
Submitted:
30 January 2024
Posted:
01 February 2024
You are already at the latest version
Abstract
Salmonella enterica serovar Typhi, is one of the major infection spreading from consuming contaminated food and water. The poor sanitation is one the major cause for successful infection. S. typhi is responsible for causing typhoid fever, which is a growing public health concern in developing countries. This bacterium has acquired antimicrobial resistance to many promising which is a growing health concern for community. This study is aim to elucidate the molecular complexity of flouroquinolone resistance in S. Typhi isolates (n=286). Genomic data was downloaded from NCBI and analysis was done by Resfinder, MLST, and Pointfinder. Genomic analysis unveils a complex resistance profile, featuring antimicrobial resistance genes (ARG) including blaCTXM-15, blaTEM-1D, catA1, dfrA7, qnrS1, sul1, sul2, and tetA(B). The investigation focuses on mutations in DNA gyrA, parC, and qnrS1 genes which plays an important role in quinolone resistance. Mutation analysis reveals mutations such as DNA gyrA_S83F, DNA gyrA_D87N, DNA gyrA_S83Y, DNA gyrB_S464F, parC_S80I, and parE_L416F with the presence of qnrS1 gene produces resistance against quinolones. The Docking studies was conducted by “Autodock vina” and visualize by “Discovery studio” which demonstrate that these mutations leads to ciprofloxacin resistance, indicating reduced binding affinity, and altered solvent accessibility which indicates the structural changes at mutation sites. This study provides crucial insights into the molecular mechanisms driving flouroquinolone resistance in S. typhi, guiding future strategies to combat antibiotic resistance. Further validation through experimental mutagenesis is recommended, for targeted therapeutic interventions against the mounting threat of antibiotic-resistant S. typhi.
Keywords:
Flouroquinolone resistance
; Docking analysis
; Antimicrobial resistance
; Salmonella typhi
; Mutation analysis
1. Introduction:
Salmonella enterica serovar Typhi is a Gram-negative bacterium causing typhoid fever in humans, transmitted through contaminated food or water [1]. It exhibits human specificity, causing systemic infection characterized by prolonged fever, headache, nausea, loss of appetite, and abdominal discomfort, and potential life-threatening complications [2,3]. The primary treatment for typhoid fever includes the use of antibiotics to inhibit the Salmonella Typhi bacteria. Commonly prescribed antibiotics include ciprofloxacin, ceftriaxone, and azithromycin. In severe cases, when antibiotic resistance is a concern, healthcare professionals may need to adjust the treatment plan based on individual circumstances and regional resistance patterns [4,5]
Antimicrobial resistance in S. Typhi has become a significant health concern. Over the years, strains of S. Typhi have developed resistance to multiple antibiotics which limits the effectiveness of commonly used antibiotics, such as chloramphenicol, ampicillin, and trimethoprim-sulfamethoxazole. Furthermore, Fluoroquinolones and third-generation cephalosporins has been the widely used for treatment of typhoid fever [6,7]. However, resistance to these antibiotics are also well reported. This has led to an increased reliance on azithromycin and carbapenems for treating cases of multidrug resistant (MDR) typhoid fever [8]. Surveillance and understanding the prevalence of antimicrobial resistance (AMR) patterns are crucial for determining the effective treatment options in specific regions. The spread of antimicrobial resistance (AMR) in S. Typhi is driven by numerous factors such as the overuse and misuse of antibiotics, limited healthcare access, poor sanitation, and global travel [9,10,11]. These elements contribute to the development and dissemination of resistant strains, especially in regions where typhoid fever is endemic. The transmission of resistant bacteria through contaminated water and food sources is worsen by inadequate hygiene practices. Global travel and migration further facilitate the introduction of resistant strains to new areas. The exchange of mobile genetic elements among S. Typhi strains also enhances the rapid spread of resistance [12].
Quinolones are widely use antibiotic for management of infection but S. typhi has develop resistance to this antibiotic. There are numerous factors involve at the genetic level for quinolone resistance, that includes mutations in the target genes responsible for DNA replication and repair, namely gyrase (gyrA and gyrB) and topoisomerase IV (parC and parE) [13,14,15]. These mutations result in alterations to the structure of these enzymes, particularly in the quinolone-binding regions, reducing the affinity for quinolone antibiotics. The most common mutations associated with quinolone resistance are located in the quinolone resistance-determining regions (QRDRs) of these genes. Additionally, the acquisition of plasmids or mobile genetic elements carrying quinolone resistance genes, such as qnrS genes, through horizontal gene transfer, contributes to the dissemination of resistance within bacterial populations [16,17]. Understanding the genetic mechanisms underlying quinolone resistance is crucial for surveillance efforts and the development of effective strategies to combat the spread of antibiotic resistance in Salmonella [18].
This study is aim to elucidate the genetic and molecular dynamics involves in quinolone resistance. To achieve the aim of study genetic data was extracted from databases and antimicrobial resistance profile was generated. Furthermore docking analysis was conducted elucidate the molecular dynamics involve in quinolone resistance. The findings provide valuable insights into the intricate mechanisms underlying resistance and contribute to a deeper understanding of the molecular basis of quinolone resistance, which is crucial for informing future strategies in combating antimicrobial resistance.
2. Results and Discussions:
Salmonella typhi is known for producing resistance against many promising antibiotics like Ampicillin, Broad-Spectrum Cephalosporins, Chloramphenicol, Ciprofloxacin, Sulfonamides, Trimethoprim, and Co-Trimoxazole [19,20]. The resistance is mainly acquired by the presence of plasmid, horizontal gene transfer and mobile genetic elements [21]. In this study Salmonella typhi draft genomes were downloaded with having travel history to Pakistan. The isolates were isolated from different sample type like blood (23.07%), stool (4.9%) and unknown sample type (72.02%). furthermore, 88.8% of isolates have travel history to Pakistan which is one of the alarming aspect for the prevalence of S. typhi in the local communities.
Antibiogram which shows the profile of the susceptibilities of specific pathogenic bacteria to antimicrobial agents. The analysis of data in antibiogram reveals that all isolates are multidrug resistant and shows variable range of resistance to specific antibiotic class [22,23]. The phenotypic resistance for different antibiotic classes was noted as Ampicillin (n=183), Broad-Spectrum Cephalosporins (n=108), Chloramphenicol (n=190), Ciprofloxacin (n=122), Sulfonamides(n=194), Trimethoprim (n=189), Co-Trimoxazole (n=189), Tetracycline (n=2), While, there was no resistance reported for Azithromycin, Colistin and Meropenem (Figure 1). Furthermore, the ciprofloxacin resistance belongs to quinolone class of antibiotic and has very well understood mode of action [24]. The antibiogram data also report intermediate resistance (n=161) and Sustainable (n=3) for Ciprofloxacin (Figure 2). Ciprofloxacin has very well studied mode of action. Bacterial DNA gyrase and topoisomerase IV, act as crucial enzymes involved in DNA replication and repair. By binding to these enzymes, ciprofloxacin disrupts DNA synthesis, leading to bacterial cell death [25,26,27]. This mode of action makes ciprofloxacin an effective broad-spectrum antibiotic against various bacterial infections.
The phenotypic resistance was validated by genotypic study of the draft genomes. The genotypic findings unveil the presence of multiple antimicrobial resistance genes (ARG) such as, blaCTXM-15 (55.30%), blaTEM-1D (91.79%), catA1 (97.43%), dfrA7 (96.82%), qnrS1 (54.35%), sul1 (98.46%), sul2 (65.64%) and tetA(B) (1.02%) (Figure 3). These genes are responsible for conferring resistance to a majority of antibiotics belonging to the classes of Beta-lactamases, Chloramphenicols, Sulphonamides, Trimethoprim, Quinolones, and Tetracycline[28,29]. Notably, no resistance was detected for carbapenems and macrolides. The identified antimicrobial resistance genes exhibit diverse modes of action, contributing to the multidrug resistance observed in Salmonella typhi isolates. The blaCTXM-15 gene, a member of the extended-spectrum beta-lactamases (ESBLs), confers resistance by hydrolyzing a broad range of beta-lactam antibiotics. blaTEM-1D, another beta-lactamase gene, similarly targets beta-lactam antibiotics [30,31]. The catA1 gene provides resistance by encoding a chloramphenicol acetyltransferase that inactivates chloramphenicol [32]. The dfrA7 gene confers resistance to trimethoprim by encoding a dihydrofolate reductase enzyme [33]. qnrS1, associated with quinolone resistance, protects DNA gyrase from inhibition by quinolone antibiotics [34]. sul1 and sul2 genes mediate resistance to sulfonamides by encoding di-hydropteroate synthase enzymes [35]. The tetA(B) gene, conferring tetracycline resistance, is associated with efflux pump mechanisms that extrude tetracycline from bacterial cells [36]. An in-depth analysis of the phenotype and genotype of isolates reveals instances where the antimicrobial resistant gene is present, but the phenotype is absent. This discrepancy may arise from factors such as the non-expression of the gene or limitations in the Antimicrobial susceptibility test. These genes are mostly acquired by plasmids, Mobile genetic elements (MGE), and horizontal gene transfer [37,38]
Flouroquinolones are widely used antibiotic for the treatment of infection caused by S. typhi but quinolone resistance is widely observed. The quinolone resistance is achieved by multiple factors such as mutation in quinolone resistance determining regions (QRDR) of DNA gyrase Subunit A (DNA gyrA), HTH-type topoisomerase IV (parC), and DNA gyrase Subunit B (DNA gyrB) [39,40]. Mutation analysis of these gene reveals mutation at DNA gyrA_S83F(n=258), DNA gyrA_D87N (n=20), DNA gyrA_S83Y (n=3), DNA gyrB_S464F (n=18), parC_S80I (n=16), parE_L416F (n=4) and no mutation detected in n=3 isolates that eventually make these isolates susceptible to ciprofloxacin (Figure 4). Furthermore, the effect of combinations of mutations and qnrS gene was investigated to elucidate the mechanism involve in ciprofloxacin resistance. The available data unveil that presence of mutation at DNA gyrA_S83F, DNA gyrA_D87N, DNA gyrA_S83Y, and DNA gyrB_S464F leads to intermediate resistance. While, mutation at DNA gyrA_S83F, and presence of qnrS1 gene leads to antibiotic resistance. Furthermore mutation at DNA gyrA S83F & D87N and parC S80I also leads to antibiotic resistance (Figure 5). In addition to that, the isolated were reported susceptible when there is no mutations at QRDR. QRDR act as a active site for quinolone and mutation leads to change in conformation of protein which also effect the binding affinity of quinolones [41].
The effect of mutation at Quinolone resistance determining region (QRDR) and its role in resistance was elucidated by docking analysis. The docking results offer valuable insights into the potential impact of specific mutations in the DNA gyrA and parC genes on the binding affinity of ciprofloxacin. The wild-type DNA gyrA exhibited a relatively strong binding affinity with ciprofloxacin, reflected by a binding energy of -7.2 Kcal/mol (Table 1). This suggests a stable and effective interaction between ciprofloxacin and the unmutated DNA gyrA, supporting the drug's efficacy against the wild-type strain. However, when mutations were introduced into the DNA gyrA gene, such as the S83F, S83Y, and D87N mutations, a consistent decrease in binding affinity was observed. The S83F mutation resulted in the lowest binding affinity among the single mutations, indicating that this specific change in the DNA gyrA gene could significantly impact the drug's binding efficacy [42,43]. The combination of S83F and D87N mutations did not further decrease the binding affinity, suggesting a saturation effect or potential structural changes that hinder effective drug binding. The solvent accessibility values provide additional context. Solvent accessibility reflects the degree to which the binding site is accessible to the surrounding solvent molecules [44,45]. In the wild-type DNA gyrA, a solvent accessibility of 2830.99 was observed, suggesting a reasonably accessible binding site. The mutations, however, showed a decrease in solvent accessibility, indicating potential alterations in the local environment, which might hinder ciprofloxacin accessibility to the binding site (Figure 6a-e).
Furthermore, the parC wild-type gene demonstrated a binding affinity of -6.7 Kcal/mol, slightly lower than that of the unmutated DNA gyrA. Despite this, it still indicates a relatively strong interaction. The introduction of the S80I mutation in parC resulted in a further decrease in binding affinity, suggesting that this mutation could confer resistance to ciprofloxacin in a manner similar to the mutations in the DNA gyrA gene (Figure 7a,b). Visualization of the docking results provided an important observation that there were no direct interactions between ciprofloxacin and the molecules at the mutation sites. This suggests that the decrease in binding affinity may be attributed to conformational changes induced by the mutations rather than direct interactions with the drug [46,47]. The decrease in binding affinity, coupled with changes in solvent accessibility and the absence of direct interactions at mutation sites, suggests that these mutations could indeed contribute to reduced effectiveness of ciprofloxacin, possibly leading to antibiotic resistance. The findings underscore the importance of understanding molecular interactions in the context of antibiotic resistance, providing valuable information for the development of more effective therapeutic strategies against resistant bacterial strains.
3. Material and Methodology:
3.1. Data Extraction:
Multi drug-resistance Salmonella enterica serovar Typhi draft genomes was extracted from NCBI database while, raw sequence reads was excluded from the study. These isolates (n=286) was exclusively from Pakistan reported from 2019 to 2023. Travel history to Pakistan and phenotypic resistance profiles was also put under consideration for corelation with genetic profile. Antibiogram of these isolates was downloaded from pathogenwatch for effective corelation with genotype.
3.2. Genomic Analysis:
The draft genomes was used to investigate the presence of antimicrobial resistance genes (ARGs), by using Resfinder 4.1 for the identification and annotation of ARGs within the genomic data[48]. The genotypic profiles, encompassing ARGs results, were then correlated with the phenotypic profiles to provide a comprehensive understanding of the relationship between genetic markers and observed resistance patterns.
3.3. Mutation analysis in DNA gyrA, and parC genes:
Quinolone resistance is a central focus of this study, prompting the investigation of mutation analysis. Protein sequences of the DNA gyrA, DNA gyrB, parE, and parC genes were extracted from annotated files. Subsequently, multiple sequence alignment was conducted using MEGA 11 to identify and characterize mutations in these genes [49]. The impact of these mutations was then assessed by considering the genotypic antimicrobial resistance gene (ARG) profile and the phenotypic resistance profile. This comprehensive approach aimed to elucidate the role of mutations in the development of quinolone resistance, providing valuable insights into the genetic mechanisms influencing bacterial response to these antibiotics.
3.4. Protein Structure Modeling:
Protein models, particularly for DNA gyrA and parC, were prepared using Swissmodel for subsequent docking analysis. The protein models were obtained and downloaded in PDB format, facilitating their utilization in docking studies. Ciprofloxacin, selected as the ligand molecule, was sourced from the DrugBank database. This ligand molecule, essential for the docking analysis, was downloaded to explore its interaction with the prepared protein models.
3.5. Docking Analysis:
Molecular dynamics involve in quinolone resistance molecular docking simulations were conducted using AutoDock Vina, complemented by docking analysis in Discovery Studio, to investigate the interactions between quinolone antibiotics and the DNA gyrase A subunit receptor and parC receptor [50]. Ligands and receptor structures were prepared, and a grid for docking was generated by using Autodock vina around the Quinolone resistance determining region. AutoDock Vina was employed for docking simulations, exploring various ligand conformations and orientations to predict binding affinities. Post-docking analysis in Discovery Studio facilitated the visualization of binding poses, identification of key binding residues, and assessment of potential interactions.
4. Conclusion:
This study unveils a complex landscape of antimicrobial resistance in Salmonella typhi, particularly emphasizing quinolone resistance. The genotypic analysis identified a plethora of antimicrobial resistance genes, including blaCTXM-15, blaTEM-1D, catA1, dfrA7, qnrS1, sul1, sul2, and tetA(B), contributing to multidrug resistance against various antibiotic classes. Furthermore, quinolone resistance was associated with mutations in quinolone resistance determining regions (QRDR) of DNA gyrA and parC genes. The mutation analysis and subsequent docking studies revealed a significant impact on the binding affinity of ciprofloxacin, with specific mutations such as DNA gyrA_S83F playing a crucial role in reducing efficacy. The absence of direct interactions at mutation sites, coupled with changes in solvent accessibility, suggests a structural basis for reduced drug effectiveness. Importantly, the study highlights instances where the presence of resistance genes does not necessarily correlate with phenotypic resistance, emphasizing the need for caution in interpreting genotypic data. To further validate and understand these findings, mutagenesis studies are recommended to experimentally confirm the impact of specific mutations on ciprofloxacin resistance. This multifaceted approach will provide a more comprehensive understanding of the mechanisms driving antibiotic resistance in Salmonella typhi, guiding the development of targeted therapeutic strategies to counteract the rising threat of resistant bacterial strains.
Author Contributions
Conceptualization, N.K. and M.Y.; Methodology, N.K.; Software, H.M. and M.W.I.; Validation, M.A.B., I.U.. and N.K.; Formal Analysis, M.Y.; Investigation, I.U, H.M., M.A.B, and M.W.I.; Data Curation, N.K. and H.M, I.U and M.W.I; Writing – Original Draft Preparation, H.M., and M.W.I.; Writing – Review & Editing, M.Y. and M.A.B.; Visualization, I.U. and M.A.B.; Supervision, N.K.; Funding Acquisition, M.Y. All authors have read and agreed to the published version of the manuscript.
Funding
Research supporting project number (RSPD2024R740), King Saud University, Riyadh, Saudi Arabia.
Institutional Review Board Statement
Not Applicable.
Informed Consent Statement
Not Applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors are grateful to King Saud University, Riyadh, Saudi Arabia for funding the work through the Researchers Supporting Project number (RSPD2023R740).
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
References
- Siourimè, S. N., Isidore, B. O. J., Traoré, Y., & Savadogo, A. (2017). Salmonella enterica Serovar Typhi and Paratyphi Responsible of Typhoid and Paratyphoid Fevers Transmitted by Environment and Food. International Journal of Sciences, 3(05), 87–96. [CrossRef]
- Kröger, C., Colgan, A., Srikumar, S., Händler, K., Sivasankaran, S. K., Hammarlöf, D. L., Canals, R., Grissom, J. E., Conway, T., Hokamp, K., & Hinton, J. C. D. (2013). An Infection-Relevant Transcriptomic Compendium for Salmonella enterica Serovar Typhimurium. Cell Host & Microbe, 14(6), 683–695. [CrossRef]
- Gordon, M. A., Graham, S. M., Walsh, A. L., Wilson, L., Phiri, A., Molyneux, E., Zijlstra, E. E., Heyderman, R. S., Hart, C. A., & Molyneux, M. E. (2008). Epidemics of InvasiveSalmonella entericaSerovar Enteritidis andS. entericaSerovar Typhimurium Infection Associated with Multidrug Resistance among Adults and Children in Malawi. Clinical Infectious Diseases, 46(7), 963–969. [CrossRef]
- Britto, C., Wong, V., Dougan, G., & Pollard, A. J. (2018). A systematic review of antimicrobial resistance in Salmonella enterica serovar Typhi, the etiological agent of typhoid. PLOS Neglected Tropical Diseases, 12(10), e0006779. [CrossRef]
- Crump, J. A., Barrett, T. J., Nelson, J., & Angulo, F. J. (2003). Reevaluating Fluoroquinolone Breakpoints for Salmonella enterica Serotype Typhi and for Non-Typhi Salmonellae. Clinical Infectious Diseases, 37(1), 75–81. [CrossRef]
- Akhtar, S., Sarker, M. R., Jabeen, K., Sattar, A., Qamar, A., & Fasih, N. (2014). Antimicrobial resistance in Salmonella entericaserovar typhi and paratyphi in South Asia-current status, issues and prospects. Critical Reviews in Microbiology, 41(4), 536–545. [CrossRef]
- Krishnan, P., Stalin, M., & Balasubramanian, S. (2009). Changing trends in antimicrobial resistance ofSalmonellaentericaserovar typhi andsalmonella entericaserovar paratyphi A in Chennai. Indian Journal of Pathology & Microbiology, 52(4), 505. [CrossRef]
- Karkey, A., Thwaites, G., & Baker, S. (2018). The evolution of antimicrobial resistance in Salmonella Typhi. Current Opinion in Gastroenterology, 34(1), 25–30. [CrossRef]
- Akinyemi, K. O., & Ajoseh, S. O. (2017). Factors contributing to the emergence and spread of antibiotics resistance in salmonella species. In InTech eBooks. [CrossRef]
- Bengtsson-Palme, J., Kristiansson, E., & Larsson, D. G. J. (2017). Environmental factors influencing the development and spread of antibiotic resistance. Fems Microbiology Reviews, 42(1). [CrossRef]
- Beceiro, A., Tomás, M., & Bou, G. (2013). Antimicrobial Resistance and Virulence: a Successful or Deleterious Association in the Bacterial World? Clinical Microbiology Reviews, 26(2), 185–230. [CrossRef]
- Silva, C., Wiesner, M., & Calva, E. (2012). The importance of mobile genetic elements in the evolution of salmonella: pathogenesis, antibiotic resistance and host adaptation. In InTech eBooks. [CrossRef]
- Dasgupta, N., Paul, D., Chanda, D. D., Chetri, S., Chakravarty, A., & Bhattacharjee, A. (2018). Observation of a New Pattern of Mutations in gyrA and parC within Escherichia coli Exhibiting Fluroquinolone Resistance. Indian Journal of Medical Microbiology, 36(1), 131–135. [CrossRef]
- Bagel, S., Hüllen, V., Wiedemann, B., & Heisig, P. (1999). Impact of gyrA and parC Mutations on Quinolone Resistance, Doubling Time, and Supercoiling Degree of Escherichia coli. Antimicrobial Agents and Chemotherapy, 43(4), 868–875. [CrossRef]
- Gopal, M., Elumalai, S., Suresh, A., Durairajpandian, V., Kannan, M. A., Selvam, E., & Srivani, S. (2016). GyrA ser83 and ParC trp106 Mutations in Salmonella enterica Serovar Typhi Isolated from Typhoid Fever Patients in Tertiary Care Hospital. Journal of Clinical and Diagnostic Research. [CrossRef]
- Tadesse, D. A., Singh, A., Zhao, S., Bartholomew, M. J., Womack, N., Ayers, S., Fields, P. I., & McDermott, P. F. (2016). Antimicrobial Resistance in Salmonella in the United States from 1948 to 1995. Antimicrobial Agents and Chemotherapy, 60(4), 2567–2571. [CrossRef]
- Kariuki, S., Gordon, M. A., Feasey, N. A., & Parry, C. M. (2015). Antimicrobial resistance and management of invasive Salmonella disease. Vaccine, 33, C21–C29. [CrossRef]
- Cuypers, W. L., Jacobs, J., Wong, V., Klemm, E. J., Deborggraeve, S., & Van Puyvelde, S. (2018). Fluoroquinolone resistance in Salmonella: insights by whole-genome sequencing. Microbial Genomics, 4(7). [CrossRef]
- Gangathraprabhu, B., Kannan, S. S., Santhanam, G., Suryadevara, N., & Murugan, M. (2020). A review on the origin of multidrug-resistant Salmonella and perspective of tailored phoP gene towards avirulence. Microbial Pathogenesis, 147, 104352. [CrossRef]
- Oo, K. M., Myat, T. O., Htike, W. W., Biswas, A., Hannaway, R. F., Murdoch, D. R., Crump, J. A., & Ussher, J. E. (2019). Molecular mechanisms of antimicrobial resistance and phylogenetic relationships of Salmonella enterica isolates from febrile patients in Yangon, Myanmar. Transactions of the Royal Society of Tropical Medicine and Hygiene, 113(10), 641–648. [CrossRef]
- Van Hoek, A. H., Mevius, D., Guerra, B., Mullany, P., Roberts, A. P., & Aarts, H. (2011). Acquired Antibiotic Resistance Genes: An Overview. Frontiers in Microbiology, 2. [CrossRef]
- Gessese, Y. A., Damessa, D. L., Amare, M. M., Bahta, Y. H., Shifera, A. D., Tasew, F. S., & Gebremedhin, E. Z. (2017). Urinary pathogenic bacterial profile, antibiogram of isolates and associated risk factors among pregnant women in Ambo town, Central Ethiopia: a cross-sectional study. Antimicrobial Resistance and Infection Control, 6(1). [CrossRef]
- Kibret, M., & Abera, B. (2014). Prevalence and antibiogram of bacterial isolates from urinary tract infections at Dessie Health Research Laboratory, Ethiopia. Asian Pacific Journal of Tropical Biomedicine, 4(2), 164–168. [CrossRef]
- Bush, N. G., Diez-Santos, I., Abbott, L., & Maxwell, A. (2020). Quinolones: mechanism, lethality and their contributions to antibiotic resistance. Molecules, 25(23), 5662. [CrossRef]
- Drlica, K., & Zhao, X. (2020). Bacterial death from treatment with fluoroquinolones and other lethal stressors. Expert Review of Anti-infective Therapy, 19(5), 601–618. [CrossRef]
- Kassab, A. E., & Gedawy, E. M. (2018). Novel ciprofloxacin hybrids using biology oriented drug synthesis (BIODS) approach: Anticancer activity, effects on cell cycle profile, caspase-3 mediated apoptosis, topoisomerase II inhibition, and antibacterial activity. European Journal of Medicinal Chemistry, 150, 403–418. [CrossRef]
- Ashley, R. E., Dittmore, A., McPherson, S. A., Turnbough, C. L., Neuman, K. C., & Osheroff, N. (2017). Activities of gyrase and topoisomerase IV on positively supercoiled DNA. Nucleic Acids Research, 45(16), 9611–9624. [CrossRef]
- Urban-Chmiel, R., Marek, A., Stępień-Pyśniak, D., Wieczorek, K., Dec, M., Nowaczek, A., & Osek, J. (2022). Antibiotic Resistance in Bacteria—A Review. Antibiotics, 11(8), 1079. [CrossRef]
- De Alcântara Rodrigues, I., Ferrari, R. G., Panzenhagen, P., Mano, S. B., & Conté-Júnior, C. A. (2020). Antimicrobial resistance genes in bacteria from animal-based foods. In Advances in Applied Microbiology (pp. 143–183). [CrossRef]
- Debabza, M., Dziri, R., Mechai, A., Bouguessa, A., Klibi, N., & Ouzari, H. (2023). Characterization of carbapenemase-producing Gram-negative bacilli: first report of blaNDM-1 in Enterobacter cloacae. Journal of Infection in Developing Countries, 17(09), 1300–1309. [CrossRef]
- Sumbana, J. J., Santona, A., Fiamma, M., Taviani, E., Deligios, M., Zimba, T. F., Lucas, G., Sacarlal, J., Rubino, S., & Paglietti, B. (2021). Extraintestinal Pathogenic Escherichia coli ST405 Isolate Coharboring blaNDM-5 and blaCTXM-15: A New Threat in Mozambique. Microbial Drug Resistance, 27(12), 1633–1640. [CrossRef]
- Obayiuwana, A., & Ibekwe, A. M. (2020). Antibiotic Resistance Genes Occurrence in Wastewaters from Selected Pharmaceutical Facilities in Nigeria. Water, 12(7), 1897. [CrossRef]
- Paulshus, E., Colque, P., Kühn, I., Tauhid, T., Hu, Y., Zhou, Y., Thorell, K., Möllby, R., Sørum, H., Sjöling, Å., & Joffré, E. (2023). Escherichia coli ST2797 Is Abundant in Wastewater and Might Be a Novel Emerging Extended-Spectrum Beta-Lactamase E. coli. Microbiology Spectrum, 11(4). [CrossRef]
- Vázquez, X., Fernández, J., Hernáez, S., Rodicio, R., & Rodicio, M. R. (2022). Plasmid-Mediated Quinolone Resistance (PMQR) in Two Clinical Strains of Salmonella enterica Serovar Corvallis. Microorganisms, 10(3), 579. [CrossRef]
- Venkatesan, M., Fruci, M., Verellen, L. A., Skarina, T., Mesa, N., Flick, R., Pham, C., Mahadevan, R., Stogios, P., & Savchenko, A. (2023). Molecular mechanism of plasmid-borne resistance to sulfonamide antibiotics. Nature Communications, 14(1). [CrossRef]
- Foong, W., Wilhelm, J., Tam, H., & Pos, K. M. (2020b). Tigecycline efflux in Acinetobacter baumannii is mediated by TetA in synergy with RND-type efflux transporters. Journal of Antimicrobial Chemotherapy, 75(5), 1135–1139. [CrossRef]
- Gajić, I., Kabić, J., Kekić, D., Jovićević, M., Milenković, M., Mitić-Ćulafić, D., Trudić, A., Ranin, L., & Opavski, N. (2022). Antimicrobial susceptibility testing: A comprehensive review of currently used methods. Antibiotics, 11(4), 427. [CrossRef]
- Van Belkum, A., Burnham, C. D., Rossen, J. W. A., Mallard, F., Rochas, O., & Dunne, W. M. (2020). Innovative and rapid antimicrobial susceptibility testing systems. Nature Reviews Microbiology, 18(5), 299–311. [CrossRef]
- Onishi, R., Shigemura, K., Osawa, K., Yang, Y., Maeda, K., Tanimoto, H., Kado, M., Fang, S., Sung, S., Miyara, T., & Fujisawa, M. (2022). Impact on quinolone resistance of plasmid-mediated quinolone resistance gene and mutations in quinolone resistance-determining regions in extended spectrum beta lactamase-producing Klebsiella pneumoniae isolated from urinary tract infection patients. Pathogens and Disease, 80(1). [CrossRef]
- Shaheen, A., Tariq, A., Iqbal, M., Mirza, O., Haque, A., Walz, T., & Rahman, M. (2021). Mutational diversity in the quinolone Resistance-Determining regions of Type-II topoisomerases of salmonella serovars. Antibiotics, 10(12), 1455. [CrossRef]
- Azargun, R., Gholizadeh, P., Sadeghi, V., Hosainzadegan, H., Tarhriz, V., Memar, M. Y., Pormohammad, A., & Eyvazi, S. (2020). Molecular mechanisms associated with quinolone resistance in Enterobacteriaceae: review and update. Transactions of the Royal Society of Tropical Medicine and Hygiene, 114(10), 770–781. [CrossRef]
- Wang, M., Cang, Z., & Wei, G. (2020). A topology-based network tree for the prediction of protein–protein binding affinity changes following mutation. Nature Machine Intelligence, 2(2), 116–123. [CrossRef]
- Teng, S., Sobitan, A., Rhoades, R., Liu, D., & Tang, Q. (2020). Systemic effects of missense mutations on SARS-CoV-2 spike glycoprotein stability and receptor-binding affinity. Briefings in Bioinformatics, 22(2), 1239–1253. [CrossRef]
- Moses, D., Ginell, G. M., Holehouse, A. S., & Sukenik, S. (2023). Intrinsically disordered regions are poised to act as sensors of cellular chemistry. Trends in Biochemical Sciences, 48(12), 1019–1034. [CrossRef]
- Potts, D. S., Bregante, D. T., Adams, J. S., Torres, C., & Flaherty, D. W. (2021). Influence of solvent structure and hydrogen bonding on catalysis at solid–liquid interfaces. Chemical Society Reviews, 50(22), 12308–12337. [CrossRef]
- Bhanja, K., Sharma, M., & Patra, N. (2023). Uncovering the structural and binding insights of dual inhibitors simultaneously targeting two distinct sites on EGFR kinase. The Journal of Physical Chemistry B, 127(50), 10749–10765. [CrossRef]
- Wang, R., & Zheng, Q. (2020). Multiple Molecular Dynamics Simulations of the Inhibitor GRL-02031 Complex with Wild Type and Mutant HIV-1 Protease Reveal the Binding and Drug-Resistance Mechanism. Langmuir, 36(46), 13817–13832. [CrossRef]
- Florensa, A. F., Kaas, R. S., Clausen, P. T. L. C., Aytan-Aktug, D., & Aarestrup, F. M. (2022). ResFinder – an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microbial Genomics, 8(1). [CrossRef]
- Tamura, K., Stecher, G., & Kumar, S. (2021). MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Molecular Biology and Evolution, 38(7), 3022–3027. [CrossRef]
- Trott, O., & Olson, A. J. (2009). AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of Computational Chemistry, 31(2), 455–461. [CrossRef]
Figure 1.
Phenotypic resistance profile of Salmonella typhi.
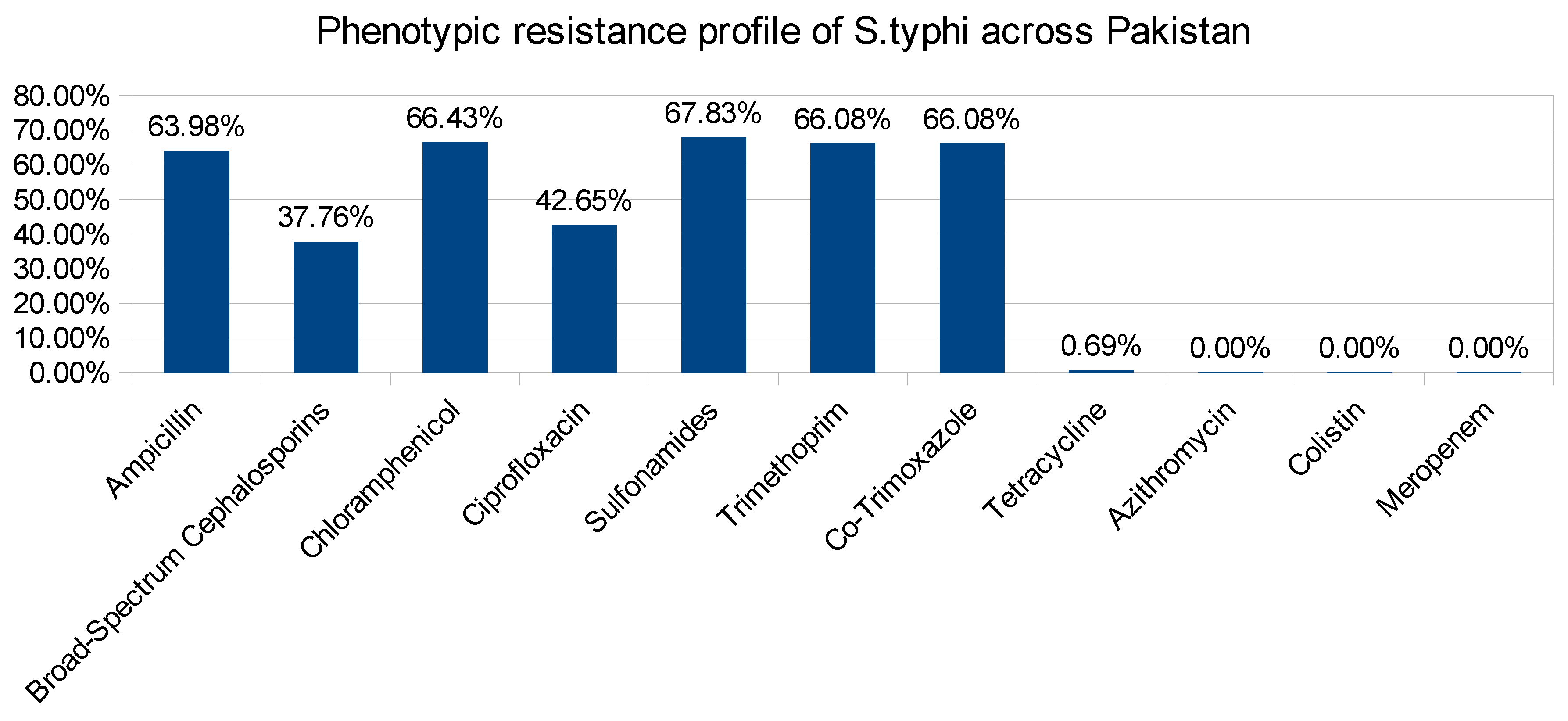
Figure 2.
Frequency of quinolone resistance in Salmonella typhi.
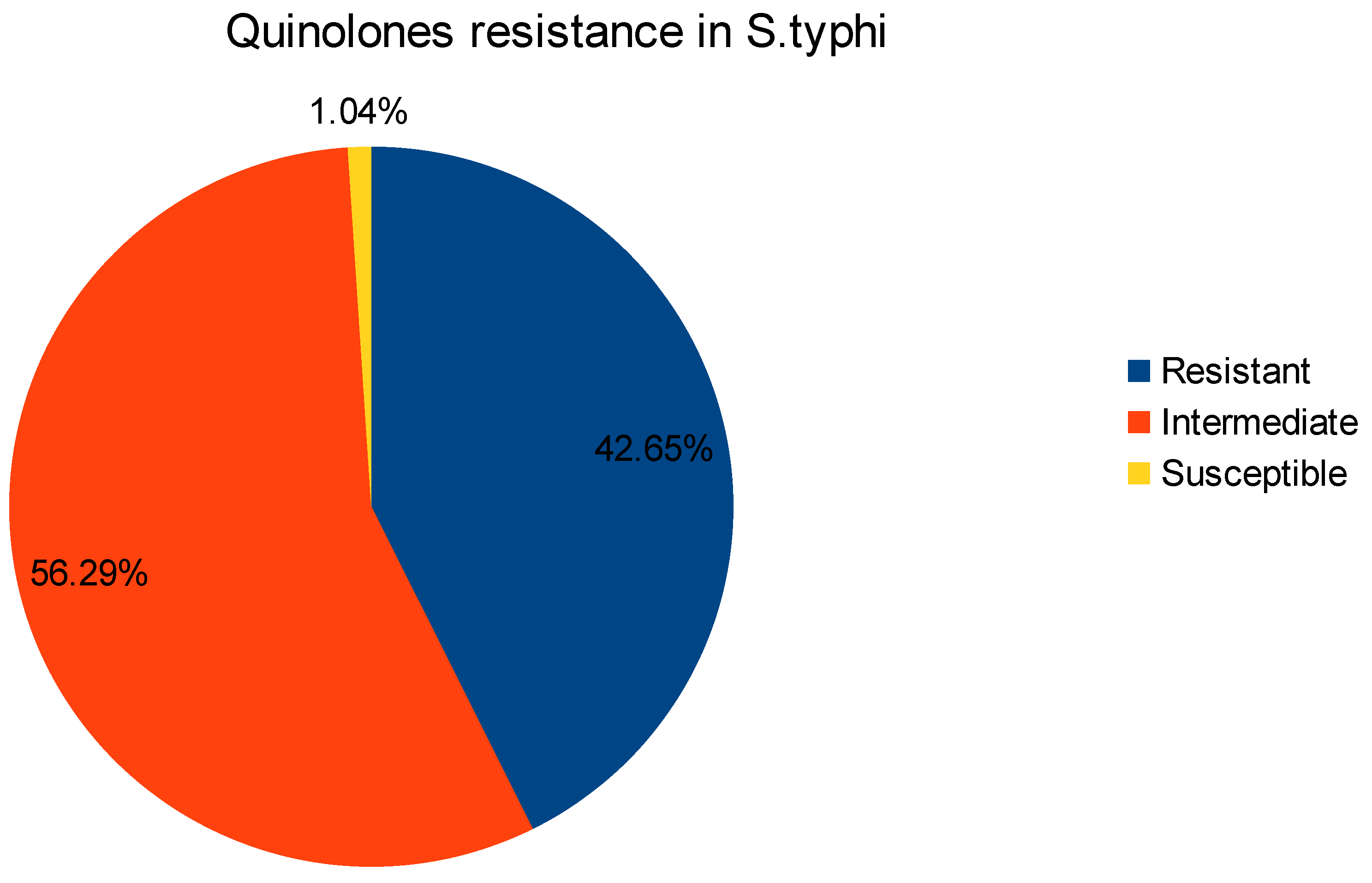
Figure 3.
Prevalence of antimicrobial resistance genes that confirms antimicrobial resistance to promising antibiotics.
Figure 3.
Prevalence of antimicrobial resistance genes that confirms antimicrobial resistance to promising antibiotics.
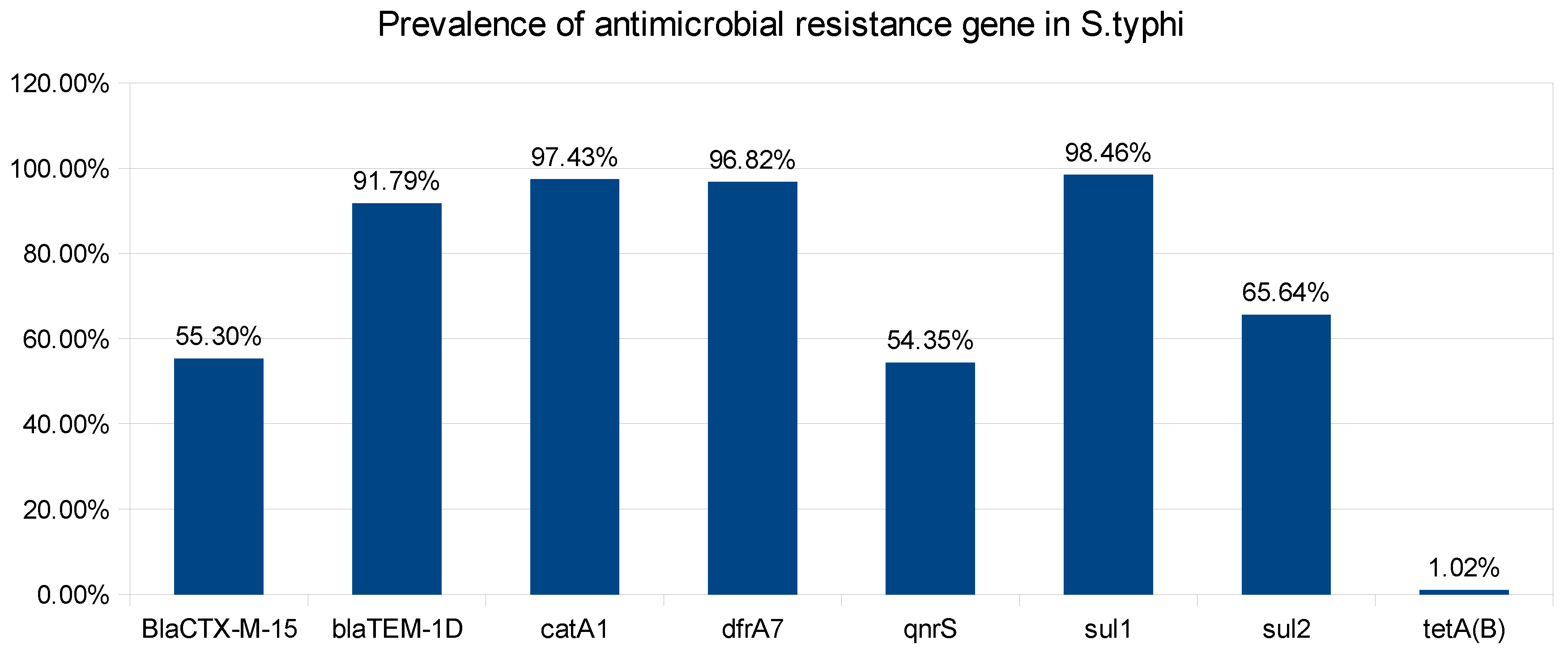
Figure 4.
Prevalence of Mutations in quinolone resistance determining genes.
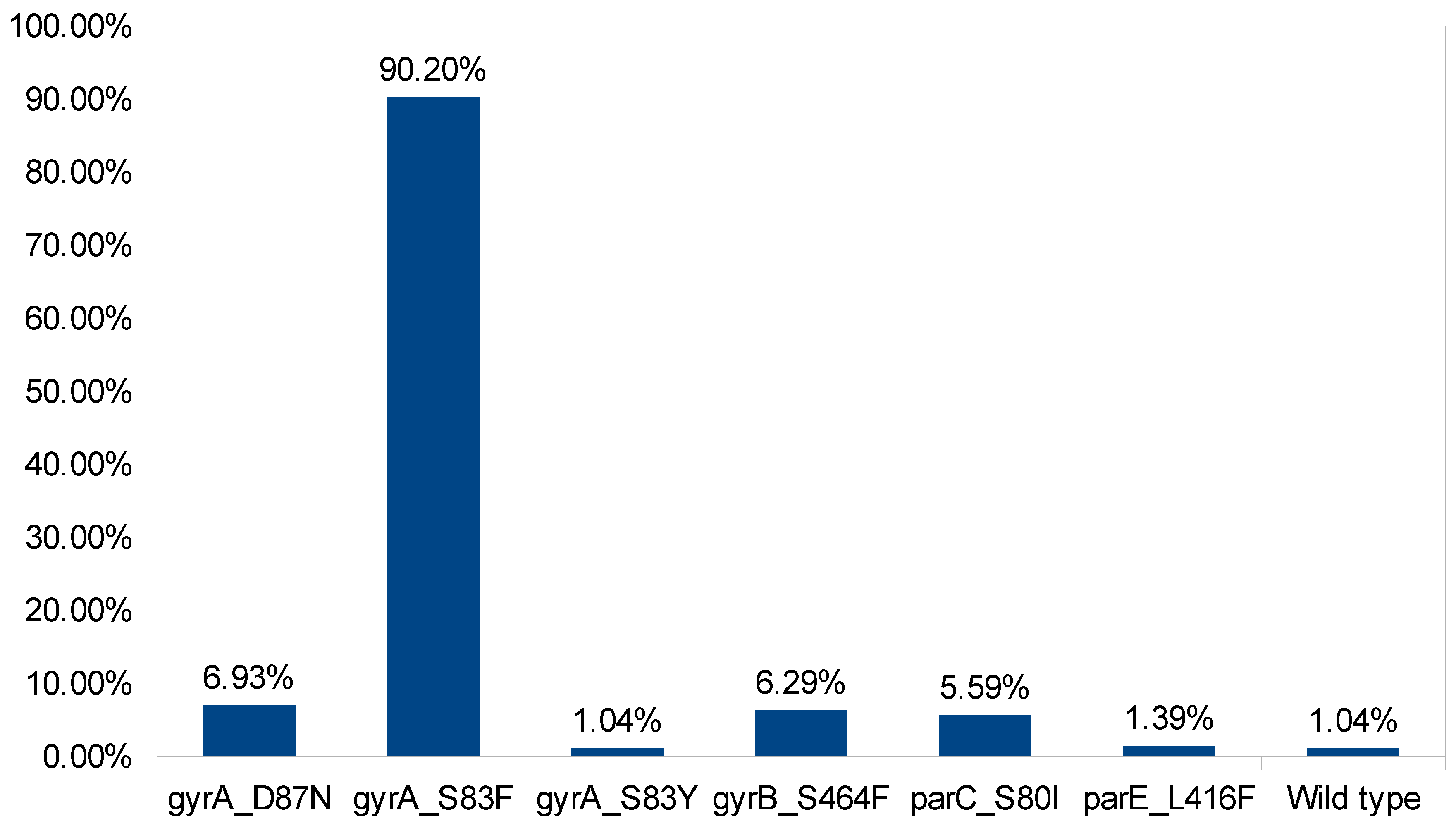
Figure 5.
Effect qnrS gene and Mutation in DNA gyrA, DNA gyrB, andParC on resistance profile of Salmonella Typhi.
Figure 5.
Effect qnrS gene and Mutation in DNA gyrA, DNA gyrB, andParC on resistance profile of Salmonella Typhi.
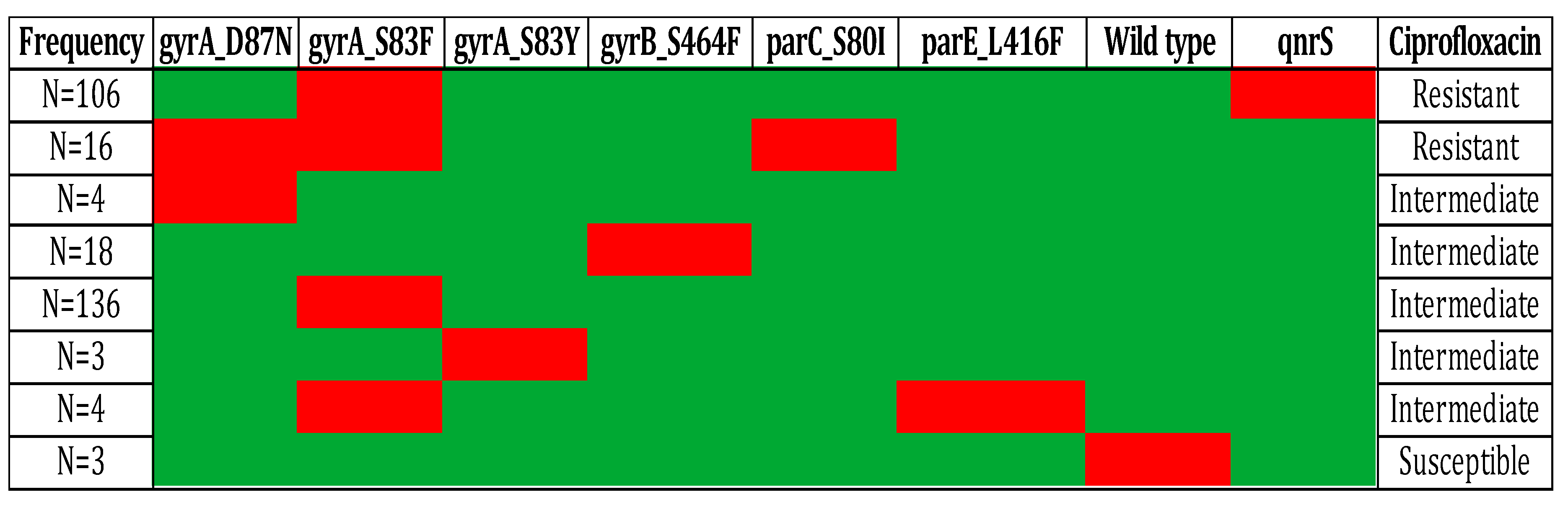
Figure 6.
(a): DNA gyrA Wild type. (b): DNA gyrA_S83F. (c): DNA gyrA_S83Y. (d): DNA gyrA_D87N. (e): DNA gyrA_S83F, D87N.
Figure 6.
(a): DNA gyrA Wild type. (b): DNA gyrA_S83F. (c): DNA gyrA_S83Y. (d): DNA gyrA_D87N. (e): DNA gyrA_S83F, D87N.
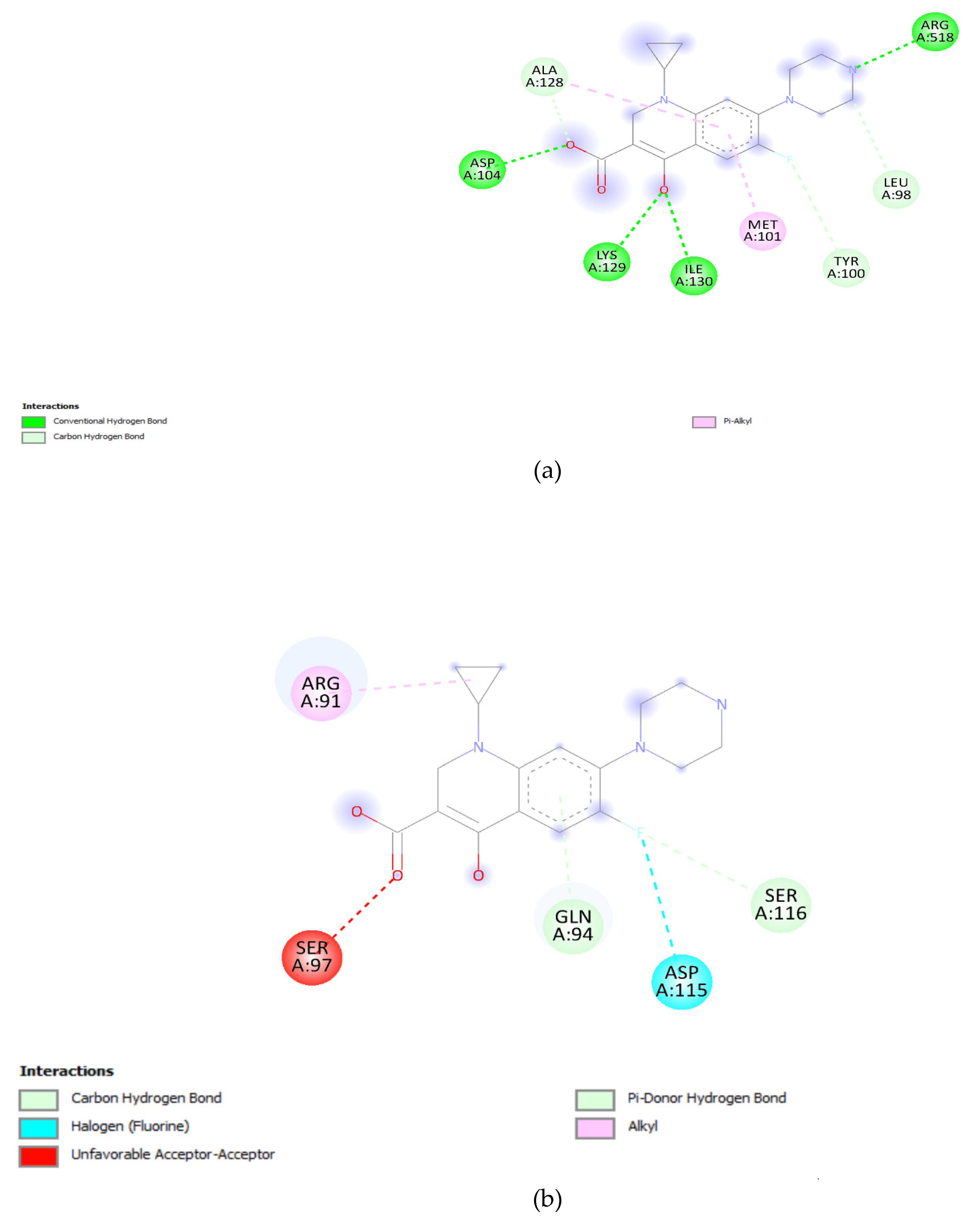
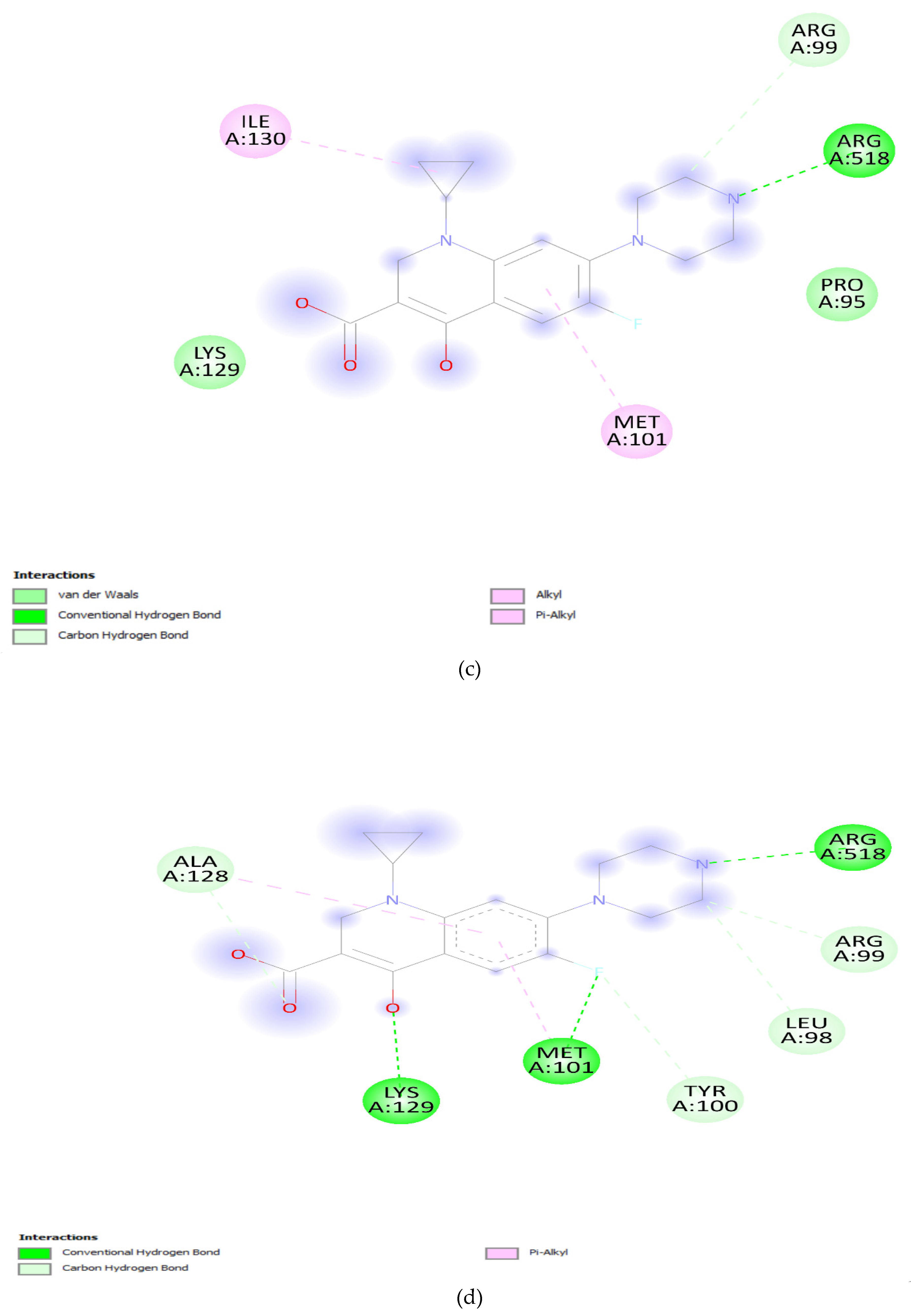
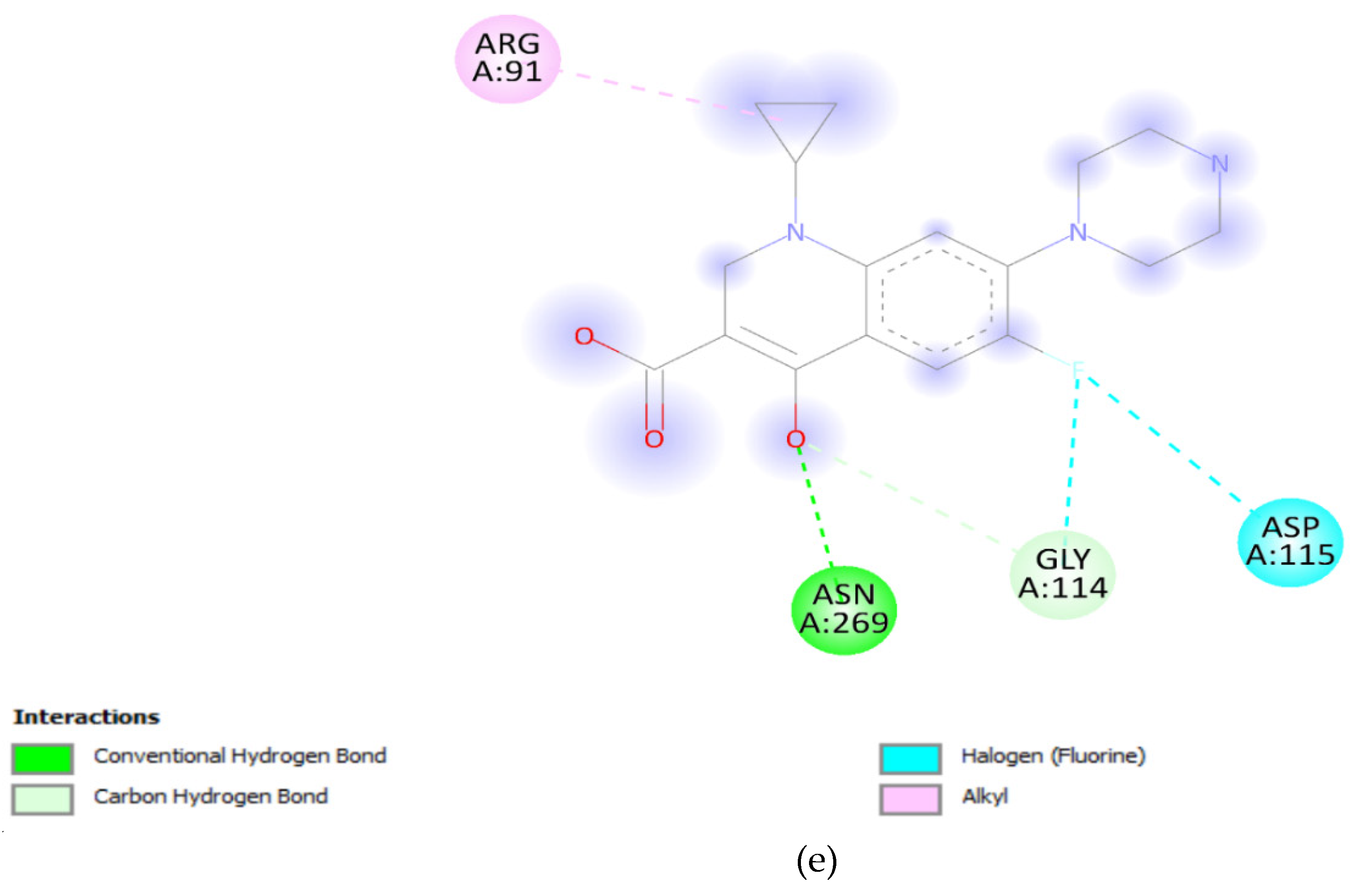
Figure 7.
(a): parC wild-type. (b): parC_S80I.
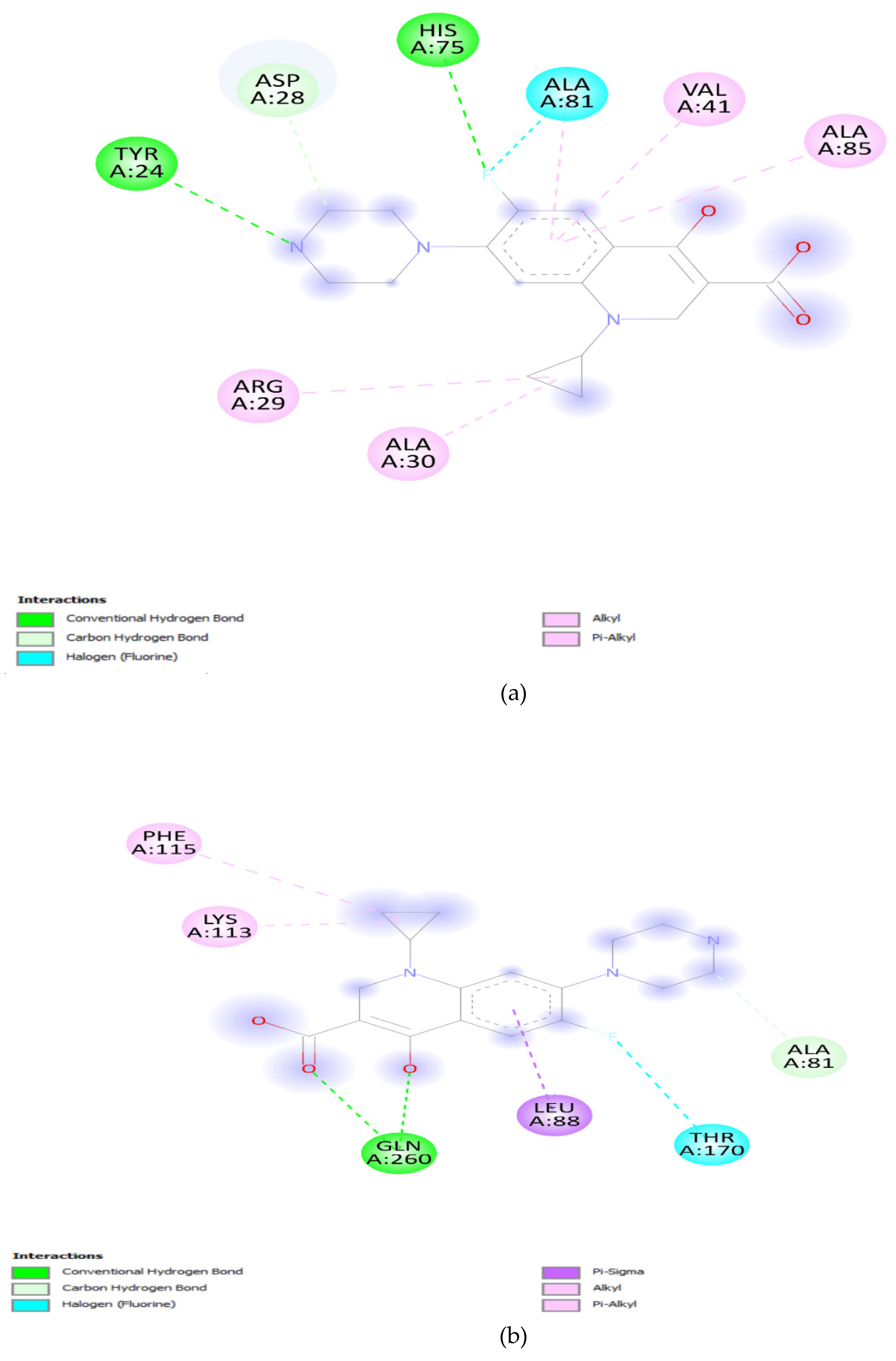

Table 1.
Effect of mutation on binding Affinity of Ciprofloxacin.
| Gene Variants | Binding Affinity in QRDR (Kcal/mol) |
Solvent Accessbility in QRDR (Ao) |
|---|---|---|
| GyrA Wild type | -7.1 | 2830.99 |
| gyrA_S83F | -6.2 | 2731.51 |
| gyrA_S83Y | -6.4 | 2741.95 |
| gyrA_D87N | -6.4 | 2756.78 |
| gyrA_S83F, D87N | -6.3 | 2758.2 |
| ParC Wild type | -6.7 | 3326.01 |
| parC_S80I | -6.0 | 3014.57 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



