
Submitted:
24 January 2024
Posted:
25 January 2024
You are already at the latest version
Abstract
The advent of CRISPR/Cas9 technology has revolutionized genome editing, enabling the attainment of once-unimaginable goals. CRISPR/Cas's groundbreaking attributes lie in its simplicity, versatility, universality, and its independence from customized DNA-protein systems, erasing the need for specialized expertise and broadening its scope of applications. Beyond its editing prowess, the discovery of novel Cas-based systems has spawned an array of additional biotechnological tools, empowering both fundamental and applied research. Researchers have harnessed CRISPR/Cas for gene regulation, deepening insights into gene expression and epigenetic changes. Precisely targeting DNA or RNA sequences, CRISPR/Cas effectively identifies and counters pathogens. Furthermore, it aids in genome imaging and sequencing, revealing genome spatial organization and chromatin dynamics. The non-editing applications of CRISPR/Cas exhibit tremendous potential across diverse domains, including diagnostics, biotechnology, and fundamental research. This article reviews and critically evaluates the primary CRISPR/Cas-based tools developed for plants and animals, underlining their transformative impact.
Keywords:
CRISPR/Cas
; Imaging
; Gene regulation
; NGS
; Viruses
; CRISPR/Cas9
; CRISPR/Cas13
; dCas9
Introduction
Since its inception in 2012, with the notable publication by Doudna and Charpentier [1], the CRISPR/Cas system has harbored the potential to catalyze an extraordinary paradigm shift in genome editing applications such as gene therapy and agricultural breeding. This technology has partly superseded meganucleases-mediated techniques like TALEN and zinc-finger proteins by obviating the need for specialized expertise in engineering bespoke DNA-binding proteins since the specificity of CRISPR/Cas target is governed by nucleic acid base pairing [2]. Originally, the CRISPR/Cas system evolved as a prokaryotic adaptive immune system to combat mobile genetic elements such as bacteriophages and plasmids. Archaea and bacteria exhibit a notable variation in the sequences of their respective Cas proteins and genomic loci compositions and structures. The growing knowledge regarding this diversity is acquired through screening of the constantly expanding databases of genomic and metagenomic data. The newest classification includes two classes, six types, and 33 subtypes. Class 1 (not discussed here) includes types I, III, IV, whereas class 2 encompasses types II, V and VI [2,3,4].
While type VI is recent discoveries, type II has been already extensively investigated: it represents the most widely exploited group of Cas proteins and contains the Cas1, Cas2, Cas4 and Cas9 genes [4,5,6]. Cas12 family coincides with the type V and it is further divided into 14 subtypes (Cas12a-Cas12n) [7], being Cas12a one of the most exploited Cas proteins (together with Cas9) for genome editing approaches. Within the Cas12 family, Cas12f (also known as Cas14) is also of particular interest due to its small size (around 550 amino acids), which represents a key step in minimizing the size of the nucleases to facilitate packaging of their genes for delivery. This nuclease is able to target both dsDNA and single-stranded DNA (ssDNA) using a single RuvC nuclease domain. Finally, worthy of note is also the discovery of Cas13a (also referred to as C2c2) belonging to type VI and isolated in Leptotrichia shahii and which is able to recognize and cleave single-stranded RNA molecules. Investigations into Cas14 and Cas13a could be economically valuable in engineering interference against plant ssDNA or RNA viruses.
The genome editing applications of CRISPR/Cas technology, such as gene therapy and agricultural breeding, have sparked significant investments and strategic partnerships across various industries including pharmaceuticals, agriculture, food, and biotechnology. In the realm of scientific review articles, the overwhelming focus on CRISPR/Cas editing applications creates a noticeable asymmetry in the literature. While the potential for genome editing using CRISPR/Cas systems cannot be understated, it is imperative to promote a more balanced exploration of non-editing applications. In this review, most of the recent non-editing – here understood as non inducing modification on the nucleotide sequence - CRISPR/Cas based tools developed in plant and animal research will be reviewed and critically discussed.
Overview of main CRISPR/Cas systems that are exploitable for both editing and non-editing uses.
The articles and works mentioned hereafter all make use of CRISPR/Cas systems to target nucleic acid portions thanks to specific recognition of their sequence. In this small overview, we will describe a few notable features of the CRISPR/Cas systems that have been used in the here reviewed articles [2] (Table 1).
CRISPR–Cas9
CRISPR/Cas9 is probably the most well-known CRISPR-system. It relies on the activity of the Cas9, a class 2 type II Cas protein (Figure 1A). These Cas proteins require the presence of a protospacer adjacent motif (hereafter referred to as PAM sequence), close to the target sequence to recognize it and contain the RuvC and HNH nuclease domains, which are involved in the cleavage of the target DNA [1,7]. SpCas9 from Streptococcus pyogenes, which was the first Cas9 to be used in non-prokaryotic cells and, still, the most widely used one, needs to recognize a NGG PAM-motif in order to fix and cleave the target sequence. The need for a PAM sequence can be considered an obstacle to the use of the CRISPR/Cas9 system to target every possible sequence and scientists have therefore already generated variants from the Cas9 that recognize another or several other PAM sequences. For example, GAT, GAA and even a binucleotidic NG can serve as a PAM sequence for the xCas9 variant. It is also possible to search living organisms for orthologues of the SpCas9 [13]. Indeed, in various species, the pattern identified as a PAM sequence for Cas9 recognition may vary, and some of them may be more adapted to specific target sequences. Additionally, in certain instances, the PAM can exhibit a certain flexibility, allowing multiple variations for a particular position. For example, the PAM sequence recognized by the Cas9 orthologue of Campylobacter jejuni is NNNVRYM, where Y can be either C or T, V can be A, C or G and M can be A or C. This CjCas9 is also one of the smallest Cas9 orthologues, being only 984 amino acids in length compared to the 1368 residues of the SpCas9 [14]. Reducing the size of the Cas9 is indeed a way to facilitate their delivery into the target cell [2].
The target sequence is specified by the guide-RNA (gRNA) or single guide-RNA (sgRNA). In the case of the Cas9 protein, the gRNA is composed of two distinct parts, often fused together in a long gRNA when engineered in laboratories. The first part is the CRISPR-RNA (crRNA) that allows targeting of the DNA portion corresponding to its complementary sequence. The other part is the trans-activating CRISPR RNA (tracrRNA), which allows the formation of the ribonucleoprotein complex. In Streptococcus pyogenes, sequences encoding diverse crRNA can be chained at the same locus of the genome, in the form of an array of crRNA spacers separated by identical direct repeats. The transcribed long pre-crRNA can then be processed into the different mature crRNA by the SpRNase III. Each crRNA may then be assembled to the the Cas9, provided that it is already binding the tracrRNA part of the gRNA [15]. In the case of sequence targeting, may it be for a genome editing purpose or not, this could allow to introduce different gRNA in a unique construct and send the Cas9 to several loci at the same time. Versions of the Cas9 protein that are deprived of their catalytic activity have been developed to be able to target DNA sequences using a specific guide without cleaving them. Generating of such protein was done by inserting two point mutations in its DNA sequence to introduce two substitutions in the proteic chain, D10A and H840A, that induce loss-of-function of the RuvC1 and HNH nuclease domains [16]. This version of Cas9 is referred to as dead-Cas9 (dCas9) (Figure 1B).
CRISPR–Cas12a
Cas12a, also referred to as Cpf1, differs from Cas9 in the sense that it cleaves DNA forming 5’ overhangs, and not blunt ends, which can be more convenient for certain applications of genome editing like the insertion of a DNA sequence at a precise position (Figure 1C). As Cas9, it requires the presence of a PAM sequence upstream of the target sequence. As it has been done for Cas9, orthologues of Cas12a have been engineered to make them specific to other PAM sequences. However, and contrary to Cas9 that requires the presence of both tracrRNA and crRNA to cleave DNA, Cas12a only requires the presence of a crRNA, that it can process on its own from an array of crRNA. This reduces the length of the required oligo to order when designing the gRNA. The locus encoding the Cas12a/Cpf1 in Francisella novicida U112 contains downstream of the FnCpf1 gene an array of several nuclease guide sequences (spacers) transcribed together as a pre-mature crRNA and interspaced by direct repeats. The Cas12a is then capable of processing each one of the crRNA with no other effector required [2,17] while Cas9 would require the presence of the tracrRNA as well [15]. Interestingly, Cas12a not only possesses a sequence-specific double-stranded DNA cleavage activity but also a sequence-independent single-stranded DNA degradation propriety, provided it is activated by the binding with high specificity of the DNA sequence specified by the gRNA [2,18]. In this review, we present a few works that made use of the CRISPR/Cas12a system to regulate gene expression and induce specific cleavage of viral DNA in infected plant cells.
CRISPR/Cas13
CRISPR/Cas13 systems form the class VI and distinguish themselves from the other CRISPR systems in the sense that they naturally recognize and cleave single-stranded RNA (ssRNA) molecules according to a gRNA (Figure 1D). Contrary to DNA targeting Cas protein, Cas13 systems do not need to recognize a PAM sequence in order to cleave its target RNA [10]. This characteristic was seen in different RNA targeting CRISPR/Cas systems [19]. Similarly to Cas12a, activation of Cas13 proteins through recognition of the target sequence induces their “collateral cleavage” activity, which allows them to also cleave nearby ssRNA specifically at uracil bases but regardless of the rest of the sequence. This property has been employed to develop a very quick and specific virus detection technique called specific high-sensitivity enzymatic reporter unlocking (SHERLOCK) [2,12]. Different Cas13 subtypes exist, namely Cas13a, Cas13b, Cas13c and Cas13d, that have been established according to phylogenetic analysis of their effector complex [20]. Furthermore, for the same subtype, different orthologues from different species have often been compared for the same experiment to find the most suited one [20,21]. Catalytically inactive (“dead”) versions of Cas13 systems, especially Cas13a orthologues, have also been engineered by replacing the arginines in positions 474 and 1046 of the two HEPN domains by alanines, to serve experimental purposes [11]. Here we show how the CRISPR/Cas13 systems have been utilized over the last decade to label RNA, modify mRNA translation in mammals, detect the presence of targeted RNA in samples, or even induce resistance against viruses in plants.
CRISPR/Cas mediated transcriptional regulation.
In eukaryotic cells, an important part of gene regulation goes through transcription factors, which are proteins that bind to specific DNA sequences, often called cis-acting regulatory elements, and activate or repress the expression of genes under the control of these sequences [22,23]. Over the past decade, a couple of artificial gene regulators based on CRISPR/Cas systems have been generated, taking advantage of the ability of Cas proteins to bind nucleic acids in a sequence-specific manner. Such systems have already been used to induce either transcriptional activation or repression of specific genes (Figure 2A-J). When used to activate transcription, the complex binds to a specific promoter region and recruits transcriptional activators to enhance gene transcription. This approach involves fusing a transcription activating domain to the Cas protein, which allows it to recruit transcriptional machinery and activate gene expression through a specific and purposely designed sgRNA. The same way, a transcriptional repressor domain can be fused to the Cas protein. When such CRISPR/Cas complex is sent to a target DNA sequence using a specific gRNAs, it binds to the promoter region and recruits transcriptional repressors, which represses gene expression. Both these approaches give access to a precise control over gene expression through the direct target of specific DNA regulatory sequences. In this context, it is often more convenient to use dCas proteins, that are deprived of their cleavage activity, instead of regular Cas, so that they are not molecular scissors anymore but rather generic RNA-guided DNA-binding proteins.
CRISPR/Cas mediated gene activation
Fusing dCas9 protein to transcription regulation effectors results in the generation of an artificial transcription factor able to pair with a specific sgRNA that can be programmed for a specific purpose. Bikard and colleagues demonstrated the potential of this molecular strategy describing (or reporting) an example of this in which they combined the ω subunit of RNA polymerase (RNAP) with a dCas9, which resulted in the up to three-fold over-activation of reporter gene in Escherichia coli [24]. Arises in this way the true possibility to use the CRISPR system for gene activation (CRISPR activation, or CRISPRa). In eukaryotic cells, one of the first successful artificial upregulation complex to be developed is referred to as dCas9-VP64 (Figure 2C). It is a fusion between the dCas9 protein and the VP64 activator, which is a synthetic homo-tetramers of four Herpes simplex VP16 transcriptional activator domains from the Herpes simplex virus. Another early developed fusion complex is the dCas9-p65, where p65 is also a transcription activating domain. The dCas9-VP64 fusion has however been more frequently and more successfully employed than its p65 fusion equivalent. Several studies have shown that dCas9-VP64 can either upregulate previously activated genes or activate silent endogenous reporters [25]. Cheng and colleagues used several sgRNAs tiled all over the promoter at the same time and obtained an over-activation of the reporter gene of up to 10 folds, proving that the expression of the target gene may depend on the copy number of activators [9]. In addition, the position of the binding sites regarding the TSS of the gene also resulted crucial since the highest activation was obtained using several sgRNA localized with 250 bp before the TSS [9]. Another strategy to increase the level of activation is to directly act on the copy number of an activator domain that the dCas9 carries. For example, Tanenbaum and colleagues developed the dCas9-SunTag Platform (Figure 2D), where the SunTag is a repetition of GCN4 proteins that is attached to the dCas9 and recruits several VP64 activator proteins, that bind to it through an anti-GCN4 antibody peptide [26,27]. dCas9-VP64 effector induced a 2-fold increase of target gene expression, using SunTag allowed to enhance the production of the targeted gene’s product by 50 folds [25,28]. Moreover, dCas9-SunTag system was adapted in Arabidopsis for targeted gene activation [29]. Furthermore a modified and more efficient version of the dCas9-SunTag Platform in which the SunTag not only recruits VP64 effectors but also TET1 effectors was developed [26,30]. Upregulation of several genes with both effectors was of 212 folds on average, while it was only of 20 folds with only TET1, and even less with only VP64 [26,30]. A similar synergy between effectors was also seen using a tripartite “VPR” activator composed of VP64, p65 and Rta (another activator) linked in tandem (Figure 2F). dCas9-VPR was indeed able to amplify the activation of endogenous genes by up to 300 folds compared to dCas9-VP64 alone [31]. dCas9-VPR system was also seen to induce high transcriptional activation in Nicotiana benthamiana [32]. A similar strategy was followed to develop a new approach called “Synergistic Activation Mediator” (SAM) which, like the previous system, combines several transcriptional activators to increase the expression level of the targeted gene (Figure 2E). The base of this complex is a dCas9-VP64 associated with a sgRNA, itself containing two copies of an MS2 phage RNA hairpin aptamer. Each one of these hairpins is bound by an RNA-binding MS2 coat protein MCP. In turn, MCP protein is fused to the p65 activator which is linked to the activation domain of the human heat shock factor 1 (HSF1). Each hairpin motif of the sgRNA is able to be bound to up to two of such activation modules, for a total of four modules per dCas9-VP64 [33]. Finally, dCas9-TV, a new CRISPRa system was specifically developed for plant species (Figure 2G). It relies on TV, an activator composed of six copies of the TALE transcription activation domain (TAD) motif and two copies of the VP64 activator. Application of this system in Arabidopsis thaliana and rice induced a strong transcriptional activation compared to the canonical dCas9-VP64 system [34].
CRISPR/Cas mediated gene repression
The first attempts to use the dCas9 for gene repression were not based on the fusion of transcriptional repressors to the dCas9 protein, but simply on the steric hamper that the protein exerts on the RNAP when bound to the target DNA (Figure 2A). This system, referred to as CRISPR interference (CRISPRi) [16], was described to be effective in bacteria. In Escherichia coli, CRISPRi displayed an up to 300-fold highly specific and revertible gene repression capacity. Its efficiency however depends on the location of the targeted site: the highest efficiency was obtained when dCas9 was sent to either the -35 box of the promoter or the beginning of the coding sequence. Initiation or elongation of the transcription by RNA-polymerase II is blocked by the presence of the dCas9 and the R-loop formed by the sgRNA-DNA interaction. CRISPRi also offers the possibility to effectively downregulate several genes and this with no crosstalk between the different inserted sgRNAs [16]. It could however only induce a mild transcription repression in eukaryotic cells. Indeed, in eukaryotes, simple steric hindrance is not sufficient to fully inhibit the activity of the RNAP. To overcome this issue, several enhancements have been developed. Indeed, in mammalian cells and as it has been done for transcription upregulation approaches, dCas9 was fused to domains that induce the recruitment of chromatin modifiers, but in this case with transcriptional downregulation activity. The regulators used as proofs of concept included the KRAB (Krüppel-associated box) domain of Kox1, the CS (chromoshadow) domain of HP1α, the WPRW domain of Hes1, and four consecutive copies of the mSin3 interaction domain (SID4X). [35,36] With this system, repression rates ranging from 90% to 99% have been observed in mammalian cells [35,36].
In plants, the application of CRISPRi is limited to a few specific instances. A transcript level reduction of about 40% was observed in A. thaliana and N. benthamiana using the two transcriptional repressors dCas9-3xSRDX (SUPERMAN Repression Domain X) and dCas9-SRDX, respectively [37,38,39] (Figure 2B).
Alternative uses of CRISPR/Cas for transcriptional regulation
One of the other approaches to induce expression regulation again relies on the in-situ assembly of regulatory platforms, each made up of one dCas9 protein associated to the sgRNA that allows the recruitment of chromatin remodelers. The core of this system is then the guide RNA since on it rely both the choice of the target and the type of regulation that is applied to this latter. The sgRNA harbors one or several hairpin structures which form a scaffold RNA (scRNA) to which the chromatin remodeler can bind (Figure 2H). The assembly of several parallel dCas9-RNA platforms allows for the simultaneous regulation of more than one target gene. Indeed, sgRNA recognizing several protospacers can be inserted at the same time to target several genes. Plus, different types of RNA binding proteins which can each detect a specific RNA hairpin configuration are made use of, which induce the recruitment of specific types of effectors. This makes it possible to recruit specific combinations of effectors to the targeted genes, to modulate their expression with precision. This approach allows then for a completely programmable regulation of each of the chosen genes in a contemporaneous and independent way. [28]
A further refinement of that method was done using ligand-inducible control of gene expression. In details, the dCas9 is engineered so that its catalytic activity is only enabled upon treatment of the cell with a specific ligand or after detection of native cellular or microenvironmental signals, allowing for a spatial and temporal control of gene function through sense input signals. To do so, two main methodologies have been developed, namely coupling dCas9 to chemical or optogenetic sensing or to ligand-sensing receptor domains. Chemically induced dimerizing domains or optogenetically inducible dimerizing domains (respectively CIDs and OIDs) have been linked to dCas9 and its corresponding regulator domain (activation or repression domain) (Figure 2I). CIDs and OIDs can dimerize when their ligands are present, recruiting the effector domain to the dCas9 binding site on the genome [40]. In this context, several CIDs and OIDs have been coupled with dCas9-effector complex, in order to obtain an induction triggered from stimuli of various kind, e.g., abscisic acid (ABA)-inducible ABI–PYL1 [41,42,43], gibberellin (GA)-inducible GID1–GAI24 [43], rapamycin-inducible FKBP–FRB [41], blue light-inducible CRY2-CIB1, Magnet pMag–nMag and phytochrome-based red light-inducible PhyB–PIF [44,45,46].
A ligand-inducible control of Cas9 is also feasible by engineering it into a split-Cas9 version (Figure 2J). In this case, the Cas9 is translated as two separate domains, N-Cas9 and C-Cas9. Each of these is fused to an estrogen receptor, which interacts with an Hsp90 chaperone. Both parts of the Cas9 are then sequestered into the cytoplasm and are catalytically inactive. Upon capture of 4-hydroxytamoxifen ligand by the ligand-binding domain ERT from estrogen receptor (ERT), the interaction between Hsp90 and ERT is disrupted, and the parts of the Cas9 can localize to the nucleus and reform the complete active ribonucleoprotein along with the RNA [47]. A dead split-Cas9 (split-dCas9) version exists and has also been fused to the previously described CID and OID [40].
When it comes to suppressing expression without inducing modifications on the genomic sequence, another strategy could be to knock down genes through the direct target of mRNA transcripts. It was shown that, apart from double-stranded DNA, Cas9 protein can recognize single-stranded RNA and cleave it. Interestingly, the presence of a PAM sequence on the ssRNA is still mandatory to activate the nuclease activity but it may be incompletely annealed with the sgRNA. Thus, introducing a mismatch in the PAM sequence allows the Cas9 to perfectly recognize the ssRNA target while forbidding the cleavage of the corresponding DNA sequence [8]. More recently, the ssRNA-specific Cas13 was used in bacteria to generate a knockdown of specific gene through degradation of their transcript RNAs [19,48], with no required recognition of any PAM sequence. The only issue regarding the use of Cas13 orthologues is their collateral sequence-unspecific cleavage of ssRNA upon recognition of the target, which could cause cellular toxicity. Introduction of R597A and R1278A mutations in the HEPN domain was however seen to suppress the collateral cleavage activity [19]. It was also possible to target and cleave gene transcripts with Cas13 orthologues in eukaryotes. shRNA interference and Cas13-mediated knockdowns both yielded comparable efficiency in terms of mRNA quantity depletion. However, Cas13 was more sequence-specific and induced almost no off-target. Furthermore, developing this technique can allow for gene knockdown in procaryotes where RNA interference does not exist [19,48]. Other techniques make use of the catalytically inactive dCas13 to target RNA and recruit RNA-modifying effectors to it. This strategy echoes the one we described earlier for modulation of gene translation. Recently, adenosine deaminase gene (ADAR2) catalytic domain has been linked to dCas13b, allowing for programmed base editing of mRNA in human cells and the engineering of target gene expression without irreversibly altering the coding DNA [10]. The same Cas13 complex was also described to yield good results in yeast and zebrafish embryos [49,50]. In plants, the Pol II promoter is used to drive the expression of both Cas12a and its crRNA. The crRNA is flanked by hammerhead and hepatitis delta virus ribozyme RNAs for precise crRNA processing. With this system, 90% gene expression decrease was achieved in A. thaliana [51].
CRISPR/Cas in depth study of gene regulation
As described until here, gene regulation can occur at a transcriptional and posttranscriptional levels, and both these are efficient pathways to target in order to develop CRISPR/Cas system-based gene regulation modulators [11,25,28]. CRISPR/Cas system can not only be used to modulate gene regulation in experimental systems but also to study the endogenous one at both these regulation levels (Table 2).
Study of gene regulation at the transcriptional level
A few years ago, Liu and colleagues developed a CRISPR affinity purification in situ of regulatory elements (CAPTURE) technique, a CRISPR-based technique to identify the DNA-binding proteins and chromatin regions that are interacting with a given locus at a given time. It relies on the use of biotinylated dCas9 protein, that is sent to a locus of interest which is specified by a sgRNA. The chromatin is then crosslinked, sonicated, and pulled down using streptavidin beads to which the biotinylated Cas9 binds. At that point, the elements that were in interaction with the locus as the crosslink was induced are all fixed to the dCas9. After reverse-crosslink, it is possible to reveal the identity of the protein fraction that has been precipitated along with the dCas9 by performing proteomic analysis. It is as well possible to have access to the distant chromatin portions that are in long-range interaction with the locus through 3C-seq technique [52,53]. The authors first confirmed the feasibility of the technique with a human telomere-specific sgRNA and then tested it on single genes. They were able to precipitate the protein factors bound to single regulatory sequences of the human β-globin genes, with almost no detectable off-target. Moreover, simultaneous use of different sgRNAs is possible, which allows the study of different regulatory sequences at the same time. The proteomic study of the pulled down proteins revealed not only already known regulators of these genes but also led to the discovery of new ones. Confirmation of the discovered interactants was done using ChIP-seq. When it comes to chromatin interactions, CAPTURE allowed to put in evidence most of the long-range interaction but also significant intra-chromosomal and short-range ones. Very recently, Wang and colleagues developed a similar technique on a vegetal specie, namely birch (Betula platyphylla). They targeted the four regions of the promoter of the BpNAC090 gene and identified 32 potentially regulating transcription factors for that gene, five of which were confirmed to specifically bind to this promoter [54].
Study of gene regulation at the post-transcriptional level.
Posttranscriptional regulation may go through the regulation of the presence and the stability of a messenger RNA (mRNA) [55], which are dependent on the post-transcription sequence modifications they bare and the effectors they are targeted by. For example, some post-transcriptional modifications are crucial for mRNA expression and stability in eukaryotes, as well as the proteins that place them, remove them or interact with them (respectively called “writers”, “erasers” and “readers”) [56]. CRISPR/Cas system-based tools can now be used to target specific RNAs and study such protein interactants. Two main types of systems have been developed to get access to the proteins that are specifically bound to a transcript [20]. The first one is to fuse the dCas13 protein with a biotin ligase that will mark with biotin every protein complex interacting with the RNA. These proteins are then purified using classical techniques such as precipitation and can be analyzed. The other method is to crosslink the cell with UV once the dCas13 has bound its target RNA to covalently bind the proteins that are in close contact with the dCas13, then precipitate the whole complex through precipitation of the dCas13. These two techniques strongly echo what was done with CAPTURE technique. Overall, these techniques take advantage of the low mismatch and off-target rate of the dCas13 that allows to target one RNA with high sequence specificity. The number of proteins caught using one of these methods can be a few hundred, which makes it a very promising approach to study RNA processing for maturation and translation. It is, however, still difficult to precipitate proteins that are only transiently bound to the RNA or to study RNAs that are present in low amounts. The efficiency of the biotin-ligase is a limit to the techniques that make use of it, and this efficiency can be influenced by the context of the experiment.
Many new genetic tools are already available to study or influence gene regulation in diverse types of prokaryotic and/or eukaryotic organisms, in a very precise and controllable manner. They all rely on the capacity of the Cas or Cas-derived proteins to bind to a target that is specified by the experimenter through the sequence of the sgRNA. In this part, we reviewed how Cas proteins have been engineered into countless bi-partite molecules, molecular complexes, or platforms in the scope of associating a functional activity to the sequence-recognition capacity. This approach is also at the basis of all the nucleic acids-imaging techniques that have been recently developed.
CRISPR/Cas system to image specific portions of nucleic acids in plants and animals.
Nucleic acid imaging is the process of visualizing and detecting nucleic acids using various imaging techniques (Figure 3). One of these techniques, the Fluorescence in-situ hybridization (FISH) uses fluorescent DNA or RNA probes carrying a fluorophore that bind to complementary sequences of nucleic acids, which can then be visualized under a fluorescent microscope [57]. Other methods make use of GFP (Green Fluorescent Protein)-tagged proteins [58]. These methods use genetically engineered cells that express a fusion protein consisting of GFP and a protein that specifically binds to nucleic acids, such as a DNA or RNA-binding protein and allows to detect the presence and location of the nucleic acid of interest. GFP-tagged proteins offer several advantages for nucleic acid imaging, including high specificity, sensitivity, and non-invasiveness. The ability of the Cas proteins to specifically bind to the sequence specified by the guide-RNA has already been widely used to observe in-situ-stained RNA and DNA. Here we review some of the diverse methods that have been developed to do so, may it be in fixed or in living cells (Table 3). Labelling of DNA sequences using CRISPR-system has also been recently reviewed by several other groups [59,60]. The Cas proteins used for that purpose are often “dead” ones, to avoid degradation of the target sequence. It is interesting to note that instead of disabling catalytic activity of the Cas9, it is also possible to use an sgRNA whose spacer is only 11 nucleotides long, which is too short to enable the Cas-protein’s catalytic activity [61]. In most of the studies and reviews mentioned here, the targeted and stained sequences were repetitive sequences, because they induce clearly visible foci of fluorescence and are therefore more suited to proofs of concepts.
One of the first approaches to stain DNA using the CRISPR/Cas system was simply to fuse a GFP to the dCas9 (Figure 3A). This type of chimerical protein was first used about a decade ago to stain telomere sequences in live human cells [62]. Cells were transfected with an activable dCas9∷eGFP fusion protein along with the corresponding sgRNA leading to telomeric repeats. The efficiency of telomere detection was evaluated by counting the number of visible focal points on the imaged loci. According to the article, a modified version of the sgRNA was developed by Chen and colleagues, leading to further improvement in the precision of telomere staining and reduction of background interference. The CRISPR-staining technique then showed an efficiency comparable to that of FISH with a quasi-absence of off-target, making it a very precise way of imaging genomes. Contrary to FISH, staining with the CRISPR system does not involve cell fixation, enabling the tracking of telomeric foci movement in various situations. Chen and colleagues also applied this technique to stain individual loci at the same time, by inserting different sgRNA in the cell. Imaging specific genome regions can be used to study chromatin conformation at these loci but was also used to test in a glance the specificity of a novel CRISPR/Cas-based method, such as CAPTURE in human cells [52]. The dCas may also carry a long tag, which can serve as a binding site for one or multiple GFP-fused proteins. Example of this is the Cas9-mediated fluorescence in situ hybridization (CASFISH) technique developed by Deng and colleagues [63], which makes use of a dCas9 harboring a HaloTag. The dCas9 is then labeled using fluorophore-conjugated Halo-ligands. The whole is incubated with the sgRNA ex situ, then used to stain specific DNA sequences. Deng and colleagues tested their technique to label repetitive sequences and single loci of cells. The ribonucleoprotein complexes were also capable of penetrating in a thin mouse brain section and staining their target repetitive sequence. The protocol duration is a mere 15 minutes, making it a viable approach for efficient identification of the presence of a specific gene sequence in a clinical assay. Furthermore, once assembled, the sgRNA-dCas9-fluorophore complex is highly stable which makes it possible to stain diverse loci at the same time, by associating each sgRNA to a different fluorophore. Pre-assembled complexes were also used to perform live imaging of specific loci through the CRISPR live-cell fluorescent in situ hybridization (LiveFISH) technique and study, among others, the recruitment of 53BP1 (p53-binding protein 1) at the double strand break (DBS) position [61]. The authors employed a sgRNA that was conjugated to a fluorophore and intentionally kept the protospacer short to prevent cutting. This allowed the use of the same Cas protein with a longer sgRNA that lacked labeling to induce a double-strand break a nearby region. Subsequently, the team was able to analyze and quantify the recruitment of 53BP1 to the DSB by examining its colocalization with the initial Cas9-sgRNA complex. By employing another Cas9-sgRNA complex labeled with a second fluorophore, they could observe a translocation between the Chr3q29 and Chr13q34 loci and investigate the kinetics of this mechanism. The main advantage of conjugating the fluorophore with the sgRNA rather than with the Cas protein is that it allows for a higher signal/background ratio compared to FISH and GFP-tagged dCas labeling [61,64]. Explanation for this is that there is no signal coming from non-specifically bound dCas9 proteins. Fu and colleagues used a sgRNA harboring 2 types of aptamers, MS2 and PP7, in a long 3’ scaffold (Figure 3C). These aptamer sequences can be bound by MCP-eGFP and PCP-mCherry fusion proteins, respectively. The use of this system with the two fluorophores allowed to independently label 2 loci of the same nucleolus [64]. The kinetic of the cellular division from the point of view of minor and major satellites in mouse fibroblast could be followed using this method. This system was also proven usable in plants, and telomeric sequences of N. benthamiana could be stained with GFP-fused MS2 proteins binding to sgRNA and observed in live cells [65]. Though incorporating the earlier reported scaffold modification to the sgRNA [62] resulted in a further reduction of the background signal in animal cells in mammals [64], it was not the case in plants [65]. Furthermore, in plants, the number of telomeric foci was lower than the one counted after staining with FISH, putatively because of the difference between the plant breading temperature and the Cas9 working one [65]. Such phenomenon had previously been noticed with GFP-fused dCas9-mediated staining of telomeres [66]. Other nucleic acid imaging methods work on the staining of extracted and fixed nuclei from cell material. In CRISPR-FISH, also called RNA-guided endonuclease - in situ labeling (RGEN-ISL), the dCas9 is again used to target and stain specific regions of the genome thanks to a given gRNA [67,68]. An advantage of this approach is that the technique can be performed using the same protocol across a broader temperature range (from 4 °C to 37 °C) and is appliable to nuclei or fixed tissues from various species. It makes use of the Alt-R CRISPR/Cas9 system (Figure 3B), in which the tracrRNA part of the sgRNA carries an ATT-550 fluorophore [69]. This allowed the visualization of satellite repeats, telomeric and centromeric regions of chromosomes with high fluorescence yield and low background in different plant species [67,68,70,71]. This kind of approach could be useful to study and compare the recruitment of certain proteins or the recruitment of specific histone marks at a specific locus. However, for now the technique has not been developed for study of single loci.
The aforementioned techniques can be categorized into two groups based on the system employed for the introduction of the Cas protein, guide RNA, and any other relevant proteins. They can first be introduced into the cell or sample as a preassembled ribonucleoprotein [61,63,63,67,71]. Other studies transformed their cells with one or more plasmid encoding these elements [52,62,64]. Fu et al. used a single vector encoding dCas9, sgRNA as well as the labeling protein MCP and PCP both fused with an eGFP [64]. Likewise, the construct to express the equivalent system in plants was introduced into the plant’s genome by Agrobacterium rhizogenes-based hairy root transformation, floral dip method and via leaf samples but only transient insertion allowed correct labeling [65].
Not only DNA can be targeted by CRISPR/Cas systems but also RNA molecules. As recently reviewed by Cao and colleagues, dead versions of different Cas13 orthologues have been used to mark stain RNA in live cells [20]. The most common method was to conjugate the dCas13 with one or more fluorophores and to send it to a specific transcript, coding or not. For example, in mammalian-cell transformed with one vector encoding the dead version of a Cas13 orthologue from Leptotrichia wadei (dLwaCas13) fused with GFP and a vector containing the guide RNA, it was possible to track the localization of transcripts to stress-granules, first in fixed cells but then in live cells [11]. A modified version of the LiveFISH technique that makes use of both dCas9 and dCas13 with diversely labeled sgRNA allowed to stain DNA and RNA at the same time in live cells [61] (Figure 3D-E).
Here we presented different techniques that, again, make use of the highly sequence specific binding propriety of the Cas proteins to stain specific portions of nucleic acids. It also offers the possibility to image live cells in a low invasive way and with reduced background. Such imaging of DNA or RNA is crucial to answer biological questions on important molecular processes that happen in the nucleus or quickly evidence a specific sequence. The Cas protein and its guide can be introduced into the cell through transient expression in a plasmid or direct transfection of the preassembled ribonucleoprotein. The latter is possible thanks to the high affinity of the Cas protein for its sgRNA. The advantage of directly transfecting the cells with sgRNA-Cas ribonucleoproteins is also that there is no need for stable integration of genetic constructions in the genomes.

Table 3.
Methods making use of CRISPR/Cas systems to evidence and image nucleic acids in a sequence-specific way.
Table 3.
Methods making use of CRISPR/Cas systems to evidence and image nucleic acids in a sequence-specific way.
| Name | Description | Organism | Type(s) of Cas protein | Advantages | Disadvantages | Performances | References |
|---|---|---|---|---|---|---|---|
| dCas9∷eGFP fusion protein | Imaging of DNA loci with a GFP-dCas9, expressed in situ along with the gRNA from transfected vectors. | Human | dCas9 | The use of an sgRNA guide with a custom scaffold reduces non-specific binding of Cas9. Possibility to label heterochromatin regions. |
Labelling of repetitive sequences as well as single loci. Tracking of telomere Dynamics in Live Cells Labelling of different positions of the same gene. Gene copy-number identification |
[62] | |
| Cas9-mediated fluorescence in situ hybridization (CASFISH) | dCas9 harbors a HaloTag flag and can be bound by fluorophores that are linked to HaloTag ligand. | Human | dCas9 | Highly stable sgRNA-dCas9-fluorophore complex High specificity Several loci can be stained at the same time: multiplexed imaging. Very quick protocol (15 minutes) Performed at room temperature |
Imaging of repetitive sequences in Detection of the allele of a certain sequence in the genome of cells at a tissue scale. Dual color Genetic diagnosis |
[63] | |
| LiveFISH | One Cas9 harbors a labeled sgRNA with a short protospacer to disable cutting and one other Cas9 harbors a normal unlabeled sgRNA | Human | Cas9, dCas9, Cas13, dCas13 | One type of Cas9 is used because the RNP complex is preassembled. Live imaging is possible. Combinable with other CRISPR/Cas system-based techniques. |
- Used to visualize and quantify the recruitment of a protein to a specific locus and study the kinetic of such recruitment. - Used to visualize translocations. - Dual labeling of DNA and RNA. |
[61] | |
| Labelling sgRNA scaffolds in animals | sgRNA carries a long 3’ scaffold that harbors aptamers, to which fluorescently labeled proteins bind. | Mouse | dCas9 | Fewer background compared to labeling with GFP-fused dCas9 because non-specific binding of dCas9 is not visible. A single vector encodes every component of the system. Live imaging is possible. Multiplex labelling |
Non-specific binding is not visible i.e., off target cannot be characterized. | Labelling of nuclear structures, repetitive sequences, and single loci. Study of chromatin dynamics during cell division. Labelling of two loci in different colors. |
[64] |
| Labelling sgRNA scaffolds in plants | sgRNA carries a long 3’ scaffold that harbors aptamers, to which fluorescently labeled proteins bind. | N. Benthamiana | dCas9 | Fewer background compared to labeling with GFP-fused dCas9 because non-specific binding of dCas9 is not visible. A single construct encodes every component of the system. This construct is inserted with A. tumefaciens-mediated transformation. Up to 2 simultaneous labeling. - Labelling efficiency is not dependent on dCas9 gene expression level |
Non-specific binding is not visible i.e., off target cannot be characterized. Lack of telomeric foci compared to FISH because of the working temperature in plants. Cannot be improved by modification of the RNA scaffold. Only repetitive sequences have yet been targeted. - Transient transformation of the construct is required. - Labelling efficiency is heavily dependent on the copy number of aptamers in the construct. |
Live imaging of telomeric repeats in plant cells. | [65] |
| RGEN-ISL/CRISPR-FISH | Imaging of loci in purified fixed nuclei using a preassembled ribonucleoprotein that contains the dCas9 and its sgRNA, for which the tracrRNA part is fused to a fluorophore for labelling. | Soybean, mouse, wheat, rye, maize, and Nicotiana benthamiana | dCas9 | No plasmid construct. No in vitro RNA synthesis. Theoretically available in any species Non disruptive technique Simple and fast Usable for repetitive sequences and single loci. |
Fixation of nuclei is required. ATT550-labeled tracrRNA and the crRNA that can bind to it must be ordered and are costly. |
Labeling of centromeric and telomeric repeats in diverse species. Optimization of sample fixation to increase labeling yield. Time-lapse-mediated study of the binding dynamics of dCas9-sgRNA-complex to DNA. |
[67,68,71] |
| mRNA imaging | dLwaCas9 is fused to GFP expressed along with the specific sgRNA using a transient vector. | Rice, mammals | dCas13 | Specific targeting of mRNA Applicable to live or fixed samples. |
Imaging of a specific gene’s to track its localization at stress-granules. |
CRISPR/Cas-system as a tool to target viruses.
Use of CRISPR/Cas system to target viruses in animals.
Inhibition of viral infection with CRISPR-Cas systems has also been explored in animal and human medicine. Numerous studies have already demonstrated the downregulation of viral infection in animal cell cultures. These studies have employed various types of Cas proteins depending on whether the targeted virus is based on DNA or RNA [77,78] For example CRISPR/Cas9 and Cas12a were shown to downregulate the infection of Green Monkey kidney cell culture by Herpes Simplex Virus 1 (HSV-1) [77]. In this study, the authors targeted HSV-1 genome with a Cas9 or Cas12a protein and a gRNA specific to a viral DNA polymerase and were able to see a clear diminution of the infection of the cell culture as well as an immunity that lasted for several days. The efficiency of the inhibition and the duration of the conferred immunity however highly depended on the protospacer that is targeted by the Cas protein. Plus, the efficiency of targeting also depends on the type of Cas used which shows that independently of their differences of PAM sequence, the Cas orthologues have diverse requirements in terms of chromatin context. On top of that, the sites targeted by Cas12a seem to display a lower mutation rate, which makes it more difficult for the virus to develop a resistance against Cas12a than against Cas9. However, Cas9 exhibited an extended duration of immunity in the observed cases. As in plants, it is possible to use other types of Cas proteins, namely Cas13, to target RNA viruses. Use of Cas13 to target viral genomic RNA or mRNA has already been extensively reviewed [79]. In this review, the authors describe different studies that were in animal cell-cultures infected by a virus, majorly with SARS-CoV-2 and influenza, whose genomic RNA and mRNAs were targeted by different Cas13 proteins. LwaCas13a, LbuCas13a, PspCas13b, and Cas13d were all found to downregulate viral infection, and in some cases conferred resistance for several days. Concerning their efficacy, Cas13b seems to be as efficient as shRNA interference, while Cas13a could induce superior efficiency. Concerning the therapeutic usage of CRISPR-Cas system in animals, the way to administer the RNP and the immune reaction it may induce come into question. Viral transfection is the most used method to deliver the protein, but also that it is important to consider the fact that the viral vector itself can induce an unwanted immune response in the body or have a cytotoxic effect in case of cell cultures. The 13d orthologue of the Cas13 could be used because of its smaller size and therefore should be prioritized. Transfection of RNP has the advantage of preventing the introduction of exogenous genetic sequences in the host’s cells, but it could be also possible to use synthetic RNAs encoding the protein and its gRNA. Still, one remaining main issue is the potential immune response induced by the RNP itself. Strategies to minimize this mainly rely on masking the bacterial antigens that are found on the protein, by either removing the antigens that can be removed without disturbing the enzyme’s activity, or by adding some antigens that downregulated the adaptive immune response. Another point that is mentioned in the article is the usage of Cas13 to diagnosticate viral infections. The SHERLOCK system uses the propriety of the Cas13 that makes it able to cut RNA randomly once it has bound its target RNA specified by the gRNA. The sample undergoes a T7 transcription to amplify the eventual RNA virus and then the Cas13 is added with a gRNA specific to the targeted virus, as well as RNA bearing fluorophore. If the target virus is bound by the Cas13, it will cut the fluorophore-RNAs. Then a kind of ELISA test is performed to separate and reveal the cut fluorophore-RNAs. The SHERLOCK technique has been further developed and its variants use different orthologues of the Cas13 to allow for simultaneous analysis of different viruses. dCas13 can also be fused to other proteins to mark viral RNA (GFP), modify its epitranscriptome (mainly m6A), base-editing, modulation of viral RNA splicing. This can be used to inhibit viral infection or to better understand host-pathogen interaction.
CRISPR/Cas-mediated resistance to viruses is a growing trend that is using different approaches to target and counter this type of pathogen. Its development requires consideration of the variety of viruses and the variety of hosts they can infect. In plants, the method is still in its embryonic phase and needs further improvement before it can be applied to crops, independently of legal restrictions. Though it still faces disadvantages compared to RNAi inhibition, it remains a promising approach. Research for the use of CRISPR-Cas systems against animal viruses, including human ones, probably benefited recently from the high needs of knowledge on human viruses due to the global pandemic and is therefore a little more advanced than it is for plants. Different aspects of the fight against virus infections have already been looked at, from the targeting of the genomic nucleic acid to the one of acid messenger RNA, to the detection of viral traces in samples. It will hopefully not take long before it is possible to do so in plants as well.
CRISPR/Cas as enrichment tool for next generation sequencing
As sequencing costs continue to decline, it is becoming increasingly difficult to avoid generating unnecessary or possibly unusable data in an experiment and, although sequencing data is easily producible in most laboratories today, the scientific community still struggles with sequence analysis. Therefore, the need to target specific genetic loci is urgent and a broadly applicable technique to screen out the signal from high abundance undesirable species before sequencing is needed [80]. Being an important support tool for sequencing, nucleic acid enrichment techniques have progressively evolved leading to the existence of a collection of methodologies applicable according to the different experimental needs (Figure 5). However, these techniques usually rely on PCR amplification to provide highly specific sequencing and are prone to allelic bias and produce non-native DNA entailing the loss of epigenetic information [80]. In the last few years, alternative tools are being developed, often in combination with the use of Nanopore sequencing, with the aim of expanding the possibilities to investigate nucleic acid features such as the observation of structural variants (SVs) at the haplotype level through long-read sequencing and detection of DNA methylation. These tools exploit the CRISPR/Cas system and its two main characteristics: the high specificity of targeting through sgRNA which also avoids long hybridization times, and the existence of a plethora of Cas proteins with the ability to perform a series of molecular actions on the nucleotide sequence of interest.
CRISPR/Cas NGS approaches based on depletion of undesired region.
The initial approaches to the application of the CRISPR/Cas technology started in 2016 in the realm of human diagnostics and focused more on lowering background noise while preserving the representational integrity of untargeted sequences than on the enrichment of the ROIs (regions of interest) [81]. In fact, despite the modest enrichment provided compared to other strategies, removing unnecessary sequences has proven to be one of the fastest and easiest workflows. The DASH protocol (Depletion of Abundant Sequences by Hybridization) involves attaching adaptors to DNA fragments and exploits the specificity of sgRNA to cleave specific DNA sequences, then sequencing the remaining intact fragments using primers compatible with the adaptors [82]. On the other hand, CUT-PCR (CRISPR-mediated, Ultrasensitive detection of Target DNA by PCR) targets and cleaves wild-type DNA sequences with intact PAM site, mutant target regions are then enriched by PCR and the process is repeated to maximize cleavage specificity prior to sequencing [83]. These technologies widely used in medical research (pros and cons are superbly reviewed by Schultzhaus and colleagues [79]) have recently been applied to plant science and metagenomics. The Cas-16S-seq method allows the sequencing of the hypervariable regions of 16S rRNA gene of the microbiota by eliminating contamination due to mitochondria and plastid 16S of the host plant [84]. The variable regions of 16S rRNA are amplified in a first PCR using universal primers with adaptors, then a specific gRNA targeting the host plant’s 16S is employed for Cas9 cleavage. After cleavage, the plant’s (rice) 16S rRNA fragments are not amplified in the second PCR and are then excluded from the sequencing. In order to eliminate repetitive sequences from a genome that is particularly rich in repetitive elements, such as that of lentil, the target specificity of the CRISPR/Cas system combined with a set of over 500,000 gRNAs was used and allowed to exclude from sequencing 40% of the reads mapping on repeats and to enhance the number of reads mapping on unique regions by more than twice as much [85]. Using such methodologies allows to focus on relevant genomic regions and to increase genotyping accuracy.
CRISPR/Cas NGS approaches based on enrichment of regions of interest (ROI)
Similarly to depletion based NGS techniques, the first advances in using CRISPR/Cas technology to selectively enrich regions of interest for sequencing purposes emerged predominantly in the field of diagnostics. High sensitivity procedures were developed taking advantage of dCas9 and its methods of isolation. In the protocol by Aalipour and colleagues [86], the allele frequency of a rare genomic alteration causing cancer is increased through a dCas9-based method. dCas9-associated sgRNAs are designed targeting mutations of interest. After incubation with DNA, the target-bound dCas9 is isolated through immunomagnetic precipitation, target DNA is then purified and analyzed through allele-specific qPCR resulting in a 21-fold enrichment. CATE-seq (CRISPR-assisted targeted enrichment-sequencing) [87] is a highly sensitive alternative approach able to reach over a 3000-fold enrichment of the target sequences. Such protocol consists in the fragmentation and subsequent specific adaptor ligation to sample DNA, targets are then bound by dCas9 and purified for allele-specific PCR or library preparation.
A few strategies are halfway between the bead-based purification of the fragments and the exploitation of the cleavage capacity of Cas9. In the ultrasensitive tool CRISDA (CRISPR–Cas9-triggered nicking endonuclease mediated Strand Displacement Amplification) [88], Cas9 cleavage activity is combined with highly specific amplification of the target site and annealing of biotin and Cy5- labeled PNA (peptide nucleic acid) probes to the amplicon to achieve attomolar sensitivity. The CRISPR-Capr approach [89] is based on enrichment of cleaved target sequences using biotinylated sgRNAs and allowed a 183-fold enrichment of a 13 kb DNA region. Another strategy includes DNA fragmentation by Cas9 and biotinylated adaptors are then used to repair DNA lesions [90]. At the end of the process, target sequences are long enough to be processed with long-read sequencing (20-30 kb). Finally, Tsai and colleagues [91] performed Cas9-mediated cleavage of a long sequence-PacBio-SMRT (Single Molecule - Real Time) DNA library. Bead-based isolation through adaptors led to a 64,000-fold enrichment of under-represented sequences.
In this context, the augmentation of genomic fragment lengths for sequencing purposes has become a crucial objective in genomic and functional research. Over the past few years, various approaches utilizing the CRISPR/Cas system have been devised and evaluated for TGSeq (Third Generation Sequencing). In the case of CISMR (CRISPR mediated isolation of specific megabase-sized regions of the genome), the isolation of the ROI through Cas9-driven cleavage at the flanking sites is combined with a pulse-gel electrophoresis step for sequence isolation and, passing through an amplification step, long-read sequencing [92]. Despite its laboriousness and time-cost this strategy proved efficient for tens-fold enrichment of sequences up to 2.3 Mb. Such strategies were followed by the more specifically Oxford Nanopore sequencing-aimed CATCH (Cas9-assisted targeting of chromosome segments) which allowed 237-fold targeted enrichment of the human BRCA1 gene through isolation of a 200 kb region [93].
In the wake of TGSeq-aimed enrichment strategies, the finding that Nanopore sequencing adapters preferentially link to Cas9-cleaved DNA rather than artificially dephosphorylated DNA ends enabled the development of a Cas9-based enrichment approach called nCATS (nanopore Cas9 Targeted-Sequencing) [94]. Before Cas9 cleavage, pre-existing DNA ends are dephosphorylated. Next, preferential ligation of the newly produced DNA ends with adaptors is performed in preparation for Nanopore sequencing. This method yields fragments that are roughly 20 kb long, which is substantially less than the two previously stated methods but still enables 300-fold target enrichment. However, the absence of an amplification step strongly reduces process times and ensures that analyses of DNA methylation states are possible. Some protocol changes were made for the detection of genomic duplications [95], the observation of gene fusion events [96] and the reduction of background reads [97]. Exploiting the possibility of obtaining native DNA, an nCATS-like approach was used to identify the causal red fruit color variant in Malus domestica (Type 1 red flesh), previously defined as an 8kb repeated mini-satellite motif upstream of the MYB10 transcription factor. The same strategy with some implementations allowed the identification of SNPs and SVs in the MYB10 sequence of Prunus salicina. Moreover, Kirov et al. were able to identify SNP and InDel variations of full-length glutenin genes using the nCATS approach in planta, providing helpful knowledge for marker design and leading to a definition of polymorphism at the single allele level [98]. Similar approaches are the ones based on “Negative Enrichment” [80,99] and the FLASH (Finding Low Abundance Sequences by Hybridization) strategy [100].Taking advantage of the recently exploited Cas9-mediated adaptor ligation approach, McDonald et al. [101] combined it with a computational pipeline for the discovery of MEIs (mobile element insertions) in repetitive genomic regions in humans. In plants, Merkulov et al. [102] developed the novel NanoCasTE pipeline to scout both genetically inherited and somatic TEIs (transposable element insertions). The EVADÉ (EVD) retrotransposon insertions were identified in Arabidopsis with a 40x sequence coverage and in an Arabidopsis mutant with only 0.2x coverage, which is much lower than the one needed for TEIs identification based on WGS (whole genome sequencing). As an additional benefit of the Cas9-mediated sequencing approaches here reported, sequencing and mapping long reads on the genome could facilitate the identification of mobile elements in highly repetitive regions like centromeres and heterochromatin. As a slight deviation from the approaches outlined above, deep sequencing of target loci is the aim of the CRISPR-DS (CRISPR-Duplex Sequencing) approach [104], which is able to reach up to 49,000-fold target enrichment by cleaving target DNA into short fragments. A focus on the sequencing of shorter fragments is the Cas9-tiling [105,106]. In this case, the ROI is divided into smaller, overlapping sub-regions (sub-ROI) long up to 25 kb and are enriched and sequenced separately. Finaly, a study proposed by Lopatriello et al. [107], the Cas9-tiling approach allowed the de novo assembly and identification of SVs of a 250-kb region on chromosome Pv05 of Phaseolus vulgaris.
Conclusions and future perspectives
Throughout this article, we tried to show how the non-editing applications of the CRISPR/Cas are vast and versatile. Some of its applications have been developed as a tool to answer biological questions, for example LiveFISH and gene regulation complexes, while others are set as a pure diagnostic or functional tool such as SHERLOCK, the virus resistance engineering systems or the ones that are used to facilitate sequencing. A couple (if not most) of them, even have their place into both these categories, like CASFISH and its derivatives. The applications of CRISPR/Cas systems may be extremely diverse, but the underlying mechanism remains the same in general: the Cas protein is sent to or recognizes a target of interest, specified by a purposely designed guide RNA, which triggers its activity or that of the effectors associated with it. The high specificity of the Cas-mediated sequence makes for the huge precision of the tools that are derived from it. The problem of off-target, inherent to the use of CRISPR/Cas proteins, however remains, and precautions must be taken, and preliminary controls performed, before making use of such techniques. Nevertheless, it is to be said that, when compared to equivalent techniques, the tools making used of CRISPR-systems where often seen less prompt to unspecific and undesired sequence binding [12,67]. We saw several times that using CRISPR/Cas-mediated tools produce clearer, more specific and therefore more reliable results than not doing so while it is possible.
If some of the here mentioned applications were developed specifically on plant systems, it is rather the exception than the norm. Indeed, plants it can be tougher organisms to begin with and constructing a stable line of transgenic plants takes some time, which makes them less suited to the premises of a new tool, which is why most of the studies we reported have been conducted in yeast, bacteria, or humans. Ther is however no doubt that the majority of them will also get their vegetal equivalent.
More generally, if the development of CRISPR/Cas systems-related technologies has opened a wide range of opportunities for the modification of genomic sequences, a deeper look reveals that this tool can also be used for non-editing purposes that are even more varied. Such understanding of the underlying genetic processes is a sine qua non to the overall goal of manipulating genes or regulatory sequences, may it be for therapeutic purposes in humans or to enhance the genetic traits of bacteria as well as domestic plants and animals. However, unlike for non-editing tools, interest in using such genome-edition could rise and/or be democratized quicker in plants than in animals including humans, for the modification of the latter’s genome is still and should remain under strict ethical rules. In overall, these techniques facilitate both theoretical research, which is a crucial building block for later practical applications and the practical applications in question. Understanding how genes function, how they are regulated, and how they interact with one another is essential to perform gene editing and regulatory sequence alteration in a meaningful and rigorous way.
To conclude, the ultimate objective of gene and regulatory sequence editing, whether for therapeutic use or genetic enhancement, necessitates a profound comprehension of gene functionality, regulation, and interactions. CRISPR-based strategies play a pivotal role in expanding this knowledge base, enabling both theoretical research and practical applications to thrive in the realm of genetic manipulation.
Funding
The study was carried out within the Agritech National Research Center and received funding from the European Union Next-Generation EU (PIANO NAZIONALE DI RIPRESA E RESILIENZA (PNRR)—MISSIONE 4 COMPONENTE 2, INVESTIMENTO 1.4—D.D. 1032 17/06/2022, CN00000022). Our study represents a position paper related to: (1) Spoke 1 “Plant and animal genetic resources and adaptation to climate changes” and a baseline for the fulfillment of the milestones within task 1.3.3 titled “Development and implementation of novel biotechnological approaches, including cisgenesis and genome editing, for accelerated precision breeding”.
Abbreviations
| 53BP1 | p53-binding protein 1 |
| ABA | Abscisic acid |
| CAPTURE | CRISPR affinity purification in-situ of regulatory elements |
| CASFISH | Cas9-mediated fluorescence in situ hybridization |
| CATCH | Cas9-assisted targeting of chromosome segments |
| CATE-seq | CRISPR-assisted targeted enrichment-sequencing |
| nCATS | Nanopore Cas9 targeted sequencing |
| CIDs | Chemically inducible dimerizing domains |
| CISMR | CRISPR mediated isolation of specific megabase-sized regions of the genome |
| CRISDA | CRISPR–Cas9-triggered nicking endonuclease mediated strand displacement amplification |
| CRISPR | Clustered regularly interspaced short palindromic repeats |
| CRISPR-DS | CRISPR-duplex sequencing |
| CRISPRa | CRISPR activation |
| CRISPRi | CRISPR inhibition |
| CS | Chromoshadow domain |
| CUT-PCR | CRISPR-mediated, Ultrasensitive detection of Target DNA by PCR |
| Cas | CRISPR associated protein |
| dCas | Dead Cas |
| LwaCas13 | Cas13a from Leptotrichia wadei |
| SpCas9 | Cas9 from Streptococcus pyogenes |
| dCas9VPR | dCas9 fused to VPC domain |
| DASH | Depletion of abundant sequences by hybridization |
| dsDNA | Double-stranded DNA |
| ssDNA | Single-stranded DNA |
| DSB | Double stranded breaks |
| ERT | Ligand-binding domain ERT from estrogen receptor |
| FISH | Fluorescence in-situ hybridization |
| LiveFISH | CRISPR live-cell fluorescent in situ hybridization |
| FLASH | Finding low abundance sequences by hybridization |
| GA | Gibberellin |
| GFP | Green fluorescent protein |
| eGFP | Enhanced green fluorescent protein |
| HEPN | Higher eukaryotes and prokaryotes nucleotide-binding |
| HNH | Histidine-asparagine-histidine endonuclease domain |
| HSV | Herpes simplex virus 1 |
| OIDs | Optogenetically inducible dimerizing domains |
| PAM | Protospacer associated motif |
| RGEN-ISL | RNA-guided endonuclease - in situ labeling |
| crRNA | CRISPR RNA |
| gRNA | Guide RNA |
| scRNA | Scaffold RNA |
| sgRNA | Short guide RNA |
| ssRNA | Single-stranded RNA |
| tracrRNA | Trans-activating crRNA |
| RNAP | RNA polymerase |
| RNP | Ribonucleoprotein |
| RNase | Ribonuclease |
| ROIs | Regions of interest |
| RuvC | Recombination UV C |
| SAM | Synergistic Activation Mediator |
| SHERLOCK | High-sensitivity enzymatic reporter unlocking |
| SID4X | mSin3 interaction domain |
| SMRT | Single molecule - real time |
| SRDX | SUPERMAN Repression Domain X |
| SVs | Structural variants |
| TAD | TALE transcription activation domain |
| TEIs | Transposable element insertions |
| TRV | Tobacco rattle virus |
| VPR | VP64, p65 and Rta |
References
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A Programmable Dual-RNA-Guided DNA Endonuclease in Adaptive Bacterial Immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Pickar-Oliver, A.; Gersbach, C.A. The next Generation of CRISPR-Cas Technologies and Applications. Nat Rev Mol Cell Biol 2019, 20, 490–507. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Iranzo, J.; Shmakov, S.A.; Alkhnbashi, O.S.; Brouns, S.J.J.; Charpentier, E.; Cheng, D.; Haft, D.H.; Horvath, P.; et al. Evolutionary Classification of CRISPR-Cas Systems: A Burst of Class 2 and Derived Variants. Nat Rev Microbiol 2020, 18, 67–83. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Koonin, E.V. Evolutionary Classification of CRISPR-Cas Systems. In Crispr; John Wiley & Sons, Ltd, 2022; pp. 13–38 ISBN 978-1-68367-379-8. [CrossRef]
- Hryhorowicz, M.; Lipiński, D.; Zeyland, J.; Słomski, R. CRISPR/Cas9 Immune System as a Tool for Genome Engineering. Arch. Immunol. Ther. Exp. 2017, 65, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Kozovska, Z.; Rajcaniova, S.; Munteanu, P.; Dzacovska, S.; Demkova, L. CRISPR: History and Perspectives to the Future. Biomedicine & Pharmacotherapy 2021, 141, 111917. [Google Scholar] [CrossRef]
- Chen, W.; Ma, J.; Wu, Z.; Wang, Z.; Zhang, H.; Fu, W.; Pan, D.; Shi, J.; Ji, Q. Cas12n Nucleases, Early Evolutionary Intermediates of Type V CRISPR, Comprise a Distinct Family of Miniature Genome Editors. Molecular Cell 2023, 83, 2768–2780.e6. [Google Scholar] [CrossRef]
- Jiang, F.; Doudna, J.A. CRISPR–Cas9 Structures and Mechanisms. Annual Review of Biophysics 2017, 46, 505–529. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, M.R.; Oakes, B.L.; Sternberg, S.H.; East-Seletsky, A.; Kaplan, M.; Doudna, J.A. Programmable RNA Recognition and Cleavage by CRISPR/Cas9. Nature 2014, 516, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.W.; Wang, H.; Yang, H.; Shi, L.; Katz, Y.; Theunissen, T.W.; Rangarajan, S.; Shivalila, C.S.; Dadon, D.B.; Jaenisch, R. Multiplexed Activation of Endogenous Genes by CRISPR-on, an RNA-Guided Transcriptional Activator System. Cell Res 2013, 23, 1163–1171. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA Editing with CRISPR-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.T.; Kellner, M.J.; Regev, A.; et al. RNA Targeting with CRISPR–Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic Acid Detection with CRISPR-Cas13a/C2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Young, J.K.; Karvelis, T.; Kazlauskas, D.; Urbaitis, T.; Jasnauskaite, M.; Grusyte, M.M.; Paulraj, S.; Wang, P.-H.; Hou, Z.; et al. A Catalogue of Biochemically Diverse CRISPR-Cas9 Orthologs. Nat Commun 2020, 11, 5512. [Google Scholar] [CrossRef]
- Nakagawa, R.; Ishiguro, S.; Okazaki, S.; Mori, H.; Tanaka, M.; Aburatani, H.; Yachie, N.; Nishimasu, H.; Nureki, O. Engineered Campylobacter Jejuni Cas9 Variant with Enhanced Activity and Broader Targeting Range. Commun Biol 2022, 5, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Zetsche, B.; Gootenberg, J.S.; Abudayyeh, O.O.; Slaymaker, I.M.; Makarova, K.S.; Essletzbichler, P.; Volz, S.E.; Joung, J.; van der Oost, J.; Regev, A.; et al. Cpf1 Is a Single RNA-Guided Endonuclease of a Class 2 CRISPR-Cas System. Cell 2015, 163, 759–771. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.S.; Ma, E.; Harrington, L.B.; Da Costa, M.; Tian, X.; Palefsky, J.M.; Doudna, J.A. CRISPR-Cas12a Target Binding Unleashes Indiscriminate Single-Stranded DNase Activity. Science 2018, 360, 436–439. [Google Scholar] [CrossRef]
- Abudayyeh, O.O.; Gootenberg, J.S.; Konermann, S.; Joung, J.; Slaymaker, I.M.; Cox, D.B.T.; Shmakov, S.; Makarova, K.S.; Semenova, E.; Minakhin, L.; et al. C2c2 Is a Single-Component Programmable RNA-Guided RNA-Targeting CRISPR Effector. Science 2016, 353, aaf5573. [Google Scholar] [CrossRef]
- Cao, H.; Wang, Y.; Zhang, N.; Xia, S.; Tian, P.; Lu, L.; Du, J.; Du, Y. Progress of CRISPR-Cas13 Mediated Live-Cell RNA Imaging and Detection of RNA-Protein Interactions. Frontiers in Cell and Developmental Biology 2022, 10. [Google Scholar] [CrossRef]
- Yu, Y.; Pan, Z.; Wang, X.; Bian, X.; Wang, W.; Liang, Q.; Kou, M.; Ji, H.; Li, Y.; Ma, D.; et al. Targeting of SPCSV-RNase3 via CRISPR-Cas13 Confers Resistance against Sweet Potato Virus Disease. Molecular Plant Pathology 2022, 23, 104–117. [Google Scholar] [CrossRef]
- Schmitz, R.J.; Grotewold, E.; Stam, M. Cis-Regulatory Sequences in Plants: Their Importance, Discovery, and Future Challenges. The Plant Cell 2022, 34, 718–741. [Google Scholar] [CrossRef]
- Hernandez-Garcia, C.M.; Finer, J.J. Identification and Validation of Promoters and Cis-Acting Regulatory Elements. Plant Science 2014, 217–218, 109–119. [Google Scholar] [CrossRef]
- Bikard, D.; Jiang, W.; Samai, P.; Hochschild, A.; Zhang, F.; Marraffini, L.A. Programmable Repression and Activation of Bacterial Gene Expression Using an Engineered CRISPR-Cas System. Nucleic Acids Research 2013, 41, 7429–7437. [Google Scholar] [CrossRef]
- La Russa, M.F.; Qi, L.S. The New State of the Art: Cas9 for Gene Activation and Repression. Molecular and Cellular Biology 2015, 35, 3800–3809. [Google Scholar] [CrossRef] [PubMed]
- Morita, S.; Horii, T.; Hatada, I. Regulation of Gene Expression Using dCas9-SunTag Platforms. In Epigenomics: Methods and Protocols; Hatada, I., Horii, T., Eds.; Methods in Molecular Biology; Springer US: New York, NY, 2023; pp. 189–195. ISBN 978-1-07-162724-2. [Google Scholar] [CrossRef]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A Protein-Tagging System for Signal Amplification in Gene Expression and Fluorescence Imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef]
- Zalatan, J.G.; Lee, M.E.; Almeida, R.; Gilbert, L.A.; Whitehead, E.H.; La Russa, M.; Tsai, J.C.; Weissman, J.S.; Dueber, J.E.; Qi, L.S.; et al. Engineering Complex Synthetic Transcriptional Programs with CRISPR RNA Scaffolds. Cell 2015, 160, 339–350. [Google Scholar] [CrossRef]
- Papikian, A.; Liu, W.; Gallego-Bartolomé, J.; Jacobsen, S.E. Site-Specific Manipulation of Arabidopsis Loci Using CRISPR-Cas9 SunTag Systems. Nature Communications 2019, 10. [Google Scholar] [CrossRef]
- Morita, S.; Horii, T.; Kimura, M.; Hatada, I. Synergistic Upregulation of Target Genes by TET1 and VP64 in the dCas9–SunTag Platform. International Journal of Molecular Sciences 2020, 21, 1574. [Google Scholar] [CrossRef]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; P R Iyer, E.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J.; et al. Highly Efficient Cas9-Mediated Transcriptional Programming. Nat Methods 2015, 12, 326–328. [Google Scholar] [CrossRef]
- Selma, S.; Bernabé-Orts, J.M.; Vazquez-Vilar, M.; Diego-Martin, B.; Ajenjo, M.; Garcia-Carpintero, V.; Granell, A.; Orzaez, D. Strong Gene Activation in Plants with Genome-Wide Specificity Using a New Orthogonal CRISPR/Cas9-Based Programmable Transcriptional Activator. Plant Biotechnology Journal 2019, 17, 1703–1705. [Google Scholar] [CrossRef] [PubMed]
- Konermann, S.; Brigham, M.D.; Trevino, A.E.; Joung, J.; Abudayyeh, O.O.; Barcena, C.; Hsu, P.D.; Habib, N.; Gootenberg, J.S.; Nishimasu, H.; et al. Genome-Scale Transcriptional Activation by an Engineered CRISPR-Cas9 Complex. Nature 2015, 517, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, D.; Xiong, X.; Yan, B.; Xie, W.; Sheen, J.; Li, J.-F. A Potent Cas9-Derived Gene Activator for Plant and Mammalian Cells. Nature Plants 2017, 3, 930–936. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A.; et al. CRISPR-Mediated Modular RNA-Guided Regulation of Transcription in Eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C.; et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Lowder, L.G.; Zhang, D.; Baltes, N.J.; Paul, J.W., III; Tang, X.; Zheng, X.; Voytas, D.F.; Hsieh, T.-F.; Zhang, Y.; Qi, Y. A CRISPR/Cas9 Toolbox for Multiplexed Plant Genome Editing and Transcriptional Regulation. Plant Physiology 2015, 169, 971–985. [Google Scholar] [CrossRef]
- Piatek, A.; Ali, Z.; Baazim, H.; Li, L.; Abulfaraj, A.; Al-Shareef, S.; Aouida, M.; Mahfouz, M.M. RNA-Guided Transcriptional Regulation in Planta via Synthetic dCas9-Based Transcription Factors. Plant Biotechnology Journal 2015, 13, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Pan, C.; Sretenovic, S.; Qi, Y. CRISPR/dCas-Mediated Transcriptional and Epigenetic Regulation in Plants. Current Opinion in Plant Biology 2021, 60, 101980. [Google Scholar] [CrossRef]
- Xu, X.; Qi, L.S. A CRISPR–dCas Toolbox for Genetic Engineering and Synthetic Biology. Journal of Molecular Biology 2019, 431, 34–47. [Google Scholar] [CrossRef]
- Bao, Z.; Jain, S.; Jaroenpuntaruk, V.; Zhao, H. Orthogonal Genetic Regulation in Human Cells Using Chemically Induced CRISPR/Cas9 Activators. ACS Synth. Biol. 2017, 6, 686–693. [Google Scholar] [CrossRef]
- Chen, F.; Hu, Y.; Vannozzi, A.; Wu, K.; Cai, H.; Qin, Y.; Mullis, A.; Lin, Z.; Zhang, L. The WRKY Transcription Factor Family in Model Plants and Crops. Critical Reviews in Plant Sciences 2017, 36, 311–335. [Google Scholar] [CrossRef]
- Gao, Y.; Xiong, X.; Wong, S.; Charles, E.J.; Lim, W.A.; Qi, L.S. Complex Transcriptional Modulation with Orthogonal and Inducible dCas9 Regulators. Nat Methods 2016, 13, 1043–1049. [Google Scholar] [CrossRef] [PubMed]
- Levskaya, A.; Weiner, O.D.; Lim, W.A.; Voigt, C.A. Spatiotemporal Control of Cell Signalling Using a Light-Switchable Protein Interaction. Nature 2009, 461, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Nihongaki, Y.; Yamamoto, S.; Kawano, F.; Suzuki, H.; Sato, M. CRISPR-Cas9-Based Photoactivatable Transcription System. Chemistry & Biology 2015, 22, 169–174. [Google Scholar] [CrossRef]
- Polstein, L.R.; Gersbach, C.A. A Light-Inducible CRISPR-Cas9 System for Control of Endogenous Gene Activation. Nat Chem Biol 2015, 11, 198–200. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.P.; Miyaoka, Y.; Gilbert, L.A.; Mayerl, S.J.; Lee, B.H.; Weissman, J.S.; Conklin, B.R.; Wells, J.A. Ligand-Binding Domains of Nuclear Receptors Facilitate Tight Control of Split CRISPR Activity. Nature Communications 2016, 7. [Google Scholar] [CrossRef]
- Tarasava, K.; Oh, E.J.; Eckert, C.A.; Gill, R.T. CRISPR-Enabled Tools for Engineering Microbial Genomes and Phenotypes. Biotechnol J 2018, 13, e1700586. [Google Scholar] [CrossRef]
- Jing, X.; Xie, B.; Chen, L.; Zhang, N.; Jiang, Y.; Qin, H.; Wang, H.; Hao, P.; Yang, S.; Li, X. Implementation of the CRISPR-Cas13a System in Fission Yeast and Its Repurposing for Precise RNA Editing. Nucleic Acids Research 2018, 46, e90. [Google Scholar] [CrossRef] [PubMed]
- Kushawah, G.; Hernandez-Huertas, L.; Abugattas-Nuñez del Prado, J.; Martinez-Morales, J.R.; DeVore, M.L.; Hassan, H.; Moreno-Sanchez, I.; Tomas-Gallardo, L.; Diaz-Moscoso, A.; Monges, D.E.; et al. CRISPR-Cas13d Induces Efficient mRNA Knockdown in Animal Embryos. Developmental Cell 2020, 54, 805–817.e7. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.; Qi, Y. Plant Gene Knockout and Knockdown by CRISPR-Cpf1 (Cas12a) Systems. In Plant Genome Editing with CRISPR Systems: Methods and Protocols; Qi, Y., Ed.; Methods in Molecular Biology; Springer: New York, NY, 2019; pp. 245–256. ISBN 978-1-4939-8991-1. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Chen, Y.; Li, M.; Zhou, F.; Li, K.; Cao, H.; Ni, M.; Liu, Y.; Gu, Z.; et al. In Situ Capture of Chromatin Interactions by Biotinylated dCas9. Cell 2017, 170, 1028–1043.e19. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, Y.; Chen, Y.; Li, M.; Shao, Z.; Zhang, M.Q.; Xu, J. CAPTURE: In Situ Analysis of Chromatin Composition of Endogenous Genomic Loci by Biotinylated dCas9. Current Protocols in Molecular Biology 2018, 123, e64. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; He, Z.; Liu, Z.; Qu, M.; Gao, C.; Wang, C.; Wang, Y. A Reverse Chromatin Immunoprecipitation Technique Based on the CRISPR–dCas9 System. Plant Physiology 2023, 191, 1505–1519. [Google Scholar] [CrossRef]
- Orphanides, G.; Reinberg, D. A Unified Theory of Gene Expression. Cell 2002, 108, 439–451. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Chen, S.; Jia, G. Detection, Regulation, and Functions of RNA N6-Methyladenosine Modification in Plants. Plant Communications 2023, 4, 100546. [Google Scholar] [CrossRef] [PubMed]
- Maslova, A.; Krasikova, A. FISH Going Meso-Scale: A Microscopic Search for Chromatin Domains. Front Cell Dev Biol 2021, 9, 753097. [Google Scholar] [CrossRef]
- Khosravi, S.; Ishii, T.; Dreissig, S.; Houben, A. Application and Prospects of CRISPR/Cas9-Based Methods to Trace Defined Genomic Sequences in Living and Fixed Plant Cells. Chromosome Res 2020, 28, 7–17. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, S.; Dreissig, S.; Schindele, P.; Wolter, F.; Rutten, T.; Puchta, H.; Houben, A. Live-Cell CRISPR Imaging in Plant Cells with a Telomere-Specific Guide RNA. In RNA Tagging: Methods and Protocols; Heinlein, M., Ed.; Methods in Molecular Biology; Springer US: New York, NY, 2020; pp. 343–356. ISBN 978-1-07-160712-1. [Google Scholar] [CrossRef]
- Wu, X.; Mao, S.; Ying, Y.; Krueger, C.J.; Chen, A.K. Progress and Challenges for Live-Cell Imaging of Genomic Loci Using CRISPR-Based Platforms. Genomics, Proteomics & Bioinformatics 2019, 17, 119–128. [Google Scholar] [CrossRef]
- Wang, H.; Nakamura, M.; Abbott, T.R.; Zhao, D.; Luo, K.; Yu, C.; Nguyen, C.M.; Lo, A.; Daley, T.P.; La Russa, M.; et al. CRISPR-Mediated Live Imaging of Genome Editing and Transcription. Science 2019, 365, 1301–1305. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Gilbert, L.A.; Cimini, B.A.; Schnitzbauer, J.; Zhang, W.; Li, G.-W.; Park, J.; Blackburn, E.H.; Weissman, J.S.; Qi, L.S.; et al. Dynamic Imaging of Genomic Loci in Living Human Cells by an Optimized CRISPR/Cas System. Cell 2013, 155, 1479–1491. [Google Scholar] [CrossRef]
- Deng, W.; Shi, X.; Tjian, R.; Lionnet, T.; Singer, R.H. CASFISH: CRISPR/Cas9-Mediated in Situ Labeling of Genomic Loci in Fixed Cells. Proceedings of the National Academy of Sciences 2015, 112, 11870–11875. [Google Scholar] [CrossRef]
- Fu, Y.; Rocha, P.P.; Luo, V.M.; Raviram, R.; Deng, Y.; Mazzoni, E.O.; Skok, J.A. CRISPR-dCas9 and sgRNA Scaffolds Enable Dual-Colour Live Imaging of Satellite Sequences and Repeat-Enriched Individual Loci. Nat Commun 2016, 7, 11707. [Google Scholar] [CrossRef] [PubMed]
- Khosravi, S.; Schindele, P.; Gladilin, E.; Dunemann, F.; Rutten, T.; Puchta, H.; Houben, A. Application of Aptamers Improves CRISPR-Based Live Imaging of Plant Telomeres. Front Plant Sci 2020, 11, 1254. [Google Scholar] [CrossRef] [PubMed]
- Dreissig, S.; Schiml, S.; Schindele, P.; Weiss, O.; Rutten, T.; Schubert, V.; Gladilin, E.; Mette, M.F.; Puchta, H.; Houben, A. Live-Cell CRISPR Imaging in Plants Reveals Dynamic Telomere Movements. The Plant Journal 2017, 91, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Ishii, T.; Schubert, V.; Khosravi, S.; Dreissig, S.; Metje-Sprink, J.; Sprink, T.; Fuchs, J.; Meister, A.; Houben, A. RNA-Guided Endonuclease – in Situ Labelling (RGEN-ISL): A Fast CRISPR/Cas9-Based Method to Label Genomic Sequences in Various Species. New Phytologist 2019, 222, 1652–1661. [Google Scholar] [CrossRef] [PubMed]
- Potlapalli, B.P.; Schubert, V.; Metje-Sprink, J.; Liehr, T.; Houben, A. Application of Tris-HCl Allows the Specific Labeling of Regularly Prepared Chromosomes by CRISPR-FISH. Cytogenet Genome Res 2020, 160, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, A.M.; Rettig, G.R.; Turk, R.; Collingwood, M.A.; Zeiner, S.A.; Quadros, R.M.; Harms, D.W.; Bonthuis, P.J.; Gregg, C.; Ohtsuka, M.; et al. Simplified CRISPR Tools for Efficient Genome Editing and Streamlined Protocols for Their Delivery into Mammalian Cells and Mouse Zygotes. Methods 2017, 121–122, 16–28. [Google Scholar] [CrossRef] [PubMed]
- Nagaki, K.; Yamaji, N. Decrosslinking Enables Visualization of RNA-Guided Endonuclease–in Situ Labeling Signals for DNA Sequences in Plant Tissues. Journal of Experimental Botany 2020, 71, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Potlapalli, B.P.; Ishii, T.; Nagaki, K.; Somasundaram, S.; Houben, A. CRISPR-FISH: A CRISPR/Cas9-Based In Situ Labeling Method. In Plant Cytogenetics and Cytogenomics: Methods and Protocols; Heitkam, T., Garcia, S., Eds.; Methods in Molecular Biology; Springer US: New York, NY, 2023; pp. 315–335. ISBN 978-1-07-163226-0. [Google Scholar] [CrossRef]
- Makarova, K.S.; Haft, D.H.; Barrangou, R.; Brouns, S.J.J.; Charpentier, E.; Horvath, P.; Moineau, S.; Mojica, F.J.M.; Wolf, Y.I.; Yakunin, A.F.; et al. Evolution and Classification of the CRISPR–Cas Systems. Nat Rev Microbiol 2011, 9, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, L.; Rajashekara, H.; Uppala, L.S.; Ambika, D.S.; Patil, B.; Shankarappa, K.S.; Nath, V.S.; Kavitha, T.R.; Mishra, A.K. Mechanisms of Microbial Plant Protection and Control of Plant Viruses. Plants 2022, 11, 3449. [Google Scholar] [CrossRef]
- Ali, Z.; Abulfaraj, A.; Idris, A.; Ali, S.; Tashkandi, M.; Mahfouz, M.M. CRISPR/Cas9-Mediated Viral Interference in Plants. Genome Biology 2015, 16, 238. [Google Scholar] [CrossRef]
- Kalinina, N.O.; Khromov, A.; Love, A.J.; Taliansky, M.E. CRISPR Applications in Plant Virology: Virus Resistance and Beyond. Phytopathology® 2020, 110, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Taliansky, M.; Samarskaya, V.; Zavriev, S.K.; Fesenko, I.; Kalinina, N.O.; Love, A.J. RNA-Based Technologies for Engineering Plant Virus Resistance. Plants 2021, 10, 82. [Google Scholar] [CrossRef] [PubMed]
- Karpov, D.S.; Demidova, N.A.; Kulagin, K.A.; Shuvalova, A.I.; Kovalev, M.A.; Simonov, R.A.; Karpov, V.L.; Snezhkina, A.V.; Kudryavtseva, A.V.; Klimova, R.R.; et al. Complete and Prolonged Inhibition of Herpes Simplex Virus Type 1 Infection In Vitro by CRISPR/Cas9 and CRISPR/CasX Systems. Int J Mol Sci 2022, 23, 14847. [Google Scholar] [CrossRef] [PubMed]
- Bayoumi, M.; Munir, M. Potential Use of CRISPR/Cas13 Machinery in Understanding Virus-Host Interaction. Front Microbiol 2021, 12, 743580. [Google Scholar] [CrossRef] [PubMed]
- Schultzhaus, Z.; Wang, Z.; Stenger, D. CRISPR-Based Enrichment Strategies for Targeted Sequencing. Biotechnology Advances 2021, 46, 107672. [Google Scholar] [CrossRef] [PubMed]
- Stevens, R.C.; Steele, J.L.; Glover, W.R.; Sanchez-Garcia, J.F.; Simpson, S.D.; O’Rourke, D.; Ramsdell, J.S.; MacManes, M.D.; Thomas, W.K.; Shuber, A.P. A Novel CRISPR/Cas9 Associated Technology for Sequence-Specific Nucleic Acid Enrichment. PLoS ONE 2019, 14, e0215441. [Google Scholar] [CrossRef] [PubMed]
- Ramani, V.; Shendure, J. Smash and DASH with Cas9. Genome Biol 2016, 17, 42. [Google Scholar] [CrossRef]
- Gu, W.; Crawford, E.D.; O’Donovan, B.D.; Wilson, M.R.; Chow, E.D.; Retallack, H.; DeRisi, J.L. Depletion of Abundant Sequences by Hybridization (DASH): Using Cas9 to Remove Unwanted High-Abundance Species in Sequencing Libraries and Molecular Counting Applications. Genome Biol 2016, 17, 41. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Yu, J.; Hwang, G.-H.; Kim, S.; Kim, H.S.; Ye, S.; Kim, K.; Park, J.; Park, D.Y.; Cho, Y.-K.; et al. CUT-PCR: CRISPR-Mediated, Ultrasensitive Detection of Target DNA Using PCR. Oncogene 2017, 36, 6823–6829. [Google Scholar] [CrossRef]
- Song, L.; Xie, K. Engineering CRISPR/Cas9 to Mitigate Abundant Host Contamination for 16S rRNA Gene-Based Amplicon Sequencing. Microbiome 2020, 8, 80. [Google Scholar] [CrossRef]
- Rossato, M.; Marcolungo, L.; De Antoni, L.; Lopatriello, G.; Bellucci, E.; Cortinovis, G.; Frascarelli, G.; Nanni, L.; Bitocchi, E.; Di Vittori, V.; et al. CRISPR/Cas9-Based Repeat Depletion for the High-Throughput Genotyping of Complex Plant Genomes; Genomics, 2022; 2022. [CrossRef]
- Aalipour, A.; Dudley, J.C.; Park, S.; Murty, S.; Chabon, J.J.; Boyle, E.A.; Diehn, M.; Gambhir, S.S. Deactivated CRISPR Associated Protein 9 for Minor-Allele Enrichment in Cell-Free DNA. Clinical Chemistry 2018, 64, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Xia, Q.; Zhang, S.; Gao, J.; Dai, W.; Wu, J.; Wang, J. CRISPR-Assisted Targeted Enrichment-Sequencing (CATE-Seq). 2019. [CrossRef]
- Zhou, W.; Hu, L.; Ying, L.; Zhao, Z.; Chu, P.K.; Yu, X.-F. A CRISPR–Cas9-Triggered Strand Displacement Amplification Method for Ultrasensitive DNA Detection. Nat Commun 2018, 9, 5012. [Google Scholar] [CrossRef]
- Lee, J.; Lim, H.; Jang, H.; Hwang, B.; Lee, J.H.; Cho, J.; Lee, J.H.; Bang, D. CRISPR-Cap: Multiplexed Double-Stranded DNA Enrichment Based on the CRISPR System. Nucleic Acids Research 2019, 47, e1–e1. [Google Scholar] [CrossRef]
- Slesarev, A.; Viswanathan, L.; Tang, Y.; Borgschulte, T.; Achtien, K.; Razafsky, D.; Onions, D.; Chang, A.; Cote, C. CRISPR/Cas9 Targeted CAPTURE of Mammalian Genomic Regions for Characterization by NGS. Sci Rep 2019, 9, 3587. [Google Scholar] [CrossRef] [PubMed]
- Tsai, Y.-C.; Greenberg, D.; Powell, J.; Höijer, I.; Ameur, A.; Strahl, M.; Ellis, E.; Jonasson, I.; Mouro Pinto, R.; Wheeler, V.C.; et al. Amplification-Free, CRISPR-Cas9 Targeted Enrichment and SMRT Sequencing of Repeat-Expansion Disease Causative Genomic Regions; Genomics, 2017; [CrossRef]
- Bennett-Baker, P.E.; Mueller, J.L. CRISPR-Mediated Isolation of Specific Megabase Segments of Genomic DNA. Nucleic Acids Research 2017, 45, e165–e165. [Google Scholar] [CrossRef]
- Gabrieli, T.; Sharim, H.; Fridman, D.; Arbib, N.; Michaeli, Y.; Ebenstein, Y. Selective Nanopore Sequencing of Human BRCA1 by Cas9-Assisted Targeting of Chromosome Segments (CATCH). Nucleic Acids Research 2018, 46, e87–e87. [Google Scholar] [CrossRef] [PubMed]
- Gilpatrick, T.; Lee, I.; Graham, J.E.; Raimondeau, E.; Bowen, R.; Heron, A.; Downs, B.; Sukumar, S.; Sedlazeck, F.J.; Timp, W. Targeted Nanopore Sequencing with Cas9-Guided Adapter Ligation. Nat Biotechnol 2020, 38, 433–438. [Google Scholar] [CrossRef]
- Watson, C.M.; Crinnion, L.A.; Hewitt, S.; Bates, J.; Robinson, R.; Carr, I.M.; Sheridan, E.; Adlard, J.; Bonthron, D.T. Cas9-Based Enrichment and Single-Molecule Sequencing for Precise Characterization of Genomic Duplications. Laboratory Investigation 2020, 100, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Stangl, C.; de Blank, S.; Renkens, I.; Westera, L.; Verbeek, T.; Valle-Inclan, J.E.; González, R.C.; Henssen, A.G.; van Roosmalen, M.J.; Stam, R.W.; et al. Partner Independent Fusion Gene Detection by Multiplexed CRISPR-Cas9 Enrichment and Long Read Nanopore Sequencing. Nat Commun 2020, 11, 2861. [Google Scholar] [CrossRef] [PubMed]
- Iyer, S.V.; Kramer, M.; Goodwin, S.; McCombie, W.R. ACME: An Affinity-Based Cas9 Mediated Enrichment Method for Targeted Nanopore Sequencing 2022, 2022.02.03.478550. [CrossRef]
- Kirov, I.; Merkulov, P.; Gvaramiya, S.; Komakhin, R.; Omarov, M.; Dudnikov, M.; Kocheshkova, A.; Soloviev, A.; Karlov, G.; Divashuk, M. Illuminating the Transposon Insertion Landscape in Plants Using Cas9-Targeted Nanopore Sequencing and a Novel Pipeline 2021, 2021.06.11.448052. [CrossRef]
- Steele, J.L.; Stevens, R.C.; Cabrera, O.A.; Bassill, G.J.; Cramer, S.M.; Guzman, F.; Shuber, A.P. Novel CRISPR-Based Sequence Specific Enrichment Methods for Target Loci and Single Base Mutations. PLoS ONE 2020, 15, e0243781. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Langelier, C.; Kuchta, A.; Batson, J.; Teyssier, N.; Lyden, A.; Caldera, S.; McGeever, A.; Dimitrov, B.; King, R.; et al. FLASH: A next-Generation CRISPR Diagnostic for Multiplexed Detection of Antimicrobial Resistance Sequences. Nucleic Acids Research 2019, 47, e83–e83. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.L.; Zhou, W.; Castro, C.P.; Mumm, C.; Switzenberg, J.A.; Mills, R.E.; Boyle, A.P. Cas9 Targeted Enrichment of Mobile Elements Using Nanopore Sequencing. Nat Commun 2021, 12, 3586. [Google Scholar] [CrossRef] [PubMed]
- Merkulov, P.; Gvaramiya, S.; Komakhin, R.; Omarov, M.; Dudnikov, M.; Kocheshkova, A.; Konstantinov, Z.; Soloviev, A.; Karlov, G.; Divashuk, M.; et al. Cas9-Targeted Nanopore Sequencing Rapidly Elucidates the Transposition Preferences and DNA Methylation Profiles of Mobile Elements in Plants; Genomics, 2021; [CrossRef]
- Nachmanson, D.; Lian, S.; Schmidt, E.K.; Hipp, M.J.; Baker, K.T.; Zhang, Y.; Tretiakova, M.; Loubet-Senear, K.; Kohrn, B.F.; Salk, J.J.; et al. Targeted Genome Fragmentation with CRISPR/Cas9 Enables Fast and Efficient Enrichment of Small Genomic Regions and Ultra-Accurate Sequencing with Low DNA Input (CRISPR-DS). Genome Res. 2018, 28, 1589–1599. [Google Scholar] [CrossRef] [PubMed]
- Bruijnesteijn, J.; van der Wiel, M.; de Groot, N.G.; Bontrop, R.E. Rapid Characterization of Complex Killer Cell Immunoglobulin-Like Receptor (KIR) Regions Using Cas9 Enrichment and Nanopore Sequencing. Frontiers in Immunology 2021, 12. [CrossRef]
- Rubben, K.; Tilleman, L.; Deserranno, K.; Tytgat, O.; Deforce, D.; Nieuwerburgh, F.V. Cas9 Targeted Nanopore Sequencing with Enhanced Variant Calling Improves CYP2D6-CYP2D7 Hybrid Allele Genotyping. PLOS Genetics 2022, 18, e1010176. [Google Scholar] [CrossRef]
- Lopatriello, G.; Maestri, S.; Alfano, M.; Papa, R.; Di Vittori, V.; De Antoni, L.; Bellucci, E.; Pieri, A.; Bitocchi, E.; Delledonne, M.; et al. CRISPR/Cas9-Mediated Enrichment Coupled to Nanopore Sequencing Provides a Valuable Tool for the Precise Reconstruction of Large Genomic Target Regions. International Journal of Molecular Sciences 2023, 24, 1076. [Google Scholar] [CrossRef]
Figure 1.
Overview of the main Cas proteins reviewed in this article. (a) Catalytically active Cas9-sgRNA ribonucleoproteic complex and how it binds DNA target., one for each DNA strand. Length of the sgRNA is indicated. (b) “dead” Cas9-sgRNA ribonucleoproteic complex. The stars represent the two mutations that induce loss of nuclease activity. (c) Catalytically active Cas12a ribonucleoproteic complex and how it binds to the target locus. Length of the sgRNA is indicated. The scissors represent the tow cleaving sites of the two RuvC nuclease domains. (d) Catalytically active Cas13 ribonucleoproteic complex and how it binds to the ssRNA. The scissors represent the cleavage site of the HEPN nuclease domain.
Figure 1.
Overview of the main Cas proteins reviewed in this article. (a) Catalytically active Cas9-sgRNA ribonucleoproteic complex and how it binds DNA target., one for each DNA strand. Length of the sgRNA is indicated. (b) “dead” Cas9-sgRNA ribonucleoproteic complex. The stars represent the two mutations that induce loss of nuclease activity. (c) Catalytically active Cas12a ribonucleoproteic complex and how it binds to the target locus. Length of the sgRNA is indicated. The scissors represent the tow cleaving sites of the two RuvC nuclease domains. (d) Catalytically active Cas13 ribonucleoproteic complex and how it binds to the ssRNA. The scissors represent the cleavage site of the HEPN nuclease domain.
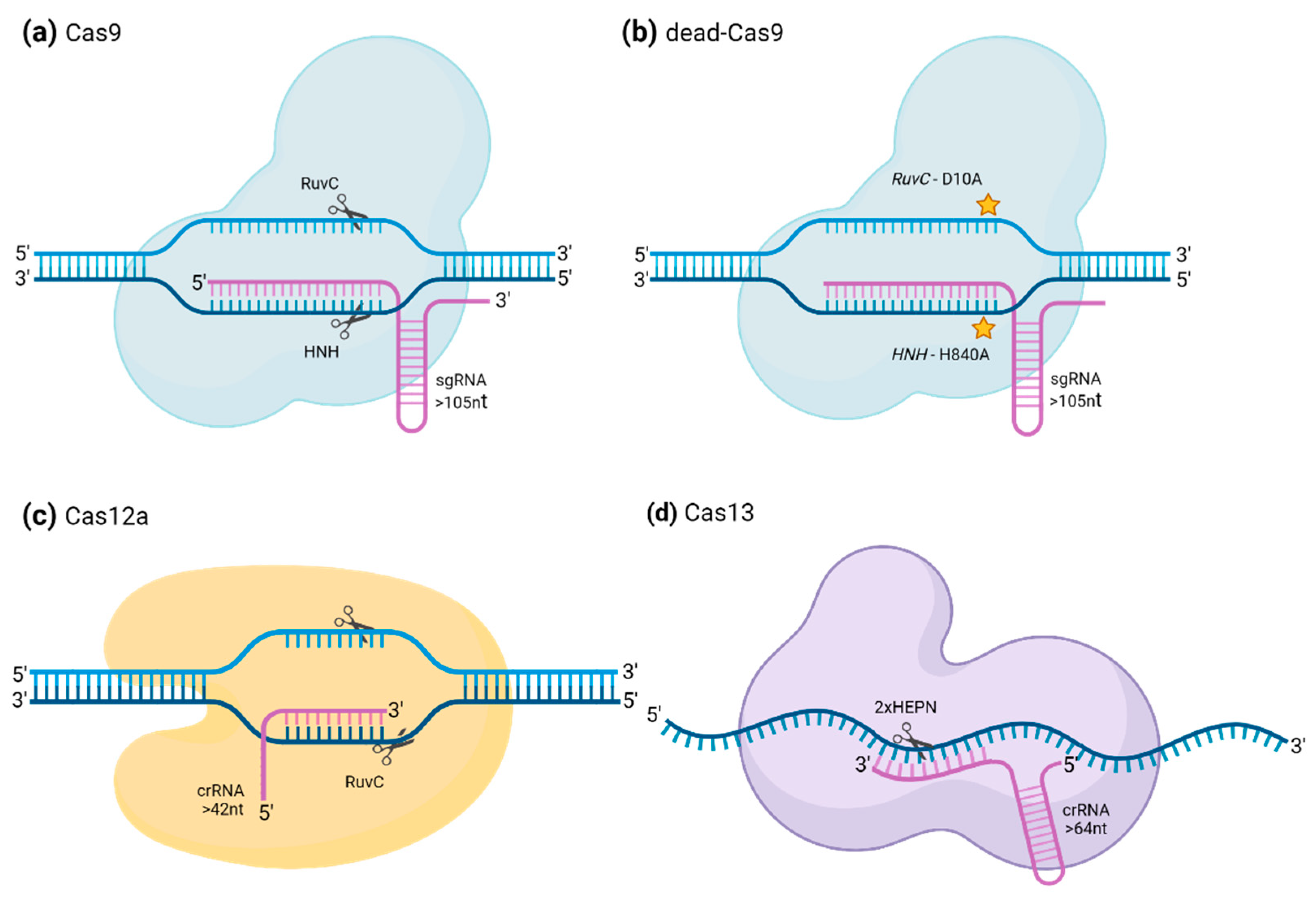
Figure 2.
CRISPR/dCas9 based systems for gene expression regulation. (a) Road blocker: dCas9 exerts steric hindrance towards RNA Polymerase by binding target DNA. (b,c) Generic activator or repressor (CRISPRa or CRISPRi): dCas9 protein is associated to a single activator or repressor. (d) SunTag: tandem array of peptides engages several copies of VP64 activator. (e) SAM: two MS2 RNA hairpins each carrying p65 and HSF1 activators. (f) VPR: VP64, p65 and Rta activators placed in a specific order to maximize gene activation. (g) TV: six copies of TALE and two of VP64 activators. (h) scRNA: any combination of activators and repressors enabled by coupling several RNA hairpins. (i) Dimerization Systems: dCas9 activity spatially and temporally controlled by sensing optical and/or chemical inputs.
Figure 2.
CRISPR/dCas9 based systems for gene expression regulation. (a) Road blocker: dCas9 exerts steric hindrance towards RNA Polymerase by binding target DNA. (b,c) Generic activator or repressor (CRISPRa or CRISPRi): dCas9 protein is associated to a single activator or repressor. (d) SunTag: tandem array of peptides engages several copies of VP64 activator. (e) SAM: two MS2 RNA hairpins each carrying p65 and HSF1 activators. (f) VPR: VP64, p65 and Rta activators placed in a specific order to maximize gene activation. (g) TV: six copies of TALE and two of VP64 activators. (h) scRNA: any combination of activators and repressors enabled by coupling several RNA hairpins. (i) Dimerization Systems: dCas9 activity spatially and temporally controlled by sensing optical and/or chemical inputs.
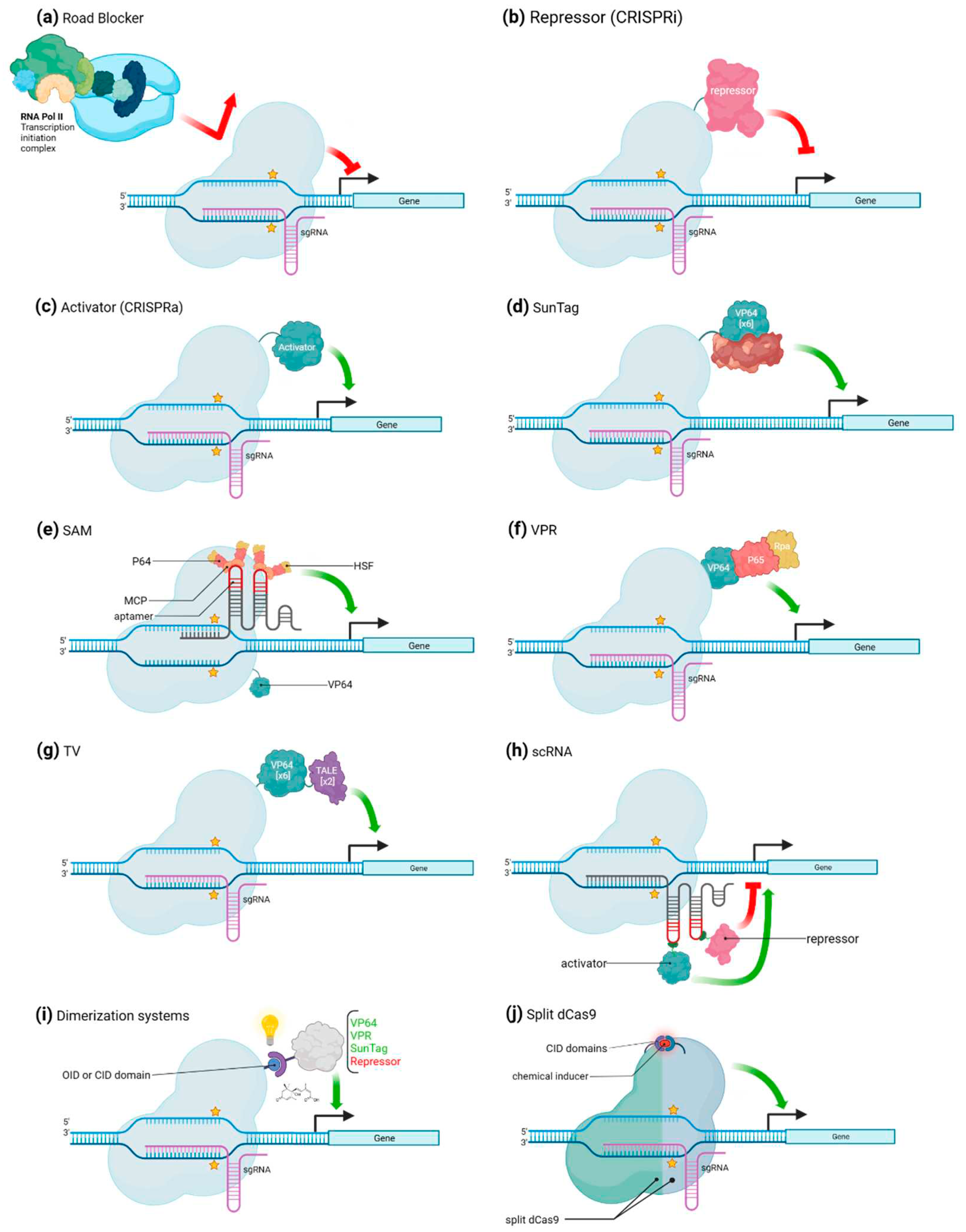
Figure 3.
CRISPR/Cas mediated imaging of nucleic acid sequences: different tools that were developed (a) dCas9 fused to GFP and its sgRNA when bound at the target DNA sequence. The stars represent the loss-of-function mutations on the two nuclease domains. (b) Alt-R CRISPR/Cas9 system with dCas9. The dCas9 is bound to DNA at the position specified by the protospacer of the crRNA part of the assembled sgRNA. The tracrRNA is bound on one side to the tail of the crRNA and to the other side is attached an ATTO fluorophore (F). (c) The sgRNA is prolonged into a scaffold harboring two aptamers to which MS2 and PP7 proteins can bind, which are themselves fused to fluorescent proteins (F). (d, e) LiveFISH. Catalytically active Cas9 guided by a short sgRNA harboring a fluorophore (F). (f) Nascent RNA labeling. Cas9 is guided to a transcribed DNA locus and is labeled through the fluorophore carried by the sgRNA. In the meantime, Cas13 is guided to a 5’ sequence of the RNA being transcribed and is labelled through the fluorophore-carrying guide.
Figure 3.
CRISPR/Cas mediated imaging of nucleic acid sequences: different tools that were developed (a) dCas9 fused to GFP and its sgRNA when bound at the target DNA sequence. The stars represent the loss-of-function mutations on the two nuclease domains. (b) Alt-R CRISPR/Cas9 system with dCas9. The dCas9 is bound to DNA at the position specified by the protospacer of the crRNA part of the assembled sgRNA. The tracrRNA is bound on one side to the tail of the crRNA and to the other side is attached an ATTO fluorophore (F). (c) The sgRNA is prolonged into a scaffold harboring two aptamers to which MS2 and PP7 proteins can bind, which are themselves fused to fluorescent proteins (F). (d, e) LiveFISH. Catalytically active Cas9 guided by a short sgRNA harboring a fluorophore (F). (f) Nascent RNA labeling. Cas9 is guided to a transcribed DNA locus and is labeled through the fluorophore carried by the sgRNA. In the meantime, Cas13 is guided to a 5’ sequence of the RNA being transcribed and is labelled through the fluorophore-carrying guide.
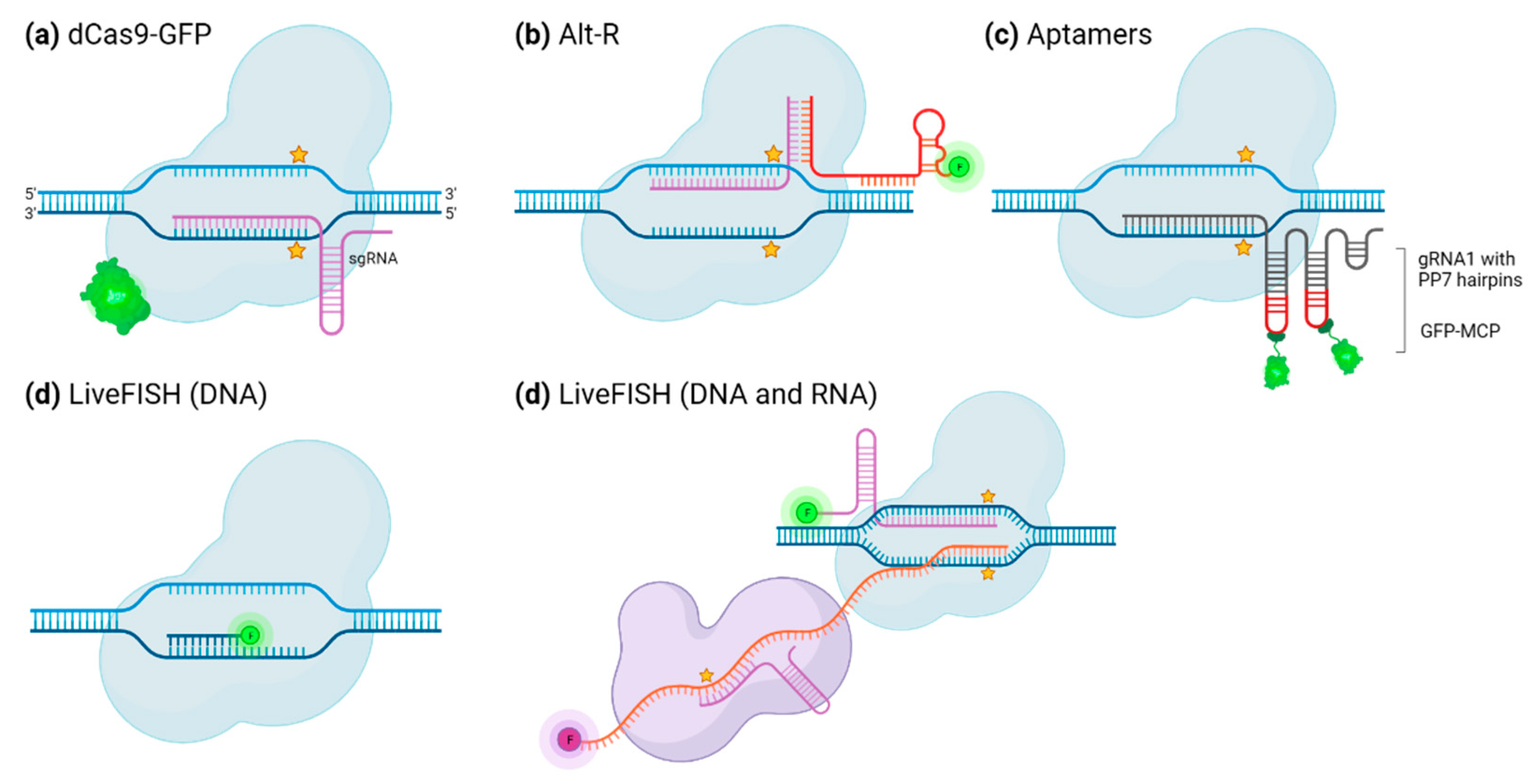
Figure 4.
CRISPR/Cas system-mediated induction of resistance against viral infection, example in vegetal organism. (a) A leaf is transfected with a vector through Agrobacterium tumefaciens-mediated agroinfiltration. (b) It is then infected with a virus. A GFP encoding gene can also be inserted into the viral genome. (c-f) Experimental designs and molecular functioning of the CRISPR/Cas-mediated resistance. to virus. The vector can by either empty (c) or harboring an sgRNA as well as the Cas9 (d) or the Cas13 (e, f).
Figure 4.
CRISPR/Cas system-mediated induction of resistance against viral infection, example in vegetal organism. (a) A leaf is transfected with a vector through Agrobacterium tumefaciens-mediated agroinfiltration. (b) It is then infected with a virus. A GFP encoding gene can also be inserted into the viral genome. (c-f) Experimental designs and molecular functioning of the CRISPR/Cas-mediated resistance. to virus. The vector can by either empty (c) or harboring an sgRNA as well as the Cas9 (d) or the Cas13 (e, f).
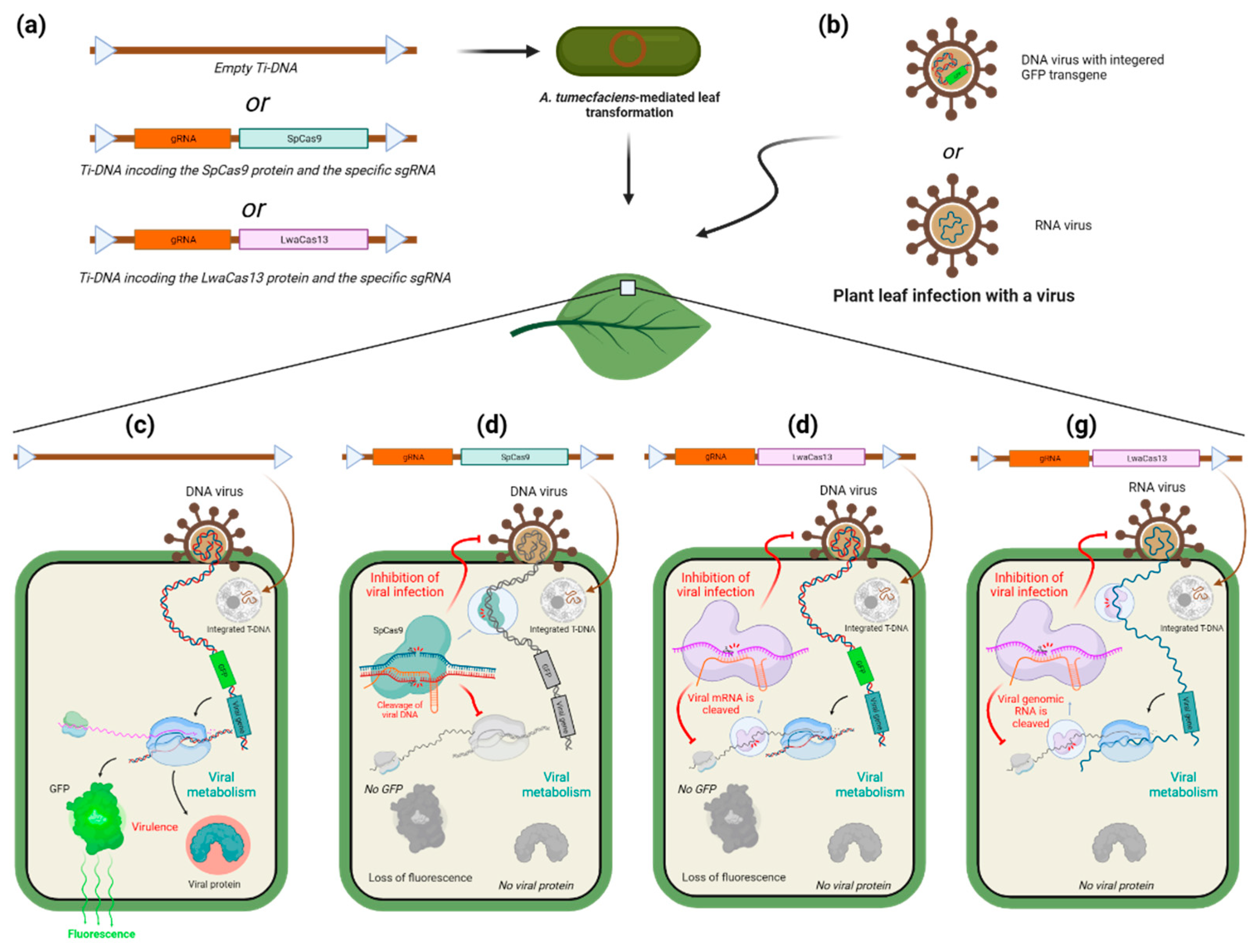
Figure 5.
Main application categories of the CRISPR/Cas system for sequencing. Methods used to deplete the samples from unwanted sequences (orange) and enrich it in desired ones by dCas9 or PFGE or bead-based isolation and nCATS (green) are shown.
Figure 5.
Main application categories of the CRISPR/Cas system for sequencing. Methods used to deplete the samples from unwanted sequences (orange) and enrich it in desired ones by dCas9 or PFGE or bead-based isolation and nCATS (green) are shown.
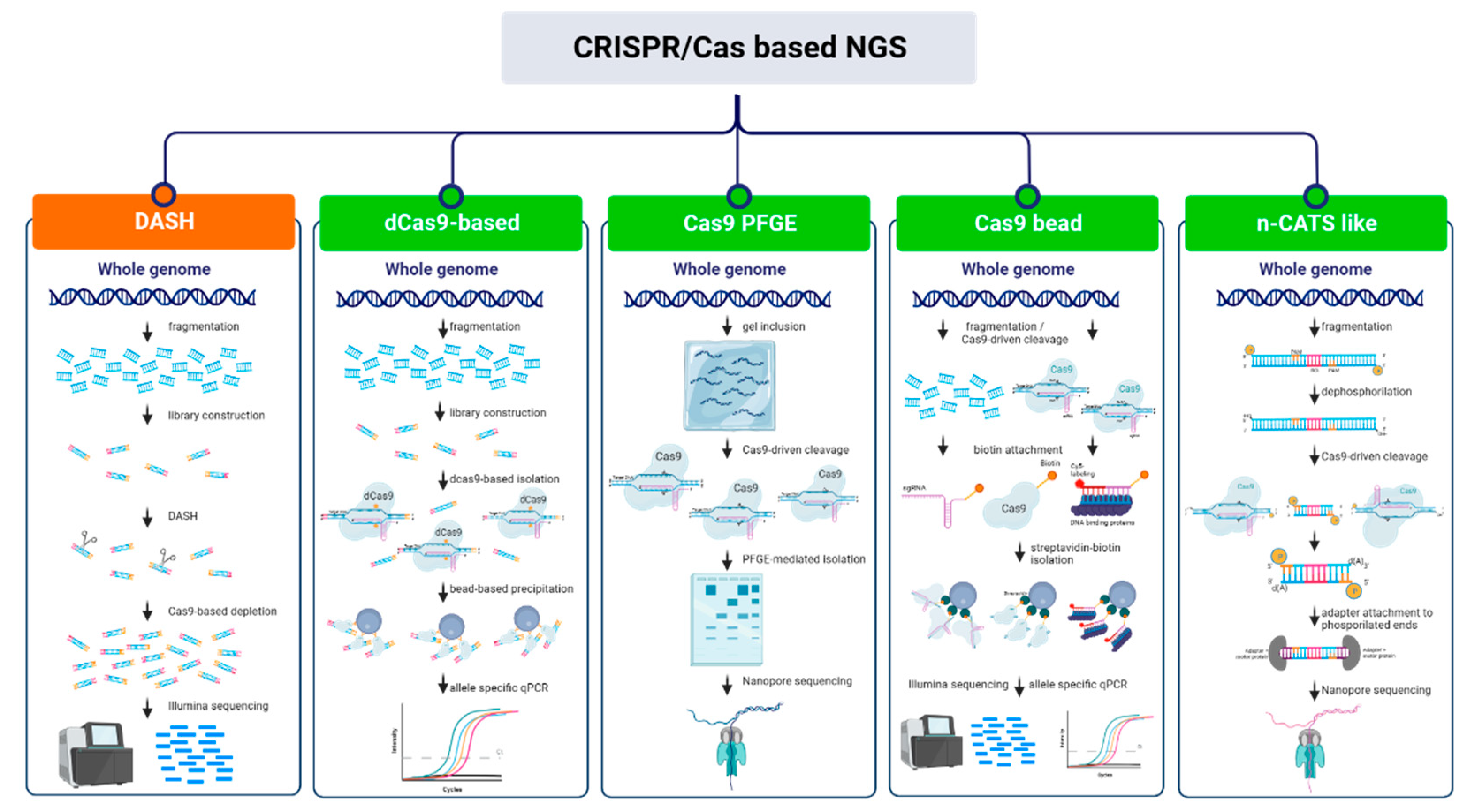
Table 1.
Cas9, Cas12a and Cas13 at a glance.
| Features | Cas9 | Cas12a | Cas13 |
|---|---|---|---|
| Other names | - | Cpf1 | C2c2 |
| Type of CRISPR/Cas system | Class 2 type II | Class 2 type V | Class 2 type VI |
| Size (aminoacids) | 1,368 (SpCas9) | 1,307 (AaCas12a) | 1389 (LshCas13a) |
| Nuclease domain | RuvC and HNH | RuvC | HEPN (×2) |
| Mutations inducing loss of function in Nuclease domain. | D10A and H840A | - | D474A and D1046A |
| sgRNA components | crRNA and tracrRNA | crRNA | crRNA |
| sgRNA crRNA processing | tracrRNA-dependent | tracrRNA-independent | - |
| sgRNA protospacer length (nucleotides) | 20 (minimum ensure DNA cleavage) | 20 | 22 - 28 |
| sgRNA total length (nucleotides) | >105 | >42 | >140 |
| Targeted nuclei acid | dsDNA (can be induced to cleave ssRNA) | dsDNA, ssDNA (not cleavable) | ssRNA |
| PAM sequence (5’-3’) | NGG (SpCas9) | T-reachTTTV (AsCas12a, LbCas12a) | None |
| Cleavage | Blunt ended double-stranded break, 3 nucleotides before PAM sequence. Each nuclease domain cleaves one strand. | PAM-distal dsDNA break with staggered 5’ and 3’ ends | Single mismatches may be tolerated. Cleavage patterns depend on features of the target sequence (like accessibility) rather than the distance from the binding site. |
| Other properties | - | Non-targeted ssDNA cleaving activity upon recognition of target sequence | Non-targeted ssRNA cleaving activity upon recognition of target sequence |
| Non-editing applications presented in this review | Modulation of gene expression and regulation Viral DNA targeting In situ DNA imaging New sequencing techniques |
Modulation of gene expression and regulation Viral DNA targeting |
In situ RNA imaging Viral RNA targeting Gene post-transcriptional regulation RNA detection techniques |
| References | [2,4,7,8] | [2,4,9] | [4,10,11,12] |
Table 2.
Types of CRISPR/Cas systems for gene expression regulation: sum up of the main features.
| Name | Description | Organism | CRISPR/Cas system | Type of regulation | Performance | References |
|---|---|---|---|---|---|---|
| VP64 | Single activator (VP16 or p65) | Mammalian cells and budding yeast | dCas9 | CRISPRa | Between 2- and 5-fold | [15,25] |
| SunTag | Tandem array of peptides which recruits several copies of VP64 | HEK293 and U2OS cells. Arabidopsis thaliana | dCas9 | CRISPRa | Up to 50-fold | [27,29] |
| VPR | Tripartite peptide composed by the VP64, p65 and Rta activators placed in a specific order to maximize gene activation | HEK293T and Neuro-2A cells. Nicotiana benthamiana | dCas9 | CRISPRa | Up to 300-fold | [31,32] |
| SAM | VP64 and sgRNA with two MS2 on turn fused to p65 and HSF1 | HEK293FT and Neuro-2a cells | dCas9 | CRISPRa | Variable | [33] |
| TV | Six copies of TAD motif and two copies of the VP64 activator | HEK293T cells. Arabidopsis thaliana and Oryza sativa | dCas9 | CRISPRa | Variable | [34] |
| Road blocker | Steric hamper due to simple bound of dCas9 | E. coli and mammalian | dCas9 | CRISPRi | Depends on organism | [16] |
| Transcriptional repressors | KRAB, CS, WPRW, SID4X, 3xSRDX and SRDX domains | Mammalian cells. Arabidopsis thaliana and Nicotiana benthamiana | dCas9 | CRISPRi | Between 40 and 99 % | [35,36,37,38,39] |
| scRNA | Differential regulation (both activation or repression) of a set of gene targets simultaneously | Human cells | dCas9 | Both | NA | [28] |
| Dimerization systems | Spatial and temporal control of gene function through sense input signals and generate functional outputs | HEK293T cells, mice and Avena sativa | dCas9 | Both | NA | [2,41,43] |
| Split dCas9 | Fusing ligand-binding domains of nuclear receptors to split Cas9 protein fragments can provide chemical control over split Cas9 activity | HEK293T cells. | dCas10 | Both | NA | [47] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



