Submitted:
30 January 2024
Posted:
31 January 2024
You are already at the latest version
Abstract
Epidemiological and clinical evidence have extensively documented the role of obesity in the develop-ment of endometrial cancer. However, the effect of fatty acids on cell growth in endometrial cancer has not been widely studied. Here we reported that palmitic acid significantly inhibited cell proliferation of endometrial cancer cells and primary cultures of endometrial cancer, and reduced tumor growth in a transgenic mouse model of endometrial cancer, in parallel with increased cellular stress and apoptosis and decreased cellular adhesion and invasion. Inhibition of cellular stress by N-acetyl-L-cysteine effec-tively reversed the effects of palmitic acid on cell proliferation, apoptosis and invasive capacity in the endometrial cancer cells. Palmitic acid increased the intracellular formation of lipid droplets in a time- and dose-dependent manner. Depletion of lipid droplets by blocking DGAT1 and DGAT2 effectively increased the ability of palmitic acid to inhibit cell proliferation and induce cleaved caspase 3 activity. Collectively, this study provides new insight into the effect of palmitic acid on cell proliferation, inva-sion and the formation of lipid droplets that may have potential clinical relevance in the treatment of obesity-driven endometrial cancer
Keywords:
palmitic acid
; endometrial cancer
; lipid droplet
; apoptosis
; invasion
; cellular stress
1. Introduction
Endometrial cancer (EC) is the fourth most common cancer in women and the most common gynecologic malignancy in the United States, with 66,200 new cases and 13,030 disease-related deaths projected in 2023 (1). Although usually detected at earlier and more favorable stages with a better prognosis, EC diagnosed at an advanced stage is prone to recurrence and lacks optimal adjuvant therapies and strategies (2). Obesity is a well-established risk factor for the development of EC and may pose a threat to treatment efficacy and prognosis (3, 4). Increased adipose tissue, particularly visceral adipose tissue, is an endocrine organ that regulates metabolism and inflammatory responses and produces bioactive substances associated with cell growth, differentiation and angiogenesis, all of which have been implicated in EC carcinogenesis (5). Recent studies have found that ECs display specific changes in different aspects of lipid metabolism, including fatty acid metabolism, under obese and lean conditions, which may be clinically relevant to EC progression and response to chemotherapy (6-9).
Cancer cells create a metabolic environment that is conducive to cancer cell growth by altering the metabolism of lipids, carbohydrates, and proteins, allowing cancer cells to effectively maintain the functionality of structures and functions in the environment (10). Fatty acids (FAs), as chemically heterogeneous compounds, are intimately involved in synthesis and metabolism of lipids, are required for energy storage, membrane structure, and signaling precursor molecules, and exhibit diverse functions in carcinogenesis, progression and chemotherapy in cancer (11). Recent studies have found that the production of monounsaturated fatty acids (MUFA) from saturated fatty acids (SFA) is an important indicator of regulating the fluidity, functionality and flexibility of biological membranes, and deregulation of the ratio of MUFA to SFA is essential for the growth of tumor cells (12-14). The pronounced imbalance of FAs in tumor cell membranes, particularly in the composition of phospholipids in biological membranes, has direct implications for determining cell proliferation, invasiveness and signaling cascades that control the proliferation of tumor cells. The fatty acid composition of membrane phospholipids is continuously remodeled by the availability of free fatty acids, the enzymatic activity of phospholipases, stress conditions, and metabolic diseases, suggesting that changes in the environmental conditions of tumor growth in vivo or by altering the proportion of ingested FAs can affect the proliferative activity of cancer cells (15-17). The high rate of fatty acid synthesis in cancer cells is initiated by the enzymatic system of fatty acid synthase (FAS) to form abundant 16-carbon palmitic acid (PA), the most widely recognized saturated long-chain fatty acid found in the human body, which represents 65% of SFA in the body, 28-32% of total serum FA as well as 10-20% of human dietary fat intake (18, 19). Biochemical pathways for MUFAs synthesis rely primarily on desaturation or elongation of PA by the action of so-called elongases and desaturases in the body in addition to dietary supplementation. Several MUFAs such as sapienic and palmitoleic acids significantly influence membrane composition and properties and change expression of growth factors and signaling cascades in breast cancer cells (13, 20). Accumulating evidence indicates that desaturases, including stearoyl-CoA desaturases-1 (SCD1), have emerged as key players in the regulation of metabolic and signaling pathways that support the biochemical and biological phenotype of cancer cells, and targeting SCD1 has the potential to develop new cancer therapeutics (21, 22). In addition to palmitoylation of PA involved in the functional regulation of multiple genes including oncogenes and tumor suppressor genes, it is generally believed that PA acts as an energy source to promote cell growth through β-oxidation, and participates in the synthesis of cancer cell membranes by functioning as a signaling molecule in cancer cells (18, 23, 24). Increased dietary PA intake or elevated circulating levels of plasma phospholipid PA have been found to be strongly associated with the risk of breast cancer (25, 26) . However, there is growing evidence that PA inhibits cancer cell proliferation, induces cellular stress and apoptosis, causes cell cycle arrest and impairs cell invasiveness, either directly or indirectly in various types of preclinical models (18, 25, 27). Importantly, PA potently inhibited cell viability and induced apoptosis along with a concomitant increase in PA composition in neuroblastoma cell membranes, without affecting the overall polyunsaturated content. The combination of PA with oleic and arachidonic acids resulted in changes in cell membrane lipidome with suppression of caspase activation and maintenance of cell viability, demonstrating an important role for membrane FA reorganization in suppressing cell growth and the potential for dietary FAs, including PA, to intervene in cancer (28, 29).
Given that PA is the most common SFA in the human diet and in the human body, and that obesity and metabolic disorders are the most important risk factors for the carcinogenesis and progression of EC, it is necessary to evaluate the function of PA in EC cell proliferation and tumor growth (4, 18). Understanding the role of PA in cell and tumor growth in EC is critical for the design of novel treatment and prevention strategies using this dietary supplementation. In this study, we aimed at characterizing the effects of PA on cell proliferation and tumor growth in EC cell lines and a transgenic mouse model of EC. Our results show that PA inhibits EC cell proliferation and invasion, increases formation of cellular lipid droplets, and reduced tumor growth under obese and lean conditions.
2. Materials and Methods
2.1. Cell culture and reagents
Two human endometrial cancer cell lines, Ishikawa (RRID: CVCL_2529) and ECC-1 (RRID: CVCL_7260), were used in this study. Both cell lines were obtained and authenticated from the Cell bank in Lineberger Cancer Center, the University of North Carolina at Chapel Hill. During the experiments, we regularly detected mycoplasma contamination in cell cultures every six months by Mycoplasma Detection Assays (InvivoGen, San Diego, CA). Ishikawa cells were maintained in DMEM/F12 medium with 10% fetal bovine serum (FBS), and ECC-1 cells were maintained in RPMI 1640 medium with 10% FBS. All media was supplemented with penicillin (100 U/ml), streptomycin (100 ug/ml) and L-glutamine (2 mM/mL). The cells were cultured in a humidified 5% CO2 incubator at 37°C. PA was obtained from Sigma (St. Louis, MO). T863 and PF-06424439 were from Cayman (Ann Arbor, Michigan). All antibodies were purchased from Cell Signaling Technology (Beverly, MA) and Abclonal Science (Woburn, MA). Enhanced chemiluminescence western blotting detection reagents were obtained from Amersham (Arlington Heights, IL). All other chemicals were purchased from Thermo Fisher Scientific (Waltham, MA).
2.2. Preparation of BSA-bound PA
PA was prepared as described previously (30). Briefly, 0.053 g of PA (Sigma-Aldrich, P0500) powder was dissolved in 210μl NaOH solution (0.2M) by heating at 75℃ in a shaking water bath for 15 minutes until the PA-NaOH solution turned clear. 840μl of 30% fatty-acid-free BSA (Sigma-Aldrich, A9576) was then added, and the combination was vortexed for 10 seconds followed by a further 10-minute incubation at 55℃. The final concentration of PA in the BSA-PA complex was 20mM at a ratio of PA: BSA=5:1. The BSA-bound PA was filtered through a 0.22-μm filter and stored as aliquots in a −20 °C freezer. When cells were treated with PA, control cells were treated with an equal volume of BSA solution.
2.3. Primary Culture of Human-Derived EC
Collection of human EC tissue was approved by the Institutional Review Board of the University of North Carolina at Chapel Hill and performed at UNC Hospital. After informed consent was obtained from all individual patients, seventeen tumor tissues were collected from patients with EC at the time of hysterectomy (Supplemental table). The pathological diagnosis of EC was made by pathologists in UNC-CH. Cube samples of 5 x 5 mm were placed in DMEM/F12 culture medium containing antibiotics and transferred to the laboratory for primary cultures. The tissues were washed with PBS three times and then digested in 0.5% collagenase IA, 0.1% DNase, 100 U/ml penicillin and streptomycin for 30-60 min in a 37°C water bath with shaking. After washing twice with PBS, the tumor cells (2 x 104/per well) were seeded into 96-well plates, and cell proliferation was measured by MTT assay 72 hours after treatment with PA.
2.4. Cell viability assay
Cell viability was assessed using MTT assay. In brief, the Ishikawa and ECC-1 cells were plated at a concentration of 6.0 x 103 cells/well and 4.0 x 103 cells/well, respectively, and cultured for 24 hours. The cells were then treated with various concentrations of PA for 72 hours. MTT (5 mg/ml) was added into each well for 1 hour. Subsequently, the culture medium was discarded, and 100 µL dimethyl sulfoxide (DMSO) was added into each well to terminate the reaction. The optical density (OD) value of each sample was detected at a wavelength of 562nm with a microplate reader (Tecan, Morrisville, NC). The effect of PA on cell proliferation was calculated as a percentage of control. AAT Bioquest calculator was used to determine IC50 values. Each experiment was performed at least three times to assess for consistency of results.
2.5. Colony assays
The Ishikawa and ECC-1 cells were harvested from a stock culture and plated at a density of 400 cells/well in six well plates. After attachment of cells to the wells (approximately 2 to 3 hours after incubation), the cells were treated with various concentrations of PA. Thirty-six hours after PA treatment, the media was replaced with fresh media, and the medium was changed every 3 days. After culturing for 10-12 days, each well was washed with PBS and stained with a mixture of 6.0% glutaraldehyde and 0.5% crystal violet. The clones were imaged and quantified using Image J software (National Institutes of Health, Bethesda, MD).
2.6. Glucose uptake assay
Glucose uptake was determined by 2-NBDG Glucose Uptake Assay (BioVision, Milpitas, CA). Briefly, the Ishikawa and ECC-1 cells on 96 well plates were treated with different concentrations of PA for 16-18 hours. The cells were washed with HBSS and incubated with glucose free medium with 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxy-D-glucose (2-NBDG, 100 ug/ml) for 15 minutes. After washing the plates with Hanks' Balanced Salt Solution (HBSS) twice, the fluorescence intensity (Ex/Em =465/540) of cellular 2-NBDG in each well was measured using a Tecan plate reader. The experiments were performed in triplicate and repeated three times.
2.7. Lactate assay
The Ishikawa and ECC-1 cells were cultured in six well plates at 2.5 x 105 cells/well overnight and then treated with different concentrations of PA for 24 hours. The media was collected to assess lactate production, using the BioVision lactate assay kit (Milpitas, CA) and following the manufacturer’s instructions. The OD values were determined at 450 nm with a Tecan plate reader. The lactate levels in each well were normalized to the cell number.
2.8. ATP assay
The luminometric ATP assay kit (AAT bioquest, Sunnyvale, CA) was used to investigate the effects of PA on cellular production of ATP in EC cells. The Ishikawa and ECC-1 cells were seeded at 5 × 103/well in 96 well plates overnight, and then treated with different dosages of PA for 24 hours. 100 µl ATP detecting solution was added into each well, mixed and then incubated for 20 minutes at room temperature. The luminescence intensity was immediately measured on a Tecan plate reader. The ATP levels were normalized according to the viable cell counts measured by the MTT assays. The experiments were performed in triplicate and repeated three times.
2.9. Cleaved caspase 3, 8 and 9 ELISA assays
The Ishikawa and ECC-1 cells were plated in 6-well plates at the concentration of 2.5 x 105 cells/well and 3.5 x 105 cells/well, respectively. After 24 hours, cells were exposed to different concentrations of PA for 14 hours. Protein concentrations were determined via BCA assay (Thermo Fisher). 150-180 µl of 1X caspase lysis buffer was added into each well, and reaction buffer with caspase 3, 8 and 9 substrates were added to the lysis buffer in a new black 96-well plate at 37°C for 20 minutes. The fluorescence intensity for cleaved caspase 3 (Ex/Em=400/505), cleaved caspase 8 (Ex/Em=376/482), cleaved caspase 9 (Ex/Em=341/441) were recorded using a Tecan microplate reader. These assays were repeated three times to assess for consistency of results.
2.10. Reactive oxygen species (ROS) assay
The Fluorometric Intracellular Total ROS Activity Assay Kit (AAT Bioquest, Sunnyvale, CA) was used to detect alterations in the production of ROS. The Ishikawa and ECC-1 cells (6.0 × 103 cells/well) were seeded into black 96-well plates. After 24 hours, the cells were treated with PA and allowed to incubate for 12 hours at 37°C to induce ROS generation. DCFH-DA (15 µM) was then applied to the cells and allowed to incubate for 30 minutes. The fluorescence intensity was measured at Ex/Em 485/530nm using a Tecan microplate reader. All experiments were performed at least three times to assess for consistency of response.
2.11. Mitochondrial membrane potential assays
Mitochondrial membrane potential was analyzed with specific fluorescent probes for JC-1 and TMRE (AAT Bioquest, Sunnyvale, CA). The Ishikawa and ECC-1 cells were plated in 96 well plates overnight and treated with different concentrations of PA for 14 hours. The cells were then treated with 2 µM JC-1 or 800 µM TMRE for 30 minutes at 37°C. The microplate reader measured fluorescence intensity at Ex/ Em=480/590 nm for the JC-1 assay and Ex/Em=549/575 nm for the TMRE assay, respectively. Each experiment was repeated three times to assess for consistency of results.
2.12. Adhesion assay
Each well in a 96-well plate was coated with 100 µl laminin-1 (10 ug/ml) and incubated at 37°C for 1 hour. The fluid was then aspirated and 200μl of blocking buffer was added to each well for 45 to 60 minutes at 37°C and then washed with PBS while chilling on ice. To each well, 1.2 x 104 cells were added with varying concentrations of PA. The plate was then allowed to incubate at 37°C for 1.5-2 hours. After this period, the medium was aspirated, and cells were fixed by adding 100 µl of 5% glutaraldehyde and incubating for 30 minutes at room temperature. Adhered cells were then washed with PBS and stained with 100 µl of 0.1% crystal violet for 30 minutes. The cells were then washed repeatedly with water, and 100 µl of 10% acetic acid was added to each well to solubilize the dye. The plate was shaken for 5 minutes, and absorbance was measured at 562 nm using a Tecan microplate reader. Each experiment was repeated three times for consistency of response.
2.13. Transwell assay
Cell invasiveness was quantified by a modified Boyden chamber assay (Corning Inc. Corning, NY). The Ishikawa and ECC-1 cells were starved for 12-14 hours, and then seeded on top of a Matrigel invasion chamber (8-mm pore size) with a density of 3 x 104. Different concentrations of PA were added to the top chambers. The lower chambers were filled with regular medium. The plates were cultured for 4-6 hours to allow cell invasion into the lower chambers. After removing the top chambers and washing the lower chambers with PBS, 100 µl of calcein AM solution (Invitrogen, Carlsbad, CA) was applied to the lower chambers and incubated for 30-60 minutes at 37°C. The fluorescence intensity of the lower chambers was measured by a Tecan reader at Ex/Em 485/520 nm. Each experiment was repeated three times.
2.14. Wound healing assay
The Ishikawa and ECC-1 cells were plated at 4.0 × 105 cells/well in six well plates and cultured overnight or until the percentage of cells was greater than 80%. A uniform wound was created by a 20 μl pipette. The cells were washed and then treated with different concentrations of PA for 48 hours. Photos were taken at 24 and 48 hours after scratching, and the width of wound was measured and analyzed with ImageJ software (National Institutes of Health, Bethesda, MD). Percent closure of the scratch was measured by comparing to control cells.
2.15. Fatty Acid Oxidation (FAO) Assay
FAO activity was detected by FAO enzyme assay (Biomedical Research Service, State University of New York at Buffalo). The Ishikawa and ECC-1 cells were plated in 6 well plates with a density of 2.5 × 105 cells/well and cultured overnight in 37℃, and then were treated with different concentrations of PA for 24 hours. Harvested cells with ice-cold 1X Sample Buffer and collected the supernatant after centrifugation. After equalizing the concentrations of protein, 20ul of each sample was treated with 50 µl of FAO Assay Solution and 50 µl control solution. The mixed solutions were added 96-well plate. The plate was kept in a non-CO2 incubator at 37°C for 60 minutes. The optical density (OD) value was measured by a Tecan reader at a wavelength of 492 nm. Blank reading was subtracted from the sample reading. Each experiment was repeated three times.
2.16. Oil Red O Staining assay
The Ishikawa and ECC-1 cells were planted in 12-well plates with the density of 1.2 × 105 cells/well and grown for 24 hours. Both cell lines were treated with varying concentrations of PA for 24 hours and then the cells were stabilized in fresh formalin for 1 hour. Oil red O (Sigma Aldrich, St. Louis, MO) and Harris Modified Hematopxylin were added to well for staining. Photos were taken a by Thermo Scientific Invitrogen EVOS Microscope. Each experiment was repeated three times for consistency of response.
2.17. Western immunoblotting
Following treatment of Ishikawa and ECC-1 cells with different concentrations of PA for 6 to 36 hours, total proteins were extracted from both cell lines using RIPA buffer (Thermo Fisher). A BCA protein assay kit (Thermo Fisher) was used to determine the protein concentration. Equal amounts of protein were separated by gel electrophoresis and transferred onto a PVDF membrane. The membranes were blocked with 5% non-fat dry milk and then incubated with a 1:1000 to 3000 dilutions of primary antibody overnight at 4°C. The appropriate secondary antibody was incubated with the membrane for 1 hour at room temperature after washing with TBS-T. Immunoblots were developed using an enhanced chemiluminescence detection buffer, and bands were visualized with the Bio-Rad Imaging System (Hercules, CA). After development, the membranes were stripped or washed, and re-probed using α-tubulin or β-actin antibodies. Each experiment was repeated at least twice to assess the consistency of the results.
2.18. Lkb1fl/flp53fl/fl transgenic mouse model of EC
In order to investigate the effect of PA on tumor growth in vivo, we utilized the Lkb1fl/flp53fl/fl mouse model of EC (31). All mice were handled according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) of the University of North Carolina at Chapel Hill (protocol # 21-209). The mice were housed at our animal facility with a 12-hour light, 12 hours dark cycle, and allowed to freely access food (50 IF/6F diet, PecoLab) and water. Six to eight-week-old female mice were injected with 5 µl of 2.5 x 1011 P.F.U of recombinant adenovirus Ad5-CMV-Cre (Transfer Vector Core, University of Iowa) in the left uterine horn. The mice were randomly divided into two groups: control group and PA treatment group (18 mice per group). After 8 weeks following injection with Ad5-CMV-Cre, the mice were treated with PA (10 mg/kg, 100 µl per mouse for oral gavage, daily) or the vehicle (100 μl, 2.5: 1 ratio of water and 0.2 N-NaOH, daily) for 4 weeks. Mice were weighed twice a week throughout the treatment. During PA treatment, the mice exhibited normal activity and behavior, and no mice died in either group. All mice were sacrificed by CO2 asphyxiation after 4 weeks of treatment. EC tumors, visceral fat tissues, liver and blood were carefully harvested and recorded. All procedures were approved by the University of North Carolina at Chapel Hill Institutional Animal Care and Use Committee (IACUC No 16-163).
2.19. Immunohistochemistry (IHC)
Endometrial tumor slides (4 µm) from the Lkb1fl/flp53fl/fl mice were first incubated with protein block solution (Dako) for 1 hour and then the primary antibodies were added overnight in a cold room. The slides were then washed and incubated with appropriate secondary antibodies at room temperature for 1 hour. After removing the secondary antibody, the specific staining was visualized using the Signal Stain Boost Immunohistochemical Detection Reagent (Cell Signaling Technology, Danvers, MA), according to the manufacturer’s instructions. Individual slides were scanned using Motic (Feasterville, PA), and digital images were analyzed for target protein expression using ImagePro software (Vista, CA).
2.20. HE Staining
Liver of EC mice were carefully harvested after sacrifice and embedded in paraffin. Stained the liver slides (4 μm) with Hematoxylin to visualize cell nucleus and then Eosin for cytoplasm. Individual slides were scanned using Motic (Feasterville, PA), and digital images were analyzed for target protein expression using ImagePro software (Vista, CA).
2.21. Serum vascular endothelial growth factor (VEGF) and triglyceride (TG) Assays
Serum VEGF and TG levels after treatment with PA were detected using a R&D VEGF ELISA Kit (Minneapolis, MN), and a BioVision Triglyceride Quantification Kit, according to the manufacturer’s directions. Each sample from the PA treatment and control groups was measured in triplicate. The plates were read at 570 nm for VEGF and TG measurements using a Tecan plate reader.
2.22. Statistical analysis
All data are reported as mean ± SD from three independent assays. Both Student’s t-test and One-way ANOVA test were used in this study. GraphPad Prism 8 (La Jolla, CA) statistical software was employed to calculate the comparisons. All tests were two-sided with p < 0.05 considered significant
3. Results
3.1. PA inhibited cell proliferation and tumor growth in EC cells and a transgenic mouse model of EC, respectively
To investigate whether PA affects EC cell proliferation, the Ishikawa and ECC-1 cells were subjected to an MTT assay. As shown in Figure 1A, both cell lines treated with different concentrations of PA showed a significant decrease in cell viability, and IC50 values for Ishikawa and ECC-1 cells were 348.2 µM and 187.3 µM, respectively, after 72 hours of treatment. When both cell lines were treated with a physiologic concentration of 250 µM PA for 72 hours (18), cell proliferation was decreased by 25.7% in Ishikawa cell and 65.1% in ECC-1 cells compared to the BSA control. Consistent with the MTT assays, the colony assays showed that after treatment with 250 µM PA for 36 hours and continued cell culture for 10–12 days, the colony formation of Ishikawa and ECC-1 cells was reduced by approximately 40.6% and 70.8%, respectively, compared to their respective control cells (p< 0.05) (Figure 1B). Since primary cell cultures from patient-derived tumors may be better predictors of anti-tumorigenic activity of cytotoxic agents than cancer cell lines, seventeen total primary cell cultures of EC were cultured with varying concentrations of PA for 72 hours. MTT assay revealed that the primary culture cells exhibited variable response to PA treatment. Nine of the seventeen primary cell cultures displayed at least 10% inhibition of cell proliferation at 250 µM PA (Figure 1C).
To determine the anti-cancer activity of PA on tumor growth, Lkb1fl/flp53fl/fl mice (18 mice/per group) were treated with 10mg/kg PA or vehicle daily through oral gavage for 4 weeks. The body weight of each mouse was measured twice a week. During the treatment period, the mice exhibited normal activity, and no significant changes in body weight (Figure 1D). There were no changes in serum triglycerides (TG) and liver weights in the mice after completion of treatment compared to controls (Figure E). However, H&E staining of liver sections showed a significant increase in ballooned hepatocytes in PA-treated mice, indicating that PA increased accumulation of fat in hepatocytes (Figure 1F). PA treatment significantly reduced tumor volume by 57.8% and tumor weight by 43.2% compared to control mice (p<0.05) (Figure 1G). Finally, IHC staining was applied to analyze the difference in cell proliferation between groups after PA treatment. PA inhibited the expression of Ki-67 by 52.4% compared to the control group (p<0.05) (Figure 1H). These results suggest that PA effectively inhibited EC cell proliferation and tumor growth in vitro and in vivo.
3.2. Effects of PA on apoptosis in EC
To assess the effects of PA on inducing apoptotic activity in EC cells, the Ishikawa and ECC-1 cell lines were treated with different concentrations of PA for 14 hours, and apoptotic activity of PA-treated cells was measured by cleaved caspase 3, caspase 8 and caspase 9 ELISA assays. As shown in Figure 2A–C, 100 µM and 250 µM treatments significantly increased cleaved caspase 3, 8, and 9 activities in both cell lines. Treatment of EC cells with 250 µM PA increased cleaved caspase 3 by 1.92 times in the Ishikawa cells and 2.35 times in the ECC-1 cells(p<0.05). Western blotting results showed that PA reduced the expression of Bcl-xL and Mcl-1 and increased cleaved PARP protein expression in a dose-dependent manner in both cell lines after treatment with PA for 24 hours (Figure 2D). To evaluate the role of mitochondrial apoptotic pathways in PA-induced apoptosis, both cell lines were pre-treated with Z-VAD-FMK (10 µM, a pan caspase inhibitor) for 1.5 hours and then treated with 250 µM PA for 14 hours for the caspase 3 assay and 72 hours for the MTT assay. Pre-treatment with Z-VAD-FMK blocked PA-induced cleaved caspase 3 activity and significantly rescued PA-induced apoptosis compared with the control-treated cells (p<0.05) (Figure 2E,F), indicating that that PA-induced apoptosis depends on the extrinsic and intrinsic mitochondrial apoptotic pathways in EC cells. To evaluate the role of PA in induction of apoptosis in vivo, the expression of Bcl-xL was examined by IHC in the endometrial tumors of Lkb1fl/flp53fl/fl mice. PA significantly reduces the expression of Bcl-xL in endometrial tumors of PA-treated mice compared with control mice (p<0.05) (Figure 2G), suggesting treatment of the mice with PA for 4 weeks was effectively in inducing apoptosis in endometrial tumors of Lkb1fl/flp53fl/fl mice.
3.3. PA induced cellular stress
To investigate the role of cellular stress on PA-induced apoptosis in EC cells, cellular ROS levels were assessed using the DCFH-DA assay. Treatment of both cell lines with 100 µM or 250 µM PA significantly increased ROS production (Figure 3A). PA increased ROS production by 1.43 times in the Ishikawa cells and 1.53 times in the ECC-1 cells, at a dose of 250 µM compared with the control groups(p<0.05). To further confirm whether the PA-induced increase in ROS is related to mitochondrial function, the JC-1 and TMRE ELISA assays were employed to detect changes of mitochondrial membrane potential (ΔΨm). In Ishikawa cells, 100 µM PA appeared to decrease ΔΨm by TMRE but not JC-1 assay; however, 250 µM PA statistically decreased ΔΨm by both JC-1 and TMRE assays in both cell lines (p<0.05). In ECC-1 cells, both 100 µM and 250 µM PA treatments induced the loss of ΔΨm in both assays after 14 hours of treatment compared to control cells(p<0.05) (Figure 3B,C). The effect of PA on the expression of ER stress-related proteins was detected by western blotting analysis. The results indicated that treatment of cells with 100 or 250 µM of PA for 24 hours up-regulated the protein expression of PERK, ATF4, BiP, IRE1-α and CLPP in both cell lines (Figure 3D). IHC staining of EC tissues demonstrated a significant increase in the expression of BCL-xL in PA-treated Lkb1fl/flp53fl/fl mice (p<0.05) (Figure 3E). These results indicate that cellular stress is also responsible, in part, for the anti-proliferative effect of PA in EC cell lines and Lkb1fl/flp53fl/fl mice.
3.4. PA reduced EC cell migration and invasion
To investigate the influence of PA on adhesion and invasion in EC cells, the laminin-1 adhesion assay, the transwell assay and the wound-healing assay were performed in Ishikawa and ECC-1 cells. In Ishikawa cells, PA reduced cell adhesion and invasion at an optimal concentration of 250 µM, while in ECC-1 cells, in addition to inhibiting cell adhesion and invasion at a dose of 250 µM PA, 100 µM PA also significantly decreased cell adhesive and invasive capacity (p<0.05) (Figure 4A,B). Similarly, the results of the wound healing assay showed that 100 µM or 250 µM PA suppressed the migration of Ishikawa and ECC-1 cells. After treatment of cells with 100 and 250 µM PA for 48 hours, the migration of Ishikawa cells was inhibited by 12.4% and 44.4%, respectively, and the migratory potential of ECC-1 cells was inhibited by 31.1% and 51.9%, respectively, compared with control cells (p<0.05) (Figure 4C). To determine whether the EMT process involved PA-mediated invasion and migration, Ishikawa and ECC-1 cells were incubated overnight with different concentrations of PA, and western blot assays were used to determine the expression of EMT and angiogenic markers. The results demonstrated that high concentrations of PA (250 µM) reduced the expression of VEGF, MMP9, Snail, Vimentin and N-Cadherin in both cell lines (Figure 4D). Although PA did not reduce serum VEGF production in Lkb1fl/flp53fl/fl mice (data not shown), IHC found that PA treatment for 4 weeks significantly decreased VEGF-receptor (VEGF-R) expression in EC tissues by 43.2% compared with untreated mice(p<0.05) (Figure 4E). Overall, our results suggest that PA has potential to inhibit adhesion and invasion in EC cells and angiogenesis in the Lkb1fl/flp53fl/fl mice.
3.5. Cellular stress modulates PA-induced migration and invasion
To verify the role of cellular stress in migration and invasion induced by PA, the Ishikawa and ECC-1 cells were pre-incubated with 0.1 mM NAC (N-acetyl-l-cysteine) for 3 hours, and then treated with 250 and 500 µM PA at the indicated times. Pre-treatment of both cell lines with NAC effectively abolished PA-associated ROS levels. Moreover, NAC partially reversed the inhibitory effect of PA on cell proliferation and attenuated PA-induced apoptosis in both cell lines (p<0.05) (Figure 5A,B). In addition, NAC was able to alleviate the effects of PA on the inhibition of wound healing and decreased invasive capacity in both cell lines(p<0.05) (Figure 5C,D). Western blotting results demonstrated that pre-treatment with NAC decreased PA-mediated downregulation of Bcl-xL, Mcl-1, Calnexin and PERK in both cell lines (Figure 5E). These results support that cellular stress pathways are a potential trigger that controls the effects of PA on cell proliferation, apoptosis and invasion in EC.
3.6. PA resulted in lipid accumulation in EC cells and increased lipogenesis in EC tissues
Given that exogenous fatty acids increase lipid droplet (LD) formations and LDs are thought to be involved in multiple metabolic processes in cancer cells, we investigated the regulatory role of PA in the formation of LDs by Oil Red O staining in the EC cell lines. PA increased LD accumulation in a dose-dependent manner in both Ishikawa and ECC-1 cells after 24 hours of treatment (p<0.05) (Figure 6A). Furthermore, treatment of both cell lines with 100 µM PA increased Oil Red O staining in a time-dependent manner compared to untreated cells, reaching a maximum at 9 hours for Ishikawa cells and at 18 hours for ECC-1 cells (p<0.05) (Figure 6B). Phosphorylation of ACC and adipose triglyceride lipase (ATGL) was observed by western blotting assay after treatment in both cell lines with 100 μM PA, in a time course fashion. PA significantly increased ACC phosphorylation and ATCL expression in both cell lines within 24 hours of 100 µM PA treatment (Figure 6C). Since LDs are the main intracellular organelles that store triglycerides (TG) and other neutral lipids, intracellular TG levels were determined by ELISA assay. Treatment of cells with 100 µM and 250 µM PA overnight also increased intracellular TG production by 20.9% and 54.1% in Ishikawa and 46.6% and 66.4% in ECC-1 cells, respectively(p<0.05), accompanied by increased the expression of ATGL, long-chain acyl-CoA synthetase-1 (ACSL-1) and fatty acid synthase (FASN) in both cell lines (Figure 6D,E). IHC results further verified that PA significantly increased the expression of phosphorylation of ACC and ATGL in EC tissues of Lkb1ff/ffp53ff/ff mice compared to untreated mice (p<0.05) (Figure 6F). Importantly, similar results were confirmed by western blotting assay that PA treatment increased the expression of FASN, Lipin-1, phosphorylated-ACC and phosphorylated-S6 in EC tissues (Figure 6G). To evaluate the influence of PA on mitochondrial fatty acid oxidation (FAO) in EC cells, both cell lines were treated with 100 µM PA in a time course manner. Interestingly, increased expression of CTP1A and activity of FAO were observed in both cell lines within 18 hours of treatment, while CPT1A expression and FAO activity were decreased when cells were treated with 100 and 250 µM PA for 24 hours (Figure 6H,I). Taken together, these results demonstrate that PA is effective in increasing LD formation and de novo fatty acid synthesis in EC cells and tissues, and support the concept of simultaneous fatty acid synthesis and FAO in PA-treated EC cells (32, 33).
Since fatty acids can stimulate cancer cell metabolic reprogramming, we next examined the effects of PA on glycolysis metabolism in EC cells. PA treatment at doses of 100 and 250 µM for 24 hours significantly reduced glucose uptake and lactate production in both cell lines (p<0.05) (Figure 6J). In further support of this, high concentrations of PA reduced the expression of Glut1, Glut4, LDHA, Hexokinase (HK) I and Hexokinase II in Ishikawa and ECC-1 cells, as assessed by western blotting analysis (Figure 6K). Finally, 250 µM PA reduced cellular ATP production by 15.3% in Ishikawa cells and 19.3% in ECC-1 cells, respectively, compared with control cells after 24 hours of treatment (p<0.05) (Figure 6L). These data support the existence of feedback regulation between PA metabolism and glycolytic activity.
3.7. Inhibition of LD formation increased the sensitivity to PA in EC cells
To investigate the role of LD formation in PA mediated growth inhibition, we selected two small inhibitors, T863 and PF-06424439, to block the functions of DGAT1 and DGAT2, which catalyze the final committed step in the mammalian TAG biosynthesis (34, 35). Ishikawa and ECC-1 cells were treated with 150 µM PA, 5 µM T863, 5 µM PF-06424439 and the combination for 24 hours. As shown in Figure 7A,B, targeting either DGAT1 by T863 or DGAT2 by PF-06424439 reduced LD formation, and T863 was shown to be more effective than PF-06424439 in reducing LD formation in both EC cells (p<0.05). Importantly, PA in combination with T863 and PF-06424439 completely blocked LD formation. MTT analysis showed that blocking DGAT1 by T863 but not DGAT2 by PF-06424439 effectively increased PA sensitivity in inhibition of cell proliferation, but the combination of T863, PF-06424439 and PA produced the greatest cytostatic effects in both cell lines (p<0.05) (Figure 7C). Consistent with our MTT results, depletion of LDs by combination treatment with T863 and PF-06424439 significantly increased cleaved caspase 3 activity in PA-treated EC cells (p<0.05) (Figure 7D). These results suggest that LDs have a cytoprotective effect on EC cells in the presence of high concentrations of PA.
3.8. Effect of PA on AKT/mTOR, MAPK and p38 pathways in EC cells and tissues
To investigate the role of the AKT/mTOR, MAPK and p38 pathways in PA-mediated inhibition of cell proliferation, the Ishikawa and ECC-1 cells were treated with different concentrations of PA for 24 hours, and western blotting assay was used to detect changes in these pathways using phosphorylated antibodies. The results showed that the expression of phosphorylated AKT, S6, 4E-BP1, p38 and p42/44 was downregulated in both cell lines following 250 μM PA treatment (Figure 8A). PA treatment for 4 weeks also reduced the expression of phosphorylated S6 and p42/44 in EC tissues of Lkb1fl/flp53fl/fl mice compared with control mice (p<0.05) (Figure 8B). These results support that PA reduced cell viability and inhibited tumor growth via effects on the AKT/mTOR, MAPK and p38 pathways.
4. Discussion
FAs have been implicated in the progression and prognosis of EC (36). However, conflicting reports exist about the effect of PA on cell proliferation, adhesion, migration, invasion, tumor growth and fatty acid metabolism in multiple cancers (18). Recent studies have demonstrated that PA has anti-tumorigenic potential through targeting multiple cell signaling and metabolic pathways in vitro and in vivo (18, 25, 37, 38). While the relationship between metabolic abnormalities and carcinogenesis and progression of EC is well established, the critical importance of FAs, including PA, for cell proliferation and tumor growth in EC has been less well-studied. Using EC cell lines and a transgenic mouse model of EC, we found that physiological concentrations of PA significantly reduced cell viability, caused cellular stress and apoptosis, decreased cell invasion and migration, and inhibited tumor growth through inhibition of the AKT/mTOR and MAKP signaling pathways in vitro and in vivo. EC cells increased intracellular fatty acid synthesis and LD formation in response to different extracellular concentrations of PA. LDs have the potential to protect EC cells from PA-induced cytostatic effects. Cellular stress was a trigger to control cell proliferation, apoptosis and invasion in PA-treated EC cells. Additionally, treatment of the primary cultures of EC cells with 250 µM PA resulted in differential inhibitory responses to PA. These differences in response to PA may be associated with different molecular classifications of EC. Overall, these results highlight the role of PA in cell viability and tumor growth in EC cells and the Lkb1fl/flp53fl/fl mouse model of EC, respectively.
Invasion and migration of EC cells are initiated and mediated by multiple signaling pathways that control the processes of EMT, including integrins and matrix-degrading enzymes, which promote cell detachment from the primary tumor and the consequent ability of cells to procure a motile phenotype through cell-matrix interactions (39, 40). Reprogramming of lipid metabolism potently changes the fatty acid composition of membrane, enhances cell membrane fluidity and promotes EC progression through EMT signaling pathways (41, 42). Increasing evidence suggests that PA has anti-invasive and anti-metastatic impacts on certain types of cancer cells (43). PA specifically improved the metastatic potential of oral cancer cells in a CD36 (a cell surface receptor of fatty acids)-dependent manner, and blocking CD36 via neutralizing antibodies almost completely inhibited metastasis in orthotopic mouse models of human oral cancer (44). Regulation of ROS-induced NF-kB and MMP9 activation by TLR4 was involved in PA-enhanced invasion of the AsPC-1 pancreatic cancer cell line (45). In gastric cancer, low concentrations of PA (50 and 100 μM) induced cell migration and invasion through CD36-dependent activation of the AKT pathway, whereas high concentrations of PA reduced cell migration and invasion possibly due to lipotoxicity (46). Interestingly, 10 or 20 μM PA exhibited an inhibitory effect on the migration and invasion in PC3 and DU145 prostate cancer cells, at the least partly through regulating PKCζ, integrin β1 and EMT processes, and abrogated M2 macrophage-induced EMT and migration of colorectal cancer cells. 100 µM PA significantly promoted cell proliferative activity, migratory capacity and EMT process in PC3 cells (37, 38, 47, 48). Treatment with different concentrations of PA from 50 to 200 µM impaired cell invasion in hepatocellular carcinoma cells by modulating membrane fluidity and glucose metabolism, and inhibited tumor growth and lung metastases in an orthotopic mouse model of hepatocellular carcinoma (49). Given the recently identified association of the dual effects of PA on invasive capacity in various cancer cells, we initially focused on the influence of PA on cell motility by employing adhesion, modified Boyden chamber and wounding healing assays in EC cells. Low concentrations of PA did not increase or decrease cell adhesion and invasion, while PA concentrations over 100 µM did reduce EC cell adhesion and invasion through inactivation of MMP9 and reduction of EMT process. IHC results confirmed that PA treatment decreased the expression of VEGF in EC tissues. Inhibition of ROS by NAC resulted in partial antagonism of PA-induced cell migration in both cell lines. Our results, together with previous observations in other laboratories, suggest that the effects of PA on migration and invasion may depend in part on tumor type and concentration, and PA-induced cellular stress is involved in cell adhesion and invasion in EC cells. The underlying regulatory mechanisms need to be explored in further experiments.
High concentrations of free FAs, particularly saturated FAs, including PA, results in imbalance between ER protein load and ER folding, ultimately triggering ER stress, apoptosis and death in cancer cells (50, 51). Palmitoylation has been linked to ER stress through regulating trafficking, localization, stability, and aggregation in ER (52). Excessive intake of PA can directly or indirectly affect the accumulation of intracellular ROS and induce cellular stress, and its effects may be related to cell type (50, 53). Exogenous PA has the ability to alter the lipid composition in the ER of MDA-MB-231 breast cancer cells, and the lipid composition in the ER appears to be involved in the inhibition of PA-induced cell proliferation and apoptosis (54). PA at 150 µM increased ROS production in HCT116 colon cancer cells and induced more ROS production in HCT116 p53-/- cells compared to p53+/+ cells, suggesting that p53 has a protective function against PA-induced cellular stress and apoptosis (55, 56). However, a recent study has confirmed that PA reduced ROS production and mitochondrial membrane potential in a dose-dependent manner in the EC RL-95-2 and HEC-1-A cell lines with concomitant changes in mitochondrial morphology (57). In our studies, we found that both the Ishikawa and ECC-1 EC cell lines exhibited different sensitivity to induction of ROS production and reduction in mitochondrial membrane potential when treated with PA above 100 µM. This difference in EC cell line responses to PA-induced cellular stress may be further delineated by characterizing the distinct lipid profile and metabolism of each cell subset as varying EC cell lines exhibit different glucose and lipid metabolic profiles (58). Our previous work showed that cell proliferation of Ishikawa and ECC-1 cells predominantly depended on glycolysis through multiple complex signaling pathways compared to other EC cell lines (59). However, by analyzing RNA-seq data of ECC-1 and Ishikawa cells from the National Institutes of Health Gene Expression Omnibus, we found that the metabolic processes of the two cell lines are not much different, but response to stimulus including response to nutrient and response to nutrient levels has significantly different (Supplemental Figures S1 and S2). In addition, changes in FA composition in ER membranes after exposure to PA may also be responsible for the different responses of the two cell lines to PA-induced cellular stress (60). Thus, multiple factors ranging from genetic background, metabolic profile to lipid composition in ER membranes may be factors affecting PA-induced cellular stress in EC cells.
Increasing evidence shows that the generation of cellular stress in response to PA treatment has been implicated as an important contributor to PA-induced apoptosis in cancer cells. Inhibition of cellular stress significantly reduces PA-induced apoptosis in cardiomyocytes, Leydig cells, pre-frontal cells and colon carcinoma cells (55, 61-63). In addition to cellular stress, PA-mediated apoptosis appears to involve a number of relevant factors and signaling pathways including p53, p62, p21, Sesn2, CD36, S6K1 and the PI3K/Akt pathway (64-67). PA can exert apoptotic effects through mitochondrial apoptotic pathways, including upregulation of CCAAT-enhancer-binding protein homologous proteins (CHOP) (68). p53 negatively regulated PA-induced apoptosis by ROS accumulation via targeting downstream genes including p21 and Sesn2 in HCT116 cells (55). Mutation of IDH1 aggravated the effects of PA on cell viability and apoptosis and consistently increased ROS production in the HCT116 cells, especially under low glucose conditions (69). We also found that PA induced apoptosis through both intrinsic and extrinsic pathways of apoptosis while inhibition of cellular stress by NAC partially attenuated apoptosis and increased cell viability in both cell lines; therefore, our results indicate that cellular stress is a main pathway for controlling PA-induced apoptosis in EC cells.
LDs are perfectly positioned in the metabolic scheme to control both the lipid acquisition and utilization for various purposes in cancer cells; and thus, LDs are emerging as novel regulators of cell growth, metabolism, invasion, inflammation, immunity, neoplastic transformation and drug resistance (70). Formation and metabolism of LDs are evolving as critical components of metabolic reprogramming in cancer cells. The accumulation of LDs is generally considered to be beneficial to the survival of cancer cells due to the fact that it can sequester inflammatory-inducing lipids such as ceramides and DAGs, safeguard polyunsaturated FAs from lipid peroxidation that causes DNA damage, and protects cancer cells under stress and oxidative stress (71, 72). Given that excess intracellular LDs have been shown to be substrates for autophagy, autophagy, especially lipophagy, has recently been implicated to be responsible for regulating LD formation by mobilizing triglycerides and cholesterol with acid hydrolases in LDs to maintain lipid homeostasis (73, 74). However, excessive FA overload loading or impeded lipid lipolysis leads to excess accumulation of LDs, eventually resulting in lipotoxicity in normal and cancer cells (72, 75, 76). In gastric cancer cells, treatment with FAs or adipocytes induced intracellular LD formation through transcriptional upregulation of DGAT2 in a C/EBPα-dependent manner (77). Similarly, treatment of HepG2 cells with 200 μM PA for 24 hours resulted in the formation of LDs and stimulated mitochondrial oxidative metabolism (78). Interestingly, Raman microscopic observations found that the average number and size of LDs in PA-treated cells were lower than those LDs in the cells treated with other FAs, suggesting that FA types may have varying influences on the formation of LDs (79). Inhibition of TAG synthesis by targeting DGAT1 and DGAT2 with specific small inhibitors effectively depleted the formation of LDs and had differential effects on cell growth depending on the type of FA in glioma cells (35, 80). Depletion of OA-induced LDs by the small inhibitors caused more growth inhibition in OA-treated cells compared with the control cells, whereas elimination of PA-induced LDs enhanced cell proliferation in PA-treated cells (35). In the current study, PA increased the formation of LDs in a time- and dose-dependent manner in the EC cell lines. Blocking the accumulation of LDs by DGTA1/2 inhibitors effectively enhanced the ability of PA to inhibit cell proliferation in both EC cell lines. Since DGAT1 and DGAT2 are enzymes that catalyze the last step in the TAG synthesis and control the biogenesis of LDs, inhibition of their functions results in increased concentration of free FAs and other lipid species in the cytoplasm, ultimately leading to higher lipotoxicity for cancer cells (81, 82). Recent studies confirmed that DGAT1-deficient cells were sensitized to lipid stress and resulted in increased caspase 3 and 7 activation, and knock-down of DGAT1 by siRNA decreased LD formation, resulted in autophagy and inhibited cell growth in prostate cancer LNCaP cells (81, 83). Thus, pharmacological inhibition of DGAT1/2 may have therapeutic potential in the treatment of cancer, including EC.
PA as an intracellular signaling molecule participates in the activation and inactivation of various signaling pathways to regulate tumor growth. Inhibition or promotion of tumor cell growth by PA often depends on multiple pathways with different functions, without any specific signaling pathway being involved (18). For example, PA inhibited cell proliferation and M2-TAM-induced migratory and invasive properties through inhibition of IL-10-STAT3-NF-κB signaling axis in colorectal cancer cells (38). In prostate cancer cells, PA targeted PI3K/Akt pathway to block prostate cancer proliferation and metastasis (67). On the other hand, treatment of neuroblastoma cells with 200 µM PA did not affect cell viability but blocks insulin-induced metabolic activation, inhibits the activation of the insulin/PI3K/Akt pathway, and activates mTOR kinase (84). Interestingly, sarcoma cells exposed to 400 µM PA promoted cell proliferation and activated the AKT/mTOR/S6 pathway through phosphorylation of PTEN at T366 (85). In addition, a recent study found that EGFR and AKT/mTOR pathways are also involved in PA metabolism in breast cancer cells (86). Our results showed that 250 µM PA significantly decreased the phosphorylated expression of AKT, S6, 4-EBP1, p38 and p42/44 in the Ishikawa and ECC-1 cells and inhibited the expression of phosphorylation of S6 and p42/44 in PA treated tumor tissues. These results support the fact that PA inhibits tumor growth involving multiple signaling pathways in EC.
FA supplements are naturally involved in membrane formation, lipid remodeling, and membrane-based signaling processes that occur in vivo and have the same potential to affect biological function as endogenously produced molecules (87). Lipidomic analysis showed that fatty acid composition of membrane and cellular lipid pool is continuously remodeled by the influence of free fatty acid availability, including endogenous synthesis and exogenous uptake (15, 16). PA is able to alter the fatty acid composition of cancer cell membranes and cellular lipid pools, ultimately leading to changes in cell proliferation and tumor growth. However, changes in membrane FA composition by PA may have bidirectional effects, depending on the concomitant presence of other types of fatty acids, the metabolic phenotype, and the duration and dose of PA exposure (28). High-dose of PA (300 mg/kg) treatment for 4 weeks did not influence body weight and serum TG concentration in KK-Ay Mice with genetic type 2 diabetes, while 100 mg/kg PA treatment for 4 weeks dramatically increased serum TG content and liver weight in C57BL/6N mice (88, 89). Hepatomegaly in PA-treated mice is an effective indicator showing increased activity of TG synthesis in the liver due to excess FAs (88). Our data revealed that 250 µM PA treatment substantially increased TG concentration in both ECC-1 and Ishikawa cells, yet treating the mice with PA for 4 weeks did not increase mouse body weight. Although serum TG concentration and liver weight have trend to increase compared with control mice, there were no statistics significant between the two groups. H&E staining showed that balloon-shaped hepatocytes appeared in PA-treated LKB1 p53 mice, suggesting that this may be connected to the duration of PA treatment or the dose of PA. Thus, the effect of different doses and different treatment durations of PA as well as the combined use of PA with other FAs on the balance of lipid pool and the composition of cell membrane deserves further study in our EC cell lines and LKB1fl/fl p53fl/fl mouse model of EC.
Primary cell cultures from tumor tissues are progressively being used as a reliable tool in molecular biology, cancer biology, metabolism, toxicity testing and drug screening (90). In current study, seventeen primary cell cultures from human EC tissues were used to examine the effect of different concentrations of PA on cell proliferation and showed that these primary cultures responded differently to 250 and 500 µM PA treatments in the MTT assay. Likewise, the Ishikawa and ECC-1 cells exhibited different sensitivities to 50, 100, 250 and 500 µM PA treatments. Given that RNA-seq data showed that Ishikawa and ECC-1 cell lines had different abilities to respond to nutrients and nutrient levels, we hypothesized that the FA composition of the cell membranes before PA treatment or the alternations of FA composition in the cell membranes after PA treatment might be responsible for the difference in the sensitivity of primary cell cultures and EC cell lines to PA. Indeed, the balance between FA types in the cell membrane is essential to the initiation of the “predisposition” of the signals that control the growth of tumor cells. The balance of FA composition is not the same from tissue to tissue in the body, suggesting that different tissue cells may respond to FA treatment differently (12, 14). Cancer cells can efficiently self-adapt by reorganizing their biological membranes to maintain proliferation, evade apoptosis, and resist anticancer drug treatment. PA rapidly increased the proportion of PA in NB100 membrane after 1-hour incubation (28). More importantly, analysis of membrane lipidomics before and after FA treatments may help to clarify the connection between lipidome and sensitivity to FAs and is a fundamental step toward understanding the impact of FA on tumor growth in the clinic (16).
5. Conclusions
There is limited data on the effect of FAs on cell proliferation and tumor growth in EC. We report for the first time that PA exhibited anti-proliferative and anti-tumorigenic activities in EC cells and the Lkb1fl/flp53fl/fl mouse model of EC through induction of cellular stress and apoptosis and inhibition of AKT/mTOR and MAPK pathways. Inhibition of cellular stress effectively reversed PA-mediated effects on decreasing EC cell proliferation, inducing apoptosis and stimulating invasion. In addition, inhibition of LD formation by targeting DGAT1/2 increased the cytotoxic effects of PA. Thus, PA treatment in combination with targeting the formation of LDs may provide new insights into the treatment of EC. Future studies are warranted to further explore the anti-tumorigenic mechanisms of PA as to lay the basis for future clinical investigations of this novel dietary supplement in obesity-driven EC.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, VLB and C.Z; methodology and experimental design, ZZ, XZ, JW, YF, TH, NAN, WCB, HS, LB and JOD, Data collection and interpretation, ZZ, XZ, JW, WK, and YY; Prepared figures, ZZ and CZ; Drafted manuscript, CZ and VLB.
Funding
This work is supported by: Endometrial Cancer Molecularly Targeted Therapy Consortium and NIH/NCI - R37CA226969.
Institutional Review Board Statement
The protocol for mouse study was approved by the UNC-CH Institutional Animal Care and Use Committee (IACUC, #20-219).
Informed Consent Statement
The protocol for human EC sample study was approved by the Institutional Review Board (IRB) of the University of North Carolina at Chapel Hill (UNC-CH). All patients provided written consent.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation. RNA sequencing data located in the National Institutes of Health Gene Expression Omnibus (www.ncbi.nlm.nih.gov/geo/), (accession number GSE151207).
Acknowledgments
not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.C.; Huang, Y.; Hou, J.Y.; Khoury-Collado, F.; Melamed, A.; St Clair, C.M.; et al. Toxicity after adjuvant therapy for stage III uterine cancer. Gynecol. Oncol. 2020, 159, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Crosbie, E.J.; Kitson, S.J.; McAlpine, J.N.; Mukhopadhyay, A.; Powell, M.E.; Singh, N. Endometrial cancer. Lancet 2022, 399, 1412–1428. [Google Scholar] [CrossRef] [PubMed]
- Onstad, M.A.; Schmandt, R.E.; Lu, K.H. Addressing the Role of Obesity in Endometrial Cancer Risk, Prevention, and Treatment. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2016, 34, 4225–4230. [Google Scholar] [CrossRef]
- Michalczyk, K.; Niklas, N.; Rychlicka, M.; Cymbaluk-Płoska, A. The Influence of Biologically Active Substances Secreted by the Adipose Tissue on Endometrial Cancer. Diagnostics 2021, 11, 494. [Google Scholar] [CrossRef] [PubMed]
- Saidi, S.A.; Holland, C.M.; Kreil, D.P.; MacKay, D.J.C.; Charnock-Jones, D.S.; Print, C.G.; et al. Independent component analysis of microarray data in the study of endometrial cancer. Oncogene 2004, 23, 6677–6683. [Google Scholar] [CrossRef] [PubMed]
- Njoku, K.; Sutton, C.J.J.; Whetton, A.D.; Crosbie, E.J. Metabolomic Biomarkers for Detection, Prognosis and Identifying Recurrence in Endometrial Cancer. Metabolites 2020, 10, 314. [Google Scholar] [CrossRef]
- Altadill, T.; Dowdy, T.M.; Gill, K.; Reques, A.; Menon, S.S.; Moiola, C.P.; et al. Metabolomic and Lipidomic Profiling Identifies The Role of the RNA Editing Pathway in Endometrial Carcinogenesis. Sci. Rep. 2017, 7, 8803. [Google Scholar] [CrossRef] [PubMed]
- Jové, M.; Gatius, S.; Yeramian, A.; Portero-Otin, M.; Eritja, N.; Santacana, M.; et al. Metabotyping human endometrioid endometrial adenocarcinoma reveals an implication of endocannabinoid metabolism. Oncotarget 2016, 7, 52364–52374. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, C.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef] [PubMed]
- Röhrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef] [PubMed]
- Bermúdez, M.A.; Pereira, L.; Fraile, C.; Valerio, L.; Balboa, M.A.; Balsinde, J. Roles of Palmitoleic Acid and Its Positional Isomers, Hypogeic and Sapienic Acids, in Inflammation, Metabolic Diseases and Cancer. Cells. 2022, 11. [Google Scholar] [CrossRef] [PubMed]
- Dierge, E.; Feron, O. Dealing with saturated and unsaturated fatty acid metabolism for anticancer therapy. Curr Opin Clin Nutr Metab Care 2019, 22, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Maulucci, G.; Cohen, O.; Daniel, B.; Ferreri, C.; Sasson, S. The Combination of Whole Cell Lipidomics Analysis and Single Cell Confocal Imaging of Fluidity and Micropolarity Provides Insight into Stress-Induced Lipid Turnover in Subcellular Organelles of Pancreatic Beta Cells. Molecules 2019, 24, 3742. [Google Scholar] [CrossRef]
- Ferreri, C.; Sansone, A.; Ferreri, R.; Amézaga, J.; Tueros, I. Fatty Acids and Membrane Lipidomics in Oncology: A Cross-Road of Nutritional, Signaling and Metabolic Pathways. Metabolites 2020, 10, 345. [Google Scholar] [CrossRef] [PubMed]
- Van der Paal, J.; Neyts, E.C.; Verlackt, C.C.W.; Bogaerts, A. Effect of lipid peroxidation on membrane permeability of cancer and normal cells subjected to oxidative stress. Chem Sci. 2016, 7, 489–498. [Google Scholar] [CrossRef] [PubMed]
- Fatima, S.; Hu, X.; Gong, R.H.; Huang, C.; Chen, M.; Wong, H.L.X.; et al. Palmitic acid is an intracellular signaling molecule involved in disease development. Cell. Mol. Life Sci. CMLS 2019, 76, 2547–2557. [Google Scholar] [CrossRef] [PubMed]
- Murru, E.; Manca, C.; Carta, G.; Banni, S. Impact of Dietary Palmitic Acid on Lipid Metabolism. Front Nutr. 2022, 9, 861664. [Google Scholar] [CrossRef] [PubMed]
- Waki, M.; Ide, Y.; Ishizaki, I.; Nagata, Y.; Masaki, N.; Sugiyama, E.; et al. Single-cell time-of-flight secondary ion mass spectrometry reveals that human breast cancer stem cells have significantly lower content of palmitoleic acid compared to their counterpart non-stem cancer cells. Biochimie 2014, 107 Pt A, 73–77. [Google Scholar] [CrossRef]
- Igal, R.A. Roles of StearoylCoA Desaturase-1 in the Regulation of Cancer Cell Growth, Survival and Tumorigenesis. Cancers 2011, 3, 2462–2477. [Google Scholar] [CrossRef] [PubMed]
- Koltun, D.O.; Vasilevich, N.I.; Parkhill, E.Q.; Glushkov, A.I.; Zilbershtein, T.M.; Mayboroda, E.I.; et al. Orally bioavailable, liver-selective stearoyl-CoA desaturase (SCD) inhibitors. Bioorganic Med. Chem. Lett. 2009, 19, 3050–3053. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Fatima, S.; Chen, M.; Xu, K.; Huang, C.; Gong, R.H.; et al. Toll-like receptor 4 is a master regulator for colorectal cancer growth under high-fat diet by programming cancer metabolism. Cell Death Dis. 2021, 12, 791. [Google Scholar] [CrossRef] [PubMed]
- Fatima, S.; Hu, X.; Huang, C.; Zhang, W.; Cai, J.; Huang, M.; et al. High-fat diet feeding and palmitic acid increase CRC growth in β2AR-dependent manner. Cell Death Dis. 2019, 10, 711. [Google Scholar] [CrossRef] [PubMed]
- Bojková, B.; Winklewski, P.J.; Wszedybyl-Winklewska, M. Dietary Fat and Cancer-Which Is Good, Which Is Bad, and the Body of Evidence. Int. J. Mol. Sci. 2020, 21, 4114. [Google Scholar] [CrossRef] [PubMed]
- Sczaniecka, A.K.; Brasky, T.M.; Lampe, J.W.; Patterson, R.E.; White, E. Dietary intake of specific fatty acids and breast cancer risk among postmenopausal women in the VITAL cohort. Nutr. Cancer 2012, 64, 1131–1142. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Zeng, X.; Liu, Z.; Chen, X.; Li, L.; Luo, R.; et al. Mesenchymal stromal cells protect hepatocytes from lipotoxicity through alleviation of endoplasmic reticulum stress by restoring SERCA activity. J. Cell. Mol. Med. 2021, 25, 2976–2993. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, A.; Chatgilialoglu, A.; Polito, L.; Ferreri, C. Membrane lipidome reorganization correlates with the fate of neuroblastoma cells supplemented with fatty acids. PLoS ONE 2013, 8, e55537. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, C.; Sansone, A.; Chatgilialoglu, C.; Ferreri, R.; Amézaga, J.; Burgos, M.C.; et al. Critical Review on Fatty Acid-Based Food and Nutraceuticals as Supporting Therapy in Cancer. Int J Mol Sci. 2022, 23, 6030. [Google Scholar] [CrossRef]
- Cousin, S.P.; Hügl, S.R.; Wrede, C.E.; Kajio, H.; Myers, M.G.; Jr Rhodes, C.J. Free fatty acid-induced inhibition of glucose and insulin-like growth factor I-induced deoxyribonucleic acid synthesis in the pancreatic beta-cell line INS-1. Endocrinology 2001, 142, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Kong, W.; Zhang, L.; Han, J.; Clark, L.H.; Yin, Y.; et al. Reversal of obesity-driven aggressiveness of endometrial cancer by metformin. Am J Cancer Res. 2019, 9, 2170–2193. [Google Scholar] [PubMed]
- Corbet, C.; Pinto, A.; Martherus, R.; Santiago de Jesus, J.P.; Polet, F.; Feron, O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Temkin, S.M.; Hawkridge, A.M.; Guo, C.; Wang, W.; Wang, X.Y.; et al. Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett. 2018, 435, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Li, L.; Lian, J.; Watts, R.; Nelson, R.; Goodwin, B.; et al. Roles of Acyl-CoA:Diacylglycerol Acyltransferases 1 and 2 in Triacylglycerol Synthesis and Secretion in Primary Hepatocytes. Arterioscler Thromb Vasc Biol. 2015, 35, 1080–1091. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Shah, N.; Almohaisin, M.I.; Saha, S.; Lu, F. Assessing fatty acid-induced lipotoxicity and its therapeutic potential in glioblastoma using stimulated Raman microscopy. Sci Rep. 2021, 11, 7422. [Google Scholar] [CrossRef] [PubMed]
- Mozihim, A.K.; Chung, I.; Said, N.; Jamil, A.H.A. Reprogramming of Fatty Acid Metabolism in Gynaecological Cancers: Is There a Role for Oestradiol? Metabolites 2022, 12, 350. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Jiao, W.; Xu, Y.; Hou, L.; Li, H.; Shao, J.; et al. Palmitic acid inhibits prostate cancer cell proliferation and metastasis by suppressing the PI3K/Akt pathway. Life Sci. 2021, 286, 12. [Google Scholar] [CrossRef] [PubMed]
- de Araujo Junior, R.F.; Eich, C.; Jorquera, C.; Schomann, T.; Baldazzi, F.; Chan, A.B.; et al. Ceramide and palmitic acid inhibit macrophage-mediated epithelial-mesenchymal transition in colorectal cancer. Mol. Cell. Biochem. 2020, 468, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Makker, A.; Goel, M.M. Tumor progression, metastasis, and modulators of epithelial-mesenchymal transition in endometrioid endometrial carcinoma: an update. Endocr.-Relat. Cancer 2016, 23, R85–R111. [Google Scholar] [CrossRef] [PubMed]
- Colas, E.; Pedrola, N.; Devis, L.; Ertekin, T.; Campoy, I.; Martínez, E.; et al. The EMT signaling pathways in endometrial carcinoma. Clin. Transl. Oncol. Off. Publ. Fed. Span. Oncol. Soc. Natl. Cancer Inst. Mex. 2012, 14, 715–720. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Lei, H.; Yi, T.; Su, P.; Tang, S.; Tong, Y. Lipid reprogramming induced by the TFEB-ERRα axis enhanced membrane fluidity to promote EC progression. J. Exp. Clin. Cancer Res. CR 2022, 41, 28. [Google Scholar] [CrossRef] [PubMed]
- Troisi, J.; Sarno, L.; Landolfi, A.; Scala, G.; Martinelli, P.; Venturella, R.; et al. Metabolomic Signature of Endometrial Cancer. J Proteome Res. 2018, 17, 804–812. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Yu, X.; Peng, C.; Liu, N.; Chen, W.; Xu, H.; et al. Activation of SREBP-1c alters lipogenesis and promotes tumor growth and metastasis in gastric cancer. Biomed. Pharmacother. = Biomed. Pharmacother. 2020, 128, 25. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Avgustinova, A.; Mejetta, S.; Martín, M.; Castellanos, A.; Attolini, C.S.; et al. Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 2017, 541, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Binker-Cosen, M.J.; Richards, D.; Oliver, B.; Gaisano, H.Y.; Binker, M.G.; Cosen-Binker, L.I. Palmitic acid increases invasiveness of pancreatic cancer cells AsPC-1 through TLR4/ROS/NF-κB/MMP-9 signaling pathway. Biochem. Biophys. Res. Commun. 2017, 484, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.; Fan, Z.; Wang, Z.; Dai, Q.; Xiang, Z.; Yuan, F.; et al. CD36 mediates palmitate acid-induced metastasis of gastric cancer via AKT/GSK-3β/β-catenin pathway. J. Exp. Clin. Cancer Res. CR 2019, 38, 019–1049. [Google Scholar] [CrossRef] [PubMed]
- Maly, I.V.; Hofmann, W.A. Effect of Palmitic Acid on Exosome-Mediated Secretion and Invasive Motility in Prostate Cancer Cells. Molecules 2020, 25, 2722. [Google Scholar] [CrossRef] [PubMed]
- Landim, B.C.; de Jesus, M.M.; Bosque, B.P.; Zanon, R.G.; da Silva, C.V.; Góes, R.M.; et al. Stimulating effect of palmitate and insulin on cell migration and proliferation in PNT1A and PC3 prostate cells: Counteracting role of metformin. The Prostate 2018, 78, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Ding, Y.; Wang, Y.; Wang, Z.; Yin, X.; Yan, G.; et al. Functional lipidomics: Palmitic acid impairs hepatocellular carcinoma development by modulating membrane fluidity and glucose metabolism. Hepatology 2017, 66, 432–448. [Google Scholar] [CrossRef] [PubMed]
- Farhat, D.; Ghayad, S.E.; Icard, P.; Le Romancer, M.; Hussein, N.; Lincet, H. Lipoic acid-induced oxidative stress abrogates IGF-1R maturation by inhibiting the CREB/furin axis in breast cancer cell lines. Oncogene 2020, 39, 3604–3610. [Google Scholar] [CrossRef] [PubMed]
- Gentric, G.; Mieulet, V.; Mechta-Grigoriou, F. Heterogeneity in Cancer Metabolism: New Concepts in an Old Field. Antioxid Redox Signal 2017, 26, 462–485. [Google Scholar] [CrossRef] [PubMed]
- Pagliassotti, M.J.; Kim, P.Y.; Estrada, A.L.; Stewart, C.M.; Gentile, C.L. Endoplasmic reticulum stress in obesity and obesity-related disorders: An expanded view. Metabolism 2016, 65, 1238–1246. [Google Scholar] [CrossRef] [PubMed]
- Munir, R.; Lisec, J.; Swinnen, J.V.; Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br J Cancer 2019, 120, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.M.; Colombo, I.; Montorfano, G.; Zava, S.; Corsetto, P.A. Exogenous Fatty Acids Modulate ER Lipid Composition and Metabolism in Breast Cancer Cells. Cells 2021, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Luo, H.; Zhang, N.; Wang, Y.; Li, Y.; Huang, H.; et al. Loss of p53 Sensitizes Cells to Palmitic Acid-Induced Apoptosis by Reactive Oxygen Species Accumulation. Int. J. Mol. Sci. 2019, 20, 6268. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Lu, Y.C.; Wang, J.; Wang, L.; Yu, H.; Li, Y.F.; et al. Type 2 Diabetes Promotes Cell Centrosome Amplification via AKT-ROS-Dependent Signalling of ROCK1 and 14-3-3σ. Cell Physiol Biochem. 2018, 47, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.S.; Huang, S.M.; Wang, Y.C. Palmitate Enhances the Efficacy of Cisplatin and Doxorubicin against Human Endometrial Carcinoma Cells. Int J Mol Sci. 2021, 23, 80. [Google Scholar] [CrossRef] [PubMed]
- Byrne, F.L.; Poon, I.K.; Modesitt, S.C.; Tomsig, J.L.; Chow, J.D.; Healy, M.E.; et al. Metabolic vulnerabilities in endometrial cancer. Cancer Res. 2014, 74, 5832–5845. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Zhang, L.; Guo, H.; Wysham, W.Z.; Roque, D.R.; Willson, A.K.; et al. Glucose promotes cell proliferation, glucose uptake and invasion in endometrial cancer cells via AMPK/mTOR/S6 and MAPK signaling. Gynecol Oncol. 2015, 138, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, A.M.; Colombo, I.; Montorfano, G.; Zava, S.; Corsetto, P.A. Exogenous Fatty Acids Modulate ER Lipid Composition and Metabolism in Breast Cancer Cells. Cells 2021, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Guan, G.; Lei, L.; Lv, Q.; Gong, Y.; Yang, L. Curcumin attenuates palmitic acid-induced cell apoptosis by inhibiting endoplasmic reticulum stress in H9C2 cardiomyocytes. Hum Exp Toxicol. 2019, 38, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Wen, D.; Wang, F.; Wang, C.; Yang, L. Curcumin protects against palmitic acid-induced apoptosis via the inhibition of endoplasmic reticulum stress in testicular Leydig cells. Reprod Biol Endocrinol. 2019, 17, 71. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Li, F.; Cai, M.; Hu, J.; Wang, Q.; Lou, S. Interactions between Endoplasmic Reticulum Stress and Autophagy: Implications for Apoptosis and Neuroplasticity-Related Proteins in Palmitic Acid-Treated Prefrontal Cells. Neural Plast. 2021, 2021, 8851327. [Google Scholar] [CrossRef] [PubMed]
- Pardo, V.; González-Rodríguez, Á.; Muntané, J.; Kozma, S.C.; Valverde, Á.M. Role of hepatocyte S6K1 in palmitic acid-induced endoplasmic reticulum stress, lipotoxicity, insulin resistance and in oleic acid-induced protection. Food Chem Toxicol. 2015, 80, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.P.; Kuo, W.W.; Baskaran, R.; Day, C.H.; Chen, R.J.; Wen, S.Y.; et al. Acute hypoxic preconditioning prevents palmitic acid-induced cardiomyocyte apoptosis via switching metabolic GLUT4-glucose pathway back to CD36-fatty acid dependent. J Cell Biochem. 2018, 119, 3363–3372. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Zhou, C.; Guo, X.; Ding, Y.; Ma, S.; Gong, X. Palmitate impairs the autophagic flux to induce p62-dependent apoptosis through the upregulation of CYLD in NRCMs. Toxicology, 2022; 465, 153032. [Google Scholar]
- Zhu, S.; Jiao, W.; Xu, Y.; Hou, L.; Li, H.; Shao, J.; et al. Palmitic acid inhibits prostate cancer cell proliferation and metastasis by suppressing the PI3K/Akt pathway. Life Sci. 2021, 286, 120046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, Y.; Han, D.; Lu, J.; Yin, S.; Hu, H.; et al. Combination of Palmitic Acid and Methylseleninic Acid Induces Mitochondria-Dependent Apoptosis via Attenuation of the IRE1α Arm and Enhancement of CHOP in Hepatoma. ACS Omega 2021, 6, 15708–15715. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Sun, C.; Gu, Y.; Gao, X.; Zhao, Y.; Yuan, Y.; et al. Mutation of IDH1 aggravates the fatty acid-induced oxidative stress in HCT116 cells by affecting the mitochondrial respiratory chain. Mol Med Rep. 2019, 19, 2509–2518. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.L.S.; Barreto, E.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Dis. 2020, 11, 105. [Google Scholar] [CrossRef] [PubMed]
- Cruz, A.L.S.; Barreto, E.A.; Fazolini, N.P.B.; Viola, J.P.B.; Bozza, P.T. Lipid droplets: platforms with multiple functions in cancer hallmarks. Cell Death Disease 2020, 11, 020–2297. [Google Scholar] [CrossRef] [PubMed]
- Petan, T.; Jarc, E.; Jusović, M. Lipid Droplets in Cancer: Guardians of Fat in a Stressful World. Molecules 2018, 23, 19741. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; et al. Autophagy regulates lipid metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Czaja, M.J. Regulation of lipid droplets by autophagy. Trends Endocrinol. Metab. 2011, 22, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Stockwell, B.R.; Friedmann Angeli, J.P.; Bayir, H.; Bush, A.I.; Conrad, M.; Dixon, S.J.; et al. Ferroptosis: A Regulated Cell Death Nexus Linking Metabolism, Redox Biology, and Disease. Cell 2017, 171, 273–285. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Wang, H.; Ren, L.; Lu, Z.; Zheng, Q.; Ding, L.; et al. Adding fuel to the fire: The lipid droplet and its associated proteins in cancer progression. Int J Biol Sci. 2022, 18, 6020–6034. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, T.; Lu, Y.X.; Wang, J.X.; Yu, F.H.; Yang, M.Z.; et al. Obesity promotes gastric cancer metastasis via diacylglycerol acyltransferase 2-dependent lipid droplets accumulation and redox homeostasis. Redox Biol. 2020, 36, 29. [Google Scholar] [CrossRef] [PubMed]
- Eynaudi, A.; Díaz-Castro, F.; Bórquez, J.C.; Bravo-Sagua, R.; Parra, V.; Troncoso, R. Differential Effects of Oleic and Palmitic Acids on Lipid Droplet-Mitochondria Interaction in the Hepatic Cell Line HepG2. Front Nutr. 2021, 8, 901. [Google Scholar] [CrossRef] [PubMed]
- Paramitha, P.N.; Zakaria, R.; Maryani, A.; Kusaka, Y.; Andriana, B.B.; Hashimoto, K.; et al. Raman Study on Lipid Droplets in Hepatic Cells Co-Cultured with Fatty Acids. Int. J. Mol. Sci. 2021, 22. [Google Scholar] [CrossRef]
- Naik, R.; Obiang-Obounou, B.W.; Kim, M.; Choi, Y.; Lee, H.S.; Lee, K. Therapeutic strategies for metabolic diseases: Small-molecule diacylglycerol acyltransferase (DGAT) inhibitors. ChemMedChem 2014, 9, 2410–2424. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Corbacho, M.J.; Obeid, L.M. A novel role for DGATs in cancer. Adv Biol Regul. 2019; 72, 89–101. [Google Scholar]
- Yuan, Y.; Shah, N.; Almohaisin, M.I.; Saha, S.; Lu, F. Assessing fatty acid-induced lipotoxicity and its therapeutic potential in glioblastoma using stimulated Raman microscopy. Sci Rep. 2021, 11, 021–86789. [Google Scholar] [CrossRef] [PubMed]
- Mitra, R.; Le, T.T.; Gorjala, P.; Goodman, O.B., Jr. Positive regulation of prostate cancer cell growth by lipid droplet forming and processing enzymes DGAT1 and ABHD5. BMC Cancer 2017, 17, 017–3589. [Google Scholar] [CrossRef] [PubMed]
- Calvo-Ochoa, E.; Sánchez-Alegría, K.; Gómez-Inclán, C.; Ferrera, P.; Arias, C. Palmitic acid stimulates energy metabolism and inhibits insulin/PI3K/AKT signaling in differentiated human neuroblastoma cells: The role of mTOR activation and mitochondrial ROS production. Neurochem Int. 2017, 110, 75–83. [Google Scholar] [CrossRef] [PubMed]
- Bai, D.; Wu, Y.; Deol, P.; Nobumori, Y.; Zhou, Q.; Sladek, F.M.; et al. Palmitic acid negatively regulates tumor suppressor PTEN through T366 phosphorylation and protein degradation. Cancer Lett. 2021, 496, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Küçüksayan, E.; Sansone, A.; Chatgilialoglu, C.; Ozben, T.; Tekeli, D.; Talibova, G.; et al. Sapienic Acid Metabolism Influences Membrane Plasticity and Protein Signaling in Breast Cancer Cell Lines. Cells 2022, 11, 225. [Google Scholar] [CrossRef] [PubMed]
- Ferreri, C.; Sansone, A.; Chatgilialoglu, C.; Ferreri, R.; Amezaga, J.; Burgos, M.C.; et al. Critical Review on Fatty Acid-Based Food and Nutraceuticals as Supporting Therapy in Cancer. Int. J. Mol. Sci. 2022, 23, 6030. [Google Scholar] [CrossRef] [PubMed]
- Ruan, J.S.; Lin, J.K.; Kuo, Y.Y.; Chen, Y.W.; Chen, P.C. Chronic palmitic acid-induced lipotoxicity correlates with defective trafficking of ATP sensitive potassium channels in pancreatic β cells. J Nutr Biochem. 2018, 59, 37–48. [Google Scholar] [CrossRef]
- Yang, Z.H.; Miyahara, H.; Hatanaka, A. Chronic administration of palmitoleic acid reduces insulin resistance and hepatic lipid accumulation in KK-Ay Mice with genetic type 2 diabetes. Lipids Health Dis. 2011, 10, 10–120. [Google Scholar] [CrossRef] [PubMed]
- Miserocchi, G.; Mercatali, L.; Liverani, C.; De Vita, A.; Spadazzi, C.; Pieri, F.; et al. Management and potentialities of primary cancer cultures in preclinical and translational studies. J. Transl. Med. 2017, 15, 229. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Effect of PA on cell proliferation and tumor growth in EC cells and the Lkb1fl/flp53 /fl mouse model of EC, respectively. Ishikawa and ECC-1 cells were treated with various doses of PA for 72 hours. Cell proliferation was determined by MTT assay. PA induced dose-dependent growth inhibition in both cell lines. Representative dose-response curves and IC50 values are shown (A). PA inhibited colony forming ability of Ishikawa and ECC-1 cells compared to the vehicle-treated cells (B). The effect of PA on cell proliferation was detected by MTT assay in 17 primary cultures of human endometrioid ECs (C). The mouse body weight from control and PA treatment groups was measured (D). There were no significant differences between blood triglycerides (TG) and liver weight during PA treatment compared to control mice (E). H&E staining showed that PA increased the accumulation of fat in hepatocytes compared to control mice (F). Treatment of Lkb1fl/flp53fl/fl mice with PA for 4 weeks significantly reduced tumor weights and tumor volumes (G). The expression of Ki-67 was assessed using immunohistochemistry in EC tumors from Lkb1fl/flp53fl/fl mice following PA or placebo treatment (H). *p<0.05, **p<0.01.
Figure 1.
Effect of PA on cell proliferation and tumor growth in EC cells and the Lkb1fl/flp53 /fl mouse model of EC, respectively. Ishikawa and ECC-1 cells were treated with various doses of PA for 72 hours. Cell proliferation was determined by MTT assay. PA induced dose-dependent growth inhibition in both cell lines. Representative dose-response curves and IC50 values are shown (A). PA inhibited colony forming ability of Ishikawa and ECC-1 cells compared to the vehicle-treated cells (B). The effect of PA on cell proliferation was detected by MTT assay in 17 primary cultures of human endometrioid ECs (C). The mouse body weight from control and PA treatment groups was measured (D). There were no significant differences between blood triglycerides (TG) and liver weight during PA treatment compared to control mice (E). H&E staining showed that PA increased the accumulation of fat in hepatocytes compared to control mice (F). Treatment of Lkb1fl/flp53fl/fl mice with PA for 4 weeks significantly reduced tumor weights and tumor volumes (G). The expression of Ki-67 was assessed using immunohistochemistry in EC tumors from Lkb1fl/flp53fl/fl mice following PA or placebo treatment (H). *p<0.05, **p<0.01.
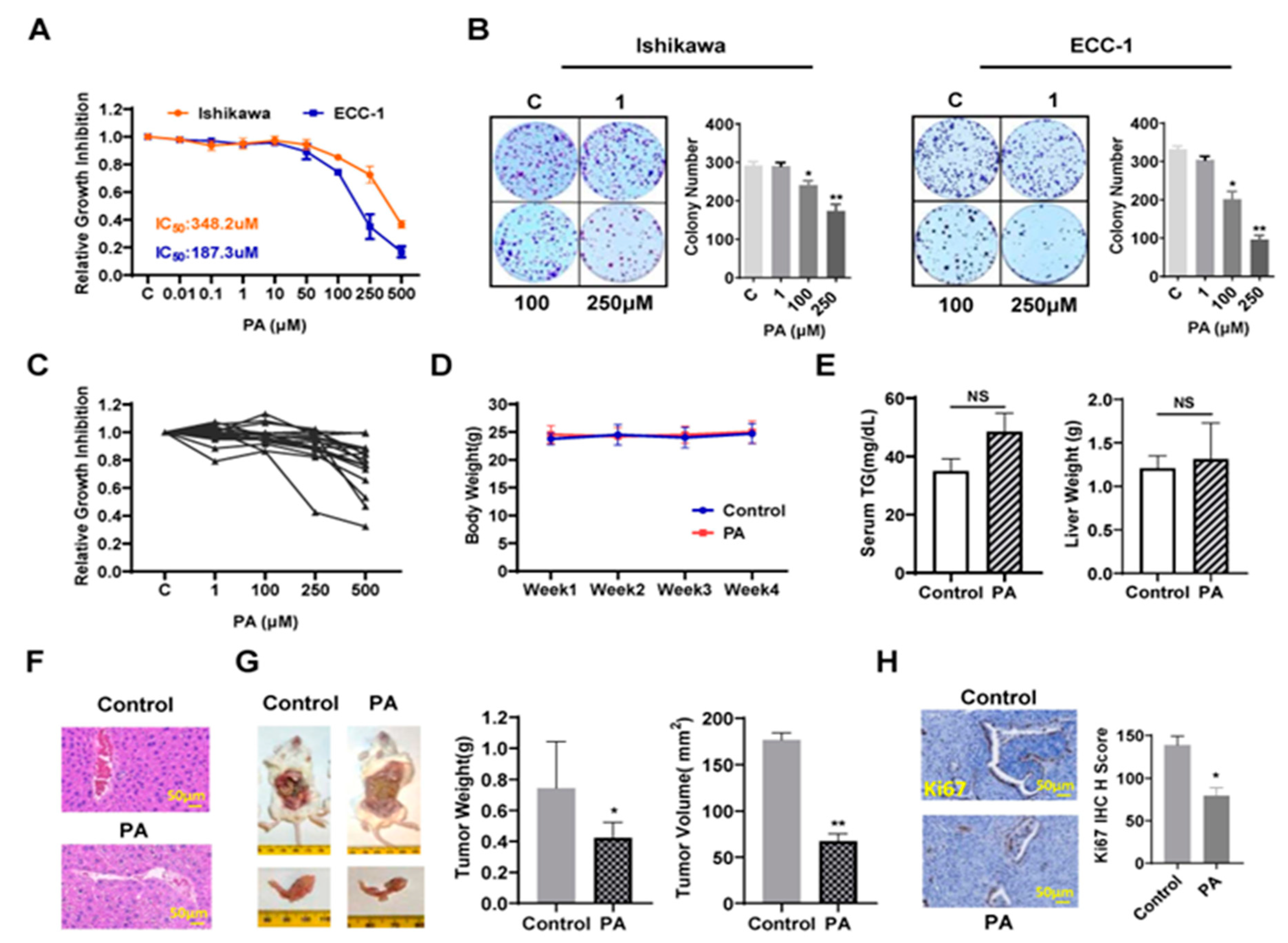
Figure 2.
PA induced apoptosis in Ishikawa and ECC cells and Lkb1fl/flp53fl/fl mice. Cleaved caspase 3, 8 and 9 activities were assayed by ELISA assay. PA induced the activity of the cleaved caspases 3, 8 and 9 in both cell lines after treatment with PA for 12-14 hours (A-C). Ishikawa and ECC-1 cells were cultured with different concentrations of PA for 14 hours, and western blotting analysis showed PA reduced the expression of Bcl-xL and Mcl-1 and induced cleaved PARP expression in both cell lines (D). Pre-treatment with Z-VAD-FMK blocked PA-induced cleaved caspase 3 activity (E) and significantly rescued cell viability from PA-treated EC cells (F). The expression of Bcl-xL in mouse EC tumors was assessed by immunohistochemistry following PA or placebo treatment in the endometrial tumors (G). *p<0.05, **p<0.01.
Figure 2.
PA induced apoptosis in Ishikawa and ECC cells and Lkb1fl/flp53fl/fl mice. Cleaved caspase 3, 8 and 9 activities were assayed by ELISA assay. PA induced the activity of the cleaved caspases 3, 8 and 9 in both cell lines after treatment with PA for 12-14 hours (A-C). Ishikawa and ECC-1 cells were cultured with different concentrations of PA for 14 hours, and western blotting analysis showed PA reduced the expression of Bcl-xL and Mcl-1 and induced cleaved PARP expression in both cell lines (D). Pre-treatment with Z-VAD-FMK blocked PA-induced cleaved caspase 3 activity (E) and significantly rescued cell viability from PA-treated EC cells (F). The expression of Bcl-xL in mouse EC tumors was assessed by immunohistochemistry following PA or placebo treatment in the endometrial tumors (G). *p<0.05, **p<0.01.
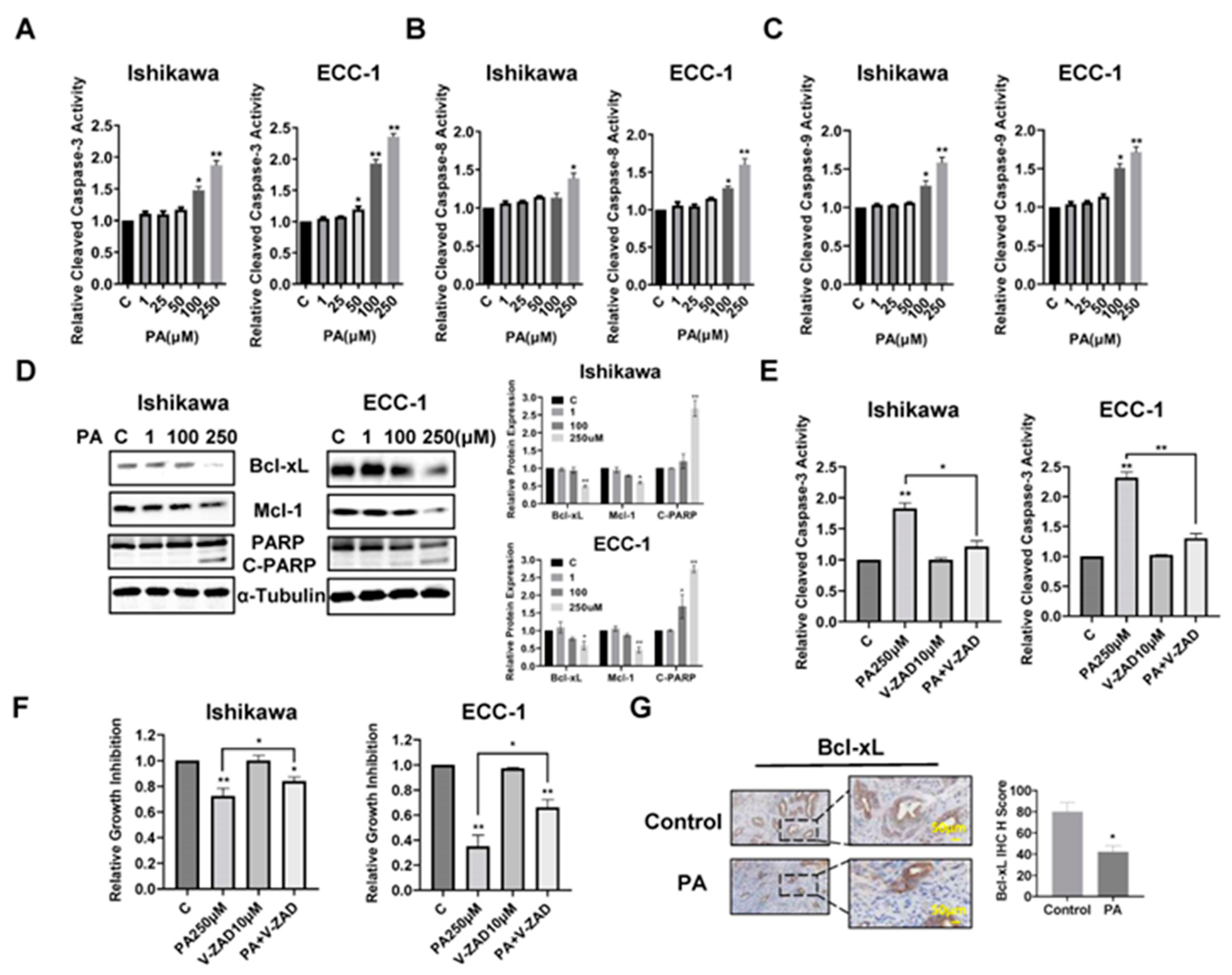
Figure 3.
PA induced ER stress in EC cells and EC tumors of Lkb1fl/flp53fl/fl mice. Ishikawa and ECC-1 cells were treated with varying concentration of PA for 14 hours. ROS, JC-1 and TMRE products were measured by ELISA assays. PA significantly increased the levels of ROS (A) and decreased mitochondrial membrane potential (B and C) in both cell lines compared to the vehicle-treated cells. PA increased the expression of cellular stress related proteins, including PERK, IRE-1α, ATF4, BiP and CLPP, in both cell lines after 24 hours of treatment (D). The expression of PERK was measured by immunohistochemistry in EC tumors from Lkb1fl/flp53fl/fl mice following PA or placebo treatment (E). *p<0.05, **p<0.01.
Figure 3.
PA induced ER stress in EC cells and EC tumors of Lkb1fl/flp53fl/fl mice. Ishikawa and ECC-1 cells were treated with varying concentration of PA for 14 hours. ROS, JC-1 and TMRE products were measured by ELISA assays. PA significantly increased the levels of ROS (A) and decreased mitochondrial membrane potential (B and C) in both cell lines compared to the vehicle-treated cells. PA increased the expression of cellular stress related proteins, including PERK, IRE-1α, ATF4, BiP and CLPP, in both cell lines after 24 hours of treatment (D). The expression of PERK was measured by immunohistochemistry in EC tumors from Lkb1fl/flp53fl/fl mice following PA or placebo treatment (E). *p<0.05, **p<0.01.
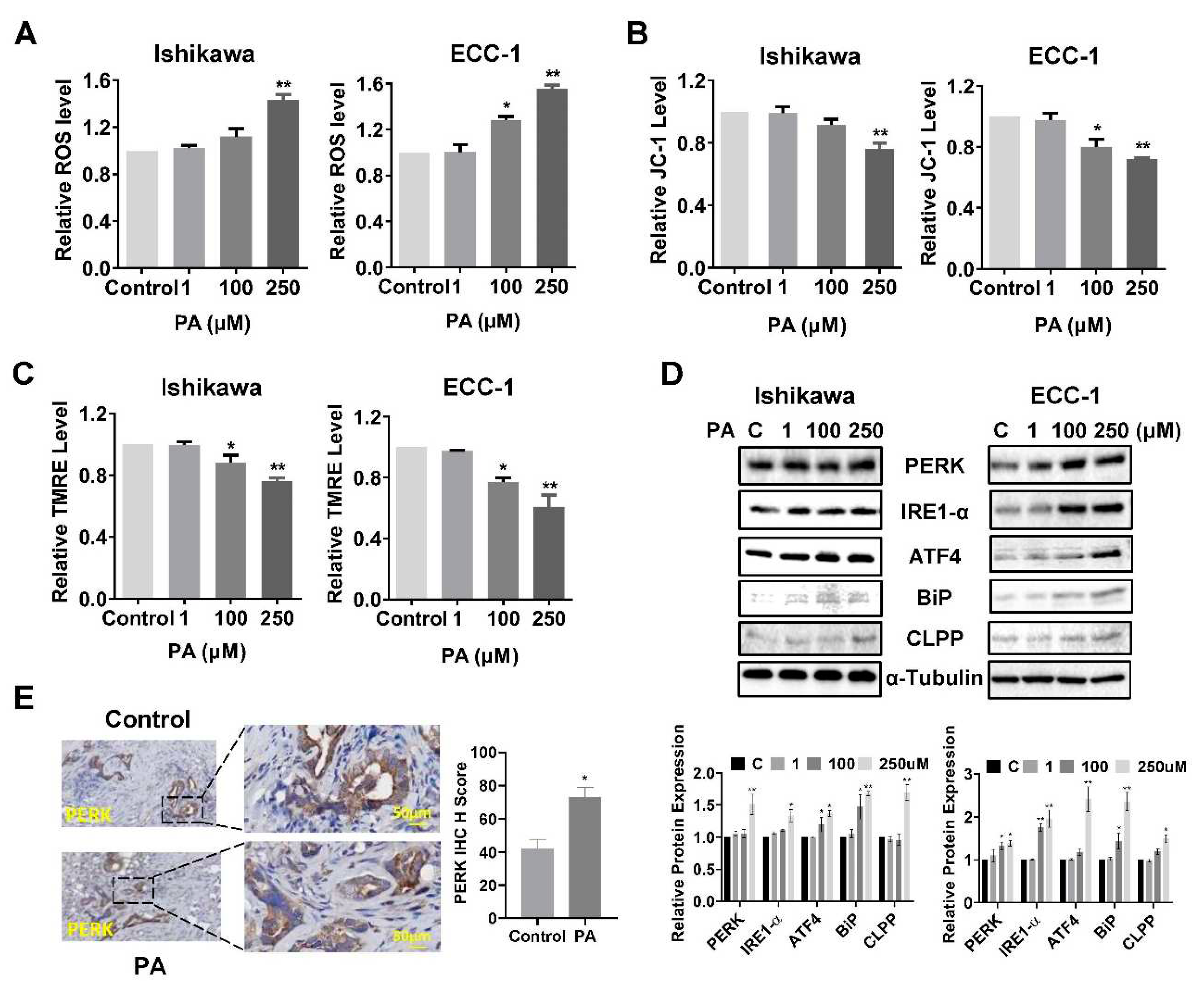
Figure 4.
PA inhibited adhesion and invasion in both EC cell lines and reduced the expression of the VEGF-R in EC tumors of Lkb1fl/flp53fl/fl mice. Adhesion was determined by laminin-1 assay in the Ishikawa and ECC-1 cells after PA treatment (A). Transwell assay was used to investigate invasive ability in both cell lines (B). Migration was measured by wound healing assay in both cell lines after treatment with PA for 48 h (C). PA decreased the expression of EMT related proteins, including Snail, VEGF, Vimentin, N-Cadherin and MMP-9, in both cell lines after 24 hours of treatment (D). Immunohistochemistry results showed that treatment of Lkb1fl/flp53fl/fl mice with PA for 4 weeks significantly reduced the expression of VEGF-R in EC tumors (E). *p<0.05, **p<0.01.
Figure 4.
PA inhibited adhesion and invasion in both EC cell lines and reduced the expression of the VEGF-R in EC tumors of Lkb1fl/flp53fl/fl mice. Adhesion was determined by laminin-1 assay in the Ishikawa and ECC-1 cells after PA treatment (A). Transwell assay was used to investigate invasive ability in both cell lines (B). Migration was measured by wound healing assay in both cell lines after treatment with PA for 48 h (C). PA decreased the expression of EMT related proteins, including Snail, VEGF, Vimentin, N-Cadherin and MMP-9, in both cell lines after 24 hours of treatment (D). Immunohistochemistry results showed that treatment of Lkb1fl/flp53fl/fl mice with PA for 4 weeks significantly reduced the expression of VEGF-R in EC tumors (E). *p<0.05, **p<0.01.
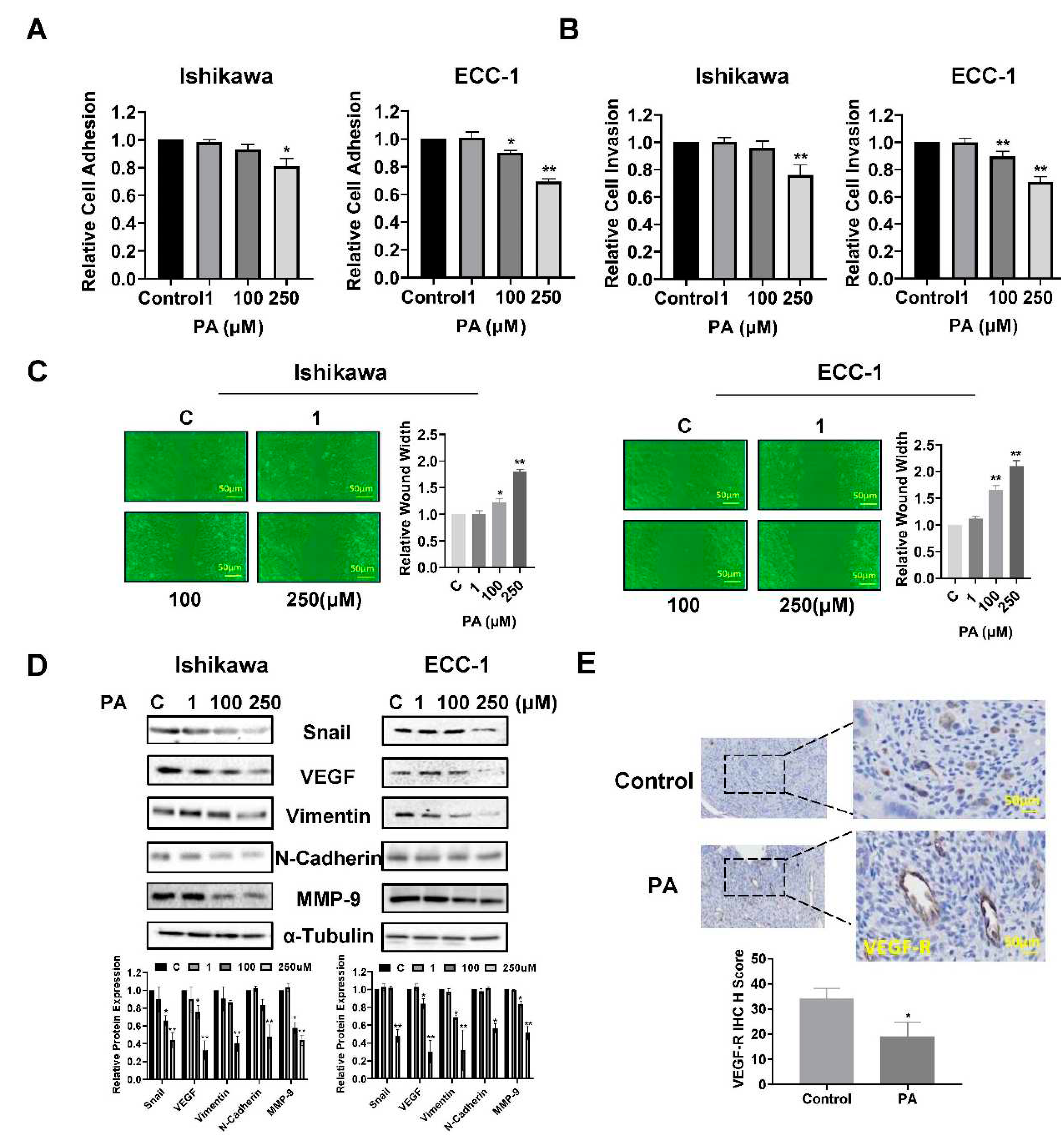
Figure 5.
Effect of cellular stress on PA-induced migration and invasion. Pre-treatment of 0.1 mM with NAC in both Ishikawa and ECC-1 cells effectively inhibited ROS production induced by PA in both cell lines (A). Inhibition of cellular stress by NAC significantly rescued cell PA-induced viability and apoptosis in both cell lines (B). Migration was assessed by wound healing assay in Ishikawa and ECC-1 cells. NAC effectively reversed PA-inhibited cell migration in both cell lines (C). Transwell assay showed that NAC was able to alleviate the effects of PA on the invasive capacity in both cell lines (D). The effect of pre-treatment with NAC on the expression of Bcl-xL, Mcl-1, Calnexin and PERK in PA-treated cells (E). *p<0.05, **p<0.01.
Figure 5.
Effect of cellular stress on PA-induced migration and invasion. Pre-treatment of 0.1 mM with NAC in both Ishikawa and ECC-1 cells effectively inhibited ROS production induced by PA in both cell lines (A). Inhibition of cellular stress by NAC significantly rescued cell PA-induced viability and apoptosis in both cell lines (B). Migration was assessed by wound healing assay in Ishikawa and ECC-1 cells. NAC effectively reversed PA-inhibited cell migration in both cell lines (C). Transwell assay showed that NAC was able to alleviate the effects of PA on the invasive capacity in both cell lines (D). The effect of pre-treatment with NAC on the expression of Bcl-xL, Mcl-1, Calnexin and PERK in PA-treated cells (E). *p<0.05, **p<0.01.
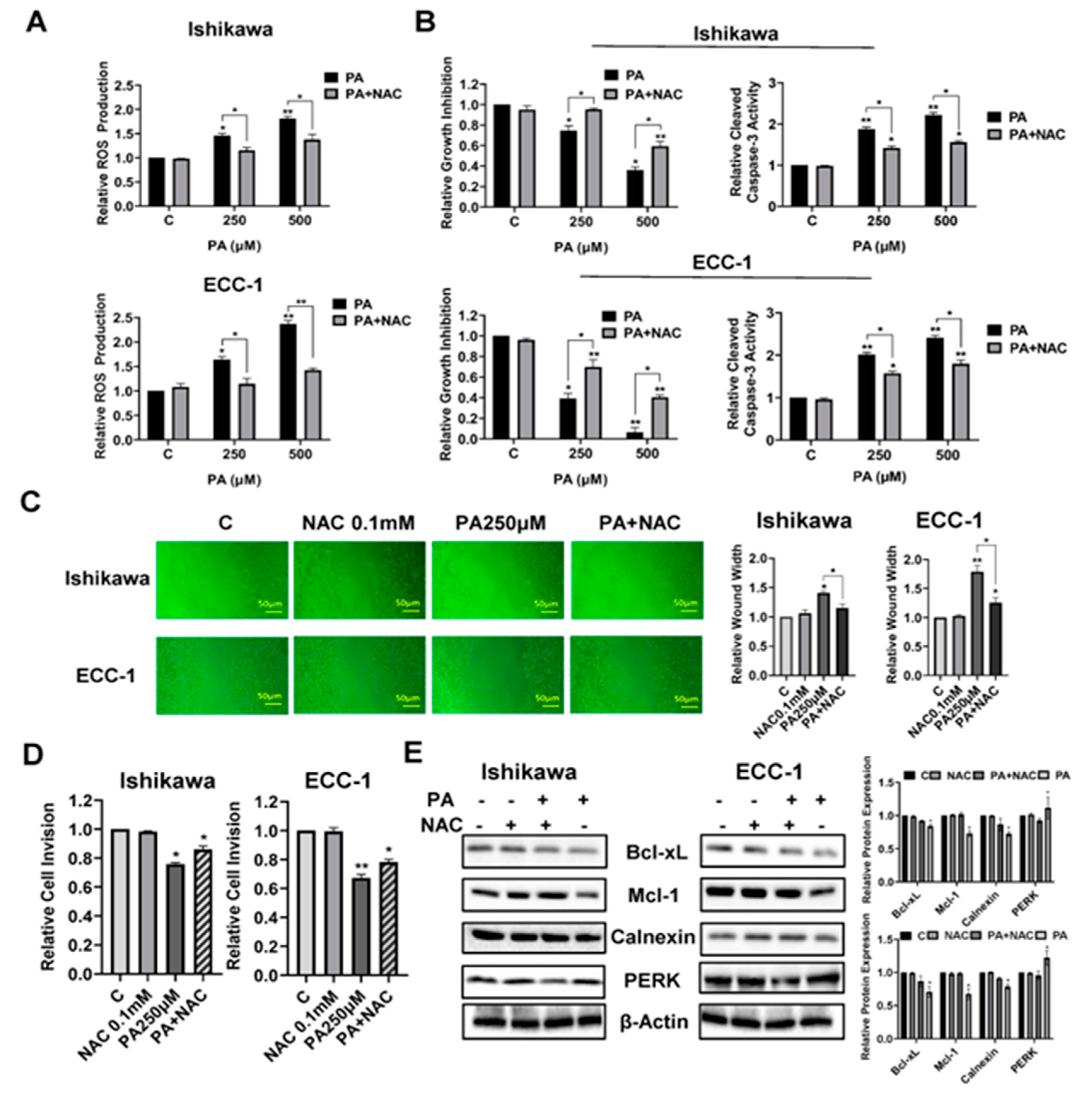
Figure 6.
PA increased lipid droplet (LDs) formation and lipogenesis. The Ishikawa and ECC-1 cells were treated with different concentrations of PA for 24 hours. PA significantly increased LD formation in a dose-dependent and time-dependent manner in both cell lines (A and B). The protein expression of p-ACC and ATGL was observed by western blotting assay (C). PA at 250 uM increased the intracellular TG levels in both cell line (D). Similarly, PA increased the protein expression of ATGL and FASN in both cell lines after 24 hours of treatment (E). The expression of p-ACC, ACSL-1 and ATGL was assessed using immunohistochemistry in EC tumors of Lkb1fl/flp53fl/fl mice following PA or placebo treatment (F). PA treatment increased the protein expression of FASN, p-ACC, Lipin-1, and inhibited the expression of p-S6 in EC tissues (G). The Changes of CPT1 and FAO activity after treatment of both cells with 100 µM PA in a time course manner (H). Treatment of cells with PA for 24 hours reduced the expression of CPT1 and activity of FAO in both cells (I). PA treatment at doses of 150 and 250 µM for 24 hours significantly reduced glucose uptake and lactate production in both cell lines (J). Western blotting results showed that PA reduced the expression of Glut1, Glut4, LDHA, Hexokinase I, Hexokinase II in Ishikawa, and ECC-1 cells (K). PA reduced cellular ATP production in EC cells compared with control cells (L). *p<0.05, **p<0.01.
Figure 6.
PA increased lipid droplet (LDs) formation and lipogenesis. The Ishikawa and ECC-1 cells were treated with different concentrations of PA for 24 hours. PA significantly increased LD formation in a dose-dependent and time-dependent manner in both cell lines (A and B). The protein expression of p-ACC and ATGL was observed by western blotting assay (C). PA at 250 uM increased the intracellular TG levels in both cell line (D). Similarly, PA increased the protein expression of ATGL and FASN in both cell lines after 24 hours of treatment (E). The expression of p-ACC, ACSL-1 and ATGL was assessed using immunohistochemistry in EC tumors of Lkb1fl/flp53fl/fl mice following PA or placebo treatment (F). PA treatment increased the protein expression of FASN, p-ACC, Lipin-1, and inhibited the expression of p-S6 in EC tissues (G). The Changes of CPT1 and FAO activity after treatment of both cells with 100 µM PA in a time course manner (H). Treatment of cells with PA for 24 hours reduced the expression of CPT1 and activity of FAO in both cells (I). PA treatment at doses of 150 and 250 µM for 24 hours significantly reduced glucose uptake and lactate production in both cell lines (J). Western blotting results showed that PA reduced the expression of Glut1, Glut4, LDHA, Hexokinase I, Hexokinase II in Ishikawa, and ECC-1 cells (K). PA reduced cellular ATP production in EC cells compared with control cells (L). *p<0.05, **p<0.01.
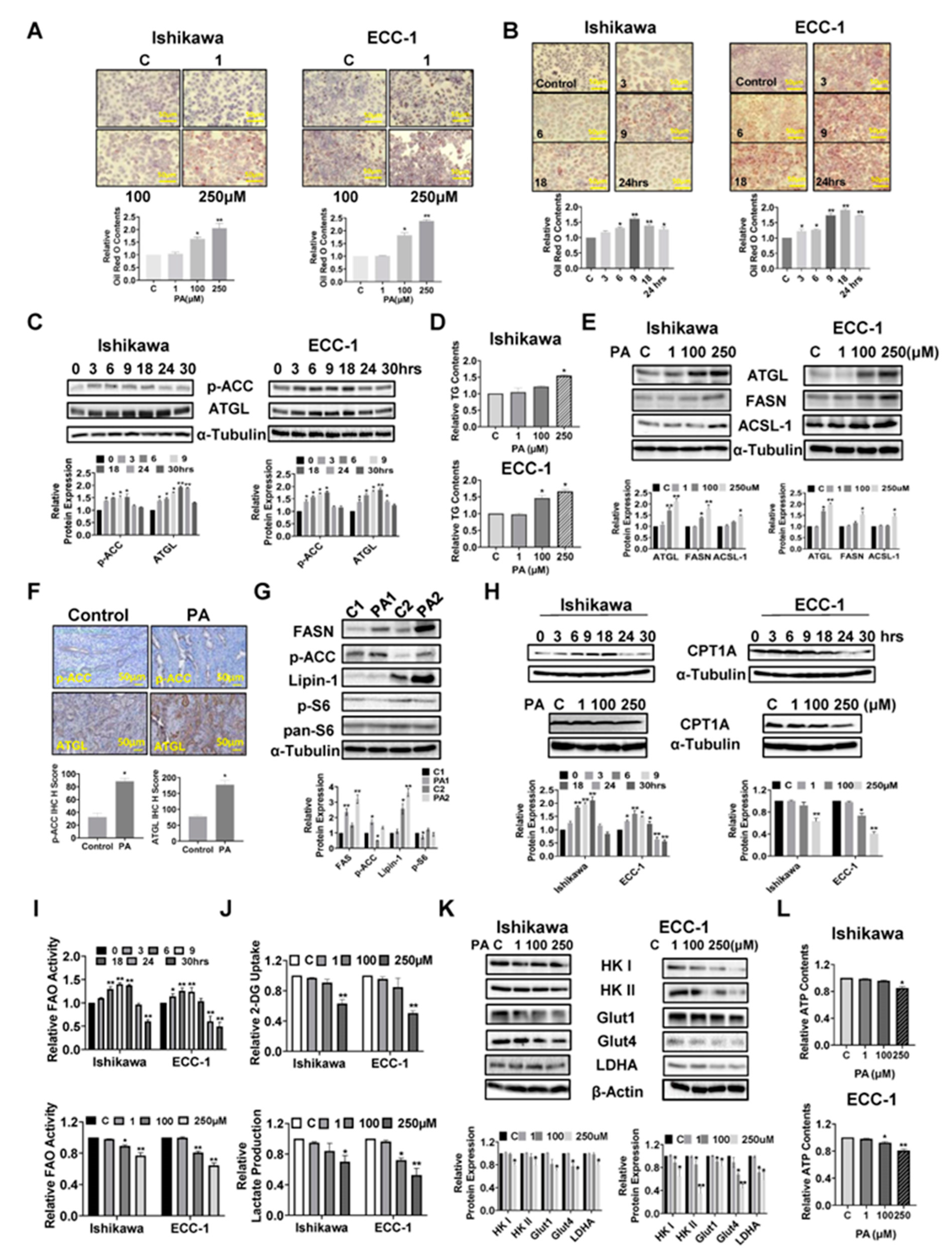
Figure 7.
Inhibition of LD formation increased the sensitivity to PA in EC cells. The Ishikawa and ECC-1 cells were treated with the DGAT1 inhibitor, T863, and the DGAT2 inhibitor, PF-06424439, and PA for 24 hours. Red and O assay showed that the combination of T863, PF-06424439 and PA completely blocked LD formation in both cell lines (A). The effect of PA T863 and PF-06424439 on the expression of DGAT1 and DGFAT2 was observed in both cell lines (B). Treatment with T863 and PF-06424439 increased PA sensitivity in inhibition of cell proliferation (C) and induction of cleaved caspase 3 activity in PA-treated EC cells (D). *p<0.05, **p<0.01.
Figure 7.
Inhibition of LD formation increased the sensitivity to PA in EC cells. The Ishikawa and ECC-1 cells were treated with the DGAT1 inhibitor, T863, and the DGAT2 inhibitor, PF-06424439, and PA for 24 hours. Red and O assay showed that the combination of T863, PF-06424439 and PA completely blocked LD formation in both cell lines (A). The effect of PA T863 and PF-06424439 on the expression of DGAT1 and DGFAT2 was observed in both cell lines (B). Treatment with T863 and PF-06424439 increased PA sensitivity in inhibition of cell proliferation (C) and induction of cleaved caspase 3 activity in PA-treated EC cells (D). *p<0.05, **p<0.01.
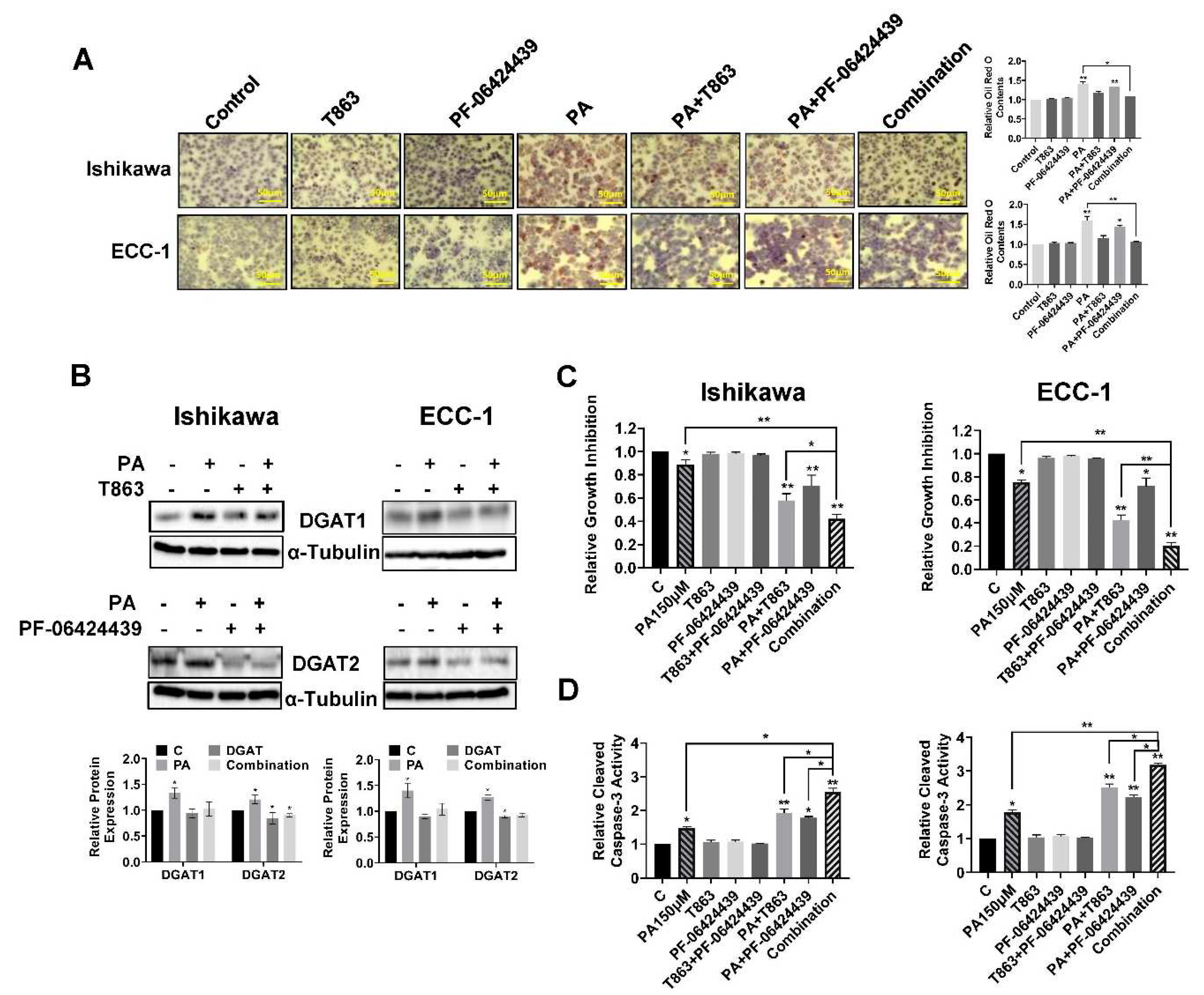
Figure 8.
Effect of PA on AKT/mTOR, MAPK and p38 pathways. The Ishikawa and ECC-1 cells were treated with different concentrations of PA for 24 hours, and western blotting results showed that the expression of phosphorylated AKT, S6, 4E-BP1, p38 and p42/44 was downregulated in both cell lines (A). The expression of p-S6 and p-42/44 was assessed using immunohistochemistry in EC tumors of Lkb1fl/flp53fl/fl mice following PA or placebo treatment (B). *p<0.05, **p<0.01.
Figure 8.
Effect of PA on AKT/mTOR, MAPK and p38 pathways. The Ishikawa and ECC-1 cells were treated with different concentrations of PA for 24 hours, and western blotting results showed that the expression of phosphorylated AKT, S6, 4E-BP1, p38 and p42/44 was downregulated in both cell lines (A). The expression of p-S6 and p-42/44 was assessed using immunohistochemistry in EC tumors of Lkb1fl/flp53fl/fl mice following PA or placebo treatment (B). *p<0.05, **p<0.01.
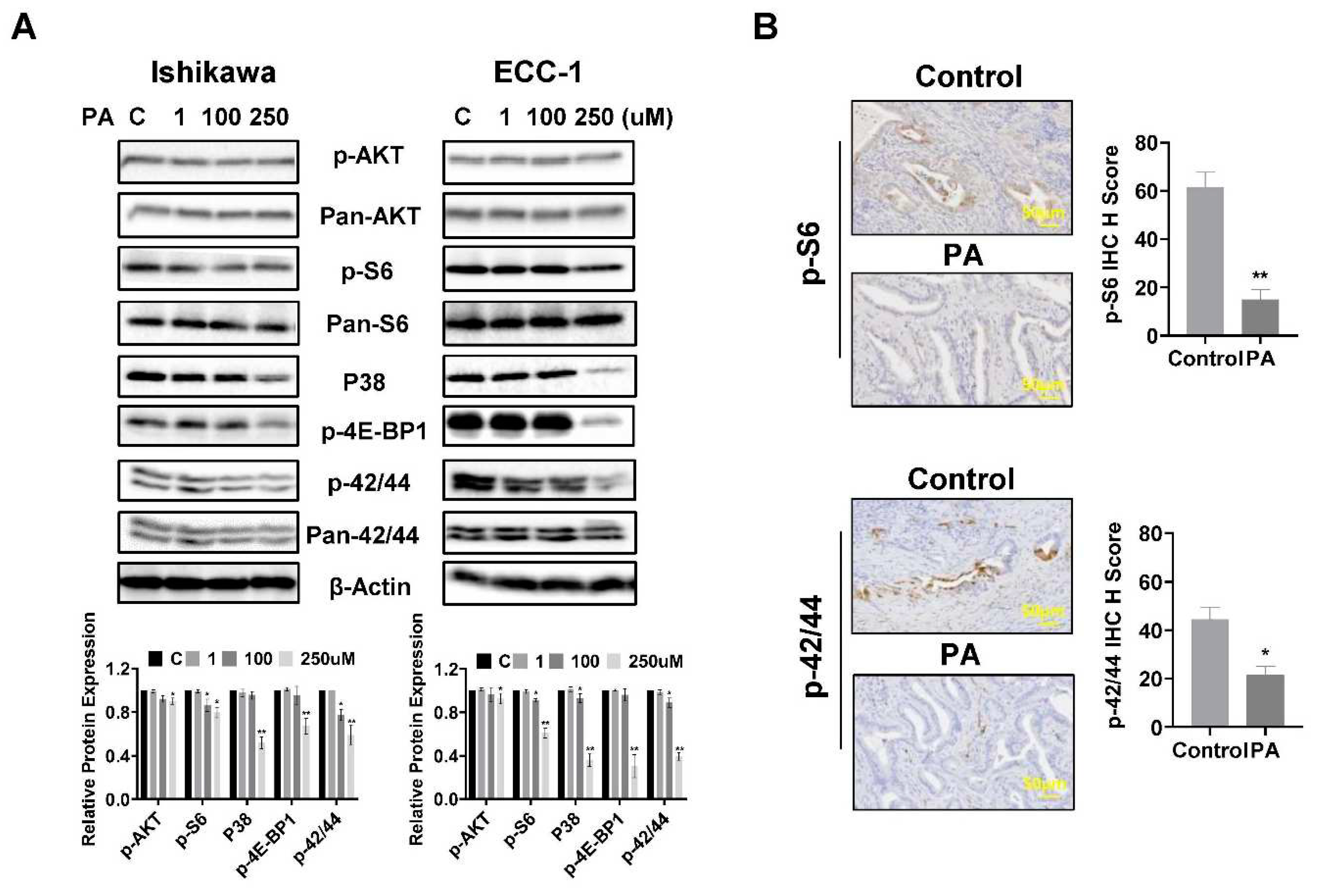
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



