Submitted:
26 January 2024
Posted:
29 January 2024
You are already at the latest version
Abstract
In "genome editing," a type of genetic engineering, DNA is added, taken away, or changed in an organism's genome. Biomedicine, biotechnology, and synthetic biology, just to name a few, have all gained a lot from how often this method is used. Before the editing process can start, a Double-Strand Breaks (DSB) must be made in the DNA at a specific gene. Researchers have made nucleases, which are sometimes called "molecular scissors," to fix this DSB. Transcription activator-like effector nucleases (TALENs), Zinc-finger nucleases (ZFNs), and homing endonucleases are all examples of protein targets that have been studied and changed. The last part gave an overview of these study projects and explained how to make Cas12a for specific uses. The applications of Cas12a have been extensively studied in recent years, but the protein still holds a lot of promise as a treatment and screening tool. In order to give a quick review of CRISPR-Cas12a and its uses, we will briefly talk about the structure and function of the different parts of the reaction pathway that lead up to the catalysis of the target DNA. Cas12a uses a multistep process to make sure that it is selecting the right DNA. This is a good trait for a device that changes the DNA because it makes it less likely that something bad will happen. Even though data shows that a new CRISPR RNAs (crRNA) molecule can stop Cas12a from randomly destroying ssDNA, it may still hurt the host cell while trying to change the genome. The action of Cas12a catalysis, which is a powerful tool for changing the genome, needs to be changed so that it can be more easily controlled and managed. Using what we know about how Cas12a works, we have made mutants that are less active on ssDNA and more active on dsDNA. So far, only Cas9 and Cas12a, which are both part of the CRISPR family, have been used to change the genome. Because of how similar and different these two endonucleases are, CRISPR can now be used for multiple scientific purposes. Cas12a is better than Cas9 because it makes double-strand breaks (DSBs) that favor Homology-Directed Repair (HDR) over Non-homologous end joining (NHEJ) and can change more than one copy of the genome at the same time. Both Cas9 and Cas12a are being changed to make them better at detecting Protospacer Adjacent Motif (PAM) than they are in their natural state. This will make it possible to start focusing on more genes.
Keywords:
CRISPR-Cas12a
; RNA guided endonucleases
; crRNA biogenesis
; Indiscriminate ssDNAse
; Endonuclease recycling.
Introduction
In "genome editing," a type of genetic engineering, DNA is added, taken away, or changed in an organism's genome. Biomedicine, biotechnology, and synthetic biology, just to name a few, have all utilized this method extensively for various applications. Before the editing process can start, a double stranded break (DSB) must be made in the DNA at a specific gene. Researchers have made nucleases, which are sometimes called "molecular scissors," to fix this DSB. TALENs, ZFNs, and homing endonucleases are all examples of protein targets that have been studied as molecular scissors and have the capability to create nicks and modify the genome. Genome editing methods have been utilized to fix genetic flaws that can lead to diseases that can be passed down from parent to child. But it is hard to change these protein structures to add new traits that are specific to DNA. When protospacer adjacent motif, the finding of the flexible Clustered Regularly Interspaced Short Palindromic Repeats-CRISPR related proteins (CRISPR-Cas) systems as molecular scissors holds enormous potential to make desired changes at protein level in the field of life sciences.
CRISPR Mediated Genome Engineering
Cas9 is now used in the growing field of CRISPR-mediated genome editing to enable various functions such as, turn off genes and make changes to the genome in specific places. Cas9 has moved the field of genome editing forward, but it has some problems, like the possibility of unexpected side effects and the fact that it is hard to move the ribonucleoprotein particle. Cas12a is close to Cas9, which gives researchers another way to change DNA at the molecular level. Several scientific studies have looked into whether or not Cas12a can be used to change the genes of certain kinds of cells. Catalytically dead Cas9 from S. pyogenes (SpdCas9) was found to be less successful than catalytically dead Cas12a from Eubacterium eligens (EedCas12a) at stopping genes from being shut in the template strand of the target DNA. Since Cas9 has trouble handling pre-crRNA, Cas12a is a better choice if you want to control more than one gene at the same time. The system's ability to handle its own crRNA has made it possible to change the genomes of native targets by putting different CRISPR RNAs on a single plasmid and delivering them all at once. This makes it possible to create gene engineering that can be on all the time or only when needed.
Cas12a-mediated multimodal gene control works, as shown by studies of cells from bacteria, plants, and mammals. Cas12a can be used to fix the problem when Cas9 shouldn't be used, like in some industrial types of Streptomyces.
CRISPR repeats and the Cas proteins that go with them give bacteria and archaea a way to defend themselves against genetic material that can move around. By showing that bacteria and archaea can get genetic material and add it to their own genomes, CRISPR-Cas showed that the global genome and bacterial genomes can share information. CRISPR screens keep track of how many times nucleic acids from outside have been found. Repetition is made up of short, similar sequences, while gaps are made up of longer patterns that don't repeat. When these Cas proteins find invader nucleic acids, they add them to the CRISPR array. Effector complexes of RNA-guided endonucleases protect prokaryotes from foreign MGEs by recognising re-infection from foreign DNA that has already been added to the CRISPR array.
Figure 1.
CRISPR Cas mediated gene editing – Insertion, deletion and disruption of genes.
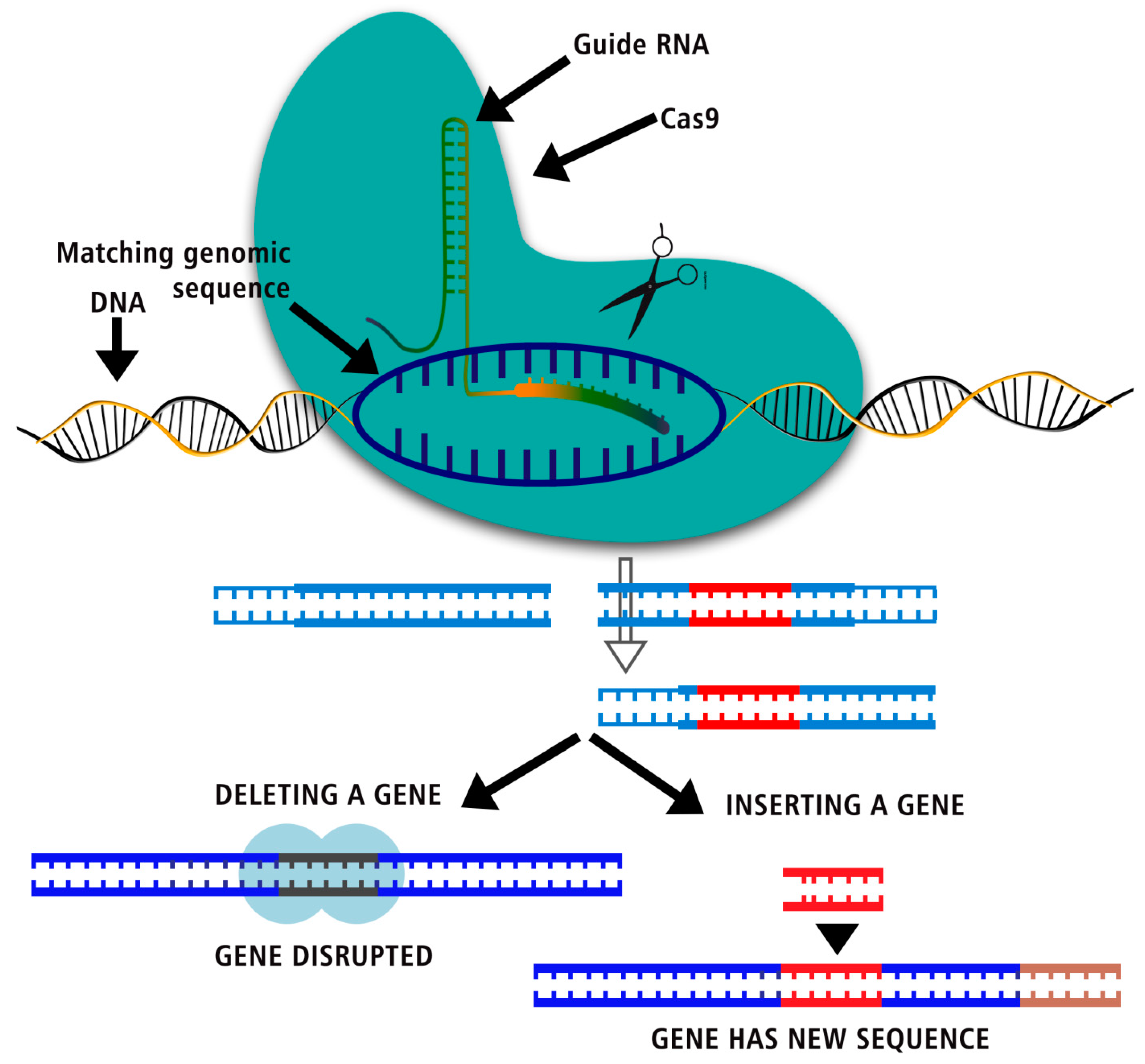
Genome Sequential Steps; Adaptation, Expression and Interference
But as tools for reading the genome get better, they can collect more data, which makes it easier to find new ways to change the DNA. Scientists are just now trying to figure out how tools like these could be used to change genes. In Class 2, there are many new people, and Cas12a is one of them. Each of the three main stages of CRISPR-Cas immunity—adaptation, expression/maturation, and inhibition—needs a different Cas protein, which is made by cas genes close to the CRISPR array. There are also other proteins that are needed for each of these things to happen. During the CRISPR-Cas adaptation process, the foreign DNA or RNA's protospacer is taken and put into the CRISPR array. The Cas1-Cas2 complex is able to do both jobs. First, the adaptation complex finds the PAM, which leads to the discovery of the protospacer. Second, a gap is added to the CRISPR array, and third, the conserved repeat sequence is copied. The PAM pattern was one of the first ways to find nucleic acids that needed to be broken down. This means that it is not a CRISPR set. During the translation and development step, the CRISPR array is turned into a long line of pre-CRISPR RNA, or pre-crRNA. From the pre-crRNA, shorter molecules of crRNA are made, each with a break and part of the repeat pattern. After the crRNA and the effector protein join to make an RNA-guided endonuclease that works, interference can happen. This endonuclease binds to the target DNA through its gap sequence when PAM is found. The DNA is then cut. This paper is about Cas12a, which is related to Cas9 and is part of the Class 2 Type V CRISPR-Cas system. Since it is made and works differently than Cas9, it has been considered as a possible new tool for changing genes.
Figure 2.
The CRISPR Cas three steps of bacterial adaptive immune system, acquisition, crRNA biogenesis and interference of viral DNA in editing process.
Figure 2.
The CRISPR Cas three steps of bacterial adaptive immune system, acquisition, crRNA biogenesis and interference of viral DNA in editing process.
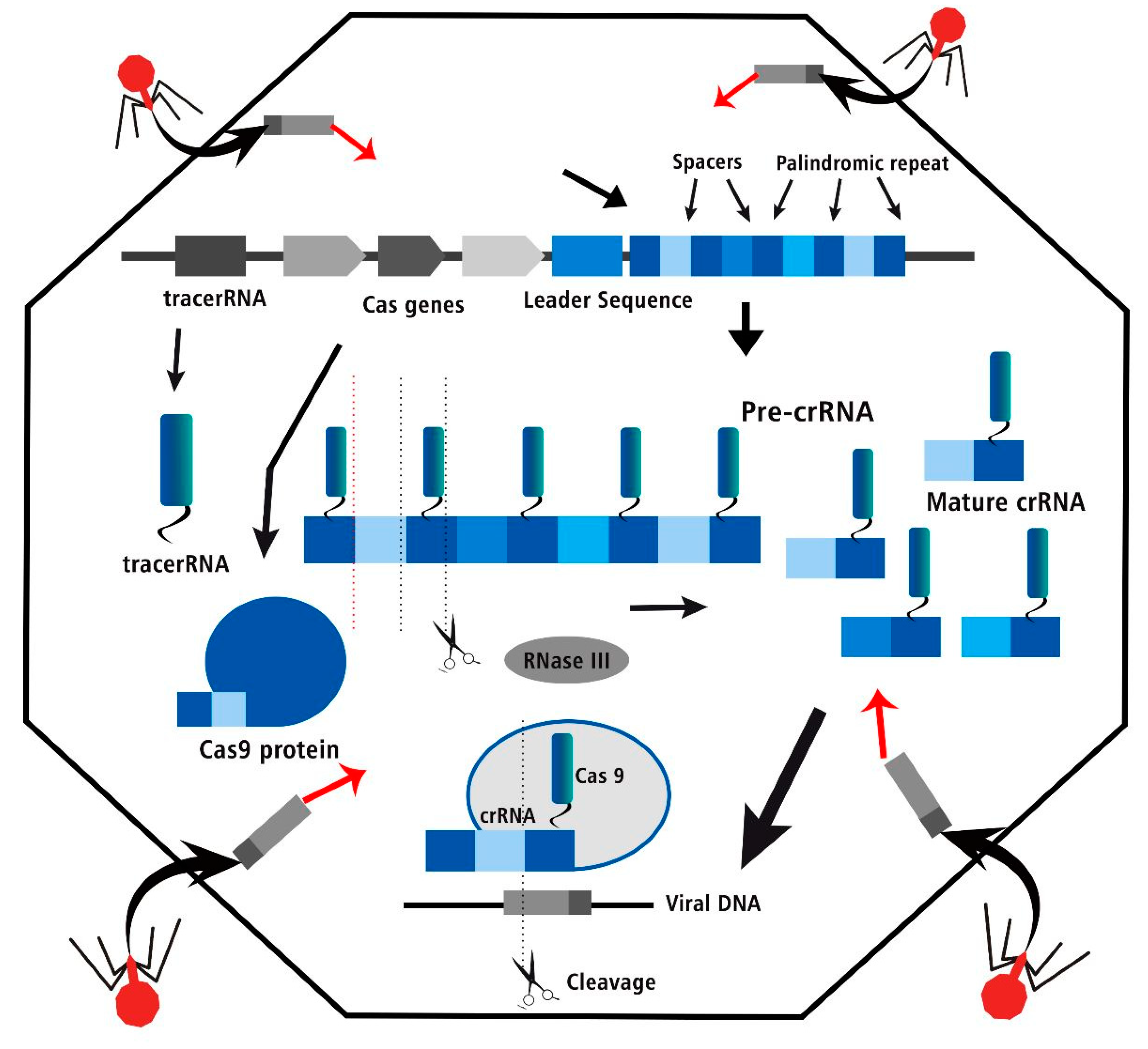
Recognition of Protospacer Adjacent Motif
The PAM is an important part of how CRISPR-Cas systems can tell the difference between their own DNA and alien nucleic acids. This lets us find a specific DNA molecule that needs to be killed. Cas12a uses a strict way of quality control to make sure that the right target spacer sequences are found. To find the PAM, the WED II-III, REC1, and PAM-interacting domains all work together. This is the first step in the process of the DNA target and the crRNA coming together. When the WED and REC1 domains find the DSDNA, the LKL region of the PI domain uses two conserved prolines to put the helix into the PAM pair. These three amino acids are at lysines 667, 671, and 677 in FnCas12a. Studies of the structure of dsDNA have shown that the helix and the long axis are at an angle of 45 degrees to each other. This makes it easy for DNA with two strands to break apart. The Watson-Crick interaction between the base pairs in the dsDNA is blocked by the three conserved lysines that come after the PAM. The non-target strand (NTS) is sent to the DNase site, and the PAM-containing strand is shown so that it can combine with the crRNA. It has been shown that Cas12a goes after spacer sequences that come a long way after a 5'T-rich PAM sequence. For LbCas12a, AsCas12a, and FnCas12a, the PAM is located upstream of the 5' end of the non-target strand and has the sequence 5'-TTTN-3'. By changing the way it makes contact with the target DNA pair, Cas12a can find non-ideal PAM sequences that contain C as well as the standard 5′-TTTN-3′ PAM.
DNA unzipping, propagation and cleavage
After the crRNA-DNA hybrid R-loop has started to form, the enzyme looks in the PAM for a seed sequence of 3-5 nucleotides to keep looking for the right target. Mismatches in the seed sequence have been linked to failed cleavage. Seed patterns make it easier for the crRNA to pair up with the DNA it wants to control. Structure studies show that the PI domain goes through a series of structural changes and takes the shape of a "rail" to make room for the nt-strand as it moves to the catalytic area, where TS and NTS are split. This structure also shows the septum, which is a barrier that keeps ds DNA from re-annealing.
By cutting t-DNA, Cas12a makes a 5′-phosphorylated product. When they enter the active site, both DNA strands must be in a 5′-3′ orientation. Structural studies have shown that the NTS is made so that its 5′-3′ polarity can enter the RuvC-Nuc pocket. This is different from the TS, which has the opposite polarity. The smFRET study showed that the TS can only reach the nuclease site in the right position if Cas12a changes its shape at the tips of the REC and NUC lobes. This could explain why NTS turns into water faster than TS. The breaking process can start even if the NTS is not in the RuvC-Nuc catalytic area. It just needs to be there. During strand separation, the PAM end of the cleavage product breaks away from the complex, but the PAM end closer to Cas12a stays connected to make a broken R-loop.
Exhibition of indiscriminate ssDNA degradation activity
Cas12a has been shown to be able to do both highly selective dsDNA cleavage and random ssDNA breakdown when it is turned on by a ssDNA that fits the crRNA guide. All Cas12a orthologs have the same ability to break down any ssDNA into its single and double bases. The structures of Cas12a before, during, and after cleavage show how structures change when random things happen. This is because of the lid area, which is part of the safety features that lets you aim accurately. Before the crRNA-DNA hybrid is made, the catalytic residues are in the space where the lid covers. During hybrid creation, the lid changes shape into a helix and binds to the hybrid assembly's crRNA. So, the active spot can be reached. After the end of the dsDNA substrate has been removed, the R-loop structure is no longer in order. This shows that the catalytic site is available. This lets the catalytic gap be wide open, so any ssDNA can be easily cut. When an RNA-DNA combination is present, Cas12a is turned on. This suggested chemistry process would show how Cas12a breaks down ssDNA. It may also be harder to use because a recent study found that non-specific nicking of target sequences, which led to mistakes in far-off parts of the target DNA, makes it harder to use.
Figure 3.
The CRISPR Cas: Target recognition and endonuclease mediated dsDNA break to delete or precise insert into the genomic sequence.
Figure 3.
The CRISPR Cas: Target recognition and endonuclease mediated dsDNA break to delete or precise insert into the genomic sequence.
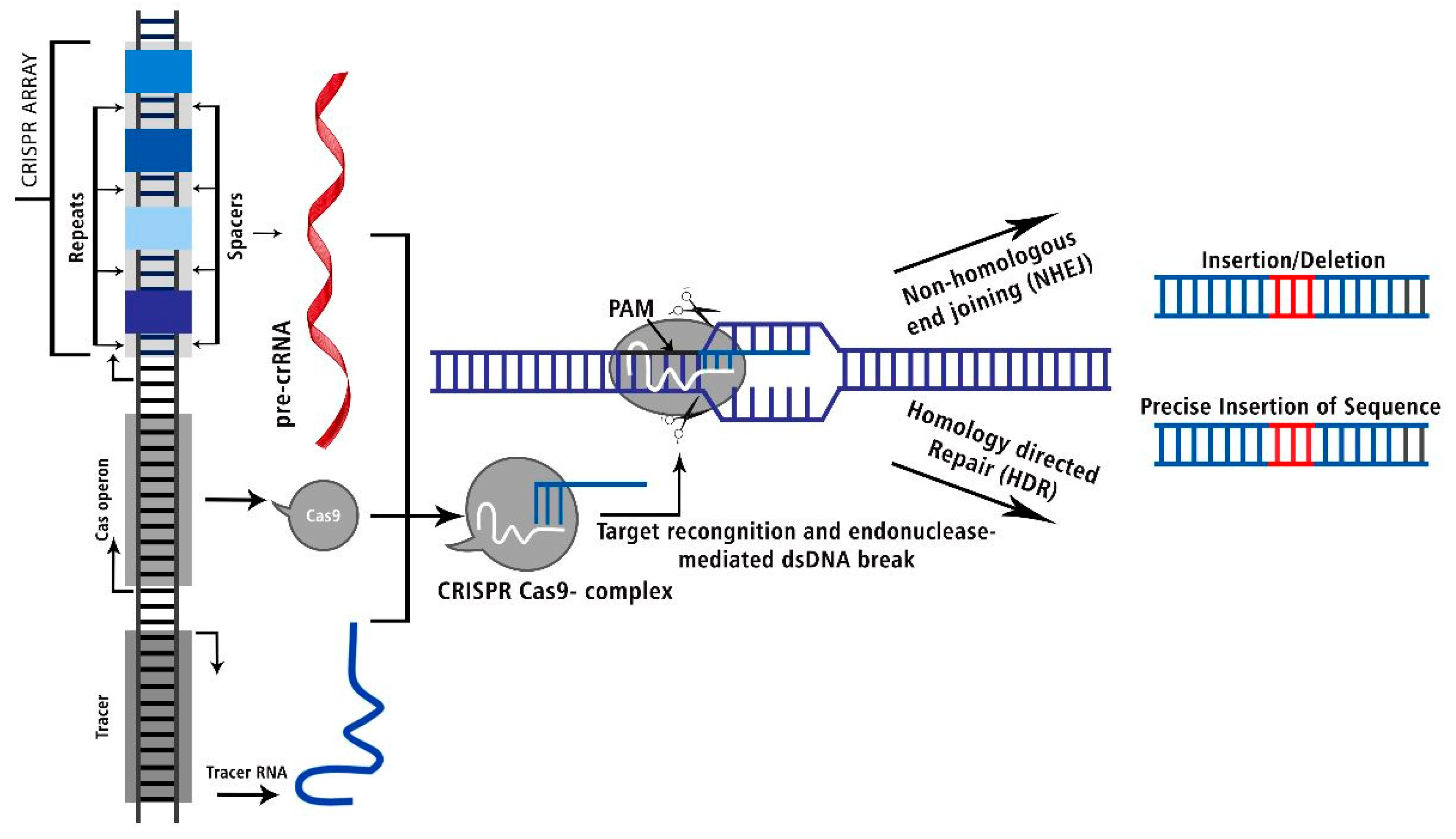
Recycling catalytic activity towards target DNAs
Randomly breaking down ssDNA could hurt the host cell because it could mess up important processes like DNA duplication, transcription, and repair. This brings up the most important question: how can we stop this dangerous chance act? Because the cell can't handle leaks that happen when ssDNA breaks randomly. Only by finding the genes in bacteria that code for Cas12a can these problems be fixed. Most bacteria only have one copy of the Cas12a gene, but a database search using a sequence of a conserved crRNA showed that some bacteria had as many as 68 copies of the crRNA. If transcription rates stay the same, there would be many more crRNAs than enzymes in a cell at any given time. Experiments show that if there are enough molecules of crRNA, the R-loop can be moved away from the enzyme and a new interference complex can be made. This shows that if a new crRNA is put in place of the cleaved R-loop, Cas12a can go back to its inactive state and stop all general activity. So, the molecule goes back to a state where the "lid" makes polar bonds and blocks entry to the catalytic area. This not only stops ssDNase from doing something wrong, but it also moves the catalytic energy it makes to other DNAs.
CRISPR-Cas System Molecular Tools to Edit Genome
CRISPR-Cas systems use the host cell's natural DNA repair systems to fix a double strand break (DSB) in a certain part of the genome. There are two ways to fix a problem: by using "homology-directed repair" (HDR) or "non-homologous end joining" (NHEJ). HDR is better than NHEJ because it doesn't directly join the broken ends of the DSB. Instead, it uses a template DNA that is homologous to the break site, such as an unbroken sister chromatid or chromosome. The user of HDR can put alien template DNA into the host genome and change it in any way they want. NHEJ makes it possible to change how genes work. On the other hand, HDR can only change a specific piece of code directly or add new DNA.
Conclusions
The last part gave an overview of these study projects and explained how to make Cas12a for specific uses. Cas12a's uses have grown a lot in recent years, but the protein still has a lot of promise as a treatment and screening tool. In order to give a quick review of CRISPR-Cas12a and its uses, we will briefly talk about the structure and function of the different parts of the reaction pathway that lead up to the catalysis of the target DNA. Cas12a uses a multistep process to make sure that it is selecting the right DNA. This is a good trait for a device that changes the DNA because it makes it less likely that something bad will happen. Even though data shows that a new crRNA molecule can stop Cas12a from randomly destroying ssDNA, it may still hurt the host cell while trying to change the genome. The action of Cas12a catalysis, which is a powerful tool for changing the genome, needs to be changed so that it can be more easily controlled and managed. Using what we know about how Cas12a works, we have made mutants that are less active on ssDNA and more active on dsDNA. So far, only Cas9 and Cas12a, which are both part of the CRISPR family, have been used to change the genome. Because of how similar and different these two endonucleases are, CRISPR can now be used for a lot more things. Cas12a is better than Cas9 because it makes double-strand breaks (DSBs) that favour HDR over NHEJ and can change more than one copy of the genome at the same time. Both Cas9 and Cas12a are being changed to make them better at detecting PAM than they are in their natural state. This will make it possible to start focusing on more genes.
Funding
This project is not-funded from any local and/ or international organization.
Authors' contributions and materials
This work was carried out in collaboration among all authors. Taha Nazir designed the study of proposed hypothesis and compile the scientific contents. Nida Taha elaborated study to make it more credible. Whereas, Hameed A Mirza managed the literature searches and citation part of the manuscript. Thus, all authors have read and approved the final manuscript for publication in this journal.
Ethical Approval and Consent to participate
All procedures performed in studies are not involving human participants. Therefore there is no need of the ethical approval of the institutional and/or national research committee and 1964 Helsinki declaration and its later amendments or comparable ethical standards. For type of studies no formal consent is required.
Animal rights
Additionally, this research studies no animals involved. The authors indicate the procedures followed are in accordance with the standards set forth in the eighth edition of Guide for the Care and Use of Laboratory Animals; published by the National Academy of Sciences, The National Academies Press, Washington, D.C.).
Consent for publication
Authors agree and grant consent to publish this article in this research journal.
Availability of data
All study information and possible research data successfully incorporated for publication.
Acknowledgments
We acknowledge the technical and scientific support of A.S. Chemical Laboratories Inc., Concord, ON L4K4M4 Canada and Advanced Multiple Inc., Mississauga ON, L5T2M9 Canada
Competing interests
The authors also declare that they are no any potential and/ or completing conflict of interest.
References
- Akbari Kordkheyli V, Rashidi M, Shokri Y, Fallahpour S, Variji A, Nabipour Ghara E, Hosseini SM. CRISPER/CAS System, a Novel Tool of Targeted Therapy of Drug-Resistant Lung Cancer. Adv Pharm Bull. 2022 Mar;12(2):262-273. Epub 2021 Apr 3. PMID: 35620343; PMCID: PMC9106967. [CrossRef] [PubMed]
- Abbott TR, Dhamdhere G, Liu Y, Lin X, Goudy L, Zeng L, Chemparathy A, Chmura S, Heaton NS, Debs R, Pande T. Development of CRISPR as an antiviral strategy to combat SARS-CoV-2 and influenza. Cell. 2020;181:865–876. [CrossRef]
- Ahi YS, Bangari S, D, Mittal KS, Adenoviral vector immunity: its implications and circumvention strategies. Curr Gene Ther. 2011;11:307–320. [CrossRef]
- Azhar M, Phutela R, Kumar M, Ansari AH, Rauthan R, Gulati S, Sharma N, Sinha D, Sharma S, Singh S, Acharya S. Rapid and accurate nucleobase detection using FnCas9 and its application in COVID-19 diagnosis. Biosens Bioelectron. 2021;183:113207. [CrossRef]
- Joung J, Ladha A, Saito M, Segel M, Bruneau R, Huang ML, Kim NG, Yu X, Li J, Walker BD, Greninger A. Point-of-care testing for COVID-19 using SHERLOCK diagnostics. MedRxiv. 2020. [CrossRef]
- Kumar A, Birnbaum MD, Moorthy BT, Singh J, Palovcak A, Patel DM, Zhang F. Insertion/deletion-activated frame-shift fluorescence protein is a sensitive reporter for genomic DNA editing. BMC Genom. 2019;20:609. [CrossRef]
- Ma L, Yin L, Li X, Chen S, Peng L, Liu G, Ye S, Zhang W, Man S. A smartphone-based visual biosensor for CRISPR-Cas powered SARS-CoV-2 diagnostics. Biosens Bioelectron. 2022;195:113646. [CrossRef]
- Testa U, Castelli G, Pelosi E. Lung cancers: molecular characterization, clonal heterogeneity and evolution, and cancer stem cells. Cancers (Basel) 2018;10(8):248. [CrossRef]
- Saito M, Shiraishi K, Kunitoh H, Takenoshita S, Yokota J, Kohno T. Gene aberrations for precision medicine against lung adenocarcinoma. Cancer Sci. 2016;107(6):713–20. [CrossRef]
- Saito M, Shiraishi K, Kunitoh H, Takenoshita S, Yokota J, Kohno T. Gene aberrations for precision medicine against lung adenocarcinoma. Cancer Sci. 2016;107(6):713–20. [CrossRef]
- Rajan A, Shrivastava S, Janhawi, Kumar A, Singh AK, Arora PK. CRISPR-Cas system: from diagnostic tool to potential antiviral treatment. Appl Microbiol Biotechnol. 2022 Sep;106(18):5863-5877. Epub 2022 Aug 26. PMID: 36008567; PMCID: PMC9411046. [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



