
Submitted:
25 January 2024
Posted:
25 January 2024
You are already at the latest version
Abstract
The World Health Organization (WHO) documents malaria as one of the leading causes of high morbidity and mortality worldwide. The disease affects millions and kills thousands of people annually. Efforts to reduce the global burden of malaria have prompted WHO to recommend prevention strategies like using anti-malarial drugs. However, these strategies have been ineffective because of anti-malarial drug resistance. The only efficacious malaria treatment is Artemisinin-based Combination Therapy (ACT). However, the extended ACTs clearance times, linked to the emergence of artemisinin monotherapy resistance recorded most recently in Africa and the Great Mekong region, pose a danger to its efficacy. Therefore, better efficacious malaria drugs are required. Since P. falciparum heat shock protein 90 (PfHsp90) is a well-characterized malaria drugs target, this study uses it to discover more efficacious malaria drugs. An in-silico approach was used to discover PfHsp90 inhibitors with pharmacological properties against Plasmodium malaria using molecular dynamics simulation (MDS) and hierarchical virtual screening. Geldanamycin (GDM), a well-known anti-PfHsp90 compound, was used to identify PfHsp90 inhibitors with pharmacological properties against Plasmodium malaria by screening it against the ZINC database via the ZINCPHARMER web server. This virtual screening process resulted in 17 hits. These ZINCPHARMER hits were subjected to drug-likeness and pharmacokinetics properties analysis in the SwissADME web server, and 9 of them satisfied the requirements. The 9 ZINC compounds were docked with PfHsp90 using the PyRx software to understand their interactions. From the molecular docking results, ZINC09060002 (-8.2 kcal/mol), ZINC72133064 (-7.8 kcal/mol), ZINC72163401 (-7.7 kcal/mol), ZINC72358537 (-8.1 kcal/mol), and ZINC72358557 (-7.6 kcal/mol) had better binding affinities to PfHsp90 than GDM (-7.5 kcal/mol). The stability of these molecularly docked protein-inhibitor complexes was assessed through MDS using GROMACS 2022. ZINC72163401, ZINC72358537, and ZINC72358557 formed stable complexes with PfHsp90. The lead compounds were subjected to in vitro validation for their inhibitory capability. They showed promising inhibition of parasite growth with IC50 values ranging between 200 – 400 ng/ml. In this regard, the three PfHsp90 inhibitors can be used as anti-malarial drugs. However, further structural optimization studies and clinical (in vivo) tests are necessary to ascertain the antimalarial activity of these compounds in humans.
Keywords:
Malaria
; Virtual Screening
; Molecular Dynamics Simulation
; Pharmacophore
; Modelling
; Ligand
Introduction
Malaria is a universal public health burden, with approximately 241 million confirmed cases and a fatality of approximately 627,000 as at 2020 (WHO, 2022). These statistics underscore an alarming rise in malaria cases and deaths compared to WHO’s 2019 report. In 2019, the WHO reported 227 million confirmed cases and approximately 558,000 deaths worldwide (WHO, 2022). Unfortunately, Africa contributes a disproportionally high portion of the global malaria burden, especially sub-Saharan Africa. In 2020, the African region contributed 95% of malaria-confirmed cases and 96% of malaria deaths (Oladipo et al., 2022). Children aged five years and below accounted for approximately 80% of all malaria deaths in the African region (Oladipo et al., 2022). Four countries within sub-Saharan Africa accounted for over half of malaria deaths globally (WHO, 2022). These shockingly high morbidity and mortality rates call for an in-depth understanding of malaria’s etiology, transmission, and pathogenesis and how malaria intervention strategies can be more effective.
Mosquitoes are the primary vectors of malaria, which primarily affects individuals in subtropical and tropical areas. Obligate protozoan parasites of the genus Plasmodium cause the ailment (Sá, Costa, & Tavares, 2022). Mosquito vectors that carry malaria transmit the parasites to people during a blood meal. Once within the host, the parasites first infect the liver cells before moving on to the red blood cells. Ramos et al. (2021) explain that by altering the infected erythrocytes to make them cytoadherent, malaria parasites cause malaria pathology at the blood stage. Malaria infection, whose incubation period is 7-14 days, can cause lethargy, fatigue, muscle aches, nausea, stomach aches, fever, and shivering (Laurens, 2020; Nadeem et al., 2022). Due to the massive loss of red blood cells, the disease can cause jaundice and anemia. Furthermore, if not appropriately treated, malaria can become lethal and cause coma, mental confusion, seizures, kidney failure, and finally, death (CDC, 2022).
To date, P. falciparum, P. knowlesi, P. ovale, P. vivax, and P. malariae are the five species of Plasmodium that can infect individuals and cause malaria. P. falciparum has the highest morbidity and fatality rates and hence poses a severe threat to public well-being in locations where malaria is endemic (Ayanful-Torgby et al., 2018; Chew et al., 2022; Laurens, 2020; Nadeem et al., 2022). Even so, the devastating effects of the other four Plasmodium species on human health should not be underrated. Currently, malaria kills approximately 400,000 individuals yearly, primarily kids in sub-Saharan Africa because they have not yet developed any immunity to the illness. It affects 200-400 million people annually (Chew et al., 2022; Gross, 2019; Wang et al., 2020). Developing tools and strategies that can interrupt the transmission of a specified malaria parasite species is a way of reducing the devastating impacts of the disease on human well-being and safeguarding the well-being of the millions of people affected by the illness yearly.
The WHO recommended malaria prevention strategies and tools that have been in use for almost two decades. Some of these strategies are malaria vaccines and the utilization of anti-malarial drugs. Even though such malaria prevention strategies and tools have reduced the global burden of the illness, the upsurge in the number of malaria’s confirmed cases and deaths from 2019 to 2020 underlies the need for more effective intervention strategies (WHO, 2022). Wang et al. (2020) outline that between 2000 and 2015, anti-malarial medications, insecticide-treated mosquito nets, and other public health initiatives helped reduce malaria cases worldwide by 50–75%. The recently developed malaria vaccine is an essential tool to alleviate the enormous socioeconomic burden that malaria causes. Despite these initiatives, malaria incidences have grown since 2015 in numerous places for various reasons (Wang et al., 2020). One of the outstanding reasons include the parasites developing resistance to anti-malarial medicines.
Anti-malarial drug resistance, which increases malaria mortality and morbidity, has become a menace that casts doubt on the effectiveness of the currently available anti-malarial medications. Only P. falciparum and P. vivax have been proven to be resistant to anti-malarial drugs currently on the market (CDC, 2018). No information is available on P. ovale or P. malariae treatment resistance. P. knowlesi, a zoonotic monkey malaria that affects individuals in Southeast Asian forested environment, is completely sensitive to chloroquine and other commonly utilized medications (CDC, 2018). P. falciparum and P. vivax are resistant to chloroquine. P. falciparum is also resistant to other anti-malarial drugs, including quinine, halofrantrine, mefloquine, and sulfadoxine/pyrimethamine (CDC, 2018). Although resistance to these medicines tends to be noticeably less geographically widespread, the impacts of such multi-drug resistant malaria can be catastrophic in various parts of the globe (CDC, 2018).
Researchers have argued that Artemisinin-based Combination Therapy (ACT) is the only efficacious malaria treatment (Maiga et al., 2021; Pousibet-Puerto et al., 2016). However, Stofberg et al. (2021) explain that the extended ACTs clearance times, which are linked to the emergence of artemisinin monotherapy resistance, have been recorded most recently in Africa and the Great Mekong region, pose a danger to its efficacy. This trend in drug resistance is comparable to the medicine chloroquine’s fate, where drug resistance first appeared in the early 2000s in the Mekong region before spreading widely to many areas, including Africa and East Asia. Genotypes of parasites resistant to the artemisinin medication have recently been discovered in Rwanda, indicating extensive artemisinin compound resistance (Tintó-Font et al., 2021). It is well known that parasite-targeted gene mutations cause the development of medication resistance (Stofberg et al., 2021). Antiparasitic drug concentrations are influenced by the proper operation of mutant genes connected to the inflow and efflux pumps; for instance, molecular chaperones such as the Hsps are responsible for the proper folding of mutated chloroquine resistance transporter (CRT). All proteins can fold naturally when under stress, including pressure from drugs, thanks to these Hsps. Therefore, the Hsps are invaluable antimalarial drug targets.
Additionally, due to their crucial function in protein quality regulation, the genes, such as the CRT gene linked to multidrug resistance, are situated in the same Hsp90 gene cluster (Zininga et al., 2015). This implies that the expression of the mutant CRT gene and the chaperone may be similarly co-regulated by cis-regulatory elements like transcription factors. Furthermore, it has been established that various molecular chaperones interact directly and indirectly with CRT through an unknown mechanism (Stofberg et al., 2021). Hsps’ chaperoning function on proteins might facilitate drug resistance during the stress response. Since the 1600s, attempts have been made to control and manage antimalaria drug resistance in vain (Chakrabarti et al., 2019). Reported malaria cases have always been in millions, as displayed in Figure 1 (Chakrabarti et al., 2019). Hsp90 can be considered a drug target whose inhibition prevents its role in protein folding and facilitates malaria treatment by reducing the level of antimalarial drug resistance that the parasite can mount within the body. In this regard, Hsp90 can be used as a target protein to discover its inhibitors and potential antimalarial drugs against Plasmodium malaria using computational approaches.
This timeline shows how P. falciparum has been mounting resistance to various antimalarial drugs over the years (Chakrabarti et al., 2019).
Hsp90 has been well-characterized in the existing literature. Understanding its functional cycle is essential to identify its active sites or binding pockets in which inhibitors can be docked. Spiegelberg et al. (2020) consider the Hsp90 an evolutionary conserved and widely expressed molecular chaperones group that accounts for around 2% of the cellular proteome. Almost all organisms possess Hsp90 proteins, which are necessary for eukaryotes to survive but optional for some eubacterial species to survive under normal circumstances (Honoré et al., 2017; Schopf et al., 2017). Hsp90 chaperone is essential in reducing stress and preserving cellular homeostasis. Schopf et al. (2017) explain that as a critical regulator of vital physiological functions, including protein folding, stress control, DNA repair, growth, signaling pathways, and immunological response, Hsp90 cooperates with a wide range of client proteins. Hsp90s are desirable pharmacological targets for numerous illnesses, including neurological conditions, cancer, and infectious illnesses like malaria, because of their crucial role in cell survival. The Hsp90 family members are ATP-dependent chaperones involved in numerous biological functions (Schopf et al., 2017). Adenosine triphosphate (ATP) binding and subsequent hydrolysis power the Hsp90, which occurs as a V-shaped homodimer functional cycle (Figure 2) (Stofberg et al., 2021).
During this cycle, Hsp90 binds to the ATP-bound state and open-dimer configuration of an unfolded substrate/client protein (Mader et al., 2020). The N5, N4, and N1 helixes in the NTD undergo a conformational shift; as a result, closing over the ATP binding pocket and serve as a lid over the cavity (Li et al., 2019). Following their association and dimerization, the NTDs undergo additional conformational changes that lead to the middle domains (MDs) attachment. The catalytic loop in the MDs is moved due to these modifications. The crucial catalytic residues Gln423, Arg419, and Asn416 that hydrolyze ATP are part of the catalytic loop in P. falciparum, which extends from residues 414 to 427 (Silva et al., 2020). The release of the appropriately folded client protein is then made possible by the dissociation of the NTDs brought on by ATP hydrolysis (Radli & Rüdiger, 2018; Rashid et al., 2020). Therefore, Hsp90’s active site is found within the NTD, where ATP can bind and initiate the folding of different client proteins. Finding compounds that can bind to Hsp90’s N-terminal ATP binding site can help curtail the protein’s function of assisting the folding of other client proteins.
In early studies, Hsp90 inhibitors were primarily used to treat cancer (Koren & Blagg, 2020). Due to the cancer cells’ insatiable addiction to Hsp90 (Koren & Blagg, 2020), these inhibitors mainly targeted Hsp90 NTD’s ATP binding pocket (Stofberg et al., 2021). Therefore, Hsp90 overexpression in cancerous cells suggests it is involved in developing various oncogenic client proteins. These characteristics make Hsp90 a promising target for anti-cancer medications (Han et al., 2018). In P. falciparum, similar to other eukaryotes, all four Hsp90s may be necessary for parasite persistence and changes in erythrocytic stage transitions (Stofberg et al., 2021). It ought to be highlighted, nonetheless, that several investigations on parasite Hsp90s utilize renowned human Hsp90 inhibitors to assess the role and necessity of those particular proteins. Since it has not been meticulously established that the impact of such inhibitors on parasite growth is solely due to the protein of interest and no other target proteins, care must be used in interpreting such results. Despite this drawback, Hsp90 proteins are desirable therapeutic targets for several reasons. First, Hsp90s are ATPases with differing activity levels in various organisms (Whitesell et al., 2019), and diseased cells have higher ATP hydrolysis rates, making them more vulnerable to ATPase inhibitors (Stofberg et al., 2021).
Similarly, P. falciparum Hsp90 is more susceptible to inhibition than the human homolog due to its increased ATPase activity (Stofberg et al., 2021). Second, in various organisms, Hsp90s interact with various co-chaperones. These different co-chaperone interactions that are species-specific could also be used for selective inhibition (Mak et al., 2021). Lastly, it has been established that the minor variations in amino acid residues among Hsp90 proteins from various organisms and cellular compartments resulted in specific structural variances (Park et al., 2020). Identifying drug targets based on distinctive Hsp90 protein conformations has become easier because of the elucidation of various Hsp90 crystal structures (Huck et al., 2019). High-resolution crystal structures have revealed structural information that has allowed the identification and screening of new Hsp90 inhibitors. Numerous crystal structures of various Hsp90 NTDs in complex with inhibitors and nucleotides have been resolved (Que et al., 2018). Additionally, Hsp90’s complete structural characterization has prompted the creation of inhibitors targeting its MD (Silva et al., 2020; Zhang et al., 2018; Mak et al., 2021) and CTD (Bopp et al., 2016). Figure 3 (Dutta et al., 2022) shows PfHsp90’s domain organization and structure. Through targeting Hsp90, some inhibitors have been demonstrated to be efficient at reducing the parasite’s development (Posfai et al., 2018; Wang et al., 2016; Zininga & Shonhai, 2019). Therefore, PfHsp90 was used as the target protein to determine novel PfHsp90 inhibitors with pharmacological properties against Plasmodium malaria.
One of the first Hsp90 inhibitors discovered was geldanamycin (GDM), a benzoquinone ansamycin molecule naturally generated by Streptomyces hygroscopicus (Stofberg et al., 2021). GDM was initially believed to be an antibiotic that inhibited kinases, but it was later discovered that it had a high degree of selectivity in its binding to Hsp90 (Stofberg et al., 2021). Some of these initial studies targeted Hsp90 in tumor cells and eventually adapted their findings to treat other illnesses, such as malaria. As a result, it was demonstrated that some cancer treatments and inhibitors have strong anti-plasmodial efficacy (Posfai et al., 2018). GDM competitively binds to the PfHsp90’s ATPase domain (Stofberg et al., 2021). The unfolded client protein is then degraded due to GDM’s ability to prevent PfHsp90-client protein interaction (Stofberg et al., 2021). In a study at an IC50 similar to the well-known anti-malarial chloroquine (20 nM and 15 nM, respectively), GDM suppressed in vitro parasite development (Stofberg et al., 2021). PfHsp90 may be crucial for the growth of parasites because its inhibition causes a stage evolution arrest for intra-erythrocytic parasite phases, primarily the transition from the ring to the trophozoite stages (Stofberg et al., 2021). Furthermore, GDM has been noted to be similarly effective against strains that are sensitive to and resistant to chloroquine (Stofberg et al., 2021). Compared to human HSPC2, independent research found GDM to be extra efficacious at decreasing PfHsp90’s ATPase action (Stofberg et al., 2021). This shows that GDM is more selective in abrogating PfHsp90’s enzymatic role than its human counterpart. Therefore, it was used as the reference ligand during virtual screening to determine novel PfHsp90 inhibitors with pharmacological properties against Plasmodium malaria. Figure 4 shows the interaction between PfHsp90 and GDM.
Several researchers have resorted to the use of in-silico means to design and develop drugs, shying away from the traditional techniques that are expensive and time-consuming. Some of the most common computational methods used in drug design and discovery include drug design visualization, hierarchical virtual screening, molecular dynamics simulations, and molecular docking (Onyango, 2023). These in-silico methods have been applied by numerous researchers to create drug candidates. For instance, Onyango et al. (2022) used silico techniques to identify prospective anti-SARS-CoV-2 main protease (Mpro) medicines. Similarly, Mengist et al. (2021) discovered 15 effective anti-viral Mpro compounds using in-silico methods, including chloroquine, cilexetil, dipyridamole, hydroxychloroquine, and candesartan. Computational techniques are crucial in drug discovery to find potential antimalarial drug candidates.
Furthermore, in-silico techniques can be used to test the anti-plasmodial potential of synthetic molecules in vivo through molecular binding. For instance, Tahghighi et al. (2020) used in-silico methods to ascertain that the synthetic derivatives of 1-(heteroaryl)-2-((5-nitroheteroaryl) methylene) hydrazine that showed anti-plasmodial activities in vitro, also possessed the same capability in vivo. From the molecular docking results in the study, the authors verified Pf lactate dehydrogenase (LDH) inhibition and the inhibitory effect on the haemozoin formation for the studied compounds (Tahghighi et al., 2020). In a similar study, Sachdeva et al. (2020) used in-silico approaches to assess the capability of repurposing approved antimalarial medicines against COVID-19. The researchers discovered that the antimalarial drug doxycycline (DOX) could be an ideal candidate for repurposing for COVID-19 because it bound effectively to the spike protein of SARS-CoV-2 (Sachdeva et al., 2020). These studies prove that in-silico methodologies are ideal alternatives to drug design, development, and discovery.
Using computational approaches to discover drug candidates often occur during the pre-clinical phase of drug design and development. After discovering potential drug candidates, undertaking further in vitro and in vivo validation of their therapeutic capacity is essential. Sachdeva et al. (2020) recommended further in vitro and in vivo studies to ascertain the actual potential of DOX against COVID-19. In most cases, the in vitro drug sensitivity assays are utilized depending on the illness and pathogen of interest. Sinha et al. (2017) undertook a systematic review pointing out the various drug sensitivity assay utilized for antimalarial drug efficacy testing targeting different stages of the parasite’s development. Some of those assays include blood stages assays (Schizont maturation, microscopic assay, radioisotopic assay, and enzymatic assay), gametocytes stage assays (oxido-reduction indicator, Alamar blue, and SMFA), liver stage assays (infrared fluorescence detection method), and HTS (fluorescence-based assay and in vitro beta-hematin formation assay) (Sinha et al., 2017).
One of the most common antimalarial drug sensitivity assays is the SYBR green assay. It is considered the GOLD standard for in vitro malaria drug sensitivity testing because of its reliability as a drug screening and surveillance tool (Cheruiyot et al., 2016). It is also described as a simple and cost-effective methodology that has been utilized to determine the 50% inhibitory concentrations (IC50) of clinical isolates (Cheruiyot et al., 2016). Researchers have used this assay in their studies, including Traoré et al. (2019) when assessing the susceptibility of P. falciparum isolates to antimalarial medicines in Mali. Similarly, Duan et al. (2022) determined susceptibilities of P. falciparum isolates to 11 antimalarial medicines using the SYBR green assay. The antimalarial drugs included PND, lumefantrine (LMF), quinine (QN), artemether (AM), DHA, artesunate (AS), pyrimethamine (PY), NQ, mefloquine (MFQ), PPQ, and chloroquine (CQ) (Duan et al., 2022). Therefore, this assay determined the inhibitory capability of the selected PfHsp90 inhibitors.
Methodology
Design and Software
A computer-based design was used to find PfHsp90 inhibitors. A computer with the following specifications was used in this research: 11th Gen Intel(R) Core(TM) i7-11800H @ 2.30GHz. Software like Bio Edit, MEGA 11, BIOVIA Discovery Studio (BDS) 2021, PYRX, and GROMACS were downloaded and installed into the computer. Web-based servers and databases like PDB, PubChem, and SwissADME were also used.
PfHsp90 Structure Retrieval and Preparation
The 3D structure of PfHsp90 was retrieved from PDB database (https://www.rcsb.org/). PfHsp90’s domain of interest (NTD) was retrieved using PDB ID 3K60 and downloaded in the PDB format. This 3D structure was essential during molecular docking. It was prepared for docking after retrieval using BDS 2021. All side chains and bound ligands were removed, leaving only the A chain of the target protein. Similarly, all heteroatoms and water molecules were removed as well. These compounds were removed because they do not participate in the interaction between the ligands of interest and PfHsp90. Deleting them presents a desirable pose search and eases computations that would otherwise prove challenging if such compounds clouded the target protein’s binding pocket. Another preparation step involved adding polar hydrogens to aid in locating hydrogen bond interactions in the 3D structure. The hydrogen bond interactions are essential to ascertain the ligands’ binding affinity to PfHsp90. The prepared 3D structure of PfHsp90 was saved as a .pdb file.
Retrieval of Geldanamycin (GDM) Structures
The PubChem library database (https://pubchem.ncbi.nlm.nih.gov/) was used to retrieve the 2D and 3D structures of GDM, an inhibitor of interest. The 3D structure of the ligand will be crucial during virtual screening to classify structurally similar compounds with antimalarial properties or activities.
Pharmacophore-Based Virtual Screening
The 3D structure of GDM was used to locate active compounds with similar structures that can inhibit PfHsp9. The ZINCPHARMER web server (http://zincpharmer.csb.pitt.edu/pharmer.html) was used in the process. These active compounds were subjected to further processes to ascertain whether or not they can be used as antimalarial compounds or drugs.
Drug-Likeness and Pharmacokinetics Test
The compounds retrieved from the virtual screening process were subjected to a drug-likeness test to determine their drug-ability and pharmacokinetics analysis to determine their oral bioavailability. SwissADME web tool (http://www.swissadme.ch/) was used to perform the drug-likeness test and pharmacokinetics analysis. The SMILES of the virtual screening hits was copy-pasted into the SwissADME web server. The various drug-likeness filters that were utilized include Muegge, Egan, Veber, Ghose, and Lipinski’s Rule. These filters assisted in selecting the compounds with desirable drug properties. Similarly, pharmacokinetic results were analyzed in the form of bioavailability radars and the Brain Or IntestinaL EstimateD permeation (BOILED-Egg) diagram. The molecules that satisfied all the bioavailability and permeation and at least four drug-likeness filters’ requirements were selected for docking studies.
Molecular Docking
The selected molecules, potential PfHsp90 inhibitors, were docked with PfHsp90 using Autodock Vina, an inbuilt tool within the PyRx software. The format of PfHsp90 was converted from .pdb to .pdbqt using the PyRx software. The .pdbqt format is the desirable molecular docking format. The chosen ligand compounds were prepared for molecular docking by minimizing their energies and converting them to a .pdbqt format using the PyRx software. Molecular docking was then performed, and all protein-ligand complexes with the lowest binding energies were chosen as the final potential candidate drugs. This molecular docking process was also undertaken using the reference ligand, GDM. Its binding energy was compared to those of the final potential drug candidates to ascertain whether or not the selected drug candidates preferentially bind to PfHsp90.
Molecular Dynamics Simulation (MDS)
The docked complexes of the final drug candidates and PfHsp90 were subjected to MDS to confirm the docking outcomes and perform an in-depth examination of the behavior of the ligands within the target proteins’ binding pocket. GROMACS 2022 was used during the MDS process. The GROMACS MDS files, which include topology files for the ligand and protein and parameter files for the ligand, were first generated using Charmm 36 Force Field from the CHARMM-GUI web server. The CHARMM-GUI web server’s default settings was preferred, including water box size options, number of ions to be added to the protein-ligand complexes, method of ions addition (Monte-Carlo ion placing method), and system temperature (300.00K). During the GROMACS energy minimization process, the number of steps was set at 5000. The minimized system was equilibrated via a 100ps run. The final MDS run was set at 100ns. After the last run, the number of hydrogen bonds, root mean square fluctuation (RMSF), and root mean square deviation (RMSD) were calculated using GROMACS.
In Vitro Validation of PfHsp90 Inhibitors
The final PfHsp90 inhibitors underwent an in vitro validation to ascertain their inhibitory capability. Their antimalarial activity was measured using the SYBR green assay. The inhibitors were dissolved in 300ul Dimethyl Sulfoxide (DMSO) to a 200ng/ml concentration. This initial concentration was then serially diluted to 11 dilutions across a 96 well plate (12 columns by 8 rows) using a specific growth media for Plasmodium parasites whose preparation is described in WWARN procedure INV02 . 150ul of each dilution was then pipetted into another plate. 150ul of a 1% parasitemic sample was added to each well across the drug coated plate. The dosed plate with parasites was then incubated in a closed system at 37oC for 3 days, then removed from the incubator and 150ul lysis buffer containing a DNA intercalating dye (SYBR green 1) added to the wells. The plate was then incubated at room temperature. Thereafter, the Tecan machine was used to read the fluorescence/absorption from surviving parasites. Wavelength readings from this assay were used to describe the inhibition concentration 50 (IC50). Microsoft Excel was used for analysis, translating the figures read from the Tecan machine to a graph. The R2 value generated was a significant value in interpreting the graphs. An R2 value closer to 1 suggests that the regression line is a perfect fit for the data and can be used to calculate the IC50 values.
Results
PfHsp90 Structure Retrieval and Preparation
The 3D structure of the NTD of PfHsp90 was retrieved from PDB database using PDB ID 3K60. It was loaded into BDS 2021 and prepared for molecular docking. It was discovered that the NTD of PfHsp90 contains two chains, A and B, as shown in Figure 5. Several active sites of the NTD were within chain A. Therefore, during preparation, chain B was deleted, leaving behind a prepared chain A of PfHsp90 NTD, also presented in Figure 5.
Retrieval of Geldanamycin (GDM) Structures
Pharmacophore-Based Virtual Screening
The 3D structure of GDM was loaded into the ZINCPHARMER web server. Four of GDM’s features, namely two hydrogen donors (interacting with GLU 33 and ASN 37 and one freely available) and two hydrogen acceptors (interacting with SER 36 and GLY 123) that are integral in the interaction between GDM and PfHsp90 were selected to act as the preferred pharmacophore features. The virtual screening process was then initiated, resulting in 17 hits (Table 2). The 17 hits were downloaded and saved in a .sdf file.
Drug-Likeness and Pharmacokinetics Test
The hits were then subjected to a drug-likeness test and pharmacokinetics analysis using the SwissADME web server (http://www.swissadme.ch/). The drug-likeness test results are as displayed in Table 3. 11 of the 17 hits satisfied at least four of the five drug-likeness filters. Therefore, they were selected for further pharmacokinetics analysis. Three other hits were excluded because of their unfavorable pharmacokinetics properties as evident in Figure 7. 9 hits (Figure 8) were selected for docking studies.
Molecular Docking
The prepared PfHsp90 (PDB ID 3K60) was loaded into the PYRX software as a .pdb file and converted into a .pdbqt molecule for docking purposes. A file containing GDM and the 9 lead compounds in the .sdf format was then loaded into the PYRX software. Energy of all the 10 ligands was minimized. The ligands were then converted into the preferred .pdbqt format. Docking was then done and the results confirmed that only five ligands had better binding affinities than GDM. The binding affinity of GDM to PfHsp90 was -7.5 kcal/mol. ZINC72163401 (-7.7 kcal/mol), ZINC72133064 (-7.8 kcal/mol), ZINC09060002 (-8.2 kcal/mol), ZINC72358557 (-7.6 kcal/mol), and ZINC72358537 (-8.1 kcal/mol) had better binding affinities to PfHsp90 than GDM. The other 4 molecules had -6.7 kcal/mol (ZINC92700801), -7.1 kcal/mol (ZINC63526364), -7.0 kcal/mol (ZINC91416974), and -7.1 kcal/mol (ZINC72358880). Therefore, the five ligands with better binding affinities than GDM were considered for molecular dynamics simulation. Figure 9 shows the interaction between PfHsp90 and the 6 ligands.
Molecular Dynamics Simulation (MDS)
The docked complexes were loaded into the CHARMM-GUI web server as .pdb files to obtain the topology and parameter files. The files were then used to run minimization, equilibration, and production steps of MDS in GROMACS 2022. The root-mean-square deviation (RMSD), root-mean-square fluctuation (RMSF), and hydrogen bonds analysis was done. As shown in Figure 10, three (ZINC72163401, ZINC72358537, and ZINC72358557) of the five ZINC database compounds do not experience major deviations. The conformational change of their backbone atoms in complex with PfHsp90 is comparable to the conformations of GDM in complex with the target protein over 100ns MDS. ZINC72163401 and ZINC72358557 have negligible deviation distance of approximately 0.3nm within PfHsp90’s binding pocket throughout the 100ns MDS. As for ZINC72358537, before 25ns and after 85ns, its deviation distance is almost 4nm. The deviation distance is below 0.25nm between 25ns and 85ns. ZINC09060002 and ZINC72133064 have high RMSDs throughout the 100ns MDS, indicating increased risk of conformational changes in complex with PfHsp90. These results confirmed that only ZINC72163401 and ZINC72358557 are tightly bound in PfHsp90’s active site and cannot affect the overall topology of the target protein.
The RMSF was calculated to indicate individual residue flexibility, or how much a specific residue moves (fluctuates) during the 100ns simulation. It is a structural indication of which amino acids in a protein or atoms in a chemical compound contribute the most to a molecular motion. In the process, the RMSF measures the movement of a specific atom, or group of atoms, in relation to the reference structure, averaged over the number of atoms (Martínez, 2015). Figure 11 shows the RMSF values of the five ZINC database compounds relative to the reference ligand, GDM. GDM fluctuates within a distance of 0.175nm, with the residue at around position 3450 attaining the lowest fluctuation of approximately 0.025nm and the atom at position 3505 reaching a maximum fluctuation of almost 0.2nm. ZINC72358537 and ZINC72163401 also fluctuates within this range, indicating low divergence from the average position and therefore low structural mobility. Even though ZINC72358557 and ZINC72133064 have atoms that attain a lowest fluctuation of 0.025nm, similar to GDM, some of their atoms surpass the maximum fluctuation threshold of 0.2nm to approximately 0.25nm. Regardless, this 0.05nm difference is low, making them within the desired RMSF levels. As for ZINC09060002, its highest fluctuation surpasses 0.4nm, indicating increased risk of high divergence from the average position and therefore high structural mobility when compared to GDM. These results confirmed that ZINC72163401, ZINC72358537, ZINC72133064, and ZINC72358557 do not undergo high divergence from the average positions.
Hydrogen bonds are essential in protein-ligand complexes because they are considered facilitators of protein-ligand binding. They are believed to increase the affinity with which a ligand binds to a target protein. Figure 12 shows the number of bonds between PfHsp90 and the six ligands. As the reference, PfHsp90-GDM complex forms 1-3 hydrogen bonds in the course of the 100ns simulation. ZINC09060002 forms a maximum of 4 hydrogen bonds with PfHsp90, ZINC72133064 forms 3, ZINC72163401 forms 4, ZINC72358537 forms 4, and ZINC72358557 forms 6. These results confirmed that all the five ZINC database compounds bind strongly to PfHsp90 when compared with GDM.
The number of hydrogen bonds between PfHsp90 (PDB ID: 3K60) with GDM as reference ligand and top-five ZINC database compounds as a function of 100ns simulation time. ZINC09060002 (Black), ZINC72133064 (Red), ZINC72163401 (Green), ZINC72358537 (Blue), ZINC72358557 (Yellow), and GDM (Brown).
In Vitro Validation of PfHsp90 Inhibitors
The in vitro activities of PfHsp90 inhibitors were analyzed against field isolates of the parasite, P. falciparum. Figure 13 shows promising inhibition of parasite growth, with IC50 values of 271.54 ng/ml (ZINC72163401), 384.70 ng/ml (ZINC72358537), and 241.66 ng/ml (ZINC72358557).
Discussion
While several licensed treatment regimens exist, malaria still causes considerable fatality because of antimalarial drug resistance, heightening the need for novel chemotherapeutic interventions (Su et al., 2019; Varo et al., 2020). Targeting a distinct parasitic pathway that differs significantly from the host’s is a valuable means to prevent the spread of the malarial parasite. Since PfHsp90 facilitates parasite growth during its asexual blood phase (Posfai et al., 2018), Plasmodium falciparum uses it to cause malaria. In this regard, inhibiting this protein may hold promise for a Plasmodium falciparum malaria cure.
Currently, there is a paradigm shift from traditional to computational approaches of novel drug design and discovery. In-silico approaches are gaining traction as options for drug discovery. Numerous studies have used in-silico approaches in the discovery of novel therapeutic compounds against malaria, which have progressed to clinical trials (Ashton et al., 2019; White et al., 2018). P. falciparum dihydroorotate dehydrogenase (PfDHODH) inhibitor (DSM265) has been reported to possess antimalarial activity, which has progressed through phase 2 of clinical trials (Alzain et al., 2022; Murphy et al., 2018; Phillips et al., 2015; Sulyok et al., 2017).
Alzain et al. (2022) discovered Z1481646084, Z24317941, and Z951873618 as potential antimalarial compounds inhibiting PfDHODH using in-silico means without subjecting them to in vitro and in vivo validation. Now that the use of in-silico methods to find novel antimalarial compounds is limited, and the existing in-silico studies focus mainly on P. falciparum DHODH and no other parasite enzymes or proteins like PfHsp90, there is need to widen the scope of antimalarial drugs discovery. This study sought to discover PfHsp90 inhibitors as antimalaria drugs.
Virtual screening process was performed using GDM as the ligand to identify the best scoring natural compounds with the ability to inhibit PfHsp90. Due to GDM’s vast stereochemical properties that could limit the number of ZINCPHARMER hits obtained, a pharmacophore model was developed using some of its pharmacophore features. The pharmacophore-based virtual screening yielded 17 hits (Table 2). All the ZINCPHARMER hits were subjected to other in-silico processes because of their low RMSD values (below 1 Å), which suggests that all the hits are structurally similar to GDM. Alzain et al. (2022) and Oduselu et al. (2023) performed pharmacophore-based virtual screening and focused on RMSD values of their hits to determine the most suitable inhibitors against different P. falciparum proteins. Usually, more similar compounds or proteins have small RMSD values (Oduselu et al., 2023). Therefore, RMSD values below 1 Å found in this current study suggests the suitability of all the ZINCPHARMER hits as potential PfHsp90 inhibitors. However, this is subject to confirmation via other in-silico processes like drug-likeness test, ADMET property analysis, molecular docking, and MDS.
Even though several studies perform molecular docking of lead compounds to target proteins before drug-likeness and ADMET property analysis (Alzain et al., 2022; Gao et al., 2023; Oduselu et al., 2023; Onyango et al., 2022), drug-likeness and ADMET property analysis were done before molecular docking to get rid of all lead compounds with unsuitable drug characteristics. This approach is consistent with other studies (Oduselu et al., 2023; Onyango et al., 2022). The drug-likeness test and ADMET properties analysis yielded 9 potential PfHsp90 inhibitors with drug-likeness and ADMET characteristics for further molecular docking (Figure 8).
After determining the 9 potential PfHsp90 inhibitors, our intent to determine the stability of the complexes they form with PfHsp90 necessitated two in-silico process, molecular docking and MDS. These two in-silico approaches are common in drug design and discovery (Onyango, 2023). Regarding the binding affinity of GDM to PfHsp90 (-7.5 kcal/mol), only five of the nine PfHsp90 inhibitors had better binding affinities: ZINC72163401 (-7.7 kcal/mol), ZINC72133064 (-7.8 kcal/mol), ZINC09060002 (-8.2 kcal/mol), ZINC72358557 (-7.6 kcal/mol), and ZINC72358537 (-8.1 kcal/mol) (Figure 9). This means that five PfHsp90 inhibitors bind more strongly to PfHsp90 than GDM.
Despite the positive results of the molecular docking, which validated this study’s design rationale, additional MDS studies were performed to confirm and validate the stability of PfHsp90-ligands complexes. To identify, analyze, and provide insights for future lead optimization, six molecular dynamic simulation tests were done. The stability of the ligand when complexed with PfHsp90 and its binding pocket is shown by the ligands’ RMSD analysis. The RMSD was used to evaluate the structural changes of the six PfHsp90-ligands complexes. In this study, a stable ligand-protein interaction was indicated by the plot of ligand RMSD vs time (100 ns), which was within the average limits . This finding is true for all the lead compounds except ZINC09060002 and ZINC72133064 that did not demonstrate the required stability, evidenced by the high RMSD values reaching 4nm (Figure 10).
ZINC72163401, ZINC72358537, and ZINC72358557, with average RMSD value of 0.25nm suggested that the conformation of the complex formed between these candidate drugs and PfHsp90 remained relatively stable throughout the 100ns simulation. Usually, low RMSD values regarding the true binding pose between a ligand and target protein suggest low binding energy or high binding affinity that facilitates stability (Zheng et al., 2022). Oduselu et al., (2023) concluded that protein-ligand complexes that deviate at distances between 1.5 and 4.0 Å, with an average RMSD of less than 3 Å remain relatively stable throughout the MD simulation. Similarly, Alzain et al. (2022) found out that the ligand and protein RMSD in their study remained between 1.125 and 2.25 Ǻ, indicating that the conformations attained from MDS were structurally stable and ideal for further in-silico assessment. These findings are consistent with those of the current study, proving that ZINC72163401, ZINC72358537, and ZINC72358557 form stable complexes with PfHsp90.
Further RMSF and hydrogen bonds analysis confirmed the stability of the complexes formed between ZINC72163401, ZINC72358557, and ZINC72358537 with PfHsp90. The fluctuations were within RMSF values of 0.2nm, which is acceptable when compared with the RMSF value of the reference ligand (GDM). There were no major fluctuations to indicate that the inhibitors’ atoms shift from their average positions during the 100ns simulation (Figure 11). This finding is consistent with Oduselu et al.’s (2023) and Razzaghi-Asl et al.’s (2022) studies that ensured RMSF values of their protein-ligand complexes are within acceptable levels to infer their stability.
The hydrogen bond analyses (Figure 12) demonstrated that the potential PfHsp90 inhibitors maintained stable conformation in PfHsp90’s active site during the 100ns simulation, signifying their inhibitory capability. Hydrogen bond formation between a ligand and target protein is essential in complexes’ stability because it increases the binding affinity between the two molecules (Oduselu et al., 2023). Oduselu et al. (2023) discovered that the nine hydrogen bonds between ASP 430 as the acceptor and CSMS00081585868 as the donor increased their binding affinity and inhibitory potential. This finding suggests that ZINC09060002, ZINC72163401, and ZINC72358537 that forms a maximum of 4 hydrogen bonds with PfHsp90 and ZINC72358557 that forms 6, all higher than the 3 hydrogen bonds formed between PfHsp90 and GDM, are bound more tightly to the target protein and might possess better inhibitory capability than GDM.
Since in vitro validation of the inhibitory potential and capability of the lead compounds was also a primary objective of this study, ascertaining their stability in complex with their target proteins was not enough. In vitro validation has become a popular approach following MDS in recent drug design and discovery processes (Onyango, 2023). Cheng et al. (2023), Kant et al. (2022), and Ornnork et al. (2020) performed in vitro validation of the lead compounds they discovered in their respective studies. Even though they employed different assays in their studies, they had a common goal that this project shares. The point-to-point calculation found the IC50 values of the three lead compounds to be within 200 – 400 ng/ml (Figure 13). These IC50 values can be compared to that of chloroquine to assess their effectiveness levels. Chloroquine is preferred for this comparison because it is utilized to treat susceptible infections with P. falciparum, P. ovale, P. vivax, and P. malariae. It is also characterized by low toxicity, extended duration of action, rapid onset, and high tolerance in humans (Zhou et al., 2020).
According to Agarwal et al. (2017), the highest concentration of chloroquine utilized for analysis of chemosensitivity against the 3D strain of P. falciparum is 50nM. Using the formula (nM) = (ng/mL)/(MW in KD), the IC50 value of chloroquine was converted to 25.793 ng/ml. The IC50 values of the three PfHsp90 inhibitors are extremely high compared to that of chloroquine. Therefore, the inhibitors are required in higher concentrations than chloroquine to treat malaria. Even though this is not ideal, at those particular high concentrations, these three PfHsp90 inhibitors might still be effective against malaria. ZINC72163401, ZINC72358537, and ZINC72358557 can act as antimalarial drugs against Plasmodium malaria. However, further structural optimization studies and clinical testing through in vivo approaches should be performed to ascertain the efficacy of PfHsp90 inhibitors as antimalarial drugs.
Conclusion
The high mortality and morbidity rates caused by malaria necessitates the design and development of new antimalarial medicines. In this study, a thorough in-silico drug discovery pipeline to discover PfHsp90 inhibitors was developed. Virtual screening for small molecules by both pharmacophore approaches and docking that identified only five potential PfHsp90 inhibitors was performed. After molecular dynamics simulation, three potent PfHsp90 inhibitors, ZINC72163401, ZINC72358537, and ZINC72358557, with good activities against P. falciparum parasites were identified. The three potential antimalarial drugs were subjected to in vitro validation using the SYBR Green assay. They exhibited potent inhibitory activity against P. falciparum with IC50 values ranging between 200 and 400 ng/ml. The effective antimalarial action and low cytotoxicity of these inhibitors mainly suggested that they may be considered for further structural optimization studies and successive clinical validations. This study offers a valuable in-silico blueprint for the rational discovery of novel PfHsp90 inhibitors for Plasmodium falciparum malaria treatment.
Authors’ Contributions
HO, GG, PO, and JM conceptualized the study. HO collected data, analyzed, and drafted the original manuscript. GG, PO, and JM offered the much-required technical support. PO supervised the project and gave technical expert advice. All authors read and approved the manuscript for publication.
Data Availability Statement
Data supporting the findings of this study is available upon request from the corresponding author.
Conflicts of Interests
The authors declare no conflict of interest, no financial or personal relationships that may have inappropriately influenced the writing of this article.
References
- Agarwal, P., Anvikar, A. R., Pillai, C. R., & Srivastava, K. (2017). In vitro susceptibility of Indian Plasmodium falciparum isolates to different antimalarial drugs & antibiotics. The Indian Journal of Medical Research, 146(5), 622-628. [CrossRef]
- Alzain, A. A., Ahmed, Z. A. M., Mahadi, M. A., & Elbadwi, F. A. (2022). Identification of novel Plasmodium falciparum dihydroorotate dehydrogenase inhibitors for malaria using in silico studies. Scientific African, 16, e01214. [CrossRef]
- Ashton, T. D., Devine, S. M., Mohrle, J. J., Laleu, B., Burrows, J. N., Charman, S. A.,... & Sleebs, B. E. (2019). The development process for discovery and clinical advancement of modern antimalarials. Journal of Medicinal Chemistry, 62(23), 10526-10562. [CrossRef]
- Ayanful-Torgby, R., Quashie, N. B., Boampong, J. N., Williamson, K. C., & Amoah, L. E. (2018). Seasonal variations in Plasmodium falciparum parasite prevalence assessed by varying diagnostic tests in asymptomatic children in southern Ghana. PloS One, 13(6), e0199172. [CrossRef]
- Bopp, B., Ciglia, E., Ouald-Chaib, A., Groth, G., Gohlke, H., & Jose, J. (2016). Design and biological testing of peptidic dimerization inhibitors of human Hsp90 that target the C-terminal domain. Biochimica et Biophysica Acta (BBA)-General Subjects, 1860(6), 1043-1055. [CrossRef]
- CDC. (2018). Drug resistance in the malaria-endemic world. Center for Disease Control and Prevention. https://www.cdc.gov/malaria/malaria_worldwide/reduction/drug_resistance.html.
- CDC. (2022) About malaria. Center for Disease Control and Prevention. https://www.cdc.gov/malaria/about/faqs.html#:~:text=Symptoms%20of%20malaria%20include%20fever,loss%20of%20red%20blood%20cells.
- Chakrabarti, A., Singh, V., Singh, S. (2019). Management and control of antimalarial drug resistance. In: Mandal, S., Paul, D. (eds) Bacterial Adaptation to Co-resistance. Springer, Singapore. [CrossRef]
- Cheng, Z., Bhave, M., Hwang, S. S., Rahman, T., & Chee, X. W. (2023). Identification of Potential p38γ Inhibitors via In Silico Screening, In Vitro Bioassay and Molecular Dynamics Simulation Studies. International Journal of Molecular Sciences, 24(8), 7360. [CrossRef]
- Cheruiyot, A. C., Auschwitz, J. M., Lee, P. J., Yeda, R. A., Okello, C. O., Leed, S. E.,... & Johnson, J. D. (2016). Assessment of the Worldwide antimalarial resistance network standardized procedure for in vitro malaria drug sensitivity testing using SYBR green assay for field samples with various initial parasitemia levels. Antimicrobial Agents and Chemotherapy, 60(4), 2417-2424. [CrossRef]
- Chew, M., Ye, W., Omelianczyk, R. I., Pasaje, C. F., Hoo, R., Chen, Q.,... & Preiser, P. (2022). Selective expression of variant surface antigens enables Plasmodium falciparum to evade immune clearance in vivo. Nature Communications, 13(1), 1-12. [CrossRef]
- Duan, M., Bai, Y., Deng, S., Ruan, Y., Zeng, W., Li, X.,... & Cui, L. (2022). Different In Vitro Drug Susceptibility Profile of Plasmodium falciparum Isolates from Two Adjacent Areas of Northeast Myanmar and Molecular Markers for Drug Resistance. Tropical Medicine and Infectious Disease, 7(12), 442. [CrossRef]
- Dutta, T., Singh, H., Edkins, A. L., & Blatch, G. L. (2022). Hsp90 and associated co-chaperones of the malaria parasite. Biomolecules, 12(8), 1018. [CrossRef]
- Gao, M., Kang, D., Liu, N., & Liu, Y. (2023). In Silico Discovery of Small-Molecule Inhibitors Targeting SARS-CoV-2 Main Protease. Molecules, 28(14), 5320. [CrossRef]
- Gross, M. (2019). Fresh efforts needed against malaria. Current Biology, 29(9), R301-R303. [CrossRef]
- Han, J., Goldstein, L. A., Hou, W., Chatterjee, S., Burns, T. F., & Rabinowich, H. (2018). HSP90 inhibition targets autophagy and induces a CASP9-dependent resistance mechanism in NSCLC. Autophagy, 14(6), 958-971. [CrossRef]
- Honoré, F. A., Méjean, V., & Genest, O. (2017). Hsp90 is essential under heat stress in the bacterium Shewanella oneidensis. Cell Reports, 19(4), 680-687. [CrossRef]
- Huck, J. D., Que, N. L., Immormino, R. M., Shrestha, L., Taldone, T., Chiosis, G., & Gewirth, D. T. (2019). NECA derivatives exploit the paralog-specific properties of the site 3 side pocket of Grp94, the endoplasmic reticulum Hsp90. Journal of Biological Chemistry, 294(44), 16010-16019. [CrossRef]
- In vitro Module, WWARN. 2011. Preparation of complete medium for malaria culture. WWARN procedure.
- Kant, V., Kumar, P., Ranjan, R., Kumar, P., Mandal, D., & Vijayakumar, S. (2022). In silico screening, molecular dynamic simulations, and in vitro activity of selected natural compounds as an inhibitor of Leishmania donovani 3-mercaptopyruvate sulfurtransferase. Parasitology Research, 121(7), 2093-2109. [CrossRef]
- Koren, J., & Blagg, B. S. (2020). The right tool for the job: An overview of Hsp90 inhibitors. HSF1 and Molecular Chaperones in Biology and Cancer, 135-146. [CrossRef]
- Laurens, M. B. (2020). RTS, S/AS01 vaccine (Mosquirix™): An overview. Human Vaccines & Immunotherapeutics, 16(3), 480-489. [CrossRef]
- Li, L., Wang, L., You, Q. D., & Xu, X. L. (2019). Heat shock protein 90 inhibitors: An update on achievements, challenges, and future directions. Journal of Medicinal Chemistry, 63(5), 1798-1822. [CrossRef]
- Mader, S. L., Lopez, A., Lawatscheck, J., Luo, Q., Rutz, D. A., Gamiz-Hernandez, A. P.,... & Kaila, V. R. (2020). Conformational dynamics modulate the catalytic activity of the molecular chaperone Hsp90. Nature Communications, 11(1), 1-12. [CrossRef]
- Maiga, F. O., Wele, M., Toure, S. M., Keita, M., Tangara, C. O., Refeld, R. R.,... & Shaffer, J. G. (2021). Artemisinin-based combination therapy for uncomplicated Plasmodium falciparum malaria in Mali: a systematic review and meta-analysis. Malaria Journal, 20(1), 1-13. [CrossRef]
- Mak, O. W., Sharma, N., Reynisson, J., & Leung, I. K. (2021). Discovery of novel Hsp90 C-terminal domain inhibitors that disrupt co-chaperone binding. Bioorganic & Medicinal Chemistry Letters, 38, 127857. [CrossRef]
- Martínez, L. (2015). Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PloS One, 10(3), e0119264. [CrossRef]
- Mengist, H. M., Dilnessa, T., & Jin, T. (2021). Structural basis of potential inhibitors targeting SARS-CoV-2 main protease. Frontiers in Chemistry, 9, 622898. [CrossRef]
- Murphy, S. C., Duke, E. R., Shipman, K. J., Jensen, R. L., Fong, Y., Ferguson, S.,... & Kublin, J. G. (2018). A randomized trial evaluating the prophylactic activity of DSM265 against preerythrocytic Plasmodium falciparum infection during controlled human malarial infection by mosquito bites and direct venous inoculation. The Journal of Infectious Diseases, 217(5), 693-702. [CrossRef]
- Nadeem, A. Y., Shehzad, A., Islam, S. U., Al-Suhaimi, E. A., & Lee, Y. S. (2022). Mosquirix™ RTS, S/AS01 vaccine development, immunogenicity, and efficacy. Vaccines, 10(5), 713. 13. [CrossRef]
- Oduselu, G. O., Afolabi, R., Ademuwagun, I., Vaughan, A., & Adebiyi, E. (2023). Structure-based pharmacophore modeling, virtual screening, and molecular dynamics simulation studies for identification of Plasmodium falciparum 5-aminolevulinate synthase inhibitors. Frontiers in Medicine, 9, 1022429. [CrossRef]
- Oladipo, H. J., Tajudeen, Y. A., Oladunjoye, I. O., Yusuff, S. I., Yusuf, R. O., Oluwaseyi, E. M.,... & El-Sherbini, M. S. (2022). Increasing challenges of malaria control in sub-Saharan Africa: Priorities for public health research and policymakers. Annals of Medicine and Surgery, 81, 104366. [CrossRef]
- Onyango, H., Odhiambo, P., Angwenyi, D., & Okoth, P. (2022). “In silico identification of new anti-SARS-CoV-2 main protease (Mpro) molecules with pharmacokinetic properties from natural sources using molecular dynamics (MD) simulations and hierarchical virtual screening”, Journal of Tropical Medicine, vol. 2022, Article ID 3697498, 1-22. [CrossRef]
- Onyango, O. H. (2023). In silico models for anti-COVID-19 drug discovery: A systematic review. Advances in Pharmacological and Pharmaceutical Sciences, vol. 2023, Article ID 4562974, 15 pages, 2023. [CrossRef]
- Ornnork, N., Kiriwan, D., Lirdprapamongkol, K., Choowongkomon, K., Svasti, J., & Eurtivong, C. (2020). Molecular dynamics, MM/PBSA and in vitro validation of a novel quinazoline-based EGFR tyrosine kinase inhibitor identified using structure-based in silico screening. Journal of Molecular Graphics and Modelling, 99, 107639. [CrossRef]
- Park, H. K., Yoon, N. G., Lee, J. E., Hu, S., Yoon, S., Kim, S. Y.,... & Kang, B. H. (2020). Unleashing the full potential of Hsp90 inhibitors as cancer therapeutics through simultaneous inactivation of Hsp90, Grp94, and TRAP1. Experimental & Molecular Medicine, 52(1), 79-91. [CrossRef]
- Phillips, M. A., Lotharius, J., Marsh, K., White, J., Dayan, A., White, K. L.,... & Charman, S. A. (2015). A long-duration dihydroorotate dehydrogenase inhibitor (DSM265) for prevention and treatment of malaria. Science Translational Medicine, 7(296), 296ra111-296ra111. [CrossRef]
- Pousibet-Puerto, J., Salas-Coronas, J., Sánchez-Crespo, A., Molina-Arrebola, M. A., Soriano-Pérez, M. J., Giménez-López, M. J.,... & Cabezas-Fernández, M. T. (2016). Impact of using artemisinin-based combination therapy (ACT) in the treatment of uncomplicated malaria from Plasmodium falciparum in a non-endemic zone. Malaria Journal, 15(1), 1-7. [CrossRef]
- Posfai, D., Eubanks, A. L., Keim, A. I., Lu, K. Y., Wang, G. Z., Hughes, P. F.,... & Derbyshire, E. R. (2018). Identification of Hsp90 inhibitors with anti-Plasmodium activity. Antimicrobial Agents and Chemotherapy, 62(4), e01799-17. [CrossRef]
- Que, N. L., Crowley, V. M., Duerfeldt, A. S., Zhao, J., Kent, C. N., Blagg, B. S., & Gewirth, D. T. (2018). Structure based design of a Grp94-selective inhibitor: Exploiting a key residue in Grp94 to optimize paralog-selective binding. Journal of Medicinal Chemistry, 61(7), 2793-2805. [CrossRef]
- Radli, M., & Rüdiger, S. G. (2018). Dancing with the diva: Hsp90–client interactions. Journal of Molecular Biology, 430(18), 3029-3040. [CrossRef]
- Ramos, S., Ademolue, T. W., Jentho, E., Wu, Q., Guerra, J., Martins, R.,... & Soares, M. P. (2021). A hypometabolic defense strategy against plasmodium infection. BioRxiv, 1-62. [CrossRef]
- Rashid, S., Lee, B. L., Wajda, B., & Spyracopoulos, L. (2020). Nucleotide binding and active site gate dynamics for the Hsp90 chaperone ATPase domain from benchtop and high field 19F NMR spectroscopy. The Journal of Physical Chemistry B, 124(15), 2984-2993. [CrossRef]
- Sá, M., Costa, D. M., & Tavares, J. (2022). Imaging infection by vector-borne protozoan parasites using whole-mouse bioluminescence. Bioluminescence: Methods and Protocols, Volume 1, 2524, 353-366. [CrossRef]
- Sachdeva, C., Wadhwa, A., Kumari, A., Hussain, F., Jha, P., & Kaushik, N. K. (2020). In silico potential of approved antimalarial drugs for repurposing against COVID-19. Omics: A Journal of Integrative Biology, 24(10), 568-580.
- Schopf, F. H., Biebl, M. M., & Buchner, J. (2017). The HSP90 chaperone machinery. Nature Reviews Molecular Cell Biology, 18(6), 345-360. [CrossRef]
- Silva, N. S., Torricillas, M. S., Minari, K., Barbosa, L. R., Seraphim, T. V., & Borges, J. C. (2020). Solution structure of Plasmodium falciparum Hsp90 indicates a high flexible dimer. Archives of Biochemistry and Biophysics, 690, 108468. [CrossRef]
- Sinha, S., Sarma, P., Sehgal, R., & Medhi, B. (2017). Development in assay methods for in vitro antimalarial drug efficacy testing: A systematic review. Frontiers in Pharmacology, 8, 754. [CrossRef]
- Spiegelberg, D., Abramenkovs, A., Mortensen, A. C. L., Lundsten, S., Nestor, M., & Stenerlöw, B. (2020). The HSP90 inhibitor Onalespib exerts synergistic anti-cancer effects when combined with radiotherapy: an in vitro and in vivo approach. Scientific Reports, 10(1), 1-11. [CrossRef]
- Stofberg, M. L., Caillet, C., de Villiers, M., & Zininga, T. (2021). Inhibitors of the Plasmodium falciparum Hsp90 towards Selective Antimalarial Drug Design: The Past, Present and Future. Cells, 10(11), 2849. [CrossRef]
- Su, X. Z., Lane, K. D., Xia, L., Sá, J. M., & Wellems, T. E. (2019). Plasmodium genomics and genetics: New insights into malaria pathogenesis, drug resistance, epidemiology, and evolution. Clinical Microbiology Reviews, 32(4), 10-1128. [CrossRef]
- Sulyok, M., Rückle, T., Roth, A., Mürbeth, R. E., Chalon, S., Kerr, N.,... & Mordmüller, B. (2017). DSM265 for Plasmodium falciparum chemoprophylaxis: a randomised, double blinded, phase 1 trial with controlled human malaria infection. The Lancet Infectious Diseases, 17(6), 636-644. [CrossRef]
- Tahghighi, A., Mohamadi-Zarch, S. M., Rahimi, H., Marashiyan, M., Maleki-Ravasan, N., & Eslamifar, A. (2020). In silico and in vivo anti-malarial investigation on 1-(heteroaryl)-2-((5-nitroheteroaryl) methylene) hydrazine derivatives. Malaria Journal, 19(1), 1-12.
- Tintó-Font, E., Michel-Todó, L., Russell, T. J., Casas-Vila, N., Conway, D. J., Bozdech, Z.,... & Cortés, A. (2021). A heat-shock response regulated by the PfAP2-HS transcription factor protects human malaria parasites from febrile temperatures. Nature Microbiology, 6(9), 1163-1174. [CrossRef]
- Traoré, K., Diakité, S. A., Bah, S., Konaté, D. S., Dabitao, D., Sanogo, I.,... & Diakité, M. (2019). Susceptibility of Plasmodium falciparum isolates to antimalarial drugs in a highly seasonal malaria endemic village in Mali. [CrossRef]
- Varo, R., Chaccour, C., & Bassat, Q. (2020). Update on malaria. Medicina Clínica (English Edition), 155(9), 395-402. [CrossRef]
- Wang, T., Mäser, P., & Picard, D. (2016). Inhibition of Plasmodium falciparum Hsp90 contributes to the anti-malarial activities of aminoalcohol-carbazoles. Journal of Medicinal Chemistry, 59(13), 6344-6352. [CrossRef]
- Wang, L. T., Pereira, L. S., Flores-Garcia, Y., O’Connor, J., Flynn, B. J., Schön, A.,... & Seder, R. A. (2020). A potent anti-malarial human monoclonal antibody targets circumsporozoite protein minor repeats and neutralizes sporozoites in the liver. Immunity, 53(4), 733-744. [CrossRef]
- White, J., Dhingra, S. K., Deng, X., El Mazouni, F., Lee, M. C., Afanador, G. A.,... & Phillips, M. A. (2018). Identification and mechanistic understanding of dihydroorotate dehydrogenase point mutations in Plasmodium falciparum that confer in vitro resistance to the clinical candidate DSM265. ACS Infectious Diseases, 5(1), 90-101. [CrossRef]
- Whitesell, L., Robbins, N., Huang, D. S., McLellan, C. A., Shekhar-Guturja, T., LeBlanc, E. V.,... & Cowen, L. E. (2019). Structural basis for species-selective targeting of Hsp90 in a pathogenic fungus. Nature Communications, 10(1), 1-17. [CrossRef]
- WHO. (2022). Malaria. World Health Organization. https://www.who.int/news-room/fact-sheets/detail/malaria.
- Zhang, F. Z., Ho, D. H. H., & Wong, R. H. F. (2018). Triptolide, a HSP90 middle domain inhibitor, induces apoptosis in triple manner. Oncotarget, 9(32), 22301-22315. [CrossRef]
- Zheng, L., Meng, J., Jiang, K., Lan, H., Wang, Z., Lin, M.,... & Mu, Y. (2022). Improving protein–ligand docking and screening accuracies by incorporating a scoring function correction term. Briefings in Bioinformatics, 23(3), bbac051. [CrossRef]
- Zhou, W., Wang, H., Yang, Y., Chen, Z. S., Zou, C., & Zhang, J. (2020). Chloroquine against malaria, cancers and viral diseases. Drug Discovery Today, 25(11), 2012-2022. [CrossRef]
- Zininga, T., Makumire, S., Gitau, G. W., Njunge, J. M., Pooe, O. J., Klimek, H.,... & Shonhai, A. (2015). Plasmodium falciparum Hop (PfHop) interacts with the Hsp70 chaperone in a nucleotide-dependent fashion and exhibits ligand selectivity. PLoS One, 10(8), e0135326. [CrossRef]
- Zininga, T., & Shonhai, A. (2019). Small molecule inhibitors targeting the heat shock protein system of human obligate protozoan parasites. International Journal of Molecular Sciences, 20(23), 5930. [CrossRef]
Figure 1.
Antimalarial drug resistance.
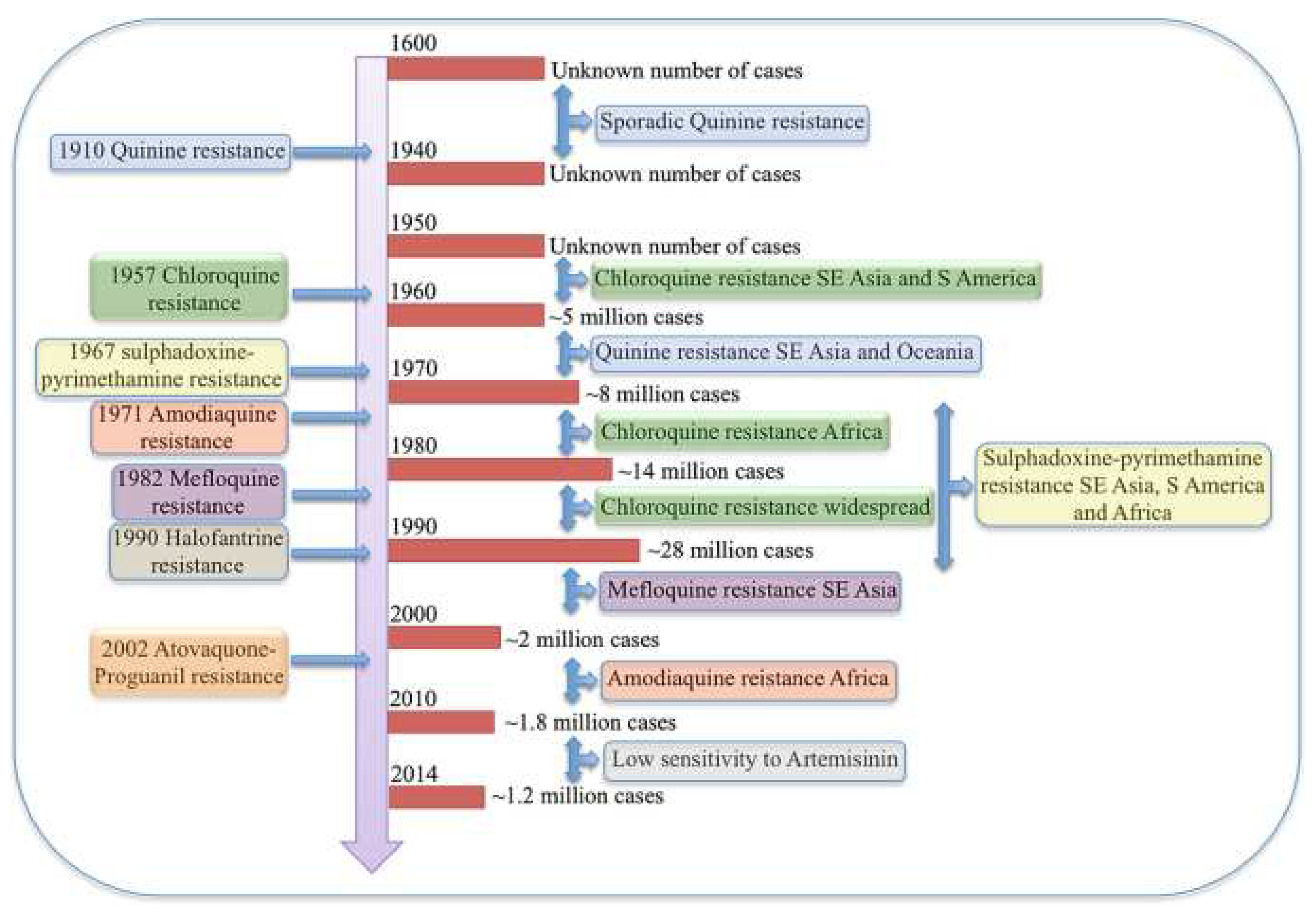
Figure 2.
Hsp90 domain organization and functional cycle. (A) Schematic representation of the V-shaped domain organization of the Hsp90 protein. (B) Schematic representation of the Hsp90 functional cycle begins with the binding of ATP to Hsp90, provoking the association of the protein with a client/unfolded protein. Consequently, a closed conformation is adopted following the NTD dimerization after the lid region of Hsp90 closes over the ATP binding pocket. ATP hydrolysis occurs after repositioning the catalytic loop when the MDs associate. The correctly folded client protein is released upon ATP hydrolysis. This protein folding and ATP hydrolysis cycle reoccur after the Hsp90 homodimer regains its unbound open configuration and is primed. Aha1, HOP, and p23 are other co-chaperones that modulate this functional cycle (Stofberg et al., 2021).
Figure 2.
Hsp90 domain organization and functional cycle. (A) Schematic representation of the V-shaped domain organization of the Hsp90 protein. (B) Schematic representation of the Hsp90 functional cycle begins with the binding of ATP to Hsp90, provoking the association of the protein with a client/unfolded protein. Consequently, a closed conformation is adopted following the NTD dimerization after the lid region of Hsp90 closes over the ATP binding pocket. ATP hydrolysis occurs after repositioning the catalytic loop when the MDs associate. The correctly folded client protein is released upon ATP hydrolysis. This protein folding and ATP hydrolysis cycle reoccur after the Hsp90 homodimer regains its unbound open configuration and is primed. Aha1, HOP, and p23 are other co-chaperones that modulate this functional cycle (Stofberg et al., 2021).
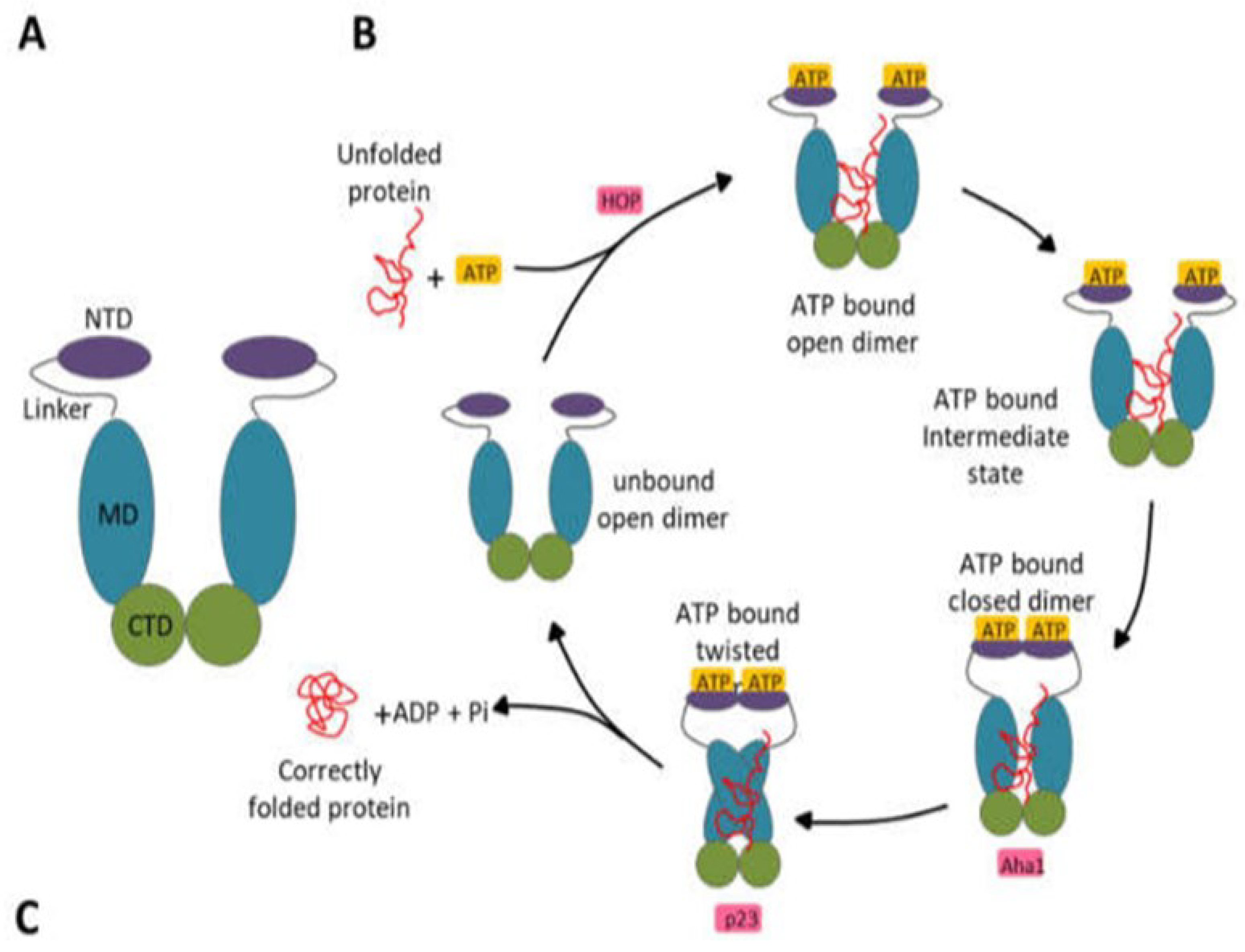
Figure 3.
PfHsp90 domain organization and structure view. (A) Schematic model of the different PfHsp90 domains labeled NTD (N-Terminal Domain), L (Linker Region), MD (Middle Domain), and CTD (C-Terminal Domain). (B) Cartoon representation of PfHsp90 proteins. Red spheres represent ATP bound to NTD. The purple and blue colors in the model depict the two PfHsp90 monomers. (C) Surface representation of PfHsp90’s NTD with 60% transparency and colored according to element type (Dutta et al., 2022).
Figure 3.
PfHsp90 domain organization and structure view. (A) Schematic model of the different PfHsp90 domains labeled NTD (N-Terminal Domain), L (Linker Region), MD (Middle Domain), and CTD (C-Terminal Domain). (B) Cartoon representation of PfHsp90 proteins. Red spheres represent ATP bound to NTD. The purple and blue colors in the model depict the two PfHsp90 monomers. (C) Surface representation of PfHsp90’s NTD with 60% transparency and colored according to element type (Dutta et al., 2022).
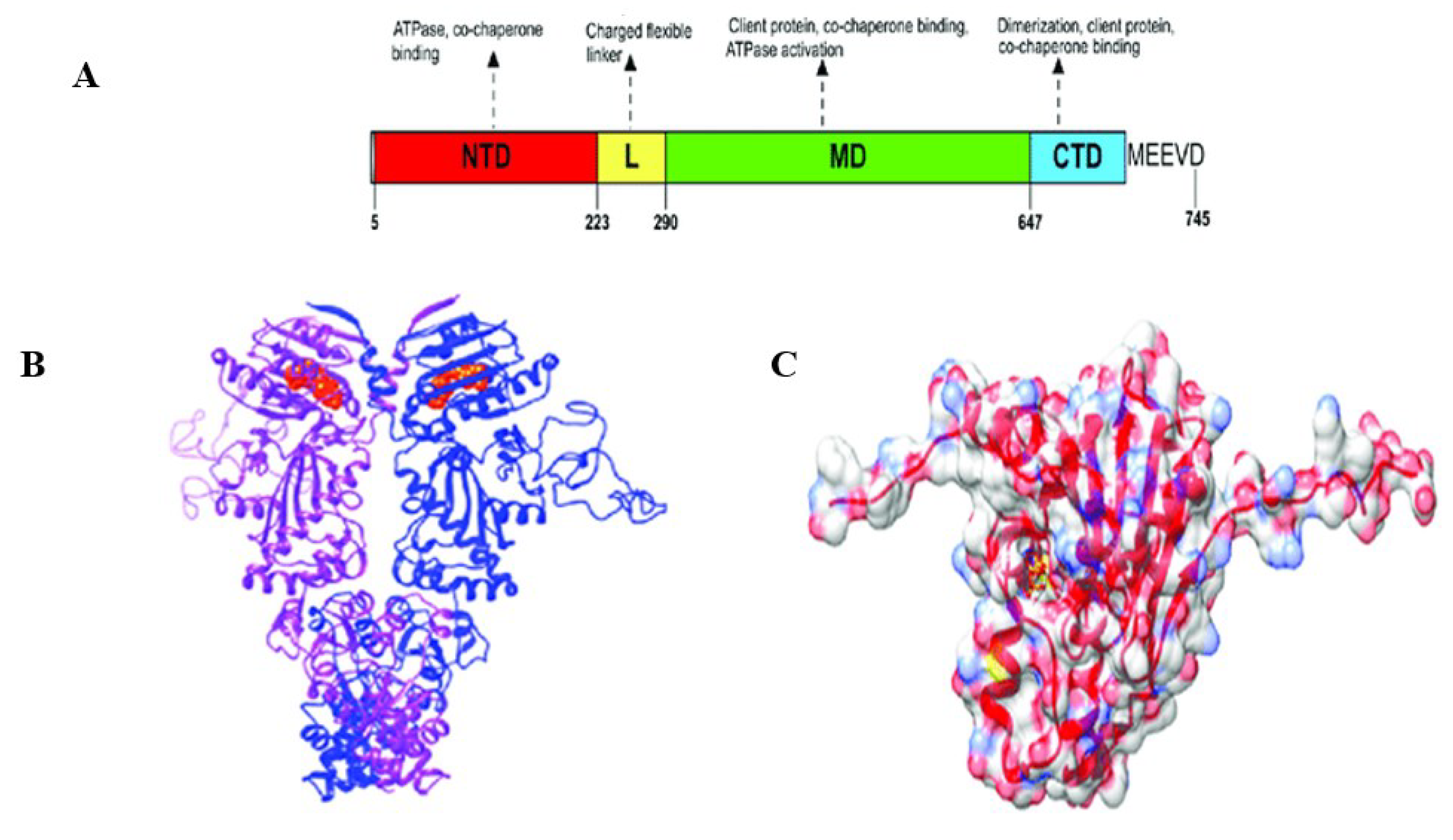
Figure 4.
Geldanamycin (GDM) as a PfHsp90 inhibitor. The green stick-like structure represents GDM. The dotted lines of different colors display the interaction points between GDM and PfHsp90. The structure is available in the PDB database (https://www.rcsb.org/) and can be retrieved using the PDB ID 1YET. BIOVIA Discovery Studio 2021 was used to find the suitable binding pocket pose and show the points of interaction between the two molecules.
Figure 4.
Geldanamycin (GDM) as a PfHsp90 inhibitor. The green stick-like structure represents GDM. The dotted lines of different colors display the interaction points between GDM and PfHsp90. The structure is available in the PDB database (https://www.rcsb.org/) and can be retrieved using the PDB ID 1YET. BIOVIA Discovery Studio 2021 was used to find the suitable binding pocket pose and show the points of interaction between the two molecules.
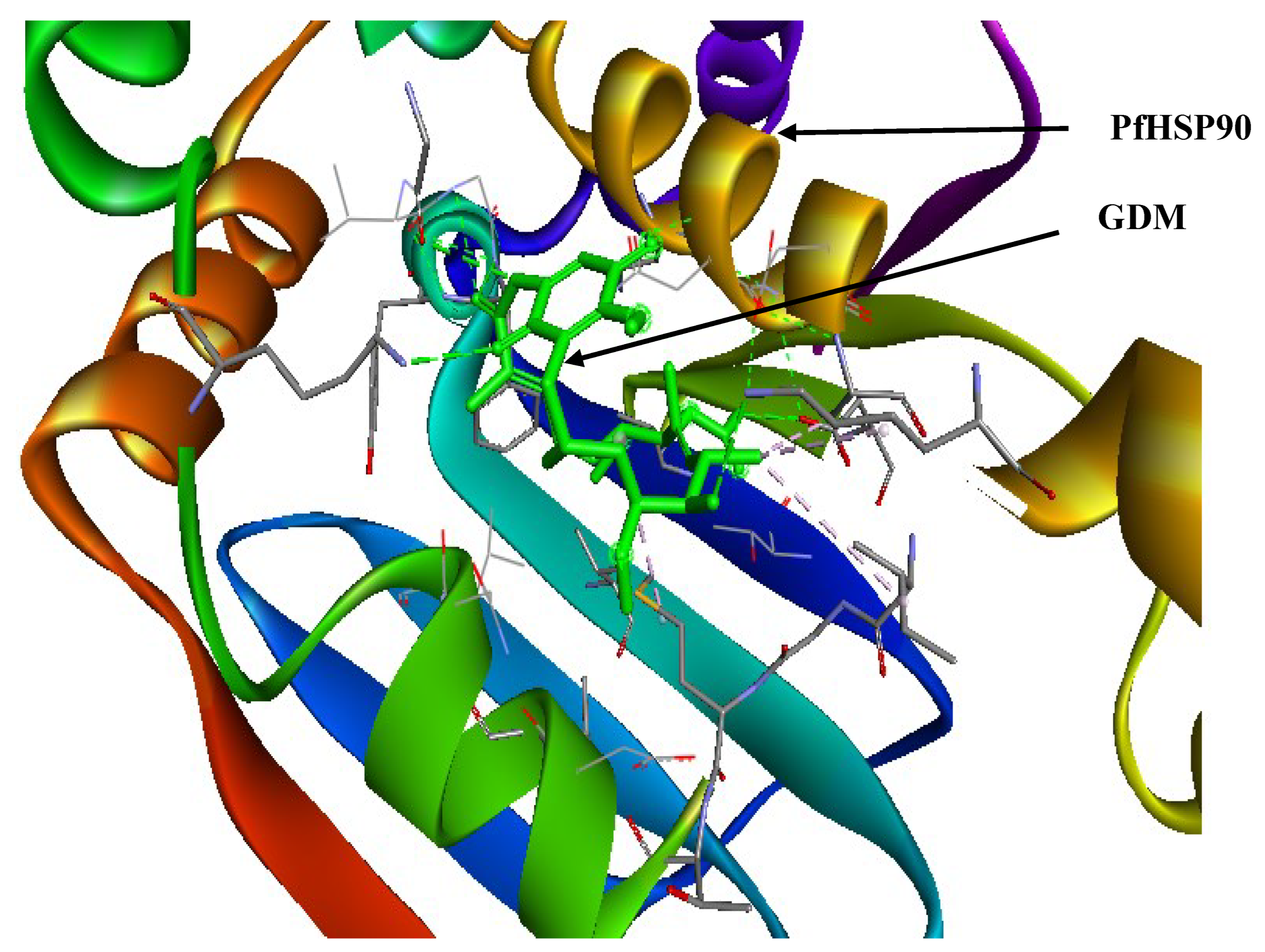
Figure 5.
The 3D structure of prepared PfHsp90 NTD. PfHsp90 NTD retrieved from PDB, ID 3K60. All heteroatoms and water molecules removed and polar hydrogens added. The two chains, A and B, are indicated.
Figure 5.
The 3D structure of prepared PfHsp90 NTD. PfHsp90 NTD retrieved from PDB, ID 3K60. All heteroatoms and water molecules removed and polar hydrogens added. The two chains, A and B, are indicated.

Figure 6.
2D and 3D Structures of GDM. (A) 2D structure. (B) 3D structure.
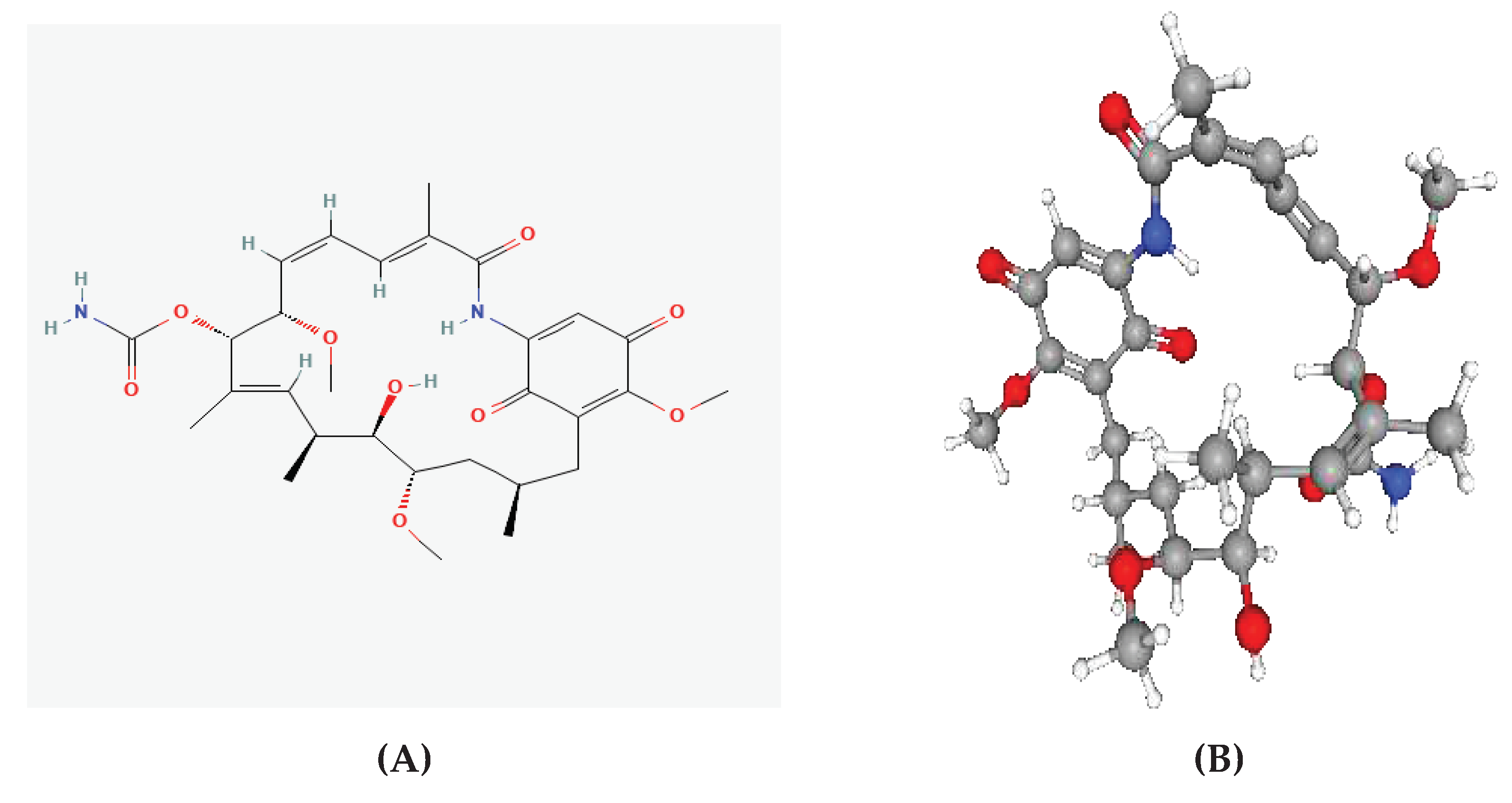
Figure 7.
BOILED-Egg Analysis (GDM). Boiled egg prediction of blood brain barrier permeability and gastrointestinal absorption for the 17 hits. 4 molecules (15, 7, 2, and 16) are out of range, thus excluded. The other molecules are P-glycoprotein (P-gp) substrate, indicated by the blue dot, depicting their ease of excretion from the body.
Figure 7.
BOILED-Egg Analysis (GDM). Boiled egg prediction of blood brain barrier permeability and gastrointestinal absorption for the 17 hits. 4 molecules (15, 7, 2, and 16) are out of range, thus excluded. The other molecules are P-glycoprotein (P-gp) substrate, indicated by the blue dot, depicting their ease of excretion from the body.
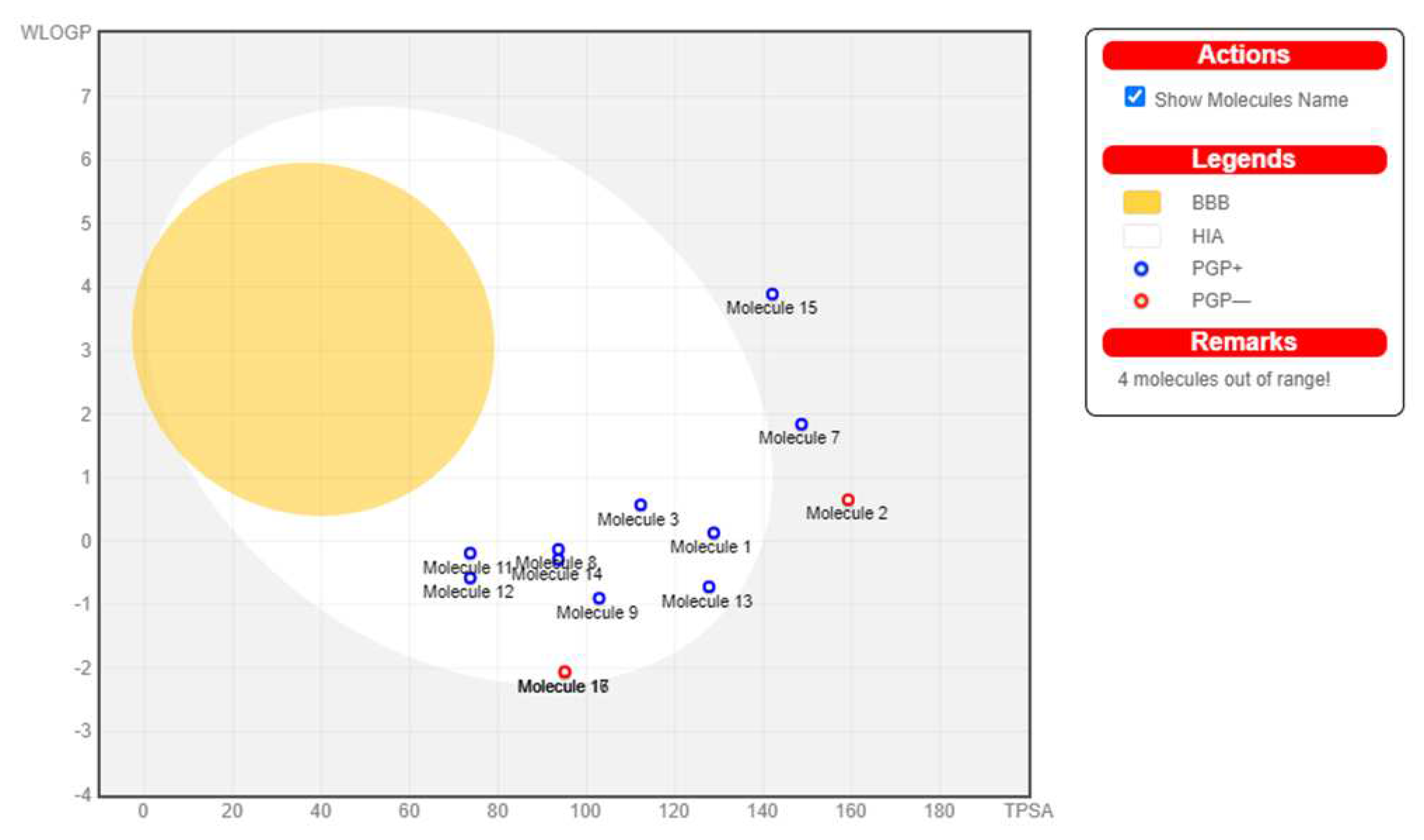
Figure 8.
The Structures and Oral Bioavailability Radars of the 9 Hits (GDM). (A) ZINC09060002, (B) ZINC63526364, (C) ZINC72133064, (D) ZINC72163401, (E) ZINC72358537, (F) ZINC72358557, (G) ZINC72358880, (H) ZINC91416974, (I) ZINC92700801.
Figure 8.
The Structures and Oral Bioavailability Radars of the 9 Hits (GDM). (A) ZINC09060002, (B) ZINC63526364, (C) ZINC72133064, (D) ZINC72163401, (E) ZINC72358537, (F) ZINC72358557, (G) ZINC72358880, (H) ZINC91416974, (I) ZINC92700801.
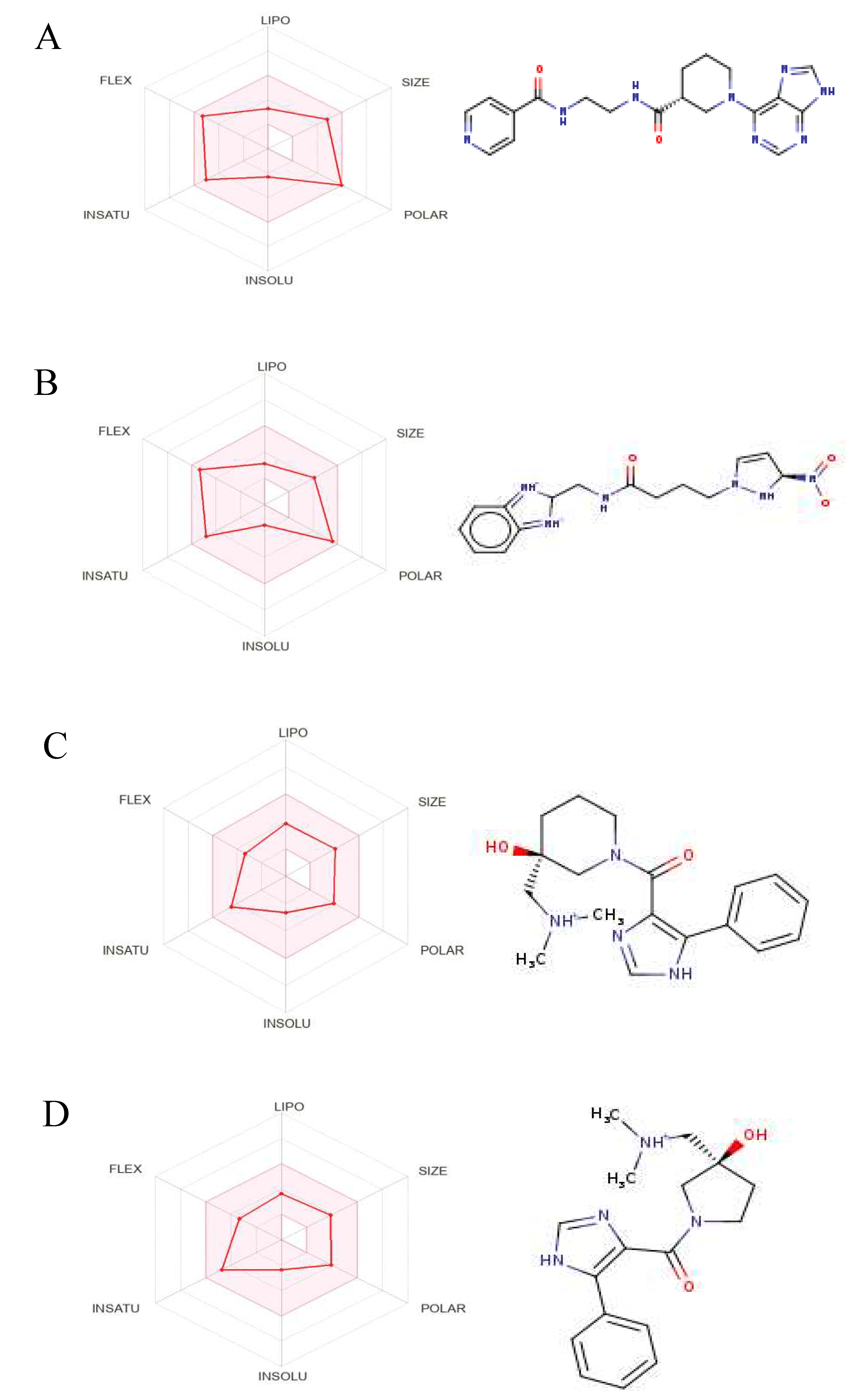
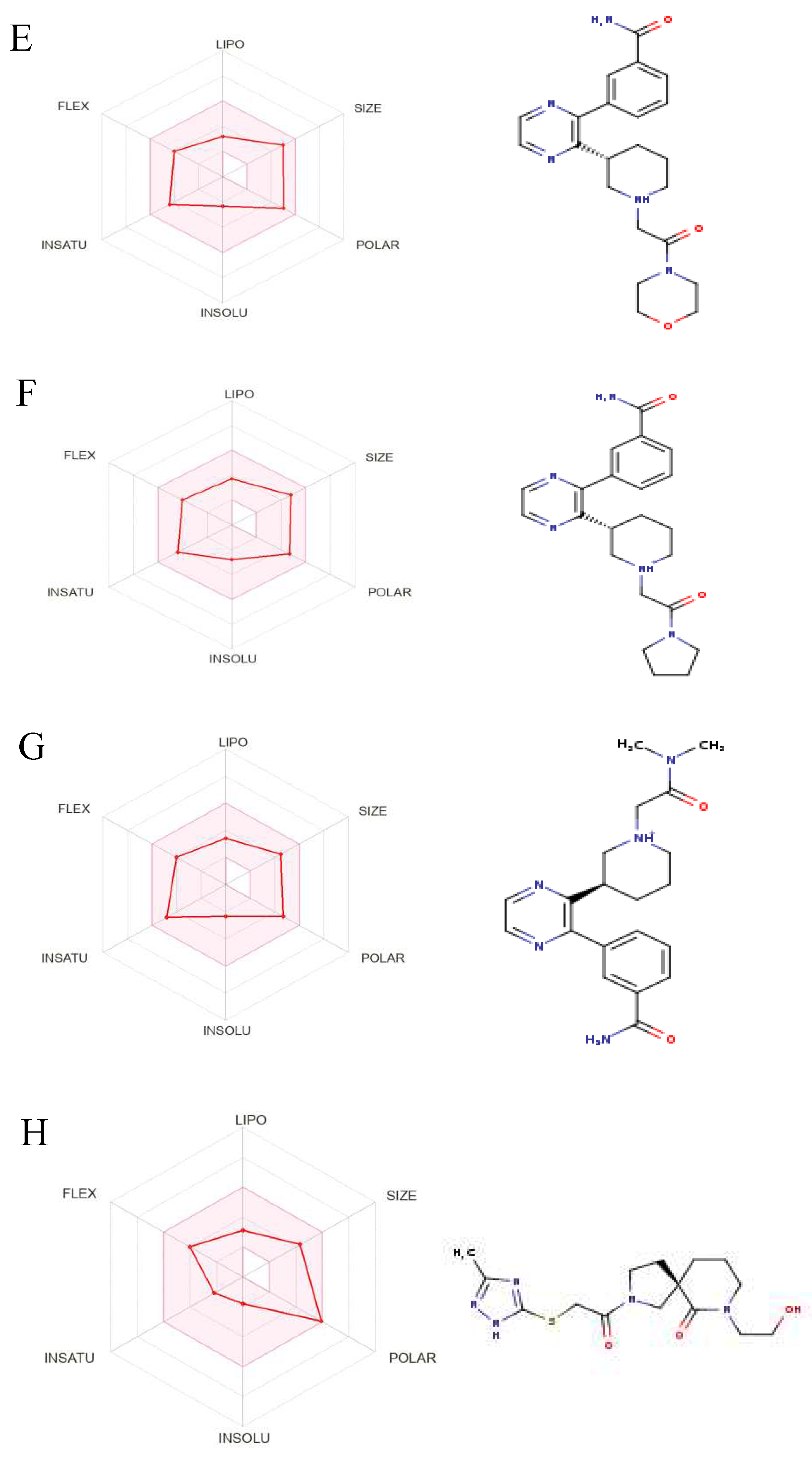
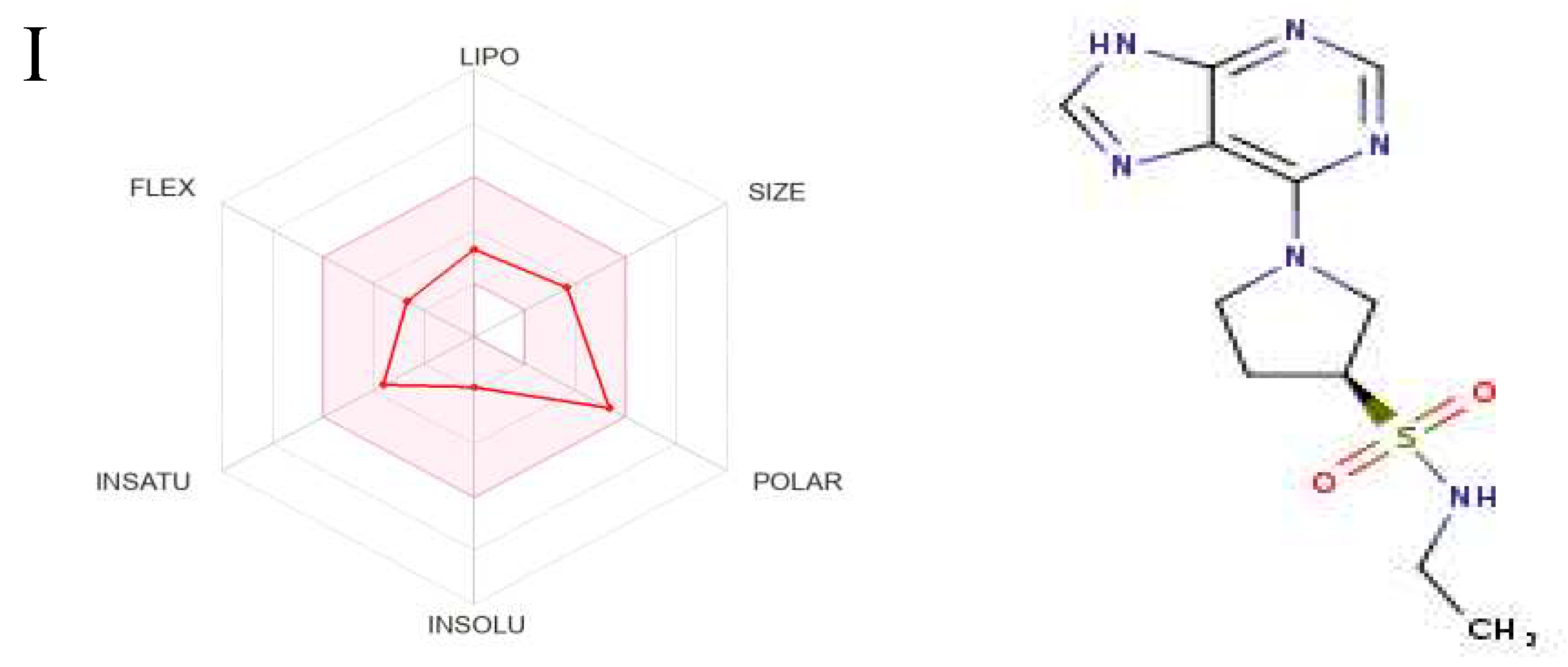
Figure 9.
3D and 2D Interactions of PfHsp90 and the 6 Ligands. (A) PfHsp90 and GDM, with binding affinity of -7.5 kcal/mol. (B) PfHsp90 and ZINC09060002, with binding affinity of -8.2 kcal/mol. (C) PfHsp90 and ZINC72133064, with binding affinity of -7.8 kcal/mol. (D) PfHsp90 and ZINC72163401, with binding affinity of -7.7 kcal/mol. (E) PfHsp90 and ZINC72358537, with binding affinity of -8.1 kcal/mol. (F) PfHsp90 and ZINC72358557, with binding affinity of -7.6 kcal/mol.
Figure 9.
3D and 2D Interactions of PfHsp90 and the 6 Ligands. (A) PfHsp90 and GDM, with binding affinity of -7.5 kcal/mol. (B) PfHsp90 and ZINC09060002, with binding affinity of -8.2 kcal/mol. (C) PfHsp90 and ZINC72133064, with binding affinity of -7.8 kcal/mol. (D) PfHsp90 and ZINC72163401, with binding affinity of -7.7 kcal/mol. (E) PfHsp90 and ZINC72358537, with binding affinity of -8.1 kcal/mol. (F) PfHsp90 and ZINC72358557, with binding affinity of -7.6 kcal/mol.
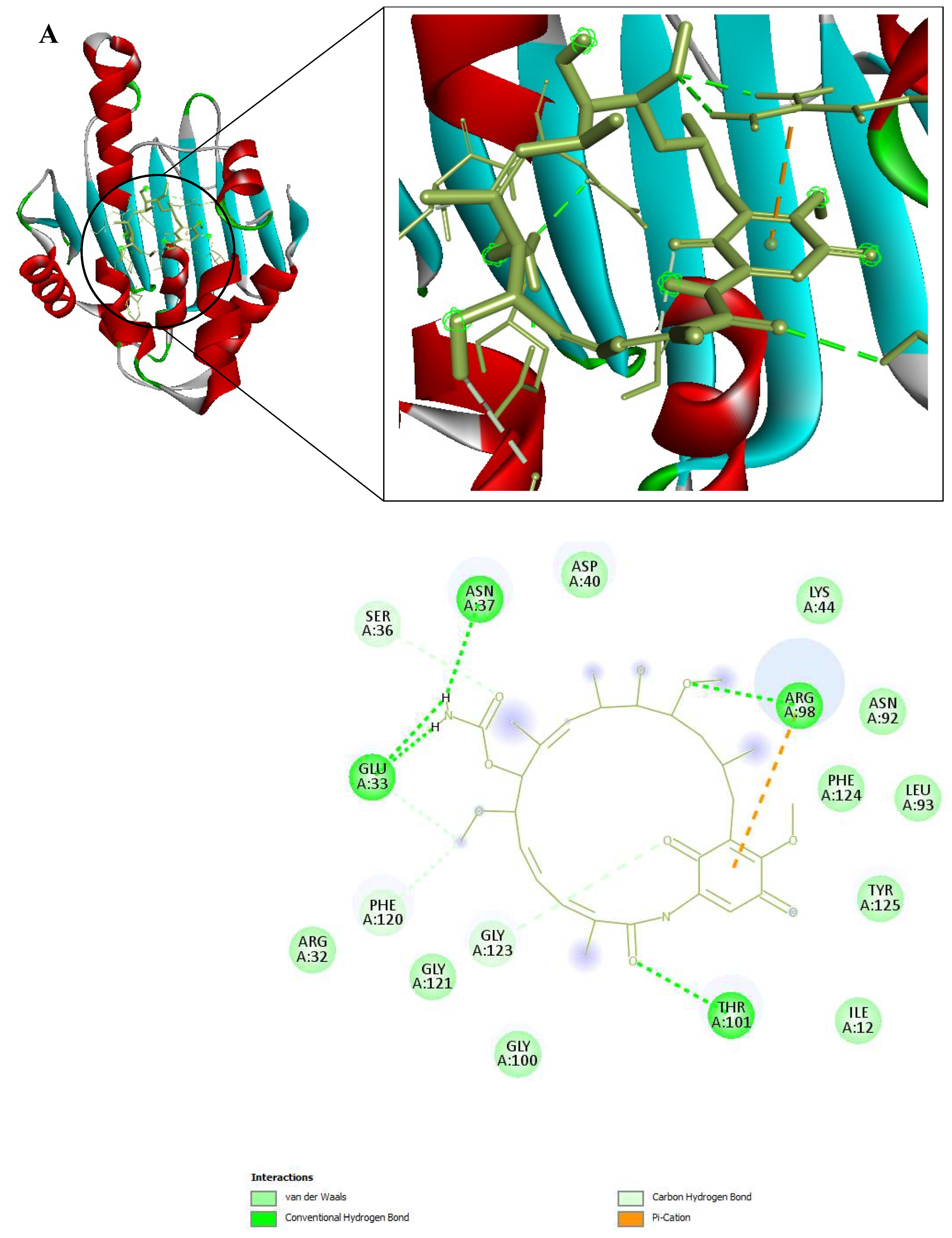
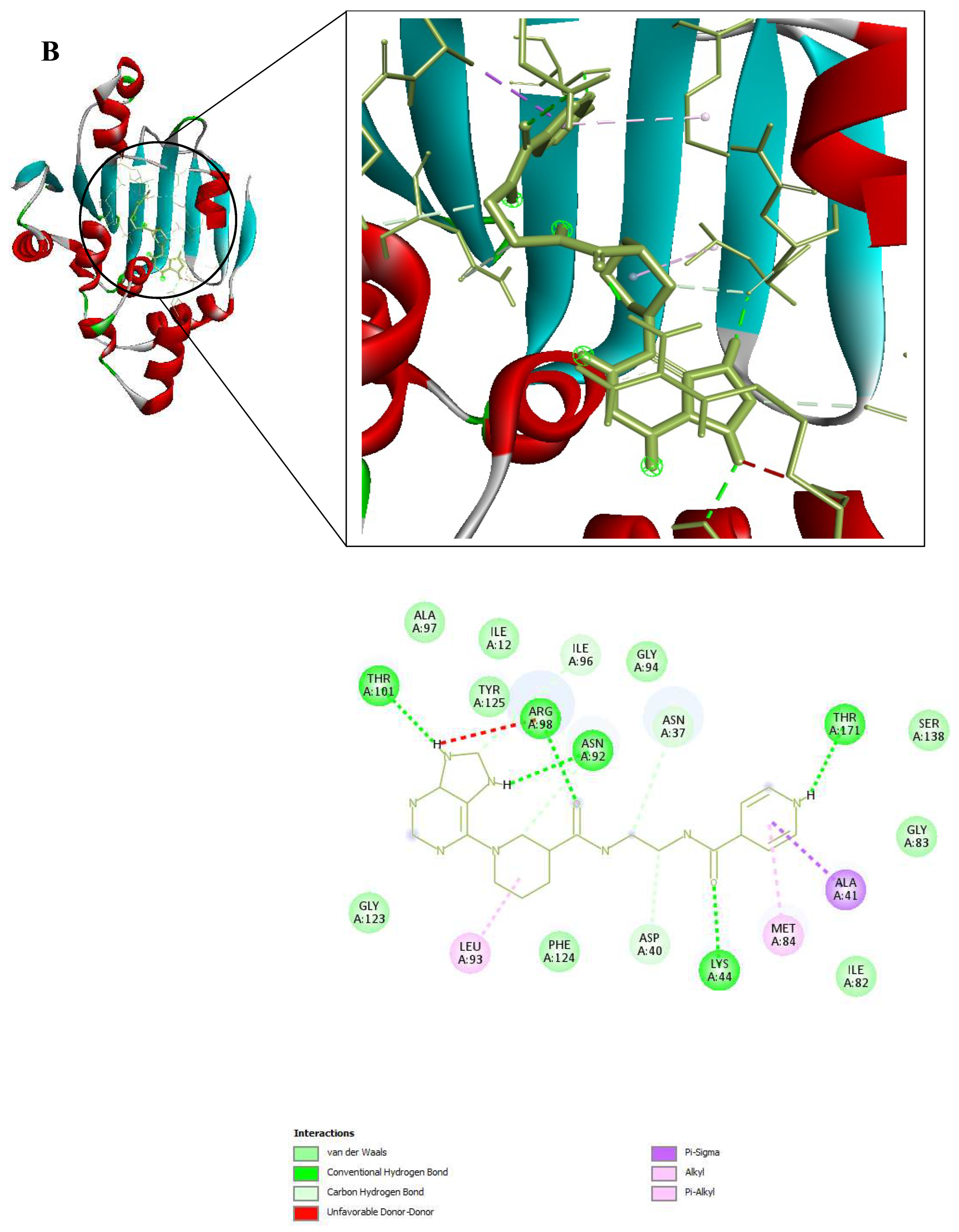
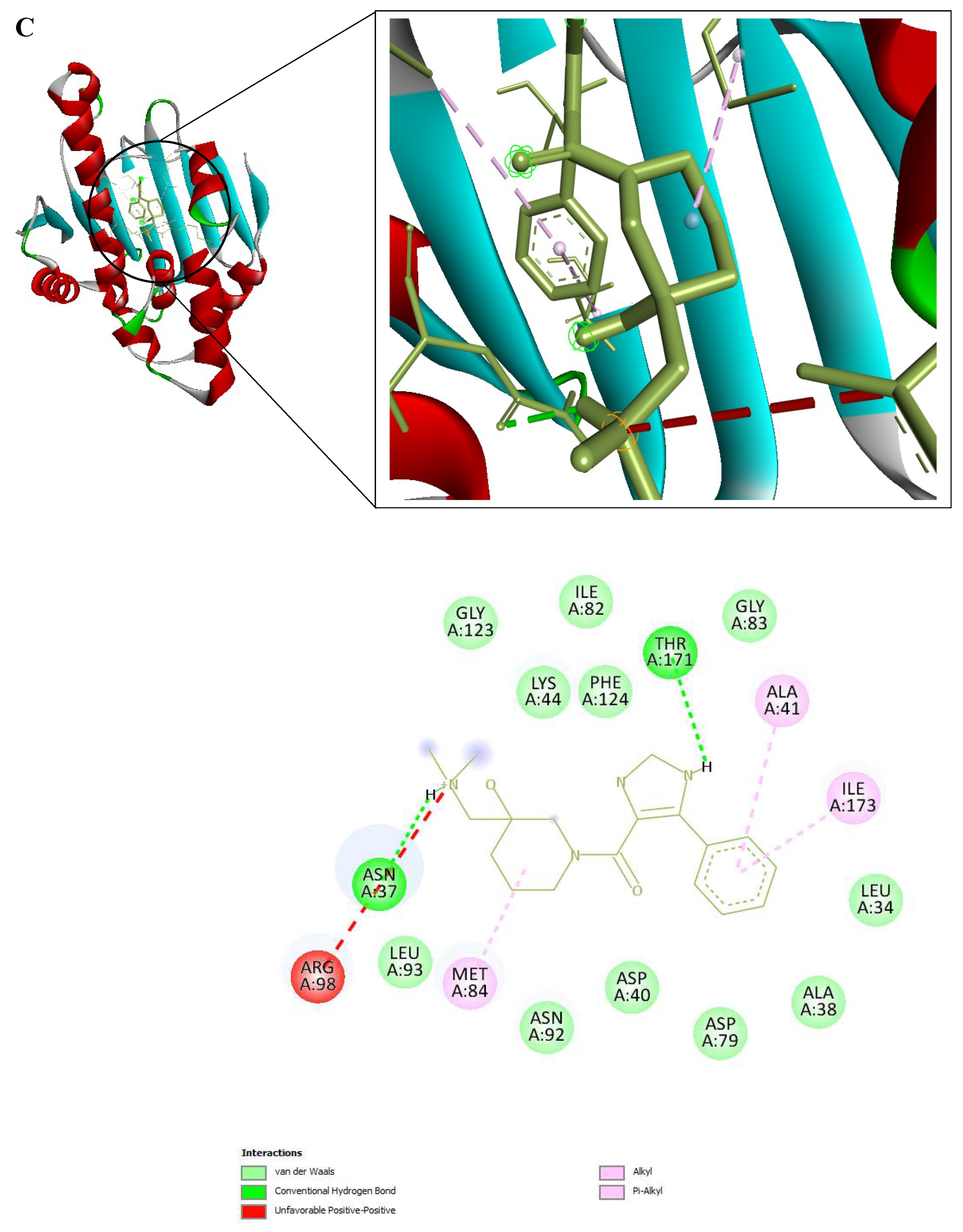
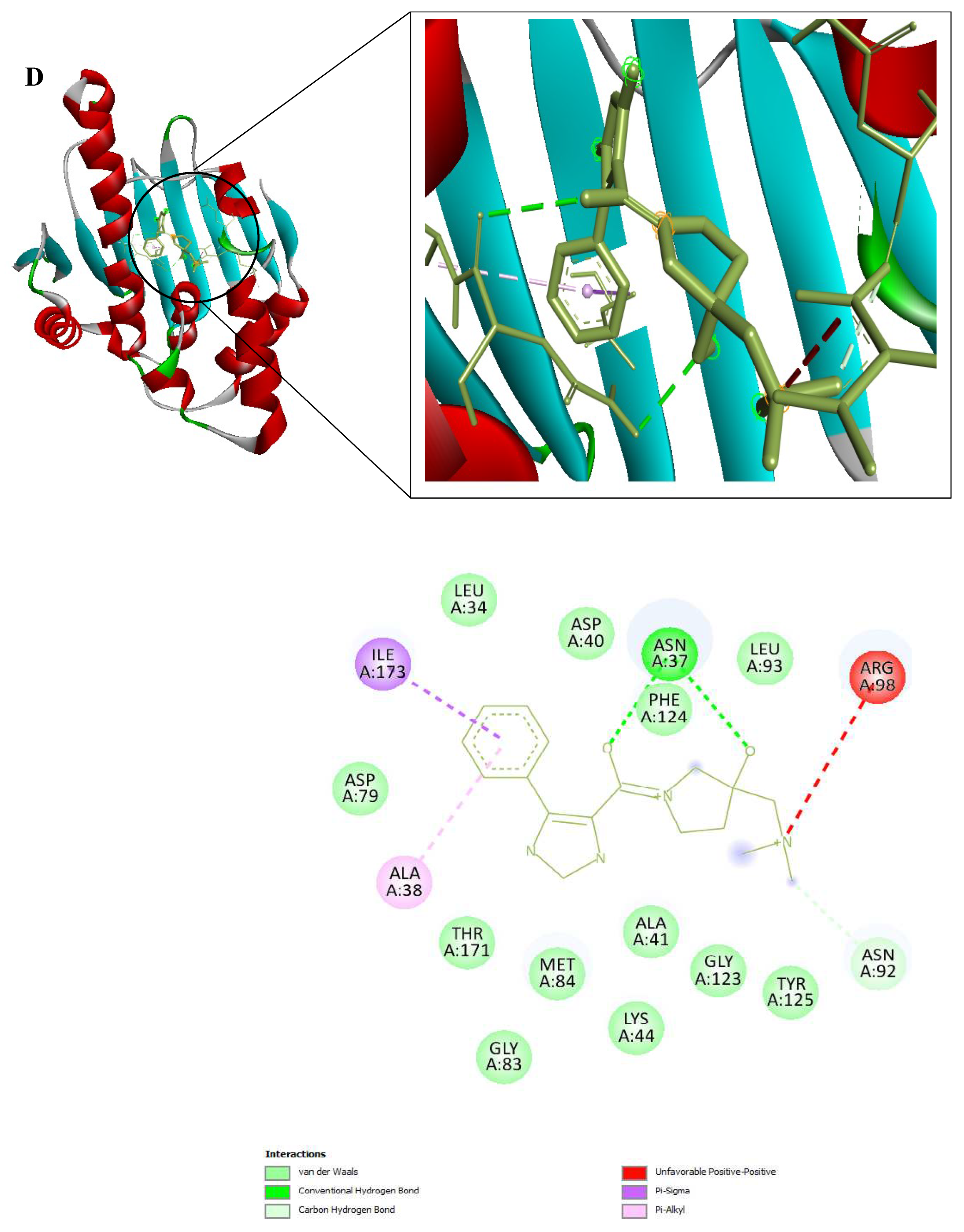

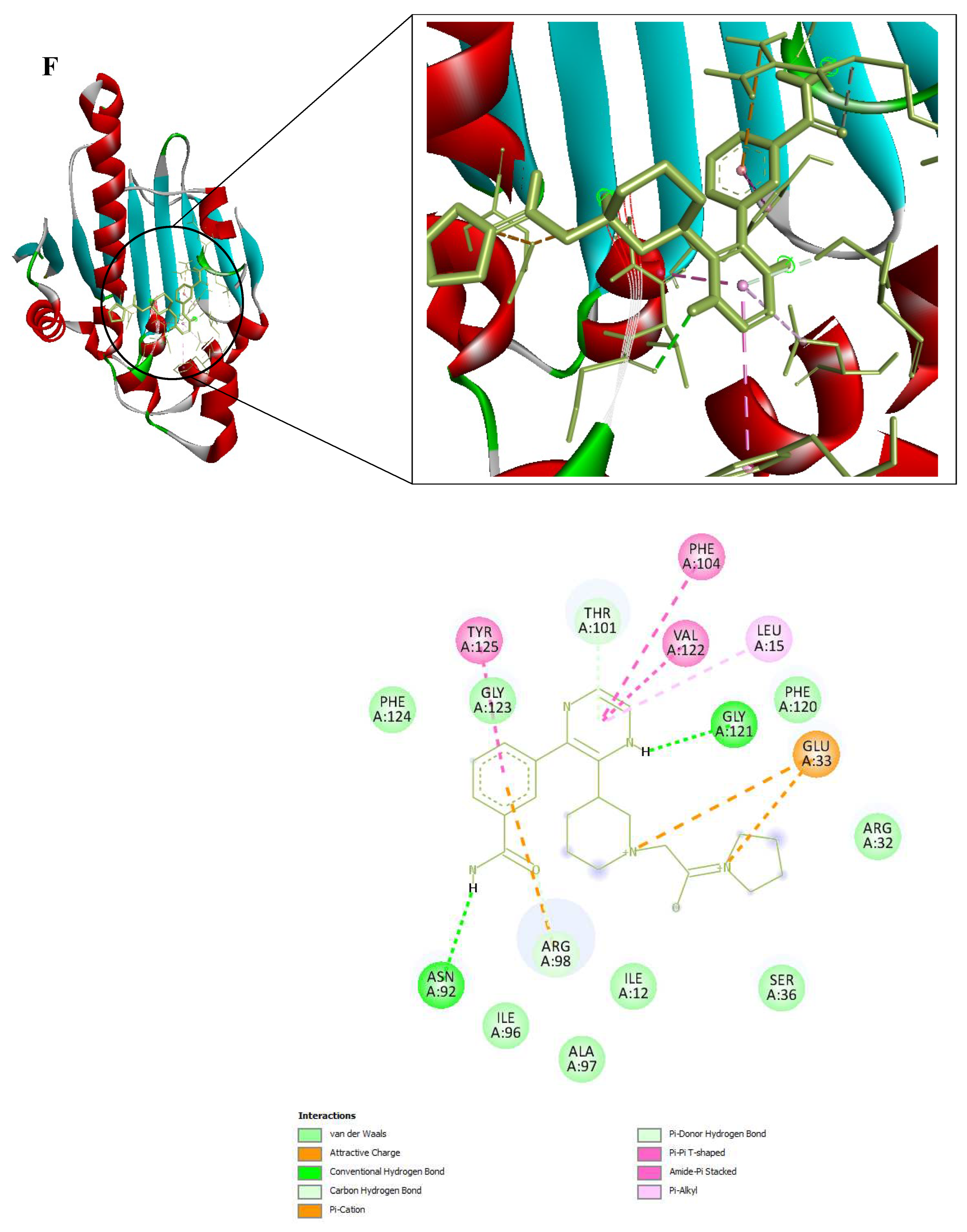
Figure 10.
RMSD plot of PfHsp90. (PDB ID: 3K60) with GDM as reference ligand and top-five ZINC database compounds as a function of 100ns simulation time. ZINC09060002 (Black), ZINC72133064 (Red), ZINC72163401 (Green), ZINC72358537 (Blue), ZINC72358557 ((Yellow), and GDM (Brown).
Figure 10.
RMSD plot of PfHsp90. (PDB ID: 3K60) with GDM as reference ligand and top-five ZINC database compounds as a function of 100ns simulation time. ZINC09060002 (Black), ZINC72133064 (Red), ZINC72163401 (Green), ZINC72358537 (Blue), ZINC72358557 ((Yellow), and GDM (Brown).
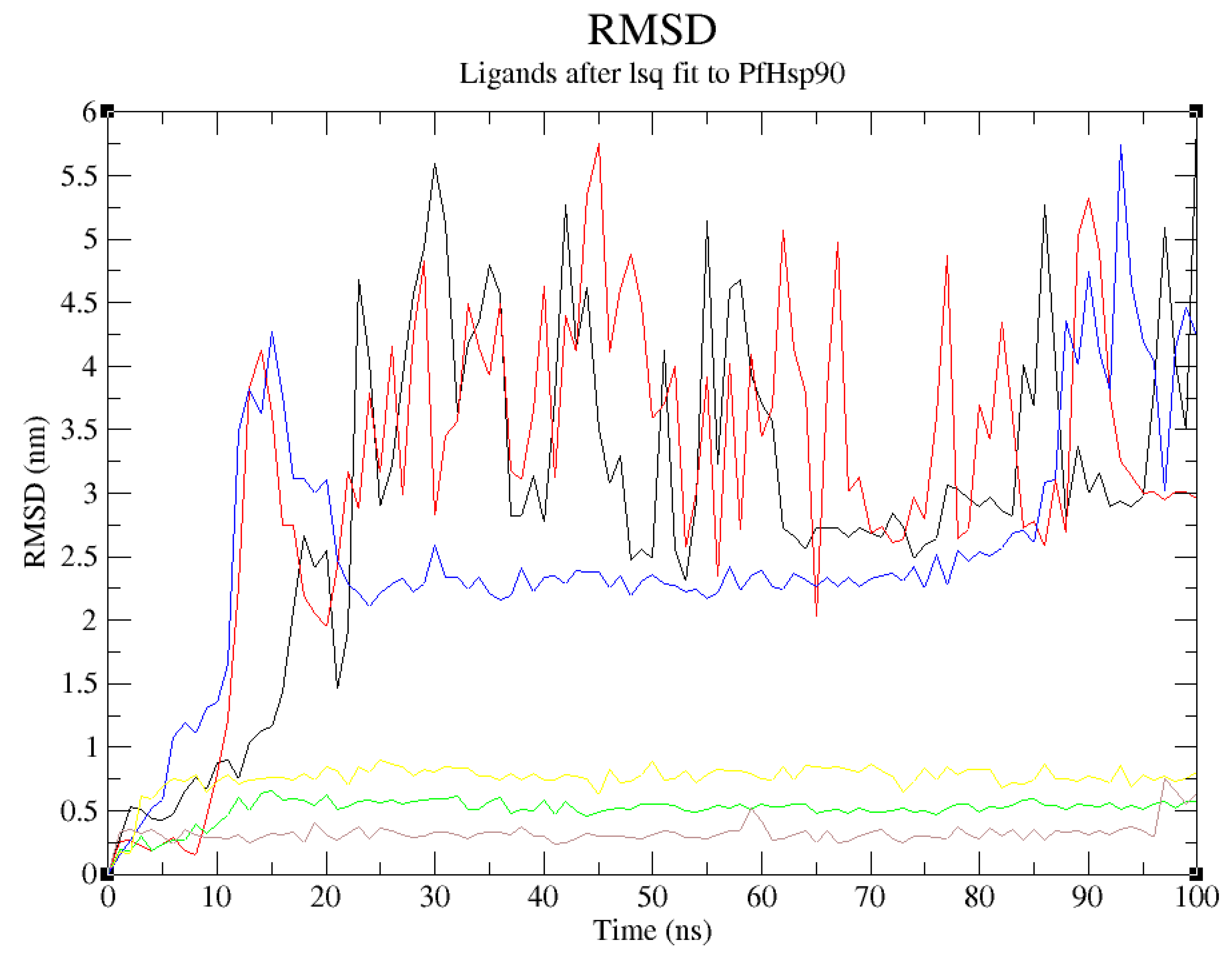
Figure 11.
RMSF plot of GDM as reference ligand and top-five ZINC database compounds. ZINC09060002 (Black), ZINC72133064 (Red), ZINC72163401 (Green), ZINC72358537 (Blue), ZINC72358557 (Yellow), and GDM (Brown).
Figure 11.
RMSF plot of GDM as reference ligand and top-five ZINC database compounds. ZINC09060002 (Black), ZINC72133064 (Red), ZINC72163401 (Green), ZINC72358537 (Blue), ZINC72358557 (Yellow), and GDM (Brown).
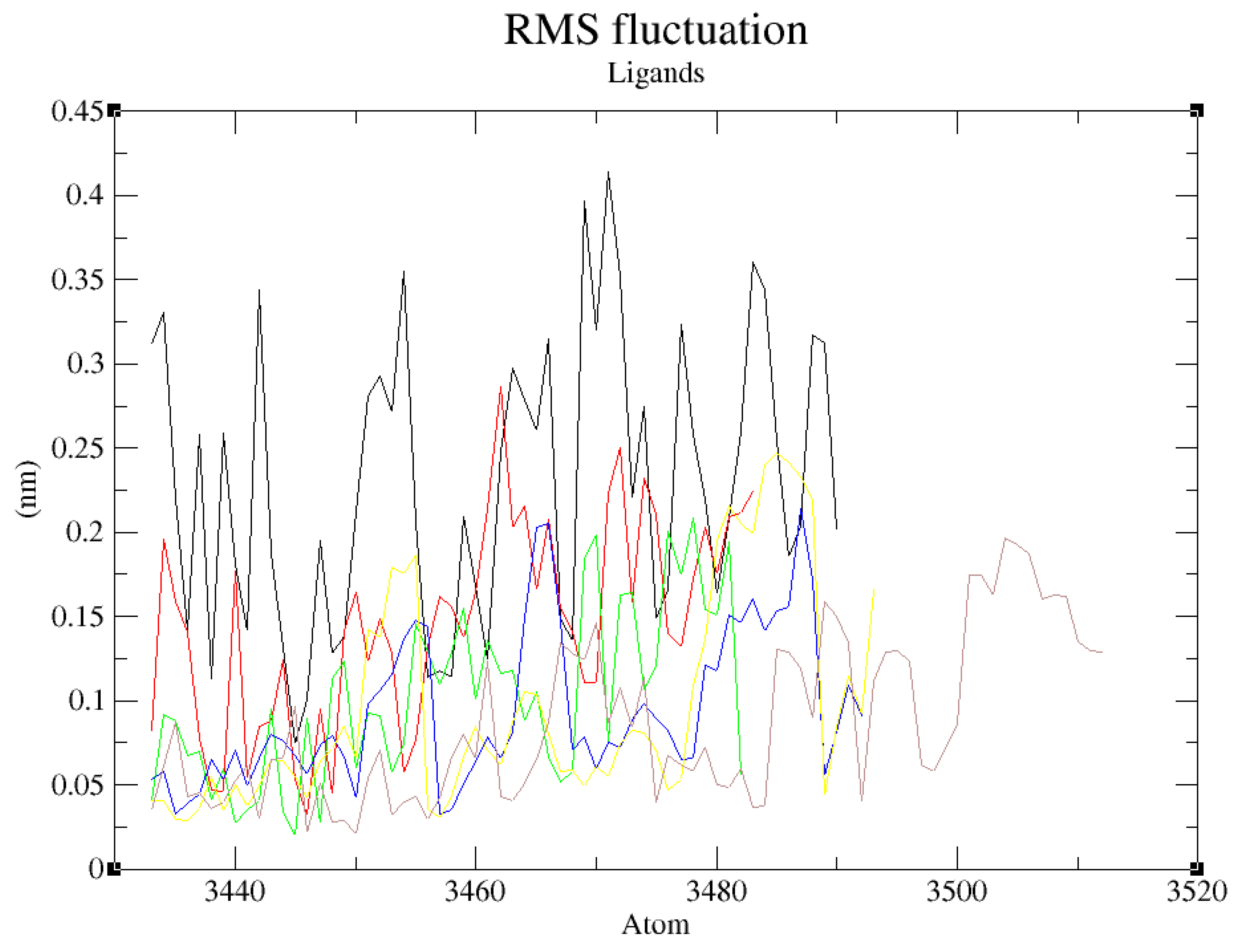
Figure 12.
Number of Hydrogen Bonds Plot.
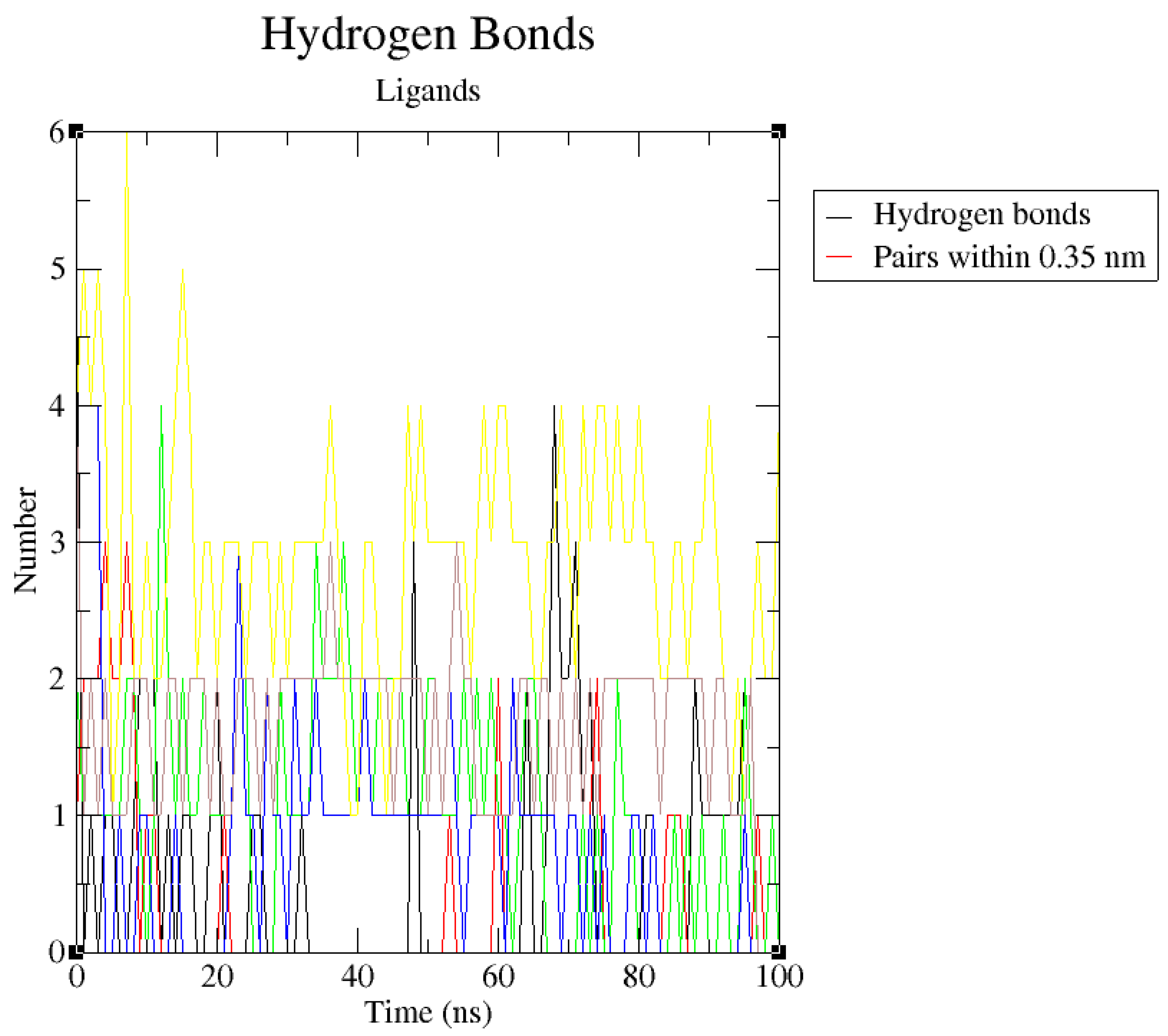
Figure 13.
Point to Point Plot. IC50 value determination of PfHsp90 inhibitors using point-to-point calculation.
Figure 13.
Point to Point Plot. IC50 value determination of PfHsp90 inhibitors using point-to-point calculation.
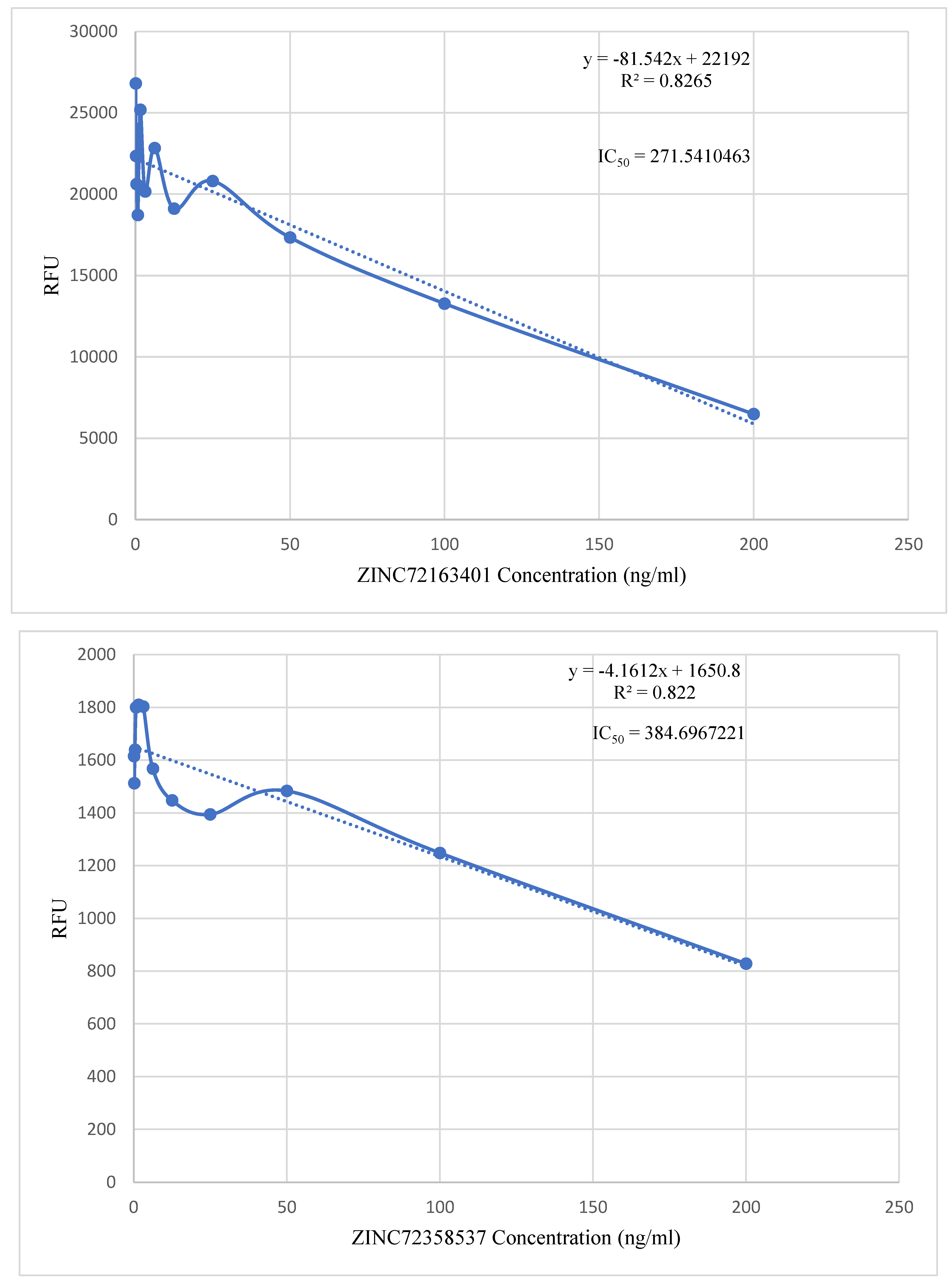
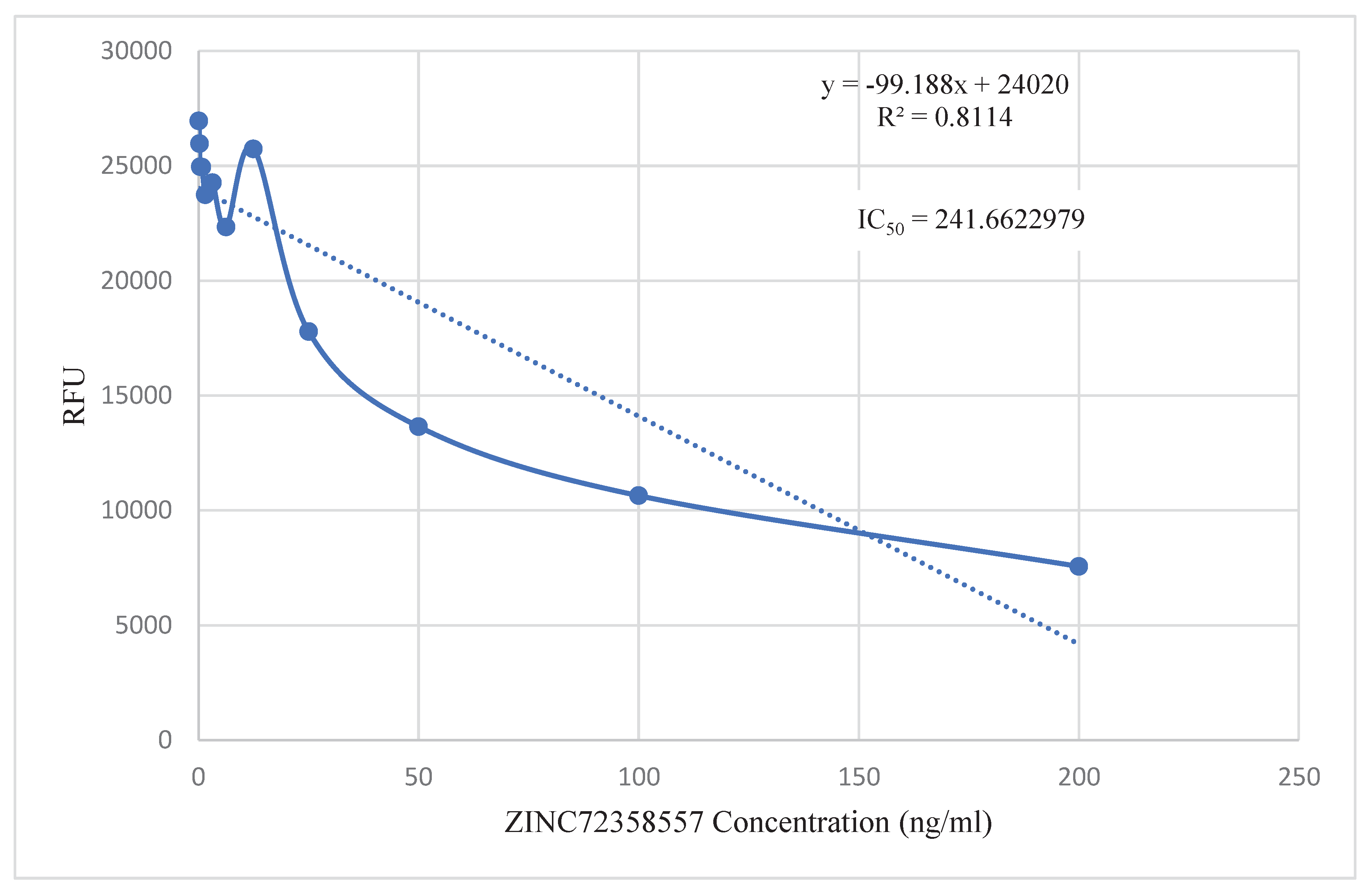

Table 1.
Basic Information on GDM.
| Molecule | Name | PubChem CID | Molecular Formular (MF) | Molecular Weight (MW) |
|---|---|---|---|---|
| 1. | Geldanamycin (GDM) | 5288382 | C29H40N2O9 | 560.6 |
Table 2.
Basic Information on the Virtual Screening Results using GDM as Ligand.
| No. | Molecule | RMSD | Mass | RBnds |
|---|---|---|---|---|
| 1 | ZINC09060002 | 0.363 | 394 | 4 |
| 2 | ZINC32537723 | 0.380 | 305 | 5 |
| 3 | ZINC92700801 | 0.291 | 296 | 5 |
| 4 | ZINC71617232 | 0.257 | 244 | 6 |
| 5 | ZINC63526364 | 0.342 | 332 | 6 |
| 6 | ZINC71617229 | 0.270 | 244 | 6 |
| 7 | ZINC22325332 | 0.352 | 473 | 7 |
| 8 | ZINC72358557 | 0.332 | 394 | 7 |
| 9 | ZINC72358537 | 0.331 | 410 | 7 |
| 10 | ZINC77271253 | 0.240 | 300 | 7 |
| 11 | ZINC72133064 | 0.279 | 329 | 8 |
| 12 | ZINC72163401 | 0.410 | 315 | 8 |
| 13 | ZINC91416974 | 0.373 | 353 | 8 |
| 14 | ZINC72358880 | 0.330 | 368 | 9 |
| 15 | ZINC32796276 | 0.300 | 453 | 10 |
| 16 | ZINC70981147 | 0.245 | 336 | 15 |
| 17 | ZINC70981147 | 0.249 | 336 | 15 |
Table 3.
Drug-Likeness Test Results of the 17 Molecules.
| No. | Molecule | Lipinski | Ghose | Veber | Egan | Muegge | Drug-Like? |
|---|---|---|---|---|---|---|---|
| 1 | ZINC09060002 | Yes | Yes | Yes | Yes | Yes | Yes |
| 2 | ZINC32537723 | Yes | Yes | No | No | No | No |
| 3 | ZINC92700801 | Yes | Yes | Yes | Yes | Yes | Yes |
| 4 | ZINC71617232 | Yes | No | Yes | No | Yes | No |
| 5 | ZINC63526364 | Yes | No | Yes | Yes | Yes | Yes |
| 6 | ZINC71617229 | Yes | No | Yes | No | Yes | No |
| 7 | ZINC22325332 | Yes | No | No | No | Yes | No |
| 8 | ZINC72358557 | Yes | Yes | Yes | Yes | Yes | Yes |
| 9 | ZINC72358537 | Yes | No | Yes | Yes | Yes | Yes |
| 10 | ZINC77271253 | Yes | No | Yes | No | Yes | No |
| 11 | ZINC72133064 | Yes | Yes | Yes | Yes | Yes | Yes |
| 12 | ZINC72163401 | Yes | No | Yes | Yes | Yes | Yes |
| 13 | ZINC91416974 | Yes | No | Yes | Yes | Yes | Yes |
| 14 | ZINC72358880 | Yes | Yes | Yes | Yes | Yes | Yes |
| 15 | ZINC32796276 | Yes | Yes | No | No | Yes | No |
| 16 | ZINC70981147 | Yes | No | Yes | Yes | Yes | Yes |
| 17 | ZINC70981147 | Yes | No | Yes | Yes | Yes | Yes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



