
Submitted:
19 January 2024
Posted:
25 January 2024
You are already at the latest version
Abstract
The receptor Tyrosine Kinase (RTK) pathway and the downstream related Mitogen Activated Protein Kinase (MAPK) cascades are crucial signal transduction pathways that preside over various cellular processes, including proliferation, differentiation, apoptosis, and responses to stressors. They are well described in solid malignancies, where many of the involved factors are used as prognostic molecular markers or targets for precision therapy. Germ cell tumors (GCTs) are relatively rare. However, they are the most diagnosed malignancies occurring in the testis (TGCT) among men aged between 15 and 40 years. Despite high aneuploidy and a paucity of somatic mutations, several genomic and transcriptomic assays have identified few significantly mutated somatic genes, primarily KIT and K-RAS, so affecting, at various levels, the KIT–RAS–RAF–MEK–ERK cascade. This review focuses on the most recent investigations about this signaling pathway in TGCTs concerning their development, natural history, response to standard chemotherapy protocols (i.e., cisplatin-based), and on the efforts being made to identify targetable markers for possible precision medicine approaches.
Keywords:
Testicular Germ Cell Tumors (TGCT)
; KIT
; MAPK
; cancer
1. INTRODUCTION
1.1. Germ cell origin and prenatal development
Germ cells are the unique cells that transmit their genome to the new organism through the process of fertilization. Their genetic material is kept in a genetically stable and epigenetically reprogrammable state to allow the specification and the development of new gametes that will be specified when the embryo will form. The specification of the germline in mammalian embryos is inductive and is governed by a sophisticated core regulatory circuitry of transcription factors that are locally activated by a particular combination of growth factors. The genetically determined transcriptional program controlling the specification and determination of the germline in mammals has been the subject of extensive research during the past fifteen years. The work done on mouse models has yielded much of the current knowledge about mammalian germline development. However, significant species-specific variations in the transcription factor network between mouse and human primordial germ cell (mPGC and hPGC) development have been identified [1,2].
Recent advances in pluripotent stem cell technology have made it possible to overcome the ethical and technical obstacles in studying germline specification in humans by creating surrogate cell culture models. In fact, human primordial germ cell-like cells (hPGCLCs) derived from pluripotent stem cells have allowed to investigate this process in vitro [3]
By exploiting in vitro derivation of hPGCLCs it has been demonstrated that a lineage-primed progenitor expressing the transcription factor TFAP2A is most likely the source of hPGCs [3]. Around day post conception (dpc) 11, in the implanted blastocyst, epiblast cells with a transitional pluripotent state, that shares traits with pre- (ground state naive pluripotency) and post-implantation (primed state) epiblasts, are selected for TFAP2A expression under the influence of the growth factor BMP4 [4]. TFAP2A+ progenitors that express the mesodermal/endodermal transcription factors GATA3, EOMES, and BRACHYURY (also defined as T) transiently possess the ability to differentiate toward both the germ cell and the somatic cell fates. Extended exposure to BMP4 in the progenitors activates the expression of TFAP2C, that is located upstream of BLIMP1 (PRDM1) to control SOX17 expression and germ cell fate determination [3]. Fully specified SOX17/NANOS3 expressing PGCs require then the action of additional factors, such as PRDM14, that cooperates with TFAP2C and PRDM1, to upregulate and maintain germ pluripotency [2].
Following specification, PGCs reach the gonadal anlage by migrating through the hindgut and the dorsal mesentery [5]. While PGCs translocation into the gut is an active mechanism in the mouse, it may be passive in the human embryo [5]. It has recently been demonstrated that several PGCs are present in the human embryo at approximately 35 dpc. They have been identified along Schwann cells and autonomic nerve fibers from the dorsal mesentery to the gonad, where they appear to be transported by nerve fibers [6]. A surge of Stem Cell Factor/Kit Ligand (SCF/KL) expression by the surrounding cells is essential to guide germ cells during their migration [6]. Late PGCs that remain in the midline structures (dorsal mesentery) or mistake the correct migratory route should undergo cell death by apoptotic pathway [7].The growth factors that regulate the survival and/or proliferation of mPGCs in vitro have been shown to be active also on hPGCs [8].
In particular the Kit/KL system, largely conserved in mammals, regulates PGCs growth both in mouse and human PGCs and loss of function mutations at the White Spotting (W) and Steel loci (Sl), which encode the Kit receptor and its ligand (KL), respectively, lead to germ cell loss [9]. Interestingly, both mouse and human PGCs are prone to be reprogrammed in vitro giving rise to pluripotent embryonic germ cells (mEGCs and hEGCs, respectively), when cultured in the presence of a variety of compounds and growth factors, including LIF, bFGF and KL [8,10,11]. The ability of PGCs to be reprogrammed is shared by both sexes during the migratory period, but only male germ cells retain such ability for few time after gonadal colonization [12]. mEGCs, similarly to embryonic stem cells (ESCs) undergo malignant transformation, when transplanted in mice, giving rise to teratomas (TEs) with embryonal carcinoma components (ECs) [13], indicating that PGC latent pluripotency can lead to tumorigenesis in vivo.
1.2. Testicular Germ Cell Tumors
TGCTs are the most common malignancy in European young adult males [14]. Human TGCTs have recently been classified into two groups: tumors originating from a pre-existing in situ lesion, defined as germ cell neoplasia in situ (GCNIS, type I), and tumors that develop independently from GCNIS (type II) [15,16]. The GCT class unrelated to GCNIS is rare, self-limiting and typical of elderly men [16]. It includes spermatocytic tumors (STs), which are thought to originate from spermatogonia or spermatocytes, pre-pubertal yolk sac tumors (YSTs,) pre-pubertal teratomas (TEs), and mixed pre-pubertal TEs and YSTs [17]. TGCTs originating from GCNIS occur after puberty in young males [18,19] and can be classified into two groups on the basis of their morphological, immunohistochemical and molecular evidences: pure classic seminomas (SEs) and non-seminomatous germ cell tumors (NSEs) [16,20]. SEs are less aggressive compared to non-SEs, that include tumors with different malignancy grades and histological components such as embryonal carcinoma (EC) (Figure 1), trophoblastic tumors (TTs), post-pubertal YSTs, post-pubertal TEs, and teratomas with somatic type malignancy.
The biological causes of the initial malignant transformation from a precursor to GCNIS cells are still unclear. The initial transformation most likely occurs in utero during the early development of the germline in embryonic germ cells: PGCs or gonocytes [21]. Faulty epigenetic remodeling, unrestricted proliferation, and defects in apoptosis or in meiotic commitment can be all potential mechanisms that drive germ cell neoplastic transformation during gametogenesis [22]. Studies have demonstrated the important role of the MAPK pathway in controlling spermatogonia proliferation [23,24] and in driving male and female mouse teratocarcinomas from late PGCs [21]. However, multiple factors likely contribute to the development of TGCTs and that both genetic and environmental factors play a pivotal role in tumorigenesis [25].
1.3. Genetics of testicular germ cell tumors
The heritability of TGCTs is among the highest of any malignancies, suggesting that the contribution of genetic factors to TGCT may be relatively more important than for other cancers. Aneuploidy as whole-chromosome or whole-arm DNA imbalance is a hallmark of human cancers, and aneuploidy is nearly universal in GCTs [26,27]. Somatic genomic profiling studies have identified gain of the short arm of chromosome 12, typically as an isochromosome (i12p), as pathognomonic for TGCTs being present in more than 80% of cases. However, the gene(s) on 12p critical to GCT have not yet been defined [26].
The small number of large families has limited the search by genetic linkage analysis for familial TGCT susceptibility genes. Up to date, only one moderate-penetrance predisposition gene, checkpoint kinase 2 (CHEK2), a tumor suppressor gene involved in DNA double-strand break repair, cell cycle regulation, and cellular apoptosis has been identified [28] [29], and 78 low-to-moderate-risk single nucleotide polymorphisms, identified in genome-wide-associated studies, which account for 44% of familial risk [30]. Many of these genes encode proteins with a function in biological pathways relevant to TGCT susceptibility, including those involved in male germ cell specification and migration, sex determination and maturation, and regulation of mitosis [30]. For several target genes, findings from murine models support their direct role in the development of TGCT or TGCT-related phenotypes. For example, deletion variants at the Steel locus on the murine 129/Sv background are associated with increased incidence of TGCT; loss of Dmrt1, which plays a critical role in sex determination and maintenance of the male somatic niche, on the murine 129/Sv background leads to an over 90% incidence of testicular TEs, due to a lack of ability to silence regulators of pluripotency [31,32].
1.4. Epigenetics of Testicular germ cell tumors
During germline development, male cells undergo chromatin remodeling processes that lead to the formation of unique epigenome different from their somatic counterparts. Epigenetic mechanisms that include DNA methylation, posttranslational modifications of histones, and noncoding RNA (miRNA), occur more frequently than genetic mutations and are important for regulating gene expression during male germ cell development. The first epigenetic reprogramming in male germ cells takes place in utero in developing PGCs. They undergo rapid genome-wide erasure of original DNA methylation, including the genomic imprints that are necessary for the development of two types of germ cells accordingly to the sex of the child. In the mouse embryo DNA demethylation starts early in PGCs at embryonic day (E)6.5, at the time of their specification and is almost completed after gonadal colonization at about E13.5 [33]. At this embryonal age the overall DNA methylation status is less than 10% [34]. DNA methylation state in hPGCs is generally similar to that in mice at comparable developmental stages, suggesting the evolutionary conservation of these reprogramming dynamics in these two species. Similarly to mPGCs, global erasure of DNA methylation in hPGCs is apparently completed by Wk10-11, around the time of sex determination [35]. Most of DNA re-methylation is initiated before birth, in mitotically arrested prospermatogonia [36]. DNMT3 family members play an essential role in germ cell de novo DNA methylation [37]. Histone modifications occur in mPGCs after the first week of development (for a review see [38]) and after birth during the spermatogenetic process in spermatocytes with a global enrichment in H3K4me3 at the leptotene stage of prophase [39].
Altered epigenetic modifications (e.g., DNA methylation/demethylation, histone modifications, and non-coding RNAs) have been identified in TGCTs [40,41] with distinct genome-wide DNA methylation patterns in different types of TGCTs. For instance, the genome of GCNIS, gonadoblastoma and seminomas have been shown to be highly unmethylated like that of fetal gonocytes, despite of the expression of DNMT1 and, to a lower extent, of DNMT3B and 3L [42,43].
Instead, in non-seminomatous germ cell tumors (NSGCTs) hypermethylation pattern has been identified [44]. ECs show a DNA methylome like that of ESCs, while non-embryonal carcinomas tumor methylome resembles that of extraembryonic lineages[45,46]. Smiraglia et al. have proposed a model where SEs arise from GCNIS cells derived from PGCs which have undergone global demethylation, while non-seminomas resulted from GCNIS cells that have passed through the de novo methylation [47].
The methylation status of specific genes could be considered as potential biomarker for TGCTs. For example, DNA repair genes MGMT, RASSF1A, and BRCA1, and the transcriptional repressor gene HIC1, were frequently methylated in the NSGCTs.
Notably, RASSF1A encodes a splice variant of human RAS effector homologue, which interacts with the XPA protein and functions as a negative regulator of cell growth [48]. RASSF1A has been shown to be inactivated by promoter methylation in a variety of tumor types including TGCTs [49,50].
Histone modifications associated with chromatin structure repression like H3K9me2 and H3K27me3 have been found to be expressed at low levels in GCNIS cells in contrast to H3K4me1, H3K4me2/3, H3K9ac and a histone variant H2A.Z that are associated with permissive chromatin structure.
Lastly, it has been discovered that miRNAs are important in TGCTs and offer new potential targets for treatment. Among them, the miR-371–373 cluster and the miR-302 cluster are found overexpressed in TGCTs. Members of the miR-371–373 cluster are the prevalent miRNAs in human embryonic stem cells and are involved in self-renewal processes and in maintaining the pluripotency status of ESCs [51].
The miR-302 cluster plays a critical role in the regulation of the cell cycle in embryonic and pluripotent stem cells. High expression of miR-302s in TGCTs suggest that they act as oncogenes in TGCTs. Interestingly, miR-302s has been shown to induce the expression of SPRY4 and activation of MAPK/ERK pathway while it inhibited apoptosis via increasing the expression of survivin [52].
1.5. MAPK signaling pathway in cancer.
MAPK cascade is a key signaling pathway involved in the transduction of extracellular signals to cellular responses like proliferation, differentiation, apoptosis, and stress responses. In mammalian cells, four leading MAPK families have been characterized: extracellular signal-regulated kinases (ERK1 and ERK2), Jun N-terminal kinases (JNK1, JNK2, and JNK3), p38 kinase, and ERK5 [53,54]. JNK and p38 MAPK pathways are mainly related to cell stress and apoptosis, while the ERK1/2 and ERK5 MAPK signaling pathway is preferably linked to cell proliferation and differentiation [53,55,56,57,58] . A dual phosphorylation event on MAPK threonine and tyrosine residues activates the phosphorylation of targets on serine and threonine residues within a consensus PXT/SP motif (where the X residue depends on the different MAPK) [59,60] .
Activation of the MAPK pathway is based upon sequential activation of several layers of protein kinases (three to five) known as MAPK kinase kinase kinase (MAP4K), MAPK kinase kinase (MAP3K), MAPK kinase (MAPKK), MAPK and MAPK activated protein kinases (MAPKAPK). The first three central layers are considered an essential core unit, while the last two layers have been identified in some cascades and can vary among cells and stimuli [53].
Dysregulated MAPK signaling is involved in diverse human diseases, primarily cancer [61,62,63,64,65,66,67] and it is generally caused by activating mutations in the receptors or in the downstream signaling effectors that activate the pathway in the absence of appropriate stimuli. Correspondingly, constitutive receptor tyrosine kinase (RTK) pathway activity is prevalent in many cancer types, mainly due to activating mutations in the constituents of the RAS-RAF-MEK axis [68,69].
Among all MAPK signal transduction pathways, the RAS/RAF/MAPKK (MEK)/ERK pathway is the most important signaling cascade and plays a crucial role in the survival and development of tumor cells [53]. Perturbation in the RAS/ERK pathway represents a primer for the development of several cancer types [70,71]. As reported by several authors, the frequency of mutations in the RAS gene (mainly K-RAS) is about 30% of all cancer types [72] and 10% of all patients with cancer [73]; RAF mutations (particularly B-RAF) have been identified in about 8% of all cancer types [74]. The rate of extracellular signal-regulated kinase kinase (MEK) mutations is low (~1%), and only a few major pathogenic mutations in ERKs have been stated [53,71,72,73,74].
The RAF family (which includes A-RAF, B-RAF, and C-RAF or RAF1 gene isoforms) displays serine/threonine protein kinase activity after binding to RAS-guanosine triphosphate (GTP). Upon activation, C-RAF interacts with MEK and phosphorylates S218 and S222 residues of the MEK catalytic domain. Thus, when multiple kinases act on MEK, activated MEK directly catalyzes the phosphorylation of Tyr and Thr residues of ERKs, the last Ser/Thr protein kinase of the cascade. Once activated, ERK1/2 moves from the cytoplasm to the nucleus influencing the activity of several transcription factors (such as proto-oncogenes c-FOS, c-JUN, ETS domain-containing protein ELK-1, c-MYC, cyclic AMP-dependent transcription factor ATF2, etc.), modulating cell cytoskeletal components such as microtubule-associated protein (MAP), cell metabolism and cell fate. The downstream effector ERK1/2 becomes abnormally phosphorylated and activated in many human cancers, implying that their activity is critical for tumorigenesis [62,75]. B-RAF mutation occurs in 27-70% of malignant melanomas, 40-70% of papillary or anaplastic thyroid tumors, 30% of ovarian carcinomas, 5-22% of colorectal cancer, and small percentages of carcinomas from other sites [76]. The most frequent B-RAF mutation entails thymine:adenine transversion at codon 600 (B-RAFV600E), with the replacement of a valine by glutamic acid into the polypeptide chain, causing an increase of its kinase activity. Intriguingly, however, oncogenic mutations in ERK1/2 have not yet been identified, and activating ERK mutations have not been shown to play a causative role in cancer [77]. With a negative feedback regulatory loop, cytoplasmic ERK1/2 can phosphorylate other protein kinases upstream of the ERK pathway, such as SOS, C-RAF, and MEK [53]. Upstream to the RAF-MEK-ERK pathway is RAS. It was first unearthed as a family of viral oncogenes, H-RAS (Harvey’s Sarcoma) and K-RAS (Kirtsten’s Sarcoma) (another member, N-RAS, was discovered in neuroblastoma). Their products are small G proteins that after the stimulation of growth factors, cytokines or other activators (PKC, SRC, etc.) can switch from an active to an inactive conformation by swapping GTP and GDP, respectively, thus controlling signal transduction [77].
1.6. ERK/MAPK signaling in tumor invasion and metastasis.
The ERK/MAPK signaling pathway also plays an essential role in tumor invasion and metastasis by phosphorylating nuclear and cytoplasmic targets. Accordingly, blocking the ERK/MAPK signaling pathway may inhibit the role of extracellular signals that promote cell movement, inhibiting tumor invasion and metastasis [78]. Experimental models using human ovarian cancer cells demonstrated that the activated ERK signaling pathway promoted the proliferation and migration of ovarian cancer ascites cells [28,79]. ERK activation can mediate tumor cell migratory activity by the up-regulation of genes involved in invasion, cell adhesion, extracellular matrix production and degradation, and scaffold proteins that play essential roles in cell adhesion, transcription, and cytoskeletal organization [80,81,82] or matrix degradation [83,84] . Indeed, MEK inhibitors have been shown to block migration and invasion of many cancer cell types [85], including SK-OV-3 human ovarian carcinoma cells [86].
Downstream to activated ERKs is the ribosomal S6 kinases (p90-RSKs) family of kinases that have been involved in the regulation of tumor metastasis [80]. RSK mechanism of action depends both on the isoform and the cancer type. While some isoforms promote cell motility and invasion by altering transcription and integrin activity, others impair cell motility and invasion by affecting the actin cytoskeleton [80]. However, despite the variance in RSK-mediated outcomes, chemical inhibition of this group of kinases has proven to be effective in blocking invasion and metastasis of several solid tumors in pre-clinical models [80].
1.7. Alterations of MAPK pathway in TGCTs
Although TGCTs have a paucity of somatic mutations, three genes of significance have been identified by DNA exome sequencing: KIT (18–25%), K-RAS (14%), and N-RAS (4%), all of which are observed exclusively in tumors with seminomatous components (either pure SE or as a component of mixed NSGCT) [87,88].
KIT locus (chromosome 5) encodes for a type III receptor tyrosine kinase that dimerizes upon binding to its cognate ligand KL. Such event leads to the activation of KIT intrinsic tyrosine kinase activity and the phosphorylation of key tyrosine residues within the receptor. The resulting phosphor-tyrosine residues serve as docking sites for molecules containing SH2 and other phosphor-tyrosine-binding domains. Among the major signaling pathways activated by the KL/KIT, the RAS-RAF-MEK-ERK, and PI3K/AKT pathways are involved in TGCT (Figure 2).
Stimulation of KIT signaling through activating mutations and genomic amplification has been extensively demonstrated in TGCTs [88,89].
Activatory mutations reside within the activation loop of the kinase 2 domain, in the juxtamembrane domain, and in the protein tyrosine kinase 1 domain of the KIT gene. In an extended genomic analysis of a large cohort of patients affected by TGCTs, 8.1% of cases (28 SEs and 18 mixed/NSEs) resulted in somatic KIT mutations. Exons 17 (67.3%), 11 (22.4%), and 13 (6.1%) were the most affected exons. KIT-mutant cases were enriched for oncogenic RAS/MAPK pathway alterations compared to KIT-wildtype cases (34.8% vs 19.2%, P = 0.02). Among KIT-mutant cases, concurrent mutations were noted in K-RAS (21.7%), RRAS2 (11.8%), CBL (6.5%), N-RAS (4.3%), MAP2K1 (2.2%), and RAC1 (2.2%), while mutations in K-RAS, RRAS2, and N-RAS were mutually exclusive [90].
The KL locus maps to chromosome 12 in humans, and its gene product controls the survival and proliferation of both PGCs and postnatal male germ cells [9,24]. As previously anticipated, the KL locus acts as a modifier of TGCT susceptibility in the 129/Sv strain of mice mimicking pediatric TGCTs, human teratomas that develop from PGCs [21,91]. Indeed, among the most statistically significant mutated loci in TGCT patients is the KL rs4474514 variant, which is associated with TGCT of the adolescent age [89,92].
Mutations of K-RAS and N-RAS account for about 26% and 4% of TGCTs, respectively, as reported from four studies in the cBioportal database [93]. The most recurrent K-RAS defects were amplifications and mutations at codon 12 [93,94]. K-RAS and N-RAS mutations were independently identified both in SEs and in NSEs. However, both mutations were also found to concomitantly occur in GCT patients carrying both SE and NSE components [95,96]. Accordingly, another study reported RAS mutations in 16% of SEs and in 15% of NSEs [97]. In an Indian TGCT patient set, K-RAS mutation was identified in 17% of patients, the majority of whom were affected by mixed GCT (13%) while the remaining were represented by pure seminomas (4%). Metastatic tumors were shown to carry more K-RAS mutations than primary tumors (100% vs. 13%) [50]. B-RAF mutations are rare in TGCTs [93]. B-RAF missense mutations (1796T>A mutations) were found in 9% of NSEs within the EC component but not in SEs [96] while other studies did not detect any B-RAF defect in the TGCT cohorts studied [98,99]. B-RAF mutated GCTs were reported to occur at a high percentage (24%) within a chemotherapy-refractoriness cohort showing platinum-resistance and to correlate to low or absent mismatch repair protein (MMR) expression and high microsatellite instability (MSI) [100]. These tumors are frequently associated with a mediastinal primary onset and with a trend to more prolonged progression-free survival [100]. In agreement, another study reported a positive correlation between MSI and the presence of a mutated B-RAF allele in ECs [96]. While B-RAF mutations were mutually exclusive with any RAS mutations [50], constitutively activated ERKs were detected in almost all tumors tested [96]. ]. A summary of the mutated somatic genes in the cKIT–RAS–RAF–MEK–ERK cascade in TGCTs is reported in Table 1.
1.8. Prognosis and response to treatment
Cisplatin-based chemotherapy is the elective treatment for TGCT [102]. Previous reports have shown that cisplatin activates the MAPK pathway in several cancer types. However, it has not been established yet whether this activation is linked to apoptosis or to its prevention [[103] and references therein]. Functional assays using MEK inhibitors indicated that MEK and ERK phosphorylation is necessary for caspase-3 activation by cisplatin either in p53wt or in p53null human teratocarcinoma cell lines [104]. Cisplatin resistance is determined by several biological mechanisms [27,105], among which the acquisition of DNA repair proficiency of cancer cells plays an important role in cisplatin-resistant TCGT cell lines [106]. Platinum refractoriness has been associated with the NSE subtypes and those that originate in extragonadal sites [107]. A recent study on the genomic landscape of platinum resistant and sensitive testicular cancers has shown that resistant tumors are enriched for somatic WNT/CTNNB1 pathway mutations that were detected in both platinum resistant and metastatic TGCTs, while platinum sensitive were associated to mutated KIT alleles [88]. The RAS/RAF/MAPK-activated pathway plays an important role in this context, and kinase inhibitors that insist on this pathway can reverse platinum resistance in several cancer cell lines [108,109]. Interestingly, two independent studies reported the occurrence of B-RAF V600E mutations and MSI in unselected GCTs from cisplatin-resistant or relapsed NSE patient cohorts with an increased frequency [100,110] .
2. CONCLUSIONS
Improved understanding of the genomic drivers of GCT development and heritability has the potential to yield valuable insight into GCT pathogenesis, helps patients at higher risk for GCT identification, and aids in the development of novel therapeutic approaches to benefit the cohort of patients with chemo-resistant disease. The RAS/RAF/MEK/ERK, as well as the KIT-activated pathways, have been demonstrated to be efficiently druggable, providing new therapy tools to limit tumor progression, particularly in patients that develop resistance to platinum-based treatments. However, despite the knowledge of this molecular pathway, no targeted therapies aimed at inhibiting MAPKs or KIT are approved for TGCT treatment.
Understanding the molecular mechanisms underlying GCT resistance to cisplatin is, therefore, essential to identify new targeted therapies.
Author Contributions
Conceptualization: S.D, A.O. and P.G.; writing: S.D, A.O. and P.G.; revision and editing: S.D., E.G, A.C., A.O. and P.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by PRIN2022 to S.D.: Understanding the Origin and Behavior of Ectopic LipIds eXcess depots: the OBELIX study and by RSA 2021 to P.G.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Irie, N.; Weinberger, L.; Tang, W.W.; Kobayashi, T.; Viukov, S.; Manor, Y.S.; Dietmann, S.; Hanna, J.H.; Surani, M.A. SOX17 is a critical specifier of human primordial germ cell fate. Cell 2015, 160, 253–268. [Google Scholar] [CrossRef]
- Tang, W.W.; Kobayashi, T.; Irie, N.; Dietmann, S.; Surani, M.A. Specification and epigenetic programming of the human germ line. Nat Rev Genet 2016, 17, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Liu, W.; Zimmerman, J.; Pastor, W.A.; Kim, R.; Hosohama, L.; Ho, J.; Aslanyan, M.; Gell, J.J.; Jacobsen, S.E.; et al. The TFAP2C-Regulated OCT4 Naive Enhancer Is Involved in Human Germline Formation. Cell Rep 2018, 25, 3591–3602. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Sun, N.; Hou, L.; Kim, R.; Faith, J.; Aslanyan, M.; Tao, Y.; Zheng, Y.; Fu, J.; Liu, W.; et al. Human Primordial Germ Cells Are Specified from Lineage-Primed Progenitors. Cell Rep 2019, 29, 4568–4582 e4565. [Google Scholar] [CrossRef] [PubMed]
- Freeman, B. The active migration of germ cells in the embryos of mice and men is a myth. Reproduction 2003, 125, 635–643. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Runyan, C.; Shoemaker, A.; Surani, A.; Wylie, C. Steel factor controls primordial germ cell survival and motility from the time of their specification in the allantois, and provides a continuous niche throughout their migration. Development 2009, 136, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Oosterhuis, J.W.; Stoop, H.; Honecker, F.; Looijenga, L.H. Why human extragonadal germ cell tumours occur in the midline of the body: old concepts, new perspectives. Int J Androl 2007, 30, 256–263. [Google Scholar] [CrossRef]
- Shamblott, M.J.; Axelman, J.; Wang, S.; Bugg, E.M.; Littlefield, J.W.; Donovan, P.J.; Blumenthal, P.D.; Huggins, G.R.; Gearhart, J.D. Derivation of pluripotent stem cells from cultured human primordial germ cells. Proc Natl Acad Sci U S A 1998, 95, 13726–13731. [Google Scholar] [CrossRef] [PubMed]
- Dolci, S.; Williams, D.E.; Ernst, M.K.; Resnick, J.L.; Brannan, C.I.; Lock, L.F.; Lyman, S.D.; Boswell, H.S.; Donovan, P.J. Requirement for mast cell growth factor for primordial germ cell survival in culture. Nature 1991, 352, 809–811. [Google Scholar] [CrossRef]
- Matsui, Y.; Zsebo, K.; Hogan, B.L. Derivation of pluripotential embryonic stem cells from murine primordial germ cells in culture. Cell 1992, 70, 841–847. [Google Scholar] [CrossRef]
- Resnick, J.L.; Bixler, L.S.; Cheng, L.; Donovan, P.J. Long-term proliferation of mouse primordial germ cells in culture. Nature 1992, 359, 550–551. [Google Scholar] [CrossRef] [PubMed]
- Donovan, P.J.; de Miguel, M.P. Turning germ cells into stem cells. Curr Opin Genet Dev 2003, 13, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Conway, A.E.; Lindgren, A.; Galic, Z.; Pyle, A.D.; Wu, H.; Zack, J.A.; Pelligrini, M.; Teitell, M.A.; Clark, A.T. A self-renewal program controls the expansion of genetically unstable cancer stem cells in pluripotent stem cell-derived tumors. Stem Cells 2009, 27, 18–28. [Google Scholar] [CrossRef] [PubMed]
- De Felici, M.; Dolci, S. From testis to teratomas: a brief history of male germ cells in mammals. Int J Dev Biol 2013, 57, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Dolci, S.; Campolo, F.; De Felici, M. Gonadal development and germ cell tumors in mouse and humans. Semin Cell Dev Biol 2015, 45, 114–123. [Google Scholar] [CrossRef]
- Oosterhuis, J.W.; Looijenga, L.H. Testicular germ-cell tumours in a broader perspective. Nat Rev Cancer 2005, 5, 210–222. [Google Scholar] [CrossRef] [PubMed]
- Moch, H.; Cubilla, A.L.; Humphrey, P.A.; Reuter, V.E.; Ulbright, T.M. The 2016 WHO Classification of Tumours of the Urinary System and Male Genital Organs-Part A: Renal, Penile, and Testicular Tumours. Eur Urol 2016, 70, 93–105. [Google Scholar] [CrossRef]
- Reuter, V.E. Origins and molecular biology of testicular germ cell tumors. Mod Pathol 2005, 18 Suppl 2, S51–60. [Google Scholar] [CrossRef]
- Skakkebaek, N.E.; Berthelsen, J.G.; Giwercman, A.; Muller, J. Carcinoma-in-situ of the testis: possible origin from gonocytes and precursor of all types of germ cell tumours except spermatocytoma. Int J Androl 1987, 10, 19–28. [Google Scholar] [CrossRef]
- Almstrup, K.; Sonne, S.B.; Hoei-Hansen, C.E.; Ottesen, A.M.; Nielsen, J.E.; Skakkebaek, N.E.; Leffers, H.; Rajpert-De Meyts, E. From embryonic stem cells to testicular germ cell cancer-- should we be concerned? Int J Androl 2006, 29, 211–218. [Google Scholar] [CrossRef]
- Guida, E.; Tassinari, V.; Colopi, A.; Todaro, F.; Cesarini, V.; Jannini, B.; Pellegrini, M.; Botti, F.; Rossi, G.; Rossi, P.; et al. MAPK activation drives male and female mouse teratocarcinomas from late primordial germ cells. J Cell Sci 2022, 135. [Google Scholar] [CrossRef] [PubMed]
- Barchi, M.; Guida, E.; Dolci, S.; Rossi, P.; Grimaldi, P. Endocannabinoid system and epigenetics in spermatogenesis and testicular cancer. Vitam Horm 2023, 122, 75–106. [Google Scholar] [CrossRef] [PubMed]
- Tassinari, V.; Campolo, F.; Cesarini, V.; Todaro, F.; Dolci, S.; Rossi, P. Fgf9 inhibition of meiotic differentiation in spermatogonia is mediated by Erk-dependent activation of Nodal-Smad2/3 signaling and is antagonized by Kit Ligand. Cell Death Dis 2015, 6, e1688. [Google Scholar] [CrossRef] [PubMed]
- Dolci, S.; Pellegrini, M.; Di Agostino, S.; Geremia, R.; Rossi, P. Signaling through extracellular signal-regulated kinase is required for spermatogonial proliferative response to stem cell factor. J Biol Chem 2001, 276, 40225–40233. [Google Scholar] [CrossRef] [PubMed]
- Di Siena, S.; Campolo, F.; Rossi, P.; Jannini, E.A.; Dolci, S.; Pellegrini, M. UV and genotoxic stress induce ATR relocalization in mouse spermatocytes. Int J Dev Biol 2013, 57, 281–287. [Google Scholar] [CrossRef] [PubMed]
- Litchfield, K.; Summersgill, B.; Yost, S.; Sultana, R.; Labreche, K.; Dudakia, D.; Renwick, A.; Seal, S.; Al-Saadi, R.; Broderick, P.; et al. Whole-exome sequencing reveals the mutational spectrum of testicular germ cell tumours. Nat Commun 2015, 6, 5973. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Weiner, A.; Zack, T.; O'Donnell, E.; Guerriero, J.L.; Bernard, B.; Reddy, A.; Han, G.C.; AlDubayan, S.; Amin-Mansour, A.; Schumacher, S.E.; et al. Genomic evolution and chemoresistance in germ-cell tumours. Nature 2016, 540, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.B.; Lin, X.P.; Xu, Y.; Shen, Z.F.; Pan, W.W. DAXX promotes ovarian cancer ascites cell proliferation and migration by activating the ERK signaling pathway. J Ovarian Res 2018, 11, 90. [Google Scholar] [CrossRef] [PubMed]
- Martin, F.C.; Conduit, C.; Loveland, K.L.; Thomas, B.; Lewin, J.; Tran, B. Genetics of testicular cancer: a review. Curr Opin Urol 2022, 32, 481–487. [Google Scholar] [CrossRef]
- Pluta, J.; Pyle, L.C.; Nead, K.T.; Wilf, R.; Li, M.; Mitra, N.; Weathers, B.; D'Andrea, K.; Almstrup, K.; Anson-Cartwright, L.; et al. Identification of 22 susceptibility loci associated with testicular germ cell tumors. Nat Commun 2021, 12, 4487. [Google Scholar] [CrossRef]
- Krentz, A.D.; Murphy, M.W.; Zhang, T.; Sarver, A.L.; Jain, S.; Griswold, M.D.; Bardwell, V.J.; Zarkower, D. Interaction between DMRT1 function and genetic background modulates signaling and pluripotency to control tumor susceptibility in the fetal germ line. Dev Biol 2013, 377, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Krentz, A.D.; Murphy, M.W.; Kim, S.; Cook, M.S.; Capel, B.; Zhu, R.; Matin, A.; Sarver, A.L.; Parker, K.L.; Griswold, M.D.; et al. The DM domain protein DMRT1 is a dose-sensitive regulator of fetal germ cell proliferation and pluripotency. Proc Natl Acad Sci U S A 2009, 106, 22323–22328. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Mikkelsen, T.S.; Gu, H.; Wernig, M.; Hanna, J.; Sivachenko, A.; Zhang, X.; Bernstein, B.E.; Nusbaum, C.; Jaffe, D.B.; et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008, 454, 766–770. [Google Scholar] [CrossRef] [PubMed]
- Popp, C.; Dean, W.; Feng, S.; Cokus, S.J.; Andrews, S.; Pellegrini, M.; Jacobsen, S.E.; Reik, W. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 2010, 463, 1101–1105. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Yan, L.; Guo, H.; Li, L.; Hu, B.; Zhao, Y.; Yong, J.; Hu, Y.; Wang, X.; Wei, Y.; et al. The Transcriptome and DNA Methylome Landscapes of Human Primordial Germ Cells. Cell 2015, 161, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Matsui, Y. Epigenetic events in mammalian germ-cell development: reprogramming and beyond. Nat Rev Genet 2008, 9, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Hata, K.; Okano, M.; Lei, H.; Li, E. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development 2002, 129, 1983–1993. [Google Scholar] [CrossRef]
- Kota, S.K.; Feil, R. Epigenetic transitions in germ cell development and meiosis. Dev Cell 2010, 19, 675–686. [Google Scholar] [CrossRef]
- Kimmins, S.; Sassone-Corsi, P. Chromatin remodelling and epigenetic features of germ cells. Nature 2005, 434, 583–589. [Google Scholar] [CrossRef]
- Messerschmidt, D.M.; Knowles, B.B.; Solter, D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev 2014, 28, 812–828. [Google Scholar] [CrossRef]
- Fendler, A.; Stephan, C.; Yousef, G.M.; Kristiansen, G.; Jung, K. The translational potential of microRNAs as biofluid markers of urological tumours. Nat Rev Urol 2016, 13, 734–752. [Google Scholar] [CrossRef] [PubMed]
- Netto, G.J.; Nakai, Y.; Nakayama, M.; Jadallah, S.; Toubaji, A.; Nonomura, N.; Albadine, R.; Hicks, J.L.; Epstein, J.I.; Yegnasubramanian, S.; et al. Global DNA hypomethylation in intratubular germ cell neoplasia and seminoma, but not in nonseminomatous male germ cell tumors. Mod Pathol 2008, 21, 1337–1344. [Google Scholar] [CrossRef] [PubMed]
- Kristensen, D.G.; Nielsen, J.E.; Jorgensen, A.; Skakkebaek, N.E.; Rajpert-De Meyts, E.; Almstrup, K. Evidence that active demethylation mechanisms maintain the genome of carcinoma in situ cells hypomethylated in the adult testis. Br J Cancer 2014, 110, 668–678. [Google Scholar] [CrossRef] [PubMed]
- Landero-Huerta, D.A.; Vigueras-Villasenor, R.M.; Yokoyama-Rebollar, E.; Arechaga-Ocampo, E.; Rojas-Castaneda, J.C.; Jimenez-Trejo, F.; Chavez-Saldana, M. Epigenetic and risk factors of testicular germ cell tumors: a brief review. Front Biosci (Landmark Ed) 2017, 22, 1073–1098. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Pelizzola, M.; Dowen, R.H.; Hawkins, R.D.; Hon, G.; Tonti-Filippini, J.; Nery, J.R.; Lee, L.; Ye, Z.; Ngo, Q.M.; et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 2009, 462, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zhang, J.; Liu, Y.; Liu, S.; Liu, Z.; Duan, Z.; Wang, Z.; Zhu, B.; Guo, Y.L.; Tian, Z. DNA methylation footprints during soybean domestication and improvement. Genome Biol 2018, 19, 128. [Google Scholar] [CrossRef] [PubMed]
- Smiraglia, D.J.; Szymanska, J.; Kraggerud, S.M.; Lothe, R.A.; Peltomaki, P.; Plass, C. Distinct epigenetic phenotypes in seminomatous and nonseminomatous testicular germ cell tumors. Oncogene 2002, 21, 3909–3916. [Google Scholar] [CrossRef] [PubMed]
- Burbee, D.G.; Forgacs, E.; Zochbauer-Muller, S.; Shivakumar, L.; Fong, K.; Gao, B.; Randle, D.; Kondo, M.; Virmani, A.; Bader, S.; et al. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst 2001, 93, 691–699. [Google Scholar] [CrossRef]
- Dammann, R.; Yang, G.; Pfeifer, G.P. Hypermethylation of the cpG island of Ras association domain family 1A (RASSF1A), a putative tumor suppressor gene from the 3p21.3 locus, occurs in a large percentage of human breast cancers. Cancer Res 2001, 61, 3105–3109. [Google Scholar]
- Ahmad, F.; Surve, P.; Natarajan, S.; Patil, A.; Pol, S.; Patole, K.; Das, B.R. Aberrant epigenetic inactivation of RASSF1A and MGMT gene and genetic mutations of KRAS, cKIT and BRAF in Indian testicular germ cell tumours. Cancer Genet 2020, 241, 42–50. [Google Scholar] [CrossRef]
- Ghasemi, M.; Samaei, N.M.; Mowla, S.J.; Shafiee, M.; Vasei, M.; Ghasemian, N. Upregulation of miR-371-373 cluster, a human embryonic stem cell specific microRNA cluster, in esophageal squamous cell carcinoma. J Cancer Res Ther 2018, 14, S132–S137. [Google Scholar] [CrossRef]
- Das, M.K.; Evensen, H.S.F.; Furu, K.; Haugen, T.B. miRNA-302s may act as oncogenes in human testicular germ cell tumours. Sci Rep 2019, 9, 9189. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.J.; Pan, W.W.; Liu, S.B.; Shen, Z.F.; Xu, Y.; Hu, L.L. ERK/MAPK signalling pathway and tumorigenesis. Exp Ther Med 2020, 19, 1997–2007. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tournier, C. Regulation of cellular functions by the ERK5 signalling pathway. Cell Signal 2006, 18, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Kolch, W.; Calder, M.; Gilbert, D. When kinases meet mathematics: the systems biology of MAPK signalling. FEBS Lett 2005, 579, 1891–1895. [Google Scholar] [CrossRef]
- Kolch, W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol 2005, 6, 827–837. [Google Scholar] [CrossRef] [PubMed]
- Kyriakis, J.M.; Avruch, J. Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 2001, 81, 807–869. [Google Scholar] [CrossRef]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef]
- Songyang, Z.; Lu, K.P.; Kwon, Y.T.; Tsai, L.H.; Filhol, O.; Cochet, C.; Brickey, D.A.; Soderling, T.R.; Bartleson, C.; Graves, D.J.; et al. A structural basis for substrate specificities of protein Ser/Thr kinases: primary sequence preference of casein kinases I and II, NIMA, phosphorylase kinase, calmodulin-dependent kinase II, CDK5, and Erk1. Mol Cell Biol 1996, 16, 6486–6493. [Google Scholar] [CrossRef]
- Chen, Z.; Gibson, T.B.; Robinson, F.; Silvestro, L.; Pearson, G.; Xu, B.; Wright, A.; Vanderbilt, C.; Cobb, M.H. MAP kinases. Chem Rev 2001, 101, 2449–2476. [Google Scholar] [CrossRef]
- Porter, A.C.; Vaillancourt, R.R. Tyrosine kinase receptor-activated signal transduction pathways which lead to oncogenesis. Oncogene 1998, 17, 1343–1352. [Google Scholar] [CrossRef] [PubMed]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y. The EGFR family and its ligands in human cancer. signalling mechanisms and therapeutic opportunities. Eur J Cancer 2001, 37 Suppl 4, S3–8. [Google Scholar] [CrossRef]
- Bache, K.G.; Slagsvold, T.; Stenmark, H. Defective downregulation of receptor tyrosine kinases in cancer. EMBO J 2004, 23, 2707–2712. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: the next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Zandi, R.; Larsen, A.B.; Andersen, P.; Stockhausen, M.T.; Poulsen, H.S. Mechanisms for oncogenic activation of the epidermal growth factor receptor. Cell Signal 2007, 19, 2013–2023. [Google Scholar] [CrossRef] [PubMed]
- Klein, S.; Levitzki, A. Targeting the EGFR and the PKB pathway in cancer. Curr Opin Cell Biol 2009, 21, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Weinberg, R.A. Ras oncogenes: split personalities. Nat Rev Mol Cell Biol 2008, 9, 517–531. [Google Scholar] [CrossRef] [PubMed]
- Bollag, G.; Tsai, J.; Zhang, J.; Zhang, C.; Ibrahim, P.; Nolop, K.; Hirth, P. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov 2012, 11, 873–886. [Google Scholar] [CrossRef]
- Garcia-Gomez, R.; Bustelo, X.R.; Crespo, P. Protein-Protein Interactions: Emerging Oncotargets in the RAS-ERK Pathway. Trends Cancer 2018, 4, 616–633. [Google Scholar] [CrossRef]
- Khotskaya, Y.B.; Holla, V.R.; Farago, A.F.; Mills Shaw, K.R.; Meric-Bernstam, F.; Hong, D.S. Targeting TRK family proteins in cancer. Pharmacol Ther 2017, 173, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Maik-Rachline, G.; Hacohen-Lev-Ran, A.; Seger, R. Nuclear ERK: Mechanism of Translocation, Substrates, and Role in Cancer. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337 e310. [Google Scholar] [CrossRef]
- Holderfield, M.; Deuker, M.M.; McCormick, F.; McMahon, M. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer 2014, 14, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Pouyssegur, J. Targeting the ERK signaling pathway in cancer therapy. Ann Med 2006, 38, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef] [PubMed]
- Terrell, E.M.; Morrison, D.K. Ras-Mediated Activation of the Raf Family Kinases. Cold Spring Harb Perspect Med 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Burotto, M.; Chiou, V.L.; Lee, J.M.; Kohn, E.C. The MAPK pathway across different malignancies: a new perspective. Cancer 2014, 120, 3446–3456. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Ye, W.; Wu, J.; Liu, L.; Yang, L.; Gao, L.; Chen, B.; Zhang, F.; Yang, H.; Li, Y. Sp1-CD147 positive feedback loop promotes the invasion ability of ovarian cancer. Oncol Rep 2015, 34, 67–76. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Ramos, J.W. RSK isoforms in cancer cell invasion and metastasis. Cancer Res 2013, 73, 6099–6105. [Google Scholar] [CrossRef]
- Gialeli, C.; Theocharis, A.D.; Karamanos, N.K. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J 2011, 278, 16–27. [Google Scholar] [CrossRef] [PubMed]
- Maeda-Yamamoto, M.; Suzuki, N.; Sawai, Y.; Miyase, T.; Sano, M.; Hashimoto-Ohta, A.; Isemura, M. Association of suppression of extracellular signal-regulated kinase phosphorylation by epigallocatechin gallate with the reduction of matrix metalloproteinase activities in human fibrosarcoma HT1080 cells. J Agric Food Chem 2003, 51, 1858–1863. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Gao, J.; Wang, Y.; Yang, J.; Zhang, W.; Meng, K.; Sun, Y.; Li, Y.; He, Q.Y. RNF128 Promotes Invasion and Metastasis Via the EGFR/MAPK/MMP-2 Pathway in Esophageal Squamous Cell Carcinoma. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Horiuchi, H.; Kawamata, H.; Furihata, T.; Omotehara, F.; Hori, H.; Shinagawa, Y.; Ohkura, Y.; Tachibana, M.; Yamazaki, T.; Ajiki, T.; et al. A MEK inhibitor (U0126) markedly inhibits direct liver invasion of orthotopically inoculated human gallbladder cancer cells in nude mice. J Exp Clin Cancer Res 2004, 23, 599–606. [Google Scholar] [PubMed]
- Basu, M.; Mukhopadhyay, S.; Chatterjee, U.; Roy, S.S. FGF16 promotes invasive behavior of SKOV-3 ovarian cancer cells through activation of mitogen-activated protein kinase (MAPK) signaling pathway. J Biol Chem 2014, 289, 1415–1428. [Google Scholar] [CrossRef] [PubMed]
- Todaro, F.; Campolo, F.; Barrios, F.; Pellegrini, M.; Di Cesare, S.; Tessarollo, L.; Rossi, P.; Jannini, E.A.; Dolci, S. Regulation of Kit Expression in Early Mouse Embryos and ES Cells. Stem Cells 2019, 37, 332–344. [Google Scholar] [CrossRef]
- Loveday, C.; Litchfield, K.; Proszek, P.Z.; Cornish, A.J.; Santo, F.; Levy, M.; Macintyre, G.; Holryod, A.; Broderick, P.; Dudakia, D.; et al. Genomic landscape of platinum resistant and sensitive testicular cancers. Nat Commun 2020, 11, 2189. [Google Scholar] [CrossRef]
- McIntyre, A.; Summersgill, B.; Grygalewicz, B.; Gillis, A.J.; Stoop, J.; van Gurp, R.J.; Dennis, N.; Fisher, C.; Huddart, R.; Cooper, C.; et al. Amplification and overexpression of the KIT gene is associated with progression in the seminoma subtype of testicular germ cell tumors of adolescents and adults. Cancer Res 2005, 65, 8085–8089. [Google Scholar] [CrossRef]
- Shen, H.; Shih, J.; Hollern, D.P.; Wang, L.; Bowlby, R.; Tickoo, S.K.; Thorsson, V.; Mungall, A.J.; Newton, Y.; Hegde, A.M.; et al. Integrated Molecular Characterization of Testicular Germ Cell Tumors. Cell Rep 2018, 23, 3392–3406. [Google Scholar] [CrossRef]
- Pierpont, T.M.; Lyndaker, A.M.; Anderson, C.M.; Jin, Q.; Moore, E.S.; Roden, J.L.; Braxton, A.; Bagepalli, L.; Kataria, N.; Hu, H.Z.; et al. Chemotherapy-Induced Depletion of OCT4-Positive Cancer Stem Cells in a Mouse Model of Malignant Testicular Cancer. Cell Rep 2017, 21, 1896–1909. [Google Scholar] [CrossRef] [PubMed]
- Poynter, J.N.; Hooten, A.J.; Frazier, A.L.; Ross, J.A. Associations between variants in KITLG, SPRY4, BAK1, and DMRT1 and pediatric germ cell tumors. Genes Chromosomes Cancer 2012, 51, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2012, 2, 401–404. [Google Scholar] [CrossRef] [PubMed]
- Roelofs, H.; Mostert, M.C.; Pompe, K.; Zafarana, G.; van Oorschot, M.; van Gurp, R.J.; Gillis, A.J.; Stoop, H.; Beverloo, B.; Oosterhuis, J.W.; et al. Restricted 12p amplification and RAS mutation in human germ cell tumors of the adult testis. Am J Pathol 2000, 157, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Boublikova, L.; Bakardjieva-Mihaylova, V.; Skvarova Kramarzova, K.; Kuzilkova, D.; Dobiasova, A.; Fiser, K.; Stuchly, J.; Kotrova, M.; Buchler, T.; Dusek, P.; et al. Wilms tumor gene 1 (WT1), TP53, RAS/BRAF and KIT aberrations in testicular germ cell tumors. Cancer Lett 2016, 376, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Sommerer, F.; Hengge, U.R.; Markwarth, A.; Vomschloss, S.; Stolzenburg, J.U.; Wittekind, C.; Tannapfel, A. Mutations of BRAF and RAS are rare events in germ cell tumours. Int J Cancer 2005, 113, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Hacioglu, B.M.; Kodaz, H.; Erdogan, B.; Cinkaya, A.; Tastekin, E.; Hacibekiroglu, I.; Turkmen, E.; Kostek, O.; Genc, E.; Uzunoglu, S.; et al. K-RAS and N-RAS mutations in testicular germ cell tumors. Bosn J Basic Med Sci 2017, 17, 159–163. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, A.; Summersgill, B.; Spendlove, H.E.; Huddart, R.; Houlston, R.; Shipley, J. Activating mutations and/or expression levels of tyrosine kinase receptors GRB7, RAS, and BRAF in testicular germ cell tumors. Neoplasia 2005, 7, 1047–1052. [Google Scholar] [CrossRef]
- Carcano, F.M.; Lengert, A.H.; Vidal, D.O.; Scapulatempo Neto, C.; Queiroz, L.; Marques, H.; Baltazar, F.; Berardinelli, G.N.; Martinelli, C.M.; da Silva, E.C.; et al. Absence of microsatellite instability and BRAF (V600E) mutation in testicular germ cell tumors. Andrology 2016, 4, 866–872. [Google Scholar] [CrossRef]
- Honecker, F.; Wermann, H.; Mayer, F.; Gillis, A.J.; Stoop, H.; van Gurp, R.J.; Oechsle, K.; Steyerberg, E.; Hartmann, J.T.; Dinjens, W.N.; et al. Microsatellite instability, mismatch repair deficiency, and BRAF mutation in treatment-resistant germ cell tumors. J Clin Oncol 2009, 27, 2129–2136. [Google Scholar] [CrossRef]
- Solomon, H.; Madar, S.; Rotter, V. Mutant p53 gain of function is interwoven into the hallmarks of cancer. J Pathol 2011, 225, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Einhorn, L.H.; Donohue, J. Cis-diamminedichloroplatinum, vinblastine, and bleomycin combination chemotherapy in disseminated testicular cancer. Ann Intern Med 1977, 87, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Achkar, I.W.; Abdulrahman, N.; Al-Sulaiti, H.; Joseph, J.M.; Uddin, S.; Mraiche, F. Cisplatin based therapy: the role of the mitogen activated protein kinase signaling pathway. J Transl Med 2018, 16, 96. [Google Scholar] [CrossRef] [PubMed]
- Schweyer, S.; Soruri, A.; Meschter, O.; Heintze, A.; Zschunke, F.; Miosge, N.; Thelen, P.; Schlott, T.; Radzun, H.J.; Fayyazi, A. Cisplatin-induced apoptosis in human malignant testicular germ cell lines depends on MEK/ERK activation. Br J Cancer 2004, 91, 589–598. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Chang, J.Y. New Insights into Mechanisms of Cisplatin Resistance: From Tumor Cell to Microenvironment. Int J Mol Sci 2019, 20. [Google Scholar] [CrossRef] [PubMed]
- Caggiano, C.; Cavallo, F.; Giannattasio, T.; Cappelletti, G.; Rossi, P.; Grimaldi, P.; Feldman, D.R.; Jasin, M.; Barchi, M. Testicular Germ Cell Tumors Acquire Cisplatin Resistance by Rebalancing the Usage of DNA Repair Pathways. Cancers (Basel) 2021, 13. [Google Scholar] [CrossRef] [PubMed]
- Bokemeyer, C.; Nichols, C.R.; Droz, J.P.; Schmoll, H.J.; Horwich, A.; Gerl, A.; Fossa, S.D.; Beyer, J.; Pont, J.; Kanz, L.; et al. Extragonadal germ cell tumors of the mediastinum and retroperitoneum: results from an international analysis. J Clin Oncol 2002, 20, 1864–1873. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Chun, J.; Pan, C.; Li, D.; Lin, R.; Alesi, G.N.; Wang, X.; Kang, H.B.; Song, L.; Wang, D.; et al. MAST1 Drives Cisplatin Resistance in Human Cancers by Rewiring cRaf-Independent MEK Activation. Cancer Cell 2018, 34, 315–330. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.R.; Chua, K.N.; Sim, W.J.; Ng, H.C.; Bi, C.; Ho, J.; Nga, M.E.; Pang, Y.H.; Ong, W.R.; Soo, R.A.; et al. MEK Inhibition Overcomes Cisplatin Resistance Conferred by SOS/MAPK Pathway Activation in Squamous Cell Carcinoma. Mol Cancer Ther 2015, 14, 1750–1760. [Google Scholar] [CrossRef]
- Mayer, F.; Wermann, H.; Albers, P.; Stoop, H.; Gillis, A.J.; Hartmann, J.T.; Bokemeyer, C.C.; Oosterhuis, J.W.; Looijenga, L.H.; Honecker, F. Histopathological and molecular features of late relapses in non-seminomas. BJU Int 2011, 107, 936–943. [Google Scholar] [CrossRef]
Figure 1.
BRafV600E embryonal carcinoma disrupting gonadal architecture of a 20 days post-partum mouse testis. Red arrow points to Sox2 and Oct4 positive teratocarcinoma cells. Black arrow points to a normal tubule. Methods and details are found in [20].
Figure 1.
BRafV600E embryonal carcinoma disrupting gonadal architecture of a 20 days post-partum mouse testis. Red arrow points to Sox2 and Oct4 positive teratocarcinoma cells. Black arrow points to a normal tubule. Methods and details are found in [20].
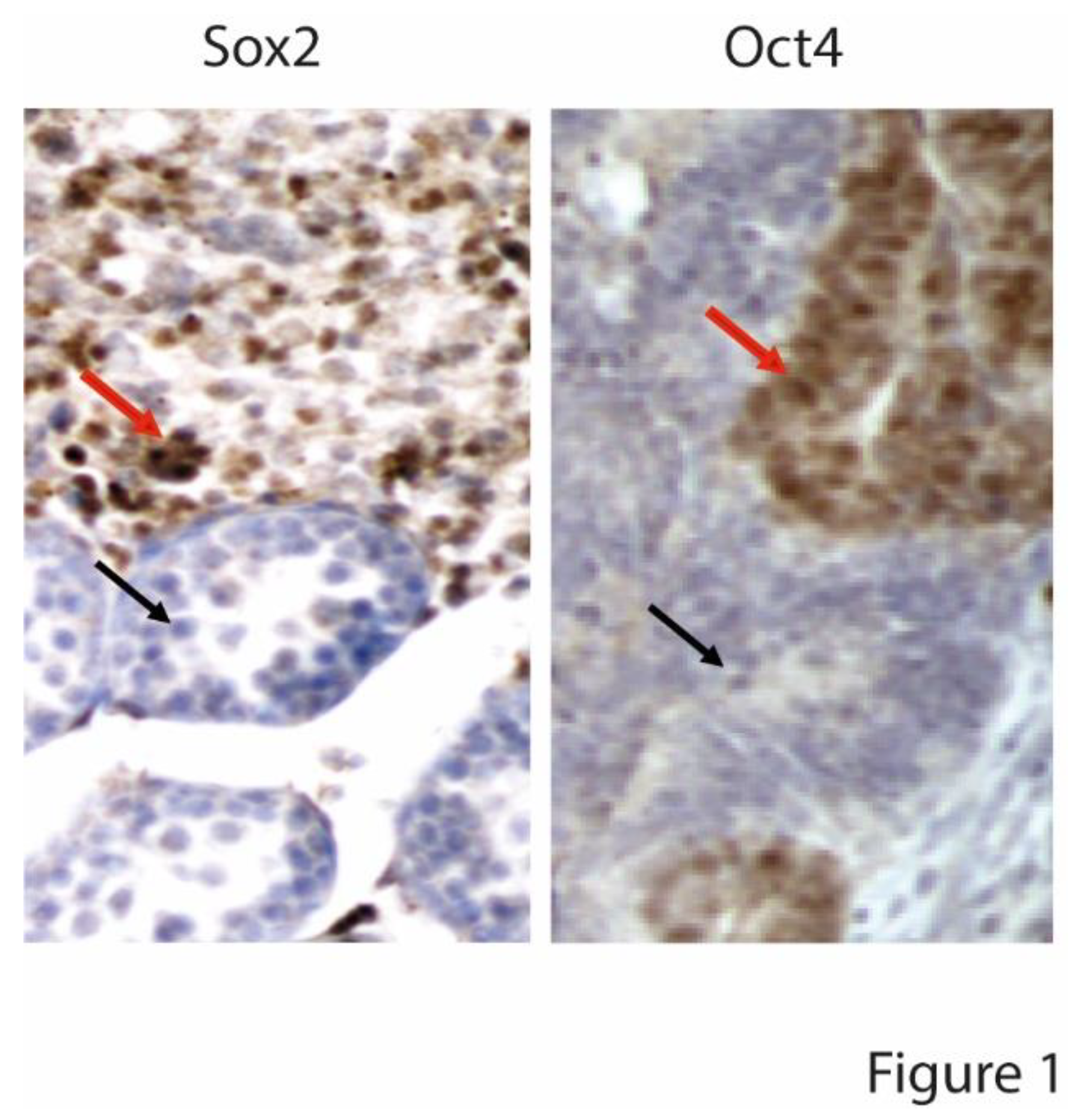
Figure 2.
Schematic representation of deranged ERK-MAPK pathway in TGCTs.
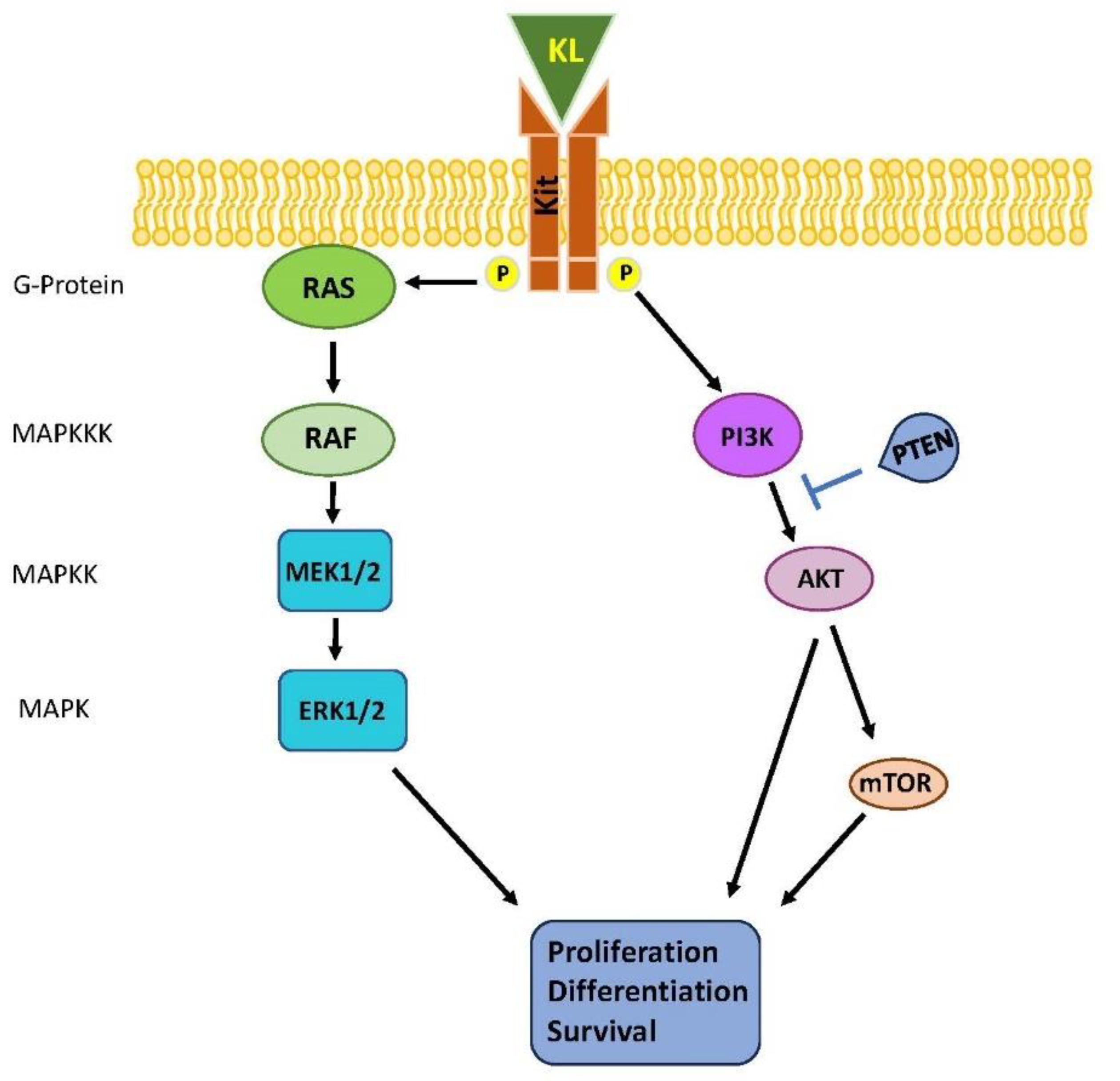

Table 1.
Frequency of mutated somatic genes in the cKIT–RAS–RAF–MEK–ERK cascade in TGCTs.
| KIT | RAS | RAF | MEK/ERK | ||
| Sommerer et al., 2005 [96] |
SE | 7% 9% |
0% 9% |
Activated in all (but not mutated) | |
|
NSE | |||||
| Solomon et al., 2011 [101] | SE | 19% 2% |
5-7% 0% |
1% 2% |
|
|
NSE | |||||
| Hacioglu et al., 2017 [97] |
SE | 29% 26% |
|||
|
NSE | |||||
| Shen et al., 2018 [90] |
SE | 18-25% 2% |
KRAS:14% NRAS: 4% |
||
| NSE | low | ||||
| Ahmad et al., 2019 [50] |
SE | 0% | 4% 31% |
0% | |
|
NSE | |||||
SE= seminomatous NSE= mixed/ Non-seminomatous.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



