
Submitted:
26 January 2024
Posted:
26 January 2024
You are already at the latest version
Abstract
PitNETs, although typically benign, comprise a small percentage of therapy-resistant aggressive, occasionally metastatic tumors, underpinning a need to identify novel therapeutic targets. PitNETs carry few mutations but associate with conditions that promote alternative splicing as an oncogenic alternative and express the TrkA neurotrophin receptor, which exhibits oncogenic alternative TrkAIII splicing in other tumors. Alternative TrkAIII splicing was assessed by RT-PCR in 53 PitNETs and compared to HIF1a, HIF2a, SF3B1, SRSF2, U2AF1 and JCPyV large T antigen mRNA expression, unconventional Xbp1 splicing, SF3B1 mutation, and TrkA isoform(s) immunoreactivity by confocal immunofluorescence (IF). Alternative TrkAIII mRNA splicing was detected in all invasive PitNET phenotypes, was significantly elevated in invasive compared to non-invasive PIT1 PitNETs, relatively high in invasive and non-invasive SF1 and TPIT PitNETs, and was associated with IF consistent with intracellular TrkAIII activation but not with hotspot SF3B1 mutations, altered SF3B1, SRSF2 and U2AF1 splice factor mRNA expression, JCPyV large T antigen expression or unconventional Xbp1 splicing. A potential role for hypoxia in TrkAIII mRNA splicing was supported by significant elevation of HIF2a mRNA expression in invasive PIT1 PitNETs. These data demonstrate that alternative TrkAIII splicing, potentially promoted by hypoxia, occurs frequently in PitNETs and may associate with IF consistent with intracellular TrkAIII activation, supporting a potential role for TrkAIII in PitNET pathogenesis and progression, identifying TrkAIII as a novel potential target in refractory PitNETs.
Keywords:
PitNETs
; alternative splicing
; TrkAIII splice variant
; HIF2a
; Splice factors SF3B1
; U2AF and SRSF2
; hotspot SF3B1 mutation
; Xbp1
; JCPyV large T antigen
1. Introduction
Pituitary neuroendocrine tumors (PitNETs) originate from cells of the anterior pituitary and are classified by immunohistochemistry according to pituitary transcription factor and hormone expression [1,2,3,4,5]. Three lineages of origin and several morphofunctional phenotypes can be identified: functioning lactotroph, somatotroph and thyrotroph (PIT1-derived), corticotroph (TPIT-derived) as well as gonadotroph (SF1-derived) silent/non-functioning PIT1, SF1 or TPIT sub-types, pluri-hormonal tumors and “null cell” PitNETs [1,2]. PitNETS are recognized as functioning when associated with bio-clinical evidence of hormone hypersecretion. Although typically benign, about 40% of PitNETs are invasive towards surrounding structures and a small percentage, mainly lactotroph and corticotroph PitNETs, progress to aggressive, therapy-resistant/refractory, occasionally metastatic tumors [2,6,7]. Despite the lack of specific molecular markers, aggressive and metastatic PitNETs share a number of bio-clinical features [8] and, according to current guidelines, are both treated first with temozolomide [6,7]. However, primary or secondary resistance is frequent [7], underpinning a need to identify novel therapeutic targets for this particular subgroup, which continues to represent a major therapeutic challenge.
In general, PitNETs are sporadic tumors that carry few oncogenic mutations but exhibit transcriptomic and epigenetic signatures, chromosomal alterations [5,9,10,11], and associate with conditions reported to promote alternative splicing, including hypoxia [12], oxidative stress [13], somatic mutations in the splice factor SF3B1 [14,15,16], de-regulated splicing machinery [17], and neurotropic JCPyV polyomavirus infection [18,19]. Alternative splicing is now considered to be a hallmark of cancer and is frequently involved in oncogene and oncogene signaling pathway activation in tumor types that carry few oncogene mutations [20,21,22]. This raises the possibility that oncogenic alternative splicing may participate in PitNET pathogenesis and progression. Within this context, normal pituitary cells and PitNETs express the neurotrophin receptor tropomyosin-related kinase TrkA [9,23,24]. Although an oncogenic alternative TrkAIII splicing has been documented in neuroblastomas (NBs), MCPyV positive Merkel cell carcinomas, cutaneous malignant melanomas and acute myeloid leukaemia [25,26,27,28,29], this has not yet been studied in PitNETs.
The oncogenic alternative TrkAIII splice variant (GeneBank OP866787.1), originally discovered in human NBs and correlated with post-therapeutic relapse and advanced stage metastatic disease [25,28], is characterized by NTRK1/TrkA exons 6, 7 and 9 skipping. This results in expression of a variant receptor devoid of the extracellular D4 IG-C1 domain and several N-glycosylation sites, required for fully spliced (fs-) TrkA cell surface expression and prevention of ligand-independent activation [30,31,32]. These omissions result in intracellular TrkAIII re-localization to pre-Golgi membranes, centrosomes and mitochondria [33,34,35]. Consistent with an oncogenic function, TrkAIII transforms NIH3T3 cells, and in NB models exhibits ligand-independent cell cycle, stress-regulated and drug-induced intracellular activation [25,35,36] and promotes primary and metastatic tumorigenicity in a manner similar to TrkT3 TrkA-fusion oncogene [25], confirming oncogenic equivalence to TrkA-fusion and engineered D4 domain-deleted TrkA oncogenes [31,37,38]. In NB cells, alternative TrkAIII splicing is promoted by hypoxia, by agents that cause ER, Ca2+, redox and nutrient stresses, and by SV40 polyomavirus small t-antigen [25,31,36]. Intracellular, ligand-independent TrkAIII activation induces pro-survival IP3K/Akt signaling, increases Bcl-2, Mcl-1 and SOD2 expression, promotes a pro-angiogenic MMP-9/VEGF/Tsp1 expression equilibrium, causes centrosome amplification, induces stress-regulated metabolic adaptation, modifies the unfolded protein response (UPR) and promotes a more stem cell-like, anaplastic NB phenotype [25,30].
In this study, therefore, considering that PitNETs exhibit low oncogene mutation rates, associate with conditions that may promote alternative splicing and often express TrkA, we assessed whether alternative TrkAIII splicing and expression may represent a potentially targetable oncogenic participant in PitNET pathogenesis and progression.
2. Materials and Methods
2.1. Patients and Tumors
Fifty-three patients affected by PitNETs were operated on for medical reasons at the Neuromed Institute (Pozzili, Italy). All patients were characterized before surgery for bio-clinical evidence of hormone hypersecretion and by Magnetic nuclear Imaging (MRI) for macroscopic tumor characteristics. Twenty-four patients were clinically diagnosed with functioning PitNETs (6 prolactinomas, 13 acromegaly, 2 central hyperthyroidism and 4 Cushing’s disease) and the remaining 29 with clinically non-functioning tumors. With one exception, a young female with a microprolactinoma, all other patients had macro-tumors (maximal diameter > 10 mm). Tumor invasiveness towards surrounding structures (cavernous sinus/sphenoid sinus/bone/dura) was defined according to pre-operative MRI and intra-operative findings. Overall, 26/53 PitNETs were invasive (49%), and among 6 reccurrent tumors, 4 were invasive of which 2 were aggressive and 1 metastatic. Tumors were classified for routine pathological diagnosis by immunohistochemistry (IHC) using an automatic VENTANA Benchmark ultra XT IHC/ISH System, as directed (Roche Diagnostics Int.; Rotkeuz, Switzerland), using antibodies against pituitary hormones and transcription factors, and Ki67 and a VENTANA Ultraview DAB detection kit, as directed (Roche Diagnostics Int.; Rotkeuz, Switzerland), according to European Pituitary Pathology Group proposals [39].
The PitNETs in this study were then classified according to their lineage of origin as: 24 PIT1, 24 SF1, and 5 TPIT-positive tumors, and patient’s and PitNET details are provided in Table 1. For molecular studies, surgically removed PitNET tissues were immediately placed in RNAlaterTM nucleic acid stabilizing solution, as directed by the manufacturer (Ambion®, Life Technologies, Monza, Italy), and frozen at -80°C, prior to nucleic acid purification. In some cases, slide mounted 4mm FFPE PitNET tissue sections were also provided for confocal immunofluorescence analysis. This study was approved by the Neuromed Institute Internal Review Board (Biopit 270423) and performed according to Helsinki declarations. Written informed consent was obtained from patients, with the exception of a minority of archived RNAs from patients lost to follow-up.
2.2. Antibodies and Reagents
Mouse monoclonal anti-human TrkA carboxyl-terminus (cod. B3, 200 µg/mL) antibody was from Santa Cruz Biotechnology (Dallas, TX, USA) and recognises both fs-TrkA and TrkAIII [25,26,27]. Rabbit monoclonal anti-human Y490-phosphorylated TrkA antibody (cod. 9141; 36 µg/mL) was from Cell Signalling Technology (Danvers, MA, USA) and recognises both phosphorylated fs-TrkA and TrkAIII [22,23,24]. Secondary Alexa Flour 488-labeled donkey anti-rabbit and Alexa Fluor donkey anti-mouse antibodies were from Life Technologies (1 mg/mL) (Fortis, Waltham, MA, USA). ProlongTM Gold anti-fade reagent with DAPI was from Invitrogen (Thermo-Fisher Scientific, Waltham, MA, USA).
2.3. RNA Extraction and Reverse Transcriptase Polymerase Chain Reaction
Total RNAs were extracted from tissues using Trizol, according to the manufacturer’s instructions (Life Technologies, Monza, Italy). Briefly, tumor tissues were homogenized in 1 mL of Trizol and resulting supernatants were mixed with chloroform and centrifuged to obtain phase separation. The upper phase was recovered and washed in isopropanol, RNAs were then precipitated in 75% ethanol, centrifuged at 14,000 x g in an eppendorf microfuge at 4°C, and RNA pellets resuspended in 20ml of RNase/DNAse-free water. RNA purity and concentrations were evaluated in a nanodrop spectrophotometer, as directed (Thermo Fisher Scientific, CA, USA). Purified RNAs were reverse-transcribed using a Wonder RT transcription kit, as directed (Euroclone, Pero, Italy), and reverse transcription reactions, at various dilutions, were subjected to RT-PCR, using the primers and conditions detailed in Table 2. All RT-PCRs were performed in duplicate and repeated at least 2 times. For densitometric analysis, 1.5% agarose gels were digitally photographed and images analysed by Image J software (ImageJ bundled with Java 1.8.0_172), with inter-gel comparisons performed using common 18S rRNA RT-PCR product and DNA ladder standards, where appropriate.
2.4. Tumor DNA purification
Tumor DNA (tDNA) was extracted from 8 PRL PitNETs using Quick-DNA Miniprep Plus Kit, as directed (Zymo Research). DNA quality was checked by 0.8% agarose gel electrophoresis and PCR amplification for the house keeping gene GAPDH [40].
2.5. DNA Sequencing
For DNA sequencing, TrkA exon 1-8, TrkA exon 8-17 and SF3B1 RT-PCR products (cDNA and tDNA) were purified from ethidium bromide-stained agarose gels, using a Jet Quick gel extraction spin kit, as directed (Genomed, Harrow, UK), cleaned using a EuroSAP PCR enzymatic Clean-Up kit, as directed (Euroclone, Milan, Italy), and PCR amplified using the primers detailed in Table 2, and BigDye Terminator V.2.1. Cycle Sequencing kit, as directed (Thermo-Fisher Scientific, CA, Cells 2023, 12, 237 5 of 21 USA). Re-amplified products were sequenced by double-stranded Sanger sequencing, in a mono-capillary DNA sequencer (Genetic Analyzer 3500, Thermo-Fischer Scientific, CA, USA).
2.6. Indirect IF
FFPE sections (4 µm) were de-paraffinized, re-hydrated and processed for antigen retrieval by incubation in 0.01 M sodium citrate buffer (pH 6.0) for 20 min at 98 ◦C. Sections were blocked in blocking solution (1x PBS, 5% BSA, 0.1% Tx100), incubated overnight at 4°C with mouse monoclonal anti-human TrkA (B3, 1:100 dilution in 1 x PBS, 1% BSA, 0.1% Tx100) and rabbit monoclonal anti-human Y490-phopsphorylated TrkA (pY490-TrkA, 1:100 dilution, in 1 x PBS, 1% BSA, 0.1% Tx100) primary antibodies, washed extensively in PBS, and then incubated with appropriate fluorochrome-conjugated Alexa Fluor secondary antibodies (diluted 1:1000 in 1 x PBS) for 2h at 37°C. Slides were then washed, counterstained with Bisbenzimide nuclear dye (Hoechst, Thermo Fisher Scientific, CA, USA) and images acquired under scanning confocal microscopy (Leica TCS SP5 II).
2.7. Statistical Analysis
Data are expressed as median [range] and were statistically analysed using non-parametric tests: Mann-Whitney U and Kruskall Wallis for comparison of continuous variables between 2 or 3 groups, respectively, and Spearman’s correlation. P values < 0.05 were considered significant.
3. Results
Individual patient’s and tumor data are detailed in
Table 1
and the relative identifying case numbers will be used throughout the Results section. Overall, the majority of PIT1 PitNETs were functioning (21/24), and comprised of 6 functioning and 1 silent lactotroph (PRL), 9 functioning and 1 silent somatotroph (GH), 4 functioning mixed PRL/GH, 2 functioning thyrotroph (TSH) and 1 hormone-negative PIT1 tumors. In this group, 11/24 tumors were invasive (45.8%). In contrast, all SF1/gonadotroph PitNETs were clinically non-functioning, and comprised 18 hormone-positive (FSH and/or LH) and 6 pure SF1 tumors. In this group, 14/24 tumors were invasive (58.3%). The majority of TPIT PitNETs were functioning (4/5), with a single silent ACTH-secreting tumor and 3/5 TPIT PitNETs were invasive. As shown in
Table 1
, the TrkAIII mRNA splice variant was detected in all but 3 PitNETs (n.12, 16 and 39).
3.1. TrkAIII was the only in-frame alternative TrkA splice variant expressed in PitNETs
RT-PCR using primers spanning NTRK1/TrkA exons 1 to 8 detected 3 products in PitNET cDNAs, sequence characterized as fully spliced (fs-)TrkA, exons 6 and 7 skipped TrkAIII and exons 2-7 skipped D2-7TrkA (Figure 1a and b). RT-PCR using primers spanning NTRK1/TrkA exons 8-17 in PitNETs RT-PCR-positive for exon 1-8 TrkAIII, detected single products that were sequence characterized as fs-TrkA exons 8-17 (Figure 1c). TrkAIII and fs-TrkA were the only in-frame splice variant mRNAs expressed in PitNETs, the D2-7TrkA splice variant was sequence characterized as a nonsense mRNA, exhibiting a frame-shift at the novel exon 1/8 splice junction resulting in a premature down-stream stop codon (TGA) at position 1039-1041 (fs-NTRK1/TrkA numeration) (this study) and [26,27].
To better evaluate the percentage ratios of TrkAIII to fs-TrkA mRNA expression, specific primers spanning NTRK1/TrkA exons 5 to 8 were employed to generate smaller 452 bp fs-TrkA and 176 bp TrkAIII amplicons. This primer set detected TrkAIII mRNA in all invasive PitNETs, regardless of lineage, and also in the majority (85.7%) of non-invasive PitNETs (10/13 PIT1, 12/13 SF1 and 2/2 TPIT). However, it is noteworthy that exclusive TrkAIII mRNA expression was detected in 6 invasive PitNETs (2/11 PIT1, 1/12 SF1 and 3/3 TPIT) and in 1 of the 2 non-invasive TPIT PitNETs, whereas exclusive fs-TrkA mRNA expression was detected in 3 non-invasive PIT1 and 1 non-invasive SF1 PitNETs but not in any invasive or TPIT PitNET.
Relative levels of TrkAIII and fs-TrkA mRNA expression in different PitNET subgroups were evaluated as percentage densitometric RT-PCR product ratios in each individual PitNET, with exclusive TrkAIII mRNA expression indicated as 100% (Figure 2). Overall, as a densitometric percentage of total TrkAIII and fs-TrkA RT-PCR products, TrkAIII ranged from 21.7% to 100% in invasive and from 0% to 91.8% in non-invasive PitNETs, and was not significantly different between combined invasive PIT1, SF1 and TPIT PitNETs compared to non-invasive counterparts (median 61.1% versus 43%, p = 0.168). When separated according to lineage, however, TrkAIII to fs-TrkA expression ratios in invasive PIT1 PitNETs, which ranged from 34.4% to 100% (median 60.2%), was significantly higher (P = 0.035) than in non-invasive counterparts, which ranged from 0% to 59% (median 32.3%). In contrast, invasive and non-invasive SF1 PitNETs exhibited similar densitometric TrkAIII to fs-TrkA ratios (median 52.1% vs 49.2%) which was not significantly different (p = 0.238). Although TrkAIII as a densitometric ratio to fs-TrkA was remarkably high in all TPIT PitNETs, this group was too small for statistical comparison of invasive and non-invasive tumors. Statistical analysis also confirmed a significant difference in TrkAIII to fs-TrkA RT-PCR densitometric ratios in PitNETs according to their lineage of origin (PIT1, SP1 and TPIT; p = 0.007). It is noteworthy that exclusive TrkAIII mRNA expression characterized aggressive invasive PIT1 PitNET (case n.1), whereas a fs-TrkA to TrkAIII expression ratio of 65 to 35% characterized the invasive metastatic immunotherapy responsive Pit1 PitNET [41] (case n.11).
3.2. TrkAIII mRNA expression associates with IF immunoreactivity consistent with intracellular TrkAIII activation.
Due to limited tissue availability, immunoreactivity for phosphorylated and non-phosphorylated TrkA isoforms was assessed in a representative subgroup of 7 PitNETs including invasive (n. 25,30,37,49) and non-invasive (n. 17, 41,53) samples. As demonstrated in Figure 3, antibodies that recognize both fs-TrkA and TrkAIII [21,22,23], detected overlapping immunoreactivity for TrkA and phosphorylated TrkA isoform(s) in all 3 invasive SF1 PitNETs, exhibiting either predominant fs-TrkA mRNA expression (n.25 and 30) or approximately equal fs-TrkA and TrkAIII mRNA expression (n.37). In contrast, little evidence of overlapping TrkA and phosphorylated TrkA immunoreactivity characterized non-invasive SF1 (n.41) and PIT1 (n.17) PitNET samples, despite detection of similar levels of fs-TrkA and TrkAIII or predominant fs-TrkA mRNA expression, respectively. In TPIT samples, overlapping TrkA and phosphorylated TrkA isoform(s) immunoreactivity characterized both invasive (n. 49) and non-invasive (n.53) cases, in association with exclusive TrkAIII and similar levels of TrkAIII and fs-TrkA mRNA expression, respectively. Both of these TPIT PitNETs displayed the highest levels of TrkA isoform(s) phosphorylation. Overall, these data demonstrate that immunoreactivity consistent with expression and phosphorylation of intracellular TrkA isoform(s), and TrkAIII in particular, characterizes a subset of PitNETs, in association with exclusive and predominant TrkAIII mRNA expression. This is particularly evident in invasive and in TPIT PitNETs.
3.3. Enhanced alternative TrkAIII splicing in invasive PIT1 PitNETs associates with increased HIF2a mRNA expression
Potential involvement of hypoxia in PitNET alternative TrkAIII mRNA splicing was assessed by comparing HIF2a and HIF1a mRNA expression in 50 PitNETs, for which sufficient RNA was available. As demonstrated in Figure 4, densitometric RT-PCR analysis detected significantly higher HIF2a mRNA expression in combined invasive compared to non-invasive PIT1, SF1 and TPIT PitNETs (p = 0.0028). When grouped according to lineage significantly higher HIF-2a mRNA expression characterized invasive compared to non-invasive PIT1 PitNETs (p = 0.0198) but not SF-1 PitNETs (p = 0.238). HIF2a expression also appeared to be higher in invasive compared to non-invasive TPIT PitNETs but were too few for statistical analysis. In comparisons of the 3 PIT1, SF1 and TPIT lineage groups, HIF2a mRNA expression in combined invasive and non-invasive PitNETs did not exhibit statistical significance (p = 0.849). In contrast, differences in HIF1a mRNA expression in combined invasive compared to non-invasive PIT1, SF1 and TPIT PitNETs were not significant (p = 0.168) nor was HIF1a mRNA expression significantly different between invasive and non-invasive PitNETs when grouped into PIT1 or SF1 lineages (p = 0.78 and p = 0.15, respectively). HIF1a mRNA expression was also similar in invasive and non-invasive TPIT PitNETs, which were too few for statistical analysis (Figure 4A and 4B). In analyses of correlation between HIF2a mRNA expression and percentage TrkAIII to fs-TrkA mRNA expression ratios in individual invasive and non-invasive PitNETs combined or grouped according to PIT1 or SF-1 lineages failed to provide statistical significance for a direct correlation. Therefore, in addition to higher levels of TrkAIII splicing, indicated by TrkAIII/fs-TrkA densitometric ratios, invasive PitNETs were also associated with significantly higher levels of HIF2a mRNA expression. However, no significant quantitative correlation was detected between these two variables.
3.3. Alternative TrkAIII splicing in PitNETs does not associate with hotspot SF3B1 mutations or de-regulated SF3B1, SRSF2, U2AF1 expression.
Considering reports that lactotroph PitNETs associate with somatic hotspot SF3B1 mutations [11,12,13], hotspot SF3B1 mutations: c.1866 G > T, c. 1873 C > T, c. 1874 G > A, c.1986 C > G, c.1996 A > C, c.2098 A > G (cDNA/tDNA) and c.2225 G > A (cDNA only) were searched for in tumor DNAs (tDNA) from 6 and cDNAs from 22 lactotroph PitNETs exhibiting TrkAIII mRNA expression. None of these hotspot SF3B1 mutations were detected in any of the tDNAs or cDNAs analyzed (Figure 5).
In addition, SF3B1, U2AF1 and SRSF2 splice factor mRNA expression was also evaluated by densitometric RT-PCR in PitNET samples, but failed to detect significant differences in either SF3B1, U2AF1 or SRSF2 expression between invasive and non-invasive combined PIT1, SF1 and TPIT PitNETs or in PitNETs grouped according to PIT1 and SF-1 lineage, and did not correlate with the significantly enhanced alternative TrkAIII splicing in invasive compared to non-invasive PIT1 PitNETs (Figure 6).
3.4. PitNET alternative TrkAIII splicing does not associate with unconventional Xbp1 splicing or JCPyV large T antigen mRNA expression
Because UPR activators promote alternative TrkAIII splicing [25,30,33,36], RT-PCR analysis of unconventional Xbp1 splicing was evaluated as an index of UPR activation [42]. Unconventional Xbp1 splicing was detected in DTT-treated but not untreated SH-SY5Y controls but was not detected in any of the 50 PitNET cDNAs analyzed, representative examples of which are displayed in Figure 7.
In addition, the potential role of viral infection in promoting alternative TrkAIII splicing was also assessed, considering that JCPyV polyomavirus infection has been implicated in PitNET pathogenesis [18,19]. RT-PCR analysis failed to detect JCPyV large T-antigen mRNA expression in 45 PitNETs analyzed in this study (data not shown), suggesting that JCPyV involvement in PitNET alternative TrkAIII splicing is unlikely.
4. Discussion
In this study, we report that alternative TrkA splicing, restricted to NTRK1/TrkA exons 1 to 8, is a very frequent event in PitNETs. PitNETs were found to express three alternative TrkA splice variants fs-TrkA, TrkAIII and D2-8TrkA, of which TrkAIII was the only potentially oncogenic in-frame tyrosine kinase-domain-encoding alternative mRNA to fs-TrkA. Overall, alternative TrkAIII splicing was significantly elevated in invasive compared to non-invasive PitNETs, regardless of their lineage of origin, but was not restricted to invasive cases. Indeed, TrkAIII splicing was also pronounced in non-invasive SF1 and TPIT PitNETs. The restriction of alternative TrkA splicing in PitNETs to NTRK1/TrkA exons 1-8, extends previous reports of NTRK1/TrkA exons 1-8-restricted alternative TrkA splicing in NBs, MCPyV positive Merkel call carcinomas and cutaneous malignant melanomas [25,26,27,28], and confirms that NTRK1/TrkA exons 2 to 7 are more susceptible to alterative splicing. PitNETs, however, in contrast to MCPyV positive Merkel cell carcinomas and cutaneous malignant melanomas that express several alternative exon 2-7 TrkA splice variants [26,27], expressed only TrkAIII and D2-7TrkA splice variants, of which TrkAIII mRNA was unique in encoding an in-frame tyrosine kinase-domain. Furthermore, PitNETs were unique amongst these tumor types in exhibiting frequent predominant TrkAIII over fs-TrkA mRNA expression and occasionally expressed TrkAIII in the absence of fs-TrkA mRNA.
A role for alternative TrkA III splicing in PitNETs in different stages of pathogenesis and progression was supported by TrkAIII mRNA detection in both invasive and non-invasive PitNETs of all lineages, and the association with intracellular phosphorylated TrkA isoform(s) immunoreactivity in a subset of tumors exhibiting predominant or exclusive TrkAIII mRNA expression, consistent with intracellular TrkAIII activation. An association between alternative TrkAIII splicing and tumor invasiveness was also supported by a predominance of TrkAIII over fs-TrkA mRNA expression in many invasive PitNETs, with exclusive fs-TrkA or exclusive TrkAIII mRNA expression restricted to a subset of non-invasive or invasive cases, respectively. Expressed as a ratio to fs-TrkA mRNA, TrkAIII mRNA was also significantly elevated in invasive compared to non-invasive PIT1 PitNETs, although this appeared to depend on the lineage of origin and was significant only in PIT1 tumors. In the SF1 subgroup, no significant difference was found in TrkAIII splicing between invasive and non-invasive cases, and immunoreactivity consistent with intracellular TrkAIII activation was detected in the presence of TrkAIII mRNA expression, regardless of invasive characteristics. The TPIT subgroup of PitNETs was too small to compare invasive and non-invasive cases, but TrkAIII mRNA was detected in all samples, was exclusive in the invasive cases, and associated with the strongest evidence of intracellular TrkA isoform(s) phosphorylation. Such findings may register within the pathogenic differences reported in PitNETs according to their cellular origin [5,10,43]. It is noteworthy also that fs-TrkA and/or TrkAIII mRNA expression in PitNETs was not always associated with positive immunoreactivity for TrkA and/or phosphorylated TrkA isoform(s). Indeed, heterogeneous TrkA expression characterizes normal endocrine pituitary cell types and TrkA expression has been reported in PitNETs but its biological significance is poorly understood [24,44]
Alternative TrkAIII splicing is promoted by hypoxia in NB cells [25,36], and PitNETs may exhibit activated hypoxia responses, which in turn may promote invasion, as reported for the HIF1a-RSUME-VEGF pathway [10,43,44,46]. Increased VEGF and angiogenesis are relevant in the progression of PitNETs and represent a current target for the treatment of refractory aggressive and metastatic cases [6,7]. Considering the unavailability of protein extracts, assessment of potential hypoxia involvement in PitNET alternative TrkAIII mRNA splicing was limited to RT-PCR comparisons with HIF1a and HIF2a mRNA expression. In support of a potential role for hypoxia, significantly elevated alternative TrkAIII splicing in invasive PIT1 PitNETs was associated with significantly higher HIF2a but not HIF1a mRNA expression, identifying HIF2a as a novel potential marker of invasive PIT1 behavior. Reports that NB cells exhibit HIF2a transcriptional sensitivity to hypoxia [47] and exhibit hypoxia promoted alternative TrkAIII splicing [25,36], suggests similarity between these tumor types, which may reflect a common neural crest cell origin for NBs [48] and some PitNETs, [49–53]. The lack of strict significant correlation between HIF2a mRNA levels and the proportion of alternative TrkAIII mRNA splicing in individual PIT1 PitNETs, however, does not preclude hypoxia participation in PitNET alternative TrkAIII mRNA splicing, as HIF1a mRNA expression was also detected in all PitNET lineages, hypoxia also regulates HIF1a and HIF2a protein expression at the post-transcriptional level [54], and HIF1a forms part of the PitNET hypoxia response [6,55].
With respect to alternative conditions that could promote alternative TrkAIII splicing in PitNETs, lactotroph-associated hotspot SF3B1 splice factor mutations [14,15,16,17] were not detected in any PitNET subtype exhibiting alternative TrkAIII mRNA splicing. Furthermore, altered SF3B1, SRSF2 or U2AF1 splice factor mRNA expression, previously implicated in PitNET pathogenesis and progression [17], also did not appear to associate with differences in alternative TrkAIII mRNA splicing in invasive and non-invasive combined PIT1, SF1 and TPIT PitNETs, or individual Pit1 and SF1 PitNET lineages. However, the splicing machinery is complex and other components dysregulated in PitNETs and aggressive PitNET subtypes [17], could potentially be involved. Unconventional Xbp-1 splicing, as an index of UPR activation [42], was also not detected in PitNETs exhibiting alternative TrkAIII mRNA splicing, suggesting that conditions that activate the UPR are unlikely to underpin PitNET alternative TrkAIII splicing. This was unexpected considering that hypoxia has been reported to activate the UPR [56]. Whether this reflects defective IRE1/Xbp1 activation in PitNETs, UPR modification by TrkAIII [30], or an alternative mechanism remains to be elucidated. Finally, a potential role for JCPyV infection in alternative TrkAIII splicing, previously implicated in PitNET pathogenesis [18,19], is also unlikely as JCPyV large T antigen mRNA expression was not detected in any of the PitNETs exhibiting alternative TrkAIII mRNA splicing analyzed.
5. Conclusions
Overall, the data demonstrate that alternative TrkAIII mRNA splicing, potentially promoted by hypoxia, occurs frequently in both invasive and non-invasive PitNETs of all lineages, but is significantly more pronounced in invasive compared to non-invasive cases and can associate with immunoreactivity consistent with intracellular TrkAIII activation. Increased alternative TrkAIII mRNA splicing was significantly associated with invasiveness and HIF2a mRNA expression in the PIT1 but not SF1 lineage, and although less represented due to lower incidence, TPIT PitNETs appeared to be especially prone to complete TrkAIII mRNA splicing associated with intracellular TrkA isoform(s) activation, also consistent with TrkAIII. We conclude, therefore, that alternative TrkAIII mRNA splicing that results in intracellular expression and activation of the TrkAIII oncoprotein is likely to participate in PitNET pathogenesis and progression, identifying TrkAIII as a novel potential oncogenic target in refractory PitNETs, for clinically approved Trk inhibitors [57,58].
Author Contributions
M.LJ.R., A.R.F., A.R.M. designed this study; M.S., F.C., V.Z., L.C., M.R., and I.M. performed molecular analyses; A.R.M., R.M., and G.C. performed confocal immunofluorescence. M.LJ.R., T.F and V.E were responsible for clinical characterization and management of the patients. F.G. and A.A. were responsible for the pathological characterization of the tumors and proper sample collection. M.LJ.R, T.F., S.G., and M.C analyzed the clinical data M.LJ.R. and A.R.M. drafted the manuscript; A.R.F. and A.R.M revised and validated the manuscript. All authors provided multidisciplinary expertise and agreed to the publication of this manuscript.
Funding
This research was funded by intramural grants from the Department of Biotechnological and Applied Clinical Sciences of the University of L’Aquila: “DISCAB GRANTS 2023”: 07_DG-2023_01 and 07_DG-2023_06 (L’Aquila, AQ) with a contribution from Neuromed IRCCS (Pozzilli, IS) and the “Carlo Ferri” foundation for the prevention of cancer (Monterotondo (RM).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee (Neuromed, BioPit 270423).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study except for a few cases of archived material, lost to follow-up.
Data Availability Statement
The data presented in this study are available from the corresponding author upon reasonable request.
Acknowledgments
The first two authors (M.S. and M.L.J.R) and the last two authors (A.R.F and A.R.M) contributed equally to this manuscript. The authors wish to thank Sabrina Staffieri and Maria-Antonietta Oliva for excellent technical assistance in samples handling and diagnostic immunohistochemical studies.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Osamura, R.Y.; Grossman, A.; Korbonits, M.; Kovacs, K.; Lopes, M.B.S.; Matsuno, A.; Trouillas, J. Pituitary adenoma. In: WHO Classification of Tumours of Endocrine Organs. Editors. Lloyd, R.V.; Osamura, R.Y.; Koppel, G.R. WHO, OMS, International Agency for Research on Cancer. Lyon France, 2017 pp. 14–18.
- Trouillas, J.; Jaffrain-Rea, M.L.; Vasiljevic, A.; Raverot, G.; Rioncaroli, F.; Villa, C. How to classify pituitary neuroendocrine tumors (PitNET)s in 2020. Cancers 2020, 12, 514. [CrossRef]
- Asa, S.L.; Mete, O.; Perry, A.; Osamura, R.Y. Overview of the 2022 WHO Classification of Pituitary Tumors. Endocr Pathol 2022, 33, 6-26. [CrossRef]
- Tsukamoto,T.; Miki,Y. Imaging of pituitary tumors: an update with the 5th WHO classifications: part 1. Pituitary neuroendocrine tumor (PitNET) pituitary adenoma. Jap J Radiol 2023, 41, 789-806. [CrossRef]
- Neou, M., Villa, C., Armignacco, R., Jouinot, A., Raffin-Sanson, M.L., Septier, A., Letoumeur, F., Diry, S., Diedisheim, M., Izac, B., Gaspar, C., Perlemione, K., Verjus,, V., Bernier M., Boulin, A., Emile, J.F., Bertagna, X., Jaffrezic, F., Laloe, D., Baussart, B., Bertherat, J., Gaillard, S., Assie, G. Pangenomic classification of pituitary neuroendocrine tumors. Cancer Cell 2020, 37, 123-134. [CrossRef]
- Raverot, G.; Ilie, M.D.; Lasolle, H.; Amodru, V.; Trouillas, J.; Castinetti, F.; Brue, T. Aggressive pituitary tumours and pituitary carcinomas. Nat Rev Endocrinol 2021, 17, 671-684. [CrossRef]
- Burman P, Trouillas J, Losa M, McCormack A, Petersenn S, Popovic V, Theodoropoulou M, Raverot G, Dekkers OM; ESE survey collaborators. Aggressive pituitary tumours and carcinomas, characteristics and management of 171 patients. Eur J Endocrinol 2022, 187, 593-605. [CrossRef]
- Trouillas, J.; Jaffrain-Rea, M.L.; Vasiljevic, A.; Dekkers, O.; Popovic, V.; Wierinckx, A.; McCormack, A.; Petersenn, S.; Burman, P.; Raverot, G., Villa, C. Are aggressive pituitary tumors and carcinomas two sides of the same coin? Pathologists reply to clinician's questions. Rev Endocr Metab Disord 2020, 21, 243-251. [CrossRef]
- Guaraldi, F.; Morandi, L.; Zoli, M.; Mazzatenta, D.; Righi, A.; Evangelisti, S.; Ambrosi, F.; Tonon, C.; Giannini, C.; Lloyd, R.V.; Asioli, S. Epigenomic and somatic mutations in pituitary tumors with clinical and pathological correlations in 111 patients. Clin Endocrinol (Oxf) 2022, 97, 763-772. [CrossRef]
- Srirangam Nadhamuni, V.; Korbonits, M. Novel Insights into Pituitary Tumorigenesis: Genetic and Epigenetic Mechanisms. Endocr Rev 2020, 41, 821-846. [CrossRef]
- Melmed, S.; Kaiser, U.B.; Lopes, M.B.; Bertherat, J.; Syro, L.V.; Raverot, G.; Reincke, M.; Johannsson, G.; Beckers, A.; Fleseriu, M.; Giustina, A.; Wass, J.A.H.; Ho, K.K.Y. Clinical Biology of the Pituitary Adenoma. Endocr Rev 2022, 43, 1003-1037. [CrossRef]
- Bao, Y.; Yoshida, D.; Morimoto, M.D.; Teramoto, A. Expression of laminin B2: a novel marker of hypoxia in pituitary adenomas. Endocr Pathol 2006, 17, 251-261. [CrossRef]
- Zhou, Y.; Zhang, A.; Fang, C.; Yuan, L.; Shao, A.; Xu, Y.; Zho,u D. Oxidative stress in pituitary neuroendocrine tumors: affecting the tumor microenvironment and becoming a new target for pituitary neuroendocrine tumor therapy. CNS Neurosci Ther 2023, 29, 2744-2759. [CrossRef]
- Li, C.; Xie, W.; Rosenblum, J.S.; Zhou, J.; Guo, J.; Miao, Y.; Shen,Y.; Wang, H.; Gong, L.; Li, M.; Zhao, S.; Cheng, S.; Zhu, H.; Jiang, T.; Ling, S.; Wang, F.; Zhang, H.; Zhang, M.; Qu, Y.; Zhang, Q.; Li, G.; Wang, J.; Ma, J.; Zhuang, Z.; Zhang, Y. Somatic SF3B1 hotspot mutation in prolactinomas. Nature Commun 2020, 11, 2056. [CrossRef]
- Torres-Moran, M.; Franco-Alvarez, A.; Rebollar-Vega, R.G.; Hernandez-Ramirez, L.C. Hotspots of somatic genetic variation in pituitary neuroendocrine tumors. Cancers (Basel) 2023, 15, 5685. [CrossRef]
- Simon, J.; Perez-Rivas, L.G.; Zhao, Y.; Chasseloup, F.; Lasolle, H., Cortet, C.; Descotes, F.; Villa, C., Baussart, B.; Burman, P., Maiter, D.; von Selzam, V.; Rotermund, R.; Flitsch J.; Thorsteinsdottir, J.; Jouanneau, E.; Buchfelder, M.; Chanson, P.; Raverot, G.; Theodoropoulou, M. Prevalence and clinical correlations of SF3B1 variants in lactotroph tumours. Eur J Endocrinol 2023, 189, 372-378. [CrossRef]
- Vazquez-Borrego, M.C.; Fuentes-Fayos, A.C.; Venegas-Moreno, E.; Rivero-Cortes, E.; Dios, E.; Moreno-Moreno, P.; Madrazo-Atutxa, A.; Remon, P.; Solivera, J.; Wildemberg, L.E.; Kasuli, L.; Lopez-Fernandez, J.M.; Gadelha, M.R.; Galvez-Moreno, M.A.; Soto-Moreno, A.; Gahete, M.D.; Castano, J.P.; Luque, R.M. Splicing machinery is dysregulated in pituitary neuroendocrine tumors and is associated with aggressive features. Cancers (Basel) 2019, 11, 1439. [CrossRef]
- Gordon, J.; Del Valle, L.; Otte, J.; Khalili, K. Pituitary neoplasia induced by expression of human neurotropic polyomavirus, JCV, early genome in transgenic mice. Oncogene 2000, 19, 4840-4846. [CrossRef]
- Del Valle, L.; Khalili, K. Induction of brain tumors by the archetypal strain of human neurotropic JCPyV in a transgenic mouse model. Viruses 2021, 13, 162. [CrossRef]
- Farina, A.R., Cappabianca, L., Sebastiano, M., Zelli, V., Guadagn,i S., Mackay, A.R. Hypoxia-induced alternative splicing: The 11th hallmark of cancer. J Clin Exp Cancer Res 2020, 39:110. [CrossRef]
- Siddaway R, Milos S, Kumaran Anguraj Vadivel A, Dobson THW, Swaminathan J, Ryall S, Pajovic S, Patel PG, Nazarian J, Becher O, Brudno M, Ramani A, Gopalahrishnan V, Hawkins C. Splicing is an alternative oncogenic pathway activation mechanism in glioma. Nature Commun 2022, 13, 588. [CrossRef]
- Bonomi, S.; Gallo, S.; Catillo, M.; Pignataro, D.; Biamonti, G.; Ghigna, C. Oncogenic alternative splicing switches: role in cancer progression and prospects for therapy. Int J Cell Biol 2013, 2013, 962038. [CrossRef]
- Patterson, J.C.; Childs, G.V. Nerve growth factor and its receptor in the anterior pituitary. Endocrinology 1994, 135, 1689-9166. [CrossRef]
- Assimakopoulou, M.; Zolota, V.; Chondrogianni, C.; Gatzounis, G.; Varakis, J. p75 and TrkC neurotrophin receptors demonstrate a different immunoreactivity profile in comparison to TrkA and TrkB receptors in human normal pituitary gland and adenomas. Neuroendocrinology 2008, 88, 127-134. [CrossRef]
- Tacconelli, A., Farina, A.R., Cappabianca, L., DeSantis, G., Tessitore, A., Vetuschi, A., Sferra, R., Rucci, N., Argenti, B., Screpanti, I., Gulino, A, Mackay, A.R. TrkA alternative splicing: A regulated tumor-promoting switch in human neuroblastoma. Cancer Cell 2004, 6, 347–360. [CrossRef]
- Cappabianca, L., Guadagni, S., Maccarone, R., Sebastiano, M., Chiominto, A., Farina, A.R., Mackay, A.R. A pilot study of alternative TrkAIII splicing in Merkel cell carcinoma: A potential oncogenic mechanism and novel therapeutic target. J Exp Clin Cancer Res 2019, 38, 424. [CrossRef]
- Cappabianca, L., Zelli, V., Pellegrini, C., Sebastiano, M., Maccarone, R., Clementi, M., Chiominto, A., Ruggeri, P., Cardelli, L., Ruggieri, M., Sbaffone, M., Fargnoli, M.C., Guadagni, S., Farina, A.R., Mackay, A.R. The alternative TrkAIII splice variant, a targetable oncogenic participant in human cutaneous malignant melanoma. Cells 2023, 12, 237. [CrossRef]
- Schramm, A., Schowe, B., Fielitz, K., Hweilman, M., Martin, M., Marschall, T., Koster, J., Vandenstompele, J., Vermeulen, J., de Pterter, K., Koster, J., Versteeg, R., Noguera, R., Speleman, F., Rahmann, S., Eggert, A., Morik, K., Schulte, J.H. Exon-level expression analyses identify MYCN and NTRK1 as major determinants of alternative exon usage and robustly predict primary neuroblastoma outcome. Br J Cancer 2012, 107, 1409-1417. [CrossRef]
- Lebedev, T.D., Vagapova, E.R., Popenko, V.I., Leonova, O.G., Spirin, P.V., Prassolov, V.S. Two receptors, two isoforms, two cancers: comprehensive analysis of KIT and TrkA expression in neuroblastoma and acute myeloid leukemia. Front Oncol 2019, 9, 1046. [CrossRef]
- Farina, A.R., Di Ianni, N., Cappabianca, L., Ruggeri, P., Gneo, L., Pellegrini, C., Fargnoli, M.C., Mackay, A.R. The oncogenic neurotrophin receptor tropomyosin-related kinase variant, TrkAIII. J Clin Exp Cancer Res 2018, 37, 119. [CrossRef]
- Arevalo, J.C., Conde, B., Hempstead, B.L., Chao, M.V., Martin-Zanca, D., Perez, P. TrkA immunoglobulin-like ligand binding domains inhibit spontaneous activation of the receptor. Mol Cell Biol 2000, 20, 5908–5916. [CrossRef]
- Watson, F.L., Porcionatto, M.A., Battacharyya, A., Stiles, C.D., Segal, R.A. TrkA glycosylation regulates receptor localization and activity. J Neurobiol 1999, 39, 323-336. [CrossRef]
- Farina, A.R.; Cappabianca, L.; Ruggeri, P.; Gneo, L.; Maccarone, R.; Mackay, A.R. Retrograde TrkAIII transport from ERGIC to ER: A re-localization mechanism for oncogenic activity. Oncotarget 2015, 34, 35636-35651. [CrossRef]
- Farina, A.R.; Tacconelli, A.; Cappabianca L.; Cea, G.; Panella, S.; Chioda, A.; Romanelli, A.; Pedone, C.; Gulino, A.; Mackay, A. R. The alternative TrkA splice variant targets the centrosome and promotes genetic instability. Mol Cell Biol 2009, 17, 4182-4130. [CrossRef]
- Farina, A.R.; Cappabianca, L.; Gneo, L.; Ruggeri, P.; Mackay, A.R. TrkAIII signals endoplasmic reticulum stress to the mitochondria in neuroblastoma cells, resulting in glycolytic metabolic adaptation. Oncotarget 2017, 9, 8368-8390. [CrossRef]
- Cappabianca, L.; Sebastiano, M.; Ruggieri, M.; Sbaffone, M.; Zelli, V.; Farina, A.R.; Mackay, A.R. Doxorubicin-induced TrkAIII activation: A selection mechanism for resistant dormant neuroblastoma cells. Int J Mol Sci 2022, 18, 10895. [CrossRef]
- Shulman, D.S., DuBois, S.G. The evolving diagnostic and treatment landscape of NTRK-fusion driven pediatric cancers. Pediatric Drugs 2020, 22, 189-197. [CrossRef]
- Rohrberg, K.S., Lassen, U. Detecting and targeting NTRK fusions in cancer in the era of tumor agnostic oncology. Drugs 2021, 81, 445-452. [CrossRef]
- Villa C, Vasiljevic A, Jaffrain-Rea ML, Ansorge O, Asioli S, Barresi V, Chinezu L, Gardiman MP, Lania A, Lapshina AM, Poliani L, Reiniger L, Righi A, Saeger W, Soukup J, Theodoropoulou M, Uccella S, Trouillas J, Roncaroli F. A standardised diagnostic approach to pituitary neuroendocrine tumours (PitNETs): a European Pituitary Pathology Group (EPPG) proposal. Virchows Arch 2019, 475, 687-692. [CrossRef]
- Singh R, Green MR. Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science 1993, 259, 365-368. [CrossRef]
- Feola, T.; Carbonara, F.; Verrico, M.; Di Crescenzo, R.M.; Gianno, F.; Colonnese, C.; Arcella, A.; de Alcubierre, D.; Tomao, S.; Esposito, V.; Giangaspero, F.; Minniti, G.; Jaffrain-Rea, M.L. Immunotherapy for Aggressive and Metastatic Pituitary Neuroendocrine Tumors (PitNETs): State-of-the Art. Cancers (Basel) 2022, 14, 4093. [CrossRef]
- Yoshida, H. Unconventional splicing of XBP-1 mRNA in the unfolded protein response. Antioxid Redoc Signal 2007, 9, 2323-2333. [CrossRef]
- Yang, Q.; Li X. Molecular Network Basis of Invasive Pituitary Adenoma: A Review. Front Endocrinol (Lausanne) 2019, 10, 7. [CrossRef]
- Artico, M.; Bianchi, E.; Magliulo, G.; De Vincentiis, M.; De Santis, E.; Orlandi, A.; Santoro, A.; Pastore, F.S.; Giangaspero, F.; Caruso, R.; Re, M.; Fumagalli, L. Neurotrophins, their receptors and KI-67 in human GH-secreting pituitary adenomas: an immunohistochemical analysis. Int J Immunopathol Pharmacol 2012, 25, 117-125. [CrossRef]
- Tebani, A., Jotanovic, J., Hekmati, N., Sivertsson, A., Gudjonsson, O., Engstrom, B.E., Wikstrom, J., Uhlen, M., Casar-Borota, O., Ponten, F. Annotation of pituitary neuroendocrine tumor with genome-wide expression analysis. Acta Neuropathol Commun 2021, 9, 181. [CrossRef]
- Lucia, K.; Wu, Y.; Garcia, J.M.; Barlier, A.; Buchfelder, M.; Saeger, W.; Renner, U.; Stalla, G.K.; Theodoropoulou, M. Hypoxia and the hypoxia inducible factor 1α activate protein kinase A by repressing RII beta subunit transcription. Oncogene 2020, 39, 3367-3380. [CrossRef]
- Hamidian, A., von Stedingk, K., Thoren, M.M., Mohlin, S., Pahlman, S. Differential regulation of HIF-1a and HIF-2a in neuroblastoma, estrogen related receptor alpha (ERRa) regulates HIF2A transcription and correlates to poor outcome. BBRC 2015, 461, 560-567. [CrossRef]
- Johnsen, J.I., Dyberg, C., Wickstrom, M. Neuroblastoma-a neural crest derived embryonal malignancy. Front Mol Neurosci 2019, 12, 9. [CrossRef]
- Ferrand, R., Pearse, A.G.E., Polak, J.M., Le Douarin, N.M. Immunohistochemical studies on the development of avian embryo pituitary corticotrophs under normal and experimental conditions. Histochemistry 1974, 38, 133-141. [CrossRef]
- Eagleson, G.W., Jenks, B.G., Van Overbeeke, A.P. The pituitary adrenocorticotropes originate from neural ridge tissue in xenopus laevis. J Embryol Exp Morph 1986, 95, 1-14. [CrossRef]
- Ueharu, H., Yoshida, S., Kikkawa, T., Kanno, N., Higuchi, M., Kato, T., Osumi, N., Kato, Y. Gene tracing analysis reveals the contribution of neural crest-derived cells in pituitary development. J Anat 2017, 230, 373-380. [CrossRef]
- Ueharu, H., Yoshida, S., Kanno, N., Horiguchi, K., Nishimura, N., Kato, T., Kato, Y. SOX10-positive cells emerge in the rat pituitary gland during late embryogenesis and start to express S100β. Cell Tissue Res 2018, 372, 77-90. [CrossRef]
- Duan, S., Sawyer, T.W., Sontz, R.A., Wieland, B.A., Diaz, A.F., Merchant, J.L. GFAP-directed inactivation of men1 exploits glail cell plasticity in favor of neuroendocrine reprogramming. Cell Mol Gastroent Hepatol 2022, 14, 1025-1051. [CrossRef]
- Albanese, A.; Daly, L.A.; Mennerich, D.; Kietzmann, T.; Sée, V. The Role of Hypoxia-Inducible Factor Post-Translational Modifications in Regulating Its Localisation, Stability, and Activity. Int J Mol Sci 2020, 22, 268. [CrossRef]
- Gupta, P.; Dutta, P. Landscape of Molecular Events in Pituitary Apoplexy. Front Endocrinol (Lausanne) 2018, 9, 107. [CrossRef]
- Chipurupalli, S., Kannan, E., Tergaonkar, V., D’Andrea, R., Robinson, N. Hypoxia induced ER stress response as an adaptive mechanism in cancer. Int J Mol Sci 2019, 20, 749. [CrossRef]
- Cocco, E., Scaltrtiti, M., Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol 2018, 15, 731-747. [CrossRef]
- Treis, D., Umapoathy, G., Fransson, S., Guan, J., Mendoza-Garcia, P., Siaw, J.T., Wessman, S., Gordon Murkes, L., Stenman, J.J.E., Djos, A., Elfman, L.H.M., Inge Johnsen, J., Hallberg, B., Palmer, R.H., Martinsson, T., Kogner, P. Sustained response to entrectinib in an infant with a germline ALKAL2 variant and refractory metastatic neuroblastoma with chromosomal 2p gain and anaplastic lymphoma kinase and tropomyosin receptor kinase activation. JCO Precision Oncol 2022, 6. [CrossRef]
Figure 1.
a) Schematic diagram of chromosomal NTRK1/TrkA gene localization, fs-TrkA, TrkAIII and D2-7TrkA exon structures and exon 1-8 and 8-17 RT-PCR amplicons, in bases pairs (bp). b) Representative RT-PCRs demonstrating fs-TrkA, TrkAIII and D2-7 TrkA products generated using primers spanning TrkA exons 1 to 8, and fs-TrkA products generated using primers spanning TrkA exons 8 to 17, in cDNAs from invasive PIT1 PitNETs (cases nr.1, 3 and 6), and non-invasive PIT1 PitNETs (cases nr.20 and 22). c) Representative DNA sequences demonstrating the presence of the fs-TrkA exon 6-7, TrkAIII exon 5- 8 and Dex2-7 TrkA exon 1- 8 splice junctions in purified RT-PCRproducts in cDNA from an invasive PIT1 PitNET (case nr.3).
Figure 1.
a) Schematic diagram of chromosomal NTRK1/TrkA gene localization, fs-TrkA, TrkAIII and D2-7TrkA exon structures and exon 1-8 and 8-17 RT-PCR amplicons, in bases pairs (bp). b) Representative RT-PCRs demonstrating fs-TrkA, TrkAIII and D2-7 TrkA products generated using primers spanning TrkA exons 1 to 8, and fs-TrkA products generated using primers spanning TrkA exons 8 to 17, in cDNAs from invasive PIT1 PitNETs (cases nr.1, 3 and 6), and non-invasive PIT1 PitNETs (cases nr.20 and 22). c) Representative DNA sequences demonstrating the presence of the fs-TrkA exon 6-7, TrkAIII exon 5- 8 and Dex2-7 TrkA exon 1- 8 splice junctions in purified RT-PCRproducts in cDNA from an invasive PIT1 PitNET (case nr.3).
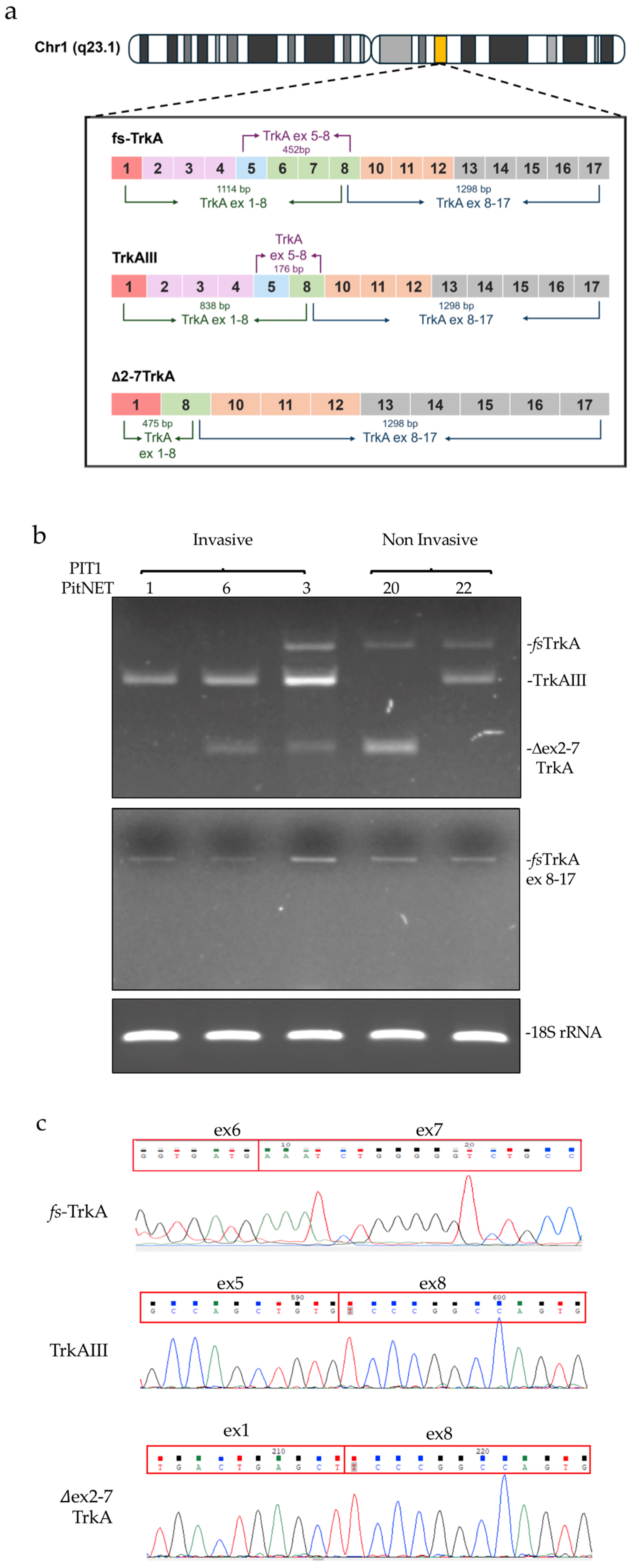
Figure 2.
a) RT-PCR demonstrating relative levels of TrkAIII to fs-TrkA expression in RNAs from representative invasive and non-invasive PIT1, SF1 and TPIT PitNETs. b) Box plots demonstrating densitometric comparisons of the percentage TrkAIII to fs-TrkA ratios in combined invasive (grey) and combined non-invasive (white) PIT1, SF1 and TPIT PitNETs, and in PitNETs grouped into PIT1, SF1 and TPIT lineages (* = p < 0.05). Of note, exclusive TrkAIII expression was observed in all invasive TPIT cases.
Figure 2.
a) RT-PCR demonstrating relative levels of TrkAIII to fs-TrkA expression in RNAs from representative invasive and non-invasive PIT1, SF1 and TPIT PitNETs. b) Box plots demonstrating densitometric comparisons of the percentage TrkAIII to fs-TrkA ratios in combined invasive (grey) and combined non-invasive (white) PIT1, SF1 and TPIT PitNETs, and in PitNETs grouped into PIT1, SF1 and TPIT lineages (* = p < 0.05). Of note, exclusive TrkAIII expression was observed in all invasive TPIT cases.
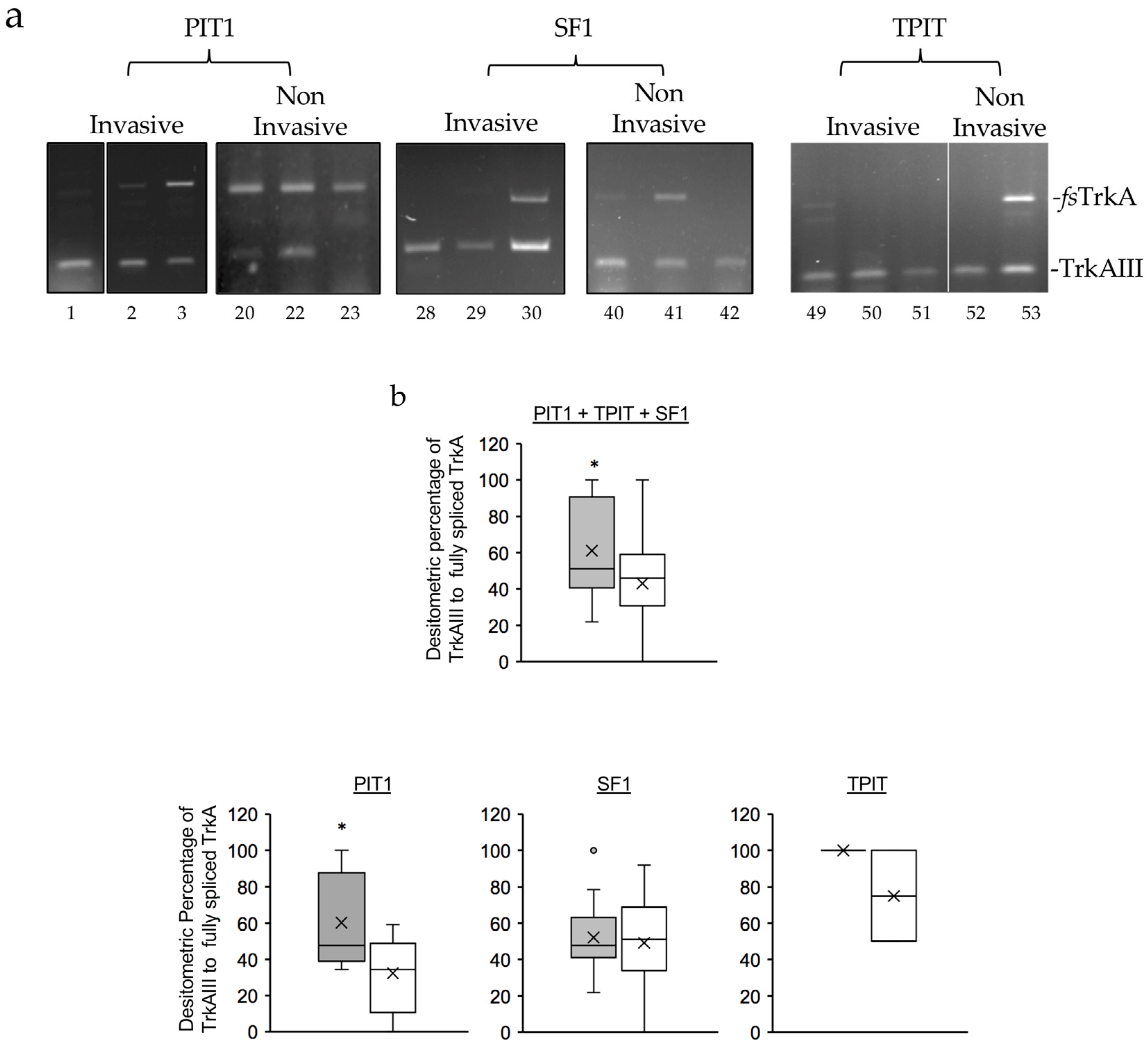
Figure 3.
Confocal IF micrographs demonstrating overlapping (orange/yellow) immunoreactivity for TrkA (red) and Y490 phosphorylated TrkA (green) isoform(s) in invasive SF1 PitNETs n. 25, n. 30 and n. 37, invasive TPIT PitNET n. 49, and in non-invasive SF1 PitNET n. 41, TPIT PitNET n. 53 and PIT1 PitNET n.17, plus background immunoreactivity to secondary antibodies (bottom right panels), nuclei are stained with dapi (Blue) (bar = 100mm) .
Figure 3.
Confocal IF micrographs demonstrating overlapping (orange/yellow) immunoreactivity for TrkA (red) and Y490 phosphorylated TrkA (green) isoform(s) in invasive SF1 PitNETs n. 25, n. 30 and n. 37, invasive TPIT PitNET n. 49, and in non-invasive SF1 PitNET n. 41, TPIT PitNET n. 53 and PIT1 PitNET n.17, plus background immunoreactivity to secondary antibodies (bottom right panels), nuclei are stained with dapi (Blue) (bar = 100mm) .
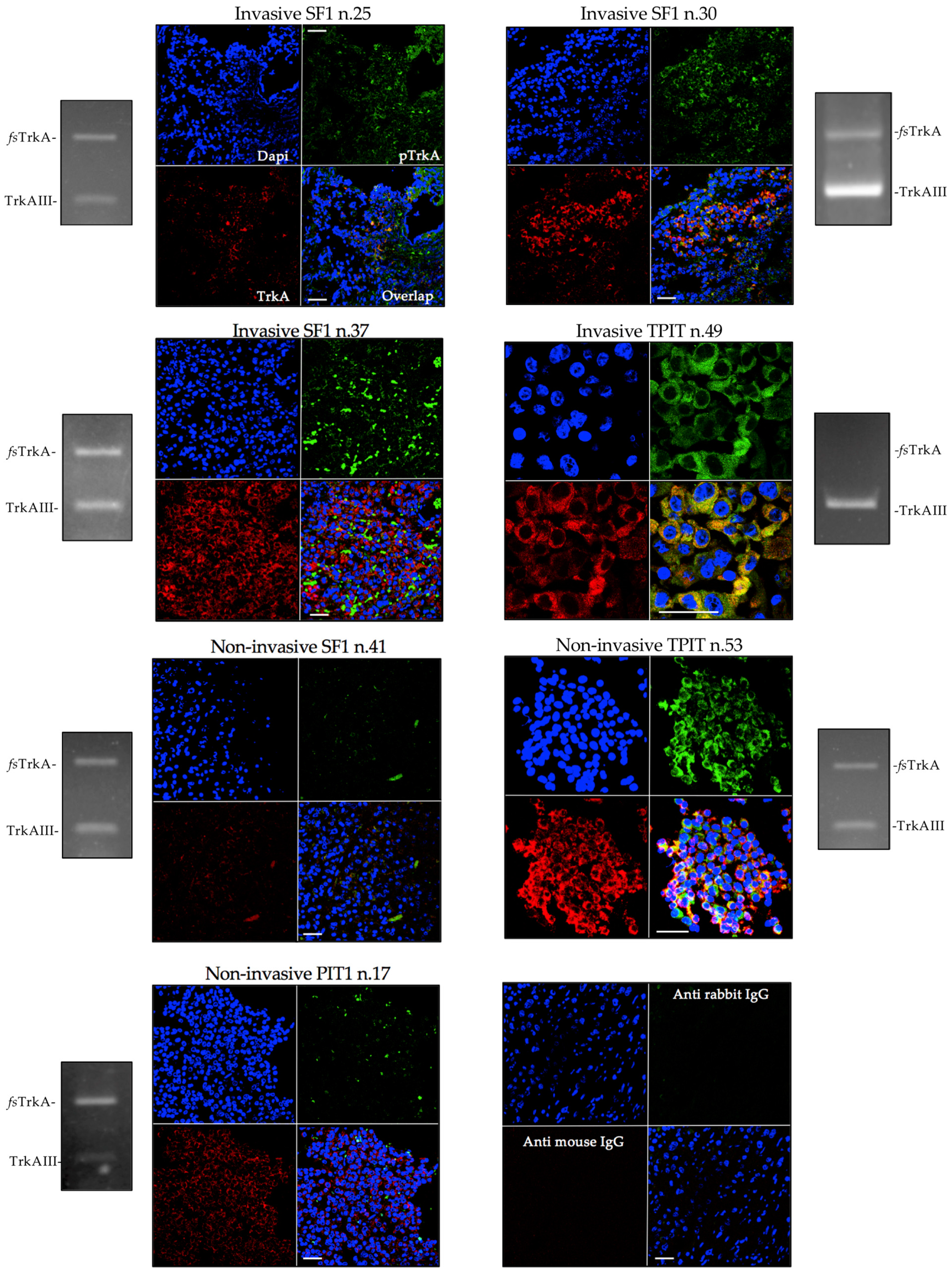
Figure 4.
a) RT-PCR demonstrating relative levels of HIF-1a (150bp) and Hif-2a (121bp) expression in representative samples of invasive and non-invasive PIT1, SF1 and TPIT cases, with products run on the same gel for comparative purposes. b) Box plots demonstrating densitometric comparisons of HIF-1a and Hif-2a RT-PCRs in invasive (grey) and non-invasive (white) combined PIT1, SF1 and TPIT PitNETs and in PitNETs grouped into PIT1, SF1 and TPIT lineages (* p < 0.05).
Figure 4.
a) RT-PCR demonstrating relative levels of HIF-1a (150bp) and Hif-2a (121bp) expression in representative samples of invasive and non-invasive PIT1, SF1 and TPIT cases, with products run on the same gel for comparative purposes. b) Box plots demonstrating densitometric comparisons of HIF-1a and Hif-2a RT-PCRs in invasive (grey) and non-invasive (white) combined PIT1, SF1 and TPIT PitNETs and in PitNETs grouped into PIT1, SF1 and TPIT lineages (* p < 0.05).
Figure 5.
Representative: a) SF3B1cDNA sequences in PIT1 PitNET 1, demonstrating the absence of PitNET-associated hotspot SF3B1 c.1886, c.1873. c.1874, c.1986, c.1996, c.2098 and c.2225 mutations (red hatched boxes identify hotspot mutation sites).
Figure 5.
Representative: a) SF3B1cDNA sequences in PIT1 PitNET 1, demonstrating the absence of PitNET-associated hotspot SF3B1 c.1886, c.1873. c.1874, c.1986, c.1996, c.2098 and c.2225 mutations (red hatched boxes identify hotspot mutation sites).
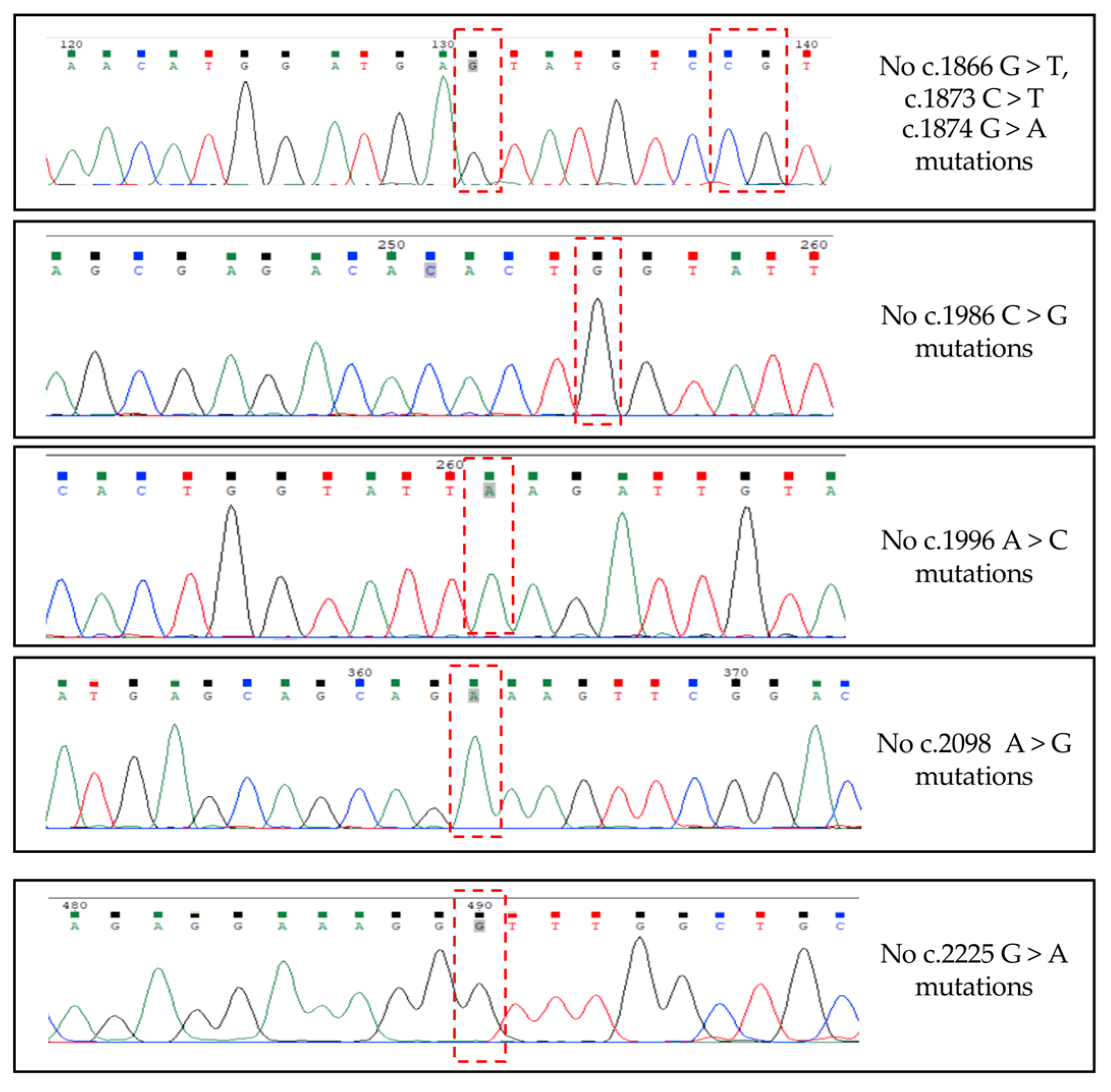
Figure 6.
a) RT-PCR demonstrating relative levels of SF3B1 (693bp), U2AF1 (606bp) and SRSF2 (408bp) RT-PCR products in representative invasive and non-invasive PIT1, SF1 and TPIT PitNET cDNAs, with products run on the same gel for comparative purposes. b) Box plots demonstrating densitometric comparisons of SF3B1, U2AF1 and SRSF2 RT-PCRs in invasive (grey) and non-invasive (white) combined PIT1, TPIT and SF-1 PitNETs (PIT1+TPIT+SF1) and in PitNETs grouped according to PIT1, SF1 and TPIT lineage (Mann-Whitney p values are provided in brackets).
Figure 6.
a) RT-PCR demonstrating relative levels of SF3B1 (693bp), U2AF1 (606bp) and SRSF2 (408bp) RT-PCR products in representative invasive and non-invasive PIT1, SF1 and TPIT PitNET cDNAs, with products run on the same gel for comparative purposes. b) Box plots demonstrating densitometric comparisons of SF3B1, U2AF1 and SRSF2 RT-PCRs in invasive (grey) and non-invasive (white) combined PIT1, TPIT and SF-1 PitNETs (PIT1+TPIT+SF1) and in PitNETs grouped according to PIT1, SF1 and TPIT lineage (Mann-Whitney p values are provided in brackets).
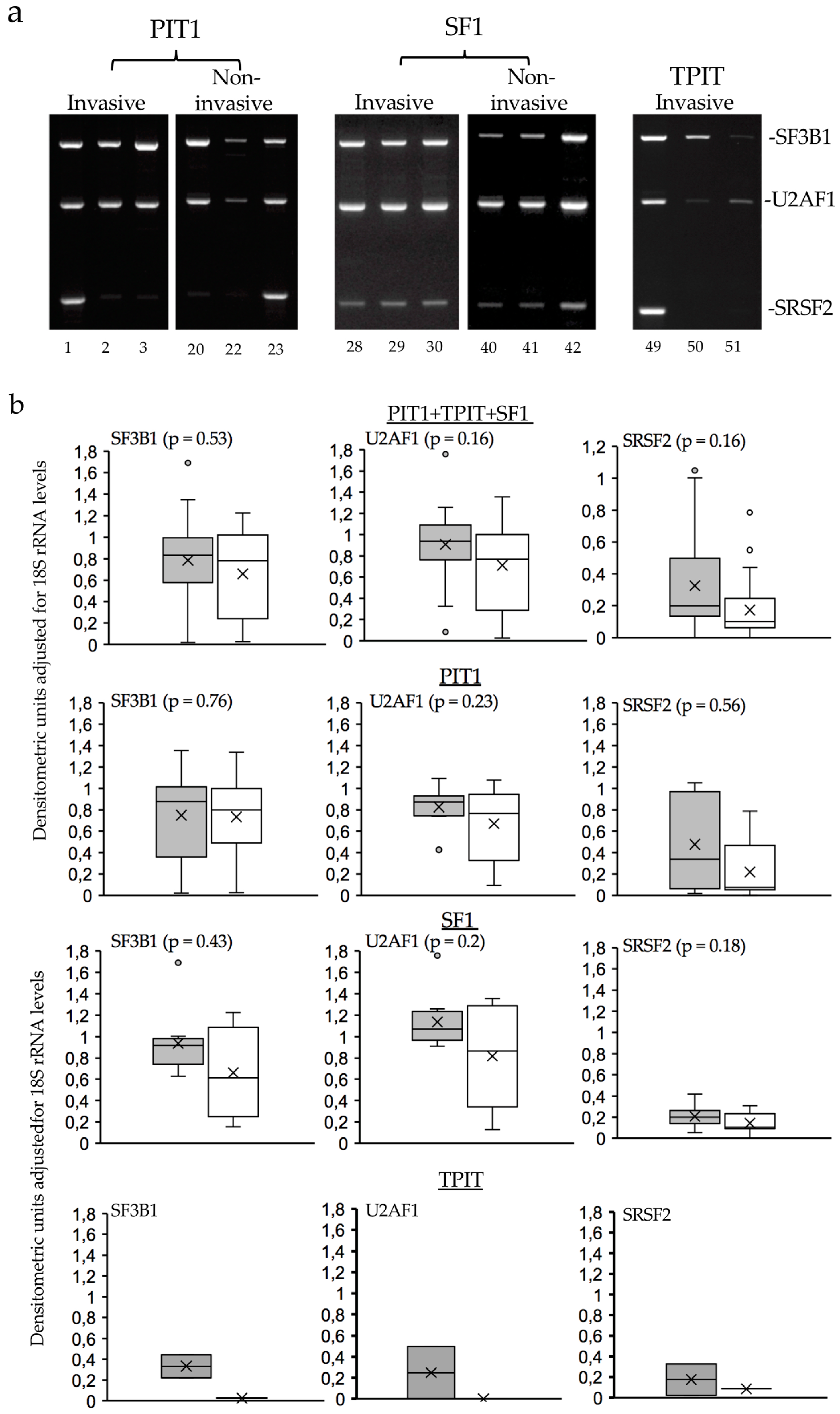
Figure 7.
RT-PCR demonstrating unconventional Xbp1 splicing (spliced Xbp1) in DTT-treated but not untreated (CON) SH-SY5Y cells nor in representative PIT1, SF1 and TPIT PitNET cDNAs.
Figure 7.
RT-PCR demonstrating unconventional Xbp1 splicing (spliced Xbp1) in DTT-treated but not untreated (CON) SH-SY5Y cells nor in representative PIT1, SF1 and TPIT PitNET cDNAs.
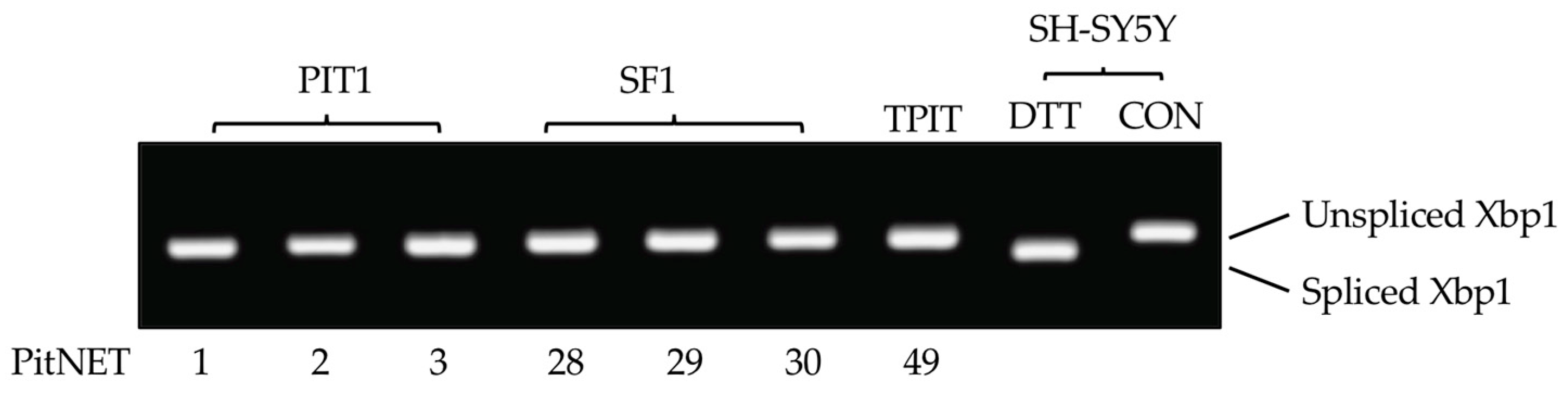

Table 1.
Individual Patient’s details, grouped according to PIT1, SF1, TPIT PitNET lineages, including: age at surgery; sex; positivity for Prolactin (PRL), Growth Hormone (GH), Thyroid stimulating hormone (TSH), Follicle Stimulating Hormone (FSH), Luteinizing Hormone (LH) and Adrenocorticotropic Hormone (ACTH) immunostaining; Ki67 % proliferation index (n/a, not available); functioning (F) or non-functioning (NF) clinical status; recurrent tumors (Rec) with associated aggressive (A) and metastatic (M) cases.
Table 1.
Individual Patient’s details, grouped according to PIT1, SF1, TPIT PitNET lineages, including: age at surgery; sex; positivity for Prolactin (PRL), Growth Hormone (GH), Thyroid stimulating hormone (TSH), Follicle Stimulating Hormone (FSH), Luteinizing Hormone (LH) and Adrenocorticotropic Hormone (ACTH) immunostaining; Ki67 % proliferation index (n/a, not available); functioning (F) or non-functioning (NF) clinical status; recurrent tumors (Rec) with associated aggressive (A) and metastatic (M) cases.
| PIT1 PitNETs | |||||||||||||||||||||||||||||||
| INVASIVE (n = 11) | NON-INVASIVE (n= 13) | ||||||||||||||||||||||||||||||
| Pt | Age | Sex | IHC |
Clinical Status |
Rec |
Ki67 (%) |
Pt | Age | Sex | IHC |
Clinical Status |
Rec |
Ki67 (%) |
||||||||||||||||||
| 1 | 53 | F | PRL | F | Y(A) | ≥3 | 12* | 37 | F | GH | F | N | ≥3 | ||||||||||||||||||
| 2 | 19 | M | GH | F | N | <3 | 13 | 52 | F | GH | F | N | <3 | ||||||||||||||||||
| 3 | 16 | M | PRL | NF | N | ≥3 | 14 | 52 | M | GH | F | N | <3 | ||||||||||||||||||
| 4 | 18 | F | GH | NF | N | ≥3 | 15 | 34 | F | PRL | F | N | n/a | ||||||||||||||||||
| 5 | 74 | M | TSH | F | N | ≥3 | 16* | 49 | M | GH/PRL | F | N | <3 | ||||||||||||||||||
| 6 | 37 | M | TSH | F | N | ≥3 | 17 | 40 | F | GH/PRL | F | N | ≥3 | ||||||||||||||||||
| 7 | 25 | F | GH | F | N | ≥3 | 18 | 55 | M | GH/PRL | F | N | ≥3 | ||||||||||||||||||
| 8 | 21 | M | PRL | F | N | n/a | 19 | 36 | F | PRL | F | N | n/a | ||||||||||||||||||
| 9 | 76 | F | GH | F | N | <3 | 20 | 26 | M | PRL | F | N | n/a | ||||||||||||||||||
| 10 | 14 | M | GH/PRL | F | Y | ≥3 | 21* | 50 | M | GH | F | N | ≥3 | ||||||||||||||||||
| 11 | 62 | M | Pit1 only | NF | Y(M) | ≥3 | 22 | 43 | F | GH/PRL | NF | N | <3 | ||||||||||||||||||
| 23 | 49 | M | PRL | F | N | n/a | |||||||||||||||||||||||||
| 24 | 32 | F | GH | F | N | <3 | |||||||||||||||||||||||||
| SF1 PitNETs | |||||||||||||||||||||||||||||||
| INVASIVE (n = 12) | NON-INVASIVE (n = 12) | ||||||||||||||||||||||||||||||
| Pt | Age | Sex | IHC | Clinical status | Rec |
Ki67 (%) |
Pt | Age | Sex | IHC | Clinical status | Rec |
Ki67 (%) |
||||||||||||||||||
| 25 | 45 | M | FSH/LH | NF | N | ≥3 | 37 | 68 | M | FSH/LH | NF | N | ≥3 | ||||||||||||||||||
| 26 | 56 | M | FSH/LH | NF | N | ≥3 | 38 | 71 | F | SF1 only | NF | Y | <3 | ||||||||||||||||||
| 27 | 73 | F | FSH/LH | NF | N | ≥3 | 39* | 71 | M | FSH/LH | NF | N | <3 | ||||||||||||||||||
| 28 | 49 | F | SF1 only | NF | N | ≥3 | 40 | 67 | M | SF1 only | NF | Y | ≥3 | ||||||||||||||||||
| 29 | 55 | F | SF1 only | NF | N | ≥3 | 41 | 61 | M | FSH/LH | NF | N | <3 | ||||||||||||||||||
| 30 | 48 | M | FSH/LH | NF | N | ≥3 | 42 | 46 | M | FSH/LH | NF | N | ≥3 | ||||||||||||||||||
| 31 | 53 | M | FSH/LH | NF | N | ≥3 | 43 | 75 | M | FSH/LH | NF | N | <3 | ||||||||||||||||||
| 32 | 47 | F | FSH/LH | NF | N | <3 | 44 | 74 | M | SF1 only | NF | N | <3 | ||||||||||||||||||
| 33 | 69 | M | FSH/LH | NF | Y | <3 | 45 | 66 | M | FSH/LH | NF | N | <3 | ||||||||||||||||||
| 34 | 70 | F | FSH/LH | NF | N | <3 | 46 | 39 | M | FSH/LH | NF | N | <3 | ||||||||||||||||||
| 35 | 55 | M | FSH/LH | NF | N | ≥3 | 47 | 46 | M | FSH/LH | NF | N | ≥3 | ||||||||||||||||||
| 36 | 73 | M | SF1 only | NF | N | ≥3 | 48 | 69 | F | FSH/LH | NF | N | <3 | ||||||||||||||||||
| TPIT PitNETs | |||||||||||||||||||||||||||||||
| INVASIVE (n = 3) | NON-INVASIVE (n = 2) | ||||||||||||||||||||||||||||||
| Pt | Age | Sex | IHC | Clinical status | Rec |
Ki67 (%) |
Pt | Age | Sex | IHC | Clinical status | Rec |
Ki67 (%) |
||||||||||||||||||
| 49 | 57 | M | ACTH | F | Y(A) | ≥3 | 52 | 78 | F | ACTH | F | N | <3 | ||||||||||||||||||
| 50 | 52 | F | ACTH | NF | N | ≥3 | 53 | 36 | F | ACTH | F | N | ≥3 | ||||||||||||||||||
| 51 | 26 | F | ACTH | F | N | ≥3 | |||||||||||||||||||||||||
* PitNETs that do not express TrkAIII mRNA.
Table 2.
RT-PCR primers and conditions used in this study.
| Target | Sequence | Denat | Ann | Ext | Amplicon |
|---|---|---|---|---|---|
| 18S rRNA**** |
F: 5’-AAACGGCTACCACATCCAAG-3’ R: 5’-CCTCGAAAGAGTCCTGTATTG-3’ |
30s - 94°C | 30s - 58°C | 30s-72°C | 100bp |
| TrkA ex 8-17* |
F: 5’-AACCCCTTCGGCCAGGCCTCC-3’ R: 5’-CTAGCCCAGGACATCCAGGTA-3’ |
1 min - 94°C | 30s - 65°C | 1 min -72°C | 1298bp TrkA |
| TrkA ex 1-8* |
F: 5’-ATGCTGCGAGGCGGACGGCGC-3’ R: 5’-GGAGGCCTGGCCGAAGGGGTT-3’ |
1 min -94°C | 30s - 68°C | 1 m- 72°C | 1114bp TrkA, 838bp TrkAIII, 475bp Dex2-7 TrkA |
| TrkA ex 5-8* |
F: 5’-AGAAGCTGCAGTGTCATGGG-3’ R: 5’-ATTGAGCACGGAGCCATTGA-3’ |
40s - 94°C | 30s - 58°C | 40s -72°C | 452bp TrkA 176bp TrkAIII |
| SRSF2*** |
F: 5’-CTCCCGATGTGGAGGGTATG-3’ R: 5’-GAGATCGGCTGCGAGACC-3’ |
40s - 94°C | 30s - 58°C | 40 s - 72°C | 408 bp |
| SF3B1** |
F: 5’-TGTGCATAAGATCCTCGTGGT-3’ R: 5’-ACACCATCTGTCCCACAACA-3’ |
40s - 94°C | 30s - 58°C | 4s - 72°C | 693 bp |
| SF3B1 ( tDNA) |
F: 5’-TAGGCTGCTGGTCTGGCTAC-3’ R: 5’-ATGGCACAGCCCATAAGAATAG-3’ |
30s - 95°C | 30s - 60°C | 1m -72°C | 233 bp |
| U2AF1** |
F: 5’-CGGAGTATCTGGCCTCCATC-3’ R: 5’-GCAGCTCTCTGGAAATGGGCT-3’ |
40s - 94°C | 30s - 60°C | 40s -72°C | 606 bp |
| HIF-1a** |
F: 5’-TTCACCTGAGCCTAATAGTCC-3’ R: 5’-AAGTCTAAATCTGTGTCCTG-3’ |
30s - 94°C | 30s - 50°C | 30s - 72°C | 150 bp |
| HIF-2a*** |
F: 5’-AGCCTCCATCTGCCATCAGTC-3’ R: 5’-CTTGCCATGCCTGACACCTTG-3’ |
30s - 94°C | 30s - 55°C | 30s - 72°C | 121 bp |
| JCPyV T-Ag* |
F: 5’-ATATTATGACCCCCAAAACCATG-3’ R: 5’-GGTAGAAGACCCTAAGGACTTTCC-3’ |
40s -94°C | 30s - 58°C | 40s - 68°C | 189 bp |
RT-PCR: 40 cycles; * non-diluted cDNA (50ng), ** 1:10 (5ng); ***1:100 (0.5 ng), and **** 1:1000 (0.05ng) cDNA dilutions.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



