Submitted:
30 January 2024
Posted:
30 January 2024
You are already at the latest version
Abstract
Although a disintegrin-like and metalloproteinase with thrombospondin type-1 motifs 13 (ADAMTS13) serve as the cleaving protease of vWF-multimers, its role in liver ischemia-reperfusion injury (IRI), as well as partial liver transplantation (pLT), remains unknown. First, ADAMTS13 deficient mice were subjected to IRI to investigate the impact on hepatic IRI. Hepatic IRI significantly decreased serum ADAMTS13 activity, while simultaneously up-regulated intrahepatic vWF expression. Ablation of ADAMTS13 aggravated liver IRI by promoting platelet aggregation and disturbing microcirculation in the liv-er accompanied by exacerbated transaminase release and enhanced pro-inflammatory cytokine/chemokines levels. Adjunctive supplementation of recombinant ADAMTS13 (rADAMTS13) improved hepatic microcirculatory failure observed in ADAMTS13-null mice. Immunohistochemistry revealed massive platelet aggregation within hepatic micro-vasculatures in ADAMTS13-null mice, which was attenuated by rADAMTS13 treatment. Consequently, peripheral platelet counts, transaminase release, and pro-inflammatory cytokines/chemokines levels deteriorated in ADAMTS13-null mice, while ameliorated by rADAMTS13 administration. Notably, rADAMTS13 improved IRI-induced pathologies even in WT mice compared with those without. Next, we tested rADAMTS13 in the rat pLT model. Similarly, in rat pLT, ADAMTS13 supplementation attenuated the aforementioned pathologies observed in the experimental mouse model. In conclusion, ADAMTS13 plays a pivotal role in alleviating both hepatic microcirculation and inflammatory responses, thus providing a novel therapeutic approach against LT-related stress.
Keywords:
ADAMTS13
; von Willebrand factor
; hepatic stellate cell
; thrombocytopenia
; liver transplantation
1. Introduction
Hepatic ischemia/reperfusion injury (IRI) is often encountered in various clinical situations, including liver transplantation (LTx), liver resection, and trauma/shock conditions followed by resuscitation. Hepatic IRI remains the leading cause of post-hepatectomy liver failure[1,2,3] and causes more than 10% of early allograft dysfunction in LTx[4,5].
In the meanwhile, living-donor liver transplantation (LDLT) has also been developed and spread to the world from the first successful LDLT in 1989[6] as a therapeutic alternative to deceased donor liver transplantation (DDLT) in the era of critical shortage of donor organs. LDLT has several inherent disadvantages compared with DDLT. For example, liver graft volume is inevitably small in adult-to-adult LDLT, i.e. small-for-size syndrome (SFSS)[7], which increases the portal venous pressure and subsequent endothelial injury. Additionally, there has been a recent transition towards the use of smaller grafts than before to increase the future remnant volume of donor livers to maximize donor safety[8]. Hence, the grafts from living donors may presumably be more susceptible to IRI than those from deceased donors.
Multimeric von-Willebrand factor (vWF) is a well-known central player not only in physiological hemostasis but also in pathological intravascular coagulation under diverse disease conditions. Much attention has been paid to its counteracting partner, a disintegrin-like and metalloproteinase with thrombospondin type-1 motifs 13 (ADAMTS13), as a key player in various coagulation disorders, including thrombotic microangiopathy (TMA)[9,10,11], thrombotic thrombocytopenic purpura (TTP)[12,13], cerebral[14], and myocardial infarction[15,16,17]. ADAMTS13 was originally discovered in 2001, whose congenital deficiency causes TTP[12,18,19]. Moreover, its acquired deficiency has been shown to induce various types of microangiopathies, such as hemolytic uremic syndrome (HUS)[12,18,19], atherosclerosis[20,21], etc. Under physiological conditions, ADAMTS13 cleaves highly prothrombotic unusually-large vWF multimers (UL-vWFM) into appropriate size, thereby preventing excessive platelet aggregation that may occlude microvessels. Thus, ADAMTS13 plays a pivotal role in maintaining microcirculatory blood flow without interfering with primary hemostasis.
Meanwhile, it has been shown that ADAMTS13 was predominantly produced by the liver[13,22,23], especially from hepatic stellate cells (HSCs)[24], although it can also be synthesized modestly in other tissues[25,26,27]. Therefore, we assumed that TMA-like pathologies might unavoidably occur in critical liver diseases potentially through the following mechanisms- i.e. ADAMTS13 production by HSCs may be decreased by liver damage, whereas highly-reactive UL-vWFM may be over-expressed from injured sinusoidal/endothelial cells. Consequently, the resulting imbalance between UL-vWFM and ADAMTS13 may lead to excessive platelet aggregation locally and systemically. Then, impaired hepatic microcirculation further accelerates UL-vWFM upregulation and ADAMTS13 downregulation. This would, in turn, be followed by much further platelet aggregation, hyper-coagulation, leukocyte adhesion/activation, and a cytokines/chemokines storm as a “vicious cycle”. However, the role of ADAMTS13 on hepatic IRI as well as LT-induced stress remains fully understood.
In this study, we first aimed to investigate the putative roles of ADAMTS13 in hepatic IRI utilizing ADAMTS13 deficient mice, and then elucidate the therapeutic potential of ADAMTS13 supplementation against a partial LT model.
2. Materials and Methods
2.1. Animals
Male ADAMTS13 knockout (KO) mice[28] (129/+Ter/SvJcl-TgH NCVC) were purchased from the National Cerebral and Cardiovascular Center Research Institute (Osaka, Japan), and corresponding wild-type (WT) mice were obtained from Japan CLEA (Osaka, Japan). The data of biochemical exams of blood from WT or ADAMTS13 KO mice are provided in Supplemental Table S1. The protein and mRNA expressions are evaluated in each genotype (Suppl. Figure S1). Male LEW/CrlCrlj rats were purchased from Charles River Laboratories (Yokohama, Japan). All animals were maintained under specific pathogen-free conditions with a 12-hour day/night rhythm and with ad libitum access to food and water. They all received humane care according to the Guide for the Care and Use of Laboratory Animals (National Institutes of Health Publication 8th, revised, 2011). All experimental protocols were approved by the Animal Research Committee of Kyoto University (MedKyo 15210).
2.2. Mouse warm IRI
We used an established mouse model of 70% partial warm hepatic IRI as previously described[29]. To investigate the role of ADAMTS13 in hepatic IRI, WT and ADAMTS13-deficient mice were pretreated with vehicle (phosphate-buffered saline: PBS) or recombinant ADAMTS13 (W688X, kindly provided by Kaketsuken, Kumamoto, Japan) before ischemic insult and before reperfusion (20 U/body). W688X is a C-terminal truncated protein (97 kDa molecular weight) containing the cysteine-rich/spacer domain that is functionally essential for cleaving UL-vWFMs[30]. Then, mice were subjected to warm IRI and sacrificed to collect blood and tissue samples at 2, 6, and 24 hours after reperfusion.
2.3. Rat partial liver transplantation
To mimic small-for-size grafts in adult-to-adult living-donor partial liver transplantation (LDLT), we developed a rat model of orthotopic 20% (partial liver transplantation) pLTx using a right lobe graft, as described in Supplemental materials. After 4-hour cold storage in histidine-tryptophan-ketoglutarate (HTK, Dr. Franz Köhler Chemie, Alsbach-Hähnlein, Germany) solution at 4°C, partial liver grafts were transplanted into recipients orthotopically (Suppl. Figure S4). To investigate the therapeutic impact of W688X in rat 20% pLTx model, the recipients were randomly assigned into the following 2 groups: A group treated with vehicle, phosphate-buffered saline (PBS) or a group treated with W688X. In the latter, W688X (200 U/body in 1.5ml PBS) was administered via the penile vein twice, just before clamping major vessels in the recipient and just before reperfusion during the recipient operation. In the vehicle group, the same volume of vehicle, 1.5ml PBS, was injected in the same manner. In the 20% pLTx model with 4-hour cold storage in HTK, the survival rate was investigated for 7 days after transplantation. After pLTx, the recipients were sacrificed to collect blood and liver tissue samples at 2, 6, and 24 hours after reperfusion.
2.4. Liver enzymes and platelet counts
Serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), and lactate hydrogenase (LDH) levels were measured using standard spectrophotometric methods with an automated clinical analyzer (JCA-BM9030, JEOL Ltd., Tokyo, Japan). Platelet counts were quantified using a Becton Dickinson QBC II Plus 4452 Automatic Blood Cell Counter.
2.5. Histology
Paraffin-embedded sections (4 µm thick) of liver tissues were stained with hematoxylin and eosin. The severity of liver IRI (necrosis, sinusoidal congestion, and vacuolization) was graded according to Suzuki’s criteria on a scale in a blinded manner[31].
2.6. Quantitative analysis of hepatic microcirculation
Hepatic microcirculation was assessed by a laser Doppler flowmetry (O2C®, LEA Medizintechnik GmbH, Giessen, Germany)[32,33]. The probe was placed on the liver surface (touched but without any pressure), and at least four independent points were randomly selected and recorded. The measurement was performed at 2 mm (mouse) or 8 mm (rat) depth from the liver surface, and the average value was calculated from four independent observations per animal. Relative changes in tissue blood flow to the pre-ischemic value were calculated.
2.7. Measurement of plasma ADAMTS13 activity
We employed the FRETS-VWF73 method, as described previously[34]. Briefly, plasma samples were diluted in reaction buffer (5mM bis-Tris, 25mM CaCl2, and 0.05% Tween-20, pH 6.0), then 4μM FRETS-VWF73 (PEPTIDE INSTITUTE, INC., Osaka, Japan) substrate solution and 10μL protease inhibitor cocktail (P8340, Sigma-Aldrich Inc., St. Louis, MO, USA) were added. After incubation, the emitted fluorescence intensity was measured using a fluorescence spectrophotometer (Fluoroskan Ascent FL, Thermo Labsystems, Helsinki, Finland) with excitation and emission at 355 and 460 nm, respectively. Plasma from naïve WT mice or LEW/CrlCrlj rats was used as the standard.
2.8. Immunofluorescence
After deparaffinization of liver sections, the antigen retrieval was performed with citrate buffer (10 mM, pH 6.0). After blocking with Protein Block Serum-Free (X0909, DAKO, Tokyo, Japan) for 30 minutes, the sections were incubated with primary rabbit anti-mouse CD42b (bs-2347R, Boston, MA, USA) or rabbit anti-rat CD41 antibody at 1:200 dilution overnight at 4°C. Subsequently, the sections were reacted with Alexa Fluor 594-conjugated goat anti-rabbit IgG (H+L) secondary antibody (Life Technologies Japan Ltd., Tokyo, Japan). The CD42b-positive area was quantified using Image J software (NIH, USA)[35]. Negative control slides were prepared by incubation with normal rabbit IgG (sc-2027, SantaCruz, CA, USA) instead of the primary antibody (Supplemental Figure S3).
2.9. Real-time reverse-transcription polymerase chain reaction (RT-PCR)
Total RNA was extracted from liver tissue using an RNeasy Kit (Qiagen, Venlo, Netherlands), and complementary DNA was prepared using an Omniscript RT kit (Qiagen). Quantitative PCR was performed with StepOnePlus™ Real-Time PCR System (Applied Biosystems, Tokyo, Japan) with Fast SYBR Green Master Mix or TaqMan Master Mix. Amplification conditions were as follows: 95°C for 20 seconds (sec), 95°C for 3 sec, followed by 45 cycles of 95°C for 15 sec and 60°C for 30 sec. Primers used to amplify specific gene fragments are listed in Supplementary Table S2 [28,29]. Target gene expressions were calculated relative to the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), or β-actin.
2.10. Immunohistochemistry
Paraffin sections were pretreated with 3% H2O2 in methanol for 10 minutes, and then subjected to antigen retrieval in citrate buffer (10mM, pH 6.0) by microwave. After blocking with 10% rabbit serum for 20 min, the sections were incubated with primary rat anti-mouse CD68 (FA-11; AbD Serotec, Kidlington, UK) or Ly-6G (RB6-8C5; Tonbo Biosciences, Irvine, CA, USA) antibodies at 1:100 dilution or anti-vWF antibody (ab6994, Abcam, Cambridge, UK) overnight at 4°C. Subsequently, the sections were reacted with biotinylated rabbit anti-rat IgG. The sections were then incubated with VECTASTAIN Elite ABC Kit (Vector Laboratories, Burlingame, CA, USA), stained with a Liquid DAB Substrate Chromogen System (DAKO), and then observed with a BZ-9000 fluorescence microscope (Keyence, Osaka, Japan).
2.11. Apoptosis assay
Terminal deoxynucleotidyl transferase-mediated dNTP nick-end labeling (TUNEL) assays were performed for the detection of apoptosis in 4-µm-thick paraffin sections with an In Situ Apoptosis Detection Kit (Takara Bio, Shiga, Japan) according to the manufacturer’s protocol. The negative control was prepared by omitting terminal deoxynucleotidyl transferase. TUNEL-positive cells were counted in at least five high-powered fields/sections under a light microscope.
2.12. Statistical analysis
All data are expressed as means ± standard errors of the means (SEMs). Differences among the experimental groups were analyzed with a two-way analysis of variance (ANOVA) followed by Bonferroni’s post-hoc tests for time-dependent parameters or with Student’s t-tests for unpaired data sets. Survival rates were estimated with the Kaplan-Meier method and a Mantel-Cox log-rank analysis. All statistical calculations were performed using Prism 5 (GraphPad Software Inc., La Jolla, CA, USA). Differences with P values < 0.05 were considered statistically significant.
3. Results
3.1. Liver IRI down-regulates ADAMTS13 and provokes imbalance between ADAMTS13 and vWF
First, we investigated whether hepatic IRI influenced the expression of ADAMTS13 in WT mice after the injury. As shown in Figure 1A, hepatic IRI decreased plasma ADAMTS13 activity down to 31.7 % of the physiological level 2 hours after reperfusion, although the remaining 30% of hepatic lobes were not exposed to IRI. Also, a 70% reduction of plasma ADAMTS13 activity persisted for 24 hours after reperfusion, indicating the deleterious damages to HSCs after hepatic IRI. Figure 1B depicts chronological alterations of hepatic ADAMTS13 and vWF mRNA expressions in WT after hepatic IRI, with or without W688X administration. Of note, ADAMTS13 was down-regulated from the onset of ischemia and suppressed even after reperfusion. In contrast, vWF mRNA was up-regulated during ischemia, reaching up to twice the pre-ischemic value at the end of 90 minutes ischemia. A 5-fold increase was then observed 24 hours after reperfusion, reflecting the damage to sinusoidal endothelial cells (SECs) by reperfusion injury. Of interest, rADAMTS13 administration completely normalized IR-induced upregulation of vWF mRNA down to the pre-ischemic level. These results indicate that HSCs and SECs are susceptible to hepatic IRI, resulting in a detrimental imbalance between vWF and ADAMTS13 expressions.
3.2. ADAMTS13-null mutation results in massive platelet aggregation within hepatic sinusoids and impairment of hepatic microcirculation during IRI
As shown in Figure 2A, immunohistochemical staining for CD42b revealed that ADAMTS13-deficiency significantly increased platelet aggregation within hepatic sinusoids after IRI compared with WT both at 6 (1.1 ± 0.2 vs. 0.4 ± 0.1%, p<0.05) and 24 hours (2.2 ± 0.5 vs. 0.6 ± 0.2%, p<0.001). These deleterious alterations in KO were, however, significantly ameliorated by rADAMTS13 administration both at 6 (1.1 ± 0.2 vs. 0.3 ± 0.1%, p<0.05) and 24 hours (2.2 ± 0.5 vs. 0.6 ± 0.1%, p<0.05). Peripheral platelet count in KO + vehicle (16.6 ± 3.2 × 104/μL) was significantly lower than that in WT + vehicle (37.2 ± 1.4 × 104/μL, p<0.001) at 24 hours after reperfusion, suggesting coexistence of TMA-like pathology in hepatic IRI. In contrast, W688X supplementation into KO mice significantly ameliorated severe thrombocytopenia at 6 hours (31.4 ± 2.5 vs. 48.0 ± 3.8 × 104/μL, p<0.001) and 24 hours (16.6 ± 3.2 vs. 44.5 ± 4.5 × 104/μL, p<0.001) after reperfusion. Similarly, W688X treatment significantly improved thrombocytopenia after hepatic IRI even in WT both at 6 (41.1 ± 3.6 vs. 53.1 ± 3.6 × 104/μL, p<0.05) and 24 hours (37.2 ± 1.4 vs. 49.0 ± 2.3 × 104/μL, p<0.05, Figure 2B). Of note, CD42b-positive stains increased as peripheral platelet count decreased, suggesting that circulating platelets aggregated into the injured microvasculature of the liver during IRI. Consequently, hepatic microcirculation in ADAMTS13 KO mice treated with vehicle remarkably fell to 38.4 ± 5.5 % of the pre-ischemic value at 24 hours after reperfusion, which was significantly lower than in WT treated with vehicle (67.5 ± 4.1 %, p<0.01, Figure 2C). Supplementation of W688X, however, significantly ameliorated hepatic microcirculation both in WT and KO at 2 hours after reperfusion (p<0.05 and p<0.01, respectively). Interestingly, hepatic microcirculation was well correlated with plasma ADAMTS13 activity (Supple Figure S2), suggesting the pivotal role of ADAMTS13 in the maintenance of hepatic microcirculation.
3.3. ADAMTS13 ameliorates IR-induced parenchymal damage
Next, we evaluated whether ADAMTS13 might affect IR-related hepatocellular damage. As shown in Figure 3A–C, serum AST, ALT, and LDH release were significantly higher in the KO + vehicle than in the WT + vehicle. In contrast, W688X supplementation in KO significantly reduced AST, ALT, and LDH levels. Moreover, W688X administration significantly ameliorated those liver enzymes both at 6 and 24 hours in WT. Consistently, histopathological evaluation (Figure 4D,E) showed severe lobular edema, congestion, ballooning, and massive necrosis all over the lobules in KO + vehicle as compared with WT + vehicle (Suzuki’s score at 6 hours: 9.7± 0.3 vs. 7.3 ± 0.5, respectively, p<0.01). While W688X administration significantly improved hepatocellular injury (7.3 ± 0.5 in WT + vehicle vs. 4.7 ± 1.0 in WT + W688X at 6 hours, p<0.01; 9.7 ± 0.3 vs. 4.5 ± 0.6 at 24 hours, p<0.001; 8.7 ± 0.5 in KO + vehicle vs.7.5 ± 0.2 in KO +W688X at 6 hours, p<0.05). Since apoptotic cell death is a major pathology deteriorating liver tissue after hepatic IRI, we next evaluated apoptosis in liver tissues by TUNEL staining at 6 and 24 hours after reperfusion. As shown in Figure 3F, TUNEL-positive cells were significantly reduced by W688X treatment in both WT and KO at 24 hours (65.1 ± 6.7 in WT + vehicle vs. 18.6 ± 5.2 in WT + rADAMTS13, p<0.01; 78.0 ± 10.3 in KO + vehicle vs. 39.1 ± 9.7 in KO + rADAMTS13, p<0.01).
3.4. ADAMTS13 attenuates the infiltration of macrophages and neutrophils after hepatic IRI
W688X supplementation significantly attenuated infiltration of CD68-positive macrophages to IR-exposed liver tissues in both KO and WT than in those without (p<0.05 for WT at 24 hours; p<0.01 for KO at 6 hours; p<0.001 for KO at 24 hours, Figure 4A). Moreover, infiltration of neutrophils, shown as Ly6G-positive cells in Figure 4B, was also significantly alleviated by W688X supplementation than in those without (p<0.001 for WT at 24 hours; p<0.05 for KO at 6 hours).
3.5. Influence of ADAMTS13 on pro-inflammatory cytokines and chemokines
We examined the effects of ADAMTS13 deficiency and W688X supplementation on the expression of pro- and anti-inflammatory cytokines (TNF-α, IL-1β, IL-6, and IL-10) and chemokine ligands (CXCL-2 and CXCL-10) in the liver tissues by quantitative RT-PCR (Figure 5). Notably, six hours after reperfusion, ADAMTS13 deficiency significantly increased the expression of IL-1β (p<0.05), IL-6 (p<0.01) and CXCL-10 (P <0.05) compared with those in WT mice. In contrast, W688X treatment significantly decreased TNF-α, IL-1β, IL-6, CXCL-2, and CXCL-10 expressions both in WT and KO mice (TNF-α: p<0.001 for WT and KO; IL-1β: p<0.001 for WT and KO; IL-6: p<0.05 for WT and p<0.001 for KO; CXCL-2: p<0.01 for WT and KO; CXCL-10: p<0.001 for WT and KO), indicating the anti-inflammatory potential of ADAMTS13.
3.6. ADAMTS13 suppresses LT stress-induced vWF upregulation and platelet aggregation within hepatic sinusoids and attenuates hepatic microcirculation
Having demonstrated the critical role of ADATS13 in liver IRI. Next, we sought to validate its efficacy utilizing a more clinically relevant rat partial LT model. As shown in Figure 6A, massive expression of vWF in sinusoidal space was observed at 2, 6, and 24 hours after transplantation. In contrast, W688X treatment suppressed vWF expressions in sinusoidal space, indicating the protective effect of ADAMTS13 on SECs (p=0.012 between the groups by 2-way ANOVA; 8.32 ± 0.42 vs. 1.97 ± 0.12 at 2 hours, p<0.001; 16.0 ± 0.71 vs. 3.44 ± 0.18 at 6 hours, p<0.001; 6.57 ± 0.42 vs. 0.68 ± 0.04, at 24 hours, p<0.001 by Bonferroni’s post-test). CD41 immunofluorescence of the partial liver allografts revealed massive platelet aggregation in hepatic sinusoids in the vehicle treatment group. While supplementation of W688X significantly alleviated platelet aggregation as manifested in Figure 6B. In line with these results, peripheral platelet counts in the vehicle group decreased down to half or less by 2 hours after reperfusion and persisted at least up to 24 hours after reperfusion. Supplementation of W688X significantly ameliorated thrombocytopenia at 2, 6, and 24 hours after reperfusion (p<0.025 between the groups by 2-way ANOVA: 39.4 ± 1.9 ×104 vs. 56.9 ± 0.8 ×104/μL at 2 hours, p<0.05; 36.9 ± 1.6 ×104 vs. 55.6 ± 0.6 ×104/μL at 6 hours, p<0.01; 34.0 ± 3.0 ×104 vs. 57.7 ± 1.0 ×104/μL at 24 hours, p<0.001 by Bonferroni’s post-test). In the vehicle group, LDH release reached the peak at 6 hours after reperfusion, while W688X treatment significantly attenuated such LDH release (Figure 6D, p<0.0001 between the groups by 2-way ANOVA; 4829 ± 188 U/L vs. 1680 ± 64 U/L at 6 hours, p<0.001 by Bonferroni’s post-test).
As shown in Figure 6E, by administration of W688X, the hepatic blood flow of the partial liver allograft was significantly improved from only 20 minutes to 24 hours after reperfusion (p<0.021 between the groups by 2-way ANOVA; 114 ± 2.5 A.U. vs. 170 ± 2.9 A.U at 20 minutes, p<0.01; 105 ± 2.5 A.U. vs. 181 ± 2.9 A.U. at 40 minutes, p<0.001; 105 ± 7.8 A.U. vs. 177 ± 15.4 A.U. at 2 hours, p<0.001; 115 ± 7.0 A.U. vs. 184 ± 2.5 A.U. at 6 hours, p<0.001; 152 ± 4.2 A.U. vs. 230 ± 2.4 A.U. at 24 hours, p<0.001 by Bonferroni’s post-test). Collectively, ADAMTS13 protected partial grafts from otherwise fulminant TMA-like microcirculation disturbance.
3.7. ADAMTS13 protects partial liver allografts from LT-related tissue damage
Figure 7A shows representative tissue sections of the partial liver allografts at 2, 6, and 24 hours after reperfusion. Liver tissues in the vehicle group exhibited cytoplasmic vacuolization, sinusoidal congestion, and massive cellular infiltration, whereas in the W688X group, these damages were significantly alleviated, reaching statistical significance in Suzuki’s score, as summarized in Figure 7B (5.83 ± 0.13 vs. 3.67 ± 0.17 at 2 hours, p<0.001; 6.67 ± 0.17 vs. 3.83 ± 0.13 at 6 hours, p<0.001; 7.83 ± 0.13 vs. 4.50 ± 0.17 at 24 hours, p<0.001 by Bonferroni’s post-test). As parameters for hepatocellular damage, we measured transaminase release at 2, 6, and 24 hours after reperfusion. Consistent with histological assessment, serum AST was significantly lower in W688X group (p<0.014 between the groups by 2-way ANOVA; 1211 ± 41 vs. 693 ± 16 IU/L at 6 hours, p<0.001; 1226 ± 69 vs. 865 ± 29 IU/L at 24 hours, p<0.05 by Bonferroni’s post-test). Similarly, serum ALT also exhibited a significant reduction in W688X group, indicating significantly less hepatocellular damage achieved by rADAMTS13 supplementation (p<0.015 between the groups by 2-way ANOVA; 989 ± 77 vs. 461 ± 12 IU/L at 6 hours, p<0.001; 791 ± 59 vs. 378 ± 10 IU/L at 24 hours, p<0.05 by Bonferroni’s post-test). Importantly, W688X treatment significantly improved an otherwise poor 7-day graft survival rate (Figure 7D).
3.8. ADAMTS13 suppress tissue inflammatory cytokines and endothelin-1
To reveal inflammatory cytokines expression, we quantified relative IL-1β, IL-6, and TNF-α mRNA expression in both groups at 2, 6, and 24 hours after reperfusion (Figure 8). In Group-W688X, the expression of IL-1β was significantly lower than in Group-vehicle at 2 and 6 hours after reperfusion (p<0.001 between the groups by 2-way ANOVA; 2.00 ± 0.05 vs. 0.50 ± 0.04, at 2 hours, p<0.001; 0.65 ± 0.05 vs. 0.21 ± 0.01 at 6 hours, p<0.01 by Bonferroni’s post-test). In Group-W688X, the expression of IL6 and TNF-α were significantly lower than in Group-vehicle at 2 hours after reperfusion (IL-6; p<0.05 between the groups by 2-way ANOVA: 638 ± 67 vs. 292 ± 21, p<0.01 by Bonferroni’s post-test, TNF-α; p<0.001 between the groups by 2-way 2 ANOVA: 3.55 ± 0.09 vs. 1.46 ± 0.06, p<0.001 by Bonferroni’s post-test). These results indicated that ADAMTS13 possessed a powerful anti-inflammatory effect on liver IRI. Similarly, in Group-W688X, the expression of endothelin (ET1) was significantly lower than in Group-vehicle at 2 hours after reperfusion (p=0.001 between the groups by 2-way 2ANOVA, 7.07 ± 0.17 vs. 3.73 ± 0.17, p<0.001 by Bonferroni’s post-test).
4. Discussion
In the present study, we demonstrated that hepatic IRI-induced TMA-like pathology is characterized by a significant reduction of ADAMTS13 activity and simultaneous up-regulation of vWF both in mouse warm IRI model and rat pLT model. The imbalance between prothrombotic vWF and its counter-acting partner, ADAMTS13, reached up to 5-fold higher for mRNA expression at 24 hours after reperfusion, compared with the physiological, pre-ischemic level in mouse hepatic IRI model. Consequently, plasma ADAMTS13 activity was rapidly decreased just after the onset of hepatic IR, which persisted for at least 24 hours without any recovery (Figure 1A). In contrast, vWF upregulation in SECs started from the ischemic phase, with a burst observed by 24 hours after reperfusion, representing further enhancement of SECs damage upon reperfusion (Figure 1B). These results indicate the high vulnerability of HSCs and SECs to hepatic IRI, both of which are main components of the sinusoidal wall, thus resulting in severe microangiopathy therein. Importantly, ADAMTS13 is exclusively produced by HSCs[22,24], and the liver contains an extremely large vascular bed, which, once injured, potentially expresses and releases vast amounts of UL-vWFM[36,37]. Thus, critical liver damage, including hepatic IRI, inevitably increases the vWF/ADAMTS13 ratio to varying degrees, thereby resulting in TMA-like pathology. Because these features appear to be unique to the liver, we would like to propose a new concept of liver damage-induced TMA-like pathology in critical liver diseases, i.e., “hepatic TMA”.
To elucidate the putative role of ADAMTS13 and the therapeutic impact of rADAMTS13 on critical liver damage, we first employed a combination of ADAMTS13-deficient mice and rADAMTS13 in a standardized model of acute liver damage, 70% partial hepatic IRI. As expected, ADAMTS13-deficiency provoked massive platelet aggregation within hepatic microvasculatures after IRI, resulting in microcirculatory impairment and subsequent parenchymal injuries, whereas rADAMTS13 administration significantly attenuated all these liver damages (Figure 2 and Figure 3). Based on these results, the following deteriorating process seems to be involved in hepatic IRI: First, IRI results in significant up-regulation of UL-vWFM from SECs and simultaneous down-regulation of ADAMTS13 from HSCs; massive platelet aggregation then occurs locally and systemically, eliciting microcirculatory disturbance and parenchymal inflammation, thereby falling into apoptosis/necrosis in hepatic parenchyma. The latter parts of this cascade further exacerbate the imbalance of vWF-multimers/ADAMTS13. Consequently, such a “vicious cascade” is enormously amplified, finally resulting in irreversible tissue damage, unless appropriately treated. From this perspective, rADAMTS13 supplementation could successfully eliminate such a “vicious cascade” in TMA-like pathology, thereby significantly mitigating hepatic IRI.
In addition to the aforementioned protective benefits based on the improvement of hepatic microangiopathies, significant attenuation of the inflammatory response in injured hepatic tissue was also observed. ADAMTS13 administration suppressed the infiltration of macrophages and neutrophils into the livers (Figure 4), and pro-inflammatory cytokines and chemokines were all significantly suppressed by rADAMTS13 (Figure 5). These results demonstrated a significant potential of ADAMTS13 to alleviate hepatic inflammation. Indeed, it may be difficult to deny that these anti-inflammatory responses mostly resulted from the amelioration of reperfusion injury itself. However, these anti-inflammatory effects were more pronounced than expected from other parameters, such as lowered transaminase release or improved microcirculation. Although there has been little evidence, De Meyer et al. also suggested the anti-inflammatory potential of ADAMTS13 in a mouse model of heart transplant[15]. Taken together, our data suggest that ADAMTS13 per se possesses powerful anti-inflammatory potential on hepatic IRI.
Although adult-to-adult LDLT has emerged as a life-saving option for end-stage liver diseases in the era of critical donor shortage, this procedure incorporates an unavoidable disadvantage of insufficient volume of liver allografts. The possible causative factors of TMA-like disorders have been reported after LTx recipients in clinical situations [38,39,40]. Hori et al. reported seven cases of TMA-like disorder after living-donor liver transplantation[41]. The vWF/ADAMTS13 ratio in these patients increased up to 11.0 ± 2.4 (range, 7.8–14.6), coupled with extremely high peri-operative mortality of up to 71.4% (loss of five out of seven patients), despite various intensive treatments, including repeated plasma exchange. Additionally, the vWF/ADAMTS13 ratio is a significant predictor determining the severity of TMA[42,43,44,45]. However, to date, the only effective treatment for this lethal condition is plasma exchange[46,47,48,49] (and conversion of suspected drugs in some cases[50,51]). Thus, the high mortality rates in patients with hepatic TMA represent an “unmet medical need” in this field. Hence, we adopted the rat 20% pLTx (CIT 4 hours) model to elucidate the specific pathophysiological mechanism of TMA-like disorder after LDLT. Similar to the mouse experimental results, ADAMTS13 activity decreased less than 30% at 2, 6, and 24 hours after reperfusion (Suppl. Figure S5), and massive expression of vWF in sinusoidal space was observed at 2, 6, and 24 hours after reperfusion in vehicle pLT group. Consequently, the imbalance between UL-vWF and ADAMTS13 activity causes the massive platelet aggregation and forms a large amount of platelet thrombi. as evident byCD41 immunofluorescence of the partial liver allograft showing that massive platelet aggregation in sinusoidal space especially at 6 and 24 hours after reperfusion. The rADAMTS13 supplementation improved the imbalance between vWF and ADAMTS13 and suppressed the massive platelet aggregation in the partial liver allograft. As a result, hepatic microcirculation was dramatically improved from 20 minutes to 24 hours after reperfusion (Figure 6). Consequently, as shown in Figure 7, the liver damage was ameliorated in the aspect of biochemical and histopathological examinations. Importantly, ADAMTS13 treatment improved rat 20% pLTx model’s 7-day survival rate from 40% to 80%, indicating the critical role of ADAMTS13 in pLT-induced TMA-like pathology.
In the treatment of diverse coagulation disorders including TMA, disseminated intravascular coagulation (DIC), or various organ infarctions/embolizations, developing new therapeutic intervention is generally accompanied by bleeding complications as potentially adverse effects, thus hindering its translation into clinical practice[52,53,54]. In this regard, rADAMTS13 has an advantage compared with other agents in this field; no bleeding tendency. Consistent with a recent report[15], any mice or rats treated with rADAMTS13 did not suffer from bleeding complications during and after surgery, although plasma ADAMTS13 activity was relatively higher in W688X treated groups; 1000% in mice 2 hours after surgery, 200% in rats from 2 hours to 24 hours after reperfusion (Suppl. Figures S2 and S5). These results may imply the valuable safety of ADAMTS13 supplementation as a new therapeutic strategy in the treatment of TMA, IRI, and critical liver conditions. This unique and unobtainable feature of rADAMTS13 may facilitate its clinical translation.
In conclusion, severe liver damages inevitably deteriorate the balance between prothrombotic vWF-multimers and its cleaving protease, ADAMTS13, thereby causing microcirculatory impairment locally and systemically by forming platelets thrombi. This, in turn, results in long-lasting IRI of the liver, further enhancing the imbalance of vWF/ADAMTS13. We designate this liver damage-initiated TMA-like pathology as “hepatic TMA”. Administration of rADAMTS13 could disrupt the pathological cascades therein, thus improving severe conditions of “hepatic TMA”. Given its strong anti-inflammatory potential and valuable safety, rADAMTS13 supplementation may be a novel therapeutic intervention against critical liver diseases.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
This work was supported by the Scientific Research B (No.26293287) from the Japan Society for the Promotion of Science (JSPS).
Conflicts of Interest
The author declares no conflicts of interest.
References
- Balzan, S.; Belghiti, J.; Farges, O.; Ogata, S.; Sauvanet, A.; Delefosse, D.; Durand, F. The “50-50 criteria” on postoperative day 5: an accurate predictor of liver failure and death after hepatectomy. Ann Surg 2005, 242, 824-8, discussion 828-9. [Google Scholar] [CrossRef]
- Rahbari, N.N.; Reissfelder, C.; Koch, M.; Elbers, H.; Striebel, F.; Büchler, M.W.; Weitz, J. The predictive value of postoperative clinical risk scores for outcome after hepatic resection: a validation analysis in 807 patients. Ann Surg Oncol 2011, 18, 3640–9. [Google Scholar] [CrossRef]
- Paugam-Burtz, C.; Janny, S.; Delefosse, D.; Dahmani, S.; Dondero, F.; Mantz, J.; Belghiti, J. Prospective validation of the “fifty-fifty” criteria as an early and accurate predictor of death after liver resection in intensive care unit patients. Ann Surg 2009, 249, 124–8. [Google Scholar] [CrossRef]
- D.G. Farmer, F. Amersi, J. Kupiec-Weglinski, and R.W. Busuttil.
- Hirao, H.; Nakamura, K.; Kupiec-Weglinski, J.W. Liver ischaemia-reperfusion injury: a new understanding of the role of innate immunity. Nature reviews. Gastroenterology & hepatology 2022, 19, 239–256. [Google Scholar]
- Strong, R.W.; Lynch, S.V.; Ong, T.H.; Matsunami, H.; Koido, Y.; Balderson, G.A. Successful liver transplantation from a living donor to her son. The New England journal of medicine 1990, 322, 1505–7. [Google Scholar] [CrossRef] [PubMed]
- Riddiough, G.E.; Christophi, C.; Jones, R.M.; Muralidharan, V.; Perini, M.V. A systematic review of small for size syndrome after major hepatectomy and liver transplantation. HPB : the official journal of the International Hepato Pancreato Biliary Association 2020, 22, 487–496. [Google Scholar] [CrossRef]
- Uemura, T.; Wada, S.; Kaido, T.; Mori, A.; Ogura, Y.; Yagi, S.; Fujimoto, Y.; Ogawa, K.; Hata, K.; Yoshizawa, A.; Okajima, H.; Uemoto, S. How far can we lower graft-to-recipient weight ratio for living donor liver transplantation under modulation of portal venous pressure? Surgery 2016, 159, 1623–1630. [Google Scholar] [CrossRef] [PubMed]
- Coppo, P.; Veyradier, A.; Durey, M.A.; Fremeaux-Bacchi, V.; Scrobohaci, M.L.; Amesland, F.; Bussel, A. [Pathophysiology of thrombotic microangiopathies: current understanding]. Ann Med Interne (Paris) 2002, 153, 153–66. [Google Scholar] [PubMed]
- Rieger, M.; Ferrari, S.; Hovinga, J.A.K.; Konetschny, C.; Herzog, A.; Koller, L.; Weber, A.; Remuzzi, G.; Dockal, M.; Plaimauer, B.; Scheiflinger, F. Relation between ADAMTS13 activity and ADAMTS13 antigen levels in healthy donors and patients with thrombotic microangiopathies (TMA). Thromb Haemost 2006, 95, 212–20. [Google Scholar] [PubMed]
- Obert, V.B.; Houllier, A.; Meyer, D.; Girma, J.P. Specific von Willebrand factor-cleaving protease in thrombotic microangiopathies: a study of 111 cases. Blood 2001, 98, 1765–72. [Google Scholar]
- Bianchi, V.; Robles, R.; Alberio, L.; Furlan, M.; Lämmle, B. Von Willebrand factor-cleaving protease (ADAMTS13) in thrombocytopenic disorders: a severely deficient activity is specific for thrombotic thrombocytopenic purpura. Blood 2002, 100, 710–3. [Google Scholar] [CrossRef]
- Levy, G.G.; Nichols, W.C.; Lian, E.C.; Foroud, T.; McClintick, J.N.; McGee, B.M.; Yang, A.Y.; Siemieniak, D.R.; Stark, K.R.; Gruppo, R.; Sarode, R.; Shurin, S.B.; Chandrasekaran, V.; Stabler, S.P.; Sabio, H.; Bouhassira, E.E.; Upshaw, J.D.; Ginsburg, D.; Tsai, H.M. Mutations in a member of the ADAMTS gene family cause thrombotic thrombocytopenic purpura. Nature 2001, 413, 488–94. [Google Scholar] [CrossRef]
- Zhao, B.Q.; Chauhan, A.K.; Canault, M.; Patten, I.S.; Yang, J.J.; Dockal, M.; Scheiflinger, F.; Wagner, D.D. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood 2009, 114, 3329–34. [Google Scholar] [CrossRef] [PubMed]
- Meyer, D.S.F.; Savchenko, A.S.; Haas, M.S.; Schatzberg, D.; Carroll, M.C.; Schiviz,, A.; Dietrich, B.; Rottensteiner, H.; Scheiflinger, F.; Wagner, D.D. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood 2012, 120, 5217–23. [Google Scholar] [CrossRef]
- Doi, M.; Matsui, H.; Takeda, H.; Saito, Y.; Takeda, M.; Matsunari, Y.; Nishio, K.; Shima, M.; Banno, F.; Akiyama, M.; Kokame, K.; Miyata, T.; Sugimoto, M. ADAMTS13 safeguards the myocardium in a mouse model of acute myocardial infarction. Thromb Haemost 2012, 108, 1236–8. [Google Scholar] [CrossRef]
- Gandhi, C.; Motto, D.G.; Jensen, M.; Lentz, S.R.; Chauhan, A.K. ADAMTS13 deficiency exacerbates VWF-dependent acute myocardial ischemia/reperfusion injury in mice. Blood 2012, 120, 5224–30. [Google Scholar] [CrossRef] [PubMed]
- Remuzzi, G.; Galbusera, M.; Noris, M.; Canciani, M.T.; Daina, E.; Bresin, E.; Contaretti, S.; Caprioli, J.; Gamba, S.; Ruggenenti, P.; Perico, N.; Mannucci, P.M.; I.R.o.R.a.F.H.T.T.t.p.h.u. syndrome, von Willebrand factor cleaving protease (ADAMTS13) is deficient in recurrent and familial thrombotic thrombocytopenic purpura and hemolytic uremic syndrome. Blood 2002, 100, 778–85. [Google Scholar] [CrossRef]
- Zheng, X.; Majerus, E.M.; Sadler, J.E. ADAMTS13 and TTP. Curr Opin Hematol 2002, 9, 389–94. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, C.; Khan, M.M.; Lentz, S.R.; Chauhan, A.K. ADAMTS13 reduces vascular inflammation and the development of early atherosclerosis in mice. Blood 2012, 119, 2385–91. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.Y.; Tohyama, J.; Bauer, R.C.; Cao, N.N.; Rader, D.J.; Zheng, X.L. Genetic ablation of Adamts13 gene dramatically accelerates the formation of early atherosclerosis in a murine model. Arterioscler Thromb Vasc Biol 2012, 32, 1817–23. [Google Scholar] [CrossRef]
- Soejima, K.; Mimura, N.; Hirashima, M.; Maeda, H.; Hamamoto, T.; Nakagaki, T.; Nozaki, C. A novel human metalloprotease synthesized in the liver and secreted into the blood: possibly, the von Willebrand factor-cleaving protease? J Biochem 2001, 130, 475–80. [Google Scholar] [CrossRef]
- Zheng, X.; Chung, D.; Takayama, T.K.; Majerus, E.M.; Sadler, J.E.; Fujikawa, K. Structure of von Willebrand factor-cleaving protease (ADAMTS13), a metalloprotease involved in thrombotic thrombocytopenic purpura. J Biol Chem 2001, 276, 41059–63. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Tatsumi, K.; Matsumoto, M.; Fujimoto, M.; Matsuyama, T.; Ishikawa, M.; Iwamoto, T.A.; Mori, T.; Wanaka, A.; Fukui, H.; Fujimura, Y. Localization of ADAMTS13 to the stellate cells of human liver. Blood 2005, 106, 922–4. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Murata, M.; Matsubara, Y.; Uchida, T.; Ishihara, H.; Shibano, T.; Ashida, S.; Soejima, K.; Okada, Y.; Ikeda, Y. Detection of von Willebrand factor-cleaving protease (ADAMTS-13) in human platelets. Biochem Biophys Res Commun 2004, 313, 212–6. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.; Nolasco, L.; Tao, Z.; Dong, J.F.; Moake, J. Human endothelial cells synthesize and release ADAMTS-13. J Thromb Haemost 2006, 4, 1396–404. [Google Scholar] [CrossRef]
- Manea, M.; Kristoffersson, A.; Schneppenheim, R.; Saleem, M.A.; Mathieson, P.W.; Mörgelin, M.; Björk, P.; Holmberg, L.; Karpman, D. Podocytes express ADAMTS13 in normal renal cortex and in patients with thrombotic thrombocytopenic purpura. Br J Haematol 2007, 138, 651–62. [Google Scholar] [CrossRef]
- Banno, F.; Kokame, K.; Okuda, T.; Honda, S.; Miyata, S.; Kato, H.; Tomiyama, Y.; Miyata, T. Complete deficiency in ADAMTS13 is prothrombotic, but it alone is not sufficient to cause thrombotic thrombocytopenic purpura. Blood 2006, 107, 3161–6. [Google Scholar] [CrossRef]
- Hirao, H.; Uchida, Y.; Kadono, K.; Tanaka, H.; Niki, T.; Yamauchi, A.; Hata, K.; Watanabe, T.; Terajima, H.; Uemoto, S. The protective function of galectin-9 in liver ischemia and reperfusion injury in mice. Liver Transpl 2015. [Google Scholar] [CrossRef]
- Soejima, K.; Matsumoto, M.; Kokame, K.; Yagi, H.; Ishizashi, H.; Maeda, H.; Nozaki, C.; Miyata, T.; Fujimura, Y.; Nakagaki, T. ADAMTS-13 cysteine-rich/spacer domains are functionally essential for von Willebrand factor cleavage. Blood 2003, 102, 3232–7. [Google Scholar] [CrossRef]
- Suzuki, S.; Toledo-Pereyra, L.H.; Rodriguez, F.J.; Cejalvo, D. Neutrophil infiltration as an important factor in liver ischemia and reperfusion injury. Modulating effects of FK506 and cyclosporine. Transplantation 1993, 55, 1265–72. [Google Scholar]
- Nagai, K.; Yagi, S.; Afify, M.; Bleilevens, C.; Uemoto, S.; Tolba, R.H. Impact of venous-systemic oxygen persufflation with nitric oxide gas on steatotic grafts after partial orthotopic liver transplantation in rats. Transplantation 2013, 95, 78–84. [Google Scholar] [CrossRef]
- Li, C.H.; Ge, X.L.; Pan, K.; Wang, P.F.; Su, Y.N.; Zhang, A.Q. Laser speckle contrast imaging and Oxygen to See for assessing microcirculatory liver blood flow changes following different volumes of hepatectomy. Microvasc Res 2017, 110, 14–23. [Google Scholar] [CrossRef]
- Kokame, K.; Nobe, Y.; Kokubo, Y.; Okayama, A.; Miyata, T. FRETS-VWF73, a first fluorogenic substrate for ADAMTS13 assay. Br J Haematol 2005, 129, 93–100. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012, 9, 671–5. [Google Scholar] [CrossRef]
- Baruch, Y.; Neubauer, K.; Ritzel, A.; Wilfling, T.; Lorf, T.; Ramadori, G. Von Willebrand gene expression in damaged human liver. Hepatogastroenterology 2004, 51, 684–8. [Google Scholar]
- Hulstein, J.J.; van Runnard Heimel, P.J.; Franx, A.; Lenting, P.J.; Bruinse, H.W.; Silence, K.; de Groot, P.G.; Fijnheer, R. Acute activation of the endothelium results in increased levels of active von Willebrand factor in hemolysis, elevated liver enzymes and low platelets (HELLP) syndrome. J Thromb Haemost 2006, 4, 2569–75. [Google Scholar] [CrossRef] [PubMed]
- Rerolle, J.P.; Akposso, K.; Lerolle, N.; Mougenot, B.; Ponnelle, T.; Rondeau, E.; Sraer, J.D. Tacrolimus-induced hemolytic uremic syndrome and end-stage renal failure after liver transplantation. Clinical transplantation 2000, 14, 262–5. [Google Scholar] [CrossRef] [PubMed]
- Ramasubbu, K.; Mullick, T.; Koo, A.; Hussein, M.; Henderson, J.M.; Mullen, K.D.; Avery, R.K. Thrombotic microangiopathy and cytomegalovirus in liver transplant recipients: a case-based review. Transplant infectious disease : an official journal of the Transplantation Society 2003, 5, 98–103. [Google Scholar] [CrossRef] [PubMed]
- Shindoh, J.; Sugawara, Y.; Akamatsu, N.; Kaneko, J.; Tamura, S.; Yamashiki, N.; Aoki, T.; Sakamoto, Y.; Hasegawa, K.; Kokudo, N. Thrombotic microangiopathy after living-donor liver transplantation. American journal of transplantation : official journal of the American Society of Transplantation and the American Society of Transplant Surgeons 2012, 12, 728–36. [Google Scholar] [CrossRef] [PubMed]
- Hori, T.; Kaido, T.; Oike, F.; Ogura, Y.; Ogawa, K.; Yonekawa, Y.; Hata, K.; Kawaguchi, Y.; Ueda, M.; Mori, A.; Segawa, H.; Yurugi, K.; Takada, Y.; Egawa, H.; Yoshizawa, A.; Kato, T.; Saito, K.; Wang, L.; Torii, M.; Chen, F.; Baine, A.M.; Gardner, L.B.; Uemoto, S. Thrombotic microangiopathy-like disorder after living-donor liver transplantation: a single-center experience in Japan. World J Gastroenterol 2011, 17, 1848–57. [Google Scholar] [CrossRef] [PubMed]
- Green, D.; Tian, L.; Greenland, P.; Liu, K.; Kibbe, M.; Tracy, R.; Shah, S.; Wilkins, J.T.; Huffman, M.D.; Liao, Y.; Jones, D.L.; McDermott, M.M. Association of the von Willebrand Factor-ADAMTS13 Ratio With Incident Cardiovascular Events in Patients With Peripheral Arterial Disease. Clin Appl Thromb Hemost 2016. [Google Scholar] [CrossRef] [PubMed]
- Qu, L.; Jiang, M.; Qiu, W.; Lu, S.; Zhao, Y.; Xia, L.; Ruan, C. Assessment of the Diagnostic Value of Plasma Levels, Activities, Their Ratios of von Willebrand Factor and ADAMTS13 in Patients with Cerebral Infarction. Clin Appl Thromb Hemost 2016, 22, 252–9. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, S.; de Vries, P.S.; Boender, J.; Sonneveld, M.A.; Hoorn, E.J.; Hofman, A.; de Maat, M.P.; Franco, O.H.; Ikram, M.A.; Leebeek, F.W.; Dehghan, A. von Willebrand Factor, ADAMTS13 Activity, Decline in Kidney Function: A Population-Based Cohort Study. Am J Kidney Dis 2016, 68, 726–732. [Google Scholar] [CrossRef]
- Sonneveld, M.A.; Franco, O.H.; Ikram, M.A.; Hofman, A.; Kavousi, M.; de Maat, M.P.; Leebeek, F.W. Von Willebrand Factor, ADAMTS13, the Risk of Mortality: The Rotterdam Study. Arterioscler Thromb Vasc Biol 2016, 36, 2446–2451. [Google Scholar] [CrossRef]
- Caprioli, J.; Remuzzi, G.; Noris, M. Thrombotic microangiopathies: from animal models to human disease and cure. Contrib Nephrol 2011, 169, 337–50. [Google Scholar] [PubMed]
- Darmon, M.; Azoulay, E.; Thiery, G.; Ciroldi, M.; Galicier, L.; Parquet, N.; Veyradier, A.; Le Gall, J.R.; Oksenhendler, E.; Schlemmer, B. Time course of organ dysfunction in thrombotic microangiopathy patients receiving either plasma perfusion or plasma exchange. Crit Care Med 2006, 34, 2127–33. [Google Scholar] [CrossRef] [PubMed]
- Federici, L.; Klouche, K.; Amigues, L.; Kanouni, T.; Lopez-Martinez, E.; Latry, P.; Beraud, J.J.; Rossi, J.F. Outcome and prognosis of severe thrombotic microangiopathies treated by plasma exchange. Med Sci Monit 2006, 12, CR302–7. [Google Scholar]
- Nguyen, T.C.; Han, Y.Y. Plasma exchange therapy for thrombotic microangiopathies. Organogenesis 2011, 7, 28–31. [Google Scholar] [CrossRef]
- Pisoni, R.; Ruggenenti, P.; Remuzzi, G. Drug-induced thrombotic microangiopathy: incidence, prevention and management. Drug Saf 2001, 24, 491–501. [Google Scholar] [CrossRef]
- Zakarija, A.; Bennett, C. Drug-induced thrombotic microangiopathy. Semin Thromb Hemost 2005, 31, 681–90. [CrossRef]
- Urokinase: new indication. Thrombosed venous and dialysis catheters: when heparin fails. Urokinase clears about 75% of thrombosed venous catheters intended for long-term use with a dose-related increase in the bleeding risk. Prescrire Int 2009, 18, 118.
- Singer, J.O.C.; Berkefeld, M.W.; Lorenz, J.; Fiehler, G.W.; Albers, M.G.; Lansberg, A.; Kastrup, A.; Rovira, D.S.; Liebeskind, A.; Gass, C.; et al. Investigators, Risk of symptomatic intracerebral hemorrhage in patients treated with intra-arterial thrombolysis. Cerebrovasc Dis 2009, 27, 368–74. [Google Scholar] [CrossRef] [PubMed]
- Vora, N.A.; Gupta, R.; Thomas, A.J.; Horowitz, M.B.; Tayal, A.H.; Hammer, M.D.; Uchino, K.; Wechsler, L.R.; Jovin, T.G. Factors predicting hemorrhagic complications after multimodal reperfusion therapy for acute ischemic stroke. AJNR Am J Neuroradiol 2007, 28, 1391–4. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Plasma ADAMTS13 activity during IRI, and imbalance between ADAMTS13 and vWF expressions induced by hepatic IRI in WT mice. (A) Time-dependent alteration of plasma ADAMTS13 activity in wild type (WT) mice after 70% partial hepatic IRI (n = 6 at each time point). (B) Chronological alterations of ADAMTS13 and vWF mRNA expressions in liver tissues during hepatic IRI (blue solid line: ADAMTS13 with vehicle, red solid line: vWF with vehicle, blue dotted line: ADAMTS13 with rADAMTS13, red dotted line: vWF with rADAMTS13). Data are represented as means ± SEM. mRNA expression of vWF: 2.21 ± 0.35 in WT + vehicle vs. 1.08 ± 0.07 in WT + rADAMTS13 at the end of ischemia, †p<0.01; 4.45 ± 0.76 in WT + vehicle vs. 1.23 ± 0.11 in WT + rADAMTS13 at 24 hours, §p<0.001. 2-way ANOVA followed by Bonferroni’s post-hoc tests (n = 6 / group).
Figure 1.
Plasma ADAMTS13 activity during IRI, and imbalance between ADAMTS13 and vWF expressions induced by hepatic IRI in WT mice. (A) Time-dependent alteration of plasma ADAMTS13 activity in wild type (WT) mice after 70% partial hepatic IRI (n = 6 at each time point). (B) Chronological alterations of ADAMTS13 and vWF mRNA expressions in liver tissues during hepatic IRI (blue solid line: ADAMTS13 with vehicle, red solid line: vWF with vehicle, blue dotted line: ADAMTS13 with rADAMTS13, red dotted line: vWF with rADAMTS13). Data are represented as means ± SEM. mRNA expression of vWF: 2.21 ± 0.35 in WT + vehicle vs. 1.08 ± 0.07 in WT + rADAMTS13 at the end of ischemia, †p<0.01; 4.45 ± 0.76 in WT + vehicle vs. 1.23 ± 0.11 in WT + rADAMTS13 at 24 hours, §p<0.001. 2-way ANOVA followed by Bonferroni’s post-hoc tests (n = 6 / group).
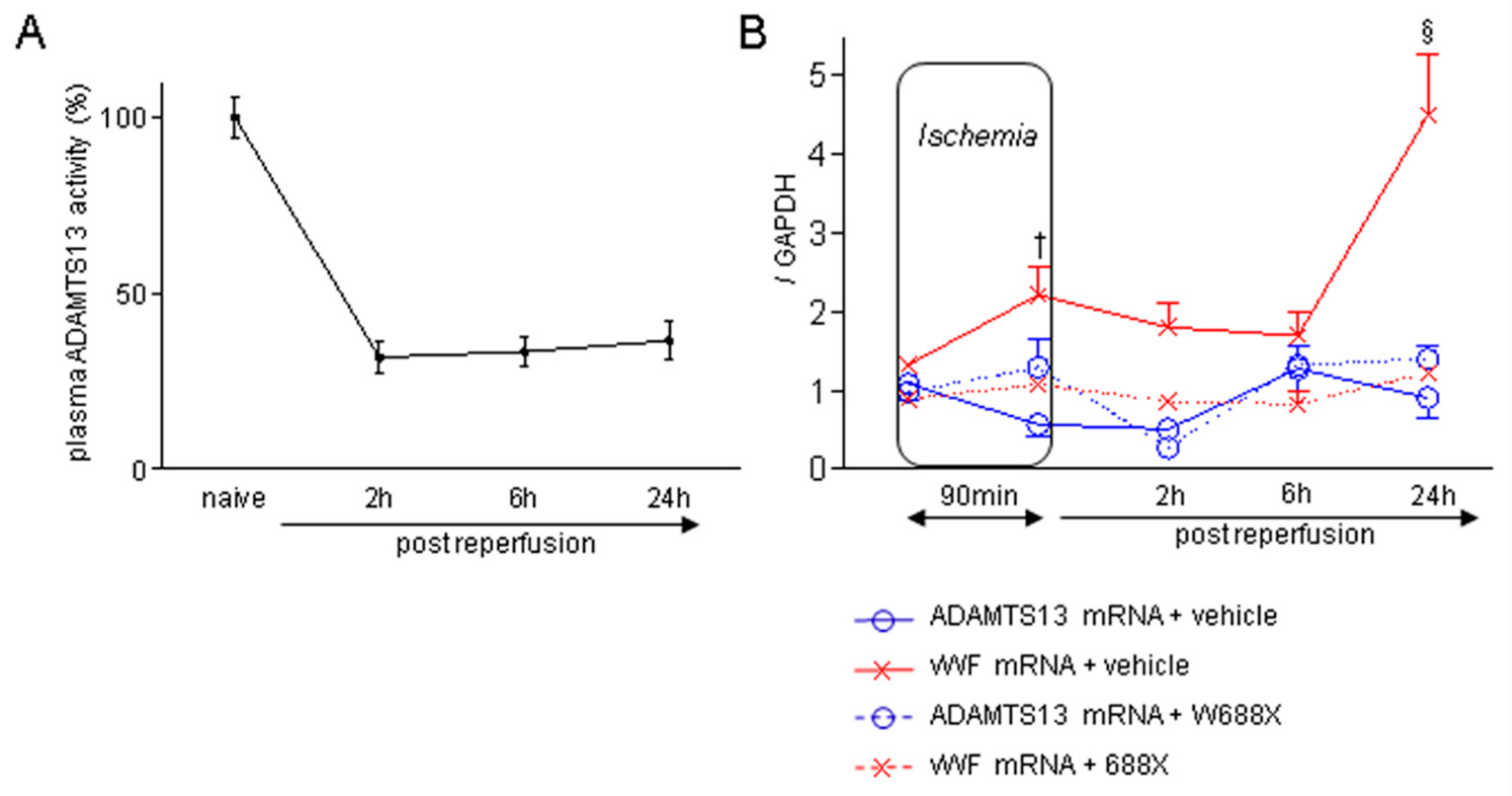
Figure 2.
Platelet aggregation within hepatic microvasculature and subsequent alteration of hepatic microcirculation after IRI. (A) Representative tissue sections of the ischemic liver lobes stained by immunofluorescence for CD42b (original magnification × 100). A scale bar in the right lower panel represents 100 µm. (B) CD-42b positive area quantified by Image J software. Data are represented as mean ± SEM. *p <0.05, ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n= 4 / group). (C) Peripheral platelet counts during hepatic IRI. Data are represented as means ± SEM. *p<0.05, §p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n= 8 / group) (D) Hepatic microcirculation before and after reperfusion assessed by a laser Doppler flowmetry. Data are provided in the relative change to pre-ischemic value (%-decrease, Mean ± SEM). *p<0.05, †p<0.01, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n= 8 / group).
Figure 2.
Platelet aggregation within hepatic microvasculature and subsequent alteration of hepatic microcirculation after IRI. (A) Representative tissue sections of the ischemic liver lobes stained by immunofluorescence for CD42b (original magnification × 100). A scale bar in the right lower panel represents 100 µm. (B) CD-42b positive area quantified by Image J software. Data are represented as mean ± SEM. *p <0.05, ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n= 4 / group). (C) Peripheral platelet counts during hepatic IRI. Data are represented as means ± SEM. *p<0.05, §p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n= 8 / group) (D) Hepatic microcirculation before and after reperfusion assessed by a laser Doppler flowmetry. Data are provided in the relative change to pre-ischemic value (%-decrease, Mean ± SEM). *p<0.05, †p<0.01, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n= 8 / group).
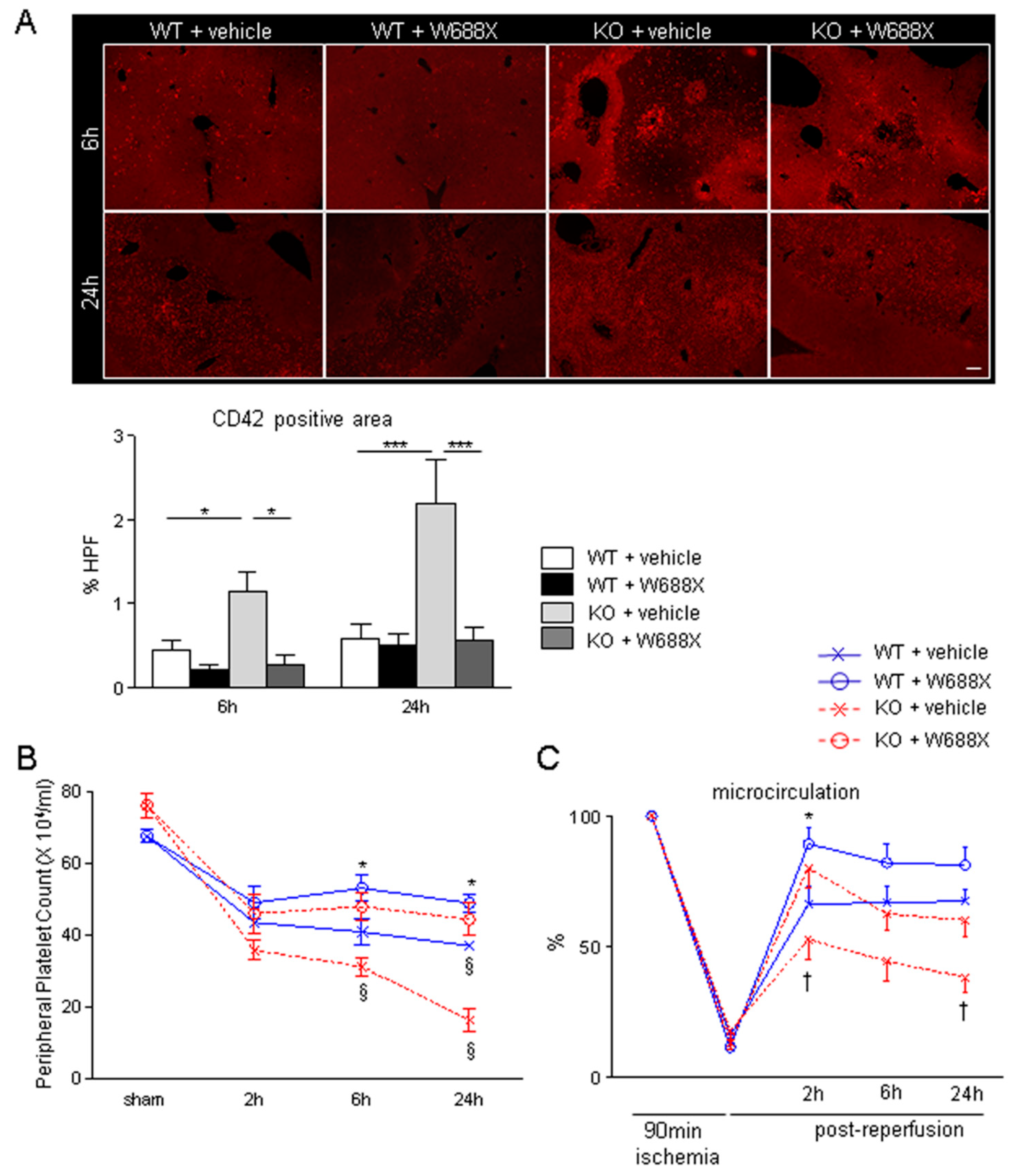
Figure 3.
ADAMTS13-null mutation exacerbates hepatocellular damage, while ADAMTS13 supplementation attenuates IR-induced injury. Serum AST (A), ALT (B), and LDH (C) release after IRI. (D) Representative H&E staining IR-stressed livers. Original magnification, x100. A scale bar in the right lower panel represents 100 µm. (E) Suzuki’s histological grading of liver IRI. (F) Representative TUNEL images. A scale bar in the right lower panel indicates 50 µm. Data are represented as mean ± SEM. *p<0.05, †p<0.01, §p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n= 8 / group).
Figure 3.
ADAMTS13-null mutation exacerbates hepatocellular damage, while ADAMTS13 supplementation attenuates IR-induced injury. Serum AST (A), ALT (B), and LDH (C) release after IRI. (D) Representative H&E staining IR-stressed livers. Original magnification, x100. A scale bar in the right lower panel represents 100 µm. (E) Suzuki’s histological grading of liver IRI. (F) Representative TUNEL images. A scale bar in the right lower panel indicates 50 µm. Data are represented as mean ± SEM. *p<0.05, †p<0.01, §p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n= 8 / group).
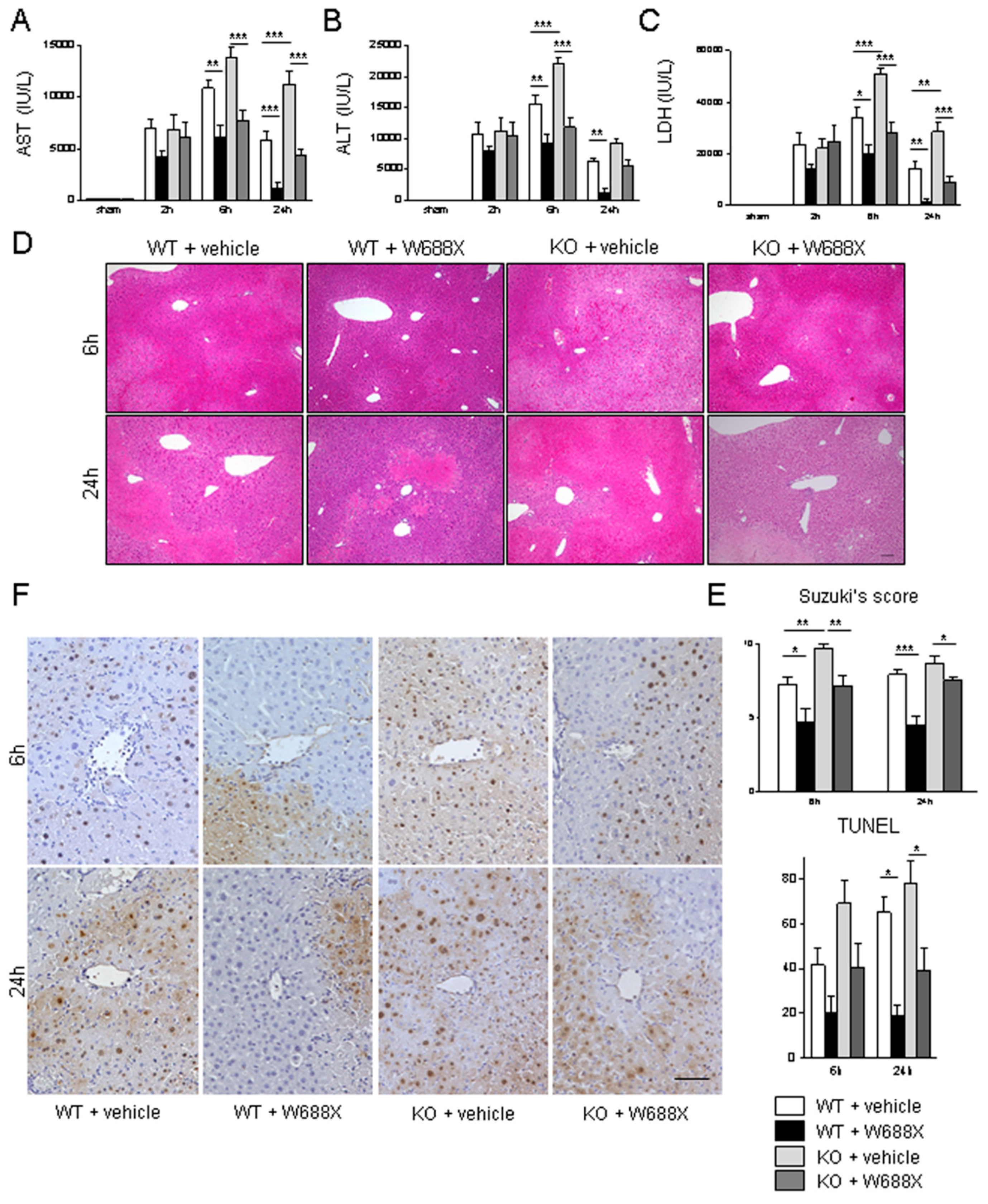
Figure 4.
Assessment of the infiltration of macrophage and neutrophil into the livers. (A) Representative liver sections stained by CD68 (A) and Ly6G (B) (original magnification × 400). A scale bar in the right lower panel represents 50 µm. Data were provided as mean and SEM. *p<0.05, **p<0.01, ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n= 5 / group).
Figure 4.
Assessment of the infiltration of macrophage and neutrophil into the livers. (A) Representative liver sections stained by CD68 (A) and Ly6G (B) (original magnification × 400). A scale bar in the right lower panel represents 50 µm. Data were provided as mean and SEM. *p<0.05, **p<0.01, ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n= 5 / group).
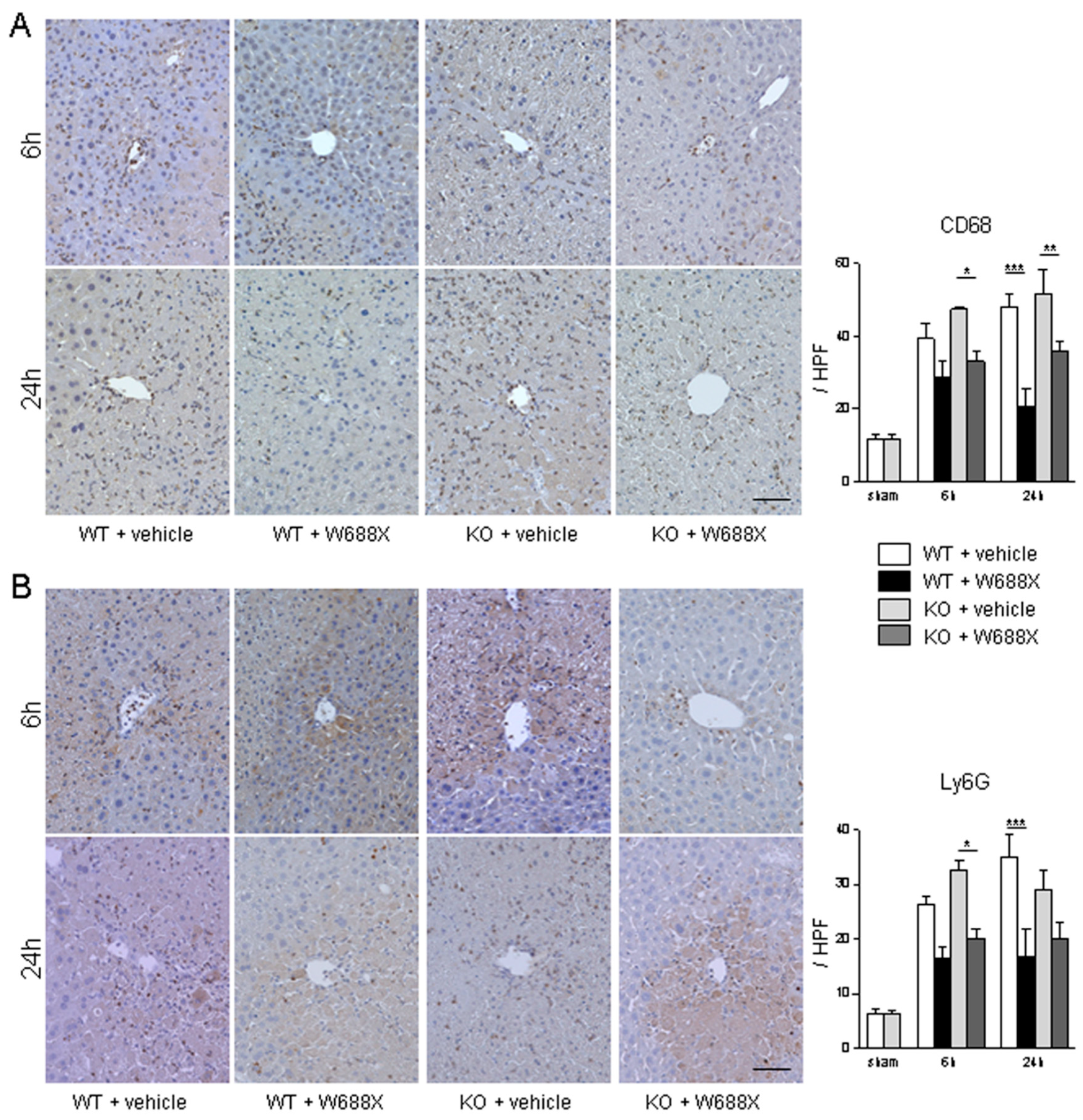
Figure 5.
Anti-inflammatory effect of ADAMTS13 quantified by the RT-PCR detection of gene expression of pro-inflammatory cytokines and chemokines in IRI liver. The induction ratios of cytokines (A-D) and chemokines (E, F). Data were normalized to GAPDH gene expression. Data were provided as mean and SEM. *p<0.05, **p<0.01, ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n = 8 / group).
Figure 5.
Anti-inflammatory effect of ADAMTS13 quantified by the RT-PCR detection of gene expression of pro-inflammatory cytokines and chemokines in IRI liver. The induction ratios of cytokines (A-D) and chemokines (E, F). Data were normalized to GAPDH gene expression. Data were provided as mean and SEM. *p<0.05, **p<0.01, ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n = 8 / group).
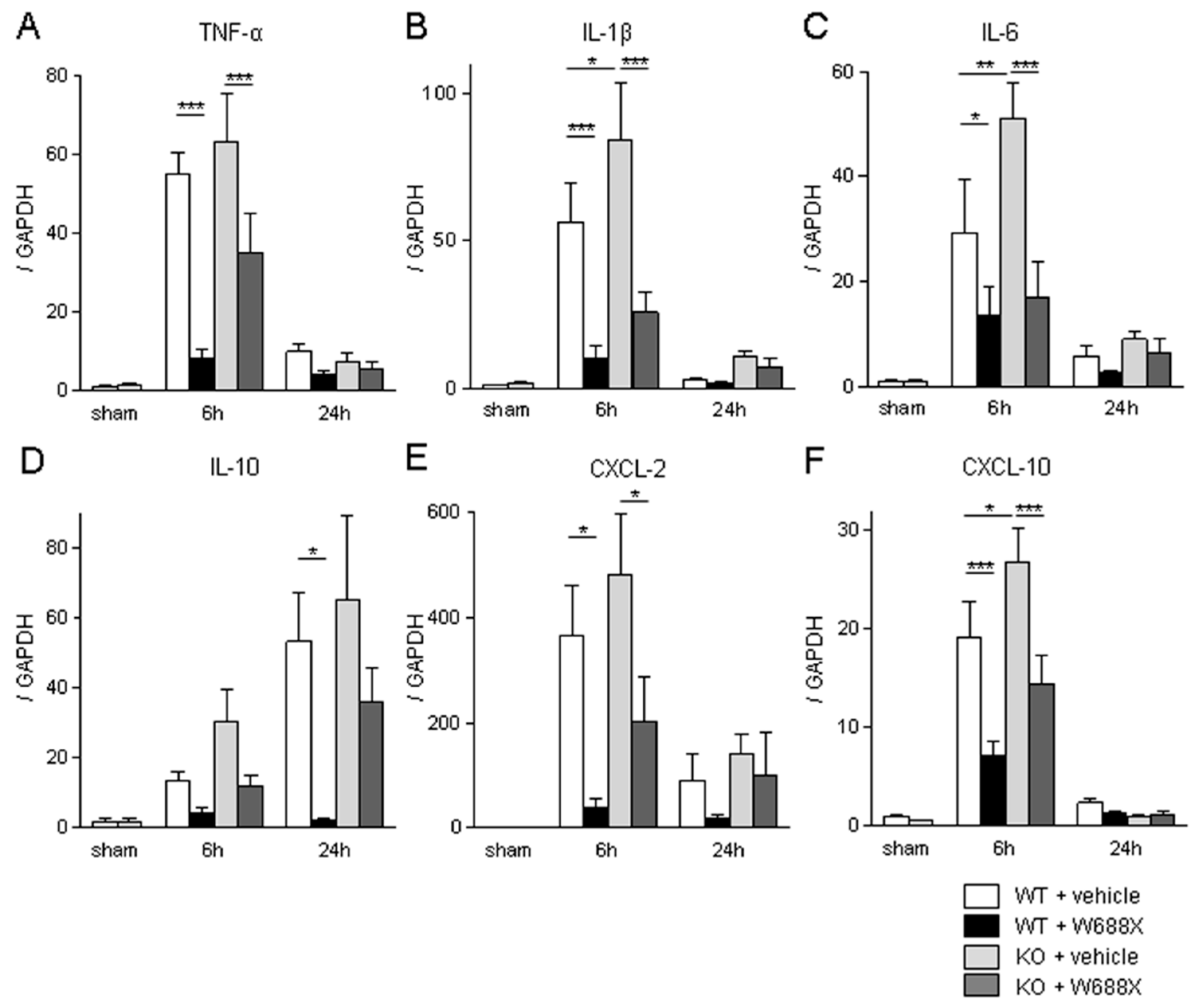
Figure 6.
ADAMTS13 supplementation down-regulates LT stress-induced vWF expressions and platelet aggregation. (A) Representative liver sections stained by vWF (original magnification x200). A scale bar in each panel represents 100μm. (B) representative immunofluorescence for CD41 (original magnification x100). A scale bar in each panel represents 100μm. Perioperative platelet count (C), LDH release (D), and microcirculation after partial LT. Data were provided as mean and SEM. *p<0.05, **p<0.01, ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n = 7 / group).
Figure 6.
ADAMTS13 supplementation down-regulates LT stress-induced vWF expressions and platelet aggregation. (A) Representative liver sections stained by vWF (original magnification x200). A scale bar in each panel represents 100μm. (B) representative immunofluorescence for CD41 (original magnification x100). A scale bar in each panel represents 100μm. Perioperative platelet count (C), LDH release (D), and microcirculation after partial LT. Data were provided as mean and SEM. *p<0.05, **p<0.01, ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n = 7 / group).
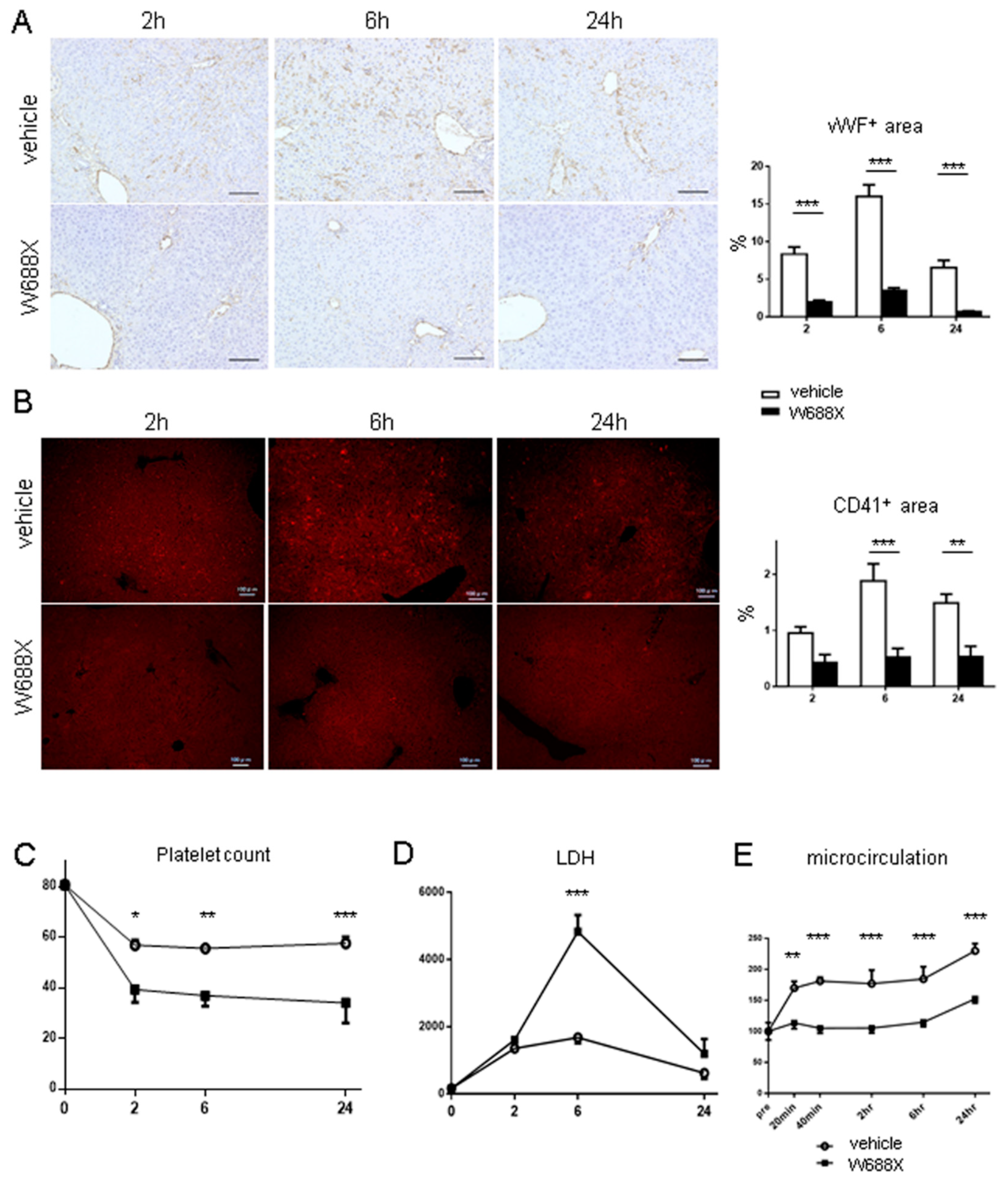
Figure 7.
ADAMTS13 treatment ameliorates LT-related hepatocellular damage and graft survival. (A) representative H&E staining (original magnification x200). (B) Quantification of Suzuki’s histological scores. Data were provided as mean and SEM. ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n = 8 / group). (C) Tissue damage measured by serum AST and ALT. (D) Survival rates using the log-rank test (n = 10 / group).
Figure 7.
ADAMTS13 treatment ameliorates LT-related hepatocellular damage and graft survival. (A) representative H&E staining (original magnification x200). (B) Quantification of Suzuki’s histological scores. Data were provided as mean and SEM. ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n = 8 / group). (C) Tissue damage measured by serum AST and ALT. (D) Survival rates using the log-rank test (n = 10 / group).
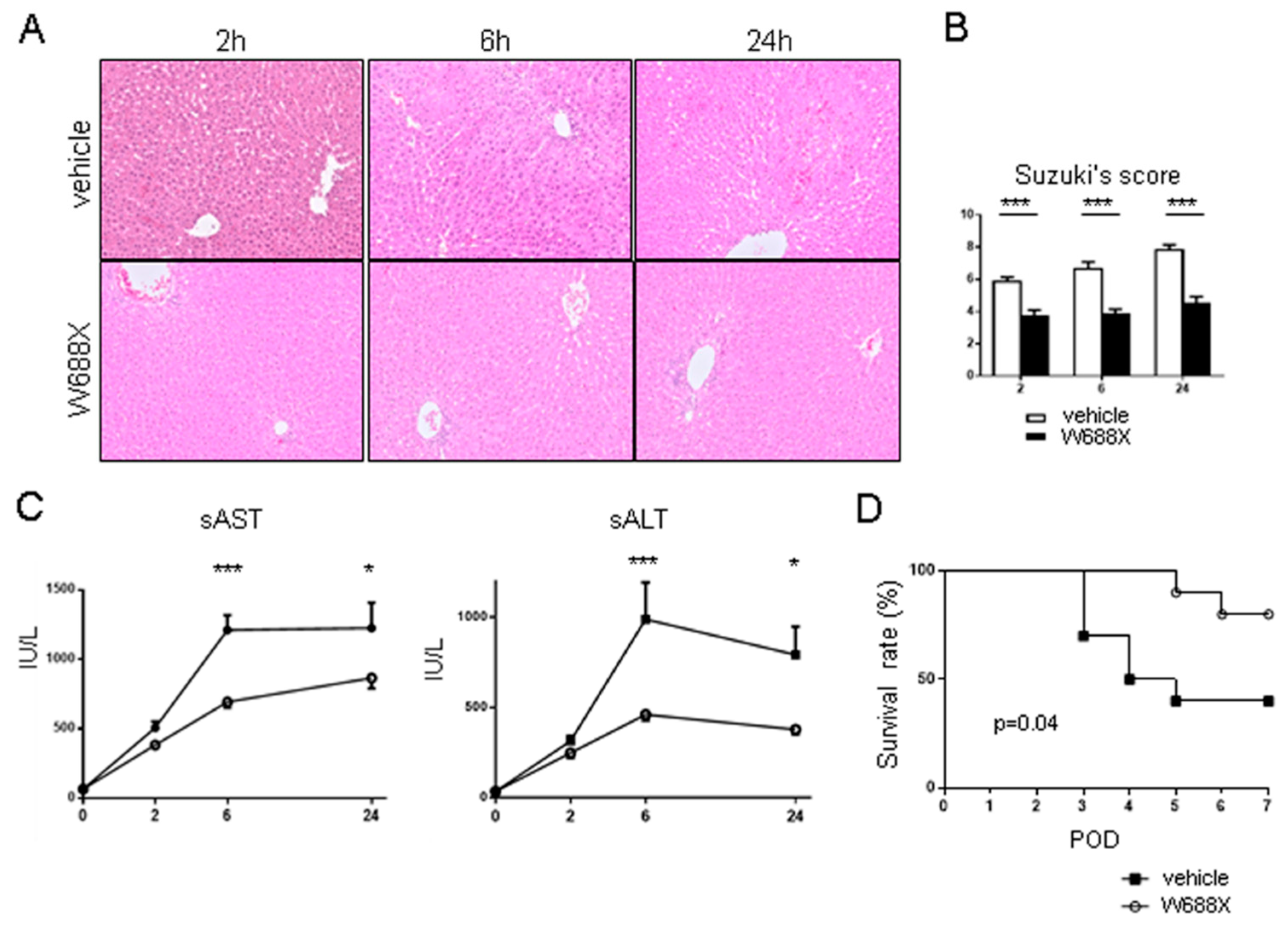
Figure 8.
Inflammatory cytokines and endothelin-1. The induction ratios of cytokines (TNF-α, IL-1β, IL-6 and endothelin-1. Data were normalized to β-actin expression. Data were shown as mean and SEM. *p<0.05, ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n = 7 / group).
Figure 8.
Inflammatory cytokines and endothelin-1. The induction ratios of cytokines (TNF-α, IL-1β, IL-6 and endothelin-1. Data were normalized to β-actin expression. Data were shown as mean and SEM. *p<0.05, ***p<0.001, 2-way ANOVA followed by Bonferroni’s post-hoc tests (n = 7 / group).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



