon carcinoma), MDA-MB-231 (breast), PC-3 (prostate) were determined by an MTS method (Table 3). The effect of the two compounds on EGFR kinase was assessed using the EGFR Kinase Enzyme System (Promega) (Table 3). Cytotoxicity results showed that compound 1A displayed good antiproliferative activity in MDA-MB-231 (breast) cells with an IC50 value of 16.18±1.42 μM. However, it was found to be ineffective on other cell lines. To our disappointment, compound 1A2 exerted no potencies in the five cancer cell lines (IC50s>40 μM). Both compounds show little ability to inhibit the EGFR kinase, with compound 1A2 displayed relatively higher inhibition percentage than compound 1A at 40 μM concentration.
3. Experimental Section
3.1. Materials and physical measurements
NMR spectra were recorded on Bruker DRX-400 instrument with TMS as internal standard. The Chemical shifts (δ ppm) are calibrated with reference to the deuterated solvent DMSO (δ 2.50 for 1H NMR and 39.52 for 13C NMR); CD3OD (δ 3.31 for 1H NMR and 49.00 for 13C NMR), and coupling constants (J) are given in Hz. Silica gel column chromatography (200-300 mesh) was performed with various combinations of dichloromethane and methanol as eluent to purify compounds. ESI-MS analysis was determined on API QSTAR Pulsari spectrometer. Infrared spectra were recorded on a FT-IR spectrometer with KBr pellets. Yields refer to purified, dried and spectroscopically (1H NMR) homogeneous material. All the commercial chemicals were used without further purification.
3.2. Crystal structural determination
The crystal data for compound 1A2a was collected at 100(2) K on APEX DUO using Cu Kα radiation (λ = 1.54178Å). Data absorption corrections were applied using Semi-empirical from equivalents methods. This structure was solved using refinement method of full⁃matrix least⁃squares against F2 with all non⁃hydrogen atoms anisotropic and hydrogen atoms isotropic. The final R1 values were 0.0932 with I > 2σ(I). Maximum and minimum transmission factors were 0.73 and 0.29, respectively. 18242 unique reflections were measured giving 4496 independent reflections (Rint = 0.0875). An ORTEP representation of the coordination environment of the compound 1A2a including the atom labeling scheme is shown in Figure 3. The diffraction data is listed in . The selected bond, interatomic distances and torsion angles is listed in Table 2.
3.3. Cancer Cell Growth Inhibition Assay
The human tumor cell lines A549, HepG2, HCT116, MDA-MB-231, and PC-3 which were obtained from ATCC (Manassas, VA, USA) were cultured in Dulbecco's modified Eagle's medium (DMEM) or RMPI-1640 containing 10% heat-inactivated fetal bovine serum (FBS) at 37 oC in 5% CO2, then seeded in 96-well plates at the concentration of 3000-5000 cells per well for 12-24 h when they were growing exponentially. The cells were treated with all compounds in different concentrations in triplicates, and incubated for another 48 h. A 20 μL of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfopheny)-2H-tetrazolium, inner salt (MTS, 1.9 mg/mL) and 100 μL culture medium were added to all wells and stained for 2-4 h at 37oC. The absorbance was measured with a MULTISKAN FC reader at λ = 492 nm. Data from triplicate experiments were expressed as mean ± SD. The IC50 value of each compound was calculated using Reed and Muench’s method.
3.4. Cell-free EGFR Kinase Assay
The inhibitory percentage against EGFR kinase of compounds 1A, 1A2 at 40 μM was measured using the EGFR Kinase Enzyme System (Promega). Operation steps strictly followed instructions from the manufacturer. EGFR (3 units/reaction), reaction buffer A, MnCl2, DTT and peptide substrate were added to compound and incubated at 30 °C for 10 min followed by addition of ATP and an hour incubation at 30 °C. ADP-Glo™ Reagent was then added and incubated for another 40 min at 30 °C. A final 30 min incubation was needed after the addition of the Kinase Detection Reagent. Luminescence was measured using FlexStation 3.
3.5. Synthesis of Methyl 2-(N-(5-nitropyrimidin-2-yl)amino)pyridine-4-carboxylate hydrochloride (2)
To a solution of methyl 2-aminopyridine-4-carboxylate (1) (10.0 g, 65.8 mmol) in DMF (150 mL) was added 2-chloro-5-nitropyrimidine (10.5 g, 65.8 mmol) at 0 oC. Then mixture was warmed to room temperature and stirred overnight under nitrogen atmosphere. Upon completion of the reaction as indicated by TLC, the mixture was vacuum-filtered,and the filter cake was purified via silica gel column chromatography with DCM and MeOH as the eluent (10: 1 DCM: MeOH) to afford compound 2 as a yellow solid (13.0 g, 63%). 1H NMR (400 MHz, DMSO-d6) δ 11.17 (br s, 1 H), 10.12 (br s, 1 H), 9.85 (s, 2 H), 8.67 (d, J = 7.4 Hz, 1H), 8.04 (s, 1 H), 7.27 (d, J = 7.3 Hz, 1H), 3.95 (s, 3 H); IR (KBr) νmax (cm-1): 3430, 3234, 3033, 2958, 2852, 1732, 1671, 1606, 1531, 1418, 1349, 1277, 1116, 867, 765, 648, 532, 429; ESI-MS displays a peak at m/z 276.1, calcd for [M + H]+: 276.2282.
3.6. Synthesis of Methyl-2-(N-(5-aminopyrimidin-2-yl)amino)pyridine-4-carboxylate hydrochloride (3)
To a solution of compound 2 (13.0 g, 41.7 mmol) in MeOH (100 mL) was added Raney Ni (3.0 g). The reaction mixture was degassed and back filled with hydrogen for three times, then attached to a hydrogen source, stirred vigorously at room temperature overnight. The catalyst was removed by filtration and the filtrate was concentrated to give a crude material which was purified by flash column chromatography (silica, 10: 1 DCM: MeOH) to yield compound 3 as a yellow solid (5.2 g, 44.2%). 1H NMR (400 MHz, CD3OD) δ 8.77 (d, J = 7.4 Hz, 1H), 8.31 (s, 2 H), 7.72 (d, J = 1.6 Hz, 1 H), 7.32 (dd, J1 = 7.4 Hz, J2 = 1.6 Hz, 1 H), 4.00 (s, 3 H); IR (KBr) νmax (cm-1): 3414, 3236, 3119, 1723, 1674, 1627, 1583, 1441, 1301, 1204, 996, 831, 756, 721, 669, 553; ESI-MS displays a peak at m/z 246.4, calcd for [M + H]+: 246.2453.
3.7. Synthesis of 1-(4-chloro-3-(trifluoromethyl)phenyl)-3-(2-((4-(methyloxycarbonyl)pyridine-2-yl)amino)Pyrimidin-5-yl)urea (4)
To a solution of compound 3 (5.2 g, 18.4 mmol) and Et3N (3.7 g, 36.8 mmol) in DCM (100 mL) was added 1-chloro-4-isocyanato-2-(trifluoromethyl)benzene (7.0 g, 31.8 mmol) at 0 oC. Then the reaction progressed at room temperature overnight. Upon completion the reaction, the mixture was washed with brine (30 mL x 3), dried over Na2SO4, and evaporated under vacuum to give a crude material, which was purified via silica gel column chromatography with DCM and MeOH as the eluent (10: 1 DCM: MeOH) to afford compound 4 as a yellow solid (5.0 g, 58%). 1H NMR (400 MHz, CD3OD) δ 9.34 (s, 1 H), 9.25-9.10 (m, 1 H), 8.38 (s, 2 H), 8.07 (s, 1 H), 7.84 (d, J = 6.7 Hz, 1 H), 7.75 (dd, J1 = 8.7 Hz, J2 = 2.3 Hz, 1 H), 7.58 (d, J = 8.8 Hz, 1 H), 4.06 (s, 3 H); IR (KBr) νmax (cm-1): 3387, 3344, 3214, 3120, 2928, 1735, 1679, 1583, 1556, 1435, 1334, 1200, 1131, 902, 836, 759, 722; ESI-MS displays a peak at m/z 467.3, calcd for [M + H]+: 467.8090.
3.8. Synthesis of 1-(4-chloro-3-(trifluoromethyl)phenyl)-3-(2-(4-carboxypyridine-2-yl)amino)Pyrimidin-5-yl)urea (5)
To a solution of compound 4 (5.0 g, 10.7 mmol) in MeOH (100 mL) was added 2 N LiOH (10.7 mL, 21.4 mmol). The mixture was stirred at room temperature overnight. After consumption of the starting material, the mixture was acidified with 2 N HCl to pH 6-7, then concentrated to afford compound 5 as a yellow solid, which was used in the next step without further purification. LC-MS retention time 3.907 minute. ESI-MS displays a peak at m/z 453.0, calcd for [M + H]+: 452.7745.
3.9. Synthesis of 1-(4-chloro-3-(trifluoromethyl)phenyl)-3-(2-((4-(methylaminocarbonyl)pyridine-2-yl)amino)Pyrimidin-5-yl)urea (1A)
A flask was charged with HATU (7.5 g, 19.7 mmol) and dissolved in DCM/DMF (1: 1) (60 mL), followed by immersion in an ice bath at 0 oC, then compound 5 (5.9 g, 13.0 mmol), methylamine hydrochloride (3.5 g, 51.8 mmol) and DIEA (10.1 g, 78.1 mmol) were added to the cold solution. The mixture was flushed with nitrogen, progressed at room temperature overnight. Upon completion the reaction, the mixture was concentrated in vacuo, diluted with AcOEt (300 mL), washed with Saturated Na2CO3 (30 mL x 2) and Saturated NaCl (50 mL x 3), respectively. The organic phase was dried over Na2SO4, concentrated. The crude material was purified by silica gel column chromatography (10: 1 DCM: MeOH) to afford compound 1A as a yellow solid (2.0 g, 33.0%). 1H NMR (400 MHz, DMSO-d6) δ 12.47 (s, 1H), 8.75-8.59 (m, J = 4.6 Hz, 1H), 8.43 (d, J = 5.1 Hz, 1H), 8.34 (s, 1H), 8.07 (s, 2H), 7.85 (d, J = 2.4 Hz, 1H), 7.79 (d, J = 8.5 Hz, 1H), 7.62 (dd, J = 8.5, 2.4 Hz, 1H), 7.42 (dd, J = 5.2, 1.4 Hz, 1H), 5.51 (s, 2H), 2.77 (d, J = 4.5 Hz, 3H); 13C NMR (101 MHz, DMSO) δ 165.13, 152.77, 151.99, 150.22, 148.57, 143.91, 142.11, 140.15, 139.13, 135.44, 132.17, 129.32 (q, J = 20.5), 128.91 (q, J = 7.4), 127.02 (q, J = 35.3), 124.02, 121.30, 116.44, 110.91, 26.29; IR (KBr) νmax (cm-1): 3440, 3310, 3219, 3124, 1635, 1563, 1506, 1428, 1410, 1314, 1283, 1229, 1131, 1030, 943, 897, 824, 704, 657; ESI-MS displays a peak at m/z 466.0, calcd for [M + H]+: 466.8242.
3.10. Synthesis of 1-((5-amino)Pyrimidin-2-yl)-1-(4-chloro-3-(trifluoromethyl)phenyl)-3-((4-(methylaminocarbonyl)pyridine-2-yl)urea (1A2)
A frame-dried round-bottomed flask was charged compound 1A (932 mg, 2.0 mmol, 1.0 equiv) and sodium hydride (NaH) (240 mg, 6.0 mmol, 3.0 equiv, 60% dispersion in mineral oil), cooled to 0 °C, then DMF (15 mL) was added via syringe all at once. The resulting suspension was allowed to warm to room temperature over 5 min. Upon completion of the reaction, as indicated by TLC, water (30 mL) was added to quench the reaction and extracted with DCM (3 x 30 mL). The combined organic phase was dried over anhydrous Na2SO4 filtered, evaporated. The crude material was purified by silica gel column chromatography (10: 1 DCM: MeOH) to afford compound 1A2 as a brown solid (755 mg, 81%). 1H NMR (400 MHz, DMSO-d6) δ 9.17 (s, 1H), 8.76 (d, J = 4.4 Hz, 1H), 8.27 (s, 2H), 8.20 (s, 2H), 7.93 (d, J = 6.6 Hz, 1H), 7.81 (br s, 1H), 7.50 (br s, 1H), 6.76 (d, J = 6.5 Hz, 1H), 5.99 (s, 2H), 2.78 (d, J = 4.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 164.60, 159.90, 158.45, 148.22, 143.96, 142.96, 142.77, 140.69, 138.67, 131.46, 126.31 (q, J = 120.6), 124.32, 122.67, 121.60, 121.26, 117.49, 116.62 (q, J = 23.7), 105.72, 26.35; ESI-MS displays a peak at m/z 466.0, calcd for [M + H]+: 466.8242; IR (KBr) νmax (cm-1): 3433, 3350, 3226, 1680, 1663, 1561, 1482, 1439, 1307, 1235, 1133, 1038, 827, 741, 683, 663, 606, 584, 504.
3.11. Synthesis of 1-((5-amino)Pyrimidin-2-yl)-1-(4-chloro-3-(trifluoromethyl)phenyl)-3-((4-(methylaminocarbonyl)pyridine-2-yl)urea dichloromethane solvate (1A2a)
Crystallization 1A2 from mixed solvent of methanol and dichloromethane afforded 1A2a as a dichloromethane solvate at the stoichiometric ratio 1:1 as a brown triclinic crystal. Crystal data. C19H15ClF3N7O2•CH2Cl2 1A2a, M = 550.76, triclinic, space group P1, a = 9.8517(7) Å, b = 10.2973(7) Å, c = 11.4712(8) Å, α = 86.802(3)°, β = 81.486(3)°, γ = 84.921(3)°, V = 1145.29(14) Å3, Z = 2, Dcalc = 1.597 g/cm3, μ(CuKα) = 4.164 mm-1, T = 100.(2) K, F(000) = 560, crystal size = 0.340 x 0.270 x 0.080 mm3, intensities of 18242 reflections (4496 independent, Rint = 0.0875).
4. Conclusions
In conclusion, a sorafenib analog 1A with good antiproliferative activity in MDA-MB-231 (breast) cells was designed and synthesized. Base-mediated (NaH) arrangement of compound 1A delivered compound 1A2 which was crystallized from MeOH/DCM affording compound 1A2a as a dichloromethane solvate at the stoichiometric ratio 1:1. The structure of compound 1A2a was corroborated by the single crystal X-ray diffraction study. Both compounds 1A and 1A2 show little ability to inhibit the EGFR kinase, with compound 1A2 displayed relatively higher inhibition percentage than compound 1A. Further biological activity and mechanisms investigation is ongoing in the laboratory to broaden their applications.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/xxx/s1, Figure S2: General Information ; Figure S2: 1H NMR spectrum (400 MHz) of compound 2 in DMSO-d6; Figure S3: 1H NMR spectrum (400 MHz) of compound 3/4 in CD3OD; Figure S4: 1H/13C NMR spectrum (400 MHz) of compound 1A in DMSO-d6; Figure S5: 1H NMR spectrum (400 MHz) of compound 1A2 in DMSO-d6; Figure S6: IR spectrum of 2/3; Figure S7: IR spectrum of 4/1A2; Figure S8: IR/ ESI-MS spectrum of 1A; Figure S9: ESI-MS spectrum of 1A2.
Author Contributions
Qunying Yu designed and performed the research, analyzed the data and drafted the manuscript.
Funding
Please add: This work was financially supported by Jiujiang Programs for Science and Technology Development (S2021QNZZ037).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article and supplementary materials.
Conflicts of Interest
The author declare no conflicts of interest.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A,; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA-Cancer J. Clin. 2021, 71, 209–249. [CrossRef]
- Soerjomataram, I.; Bray, F. Planning for tomorrow: global cancer incidence and the role of prevention 2020-2070. Nat. Rev. Clin. Oncol. 2021, 18, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Scudellari, M. Drug development: Try and try again. Nature, 2014, 516, S4–S6. [CrossRef]
- Kudo, M. Systemic Therapy for Hepatocellular Carcinoma: Latest Advances, Cancers, 2018, 10, 412–427. [CrossRef]
- Wilhelm, S. M.; Adnane, L.; Newell, P.; Villanueva, A.; Llovet, J. M.; Lynch, M. Mol. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling, Cancer Ther. 2008, 7, 3129–3140. [CrossRef]
- Chen, L.; Fu, W.; Zheng, L.; Liu, Z.; Liang, G. Recent Progress of Small-Molecule Epidermal Growth Factor Receptor (EGFR) Inhibitors against C797S Resistance in Non-Small-Cell Lung Cancer, J. Med. Chem. 2018, 61, 4290–4300. [CrossRef]
Figure 1.
Third-generation of EGFR tyrosine kinase inhibitors with 2-amino-pyrimidine core.
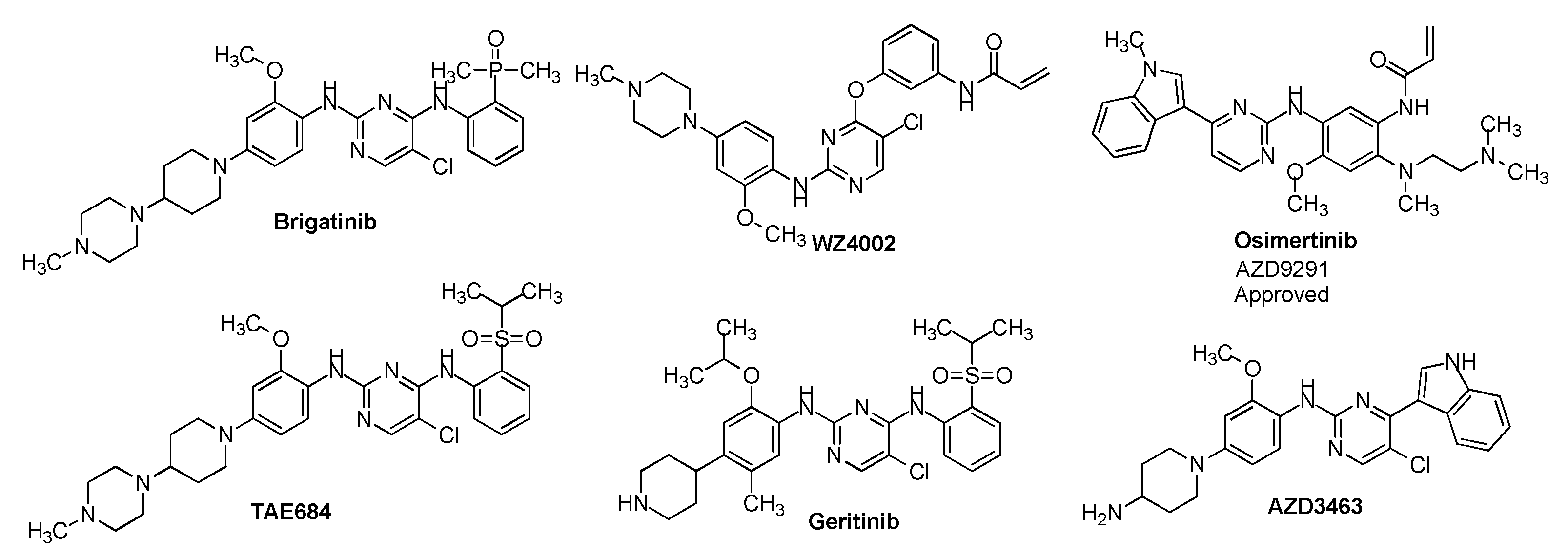
Figure 2.
Design of Solafenib analogue utilizing a scaffold hopping strategy and its rearrangement product.
Figure 2.
Design of Solafenib analogue utilizing a scaffold hopping strategy and its rearrangement product.
Scheme 1.
Synthetic Route to the Molecule 1A and 1A2. Reagents and conditions: (a) 1a (1.0 equiv), DMF, 0 oC, than added 1 (1.0 equiv), than rt (overnight); (b) H2, Reny Ni, MeOH, rt, overnight; (c) 3 (1.0 equiv), Et3N (2.0 equiv), DCM, 0 oC, than added 3a (1.5 equiv), than rt (overnight); (d) 4 (1.0 equiv), MeOH, 2 N LiOH (2.0 equiv), rt (overnight); (e) 5 (1.0 equiv), DMF, HATU (1.5 equiv), Methylamine hydrochloride (4.0 equiv), DIEA (6.0 equiv), rt (overnight); (f) 1A (1.0 equiv), DMF, 0 oC, NaH (3.0 equiv), than rt; DIEA = N,N-Ethyldiisopropylamine, HATU = 2-(7-Azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate.
Scheme 1.
Synthetic Route to the Molecule 1A and 1A2. Reagents and conditions: (a) 1a (1.0 equiv), DMF, 0 oC, than added 1 (1.0 equiv), than rt (overnight); (b) H2, Reny Ni, MeOH, rt, overnight; (c) 3 (1.0 equiv), Et3N (2.0 equiv), DCM, 0 oC, than added 3a (1.5 equiv), than rt (overnight); (d) 4 (1.0 equiv), MeOH, 2 N LiOH (2.0 equiv), rt (overnight); (e) 5 (1.0 equiv), DMF, HATU (1.5 equiv), Methylamine hydrochloride (4.0 equiv), DIEA (6.0 equiv), rt (overnight); (f) 1A (1.0 equiv), DMF, 0 oC, NaH (3.0 equiv), than rt; DIEA = N,N-Ethyldiisopropylamine, HATU = 2-(7-Azabenzotriazol-1-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate.
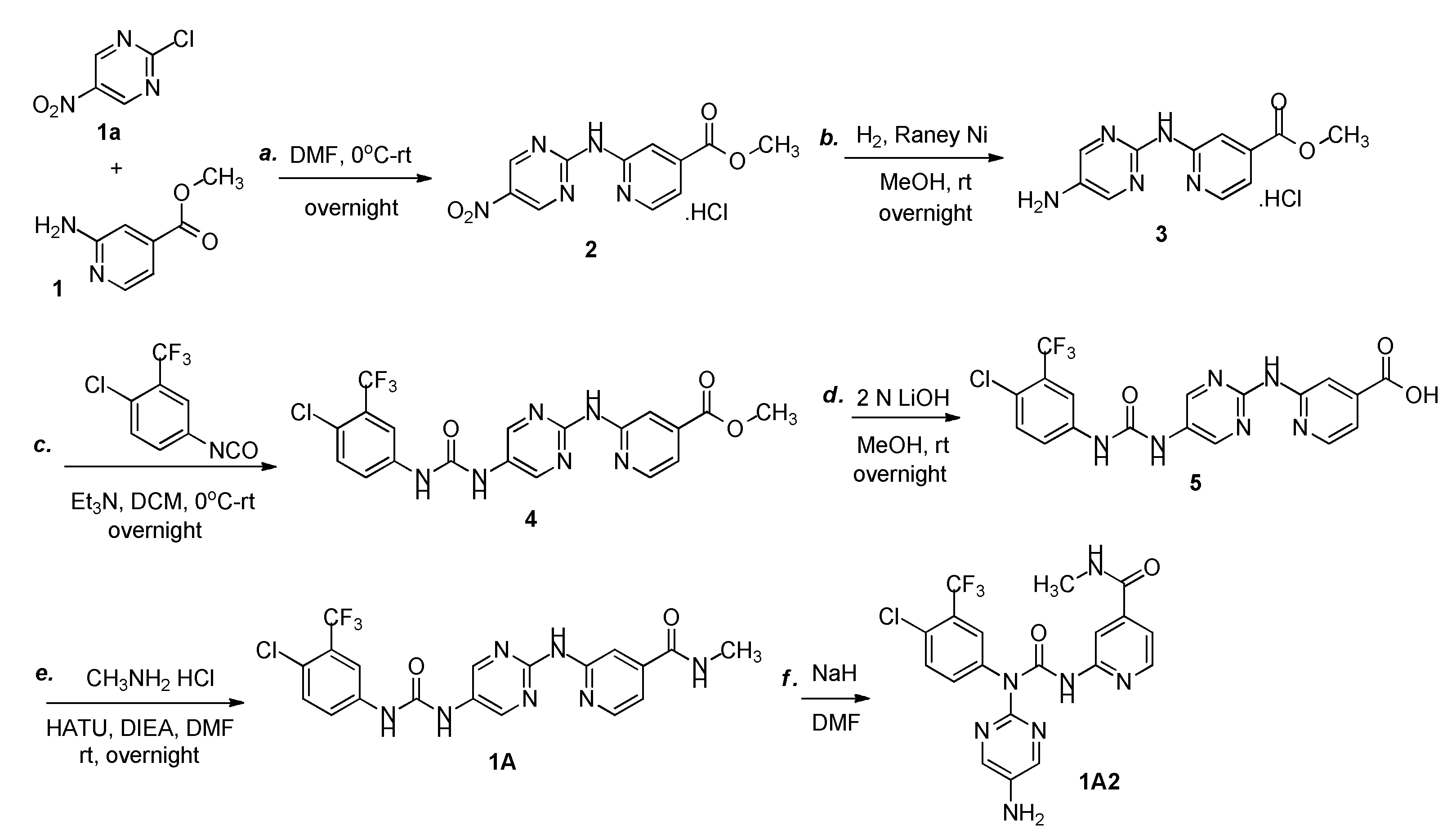
Figure 3.
An ORTEP view the asymmetric unit of compound 1A2a with displacement ellipsoids drawn at the 30% probability level and intramolecular hydrogen bond interaction.
Figure 3.
An ORTEP view the asymmetric unit of compound 1A2a with displacement ellipsoids drawn at the 30% probability level and intramolecular hydrogen bond interaction.
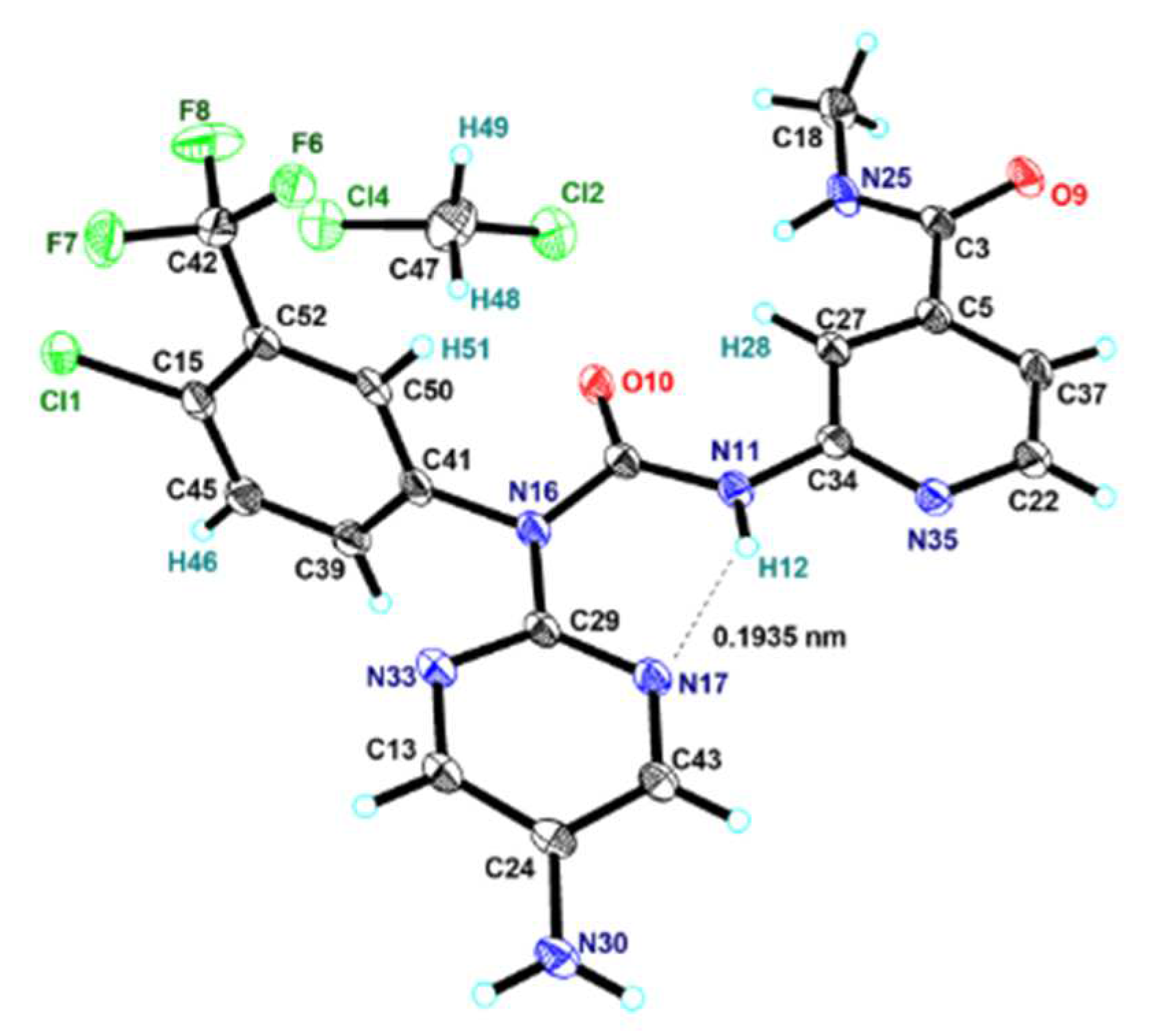
Figure 4.
The pack drawing of compound 1A2a with hydrogen-bonds shown as dashed lines.
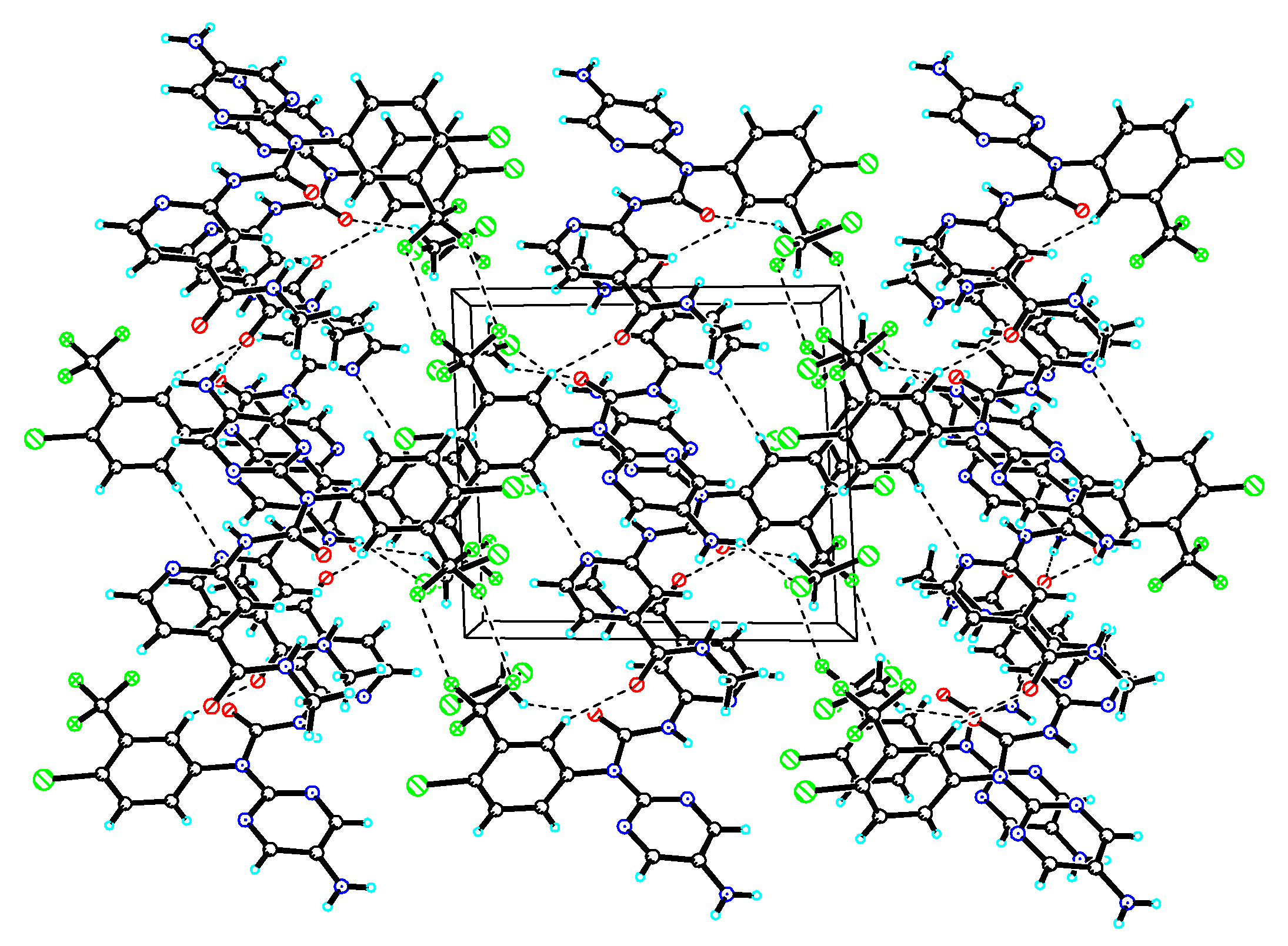

Table 1.
Crystallographic Data of compound 1A2a.
| Formula | C19H15ClF3N7O2•CH2Cl2 | F(000) | 560 |
|---|---|---|---|
| Formula weight | 550.76 | Crystal size / mm | 0.340×0.270×0.080 |
| Temperature / K | 100(2) | θ range / (o) | 3.90~72.42 |
| Crystal system | Triclinic | Index ranges | -12<=h<=10, -12<=k<=12, -14<=l<=14 |
| Space group | P1 | Reflections collected, unique | 18242 |
| a / nm | 0.98517(7) | Rint | 0.0875 |
| b / nm | 1.02973(7) | Data, restraint, parameter | 4496, 0, 317 |
| c / nm | 1.14712(8) | Goodness of fit on F2 | 1.085 |
| V / nm3 | 1.14529(14) | Final R indices [I>2σ(I)] | R1=0.0932, wR2=0.2667 |
| Z | 2 | R1 (all data) | 0.1097, |
| Dc / (g●cm-3) | 1.597 | wR2(all data) | 0.2854 |
| μ / mm-1 | 4.164 | (Δρ)max, (Δρ)min/ (e●nm-3) | 773, -1183 |
Table 2.
Selected bond, interatomic distances (nm) and torsion angles (o) for compound 1A2a.
| N16-C41 | 0.1444 | N11-H12 | 0.0880 | F8-C42 | 0.1339 |
| N16-C29 | 0.1415 | N11-C34 | 0.1391 | F7-C42 | 0.1348 |
| N16-C36 | 0.1409 | N11-C36 | 0.1366 | F6-C42 | 0.1313 |
| C29-N33 | 0.1330 | C34-N35 | 0.1346 | N17-C43 | 0.1341 |
| C13-N33 | 0.1338 | C22-N35 | 0.1336 | N17-C29 | 0.1335 |
| F7-H51 | 0.2308 | O10-H48 | 0.2215 | Cl1-C42 | 0.3104 |
| O10-C41 | 0.2604 | O10-H28 | 0.2243 | N33-C41 | 0.2641 |
| H12-N17 | 0.1935 | N11-N17 | 0.2647 | ||
| F6-C42-C52-C50 | 114.624 | F8-C42-C52-C15 | 57.378 | N16-C29-N33-C13 | 176.909 |
| F7-C42-C52-C15 | 175.694 | N16-C41-C50-C52 | 179.566 | N30-C24-C43-N17 | 176.285 |
| C36-N16-C41-C39 | 103.99 | C36-N16-C29-N33 | 168.588 | C29-N16-C36-O10 | 179.692 |
| C29-N16-C41-C50 | 99.556 | C41-N16-C29-N17 | 176.294 | C29-N16-C36-N11 | 0.717 |
| C45-C15-C52-C42 | 177 | H12-N11-C34-N35 | 6.627 | H12-N11-C36-O10 | 175.811 |
Table 3.
Human Cancer Cell Growth Inhibition and percentage of EGFR kinase inhibition of compounds 1A and 1A2.
Table 3.
Human Cancer Cell Growth Inhibition and percentage of EGFR kinase inhibition of compounds 1A and 1A2.
| compd | IC50(μM)a | EGFR(%)b | ||||
|---|---|---|---|---|---|---|
| A549 | HepG2 | HCT116 | MDA-MB-231 | PC-3 | ||
| 1A | >40 | >40 | >40 | 16.18±1.42 | >40 | 13.55±0.50 |
| 1A2 | >40 | >40 | >40 | >40 | >40 | 34.04±1.72 |
Note: Data from triplicate experiments were expressed as mean ± SD. a IC50 values represent the concentration needed to inhibit cancer cell line proliferation by 50%.bPercentage of kinase inhibition obtained at 40 μM of the test compounds.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



