
Submitted:
27 January 2024
Posted:
29 January 2024
You are already at the latest version
Abstract
Alveolar Rhabdomyosarcoma (ARMS), an invasive subtype of Rhabdomyosarcoma (RMS), is associated with chromosomal translocation events resulting in one of two oncogenic fusion genes PAX3-FOXO1 or PAX7-FOXO1. ARMS patients exhibit an overexpression of the pleiotropic cytokine Transforming growth factor beta (TGF-β). This overexpression of TGF-β1 causes an increased expression of a downstream transcription factor called SNAIL, which promotes Epithelial to Mesenchymal Transition (EMT). Overexpression of TGF-β also inhibits myogenic differentiation, making ARMS patients highly resistant to chemotherapy. In this review, we first describe different types of RMS and then focus on ARMS and the impact of TGF-β in this tumor type. We next highlight current chemotherapy strategies including a combination of FDA approved drugs vincristine, actinomycin D, and cyclophosphamide (VAC), Cabozantinib, Bortezomib, Vinorelbine, AZD 1775 and cisplatin. Lastly, we discuss chemotherapy agents that target the differentiation of tumor cells in ARMS which include all trans retinoic acid (ATRA) and 5-Azacytidine. Improving our understanding of the role of signaling pathways, such as TGF-β1, in the development of ARMS tumor cells differentiation will help inform more tailored drug administration in the future.
Keywords:
Rhabdomyosarcoma
; Combination-therapy
; all trans retinoic acid
; TGF-beta1
Introduction
Rhabdomyosarcoma overview
Rhabdomyosarcoma (RMS) is a highly recurrent pediatric soft tissue sarcoma accounting for approximately 50% of the childhood cancer cases, and has a higher incidence in males than females with a male/female ratio of 1.4:1 (1-3). RMS is considered as a high grade, malignant neoplasm wherein the cells resemble myoblasts, express myogenic markers, and are suspected to be derived from muscle progenitor cells (3-5). The expression of myogenic factors such as MyoD strongly supports the hypothesis that RMS originates from a mesenchymal cell lineage that gives rise to myoblasts (Keller & Guttridge, 2013; Martin-Giacalone et al., 2021; Morita & Hayashi, 2022). In addition to occurring within skeletal muscle of the extremities, due to its totipotent origin, RMS can also occur in regions such as the head, neck, genitourinary organs, abdomen, and bile ducts (2, 6, 7). According to the World Health Organization (WHO), there are four classifications of RMS, namely alveolar, embryonal, spindle cell/sclerosing and pleomorphic RMS (7) (Figure 1). However, histologically alveolar (~60-70% of the cases) and embryonal (~20-30% of the cases) RMS are considered as the two major subtypes (7, 8). Due to its high tendency to metastasize, alveolar rhabdomyosarcoma (ARMS) is considered an invasive subtype largely found at the extremities of adolescents (9). Its name is derived from its histological features, as it exhibits a fibrovascular septum of connective tissue with neoplastic cells attached, vaguely reminiscent of the alveolar spaces in the lungs (2, 10). The cells themselves can be characterized as uniformly polygonal with an oval/round nucleus that are hyperchromatic (2, 11).
Tumour Growth and Differentiation in ARMS
PAX3/7 and FOXO1
Epithelial to mesenchymal (EMT) is a multi-step process. During EMT epithelial cells are reprogrammed into a mesenchymal phenotype decreasing cell-to-cell adhesion, cytoskeleton remodeling, basement membrane invasion, acquisition of motility, and a lack of cell polarity (Sannino et al., 2017). This results in normally cohesive cells to shift from their rigid epithelial organization and metastasize to other loci contributing to cancer progression and drug resistance (12). The commitment of cells to the EMT program is orchestrated by specific transcription factors, therefore making transcription factors a major focus of cancer research.
Significantly, ~80% of the ARMS cases manifest due to a chromosomal translocation between paired box protein PAX3 gene found on chromosome 2 or PAX7 gene found on chromosome 1, and forkhead box protein FOXO1 gene on chromosome 13. It consequently results in fusion genes (PAX 3/7-FOXO1) that are considered major drivers of oncogenic activity (7, 13) (Figure 2). The N-terminal DNA binding domain of PAX3 or PAX7 is fused to the C-terminal transactivation domain of FOXO1 (5, 14).
Transcription factors, known as myogenic factors, not only regulate the physiological process of muscle formation (i.e. myogenesis), but also play an important role in pathologic myogenic differentiation. Accordingly, aberrations within these pathways can lead to development of RMS (15, 16). Previous studies suggest that the development of RMS correlates with a differentiation defect in either stem cells or early progenitor cells such as mesenchymal stem cells (15, 17). ARMS not only has the presence of the PAX3/PAX7 and FOXO1 fusion gene but also display increased levels of MET; a receptor tyrosine kinase (RTK) known to be linked with the metastatic potential of RMS cells (15, 18). Furthermore, recent studies have shown that aggressive ARMS tumors exhibit high expressions of SNAIL which coincidently has a positive correlation to PAX3/7-FOXO1 (15, 19-21). SNAIL has thus become a prominent research focus as it plays a central role in EMT (15, 19).
SNAIL is a zinc finger transcription factor that usually acts as a gene repressor making it a crucial regulator of ARMS growth (20, 21). It is known to be a master regulator of growth and metastasis as it involves the induction of EMT by binding to E-box sequences in the promoter region on genes, and, affects the EZRIN cytoskeleton protein and AKT serine/threonine kinase levels respectively (16, 20, 21). In ARMS the PAX3/7-FOXO1 fusion gene not only alters the myogenic program and maintains the proliferative state while blocking terminal differentiation, but also results in an overexpression of TGF-β (22, 23). This overexpression of TGF-β results in an upregulation of its two major downstream pathways further allowing SNAIL to promote EMT (Figure 3). Additionally, SNAIL also regulates the microRNA transcriptome either directly or indirectly through binding their promoter or regulatory regions in RMS cells (16, 19, 21). To this end, gene ontology analysis revealed that SNAIL-miRNA axis does in fact play a role in differentiation, reorganization of actin skeleton and migration (16, 19-21).
Transforming Growth Factor-β
In addition to the PAX 3/7-FOXO1 fusion gene, ARMS cells also exhibit an increased expression of TGF-β family proteins, in particular TGF-β1 (24, 25). TGF-β1, a member of the transforming growth factor (TGF) superfamily, is a pleiotropic cytokine that is secreted by fibroblasts and epithelial cells (26-28). It is encoded by 33 genes in mammals with the capability to function as both a homo- and heterodimer (27, 29). In addition to the function of TGF-β in cellular development, different members of the TGF-β superfamily are intensely studied for its widespread role in various diseases, including cancer (29, 30). TGF-β is known to strongly inhibit the proliferation of many cell types, including endothelial, epithelial and immune cells, while playing a prominent role in controlling the differentiation of cell lineages during development (29, 31, 32). Additionally, TGF-β family proteins are involved in other cellular functions such as promoting/protecting against cell death, cell motility and invasion (29, 31, 33).
As shown in Figure 4, the TGF-β receptor complex transduces signals to regulate context-dependent transcription through two major downstream signalling pathways: (1) SMAD pathways (canonical) and (2) non-SMAD pathways (non-canonical) (Peng et al., 2022b). The canonical TGF-β pathway transfers signals via SMAD2/3, and once phosphorylated by TβR I it regulates gene transcription by translocating into the nucleus (Peng et al., 2022b; Yeganeh et al., 2013). On the contrary, the non-canonical pathway refers to the activation of downstream cascades by TGF-β via phosphorylation, acetylation, protein-protein interactions, ubiquitination and sumoylation as well as involves the MAPK/ERK kinases (32).
TGF-β plays a dual role in cancer (23) (Figure 5). Under normal expression TGF-β has a tumour suppressor effect and promotes differentiation. Conversely, when overexpressed, as in the case of ARMS, TGF-β is known to decrease differentiation and inhibit myogenic differentiation (23). The inhibition of myogenic differentiation is considered a negative effect, as undifferentiated cells are more resistant to chemotherapy. This supports the notion that ARMS is not only an invasive subtype but also highly resistant to therapy (23). Overexpression also results in an over-activation of the downstream cascades resulting in the induction of EMT and cancer progression.
Thus, using chemotherapy agents that both increase differentiation and kills ARMS tumor cells could be advantageous for better treatment of this malignant tumor.
ARMS Chemotherapy Drugs and Their Impact in the Tumor Cells Differentiation
Currently, a multiple approaches consisting of a combination of surgery, chemotherapy and/or radiotherapy are being used to treat at risk ARMS patients (34). Due to the shortage of efficacious treatment options there has been insufficient advancements when it comes to treatment options, thus hindering improvement in the outcome of metastatic or relapsed ARMS (35). Intravenous (IV) administration of Vincristine, actinomycin D, and cyclophosphamide (VAC) are considered the classic chemotherapy drugs in North America with only the duration and dosage of each being modified over the last four decades (34). Multiple studies have been conducted to improve treatment plans using a variety of chemotherapeutic agents that target characteristic features of ARMS, including inhibition of myogenic differentiation. Thus, many of these drug strategies focus on the ability to ‘rehabilitate’ differentiation.
Vincristine, Actinomysin D and Cyclophosphamide (VAC)
The VAC regimen consisting of FDA approved drugs; vincristine, actinomycin D, and cyclophosphamide is the standard chemotherapy combination with a response rate of ~70-80% in ARMS patients (36). Vincristine is an anti-tumour vinca alkaloid considered to be a potent microtubule inhibitor (37, 38). It acts by irreversibly binding to and stabilizing tubulin therefore, interfering with microtubule polymerization, inherently preventing mitosis and inhibiting cell growth (39). Actinomycin D (ActD) is a known transcription inhibitor, it binds guanine residues and inhibits the function of DNA-dependent RNA polymerase therefore preventing RNA synthesis (40). This characteristic permits it to act a cytotoxic inducer of apoptosis against tumor cells (40). A study also showed that when administered in low controlled doses, actinomycin D promotes myogenic differentiation while inhibiting proliferation thereby making cells less resistant to chemotherapy (41). Cyclophosphamide is a nitrogen mustard drug that has the ability to alkylate DNA; it does so by metabolizing into it’s active form phosphoramide (42). Phosphoramide has the capability of forming cross-linkages at the guanine N-7 position, both in between and within DNA strands, thus leading to programmed cell death (42).
All three of these drugs act differentially to collective halt tumour growth. This is achieved by either preventing cell division, stopping metastasis or facilitating cell death, therefore proving to be an effective chemotherapy standard for ARMS patients. (Figure 6)
Cabozantinib (XL184)
As discussed above, in ARMS MET signalling promotes growth and proliferation, inhibits myogenic differentiation, and increases its metastatic potential (35). The receptor tyrosine kinase inhibitor, Cabozantinib (XL184), upstream of the MET pathway, is used to counteract and impair tumor cell proliferation and angiogenesis, promoting myogenic differentiation (35). Cabozantinib is administered orally and is currently in phase II clinical trials with a recommended dosage of 40 mg/m2/day (43) (Figure 7).
Bortezomib
Another characteristic feature of ARMS is that it exhibits decreased levels of apoptosis and is successful in evading cell cycle arrest (34). In vitro studies have shown that when treated with Bortezomib, a protease inhibitor that functions by inhibiting the 26S proteosome, ARMS cell lines express increased levels of apoptosis and cell cycle arrest (35).
Vinorelbine
A recent clinical trial was conducted to test the effectiveness of Vinorelbine, a second generation semisynthetic vinca alkaloid with antimitotic and anticancer properties (9). Vinorelbine acts by inhibiting microtubule dynamics by binding microtubular proteins in the mitotic spindle preventing chromosomal segregation and triggering the cancerous cells to undergo apoptosis (9). The results of the trial indicated that Vinorelbine had significant affects both when used as a single agent and in combination, therefore making it an effective chemotherapeutic drug (9). The recommended combination therapy is a low dose of cyclophosphamide ~25mg/m2 per day for 28 days with the administration of 25mg/m2 of vinorelbine on days 1, 8 and 15 (44).
AZD1775
Wee1 kinase is a cell cycle regulator and it acts by the inhibition of cyclin dependent kinase 1 and phosphorylation (35). AZD1775 is a selective tyrosine kinase inhibitor which has been reported to inhibit the growth of several sarcoma cell lines, however it’s role is ARMS is still misunderstood (35).
A recent study illustrated that AZD1775 could serve as a viable therapeutic agent in combination with conventional chemotherapy (35). The Wee 1 kinase is a cell cycle regulator that specifically maintains the cell in the G2/M phase therefore providing sufficient time to repair DNA prior to undergoing mitosis (Kahen et al., 2016). When inhibited by AZD1775 however, the CDK1/2 activity takes over unchecked, allowing the cells to prematurely progress through the G2/M phase and undergo mitosis. The outcome of this leads to DNA strand breaks, mitotic dysfunction and cell death (35).
Previous literature has also shown that when used in combination with gemcitabine, AZD1775 lead to not only a delay in tumor growth but also smaller tumors in osteosarcoma cells (35). It is also hypothesized that if combined with conventional chemotherapy, they could work synergistically to make the DNA damage more potent (35). These findings indicate potential of this agent for improved treatment strategies for ARMS, once evaluated more in future studies(35).
Entinostat, Panobinopstat and Vorinostat
Histone deacetylase (HDAC) is an epigenetic marker that when inhibited has antitumour effects, as has been demonstrated using an RMS model (34). Agents such as entinostat, panobinopstat and vorinostat are known HDAC inhibitors and have been reported to delay tumor growth in RMS xenografts (34). In addition to disrupting transcriptional complexes leading to suppression of key oncogenic genes, HDAC inhibitors also have the capability to induce transcriptional stress resulting in their terminal differentiation or apoptotic cell death (34) (Figure 8).
Critotinib, Bevacizumab (mAb) and Regorafenib
The constitutive activation of receptor tyrosine kinases (RTK) such as ALK, MET, VEGFR is known to promote tumor progression in ARMS, by reprogramming many intracellular pathways, such as those involved in differentiation (34). Currently, there are two main strategies for targeting RTK’s: small molecule kinase inhibitors and immunotherapy-like monoclonal antibodies. Critotinib, Bevacizumab (mAb) and Regorafenib namely are a few examples of RTK inhibitors that have successfully induced tumor regression in preclinical models (34).
All Trans Retinoic Acid (ATRA)
All trans retinoic acid (ATRA) is an important metabolite of vitamin A mediating functions of growth and development (45). It plays a crucial role in cell proliferation, cell differentiation, apoptosis and embryonic development (46). ATRA primarily exerts its effects through its interactions with nuclear retinoic acid receptors (RARs), which are transcription factors (47). RARs form heterodimers with retinoid X receptors (RXRs) and regulate the transcription of target genes (48, 49). The RAR signalling pathway plays a crucial role in mediating the differentiation-promoting effects of ATRA in ARMS (50). By activating RARs, ATRA can modulate the expression of genes associated with myogenic differentiation; the process by which ARMS cells transform into more mature, muscle-like cells.
ATRA exerts differentiation-inducing effects in ARMS through various mechanisms. For instance, it can act by upregulating MYOD1, a crucial myogenic regulator, promoting commitment to the muscle lineage and facilitating ARMS cell differentiation (51). Moreover, ATRA also targets the hallmark genetic abnormality in ARMS, inhibiting the PAX3-FOXO1 fusion protein's expression and activity, thus disrupting oncogenic signaling and facilitating differentiation (49). Finally, ATRA induces epigenetic changes, such as DNA methylation and histone modifications, influencing the expression of genes related to myogenic differentiation in ARMS cells (52). These mechanisms collectively contribute to ATRA's potential as a therapeutic agent for ARMS differentiation.
Cisplatin
Cisplatin, is a toxic antineoplastic agent, but one of the most heavily employed agents that first came to use in the 1970’s. It operates by inducing DNA damage and subsequent cell cycle arrest, rather than directly promoting differentiation in cancer cells (53). It forms covalent bonds with DNA, leading to the formation of DNA crosslinks and adducts, triggering DNA repair responses and, in ARMS cells, potentially causing apoptosis rather than differentiation (54). Cisplatin is often administered in combination with other chemotherapeutic agents to enhance treatment efficacy and inhibit tumor growth. Additionally, it can act as a radiosensitizer, increasing the sensitivity of cancer cells to radiation therapy, which is commonly part of ARMS treatment (55). The utilization of various therapeutic approaches, including cisplatin, aims to achieve comprehensive control of ARMS, with treatment plans tailored to individual patients in consultation with medical oncologists.
5-Azacytidine
5. -Azacytidine is a demethylating agent which functions by incorporating into DNA during replication and inhibiting DNA methyltransferase activity, resulting in DNA demethylation (56). In ARMS, hypermethylation of specific genes can lead to their silencing, including those associated with differentiation (57). 5-Azacytidine treatment can reverse this process, allowing previously silenced genes linked to myogenic differentiation to be re-expressed. This demethylation also contributes to the restoration of myogenic regulatory factors (MRFs) such as MyoD and myogenin, which are crucial for myogenic differentiation (58). Furthermore, 5-Azacytidine can induce cell cycle arrest and cellular senescence, promoting a more mature cellular state in ARMS (59). However, it is important to note that 5-Azacytidine's effectiveness often varies among patients and is often used in the context of personalized treatment plans for ARMS, typically under clinical trial conditions.
The following table summarizes the key molecular targets of the drugs discussed.

Table 1.
Chemotherapy drugs used for ARMS and their molecular targets.
| Drug/Compound | Molecular Target | Reference |
|---|---|---|
| Vincristine, Actinomycin D, and Cyclophosphamide (VAC) | Microtubule Polymerization, Guanine nucleotide in DNA, cross-linkages with guanine N-7 respectively | (40, 42) |
| Cabozantinib (XL184) | Tyrosine Kinase (MET) | (35) |
| Bortezomib | 26s proteosome | (35) |
| Vinorelbin | Microtubular Proteins | (9) |
| AZD1775 | Wee 1 | (35) |
| Entinostat, Panobinopstat and Vorinostat | Histone Deacetylase (HDAC) | (34) |
| Critotinib, Bevacizumab (mAb) and Regorafenib | Receptor Tyrosine Kinae (RTK) | (34) |
| All Trans Retinoic Acid (ATRA) | retinoic acid receptors (RARs) | (20) |
| Cisplatin | DNA | (53) |
| 5-Azacytidine | DNA methyltransferase | (56) |
Conclusion and Future Direction
Targeting tumor cells using multiple targeted strategies is becoming a more popular approach to providing chemotherapy. As an example, in addition to the current therapies there is ongoing investigations exploring if simultaneous targeting of differentiation pathways and other chemotherapy agents could be more effective in ARMS therapy. Since ARMS cells show a decrease in myogenic differentiation this is becoming a major focus of future studies. As mentioned above, the TGF-β pathway could be an important research target for better chemotherapeutic outputs in ARMS because TGF-β has been observed to be involved in decreased differentiation in ARMS tumor cells. Future studies should focus on multiple molecularly targeted therapies since the addition of cytotoxic agents to the current chemotherapy regime has depicted minimal success (Skapek et al., 2019). As an example, our team has recently designed a specific ATRA encapsulated in bioengineered MESH material (60) that could be used in the future for combination targeted therapy for ARMS. Bearing in mind the ground-breaking findings that point towards an overexpression of TGF-β in ARMS, it is safe to say that future research should invest greater timein understanding this pathway and its role in ARMS. Developing new treatment therapies that specifically target TGF-β and its effect on ARMS differentiation and lead to its downregulation would present a promising advancement in improving the outcome of ARMS patients.
Author Contributions
BB: RI, AE, and SCDR participated the literature review, first draft preparation, figure design. SCDR and SG supervised BB, RI, and AE. BL led the differentiation section and final draft preparation. JWG led muscular differentiation sections and final draft preparation. SG led the team, led the cancer biology and TGF section.
Acknowledgments
Rosa Iranpour was supported by a Cancer Care Manitoba (CCMB) Summer Studentship. Simone C da Silva Rosa was supported by a Canadian Institutes of Health and Research (CIHR) Postdoctoral Fellowship (CIHR-FRN-176508).
Conflicts of Interest
The authors do not have any conflict of interest.
References
- Aghaei M, Nasimian A, Rahmati M, Kawalec P, Machaj F, Rosik J, et al. The Role of BiP and the IRE1alpha-XBP1 Axis in Rhabdomyosarcoma Pathology. Cancers (Basel). 2021, 13.
- Eguia-Aguilar P, Lopez-Martinez B, Retana-Contreras C, Perezpena-Diazconti M. Alveolar rhabdomyosarcoma: Origin and prognostic implications of molecular findings. Bol Med Hosp Infant Mex. 2016, 73, 405–410.
- Stefanek E, Samiei E, Kavoosi M, Esmaeillou M, Roustai Geraylow K, Emami A, et al. A bioengineering method for modeling alveolar Rhabdomyosarcoma and assessing chemotherapy responses. MethodsX. 2021, 8, 101473. [Google Scholar] [CrossRef] [PubMed]
- Nhung TH, Minh VL, Tuyet TT, Cuong TM, Lam NL, Trang HT, et al. Orbital rhabdomyosarcoma in a 19-year-old male patient: A case report and literature review. Radiol Case Rep. 2023, 18, 2744–2749. [Google Scholar] [CrossRef]
- Skapek SX, Ferrari A, Gupta AA, Lupo PJ, Butler E, Shipley J, et al. Rhabdomyosarcoma. Nat Rev Dis Primers. 2019, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- Emami A, Shojaei S, da Silva Rosa SC, Aghaei M, Samiei E, Vosoughi AR, et al. Mechanisms of simvastatin myotoxicity: The role of autophagy flux inhibition. Eur J Pharmacol. 2019, 862, 172616. [Google Scholar] [CrossRef] [PubMed]
- Martin-Giacalone BA, Weinstein PA, Plon SE, Lupo PJ. Pediatric Rhabdomyosarcoma: Epidemiology and Genetic Susceptibility. J Clin Med. 2021, 10. [Google Scholar]
- Moghadam AR, da Silva Rosa SC, Samiei E, Alizadeh J, Field J, Kawalec P, et al. Autophagy modulates temozolomide-induced cell death in alveolar Rhabdomyosarcoma cells. Cell Death Discov. 2018, 4, 52. [Google Scholar] [CrossRef]
- Allen-Rhoades W, Lupo PJ, Scheurer ME, Chi YY, Kuttesch JF, Venkatramani R, et al. Alveolar rhabdomyosarcoma has superior response rates to vinorelbine compared to embryonal rhabdomyosarcoma in patients with relapsed/refractory disease: A meta-analysis. Cancer Med. 2023, 12, 10222–10229. [Google Scholar] [CrossRef]
- Lewandowski D, Szewczyk A, Radzka J, Dubinska-Magiera M, Kazimierczak W, Daczewska M, et al. The natural origins of cytostatic compounds used in rhabdomyosarcoma therapy. Adv Clin Exp Med. 2023, 32, 1179–1191. [Google Scholar] [CrossRef]
- Nelson AC, Singh C, Pambuccian SE. Cytological diagnosis of metastatic alveolar rhabdomyosarcoma in the ascitic fluid: Report of a case highlighting the diagnostic difficulties. Cytojournal. 2012, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Sannino G, Marchetto A, Kirchner T, Grunewald TGP. Epithelial-to-Mesenchymal and Mesenchymal-to-Epithelial Transition in Mesenchymal Tumors: A Paradox in Sarcomas? Cancer Res. 2017, 77, 4556–4561. [Google Scholar] [CrossRef] [PubMed]
- Lagutina IV, Valentine V, Picchione F, Harwood F, Valentine MB, Villarejo-Balcells B, et al. Modeling of the human alveolar rhabdomyosarcoma Pax3-Foxo1 chromosome translocation in mouse myoblasts using CRISPR-Cas9 nuclease. PLoS Genet. 2015, 11, e1004951. [Google Scholar]
- Azorsa DO, Bode PK, Wachtel M, Cheuk ATC, Meltzer PS, Vokuhl C, et al. Immunohistochemical detection of PAX-FOXO1 fusion proteins in alveolar rhabdomyosarcoma using breakpoint specific monoclonal antibodies. Mod Pathol. 2021, 34, 748–757. [Google Scholar] [CrossRef]
- Skrzypek K, Kusienicka A, Trzyna E, Szewczyk B, Ulman A, Konieczny P, et al. SNAIL is a key regulator of alveolar rhabdomyosarcoma tumor growth and differentiation through repression of MYF5 and MYOD function. Cell Death Dis. 2018, 9, 643. [Google Scholar] [CrossRef]
- Skrzypek K, Nieszporek A, Badyra B, Lasota M, Majka M. Enhancement of myogenic differentiation and inhibition of rhabdomyosarcoma progression by miR-28-3p and miR-193a-5p regulated by SNAIL. Mol Ther Nucleic Acids. 2021, 24, 888–904. [Google Scholar] [CrossRef]
- Charytonowicz E, Cordon-Cardo C, Matushansky I, Ziman M. Alveolar rhabdomyosarcoma: Is the cell of origin a mesenchymal stem cell? Cancer Lett. 2009, 279, 126–136. [Google Scholar] [CrossRef]
- Miekus K, Lukasiewicz E, Jarocha D, Sekula M, Drabik G, Majka M. The decreased metastatic potential of rhabdomyosarcoma cells obtained through MET receptor downregulation and the induction of differentiation. Cell Death Dis. 2013, 4, e459. [Google Scholar] [CrossRef]
- Ramadan F, Saab R, Hussein N, Clezardin P, Cohen PA, Ghayad SE. Non-coding RNA in rhabdomyosarcoma progression and metastasis. Front Oncol. 2022, 12, 971174. [Google Scholar] [CrossRef]
- Skrzypek K, Kot M, Konieczny P, Nieszporek A, Kusienicka A, Lasota M, et al. SNAIL Promotes Metastatic Behavior of Rhabdomyosarcoma by Increasing EZRIN and AKT Expression and Regulating MicroRNA Networks. Cancers (Basel). 2020, 12. [Google Scholar]
- Skrzypek K, Adamek G, Kot M, Badyra B, Majka M. Progression and Differentiation of Alveolar Rhabdomyosarcoma Is Regulated by PAX7 Transcription Factor-Significance of Tumor Subclones. Cells. 2021, 10. [Google Scholar]
- Laubscher D, Gryder BE, Sunkel BD, Andresson T, Wachtel M, Das S, et al. BAF complexes drive proliferation and block myogenic differentiation in fusion-positive rhabdomyosarcoma. Nat Commun. 2021, 12, 6924. [Google Scholar] [CrossRef]
- Schmitt-Ney M, Camussi G. The PAX3-FOXO1 fusion protein present in rhabdomyosarcoma interferes with normal FOXO activity and the TGF-beta pathway. PLoS ONE. 2015, 10, e0121474. [Google Scholar]
- Petragnano F, Pietrantoni I, Camero S, Codenotti S, Milazzo L, Vulcano F, et al. Clinically relevant radioresistant rhabdomyosarcoma cell lines: Functional, molecular and immune-related characterization. J Biomed Sci. 2020, 27, 90. [Google Scholar]
- Wang S, Guo L, Dong L, Guo L, Li S, Zhang J, et al. TGF-beta1 signal pathway may contribute to rhabdomyosarcoma development by inhibiting differentiation. Cancer Sci. 2010, 101, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Dalvand A, da Silva Rosa SC, Ghavami S, Marzban H. Potential role of TGFBeta and autophagy in early crebellum development. Biochem Biophys Rep. 2022, 32, 101358. [Google Scholar]
- Morikawa M, Derynck R, Miyazono K. TGF-beta and the TGF-beta Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb Perspect Biol. 2016, 8.
- Siapoush S, Rezaei R, Alavifard H, Hatami B, Zali MR, Vosough M, et al. Therapeutic implications of targeting autophagy and TGF-beta crosstalk for the treatment of liver fibrosis. Life Sci. 2023, 329, 121894. [Google Scholar] [CrossRef]
- Derynck R, Budi EH. Specificity, versatility, and control of TGF-beta family signaling. Sci Signal. 2019, 12.
- Alizadeh J, Glogowska A, Thliveris J, Kalantari F, Shojaei S, Hombach-Klonisch S, et al. Autophagy modulates transforming growth factor beta 1 induced epithelial to mesenchymal transition in non-small cell lung cancer cells. Biochim Biophys Acta Mol Cell Res. 2018, 1865, 749–768. [Google Scholar] [CrossRef]
- Esmaeilzadeh A, Mohammadi V, Elahi R. Transforming growth factor beta (TGF-beta) pathway in the immunopathogenesis of multiple sclerosis (MS); molecular approaches. Mol Biol Rep. 2023, 50, 6121–6131. [Google Scholar] [CrossRef] [PubMed]
- Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-beta signal transduction for fibrosis and cancer therapy. Mol Cancer. 2022, 21, 104. [Google Scholar] [CrossRef] [PubMed]
- Tzavlaki K, Moustakas A. TGF-beta Signaling. Biomolecules. 2020, 10.
- Chen C, Dorado Garcia H, Scheer M, Henssen AG. Current and Future Treatment Strategies for Rhabdomyosarcoma. Front Oncol. 2019, 9, 1458. [Google Scholar] [CrossRef]
- Kahen E, Yu D, Harrison DJ, Clark J, Hingorani P, Cubitt CL, et al. Identification of clinically achievable combination therapies in childhood rhabdomyosarcoma. Cancer Chemother Pharmacol. 2016, 78, 313–323. [Google Scholar] [CrossRef]
- Makimoto, A. Optimizing Rhabdomyosarcoma Treatment in Adolescents and Young Adults. Cancers (Basel). 2022, 14. [Google Scholar] [CrossRef]
- George P, Journey LJ, Goldstein MN. Effect of vincristine on the fine structure of HeLa cells during mitosis. J Natl Cancer Inst. 1965, 35, 355–375. [Google Scholar]
- Gidding CE, Kellie SJ, Kamps WA, de Graaf SS. Vincristine revisited. Crit Rev Oncol Hematol. 1999, 29, 267–287. [Google Scholar] [CrossRef]
- Awosika AO, Below J, J MD. Vincristine. StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Jameshia Below declares no relevant financial relationships with ineligible companies. Disclosure: Joe M Das declares no relevant financial relationships with ineligible companies.2023.
- Lu DF, Wang YS, Li C, Wei GJ, Chen R, Dong DM, et al. Actinomycin D inhibits cell proliferations and promotes apoptosis in osteosarcoma cells. Int J Clin Exp Med. 2015, 8, 1904–1911. [Google Scholar]
- Marchal JA, Prados J, Melguizo C, Fernandez JE, Velez C, Alvarez L, et al. Actinomycin D treatment leads to differentiation and inhibits proliferation in rhabdomyosarcoma cells. J Lab Clin Med. 1997, 130, 42–50. [Google Scholar] [CrossRef]
- Ogino MH, Tadi P. Cyclophosphamide. StatPearls. Treasure Island (FL) ineligible companies. Disclosure: Prasanna Tadi declares no relevant financial relationships with ineligible companies.2023.
- Chuk MK, Widemann BC, Minard CG, Liu X, Kim A, Bernhardt MB, et al. A phase 1 study of cabozantinib in children and adolescents with recurrent or refractory solid tumors, including CNS tumors: Trial ADVL1211, a report from the Children's Oncology Group. Pediatr Blood Cancer. 2018, 65, e27077. [Google Scholar] [CrossRef] [PubMed]
- Casanova M, Ferrari A, Bisogno G, Merks JH, De Salvo GL, Meazza C, et al. Vinorelbine and low-dose cyclophosphamide in the treatment of pediatric sarcomas: Pilot study for the upcoming European Rhabdomyosarcoma Protocol. Cancer. 2004, 101, 1664–1671. [Google Scholar] [CrossRef] [PubMed]
- Liang C, Qiao G, Liu Y, Tian L, Hui N, Li J, et al. Overview of all-trans-retinoic acid (ATRA) and its analogues: Structures, activities, and mechanisms in acute promyelocytic leukaemia. Eur J Med Chem. 2021, 220, 113451. [Google Scholar] [CrossRef] [PubMed]
- Ni X, Hu G, Cai X. The success and the challenge of all-trans retinoic acid in the treatment of cancer. Crit Rev Food Sci Nutr. 2019;59(sup1):S71-S80.
- Szymanski L, Skopek R, Palusinska M, Schenk T, Stengel S, Lewicki S, et al. Retinoic Acid and Its Derivatives in Skin. Retinoic Acid and Its Derivatives in Skin. Cells. 2020, 9. [Google Scholar]
- le Maire A, Teyssier C, Balaguer P, Bourguet W, Germain P. Regulation of RXR-RAR Heterodimers by RXR- and RAR-Specific Ligands and Their Combinations. Cells. 2019, 8.
- O'Brien E, Tse C, Tracy I, Reddin I, Selfe J, Gibson J, et al. Pharmacological EZH2 inhibition combined with retinoic acid treatment promotes differentiation and apoptosis in rhabdomyosarcoma cells. Clin Epigenetics. 2023, 15, 167. [Google Scholar] [CrossRef] [PubMed]
- Williams AP, Waters AM, Stewart JE, Atigadda VR, Mroczek-Musulman E, Muccio DD, et al. A novel retinoid X receptor agonist, UAB30, inhibits rhabdomyosarcoma cells in vitro. J Surg Res. 2018, 228, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Chen J, Li Q. Implication of retinoic acid receptor selective signaling in myogenic differentiation. Sci Rep. 2016, 6, 18856. [CrossRef]
- Gudas, LJ. Retinoids induce stem cell differentiation via epigenetic changes. Semin Cell Dev Biol. 2013, 24, 701–705. [Google Scholar] [CrossRef]
- Qi L, Luo Q, Zhang Y, Jia F, Zhao Y, Wang F. Advances in Toxicological Research of the Anticancer Drug Cisplatin. Chem Res Toxicol. 2019, 32, 1469–1486. [Google Scholar] [CrossRef]
- Makovec, T. Cisplatin and beyond: Molecular mechanisms of action and drug resistance development in cancer chemotherapy. Radiol Oncol. 2019, 53, 148–158. [Google Scholar] [CrossRef]
- arrabi A, Perrin D, Kavoosi M, Sommer M, Sezen S, Mehrbod P; et al. Rhabdomyosarcoma: Current Therapy, Challenges, and Future Approaches to Treatment Strategies. Cancers (Basel). 2023, 15.
- Jin S, Cojocari D, Purkal JJ, Popovic R, Talaty NN, Xiao Y, et al. 5-Azacitidine Induces NOXA to Prime AML Cells for Venetoclax-Mediated Apoptosis. Clin Cancer Res. 2020, 26, 3371–3383. [Google Scholar] [CrossRef] [PubMed]
- Mahoney SE, Yao Z, Keyes CC, Tapscott SJ, Diede SJ. Genome-wide DNA methylation studies suggest distinct DNA methylation patterns in pediatric embryonal and alveolar rhabdomyosarcomas. Epigenetics. 2012, 7, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Zhu Q, Liang F, Cai S, Luo X, Duo T, Liang Z, et al. KDM4A regulates myogenesis by demethylating H3K9me3 of myogenic regulatory factors. Cell Death Dis. 2021, 12, 514. [Google Scholar] [CrossRef] [PubMed]
- Filip K, Lewinska A, Adamczyk-Grochala J, Marino Gammazza A, Cappello F, Lauricella M; et al. 5-Azacytidine Inhibits the Activation of Senescence Program and Promotes Cytotoxic Autophagy during Trdmt1-Mediated Oxidative Stress Response in Insulinoma beta-TC-6 Cells. Cells. 2022, 11.
- Mirani B, Pagan E, Shojaei S, Duchscherer J, Toyota BD, Ghavami S, et al. A 3D bioprinted hydrogel mesh loaded with all-trans retinoic acid for treatment of glioblastoma. Eur J Pharmacol. 2019, 854, 201–212. [Google Scholar] [CrossRef]
Figure 1.
Subtypes of Rhabdomyosarcoma. Rhabdomyosarcoma can be differentiated into four subtypes: (1) Embryonal Rhabdomyosarcoma occurs primarily in the head, neck and bladder regions. (2) Alveolar Rhabdomyosarcoma (ARMS) is predominantly found in the trunk, arms and legs, characterized by polygonal cells with hyperchromatic nuclei. (3) Spindle Rhabdomyosarcoma (SRMS) occurs in the Para testicular, head and neck regions. (4) Pleomorphic Rhabdomyosarcoma (PRMS) is localized to the deep soft tissues of the body extremities. Created with BioRender.com, 22 June 2023.
Figure 1.
Subtypes of Rhabdomyosarcoma. Rhabdomyosarcoma can be differentiated into four subtypes: (1) Embryonal Rhabdomyosarcoma occurs primarily in the head, neck and bladder regions. (2) Alveolar Rhabdomyosarcoma (ARMS) is predominantly found in the trunk, arms and legs, characterized by polygonal cells with hyperchromatic nuclei. (3) Spindle Rhabdomyosarcoma (SRMS) occurs in the Para testicular, head and neck regions. (4) Pleomorphic Rhabdomyosarcoma (PRMS) is localized to the deep soft tissues of the body extremities. Created with BioRender.com, 22 June 2023.
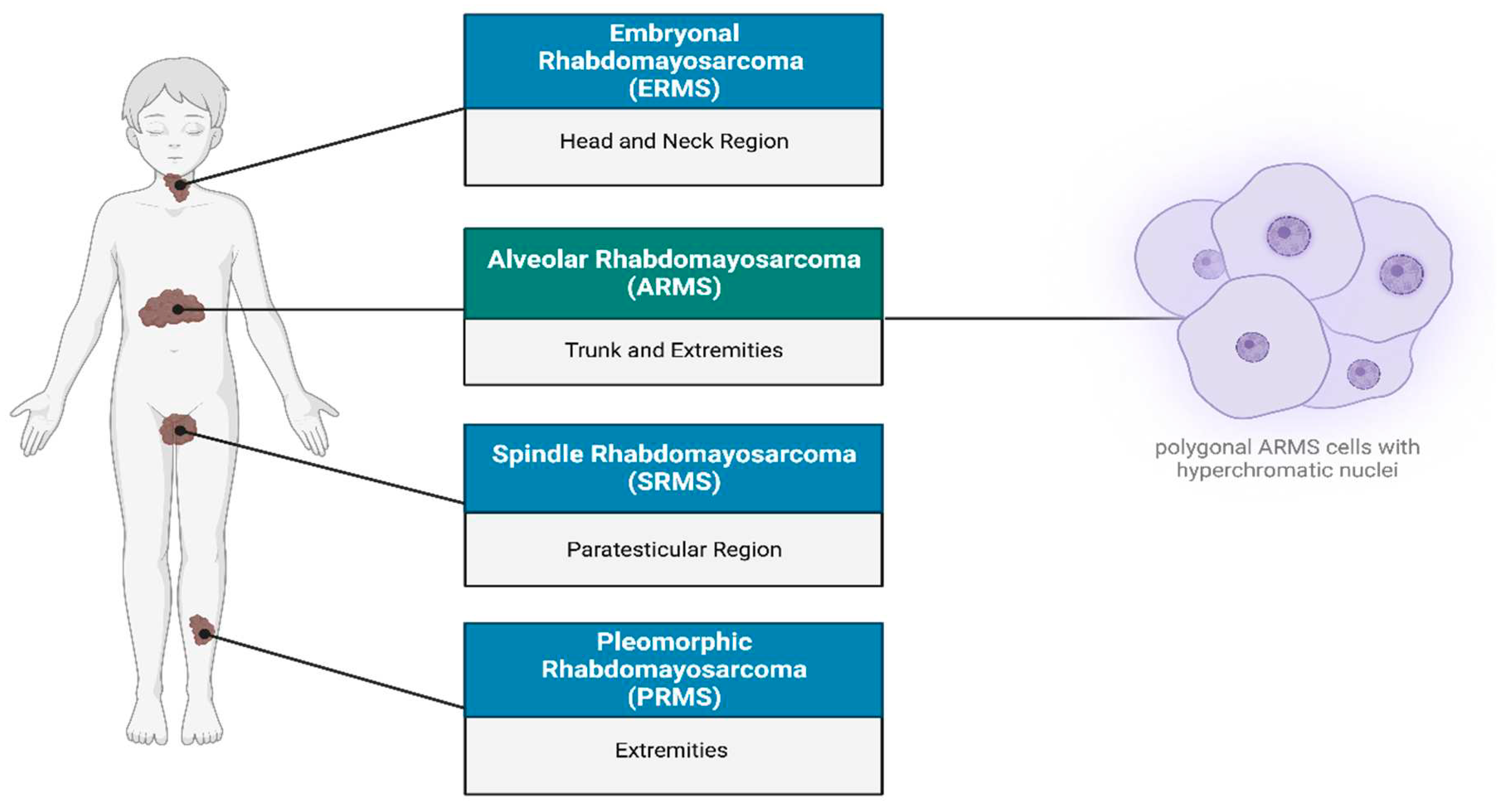
Figure 2.
PAX3/7-FOXO1 Fusion Gene. The Majority of ARMS cases are caused by a chromosomal translocation event between the N-terminal DNA Binding Domain of the PAX3 or PAX7 and the C-terminal Transactivation Domain of the FOXO1 gene. These events result in a fusion gene PAX3/7-FOXO1, that is found to be an oncogenic driver. The PAX3/7-FOXO1 fusion gene retains the DNA binding activity of PAX3/7 and is therefore capable of initiating transcription. Created with BioRender.com, 27 June 2023.
Figure 2.
PAX3/7-FOXO1 Fusion Gene. The Majority of ARMS cases are caused by a chromosomal translocation event between the N-terminal DNA Binding Domain of the PAX3 or PAX7 and the C-terminal Transactivation Domain of the FOXO1 gene. These events result in a fusion gene PAX3/7-FOXO1, that is found to be an oncogenic driver. The PAX3/7-FOXO1 fusion gene retains the DNA binding activity of PAX3/7 and is therefore capable of initiating transcription. Created with BioRender.com, 27 June 2023.
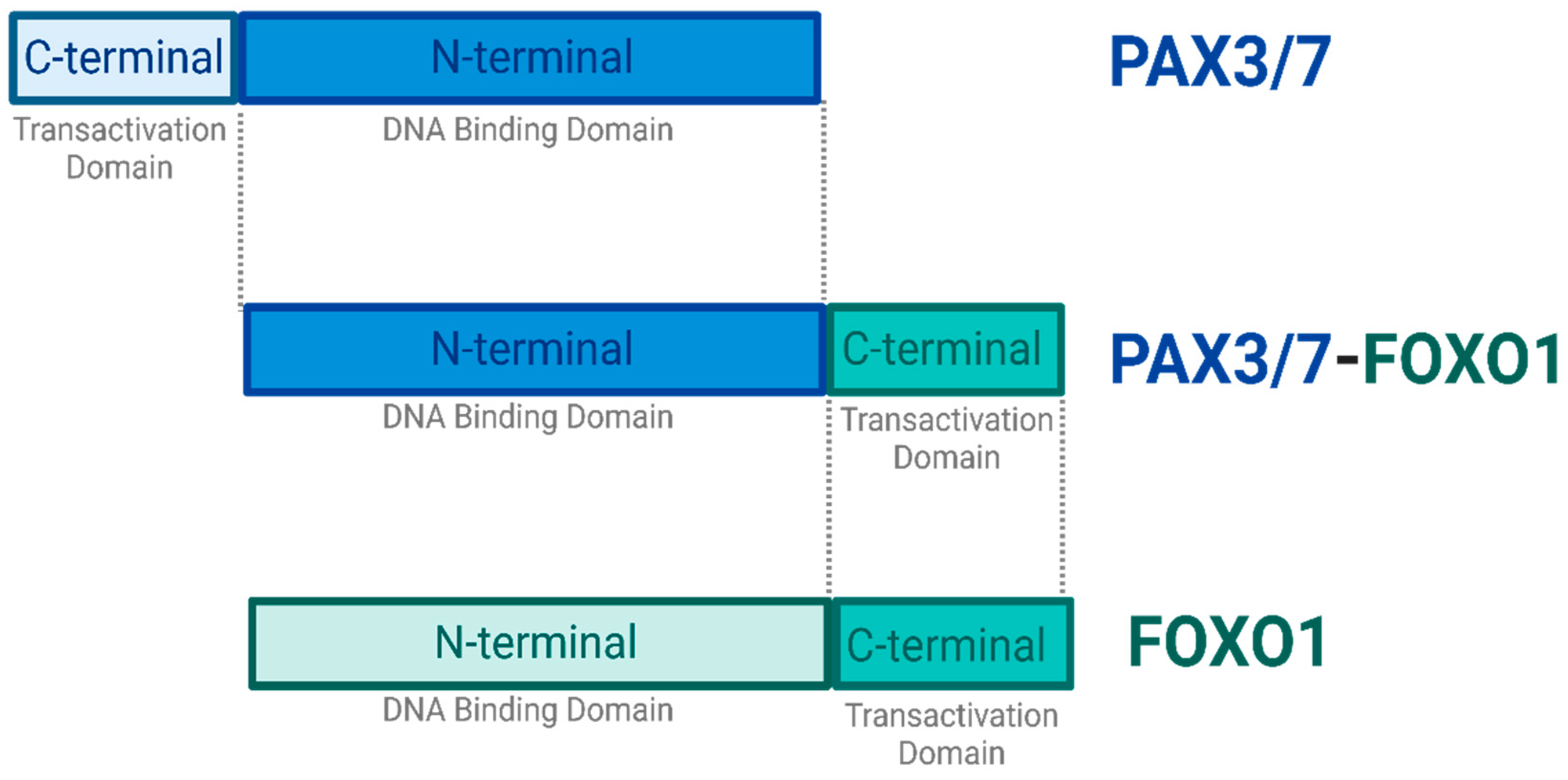
Figure 3.
TGF-β induces EMT via SNAIL Transcription Factor in AMRS. Overexpression of TGF-β in AMRS leads to phosphorylation and activation of its two major downstream pathways. This leads to SNAIL, a transcription factor binding to the E-box sequence in the promotor region of genes resulting in the epithelial to mesenchymal transition (EMT). Created with BioRender.com, 28 June 2023.
Figure 3.
TGF-β induces EMT via SNAIL Transcription Factor in AMRS. Overexpression of TGF-β in AMRS leads to phosphorylation and activation of its two major downstream pathways. This leads to SNAIL, a transcription factor binding to the E-box sequence in the promotor region of genes resulting in the epithelial to mesenchymal transition (EMT). Created with BioRender.com, 28 June 2023.
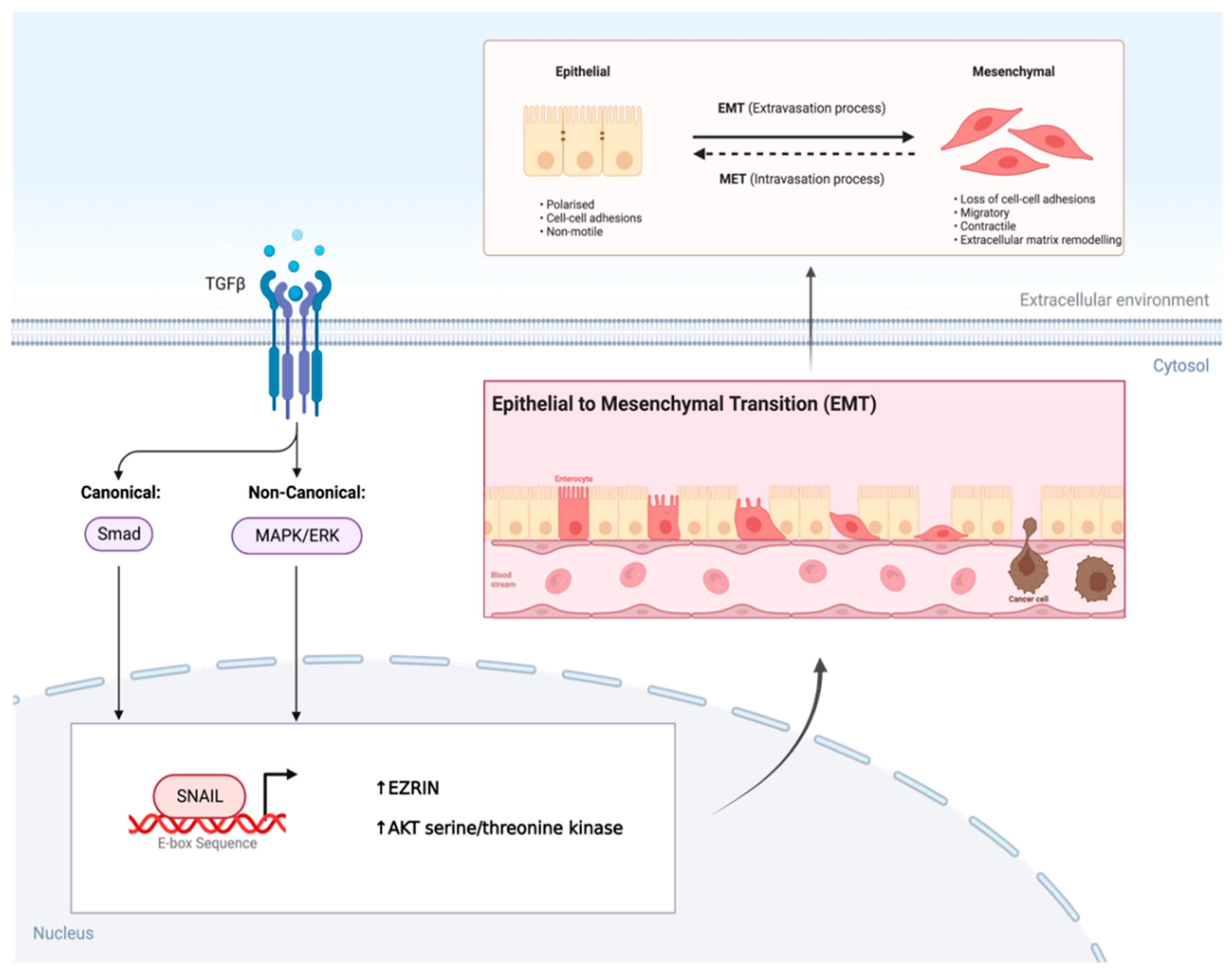
Figure 4.
Canonical and Non-Canonical Downstream Pathways of TGF-β. In the canonical pathway the SMAD 2/3 are phosphorylated via TGF-β thus translocating into the nucleus and regulating gene transcription. The non-canonical pathway activates downstream cascades like MAPK/ERK via various post translational modifications. Both these pathways ultimately result in epithelial to mesenchymal transition (EMT) therefore promoting cancer. Created with BioRender.com, 28 June 2023.
Figure 4.
Canonical and Non-Canonical Downstream Pathways of TGF-β. In the canonical pathway the SMAD 2/3 are phosphorylated via TGF-β thus translocating into the nucleus and regulating gene transcription. The non-canonical pathway activates downstream cascades like MAPK/ERK via various post translational modifications. Both these pathways ultimately result in epithelial to mesenchymal transition (EMT) therefore promoting cancer. Created with BioRender.com, 28 June 2023.
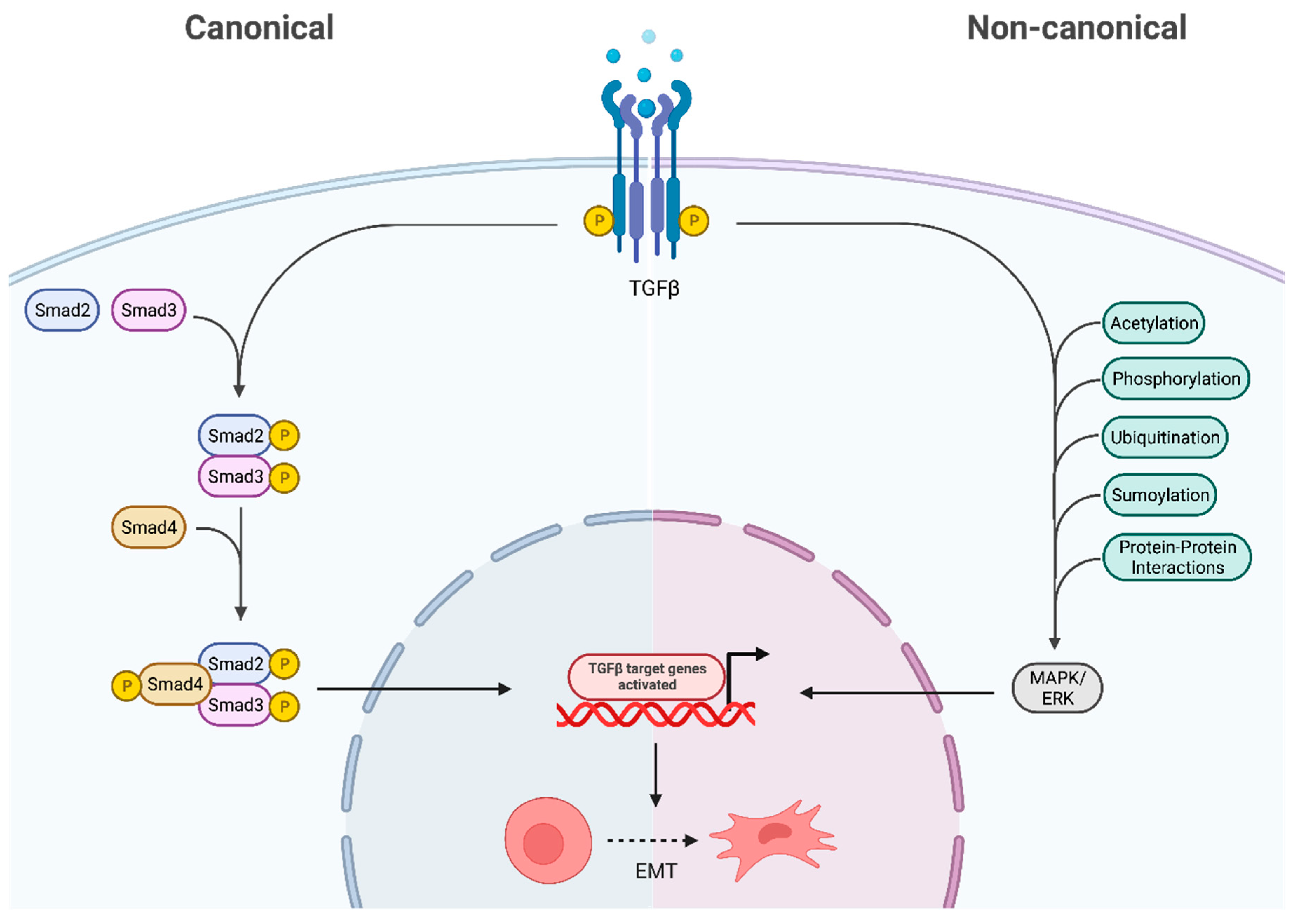
Figure 5.
Dual role of TGF-β. TGF-β under normal circumstances plays a crucial role as a tumor suppressor by regulating cell cycle arrest and inducing apoptosis (Left). However, when overexpressed TGF-β promotes cancer progression and metastasis via EMT induction (Right). Created with BioRender.com, 22 June 2023.
Figure 5.
Dual role of TGF-β. TGF-β under normal circumstances plays a crucial role as a tumor suppressor by regulating cell cycle arrest and inducing apoptosis (Left). However, when overexpressed TGF-β promotes cancer progression and metastasis via EMT induction (Right). Created with BioRender.com, 22 June 2023.
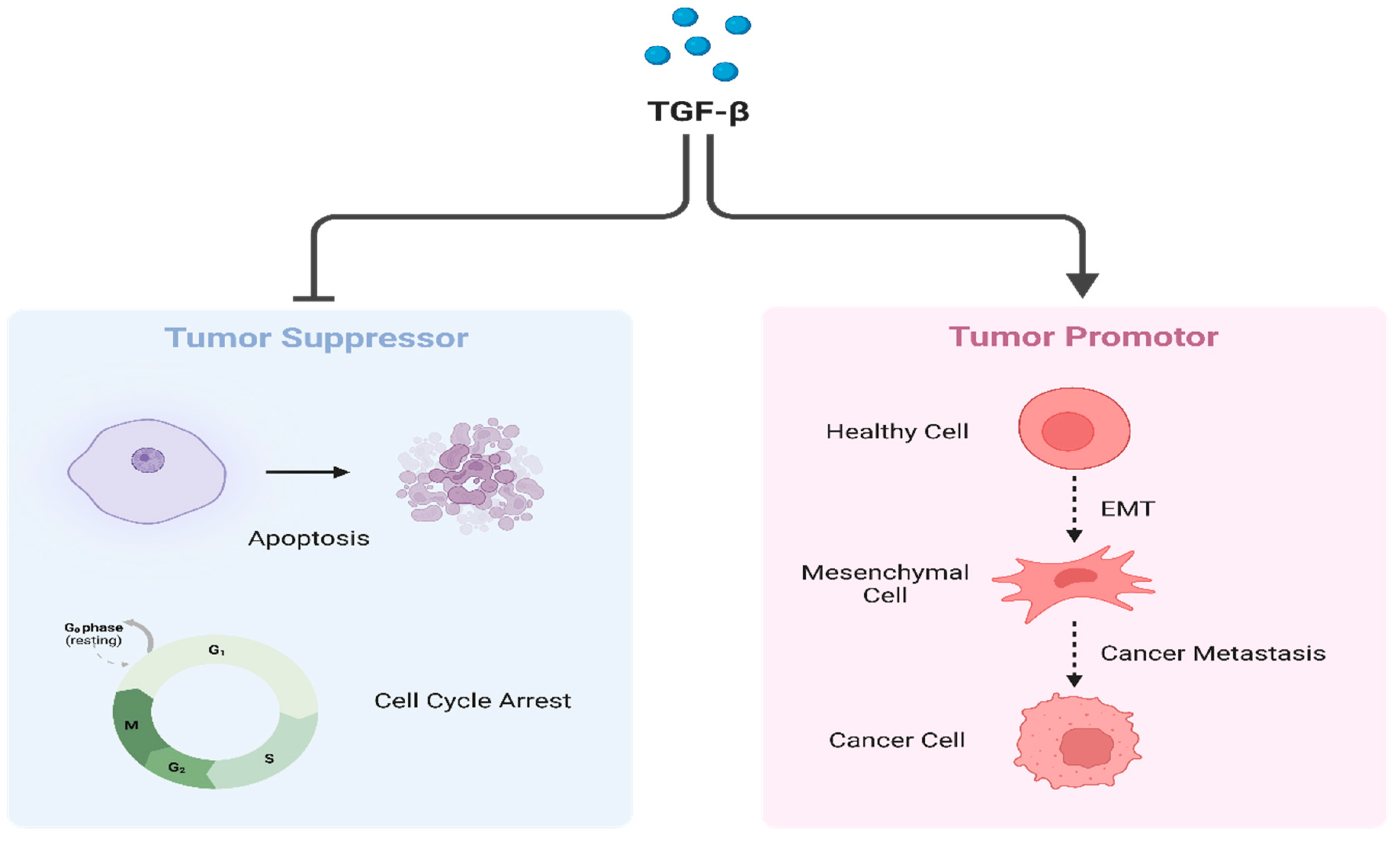
Figure 6.
VAC Mechanism of Action. Vincristine (Group6-3, 2011), Actinomycin D (Actinomycin D, n.d.) and Cyclophosphamide (“Cyclophosphamide,” 2023) work in unison to prevent tumour growth. Created with BioRender.com, 12 July 2023.
Figure 6.
VAC Mechanism of Action. Vincristine (Group6-3, 2011), Actinomycin D (Actinomycin D, n.d.) and Cyclophosphamide (“Cyclophosphamide,” 2023) work in unison to prevent tumour growth. Created with BioRender.com, 12 July 2023.
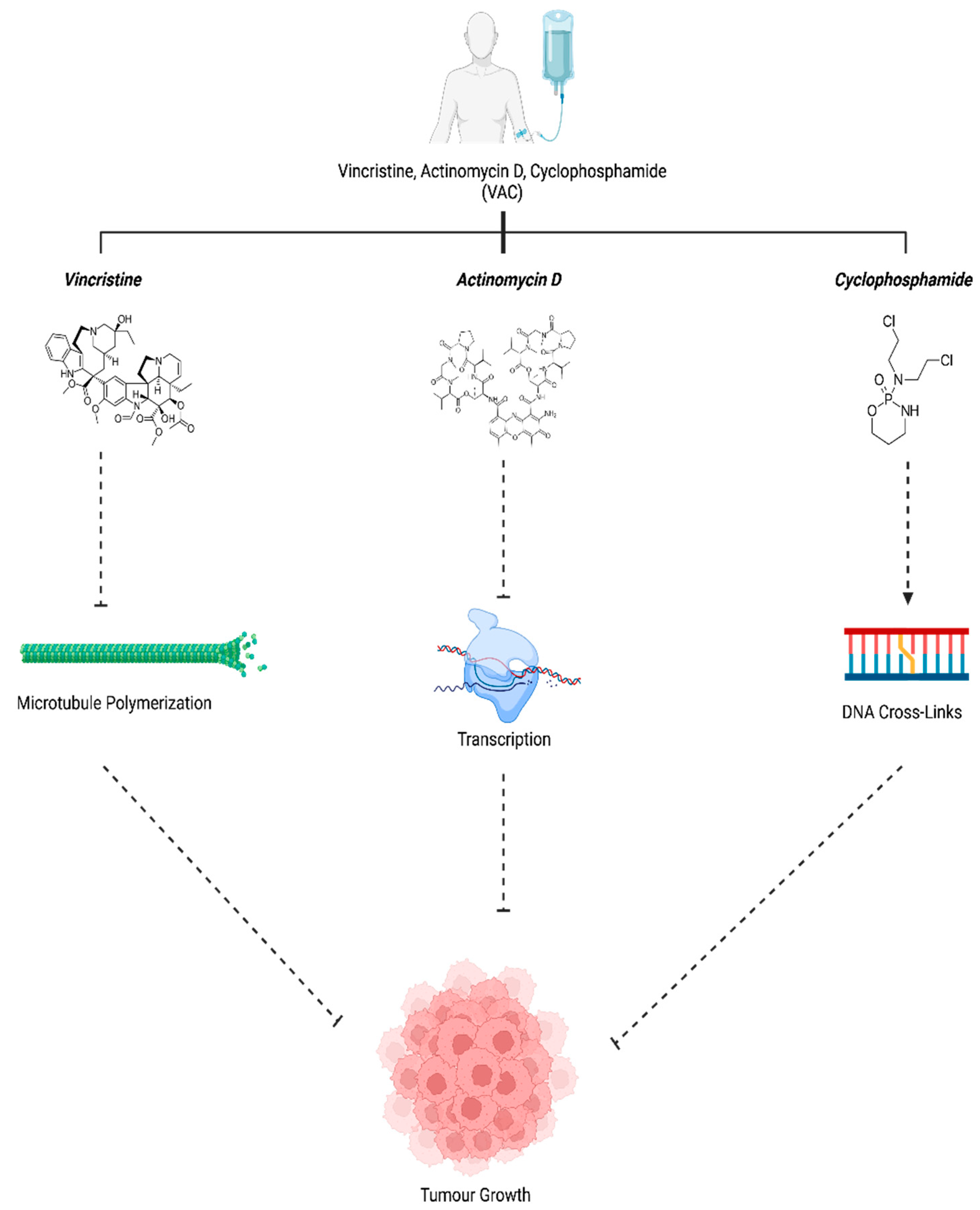
Figure 7.
Mechanism of action of Cabozantinib (XL184). Cabozantinib is a Receptor Tyrosine Kinase (RTK) inhibitor which counteracts the affect of MET thereby allowing myogenic differentiation. Created with BioRender.com, 12 July 2023.
Figure 7.
Mechanism of action of Cabozantinib (XL184). Cabozantinib is a Receptor Tyrosine Kinase (RTK) inhibitor which counteracts the affect of MET thereby allowing myogenic differentiation. Created with BioRender.com, 12 July 2023.
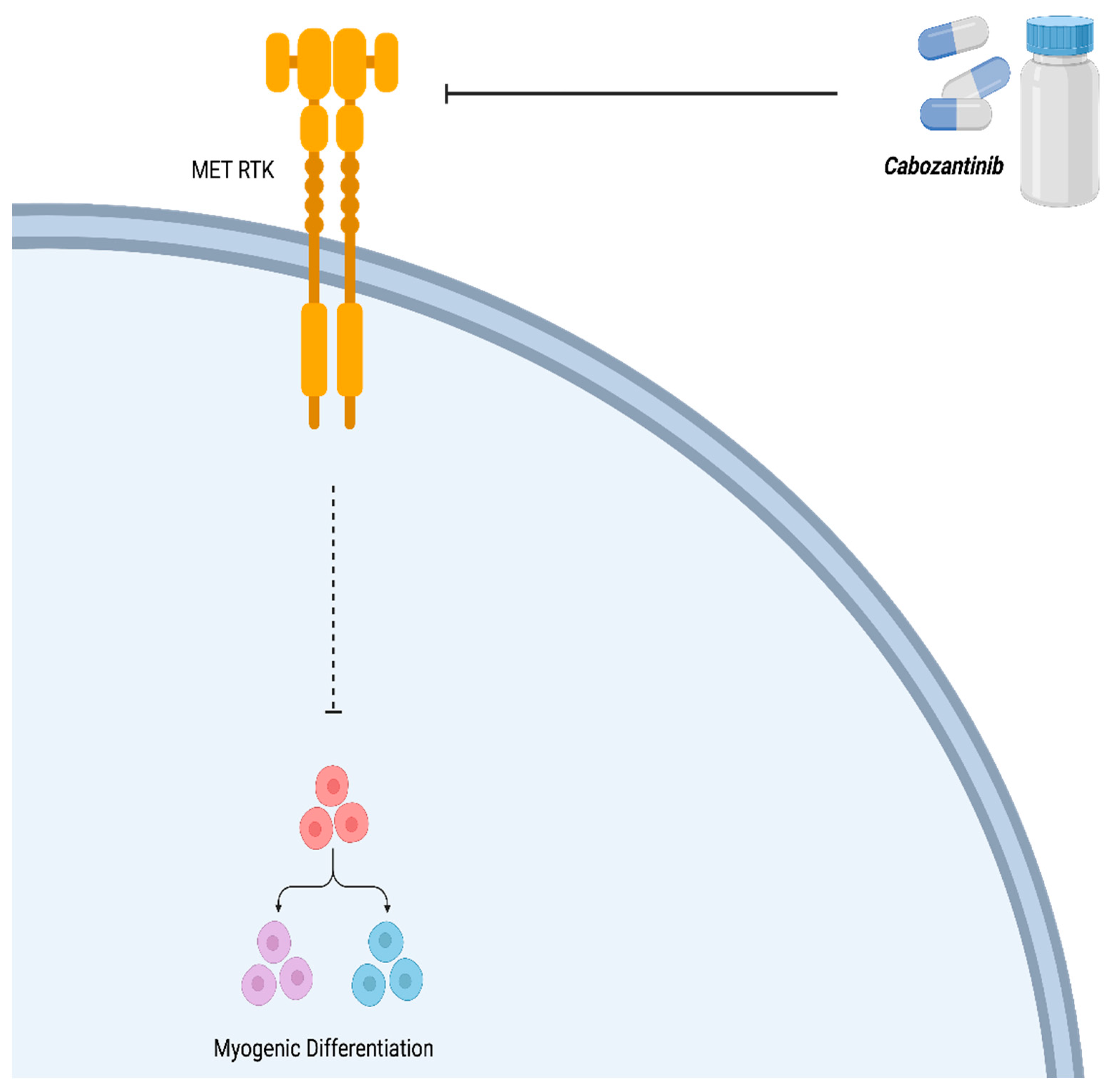
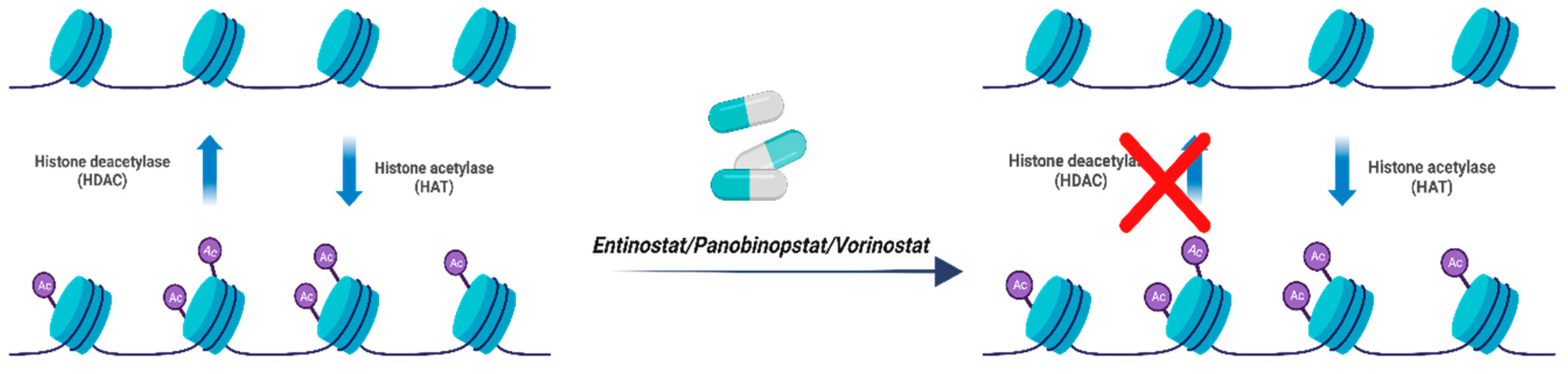
Figure 8.
Mechanism of Action of Inhibitors of Histone Deacetylase’s (HDAC). Entinostat, panobinopstat and vorinostat are agents that inhibit histone deacetylation resulting in delayed tumour growth and apoptosis. Created with BioRender.com, 22 June 2023.
Figure 8.
Mechanism of Action of Inhibitors of Histone Deacetylase’s (HDAC). Entinostat, panobinopstat and vorinostat are agents that inhibit histone deacetylation resulting in delayed tumour growth and apoptosis. Created with BioRender.com, 22 June 2023.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



