
Submitted:
31 January 2024
Posted:
31 January 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Glycosylation, a prevalent post-translational modification, plays a pivotal role in regulating intricate cellular processes by covalently attaching glycans to macromolecules. Dysregulated glycosylation is linked to a spectrum of diseases, encompassing cancer, neurodegenerative disorders, congenital disorders, infections, and inflammation. This review delves into the intricate interplay between glycosylation and protein conformation, with a specific focus on the profound impact of N-glycans on the selection of distinct protein conformations, characterized by distinct interactomes – namely protein assemblies - under normal and pathological conditions across various diseases. We begin by examining the spike protein of the SARS virus, illustrating how N-glycans regulate the infectivity of pathogenic agents. Subsequently, we utilize the prion protein and the chaperone glucose-regulated protein 94 as examples, exploring instances where N-glycosylation transforms physiological protein structures into disease-associated forms. Unraveling these connections provides valuable insights into potential therapeutic avenues and a deeper comprehension of the molecular intricacies that underlie disease conditions. This exploration of glycosylation's influence on protein conformation effectively bridges the gap between the glycome and disease, offering a comprehensive perspective on the therapeutic implications of targeting conformational mutants and their pathologic assemblies in various diseases. The goal is to unravel the nuances of these post-translational modifications, shedding light on how they contribute to the intricate interplay between protein conformation, assembly and disease.
Keywords:
N-glycosylation
; disease
; conformational mutant
; aberrant protein assembly
; energy landscape
; SARS-CoV-2 spike protein
; glucose regulated protein 94 (GRP94)
; prion protein
; disease-associated protein conformation
; protein dynamics
; protein assembly mutation
; gain-of-function conformational change
1. Introduction
Glycosylation stands as a fundamental and pervasive post-translational modification crucial to various cellular processes. This complex process involves the covalent attachment of carbohydrates, collectively known as glycans, to macromolecules, shaping their structure and function. Orchestrated by a diverse set of enzymes—glycosyltransferases and glycosidases—encoded by a significant fraction of the human genome, glycosylation plays a central role in cellular regulation [1,2,3,4].
The importance of glycosylation is underscored by the abundance of proteins dedicated to shaping glycans. Over 750,000 glycosyltransferase sequences, distributed across all kingdoms, have been identified, and this number is steadily growing [5]. These enzymes, categorized into more than 110 glycosyltransferase families in the Carbohydrate-Active Enzymes (CAZy) database (http://www.cazy.org), exemplify the diversity and complexity of glycan modification. Glycosidases, comprising over 870,000 members in more than 170 CAZy families, contribute to the dynamic turnover of glycans [5].
There are a variety of building blocks for the generation of glycans [6,7,8]. In mammals, glycans are constructed from a combination of 10 monosaccharides — D-glucose (Glc), D-galactose (Gal), N-acetyl-D-glucosamine (GlcNAc), N-acetyl-D-galactosamine (GalNAc), L-fucose (Fuc), D-glucuronic acid (GlcA), D-mannose (Man), N-acetylneuraminic acid (Neu5Ac), D-xylose (Xyl) and D-ribose (Rib). These monosaccharides form linear or branched structures through α- or β-glycosidic bonds, constituting the glycan repertoire observed in various cellular components [6,7,8].
Glycosylation extends its influence across multiple macromolecules, including proteins, lipids, and nucleic acids [9]. The complement of glycan structures produced by cells is referred to as the glycome (in analogy to the genome, transcriptome, proteome, lipidome and metabolome). Glycans attached to lipid molecules form glycolipids. These are important components of cell membranes and are involved in cell recognition and signaling. Recently, it has been discovered that RNA molecules, particularly small non-coding RNAs, can undergo glycosylation, uncovering new roles for glycosylation in RNA function [9]. Glycoproteins, however, formed by the attachment of glycans to proteins, constitute a major part of the glycome [9].
Protein glycosylation is characterized by two major linkages: N-linked glycosylation, involving the amide nitrogen of asparagine residues, and O-linked glycosylation, linking carbohydrates to the hydroxyl oxygen of serine, threonine, or tyrosine residues. N-glycans are attached to asparagine residues within specific sequons (i.e, a consensus Asn-XSer/Thr where X is any amino acid except proline) [10]. In contrast, O-glycans are commonly linked to serine or threonine residues, include variations such as O-linked GlcNAc and mucin-type O-glycans. Unlike N-glycans, O-GalNAc-linked glycans lack a specific glycoside amino acid sequon [10].
Protein glycosylation is a non-templated process and is mediated by enzymes known as glycosyltransferases, responsible for the initiation or elongation of glycans, and oligosaccharyltransferases, responsible for the addition of whole carbohydrate chains [1,3,4]. In the context of biological processes, a template is often a molecule or a sequence of information that guides the assembly or formation of a particular structure (i.e., DNA to protein). In contrast, non-templated processes lack a strict template-based guidance system. In this sense, glycosylation, exhibits a degree of flexibility in the generation of products, a process influenced by the cellular milieu and its response to intracellular and extracellular cues. In cells, the complex interplay between glycosyltransferases or oligosaccharyltransferases, carbohydrate transporters - proteins responsible for the movement of carbohydrates across cell membranes - and glycosidases - a class of enzymes that catalyze the hydrolysis of glycosidic bonds- fine-tunes the glycan structures observed on individual proteins and regulates glycoprotein function. Together, these enzymes initiate or elongate glycans, shaping the glycan structures observed on individual proteins.
The resulting glycoproteins display diverse "glycoforms," featuring variations in glycosylation site occupancy and the assembled glycan structure [11,12,13,14]. Consequently, the glycoproteome is shaped not only by the glycoproteins themselves but also by macroheterogeneity, characterized by variations in glycosylation site occupancy (i.e., structural diversity influenced by the presence or absence of glycans at specific glycosylation sites), and microheterogeneity, which involves variations in glycan structure (i.e., structural diversity reflecting different glycosylation patterns at individual glycosylation sites) within glycoproteins. For instance, N-Glycans share a common pentasaccharide core (Man3-GlcNAc2) and variable distal compositions categorized into three subtypes: high mannose, hybrid, and complex types. Complex structures can be further classified as bi-, tri-, and tetra-antennary, indicating the number of carbohydrate branches originating from the trimannosyl core [11,12,13,14].
It is also crucial to note that the glycome is highly responsive to the dynamic cellular environment and shaped by intra- and extra-cellular conditions, making it context-dependent [15]. Glycan assembly and the formation of glycoconjugates are regulated by numerous factors, including the availability of glycans and cofactors, as well as the glycosyltransferases and glycosidases necessary to catalyze such reactions. Further, each of these elements must be localized appropriately within the cell and secretory apparatus. For example, viral envelope glycoproteins such as the HIV-1 envelope protein (Env) and SARS-CoV-2 spike glycoprotein contain higher degrees of N-glycan processing on native virions than when individual viral proteins are ectopically expressed in cell lines [16].
Equally important is the recognition that different cells, tissues, organs, and organisms have distinct glycomes and varying vulnerabilities to changes in glycosylation mechanisms [9,17]. While mice lacking ST3GAL5—a gene encoding a protein catalyzing the formation of ganglioside GM3—seem to exhibit only moderate phenotypes, humans with similar defects suffer from severe multisystem disease, such as the Amish infantile epilepsy syndrome—an autosomal recessive, infantile-onset symptomatic epilepsy associated with developmental stagnation and blindness [4]. Mice inherently express glycan epitopes that are not typically observed in humans, including α-Gal and Neu5Gc epitopes [18,19]. Similarly, defects in glycosylation pathways that manifest as "limited phenotypes" in cultured cells can yield severe consequences in intact organisms, even in the form of hypomorphic alleles in humans [4]. For instance, the absence of MGAT1 - a gene encoding for a transferase enzyme essential for the synthesis of hybrid and complex N-glycans - results in embryonic lethality in mice but has minimal phenotypic effects in Arabidopsis plants and restricted phenotypes in Drosophila [4]. In addition to highlighting the complexity of the glycome, these instances underscore the imperative need to investigate the impact of glycosylation in native biological states.
To unravel the intricacy of the glycome, there has been a notable surge in analytical methods [20,21,22,23]. These approaches enable mapping glycosylation events on a cellular, tissue, and organism scale, providing insights into their functional roles in biological processes. Resources and repositories were created to catalogue such diversity [24,25,26,27]. They include Glycosciences.DB (contains the data supplemented with NMR spectra, 3D structures and analytical tools) [28]; UniCarbKB (contains eukaryotic glycans supplemented with NMR, MS and HPLC data) [24]; KEGG Glycan (glycan-related data from the Kyoto Encyclopedia of Genes and Genomes) [29]; Japan Consortium for Glycobiology and Glycotechnology (JCGG/ACGG collection of databases on glycoproteins and glycome-associated diseases supplemented with analytical data) [30] and CSDB (the Carbohydrate Structure Database, data (structural, taxonomical, bibliographical, NMR spectroscopic, etc.) on glycans of bacterial, fungal and plant origins) [31]. The N-GlycositeAtlas database contains more than 30,000 glycosite-containing peptides (representing > 14,000 N-glycosylation sites) from more than 7200 N-glycoproteins from different biological sources including human-derived tissues, body fluids and cell lines from over 100 studies [32]. The relatively new field of glycoinformatics has emerged to provide scientists with various means of accessing, processing and handling all sorts of carbohydrate-related data. The broad usage of glycomic databases and associated software tools has been recently reported [9,33,34].
These combined studies have begun to decipher the extent of protein glycosylation in health and disease, highlighting the large proportion of proteins undergoing glycosylation. It is estimated that more than 50% of mammalian proteins are glycosylated, emphasizing the prevalence and significance of this modification. The biological processes in which glycosylated proteins are involved are diverse and essential for normal cellular function. For details on how N-linked glycosylation plays an essential role in the folding and the quality control of proteins in the ER, we refer the reader to excellent papers [35,36,37]. Beyond these functions, key biological processes in which glycosylation plays a crucial role are cellular recognition and signaling, immune cell recognition, antigen presentation, morphogenesis, and the regulation of cell growth, proper neuronal development and function, processes important for extracellular matrix structure and integrity [4,20,38,39,40,41]. It thus comes as no surprise that defects in protein glycosylation are associated with a variety of diseases. We direct the reader to excellent reviews on glycosylation in cancer [42,43,44,45,46,47], neurodegenerative disorders, including Alzheimer’s disease, Parkinson’s disease, autism spectrum disorder, and schizophrenia [48,49,50], congenital disorders [51,52], infection and inflammation [53,54,55,56,57]. To understand how the nature and conformation of the glycan can drastically change the interaction of a protein with another in the context of health and disease, we direct the reader to several review articles [46,58,59,60]. Among the well-studied and understood examples are antibodies, where changes in glycan composition at specific sites are known to influence the antigen-binding affinity of monoclonal antibodies, with fucosylation reducing this interaction [61,62,63,64,65]; the cancer cell glycocalyx—a layer of multifunctional glycans that covers the cell surface—where abundant glycosylation, including sialylation, fucosylation, O-glycan truncation, and N- and O-linked glycan branching has an impact on cell adhesion, and promotion of cancer migration and invasion [18,66,67].
In this review, we delve into the intricate interplay between glycosylation and protein conformation, emphasizing the profound impact of N-glycans on proteins in various diseases. What sets this exploration apart is our focused examination of proteins wherein the presence or absence of specific glycan chains directly shapes the distribution of protein conformations on the energy landscape, dictating the mechanisms of interconversion among these conformations. Importantly, we highlight the role of protein conformational changes and their resulting pathologic functions in understanding the impact of N-glycans. These changes are intricately linked to how the affected protein assembles and interacts with other proteins. This unique focus allows us to unravel the connections between glycosylation and aberrant protein structures, shedding light on the therapeutic implications of targeting conformational and assembly mutants associated with disease. Unlike previous reviews that broadly discuss the role of glycosylation in diseases, our exploration navigates the landscape where the glycome intersects with protein conformation. By doing so, we aim to provide distinctive insights into potential therapeutic avenues and a deeper understanding of the molecular intricacies of how N-glycans impact disease conditions.
2. Glycosylation and Protein Conformation
The attachment of N-glycans to specific asparagine residues in a protein’s polypeptide chain introduces bulky and flexible carbohydrate moieties which can have a dramatic effect on a protein, both locally and distally. For example, Lee et al. performed computational analyses and atomistic molecular dynamics simulations of six glycosylated and deglycosylated protein pairs sourced from the Protein Data Bank [68]. They found that residues being most impacted upon glycosylation were not necessarily near the glycosylated sites, implying that the impact of glycosylation is not localized but rather allosterically propagated to other regions of the protein. Overall the impact was a decrease in the dynamic movements of the proteins under investigation.
While the study provides valuable insights, a potential limitation arises from the current PDB glycoprotein library. Published crystal structures of naturally occurring glycoproteins tend to be derived from proteins that either had their glycosylation sites mutated, had their glycans partially or completely degraded, prior to crystallization, or where the protein was generated using non-native protein expression systems. Recent advancements, such as the limited deglycosylation assay developed by Coutinho et al., address this limitation by offering a systems-wide approach to analyze glycoprotein conformational changes upon exposure of cells to a stressor [69]. This method involves native protein-level N-deglycosylation, trypsin digestion, glycopeptide enrichment, peptide-level N-deglycosylation, and quantitative mass spectrometry-based analysis. Applied to LLC-MK2 epithelial cells exposed to dithiothreitol to induce endoplasmic reticulum stress without impacting protein abundance, the study identified 1145 deglycopeptides. Of these, 1001 peptides, corresponding to 433 unique source glycoproteins, had one or more N-glycosylation sequence motifs. The majority were proteins associated with cellular membranes and were annotated to molecular functions such as binding of cell adhesion molecules, transmembrane transporters and glycosyltransferases activity. Their known participation in biological processes were related to cell adhesion, integrin-mediated cell signaling, glycosylation, secretion and vesicle-mediated transport as biological processes.
Notably, the study revealed that N-glycosites undergoing the highest conformational changes under such minor/moderate ER stress induction were located on loops. Protein loops are inherently flexible regions that readily undergo conformational changes compared to structured elements. The observation that N-glycosites on loops exhibit the highest conformational changes suggests their sensitivity and responsiveness to environmental cues, such as the cellular stress environment. Indeed, loops, along with disordered regions, have recently emerged as crucial "connectors" for relaying information among structured regions of proteins, including secondary structures and domains, thereby shaping protein structural and dynamic profiles and playing key roles in allosteric communication [70]. Glycosylation of these regions can be expected to have a profound effect on the functional motions of PTM-modified proteins. This functional adaptation may involve proteins undergoing conformational changes as part of their response to changing cellular conditions. Additionally, such changes could influence interactions with other molecules, cellular localization, or participation in signaling or other protein pathways. In summary, the dynamic interplay between glycosylation and protein conformation extends beyond the immediate glycosylation sites, influencing protein dynamics and adding an additional layer of complexity to the mechanisms of functional adaptations.
3. N-glycans’ Effect on Pathologic Protein Conformations in Disease
Considering the allosteric effects of N-glycans in regulating protein conformation, with potential implications in its assembly and function, it is of no surprise that dysregulated N-glycosylation has been implicated in several disease-associated human proteins. Furthermore, these glycans may play a pivotal role in modulating the conformation of pathogen-associated proteins, influencing their infectivity within human cells. In the upcoming sections, we delve into specific proteins to illustrate both scenarios, highlighting instances where glycosylation facilitates cellular transformation and enhances the infectivity of pathogenic agents (see section 3.1). Additionally, we examine cases where protein glycosylation transforms a physiological protein into a pathogenic, disease-causing form (see sections 3.2 and 3.3). The goal is to unravel the nuances of these post-translational modifications, shedding light on how they contribute to the intricate interplay between protein conformation and disease.
3.1. Severe Acute Respiratory Syndrome SARS Proteins
Coronavirus Disease 2019 (COVID-19) is a highly transmissible viral infection caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The global impact of COVID-19 has been devastating, resulting in over 6 million deaths worldwide. Initially reported in Wuhan, Hubei Province, China, in late December 2019, SARS-CoV-2 swiftly spread across the globe, prompting the World Health Organization to declare it a global pandemic on March 11, 2020. In 2020, COVID-19 ranked as the third leading cause of death in the United States after heart disease and cancer, accounting for approximately 375,000 deaths [71].
Few glycoproteins have captured as much attention or undergone more detailed investigations than the SARS proteins. Studies have unveiled how glycans can function as shields preventing recognition of viral proteins by the immune system [72], or how they may serve as activators for the lectin pathway [73]. Perhaps most crucially, these investigations have provided significant insights into how glycans can modify and regulate protein conformation, dynamics, and interactions, a topic we will briefly explore below.
Structurally and phylogenetically, SARS-CoV-2 shares similarities with SARS-CoV and MERS-CoV, featuring four main structural proteins: spike (S), envelope (E) glycoprotein, nucleocapsid (N), and membrane (M) protein, along with 16 nonstructural proteins and 5-8 accessory proteins [74]. The surface spike (S) glycoprotein, resembling a crown, is a threefold symmetric homo-trimer [75], where each protein contains approximately 1200 residues (Figure 1A). The spike is positioned on the virion’s outer surface. It undergoes cleavage into an amino N-terminal S1 subunit, facilitating virus incorporation into the host cell. The carboxyl C-terminal S2 subunit includes a fusion peptide (FP), a transmembrane domain (TM), and a cytoplasmic domain crucial for virus-cell membrane fusion [76]. The S1 subunit is further divided into a receptor-binding domain (RBD) and a N-terminal domain (NTD), playing roles in viral entry and serving as a potential target for neutralization by antisera or vaccines [77]. The RBD is pivotal in infection pathogenesis as it binds to the human angiotensin-converting enzyme 2 (ACE2) receptors in the respiratory epithelium. Following viral attachment, the spike protein S2 subunit is primed by the host transmembrane serine protease 2, facilitating cell entry and subsequent viral replication [78].
Binding of the trimeric S glycoprotein to human ACE2 is initiated by at least one protomer’s RBD switching from a ‘down’ (closed) to an ‘up’ (open) state [79,80,81] (Figure 1B). These conformational states transiently interconvert via a hinge-like motion exposing the receptor binding motif (RBM), which is composed of RBD residues S438 to Q506 [82]. The RBM is buried in the inter-protomer interface of the down S protein; therefore, binding to ACE2 relies on the stochastic interconversion between the ‘down’ and ‘up’ states. The spike S has 22 predicted N-glycosylation sites per protomer, with at least 17 experimentally demonstrated to the occupied [72,83]. Of these, 13 putative N-glycosites (N17, N61, N74, N122, N149, N165, N234, N282, N331, N343, N603, N616, and N657) with the N-X-S/T (X ≠ P) sequon and one putative N-glycosite (N334) with the N-X-C (X ≠ P) sequon are on the S1 subunit. The S2 subunit has 9 putative N-glycosites (N709, N717, N801, N1074, N1098, N1134, N1158, N1173, and N1194) with the N-X-S/T (X ≠ P) sequon [16]. Several of these sites play a crucial role in regulating the conformational movements of the protein as well as its dynamics and in turn, have an impact on binding to ACE2 and on infectivity [72,82,84,85,86,87,88,89] (Figure 1B).
MD simulations have revealed detailed information about the structural stability and the role of glycosylation for both the ‘down’ and ‘up’ states, as well as for inter-residue interactions and details of binding to ACE2. A study investigating the SARS-CoV-2 spike protein has emphasized the functional significance of glycans at N165 and N234 in regulating the ‘up’ and ‘down’ conformational states of the spike [72]. Through multiple microsecond-long, all-atom, explicitly solvated MD simulations of the full-length SARS-CoV-2 S glycoprotein with a complete glycosylation profile consistent with glycomic data, this study has unveiled a crucial structural role of N-glycans linked to N165 and N234 in modulating the conformational transitions of the RBD. When the RBD transitioned to the ‘up’ state, the glycan at N234 rotated into the resulting void, stabilizing the up conformation. Simulating the deletion of these glycans via N165A and N234A mutations resulted in a destabilizing effect on the RBD, prompting a conformational shift toward the ‘down’ state (i.e., a state unfavorable for ACE2 receptor binding). This altered RBD conformation substantially reduced binding to ACE2, as confirmed by biolayer interferometry experiments. Consequently, the specific N-glycans at positions N165 and N234 have been identified as essential structural elements for maintaining the SARS-CoV-2 spike protein in a conformation conducive to ACE2 recognition, facilitating subsequent viral entry into host cells.
To explore the pathway of the fully glycosylated SARS-CoV-2 spike protein opening its RBD, a study employed the weighted ensemble (WE) path-sampling strategy, allowing for atomistic simulations of the spike-opening process [90]. WE, as a path-sampling strategy, directs computational resources toward the transitions between stable states rather than the stable states themselves [91]. This is achieved by running multiple trajectories concurrently and periodically duplicating trajectories that transition between previously and newly visited regions of configurational space. This minimizes the time spent waiting in the initial stable state for transitions over the free energy barrier. The extensive WE MD simulations of the glycosylated SARS-CoV-2 spike head, characterizing the transition from the ’down’ to ’up’ conformation of the RBD, revealed a significant gating role for the glycan at N343. This glycan lifted and stabilized the RBD throughout the opening transition. The study also identified an ’open’ state of the spike RBD, where the N165 glycan of chain B remained the last contact with the RBD on the route to further opening of S1. In conjunction with prior studies by Casalino et al [72], this research underscored the crucial role of N343 in gating and facilitating the RBD-opening process, emphasizing the necessity of sampling functional transitions for a comprehensive understanding of mechanistic details.
Pang et al. elucidated two-dimensional free-energy landscapes depicting the opening and closing transitions of the SARS-CoV-2 S-protein, considering both glycosylated and un-glycosylated forms [92]. The study emphasized the influence of glycans on each state and their role in modifying the kinetics of spike opening. It introduced a nuanced perspective on the role of glycans, suggesting a more intricate impact than previously recognized, especially regarding the glycans at N165 and N343. According to the research, these glycans may affect both the ‘down’ and ‘up’ states, creating a local energy minimum for each. Specifically, the study proposed that these glycans could wrap around the RBM when the RBD is in the down state, effectively maintaining it in that configuration. Consequently, these two glycans were identified as contributors to stabilizing both ‘down’ and ‘up’ states, establishing a local energy minimum for each.
Glycans also contribute to infectivity, specifically the fusion peptide’s ability to capture the host cell. In the process of Spike-protein-mediated fusion, the fusion peptides need to be released from the protein core and associate with the host membrane. Successful infection depends on the transition between pre-fusion and post-fusion conformations. To mechanistically describe this pre-to-post rearrangement and understand the impact of glycans, a study conducted thousands of simulations using an all-atom model with simplified energetics [93]. These simulations revealed that the steric composition of the glycans can induce a pause during the conformational change of the Spike protein. This glycan-induced delay presents a crucial opportunity for fusion peptides to effectively capture the host cell, a process that would be inefficient in the absence of glycans. Therefore, the steric composition of both the Spike protein and glycans may guide the overall dynamics of host-membrane capture.
In sum, the SARS-CoV-2 S protein undergoes conformational changes, crucial for host cell entry (Figure 1). Key N-glycosylation sites, stabilize the ‘up’ conformation, facilitating ACE2 recognition and viral entry. Select N-glycans act in gating and stabilizing the S protein during conformational transitions. Furthermore, N-glycans actively contribute to SARS-CoV-2 infectivity by influencing the dynamics of the fusion peptide (FP). This peptide is instrumental in capturing the host cell during the spike protein-mediated fusion process. The steric composition of glycans induces strategic pauses in conformational changes, creating vital windows for efficient host cell capture. To sum up, strategically positioned N-glycans intricately regulate the conformation and dynamic movements of the S protein. This regulation, in turn, dictates its ability to attach to the ACE2 receptor, enter the cell, and initiate infection in the host.
3.2. Prion Protein
Prion diseases, or transmissible spongiform encephalopathies (TSE), are a class of fatal, infectious neurodegenerative illnesses that affect both humans and animals. Among human prion diseases are Creutzfeldt-Jakob Disease (CJD), Variant Creutzfeldt-Jakob Disease (vCJD), Gerstmann-Straussler-Scheinker Syndrome, Fatal Familial Insomnia and Kuru. Most prevalent in animals are bovine spongiform encephalopathy (BSE) (or Mad Cow Disease) in cattle, chronic wasting disease (CWD) in deer elk and moose and scrapie in sheep [94,95,96]. These diseases occur when a normally occurring protein, the prion protein, undergoes a pathogenic transformation, where the key factor driving the pathology is a conformational change. Prion proteins, occupying a middle ground between viral and human counterparts, are the subject of our second discussion exploring how N-glycans serve as modulators of protein conformation.
Prion diseases are caused by the conformational rearrangement of the endogenous cellular prion protein (PrPC) into an abnormal, toxic form (PrPSc) [97]. PrPC is a N-glycosylated protein attached to the outer leaflet of the plasma membrane through a glycosyl phosphatidylinositol (GPI) moiety [98,99]. While expressed in nearly all tissues, it is particularly abundant in neurons, which also constitute the major targets of TSE-related degeneration [100]. N-Glycosylation influences the conformational stability of PrP [101,102,103,104,105]. Changes in glycosylation patterns can impact the folding kinetics of PrP and may contribute to the transition from the normal, cellular form to the disease-associated form. Different glycoforms may exhibit variations in their susceptibility to conversion into the pathological PrPSc form, affecting the progression of prion diseases. The impact of the glycans, or the lack of, however may be strain dependent and the conversion of PrPC into PrPSc may be sustained through several pathways depending on the origin of the disease [101,102,103,104,105]. In the case of infectious TSE, it is proposed that exogenous PrPSc interacts with PrPC and acts as a template for the conversion of the latter into an extra PrPSc molecule. As for familial prion diseases, mutations within the prion protein gene would favor PrP folding in a pathogenic conformation [101,106,107,108].
Here we intend not to delve into this complexity but rather explore how changes in glycosylation may support pathogenic prion protein conformation, interaction and function. As anticipated for a membrane protein, the newly synthesized 254-amino acid PrPC undergoes cleavage of its hydrophobic N-terminal signal peptide (N-ter1–23) to facilitate targeting to the rough endoplasmic reticulum, the initial compartment of the secretory pathway. Within this compartment, co- and post-translational modifications of PrPC take place, encompassing the addition of high mannose-type N-glycans (Glc3–Man9–GlcNAc2) at positions Asn181 and Asn197 in human (huPrP) and Syrian hamster (ShPrP) PrPC (corresponding to Asn180 and Asn196 in mouse, MoPrP), formation of a unique disulfide bond, and attachment of a GPI lipidic moiety following cleavage of the hydrophobic C-terminal signal peptide. The glycosylation sites of PrPC exhibit variable occupancy, resulting in non-, mono-, or di-glycosylated forms [99,101,102,105]. The N-terminal segment (residues 23–125, after removal of the signal peptide) exhibits a high degree of flexibility. Within this segment is an octapeptide repeat (OR) domain which binds the divalent ions Cu2+ and Zn2+. The C-terminal domain (residues 126–230), which contains the glycan attachment sites, folds to a characteristic structure composed of three α-helices, numbered one through three (or A through C), and two anti-parallel β-strands flanking helix 1 (Figure 2A).
In cellulo and in vivo, studies support the role of these glycans in modulating a pathologic conformation and function of PrP [101,109,110,111] (Figure 2B). For example, since Syrian Hamster PrPC (ShPrP) contains complex-type oligosaccharides attached to Asn residues 181 and 197 (same as in humans), an early study mutated the Thr residues to Ala within the NXT consensus sites [112]. Single and double glycosylation site mutations were expressed in Tg mice deficient for mouse MoPrP, and the brains were analyzed for the distribution of mutant ShPrPC. Analysis was confined to the hippocampal region in each case. Wild-type ShPrPC was distributed almost exclusively to the dendritic trees of the CA1 to CA4 regions of Ammon’s horn and the dentate gyrus and was absent from the cell bodies of pyramidal and granule cell layers in the respective regions; additionally, it was largely absent from white matter tracts such as the corpus callosum. In contrast, mutation of either one or both glycosylation consensus sites had a profound effect on the anatomical distribution of ShPrPC. Mutation of the first glycosylation site alone or in combination with mutation of the second site resulted in low levels of mutated ShPrPC, accumulation of ShPrPC in nerve cell bodies, and little ShPrPC in the dendritic trees. When the second glycosylation site was mutated, the levels of ShPrPC(T199A) were about the same as wild-type ShPrPC; however, ShPrPC(T199A) appeared to be distributed to all neuronal compartments including the cell body, dendritic tree, and axons in the white matter. Transgenic mice inactivated at the second Asn197 site (T199A) supported prion replication upon infection, while mice mutated at the first site appeared resistant [98,112].
A recent study employed knock-in mouse models expressing cell surface PrPC with 0 or 2 N-glycans and several complementary approaches to address the impact of glycosylation on prion protein localization and function [105]. Mice expressing PrPC without glycosylation were generated through the introduction of two-point mutations at the endogenous Prnp locus using a single guide RNA. These mutations, corresponding to the substitution of asparagine to glutamine at positions 180 and 196 (in accordance with mouse PrP numbering), led to alterations in the N-glycosylation sequons. Prnp180Q/196Q mice exhibited normal expression and trafficking of PrPC with no evidence of spontaneous prion disease. However, a significant difference in susceptibility to prion infection was observed between Prnp180Q/196Q mice and WT mice. Notably, the Prnp180Q/196Q mice consistently showed more severe spongiform degeneration across all strains compared to WT mice. Additionally, upon prion infection, these mice displayed marked atrophy of the hippocampus due to severe neuronal loss, including complete loss of CA1 pyramidal neurons and the presence of numerous gemistocytic astrocytes - reflecting the enlarged and filled appearance of the cell. Gemistocytic astrocytes are often associated with certain pathological conditions. This effect persisted upon second passage. In contrast, WT mice exhibited moderate loss of hippocampal neurons. Furthermore, the cerebellum of all infected Prnp180Q/196Q mice lacked PrPSc, a notable distinction from WT mice where all three strains were present in the cerebellum. Importantly, the absence of PrPSc in the cerebellum of Prnp180Q/196Q mice was not attributed to a lack of PrPC expression, as Prnp180Q/196Q was expressed in the cerebellum at levels similar to WT PrP. The PrPSc morphology in Prnp180Q/196Q brains differed from WT mice, with most Prnp180Q/196Q brains exhibiting plaque-like deposits or dense parenchymal plaques. Consequently, Prnp180Q/196Q mice challenged with subfibrillar prion strains acquired distinct disease features, including an increase in plaques and plaque-like structures, more severe cortical spongiosis, and a noticeable absence of prions in the cerebellum.
A study by Yi and colleagues [110] explored the impact of glycosylation on various aspects of PrP, using cultured cells expressing wild-type PrP and glycosylation mutants. The study found that glycosylation significantly influenced the subcellular localization, resistance to proteolytic digestion, and aggregation ability of human PrP. Wild-type PrP and monoglycosylated mutants - N181D, N197D, and T199N/N181D/N197D - were primarily attached to the plasma membrane, while pathological mutants with altered glycosylation sites (i.e., PrP F198S) and unglycosylated PrP mutants (i.e., N181D/N197D) were mainly present in the cytoplasm. Furthermore, the study revealed that the degree of glycosylation correlated with the protein’s proteolytic resistance and aggregation ability. PrP with fewer glycosylation modifications exhibited higher aggregation propensity and a higher degree of abnormal conformers, as measured by its resistance to protease digestion. Additionally, glycosylation deficiency increased the vulnerability of the protein to stressors and enhanced its cytotoxicity.
In the context of understanding the impact of N-glycans, it is crucial to underscore the role of protein conformational changes and their resulting pathologic functions, which are intricately linked to how the affected protein assembles and interacts with other proteins. Glycosaminoglycans (GAGs), especially heparan sulfate and heparin, are considered crucial molecules for prion conversion and infection. For instance, in prion diseases, heparan sulfate, a prevalent polyanion in the brain, accelerates disease progression by facilitating the conversion and assembly of extracellular, ADAM10-cleaved PrP into parenchymal plaques [119]. In mice, di-glycosylated PrPSc demonstrated the least heparan sulfate binding, while unglycosylated PrPSc exhibited the highest heparan sulfate binding [105]. Similarly, glycans also disrupt PrP binding to heparin, with di-glycosylated PrPC exhibiting the lowest affinity for heparin binding [101]. Notably, PrPC with 2 to 3 glycans displayed low heparin affinity, while unglycosylated PrPC showed high affinity, with this affinity progressively decreasing with each additional glycan. The heparin-binding affinity of PrPC from age-matched WT and Prnp180Q/196Q mouse brain homogenates followed a similar pattern—unglycosylated PrPC exhibited higher heparin-binding affinity than glycosylated WT PrPC, a trend also observed in their ADAM10-cleaved counterparts [105].
Structural and biochemical studies have provided explanation on how these glycans may affect the prion protein. From early NMR structures of the full-length and N-terminally truncated forms of recombinant MoPrP, ShPrP, and HuPrP it became known that the entire N-terminal segment PrP23–126 is flexibly disordered and that only the C-terminal part PrP127–231 possesses a defined 3D structure [113,114]. Molecular dynamics simulations on the C-terminal region of human prion protein HuPrP(90–230), with and without the glycans, suggested the structured part of the protein, HuPrP(127–227) was stabilized overall from addition of the glycans, specifically by extensions of Helix-B (i.e., helix 2) and Helix-C (i.e., helix 3) and reduced flexibility of the linking turn containing Asn197. The stabilization appeared indirect, and not from specific interactions such as H bonds or ion pairs. Thus glycosylation at Asn197 has an allosteric effect, with impact on stabilizing a conformation of the protein [115].
Recent biochemical studies have provided additional insights into the role of glycans in the prion protein. NMR and electron paramagnetic resonance spectroscopy studies suggest that the two N-glycans play a crucial role in maintaining an intramolecular interaction with the N-terminal domain [111,116,117]. This interaction is mediated through a copper–histidine tether, bringing the C- and N-terminal domains into proximity and likely stabilizing the overall structure while reducing dynamic motion. The conformational alterations in PrP result from an interplay between two histidines on the C-terminus of the prion protein that tether the N-terminus via an N-terminally bound copper ion. Additionally, PrP glycans promote the N-C interaction, synergizing with the effect of His-Cu coordination [111,118]. A patch of negatively charged amino acids on the same protein surface as the histidines and glycans serves as a third contributor to these interactions. These interactions play a functional role in suppressing the neurotoxic activity of PrPC, as demonstrated by studies on the PrP mutant N180Q/N196Q. This mutant, where Asn residues at the glycan attachment sites were replaced with Gln residues to prevent glycosylation while preserving the polar carboxamide side chain common to both Asn and Gln, exhibited effects similar to a highly toxic deletion mutant of PrP [111,118].
Together these studies indicate the loss of glycans destabilize the prion protein structure enriching in a conformation that enable pathologic interactions. Loss of glycosylation could increase the affinity of PrPC for a particular conformer of PrPSc and of other pathologic interactors (such as heparan sulfate) determining the rate of nascent PrPSc formation and the specific patterns of PrPSc deposition (Figure 2). Intriguingly, distinct brain regions, and presumably cell types, demonstrated distinct vulnerability to these pathologic conformers. Also, the lack of glycans also increased the vulnerability of the protein to additional stressors, increasing its pathogenicity.
3.3. Glucose Regulated Protein 94 (GRP94)
Glucose-Regulated Protein 94 (GRP94), also known as endoplasmin and gp96, serves as a crucial molecular chaperone located in the endoplasmic reticulum (ER) of eukaryotic cells [120]. Its primary functions involve ensuring proper folding, maturation, and quality control of client proteins within the ER. Beyond its role in protein folding, GRP94 actively participates in various cellular processes, contributing to cellular homeostasis and overall cell function [121,122]. GRP94 forms a homodimer, and each chain comprises three domains: the N-terminal (NTD), middle, and C-terminal (CTD) (Figure 3A). Mechanistically, the chaperone undergoes ATP-driven conformational changes associated with the folding of a client protein. ATP binding at the NTD induces a shift in the chaperone to a closed conformation, inducing changes in regions critical for protein client binding. Following ATP hydrolysis, rearrangements occur in the residues of the client-binding site, leading to mechanical translation into conformational changes in the bound client [121,122].
While primarily localized to the ER, GRP94 is also found in the cytosol, at the cell surface, and extracellularly [121,123,124]. This altered distribution is often associated with and intensified in disease-related scenarios [125,126] (Figure 3B). For instance, pathogens utilize surface GRP94 to infect host cells [127,128,129]. In autoimmune diseases, overexpression of cell-surface GRP94 enhances toll-like receptor function and downstream signaling through MyD88 [130]. In cancer cells, surface GRP94 imparts an aggressive phenotype through regulating the stability of tyrosine kinases, such as HER2 and EGFR, inhibiting their internalization and enhancing their aberrant downstream signaling [131,132,133,134]. In inflammatory diseases, surface GRP94 induced a pro-inflammatory profile in macrophages [135,136]. Similarly, an extracellular GRP94 complexed with immunoglobulin Gs (IgGs) contributes to the pathogenesis of type 1 diabetes [137].
N-glycosylation plays an important role in the generation of such pathologic surface-expressed and extracellular GRP94 forms (Figure 3C). GRP94 contains six potential N-glycan acceptor sites – namely N62, N107, N217, N445, N481 and N502 - yet under normal conditions, the protein is predominantly monoglycosylated at N217 [138,139]. Cherepanova and colleagues [138] demonstrated that GRP94 can become glycosylated at all sites in cells that are exposed to oligosaccharyltransferase inhibitors or low doses of ER stress-inducing agents, and in cells with partial or complete loss of oligosaccharyltransferase complex activity. In a later study, Wen and colleagues demonstrated that glycosylation of silent sites was heterogenous. Some sites, such as N62 were enriched in with mannosylated N-glycans, such as Man9GlcNAc2 and Glc1Man9GlcNAc2 (N2H9 and N2H10), whereas N107 and N445 had a varied content of mannosylated N-glycans but also fucosylated or sialofucosylated complex-type N-glycan structures [140].
The regulation of GRP94 glycosylation, promoting uniform skipping of silent sites in nonstressed cells and efficient and rapid glycosylation of silent sites in stressed cells, raises intriguing questions. None of the GRP94 sequons have a negative flanking sequence score (i.e., surrounding amino acid sequences do not hinder glycan addition), indicating that these sites are not skipped due to suboptimal conditions. Cherepanova et al. [138] proposed instead that the existence of a mechanism restricting nascent GRP94 access to the STT3A active site, blocking cotranslational glycosylation of the silent sites, is responsible for the lack of glycosylation on the silent sites. In this proposed scenario, the N62 and N107 sites in GRP94 could enter the STT3A active site before the normal glycosylation site (N217) is incorporated into the nascent chain. However, factors associated with cellular stress may saturate pathways responsible for blocking glycosylation of the silent sites in GRP94 by OST. Consequently, the N62 and N107 silent sites could become glycosylated by OST complexes in cells exposed to stressors, potentially contributing to disease-related glycosylation patterns. One however cannot exclude that other, probably context specific, alterations in the glycosylation machinery may exist, shaping the occupancy of individual silent sites, each possibly impacting the conformation and function of the GRP94 protein.
Supportive of this notion, N-glycan occupancy at these silent sites was indeed reported in disease. In OVCAR-3 ovarian cancer cells, which exhibit high levels of EGFR at the plasma membrane [141], a study employed an unbiased, large-scale MS method to determined N-glycosylation site occupancy comparing tunicamycin (TM) treated - to inhibit the overall N-glycosylation occupancies of the cells – and untreated cells [142]. Using this method, they determined that GRP94 had four of the sites - N62, N107, N217 and N481 – occupied in OVCAR-3 cells. Out of these sites, N217 had a high occupancy (~100%), N62 occupancy was moderate (~50%), followed by N107 (<50%) and N481 (minor, ~10%). Yan and colleagues performed glycosylation site mapping by mass spectrometry and identified N62, N217, and N502 as putative N-glycosylated sites on a GRP94 variant enriched on the cell surface of MDA-MB-468 cells, an EGFR-overexpressing triple negative breast cancer cell line [133]. Here too, N217 was a high occupancy site, with N62 and N502 being partially occupied.
Biochemical, functional, and structural investigations unequivocally affirm the pathological implications of N-glycan occupancy at specific silent sites [129,133,135,136,143]. Within certain breast cancer cell subtypes, the glycosylation event at N62 emerges as a pivotal factor contributing to their aggressive phenotype, resistance to therapy, and immune evasion [133,135,144]. This transformation leads to the stabilization of a unique conformation of GRP94, with significant repercussions [133,143]. Primarily, N-glycosylation at N62 serves as a structural mediator, inducing the conversion of the GRP94 chaperone into epichaperomes—hetero-oligomeric forms tightly composed of chaperones, co-chaperones, and other factors [133,145,146]. In these epichaperomes, GRP94 adopts scaffolding functions not observed in normal cells, where GRP94 primarily participates in protein control and folding. Through this scaffolding function, GRP94 influences the assembly and connectivity of proteins crucial for maintaining a malignant phenotype, enhancing their activity. Consequently, the functions of these proteins are markedly enhanced, leading to the aberrant remodeling of dependent cellular protein networks. This provides a survival advantage to cancer cells and tumor-supporting cells within the tumor microenvironment. While the precise composition of the glycan at N62 remains unknown, evidence suggests that the modified residue, rather than the specific sugar structure, is crucial for mediating the cancer-supporting conformation of GRP94 in the N62 glycoform [133,143,147]. Despite this uncertainty, its susceptibility to deglycosylation by EndoH suggests a high mannose glycan characteristic [133]. This notion is also supported by glycoproteomics studies from Wen and colleagues which found N62 to be enriched in mannosylated N-glycans, such as Man9GlcNAc2 and Glc1Man9GlcNAc2 (N2H9 and N2H10) [140].
Among the proteins affected by this pathologic GRP94 glycoform are receptor tyrosine kinases (RTKs). In breast tumors characterized by the overexpression of RTKs like HER2 and EGFR, GRP94 with the N62 occupied site becomes enriched at the cell surface [133]. This enrichment plays a pivotal role in reducing RTK internalization and maintaining the RTK in a state conducive to constitutively enhanced downstream signaling. To validate these findings, Yan and colleagues utilized CRISPR-Cas9 to manipulate endogenous GRP94, generating homozygous clones—N62Q, N217A, and N62Q/N217A—from the MDA-MB-468 breast cancer cell line [133]. Clones expressing GRP94N62Q exhibited a significant absence of EGFR at the plasma membrane, accompanied by a lack of EGFR-signaling activity. Conversely, the GRP94N217A mutants, representing the folding form of GRP94, demonstrated no deleterious effects on EGFR levels or signaling [133]. This underscores the pathologic gain-of-function nature of N62 glycosylated GRP94 in the context of cancer. Specifically for GRP94, at the plasma membrane (where Glyc62GRP94 is located), Glyc62GRP94 may promote cancer as, by forming epichaperome platforms [133,145,147], it provides a backbone upon which oncogenic proteins and protein assemblies cluster, augmenting their pathologic function and leading to an aggressive phenotype in Glyc62GRP94-expressing cancer cells (Figure 3B).
To understand how N-glycans at N62 and N217 influence GRP94 conformation and dynamics, Castelli et al. employed MD simulation studies [143]. Their study revealed dynamic modulation of GRP94’s conformation and interactions by these glycans, impacting protein interaction mode and ATP processing essential for folding (Figure 3C). In the fully glycosylated state, sugars obstructed the N-terminal domain (NTD), impeding ATP binding. The N62 glycan favored an open, ATPase-incompetent NTD lid conformation, influencing the charged linker between the NTD and M-Domain. This glycan altered conformational ensembles, affecting the efficiency of translating nucleotide-encoded signals. Conversely, the N217 glycan had little impact on these factors. The results suggest that N62 glycosylation actively shifts GRP94 from a foldase to a protein-assembly platform, impacting the ATP-binding site’s efficiency and perturbing the charged linker’s dynamics. Ultimately, glycosylation induces GRP94 malfunction, disrupting its structural ensembles, chaperone cycle kinetics, and leading to interactome remodeling at a much larger scale than the simple local covalent modification might lead to hypothesize, amplifying dysfunction, and remodeling cellular phenotypes.
In summary, the N-glycosylation of silent sites in GRP94 during disease profoundly influences the protein’s conformation, assembly, and function (Figure 3). Under normal physiological conditions, GRP94 is glycosylated at N217, localized to the ER, and facilitates client protein folding through transient interactions (Figure 3A-C). However, N-glycosylation at N62 disrupts GRP94’s ER confinement, leading to a conformational shift that promotes stable interactions with oncoproteins at the plasma membrane, enhancing their functions and inducing aberrant remodeling of cellular protein pathways. This glycosylation transforms GRP94 from a folding chaperone to a scaffolding protein, consequently reshaping protein assembly and connectivity, resulting in systems-level dysfunction. Consequently, alterations in N-glycosylation at N62 generate a distinct protein with unique conformational, dynamic, and functional characteristics compared to normal GRP94 in healthy cells. Thus GRP94 is a unique example where N-glycosylation increases the oncogenic properties of a protein indirectly by modulating its complexation.
4. Therapeutic Implications - Targeting Conformational and Assembly Mutants in Disease
Understanding the pivotal role of N-glycans in shaping the conformation and assembly of these proteins is not only crucial for unraveling the intricate mechanisms underlying disease pathological mechanisms but also presents unique opportunities for therapeutic intervention (Figure 4). Disease specific conformations, regulated by glycosylation, are conformational mutants and as such provide a unique “target” of intervention. A conformational mutant refers to a mutant form of a protein that differs from the wild-type (normal) protein in its three-dimensional structure or conformational dynamics. Thus, the conformational and assembly mutants induced by aberrant N-glycosylation in these proteins serve as actionable targets, offering avenues for targeted interventions through antibodies, small molecules, and other innovative approaches. This exploration opens new frontiers in the development of precision therapies, highlighting the potential to modulate disease-associated protein structures and functions with a high degree of specificity. We discuss how the intricate interplay between N-glycans and protein conformational dynamics, paves the way for novel therapeutic strategies.
4.1. Targeting The viral Protein Conformations
In the preceding section, we extensively explored the mechanisms underlying SARS-CoV-2 infectivity, unraveling crucial insights that have paved the way for therapeutic advancements. Leveraging this knowledge, therapeutic strategies have been devised to manipulate glycan-mediated conformational changes in the viral spike protein, aiming to attenuate infectivity and elicit a robust immune response targeting diverse epitopes. Notable contributions include the development of mRNA vaccines encoding stabilized soluble S trimers in the prefusion conformation, fostering neutralizing antibody responses for robust host protection against viral infection [148].
Positioned on the virion’s outer surface, the spike protein undergoes cleavage into S1 and S2 subunits, where the absence of N-glycans at positions N165 and N234 on S1 significantly impacts the conformational flexibility of the receptor-binding domain (RBD). These glycans play a pivotal role in stabilizing the RBD in the "up" conformation, crucial for effective ACE2 receptor binding and viral invasion [76] (Figure 1). Casalino and colleagues [72] proposed that the introduction of N165A and N234A mutations may provide a means to deliberately control the RBD’s conformational dynamics, favoring a predominantly "down" state. This alteration could potentially yield less infectious viruses with RBDs shifted toward the closed state, making the virus more vulnerable to immune detection. An alternative approach, presented by Dodero-Rojas et al., targets the conformational change in S2 responsible for membrane fusion [93]. This highlights the potential to impede viral entry by targeting intermediates of the spike protein’s conformational change.
The critical structural role of specific N-glycans in modulating the SARS-CoV-2 spike protein’s dynamics and conformational states has also informed the rationalization of neutralizing antibody activity. With over ten thousand neutralizing antibodies discovered, the primary target is the RBD, although some also target the NTD and other non-RBD epitopes [149,150,151]. A study by Pang et al. [92] investigated the differential impact of glycans on epitope exposure, considering the spike-opening dynamics of the SARS-CoV-2 S protein and the influence on various antibodies. The analysis by Pang and colleagues involved seven antibodies with epitopes covering RBD and NTD regions. They found that epitope exposure, measured by accessible surface area, was observed to remain constant or increase significantly during the transition from the down to the up state of RBD, impacting the binding of some but not all of the evaluated antibodies. Other studies used explicit dynamic information on WT SARS-CoV-2 S protein and several mutants to reveal the intrinsic determinants of epitope presentation and the mechanisms of antibody escape [152,153,154]: in all these cases mutations modify the glycan-shield of the S-protein with a strong impact on its dynamics and recognition properties.
4.2. Correcting the Prion Protein Conformation
Many studies revealed that the conformational transition of prion protein (PrP) from a normal cellular form (PrPC) into an abnormal form (PrPSc) is a key event in the pathogenesis of prion diseases. Although the exact mechanism of transition from PrPC to PrPSc and the structure of PrPSc remain unknown, N-glycans play an important role, as detailed in prior sections above. In Papua New Guinea, a genotype, V127M129, has been identified as completely resistant to prion diseases [155,156]. To decipher the disease-resistant effect of the G127V mutant, Zheng and colleagues [157] conducted detailed structural and dynamic analyses on the G127V-mutated human PrP (residues 91–231) in comparison with wild-type PrP. Their investigations revealed distinctive alterations induced by the G127V mutation. The G127V mutant formed a stretch-strand (SS) pattern with two segments (SS1: Tyr128-Gly131; SS2: Val161-Arg164), while the WT protein formed a stable β-sheet with two strands (β1: Tyr128-Gly131; β2: Val161-Arg164). The larger hydrophobic side chain of Val127 introduced steric hindrance and prompted a notable rearrangement in the Tyr128 side chain. Consequently, this work suggests that interventions preventing the conversion of SS1 (Tyr128-Gly131) and SS2 (Val161-Arg164) segments and adjacent regions into a stable β-sheet could prevent the pathologic conformational shift of the PrP. This study therefore underscores the potential for therapeutic development by correcting specific pathologic PrP conformations, leveraging an understanding of structural differences between the wild-type and the G127V protein.
Others, have opted for designing small molecules, termed medical chaperones, that could specifically bind to and stabilize the native conformation of PrP and prevent abnormal aggregate formation [158]. Along these lines, Yamaguchi and collaborators developed a small molecule, termed MC, specifically designed to stabilize the normal conformation, PrPC [159]. Given that the normal conformation, PrPC, is highly conserved across species [160], this approach holds promise as it may be effective across various prion diseases and independent of strain or species. MC demonstrated the ability to stabilize the normal cellular prion protein, eliminate prions in infected cells, prevent the formation of drug-resistant strains, and directly inhibit the interaction between prions and abnormal aggregates. In prion-infected mice, MC extended their survival, while in macaques infected with bovine transmissible spongiform encephalopathy, MC slowed down the development of neurological and psychological symptoms and reduced the concentration of disease-associated biomarkers in cerebrospinal fluid.
4.3. Targeting Pathologic GRP94 Conformers and Assemblies
GRP94 has emerged as a key player in cancer [123,161,162]; however, clinical therapeutics targeting the protein have yet to be developed. This is largely attributed to our insufficient understanding of how to differentiate GRP94 in normal cells from that expressed by tumors and tumor-supporting cells. The identification of glycosylation on N62 of GRP94 as a mechanism for the formation of conformational mutants associated with protein assembly mutations in cancer [133,143] opens new avenues for developing therapeutics targeting disease-associated GRP94 forms. From a therapeutic perspective, the focus is on understanding the impact of N62 glycosylation on GRP94 conformation and assembly, influencing function, rather than identifying defects in the glycosylation machinery leading to impaired glycan processing of GRP94.
Yan and colleagues have shown that tumor Glyc62GRP94 (i.e., GRP94 glycosylated at N62) can be selectively targeted over the physiologic GRP94 through therapeutics such as PU-WS13 [133]. PU-WS13 is a small molecule with preferential activity for Glyc62GRP94 over GRP94 [133,143]. PU-WS13 binds to an allosteric pocket of GRP94 that only partly overlaps with the ATP-binding pocket and is not accessible in closely related paralogs of GRP94 [132,163,164,165]. PU-WS13 is inactive or shows reduced activity in Glyc62GRP94-negative but GRP94-positive cells [132,133]. PU-WS13 shows reduced binding to an N62QGRP94 mutant when compared to Glyc62GRP94. Mutations at residue N217, the site required for GRP94 folding functions, in the context of an N-glycosylated N62 residue (i.e. in N217A-Glyc62GRP94) do not interfere with PU-WS13’s binding. Deglycosylation with EndoH, which retains N-acetylglucosamine attached on Asn, reduces PU-WS13’s binding to GRP94, whereas PNGaseF, which removes the N-glycan in its entirety leaving the unmodified Asn-NH2 residue, abolishes PU-WS13’s binding to GRP94. PU-WS13 is toxic to Glyc62GRP94-expressing cancer cells but not to cancer cells with abundant GRP94 or N62QGRP94 but no Glyc62GRP94 expression. N62QGRP94 expressing clones and GRP94 KO clones displayed reduced sensitivity to PU-WS13. Conversely, the N217AGRP94 mutants (i.e. of the folding GRP94) had no deleterious effect on PU-WS13 activity. Combined, biochemical and functional studies confirm that the biological effects observed with PU-WS13 are Glyc62GRP94 specific [129,133,135,143,144].
Castelli et al. [143] employed molecular dynamics simulations to unravel the dynamic mechanisms of GRP94 glycoforms. They utilized these findings to rationalize the engagement of specific conformational states by PU-WS13, providing crucial insights into the targeting of the disease-specific, aberrantly glycosylated GRP94 variant by this small molecule. These simulations also offer structural support, elucidating how pathologically N-glycosylated GRP94 variants can be selectively targeted and influenced.
Several studies have shown that inhibition of Glyc62GRP94 by PU-WS13 was safe and active against cancers in vivo. For example, in nude mice, PU-WS13 suppressed the growth of Glyc62GRP94-dependent breast tumors without affecting GRP94 functions [133]. Even for the long-term treatment regimens that delivered 37 to 62 doses of PU-WS13 to mice over 87 days, no treatment-related toxicities were observed. In a preclinical model of 4T1 breast tumor-bearing BALB/c mice, PU-WS13 decreased CD206-expressing M2-like macrophages in the TME, reduced tumor growth and collagen content, and increased the recruitment of CD8+ cells in the TME [144]. This is of high clinical relevance as M2-macrophages are strongly associated with fast proliferation, poor differentiation and ER-negativity in human primary breast tumors [166].
PU-WS13 also alleviated inflammatory responses in primary macrophages in a model of alcohol-induced liver damage [136]; enhanced pneumococcal clearance from lung tissues and ameliorated lung pathology in a mouse model of influenza A virus infection with secondary bacterial pneumonia [129]; reduced the pro-inflammatory profile of macrophages in disease models of inflammation [135]; and showed antiviral activity against Dengue virus serotypes and Zika virus strains in multiple human cell lines in viral replication models [128], supporting Glyc62GRP94 as targets in several distinct disease states, beyond cancers [128,129,135,136].
5. Conclusions and Future Directions
In conclusion, the investigation of the intricate interplay between N-glycans, protein conformational dynamics, and disease pathogenesis not only deepens our understanding of pathophysiological mechanisms but also unveils a rich landscape for innovative therapeutic strategies. The discussions herein illuminate the potential for targeted interventions across viral infections, prion diseases, and cancer, showcasing the transformative power of comprehending and manipulating protein conformations. Importantly, while our examples are focused on these specific diseases, the principles elucidated in this exploration hold promise for impacting a myriad of other disorders where conformational mutants play a pathologic role. This profound insight not only opens new avenues for precision therapies but also underscores the importance of a multidisciplinary approach in developing effective strategies against diverse and complex diseases. As we navigate this frontier, the convergence of disciplines holds promise for a future where tailored interventions based on protein conformational nuances become a cornerstone in the treatment of intricate diseases.
Glycoform specific targeting is a new frontier in several diseases, including cancer and neurodegenerative diseases. It offers a solution to limit off-target effects by increasing the antitumor specificity. Current therapeutic strategies are based on the development of antibodies, which detect cancer-associated glycoforms of therapeutic targets, such as integrins, RTKs, and other glycoproteins and on the discovery of small molecules that interfere with specific glycosylation pathways altered in disease [45,66]. The platform we describe is unique as it targets the pathologic conformation and assembly of a protein generated by site-specific glycosylation.
By influencing protein conformations and impacting the abundance of specific forms at distinct cellular locations, glycans may significantly alter cellular networks. Distinct conformers can engage in different interactions, thereby remodeling cellular protein-protein interaction networks at a large scale by shifting the assembly of multiprotein complexes required to regulate biomolecular pathways toward protein assemblies with pathologic activities, known as protein assembly mutations. In this context, understanding the interactome – the complement of interacting partners – of the glycomutant is crucial. The interaction of a protein with different binding partners within diverse biological contexts influences the functional output of interactions. A protein may be assigned to different protein pathways in distinct cellular contexts based on the presence of potential interactors [145]. Therefore, systems-level investigations in specific disease contexts are essential to provide informed insights into cellular transformation by conformational mutants. Mapping their interactors is expected to inform not only on the identity of proteins altered by conformational mutants but also, more importantly, on the functional impact of such alterations at the network and cellular levels. This knowledge may pave the way for novel therapeutic strategies targeting disease-specific protein assemblies, particularly focusing on assembly mutants, i.e., disease-specific protein-protein interactions, which represent a promising avenue in this evolving landscape.
Author Contributions
Conceptualization, G. Colombo and G. Chiosis; writing—original draft preparation, G. Chiosis; writing—review and editing, S.S.; T.R.; C.P.; E.M.; G. Colombo; visualization, S.S.; T.R.; C.P.; E.M.; G. Colombo, G. Chiosis; funding acquisition, G. Colombo and G. Chiosis; All authors have read and agreed to the published version of the manuscript.
Funding
G. Chiosis acknowledges funding from the National Institutes of Health (R01 CA172546, P01 CA186866, R56 AG061869, RF1 AG071805, R01 AG067598, R01 AG074004, R01 AG072599, P30 CA08748); G. Colombo acknowledges funding from AIRC under IG 2022 - ID. 27139 project; from PRIN (grant 20209KYCH9); from the IMMUNO-HUB, T4-CN-02, project from the Ministero della Salute (Italy); and from Programma di ricerca CN00000013 “National Centre for HPC, Big Data and Quantum Computing. S.S. would like to acknowledge funding support from BrightFocus Foundation (Award ID: A2022020F). .
Conflicts of Interest
G.C. and S.S. are inventors on patent applications related to the epichaperome portfolio. All other authors declare no conflicts of interest.
References
- Moremen, K.W.; Tiemeyer, M.; Nairn, A.V. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol 2012, 13, 448-462. [CrossRef]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat Rev Nephrol 2019, 15, 346-366. [CrossRef]
- Schjoldager, K.T.; Narimatsu, Y.; Joshi, H.J.; Clausen, H. Global view of human protein glycosylation pathways and functions. Nat Rev Mol Cell Biol 2020, 21, 729-749. [CrossRef]
- Varki, A. Biological roles of glycans. Glycobiology 2017, 27, 3-49. [CrossRef]
- Rini, J.M.; Moremen, K.W.; Davis, B.G.; Esko, J.D. Glycosyltransferases and Glycan-Processing Enzymes. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor (NY), 2022; pp. 67-78.
- Corfield, A.P.; Berry, M. Glycan variation and evolution in the eukaryotes. Trends Biochem Sci 2015, 40, 351-359. [CrossRef]
- Lebrilla, C.B.; Liu, J.; Widmalm, G.; Prestegard, J.H. Oligosaccharides and Polysaccharides. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor (NY), 2022; pp. 33-42.
- Seeberger, P.H. Monosaccharide Diversity. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor (NY), 2022; pp. 21-32.
- Rudd, P.M.; Karlsson, N.G.; Khoo, K.H.; Thaysen-Andersen, M.; Wells, L.; Packer, N.H. Glycomics and Glycoproteomics. In Essentials of Glycobiology, 4th ed.; Varki, A., Cummings, R.D., Esko, J.D., Stanley, P., Hart, G.W., Aebi, M., Mohnen, D., Kinoshita, T., Packer, N.H., Prestegard, J.H., et al., Eds.; Cold Spring Harbor (NY), 2022; pp. 689-704.
- Chao, Q.; Ding, Y.; Chen, Z.H.; Xiang, M.H.; Wang, N.; Gao, X.D. Recent Progress in Chemo-Enzymatic Methods for the Synthesis of N-Glycans. Front Chem 2020, 8, 513. [CrossRef]
- de Haas, P.; Hendriks, W.; Lefeber, D.J.; Cambi, A. Biological and Technical Challenges in Unraveling the Role of N-Glycans in Immune Receptor Regulation. Front Chem 2020, 8, 55. [CrossRef]
- Fang, P.; Ji, Y.; Oellerich, T.; Urlaub, H.; Pan, K.T. Strategies for Proteome-Wide Quantification of Glycosylation Macro- and Micro-Heterogeneity. Int J Mol Sci 2022, 23. [CrossRef]
- Schmaltz, R.M.; Hanson, S.R.; Wong, C.H. Enzymes in the synthesis of glycoconjugates. Chem Rev 2011, 111, 4259-4307. [CrossRef]
- Zacchi, L.F.; Schulz, B.L. N-glycoprotein macroheterogeneity: biological implications and proteomic characterization. Glycoconj J 2016, 33, 359-376. [CrossRef]
- Springer, S.A.; Gagneux, P. Glycan evolution in response to collaboration, conflict, and constraint. J Biol Chem 2013, 288, 6904-6911. [CrossRef]
- Gong, Y.; Qin, S.; Dai, L.; Tian, Z. The glycosylation in SARS-CoV-2 and its receptor ACE2. Signal Transduct Target Ther 2021, 6, 396. [CrossRef]
- Otaki, M.; Hirane, N.; Natsume-Kitatani, Y.; Nogami Itoh, M.; Shindo, M.; Kurebayashi, Y.; Nishimura, S.I. Mouse tissue glycome atlas 2022 highlights inter-organ variation in major N-glycan profiles. Sci Rep 2022, 12, 17804. [CrossRef]
- Lumibao, J.C.; Tremblay, J.R.; Hsu, J.; Engle, D.D. Altered glycosylation in pancreatic cancer and beyond. J Exp Med 2022, 219. [CrossRef]
- Saleh, F.M.; Chandra, P.K.; Lin, D.; Robinson, J.E.; Izadpanah, R.; Mondal, D.; Bollensdorff, C.; Alt, E.U.; Zhu, Q.; Marasco, W.A.; et al. A New Humanized Mouse Model Mimics Humans in Lacking alpha-Gal Epitopes and Secreting Anti-Gal Antibodies. J Immunol 2020, 204, 1998-2005. [CrossRef]
- Riley, N.M.; Hebert, A.S.; Westphall, M.S.; Coon, J.J. Capturing site-specific heterogeneity with large-scale N-glycoproteome analysis. Nat Commun 2019, 10, 1311. [CrossRef]
- Yang, X.; Wang, Z.; Guo, L.; Zhu, Z.; Zhang, Y. Proteome-Wide Analysis of N-Glycosylation Stoichiometry Using SWATH Technology. J Proteome Res 2017, 16, 3830-3840. [CrossRef]
- Bagdonaite, I.; Malaker, S.A.; Polasky, D.A.; Riley, N.M.; Schjoldager, K.; Vakhrushev, S.Y.; Halim, A.; Aoki-Kinoshita, K.F.; Nesvizhskii, A.I.; Bertozzi, C.R.; et al. Glycoproteomics. Nature Reviews Methods Primers 2022, 2, 48. [CrossRef]
- Hu, Y.; Pan, J.; Shah, P.; Ao, M.; Thomas, S.N.; Liu, Y.; Chen, L.; Schnaubelt, M.; Clark, D.J.; Rodriguez, H.; et al. Integrated Proteomic and Glycoproteomic Characterization of Human High-Grade Serous Ovarian Carcinoma. Cell Rep 2020, 33, 108276. [CrossRef]
- Campbell, M.P.; Peterson, R.; Mariethoz, J.; Gasteiger, E.; Akune, Y.; Aoki-Kinoshita, K.F.; Lisacek, F.; Packer, N.H. UniCarbKB: building a knowledge platform for glycoproteomics. Nucleic Acids Res 2014, 42, D215-221. [CrossRef]
- Li, X.; Xu, Z.; Hong, X.; Zhang, Y.; Zou, X. Databases and Bioinformatic Tools for Glycobiology and Glycoproteomics. Int J Mol Sci 2020, 21. [CrossRef]
- Toukach, P.V.; Egorova, K.S. Carbohydrate structure database merged from bacterial, archaeal, plant and fungal parts. Nucleic Acids Res 2016, 44, D1229-1236. [CrossRef]
- Scherbinina, S.I.; Toukach, P.V. Three-Dimensional Structures of Carbohydrates and Where to Find Them. Int J Mol Sci 2020, 21. [CrossRef]
- Bohm, M.; Bohne-Lang, A.; Frank, M.; Loss, A.; Rojas-Macias, M.A.; Lutteke, T. Glycosciences.DB: an annotated data collection linking glycomics and proteomics data (2018 update). Nucleic Acids Res 2019, 47, D1195-D1201. [CrossRef]
- Aoki-Kinoshita, K.F.; Kanehisa, M. Glycomic Analysis Using KEGG GLYCAN. In Glycoinformatics, Lütteke, T., Frank, M., Eds.; Springer New York: New York, NY, 2015; pp. 97-107.
- Maeda, M.; Fujita, N.; Suzuki, Y.; Sawaki, H.; Shikanai, T.; Narimatsu, H. JCGGDB: Japan Consortium for Glycobiology and Glycotechnology Database. In Glycoinformatics, Lütteke, T., Frank, M., Eds.; Springer New York: New York, NY, 2015; pp. 161-179.
- Toukach, P.V.; Egorova, K.S. Source files of the Carbohydrate Structure Database: the way to sophisticated analysis of natural glycans. Scientific Data 2022, 9, 131. [CrossRef]
- Sun, S.; Hu, Y.; Ao, M.; Shah, P.; Chen, J.; Yang, W.; Jia, X.; Tian, Y.; Thomas, S.; Zhang, H. N-GlycositeAtlas: a database resource for mass spectrometry-based human N-linked glycoprotein and glycosylation site mapping. Clin Proteomics 2019, 16, 35. [CrossRef]
- Abrahams, J.L.; Taherzadeh, G.; Jarvas, G.; Guttman, A.; Zhou, Y.; Campbell, M.P. Recent advances in glycoinformatic platforms for glycomics and glycoproteomics. Curr Opin Struct Biol 2020, 62, 56-69. [CrossRef]
- Egorova, K.S.; Toukach, P.V. Glycoinformatics: Bridging Isolated Islands in the Sea of Data. Angew Chem Int Ed Engl 2018, 57, 14986-14990. [CrossRef]
- Breitling, J.; Aebi, M. N-linked protein glycosylation in the endoplasmic reticulum. Cold Spring Harb Perspect Biol 2013, 5, a013359. [CrossRef]
- Hanson, S.R.; Culyba, E.K.; Hsu, T.L.; Wong, C.H.; Kelly, J.W.; Powers, E.T. The core trisaccharide of an N-linked glycoprotein intrinsically accelerates folding and enhances stability. Proc Natl Acad Sci U S A 2009, 106, 3131-3136. [CrossRef]
- Xu, C.; Ng, D.T. Glycosylation-directed quality control of protein folding. Nat Rev Mol Cell Biol 2015, 16, 742-752. [CrossRef]
- Ohtsubo, K.; Marth, J.D. Glycosylation in cellular mechanisms of health and disease. Cell 2006, 126, 855-867. [CrossRef]
- Wolfert, M.A.; Boons, G.J. Adaptive immune activation: glycosylation does matter. Nat Chem Biol 2013, 9, 776-784. [CrossRef]
- Dzobo, K.; Dandara, C. The Extracellular Matrix: Its Composition, Function, Remodeling, and Role in Tumorigenesis. Biomimetics (Basel) 2023, 8. [CrossRef]
- Pinho, S.S.; Alves, I.; Gaifem, J.; Rabinovich, G.A. Immune regulatory networks coordinated by glycans and glycan-binding proteins in autoimmunity and infection. Cellular & Molecular Immunology 2023, 20, 1101-1113. [CrossRef]
- Stowell, S.R.; Ju, T.; Cummings, R.D. Protein glycosylation in cancer. Annu Rev Pathol 2015, 10, 473-510. [CrossRef]
- Taniguchi, N.; Kizuka, Y. Glycans and cancer: role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv Cancer Res 2015, 126, 11-51. [CrossRef]
- Chandler, K.B.; Costello, C.E.; Rahimi, N. Glycosylation in the Tumor Microenvironment: Implications for Tumor Angiogenesis and Metastasis. Cells 2019, 8. [CrossRef]
- Costa, A.F.; Campos, D.; Reis, C.A.; Gomes, C. Targeting Glycosylation: A New Road for Cancer Drug Discovery. Trends Cancer 2020, 6, 757-766. [CrossRef]
- Mereiter, S.; Balmana, M.; Campos, D.; Gomes, J.; Reis, C.A. Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading? Cancer Cell 2019, 36, 6-16. [CrossRef]
- Thomas, D.; Rathinavel, A.K.; Radhakrishnan, P. Altered glycosylation in cancer: A promising target for biomarkers and therapeutics. Biochim Biophys Acta Rev Cancer 2021, 1875, 188464. [CrossRef]
- Haukedal, H.; Freude, K.K. Implications of Glycosylation in Alzheimer's Disease. Front Neurosci 2020, 14, 625348. [CrossRef]
- Pradeep, P.; Kang, H.; Lee, B. Glycosylation and behavioral symptoms in neurological disorders. Transl Psychiatry 2023, 13, 154. [CrossRef]
- Zhao, J.; Lang, M. New insight into protein glycosylation in the development of Alzheimer's disease. Cell Death Discov 2023, 9, 314. [CrossRef]
- Conroy, L.R.; Hawkinson, T.R.; Young, L.E.A.; Gentry, M.S.; Sun, R.C. Emerging roles of N-linked glycosylation in brain physiology and disorders. Trends Endocrinol Metab 2021, 32, 980-993. [CrossRef]
- Paprocka, J.; Jezela-Stanek, A.; Tylki-Szymanska, A.; Grunewald, S. Congenital Disorders of Glycosylation from a Neurological Perspective. Brain Sci 2021, 11. [CrossRef]
- Groux-Degroote, S.; Cavdarli, S.; Uchimura, K.; Allain, F.; Delannoy, P. Glycosylation changes in inflammatory diseases. Adv Protein Chem Struct Biol 2020, 119, 111-156. [CrossRef]
- Kissel, T.; Toes, R.E.M.; Huizinga, T.W.J.; Wuhrer, M. Glycobiology of rheumatic diseases. Nat Rev Rheumatol 2023, 19, 28-43. [CrossRef]
- Loke, I.; Kolarich, D.; Packer, N.H.; Thaysen-Andersen, M. Emerging roles of protein mannosylation in inflammation and infection. Mol Aspects Med 2016, 51, 31-55. [CrossRef]
- Ugonotti, J.; Chatterjee, S.; Thaysen-Andersen, M. Structural and functional diversity of neutrophil glycosylation in innate immunity and related disorders. Mol Aspects Med 2021, 79, 100882. [CrossRef]
- Lin, B.; Qing, X.; Liao, J.; Zhuo, K. Role of Protein Glycosylation in Host-Pathogen Interaction. Cells 2020, 9. [CrossRef]
- Wang, S.H.; Wu, T.J.; Lee, C.W.; Yu, J. Dissecting the conformation of glycans and their interactions with proteins. J Biomed Sci 2020, 27, 93. [CrossRef]
- Varki, A.; Freeze, H.H. Glycans in Acquired Human Diseases. In Essentials of Glycobiology, Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press Copyright © 2009, The Consortium of Glycobiology Editors, La Jolla, California.: Cold Spring Harbor (NY), 2009.
- Zhou, J.Y.; Cobb, B.A. Glycans in Immunologic Health and Disease. Annual Review of Immunology 2021, 39, 511-536. [CrossRef]
- Barb, A.W. Fc gamma receptor compositional heterogeneity: Considerations for immunotherapy development. J Biol Chem 2021, 296, 100057. [CrossRef]
- Saporiti, S.; Parravicini, C.; Pergola, C.; Guerrini, U.; Rossi, M.; Centola, F.; Eberini, I. IgG1 conformational behavior: elucidation of the N-glycosylation role via molecular dynamics. Biophys J 2021, 120, 5355-5370. [CrossRef]
- Subedi, G.P.; Hanson, Q.M.; Barb, A.W. Restricted motion of the conserved immunoglobulin G1 N-glycan is essential for efficient FcgammaRIIIa binding. Structure 2014, 22, 1478-1488. [CrossRef]
- Sun, Y.; Izadi, S.; Callahan, M.; Deperalta, G.; Wecksler, A.T. Antibody-receptor interactions mediate antibody-dependent cellular cytotoxicity. J Biol Chem 2021, 297, 100826. [CrossRef]
- Chen, B.; Liu, W.; Li, Y.; Ma, B.; Shang, S.; Tan, Z. Impact of N-Linked Glycosylation on Therapeutic Proteins. Molecules 2022, 27. [CrossRef]
- Pinho, S.S.; Reis, C.A. Glycosylation in cancer: mechanisms and clinical implications. Nat Rev Cancer 2015, 15, 540-555. [CrossRef]
- Buffone, A.; Weaver, V.M. Don't sugarcoat it: How glycocalyx composition influences cancer progression. J Cell Biol 2020, 219. [CrossRef]
- Lee, H.S.; Qi, Y.; Im, W. Effects of N-glycosylation on protein conformation and dynamics: Protein Data Bank analysis and molecular dynamics simulation study. Sci Rep 2015, 5, 8926. [CrossRef]
- Mule, S.N.; Rosa-Fernandes, L.; Coutinho, J.V.P.; Gomes, V.M.; Macedo-da-Silva, J.; Santiago, V.F.; Quina, D.; de Oliveira, G.S.; Thaysen-Andersen, M.; Larsen, M.R.; et al. Systems-wide analysis of glycoprotein conformational changes by limited deglycosylation assay. J Proteomics 2021, 248, 104355. [CrossRef]
- Papaleo, E.; Saladino, G.; Lambrughi, M.; Lindorff-Larsen, K.; Gervasio, F.L.; Nussinov, R. The Role of Protein Loops and Linkers in Conformational Dynamics and Allostery. Chemical Reviews 2016, 116, 6391-6423. [CrossRef]
- Cascella, M.; Rajnik, M.; Aleem, A.; Dulebohn, S.C.; Di Napoli, R. Features, Evaluation, and Treatment of Coronavirus (COVID-19). In StatPearls; Treasure Island (FL), 2023.
- Casalino, L.; Gaieb, Z.; Goldsmith, J.A.; Hjorth, C.K.; Dommer, A.C.; Harbison, A.M.; Fogarty, C.A.; Barros, E.P.; Taylor, B.C.; McLellan, J.S.; et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent Sci 2020, 6, 1722-1734. [CrossRef]
- Malaquias, M.A.S.; Gadotti, A.C.; Motta-Junior, J.D.S.; Martins, A.P.C.; Azevedo, M.L.V.; Benevides, A.P.K.; Cezar-Neto, P.; Panini do Carmo, L.A.; Zeni, R.C.; Raboni, S.M.; et al. The role of the lectin pathway of the complement system in SARS-CoV-2 lung injury. Transl Res 2021, 231, 55-63. [CrossRef]
- Jiang, S.; Hillyer, C.; Du, L. Neutralizing Antibodies against SARS-CoV-2 and Other Human Coronaviruses: (Trends in Immunology 41, 355-359; 2020). Trends Immunol 2020, 41, 545. [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260-1263. [CrossRef]
- Du, L.; He, Y.; Zhou, Y.; Liu, S.; Zheng, B.J.; Jiang, S. The spike protein of SARS-CoV--a target for vaccine and therapeutic development. Nat Rev Microbiol 2009, 7, 226-236. [CrossRef]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLoS Pathog 2018, 14, e1007236. [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271-280.e278. [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327-330. [CrossRef]
- Lu, M.; Uchil, P.D.; Li, W.; Zheng, D.; Terry, D.S.; Gorman, J.; Shi, W.; Zhang, B.; Zhou, T.; Ding, S.; et al. Real-Time Conformational Dynamics of SARS-CoV-2 Spikes on Virus Particles. Cell Host Microbe 2020, 28, 880-891 e888. [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 183, 1735. [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215-220. [CrossRef]
- Tian, Y.; Parsons, L.M.; Jankowska, E.; Cipollo, J.F. Site-Specific Glycosylation Patterns of the SARS-CoV-2 Spike Protein Derived From Recombinant Protein and Viral WA1 and D614G Strains. Front Chem 2021, 9, 767448. [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M., Jr.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586-1592. [CrossRef]
- Fan, X.; Cao, D.; Kong, L.; Zhang, X. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat Commun 2020, 11, 3618. [CrossRef]
- Harbison, A.M.; Fogarty, C.A.; Phung, T.K.; Satheesan, A.; Schulz, B.L.; Fadda, E. Fine-tuning the spike: role of the nature and topology of the glycan shield in the structure and dynamics of the SARS-CoV-2 S. Chem Sci 2022, 13, 386-395. [CrossRef]
- Ray, D.; Le, L.; Andricioaei, I. Distant residues modulate conformational opening in SARS-CoV-2 spike protein. Proc Natl Acad Sci U S A 2021, 118. [CrossRef]
- Zimmerman, M.I.; Porter, J.R.; Ward, M.D.; Singh, S.; Vithani, N.; Meller, A.; Mallimadugula, U.L.; Kuhn, C.E.; Borowsky, J.H.; Wiewiora, R.P.; et al. SARS-CoV-2 simulations go exascale to predict dramatic spike opening and cryptic pockets across the proteome. Nat Chem 2021, 13, 651-659. [CrossRef]
- Fallon, L.; Belfon, K.A.A.; Raguette, L.; Wang, Y.; Stepanenko, D.; Cuomo, A.; Guerra, J.; Budhan, S.; Varghese, S.; Corbo, C.P.; et al. Free Energy Landscapes from SARS-CoV-2 Spike Glycoprotein Simulations Suggest that RBD Opening Can Be Modulated via Interactions in an Allosteric Pocket. Journal of the American Chemical Society 2021, 143, 11349-11360. [CrossRef]
- Sztain, T.; Ahn, S.H.; Bogetti, A.T.; Casalino, L.; Goldsmith, J.A.; Seitz, E.; McCool, R.S.; Kearns, F.L.; Acosta-Reyes, F.; Maji, S.; et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat Chem 2021, 13, 963-968. [CrossRef]
- Zhang, B.W.; Jasnow, D.; Zuckerman, D.M. The “weighted ensemble” path sampling method is statistically exact for a broad class of stochastic processes and binning procedures. The Journal of Chemical Physics 2010, 132, 054107. [CrossRef]
- Pang, Y.T.; Acharya, A.; Lynch, D.L.; Pavlova, A.; Gumbart, J.C. SARS-CoV-2 spike opening dynamics and energetics reveal the individual roles of glycans and their collective impact. Commun Biol 2022, 5, 1170. [CrossRef]
- Dodero-Rojas, E.; Onuchic, J.N.; Whitford, P.C. Sterically confined rearrangements of SARS-CoV-2 Spike protein control cell invasion. Elife 2021, 10. [CrossRef]
- Geschwind, M.D. Prion Diseases. Continuum (Minneap Minn) 2015, 21, 1612-1638. [CrossRef]
- Collinge, J. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 2016, 539, 217-226. [CrossRef]
- Johnson, R.T. Prion diseases. The Lancet Neurology 2005, 4, 635-642. [CrossRef]
- Westergard, L.; Christensen, H.M.; Harris, D.A. The cellular prion protein (PrPC): Its physiological function and role in disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2007, 1772, 629-644. [CrossRef]
- Ermonval, M.; Mouillet-Richard, S.; Codogno, P.; Kellermann, O.; Botti, J. Evolving views in prion glycosylation: functional and pathological implications. Biochimie 2003, 85, 33-45. [CrossRef]
- Xiao, X.; Yuan, J.; Haik, S.; Cali, I.; Zhan, Y.; Moudjou, M.; Li, B.; Laplanche, J.L.; Laude, H.; Langeveld, J.; et al. Glycoform-selective prion formation in sporadic and familial forms of prion disease. PLoS One 2013, 8, e58786. [CrossRef]
- Mallucci, G.; Dickinson, A.; Linehan, J.; Klohn, P.C.; Brandner, S.; Collinge, J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 2003, 302, 871-874. [CrossRef]
- Aguilar-Calvo, P.; Callender, J.A.; Sigurdson, C.J. Short and sweet: How glycans impact prion conversion, cofactor interactions, and cross-species transmission. PLOS Pathogens 2021, 17, e1009123. [CrossRef]
- Aguilar-Calvo, P.; Xiao, X.; Bett, C.; Erana, H.; Soldau, K.; Castilla, J.; Nilsson, K.P.; Surewicz, W.K.; Sigurdson, C.J. Post-translational modifications in PrP expand the conformational diversity of prions in vivo. Sci Rep 2017, 7, 43295. [CrossRef]
- Angers, R.C.; Kang, H.E.; Napier, D.; Browning, S.; Seward, T.; Mathiason, C.; Balachandran, A.; McKenzie, D.; Castilla, J.; Soto, C.; et al. Prion strain mutation determined by prion protein conformational compatibility and primary structure. Science 2010, 328, 1154-1158. [CrossRef]
- Nakic, N.; Tran, T.H.; Novokmet, M.; Andreoletti, O.; Lauc, G.; Legname, G. Site-specific analysis of N-glycans from different sheep prion strains. PLoS Pathog 2021, 17, e1009232. [CrossRef]
- Sevillano, A.M.; Aguilar-Calvo, P.; Kurt, T.D.; Lawrence, J.A.; Soldau, K.; Nam, T.H.; Schumann, T.; Pizzo, D.P.; Nystrom, S.; Choudhury, B.; et al. Prion protein glycans reduce intracerebral fibril formation and spongiosis in prion disease. J Clin Invest 2020, 130, 1350-1362. [CrossRef]
- Corsaro, A.; Thellung, S.; Villa, V.; Nizzari, M.; Florio, T. Role of prion protein aggregation in neurotoxicity. Int J Mol Sci 2012, 13, 8648-8669. [CrossRef]
- Poggiolini, I.; Saverioni, D.; Parchi, P. Prion protein misfolding, strains, and neurotoxicity: an update from studies on Mammalian prions. Int J Cell Biol 2013, 2013, 910314. [CrossRef]
- Salamat, M.K.; Dron, M.; Chapuis, J.; Langevin, C.; Laude, H. Prion propagation in cells expressing PrP glycosylation mutants. J Virol 2011, 85, 3077-3085. [CrossRef]
- Cancellotti, E.; Mahal, S.P.; Somerville, R.; Diack, A.; Brown, D.; Piccardo, P.; Weissmann, C.; Manson, J.C. Post-translational changes to PrP alter transmissible spongiform encephalopathy strain properties. EMBO J 2013, 32, 756-769. [CrossRef]
- Yi, C.W.; Wang, L.Q.; Huang, J.J.; Pan, K.; Chen, J.; Liang, Y. Glycosylation Significantly Inhibits the Aggregation of Human Prion Protein and Decreases Its Cytotoxicity. Sci Rep 2018, 8, 12603. [CrossRef]
- Schilling, K.M.; Jorwal, P.; Ubilla-Rodriguez, N.C.; Assafa, T.E.; Gatdula, J.R.P.; Vultaggio, J.S.; Harris, D.A.; Millhauser, G.L. N-glycosylation is a potent regulator of prion protein neurotoxicity. J Biol Chem 2023, 299, 105101. [CrossRef]
- DeArmond, S.J.; Sanchez, H.; Yehiely, F.; Qiu, Y.; Ninchak-Casey, A.; Daggett, V.; Camerino, A.P.; Cayetano, J.; Rogers, M.; Groth, D.; et al. Selective neuronal targeting in prion disease. Neuron 1997, 19, 1337-1348. [CrossRef]
- Giachin, G.; Biljan, I.; Ilc, G.; Plavec, J.; Legname, G. Probing early misfolding events in prion protein mutants by NMR spectroscopy. Molecules 2013, 18, 9451-9476. [CrossRef]
- Riek, R.; Hornemann, S.; Wider, G.; Glockshuber, R.; Wuthrich, K. NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett 1997, 413, 282-288. [CrossRef]
- Zuegg, J.; Gready, J.E. Molecular dynamics simulation of human prion protein including both N-linked oligosaccharides and the GPI anchor. Glycobiology 2000, 10, 959-974. [CrossRef]
- Schilling, K.M.; Tao, L.; Wu, B.; Kiblen, J.T.M.; Ubilla-Rodriguez, N.C.; Pushie, M.J.; Britt, R.D.; Roseman, G.P.; Harris, D.A.; Millhauser, G.L. Both N-Terminal and C-Terminal Histidine Residues of the Prion Protein Are Essential for Copper Coordination and Neuroprotective Self-Regulation. Journal of Molecular Biology 2020, 432, 4408-4425. [CrossRef]
- Evans, Eric G.B.; Pushie, M.J.; Markham, Kate A.; Lee, H.-W.; Millhauser, Glenn L. Interaction between Prion Protein's Copper-Bound Octarepeat Domain and a Charged C-Terminal Pocket Suggests a Mechanism for N-Terminal Regulation. Structure 2016, 24, 1057-1067. [CrossRef]
- Spevacek, Ann R.; Evans, Eric G.B.; Miller, Jillian L.; Meyer, Heidi C.; Pelton, Jeffrey G.; Millhauser, Glenn L. Zinc Drives a Tertiary Fold in the Prion Protein with Familial Disease Mutation Sites at the Interface. Structure 2013, 21, 236-246. [CrossRef]
- Aguilar-Calvo, P.; Sevillano, A.M.; Bapat, J.; Soldau, K.; Sandoval, D.R.; Altmeppen, H.C.; Linsenmeier, L.; Pizzo, D.P.; Geschwind, M.D.; Sanchez, H.; et al. Shortening heparan sulfate chains prolongs survival and reduces parenchymal plaques in prion disease caused by mobile, ADAM10-cleaved prions. Acta Neuropathol 2020, 139, 527-546. [CrossRef]
- Marzec, M.; Eletto, D.; Argon, Y. GRP94: An HSP90-like protein specialized for protein folding and quality control in the endoplasmic reticulum. Biochim Biophys Acta 2012, 1823, 774-787. [CrossRef]
- Ansa-Addo, E.A.; Thaxton, J.; Hong, F.; Wu, B.X.; Zhang, Y.; Fugle, C.W.; Metelli, A.; Riesenberg, B.; Williams, K.; Gewirth, D.T.; et al. Clients and Oncogenic Roles of Molecular Chaperone gp96/grp94. Curr Top Med Chem 2016, 16, 2765-2778. [CrossRef]
- Eletto, D.; Dersh, D.; Argon, Y. GRP94 in ER quality control and stress responses. Semin Cell Dev Biol 2010, 21, 479-485. [CrossRef]
- Lee, A.S. Glucose-regulated proteins in cancer: molecular mechanisms and therapeutic potential. Nat Rev Cancer 2014, 14, 263-276. [CrossRef]
- Wiersma, V.R.; Michalak, M.; Abdullah, T.M.; Bremer, E.; Eggleton, P. Mechanisms of Translocation of ER Chaperones to the Cell Surface and Immunomodulatory Roles in Cancer and Autoimmunity. Front Oncol 2015, 5, 7. [CrossRef]
- Altmeyer, A.; Maki, R.G.; Feldweg, A.M.; Heike, M.; Protopopov, V.P.; Masur, S.K.; Srivastava, P.K. Tumor-specific cell surface expression of the-KDEL containing, endoplasmic reticular heat shock protein gp96. Int J Cancer 1996, 69, 340-349. [CrossRef]
- Booth, C.; Koch, G.L. Perturbation of cellular calcium induces secretion of luminal ER proteins. Cell 1989, 59, 729-737. [CrossRef]
- Martins, M.; Custodio, R.; Camejo, A.; Almeida, M.T.; Cabanes, D.; Sousa, S. Listeria monocytogenes triggers the cell surface expression of Gp96 protein and interacts with its N terminus to support cellular infection. J Biol Chem 2012, 287, 43083-43093. [CrossRef]
- Rothan, H.A.; Zhong, Y.; Sanborn, M.A.; Teoh, T.C.; Ruan, J.; Yusof, R.; Hang, J.; Henderson, M.J.; Fang, S. Small molecule grp94 inhibitors block dengue and Zika virus replication. Antiviral Res 2019, 171, 104590. [CrossRef]
- Sumitomo, T.; Nakata, M.; Nagase, S.; Takahara, Y.; Honda-Ogawa, M.; Mori, Y.; Akamatsu, Y.; Yamaguchi, M.; Okamoto, S.; Kawabata, S. GP96 Drives Exacerbation of Secondary Bacterial Pneumonia following Influenza A Virus Infection. mBio 2021, 12, e0326920. [CrossRef]
- Liu, B.; Dai, J.; Zheng, H.; Stoilova, D.; Sun, S.; Li, Z. Cell surface expression of an endoplasmic reticulum resident heat shock protein gp96 triggers MyD88-dependent systemic autoimmune diseases. Proc Natl Acad Sci U S A 2003, 100, 15824-15829. [CrossRef]
- Li, X.; Sun, L.; Hou, J.; Gui, M.; Ying, J.; Zhao, H.; Lv, N.; Meng, S. Cell membrane gp96 facilitates HER2 dimerization and serves as a novel target in breast cancer. Int J Cancer 2015, 137, 512-524. [CrossRef]
- Patel, P.D.; Yan, P.; Seidler, P.M.; Patel, H.J.; Sun, W.; Yang, C.; Que, N.S.; Taldone, T.; Finotti, P.; Stephani, R.A.; et al. Paralog-selective Hsp90 inhibitors define tumor-specific regulation of HER2. Nat Chem Biol 2013, 9, 677-684. [CrossRef]
- Yan, P.; Patel, H.J.; Sharma, S.; Corben, A.; Wang, T.; Panchal, P.; Yang, C.; Sun, W.; Araujo, T.L.; Rodina, A.; et al. Molecular Stressors Engender Protein Connectivity Dysfunction through Aberrant N-Glycosylation of a Chaperone. Cell Rep 2020, 31, 107840. [CrossRef]
- Chavany, C.; Mimnaugh, E.; Miller, P.; Bitton, R.; Nguyen, P.; Trepel, J.; Whitesell, L.; Schnur, R.; Moyer, J.; Neckers, L. p185erbB2 binds to GRP94 in vivo. Dissociation of the p185erbB2/GRP94 heterocomplex by benzoquinone ansamycins precedes depletion of p185erbB2. J Biol Chem 1996, 271, 4974-4977. [CrossRef]
- Chaumonnot, K.; Masson, S.; Sikner, H.; Bouchard, A.; Baverel, V.; Bellaye, P.S.; Collin, B.; Garrido, C.; Kohli, E. The HSP GRP94 interacts with macrophage intracellular complement C3 and impacts M2 profile during ER stress. Cell Death Dis 2021, 12, 114. [CrossRef]
- Ratna, A.; Lim, A.; Li, Z.; Argemi, J.; Bataller, R.; Chiosis, G.; Mandrekar, P. Myeloid Endoplasmic Reticulum Resident Chaperone GP96 Facilitates Inflammation and Steatosis in Alcohol-Associated Liver Disease. Hepatol Commun 2021, 5, 1165-1182. [CrossRef]
- Pagetta, A.; Tramentozzi, E.; Tibaldi, E.; Cendron, L.; Zanotti, G.; Brunati, A.M.; Vitadello, M.; Gorza, L.; Finotti, P. Structural insights into complexes of glucose-regulated Protein94 (Grp94) with human immunoglobulin G. relevance for Grp94-IgG complexes that form in vivo in pathological conditions. PLoS One 2014, 9, e86198. [CrossRef]
- Cherepanova, N.A.; Venev, S.V.; Leszyk, J.D.; Shaffer, S.A.; Gilmore, R. Quantitative glycoproteomics reveals new classes of STT3A- and STT3B-dependent N-glycosylation sites. J Cell Biol 2019, 218, 2782-2796. [CrossRef]
- Wearsch, P.A.; Nicchitta, C.V. Purification and partial molecular characterization of GRP94, an ER resident chaperone. Protein Expr Purif 1996, 7, 114-121. [CrossRef]
- Wen, P.; Chen, J.; Zuo, C.; Gao, X.; Fujita, M.; Yang, G. Proteome and Glycoproteome Analyses Reveal the Protein N-Linked Glycosylation Specificity of STT3A and STT3B. Cells 2022, 11. [CrossRef]
- Gottschalk, N.; Kimmig, R.; Lang, S.; Singh, M.; Brandau, S. Anti-epidermal growth factor receptor (EGFR) antibodies overcome resistance of ovarian cancer cells to targeted therapy and natural cytotoxicity. Int J Mol Sci 2012, 13, 12000-12016. [CrossRef]
- Sun, S.; Zhang, H. Large-Scale Measurement of Absolute Protein Glycosylation Stoichiometry. Anal Chem 2015, 87, 6479-6482. [CrossRef]
- Castelli, M.; Yan, P.; Rodina, A.; Digwal, C.S.; Panchal, P.; Chiosis, G.; Moroni, E.; Colombo, G. How aberrant N-glycosylation can alter protein functionality and ligand binding: An atomistic view. Structure 2023, 31, 987-1004.e1008. [CrossRef]
- Bouchard, A.; Sikner, H.; Baverel, V.; Garnier, A.R.; Monterrat, M.; Moreau, M.; Limagne, E.; Garrido, C.; Kohli, E.; Collin, B.; et al. The GRP94 Inhibitor PU-WS13 Decreases M2-like Macrophages in Murine TNBC Tumors: A Pharmaco-Imaging Study with (99m)Tc-Tilmanocept SPECT. Cells 2021, 10. [CrossRef]
- Rodina, A.; Xu, C.; Digwal, C.S.; Joshi, S.; Patel, Y.; Santhaseela, A.R.; Bay, S.; Merugu, S.; Alam, A.; Yan, P.; et al. Systems-level analyses of protein-protein interaction network dysfunctions via epichaperomics identify cancer-specific mechanisms of stress adaptation. Nature Communications 2023, 14, 3742. [CrossRef]
- Chiosis, G.; Digwal, C.S.; Trepel, J.B.; Neckers, L. Structural and functional complexity of HSP90 in cellular homeostasis and disease. Nature Reviews Molecular Cell Biology 2023, 24, 797-815. [CrossRef]
- Rodina, A.; Wang, T.; Yan, P.; Gomes, E.D.; Dunphy, M.P.; Pillarsetty, N.; Koren, J.; Gerecitano, J.F.; Taldone, T.; Zong, H.; et al. The epichaperome is an integrated chaperome network that facilitates tumour survival. Nature 2016, 538, 397-401. [CrossRef]
- Barouch, D.H. Covid-19 Vaccines - Immunity, Variants, Boosters. N Engl J Med 2022, 387, 1011-1020. [CrossRef]
- Corti, D.; Purcell, L.A.; Snell, G.; Veesler, D. Tackling COVID-19 with neutralizing monoclonal antibodies. Cell 2021, 184, 4593-4595. [CrossRef]
- Raybould, M.I.J.; Kovaltsuk, A.; Marks, C.; Deane, C.M. CoV-AbDab: the coronavirus antibody database. Bioinformatics 2021, 37, 734-735. [CrossRef]
- Voss, W.N.; Hou, Y.J.; Johnson, N.V.; Delidakis, G.; Kim, J.E.; Javanmardi, K.; Horton, A.P.; Bartzoka, F.; Paresi, C.J.; Tanno, Y.; et al. Prevalent, protective, and convergent IgG recognition of SARS-CoV-2 non-RBD spike epitopes. Science 2021, 372, 1108-1112. [CrossRef]
- Serapian, S.A.; Marchetti, F.; Triveri, A.; Morra, G.; Meli, M.; Moroni, E.; Sautto, G.A.; Rasola, A.; Colombo, G. The Answer Lies in the Energy: How Simple Atomistic Molecular Dynamics Simulations May Hold the Key to Epitope Prediction on the Fully Glycosylated SARS-CoV-2 Spike Protein. The Journal of Physical Chemistry Letters 2020, 11, 8084-8093. [CrossRef]
- Triveri, A.; Serapian, S.A.; Marchetti, F.; Doria, F.; Pavoni, S.; Cinquini, F.; Moroni, E.; Rasola, A.; Frigerio, F.; Colombo, G. SARS-CoV-2 Spike Protein Mutations and Escape from Antibodies: A Computational Model of Epitope Loss in Variants of Concern. Journal of Chemical Information and Modeling 2021, 61, 4687-4700. [CrossRef]
- Triveri, A.; Casali, E.; Frasnetti, E.; Doria, F.; Frigerio, F.; Cinquini, F.; Pavoni, S.; Moroni, E.; Marchetti, F.; Serapian, S.A.; et al. Conformational Behavior of SARS-Cov-2 Spike Protein Variants: Evolutionary Jumps in Sequence Reverberate in Structural Dynamic Differences. Journal of Chemical Theory and Computation 2023, 19, 2120-2134. [CrossRef]
- Mead, S.; Whitfield, J.; Poulter, M.; Shah, P.; Uphill, J.; Campbell, T.; Al-Dujaily, H.; Hummerich, H.; Beck, J.; Mein, C.A.; et al. A novel protective prion protein variant that colocalizes with kuru exposure. N Engl J Med 2009, 361, 2056-2065. [CrossRef]
- Asante, E.A.; Smidak, M.; Grimshaw, A.; Houghton, R.; Tomlinson, A.; Jeelani, A.; Jakubcova, T.; Hamdan, S.; Richard-Londt, A.; Linehan, J.M.; et al. A naturally occurring variant of the human prion protein completely prevents prion disease. Nature 2015, 522, 478-481. [CrossRef]
- Zheng, Z.; Zhang, M.; Wang, Y.; Ma, R.; Guo, C.; Feng, L.; Wu, J.; Yao, H.; Lin, D. Structural basis for the complete resistance of the human prion protein mutant G127V to prion disease. Sci Rep 2018, 8, 13211. [CrossRef]
- Kuwata, K. Logical design of medical chaperone for prion diseases. Curr Top Med Chem 2013, 13, 2432-2440. [CrossRef]
- Yamaguchi, K.; Kamatari, Y.O.; Ono, F.; Shibata, H.; Fuse, T.; Elhelaly, A.E.; Fukuoka, M.; Kimura, T.; Hosokawa-Muto, J.; Ishikawa, T.; et al. A designer molecular chaperone against transmissible spongiform encephalopathy slows disease progression in mice and macaques. Nat Biomed Eng 2019, 3, 206-219. [CrossRef]
- Lysek, D.A.; Schorn, C.; Nivon, L.G.; Esteve-Moya, V.; Christen, B.; Calzolai, L.; von Schroetter, C.; Fiorito, F.; Herrmann, T.; Guntert, P.; et al. Prion protein NMR structures of cats, dogs, pigs, and sheep. Proc Natl Acad Sci U S A 2005, 102, 640-645. [CrossRef]
- Duan, X.; Iwanowycz, S.; Ngoi, S.; Hill, M.; Zhao, Q.; Liu, B. Molecular Chaperone GRP94/GP96 in Cancers: Oncogenesis and Therapeutic Target. Front Oncol 2021, 11, 629846. [CrossRef]
- Wu, B.X.; Hong, F.; Zhang, Y.; Ansa-Addo, E.; Li, Z. GRP94/gp96 in Cancer: Biology, Structure, Immunology, and Drug Development. Adv Cancer Res 2016, 129, 165-190. [CrossRef]
- Gewirth, D.T. Paralog Specific Hsp90 Inhibitors - A Brief History and a Bright Future. Curr Top Med Chem 2016, 16, 2779-2791. [CrossRef]
- Patel, H.J.; Patel, P.D.; Ochiana, S.O.; Yan, P.; Sun, W.; Patel, M.R.; Shah, S.K.; Tramentozzi, E.; Brooks, J.; Bolaender, A.; et al. Structure-activity relationship in a purine-scaffold compound series with selectivity for the endoplasmic reticulum Hsp90 paralog Grp94. J Med Chem 2015, 58, 3922-3943. [CrossRef]
- Shrestha, L.; Patel, H.J.; Chiosis, G. Chemical Tools to Investigate Mechanisms Associated with HSP90 and HSP70 in Disease. Cell Chem Biol 2016, 23, 158-172. [CrossRef]
- Sousa, S.; Brion, R.; Lintunen, M.; Kronqvist, P.; Sandholm, J.; Monkkonen, J.; Kellokumpu-Lehtinen, P.L.; Lauttia, S.; Tynninen, O.; Joensuu, H.; et al. Human breast cancer cells educate macrophages toward the M2 activation status. Breast Cancer Res 2015, 17, 101. [CrossRef]
Figure 1.
A. Schematic illustrating the primary structure of the full-length SARS-CoV-2 Spike (S) protein with color-coded domains: N-terminal domain (NTD), receptor binding domain (RBD), fusion peptide (FP), heptad repeat 1 and 2 (HR1, HR2) domain, transmembrane domain (TM), and cytoplasmic tail (CT). The S protein comprises two subunits, S1 and S2. Asparagine residues crucial for N-glycosylation regulation of S binding to the angiotensin-converting enzyme 2 (ACE2) receptor on the host cell are highlighted based on their sequence position. B. Illustration depicting the mechanism of trimeric S glycoprotein binding to human ACE2, initiated by at least one protomer’s RBD switching from a ’down’ (closed) state (unfavorable for ACE2 binding) to an ’up’ (open) state (favorable for ACE2 binding). The S protein is represented by cartoons, with individual protomers colored in cyan, red, and gray. Both the ’down’ closed state (PDB: 7ZH2) and ’up’ open state (PDB: 7ZH5) of the S protein RBD are shown. Glycans are depicted in blue using Van der Waals representation. N-glycans at positions N165 and N234 are identified as essential structural elements for maintaining the SARS-CoV-2 spike protein in a conformation conducive to ACE2 recognition, facilitating subsequent viral entry into host cells. The glycan at N343 lifts and stabilizes the RBD throughout the opening transition. Visualizations of the S protein were generated using Visual Molecular Dynamics (v1.9.4a55).
Figure 1.
A. Schematic illustrating the primary structure of the full-length SARS-CoV-2 Spike (S) protein with color-coded domains: N-terminal domain (NTD), receptor binding domain (RBD), fusion peptide (FP), heptad repeat 1 and 2 (HR1, HR2) domain, transmembrane domain (TM), and cytoplasmic tail (CT). The S protein comprises two subunits, S1 and S2. Asparagine residues crucial for N-glycosylation regulation of S binding to the angiotensin-converting enzyme 2 (ACE2) receptor on the host cell are highlighted based on their sequence position. B. Illustration depicting the mechanism of trimeric S glycoprotein binding to human ACE2, initiated by at least one protomer’s RBD switching from a ’down’ (closed) state (unfavorable for ACE2 binding) to an ’up’ (open) state (favorable for ACE2 binding). The S protein is represented by cartoons, with individual protomers colored in cyan, red, and gray. Both the ’down’ closed state (PDB: 7ZH2) and ’up’ open state (PDB: 7ZH5) of the S protein RBD are shown. Glycans are depicted in blue using Van der Waals representation. N-glycans at positions N165 and N234 are identified as essential structural elements for maintaining the SARS-CoV-2 spike protein in a conformation conducive to ACE2 recognition, facilitating subsequent viral entry into host cells. The glycan at N343 lifts and stabilizes the RBD throughout the opening transition. Visualizations of the S protein were generated using Visual Molecular Dynamics (v1.9.4a55).
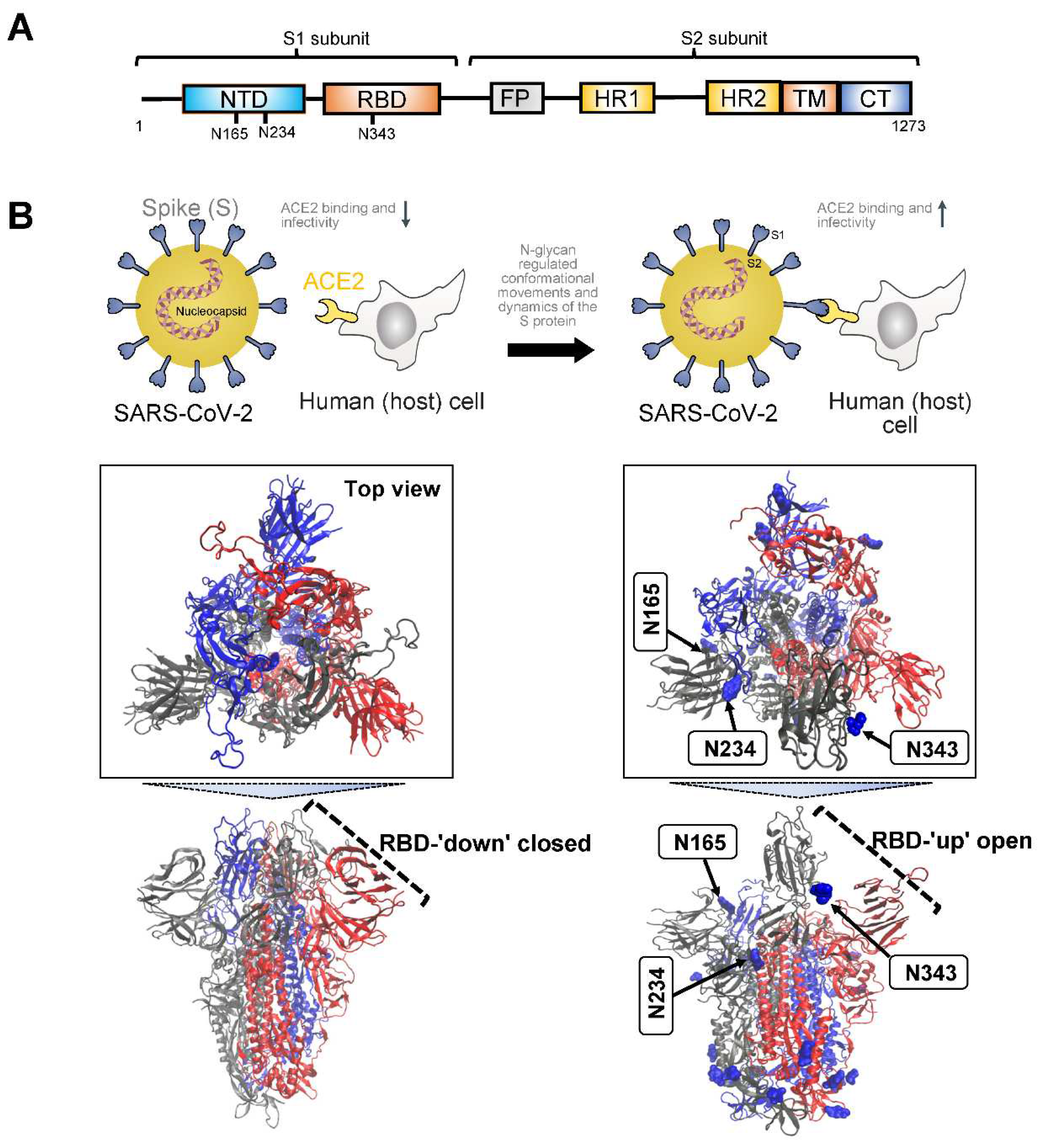
Figure 2.
A. Schematic illustrating the primary structure of the full-length prion (PrPc) protein. The N-terminal segment (residues 23–125, after removal of the signal peptide) exhibits a high degree of flexibility. Within this segment is an octapeptide repeat (OR) domain which binds divalent ions. The C-terminal domain (residues 126–230) folds to a characteristic structure composed of three α-helices, numbered one through three (or A through C), and two anti-parallel β-strands flanking helix 1. PrPc is modified by N-glycans at positions Asn181 and Asn197 in the human protein (corresponding to Asn180 and Asn196 in the mouse protein). Several histidine residues implicated in regulating PrPc stability are also indicated. B. PrPC is attached to the outer leaflet of the plasma membrane through a glycosyl phosphatidylinositol (GPI) moiety. Loss of glycans destabilize the prion protein structure enriching in a conformation that enable pathologic interactions (such as with heparane sulfate). The fewer glycosylation modifications PrP undergoes, the more likely it is to be located in the cytoplasm and the stronger are its proteolytic resistance, toxicity and aggregation ability. C. Top, Cartoon showing how glycans stabilize the PrP protein. Glycosylation at Asn197 may have an allosteric effect, with impact on participate in stabilizing a stable conformation of the protein. N181 may act in concert with other residues to anchor the disordered NTD to its regulatory CDT. Specifically, Histidine residues, acting through copper coordination, glycans at Asn181, and acidic residues (see acidic patch) may act in concert to anchor the toxic effector N-terminal domain to its regulatory site on the C-terminal domain. Bottom, ribbon representation of the PrPC showing the position of the two glycans (sticks). AlphaFold generated structure using the human prion protein sequence (UNIPROT ID: P04156).
Figure 2.
A. Schematic illustrating the primary structure of the full-length prion (PrPc) protein. The N-terminal segment (residues 23–125, after removal of the signal peptide) exhibits a high degree of flexibility. Within this segment is an octapeptide repeat (OR) domain which binds divalent ions. The C-terminal domain (residues 126–230) folds to a characteristic structure composed of three α-helices, numbered one through three (or A through C), and two anti-parallel β-strands flanking helix 1. PrPc is modified by N-glycans at positions Asn181 and Asn197 in the human protein (corresponding to Asn180 and Asn196 in the mouse protein). Several histidine residues implicated in regulating PrPc stability are also indicated. B. PrPC is attached to the outer leaflet of the plasma membrane through a glycosyl phosphatidylinositol (GPI) moiety. Loss of glycans destabilize the prion protein structure enriching in a conformation that enable pathologic interactions (such as with heparane sulfate). The fewer glycosylation modifications PrP undergoes, the more likely it is to be located in the cytoplasm and the stronger are its proteolytic resistance, toxicity and aggregation ability. C. Top, Cartoon showing how glycans stabilize the PrP protein. Glycosylation at Asn197 may have an allosteric effect, with impact on participate in stabilizing a stable conformation of the protein. N181 may act in concert with other residues to anchor the disordered NTD to its regulatory CDT. Specifically, Histidine residues, acting through copper coordination, glycans at Asn181, and acidic residues (see acidic patch) may act in concert to anchor the toxic effector N-terminal domain to its regulatory site on the C-terminal domain. Bottom, ribbon representation of the PrPC showing the position of the two glycans (sticks). AlphaFold generated structure using the human prion protein sequence (UNIPROT ID: P04156).
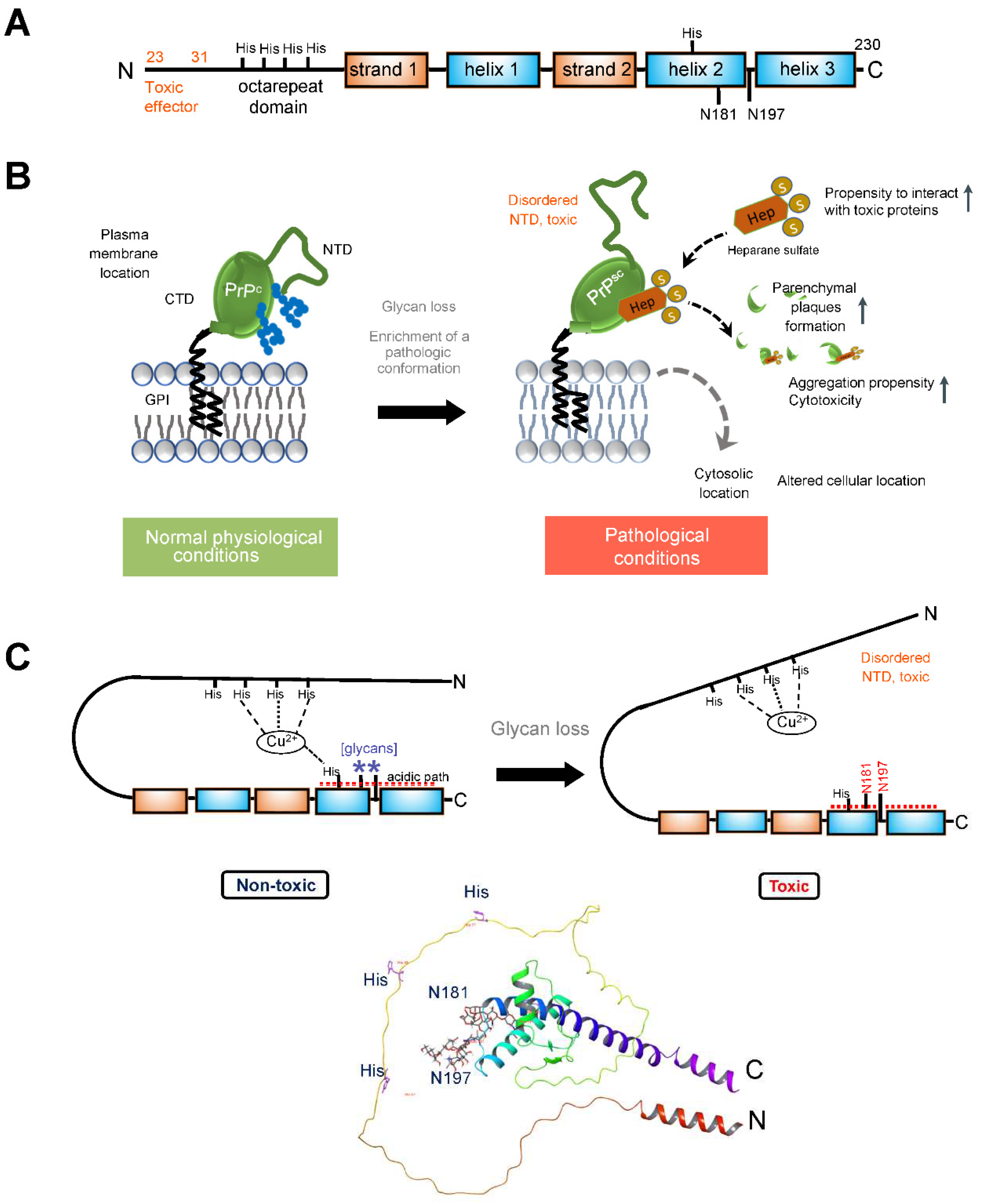
Figure 3.
A. Schematic illustrating the primary structure of the full-length GRP94 protein with color-coded domains: N-terminal domain (NTD), middle domain, and C-terminal domain (CTD). The right-side figure depicts the 3D structure of the GRP94 dimer, with individual protomers colored in green and red, respectively. An unstructured loop is colored in light blue. The location of two key regulatory N-glycans, on asparagine residues N217 and N62, is indicated by an arrow. B. GRP94 glycosylation at N62 is a pivotal factor turning GRP94 from a folding chaperone to a pathologic protein. Primarily, N-glycosylation at N62 serves as a structural mediator, inducing the conversion of the GRP94 chaperone into epichaperomes—hetero-oligomeric forms tightly composed of chaperones, co-chaperones, and other factors. In these epichaperomes, GRP94 adopts scaffolding functions not observed in normal cells, where GRP94 primarily participates in protein control and folding. Through this scaffolding function, GRP94 influences the assembly and connectivity of proteins crucial for maintaining a malignant phenotype, enhancing their activity. Ultimately, N62 glycosylation induces GRP94 malfunction, disrupting its structural ensembles, chaperone cycle kinetics, and leading to interactome remodeling at a much larger scale than the simple local covalent modification might lead to hypothesize, amplifying dysfunction, and remodeling cellular phenotypes. C. The figure illustrates the structural impact of N62 glycan on GRP94. The absence of the N62 glycan favors a GRP94 conformation that is favorable for folding GRP94 function, as observed in the conformation of the lid of the ATP binding site (purple) and a more open and flexible protein client binding site. In the N62 glycosylated GRP94, the glycan pulls the ATP-lid into an open conformation (yellow), favoring a more closed and inaccessible protein client binding site, thus unfavorable for folding. Through its impact on the ATP-binding site’s efficiency and by perturbing GRP94’s structural dynamics, N62 glycosylation thus actively shifts GRP94 from a foldase to a protein-assembly platform. .
Figure 3.
A. Schematic illustrating the primary structure of the full-length GRP94 protein with color-coded domains: N-terminal domain (NTD), middle domain, and C-terminal domain (CTD). The right-side figure depicts the 3D structure of the GRP94 dimer, with individual protomers colored in green and red, respectively. An unstructured loop is colored in light blue. The location of two key regulatory N-glycans, on asparagine residues N217 and N62, is indicated by an arrow. B. GRP94 glycosylation at N62 is a pivotal factor turning GRP94 from a folding chaperone to a pathologic protein. Primarily, N-glycosylation at N62 serves as a structural mediator, inducing the conversion of the GRP94 chaperone into epichaperomes—hetero-oligomeric forms tightly composed of chaperones, co-chaperones, and other factors. In these epichaperomes, GRP94 adopts scaffolding functions not observed in normal cells, where GRP94 primarily participates in protein control and folding. Through this scaffolding function, GRP94 influences the assembly and connectivity of proteins crucial for maintaining a malignant phenotype, enhancing their activity. Ultimately, N62 glycosylation induces GRP94 malfunction, disrupting its structural ensembles, chaperone cycle kinetics, and leading to interactome remodeling at a much larger scale than the simple local covalent modification might lead to hypothesize, amplifying dysfunction, and remodeling cellular phenotypes. C. The figure illustrates the structural impact of N62 glycan on GRP94. The absence of the N62 glycan favors a GRP94 conformation that is favorable for folding GRP94 function, as observed in the conformation of the lid of the ATP binding site (purple) and a more open and flexible protein client binding site. In the N62 glycosylated GRP94, the glycan pulls the ATP-lid into an open conformation (yellow), favoring a more closed and inaccessible protein client binding site, thus unfavorable for folding. Through its impact on the ATP-binding site’s efficiency and by perturbing GRP94’s structural dynamics, N62 glycosylation thus actively shifts GRP94 from a foldase to a protein-assembly platform. .
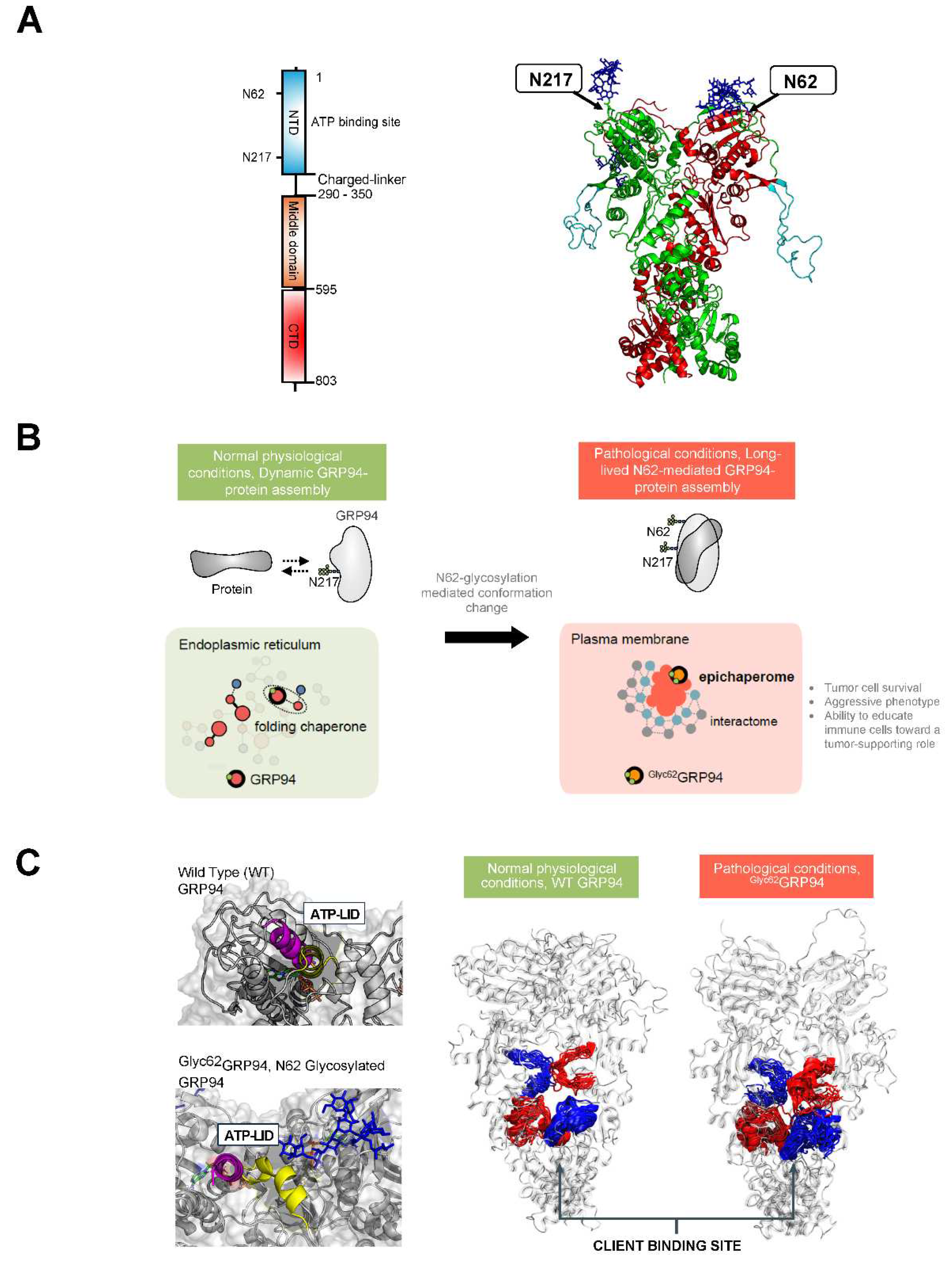
Figure 4.
The schematic illustrates the central role of N-glycans in orchestrating protein conformation and assembly dynamics, illustrating their profound impact on disease. The figure emphasizes that glycosylation-induced conformational mutants, characterized by altered three-dimensional structures or conformational dynamics compared to wild-type proteins, serve as unique targets for therapeutic intervention. Importantly, the figure highlights how glycan-mediated remodeling extends beyond individual protein structures, influencing the interactome of the protein and reshaping functional pathways at the systems level. This intricate interplay at the molecular and systems levels ultimately contributes to the reshaping of cellular phenotypes. The schematic not only highlights how N-glycosylation provides actionable targets for precision therapies but also underscores the transformative potential of modulating glycan-regulated protein structures and functions in a systemic context.
Figure 4.
The schematic illustrates the central role of N-glycans in orchestrating protein conformation and assembly dynamics, illustrating their profound impact on disease. The figure emphasizes that glycosylation-induced conformational mutants, characterized by altered three-dimensional structures or conformational dynamics compared to wild-type proteins, serve as unique targets for therapeutic intervention. Importantly, the figure highlights how glycan-mediated remodeling extends beyond individual protein structures, influencing the interactome of the protein and reshaping functional pathways at the systems level. This intricate interplay at the molecular and systems levels ultimately contributes to the reshaping of cellular phenotypes. The schematic not only highlights how N-glycosylation provides actionable targets for precision therapies but also underscores the transformative potential of modulating glycan-regulated protein structures and functions in a systemic context.
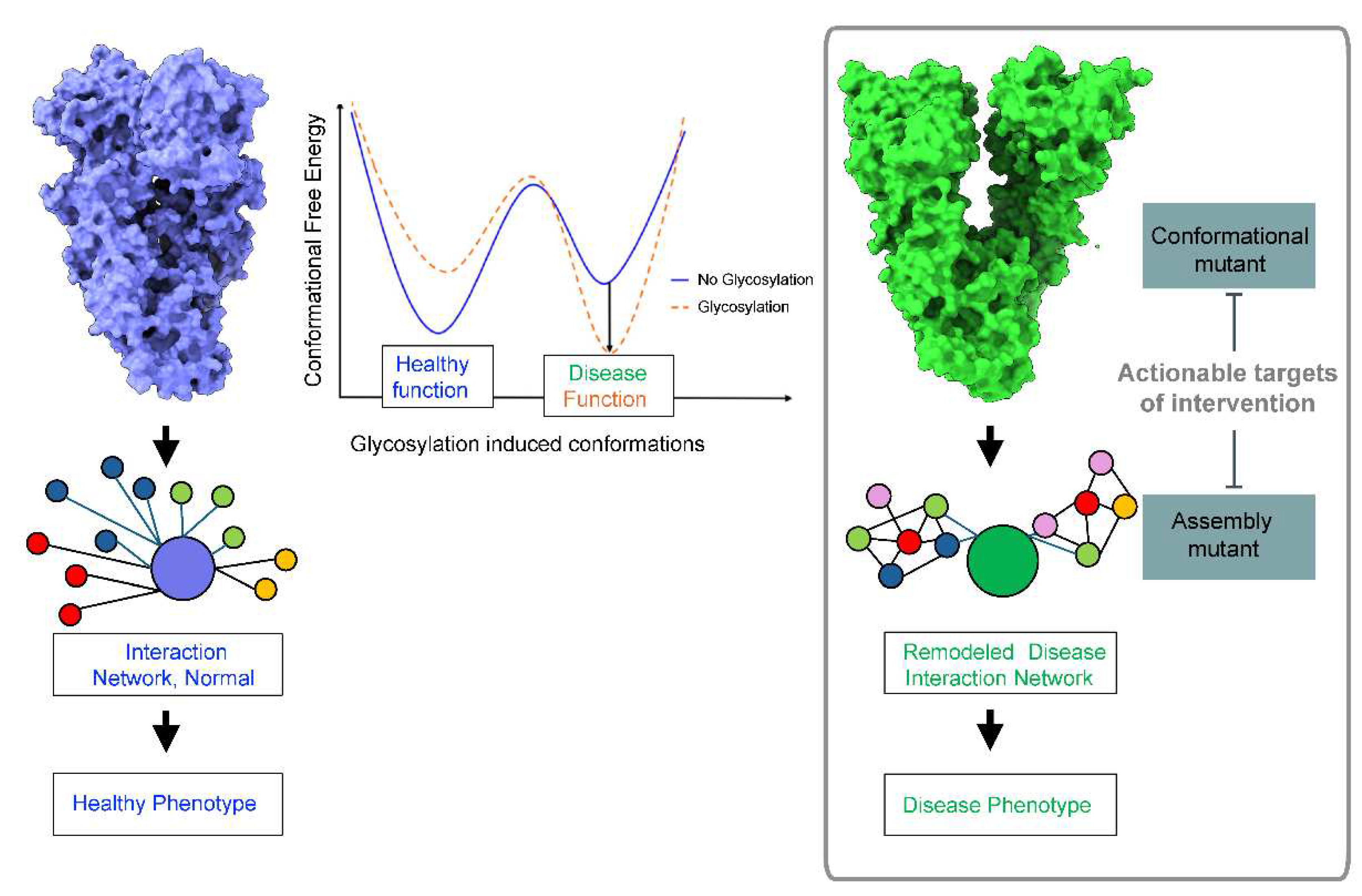
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



