
Submitted:
26 January 2024
Posted:
30 January 2024
You are already at the latest version
Abstract
In the era of large data, the cloud is increasingly used as a computing environment, necessitating the development of cloud-compatible pipelines that can provide uniform analysis across disparate biological datasets. The WDL Analysis Research Pipelines (WARP) repository is a GitHub repository of open-source, cloud-optimized WDL workflows for biological data processing that are semantically versioned, tested, and documented. A companion repository, WARP-Tools, hosts Docker containers and custom tools used in WARP workflows.
Keywords:
WARP
; WDL
; pipelines
; FAIR pipelines
; cloud pipelines
; sequencing analysis
1. Introduction
With the advent of efficient sequencing technology, the scientific community is producing petabytes of data every day (Papageorgiou et al., 2018). These data are prepared to answer diverse biological questions, requiring a range of sequencing approaches. To effectively combine these disparate datasets and turn them into meaningful insights, researchers need analysis tools and data that adhere to Findable, Accessible, Interoperable, and Reusable (FAIR) practices (Wilkinson et al., 2016; Khan et al. 2019). To this end, the cloud is increasingly used as a computing environment; it allows for efficient resource sharing and scalability of computing resources (Ewels et al., 2020; Schatz et al., 2021). However, the migration to cloud computing requires new pipelines that are optimized to harness the expanse of cloud resources. While the number of cloud-optimized pipelines is growing, many are developed to meet niche research needs and may lack the robust systems for documentation, versioning, and testing which are crucial to FAIR.
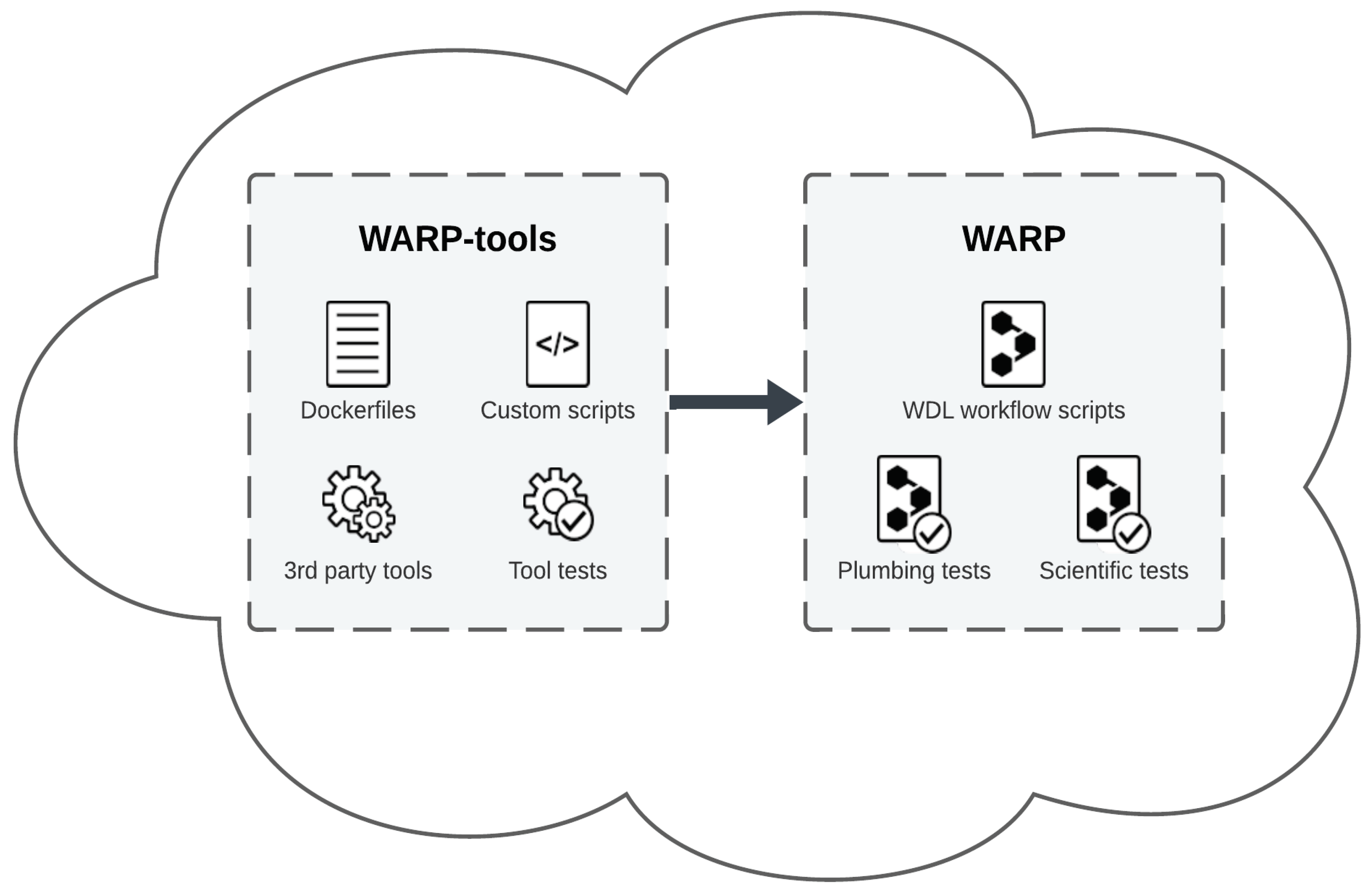
The WDL Analysis Research Pipelines (WARP) repository (Degatano et al., 2021) in GitHub is a collection of scalable, cloud-optimized pipelines written in the Workflow Description Language (WDL) and designed for processing a broad range of “omic” datasets (Figure 1; Supplementary Table 1). WARP’s pipelines are developed, tested, and scientifically vetted in collaboration with community scientists and global consortia. The repository has an open-access infrastructure under a BSD-3 license that includes all workflow code, and reference files, as well as a companion repository (WARP-tools) containing Dockerfiles and custom tools used in WARP pipelines (Figure 1). Each pipeline is semantically versioned, documented, publicly released in WARP and Dockstore (O'Connor et al., 2017), and available to run in cloud bioinformatics platforms such as Terra (https://app.terra.bio).
2. Usage
WARP pipelines are made available to the entire research community from the WARP releases page, Dockstore, and Terra. When a pipeline is released, it is first tagged with its version number and packaged on the WARP releases page, where it can be discovered using a command-line search tool—Wreleaser (https://github.com/broadinstitute/warp/tree/master/wreleaser). Each release download includes the workflow, its subtasks, and example configuration files that can be used to run the pipeline.
Since WARP pipelines are written in WDL, they can be deployed both locally and in the cloud using a portable execution engine like Cromwell (https://cromwell.readthedocs.io/en/develop/). The majority of the pipelines use public testing data hosted in Google bucket storage and accessible via the associated Terra workspace, and public Docker images which are pulled from cloud repositories like Azure Container Registry (https://azure.microsoft.com/en-us/products/container-registry) and Google Container Registry (https://cloud.google.com/container-registry). By using public cloud resources and only requiring an execution engine to run, WARP ensures pipelines are portable and able to run on the research community’s wide variety of computing platforms.
After release, each tagged pipeline is automatically pushed to Dockstore, where it can be downloaded, run, or exported to cloud-based analysis platforms like Terra. The WARP team maintains a dedicated Terra workspace for each pipeline; the workspaces are preloaded with the pipeline workflow, example data, and instructions for running the workflow (Supplementary Table 1).
Broad’s Genomics Platform and global research groups, including the Human Cell Atlas project, the Brain Research through Advancing Innovative Neurotechnologies (BRAIN) Cell Census Network (Ament et al., 2023; Hawrylycz et al., 2023), the BRAIN Initiative Cell Atlas Network, and the National Cancer Institute, have used WARP pipelines for large-scale data processing. According to GREV (https://hanadigital.github.io/grev/), WARP pipeline releases have been downloaded more than 13,000 times. Additionally, the pipelines are accessible to any researchers or tool developers interested in running them on individual or consortia data. Pipelines are developed in collaboration with the consortia and individual scientists. While the WARP team is still limited in its ability to accept pipeline contributions due to capacity constraints, anyone with a GitHub account can make suggestions to the existing pipeline code, or collaborate with WARP to add a new pipeline, by following WARP contribution guidelines (https://broadinstitute.github.io/warp/docs/contribution/README).
3. Implementation
The WARP infrastructure is designed to enable rigorous pipeline testing, ensuring that all updates to workflow code function appropriately, produce accurate and consistent scientific outputs, and are appropriately reviewed. To this end, the repository contains three branches: a develop branch for initial pipeline changes and testing, a staging branch for scientific testing on full-size example data, and a master branch for pipeline releases. All pipeline contributions start with a pull request (PR) to the develop branch. No changes may be merged to any of the three WARP branches without a mandatory review from a minimum of two reviewers. Some changes require an additional review from the scientific owner of each of the affected pipelines, depending on the nature of the introduced changes.
The WARP repository encourages community contributions and updates by implementing syntax linters and tests that remove overhead from the pipeline developer. Upon a PR, these automated tests, known as “smart tests,” identify updates to workflow code and validate WDL syntax using WOMtool (https://cromwell.readthedocs.io/en/stable/WOMtool/). If changes are made to any code shared across pipelines, such as task WDL code or Docker container code, the smart tests identify all pipelines affected by the change, requiring only affected pipelines to be further validated with engineering and scientific tests.
Smart tests also check for pipeline versioning and changelog updates, ensuring that pipeline development, particularly changes that affect scientific outputs, are well-documented. Each pipeline’s workflow is semantically versioned, a system that provides researchers insight into how pipeline changes may affect outputs and whether changes necessitate data reprocessing. In WARP, a patch change represents engineering updates, such as memory changes and variable name changes, that do not affect scientific outputs. A minor change reflects changes to outputs within a level of reasonable noise, such that the adjustment does not fundamentally affect the scientific outputs and does not necessitate any data reprocessing. A major change alters outputs in a way that may require reprocessing. This versioning allows researchers to know exactly how each pipeline’s version affects downstream analysis.
In addition to smart tests, engineering, and scientific tests verify that all pipeline tasks function appropriately and produce accurate outputs. The engineering tests use small, customized example data to effectively test all pipeline components, including optional pipeline tasks. For example, engineering tests for the variant discovery pipeline Whole Genome Germline Single Sample (Supplementary Table 1) use example data covering specific SNPs to facilitate testing the optional genomic contamination and fingerprinting tasks.
After approved changes are merged to the WARP staging branch, scientific tests use full-size example data to confirm that pipeline changes produce outputs that exactly match selected scientific reference datasets. Multiple reference datasets are chosen to catch special data edge cases, like high contamination or low coverage.
4. Discussion
The WARP repository is a scientifically vetted resource that employs pipeline development best practices to help maximize the use of community data (Wilkinson et al., 2016). The WARP team continuously implements feedback and explores ways where possible to address current limitations.
One limitation is the exclusive use of WDL for defining pipelines in WARP. WDL is a powerful tool for describing complex, portable, and scalable workflows, but it is dependent on execution engines like Cromwell or other WDL-supporting engines. This requirement may present a hurdle for users more comfortable with different languages or those who have already developed pipelines in other formats. Moreover, this exclusive use of WDL necessitates an understanding of the language and its execution environments. These factors might deter potential users and limit the repository's accessibility to a broader user base. This limitation underscores the importance of community feedback to broaden the repository's accessibility and make it more adaptable to various user needs.
Currently, WARP contributions are primarily sourced from direct collaborators, but the team aims to expand its interaction to the wider community. Anyone can currently contribute to pipeline code, though the team is still limited in the number of contributions that can be accepted and prioritized due to capacity constraints.
Overall, the WARP repository provides a model for cloud-optimized pipeline best practices that can be reused by the community (Figure 1). As data needs continue to evolve, the WARP team will optimize pipelines to meet the demand and work to ensure that pipelines are readily accessible to help foster biological discovery.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
This work has been supported by the NIH project 1U24MH114827-01.
Acknowledgments
We thank members of the Data Sciences Platform, especially those who have given feedback and suggestions to improve WARP or promoted its vision: Yossi Farjoun, Laura Gauthier, Chris Kachulis, Tim Tickle, and Charlotte Tolonen. We thank Chengchen (Rex) Wang for his work developing the WARP documentation site. We also want to thank all Pipelines Team alumni for their contributions: Ambrose Carr, Nikolas Barkas, Jishu Xu, Polina Shpilker, Trevyn Langsford, Ariel Schwartz, Philip Shapiro, Jason Rose, Sophie Crennan, and Cheyenne Gold. We also thank DSP leadership for their support including: Anthony Philippakis, Clare Bernard, Eric Banks, Kyle Vernest, Kathleen Tibbetts, and Akum Shergill.
Conflicts of Interest
none declared.
References
- Ament SA, Adkins RS, Carter R et al. (2023) The Neuroscience Multi-Omic Archive: a BRAIN Initiative resource for single-cell transcriptomic and epigenomic data from the mammalian brain. Nucleic Acids Res, 6;51(D1):D1075-D1085. [CrossRef]
- Degatano K, Grant G, Khajouei F et al. (2021). Introducing WARP: A collection of cloud-optimized workflows for biological data processing and reproducible analysis [version 1; not peer reviewed]. In F1000Research (Vol. 10, Issue 705). [CrossRef]
- Ewels PA, Peltzer A, Fillinger S et al. (2020). The nf-core framework for community-curated bioinformatics pipelines. Nature Biotechnology, 38(3), 276–278. [CrossRef]
- Hawrylycz M, Martone ME, Ascoli GA et al. (2023) A guide to the BRAIN Initiative Cell Census Network data ecosystem. PLOS Biology 21(6): e3002133. [CrossRef]
- Khan, F. Z., Soiland-Reyes, S., Sinnott, R. O., Lonie, A., Goble, C., & Crusoe, M. R. (2019). Sharing interoperable workflow provenance: A review of best practices and their practical application in CWLProv. GigaScience, 8(11), giz095. [CrossRef]
- Papageorgiou L, Eleni P, Raftopoulou S et al. (2018). Genomic big data hitting the storage bottleneck. EMBnet.journal, 24(e910). [CrossRef]
- Schatz MC, Philippakis AA, Afgan E et al. (2021). Inverting the model of genomics data sharing with the NHGRI Genomic Data Science Analysis, Visualization, and Informatics Lab-space (AnVIL). BioRxiv, 2021.04.22.436044. [CrossRef]
- Wilkinson MD, Dumontier M, Aalbersberg IJ et al. (2016). The FAIR Guiding Principles for scientific data management and stewardship. Scientific Data, 3(1), 160018. [CrossRef]
Figure 1.
An illustrated overview of the WARP and WARP-tools repositories. The WARP repository hosts a collection of cloud-based WDL workflow scripts, or pipelines (Supplementary Table 1), for processing high-throughput sequencing data along with plumbing and scientific tests used to validate pipeline releases. The WARP-tools repository offers a suite of Dockerfiles, custom scripts, third-party tools, and tool tests essential for data preprocessing and analysis performed within WARP pipelines. Together, these repositories provide a collection of cloud-optimized pipelines for genomic data processing that are accessible and FAIR.
Figure 1.
An illustrated overview of the WARP and WARP-tools repositories. The WARP repository hosts a collection of cloud-based WDL workflow scripts, or pipelines (Supplementary Table 1), for processing high-throughput sequencing data along with plumbing and scientific tests used to validate pipeline releases. The WARP-tools repository offers a suite of Dockerfiles, custom scripts, third-party tools, and tool tests essential for data preprocessing and analysis performed within WARP pipelines. Together, these repositories provide a collection of cloud-optimized pipelines for genomic data processing that are accessible and FAIR.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



