
Submitted:
31 January 2024
Posted:
31 January 2024
You are already at the latest version
Abstract
The studies regarding macrophage lncRNAs described in this review can help provide valuable information for developing treatments for various pathological conditions involving macrophages. Over the past century, molecular biology's focus has transitioned from proteins to DNA, and now to RNA. Once considered merely a genetic information carrier, RNA is now recognized as both a vital element in early cellular life and a regulator in complex organisms. Long noncoding RNAs (lncRNAs), which are over 200 bases long but do not code for proteins, play roles in gene expression regulation and signal transduction by inducing epigenetic changes or interacting with various proteins and RNAs. These interactions exhibit a range of functions in various cell types including macrophages. Notably, some macrophage lncRNAs influence the activation of NF-κB, a crucial transcription factor governing immune and inflammatory responses. Macrophage NF-κB is instrumental in the progression of various pathological conditions including sepsis, atherosclerosis, cancer, autoimmune disorders, and hypersensitivity. It orchestrates gene expression related to immune responses, inflammation, cell survival, and proliferation. Consequently, its malfunction is a key contributor to the onset and development of these diseases. This review summarizes the role of lncRNAs that regulate NF-κB in macrophage activation and inflammation, especially focusing on their implications in human diseases and their potential as therapeutic targets
Keywords:
human diseases
; inflammation
; lncRNA
; macrophage
; NF-κB
1. Introduction
Inflammation, a natural and complex biological response, is crucial in defending against infection [1,2]. Yet, chronic or uncontrolled inflammation can be harmful, contributing various disease developments [3]. Diet, exercise, smoking, and stress, as lifestyle factors, can impact bodily inflammation levels, underscoring a healthy lifestyle’s importance in preventing and managing diseases [4]. In some instances, medical treatments aim at targeting inflammation to ease symptoms and control disease progression. Macrophages, innate immune cells with antigen-presenting capabilities, are central in the inflammatory responses, significantly impacting acute and chronic inflammation [5]. Notably, macrophages exhibit a dual role: pro-inflammatory M1 macrophages drive inflammation, whereas anti-inflammatory M2 macrophages aid in tissue repair and resolving inflammation [6,7]. Imbalance in macrophage function or M1 and M2 polarization can result in immune-related diseases like chronic inflammation, cancer, hypersensitivity, and autoimmune disorders [8,9]. Therefore, targeting macrophage activity modulation emerges as a promising approach in developing new therapies for these diseases.
The nuclear factor kappa-light-chain-enhancer of activated B cell (NF-κB) is pivotal in regulating inflammation, particularly in macrophages. NF-κB, comprising p50, p52, p65 (RelA), RelB, and c-Rel subunits, activates genes related to immune response, inflammation, and cell survival by forming homodimers and heterodimers [10]. Activation of NF-κB involves the nuclear translocation of the p65/p50 heterodimer, whereas the p50 homodimer acts as an inhibitor [11]. NF-κB activation occurs via two primary pathways: the canonical (classical) and the non-canonical (alternative) [12]. Differing in activation mechanisms and stimuli, these pathways enable precise and dynamic NF-κB regulation. While the canonical pathway primarily mediates rapid and transient NF-κB activation in response to pro-inflammatory stimuli, the non-canonical pathway is involved in the regulation of adaptive immune responses, lymphoid organ development, and maintenance of immune homeostasis. Induced by cytokines like tumor necrosis factor (TNF)-α and and interleukin (IL)-1, the canonical pathway involves TNF receptor-associated factor 6 (TRAF6) and the IKK complex (IKKα, IKKβ, IKKγ), leading to IκB phosphorylation. Subsequent degradation of IκB liberates p65/p50 heterodimer for nuclear translocation. Initiated by TNF receptor superfamily members like lymphotoxin β receptor (LTβR) or B cell-activating factor receptor (BAFF-R), the non-canonical pathway is then mediated by NF-κB inducing kinase (NIK) and IKKα for phosphorylation and processing of p100 to generate p52. The resulting RelB/p52 heterodimers then translocate to nucleus for transcriptional activation [12]. Dysregulation in either pathway can lead to numerous inflammatory and immune-related diseases.
Long noncoding RNAs (lncRNAs) are known to regulate chromatin organization, gene transcription, RNA splicing, and epigenetic modifications [13,14,15,16]. Additionally, lncRNAs influence cell signaling and post-transcriptional regulation by interacting with signaling mediators and sponging microRNAs (miRNAs) [17,18,19,20,21]. With the growing identification of lncRNAs involved in macrophage activity, an increasing number are recognized for their regulation of NF-κB activity in macrophages. This review specifically addresses the roles of lncRNAs in modulating NF-κB activity within macrophages and their consequential impact on the development and progression of various human diseases.
2. Sepsis
Sepsis is a life-threatening condition marked by a dysregulated immune response to infection, leading to widespread inflammation, organ dysfunction, and potentially organ failure and death [22]. Often caused by bacteria, viruses, fungi, or other pathogens, a prime stimulant is lipopolysaccharide (LPS), an endotoxin found in the cell wall of gram-negative bacteria [23]. The systemic inflammatory response, particularly post-bloodstream infection, triggers an excessive release of pro-inflammatory cytokines and chemokine. These mediators damage endothelial cells lining the blood vessels causing permeability increase, fluid leakage, tissue swelling, and edema [24]. Persistent inflammation can impact multiple organs and systems, with cytokines like TNF-α being key mediators of sepsis [25]. Commonly affected organs include the lungs, heart, kidneys, liver, and even the central nervous system.
NF-κB-mediated inflammation in macrophages, followed by M1 polarization, is pivotal in the pathogenesis of sepsis [26]. Pattern recognition receptors PRRs, including Toll-like receptors (TLRs), allow macrophages to identify pathogen-associated molecular patterns on invading microorganisms [23]. Activation of NF-κB, triggered by LPS interaction with TLR4 via the canonical pathway, upregulates numerous pro-inflammatory genes, such as cytokines, chemokines, adhesion molecules, and enzymes like inducible nitric oxide synthase [27]. NF-κB serves as a primary target for immunomodulatory therapies aimed at regulating inflammation and reestablishing immune equilibrium in many diseases including sepsis (Figure 1). Such strategies might involve inhibiting NF-κB activation or employing targeted therapies to neutralize specific pro-inflammatory cytokines.
The lncRNA nuclear paraspeckle assembly transcript 1 (NEAT1), confined to the nucleus, is instrumental in forming paraspeckles, subnuclear structures involved in antiviral responses [28]. NEAT1 levels notably increase in the sera of sepsis patients and in mouse sepsis models [29,30,31,32]. Induced by LPS, NEAT1 in the human monocytic leukemia cell line THP-1 enhances inflammatory responses by sponging miR-17-5p, thereby stabilizing TLR4 mRNA (the miR-17-5p/TLR4 axis) (Table 1) [32]. In Kupffer cells and the murine macrophages, LPS-induced NEAT1 promotes inflammatory activities via the Let-7q/TLR4 axis (Figure 2) [29,30,31,33]. Additionally, NEAT1 also facilitates M1 polarization in macrophages through the miR-125a-5p/TRAF6/TGF-β-activated kinase 1 (TAK1) axis [34], underscoring its potential as a therapeutic target for sepsis.
Metastasis-associated lung adenocarcinoma transcript 1 (MALAT1), a multifunctional lncRNAs in macrophages, is extensively studied. In late-onset sepsis patients, increased blood MALAT1 levels correlate with disease severity [35]. MALAT1 expression rises in activated macrophages, particularly post-LPS treatment [35,36]. Animal studies show that MALAT1 expression surges with sepsis induction; reducing MALAT1 lessens inflammation and mortality [35,37,38,39], possibly by suppressing M1 and enhancing M2, macrophage polarization [39]. In an LPS-induced septic lung injury model, intravenous MALAT1-specific small interfering RNA (siRNA) decreases inflammatory cytokines and immune cells in bronchoalveolar lavage fluid by inhibiting the p38 mitogen-activated protein kinase (MAPK)/p65 pathway [38]. However, contradicting reports suggest MALAT1 decreases in sepsis, acting as a negative feedback regulator that inhibits NF-κB DNA-binding activity [36,40]. These conflicting findings underscore that need for future research on MALAT1’s role in sepsis.
In sepsis patients, the lncRNA colorectal neoplasia differentially expressed (CRNDE) exhibits elevated expression in peripheral blood, with higher levels correlating to improved survival rates [41]. CRNDE intensifies LPS-induced NF-κB activation and subsequent proinflammatory cytokine release in THP-1 cells via the miR-181-5p/TLR4 axis [41]. Beyond sepsis, CRNDE dysregulation influences macrophage activity in various diseases. For instance, CRNDE levels are higher in the bone marrow of acute myeloid leukemia (AML) patients and inversely correlate with survival rates [42,43]. Elevated CRNDE is also observed in peripheral blood mononuclear cells (PBMCs) from patients with immunoglobulin A nephropathy, where it stabilizes NLR family pyrin domain containing 3 (NLRP3), promoting inflammasome-related proteins and proinflammatory cytokine expression [44]. These findings suggest CRNDE’s role in regulating macrophage inflammation and inflammasome formation across multiple diseases, including sepsis.
Increased levels of lncRNA plasmacytoma variant translocation 1 (PVT1) in sepsis patients correlate with heightened pro-inflammatory mediators and survival rates [45,46]. LPS induces PVT1 expression in THP-1 cells, which in turn amplifies NF-κB activity via p38 stimulation [47]. Elevated PVT1 expression, promoting M1 polarization through the miR-29a/high mobility group box 1 (HMGB1) axis, is observed in heart-infiltrating macrophages of septic mice [48]. HMGB1, released from the cells, can activate TLR4 in both autocrine and paracrine manners [49]. PVT1 is also highly expressed in osteoarthritis patient’ serum and in LPS-stimulated C28/12 chondrocyte cell line, activating TLR4/NF-κB pathway via the miR-93-5p/HMGB1 axis [50]. Additionally, PVT1 levels rise in myocardial tissues and heart infiltrating macrophages during sepsis-induced myocardial injury [48].
Pan et al. reported a significant decrease in lncRNA MEG3 levels in sepsis patients has a prognostic potential [51]. In macrophages, MEG3 overexpression inhibits LPS-induced apoptosis by downregulating BAX and upregulating Bcl-2. It also suppresses inflammatory factors expression by inhibiting NF-κB signaling [51]. This suggest that the reduced MEG3 expression may exacerbate sepsis by increasing inflammation and inhibiting apoptosis in macrophages. Further research is needed to elucidate MEG3’s role in sepsis.
These findings highlight the intricate relationship between lncRNAs and NF-κB in the context of sepsis, impacting inflammatory activation and macrophage polarization. The influence of these lncRNAs on cytokine release, cell polarization, and apoptosis is notable, and their varied expression in sepsis patients suggests potential as biomarkers. Targeting these lncRNAs to regulate NF-κB activation offers promising avenues for immunomodulatory therapies to manage inflammation and restore immune balance in sepsis. However, the contrasting roles of specific lncRNAs, like MALAT1, necessitate further research. A deeper understanding of these lncRNAs' roles could lead to innovative diagnostic and therapeutic strategies, improving management and outcomes in sepsis.
3. Atherosclerosis
Macrophages are central in atherosclerosis development, marked by arterial plaque build-up. The process initiates with low-density lipoprotein (LDL) cholesterol accumulation in arterial walls, undergoing oxidation and eliciting inflammation [52,53]. Modified LDL attracts monocytes from blood, transforming into macrophages in the arterial wall. These macrophages consume oxidized-LDL (oxLDL), forming lipid-laden foam cells and creating fatty streaks, early atherosclerosis signs [54,55]. M1 macrophages exacerbate inflammation by releasing cytokines, attracting more immune cells [56,57]. Chronic inflammation leads to fibrous cap formation over plaques and extracellular matrix accumulation. Macrophages also degrade this matrix, heightening plaque instability, increasing heart attack and stroke risks [54,58]. Macrophages can also contribute to the resolution of inflammation and healing processes [59]. In atherosclerosis, inflammation resolution is overshadowed by ongoing inflammation and plaque growth.
NF-κB, activated by stimuli like oxidative stress, cytokines, and oxLDL, exacerbate atherosclerosis by promoting lipoprotein uptake, foam cells formation, and attracting more immune cells [54,60]. This activation also destabilizes plaques by encouraging matrix metalloproteinases (MMPs) secretion, increasing plaque rupture risks [61]. Chronic NF-κB activation sustains the inflammation characteristic of advanced atherosclerosis in conditions like coronary artery diseases (CADs) and myocardial infarction (MI) [62]. Given its crucial role in macrophage activation atherosclerosis progression, NF-κB present as a potential target for therapies aimed at reducing inflammation and slowing atherosclerosis progression [63].
Increased NEAT1 expression levels in PBMCs and sera of atherosclerosis patients have been noted [64,65]. NEAT1, induced by oxLDL in THP-1 cells, contributes to proinflammatory responses by enhancing p65 phosphorylation, followed by paraspeckle formation [66,67]. It is also induced in bone marrow-derived macrophages (BMDMs) treated with titanium particles and promotes NF-κB activation, NLRP3 inflammasome formation, and M1 polarization via the miR-188-5p/ Bruton’s tyrosine kinase (BTK) axis [68]. NEAT1 also stimulates proinflammatory cytokine and reactive oxygen species (ROS) production, subsequent foam cell formation by sponging miR-342-3p in THP-1 cells [67] or miR-128 in murine macrophage-like cell line RAW264.7 [69]. These reports agree with NEAT1 being expressed in activated macrophages and enhancing proinflammatory changes. One contradicting study, however, reported decreased NEAT1 in PBMCs of post-MI patients and enhanced macrophage inflammation in NEAT1-knockout mice [70].
Elevated lncRNA PVT1 levels have been detected in the serum of atherosclerosis patients [71]. Inhibiting PVT1 in animal models reduces atherosclerotic plaques by increasing HDL levels and suppressing the MAPK/NF-κB pathway and pro-atherogenic factors [71]. PVT1 is also upregulated in various cancers, particularly lung cancer, affecting key pathways such as cell proliferation, apoptosis evasion, angiogenesis, and epithelial-mesenchymal transition [72]. However, its expression pattern in TAMs or its role in NF-κB regulation within tumor microenvironment (TME) remains unexplored.
In serum samples of atherosclerosis patients and during oxLDL-induced THP-1 cell foam cell differentiation, there’s a notable increase in lncRNA small nucleolar RNA host gene (SNHG)16 and a decrease in miR-17-5p [73]. SNHG16 amplifies macrophage proliferation and pro-inflammatory responses in atherosclerosis through the miR-17-5p/NF-κB axis [73]. It also boosts NF-κB activity, as well as epithelial-mesenchymal transition and metastasis of hepatocellular carcinoma (HCC) cells via the miR-605-3p/TRAF6 axis [74]. In diabetic environment, SNHG16 enhances NF-κB activity through the miR-212-3p/p65 axis [75], indicating its role in exacerbating macrophage inflammation in various diseases including atherosclerosis.
LncRNA X-inactive specific transcript (XIST), known for its role in X-chromosome inactivation, has been found elevated in the serum of atherosclerosis patients, oxLDL-treated vascular smooth muscle cells, and the U937 human monocytic leukemia cell line [76]. XIST influences atherosclerosis by promoting proliferation and inhibiting apoptotic cell death through the miR-599/TLR4 axis [76]. This finding aligns with other studies that show apoptosis inhibition aggravates atherogenesis by increasing macrophage proliferation and plaque formation [77,78].
LncRNA H19 is found at elevated levels in the serum of atherosclerosis patients [79,80,81,82]. OxLDL stimulates H19 expression in macrophages [83], aorta vascular smooth muscle cells[79,80], and human umbilical vein endothelial cells (HUVECs) [84]. In macrophages, H19 augments oxLDL-induced lipid accumulation, ROS generation, and NF-κB activation [83,85]. Similarly, in HUVECs, H19 heightens NF-κB activation by increasing p38 and p65 activity [86]. These findings suggest that H19 could be a promising therapeutic target for atherosclerosis treatment.
In atherosclerosis and CAD patients, MALAT1 levels rise and subsequently decrease post-treatment [87,88,89]. MALAT1 impacts various macrophage processes like foam cell formation, autophagy, and pyroptosis [90,91,92]. OxLDL prompts NF-κB-dependent MALAT1 expression in THP-1 cells. MALAT1 then enhances lipid uptake and foam cell formation then by promoting scavenger receptor CD36 expression [88,90,91]. MALAT1 also enhances NF-κB activation and subsequent inflammation by sponging miR-330-5p [91]. Further, oxLDL-induced autophagy in macrophage is mediated by MALAT1, which activates MAPK/NF-κB pathway and inhibits sirtuin 1 (SIRT1), a key transcription factor deacetylase [92,93]. NLRP3 inflammasome-mediated pyroptosis, a programmed cell death as a defense mechanism against intracellular pathogens, is also influenced by MALAT1 [94]. In diabetic atherosclerosis models, a cinnamic acid derivative reduces inflammasome activation and pyroptotic by suppressing MALAT1 [95]. Extracellular vesicles (EVs) such as exosomes are crucial for cell-to-cell communication, transferring proteins and lncRNAs [96]. M1 macrophages have been found to release MALAT1-containing EVs, which regulate myocyte proliferation and angiogenesis in MI models [97]. These findings underscore MALAT1’s role in atherosclerosis, it is upregulated in activated macrophages and influence various processes including lipid uptake, foam cell formation, and cell death.
However, contrary reports exist regarding the role of MALAT1 in atherosclerosis. In atherosclerosis patients and oxLDL-treated THP-1 cells, MALAT1 levels are observed to decrease [88]. Reduced MALAT1 leads to increased lipid and total cholesterol accumulation in THP-1 cells via the miR-17-5p/ATP-binding cassette subfamily A member 1 (ABCA1) axis [88]. ABCA1 is known to facilitate cholesterol efflux, thereby reducing foam cell formation [98]. Additionally, MALAT1 deficiency in certain mouse models has been linked to accelerated macrophage inflammation and atherosclerosis [99]. Exosomal MALAT1 from oxLDL-treated HUVECs promotes a transition from M1 to M2 macrophages [100]. These findings suggest potential anti-atherogenic properties of MALAT1, highlighting the need for further research to clarify its role in atherosclerosis.
Notably, the expression levels of lncRNA HOX transcript antisense intergenic RNA (HOTAIR) are decreased in peripheral blood lymphocytes of atherosclerosis patients and oxLDL-treated RAW264.7 cells [101]. HOTAIR overexpression reduces pro-inflammatory cytokine expression while boosting anti-inflammatory cytokines, achieved by inhibiting NF-κB activity. This suppression occurs through HOTAIR’s enhancement of fragile X-related protein 1 (FXR1) levels, a protein moving between nucleus and cytoplasm and associating with polyribosomes [101,102].
These observations underscore the complex relationship between various lncRNAs and NF-κB in atherosclerosis. These lncRNAs impact crucial aspects such as lipid uptake, foam cell formation, inflammation, and cell death in macrophages. Given their link to NF-κB activation, targeting these lncRNAs for NF-κB modulation presents a promising approach in managing atherosclerosis by restoring immune equilibrium and curbing inflammatory activation. Despite the conflicting roles of some lncRNAs like MALAT1 and HOTAIR, their significant influence on macrophage function and disease progression is evident. Further research is essential to unravel the full potential of these lncRNAs in atherosclerosis treatment.

Table 1.
LncRNAs regulating macrophage NF-κB activity in diseases.
| LncRNA | Disease | Target | Function | Ref |
|---|---|---|---|---|
| Group 1. LncRNAs directly affecting NF-κB activity | ||||
| CARLR | Celiac disease | p65 | LPS-induced, regulates p65 translocation, promotes proinflammatory cytokines production | [103] |
| COX2 | Neuroinflammation | p65 | LPS-induced, promotes p65 translocation, enhances inflammasome formation, suppresses autophagy | [104] |
| COX2 | SLE | SWI/SNF/NF-κB | LPS-induced, enhances the expression of NF-κB-induced late inflammatory genes | [105] |
| MALAT1 | Sepsis | p65/p50 | LPS-induced, retrains NF-κB from promoter, regulates proinflammatory cytokine expression | [36] |
| PACER | Cancer | p50 | LPS-induced, sequesters p50 and promotes p300-mediated histone acetylation, enhances COX2 expression | [106] |
| PINT | Cancer | p65 and EZH2 | LPS-induced, bridges p65 and EZH2, activates TNFα transcription | [107] |
| Group 2. LncRNAs affecting pathways that regulate NF-κB activity | ||||
| CHRF | Cancer | miR-489/MyD88 | Silica-induced, promotes inflammatory responses and fibrosis. | [108] |
| CRNDE | Cancer | miR-181a-5p/TLR4 | LPS-induced, promotes inflammatory responses. Also increased in AML and IgA nephropathy | [41,44] |
| MALAT1 | Atherosclerosis | SIRT1/MAPK | ox-LDL-induced, promotes autophagy, reduces apoptosis | [93] |
| MIR222HG | Allergic rhinitis | miR-146a-5p/TRAF6 | Deceased in patients, causing the dominance of type 2 response. Promotes M1 and suppresses M2 polarization | [109] |
| NAIL | Ulcerative colitis | Wip1 | LPS-induced, promotes p65 phosphorylation by blocking Wip1 action, increases inflammatory response | [110] |
| NEAT1 | Osteolysis | miR-188-5p/ BTK KLF/BTK |
Induced by titanium particles, activates inflammasome and NF-κB, enhances M1 polarization | [68] |
| NEAT1 | Sepsis | miR-17-5p/TLR4 | LPS-induced, promotes inflammatory response by stabilizing TLR4 mRNA | [32] |
| NEAT1 | Sepsis | let-7a/TLR4 | LPS-induced, promotes inflammatory response by stabilizing TLR4 mRNA | [29] |
| NKILA | Asthma | IκB | Anti-inflammation by inhibiting IκB phosphorylation, induces M2 polarization | [111,112] |
| PVT1 | Sepsis | miR-29a/ HMGB1/TLR4 | LPS-induced, promotes inflammation and M1 polarization. Also increased in osteoarthritis patients | [48,50] |
| SNHG1 | Cancer/Sepsis | HMGB1/TLR4 | Enhances TLR4 signaling by interaction with HMGB1, promotes M1 polarization | [113] |
| XIST | Atherosclerosis | miR-599/TLR4 | OxLDL-induced in macrophages/vascular smooth muscle cells, promotes proliferation and suppresses apoptosis | [76] |
| Group 3. LncRNAs affecting NF-κB activity with unknown mechanism of action | ||||
| DCST1-AS1 | Cancer | p65 | Activates NF-κB signaling pathway in both cancer cells and macrophages. Promotes M2 polarization | [114] |
| FTX | Cirrhosis | NF-κB | Anti-inflammatory function. Decreased in patients, causing enhancement in inflammation | [115] |
| HOTAIR | Cancer | IκBα | LPS-induced, activates NF-κB by degrading IκB, regulates metabolic reprogramming by inducing GLUT1 | [116] |
| HOTAIR | Atherosclerosis | FXR1 | Anti-inflammation by inactivating NF-κB via FXR1. Decreased in patients and oxLDL-treated macrophages. | [101,102] |
| NEAT1 | Atherosclerosis | p65, ERK | ox-LDL-induced, regulates p65 and ERK phosphorylation, promotes TNF-α secretion. | [28,66] |
| MEG3 | Sepsis | p65 | Decreased in sepsis patients. LPS-induced, inhibits p65 phosphorylation. Downregulates inflammation and apoptosis. | [51] |
| SNHG16 | Atherosclerosis | miR-17-5p | ox-LDL-induced, promotes proliferation and inflammatory response. Also increased in cancer and diabetes | [73,74,75] |
| Group 4. LncRNAs transferred to macrophages via exosomes or EVs | ||||
| FGD5-AS1 | Cancer | P300/STAT3/NF-κB | Contained in exosomes from pancreatic cancer cells, activates STAT3/NF-κB, promotes M2 polarization | [117] |
| AP000439.2 | Cancer | STAT3, P65 | Contained in exosomes from cancer cells, promotes macrophage M2 polarization | [118] |
| GAS5 | Allergic rhinitis | mTORC1/ULK1/ATG13 | Activate NF-κB and promote M1 polarization by suppressing autophagy-dependent degradation of IKKa/b | [119] |
| HOTTIP | Cancer | - | Contained in M1-derived exosomes. Suppresses cancer growth via TLR5 activation. Promotes M1 polarization | [120] |
| MALAT1 | Acute pancreatitis | miR-181a-5p /HNGB1/TLR4 |
Carried by EVs originating from pancreatic cancer cells, promotes M1 polarization | [121] |
4. Cancer
The role of macrophage inflammation in cancer is multifaceted and contradictory. M1 macrophages, typically anti-tumorigenic, can attack tumor cells and stimulate immune responses. Conversely, M2 macrophages often aid tumor growth by supporting angiogenesis, suppressing immune responses, and facilitating tissue remodeling. [122]. Generally, tumor-associated macrophages (TAMs) exhibit an M2 phenotype, supporting tumor growth and metastasis, and contributing to an immunosuppressive tumor environment [123,124]. Given their significant impact on cancer progression, TAMs are being investigated as therapeutic targets, with strategies focusing on inhibiting their tumor-promoting functions or reprogramming them to combat tumors.
The activation of NF-κB in macrophages plays a crucial role in cancer development and progression. In TAMs, NF-κB activation leads to the production of cytokines, growth factors, and enzymes that promote tumor growth and suppress anti-tumor immune responses [125,126]. NF-κB can also alter the immune microenvironment, potentially inducing immune checkpoint molecules that weaken the immune response against tumors [127]. Additionally, NF-κB-activated macrophages can produce angiogenic factors, aiding tumor vascularization [128,129,130]. They can also stimulate matrix metalloproteinases (MMPs), breaking down extracellular matrix barriers and facilitating cancer cell spread [131,132]. Thus, macrophage NF-κB is implicated in various aspects of cancer progression and targeting macrophage NF-κB has emerged as a prominent focus in cancer treatment strategies [12].
LncRNA DC-STAMP domain containing 1-antisense 1 (DCST1-AS1) has been investigated in various cancers including gastric, colorectal, cervical, breast, glioblastoma, endometrial, and HCC [133,134,135,136,137,138,139]. In these cancers, increased DCST1-AS1 expression correlates with larger tumors and shorter survival and DCST1-AS1 promotes cancer cell proliferation and metastasis, and inhibits apoptosis, by sponging miRNAs [133,134,135,136,137,138,139]. Notably, in oral squamous cell carcinoma, DCST1-AS1 advances tumor progression by enhancing NF-κB activity in cancer cells and macrophages [114]. Elevated DCST1-AS1 in cancer cells and M2 macrophages is linked to tumor growth and cancer cell proliferation. NF-κB antagonists revealed that DCST1-AS1 enhances cancer progression and M2 macrophage polarization through NF-κB-mediated mechanisms [114].
The lncRNA FGD5 antisense RNA 1 (FGD5-AS1) shows elevated levels in non-small cell lung cancer and pancreatic cancer, correlating with metastasis and poor prognosis [117,140]. FGD5-AS1-containing exosomes from these cancers induce M2 macrophage polarization [117]. FGD5-AS1 links acetyltransferase p300, STAT3, and NF-κB, leading to acetylated STAT3/p65 complex and transcriptional activation [117,141]. STATs are crucial transcription factors in macrophage polarization, with STAT1 being integral to M1, and STAT3/6 to M2 polarization. [142]. In cervical cancer, FGD5-AS1, via the miR-129-5p/bone marrow stromal cell antigen 2 (BST2) axis, promotes tumor growth and M2 polarization [143]. BST2, a lipid raft-associated protein, is implicated in cell proliferation and immune response [144,145]. Collectively, FGD5-AS1 augments tumor growth by enhancing cancer progression and M2 macrophage polarization.
The lncRNA AP000439.2 has recently been identified as a prognostic marker for renal cell carcinoma (RCC) patient survival [146,147]. Exosomes from human RCC cell lines have been shown to induce M2 polarization in co-cultured THP-1 cells [118]. AP000439.2 promotes M2 polarization through the phosphorylation of STAT3 and NF-κB p65 subunit, which, in turn, enhances the migration potential of cocultured cancer cell lines. The impact of exosomal AP000439.2 on macrophage M2 polarization and RCC growth has been confirmed in a xenograft tumor mouse model [118].
LncRNA Five Prime to Xist (FTX), an evolutionarily conserved regulator of XIST expression, is associated with various conditions including malignancies, endometriosis, and stroke, functioning through miRNA sponging [148,149]. Liu et al. observed decreased FTX levels in cirrhosis patients, linking it to abnormal activation of CD14+ CD16+ monocytes via the miR-545/T cell immunoglobulin and mucin domain 3 (Tim-3) axis [115]. Moreover, FTX suppression in THP-1 cells increases NF-κB activity and proinflammatory cytokine expression, suggesting that a reduction in FTX might accelerate tumor progression by enhancing inflammation in the tumor microenvironment (TME)
LncRNA HOTAIR, known for its role in gene regulation and epigenetic modifications, is implicated in various human diseases [150]. It is often overexpressed in cancer, contributing to tumor progression, metastasis, and poor prognosis by altering gene expression related to the cell cycle, apoptosis, and metastasis [150,151]. HOTAIR is also associated with central nervous system disorders, fibrosis, and inflammatory conditions, impacting cellular processes and immune responses [152,153,154,155]. It regulates glucose transporter isoform 1 (GLUT1) expression in human neuroblastoma cells and macrophages by stimulating NF-κB activity, suggesting a role in metabolic reprogramming in cancer [116,156]. In addition, inflammatory activation of macrophages triggers HOTAIR expression, which then promotes NF-κB activation and cytokine gene expression by aiding in the degradation of IκBα [157]. HOTAIR's expression pattern in cancer tissue macrophages remains unexplored and warrants future investigation.
Elevated levels of lncRNA HOXA transcript at the distal tip (HOTTIP) have been observed in AML patients and cell lines, such as U937 and THP-1 [158]. HOTTIP facilitates cell proliferation via the miR-608/DET1- and DDB1-associated 1 (DDA1) axis, with DDA1 being a gene known for its oncogenic properties [158,159]. In squamous cell carcinoma, M1-derived exosomes containing HOTTIP inhibit cancer cell proliferation and induce apoptosis by activating the TLR5/NF-κB pathway [120]. Additionally, exosomal HOTTIP influences the M1 polarization of circulating monocytes [120]. The comprehensive role of HOTTIP in cancer progression remains an area for future exploration.
Cyclooxygenase (COX)2, linked with inflammation in immune cells, is implicated in several cancers [160]. LncRNA p50-associated COX2 extragenic RNA (PACER), located upstream of the COX2 promoter, regulates COX2 expression [106]. PACER, through its association with p50, facilitates p65/p50 heterodimer binding to the COX2 promoter, recruiting p300 histone acetyltransferase [106]. Its expression is upregulated in various cancer tissues, influencing COX2 and PGE2 synthesis and cancer cell proliferation, migration, and invasion [161,162,163]. PACER's role extends beyond cancer, regulating COX2 in cells infected with Mycoplasma pneumoniae and showing altered expression in bipolar disorder patients [164,165].
LncRNA cardiac hypertrophy-related factor (CHRF) functions as an oncogene, promoting migration and invasion in various tumor types [166]. In a silica-induced pulmonary fibrosis mouse model, CHRF activates inflammatory and fibrotic pathways via the miR-489/MyD88 and miR-489/SMAD3 axes, with SMAD3 being an adaptor in receptor-regulated signaling [108,167]. CHRF's pro-inflammatory effects are also observed in LPS-induced acute lung injury [168]. However, its specific role in macrophage inflammation within the TME remains unclear, necessitating further research.
LncRNA SNHG1, commonly overexpressed in various cancers as an oncogene, affects cellular signaling via interactions with miRNAs and signaling regulators [169]. In cholangiocarcinoma cell lines, SNHG1 is associated with increased proliferation and invasion, mediated by NF-κB activation through the miR-140/TLR4 axis, contributing to an inflammatory TME [170]. In an LPS-induced acute lung injury model, SNHG1 is upregulated in M1 polarized THP-1 cells, enhancing NF-κB activation and inflammation through interaction with HMGB1 [113]. However, SNHG1's specific role in macrophage-related TME remains unexplored.
These findings highlight the intricate relationship between macrophages, lncRNAs, and NF-κB in cancer, affecting cell proliferation, invasion, inflammation, and macrophage polarization. The dichotomy of macrophages, especially TAMs, underscores their potential as therapeutic targets. Their influence extends to inflammation, TME modulation, angiogenesis, and immunosuppression, making them key in the interplay between cancer cells and the immune system. Understanding lncRNA-driven macrophage NF-κB regulation is essential for developing targeted cancer therapies. Despite advances, many aspects of lncRNA functions in cancer and inflammation require further exploration, presenting exciting opportunities for future research and potential therapeutic interventions.
5. Autoimmunity and hypersensitivity
Increased GAS5 levels have been found in exosomes from nasal mucus of allergic rhinitis patients and a mouse model, promoting macrophage NF-κB activation and M1 polarization.[119,171]. GAS5 influences this by inhibiting mTORC1/ULK1/ATG13-mediated autophagy, leading to NF-κB activation [119]. Specifically, GAS5's activation of NF-κB signaling occurs through the suppression of autophagy-dependent degradation of IKKα/β in macrophages [119]. GAS5 also enhances M1 macrophage polarization, observed in hyperglycemia-induced differentiation in diabetics and in pneumonia-affected children's macrophages [172,173]. It suppresses the Janus kinase 2/STAT3 pathway, promoting M1 while inhibiting M2 polarization via the miR-455-5p/SOCS3 axis [173]. In diabetes, hyperglycemia-induced GAS5 expression shifts polarization from M2 to M1 by upregulating STAT1 [172]. This shift is significant since M1 macrophage-driven inflammation worsens diabetic wounds, suggesting that reducing GAS5 could facilitate wound healing by promoting an M1 to M2 transition. On the other hand, microglia exhibit increased GAS5 expression in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE) mouse models [174]. GAS5 hinders M2 polarization by recruiting the polycomb repressive complex 2 (PRC2), suppressing topoisomerase-related function 4 transcription, crucial for M2 polarization [174]. Ito et al. found that reducing GAS5 expression leads to M2b polarization, while increasing it has the opposite effect [175]. GAS5 also plays a role in regulating NLRP3 inflammasome formation in cardiomyocytes [176]. In contrast, GAS5 expression levels are decreased in various cancer tissues [177,178]. This decrease is attributed to GAS5's ability to induce apoptosis and inhibit tumor proliferation and metastasis through interactions with miRNAs and proteins. The M1-enhancing property of GAS5 might contribute to its reduced expression in cancer tissues, necessitating further investigation. Overall, research indicates that GAS5 boosts NF-κB-mediated inflammation and shifts macrophage polarization towards M1, while inhibiting GAS5 has the opposite effect in various diseases.
LncRNA-Cox2, located near the COX2 gene [179], is linked with chronic inflammatory diseases and innate immune activation [180,181]. In Systemic Lupus Erythematosus (SLE) patients, elevated serum lncRNA-Cox2 levels correlate with neurological symptoms [182]. It is highly induced in an NF-κB-dependent manner in activated macrophages and interacts with the switch/sucrose nonfermentable (SWI/SNF) chromatin remodeling complex [179,183]. This interaction enhances the expression of NF-κB-induced late inflammatory genes [105]. In neuroinflammation, lncRNA-Cox2 promotes NLRP3-inflammasome formation and hinders autophagy by binding and facilitating the p65 subunit's nuclear transport [104]. However, its role in inflammation is subject to debate, as it appears to suppress inflammation in M1 macrophages and BMDMs in septic mouse models [183,184], indicating a complex influence in inflammatory contexts.
During allergic disease development, a type 2 immune response led by Th2 cells, eosinophils, basophils, and M2 macrophages is predominant [185]. In allergic rhinitis clinical samples and animal models, a downregulation of lncRNA miR222 host gene (MIR222HG) was observed, linking it to the type 2 response [109]. MIR222HG is upregulated in M1 macrophages but downregulated in M2 macrophages, with its overexpression reducing M2 polarization and allergic inflammation. MIR222HG activates the NF-κB pathway via the miR-146a-5p/TRAF6 axis [109]. This suggests that MIR222HG downregulation in allergic rhinitis contributes to pathogenesis by favoring M2 macrophage polarization, and its overexpression might alleviate the condition.
Celiac disease, characterized by constant NF-κB activation, shows increased cardiac and apoptosis-related lncRNA (CARLR) expression in patient samples [103,186]. CARLR expression rises in THP-1 cells post-LPS stimulation. CARLR interacts with activated NF-κB following IκB dissociation, boosting the expression of cytokines such as IL-1β and COX2, also elevated in celiac disease samples [103]. This suggests a crucial role of CARLR in the disease's inflammatory response.
NAIL, an evolutionarily conserved lncRNA, exhibits increased expression in inflamed ulcerative colitis patient samples compared to non-inflamed ones [110]. It activates NF-κB by binding to Wip1 phosphatase, thereby removing Wip1 from its substrates, including p38 MAPK and p65, which plays a key role in the inflammatory process of ulcerative colitis.
LncRNA P53-induced transcript (PINT) is upregulated in the intestinal mucosa of ulcerative colitis patients and in inflammatory bowel disease mouse models [107]. In macrophages, LPS induces PINT expression in an NF-κB-dependent manner. Acting as a scaffold, PINT enables the binding of p65 and EZH2, a histone methyltransferase and epigenetic regulator, to NF-κB sites on target promoters [187]. This complex stimulates TNF-α expression while inhibiting other proinflammatory mediators like CCL2/7 and intercellular adhesion molecule-1.
MALAT1, with high expression in severe acute pancreatitis patients' plasma and corresponding mouse models, plays a critical role in this condition [121,188]. Its suppression reduces tissue injury and inflammation. Additionally, MALAT1 is found in serum EVs of acute pancreatitis patients, stimulating macrophages to activate TLR4/NF-κB signaling and M1 polarization via the miR-181a-5p/HMGB1 axis [121,188]. In endothelial progenitor cell-derived exosomes, MALAT1 triggers BMDM differentiation into osteoclasts, binding to miR-124 [189]. High glucose levels in macrophages also prompt MALAT1-containing exosome secretion [190], highlighting its role in cellular communication through EVs.
These research underscores the pivotal roles of various lncRNAs in autoimmune, hypersensitivity, and inflammatory conditions. These lncRNAs modulate NF-κB activity, either directly or indirectly, impacting macrophage polarization and immune responses. The intricate relationship between these lncRNAs and macrophage NF-κB dynamics opens avenues for targeted therapies in such diseases. Given NF-κB's association with inflammation, macrophage polarization, apoptosis, and pyroptosis, modulating lncRNA functions could favorably alter disease progression by influencing NF-κB activity.
6. Discussion
Research targeting lncRNAs in disease treatment shows promise, particularly through suppression or overexpression in animal models. For instance, suppressing MALAT1 with siRNA mitigates sepsis-related inflammation [37,38,39], while its overexpression exacerbates atherosclerosis severity [91]. Similarly, altering the M1/M2 macrophage polarization balance can influence disease progression. Knockdown of GAS5 in diabetic wound healing models encourages M1 to M2 transition, enhancing wound healing [172], and its suppression reduces EAE progression by inhibiting M1 polarization [174]. Reducing lncRNA-Cox2 in HCC models strengthens M2 macrophage polarization, promoting tumor growth [191].
LncRNA-targeting therapies primarily use nucleic acid-based methods like antisense oligonucleotides (ASOs), RNA interference with siRNA or shRNA, and innovative approaches like CRISPR/Cas and exosome-mediated transfer [192,193]. While clinical trials have mainly focused on miRNAs, the exploration of lncRNAs as diagnostic markers and therapeutic targets is growing. For example, MALAT1 and lncRNA prostate cancer antigen 3 are being studied as diagnostic markers for prostate cancer. Trials involving ASncmtRNA-targeting ASOs, such as Andes-1537 [194], are assessing safety and efficacy in various cancers, indicating a progressing field in lncRNA-based treatments.
The expression of lncRNAs that regulate macrophage NF-κB activity is implicated in various human diseases. Animal studies targeting these lncRNAs have yielded promising results, positioning them as potential future therapeutic targets. The primary focus will be on reducing macrophage inflammation in chronic inflammatory diseases, hypersensitivity, autoimmune disorders, and cancer. A secondary objective will involve balancing M1 and M2 macrophage polarization in these conditions. While the use of NF-κB-targeting lncRNAs as therapeutic agents is still in its early stages, the critical role of macrophage NF-κB in disease pathogenesis makes these targets particularly promising.
7. Conclusion and future directions
This review has comprehensively explored the dynamic interplay between lncRNAs, NF-κB activation, and macrophage function across various diseases, including sepsis, atherosclerosis, cancer, autoimmune, and hypersensitivity conditions. It has revealed the critical roles of different lncRNAs in modulating inflammatory responses and macrophage polarization, presenting them as potential biomarkers and therapeutic targets. The contrasting roles of certain lncRNAs, such as MALAT1 and GAS5, in different disease contexts highlight the complexity of their functions and necessitate further research for better understanding and clinical application.
Future research should focus on unraveling the detailed mechanisms of how lncRNAs influence NF-κB pathways and macrophage polarization in various diseases. The development of more specific and effective lncRNA-targeted therapies, possibly using advanced techniques like CRISPR/Cas9 and exosome-mediated delivery, is another crucial direction. Clinical trials focusing on the therapeutic potential of lncRNAs, their safety, and efficacy in human diseases will be vital. Additionally, exploring the role of lncRNAs in other immune cells and their interaction with macrophages could provide a more comprehensive understanding of immune regulation and disease pathogenesis.
Author Contributions
Conceptualization, W.H.L. and K.S.; writing—original draft preparation, J.J.S., H.S.S., J.P., and I.A.; writing—review and editing, W.H.L. and K.S.; supervision, W.H.L.; project administration, W.H.L.; funding acquisition, W.H.L. and K.S. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (Ministry of Science and ICT) (No. 2022R1A2C1010005).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data used in this article are sourced from materials mentioned in the References section.
Conflicts of Interest
The authors declare no conflict of interest.
Declaration of generative AI and AI-assisted technologies in the writing process
During the preparation of this work the authors used ChatGPT to improve the quality of English writing. After using this service, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
Abbreviations
ABCA1, ATP-binding cassette subfamily A member 1; AML, acute myeloid leukemia; ASncmtRNA, antisense noncoding mitochondrial RNA; ASO, antisense oligonucleotide; BMDM, bone marrow-derived macrophage; BST2, bone marrow stromal cell antigen 2; BTK, Bruton’s tyrosine kinase; CAD, coronary artery disease; CAS, CRISPR-associated protein; COX, cyclooxygenase; CRISPR, clustered regularly interspaced short palindromic repeats; CRNDE, colorectal neoplasia differentially expressed; DDA1, DET1- and DDB1-associated 1; EAE, experimental autoimmune encephalomyelitis; EV, extracellular vesicle; EZH2, enhancer of zeste homolog 2; FGD5-AS1, FGD5 antisense RNA 1; GAS5, growth arrest-specific 5; HCC, hepatocellular carcinoma; HMGB1, high mobility group box 1; HOTAIR, HOX transcript antisense RNA; HOTTIP, HOXA transcript at the distal tip; HUVEC, human umbilical vein endothelial cell; IL, interleukin; LDL, low-density lipoprotein; lncRNA, long noncoding RNA; LPS, lipopolysaccharide; MALAT1, metastasis-associated lung adenocarcinoma transcript 1; MAPK, mitogen-activated protein kinase; MI, myocardial infarction; miRNA, microRNA; MMP, matrix metalloproteinase; MyD88, myeloid differentiation primary response 88; NEAT1, nuclear paraspeckle assembly transcript 1; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3, NLR family pyrin domain containing 3; oxLDL, oxidized-LDL; PBMC, peripheral blood mononuclear cell; PINT, P53-induced transcript; PRC2, polycomb repressive complex 2; PVT1, plasmacytoma variant translocation 1; ROS, reactive oxygen species; shRNA, short hairpin RNA; siRNA, small interfering RNA; SMAD3, small mothers against decapentaplegic homolog 3; SOCS, suppressor of cytokine signaling; STAT, signal transducer and activator of transcription; TAK1, TGF-β-activated kinase 1; TAM, tumor-associated macrophage; TLR, Toll-like receptor; TNF-α, tumor necrosis factor-α ; TRAF6, TNF receptor-associated factor 6; ULK1, Unc51-like autophagy activating kinase 1; XIST, X-inactive specific transcript.
References
- Plytycz, B.; Seljelid, R. From inflammation to sickness: historical perspective. Arch Immunol Ther Exp (Warsz) 2003, 51, 105–109. [Google Scholar]
- Li, X.; Ma, L. From biological aging to functional decline: Insights into chronic inflammation and intrinsic capacity. Ageing Res Rev 2024, 93, 102175. [Google Scholar] [CrossRef]
- Lowe, D.B.; Storkus, W.J. Chronic inflammation and immunologic-based constraints in malignant disease. Immunotherapy 2011, 3, 1265–1274. [Google Scholar] [CrossRef]
- Furman, D.; Campisi, J.; Verdin, E.; Carrera-Bastos, P.; Targ, S.; Franceschi, C.; Ferrucci, L.; Gilroy, D.W.; Fasano, A.; Miller, G.W.; et al. Chronic inflammation in the etiology of disease across the life span. Nat Med 2019, 25, 1822–1832. [Google Scholar] [CrossRef]
- Motwani, M.P.; Gilroy, D.W. Macrophage development and polarization in chronic inflammation. Semin Immunol 2015, 27, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively Activated Macrophages. Front Immunol 2019, 10, 1084. [Google Scholar] [CrossRef]
- Tardito, S.; Martinelli, G.; Soldano, S.; Paolino, S.; Pacini, G.; Patane, M.; Alessandri, E.; Smith, V.; Cutolo, M. Macrophage M1/M2 polarization and rheumatoid arthritis: A systematic review. Autoimmun Rev 2019, 18, 102397. [Google Scholar] [CrossRef] [PubMed]
- Yunna, C.; Mengru, H.; Lei, W.; Weidong, C. Macrophage M1/M2 polarization. Eur J Pharmacol 2020, 877, 173090. [Google Scholar] [CrossRef] [PubMed]
- Funes, S.C.; Rios, M.; Escobar-Vera, J.; Kalergis, A.M. Implications of macrophage polarization in autoimmunity. Immunology 2018, 154, 186–195. [Google Scholar] [CrossRef]
- Lawrence, T.; Gilroy, D.W.; Colville-Nash, P.R.; Willoughby, D.A. Possible new role for NF-kappaB in the resolution of inflammation. Nat Med 2001, 7, 1291–1297. [Google Scholar] [CrossRef]
- Napetschnig, J.; Wu, H. Molecular basis of NF-kappaB signaling. Annu Rev Biophys 2013, 42, 443–468. [Google Scholar] [CrossRef]
- Yu, H.; Lin, L.; Zhang, Z.; Zhang, H.; Hu, H. Targeting NF-kappaB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct Target Ther 2020, 5, 209. [Google Scholar] [CrossRef]
- Herman, A.B.; Tsitsipatis, D.; Gorospe, M. Integrated lncRNA function upon genomic and epigenomic regulation. Mol Cell 2022, 82, 2252–2266. [Google Scholar] [CrossRef]
- Huang, W.; Li, H.; Yu, Q.; Xiao, W.; Wang, D.O. LncRNA-mediated DNA methylation: an emerging mechanism in cancer and beyond. J Exp Clin Cancer Res 2022, 41, 100. [Google Scholar] [CrossRef]
- Senmatsu, S.; Hirota, K. Roles of lncRNA transcription as a novel regulator of chromosomal function. Genes Genet Syst 2021, 95, 213–223. [Google Scholar] [CrossRef]
- Wang, C.; Wang, L.; Ding, Y.; Lu, X.; Zhang, G.; Yang, J.; Zheng, H.; Wang, H.; Jiang, Y.; Xu, L. LncRNA Structural Characteristics in Epigenetic Regulation. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Sun, W.; Guo, Z.; Zhang, J.; Yu, H.; Liu, B. Mechanisms of lncRNA/microRNA interactions in angiogenesis. Life Sci 2020, 254, 116900. [Google Scholar] [CrossRef] [PubMed]
- Ferrè, F.; Colantoni, A.; Helmer-Citterich, M. Revealing protein-lncRNA interaction. Brief Bioinform 2016, 17, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Khorkova, O.; Hsiao, J.; Wahlestedt, C. Basic biology and therapeutic implications of lncRNA. Adv Drug Deliv Rev 2015, 87, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, E.L.; Belhocine, M.; Dao, L.T.; Puthier, D.; Spicuglia, S. [Functions of lncRNA in development and diseases]. Med Sci (Paris) 2014, 30, 790–796. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.X.; Koirala, P.; Mo, Y.Y. LncRNA-mediated regulation of cell signaling in cancer. Oncogene 2017, 36, 5661–5667. [Google Scholar] [CrossRef]
- Pravda, J. Sepsis: Evidence-based pathogenesis and treatment. World J Crit Care Med 2021, 10, 66–80. [Google Scholar] [CrossRef] [PubMed]
- Cristofaro, P.; Opal, S.M. The Toll-like receptors and their role in septic shock. Expert Opin Ther Targets 2003, 7, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Chousterman, B.G.; Swirski, F.K.; Weber, G.F. Cytokine storm and sepsis disease pathogenesis. Semin Immunopathol 2017, 39, 517–528. [Google Scholar] [CrossRef]
- Tracey, K.J.; Beutler, B.; Lowry, S.F.; Merryweather, J.; Wolpe, S.; Milsark, I.W.; Hariri, R.J.; Fahey, T.J., 3rd; Zentella, A.; Albert, J.D.; et al. Shock and tissue injury induced by recombinant human cachectin. Science 1986, 234, 470–474. [Google Scholar] [CrossRef]
- Chen, X.; Liu, Y.; Gao, Y.; Shou, S.; Chai, Y. The roles of macrophage polarization in the host immune response to sepsis. Int Immunopharmacol 2021, 96, 107791. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.F.; Malik, A.B. NF-kappa B activation as a pathological mechanism of septic shock and inflammation. Am J Physiol Lung Cell Mol Physiol 2006, 290, L622–L645. [Google Scholar] [CrossRef]
- Naganuma, T.; Nakagawa, S.; Tanigawa, A.; Sasaki, Y.F.; Goshima, N.; Hirose, T. Alternative 3'-end processing of long noncoding RNA initiates construction of nuclear paraspeckles. EMBO J 2012, 31, 4020–4034. [Google Scholar] [CrossRef]
- Zhang, C.C.; Niu, F. LncRNA NEAT1 promotes inflammatory response in sepsis-induced liver injury via the Let-7a/TLR4 axis. Int Immunopharmacol 2019, 75, 105731. [Google Scholar] [CrossRef]
- Xia, D.; Yao, R.; Zhou, P.; Wang, C.; Xia, Y.; Xu, S. LncRNA NEAT1 reversed the hindering effects of miR-495-3p/STAT3 axis and miR-211/PI3K/AKT axis on sepsis-relevant inflammation. Mol Immunol 2020, 117, 168–179. [Google Scholar] [CrossRef]
- Wu, X.Y.; Fang, Y.; Zheng, F.X.; Zhang, Y.Z.; Li, Q.L. LncRNA NEAT1 facilitates the progression of sepsis through up-regulating TSP-1 via sponging miR-370-3p. Eur Rev Med Pharmacol Sci 2020, 24, 333–344. [Google Scholar] [CrossRef]
- Li, Y.; Guo, W.; Cai, Y. NEAT1 Promotes LPS-induced Inflammatory Injury in Macrophages by Regulating MiR-17-5p/TLR4. Open Med (Wars) 2020, 15, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Xue, J.; Qin, L.; Zhang, J.; Liu, J.; Yu, J. LncRNA NEAT1 Promotes Inflammatory Response in Sepsis via the miR-31-5p/POU2F1 Axis. Inflammation 2021, 44, 1518–1528. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Guo, Z.H. Downregulation of lncRNA NEAT1 Ameliorates LPS-Induced Inflammatory Responses by Promoting Macrophage M2 Polarization via miR-125a-5p/TRAF6/TAK1 Axis. Inflammation 2020, 43, 1548–1560. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Tang, S.; Ke, S.; Cai, J.J.; Osorio, D.; Golovko, A.; Morpurgo, B.; Guo, S.; Sun, Y.; Winkle, M.; et al. Ablation of long noncoding RNA MALAT1 activates antioxidant pathway and alleviates sepsis in mice. Redox Biol 2022, 54, 102377. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Su, Z.; Song, D.; Mao, Y.; Mao, X. The long noncoding RNA MALAT1 regulates the lipopolysaccharide-induced inflammatory response through its interaction with NF-kappaB. FEBS Lett 2016, 590, 2884–2895. [Google Scholar] [CrossRef] [PubMed]
- Dai, L.; Zhang, G.; Cheng, Z.; Wang, X.; Jia, L.; Jing, X.; Wang, H.; Zhang, R.; Liu, M.; Jiang, T.; et al. Knockdown of LncRNA MALAT1 contributes to the suppression of inflammatory responses by up-regulating miR-146a in LPS-induced acute lung injury. Connect Tissue Res 2018, 59, 581–592. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.P.; Niu, G.H.; Zhang, X.Q. Influence of lncRNA MALAT1 on septic lung injury in mice through p38 MAPK/p65 NF-kappaB pathway. Eur Rev Med Pharmacol Sci 2019, 23, 1296–1304. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Banerjee, S.; Guo, S.; Xie, N.; Ge, J.; Jiang, D.; Zornig, M.; Thannickal, V.J.; Liu, G. Long noncoding RNA Malat1 regulates differential activation of macrophages and response to lung injury. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Yang, Q.; Cao, K.; Jin, G.; Zhang, J. Hsa-miR-346 plays a role in the development of sepsis by downregulating SMAD3 expression and is negatively regulated by lncRNA MALAT1. Mol Cell Probes 2019, 47, 101444. [Google Scholar] [CrossRef]
- Wang, Y.; Xu, Z.; Yue, D.; Zeng, Z.; Yuan, W.; Xu, K. Linkage of lncRNA CRNDE sponging miR-181a-5p with aggravated inflammation underlying sepsis. Innate Immun 2020, 26, 152–161. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, Q.; Ma, J.J. High expression of lnc-CRNDE presents as a biomarker for acute myeloid leukemia and promotes the malignant progression in acute myeloid leukemia cell line U937. Eur Rev Med Pharmacol Sci 2018, 22, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Liu, B.; Shen, C.; Chu, X.; Luo, X.; Yu, L.; Ye, J.; Xiong, L.; Dan, W.; Li, J.; Zhong, L. [lncRNA CRNDE promotes proliferation and inhibits apoptosis of U937 cells by downregulating miR-136-5p and upregulating MCM5]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2021, 37, 987–995. [Google Scholar] [PubMed]
- Shen, M.; Pan, X.; Gao, Y.; Ye, H.; Zhang, J.; Chen, Y.; Pan, M.; Huang, W.; Xu, X.; Zhao, Y.; Jin, L. LncRNA CRNDE Exacerbates IgA Nephropathy Progression by Promoting NLRP3 Inflammasome Activation in Macrophages. Immunol Invest 2022, 51, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ren, H.; Liu, B. Evaluating the potency of blood long noncoding RNA PVT1 as candidate biomarker reflecting inflammation, multiple organ dysfunction, and mortality risk in sepsis patients. J Clin Lab Anal 2022, 36, e24268. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.; Xiong, X.; Du, J.; Fan, Q.; Wang, R.; Zhang, X. LncRNA PVT1 accelerates LPS-induced septic acute kidney injury through targeting miR-17-5p and regulating NF-kappaB pathway. Int Urol Nephrol 2021, 53, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Zheng, S.; Li, W.; Liao, W.; Huang, C.; Zhou, M.; Zheng, Y.; Zou, Z.; He, Z. Silencing of LncRNA-PVT1 ameliorates lipopolysaccharide-induced inflammation in THP-1-derived macrophages via inhibition of the p38 MAPK signaling pathway. Ann Palliat Med 2021, 10, 6410–6418. [Google Scholar] [CrossRef]
- Luo, Y.Y.; Yang, Z.Q.; Lin, X.F.; Zhao, F.L.; Tu, H.T.; Wang, L.J.; Wen, M.Y.; Xian, S.X. Knockdown of lncRNA PVT1 attenuated macrophage M1 polarization and relieved sepsis induced myocardial injury via miR-29a/HMGB1 axis. Cytokine 2021, 143, 155509. [Google Scholar] [CrossRef]
- Hreggvidsdottir, H.S.; Lundberg, A.M.; Aveberger, A.C.; Klevenvall, L.; Andersson, U.; Harris, H.E. High mobility group box protein 1 (HMGB1)-partner molecule complexes enhance cytokine production by signaling through the partner molecule receptor. Mol Med 2012, 18, 224–230. [Google Scholar] [CrossRef]
- Meng, Y.; Qiu, S.; Sun, L.; Zuo, J. Knockdown of exosome-mediated lnc-PVT1 alleviates lipopolysaccharide-induced osteoarthritis progression by mediating the HMGB1/TLR4/NF-kappaB pathway via miR-93-5p. Mol Med Rep 2020, 22, 5313–5325. [Google Scholar] [CrossRef]
- Pan, X.; He, L. LncRNA MEG3 expression in sepsis and its effect on LPS-induced macrophage function. Cell Mol Biol (Noisy-le-grand) 2020, 66, 131–136. [Google Scholar] [CrossRef]
- Gibson, M.S.; Domingues, N.; Vieira, O.V. Lipid and Non-lipid Factors Affecting Macrophage Dysfunction and Inflammation in Atherosclerosis. Front Physiol 2018, 9, 654. [Google Scholar] [CrossRef] [PubMed]
- Lusis, A.J. Atherosclerosis. Nature 2000, 14, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Atherosclerosis in Inflammation. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis--an inflammatory disease. N Engl J Med 1999, 340, 115–126. [Google Scholar] [CrossRef]
- Wu, J.; He, S.; Song, Z.; Chen, S.; Lin, X.; Sun, H.; Zhou, P.; Peng, Q.; Du, S.; Zheng, S.; Liu, X. Macrophage polarization states in atherosclerosis. Front Immunol 2023, 14, 1185587. [Google Scholar] [CrossRef] [PubMed]
- Momtazi-Borojeni, A.A.; Abdollahi, E.; Nikfar, B.; Chaichian, S.; Ekhlasi-Hundrieser, M. Curcumin as a potential modulator of M1 and M2 macrophages: new insights in atherosclerosis therapy. Heart Fail Rev 2019, 24, 399–409. [Google Scholar] [CrossRef]
- Park, S.H. Regulation of Macrophage Activation and Differentiation in Atherosclerosis. J Lipid Atheroscler 2021, 10, 251–267. [Google Scholar] [CrossRef]
- Tabas, I. Macrophage death and defective inflammation resolution in atherosclerosis. Nat Rev Immunol 2010, 10, 36–46. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, Y.; Nabavi, S.M.; Sahebkar, A.; Little, P.J.; Xu, S.; Weng, J.; Ge, J. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Front Cardiovasc Med 2022, 9, 925923. [Google Scholar] [CrossRef]
- Watanabe, N.; Ikeda, U. Matrix metalloproteinases and atherosclerosis. Curr Atheroscler Rep 2004, 6, 112–120. [Google Scholar] [CrossRef]
- Wilson, H.M. The intracellular signaling pathways governing macrophage activation and function in human atherosclerosis. Biochem Soc Trans 2022, 50, 1673–1682. [Google Scholar] [CrossRef]
- Taghizadeh, E.; Taheri, F.; Renani, P.G.; Reiner, Z.; Navashenaq, J.G.; Sahebkar, A. Macrophage: A Key Therapeutic Target in Atherosclerosis? Curr Pharm Des 2019, 25, 3165–3174. [Google Scholar] [CrossRef]
- Vlachogiannis, N.I.; Sachse, M.; Georgiopoulos, G.; Zormpas, E.; Bampatsias, D.; Delialis, D.; Bonini, F.; Galyfos, G.; Sigala, F.; Stamatelopoulos, K.; et al. Adenosine-to-inosine Alu RNA editing controls the stability of the pro-inflammatory long noncoding RNA NEAT1 in atherosclerotic cardiovascular disease. J Mol Cell Cardiol 2021, 160, 111–120. [Google Scholar] [CrossRef]
- Guo, J.T.; Wang, L.; Yu, H.B. Knockdown of NEAT1 mitigates ox-LDL-induced injury in human umbilical vein endothelial cells via miR-30c-5p/TCF7 axis. Eur Rev Med Pharmacol Sci 2020, 24, 9633–9644. [Google Scholar] [CrossRef] [PubMed]
- Huang-Fu, N.; Cheng, J.S.; Wang, Y.; Li, Z.W.; Wang, S.H. Neat1 regulates oxidized low-density lipoprotein-induced inflammation and lipid uptake in macrophages via paraspeckle formation. Mol Med Rep 2018, 17, 3092–3098. [Google Scholar] [CrossRef]
- Wang, L.; Xia, J.W.; Ke, Z.P.; Zhang, B.H. Blockade of NEAT1 represses inflammation response and lipid uptake via modulating miR-342-3p in human macrophages THP-1 cells. J Cell Physiol 2019, 234, 5319–5326. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Wen, Z.; Li, S.; Chen, Z.; Li, C.; Ouyang, Z.; Lin, C.; Kuang, M.; Xue, C.; Ding, Y. LncRNA Neat1 promotes the macrophage inflammatory response and acts as a therapeutic target in titanium particle-induced osteolysis. Acta Biomater 2022, 142, 345–360. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.D.; Hui, L.L.; Zhang, X.C.; Chang, Q. NEAT1 contributes to ox-LDL-induced inflammation and oxidative stress in macrophages through inhibiting miR-128. J Cell Biochem 2018. [Google Scholar] [CrossRef] [PubMed]
- Gast, M.; Rauch, B.H.; Haghikia, A.; Nakagawa, S.; Haas, J.; Stroux, A.; Schmidt, D.; Schumann, P.; Weiss, S.; Jensen, L.; et al. Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc Res 2019, 115, 1886–1906. [Google Scholar] [CrossRef] [PubMed]
- Du, H.; Zhang, H.; Yang, R.; Qiao, L.; Shao, H.; Zhang, X. Small interfering RNA-induced silencing lncRNA PVT1 inhibits atherosclerosis via inactivating the MAPK/NF-kappaB pathway. Aging (Albany NY) 2021, 13, 24449–24463. [Google Scholar] [CrossRef]
- Hakami, M.A.; Hazazi, A.; Khan, F.R.; Abdulaziz, O.; Alshaghdali, K.; Abalkhail, A.; Nassar, S.A.; Omar, B.I.A.; Almarshadi, F.; Gupta, G.; Binshaya, A.S. PVT1 lncRNA in lung cancer: A key player in tumorigenesis and therapeutic opportunities. Pathol Res Pract 2023, 253, 155019. [Google Scholar] [CrossRef] [PubMed]
- An, J.H.; Chen, Z.Y.; Ma, Q.L.; Wang, H.J.; Zhang, J.Q.; Shi, F.W. LncRNA SNHG16 promoted proliferation and inflammatory response of macrophages through miR-17-5p/NF-kappaB signaling pathway in patients with atherosclerosis. Eur Rev Med Pharmacol Sci 2019, 23, 8665–8677. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.L.; Feng, Y.; Chen, Y.Y.; Liu, J.Z.; Su, Y.; Li, P.; Huang, H.; Mao, Q.S.; Xue, W.J. SNHG16/miR-605-3p/TRAF6/NF-kappaB feedback loop regulates hepatocellular carcinoma metastasis. J Cell Mol Med 2020, 24, 7637–7651. [Google Scholar] [CrossRef]
- Huang, L.; Xiong, S.; Liu, H.; Zhang, R.; Wu, Y.; Hu, X. Silencing LncRNA SNHG16 suppresses the diabetic inflammatory response by targeting the miR-212-3p/NF-kappaB signaling pathway. Diabetol Metab Syndr 2023, 15, 119. [Google Scholar] [CrossRef]
- Yang, K.; Xue, Y.; Gao, X. LncRNA XIST Promotes Atherosclerosis by Regulating miR-599/TLR4 Axis. Inflammation 2021, 44, 965–973. [Google Scholar] [CrossRef]
- Sun, C.; Fu, Y.; Gu, X.; Xi, X.; Peng, X.; Wang, C.; Sun, Q.; Wang, X.; Qian, F.; Qin, Z.; et al. Macrophage-Enriched lncRNA RAPIA: A Novel Therapeutic Target for Atherosclerosis. Arterioscler Thromb Vasc Biol 2020, 40, 1464–1478. [Google Scholar] [CrossRef]
- Gareev, I.; Kudriashov, V.; Sufianov, A.; Begliarzade, S.; Ilyasova, T.; Liang, Y.; Beylerli, O. The role of long non-coding RNA ANRIL in the development of atherosclerosis. Noncoding RNA Res 2022, 7, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Cheng, H.; Yue, Y.; Li, S.; Zhang, D.; He, R. H19 knockdown suppresses proliferation and induces apoptosis by regulating miR-148b/WNT/beta-catenin in ox-LDL -stimulated vascular smooth muscle cells. J Biomed Sci 2018, 25, 11. [Google Scholar] [CrossRef]
- Lu, G.; Chu, Y.; Tian, P. Knockdown of H19 Attenuates Ox-LDL-induced Vascular Smooth Muscle Cell Proliferation, Migration, and Invasion by Regulating miR-599/PAPPA Axis. J Cardiovasc Pharmacol 2021, 77, 386–396. [Google Scholar] [CrossRef]
- Safaei, S.; Tahmasebi-Birgani, M.; Bijanzadeh, M.; Seyedian, S.M. Increased Expression Level of Long Noncoding RNA H19 in Plasma of Patients with Myocardial Infarction. Int J Mol Cell Med 2020, 9, 122–129. [Google Scholar] [CrossRef]
- Zhang, Z.; Gao, W.; Long, Q.Q.; Zhang, J.; Li, Y.F.; Liu, D.C.; Yan, J.J.; Yang, Z.J.; Wang, L.S. Increased plasma levels of lncRNA H19 and LIPCAR are associated with increased risk of coronary artery disease in a Chinese population. Sci Rep 2017, 7, 7491. [Google Scholar] [CrossRef]
- Han, Y.; Ma, J.; Wang, J.; Wang, L. Silencing of H19 inhibits the adipogenesis and inflammation response in ox-LDL-treated Raw264.7 cells by up-regulating miR-130b. Mol Immunol 2018, 93, 107–114. [Google Scholar] [CrossRef]
- Cao, L.; Zhang, Z.; Li, Y.; Zhao, P.; Chen, Y. LncRNA H19/miR-let-7 axis participates in the regulation of ox-LDL-induced endothelial cell injury via targeting periostin. Int Immunopharmacol 2019, 72, 496–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Che, Y.; Wang, J.; Men, K. [Effects and mechanism of knocking down lncRNA H19 to inhibit lipid accumulation in human THP-1 cells-derived macrophages]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2023, 39, 884–890. [Google Scholar] [PubMed]
- Pan, J.X. LncRNA H19 promotes atherosclerosis by regulating MAPK and NF-kB signaling pathway. Eur Rev Med Pharmacol Sci 2017, 21, 322–328. [Google Scholar]
- Qiu, S.; Sun, J. lncRNA-MALAT1 expression in patients with coronary atherosclerosis and its predictive value for in-stent restenosis. Exp Ther Med 2020, 20, 129. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Tan, L.; Yao, J.; Yang, L. Long non-coding RNA MALAT1 regulates cholesterol accumulation in ox-LDL-induced macrophages via the microRNA-17-5p/ABCA1 axis. Mol Med Rep 2020, 21, 1761–1770. [Google Scholar] [CrossRef]
- Wang, L.; Qi, Y.; Wang, Y.; Tang, H.; Li, Z.; Wang, Y.; Tang, S.; Zhu, H. LncRNA MALAT1 Suppression Protects Endothelium against oxLDL-Induced Inflammation via Inhibiting Expression of MiR-181b Target Gene TOX. Oxid Med Cell Longev 2019, 2019, 8245810. [Google Scholar] [CrossRef]
- Huangfu, N.; Xu, Z.; Zheng, W.; Wang, Y.; Cheng, J.; Chen, X. LncRNA MALAT1 regulates oxLDL-induced CD36 expression via activating beta-catenin. Biochem Biophys Res Commun 2018, 495, 2111–2117. [Google Scholar] [CrossRef]
- Shi, Z.; Zheng, Z.; Lin, X.; Ma, H. Long Noncoding RNA MALAT1 Regulates the Progression of Atherosclerosis by miR-330-5p/NF-kappaB Signal Pathway. J Cardiovasc Pharmacol 2021, 78, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; Zhang, J.; Xu, X.; Qu, Y.; Dong, H.; Dang, J.; Huo, Z.; Xu, G. LncRNA expression profile during autophagy and Malat1 function in macrophages. PLoS One 2019, 14, e0221104. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Lin, X.; Wang, L.; Sun, T.; Zhao, Q.; Ma, Q.; Zhou, Y. LncRNA MALAT1 Enhances ox-LDL-Induced Autophagy through the SIRT1/MAPK/NF-kappaB Pathway in Macrophages. Curr Vasc Pharmacol 2020, 18, 652–662. [Google Scholar] [CrossRef] [PubMed]
- Rusetskaya, N.Y.; Loginova, N.Y.; Pokrovskaya, E.P.; Chesovskikh, Y.S.; Titova, L.E. Redox regulation of the NLRP3-mediated inflammation and pyroptosis. Biomed Khim 2023, 69, 333–352. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Qiu, H.; Pei, X.; Fan, Y.; Tian, H.; Geng, J. Low-dose Sinapic Acid Abates the Pyroptosis of Macrophages by Downregulation of lncRNA-MALAT1 in Rats With Diabetic Atherosclerosis. J Cardiovasc Pharmacol 2018, 71, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Zheng, T. A Review of the Roles of Specialized Extracellular Vesicles, Migrasomes, and Exosomes in Normal Cell Physiology and Disease. Med Sci Monit 2023, 29, e940118. [Google Scholar] [CrossRef]
- Chen, B.; Luo, L.; Wei, X.; Gong, D.; Li, Z.; Li, S.; Tang, W.; Jin, L. M1 Bone Marrow-Derived Macrophage-Derived Extracellular Vesicles Inhibit Angiogenesis and Myocardial Regeneration Following Myocardial Infarction via the MALAT1/MicroRNA-25-3p/CDC42 Axis. Oxidative Medicine and Cellular Longevity 2021, 2021. [Google Scholar] [CrossRef]
- Afonso Mda, S.; Castilho, G.; Lavrador, M.S.; Passarelli, M.; Nakandakare, E.R.; Lottenberg, S.A.; Lottenberg, A.M. The impact of dietary fatty acids on macrophage cholesterol homeostasis. J Nutr Biochem 2014, 25, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Gast, M.; Rauch, B.H.; Nakagawa, S.; Haghikia, A.; Jasina, A.; Haas, J.; Nath, N.; Jensen, L.; Stroux, A.; Bohm, A.; et al. Immune system-mediated atherosclerosis caused by deficiency of long non-coding RNA MALAT1 in ApoE-/-mice. Cardiovasc Res 2019, 115, 302–314. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Han, J.; Wu, Y.; Li, S.; Wang, Q.; Lin, W.; Zhu, J. Exosomal MALAT1 derived from oxidized low-density lipoprotein-treated endothelial cells promotes M2 macrophage polarization. Mol Med Rep 2018, 18, 509–515. [Google Scholar] [CrossRef]
- Pang, J.L.; Wang, J.W.; Hu, P.Y.; Jiang, J.S.; Yu, C. HOTAIR alleviates ox-LDL-induced inflammatory response in Raw264.7 cells via inhibiting NF-kappaB pathway. Eur Rev Med Pharmacol Sci 2018, 22, 6991–6998. [Google Scholar] [CrossRef]
- Tamanini, F.; Bontekoe, C.; Bakker, C.E.; van Unen, L.; Anar, B.; Willemsen, R.; Yoshida, M.; Galjaard, H.; Oostra, B.A.; Hoogeveen, A.T. Different targets for the fragile X-related proteins revealed by their distinct nuclear localizations. Hum Mol Genet 1999, 8, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rubio, A.; Kratchmarov, R.; Sebastian, M.; Garcia-Etxebarria, K.; Garcia, L.; Irastorza, I.; Ghosh, S. Cytoplasmic Form of Carlr lncRNA Facilitates Inflammatory Gene Expression upon NF-kappaB Activation. J Immunol 2017, 199, 581–588. [Google Scholar] [CrossRef]
- Xue, Z.; Zhang, Z.; Liu, H.; Li, W.; Guo, X.; Zhang, Z.; Liu, Y.; Jia, L.; Li, Y.; Ren, Y.; et al. lincRNA-Cox2 regulates NLRP3 inflammasome and autophagy mediated neuroinflammation. Cell Death Differ 2019, 26, 130–145. [Google Scholar] [CrossRef] [PubMed]
- Hu, G.; Gong, A.Y.; Wang, Y.; Ma, S.; Chen, X.; Chen, J.; Su, C.J.; Shibata, A.; Strauss-Soukup, J.K.; Drescher, K.M.; Chen, X.M. LincRNA-Cox2 Promotes Late Inflammatory Gene Transcription in Macrophages through Modulating SWI/SNF-Mediated Chromatin Remodeling. J Immunol 2016, 196, 2799–2808. [Google Scholar] [CrossRef]
- Krawczyk, M.; Emerson, B.M. p50-associated COX-2 extragenic RNA (PACER) activates COX-2 gene expression by occluding repressive NF-kappaB complexes. Elife 2014, 3, e01776. [Google Scholar] [CrossRef]
- Ye, M.; Wang, C.; Zhu, J.; Chen, M.; Wang, S.; Li, M.; Lu, Y.; Xiao, P.; Zhou, M.; Li, X.; Zhou, R. An NF-kappaB-responsive long noncoding RNA, PINT, regulates TNF-alpha gene transcription by scaffolding p65 and EZH2. FASEB J 2021, 35, e21667. [Google Scholar] [CrossRef]
- Wu, Q.; Han, L.; Yan, W.; Ji, X.; Han, R.; Yang, J.; Yuan, J.; Ni, C. miR-489 inhibits silica-induced pulmonary fibrosis by targeting MyD88 and Smad3 and is negatively regulated by lncRNA CHRF. Sci Rep 2016, 6, 30921. [Google Scholar] [CrossRef]
- Wen, S.; Li, F.; Tang, Y.; Dong, L.; He, Y.; Deng, Y.; Tao, Z. MIR222HG attenuates macrophage M2 polarization and allergic inflammation in allergic rhinitis by targeting the miR146a-5p/TRAF6/NF-kappaB axis. Front Immunol 2023, 14, 1168920. [Google Scholar] [CrossRef]
- Akincilar, S.C.; Wu, L.; Ng, Q.F.; Chua, J.Y.H.; Unal, B.; Noda, T.; Chor, W.H.J.; Ikawa, M.; Tergaonkar, V. NAIL: an evolutionarily conserved lncRNA essential for licensing coordinated activation of p38 and NFkappaB in colitis. Gut 2021, 70, 1857–1871. [Google Scholar] [CrossRef]
- Li, Q.; Lu, L.; Li, X.; Lu, S. Long non-coding RNA NKILA alleviates airway inflammation in asthmatic mice by promoting M2 macrophage polarization and inhibiting the NF-kappaB pathway. Biochem Biophys Res Commun 2021, 571, 46–52. [Google Scholar] [CrossRef]
- Yang, T.; Li, S.; Liu, J.; Yin, D.; Yang, X.; Tang, Q. lncRNA-NKILA/NF-kappaB feedback loop modulates laryngeal cancer cell proliferation, invasion, and radioresistance. Cancer Med 2018, 7, 2048–2063. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Li, J.; Tan, Y.; Liu, Y.; Bai, C.; Gao, J.; Zhao, S.; Yao, M.; Lu, X.; Qiu, L.; Xing, L. Tanreqing Injection Attenuates Macrophage Activation and the Inflammatory Response via the lncRNA-SNHG1/HMGB1 Axis in Lipopolysaccharide-Induced Acute Lung Injury. Front Immunol 2022, 13, 820718. [Google Scholar] [CrossRef] [PubMed]
- Ai, Y.; Liu, S.; Luo, H.; Wu, S.; Wei, H.; Tang, Z.; Li, X.; Zou, C. lncRNA DCST1-AS1 facilitates oral squamous cell carcinoma by promoting M2 macrophage polarization through activating NF-κB signaling. Journal of Immunology Research 2021, 2021. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Dou, C.; Yao, B.; Xu, M.; Ding, L.; Wang, Y.; Jia, Y.; Li, Q.; Zhang, H.; Tu, K.; et al. Ftx non coding RNA-derived miR-545 promotes cell proliferation by targeting RIG-I in hepatocellular carcinoma. Oncotarget 2016, 7, 25350–25365. [Google Scholar] [CrossRef] [PubMed]
- Obaid, M.; Udden, S.M.N.; Alluri, P.; Mandal, S.S. LncRNA HOTAIR regulates glucose transporter Glut1 expression and glucose uptake in macrophages during inflammation. Sci Rep 2021, 11, 232. [Google Scholar] [CrossRef]
- He, Z.; Wang, J.; Zhu, C.; Xu, J.; Chen, P.; Jiang, X.; Chen, Y.; Jiang, J.; Sun, C. Exosome-derived FGD5-AS1 promotes tumor-associated macrophage M2 polarization-mediated pancreatic cancer cell proliferation and metastasis. Cancer Lett 2022, 548, 215751. [Google Scholar] [CrossRef]
- Shen, T.; Miao, S.; Zhou, Y.; Yi, X.; Xue, S.; Du, B.; Tang, C.; Qu, L.; Fu, D.; Jia, R.; He, H. Exosomal AP000439.2 from clear cell renal cell carcinoma induces M2 macrophage polarization to promote tumor progression through activation of STAT3. Cell Commun Signal 2022, 20, 152. [Google Scholar] [CrossRef]
- Zhu, X.; Sun, Y.; Yu, Q.; Wang, X.; Wang, Y.; Zhao, Y. Exosomal lncRNA GAS5 promotes M1 macrophage polarization in allergic rhinitis via restraining mTORC1/ULK1/ATG13-mediated autophagy and subsequently activating NF-small ka, CyrillicB signaling. Int Immunopharmacol 2023, 121, 110450. [Google Scholar] [CrossRef]
- Jiang, H.; Zhou, L.; Shen, N.; Ning, X.; Wu, D.; Jiang, K.; Huang, X. M1 macrophage-derived exosomes and their key molecule lncRNA HOTTIP suppress head and neck squamous cell carcinoma progression by upregulating the TLR5/NF-kappaB pathway. Cell Death Dis 2022, 13, 183. [Google Scholar] [CrossRef]
- Liu, J.; Niu, Z.; Zhang, R.; Peng, Z.; Wang, L.; Liu, Z.; Gao, Y.; Pei, H.; Pan, L. MALAT1 shuttled by extracellular vesicles promotes M1 polarization of macrophages to induce acute pancreatitis via miR-181a-5p/HMGB1 axis. J Cell Mol Med 2021, 25, 9241–9254. [Google Scholar] [CrossRef]
- Liu, J.; Geng, X.; Hou, J.; Wu, G. New insights into M1/M2 macrophages: key modulators in cancer progression. Cancer Cell Int 2021, 21, 389. [Google Scholar] [CrossRef]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front Immunol 2014, 5, 514. [Google Scholar] [CrossRef]
- Solinas, G.; Germano, G.; Mantovani, A.; Allavena, P. Tumor-associated macrophages (TAM) as major players of the cancer-related inflammation. J Leukoc Biol 2009, 86, 1065–1073. [Google Scholar] [CrossRef]
- Erreni, M.; Mantovani, A.; Allavena, P. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer Microenviron 2011, 4, 141–154. [Google Scholar] [CrossRef]
- Karin, M.; Greten, F.R. NF-kappaB: linking inflammation and immunity to cancer development and progression. Nat Rev Immunol 2005, 5, 749–759. [Google Scholar] [CrossRef]
- Wang, B.; Cheng, D.; Ma, D.; Chen, R.; Li, D.; Zhao, W.; Fang, C.; Ji, M. Mutual regulation of PD-L1 immunosuppression between tumor-associated macrophages and tumor cells: a critical role for exosomes. Cell Commun Signal 2024, 22, 21. [Google Scholar] [CrossRef]
- Seo, K.H.; Ko, H.M.; Choi, J.H.; Jung, H.H.; Chun, Y.H.; Choi, I.W.; Lee, H.K.; Im, S.Y. Essential role for platelet-activating factor-induced NF-kappaB activation in macrophage-derived angiogenesis. Eur J Immunol 2004, 34, 2129–2137. [Google Scholar] [CrossRef]
- Mancino, A.; Lawrence, T. Nuclear factor-kappaB and tumor-associated macrophages. Clin Cancer Res 2010, 16, 784–789. [Google Scholar] [CrossRef]
- Varney, M.L.; Olsen, K.J.; Mosley, R.L.; Bucana, C.D.; Talmadge, J.E.; Singh, R.K. Monocyte/macrophage recruitment, activation and differentiation modulate interleukin-8 production: a paracrine role of tumor-associated macrophages in tumor angiogenesis. In Vivo 2002, 16, 471–477. [Google Scholar]
- Hoesel, B.; Schmid, J.A. The complexity of NF-kappaB signaling in inflammation and cancer. Mol Cancer 2013, 12, 86. [Google Scholar] [CrossRef]
- Shang, S.; Ji, X.; Zhang, L.; Chen, J.; Li, C.; Shi, R.; Xiang, W.; Kang, X.; Zhang, D.; Yang, F.; et al. Macrophage ABHD5 Suppresses NFkappaB-Dependent Matrix Metalloproteinase Expression and Cancer Metastasis. Cancer Res 2019, 79, 5513–5526. [Google Scholar] [CrossRef]
- Chen, J.; Wu, D.; Zhang, Y.; Yang, Y.; Duan, Y.; An, Y. LncRNA DCST1-AS1 functions as a competing endogenous RNA to regulate FAIM2 expression by sponging miR-1254 in hepatocellular carcinoma. Clin Sci (Lond) 2019, 133, 367–379. [Google Scholar] [CrossRef]
- Tang, L.; Chen, Y.; Tang, X.; Wei, D.; Xu, X.; Yan, F. Long Noncoding RNA DCST1-AS1 Promotes Cell Proliferation and Metastasis in Triple-negative Breast Cancer by Forming a Positive Regulatory Loop with miR-873-5p and MYC. J Cancer 2020, 11, 311–323. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, J.; Hu, Y.; Zou, H.; Zhang, X.; Hu, X. Inhibition of lncRNA DCST1-AS1 suppresses proliferation, migration and invasion of cervical cancer cells by increasing miR-874-3p expression. J Gene Med 2021, 23, e3281. [Google Scholar] [CrossRef]
- Wang, J.; Lei, C.; Shi, P.; Teng, H.; Lu, L.; Guo, H.; Wang, X. LncRNA DCST1-AS1 Promotes Endometrial Cancer Progression by Modulating the MiR-665/HOXB5 and MiR-873-5p/CADM1 Pathways. Front Oncol 2021, 11, 714652. [Google Scholar] [CrossRef]
- Su, Y.Z.; Cui, M.F.; Du, J.; Song, B. LncRNA DCST1-AS1 regulated cell proliferation, migration, invasion and apoptosis in gastric cancer by targeting miR-605-3p. Eur Rev Med Pharmacol Sci 2020, 24, 1158–1167. [Google Scholar] [CrossRef]
- Hu, S.; Yao, Y.; Hu, X.; Zhu, Y. LncRNA DCST1-AS1 downregulates miR-29b through methylation in glioblastoma (GBM) to promote cancer cell proliferation. Clin Transl Oncol 2020, 22, 2230–2235. [Google Scholar] [CrossRef]
- Huang, L.; Dai, G. Long non-coding RNA DCST1-AS1/hsa-miR-582-5p/HMGB1 axis regulates colorectal cancer progression. Bioengineered 2022, 13, 12–26. [Google Scholar] [CrossRef]
- Lv, J.; Li, Q.; Ma, R.; Wang, Z.; Yu, Y.; Liu, H.; Miao, Y.; Jiang, S. Long Noncoding RNA FGD5-AS1 Knockdown Decrease Viability, Migration, and Invasion of Non-Small Cell Lung Cancer (NSCLC) Cells by Regulating the MicroRNA-944/MACC1 Axis. Technol Cancer Res Treat 2021, 20, 1533033821990090. [Google Scholar] [CrossRef]
- Zhai, W.; Ye, X.; Wang, Y.; Feng, Y.; Wang, Y.; Lin, Y.; Ding, L.; Yang, L.; Wang, X.; Kuang, Y.; et al. CREPT/RPRD1B promotes tumorigenesis through STAT3-driven gene transcription in a p300-dependent manner. Br J Cancer 2021, 124, 1437–1448. [Google Scholar] [CrossRef]
- Xia, T.; Zhang, M.; Lei, W.; Yang, R.; Fu, S.; Fan, Z.; Yang, Y.; Zhang, T. Advances in the role of STAT3 in macrophage polarization. Front Immunol 2023, 14, 1160719. [Google Scholar] [CrossRef]
- Liu, G.; Du, X.; Xiao, L.; Zeng, Q.; Liu, Q. Activation of fgd5-as1 promotes progression of cervical cancer through regulating bst2 to inhibit macrophage m1 polarization. Journal of Immunology Research 2021, 2021. [Google Scholar] [CrossRef]
- Neil, S.J.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Kupzig, S.; Korolchuk, V.; Rollason, R.; Sugden, A.; Wilde, A.; Banting, G. Bst-2/HM1.24 is a raft-associated apical membrane protein with an unusual topology. Traffic 2003, 4, 694–709. [Google Scholar] [CrossRef]
- Cheng, G.; Liu, D.; Liang, H.; Yang, H.; Chen, K.; Zhang, X. A cluster of long non-coding RNAs exhibit diagnostic and prognostic values in renal cell carcinoma. Aging (Albany NY) 2019, 11, 9597–9615. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Guo, K.; Tang, Z.; Feng, Y.; Cai, S.; Ye, J.; Xi, Y.; Li, J.; Liu, R.; Cai, C.; et al. Identification and experimental validation of a tumor-infiltrating lymphocytes-related long noncoding RNA signature for prognosis of clear cell renal cell carcinoma. Front Immunol 2022, 13, 1046790. [Google Scholar] [CrossRef] [PubMed]
- Chureau, C.; Prissette, M.; Bourdet, A.; Barbe, V.; Cattolico, L.; Jones, L.; Eggen, A.; Avner, P.; Duret, L. Comparative sequence analysis of the X-inactivation center region in mouse, human, and bovine. Genome Res 2002, 12, 894–908. [Google Scholar] [CrossRef] [PubMed]
- Sheykhi-Sabzehpoush, M.; Ghasemian, M.; Khojasteh Pour, F.; Mighani, M.; Moghanibashi, M.; Mohammad Jafari, R.; Zabel, M.; Dziegiel, P.; Farzaneh, M.; Kempisty, B. Emerging roles of long non-coding RNA FTX in human disorders. Clin Transl Oncol 2023, 25, 2812–2831. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, T.; Talluri, S.; Akshaya, R.L.; Dunna, N.R. HOTAIR LncRNA: A novel oncogenic propellant in human cancer. Clin Chim Acta 2020, 503, 1–18. [Google Scholar] [CrossRef]
- Xin, X.; Li, Q.; Fang, J.; Zhao, T. LncRNA HOTAIR: A Potential Prognostic Factor and Therapeutic Target in Human Cancers. Front Oncol 2021, 11, 679244. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, J.; Hu, P.; Gao, L.; Tian, S.; He, Z. Long Non-coding RNA HOTAIR in Central Nervous System Disorders: New Insights in Pathogenesis, Diagnosis, and Therapeutic Potential. Front Mol Neurosci 2022, 15, 949095. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Gao, L.; Yu, Z.H.; Hong, S.J.; Zhang, Z.W.; Qiu, Z.Z. LncRNA HOTAIR promotes renal interstitial fibrosis by regulating Notch1 pathway via the modulation of miR-124. Nephrology (Carlton) 2019, 24, 472–480. [Google Scholar] [CrossRef]
- Yu, F.; Chen, B.; Dong, P.; Zheng, J. HOTAIR Epigenetically Modulates PTEN Expression via MicroRNA-29b: A Novel Mechanism in Regulation of Liver Fibrosis. Mol Ther 2017, 25, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Price, R.L.; Bhan, A.; Mandal, S.S. HOTAIR beyond repression: In protein degradation, inflammation, DNA damage response, and cell signaling. DNA Repair (Amst) 2021, 105, 103141. [Google Scholar] [CrossRef] [PubMed]
- Ramya, V.; Shyam, K.P.; Angelmary, A.; Kadalmani, B. Lauric acid epigenetically regulates lncRNA HOTAIR by remodeling chromatin H3K4 tri-methylation and modulates glucose transport in SH-SY5Y human neuroblastoma cells: Lipid switch in macrophage activation. Biochim Biophys Acta Mol Cell Biol Lipids 2024, 1869, 159429. [Google Scholar] [CrossRef] [PubMed]
- Obaid, M.; Udden, S.M.N.; Deb, P.; Shihabeddin, N.; Zaki, M.H.; Mandal, S.S. LncRNA HOTAIR regulates lipopolysaccharide-induced cytokine expression and inflammatory response in macrophages. Sci Rep 2018, 8, 15670. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, M.F.; Li, L.J.; Ma, J.B. LncRNA HOTTIP promotes proliferation and cell cycle progression of acute myeloid leukemia cells. Eur Rev Med Pharmacol Sci 2019, 23, 2908–2915. [Google Scholar] [CrossRef]
- Cheng, L.; Yang, Q.; Li, C.; Dai, L.; Yang, Y.; Wang, Q.; Ding, Y.; Zhang, J.; Liu, L.; Zhang, S.; et al. DDA1, a novel oncogene, promotes lung cancer progression through regulation of cell cycle. J Cell Mol Med 2017, 21, 1532–1544. [Google Scholar] [CrossRef]
- Cesario, A.; Rocca, B.; Rutella, S. The interplay between indoleamine 2,3-dioxygenase 1 (IDO1) and cyclooxygenase (COX)-2 in chronic inflammation and cancer. Curr Med Chem 2011, 18, 2263–2271. [Google Scholar] [CrossRef]
- Sun, P.; Quan, J.C.; Wang, S.; Zhuang, M.; Liu, Z.; Guan, X.; Wang, G.Y.; Wang, H.Y.; Wang, X.S. lncRNA-PACER upregulates COX-2 and PGE2 through the NF-kappaB pathway to promote the proliferation and invasion of colorectal-cancer cells. Gastroenterol Rep (Oxf) 2021, 9, 257–268. [Google Scholar] [CrossRef]
- Desind, S.Z.; Iacona, J.R.; Yu, C.Y.; Mitrofanova, A.; Lutz, C.S. PACER lncRNA regulates COX-2 expression in lung cancer cells. Oncotarget 2022, 13, 291–306. [Google Scholar] [CrossRef]
- Qian, M.; Yang, X.; Li, Z.; Jiang, C.; Song, D.; Yan, W.; Liu, T.; Wu, Z.; Kong, J.; Wei, H.; Xiao, J. P50-associated COX-2 extragenic RNA (PACER) overexpression promotes proliferation and metastasis of osteosarcoma cells by activating COX-2 gene. Tumour Biol 2016, 37, 3879–3886. [Google Scholar] [CrossRef]
- Xu, C.; Deng, H.; Liu, F.; Zhao, D.; Tang, H.; Gu, H. Long Non-Coding RNA PACER Regulates Mycoplasma pneumoniae-induced Inflammatory Response through Interaction with NF-kappaB. Ann Clin Lab Sci 2022, 52, 21–26. [Google Scholar] [PubMed]
- Mola-Ali-Nejad, R.; Fakharianzadeh, S.; Maloum, Z.; Taheri, M.; Shirvani-Farsani, Z. A gene expression analysis of long non-coding RNAs NKILA and PACER as well as their target genes, NF-kappaB and cox-2 in bipolar disorder patients. Nucleosides Nucleotides Nucleic Acids 2023, 42, 527–537. [Google Scholar] [CrossRef]
- Xie, X.; Zhao, W.; Pang, J.; Xiong, X.; Wang, H.; Ma, L. Long non-coding RNA, CHRF, predicts poor prognosis of lung adenocarcinoma and promotes cell proliferation and migration. Oncol Lett 2018, 16, 6245–6252. [Google Scholar] [CrossRef]
- Wu, W.; Wang, X.; Yu, X.; Lan, H.Y. Smad3 Signatures in Renal Inflammation and Fibrosis. Int J Biol Sci 2022, 18, 2795–2806. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Ding, X.; Zhao, S.; Mou, T.; Li, R.; Cao, X. Long non-coding RNA CHRF accelerates LPS-induced acute lung injury through microRNA-146a/Notch1 axis. Ann Transl Med 2021, 9, 1299. [Google Scholar] [CrossRef] [PubMed]
- Zeng, H.; Zhou, S.; Cai, W.; Kang, M.; Zhang, P. LncRNA SNHG1: role in tumorigenesis of multiple human cancers. Cancer Cell Int 2023, 23, 198. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Du, X.; Zhang, H.; Wu, Z.; Ren, K.; Han, X. The Interaction Between lncRNA SNHG1 and miR-140 in Regulating Growth and Tumorigenesis via the TLR4/NF-kappaB Pathway in Cholangiocarcinoma. Oncol Res 2019, 27, 663–672. [Google Scholar] [CrossRef]
- Zhu, X.; Wang, X.; Wang, Y.; Zhao, Y. The regulatory network among CircHIPK3, LncGAS5, and miR-495 promotes Th2 differentiation in allergic rhinitis. Cell Death Dis 2020, 11, 216. [Google Scholar] [CrossRef]
- Hu, J.; Zhang, L.; Liechty, C.; Zgheib, C.; Hodges, M.M.; Liechty, K.W.; Xu, J. Long Noncoding RNA GAS5 Regulates Macrophage Polarization and Diabetic Wound Healing. J Invest Dermatol 2020, 140, 1629–1638. [Google Scholar] [CrossRef]
- Chi, X.; Ding, B.; Zhang, L.; Zhang, J.; Wang, J.; Zhang, W. lncRNA GAS5 promotes M1 macrophage polarization via miR-455-5p/SOCS3 pathway in childhood pneumonia. J Cell Physiol 2019, 234, 13242–13251. [Google Scholar] [CrossRef]
- Sun, D.; Yu, Z.; Fang, X.; Liu, M.; Pu, Y.; Shao, Q.; Wang, D.; Zhao, X.; Huang, A.; Xiang, Z.; et al. LncRNA GAS5 inhibits microglial M2 polarization and exacerbates demyelination. EMBO Rep 2017, 18, 1801–1816. [Google Scholar] [CrossRef]
- Ito, I.; Asai, A.; Suzuki, S.; Kobayashi, M.; Suzuki, F. M2b macrophage polarization accompanied with reduction of long noncoding RNA GAS5. Biochem Biophys Res Commun 2017, 493, 170–175. [Google Scholar] [CrossRef]
- Xu, Y.; Fang, H.; Xu, Q.; Xu, C.; Yang, L.; Huang, C. LncRNA GAS5 inhibits NLRP3 inflammasome activation-mediated pyroptosis in diabetic cardiomyopathy by targeting miR-34b-3p/AHR. Cell Cycle 2020, 19, 3054–3065. [Google Scholar] [CrossRef]
- Lin, G.; Wu, T.; Gao, X.; He, Z.; Nong, W. Research Progress of Long Non-Coding RNA GAS5 in Malignant Tumors. Front Oncol 2022, 12, 846497. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Xie, Z.; Lei, X.; Gan, R. Long non-coding RNA GAS5 in human cancer. Oncol Lett 2020, 20, 2587–2594. [Google Scholar] [CrossRef]
- Tong, Q.; Gong, A.Y.; Zhang, X.T.; Lin, C.; Ma, S.; Chen, J.; Hu, G.; Chen, X.M. LincRNA-Cox2 modulates TNF-alpha-induced transcription of Il12b gene in intestinal epithelial cells through regulation of Mi-2/NuRD-mediated epigenetic histone modifications. FASEB J 2016, 30, 1187–1197. [Google Scholar] [CrossRef] [PubMed]
- Rapicavoli, N.A.; Qu, K.; Zhang, J.; Mikhail, M.; Laberge, R.M.; Chang, H.Y. A mammalian pseudogene lncRNA at the interface of inflammation and anti-inflammatory therapeutics. Elife 2013, 2, e00762. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Amit, I.; Garber, M.; French, C.; Lin, M.F.; Feldser, D.; Huarte, M.; Zuk, O.; Carey, B.W.; Cassady, J.P.; et al. Chromatin signature reveals over a thousand highly conserved large non-coding RNAs in mammals. Nature 2009, 458, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, R.H.; Fouad, N.A.; Hefzy, E.M.; Shaker, O.G.; Ahmed, T.I.; Hussein, H.A.; Nasr, M.H.; Zaki, O.M.; Abdelghaffar, N.K.; Abdelaleem, O.O. The potential role of serum expression profile of long non coding RNAs, Cox2 and HOTAIR as novel diagnostic biomarkers in systemic lupus erythematosus. PLoS One 2022, 17, e0268176. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, S.; Aiello, D.; Atianand, M.K.; Ricci, E.P.; Gandhi, P.; Hall, L.L.; Byron, M.; Monks, B.; Henry-Bezy, M.; Lawrence, J.B.; et al. A long noncoding RNA mediates both activation and repression of immune response genes. Science 2013, 341, 789–792. [Google Scholar] [CrossRef]
- Wang, Q.; Xie, Y.; He, Q.; Geng, Y.; Xu, J. LncRNA-Cox2 regulates macrophage polarization and inflammatory response through the CREB-C/EBPbeta signaling pathway in septic mice. Int Immunopharmacol 2021, 101, 108347. [Google Scholar] [CrossRef] [PubMed]
- Ogulur, I.; Pat, Y.; Ardicli, O.; Barletta, E.; Cevhertas, L.; Fernandez-Santamaria, R.; Huang, M.; Bel Imam, M.; Koch, J.; Ma, S.; et al. Advances and highlights in biomarkers of allergic diseases. Allergy 2021, 76, 3659–3686. [Google Scholar] [CrossRef]
- Fernandez-Jimenez, N.; Castellanos-Rubio, A.; Plaza-Izurieta, L.; Irastorza, I.; Elcoroaristizabal, X.; Jauregi-Miguel, A.; Lopez-Euba, T.; Tutau, C.; de Pancorbo, M.M.; Vitoria, J.C.; Bilbao, J.R. Coregulation and modulation of NFkappaB-related genes in celiac disease: uncovered aspects of gut mucosal inflammation. Hum Mol Genet 2014, 23, 1298–1310. [Google Scholar] [CrossRef]
- Kaur, P.; Verma, S.; Kushwaha, P.P.; Gupta, S. EZH2 and NF-kappaB: A context-dependent crosstalk and transcriptional regulation in cancer. Cancer Lett 2023, 560, 216143. [Google Scholar] [CrossRef]
- Lai, L.; Wang, G.; Xu, L.; Fu, Y. CEBPB promotes gastrointestinal motility dysfunction after severe acute pancreatitis via the MALAT1/CIRBP/ERK axis. Mol Immunol 2023, 156, 1–9. [Google Scholar] [CrossRef]
- Cui, Y.; Fu, S.; Sun, D.; Xing, J.; Hou, T.; Wu, X. EPC-derived exosomes promote osteoclastogenesis through LncRNA-MALAT1. J Cell Mol Med 2019, 23, 3843–3854. [Google Scholar] [CrossRef] [PubMed]
- Shyu, K.G.; Wang, B.W.; Fang, W.J.; Pan, C.M.; Lin, C.M. Exosomal MALAT1 Derived from High Glucose-Treated Macrophages Up-Regulates Resistin Expression via miR-150-5p Downregulation. Int J Mol Sci 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Xu, Y.; Lai, Y.; He, W.; Li, Y.; Wang, R.; Luo, X.; Chen, R.; Chen, T. Long non-coding RNA cox-2 prevents immune evasion and metastasis of hepatocellular carcinoma by altering M1/M2 macrophage polarization. J Cell Biochem 2018, 119, 2951–2963. [Google Scholar] [CrossRef] [PubMed]
- Winkle, M.; El-Daly, S.M.; Fabbri, M.; Calin, G.A. Noncoding RNA therapeutics - challenges and potential solutions. Nat Rev Drug Discov 2021, 20, 629–651. [Google Scholar] [CrossRef] [PubMed]
- Arab, I.; Park, J.; Shin, J.J.; Shin, H.S.; Suk, K.; Lee, W.H. Macrophage lncRNAs in cancer development: Long-awaited therapeutic targets. Biochem Pharmacol 2023, 218, 115890. [Google Scholar] [CrossRef] [PubMed]
- Borgna, V.; Villegas, J.; Burzio, V.A.; Belmar, S.; Araya, M.; Jeldes, E.; Lobos-Gonzalez, L.; Silva, V.; Villota, C.; Oliveira-Cruz, L.; et al. Mitochondrial ASncmtRNA-1 and ASncmtRNA-2 as potent targets to inhibit tumor growth and metastasis in the RenCa murine renal adenocarcinoma model. Oncotarget 2017, 8, 43692–43708. [Google Scholar] [CrossRef]
Figure 1.
Role of lncRNAs and macrophage NF-κB in the pathogenesis of human diseases.
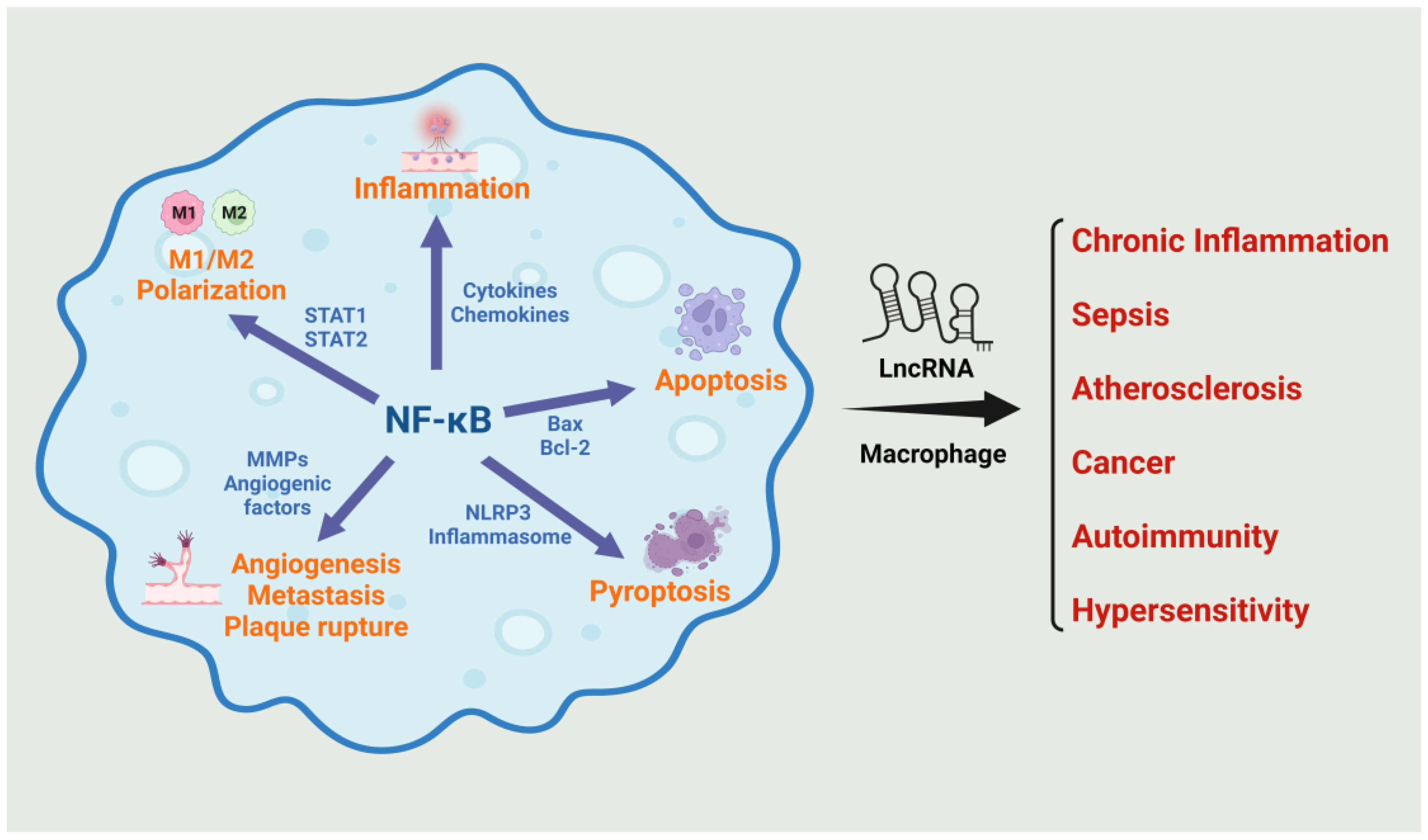
Figure 2.
Mechanisms triggered by macrophage lncRNAs in the regulation of NF-κB activity. Various lncRNAs activating (indicated in red arrows) or inhibiting (indicated in blue arrows) NF-κB signaling and their targets within the NF-κB activation pathway are shown.
Figure 2.
Mechanisms triggered by macrophage lncRNAs in the regulation of NF-κB activity. Various lncRNAs activating (indicated in red arrows) or inhibiting (indicated in blue arrows) NF-κB signaling and their targets within the NF-κB activation pathway are shown.
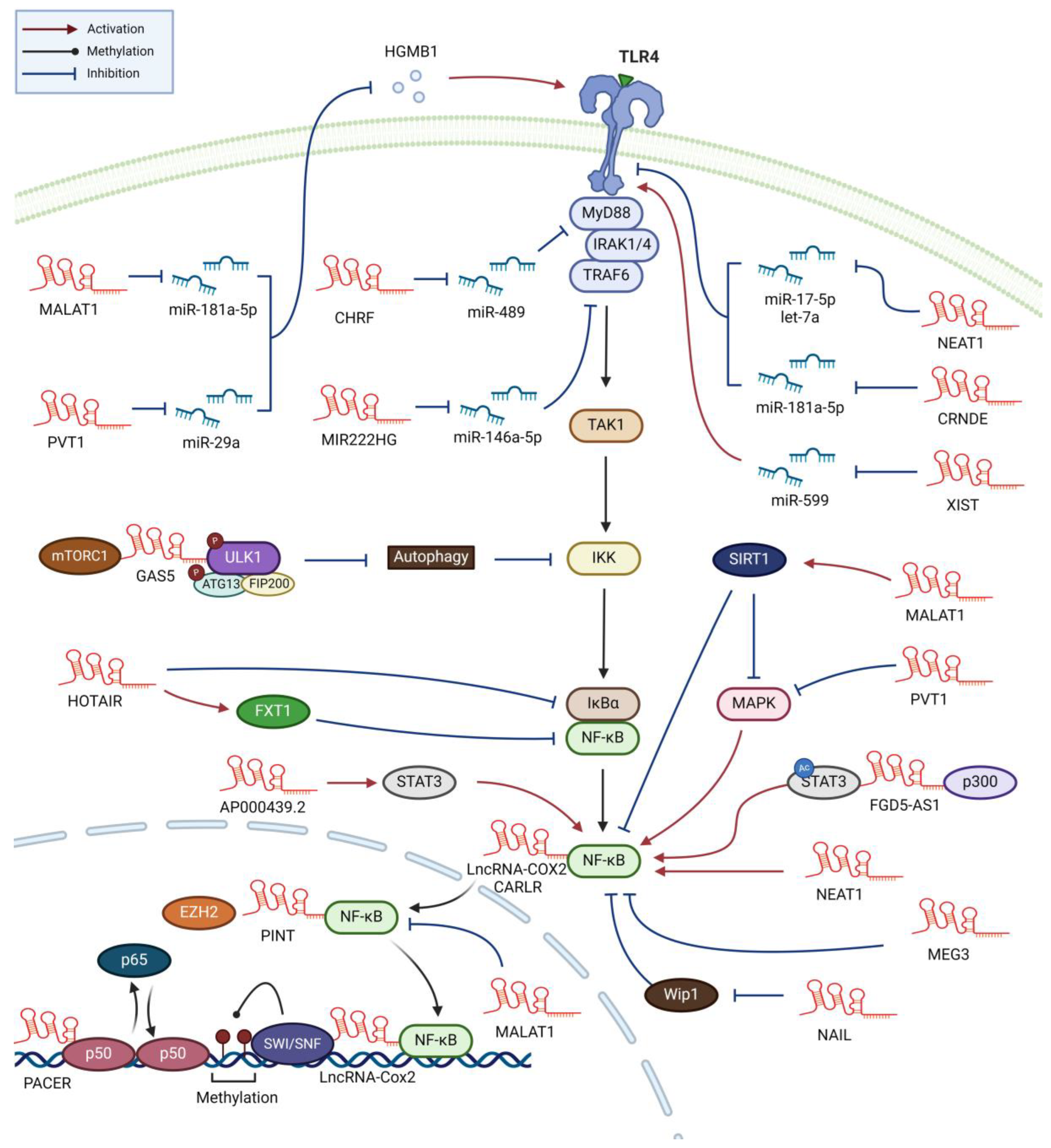
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



