
Submitted:
30 January 2024
Posted:
31 January 2024
You are already at the latest version
Abstract
Cleidocranial dysplasia (CCD) is an autosomal dominant skeletal dysplasia characterized by persistent open skull sutures whit bulging calvaria, hypoplasia or aplasia of clavicles permitting abnormal opposition of the shoulders, wide public symphysis, short middle phalanx of the fifth fingers and vertebral, craniofacial and dental anomalies. It is a rare disease, with a prevalence of 1-9/1,000,000, high penetrance, and variable expression. The responsible gene of CCD is the runt-related transcription factor 2 gene (RUNX2). We characterize the clinical, genetic and bioinformatic results of four CCD cases.
Two Mexican familial with 6 affected members, 9 asymptomatic individuals, and two sporadic cases with CCD, as well as 100 healthy controls. Genomic DNA analyses of the RUNX2 gene was performed for Sanger sequencing. Bioinformatics tools, used to predict the function, stability, and structural changes of the mutated RUNX2 proteins.
Three novel heterozygous mutations (c.651_652del; c.538_539delinsCA; c.662T>A) and a previously reported mutation (c.674G>A) were detected. In silico analysis showed that all mutations had functional, stability, and structural alterations in RUNX2 protein.
Our results show novel mutations that enrich the pool of RUNX2 gene mutations with CCD. In addition, one patient presented clinical data not previously reported that could represent an expanded phenotype of sever expression.
Keywords:
Cleidocranial dysplasia
; CCD
; RUNX2 gene
; novel mutations
1. Introduction
Cleidocranial dysplasia (CCD, OMIM119600) is a rare disease (ORPHA 1452), presenting with short stature, excessive mobility of the shoulders, impaired bone growth, skeletal and craniofacial anomalies, hypertelorism, a general midface retrusion, and a mandible prognathism. It is associated with teeth abnormalities, such as supernumerary teeth, failure of eruption and multiple dental anomalies. It is an autosomal dominant syndrome, with a prevalence of 1-9/1,000,000, high penetrance, and variable expression.1,2
CCD is caused by a heterozygous mutation of the Runt-related transcription factor 2 gene (RUNX2; OMIM 600211). The RUNX2 gene located on chromosome 6p21 encompasses a region of 223 kb with eight exons, and encodes a polypeptide of 521 amino acids (NP_001019801.3).3 This polypeptide comprises polyglutamine/alanine (Q/A) domains, Runt homologous domain (RHD), nuclear localization signal (NLS) region, proline/serine/threonine (PST)-rich region, and nuclear matrix-targeting sequence (NMTS) domain.4 RUNX2 has two promoters that result in the expression of two isoforms; the distal promoter (P1) generates the canonical isoform 1 or type II RUNX2 (NM_001024630.4) and the proximal promoter (P2) generates type I RUNX2 mRNA.5 The RUNX2 protein is a transcription factor is essential for osteoblast differentiation during intramembranous and endochondral ossification.6-8
CCD can be diagnosed with clinical and radiological evaluation, validated by molecular studies. Heterozygous loss of function RUNX2 gene, which plays an important role in osteogenesis and differentiation of precursor cells, causes CCD phenotype.9 More than 230 RUNX2 gene mutations have been associated with CCD.10,11 In the present study, we characterize the clinical, genetic and bioinformatic results of three novel and one previously reported mutation in the RUNX2 gene in two familial and in two sporadic cases with CCD and describe clinical data not previously informed.
2. Material and Methods
2.1. Patients
Two Mexican familial with 6 affected members and 9 asymptomatic individuals, and two sporadic cases (Figure 1A-D), as well as 100 healthy controls were included in this study. The probands were three males and one female. Clinical and all other relevant data of the patients are shown in Table 1. All patients agreed to participate in the study and gave their informed consent.
2.2. Direct Sequencing of RUNX2 Gene
After genomic DNA extraction using conventional methods, the RUNX2 gene was amplified by PCR. We designed primers flanking all 8 coding exons and intron-exon boundaries of the RUNX2 gene using Primer3 (http://bioinfo.ut.ee/primer3-0.4.0/). Primer sequences and PCR conditions are shown in Table 2. PCR products obtained from all affected and non-affected members of the families and 100 normal controls were analyzed through Sanger sequencing on an ABI 377 automated sequencer (PE Biosystems, Foster City, CA, USA). All DNA analyses were performed twice.
2.3. In Silico Prediction Analysis
The comparative analysis of the evolutive conservation was made with phyloP and phastCons. To assess the functional affection of the identified heterozygous variants, Polymorphism Phenotyping v2 (PolyPhen 2.0), MutationTaster (www.mutationtaster.org), PROVEAN (https://provean.jcvi.org/index.php) and SIFT tools were used. To predict protein stability changes, GROMOS96-Swiss-Pdb Viewer and I-Mutant 2.0 (https://folding.uib.es/cgi-bin/i-mutan t2.0.cgi) were used. The change in the force field energy was represented as the logarithm of the energy (Log). RUNX2 3D protein prediction was determined by alignment of the protein from the RaptorX server,12 the secondary structure changes and homology modeling were observed in Swiss-Pdb Viewer, version 4.0, using SWISS-MODEL.13
2.4. Ethical Aspects
The patients with DCC were selected under the official Mexican standard NOM-253-SSA1-2012 guidelines, all participants received oral and written information
3. Results
Analysis of the RUNX2 gene evidenced three novel mutations and one previously reported. The proband and the mother of the family 1 had the c.651_652delTA mutation in exon 4 (Figure 1A); this mutation changes the amino acid lysine to serine at position 218 and creates a premature termination 17 codons downstream of the deletion site (p.Lys218Serfs*17). The proband of the family 2 presented the c.538_539delinsCA mutation in exon 3 that generates the substitution of glutamine instead of alanine (p.Ala180Gln) (Figure 1B). The family 3 harbored the recurrent missense mutation in exon 4 c.674G>A that results in the change of glutamine by arginine, p.Arg225Gln (Figure 1C). The proband, brother, and mother of the family 4 showed the missense mutation c.662T>A in exon 4 that produces the substitution of glutamate by valine, p.Val221Glu (Figure 1D). Relevant genetic and protein data are shown in Table 1. These mutations were discarded in the RUNX2 gene in the unaffected members of the family and 100 healthy controls. No other relevant nucleotide variations or polymorphisms were detected in the rest of the analyzed exons. The novel variants were not found in ExAC, 1000 Genomes or Mutation-Taster databases.
The comparative analysis using phyloP and phastCons of evolutive conservation of the respective nucleotide in the human RUNX2 gene compared with that one of other species had a score of 4.5 to 6.0 and 1 for the c.651_652delTA, c.674G>A, c.538_539delinsCA, c.662T>A variants, respectively. This suggests that these amino acids are highly conserved, indicating a deleterious effect of these changes on the RUNX2 protein. The analysis prediction of deleterious effects in protein function is shown Table 3.
The total energy of each of the three mutants of RUNX2 protein were a high energy, with Log 18.45, 15.00, and 15.40 KJ/mol, respectively; it was a highest one compared to the wildtype (Log 14.48 KJ/mol). The deletion p.Lys218Serfs*17 showed a low energy value of 12.51 KJ/mol. These high and low energies implied an underlying damaging effect on protein structure, thereby affecting the protein stability and function. Free energy change (DDG) analysis with I-Mutant 2.0 showed negative values from -1.03 to -1.72 DGG in the three novel missense variants; it denotes a decreasing protein stability with a deleterious effect (Table 3).
4. Discussion
It is important to note that CCD has variable clinical expressiveness, and particular clinical characteristics have been described in various articles, with the following frequency of manifestations: 93% cranial, 50% facial, 89% dental, 92% clavicular, 40% chest, 40% extremities and 73% other anomalies such as height low.14-21 In this study, two sporadic and two familial cases with CCD were analyzed. Sporadic cases represent around 40%–60% of CCD patients.14,15 Particularly, it has called our attention to describe the proband of family 1, because it that presents different phenotypic characteristics than those commonly described. It is a male of 12-years-old heterozygous for c.651_652delTA in exon 4 (Figure 1A). He exhibited left cataract with detachment of the retina, low hair implantation, bushy eyebrows and eyelashes, strabismus, eyelids turned down, and a wide mouth (Table 1). These clinical characteristics had not been previously described in patients with CCD, which makes us think that it could be a more severe case and this could represent an expansion of expression of RUNX2-clinical phenotype-genotype association, in Cleidocranial dysplasia.
RUNX2 gene mutations can be identified in 71% of CCD cases being 60% of them point mutations.22 Around 50 missense mutations have been identified in the Runt domain. Similarly, the mutations observed in our patients were found inside Runt domain of the RUNX2 protein, three in exon 4 and one in exon 3 (p.Ala180Gln) of the RUNX2 gene. The recurrent point mutation p.Arg225Gln, present in our proband 3, was previously found to abolish the function of the NLS with accumulation of RUNX2 protein in the cytoplasm,14,16 while the p.Arg225Trp and the p.Arg186Thr mutations (located close to the p.Ala180Gln site of our case 2), previously reported, diminish the DNA-binding ability.23 In other hand, previous reports indicate that the p.Arg186Thr and p.Leu113Arg mutations alter the transactivation activity of osteocalcin, collagen I, bone sialoprotein and osteopontin genes,20 whereas the p.Leu62 of mouse RUNX1 homologous to p.Leu113 of human RUNX2 protein is involved in altered stabilization producing a defective DNA binding and heterodimerization with core binding factor β (CBFβ).23 Our proband 4 presented the p.Val221Glu change located on the same site of a previous patient that presented a p.Val221Gly mutation, a familial case with moderate CCD,24 our patient showed a severe phenotype while her brother and her mother were less severely affected. We consider that in the p.Ala180Gln mutation occurred an alteration in the DNA-binding due to its proximity to p.Arg186Thr; while in p.Val221Glu, due to its proximity to p.Arg225Gln and p.Arg225Trp, the alteration was due to accumulation of RUNX2 protein in the cytoplasm or DNA binding.14,16,23
The proband 1 with the c.651_652delTA (p.Lys218Serfs*17) mutation had family history and a severe phenotype. His mother and his sister had a less severe phenotype. The c.651_652delTA mutation was located near a reported small deletion CCTAdel635_638 in exon 3 of RUNX2, which led to a stop codon at 220 amino acid. This frameshift mutation caused the deletion of the NLS, affecting the accumulation of the RUNX2 protein in the nucleus.21 A 2-years-old boy with classical CCD showed a heterozygous deletion (c.593_601delCCTTGACCA, p.Thr198_Thr200del) in the Runt domain; the 3D modeling assessment demonstrated that this mutation abolished heterodimerization of the RUNX2 protein with its partner subunit, the polyomavirus enhancer-binding protein 2b (PEBP2b). The transactivation study showed that the p.Thr198_Thr200del and p.Leu199Phe mutants had significantly lower transcription activities (44% and 63% of the wild type, respectively).19 In our proband 1 the mutation led to a stop codon in 235 at the end of the NLS region, modifying the Runt and the NLS sequences and losing the PST region and the NMTS domain. This indicates that the effect produced by these mutations is of the negative dominance type. Other studies on the p.Gln67X (Q/A domain) and p.Gly462X (C-terminal PST region) mutations showed that the subcellular distribution and the transactivation activity on its downstream target genes were affected, thus lowering the protein stability, affecting the expression of bone marker genes, and the osteoblast differentiation in CCD patients.20,21 The mutation p.Lys218Sfs*17 of our proband 1 similarly would affect its nuclear distribution, the binding to DNA, or the transactivation of target genes.
No genotype-phenotype correlation is present in CCD due to the large variable clinical expression. The father with the p.Thr200Ala change showed only dental abnormalities, whereas his two children had the classic CCD. Analysis using an electrophoretic mobility shift assay revealed that p.Thr200Ala retained almost normal transcription activity with no affection of the binding to DNA or transactivation activity, possibly the mutation produced a neomorphic effect altering different functions of the Runt domain.14 A previous study exhibited a group of patients with an unaffected Runt domain that presented features of CCD and a milder short stature compared to an affected Runt domain group with partial CCD phenotype but significantly short stature.15 In contrast, other report found that short stature was more evident in the unaffected Runt domain, which was predicted to interfere with the transactivation activity of its target genes.17 In our study, two cases had short stature and other two had normal stature, all involving the Runt domain. Previous data suggest that the variations in the accumulation of the protein in the cytoplasm, the DNA binding, the transactivation activity of RUNX2 on its target genes, or the heterodimerization with CBFβ lead to impaired transcription activities and variable degrees of haploinsufficiency or protein activity,17 playing an important role in the variable clinical expression of CCD.14
In silico analysis predicted that all mutations affect the RUNX2 protein function. In addition, the force field energy of the mutant RUNX2 proteins showed significant deviations of the energy per amino acid change compared to the RUNX2 wildtype. Molecular dynamics analysis with I-mutants showed negative values; the changes with high and low energies implied a damaging effect on the protein stability and function. So, the bioinformatic analysis is in agreement with previous functional studies where changes in amino acids and the truncation mutation were involved in impaired protein stabilization and subsequent defect in DNA-binding ability, heterodimerization with CBFβ, or impaired transactivation activity.21,23 Finally, the generation of 3D structures of the wildtype and the mutant proteins revealed significant secondary structure changes with gain or loss of hydrogen bonds, the α-helix regions, and the β-leaves. These changes showed a folded protein suggesting to modify the protein-protein interaction as previously observed.19
In conclusion, the three novel mutations identified in Mexican cases with CCD enriched the pool of RUNX2 gene mutations. This study supports the published data suggesting that the stability alteration and the conformational change of RUNX2 protein play a significant role in the interaction with other proteins, affecting various functions, its transcription, thus, the variable expression of CCD.24 Besides, additional clinical features were observed in one case, such as cataract with detachment of the retina, low hair implantation, strabismus, eyelids turned down, and a wide mouth, this could represent an expansion of expression clinical phenotype-genotype in Cleidocranial dysplasia.
Contributions
JTL: LMGH: and SCC conceived and designed the experiments. JTL, LMGH, SGM, MRRV and EHZ collected blood samples and clinical data. LMGH and SGM performed the experiments. JTL, LMGH, SGM, MRRV and EHZ wrote the paper. All authors read and approved the final manuscript.
Funding
This study had no specific funding.
Data availability statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest Statement
We declare that we have no conflicts of interest.
Availability of data and materials
All relevant data used in this study have been included in the manuscript. The corresponding author can be contacted if any further information is needed.
Ethics declarations
All participants received oral and written information about the study and signed a letter of consent.
Consent for publication
All authors consent to publish.
Abbreviations
| CCD: | Cleidocranial dysplasia. |
| RUNX2: | Runt-related transcription factor 2 gene. |
| Q/A: | Polyglutamine/alanine domains. |
| RHD: | Runt homologous domain |
| NLS: | Nuclear localization signal. |
| PST-rich region: | Region, proline/serine/threonine |
| NMTS: | Nuclear matrix-targeting sequence domain. |
| PEBP2b: | Polyomavirus enhancer-binding protein 2b. |
| CBFβ: | Core Binding Factor β. |
References
- Farrow E, Nicot R, Wiss A, Laborde A, Ferri J. Cleidocranial Dysplasia: A Review of Clinical, Radiological, Genetic Implications and a Guidelines Proposal. J Craniofac Surg 2018, 29, 382–9. [CrossRef]
- Mundlos S. Cleidocranial dysplasia: clinical and molecular genetics. J Med Genet 1999, 36, 177–82. [CrossRef]
- Jaruga A, Hordyjewska E, Kandzierski G, Tylzanowski P. Cleidocranial dysplasia and RUNX2-clinical phenotype-genotype correlation. Clin Genet 2016, 90, 393–402. [CrossRef]
- Kanno T, Kanno Y, Chen LF, Ogawa E, Kim WY, Ito Y. Intrinsic transcriptional activation-inhibition domains of the polyomavirus enhancer binding protein 2/core binding factor alpha subunit revealed in the presence of the beta subunit. Mol Cell Biol 1998, 18, 2444–54. [CrossRef]
- Liu JC, Lengner CJ, Gaur T, Lou Y, Hussain S, Jones MD, Borodic B, Colby JL, Steinman HA, van Wijnen AJ, Stein JL, Jones SN, Stein GS, Lian JB. Runx2 protein expression utilizes the Runx2 P1 promoter to establish osteoprogenitor cell number for normal bone formation. J Biol Chem 2011, 286, 30057–70. [CrossRef]
- Choi KY, Lee SW, Park MH, Bae YC, Shin HI, Nam S, Kim YJ, Kim HJ, Ryoo HM. Spatio-temporal expression patterns of Runx2 isoforms in early skeletogenesis. Exp Mol Med 2002, 34, 426–33. [CrossRef] [PubMed]
- Stein GS, Lian JB, van Wijnen AJ, Stein JL, Montecino M, Javed A, Zaidi SK, Young DW, Choi JY, Pockwinse SM. Runx2 control of organization, assembly and activity of the regulatory machinery for skeletal gene expression. Oncogene 2004, 23, 4315–29. [CrossRef] [PubMed]
- Komori T. Whole Aspect of Runx2 Functions in Skeletal Development. Int J Mol Sci 2022, 23, 5776. [CrossRef] [PubMed]
- Kalayci Yigin A, Duz MB, Seven M. Rare Findings in Cleidocranial Dysplasia Caused by RUNX Mutation. Glob Med Genet 2021, 9, 23–8. [CrossRef]
- HGMD® Professional 2020.4 [Internet] - The Human Gene Mutation Database, QIAGEN Digital Insights. Redwood, US: QIAGEN; 2020. Available from: https://portal.biobaseinternational.com/hgmd/pro/start.php. [access in 2021 May 18].
- Stenson PD, Mort M, Ball EV, Chapman M, Evans K, Azevedo L, Hayden M, Heywood S, Millar DS, Phillips AD, Cooper DN. The Human Gene Mutation Database (HGMD®): optimizing its use in a clinical diagnostic or research setting. Hum Genet 2020, 139, 1197–207. [CrossRef]
- Källberg M, Margaryan G, Wang S, Ma J, Xu J. RaptorX server: a resource for template-based protein structure modeling. Methods Mol Biol 2014, 1137, 17–27. [CrossRef]
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [CrossRef] [PubMed]
- Zhou G, Chen Y, Zhou L, Thirunavukkarasu K, Hecht J, Chitayat D, Gelb BD, Pirinen S, Berry SA, Greenberg CR, Karsenty G, Lee B. CBFA1 mutation analysis and functional correlation with phenotypic variability in cleidocranial dysplasia. Hum Mol Genet 1999, 8, 2311–6. [CrossRef] [PubMed]
- Yoshida T, Yoshida T, Kanegane H, Osato M, Yanagida M, Miyawaki T, Ito Y, Shigesada K. Functional analysis of RUNX2 mutations in Japanese patients with cleidocranial dysplasia demonstrates novel genotype-phenotype correlations. Am J Hum Genet 2002, 71, 724–38. [CrossRef] [PubMed]
- Quack I, Vonderstrass B, Stock M, Aylsworth AS, Becker A, Brueton L, Lee PJ, Majewski F, Mulliken JB, Suri M, Zenker M, Mundlos S, Otto F. Mutation analysis of core binding factor A1 in patients with cleidocranial dysplasia. Am J Hum Genet 1999, 65, 1268–78. [CrossRef]
- Wang GX, Sun RP, Song FL. A novel RUNX2 mutation (T420I) in Chinese patients with cleidocranial dysplasia. Genet Mol Res 2010, 9, 41–7. [CrossRef] [PubMed]
- Chen T, Hou J, Hu LL, Gao J, Wu BL. A novel small deletion mutation in RUNX2 gene in one Chinese family with cleidocranial dysplasia. Int J Clin Exp Pathol 2014, 7, 2490–5.
- Matsushita M, Kitoh H, Kaneko H, Mishima K, Itoh Y, Tokita Y, Ishiguro N. A novel in-frame deletion of the RUNX2 gene causes a classic form of cleidocranial dysplasia. J Bone Miner Metab 2014, 32, 96–9. [CrossRef]
- Zhang X, Liu Y, Wang X, Sun X, Zhang C, Zheng S. Analysis of novel RUNX2 mutations in Chinese patients with cleidocranial dysplasia. PLoS One 2017, 12, e0181653. [CrossRef]
- Jung YJ, Bae HS, Ryoo HM, Baek SH. A novel RUNX2 mutation in exon 8, G462X, in a patient with Cleidocranial Dysplasia. J Cell Biochem 2018, 119, 1152–62. [CrossRef] [PubMed]
- Ott CE, Leschik G, Trotier F, Brueton L, Brunner HG, Brussel W, Guillen-Navarro E, Haase C, Kohlhase J, Kotzot D, Lane A, Lee-Kirsch MA, Morlot S, Simon ME, Steichen-Gersdorf E, Tegay DH, Peters H, Mundlos S, Klopocki E. Deletions of the RUNX2 gene are present in about 10% of individuals with cleidocranial dysplasia. Hum Mutat 2010, 31, E1587–93. [CrossRef]
- Nagata T, Werner MH. Functional mutagenesis of AML1/RUNX1 and PEBP2 beta/CBF beta define distinct, non-overlapping sites for DNA recognition and heterodimerization by the Runt domain. J Mol Biol 2001, 308, 191–203. [CrossRef] [PubMed]
- Tessa A, Salvi S, Casali C, Garavelli L, Digilio MC, Dotti MT, Di Giandomenico S, Valoppi M, Grieco GS, Comanducci G, Bianchini G, Fortini D, Federico A, Giannotti A, Santorelli FM. Six novel mutations of the RUNX2 gene in Italian patients with cleidocranial dysplasia. Hum Mutat 2003, 22, 104. [CrossRef]
Figure 1.
(A-D) Pedigree of the families 1 to 4 with the partial electropherogram of the healthy control and of the c.651_652delTA (p.Lys218Sfs*17), c.538_539delinsCA (p.Ala180Gln), c.674G>A (p.Arg225Gln) and c.662T>A (p.Val221Glu) mutations, respectively.
Figure 1.
(A-D) Pedigree of the families 1 to 4 with the partial electropherogram of the healthy control and of the c.651_652delTA (p.Lys218Sfs*17), c.538_539delinsCA (p.Ala180Gln), c.674G>A (p.Arg225Gln) and c.662T>A (p.Val221Glu) mutations, respectively.
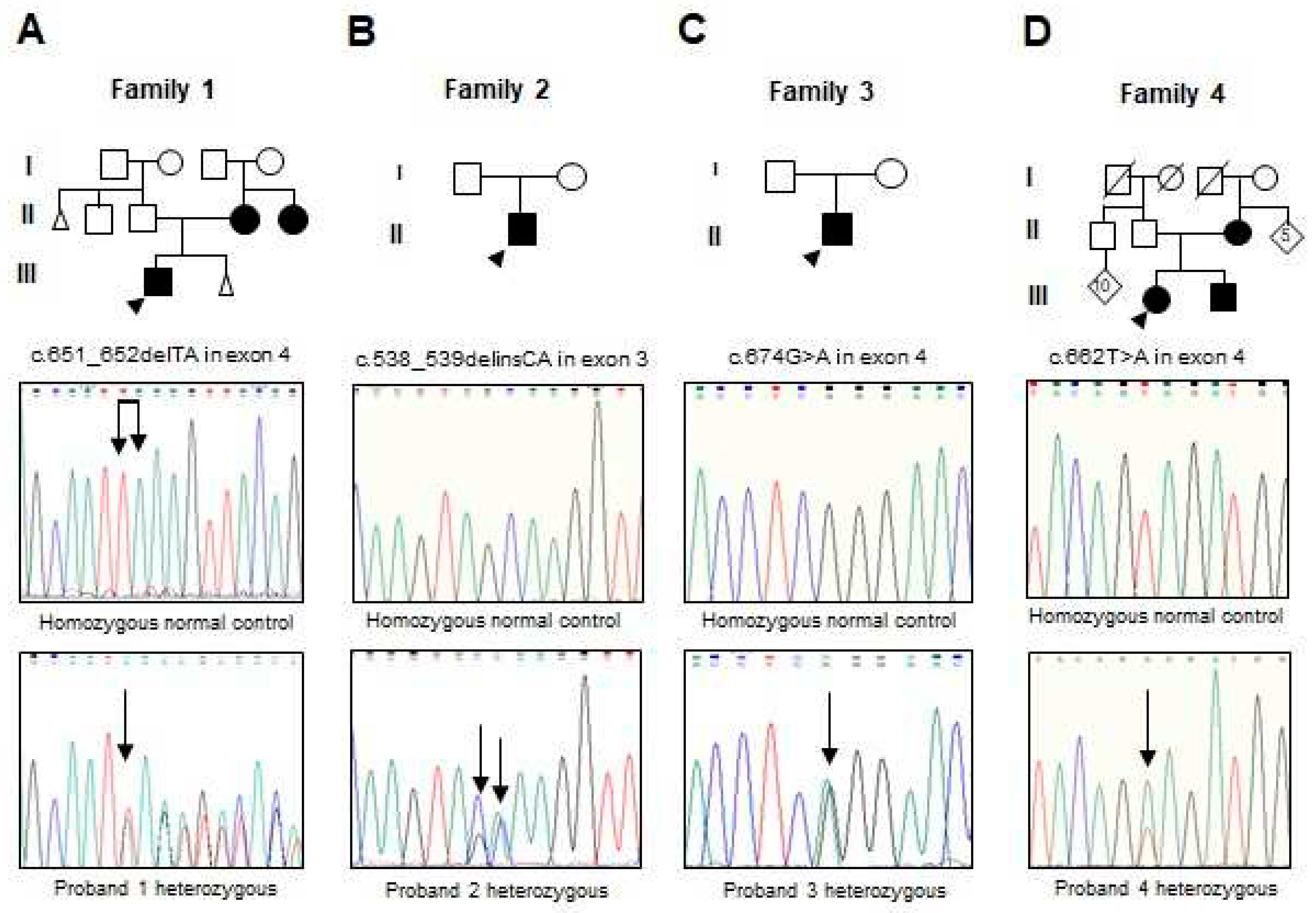
Figure 2.
Protein-structure prediction of p.Lys218Sfs*17 and p.Ala180Gln-mutated human RUNX2. The α-helix (red), the β-leaf (green), coils, and loops (blue) are shown. (A) The wild-type (Wt) p.Lys218 protein loses the nuclear localization site, proline-serine-threonine-rich domain, nuclear matrix targeting signal, and the conserved repression motif (white) in the p.Lys218Sfs*17 mutated (Mut) protein. (B) Wild-type Ala180 protein adopts a folded conformation in the (C) p.Gln180 mutated protein. The wildtype p.Ala180 (blue) without bridges of hydrogens, in the mutated p.Gln180 (blue) gain a bridge of hydrogen bonds with p.Gln178 (yellow).
Figure 2.
Protein-structure prediction of p.Lys218Sfs*17 and p.Ala180Gln-mutated human RUNX2. The α-helix (red), the β-leaf (green), coils, and loops (blue) are shown. (A) The wild-type (Wt) p.Lys218 protein loses the nuclear localization site, proline-serine-threonine-rich domain, nuclear matrix targeting signal, and the conserved repression motif (white) in the p.Lys218Sfs*17 mutated (Mut) protein. (B) Wild-type Ala180 protein adopts a folded conformation in the (C) p.Gln180 mutated protein. The wildtype p.Ala180 (blue) without bridges of hydrogens, in the mutated p.Gln180 (blue) gain a bridge of hydrogen bonds with p.Gln178 (yellow).
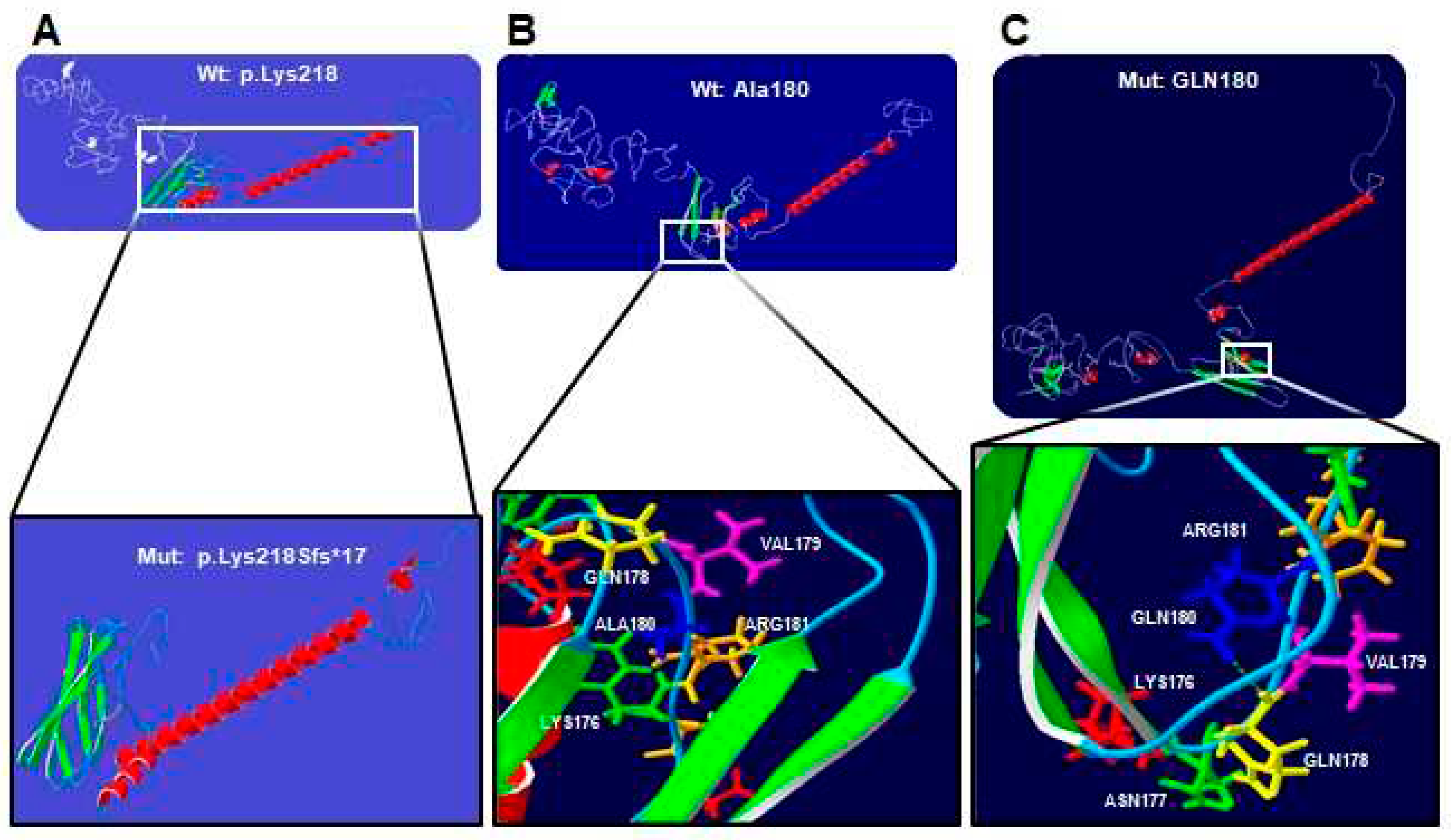
Figure 3.
Protein-structure prediction of p.Val221Glu and p.Arg225Gln-mutated human RUNX2. Wild-type protein acquires a folded conformation in the mutated proteins The α-helix (red), the β-leaf (green), and coils/loops (blue) are shown. (A) The p.Val221 (red) has two bridges of hydrogens bond with p.Phe197 (yellow), while (B) p.Glu221 (red) loses the two hydrogen bonds with p.Phe197, but acquires a covalent bonds between them; additionally, p.Phe197 gains two hydrogens bonds with p.Val219 (blue). (C) The p.Arg225 (blue) has two bridges of hydrogens bonds with p.Gln226 (pink), while (D) p.Gln225 (blue) loses its two hydrogen bonds.
Figure 3.
Protein-structure prediction of p.Val221Glu and p.Arg225Gln-mutated human RUNX2. Wild-type protein acquires a folded conformation in the mutated proteins The α-helix (red), the β-leaf (green), and coils/loops (blue) are shown. (A) The p.Val221 (red) has two bridges of hydrogens bond with p.Phe197 (yellow), while (B) p.Glu221 (red) loses the two hydrogen bonds with p.Phe197, but acquires a covalent bonds between them; additionally, p.Phe197 gains two hydrogens bonds with p.Val219 (blue). (C) The p.Arg225 (blue) has two bridges of hydrogens bonds with p.Gln226 (pink), while (D) p.Gln225 (blue) loses its two hydrogen bonds.
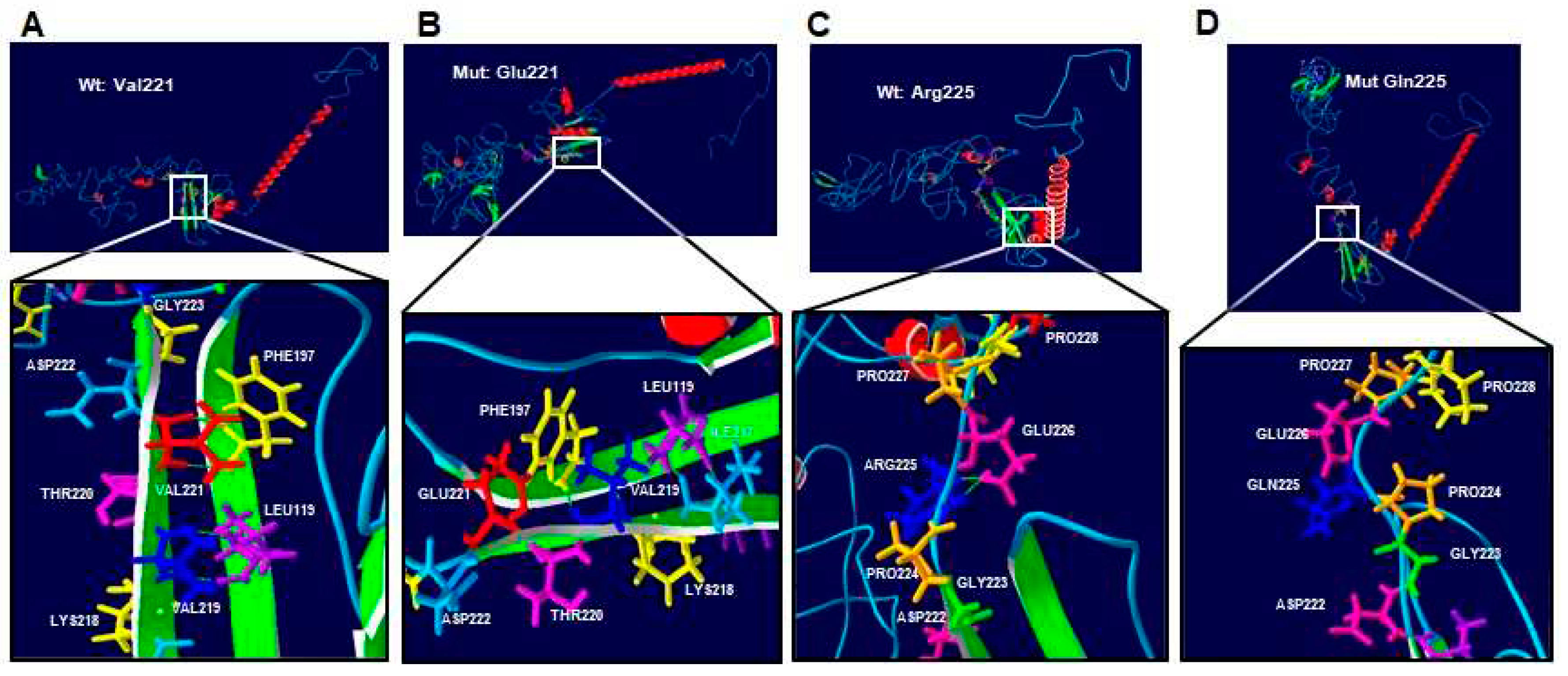

Table 1.
Relevant clinical and RUNX2 gene mutations data of the four proband with cleidocranial dysplasia.
Table 1.
Relevant clinical and RUNX2 gene mutations data of the four proband with cleidocranial dysplasia.
| Features | Case 1 | Case 2 | Case 3 | Case 4 |
|---|---|---|---|---|
| Age (y) | 12 | 1 | 7 | 29 |
| Sex | M | M | M | F |
|
Cranial abnormalities |
Low hair implantation, wormian bones, mild open fontanelle/ sutures. |
Prominence frontal and parietal, broad fontanelle and sutures. | Frontal prominent, broad sutures and fontanelles. | Brachycephaly, broad forehead with medium cleft, cranial bone thickening, wormian bones, headache. |
|
Facial abnormalities |
Populated eyelashes/ eyebrows, strabismus, hypertelorism, eyelids turned down, anteverted nostrils, long philtrum, wide mouth. | Hypertelorism. | Midface hypoplasia, hypertelorism, wide/ low nasal bridge, bulbous nasal tip, long philtrum. | Midface asymmetry and hypoplasia, nasal deviation, hypertelorism. |
|
Dental abnormalities |
Oligodontia, supernumerary teeth. | (-) | Yellow and misaligned teeth. | Molars absent, supernumerary teeth. |
|
Clavicle abnormalities |
H | A | A | H |
|
Thorax abnormalities |
(-) | (-) | Pectum excavatum, thoraco-lumbar scoliosis. | Short and wide chest, thoracic scoliosis. |
|
Limbs abnormalities |
Pectum excavatum, thoraco-lumbar scoliosis. | (-) | (-) | Brachydactyly 4th phalanx left foot. Cubitus valgus, genu varum. |
|
Others abnormalities |
Cataract, detachment of retina. | (-) | Short stature, short neck. | Short stature, short neck with limited rotation. |
|
Phenotype expression |
Severe. | Moderate. | Severe. | Severe. |
| Family history | (+) | (-) | (-) | (+) |
| DNA change | c.651_652delTA; fs c.653_704 |
c.538,539delinsCA | c.674G>A | c.662T>A |
| Exon location | 4 | 3 | 4 | 4 |
| Protein change | p.Lys218Sfs*17 | p.Ala180Gln | p.Arg225Gln | p.Val221Glu |
|
Domain location |
Runt/ NLS. | Runt | Runt | Runt |
| Reference | Novel. | Novel. | [14,16] | Novel. |
Y: years, M: male, F: female, H: hypoplasia, A: Agenesis, (-): negative phenotype, (+): positive phenotype.
Table 2.
Primers to amplify RUNX2 gene.
| Exon number |
Primers | Size | Tm |
|---|---|---|---|
| 2 | F 5´-GGCCACTTCGCTAACTTGTG-3´ R 5´-GTAGCCTCTTACCTTGAAGG-3´ |
422pb |
56°C |
| 3 | F 5´-GGACTAGAACACTAAGTCCTG-3´ R 5´- CACTCAACTTCATCTGGATG-3´ |
419pb |
60°C |
| 4 | F 5´- CATTGCCTCCTTAGAGATGC-3´ R 5´-ATTCCTCATAGGGTCTCTGG-3´ |
280pb |
60°C |
| 5 | F 5´-GCAT GGTCAATTGTTCAGCT-3´ R 5´-CTGCCAGCGTCTATGCAAG-3´ |
273pb |
60°C |
| 6 | F 5´-GGCTGCAATGGTTGCTATAC-3´ R 5´-TGTGAGCATGGATGAGACAG-3´ |
289pb |
60°C |
| 7 | F 5´-CATAGAACATTAGAGCTGGAAGG-3´ R 5´-CTCACAAAATCGGACAGTAAC-3´ |
199pb |
60°C |
| 8 | F 5´-GGTGCATTTGAAGGTC TGTC-3´ R 5´-ATTGATA CGTGTGGGATGTGG-3´ |
677pb |
60°C |
Table 3.
Results of in silico analysis of wildtype and mutated RUNX2 proteins.
| Protein/ Amino acid ID |
PolyPhen2/ Mutation Taster/ PROVEAN/ SIFT (score) |
Log of force field energy (KJ/mol) | I-Mutant 2.0 | Secondary structure changes | |||
| Structural molding | # of α-helix | # of β-leaf | # of hydrogen bonds | ||||
| RUNX2 wildtype | - | 14.48 | - | - | 6 | 7 | NA |
| RUNX2 p.Gln180 | NA | 18.45 | NA | Folded | 5 | 11 | NA |
| RUNX2 p.Glu221 | NA | 15.00 | NA | Folded | 4 | 11 | NA |
| RUNX2 p.Gln225 | NA | 15.40 | NA | Folded | 7 | 9 | NA |
| p.Lys218Sfs*17 | 1.000/Disease causing (1.0)/ NA/ NA | 12.51 | NA | Truncated | 3 | 8 | NA |
| Ala180 | - | 5.53 | - | NA | NA | NA | 0 |
| Gln180 | 1.000/Disease causing (0.99)/ deleterious (-4.66)/ damaging (0.00) |
6.24 | -1.72 | NA | NA | NA | 1 |
| Val221 | - | 2.59 | - | NA | NA | NA | 2 |
| Glu221 | 1.000/Disease causing (0.99)/ deleterious (-5.85)/ damaging (0.00) |
7.99 | -1.69 | NA | NA | NA | 0, Gain of a covalent bond |
| Arg225 | - | 7.99 | - | NA | NA | NA | 2 |
| Gln225 | 0.980/ Disease causing (0.99)/ deleterious (-3.8)/ damaging (0.03) | 6.46 | -1.03 | NA | NA | NA | 0 |
NA: Not aplicable.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



