
Submitted:
02 February 2024
Posted:
02 February 2024
You are already at the latest version
Abstract
Rare ginsenoside compound K (CK), is an intestinal microbial metabolite with a low natural abundance that is primarily produced by physicochemical processing, side chain modification, or metabolic transformation in the gut. Moreover, CK exhibits potent biological activity compared to primary ginsenosides, which has raised concerns in the field of ginseng research and development as well as ginsenosides-related dietary supplements and natural products. Ginsenosides Rb1, Rb2, and Rc are generally used as a substrate to generate CK via several bio-conversion processes. Current research shows that CK has a wide range of pharmacological actions including boosting osteogenesis, lipid and glucose metabolism, lipid oxidation, insulin resistance, anti-inflammatory, and anti-apoptosis properties. Further research on the bioavailability and toxicology of CK can advance its medicinal application. The purpose of this review is to lay the groundwork for future clinical studies and the development of CK as a therapy for metabolic disorders. Furthermore, the toxicology and pharmacology of CK are investigated as well in this review. The findings indicate that CK primarily modulates signal-ing pathways associated with AMPK, SIRT1, PPARs, WNTs, and NF-kB. It also demonstrates a positive therapeutic effect of CK on nonalcoholic fatty liver disease, obesity, hyperlipidemia, diabetes, and its complications, as well as osteoporosis. Additionally, the analogues of CK showed more bioavailability, less toxicity, and more efficacy against disease states. Enhancing bioavailability and regulating hazardous variables are crucial for its use in clinical trials.
Keywords:
Ginsenoside compound K
; Metabolic disease
; obesity
; NAFLD
; Diabetes
; osteoporosis
1. Introduction
In recent years, metabolic disorders become a global health concern because of their rapidly increasing prevalence [1]. Global human health is severely challenged by the increasing incidence of metabolic diseases, which include type 2 diabetes (T2D), obesity, nonalcoholic fatty liver disease (NAFLD), gout, osteoporosis, hypothyroidism and hyperthyroidism [2]. The International Diabetes Federation (IDF) [3] reports that 537 million individuals worldwide had diabetes in 2021, with more than 90% of cases being type 2 diabetes. By 2045, the figure is predicted to rise to 783 million. In addition, obesity has emerged as the biggest global problem of concern for public health, with the incidence of overweight and obesity rising sharply in recent years. In 2016, there were over 650 million and over 1.9 billion obese and overweight adults worldwide, respectively, making up around 39% of the world’s population [4]. According to estimations in 2019, the worldwide incidence of gout was 0.1%–0.3% [5] and the prevalence of non-alcoholic fatty liver disease (NAFLD) was 29.62% in Asia [5]. These findings demonstrate that metabolic disorders represent a substantial challenge in human society due to the associated high mortality and morbidity; thus, understanding the pathophysiology and therapies of metabolic diseases is critical.
The pharmacological properties of Korean ginseng (Panax ginseng Meyer), which has been revered as one of the most renowned traditional Chinese herbal medicines for over two hundred years, are largely attributed to its bioactive triterpenoid saponins. These triterpenoid saponins, called ginsenosides, are divided into three categories: panaxadiol (PPD), panaxatriol (PPT), and oleanic acid [6]. More than 218 ginsenosides have been determined from multiple parts of the ginseng plant (roots, flowers, berries, and leaves) and these byproducts have become popular for research [7]. However, Ginsenoside compound K (CK) is one of the most significant among these ginsenosides due to its high uptake and absorption rate into the human gastrointestinal tract and ultimately the systemic circulation [8]. Compared to other ginsenosides, CK has superior membrane permeability and a lower molecular weight, which contribute to its increased bioavailability [9]. Major Ginsenosides undergo the transition to produce CK, which is rarely present in natural ginseng. Human gut bacteria and endophytes have been reported to use deglycosylated processes to bio-convert CK products. In the past year, yeasts have been metabolically altered, and produced by enzymes have become viable alternatives for producing CK [10]. According to current research, CK possesses pharmacological properties that include hepatoprotective, anti-inflammatory, anti-carcinogenic, anti-diabetic, anti-allergic, neuro-protective, and anti-aging activities [11].
CK is an active molecule that can reduce blood lipids and control glucose consumption [12]. Notably, CK is a regulator of PPARγ [13], and AMPK [14]. It has been demonstrated that AMPK increases glucose utilization, mobilizes lipid storage, and promotes autophagy to turnover macromolecular routes, so promoting the breakdown of biomolecules for the creation of energy [15]. PPARγ influences lipid metabolism, which enhances sensitivity to insulin and glucose metabolism [16]. AMPK and PPARγ are the fundamental targets in metabolic disorders, including nonalcoholic fatty liver disease, diabetes, osteoporosis, NAFLD, and obesity [17,18]. In addition, CK down-regulates PPAR, leptin, aP2, and C/EBP adipogenic markers, which cause obesity, diabetes, and other metabolic diseases [19]. Moreover, the Deregulation of metabolic processes associated with TP53 results in various human pathologies, such as obesity, diabetes, liver, and cardiovascular illnesses [20]. CK significantly regulates the TP53 expression in different disease states [21].
As a result, it is hypothesized that CK may be involved in metabolic illnesses by controlling inflammation and energy. However, due to the shortage of adequate information on CK regarding its use for the cure of metabolic illness, the cytotoxicity is well documented, which prevents further development of the drug. This study includes an in-depth assessment of the use of CK to treat metabolic illnesses and the signaling pathways involved, as well as an analysis of its usual negative effects and pharmacokinetics. To give direction and evidence for CK research on metabolic conditions, we investigated the Google Scholar, Web of Science, PubMed, and CNKI databases up to December 2023.
2. Physical and chemical properties
CK (20-O-β-D-glucopyranosyl-20(S)-protopanaxadiol) is a minor tetra-cyclic triterpenoid that is rarely found in natural ginseng. CK is isolated via various biotransformation processes from the protopanaxadiol saponins including ginsenoside RB1, Rb2, and Rc [22]. Several approaches for CK synthesis have been described in detail (Biotransformation of CK section). CK is a white crystalline powder with a molecular weight of 622.9 g/mol, molecular formula C36H62O8 [23], and PubChem CID 9852086. The physical and chemical properties of CK are shown in Table 1 (data collected from https://www.chemicalbook.com/).
3. Pharmacokinetics, safety, and toxicological studies of CK
The bioavailability rate of ginsenosides without conversion or modification indicates limited intestinal absorption [24]. Following oral ingestion, ginsenosides undergo several biological changes in the digestive system that result in the conversion of these molecules into deglycosylated metabolites that have more biological activity than their precursor components [25]. According to other research, some microorganisms and gut bacteria or soil fungi around ginseng roots hydrolyze ginsenosides to produce CK. It is essential to research the metabolic processes that control intestinal microbiota since it plays a crucial role in the biological transformation and therapeutic effects of CK [24]. Based on recent research on human metabolism, a high-fat diet greatly speeds up and increases the digestion of CK, and women have higher concentrations of CK than men [26]. After delivering Korean ginseng extract to ten healthy males for 36 hours, the drug levels in their blood samples were reported in additional pharmacokinetic investigation on CK [27]. The mean time to achieve the Cmax (Tmax) of CK was greater compared to Rb1 (12.20±1.81 vs. 8.70±2.63 h), and the average highest plasma concentration (Cmax) of CK was substantially greater than the mean concentration of Rb1 (8.35±3.19 vs. 3.94±1.97 ng/mL). Intestinal microflora probably converts Rb1 to CK because of the delay in CK absorption. Compared to Rb1, CK had a plasma half-life (t1/2) that was seven times shorter. The findings of this study suggest that there is a notable distinction in the pharmacokinetics of CK and Rb1. In a different study, 76 participants were given either a placebo or CK in seven individual doses taken orally (25, 50, 100, 200, 400, 600, and 800 mg) while they were fasting; the exposure to CK grew linearly between 100 and 400 mg, and the time range to attain Tmax was 1.5–6.0 h. After the seventh administration, the steady state was reached, and there were no serious adverse effects (AEs) reported. Watery stool (diarrhea) and stomachache were the most commonly reported AEs. All AEs were mild to severe, and the majority of them were cured quickly without any intervention. These findings demonstrated that CK was both safe and well-tolerated for the course of the treatment [26,28].
According to toxicity tests, CK was applied on 3T3L1 pre-adipocyte cells in a dose-dependent manner. The maximum concentration of 40µM did not affect the viability of the cells [29]. Fang et al examined Ginsenoside CK for cytotoxicity at various concentrations (0.2-10.0 μM). They observed that CK concentrations below 10 μM had no discernible impact on the survival rate of HaCaT keratinocyte cells [30]. The osteoblastic cell line MC3T3-E1 was exposed to CK at various concentrations (0.1–10 μM) and did not exhibit any appreciable toxicity [31]. When ginsenoside CK was evaluated on HepG2 cells, substantial cytotoxic effects were detected with increasing concentrations of ginsenoside CK up to 30 M compared to the control group [32]. Additionally, different CK doses (5-40 μM) were applied to HepG2 cells for a 24-hours to assess the cellular toxicity of CK. Even at dosages of 40 μM, CK did not exhibit any cellular damage [33]. However, the administration of a higher dose of CK (10 µM) significantly boosted the development of HT22 hippocampus neuron cells [34]. After the treatment with 1.25-10 μM of CK, the rate of survival of the L02 cells climbed dramatically. CK showed no toxicity at any test concentration on L02 cells [35]. Human fibroblast-like synoviocytes RA-FLS cells and murine macrophage cells were treated at the same concentrations of 0.1-5 μM for the treatment of rheumatoid arthritis. The results showed that these cells were not affected by CK at a concentration of ≤5 µM [36]. Gu et al evaluated the possible cytotoxicity of CK on MIN6 mouse pancreatic β-cell at various doses (2-32µM). CK showed minimal effect at 16 µM and decreased the cell viability at 32 µM [37]. CK has a time- and concentration-dependent mild to moderate cytotoxic impact on cancer cells. The most susceptible to CK exposure were Hk-1 cells (a cell line used to study Nasopharyngeal Carcinoma), as 41.1–88.0% of cell mortality was seen at low levels (10–20 μM) [38]. Boopathi et al reported that CK exhibits negligible cytotoxic effect on A549 lung cancer cells, Caco-2 colorectal cancer cells, and MCF-7 breast cancer cells at 12.5 μg/mL, whereas, normal cell Raw 264.7 demonstrated less toxicity at 6.25µg/ml [39]. At concentrations ranging from 8 to 64 μmol/l, compound K inhibited the growth of HT-29 cells in a dose-dependent manner; the dosage that produced 50% inhibition of growth (IC50) was 32 μmol/l [40]. Oral CK delivery to rats and mice in a toxicity trial did not result in toxicity or death at the maximal doses of 8 and 10 g/kg respectively [41]. During a beagle toxicity investigation, dogs in the 36 mg/kg group experienced considerable weight loss and reversible hepatotoxicity. There was no discernible toxicity in the animals in the 4 and 12 mg/kg groups [42]. Table 2 elucidated the cytotoxicity of CK on different cell lines.
It has been demonstrated that CK is safe and well-tolerated in both human and animal subjects. These preclinical findings imply that CK may be harmful to the liver. Although the relative weight of the kidney was high, there was no histological change, but nephrotoxicity should be noted. Abdominal pain and diarrhea were CK-related adverse events observed in clinical trials. Both clinical trials and data on AEs associated with CK are scarce. Thus, more research is required to determine the processes underlying CK-induced gastrointestinal tract irritation and CK-induced damage, particularly hepatotoxicity. We studied ADMET analysis of CK there mentioned the drug likeliness and toxicity of CK (Supporting information 1).
4. Biotransformation of CK
It has been demonstrated that the naturally occurring ginseng plant does not provide significant amounts of the minor ginsenoside CK. Therefore, several studies have concentrated on using various techniques, including hydrolysis, enzymatic biotransformation, microbial transformation, etc., to convert major ginsenosides to CK. Additionally, endophyte biotransformation is an effective technique within microbial transformation because of its low cost, excellent selectivity, accuracy, and environmental safety [10]. Chemical hydrolysis conditions led to the nonspecific cleavage of glycone moieties at position 20, which in turn caused side reactions of hydroxylation, hydration, and epimerization. Furthermore, these methods proved to increase environmental contamination [47,48]. In contrast, owing to their notable selectivity, mild reaction conditions, and environmental compatibility, enzymatic or microbial conversion modalities have emerged as the most popular ones. Figure 1 demonstrates the biotransformation of major ginsenosides to CK via several pathways using enzymatic and microbial conversion methods.
4.1. Enzymatically synthesis
One possible approach could be highly region-specific enzymatic transformation. Enzymatic techniques have been employed to produce CK from ginseng root extract, employing β-glucosidase (β-glu), β-glycosidase, and bi-composites of β-glucosidase (Table 3).
The bioconversion of ginsenoside Rb1 to CK through β-glu is an effective production method in industry [55]. Microbes isolated from the soils of ginseng farms, soybeans, tea, the gastrointestinal tract of humans, kimchi, and other fermented items can be a source of β-glu [58]. Heat-resistant β-glu yields the highest amount of CK from protopanaxadiol-type ginsenosides [59]. Qin et al. used chromatography to purify a new ginsenoside-hydrolyzing β-glu from Paecilomyces Bainier sp. 229 to increase the conversion rate of Rb1 into CK. At 45 °C and pH 3.5, the ginsenosides Rb1 and the enzyme exhibited the highest level of activity. The pathway Rb1 → Rd → F2 → CK converted roughly 84.3% of the ginsenoside Rb1 to CK one day after the incubation [49]. Furthermore, it has been observed that recombinant β-glu enzymes found in Terrabacter ginsenosidimutans sp. [50] and Lactobacillus brevis [51] can convert Rb1 into CK.
β-glycosidases are a substitute for β-glucosidases and are frequently employed in the hydrolysis of ginsenosides [60]. Particularly, PPD-type ginsenosides are hydrolyzed by β-glycosidases [61]. Noh et al [52]documented synthesizing CK from ginseng root extract by employing β-glycosidases derived from Sulfolobus solfataricus. Two transformation pathways were described by them to turn Rb1, Rb2, Rc, or Rd into CK: (1) Rb1 or Rb2 → Rd → F2 → compound K, and (2) Rc → compound Mc → compound K. Despite the strong specificity of this approach, the ginsenoside to CK conversion rate was low. Thus, recombinant β-glycosidase derived from Pyrococcus furiosus [53]and Microbacterium esteraromaticum [62] has been created to convert significant ginsenosides into minor ginsenosides. Pyrococcus furiosus was highly productive in turning Rd into CK, yielding an 83% conversion rate [53].
However, there was inadequate stability of free β-Glu, which hindered circulation and recovery, unable to recycle β-Glu and snailase, self-digestion rate, and long reaction time. This limitation can be overcome by enzymatic immobilization technology, which will also make it easier to use β-Glu in commercial production β-Glu@Cu(PTA) biocomposite reached a 49.15% conversion rate of Rb1 to CK [55]. Green synthesis of Zn-BTC co-immobilized snailase and β-glucosidase (β-Glu) resulted in the formation of β-Glu&SN@Zn-BTC biocomposite, which reached the CK conversion rate of 53.5% in 48 hours at pH 4.5. The CK concentration was 1.07 mg/mL, and 83% of all products were made up of CK [56]. Using Sna&β-Glu@H-Cu-BDC (large-sized snailase&β-glucosidase@hollow-Cu-H2BDC) biocomposite for the synthesis of the CK. Cau et al [57] reported that the total amount of CK was about 0.94 mg mL−1, and the average conversion rate of CK was around 60.12% after growing the conversion system.
4.2. Biotransformation of CK by human gut microbiota
Microbial transformation is generally regarded as a key technique for producing CK [63]. It involves using crude enzymes from Fusarium sacchari [64], Lactobassillus paralimentarius [65], Microbacterium esteraromaticum [54] Caulobacter leidyia, and Acremonium strictum [66]. For instance, Hasegawa et al. [67] thoroughly examined the metabolic conversion of CK from ginsenosides Rb1, Rb2, and Rc by gut flora. Ginsenosides Rb1, Rb2, and Rc were transformed into CK through anaerobic incubation with human gut microbiota. Among them, bacterial strains obtained from human intestinal feces, including Bacteroides sp., Bifidobacterium sp., and Eubacterium sp., successfully converted Rc into CK [68]. The composition of the gut microbiota determined the primary metabolic pathway used by intestinal bacteria to break down ginsenoside Rb1 into CK.
Compared to bacteria, fungi are easily cultured and can biotransform to replace human intestinal bacteria as a source of CK [64]. Zho et al. [69] used fungal biotransformation to efficiently manufacture CK from Panax notoginseng (PNG) saponins at a reasonable cost. The same group also showed that the fungus Paecilomyces bainier sp. 229 could efficiently convert PNG saponins into CK; this resulted in a substantially higher conversion rate of CNS to CK (82.6% vs. 35.4%) than before [70]. Furthermore, fungi that were produced from ginseng-cultivated soil, such as Fusarium moniliforme [71], A. strictum [72], A. niger [73], and F. sacchari [64], demonstrated good biotransformation of major ginsenosides into minor bioactives. Cumulative Generation of Bioactive CK from Fermented Black Ginseng using a novel Aspergillus niger KHNT-1 Strain Obtained from Korean staple food kimchi [74]. Leuconostoc strains were also isolated from kimchi, which showed good conversion of PPD-type ginsenosides to CK [75]. Microorganisms have frequently been used in the biotransformation of major ginsenosides into minor bioactive. Furthermore, a synthetic biology approach has been utilized for transformation. PPD ginsenosides could be easily converted into CK by the metabolically modified yeast expressing the heterologous UGTPg1 gene [76].
5. Mechanism of CK against metabolic diseases
The pathophysiology of metabolic disease is highly complex and is caused by multiple factors. Numerous studies have demonstrated what beneficial impacts of CK on metabolic disorders. After reviewing researches, we concluded that CK is a useful medicinal substance for treating osteoporosis, hyperlipidemia, obesity, hepatocyte steatosis, NAFLD, and diabetes and its consequences. Table 4 displays the pharmacological molecular pathways of CK in the treatment of metabolic disorders.
5.1. Obesity
Obesity is a prevalent metabolic disease, defined by adipocyte hypertrophy, which results from an imbalance between energy expenditure and food intake. Obesity has been related to additional metabolic diseases such as insulin resistance, NAFLD, T2D, dyslipidemia, cardiovascular diseases, hypertension, and cancer [90]. Adipose tissue growth results in fat storage in preexisting adipocytes and the transformation of preadipocytes into mature adipocytes, a process called adipogenesis [91]. Under adipogenic conditions, more amount of free fatty acids will be produced by hypertrophic adipocytes [92]. It shows that obesity is linked to a lipid metabolism issue; there is an indication that 43.2% of obese people have hyperlipidemia [93]. An important function of adipose tissue is to regulate energy metabolism [94]. It’s been proposed that one of the key strategies for treating obesity is to activate brown adipose tissue (BAT) and produce browning in white adipose tissue (WAT) [95].
CK might be the potential therapeutic agent against obesity via several signaling pathways (Figure 2). A study showed that C5BL/6J mice consumed a high-fat diet to induce obesity, whereas administration of CK (15, 30, 60 mg/kg) might successfully enhance the resistance to insulin and glucose tolerance, downregulate PPARγ expression, inhibit TLR4/TRAF6/TAK1/NF-κB stimulation in obese mice, and lower macrophage M1-type inflammatory cytokine levels in serum and adipose tissue in a dose-dependent manner. Furthermore, CK increased IRS1/PI3K/AKT expression which proved CK is an effective compound against obesity and early diabetes [96]. CK is a novel agonist of the glucocorticoid receptor (GR) used to treat obesity. In mice, CK was more effective than Orlistat in reducing blood lipids and weight [77]. CK treatment of 3T3-L1 adipocytes prevented lipid formation and the expression of genes particular to adipocytes (C/EBPα, leptin, aP2, and PPARγ), decreased angiogenic factors (VEGF-A and FGF-2) and MMPs (MMP-2 and MMP-9), whereas enhanced the mRNA expressions of angiogenic blockers (TSP-1, TIMP-1, and TIMP-2) [19]. CK had strong inhibitory effects on the rise of MCP-1 and TNF-α caused by the hypertrophic adipocyte supernatant. Additionally, it facilitated the expression of IL-10, prevented the induction of inflammatory macrophages, and enhanced the development of anti-inflammatory macrophages [78]. In early-stage adipogenesis, CK reduced the phosphorylation of protein kinase B (AKT), p38, and extracellular signal-regulated kinase [97]. Moreover, CK markedly elevated AMPK (AMP-activated protein kinase) and ACC (acetyl-CoA carboxylase) to suppress adipogenesis. In differentiated 3T3-L1 cells, the effect of CK on reducing PPAR-γ expression was restricted by AMPK pharmacological inhibition with dorsomorphin [79]. Therefore, by controlling lipid metabolism and energy metabolism, CK has the potential to be used as a treatment and preventive medicine for obesity.
5.2. Diabetes and related complications
Diabetes mellitus (DM) is an emerging epidemic that can be linked to hereditary and environmental factors. Diabetes has complications that require treatment, including diabetic retinopathy, nephropathy, neuropathy, infertility, and cardiovascular disease. Type I diabetes, or T1D, and type II diabetes, or T2D, are the two primary subtypes of DM. An autoimmune condition called T1D kills beta cells in the pancreas and stops insulin from being released. On the other hand, T2D is characterized by high insulin levels and cell insulin resistance. According to epidemiological studies, there will be 629 million people with diabetes worldwide by 2045, up from a total of 425 million in 2017. Due to the potential harm that diabetes mellitus (DM) can do to an individual’s quality of life, the condition needs to be controlled and managed as soon as possible [98]. To investigate the anti-diabetic activity, ICR mice were fed with CK (30mg/kg/day) for 4 weeks. Phosphoenolpyruvate carboxykinase (PEPCK) and glucose-6-phosphatase (G6Pase), two glucose-producing genes, were down-regulated after CK treatment. The results showed that CK can reduce blood sugar levels and increase insulin sensitivity in type 2 diabetes caused by a high-fat diet and fasting. This is achieved by suppressing the expression of PEPCK and G6Pase in the liver [80]. Jiang et al studied CK (30, 100, 300 mg/kg/BW) on male Wistar rats to improve insulin sensitivity. They found that CK may improve insulin resistance and hyperglycemia in diabetic rates. Additionally, studies indicated that CK could increase the expression of Glut4, PI3Kp85, InsR, IRS1, and pAkt in the skeletal muscle of diabetic rats. These findings suggest that increased insulin sensitivity, which is directly linked to the PI3K/Akt signaling pathway, mediates the hypoglycemic action of CK [81]. Another study reveals that CK has strong stimulatory effects on insulin production in MIN6 cells by upregulating GLUT2 expression (Figure 3) [37].
Despite extensive research on the antidiabetic effects of CK, Song et al studied CK on diabetic nephropathy (DN). DN mice model induced by HFD (high-fat diet) and STZ (streptozotocin) were administered CK intragastrically. The results demonstrated that CK dramatically reduced the growth of the glomerular mesangial matrix and considerably decreased the increased fasting blood glucose, serum creatinine, blood urea nitrogen, and 24-hour urine protein of the DN mice [83]. Additionally, It was observed that the expression of G6Pase and PEPCK in the liver and HepG2 was suppressed by CK. In the meantime, AMPK activation was markedly boosted upon CK administration, but FOXO1, HNF-4α, and PGC-1α expressions were significantly decreased [99]. It has been noted that diabetics with tendinopathy have a higher apoptotic tendency. Tendinopathy is a chronic illness that affects the tendons and causes a great deal of discomfort. It has a major negative influence on quality of life [100]. However, CK can effectively reduce the MMP system, inflammation, tenocyte apoptosis, and oxidative stress under hyperglycemia [84].
5.3. Osteoporosis
Osteoporosis (OP) represents an alarming clinical condition that typically manifests as rapid bone loss during menopause. It increases the possibility of a brittle fracture, which puts a great deal of strain on society. A growing number of people are experiencing OP as society ages. Hip fractures are expected to become more common worldwide by 2050 as population demographics change. OP occurs when the bone resorption (osteoclast) rate is greater than the bone formation (osteoblast) rate, leading to lowered bone density [101]. Osteoblasts are bone-decomposed cells that play a crucial role in maintaining bone homeostasis. During postmenopausal osteoporosis, the receptor activator for nuclear factor-κB ligand (RANKL) increases osteo-clastogenesis, which results in bone loss [102]. Nowadays, many medications, including calcitonin and bisphosphonates, are used to treat and prevent OP. On the other hand, documented accounts of the adverse effects of these medications in medical settings exist. As a result, to lower fracture rates and enhance patient quality of life, improved therapies for OP must be investigated [103].
Herbal supplementations have been extensively researched as potential sources for drug development because of their lesser toxicities. As an herbal product, CK showed an anti-osteoporotic effect by suppressing RANKL-induced osteoclast differentiation and ROS production by triggering NF-κB/P65 signaling pathway [85]. Additionally, CK elevated the Runx2 (master transcription factor) and β-catenin to promote osteogenic differentiation via the Wnt/ β-catenin pathway [86]. Furthermore, CK has a preventive effect on osteoarthritis via upregulating ALP, Col-1, and Runx2 in pre-osteoblastic MC3T3-E1 cell lines [31]. CK may reduce RANKL levels, boost the amount of osteoprotegerin (OPG) on AA-FLS in vitro, and prevent bone degradation in AA rats [104]. Furthermore, pretreatment with CK may prevent human CD4+ monocytes and murine RAW264.7 cells from proliferating into TRAP+ osteoclast-like cells in response to soluble RANKL (sRANKL) in a dose-dependent way. Moreover, CK inhibited the nuclear transcription factor of activated T cells (NFATc1) and RANK-associated NF-κB pathways in osteoclast progenitors. These suggest that GCK blocked osteoclastogenesis caused by RANKL through two different pathways [105]. Based on all of these data, CK appears to be a promising treatment for preventing dietary induction of OP (Figure 4).
5.4. Non-alcoholic Fatty liver disease (NAFLD)
NAFLD also known as hepatic steatosis or the formation of triglyceride in the liver which is not induced by consuming alcohol [106]. NAFLD is a broad term for a range of pathologies that include nonalcoholic fatty liver (NAFL), which is the first stage of NAFLD, nonalcoholic steatohepatitis (NASH), which is defined by the beginning of inflammation brought on by lipotoxicity, and severe NASH symptoms that include fibrosis. According to reports, the overall incidence of NAFLD might reach 15% to 18% among Asian nations and 30% in Western nations. Obesity is closely linked to this fatty liver disease. The prevalence of NAFLD is predicted to increase globally in light of the present obesity pandemic. [87]. A growing amount of clinical data indicates that NAFLD is a major risk factor for the emergence of liver cirrhosis, liver fibrosis, and liver cancer [107]. However, it is still unclear how NAFLD develops and how it leads to fibrosis and chronic liver disease. The "two-hit" theory first came forward by Day et al. in 1998. First, there is an initial metabolic change that results in insulin resistance, hyperglycemia, and hepatocyte triglyceride formation, which causes hepatic steatosis. The second hit causes the injury to worsen and progress, leading to cirrhosis, inflammation, fibrosis, and steatohepatitis [108]. On the other hand, the "multiple parallel hits" theory postulates that several parallel factors, including adipokines and cytokines secreted abnormally, dysfunctional mitochondria, stress to the endoplasmic reticulum, gut endogenous endotoxin, metabolism of lipids, lipotoxicity, oxidative stress, and genetic susceptibility cause NAFLD [109]. Still, the FDA has not approved any particular medications for NAFLD. Medicines such as atorvastatin calcium tablets and fenofibrate are commonly used to manage blood lipid levels [110]. However, using lipid-lowering medications can lead to certain negative side effects [111]. Therefore, finding novel medications that treat NAFLD with great efficacy and few adverse effects is urgent.
Various researches have shown a significant potency of CK against NAFLD (Figure 5). Chen et al. demonstrated that CK is beneficial for treating NAFLD through hepato-protective and anti-fibrotic effects [87]. Another study depicted that, CK activates AMPK and increases ACC and mononyl CoA levels in the AMPK signaling pathway to stimulate fatty acid oxidation. CK can suppress triglyceride accumulation in the liver by inhibiting lipogenic markers such as SREBP1c, SDC1, and FAS and enhancing lipolytic markers including PPAR- α, and CD36 [33]. AMPK also inhibits the formation of free fatty acids by reducing TG hydrolysis via direct phosphorylation and inactivation of hormone-sensitive lipase. Additionally, the activation of AMPK is related to elevated expression of PPAR- α, and subsequent decrease of SREBP1c and PPAR-γ activity in adipocytes and hepatocytes. Moreover, CK might be directly mediating its beneficial impacts by activating AMPK [88]. To minimize the toxicity of CK, Yue et al [112] develop natural nano-CK that acts as an mTOR inhibitor to change lipid metabolism. In steatosis hepatocytes, nano-CK can alleviate lipotoxicity and restore lipid homeostasis by encouraging lipid export and blocking DNL and lipid absorption, all of which create a feedback loop regulated by mTOR. Furthermore, CK has a definite hepatoprotective impact on sodium valproate-induced hepatotoxicity, as shown by Zhou et al [113]. These beneficial effects were mediated by reducing oxidative stress through the suppression of lipid peroxidation and the upregulation of the protective antioxidant system; controlling the peroxisome pathway through the downregulation of soluble epoxide hydrolase, and controlling iron homeostasis through the upregulation of hepcidin. In the case of hepatocellular carcinoma, CK caused a G0/G1 phase arrest, blocked cell cycle progression, and induced apoptosis through the upregulation of p21Cip1 and p27Kip1, and downregulation of cyclin D1 and cyclin-dependent kinase 4 in HepG2 cells. This was accomplished by the mitochondrial system via a modulation of the ratio of Bcl-2 to Bax [89].
Thus, as a monotherapy, in conjunction with other medications, or nanoformulation of CK may prove to be a promising therapeutic for alleviating hepatic problems associated with other metabolic diseases or diminishing fatty liver.
6. Mechanism of TP53 regulating pathway in metabolic diseases
Numerous studies have been conducted on the tumor suppressor activity of TP53, also known as p53, which has long been considered the “guardian of the genome” [114]. As a transcription factor, p53 primarily transactivates gene expression in response to genotoxic or oncogenic stress, hence exerting its tumor-suppressive properties. P53 initiates programmed cell death (apoptosis) in response to severe or protracted DNA damage, mainly by activating genes that encode pro-apoptotic proteins. P53 has been implicated in aging, innate and adaptive immunity, development, reproduction, and neuronal degeneration, among other physiological processes. P53 has been implicated in innate and adaptive immunity, development, reproduction, and neuronal degeneration, aging, among other physiological processes [115]. Furthermore, the link between p53 and metabolic disorders has emerged as an additional area of study for p53 researchers.
In 2009, Minamino and his colleagues provided the first evidence that p53 plays a role in the development of obesityT2D by demonstrating diet-induced IR in Ay transgenic mice [116]. This research demonstrated that p53 activity was inhibited by siRNA knock-down in cells or TP53 gene knock-out in mice, which reduced proinflammatory cytokines expression in the adipose tissue and alleviated senescence, ultimately averting the development of IR. Diabetes is often caused by three main factors: 1) abnormalities in glucose homeostasis; 2) functional deficits in pancreatic insulin secretion; and 3) the emergence of insulin resistance. Based on existing in vitro and in vivo data, p53 appears to be able to significantly affect each of these three pathways. The pro-inflammatory cytokines TNF-α and IFN-γ have been demonstrated to work in concert to boost apoptosis, p53 activation, and ROS generation in a pancreatic beta cell line and insulin-producing islet cells [117]. Lastly, it was discovered that ROS were also implicated in FFA-mediated beta cell death and that NAPDH oxidase 2 (NOX2) was the downstream regulator of both ROS production and p53 activation [118]. Inflammatory cytokines, ROS, and free fatty acids can all function upstream of p53 to cause p53-mediated death in pancreatic beta cells. All things considered, these scientists discovered that reduction of pancreatic beta cell activity lowers insulin output, leading to hyperglycemia and diabetes. Studies conducted in vitro and in vivo showed that elevated release of FFAs caused ROS-induced DNA damage and p53 upregulation in adipose tissue. While p53 inhibition reduced inflammation, p53 activation increased the expression of proinflammatory adipokines through the NF-κB signaling pathway, which in turn led to adipose tissue inflammation, insulin resistance, and diabetes. Obese human patients and mouse models have both shown these obesity-related alterations in p53 expression. It is believed that dysfunctional adipose tissue-related chronic inflammation fosters an environment that is conducive to tumor growth and progression [119].
Fatty liver, also known as hepatic steatosis, constitutes one of the most prevalent side effects of type 2 diabetes and obesity [120]. Yahagi et al. demonstrated that independent of the presence of obesity, p53 activation is critical to the pathophysiology of fatty liver disease [121]. Comparably, Derdak et al. showed that p53 activation increases hepatic steatosis and insulin resistance in both non-alcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD); interestingly, the same group discovered that p53 activity can be inhibited to improve the symptoms of both ALD and NAFLD [122] [123]. These investigations also provided potential mechanistic explanations, proposing that PTEN and miRNA34a are induced by p53, respectively, and that this is the cause of ALD and NAFLD formation. Another group used p53-knockout mice to confirm that p53 plays a role in the development of NAFLD, possibly via its downstream target p66shc [124]. Notably, p53 expression and the degree of steatosis are positively associated in human liver tissues, indicating that these symptoms shown in models of mice are physiologically relevant [125].
After reviewing the research, it is summarized that ginsenosides CK and TP53 gene plays a vital role in regulating metabolic diseases. However, the role of CK against the TP53 regulating pathway in metabolic diseases is unclear. We already reported that CK showed strong activity to regulate TP53 in the case of obesity and OP [126]. Thus, to better support the clinical use of CK, additional research is required to enhance the pertinent mechanisms of CK.
7. Synthesis of CK analogues and their pharmacological activity
Numerous biological actions of CK have been reported in the above sections. However, its use in medicine is limited by the poor bioavailability of CK after oral administration, which is thought to be a limiting factor. Researchers are concerned with increasing the intestinal absorption of CK by modifying the structure. In the early stages of treating asthma, CK was found to exhibit strong action against IgE. In 2019, Ren et al [127] reported couples of CK analogues were synthesized via straight forwarded methods. The produced compounds were assessed on the anti-IgE activities utilizing an in vivo airway hyper responsiveness experiment and an ovalbumin-induced asthmatic mouse model. They found that compounds T1, T2, T3, T8, and T12 (the analogues of CK) showed either superior or comparable anti-asthmatic effects compared to CK. Furthermore, Huang et al. [128] reported six derivatives of CK, among them structures 1 and 2 were highly potent to activate the LXRα (Liver X Receptor α) expression and showed lower toxicity than CK. They also demonstrated that structures 1, 2, and 4 enhanced the expression of ABCA1 (ATP-binding cassette transporter) mRNA levels. It has been documented that CK preferentially accumulates in the liver where it converts to fatty acid esters. Because the ester of CK was not eliminated by bile acid as CK was, it remained in the liver for a longer period. CK-octyl ester showed moderate detoxification and showed anti-liver cancer activity in murine-H22 cells and in vivo [129]. In addition, the novel ester prodrugs of CK (CK-butyl, and CK-octyl) have shown higher bioavailability due to their highly lipophilic properties than the CK. Even, the findings indicated that the permeability co-efficient of CK was lower than the esters [130]. These findings establish a groundwork for further alterations of CK and apply the new structures to metabolic diseases.
8. Discussion and future perspective
According to reviewed databases involving numerous studies, we summarized that CK is mostly associated with metabolic disorders, such as obesity, NAFLD, OP, diabetes mellitus, and its complications. This systemic review highlights the beneficial effects of CK against the four major metabolic diseases and the related pathways. It demonstrates the variety of ways in which CK can contribute to metabolic diseases such as enhancing IR, inhibiting glucose uptake, boosting glucose tolerance and insulin sensitivity, inhibiting bone resorption, increasing bone formation, triggering lipid synthesis, lipid uptake, boosting lipid oxidation, and blocking the inflammatory cytokines. In this study, we demonstrated that CK 1) enhances AMPK and inhibits adipogenic genes (C/EBPα, PPARγ, Ap2, leptin, and SREBP1c), lipogenic genes (FAS, FABP4, and SCD1). CK also inhibits the IKKs/ NF-kB pathway to trigger obesity mediated-inflammation. 2) Increases the expression of Glut4, PI3Kp85, InsR, IRS1, and pAkt to block the IR and increases GLUT2/PPCK/G6Phase pathway to reduce gluconeogenesis, inhibits ROS/INS and apoptosis to increase insulin sensitivity. 3) Inactivate GR that inhibits osteoclast differentiations, bone resorption and increases osteoblastic differentiation. Osteoclast production is inhibited and osteoblast development is promoted by the stimulation of the activity of genes associated with osteogenesis, such as Runx2, osterix and the suppression of the expression of genes related to osteoclasts, such as C-Fos and NFAC1. Meanwhile, CK is a novel agonist of the GR to treat obesity and OP. Furthermore, 4) ameliorates lipid accumulation by activating the AMPK/SIRT1 pathway in thev case of NAFLD. Research on CK in the future may focus on atherosclerosis, cardiovascular disease, fatty liver, hyperlipidemia, and other conditions.
Although numerous studies have established the toxicity of CK, our review revealed that the toxicity of CK is mostly dependent on the dosage and timing of administrations and the sex of subjects also affects the hepatotoxicity. Generally, doses of CK used to treat metabolic disorders in mice and rats are less than 100 mg/kg. However, doses up to 120 mg/kg in mice and rats can cause hepatotoxicity and nephrotoxicity [41]. Beagle dogs were administered 4, 12, and 36 mg/kg oral dosees for 26 weeks and did not exhibit any visible toxicity in the 4-12 mg/kg groups. 36mg/kg groups exhibited reduced body weight, enhancing plasma enzymes, and nephrotoxicity [46]. Studies showed that the dose of CK administrations did not exceed 100 mg/ml body weight ranging from for treating metabolic diseases. Additionally, in the cell lines treatments, the range of administration was 0.1-64 µg/ml showed more than 80% cell viability. Although, several cells showed cell cytotoxicity in different concentrations. Thus, it can be suggested that it is difficult for CK to cause bio-toxicity at normal doses. The development of bio-toxicity is more closely linked to an increase in CK dose than it is to an extension in administration time. As a result, the development and application of CK depend significantly on the management of doses administered.
It is well recognized that the potential for the treatment of CK is usually limited because of poor water solubility, and bioavailability and membrane permeability [131]. When it comes to medications that are poorly soluble in water, co-crystals can increase their bioavailability without altering their pharmacological action [132]. It is recommended that co-crystals of CK undergo research as an antimetabolite to increase oral bioavailability. Another recommendation is to change the chemical structure of CK or modify its dosage forms such that it dissolves better in intestinal fluids and has a higher oral bioavailability. Research on anti-metabolic disorders and other CK-related pharmacological effects should focus on comparing and examining the safety and pharmacological effects of injectable and oral CK metabolites in vivo. In addition, researchers are interested more in CK analogues due to their less cytotoxicity, more bioavailability, better membrane permeability, and higher efficacy compared to CK in various diseases. CK analogues could be drug candidate because of their physiochemical properties and pharmacological action.
To sum up, providing conceptual knowledge for the clinical use of CK as a potential anti-metabolic disease agent, the current study offers a thorough review and summary of the pharmacokinetics studies, toxicity, and physical and chemical characteristics of CK.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Methodology, M.N.M.; writing—original draft, M.N.M., R.A., & I.M.; review & editing—MRK & S.I. & M.J.I.; supervision, S.C.K & D.C.Y. All authors have read and agreed to the published version of the manuscript.
Funding
Not applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data are contained within the article.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Saklayen, M.G. The global epidemic of the metabolic syndrome. Current hypertension reports 2018, 20, 1–8. [Google Scholar] [CrossRef]
- Wu, Y.-L.; Lin, Z.-J.; Li, C.-C.; Lin, X.; Shan, S.-K.; Guo, B.; Zheng, M.-H.; Li, F.; Yuan, L.-Q.; Li, Z.-h. Epigenetic regulation in metabolic diseases: mechanisms and advances in clinical study. Signal Transduction and Targeted Therapy 2023, 8, 98. [Google Scholar] [CrossRef]
- Boulton, A. Strengthening the International Diabetes Federation (IDF). Diabetes Res Clin Pract 2020, 160, 108029. [Google Scholar] [CrossRef]
- Koenen, M.; Hill, M.A.; Cohen, P.; Sowers, J.R. Obesity, adipose tissue and vascular dysfunction. Circulation research 2021, 128, 951–968. [Google Scholar] [CrossRef]
- Singh, J.A.; Gaffo, A. Gout epidemiology and comorbidities. In Proceedings of the Seminars in arthritis and rheumatism, 2020; pp. S11-S16. [CrossRef]
- Kim, Y.-J.; Perumalsamy, H.; Markus, J.; Balusamy, S.R.; Wang, C.; Ho Kang, S.; Lee, S.; Park, S.Y.; Kim, S.; Castro-Aceituno, V. Development of Lactobacillus kimchicus DCY51T-mediated gold nanoparticles for delivery of ginsenoside compound K: in vitro photothermal effects and apoptosis detection in cancer cells. Artificial Cells, Nanomedicine, and Biotechnology 2019, 47, 30–44. [Google Scholar] [CrossRef]
- Morshed, M.N.; Ahn, J.C.; Mathiyalagan, R.; Rupa, E.J.; Akter, R.; Karim, M.R.; Jung, D.H.; Yang, D.U.; Yang, D.C.; Jung, S.K. Antioxidant Activity of Panax ginseng to Regulate ROS in Various Chronic Diseases. Applied Sciences 2023, 13, 2893. [Google Scholar] [CrossRef]
- Kang, S.; Siddiqi, M.H.; Yoon, S.J.; Ahn, S.; Noh, H.-Y.; Kumar, N.S.; Kim, Y.-J.; Yang, D.-C. Therapeutic potential of compound K as an IKK inhibitor with implications for osteoarthritis prevention: an in silico and in vitro study. In Vitro Cellular & Developmental Biology - Animal 2016, 52, 895–905. [Google Scholar] [CrossRef]
- Yang, W.; Zhou, J.; Gu, Q.; Harindintwali, J.D.; Yu, X.; Liu, X. Combinatorial Enzymatic Catalysis for Bioproduction of Ginsenoside Compound K. Journal of Agricultural and Food Chemistry 2023, 71, 3385–3397. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.L.; Hanh, N.T.Y.; Quyen, M.L.; Nguyen, Q.H.; Lien, T.T.P.; Do, K.V. Compound K Production: Achievements and Perspectives. Life 2023, 13, 1565. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.-D.; Yang, Y.-Y.; Ouyang, D.-S.; Yang, G.-P. A review of biotransformation and pharmacology of ginsenoside compound K. Fitoterapia 2015, 100, 208–220. [Google Scholar] [CrossRef] [PubMed]
- Han, G.C.; Ko, S.K.; Sung, J.H.; Chung, S.H. Compound K Enhances Insulin Secretion with Beneficial Metabolic Effects in db/db Mice. Journal of Agricultural and Food Chemistry 2007, 55, 10641–10648. [Google Scholar] [CrossRef]
- Xu, J.; Dong, J.; Ding, H.; Wang, B.; Wang, Y.; Qiu, Z.; Yao, F. Ginsenoside compound K inhibits obesity-induced insulin resistance by regulation of macrophage recruitment and polarization via activating PPARγ. Food Funct 2022, 13, 3561–3571. [Google Scholar] [CrossRef]
- Hwang, Y.C.; Oh, D.H.; Choi, M.C.; Lee, S.Y.; Ahn, K.J.; Chung, H.Y.; Lim, S.J.; Chung, S.H.; Jeong, I.K. Compound K attenuates glucose intolerance and hepatic steatosis through AMPK-dependent pathways in type 2 diabetic OLETF rats. Korean J Intern Med 2018, 33, 347–355. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nature Reviews Molecular Cell Biology 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nature Reviews Cardiology 2021, 18, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Day, E.A.; Ford, R.J.; Steinberg, G.R. AMPK as a Therapeutic Target for Treating Metabolic Diseases. Trends Endocrinol Metab 2017, 28, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Zaiou, M. Peroxisome Proliferator-Activated Receptor-γ as a Target and Regulator of Epigenetic Mechanisms in Nonalcoholic Fatty Liver Disease. Cells 2023, 12. [Google Scholar] [CrossRef]
- Park, D.; Yoon, M. Compound K, a novel ginsenoside metabolite, inhibits adipocyte differentiation in 3T3-L1 cells: Involvement of angiogenesis and MMPs. Biochemical and Biophysical Research Communications 2012, 422, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Lacroix, M.; Riscal, R.; Arena, G.; Linares, L.K.; Le Cam, L. Metabolic functions of the tumor suppressor p53: Implications in normal physiology, metabolic disorders, and cancer. Mol Metab 2020, 33, 2–22. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.E.; Lee, M.-H.; Jang, H.-M.; Im, W.-T.; Lee, J.; Kim, S.-H.; Jeon, G.J. A rare ginsenoside compound K (CK) induces apoptosis for breast cancer cells. Journal of Animal Reproduction and Biotechnology 2023, 38, 167–176. [Google Scholar] [CrossRef]
- Christensen, L.P. Ginsenosides: chemistry, biosynthesis, analysis, and potential health effects. Advances in food and nutrition research 2008, 55, 1–99. [Google Scholar] [CrossRef]
- Oh, J.-M.; Kim, E.; Chun, S. Ginsenoside compound K induces ros-mediated apoptosis and autophagic inhibition in human neuroblastoma cells in vitro and in vivo. International journal of molecular sciences 2019, 20, 4279. [Google Scholar] [CrossRef]
- Liu, T.; Zhu, L.; Wang, L. A narrative review of the pharmacology of ginsenoside compound K. Ann Transl Med 2022, 10, 234. [Google Scholar] [CrossRef]
- Xie, T.; Li, Z.; Li, B.; Sun, R.; Zhang, P.; Lv, J. Characterization of ginsenoside compound K metabolites in rat urine and feces by ultra-performance liquid chromatography with electrospray ionization quadrupole time-of-flight tandem mass spectrometry. Biomedical Chromatography 2019, 33, e4643. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, L.; Wang, Y.; Yang, G.; Huang, J.; Tan, Z.; Wang, Y.; Zhou, G.; Liao, J.; Ouyang, D. Food and sex-related impacts on the pharmacokinetics of a single-dose of ginsenoside compound K in healthy subjects. Frontiers in Pharmacology 2017, 8, 636. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-K. Pharmacokinetics of ginsenoside Rb1 and its metabolite compound K after oral administration of Korean Red Ginseng extract. Journal of ginseng research 2013, 37, 451. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Zhou, L.; Huang, J.; Wang, Y.; Yang, G.; Tan, Z.; Wang, Y.; Zhou, G.; Liao, J.; Ouyang, D. Single-and multiple-dose trials to determine the pharmacokinetics, safety, tolerability, and sex effect of oral ginsenoside compound K in healthy Chinese volunteers. Frontiers in Pharmacology 2018, 8, 965. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.H.; Kang, J.-W.; Song, Y.-S.; Kim, J.-H.; Kim, M.S.; Bak, Y.; Oh, D.-K.; Yoon, D.-Y. Compound K attenuates lipid accumulation through down-regulation of peroxisome proliferator-activated receptor γ in 3T3-L1 cells. Journal of the Korean Society for Applied Biological Chemistry 2013, 56, 141–147. [Google Scholar] [CrossRef]
- Fang, C.; Yang, H.; Zhu, C.; Fan, D.; Deng, J. Ginsenoside CK inhibits androgenetic alopecia by regulating Wnt/β-catenin and p53 signaling pathways in AGA mice. Food Frontiers 2023. [Google Scholar] [CrossRef]
- Kang, S.; Siddiqi, M.H.; Yoon, S.J.; Ahn, S.; Noh, H.-Y.; Kumar, N.S.; Kim, Y.-J.; Yang, D.-C. Therapeutic potential of compound K as an IKK inhibitor with implications for osteoarthritis prevention: an in silico and in vitro study. In Vitro Cellular & Developmental Biology-Animal 2016, 52, 895–905. [Google Scholar] [CrossRef]
- Zhang, J.; Ma, X.; Fan, D. Ginsenoside CK ameliorates hepatic lipid accumulation via activating the LKB1/AMPK pathway in vitro and in vivo. Food & Function 2022, 13, 1153–1167. [Google Scholar] [CrossRef]
- Kim, D.Y.; Yuan, H.D.; Chung, I.K.; Chung, S.H. Compound K, Intestinal Metabolite of Ginsenoside, Attenuates Hepatic Lipid Accumulation via AMPK Activation in Human Hepatoma Cells. Journal of Agricultural and Food Chemistry 2009, 57, 1532–1537. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, H.; Yang, Q.; Lan, X.; Wang, J.; Cao, Z.; Shi, X.; Li, J.; Kan, M.; Qu, X. Ginsenoside compound K ameliorates Alzheimer’s disease in HT22 cells by adjusting energy metabolism. Molecular Biology Reports 2019, 46, 5323–5332. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Zeng, X.; Rao, T.; Tan, Z.; Zhou, G.; Ouyang, D.; Chen, L. Evaluating the protective effects of individual or combined ginsenoside compound K and the downregulation of soluble epoxide hydrolase expression against sodium valproate-induced liver cell damage. Toxicology and Applied Pharmacology 2021, 422, 115555. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Kang, E.H.; Lee, E.Y.; Gong, H.S.; Kang, H.S.; Shin, K.; Lee, E.B.; Song, Y.W.; Lee, Y.J. Joint-protective effects of compound K, a major ginsenoside metabolite, in rheumatoid arthritis: in vitro evidence. Rheumatology International 2013, 33, 1981–1990. [Google Scholar] [CrossRef]
- Gu, J.; Li, W.; Xiao, D.; Wei, S.; Cui, W.; Chen, W.; Hu, Y.; Bi, X.; Kim, Y.; Li, J.; et al. Compound K, a final intestinal metabolite of ginsenosides, enhances insulin secretion in MIN6 pancreatic β-cells by upregulation of GLUT2. Fitoterapia 2013, 87, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Law, C.K.-M.; Kwok, H.-H.; Poon, P.-Y.; Lau, C.-C.; Jiang, Z.-H.; Tai, W.C.-S.; Hsiao, W.W.-L.; Mak, N.-K.; Yue, P.Y.-K.; Wong, R.N.-S. Ginsenoside compound K induces apoptosis in nasopharyngeal carcinoma cells via activation of apoptosis-inducing factor. Chinese medicine 2014, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Boopathi, V.; Nahar, J.; Murugesan, M.; Subramaniyam, S.; Kong, B.M.; Choi, S.-K.; Lee, C.-S.; Ling, L.; Yang, D.U.; Yang, D.C. In silico and in vitro inhibition of host-based viral entry targets and cytokine storm in COVID-19 by ginsenoside compound K. Heliyon 2023, 9. [Google Scholar] [CrossRef]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Zheng, J.; Yao, C.W.; Cha, J.W.; Kim, H.S.; Kim, D.H.; Bae, S.C.; Hyun, J.W. Compound K, a metabolite of ginseng saponin, inhibits colorectal cancer cell growth and induces apoptosis through inhibition of histone deacetylase activity. International Journal of Oncology 2013, 43, 1907–1914. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, T.; Wang, G.; Li, G.; Sun, C.; Jiang, Z.; Yang, J.; Li, Y.; You, Y.; Wu, X.; et al. Preclinical safety of ginsenoside compound K: Acute, and 26-week oral toxicity studies in mice and rats. Food and Chemical Toxicology 2019, 131, 110578. [Google Scholar] [CrossRef]
- Li, W.; Zhang, X.; Ding, M.; Xin, Y.; Xuan, Y.; Zhao, Y. Genotoxicity and subchronic toxicological study of a novel ginsenoside derivative 25-OCH3-PPD in beagle dogs. Journal of Ginseng Research 2019, 43, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Siddiqi, M.H.; Yoon, S.J.; Ahn, S.; Noh, H.Y.; Kumar, N.S.; Kim, Y.J.; Yang, D.C. Therapeutic potential of compound K as an IKK inhibitor with implications for osteoarthritis prevention: an in silico and in vitro study. In Vitro Cell Dev Biol Anim 2016, 52, 895–905. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-J.; Ko, W.-G.; Kim, J.-H.; Sung, J.-H.; Lee, S.-J.; Moon, C.-K.; Lee, B.-H. Induction of apoptosis by a novel intestinal metabolite of ginseng saponin via cytochrome c-mediated activation of caspase-3 protease. Biochemical Pharmacology 2000, 60, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Lim, H.K.; Kim, S.U.; Kim, Y.W.; Kim, W.T.; Chung, H.S.; Choo, M.K.; Kim, D.H.; Kim, H.S.; Shim, M.J. Induction of apoptosis by ginseng saponin metabolite in U937 human monocytic leukemia cells. Journal of food biochemistry 2005, 29, 27–40. [Google Scholar] [CrossRef]
- Li, C.; Wang, Z.; Wang, T.; Wang, G.; Li, G.; Sun, C.; Lin, J.; Sun, L.; Sun, X.; Cho, S. Repeated-dose 26-week oral toxicity study of ginsenoside compound K in Beagle dogs. Journal of ethnopharmacology 2020, 248, 112323. [Google Scholar] [CrossRef]
- Keum, Y.-S.; Park, K.-K.; Lee, J.-M.; Chun, K.-S.; Park, J.H.; Lee, S.K.; Kwon, H.; Surh, Y.-J. Antioxidant and anti-tumor promoting activities of the methanol extract of heat-processed ginseng. Cancer Letters 2000, 150, 41–48. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, J.; Li, J.; Fu, L.; Gao, J.; Du, X.; Bi, H.; Zhou, Y.; Tai, G. Highly selective biotransformation of ginsenoside Rb1 to Rd by the phytopathogenic fungus Cladosporium fulvum (syn. Fulvia fulva). Journal of Industrial Microbiology and Biotechnology 2009, 36, 721–726. [Google Scholar] [CrossRef]
- Yan, Q.; Zhou, X.-W.; Zhou, W.; Li, X.-W.; Feng, M.-Q.; Zhou, P. Purification and properties of a novel ${\beta} $-glucosidase, hydrolyzing ginsenoside Rb1 to CK, from Paecilomyces Bainier. Journal of Microbiology and Biotechnology 2008, 18, 1081–1089. [Google Scholar]
- An, D.-S.; Cui, C.-H.; Lee, H.-G.; Wang, L.; Kim, S.C.; Lee, S.-T.; Jin, F.; Yu, H.; Chin, Y.-W.; Lee, H.-K. Identification and characterization of a novel Terrabacter ginsenosidimutans sp. nov. β-glucosidase that transforms ginsenoside Rb1 into the rare gypenosides XVII and LXXV. Applied and environmental microbiology 2010, 76, 5827–5836. [Google Scholar] [CrossRef]
- Zhong, F.-L.; Dong, W.-W.; Wu, S.; Jiang, J.; Yang, D.-C.; Li, D.; Quan, L.-H. Biotransformation of gypenoside XVII to compound K by a recombinant β-glucosidase. Biotechnology letters 2016, 38, 1187–1193. [Google Scholar] [CrossRef]
- Noh, K.-H.; Son, J.-W.; Kim, H.-J.; Oh, D.-K. Ginsenoside compound K production from ginseng root extract by a thermostable β-glycosidase from Sulfolobus solfataricus. Bioscience, biotechnology, and biochemistry 2009, 73, 316–321. [Google Scholar] [CrossRef]
- Yoo, M.-H.; Yeom, S.-J.; Park, C.-S.; Lee, K.-W.; Oh, D.-K. Production of aglycon protopanaxadiol via compound K by a thermostable β-glycosidase from Pyrococcus furiosu s. Applied microbiology and biotechnology 2011, 89, 1019–1028. [Google Scholar] [CrossRef]
- Quan, L.H.; Jin, Y.; Wang, C.; Min, J.W.; Kim, Y.J.; Yang, D.C. Enzymatic transformation of the major ginsenoside Rb2 to minor compound Y and compound K by a ginsenoside-hydrolyzing β-glycosidase from Microbacterium esteraromaticum. J Ind Microbiol Biotechnol 2012, 39, 1557–1562. [Google Scholar] [CrossRef]
- Cao, S.; Yang, F.; Tian, F.; Liu, X.; Fan, D.; Wu, Z. Immobilized β-glucosidase on Cu(PTA) for the green production of rare ginsenosides CK. Process Biochemistry 2023, 133, 169–178. [Google Scholar] [CrossRef]
- Li, R.; Liu, X.; Li, X.; Tian, D.; Fan, D.; Ma, X.; Wu, Z. Co-immobilized β-glucosidase and snailase in green synthesized Zn-BTC for ginsenoside CK biocatalysis. Biochemical Engineering Journal 2022, 188, 108677. [Google Scholar] [CrossRef]
- Cao, S.; Li, R.; Tian, F.; Liu, X.; Fan, D.; Wu, Z. Construction of a hollow MOF with high sedimentation performance and co-immobilization of multiple-enzymes for preparing rare ginsenoside CK. Reaction Chemistry & Engineering 2023, 8, 2804–2817. [Google Scholar] [CrossRef]
- Tran, T.N.A.; Son, J.-S.; Awais, M.; Ko, J.-H.; Yang, D.C.; Jung, S.-K. β-Glucosidase and Its Application in Bioconversion of Ginsenosides in Panax ginseng. Bioengineering 2023, 10, 484. [Google Scholar] [CrossRef]
- Duan, Z.; Zhu, C.; Shi, J.; Fan, D.; Deng, J.; Fu, R.; Huang, R.; Fan, C. High efficiency production of ginsenoside compound K by catalyzing ginsenoside Rb1 using snailase. Chinese Journal of Chemical Engineering 2018, 26, 1591–1597. [Google Scholar] [CrossRef]
- Choi, J.-H.; Shin, K.-C.; Oh, D.-K. An L213A variant of β-glycosidase from Sulfolobus solfataricus with increased α-L-arabinofuranosidase activity converts ginsenoside Rc to compound K. PLoS One 2018, 13, e0191018. [Google Scholar] [CrossRef]
- Park, C.-S.; Yoo, M.-H.; Noh, K.-H.; Oh, D.-K. Biotransformation of ginsenosides by hydrolyzing the sugar moieties of ginsenosides using microbial glycosidases. Applied microbiology and biotechnology 2010, 87, 9–19. [Google Scholar] [CrossRef]
- Quan, L.-H.; Jin, Y.; Wang, C.; Min, J.-W.; Kim, Y.-J.; Yang, D.-C. Enzymatic transformation of the major ginsenoside Rb2 to minor compound Y and compound K by a ginsenoside-hydrolyzing β-glycosidase from Microbacterium esteraromaticum. Journal of Industrial Microbiology and Biotechnology 2012, 39, 1557–1562. [Google Scholar] [CrossRef]
- Cui, L.; Wu, S.-q.; Zhao, C.-a.; Yin, C.-r. Microbial conversion of major ginsenosides in ginseng total saponins by Platycodon grandiflorum endophytes. Journal of Ginseng Research 2016, 40, 366–374. [Google Scholar] [CrossRef]
- Han, Y.; Sun, B.; Hu, X.; Zhang, H.; Jiang, B.; Spranger, M.I.; Zhao, Y. Transformation of bioactive compounds by Fusarium sacchari fungus isolated from the soil-cultivated ginseng. Journal of agricultural and food chemistry 2007, 55, 9373–9379. [Google Scholar] [CrossRef]
- Quan, L.-H.; Kim, Y.-J.; Li, G.H.; Choi, K.-T.; Yang, D.-C. Microbial transformation of ginsenoside Rb1 to compound K by Lactobacillus paralimentarius. World Journal of Microbiology and Biotechnology 2013, 29, 1001–1007. [Google Scholar] [CrossRef]
- Cheng, L.-Q.; Kim, M.K.; Lee, J.-W.; Lee, Y.-J.; Yang, D.-C. Conversion of major ginsenoside Rb 1 to ginsenoside F 2 by Caulobacter leidyia. Biotechnology letters 2006, 28, 1121–1127. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Sung, J.-H.; Matsumiya, S.; Uchiyama, M. Main ginseng saponin metabolites formed by intestinal bacteria. Planta medica 1996, 62, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-A.; Jung, I.-H.; Park, S.-H.; Ahn, Y.-T.; Huh, C.-S.; Kim, D.-H. Comparative analysis of the gut microbiota in people with different levels of ginsenoside Rb1 degradation to compound K. PloS one 2013, 8, e62409. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Feng, M.-Q.; Li, J.-Y.; Zhou, P. Studies on the preparation, crystal structure and bioactivity of ginsenoside compound K. 2006. [CrossRef]
- Zhou, W.; Yan, Q.; Li, J.Y.; Zhang, X.C.; Zhou, P. Biotransformation of Panax notoginseng saponins into ginsenoside compound K production by Paecilomyces bainier sp. 229. Journal of applied microbiology 2008, 104, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Wang, Y.; Yan, M.; Sun, C.; Zheng, P. Screening of plant pathogenic fungi by ginsenoside compound K production. Zhongguo Zhong yao za zhi= Zhongguo Zhongyao Zazhi= China Journal of Chinese Materia Medica 2011, 36, 1596–1598. [Google Scholar] [PubMed]
- Chen, G.-T.; Yang, M.; Song, Y.; Lu, Z.-Q.; Zhang, J.-Q.; Huang, H.-L.; Wu, L.-J.; Guo, D.-A. Microbial transformation of ginsenoside Rb 1 by Acremonium strictum. Applied microbiology and biotechnology 2008, 77, 1345–1350. [Google Scholar] [CrossRef] [PubMed]
- Chi, H.; Ji, G.-E. Transformation of ginsenosides Rb1 and Re from Panax ginseng by food microorganisms. Biotechnology letters 2005, 27, 765–771. [Google Scholar] [CrossRef]
- Park, J.K.; Yang, D.U.; Arunkumar, L.; Han, Y.; Lee, S.J.; Arif, M.H.; Li, J.F.; Huo, Y.; Kang, J.P.; Hoang, V.A.; et al. Cumulative Production of Bioactive Rg3, Rg5, Rk1, and CK from Fermented Black Ginseng Using Novel Aspergillus niger KHNT-1 Strain Isolated from Korean Traditional Food. Processes 2021, 9, 227. [Google Scholar] [CrossRef]
- Quan, L.-H.; Piao, J.-Y.; Min, J.-W.; Yang, D.-U.; Lee, H.N.; Yang, D.C. Bioconversion of ginsenoside Rb1 into compound K by Leuconostoc citreum LH1 isolated from kimchi. Brazilian Journal of Microbiology 2011, 42, 1227–1237. [Google Scholar] [CrossRef]
- Yan, X.; Fan, Y.; Wei, W.; Wang, P.; Liu, Q.; Wei, Y.; Zhang, L.; Zhao, G.; Yue, J.; Zhou, Z. Production of bioactive ginsenoside compound K in metabolically engineered yeast. Cell research 2014, 24, 770–773. [Google Scholar] [CrossRef]
- Yang, S.; Liu, T.; Hu, C.; Li, W.; Meng, Y.; Li, H.; Song, C.; He, C.; Zhou, Y.; Fan, Y. Ginsenoside compound K protects against obesity through pharmacological targeting of glucocorticoid receptor to activate lipophagy and lipid metabolism. Pharmaceutics 2022, 14, 1192. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Dong, J.; Xu, J.; Qiu, Z.; Yao, F. Ginsenoside CK inhibits obese insulin resistance by activating PPARγ to interfere with macrophage activation. Microbial Pathogenesis 2021, 157, 105002. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.-M.; Chun, S. Ginsenoside CK Inhibits the Early Stage of Adipogenesis via the AMPK, MAPK, and AKT Signaling Pathways. Antioxidants 2022, 11, 1890. [Google Scholar] [CrossRef]
- Li, W.; Zhang, M.; Gu, J.; Meng, Z.-j.; Zhao, L.-C.; Zheng, Y.-n.; Chen, L.; Yang, G.-L. Hypoglycemic effect of protopanaxadiol-type ginsenosides and compound K on Type 2 diabetes mice induced by high-fat diet combining with streptozotocin via suppression of hepatic gluconeogenesis. Fitoterapia 2012, 83, 192–198. [Google Scholar] [CrossRef]
- Jiang, S.; Ren, D.; Li, J.; Yuan, G.; Li, H.; Xu, G.; Han, X.; Du, P.; An, L. Effects of compound K on hyperglycemia and insulin resistance in rats with type 2 diabetes mellitus. Fitoterapia 2014, 95, 58–64. [Google Scholar] [CrossRef]
- Yoon, S.H.; Han, E.J.; Sung, J.H.; Chung, S.H. Anti-diabetic effects of compound K versus metformin versus compound K-metformin combination therapy in diabetic db/db mice. Biological and Pharmaceutical Bulletin 2007, 30, 2196–2200. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Wei, L.; Du, Y.; Wang, Y.; Jiang, S. Protective effect of ginsenoside metabolite compound K against diabetic nephropathy by inhibiting NLRP3 inflammasome activation and NF-κB/p38 signaling pathway in high-fat diet/streptozotocin-induced diabetic mice. International Immunopharmacology 2018, 63, 227–238. [Google Scholar] [CrossRef]
- Cho, W.; Oh, H.; Abd El-Aty, A.; Hacimuftuoglu, A.; Jeong, J.H.; Jung, T.W. Therapeutic potential of ginsenoside compound K in managing tenocyte apoptosis and extracellular matrix damage in diabetic tendinopathy. Tissue and Cell 2024, 86, 102275. [Google Scholar] [CrossRef]
- Ding, L.; Gao, Z.; Wu, S.; Chen, C.; Liu, Y.; Wang, M.; Zhang, Y.; Li, L.; Zou, H.; Zhao, G. Ginsenoside compound-K attenuates OVX-induced osteoporosis via the suppression of RANKL-induced osteoclastogenesis and oxidative stress. Natural Products and Bioprospecting 2023, 13, 49. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Gu, S.; Zhou, B.; Wang, M.; Zhang, Y.; Wu, S.; Zou, H.; Zhao, G.; Gao, Z.; Xu, L. Ginsenoside compound K enhances fracture healing via promoting osteogenesis and angiogenesis. Frontiers in Pharmacology 2022, 13, 855393. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-J.; Liu, W.-J.; Wen, M.-L.; Liang, H.; Wu, S.-M.; Zhu, Y.-Z.; Zhao, J.-Y.; Dong, X.-Q.; Li, M.-G.; Bian, L.; et al. Ameliorative effects of Compound K and ginsenoside Rh1 on non-alcoholic fatty liver disease in rats. Scientific Reports 2017, 7, 41144. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.S.; Lee, K.T.; Iseli, T.J.; Hoy, A.J.; George, J.; Grewal, T.; Roufogalis, B.D. Compound K modulates fatty acid-induced lipid droplet formation and expression of proteins involved in lipid metabolism in hepatocytes. Liver International 2013, 33, 1583–1593. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Han, H.; Shi, H.; Wang, T.; Wang, B.; Zhao, J. Compound K, a metabolite of ginseng saponin, induces apoptosis of hepatocellular carcinoma cells through the mitochondria-mediated caspase-dependent pathway. Int. J. Clin. Exp. Med 2017, 10, 11146–11156. [Google Scholar]
- Awais, M.; Akter, R.; Boopathi, V.; Ahn, J.C.; Lee, J.H.; Mathiyalagan, R.; Kwak, G.-Y.; Rauf, M.; Yang, D.C.; Lee, G.S. Discrimination of Dendropanax morbifera via HPLC fingerprinting and SNP analysis and its impact on obesity by modulating adipogenesis-and thermogenesis-related genes. Frontiers in Nutrition 2023, 10. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, G.; Pandeya, P.R.; Lamichhane, R.; Rhee, S.-j.; Devkota, H.P.; Jung, H.-J. Anti-obesity potential of ponciri fructus: effects of extracts, fractions and compounds on adipogenesis in 3T3-L1 preadipocytes. Molecules 2022, 27, 676. [Google Scholar] [CrossRef]
- Su, X.; Peng, D. The exchangeable apolipoproteins in lipid metabolism and obesity. Clinica Chimica Acta 2020, 503, 128–135. [Google Scholar] [CrossRef]
- De Sereday, M.S.; Gonzalez, C.; Giorgini, D.; De Loredo, L.; Braguinsky, J.; Cobeñas, C.; Libman, C.; Tesone, C. Prevalence of diabetes, obesity, hypertension and hyperlipidemia in the central area of Argentina. Diabetes & Metabolism 2004, 30, 335–339. [Google Scholar] [CrossRef]
- Song, T.; Yang, Y.; Zhou, Y.; Wei, H.; Peng, J. GPR120: a critical role in adipogenesis, inflammation, and energy metabolism in adipose tissue. Cellular and Molecular Life Sciences 2017, 74, 2723–2733. [Google Scholar] [CrossRef] [PubMed]
- Kuryłowicz, A.; Puzianowska-Kuźnicka, M. Induction of Adipose Tissue Browning as a Strategy to Combat Obesity. International Journal of Molecular Sciences 2020, 21, 6241. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Dong, J.; Ding, H.; Wang, B.; Wang, Y.; Qiu, Z.; Yao, F. Ginsenoside compound K inhibits obesity-induced insulin resistance by regulation of macrophage recruitment and polarization via activating PPARγ. Food & Function 2022, 13, 3561–3571. [Google Scholar] [CrossRef]
- Taherkhani, S.; Suzuki, K.; Ruhee, R.T. A brief overview of oxidative stress in adipose tissue with a therapeutic approach to taking antioxidant supplements. Antioxidants 2021, 10, 594. [Google Scholar] [CrossRef]
- Shahzad, N.; Alzahrani, A.R.; Ibrahim, I.A.A.; Shahid, I.; Alanazi, I.M.; Falemban, A.H.; Imam, M.T.; Mohsin, N.; Azlina, M.F.N.; Arulselvan, P. Therapeutic strategy of biological macromolecules based natural bioactive compounds of diabetes mellitus and future perspectives: A systemic review. Heliyon 2024. [Google Scholar] [CrossRef]
- Wei, S.; Li, W.; Yu, Y.; Yao, F.; Lixiang, A.; Lan, X.; Guan, F.; Zhang, M.; Chen, L. Ginsenoside Compound K suppresses the hepatic gluconeogenesis via activating adenosine-5′ monophosphate kinase: A study in vitro and in vivo. Life sciences 2015, 139, 8–15. [Google Scholar] [CrossRef]
- Riley, G. Chronic tendon pathology: molecular basis and therapeutic implications. Expert reviews in molecular medicine 2005, 7, 1–25. [Google Scholar] [CrossRef]
- Weitzmann, M.N.; Ofotokun, I. Physiological and pathophysiological bone turnover—role of the immune system. Nature Reviews Endocrinology 2016, 12, 518–532. [Google Scholar] [CrossRef] [PubMed]
- Fischer, V.; Haffner-Luntzer, M. Interaction between bone and immune cells: Implications for postmenopausal osteoporosis. In Proceedings of the Seminars in cell & developmental biology; 2022; pp. 14–21. [Google Scholar] [CrossRef]
- Pavone, V.; Testa, G.; Giardina, S.M.C.; Vescio, A.; Restivo, D.A.; Sessa, G. Pharmacological Therapy of Osteoporosis: A Systematic Current Review of Literature. Frontiers in Pharmacology 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.; Xie, X.; Yang, Y.; Li, F. Ginsenoside compound K- a potential drug for rheumatoid arthritis. Pharmacological Research 2021, 166, 105498. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Kang, E.H.; Lee, E.Y.; Gong, H.S.; Kang, H.S.; Shin, K.; Lee, E.B.; Song, Y.W.; Lee, Y.J. Joint-protective effects of compound K, a major ginsenoside metabolite, in rheumatoid arthritis: in vitro evidence. Rheumatology International 2013, 33, 1981–1990. [Google Scholar] [CrossRef] [PubMed]
- Clark, J.M.; Brancati, F.L.; Diehl, A.M. Nonalcoholic fatty liver disease. Gastroenterology 2002, 122, 1649–1657. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.; Wang, G.; Chen, M.; Li, Y.; Tang, X.; Dai, Y. Therapeutic potential of alkaloid extract from Codonopsis Radix in alleviating hepatic lipid accumulation: insights into mitochondrial energy metabolism and endoplasmic reticulum stress regulation in NAFLD mice. Chinese Journal of Natural Medicines 2023, 21, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Zhan, Y.-T.; Su, H.-Y.; An, W. Glycosyltransferases and non-alcoholic fatty liver disease. World Journal of Gastroenterology 2016, 22, 2483. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology 2010, 52, 1836–1846. [Google Scholar] [CrossRef]
- Kumar, S.; Wong, R.; Newberry, C.; Yeung, M.; Peña, J.M.; Sharaiha, R.Z. Multidisciplinary Clinic Models: A Paradigm of Care for Management of NAFLD. Hepatology 2021, 74. [Google Scholar] [CrossRef]
- Wang, L.; Yan, Y.; Wu, L.; Peng, J. Natural products in non-alcoholic fatty liver disease (NAFLD): Novel lead discovery for drug development. Pharmacological Research 2023, 196, 106925. [Google Scholar] [CrossRef]
- Yue, C.; Li, D.; Fan, S.; Tao, F.; Yu, Y.; Lu, W.; Chen, Q.; Yuan, A.; Wu, J.; Zhao, G. Long-term and liver-selected ginsenoside C–K nanoparticles retard NAFLD progression by restoring lipid homeostasis. Biomaterials 2023, 301, 122291. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chen, L.; Zeng, X.; Liao, J.; Ouyang, D. Ginsenoside compound K alleviates sodium valproate-induced hepatotoxicity in rats via antioxidant effect, regulation of peroxisome pathway and iron homeostasis. Toxicology and Applied Pharmacology 2020, 386, 114829. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: growing ever more complex. Nature reviews cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Kung, C.P.; Murphy, M.E. The role of the p53 tumor suppressor in metabolism and diabetes. J Endocrinol 2016, 231, R61–r75. [Google Scholar] [CrossRef]
- Minamino, T.; Orimo, M.; Shimizu, I.; Kunieda, T.; Yokoyama, M.; Ito, T.; Nojima, A.; Nabetani, A.; Oike, Y.; Matsubara, H. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nature medicine 2009, 15, 1082–1087. [Google Scholar] [CrossRef]
- Kim, W.H.; Lee, J.W.; Gao, B.; Jung, M.H. Synergistic activation of JNK/SAPK induced by TNF-α and IFN-γ: apoptosis of pancreatic β-cells via the p53 and ROS pathway. Cellular signalling 2005, 17, 1516–1532. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhang, X.; Huang, X.; Lu, Y.; Tang, W.; Man, Y.; Wang, S.; Xi, J.; Li, J. NADPH oxidase 2-derived reactive oxygen species mediate FFAs-induced dysfunction and apoptosis of β-cells via JNK, p38 MAPK and p53 pathways. PloS one 2010, 5, e15726. [Google Scholar] [CrossRef] [PubMed]
- Zwezdaryk, K.; Sullivan, D.; Saifudeen, Z. The p53/Adipose-Tissue/Cancer Nexus. Front Endocrinol (Lausanne) 2018, 9, 457. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.; Shackel, N.; Gorrell, M.; McLennan, S.; Twigg, S. Diabetes and nonalcoholic fatty liver disease: a pathogenic duo. Endocrine reviews 2013, 34, 84–129. [Google Scholar] [CrossRef] [PubMed]
- Yahagi, N.; Shimano, H.; Matsuzaka, T.; Sekiya, M.; Najima, Y.; Okazaki, S.; Okazaki, H.; Tamura, Y.; Iizuka, Y.; Inoue, N. p53 involvement in the pathogenesis of fatty liver disease. Journal of Biological Chemistry 2004, 279, 20571–20575. [Google Scholar] [CrossRef] [PubMed]
- Derdak, Z.; Lang, C.H.; Villegas, K.A.; Tong, M.; Mark, N.M.; de la Monte, S.M.; Wands, J.R. Activation of p53 enhances apoptosis and insulin resistance in a rat model of alcoholic liver disease. Journal of hepatology 2011, 54, 164–172. [Google Scholar] [CrossRef]
- Derdak, Z.; Villegas, K.A.; Harb, R.; Wu, A.M.; Sousa, A.; Wands, J.R. Inhibition of p53 attenuates steatosis and liver injury in a mouse model of non-alcoholic fatty liver disease. Journal of hepatology 2013, 58, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Suzuki, T.; Oshikawa, T.; Yokoyama, H.; Shimamura, K.; Nishiyama, K.; Mataki, N.; Irie, R.; Minamino, T. p53/p66Shc-mediated signaling contributes to the progression of non-alcoholic steatohepatitis in humans and mice. Journal of hepatology 2012, 57, 837–843. [Google Scholar] [CrossRef]
- Panasiuk, A.; Dzieciol, J.; Panasiuk, B.; Prokopowicz, D. Expression of p53, Bax and Bcl-2 proteins in hepatocytes in non-alcoholic fatty liver disease. World journal of gastroenterology: WJG 2006, 12, 6198. [Google Scholar] [CrossRef]
- Kang, S.C. ALLEY PROOF. Med Sci Monit 2024, 30, e942899. [Google Scholar]
- Ren, S.; Liu, R.; Wang, Y.; Ding, N.; Li, Y. Synthesis and biological evaluation of Ginsenoside Compound K analogues as a novel class of anti-asthmatic agents. Bioorganic & Medicinal Chemistry Letters 2019, 29, 51–55. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, H.; Zhang, Y.; Li, J.; Wang, C.; Zhou, L.; Jia, Y.; Li, X. Synthesis and Biological Evaluation of Ginsenoside Compound K Derivatives as a Novel Class of LXRα Activator. Molecules 2017, 22, 1232. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Xue, J.; Zhao, X.; Wang, Z.; Li, W.; Li, X.; Zheng, Y. Octyl ester of ginsenoside compound K as novel anti-hepatoma compound: Synthesis and evaluation on murine H22 cells in vitro and in vivo. Chemical Biology & Drug Design 2018, 91, 951–956. [Google Scholar] [CrossRef]
- Zhang, B.; Zhu, X.-M.; Hu, J.-N.; Ye, H.; Luo, T.; Liu, X.-R.; Li, H.-Y.; Li, W.; Zheng, Y.-N.; Deng, Z.-Y. Absorption mechanism of ginsenoside compound K and its butyl and octyl ester prodrugs in Caco-2 cells. Journal of agricultural and food chemistry 2012, 60, 10278–10284. [Google Scholar] [CrossRef]
- Murugesan, M.; Mathiyalagan, R.; Boopathi, V.; Kong, B.M.; Choi, S.-K.; Lee, C.-S.; Yang, D.C.; Kang, S.C.; Thambi, T. Production of Minor Ginsenoside CK from Major Ginsenosides by Biotransformation and Its Advances in Targeted Delivery to Tumor Tissues Using Nanoformulations. Nanomaterials 2022, 12, 3427. [Google Scholar] [CrossRef] [PubMed]
- Emami, S.; Siahi-Shadbad, M.; Adibkia, K.; Barzegar-Jalali, M. Recent advances in improving oral drug bioavailability by cocrystals. Bioimpacts 2018, 8, 305–320. [Google Scholar] [CrossRef]
Figure 1.
Biotransformation of major ginsenosides to CK via different pathways using Enzymatic and microbial conversion methods.
Figure 1.
Biotransformation of major ginsenosides to CK via different pathways using Enzymatic and microbial conversion methods.
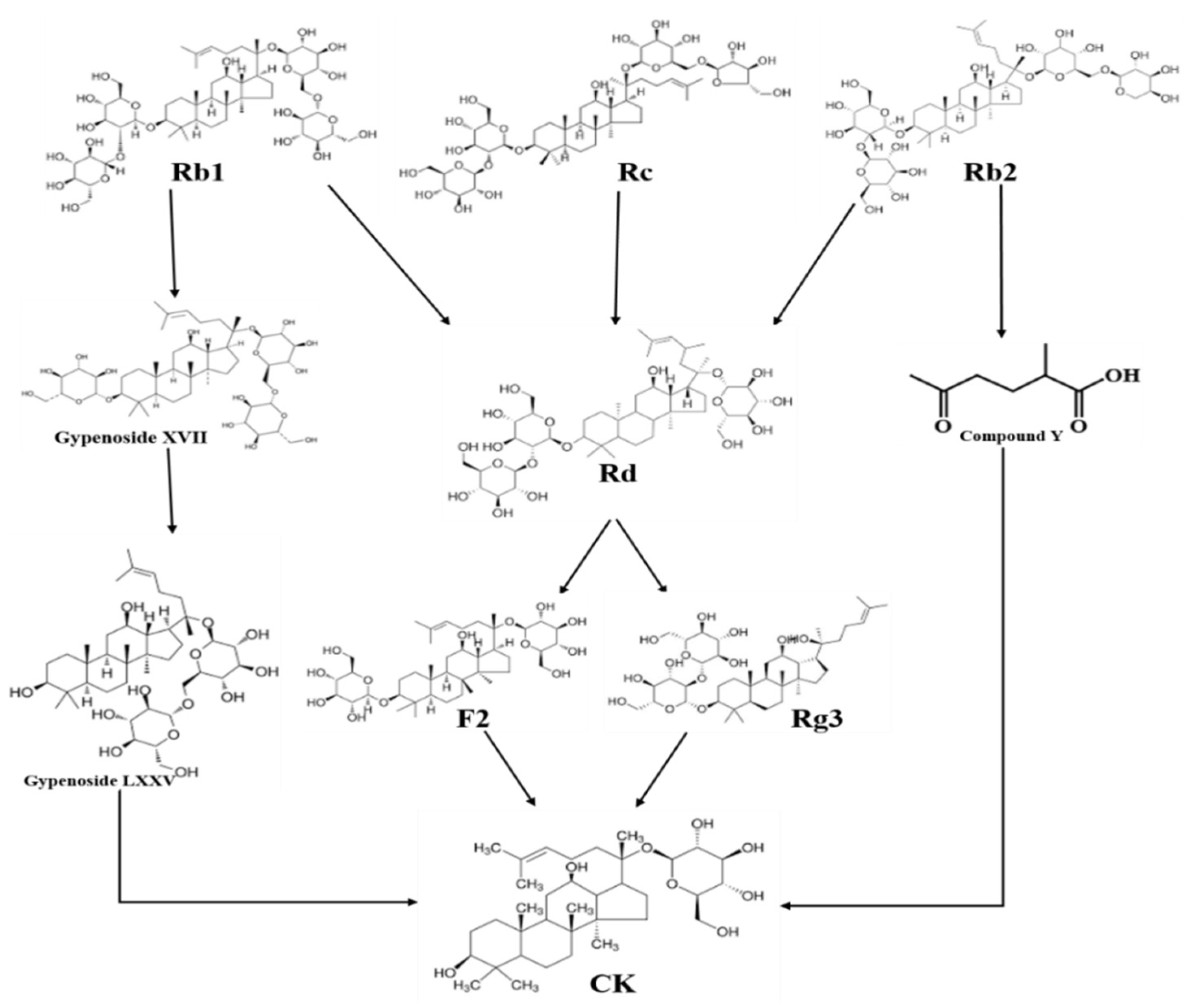
Figure 2.
The pathway involves in obesity. CK inhibits adipogenesis via AMPK/PPARγ/CEBPα, and lipogenesis via AMPK/ACC/FAS pathway. Additionally, CK increases IRS1/PI3K/AKT expression against obesity. Furthermore, CK triggers TLR4/TRAF6/TAK1/NF-κB to minimize adipose tissue.
Figure 2.
The pathway involves in obesity. CK inhibits adipogenesis via AMPK/PPARγ/CEBPα, and lipogenesis via AMPK/ACC/FAS pathway. Additionally, CK increases IRS1/PI3K/AKT expression against obesity. Furthermore, CK triggers TLR4/TRAF6/TAK1/NF-κB to minimize adipose tissue.
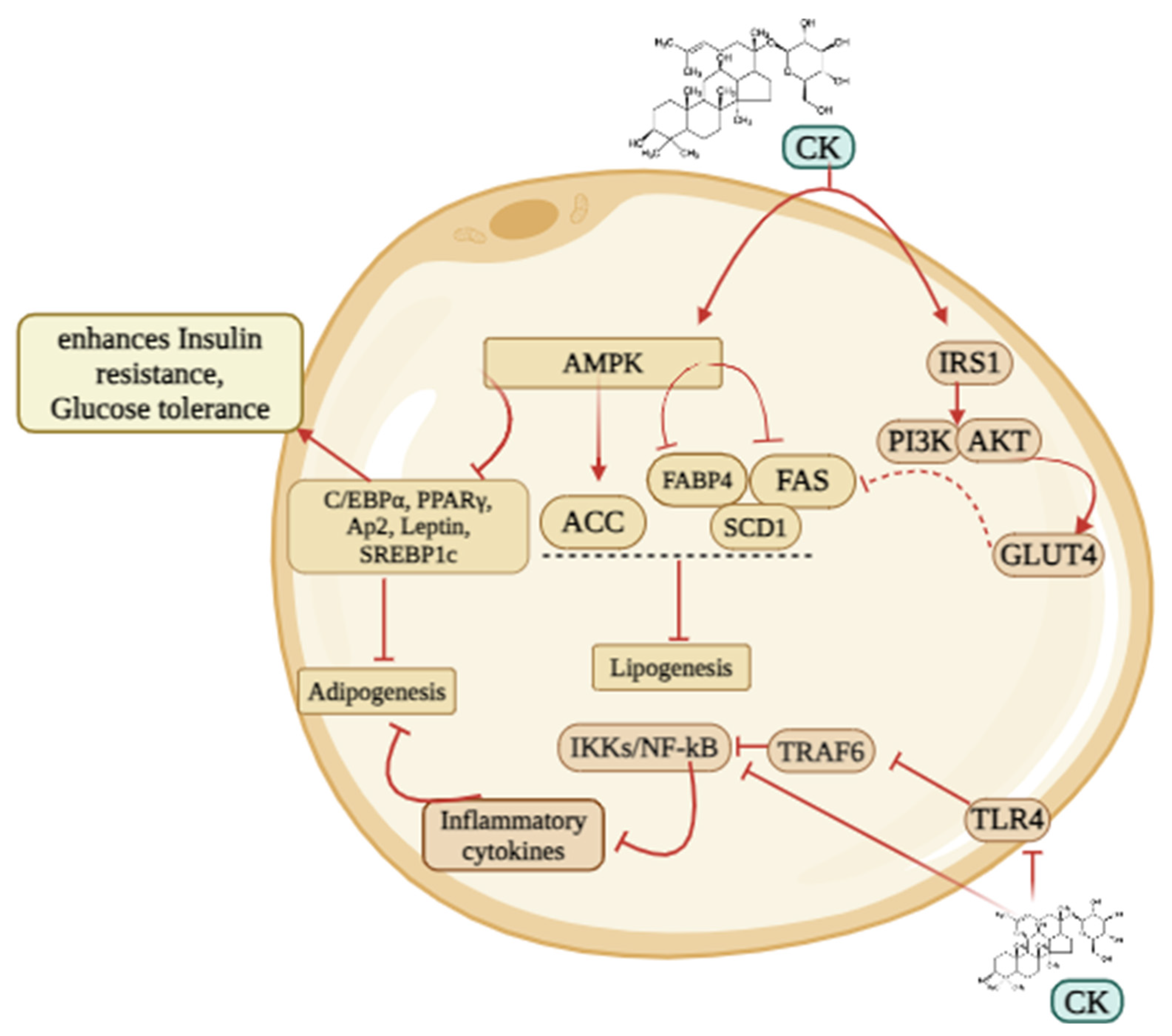
Figure 3.
The pathway involves T2D. CK triggers insulin resistance and increases insulin sensitivity to inhibit T2D by regulating several genes.
Figure 3.
The pathway involves T2D. CK triggers insulin resistance and increases insulin sensitivity to inhibit T2D by regulating several genes.
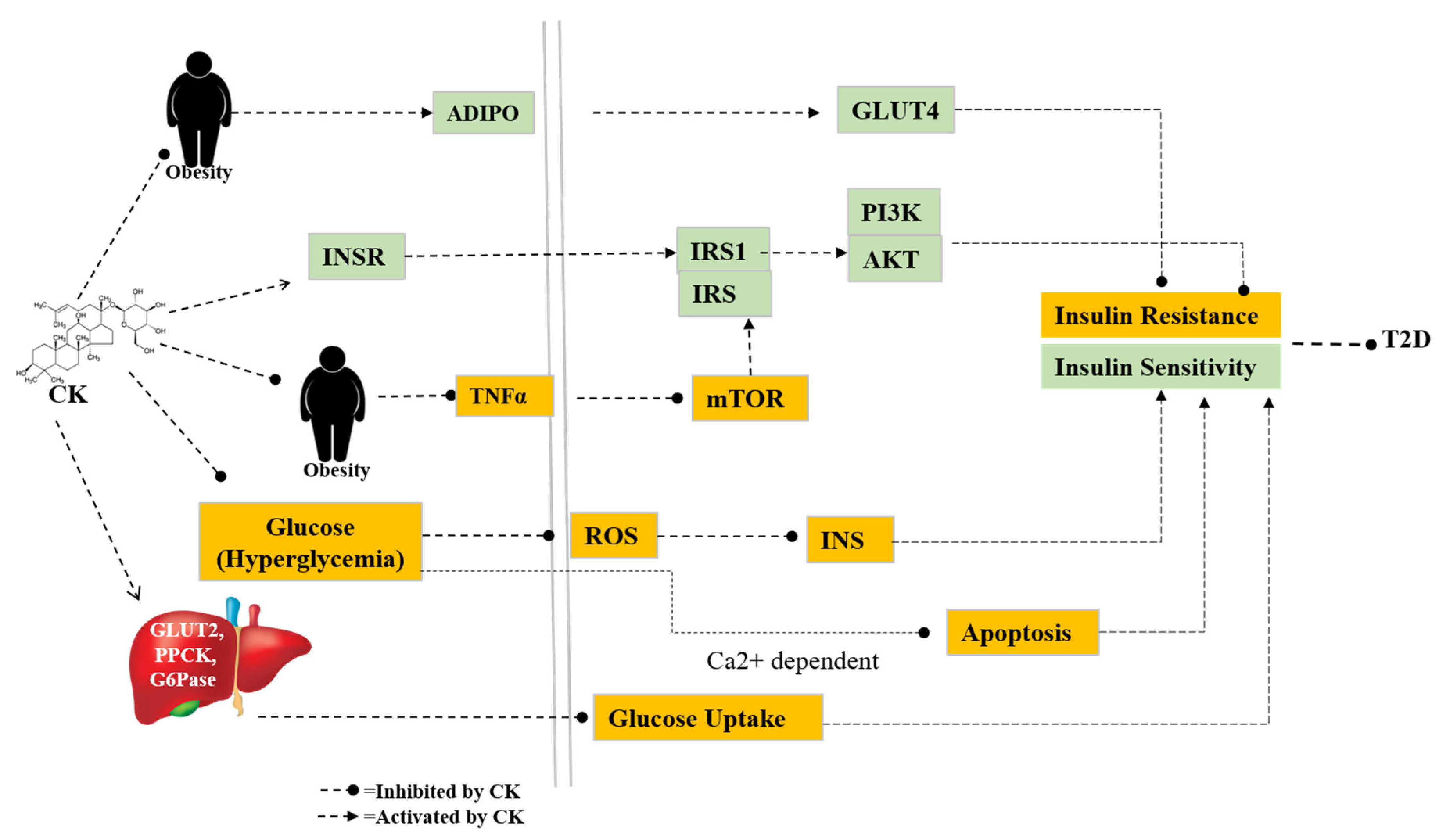
Figure 4.
CK is effective in inhibiting osteoclast by interacting with glucocorticoid receptors and this interaction triggers RANKL which attaches to RANK receptor to produce osteoclast. CK also inhibits ROS and TRAP, NFTAC1, MMPs, C-FOX, and TRAF6 to minimize bone resorption. On the other hand, CK increases the expression of preosteoblastic (Runx2, Osx) and Osteoblastic genes (ALP, COL1A1, OPN, OCN) that increase bone formation.
Figure 4.
CK is effective in inhibiting osteoclast by interacting with glucocorticoid receptors and this interaction triggers RANKL which attaches to RANK receptor to produce osteoclast. CK also inhibits ROS and TRAP, NFTAC1, MMPs, C-FOX, and TRAF6 to minimize bone resorption. On the other hand, CK increases the expression of preosteoblastic (Runx2, Osx) and Osteoblastic genes (ALP, COL1A1, OPN, OCN) that increase bone formation.
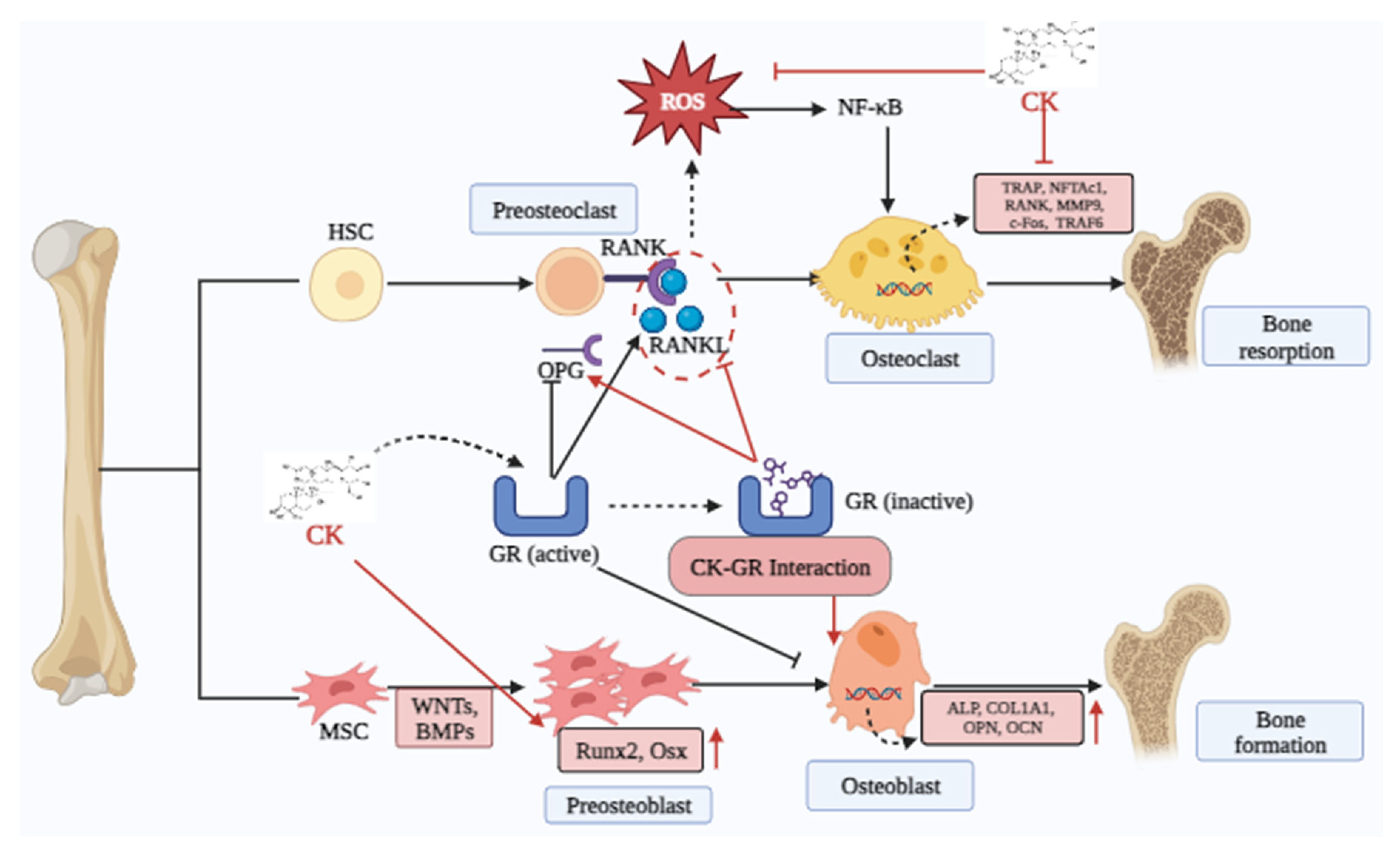
Figure 5.
Activity of CK on NAFLD pathway. CK increases the levels of the AMPK/SIRT1 pathway that increases ACC and inhibits the SREBP1/FAS/SDC1 pathway to minimize lipid synthesis. CK also PPARα to produce lipid oxidation.
Figure 5.
Activity of CK on NAFLD pathway. CK increases the levels of the AMPK/SIRT1 pathway that increases ACC and inhibits the SREBP1/FAS/SDC1 pathway to minimize lipid synthesis. CK also PPARα to produce lipid oxidation.
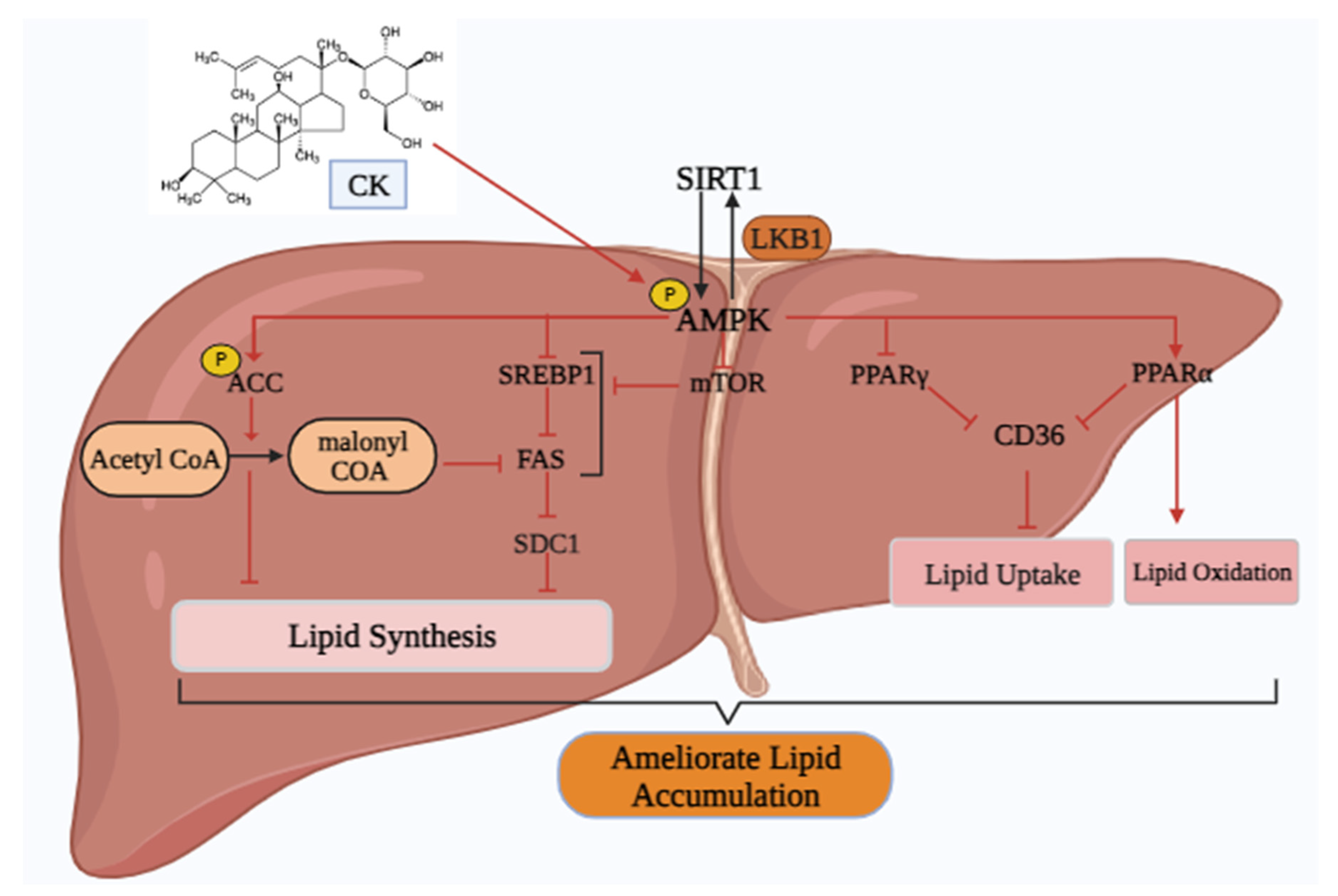

Table 1.
Physical and chemical properties of CK.
| Name | Compound K |
|---|---|
| Alias | 20-O-β-D-glucopyranosyl-20(S)-protopanaxadiol |
| CAS number | 39262-14-1 |
| Pubchem CID | 9852086 |
| Compound type | tetra-cyclic tri-terpenoid |
| Molecular formula | C36H62O8 |
| Molecular weight | 622.9 g/mol |
| Form | powder |
| color | White |
| Solubility | DMF: 10 mg/ml; DMSO: 10 mg/ml; DMSO: PBS (pH 7.2) (1:1): 0.5 mg/ml |
| Density | 1.19 |
| pka | 12.94±0.70(Predicted) |
| Melting point | 181~183℃ |
| Boiling point | 723.1±60.0 °C |
| LogP | 5.500 |
| Stability | Hygroscopic |
Table 2.
Cytotoxic studies of CK in several cell lines.
| Study model | Concentrations | Method of detection | Duration of experiment | Result | Ref. |
|---|---|---|---|---|---|
| 3T3-L1 preadipocyte cell lines | (0, 10, 20, 30, 40 µM) | MTS assay | 24 h | A high dose of CK (40 µM) did not affect cell viability | [29] |
| HaCaT keratinocytes cells | 0.2, 0.4, 0.6, 0.8, 1.0, and 10.0 μM |
MTT assay | 24 h | The survival of HaCaT cells was not significantly affected by CK doses below 10 μM. | [30] |
| MC3T3-E1 osteoblastic cell line | 0.01, 0.1, 1, 10 μM | MTT assay | 48h | CK at various doses did not exhibit any appreciable toxicity. | [43] |
| HepG2 | 1, 2, 5, 10, 15, 20, and 30 μM |
MTT assay | 24 h | As ginsenoside CK concentration increased to 30 μM, notable cytotoxic effects were seen. | [32] |
| HepG2 | 5−40 μM | CellTiter 96 AQueous One Solution Cell Proliferation Assay kit | 24h | Up to 40 μM doses, CK did not exhibit any cellular toxicity. | [33] |
| HT22 mouse hippocampal neuron cell |
2.5, 5, and 10 μM | MTT assay | 24 h | Ginsenoside CK can increase the survival of HT22 cells. | [34] |
| L02 Human liver cell line | 0.625, 1.25, 2.5, 5, 10 and 20 μM |
MTT assay | 24 h | The viability of the L02 cells appeared dramatically after treatment with CK at dosages of 1.25, 2.5, 5, or 10 μM. | [35] |
| RA-FLS and Raw 264.7 | 0.1, 0.5, 2.5, and 5 µM | MTT assay | 48 h | The ability to survive RA-FLS and RAW264.7 cells was not significantly impacted by CK at doses of ≤5µM. | [36] |
| MIN6 cell line | 2, 4, 8, 16, and 32 μM | MTT assay | 24 h | At 16 μM CK showed little toxicity on MIN6 cell | [37] |
| Hk-1 Nasopharyngeal Carcinoma cells, | 1–20μM | MTT assay | 24 h | The IC50 of CK was 11.5 on HK-1 cells | [38] |
| A549 lung cancer cells, MCF7 breast cancer cells, Caco-2 human colorectal adenocarcinoma cells, and normal RAW 264.7 cells |
0,3.125,6.25,12.5, 25 μg/mL |
MTT assay | 24 h | At 12.5 μg/mL concentration, CK showed considerable cytotoxic effect on A549 cells, MCF-7 cells, and Caco-2 cells growth. However, at 6.25 μg/mL Raw 264.7 cells showed less toxicitry. | [39] |
| HT-29 Human colon cancer cells | 8, 16, 32 and 64 μmol/l | MTT assay | 24 h | CK inhibited the growth of HT-29 cells in a dose-dependent manner. | [40] |
| HL-60 human myeloid leukemia cell line |
10, 20, 30, 50 μM | MTT assay | 72–96 h | 24.3 μM was needed to achieve 50% growth inhibition (IC50) at 96 hours. | [44] |
| U937, Jurkat, CEM-CM3, Molt4, and H9 leukemia cell lines | Did not mention | MTT assay | 96 h | The IC50 values of Compound K were as follows: 20μg/mL for U937, 26μg/mL for Jurkat, 36μg/mL for CEM-CM3, μg/mL for Molt 4, and 64μg/mL for H9. | [45] |
| Rat and mice | 8 and 10 mg/kg Respectively |
Acute oral repeated dose | 26-week | There were no indications of unusual clinical harm or death after 14 days. There were a few notable variations observed in this shift at weeks 9, 10, 12, 15, 17, 21–24, and 26. As a result, CK had a minimally negative impact on the animal’s body weight. |
[41] |
| Beagle dogs | 4, 12, or 36 mg/kg | oral doses | 26 week | No obvious toxicity was shown by the animals in the 4 and 12 mg/kg groups. The 36 mg/kg group showed elevated plasma enzyme levels, localized liver necrosis, and a decrease in body weight. |
[46] |
Table 3.
Enzymatic biotransformation method for the development of CK production.
| Enzymes | Transformation pathway | Optimum condition | Remarks | Ref. |
|---|---|---|---|---|
| β-glu from Paecilomyces Bainier sp. 229 | Rb1→Rd→F2→CK | PH=3.5, Temp=55°C, Time=48h | 84.3% ginsenoside Rb1 was converted to CK | [49] |
| β-glu from Terrabacter ginsenosidimutans | Rb1→gypenoside XVII→ gypenoside LXXV→CK |
pH 7.0 37°C |
Complete conversion of CK from Rb1 via intermediate metabolites gypenosides XVII and LXXV. | [50] |
| β-glu from Lactobacillus brevis | Rb1→gypenoside XVII→ gypenoside LXXV→CK |
pH =6.0 Temp=30°C Time=6 h |
89% molar conversion | [51] |
| β-glycosidase from S. solfataricus | Rb1/Rb2→Rd→F2→CK Rc →compound Mc → CK |
PH=5.5, Temp=85°C, Time=12h | Although good specificity, the conversion rate is low. | [52] |
| β-glucosidase from Pyrococcus furiosus | Rb1/Rb2→Rd→F2→CK | PH=5.5, Temp=95°C, Time=6h | After five hours, aglycone PPD was produced by hydrolyzing the CK. | [53] |
| β-glucosidase from Microbacterium esteraromaticum | Rb2→Compound Y→CK | PH=7, Temp=40°C, | Ginsenoside Rb2 (0.74 mg/ml) changes after 12 hours into compound Y (0.27 mg/ml) and CK (0.1 mg/ml). | [54] |
| β-glu@Cu(PTA) biocomposite | Rb1→Rd→F2→CK | PH=3, Temp=45°C, Time=24h | In these conditions, the conversion of the rare ginsenoside CK achieved 49.15%. | [55] |
| β-Glu&SN@Zn-BTC (β-Glu and snailase were co-immobilized on Zn-BTC) biocomposite | Rb1→Rd→F2→CK | PH =4.5 Temp =50°C Time =48h |
The CK conversion rate achieved 53.5%. | [56] |
| Sna&β-Glu@H-Cu-BDC biocomposite | Rb1→Rd→F2→CK | PH =4.5 Temp =50°C Time =48h |
The average conversion efficiency was about 60.12%, and the concentration of CK was roughly 0.94 mg mL−1. | [57] |
Table 4.
Pharmacological action of CK in treating metabolic disorders and its underlying molecular processes.
Table 4.
Pharmacological action of CK in treating metabolic disorders and its underlying molecular processes.
| Disease | Experimental models | Dosage form | Doses of administrations |
Mechanism | Ref. |
|---|---|---|---|---|---|
| Obesity | C57BL/6J mice | Oral | 15, 30, 60 mg/kg |
|
[13] |
| Male C57BL/6J and ob/ob (B6/JGpt-Lepem1Cd25/Gpt) mice | i.p. injection | 20 mg/kg |
|
[77] | |
| 3T3-L1 cell lines | Cell treatment | 0.05, 0.5, 5 µM |
|
[19] | |
| 3T3-L1 cell lines | Cell treatment | 20, 50 µM |
|
[78] | |
| 3T3-L1 cell lines | Cell treatment | 10-40 µM |
|
[79] | |
| Diabetes | male ICR mice | Oral | 30 mg/kg/day |
|
[80] |
| Male Wistar rats (200–250 g) | oral | 30, 100, 300 mg/kg BW |
|
[81] | |
| MIN6 cell line | Cell treatment | 2-32 μM |
|
[37] | |
| male C57BL/KsJ db/db mice | oral | CK: Metformin 1:15 |
|
[82] | |
| DN | HFD (high-fat diet)/STZ (streptozotocin)-induced DN mice model | intragastrically | 10, 20, 40 mg/kg/day |
|
[83] |
| DT | human tenocytes cell | Cell treatment | 3, 10 µM |
|
[84] |
| OP |
Raw264.7 cells Balb/C female mice |
Cell treatment, i.p. injection |
10 µM 10 mg/kg |
|
[85] |
|
bone marrow mesenchymal stem cells male Sprague Dawley (SD) rats |
Cell treatment, i.p. injection |
2.5-40 µM 10 µM |
|
[86] | |
| OA | MC3T3E1 cell lines | Cell treatment | 0.01-10 µM |
|
[31] |
| NAFLD |
SD rats, HSC-T6 cells |
i.p. injection Cell treatment |
3mg/kg/day |
|
[87] |
| HepG2 cells | Cell treatment | 20 µM |
|
[33] | |
| HuH7 cells | Cell treatment | 1 µM |
|
[88] | |
| HCC | HepG2 cells | Cell treatment | 0, 5 and 10 µmol |
|
[89] |
Abbreviations: DN=Diabetes nephropathy, DT=diabetic tendinopathy, OP=osteoporosis, OA=Osteoarthritis, NAFLD= Nonalcoholic fatty liver disease, HCC=Hepatocellular carcinoma.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



