
Submitted:
26 January 2024
Posted:
29 January 2024
You are already at the latest version
Abstract
Muscle-enriched A-type lamin-interacting protein (MLIP) is an emerging protein involved in cellular homeostasis and stress adaptation. Eukaryotic cells regulate various cellular processes, including metabolism, DNA repair, and cell cycle progression, to maintain cellular homeostasis. Disruptions in this homeostasis can lead to diseases such as cancer, characterized by uncontrolled cell growth and division. This review aims to explore for the first time the unique role MLIP may play in cancer development and progression, given its interactions with the PI3K/Akt/mTOR pathway, p53, and FOXO transcription factors, all critical regulators of cellular homeostasis and tumor suppression. We discuss the current understanding of MLIP's involvement in pro-survival pathways and its potential implications in cancer cells' metabolic remodeling and dysregulated homeostasis. Additionally, we examine the potential of MLIP as a novel therapeutic target for cancer treatment. This review aims to shed light on MLIP's potential impact on cancer biology and contribute to developing innovative therapeutic strategies.
Keywords:
MLIP
; Cancer
; PI3 kinase
; Akt
; mTOR
; Tumorigenesis
1. Introduction
Eukaryotic cells maintain cellular homeostasis through an extensive array of sensory mechanisms to respond and adapt to both intrinsic and extrinsic stimuli and insults. This involves the regulation of various cellular processes, including metabolism, DNA repair, and cell cycle progression. Disruptions to cellular homeostasis can lead to the development of various diseases, including cancer. Cancer is a devasting disease with metabolic remodeling and dysregulated homeostasis as distinctive feature of cancer 1.
Cancer is characterized by uncontrolled cell growth and division, leading to the formation of tumors2. This process is the result of a disruption in the delicate balance between cell proliferation and cell death3, which is normally maintained by cellular homeostasis. Several signaling molecules and pathways have been identified as pro-oncogenic and have therefore been targeted for the therapeutic treatment of cancer4.
In cancer cells, however, mutations or changes in the genes encoding proteins in this pathway can cause hyperactivation, resulting in uncontrolled cell growth and resistance to apoptosis (programmed cell death). For examples, the phosphoinositide 3-kinase/Protein kinase B (PI3K/Akt) / mammalian target of rapamycin (mTOR) pathway is tightly regulated in normal cells, ensuring a balance between cell growth and death. however, two Tumor suppressors p53 (p53) and Forkhead box O family (FOXO), downstream of the PI3K/Akt/mTOR pathway, respectively, are critical integrators of genomic and metabolic stresses 5–8. Both p53 and FOXO are stress-activated transcription factors that promote a pro-survival adaptive response to insult. Specifically, p53 stimulates DNA repair in response to DNA damage and FOXO regulates metabolic remodeling to maintain metabolic homeostasis. The loss of p53 and FOXO normal function is associated with tumorigenesis in a wide variety of tissues. Because of the importance of the PI3K/Akt/mTOR pathway to the propagation of tumorigenesis, a number of specific inhibitors targeting different components of this pathway have been developed.
Muscle enriched A-type Lamin-Interacting Protein (MLIP) is a novel protein of unknown structure and function, that is required for proper cardiac and skeletal muscle adaptation to stress 9–14. MLIP is a crucial mediator of cardiac adaptation through its interaction with the Akt/mTOR pro-survival pathway 11, FOXO1 14 and p53 11. Detailed comparative pathway analysis based on global gene expression differences between normal and MLIP deficient hearts has now revealed MLIP as a modulator of both p53 and FOXO activity. Given MLIP’s interaction with PI3K/Akt pathway, p53, and FOXO 15,16. this review explores the role MLIP may play in tumor formation, progression and the potential of MLIP as a new therapeutic target.
2. MLIP expression in cancer
Limited research has focused on elucidating the role of MLIP in the initiation and/or progression of cancer. Our investigation identified two primary types of cancers where MLIP's role was emphasized: breast cancer and esophageal cancer (Table 1).
Breast cancer and esophageal cancer represent significant global health challenges, with the former being one of the most prevalent cancers among women and the latter noted for its particularly low survival rates 17,18. The genetic underpinnings of these cancers are complex, and though substantial progress has been made in identifying key genetic risk factors, however a significant proportion of the genetic risk remains unexplained. Recent research has begun to shed light on this gap, with a particular focus on the role of copy number variants (CNVs) and differentially expressed genes. One gene that has emerged as a potential key player in both breast and esophageal cancer is the MLIP gene.
Expression of MLIP in different types of cancer
Breast cancer stands as one of the prevalent malignancies affecting women, with around 1 million new cases and over 400,000 reported deaths annually worldwide. In the year 2023, an estimated 297,790 women and 2,800 men are projected to be diagnosed with breast cancer19. While single nucleotide polymorphisms and mutations contribute to approximately 49% of the genetic risk associated with breast cancer20,21, Kumaran and colleagues (2017) sought to uncover the remaining 51% by identifying germline Copy Number Variants (CNVs) linked to breast cancer22. Whole genome CNV genotyping was performed on 422 cases and 348 controls. Two hundred CNVs were identified to be associated with breast cancer of which 21 CNV regions overlapped with 22 genes. MLIP was identified as 1 of 6 genes associated breast cancer risk and recurrence-free survival 22. Specifically, Kumaran and colleagues reported a loss in MLIP CNVs was associated with significant reduction of breast cancer risk and recurrence- free survival with a reported hazard ratio of 0.62 [0.4–0.94] 18.
Triple negative breast cancer (TNBC) is an aggressive subtype of breast cancer that is defined by the absence of estrogen receptor, progesterone receptor, and human epidermal growth factor receptor 2 expression. These receptors are commonly used as targets for breast cancer treatment, and the absence of these receptors in TNBC makes it more difficult to treat. Zhang and colleagues performed RNA-seq on 30 TNBC patient tumors, 15 of which had lymph node metastasis while the other 15 showed no lymph node metastasis 23. Differential gene expression analysis was performed in order to determine the key genes involved in the progression and oncogenesis of TNBC 23. The analysis revealed 2953 genes with differential expression in breast cancer tumors compared to normal control tissues and 975 genes with differential expression between 15 patients with lymph node metastasis and 15 patients without. A subset of 117 genes exhibited differential expression in both sets among those with and without lymph node metastasis in triple-negative breast cancer (TNBC), implying their involvement in TNBC oncogenesis and progression. MLIP, among the 117 genes of interest, was found to be upregulated in TNBC and exhibited a negative association with the cytotoxicity of CD8+ T cells23.
Esophageal cancer is one of the most common malignancies, ranking 7th in global morbidity and 6th in cancer-related mortality. The 5-year overall survival rate is only about 15–20%, although progress has been made in diagnosis and treatment 17. To further define prognostic mRNAs of esophageal cancer, functional enrichment analyses of lncRNA, mRNA, and miRNA of 81 tumors and 11 normal controls was performed. MLIP was identified as one of 7 risk RNAs for esophageal cancer with a hazard ratio of 1.67 (1.22-2.29, p<0.001) 24.
Finally, according to the recently found role of MLIP in cancer (Table 1), it has been suggested as a potential biomarker for triple-negative breast cancer, and esophageal cancer. However, more research is needed to fully understand MLIP's role in these cancers and its potential as a therapeutic target or diagnostic tool.
3. Molecular relationship of MLIP with pro-survival/oncogenic pathways and tumor suppressors
The intricate network of cellular signaling pathways that govern cell growth, proliferation, survival, and metabolism is often dysregulated in various cancer types, contributing to tumorigenesis and disease progression. Central to this network are the PI3K/Akt/mTOR, FOXO1, AMPK, p53, and Lamin A/C pathways, each playing critical roles in maintaining cellular homeostasis and responding to stress signals. Recently, MLIP has emerged as a key regulator within these pathways, influencing a variety of cellular processes and potentially playing a role in both cancer pathogenesis and cardiac disorders.
Primary role of AMPK function and dysfunction in cancer
Adenosine monophosphate-activated protein kinase (AMPK) serves as a pivotal enzyme governing cellular energy balance. Its primary function involves detecting shifts in cellular energy status, particularly reductions in ATP, and initiating processes that generate ATP while concurrently inhibiting ATP-consuming processes. AMPK functions as a heterotrimeric complex, comprising catalytic α subunits and regulatory β and γ subunits. The γ subunit accommodates binding sites for AMP and ATP, enabling AMPK to sense alterations in the AMP/ATP ratio and self-activate during energy depletion. AMPK activation triggers diverse downstream effects, including heightened glucose uptake, fatty acid oxidation, and mitochondrial biogenesis, along with diminished protein synthesis, lipogenesis, and gluconeogenesis. AMPK also influences autophagy, cell growth, proliferation, and inflammation25,26. In response to stressors causing ATP depletion, such as hypoxia and glucose deprivation, AMPK activity is heightened 25,26. Additionally, stimulating AMPK in skeletal muscle enhances glucose uptake and fatty acid oxidation while reducing lipid accumulation and inflammation27. These findings, combined with other research, collectively underscore the crucial role of AMPK in governing energy metabolism and cellular function.
The precise function of AMPK in cancer cells is complicated and relies on the specific context of AMPK activation. In certain instances, AMPK activation can serve as a tumor suppressor by restraining cell growth, curbing proliferation, and encouraging apoptosis. However, in other scenarios, AMPK activation might support the survival of tumor cells by facilitating metabolic adaptation to the unique conditions of the tumor microenvironment. Hence, targeting AMPK activation could be a problematic or promising approach for cancer treatment (Figure 1) 28. Additionally, research indicates that combining AMPK activation with other anticancer therapies like chemotherapy or radiation has the potential to augment their effectiveness 28,29.
The study by Cattin et al. in 2015 sheds light on the molecular mechanisms underlying the reduced glucose uptake observed in MLIP-deficient cardiac tissues11. In MLIP-deficient hearts, AMPK was reported to undergo dephosphorylation at AMPK alpha-Thr-172, a crucial step leading to the deactivation of the AMPK complex and subsequently resulting in decreased glucose uptake compared to normal cardiac tissues11. Remarkably, this deactivation of AMPK occurred despite similar activity in Liver kinase B1 (LKB1), the kinase responsible for AMPK activation30, indicating an LKB1-independent inactivation of AMPK in MLIP-deficient hearts.
Interactions between MLIP and AMPK may hold implications for cancer biology. AMPK, recognized as a metabolic tumor suppressor, hampers cell growth and proliferation during low energy conditions, thereby impeding the uncontrolled cell growth characteristic of cancer31. Consequently, the observed reduction in AMPK activation in the absence of MLIP might potentially elevate the risk of unregulated cell growth and proliferation, contributing to oncogenesis. Furthermore, the decline in AMPK levels in MLIP-deficient cardiac tissues led to the heightened activation of the Akt/mTOR pathway11,32. This pathway significantly influences cell growth, proliferation, and survival, and its dysregulation is commonly observed in various types of cancers. These findings suggest that MLIP could potentially modulate these crucial pathways, thereby influencing cancer biology11,31,32. However, it is crucial to acknowledge that these observations were made specifically in cardiac tissue, and it remains uncertain whether similar mechanisms would apply to other tissues or cancer cells. Additional research is required to directly investigate the involvement of MLIP in cancer biology.
The PI3K/Akt/mTOR pathway and MLIP
The PI3K/Akt/mTOR pathway is a key signaling pathway that regulates various cellular processes, including cell growth, proliferation, survival, and metabolism. Dysregulation of these pathways is commonly observed in many types of cancer, and its activation has been shown to contribute to cancer development and progression (Figure 2) 33–36. In cancer cells, the PI3K/Akt/mTOR pathway can become activated through several mechanisms, including mutation of genes encoding components of the pathway, activation of upstream growth factor receptors, and loss of negative regulators of the pathway34. Activation of the pathway can lead to increased cell proliferation, survival, and resistance to cell death signals, which contribute to tumor growth and progression. Targeting the PI3K/Akt/mTOR pathway (Table 2) has emerged as a promising strategy for cancer treatment35,36. Several drugs that target components of the pathway are currently being developed and tested in preclinical and clinical studies, and some have shown promising results in certain types of cancer33,36. However, targeting this pathway can also have side effects, and there is ongoing research to develop more effective and selective therapies that minimize toxicity while maximizing anti-cancer activity33,35,36.
8The documented association between MLIP and the PI3K/AKT/mTOR signaling pathway is evident in research findings that highlight MLIP's direct impact on this pathway. Specifically, the absence of MLIP leads to the selective hyperactivation of the Akt/mTOR signaling pathway in cardiac cells (Figure 3)11. Conversely, MLIP overexpression results in the inhibition of this pathway. The study demonstrates that the hyperactivation of Akt/mTOR occurs in cardiac cells when MLIP is absent11. These results suggest that a deficiency in MLIP may potentially contribute to an accelerated aging phenomenon within cardiac cells, heightening susceptibility to tumor development8
Role of MLIP in FOXO1 signaling
FOXO genes are a subgroup of the forkhead family of transcription factors that play a critical role in regulating various cellular processes, including cell cycle control, DNA repair, apoptosis, and oxidative stress response 37–40. Dysregulation of FOXO gene expression or activity has been reported to be associated with development and progression of cancer 41. There are four members of the FOXO family in mammals: FOXO1, FOXO3, FOXO4, and FOXO6. Among these, FOXO1 and FOXO3 are the most well-studied in the context of cancer (Table 3).
In normal cells, FOXO1 and FOXO3 are often activated in response to cellular stress, leading to the expression of target genes that promote cell cycle arrest, DNA repair, and apoptosis. This helps to prevent the development of cancer by eliminating cells with damaged DNA42. However, in cancer cells, the activity of FOXO1 and FOXO3 is often dysregulated 43–45. In tumors, FOXO expression or activity is often suppressed to promote cell proliferation and survival, or alternatively FOXO may be activated to promote cell migration and invasion 43–45.
FOXO1 has been found to play a role in the regulation of estrogen receptor (ER) signaling. In breast cancer, the loss of FOXO1 activity has been associated with resistance to endocrine therapy, while overexpression of FOXO1 has been shown to sensitize breast cancer cells to endocrine therapy 46. Likewise, in prostate cancer, FOXO3 has been identified as a participant in the control of androgen receptor signaling45. Reduced FOXO3 activity has been linked to resistance against androgen deprivation therapy, whereas increased FOXO3 expression has demonstrated the ability to enhance the sensitivity of prostate cancer cells to this therapy 46
Transcripts of Foxo-1 have been demonstrated to contribute to cardiac remodeling 6,47. FOXO1 acts as an inhibitor of calcineurin-mediated adverse cardiac remodeling, which promotes hypertrophic responses and contributes to heart failure 6,47. Notably, the deletion of MLIP has also been linked to the downregulation of the FOXO1 pathway 11,14. This suggests that the transcription factor FOXO-1 operates as a downstream signal of MLIP 11
Although the precise mechanism through which MLIP increases FOXO-1 expression remains unknown, FOXO-1 is acknowledged for its involvement in cell cycle arrest, apoptosis, and tumor suppression, implying a potential role of MLIP in cancer pathogenesis. The activation of FOXO1 prompts the transcription of the cyclin-dependent kinase inhibitor p27KIP1 while suppressing the transcription of cyclin D1 and D2. Both effects result in cell cycle arrest at G1. The loss of one allele of FOXO may render cells susceptible to dysregulated cell cycle events, triggering tumor formation. Activation of MLIP may mitigate the impact of FOXO haploinsufficiency on tumorigenesis 40
P53 and MLIP
The p53 gene functions as a crucial tumor suppressor, actively preventing cancer development by regulating various cellular processes, including DNA repair, cell cycle arrest, apoptosis, and senescence7,8,48–51. In response to DNA damage, p53 is activated, enabling it to pause the cell cycle for DNA repair or initiate apoptosis to eliminate damaged cells. In cancer, the p53 gene is frequently mutated or deleted, resulting in the loss of its tumor suppressor function52,53. Mutations in p53 represent one of the most prevalent genetic alterations in cancer, with up to 50% of all human cancers exhibiting p53 mutations54,55. The functional loss of p53 contributes to cancer development and progression by allowing the proliferation of damaged cells, facilitating the accumulation of additional genetic changes that can lead to cancer formation.
Beyond its role in DNA damage response, p53 also participates in the regulation of cellular metabolism56–58. P53 has been demonstrated to influence the expression of genes involved in glycolysis, oxidative phosphorylation59,60, and fatty acid metabolism56,61. P53 loss or mutation can contribute to the metabolic rewiring commonly observed in cancer cells62. MLIP deficient hearts were found to have increased activation of p5311, indicating that MLIP-deficient hearts may be experiencing genotoxic and/or metabolic stress. However, the activation of p53 is triggered by other genes and is crucial for its role as a tumor suppressor. The specific mass of p53 is less significant than the quantity of activated p53, as only the activated form can bind to DNA and initiate the expression of its target genes8. This implies a potential alternative function of MLIP, wherein it may promote tumor formation by inhibiting p53, a critical tumor suppressor gene. Alternatively, MLIP inhibition might impact p53 function by influencing other genes associated with p53 activation. Investigating such a role could provide novel insights into the impact of MLIP on cancer through potential manipulation of p53 function.
4. Conclusion, MLIP as a potential therapeutic target
MLIP is an emerging factor implicated in the regulation of key signaling pathways that govern cell growth, proliferation, survival, and metabolism, which are often dysregulated in cancer. Through its interactions with the PI3K/Akt/mTOR pathway, MLIP appears to exert an inhibitory effect11. Overexpression of MLIP leads to the downregulation of this pathway, while its loss results in the pathway's overactivation11,14,63,64. This implies that MLIP might act as a suppressor of cell growth and proliferation, two key processes that are often hyperactivated in cancer. Therefore, therapies aimed at enhancing MLIP expression or its regulatory effect on the PI3K/Akt/mTOR pathway might be beneficial for inhibiting cancer progression.
Moreover, MLIP appears to be involved in the regulation of FOXO1 signaling14, a pathway that plays a critical role in cell cycle control, apoptosis, and DNA repair - processes that are crucial for maintaining genomic integrity and preventing tumorigenesis. Dysregulation of FOXO1 signaling is often associated with cancer progression. Given that the deletion of MLIP leads to a downregulation of the FOXO1 pathway, and overexpression of MLIP is likely to have the opposite effect, therapeutics aimed at enhancing MLIP function or expression could potentially restore the normal function of FOXO1 signaling, thereby inhibiting cancer development and progression.
Additionally, MLIP's interactions with p5311, a well-known tumor suppressor gene, further underscore its potential as a therapeutic target. Given that MLIP deficient cardiomyocytes showed an increased expression of p53, it is plausible to hypothesize that MLIP could play a role in the regulation of p53, and by extension, cell cycle control and apoptosis.
However, it is essential to remember that the exact mechanisms of MLIP in these signaling pathways are not fully understood, and further research is necessary to establish MLIP as a therapeutic target. Furthermore, it's crucial to understand the potential off-target effects and safety profile of any MLIP-targeting therapies due to MLIP's role in non-cancerous cells and processes, such as cardiac function. In summary, the modulation of MLIP's function or its interactions with key signaling pathways presents a promising approach for the development of novel cancer therapeutics.
Author Contributions
All authors contributed to writing and editing this review. All authors have read and agreed to the published version of the manuscript.
Funding
All authors were supported by an Undergraduate Research Experience Program (UREP29-039-1-011) Award from QRDI.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The next Generation. Cell 2011, 144. [Google Scholar] [CrossRef] [PubMed]
- Matthews, H.K.; Bertoli, C.; de Bruin, R.A.M. Cell Cycle Control in Cancer. Nat Rev Mol Cell Biol 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Paquette, M.; El-Houjeiri, L.; Pause, A. MTOR Pathways in Cancer and Autophagy. Cancers (Basel) 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Wei, J.; Liu, P. Attacking the PI3K/Akt/MTOR Signaling Pathway for Targeted Therapeutic Treatment in Human Cancer. Semin Cancer Biol 2022, 85. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M.T. FOXOs: Signalling Integrators for Homeostasis Maintenance. Nat Rev Mol Cell Biol 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Ronnebaum, S.M.; Patterson, C. The FoxO Family in Cardiac Function and Dysfunction. Annu Rev Physiol 2010, 72, 81–94. [Google Scholar] [CrossRef]
- Vousden, K.H.; Ryan, K.M. P53 and Metabolism. Nat Rev Cancer 2009, 9, 691–700. [Google Scholar] [CrossRef] [PubMed]
- Vousden, K.H.; Prives, C. Blinded by the Light: The Growing Complexity of P53. Cell 2009, 137, 413–431. [Google Scholar] [CrossRef] [PubMed]
- Ahmady, E.; Blais, A.; Burgon, P.G. Muscle Enriched Lamin Interacting Protein (Mlip) Binds Chromatin and Is Required for Myoblast Differentiation. Cells 2021, 10, 615. [Google Scholar] [CrossRef]
- Ahmady, E.; Deeke, S.A.; Rabaa, S.; Kouri, L.; Kenney, L.; Stewart, A.F.R.; Burgon, P.G. Identification of a Novel Muscle A-Type Lamin-Interacting Protein (MLIP). J Biol Chem 2011, 286, 19702–19713. [Google Scholar] [CrossRef] [PubMed]
- Cattin, M.-E.; Wang, J.; Weldrick, J.J.; Roeske, C.L.; Mak, E.; Thorn, S.L.; DaSilva, J.N.; Wang, Y.; Lusis, A.J.; Burgon, P.G. Deletion of MLIP (Muscle-Enriched A-Type Lamin-Interacting Protein) Leads to Cardiac Hyperactivation of Akt/Mammalian Target of Rapamycin (MTOR) and Impaired Cardiac Adaptation. J Biol Chem 2015, 290, 26699–26714. [Google Scholar] [CrossRef] [PubMed]
- Cattin, M.-E.; Deeke, S.A.; Dick, S.A.; Verret-Borsos, Z.J.A.; Tennakoon, G.; Gupta, R.; Mak, E.; Roeske, C.L.; Weldrick, J.J.; Megeney, L.A.; et al. Expression of Murine Muscle-Enriched A-Type Lamin-Interacting Protein (MLIP) Is Regulated by Tissue-Specific Alternative Transcription Start Sites. 2018; 293, 19761–19770. [Google Scholar] [CrossRef]
- Lopes Abath Neto, O.; Medne, L.; Donkervoort, S.; Rodríguez-García, M.E.; Bolduc, V.; Hu, Y.; Guadagnin, E.; Foley, A.R.; Brandsema, J.F.; Glanzman, A.M.; et al. MLIP Causes Recessive Myopathy with Rhabdomyolysis, Myalgia and Baseline Elevated Serum Creatine Kinase. Brain 2021, 144, 2722–2731. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-P.; Kataoka, M.; Chen, J.; Wu, G.; Ding, J.; Nie, M.; Lin, Z.; Liu, J.; Hu, X.; Ma, L.; et al. Cardiomyocyte-Enriched Protein CIP Protects against Pathophysiological Stresses and Regulates Cardiac Homeostasis. J Clin Invest 2015, 125, 4122–4134. [Google Scholar] [CrossRef]
- Zhang, Y.; Tong, G.H.; Wei, X.X.; Chen, H.Y.; Liang, T.; Tang, H.P.; Wu, C.A.; Wen, G.M.; Yang, W.K.; Liang, L.; et al. Identification of Five Cytotoxicity-Related Genes Involved in the Progression of Triple-Negative Breast Cancer. Front Genet 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Cattin, M.E.; Wang, J.; Weldrick, J.J.; Roeske, C.L.; Mak, E.; Thorn, S.L.; DaSilva, J.N.; Wang, Y.; Lusis, A.J.; Burgon, P.G. Deletion of MLIP (Muscle-Enriched A-Type Lamin-Interacting Protein) Leads to Cardiac Hyperactivation of Akt/Mammalian Target of Rapamycin (MTOR) and Impaired Cardiac Adaptation. Journal of Biological Chemistry 2015, 290. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, Y.; Sun, Y.; Zhao, X.; Sun, X.; Gong, T.; Liang, Z.; Ma, Y.; Zhang, X. Genome-Wide Analysis of LncRNAs, MiRNAs, and MRNAs Forming a Prognostic Scoring System in Esophageal Squamous Cell Carcinoma. PeerJ 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, M.; Cass, C.E.; Graham, K.; Mackey, J.R.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Germline Copy Number Variations Are Associated with Breast Cancer Risk and Prognosis. Sci Rep 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer Facts & Stats | Incidence, Age, Survival, & More Available online:. Available online: https://www.nationalbreastcancer.org/breast-cancer-facts/ (accessed on 26 October 2023).
- Michailidou, K.; Hall, P.; Gonzalez-Neira, A.; Ghoussaini, M.; Dennis, J.; Milne, R.L.; Schmidt, M.K.; Chang-Claude, J.; Bojesen, S.E.; Bolla, M.K.; et al. Large-Scale Genotyping Identifies 41 New Loci Associated with Breast Cancer Risk. Nat Genet 2013, 45, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Fachal, L.; Dunning, A.M. From Candidate Gene Studies to GWAS and Post-GWAS Analyses in Breast Cancer. Curr Opin Genet Dev 2015, 30, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, M.; Cass, C.E.; Graham, K.; Mackey, J.R.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Germline Copy Number Variations Are Associated with Breast Cancer Risk and Prognosis. Sci Rep 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tong, G.H.; Wei, X.X.; Chen, H.Y.; Liang, T.; Tang, H.P.; Wu, C.A.; Wen, G.M.; Yang, W.K.; Liang, L.; et al. Identification of Five Cytotoxicity-Related Genes Involved in the Progression of Triple-Negative Breast Cancer. Front Genet 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, Y.; Sun, Y.; Zhao, X.; Sun, X.; Gong, T.; Liang, Z.; Ma, Y.; Zhang, X. Genome-Wide Analysis of LncRNAs, MiRNAs, and MRNAs Forming a Prognostic Scoring System in Esophageal Squamous Cell Carcinoma. PeerJ 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Salt, I.P.; Johnson, G.; Ashcroft, S.J.H.; Hardie, D.G. AMP-Activated Protein Kinase Is Activated by Low Glucose in Cell Lines Derived from Pancreatic Beta Cells, and May Regulate Insulin Release. Biochem J 1998, 335 Pt 3, 533–539. [Google Scholar] [CrossRef]
- Yan, Y.; Zhou, X.E.; Xu, H.E.; Melcher, K. Structure and Physiological Regulation of AMPK. Int J Mol Sci 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Liong, S.; Lappas, M. Activation of AMPK Improves Inflammation and Insulin Resistance in Adipose Tissue and Skeletal Muscle from Pregnant Women. J Physiol Biochem 2015, 71. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Saud, S.M.; Young, M.R.; Chen, G.; Hua, B. Targeting AMPK for Cancer Prevention and Treatment. Oncotarget 2015, 6. [Google Scholar] [CrossRef]
- Keerthana, C.K.; Rayginia, T.P.; Shifana, S.C.; Anto, N.P.; Kalimuthu, K.; Isakov, N.; Anto, R.J. The Role of AMPK in Cancer Metabolism and Its Impact on the Immunomodulation of the Tumor Microenvironment. Front Immunol 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.C.; Hardie, D.G. AMPK: Sensing Glucose as Well as Cellular Energy Status. Cell Metab 2018, 27. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Zang, M.; Guo, W. AMPK as a Metabolic Tumor Suppressor: Control of Metabolism and Cell Growth. Future Oncol 2010, 6, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Sung, M.M.; Zordoky, B.N.; Bujak, A.L.; Lally, J.S.V.; Fung, D.; Young, M.E.; Horman, S.; Miller, E.J.; Light, P.E.; Kemp, B.E.; et al. AMPK Deficiency in Cardiac Muscle Results in Dilated Cardiomyopathy in the Absence of Changes in Energy Metabolism. Cardiovasc Res 2015, 107. [Google Scholar] [CrossRef] [PubMed]
- Alzahrani, A.S. PI3K/Akt/MTOR Inhibitors in Cancer: At the Bench and Bedside. Semin Cancer Biol 2019, 59, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Ediriweera, M.K.; Tennekoon, K.H.; Samarakoon, S.R. Role of the PI3K/AKT/MTOR Signaling Pathway in Ovarian Cancer: Biological and Therapeutic Significance. Semin Cancer Biol 2019, 59, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Polivka, J.; Janku, F. Molecular Targets for Cancer Therapy in the PI3K/AKT/MTOR Pathway. Pharmacol Ther 2014, 142, 164–175. [Google Scholar] [CrossRef] [PubMed]
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu Rev Med 2016, 67, 11–28. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, V.S.; Torres, F.F.; da Silva, D.G.H. FoxO3 and Oxidative Stress: A Multifaceted Role in Cellular Adaptation. J Mol Med (Berl) 2023, 101, 83–99. [Google Scholar] [CrossRef] [PubMed]
- Tsitsipatis, D.; Klotz, L.O.; Steinbrenner, H. Multifaceted Functions of the Forkhead Box Transcription Factors FoxO1 and FoxO3 in Skin. Biochim Biophys Acta Gen Subj 2017, 1861, 1057–1064. [Google Scholar] [CrossRef]
- Hosaka, T.; Biggs, W.H.; Tieu, D.; Boyer, A.D.; Varki, N.M.; Cavenee, W.K.; Arden, K.C. Disruption of Forkhead Transcription Factor (FOXO) Family Members in Mice Reveals Their Functional Diversification. Proc Natl Acad Sci U S A 2004, 101, 2975–2980. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Tindall, D.J. Dynamic FoxO Transcription Factors. J Cell Sci 2007, 120, 2479–2487. [Google Scholar] [CrossRef] [PubMed]
- Jiramongkol, Y.; Lam, E.W.F. FOXO Transcription Factor Family in Cancer and Metastasis. Cancer Metastasis Rev 2020, 39, 681–709. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Colman, M.J.; Dansen, T.B.; Burgering, B.M.T. FOXO Transcription Factors as Mediators of Stress Adaptation. Nat Rev Mol Cell Biol 2023. [Google Scholar] [CrossRef] [PubMed]
- Freycon, C.; Lupo, P.J.; Witkowski, L.; Budd, C.; Foulkes, W.D.; Goudie, C. A Systematic Review of the Prevalence of Pathogenic or Likely Pathogenic Germline Variants in Individuals with FOXO1 Fusion-Positive Rhabdomyosarcoma. Pediatr Blood Cancer 2023, 70. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ao, X.; Ding, W.; Ponnusamy, M.; Wu, W.; Hao, X.; Yu, W.; Wang, Y.; Li, P.; Wang, J. Critical Role of FOXO3a in Carcinogenesis. Mol Cancer 2018, 17. [Google Scholar] [CrossRef] [PubMed]
- Habrowska-Górczyńska, D.E.; Kozieł, M.J.; Kowalska, K.; Piastowska-Ciesielska, A.W. FOXO3a and Its Regulators in Prostate Cancer. Int J Mol Sci 2021, 22. [Google Scholar] [CrossRef]
- Bullock, M. FOXO Factors and Breast Cancer: Outfoxing Endocrine Resistance. Endocr Relat Cancer 2016, 23, R113–R130. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.; Chen, C.; Cheng, J. The Role and Molecular Mechanism of FoxO1 in Mediating Cardiac Hypertrophy. ESC Heart Fail 2020, 7, 3497–3504. [Google Scholar] [CrossRef] [PubMed]
- Helton, E.S.; Chen, X. P53 Modulation of the DNA Damage Response. J Cell Biochem 2007, 100, 883–896. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.B.; Schumacher, B. P53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Gnanasundram, S.V.; Bonczek, O.; Wang, L.; Chen, S.; Fahraeus, R. P53 MRNA Metabolism Links with the DNA Damage Response. Genes (Basel) 2021, 12. [Google Scholar] [CrossRef]
- Albrechtsen, N.; Dornreiter, I.; Grosse, F.; Ella, K.; Wiesmüller, L.; Deppert, W. Maintenance of Genomic Integrity by P53: Complementary Roles for Activated and Non-Activated P53. Oncogene 1999, 18, 7706–7717. [Google Scholar] [CrossRef]
- Wang, Y.H.; Ho, T.L.F.; Hariharan, A.; Goh, H.C.; Wong, Y.L.; Verkaik, N.S.; Lee, M.Y.; Tam, W.L.; van Gent, D.C.; Venkitaraman, A.R.; et al. Rapid Recruitment of P53 to DNA Damage Sites Directs DNA Repair Choice and Integrity. Proc Natl Acad Sci U S A 2022, 119. [Google Scholar] [CrossRef]
- Gatz, S.A.; Wiesmüller, L. P53 in Recombination and Repair. Cell Death Differ 2006, 13, 1003–1016. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; O’Grady, S.; Crown, J. Targeting P53 for the Treatment of Cancer. Semin Cancer Biol 2022, 79, 58–67. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; McGowan, P.M.; Crown, J.; O’Connor, D.; Gallagher, W.M. P53 as a Target for the Treatment of Cancer. Cancer Treat Rev 2014, 40, 1153–1160. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, N.; Nagano, H.; Tanaka, T. The Role of Tumor Suppressor P53 in Metabolism and Energy Regulation, and Its Implication in Cancer and Lifestyle-Related Diseases. Endocr J 2019, 66, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Olovnikov, I.A.; Kravchenko, J.E.; Chumakov, P.M. Homeostatic Functions of the P53 Tumor Suppressor: Regulation of Energy Metabolism and Antioxidant Defense. Semin Cancer Biol 2009, 19, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor Suppressor P53 and Metabolism. J Mol Cell Biol 2019, 11, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Ambroise, G.; Ouchida, A.T.; Lima Queiroz, A.; Smith, D.; Gimenez-Cassina, A.; Iwanicki, M.P.; Muller, P.A.; Norberg, E.; Vakifahmetoglu-Norberg, H. Effect of Mutant P53 Proteins on Glycolysis and Mitochondrial Metabolism. Mol Cell Biol 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yu, L.; Chen, W.; Xu, Y.; Wu, M.; Todorova, D.; Tang, Q.; Feng, B.; Jiang, L.; He, J.; et al. Wild-Type P53 Promotes Cancer Metabolic Switch by Inducing PUMA-Dependent Suppression of Oxidative Phosphorylation. Cancer Cell 2019, 35, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, W.; Lian, G.; Huang, B.; Du, A.; Gong, J.; Xiao, G.; Xu, C.; Wang, H.; Xie, L. CPT1 Regulates the Proliferation of Pulmonary Artery Smooth Muscle Cells through the AMPK-P53-P21 Pathway in Pulmonary Arterial Hypertension. Mol Cell Biochem 2019, 455, 169–183. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhang, C.; Hu, W.; Feng, Z. Tumor Suppressor P53 and Its Mutants in Cancer Metabolism. Cancer Lett 2015, 356, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Yan, Y.; Long, T.; Su, Q.; Wang, Y.; Chen, K.; Yang, T.; Zhao, G.; Ma, Q.; Hu, X.; Liu, C.; et al. Cardiac ISL1-Interacting Protein, a Cardioprotective Factor, Inhibits the Transition From Cardiac Hypertrophy to Heart Failure. Front Cardiovasc Med 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Huang, Z.-P.; Young Seok, H.; Zhou, B.; Chen, J.J.-F.; Chen, J.J.-F.; Tao, Y.; Pu, W.T.; Wang, D.-Z. CIP, a Cardiac Isl1-Interacting Protein, Represses Cardiomyocyte Hypertrophy. Circ Res 2012, 110, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Kumaran, M.; Cass, C.E.; Graham, K.; Mackey, J.R.; Hubaux, R.; Lam, W.; Yasui, Y.; Damaraju, S. Germline Copy Number Variations Are Associated with Breast Cancer Risk and Prognosis. Sci Rep 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tong, G.H.; Wei, X.X.; Chen, H.Y.; Liang, T.; Tang, H.P.; Wu, C.A.; Wen, G.M.; Yang, W.K.; Liang, L.; et al. Identification of Five Cytotoxicity-Related Genes Involved in the Progression of Triple-Negative Breast Cancer. Front Genet 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Li, Y.; Sun, Y.; Zhao, X.; Sun, X.; Gong, T.; Liang, Z.; Ma, Y.; Zhang, X. Genome-Wide Analysis of LncRNAs, MiRNAs, and MRNAs Forming a Prognostic Scoring System in Esophageal Squamous Cell Carcinoma. PeerJ 2020, 2020. [Google Scholar] [CrossRef] [PubMed]
- Bou Zeid, N.; Yazbeck, V. PI3k Inhibitors in NHL and CLL: An Unfulfilled Promise. Blood Lymphat Cancer 2023, 13, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.; Hassenrück, F.; Hallek, M. Copanlisib for Treatment of B-Cell Malignancies: The Development of a PI3K Inhibitor with Considerable Differences to Idelalisib. Drug Des Devel Ther 2018, 12, 2577–2590. [Google Scholar] [CrossRef]
- Patel, K.; Danilov, A. V.; Pagel, J.M. Duvelisib for CLL/SLL and Follicular Non-Hodgkin Lymphoma. Blood 2019, 134, 1573–1577. [Google Scholar] [CrossRef]
- Langer, C.J.; Redman, M.W.; Wade, J.L.; Aggarwal, C.; Bradley, J.D.; Crawford, J.; Stella, P.J.; Knapp, M.H.; Miao, J.; Minichiello, K.; et al. SWOG S1400B (NCT02785913), a Phase II Study of GDC-0032 (Taselisib) for Previously Treated PI3K-Positive Patients with Stage IV Squamous Cell Lung Cancer (Lung-MAP Sub-Study). J Thorac Oncol 2019, 14, 1839–1846. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.; Chia, S.; Kanakamedala, H.; Hsu, W.C.; Park, J.; Chandiwana, D.; Ridolfi, A.; Yu, C.L.; Zarate, J.P.; Rugo, H.S. Effectiveness of Alpelisib + Fulvestrant Compared with Real-World Standard Treatment Among Patients with HR+, HER2-, PIK3CA-Mutated Breast Cancer. Oncologist 2021, 26, e1133–e1142. [Google Scholar] [CrossRef] [PubMed]
- Bertho, M.; Patsouris, A.; Augereau, P.; Robert, M.; Frenel, J.S.; Blonz, C.; Campone, M. A Pharmacokinetic Evaluation of Alpelisib for the Treatment of HR+, HER2-Negative, PIK3CA-Mutated Advanced or Metastatic Breast Cancer. Expert Opin Drug Metab Toxicol 2021, 17, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Sarker, D.; Ang, J.E.; Baird, R.; Kristeleit, R.; Shah, K.; Moreno, V.; Clarke, P.A.; Raynaud, F.I.; Levy, G.; Ware, J.A.; et al. First-in-Human Phase I Study of Pictilisib (GDC-0941), a Potent Pan-Class I Phosphatidylinositol-3-Kinase (PI3K) Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res 2015, 21, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Munster, P.; Aggarwal, R.; Hong, D.; Schellens, J.H.M.; Van Der Noll, R.; Specht, J.; Witteveen, P.O.; Werner, T.L.; Dees, E.C.; Bergsland, E.; et al. First-in-Human Phase I Study of GSK2126458, an Oral Pan-Class I Phosphatidylinositol-3-Kinase Inhibitor, in Patients with Advanced Solid Tumor Malignancies. Clin Cancer Res 2016, 22, 1932–1939. [Google Scholar] [CrossRef] [PubMed]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arija, J.A.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin Cancer Res 2019, 25, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Bang, Y.J.; Kang, Y.K.; Ng, M.; Chung, H.C.; Wainberg, Z.A.; Gendreau, S.; Chan, W.Y.; Xu, N.; Maslyar, D.; Meng, R.; et al. A Phase II, Randomised Study of MFOLFOX6 with or without the Akt Inhibitor Ipatasertib in Patients with Locally Advanced or Metastatic Gastric or Gastroesophageal Junction Cancer. Eur J Cancer 2019, 108, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.C.; Oliveira, M.; Howell, S.J.; Dalenc, F.; Cortes, J.; Gomez Moreno, H.L.; Hu, X.; Jhaveri, K.; Krivorotko, P.; Loibl, S.; et al. Capivasertib in Hormone Receptor-Positive Advanced Breast Cancer. N Engl J Med 2023, 388, 2058–2070. [Google Scholar] [CrossRef] [PubMed]
- Forde, K.; Resta, N.; Ranieri, C.; Rea, D.; Kubassova, O.; Hinton, M.; Andrews, K.A.; Semple, R.; Irvine, A.D.; Dvorakova, V. Clinical Experience with the AKT1 Inhibitor Miransertib in Two Children with PIK3CA-Related Overgrowth Syndrome. Orphanet J Rare Dis 2021, 16. [Google Scholar] [CrossRef] [PubMed]
- Tolcher, A.W.; Patnaik, A.; Papadopoulos, K.P.; Rasco, D.W.; Becerra, C.R.; Allred, A.J.; Orford, K.; Aktan, G.; Ferron-Brady, G.; Ibrahim, N.; et al. Phase I Study of the MEK Inhibitor Trametinib in Combination with the AKT Inhibitor Afuresertib in Patients with Solid Tumors and Multiple Myeloma. Cancer Chemother Pharmacol 2015, 75, 183–189. [Google Scholar] [CrossRef]
- Aghajanian, C.; Bell-McGuinn, K.M.; Burris, H.A.; Siu, L.L.; Stayner, L.A.; Wheler, J.J.; Hong, D.S.; Kurkjian, C.; Pant, S.; Santiago-Walker, A.; et al. A Phase I, Open-Label, Two-Stage Study to Investigate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Oral AKT Inhibitor GSK2141795 in Patients with Solid Tumors. Invest New Drugs 2018, 36, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- Myers, A.P.; Konstantinopoulos, P.A.; Barry, W.T.; Luo, W.; Broaddus, R.R.; Makker, V.; Drapkin, R.; Liu, J.; Doyle, A.; Horowitz, N.S.; et al. Phase II, 2-Stage, 2-Arm, PIK3CA Mutation Stratified Trial of MK-2206 in Recurrent Endometrial Cancer. Int J Cancer 2020, 147, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Besse, B.; Leighl, N.; Bennouna, J.; Papadimitrakopoulou, V.A.; Blais, N.; Traynor, A.M.; Soria, J.C.; Gogov, S.; Miller, N.; Jehl, V.; et al. Phase II Study of Everolimus-Erlotinib in Previously Treated Patients with Advanced Non-Small-Cell Lung Cancer. Ann Oncol 2014, 25, 409–415. [Google Scholar] [CrossRef] [PubMed]
- Besse, B.; Heist, R.S.; Papadmitrakopoulou, V.A.; Camidge, D.R.; Beck, J.T.; Schmid, P.; Mulatero, C.; Miller, N.; Dimitrijevic, S.; Urva, S.; et al. A Phase Ib Dose-Escalation Study of Everolimus Combined with Cisplatin and Etoposide as First-Line Therapy in Patients with Extensive-Stage Small-Cell Lung Cancer. Ann Oncol 2014, 25, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, L.; Bahleda, R.; Tolaney, S.M.; Kwak, E.L.; Cleary, J.M.; Pandya, S.S.; Hollebecque, A.; Abbas, R.; Ananthakrishnan, R.; Berkenblit, A.; et al. Phase I Study of Neratinib in Combination with Temsirolimus in Patients with Human Epidermal Growth Factor Receptor 2-Dependent and Other Solid Tumors. J Clin Oncol 2014, 32, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Moran, T.; Palmero, R.; Provencio, M.; Insa, A.; Majem, M.; Reguart, N.; Bosch-Barrera, J.; Isla, D.; Costa, E.C.; Lee, C.; et al. A Phase Ib Trial of Continuous Once-Daily Oral Afatinib plus Sirolimus in Patients with Epidermal Growth Factor Receptor Mutation-Positive Non-Small Cell Lung Cancer and/or Disease Progression Following Prior Erlotinib or Gefitinib. Lung Cancer 2017, 108, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Pearson, A.D.J.; Federico, S.M.; Aerts, I.; Hargrave, D.R.; DuBois, S.G.; Iannone, R.; Geschwindt, R.D.; Wang, R.; Haluska, F.G.; Trippett, T.M.; et al. A Phase 1 Study of Oral Ridaforolimus in Pediatric Patients with Advanced Solid Tumors. Oncotarget 2016, 7, 84736–84747. [Google Scholar] [CrossRef]
- García-Saenz, J.A.; Martínez-Jañez, N.; Cubedo, R.; Jerez, Y.; Lahuerta, A.; Gonzalez-Santiago, S.; Ferrer, N.; Ramos, M.; Alonso-Romero, J.L.; Anton, A.; et al. Sapanisertib plus Fulvestrant in Postmenopausal Women with Estrogen Receptor-Positive/HER2-Negative Advanced Breast Cancer after Progression on Aromatase Inhibitor. Clin Cancer Res 2022, 28, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Heudel, P.; Frenel, J.S.; Dalban, C.; Bazan, F.; Joly, F.; Arnaud, A.; Abdeddaim, C.; Chevalier-Place, A.; Augereau, P.; Pautier, P.; et al. Safety and Efficacy of the MTOR Inhibitor, Vistusertib, Combined With Anastrozole in Patients With Hormone Receptor-Positive Recurrent or Metastatic Endometrial Cancer: The VICTORIA Multicenter, Open-Label, Phase 1/2 Randomized Clinical Trial. JAMA Oncol 2022, 8, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Wunderle, L.; Badura, S.; Schleyer, E.; Brüggemann, M.; Serve, H.; Schnittger, S.; Gökbuget, N.; Pfeifer, H.; Wagner, S.; et al. A Phase I Study of a Dual PI3-Kinase/MTOR Inhibitor BEZ235 in Adult Patients with Relapsed or Refractory Acute Leukemia. BMC Pharmacol Toxicol 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Lackner, M.R.; Oudard, S.; Escudier, B.; Ralph, C.; Brown, J.E.; Hawkins, R.E.; Castellano, D.; Rini, B.I.; Staehler, M.D.; et al. Randomized Open-Label Phase II Trial of Apitolisib (GDC-0980), a Novel Inhibitor of the PI3K/Mammalian Target of Rapamycin Pathway, versus Everolimus in Patients with Metastatic Renal Cell Carcinoma. Journal of Clinical Oncology 2016, 34, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Dolly, S.O.; Wagner, A.J.; Bendell, J.C.; Kindler, H.L.; Krug, L.M.; Seiwert, T.Y.; Zauderer, M.G.; Lolkema, M.P.; Apt, D.; Yeh, R.F.; et al. Phase I Study of Apitolisib (GDC-0980), Dual Phosphatidylinositol-3-Kinase and Mammalian Target of Rapamycin Kinase Inhibitor, in Patients with Advanced Solid Tumors. Clin Cancer Res 2016, 22, 2874–2884. [Google Scholar] [CrossRef]
- Tarantelli, C.; Gaudio, E.; Arribas, A.J.; Kwee, I.; Hillmann, P.; Rinaldi, A.; Cascione, L.; Spriano, F.; Bernasconi, E.; Guidetti, F.; et al. PQR309 Is a Novel Dual PI3K/MTOR Inhibitor with Preclinical Antitumor Activity in Lymphomas as a Single Agent and in Combination Therapy. Clin Cancer Res 2018, 24, 120–129. [Google Scholar] [CrossRef] [PubMed]
- Grilley-Olson, J.E.; Bedard, P.L.; Fasolo, A.; Cornfeld, M.; Cartee, L.; Razak, A.R.A.; Stayner, L.A.; Wu, Y.; Greenwood, R.; Singh, R.; et al. A Phase Ib Dose-Escalation Study of the MEK Inhibitor Trametinib in Combination with the PI3K/MTOR Inhibitor GSK2126458 in Patients with Advanced Solid Tumors. Invest New Drugs 2016, 34, 740–749. [Google Scholar] [CrossRef]
- Munster, P.; Aggarwal, R.; Hong, D.; Schellens, J.H.M.; Van Der Noll, R.; Specht, J.; Witteveen, P.O.; Werner, T.L.; Dees, E.C.; Bergsland, E.; et al. First-in-Human Phase I Study of GSK2126458, an Oral Pan-Class I Phosphatidylinositol-3-Kinase Inhibitor, in Patients with Advanced Solid Tumor Malignancies. Clin Cancer Res 2016, 22, 1932–1939. [Google Scholar] [CrossRef] [PubMed]
- Wainberg, Z.A.; Alsina, M.; Soares, H.P.; Braña, I.; Britten, C.D.; Del Conte, G.; Ezeh, P.; Houk, B.; Kern, K.A.; Leong, S.; et al. A Multi-Arm Phase I Study of the PI3K/MTOR Inhibitors PF-04691502 and Gedatolisib (PF-05212384) plus Irinotecan or the MEK Inhibitor PD-0325901 in Advanced Cancer. Target Oncol 2017, 12, 775–785. [Google Scholar] [CrossRef] [PubMed]
- Morscher, R.J.; Brard, C.; Berlanga, P.; Marshall, L. V.; André, N.; Rubino, J.; Aerts, I.; De Carli, E.; Corradini, N.; Nebchi, S.; et al. First-in-Child Phase I/II Study of the Dual MTORC1/2 Inhibitor Vistusertib (AZD2014) as Monotherapy and in Combination with Topotecan-Temozolomide in Children with Advanced Malignancies: Arms E and F of the AcSé-ESMART Trial. Eur J Cancer 2021, 157, 268–277. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Porta, C.; Suárez, C.; Hainsworth, J.; Voog, E.; Duran, I.; Reeves, J.; Czaykowski, P.; Castellano, D.; Chen, J.; et al. Randomized Phase II Trial of Sapanisertib ± TAK-117 vs. Everolimus in Patients With Advanced Renal Cell Carcinoma After VEGF-Targeted Therapy. Oncologist 2022, 27, 1048–1057. [Google Scholar] [CrossRef] [PubMed]
- Starks, D.C.; Rojas-Espaillat, L.; Meissner, T.; Williams, C.B. Phase I Dose Escalation Study of Dual PI3K/MTOR Inhibition by Sapanisertib and Serabelisib in Combination with Paclitaxel in Patients with Advanced Solid Tumors. Gynecol Oncol 2022, 166, 403–409. [Google Scholar] [CrossRef] [PubMed]
- Jiramongkol, Y.; Lam, E.W.F. FOXO Transcription Factor Family in Cancer and Metastasis. Cancer Metastasis Rev 2020, 39, 681–709. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Regulatory Interactions Between MLIP and AMPK in Cancer Metabolism. This schematic illustrates the bidirectional regulatory relationship between Muscle-enriched A-type Lamin-interacting Protein (MLIP) and AMP-activated Protein Kinase (AMPK). MLIP is depicted as acting on the fulcrum of AMPK action as an Oncogene or Tumour suppressor.
Figure 1.
Regulatory Interactions Between MLIP and AMPK in Cancer Metabolism. This schematic illustrates the bidirectional regulatory relationship between Muscle-enriched A-type Lamin-interacting Protein (MLIP) and AMP-activated Protein Kinase (AMPK). MLIP is depicted as acting on the fulcrum of AMPK action as an Oncogene or Tumour suppressor.
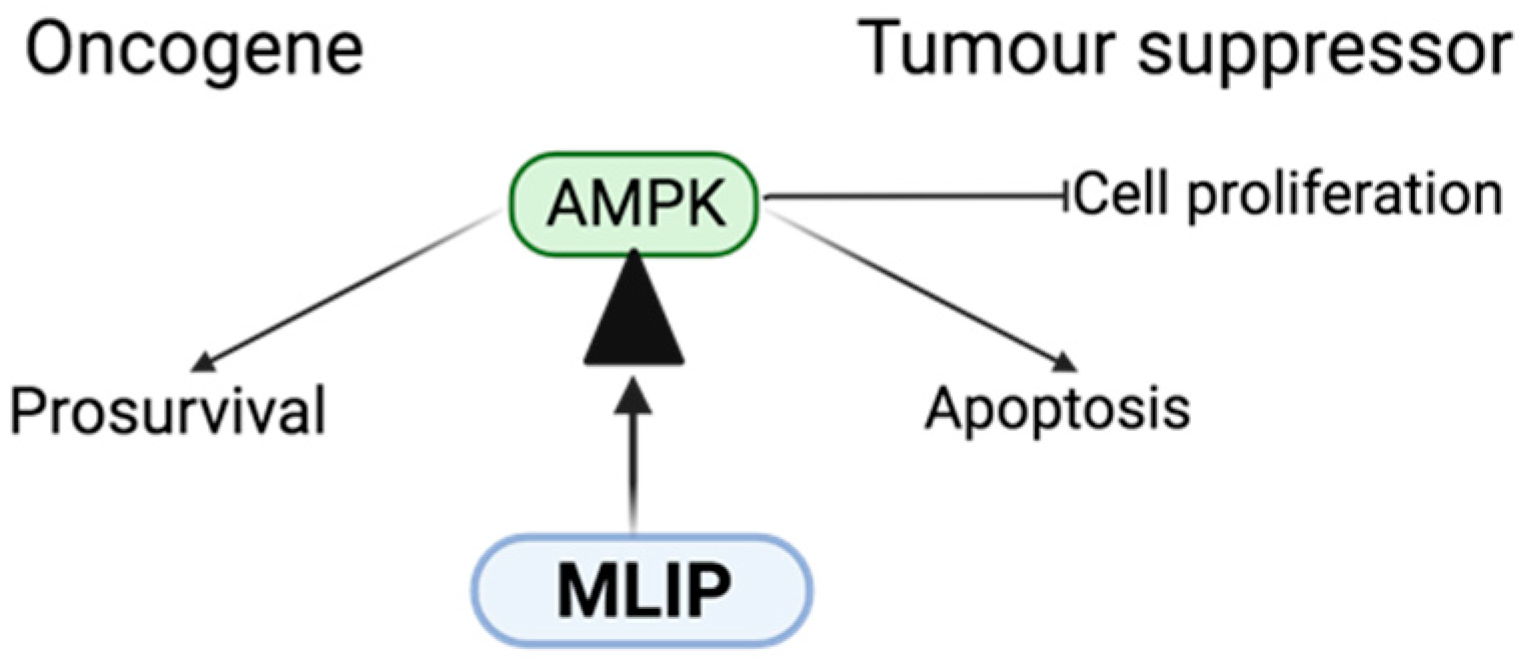
Figure 2.
Influence of MLIP on Key Signaling Pathways and Therapeutic Interventions in Cancer. This figure delineates the role of Muscle-enriched A-type Lamin-interacting Protein (MLIP) in modulating critical signaling cascades involved in cancer pathophysiology. On the left panel, MLIP interaction with the PI3K/AKT/mTOR pathway is illustrated, depicting potential points of therapeutic intervention with listed drugs. The dashed lines represent indirect effects on downstream processes such as cell cycle arrest, DNA repair, senescence, and apoptosis, which are key cellular responses in cancer therapy. On the right panel, the absence of MLIP's regulatory influence is shown to result in enhanced cell growth and division, as well as angiogenesis, contributing to tumor progression. The drugs targeting these pathways are grouped according to their mechanism of action, highlighting the potential for MLIP to serve as a pivotal point for therapeutic targeting in cancer treatment. Figure created with BioRender.com.
Figure 2.
Influence of MLIP on Key Signaling Pathways and Therapeutic Interventions in Cancer. This figure delineates the role of Muscle-enriched A-type Lamin-interacting Protein (MLIP) in modulating critical signaling cascades involved in cancer pathophysiology. On the left panel, MLIP interaction with the PI3K/AKT/mTOR pathway is illustrated, depicting potential points of therapeutic intervention with listed drugs. The dashed lines represent indirect effects on downstream processes such as cell cycle arrest, DNA repair, senescence, and apoptosis, which are key cellular responses in cancer therapy. On the right panel, the absence of MLIP's regulatory influence is shown to result in enhanced cell growth and division, as well as angiogenesis, contributing to tumor progression. The drugs targeting these pathways are grouped according to their mechanism of action, highlighting the potential for MLIP to serve as a pivotal point for therapeutic targeting in cancer treatment. Figure created with BioRender.com.
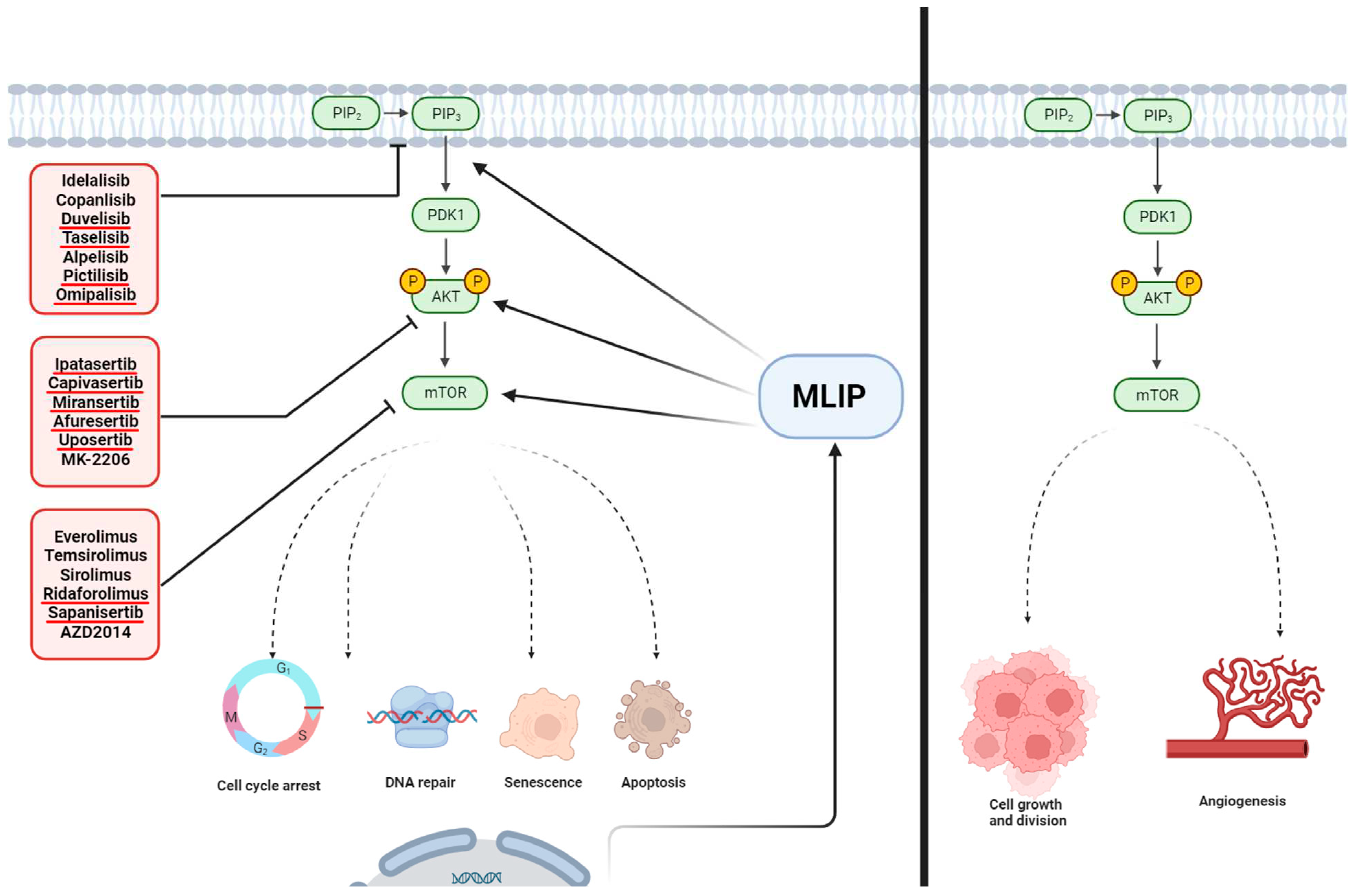
Figure 3.
Comparative Schematic of MLIP Signaling Pathways in Physiological and Pathological States. A comparative overview of the intracellular signaling pathways involving Muscle-enriched A-type Lamin-interacting Protein (MLIP) in two states: physiological (left) and pathological (right). In the physiological state, MLIP negatively regulates AKT, leading to reduced mTOR activity and an increase in AMPK activity, depicted by solid black lines. These interactions suggest a role for MLIP in maintaining cellular energy balance and potentially inhibiting cancer cell growth. In the pathological state (right), the loss of MLIP leads to increased AKT-mTOR activity, potentially promoting cell growth and proliferation. The altered signaling dynamics in the pathological state of MLIP underscore its potential as a regulatory switch in cancer metabolism and a target for therapeutic intervention. Figure created with BioRender.com.
Figure 3.
Comparative Schematic of MLIP Signaling Pathways in Physiological and Pathological States. A comparative overview of the intracellular signaling pathways involving Muscle-enriched A-type Lamin-interacting Protein (MLIP) in two states: physiological (left) and pathological (right). In the physiological state, MLIP negatively regulates AKT, leading to reduced mTOR activity and an increase in AMPK activity, depicted by solid black lines. These interactions suggest a role for MLIP in maintaining cellular energy balance and potentially inhibiting cancer cell growth. In the pathological state (right), the loss of MLIP leads to increased AKT-mTOR activity, potentially promoting cell growth and proliferation. The altered signaling dynamics in the pathological state of MLIP underscore its potential as a regulatory switch in cancer metabolism and a target for therapeutic intervention. Figure created with BioRender.com.
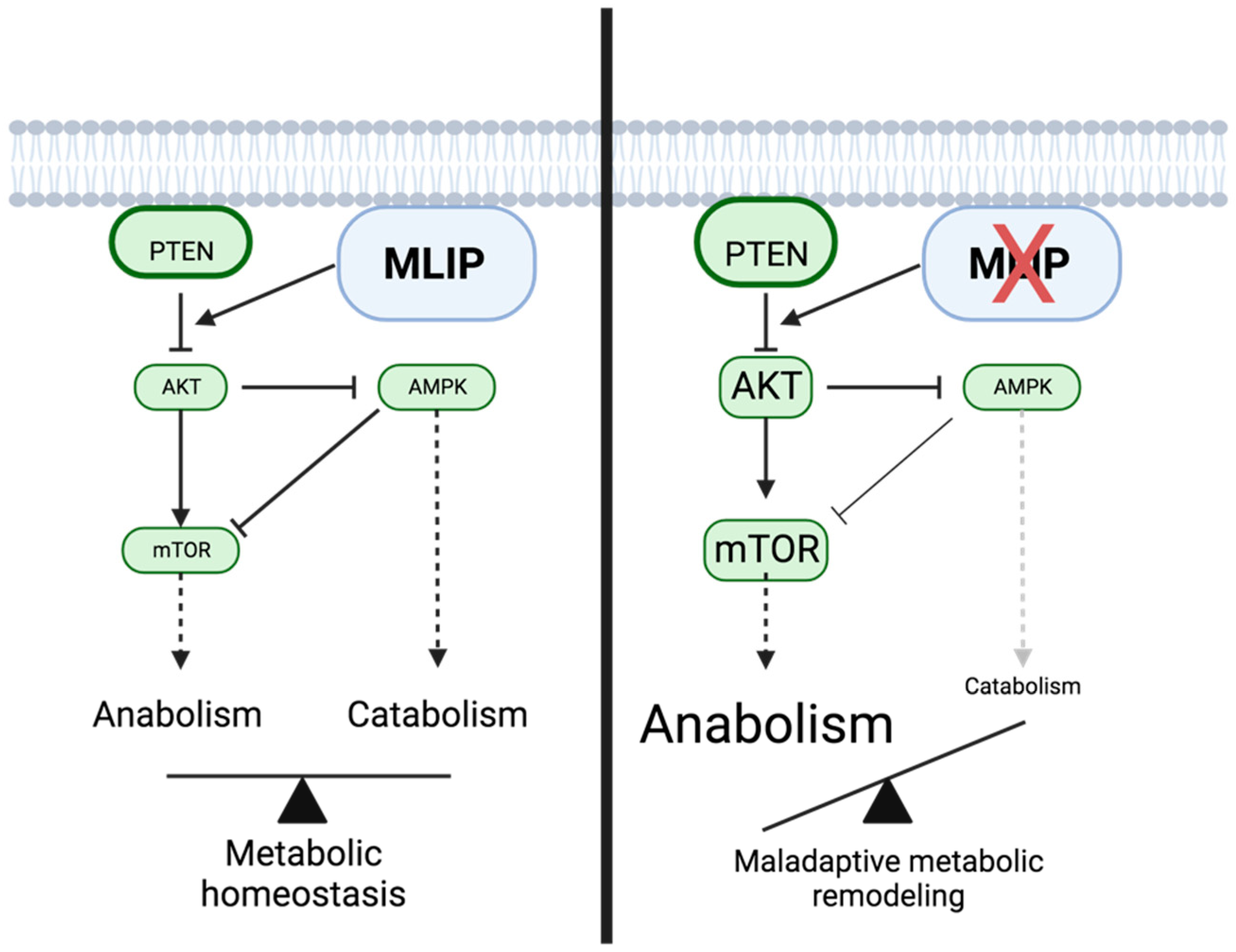

Table 1.
Association of MLIP with Various Cancer Types and Clinical Outcomes.
| Cancer | Status | Reference |
|---|---|---|
| Breast cancer | 1 out of 6 genes associated with breast cancer risk and recurrence free survival | [65] |
| Triple negative breast cancer | Upregulated in and associated with patient survival in triple-negative breast cancer | [66] |
| Esophageal cancer | One of 7 risk RNAs for esophageal cancer | [67] |
Table 2.
Overview of Targeted Therapies Acting on PI3K/AKT/mTOR Pathways in Cancer.
| Drug | Molecular Target | Status | Reference |
|---|---|---|---|
| Idelalisib (Zydelig) | PI3K | Relapsed chronic lymphocytic leukemia (CLL), follicular lymphoma, and small lymphocytic lymphoma (SLL) | [68] |
| Copanlisib (Aliqopa) | PI3K | relapsed follicular lymphoma | [69] |
| Duvelisib (Copiktra) | PI3K | relapsed or refractory CLL, SLL, and follicular lymphoma | [70] |
| Taselisib (GDC-0032) | PI3K | investigational drug clinical trials for various types of cancer, including breast cancer and lung cancer | [71] |
| Alpelisib (Piqray) | PI3K | HR-positive, HER2-negative, PIK3CA-mutated advanced or metastatic breast cancer | [72,73] |
| Pictilisib (GDC-0941) | PI3K | investigational drug clinical trials for various types of cancer, including breast cancer and non-small cell lung cancer. | [74] |
| Omipalisib (GSK2126458) | PI3K | investigational drug clinical trials for various types of cancer, including melanoma and pancreatic cancer. | [75] |
| Ipatasertib (GDC-0068) | AKT | investigational drug clinical trials for various types of cancer, including breast cancer, prostate cancer, and ovarian cancer. | [76,77] |
| Capivaserib (AZD5363) | AKT | investigational drug clinical trials for various types of cancer, including breast cancer, prostate cancer, and non-small cell lung cancer. | [78] |
| Miransertib (ARQ092) | AKT | investigational drug clinical trials for various types of cancer, including endometrial cancer, solid tumors, and proteus syndrome. | [79] |
| Afuresertib (GSK2110183) | AKT | investigational drug clinical trials for multiple myeloma and other hematologic malignancies | [80] |
| Uprosertib (GSK2141795) | AKT | investigational drug clinical trials for various types of cancer, including solid tumors and lymphomas. | [81] |
| MK-2206 | AKT | investigational drug clinical trials for various types of cancer, including breast cancer, colorectal cancer, and non-small cell lung cancer. | [82] |
| Everolimus (Afinitor, Zortress) | mTOR | advanced renal cell carcinoma (RCC), progressive neuroendocrine tumors of pancreatic origin (PNET), advanced hormone receptor-positive, HER2-negative breast cancer, renal angiomyolipoma with tuberous sclerosis complex (TSC), and subependymal giant cell astrocytoma (SEGA) associated with TSC. | [83,84] |
| Temsirolimus (Torisel) | mTOR | advanced renal cell carcinoma (RCC) | [85] |
| Sirolimus (Rapamune) | mTOR | potential anti-cancer properties in certain cancers, such as TSC-associated lymphangioleiomyomatosis (LAM). | [86] |
| Ridaforolimus (AP23573, MK-8669) | mTOR | investigational drug clinical trials for various types of cancer, including sarcomas, endometrial cancer, and other solid tumors. | [87] |
| Sapanisertib (INK128, TAK-228) | mTOR | investigational drug clinical trials for various types of cancer, including breast cancer, renal cell carcinoma, and non-Hodgkin's lymphoma. | [88] |
| AZD2014 (Vistusertib) | mTOR | investigational drug clinical trials for various types of cancer, including endometrial cancer, breast cancer, and non-small cell lung cancer. | [89] |
| Dactolisib (BEZ235) | dual PI3K/mTOR | preclinical and early-phase clinical trials for various types of solid tumors, including breast, prostate, and renal cell carcinoma. | [90] |
| Apitolisib (GDC-0980) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors, including colorectal, breast, and prostate cancer. | [91,92] |
| Bimiralisib (PQR309) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors and lymphomas. | [93] |
| Omipalisib (GSK2126458) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors and hematologic malignancies. | [94,95] |
| Gedatolisib (PF-05212384) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors and hematologic malignancies. | [96] |
| Vistusertib (AZD2014) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors and hematologic malignancies. | [97] |
| Serabelisib (INK1117, MLN0128, TAK-228) | dual PI3K/mTOR | early-phase clinical trials for various types of solid tumors and hematologic malignancies. | [98,99] |
Table 3.
Functional Roles of FOXO Transcription Factors in Cell Biology and Cancer.
| FOXO type | Role in cell biology | Role in cancer | Types of tumors | References |
|---|---|---|---|---|
| FOXO1 | Regulation of gluconeogenesis, cell proliferation, apoptosis, metabolism, inflammation, differentiation, and stress resistance. Global deletion causes embryonic cell death due to incomplete vascular development. | Tumor suppressor, regulation of cell cycle arrest, apoptosis, and DNA repair | Lymphoma, soft tissue sarcoma, acute myeloid leukemia (AML), breast cancer | [100] |
| FOXO2 | Involved in multiple important biological processes, such as cell-cycle arrest, DNA repair, apoptosis, glucose metabolism, aging, and autophagy. | Tumor suppressor, regulation of cell cycle arrest, apoptosis, and DNA repair | Not specified | [100] |
| FOXO3 | Affects lymph proliferation, widespread organ inflammation. Expression found in most tissues, including lymphocytes and myeloid cells. | Tumor suppressor, regulation of cell cycle arrest, apoptosis, and DNA repair | Neuroblastoma, breast cancer, colorectal cancer, glioblastoma, pancreatic ductal adenocarcinoma | [100] |
| FOXO4 | Required for stem cell function in multiple tissues, including maintenance of hematopoietic, neural, and muscle stem cell pools. | Tumor suppressor, regulation of cell cycle arrest and apoptosis | Not specified | [100] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



