
Submitted:
02 February 2024
Posted:
02 February 2024
You are already at the latest version
Abstract
The increasing global concern over plastic waste and its environmental impact has led to a growing interest in the development of sustainable packaging alternatives. This study focuses on the innovative use of expired dairy products as a potential resource for producing edible packaging materials. Expired dairy products, which are typically discarded, contribute to food waste and environmental pollution. By modifying these products to packaging materials, a dual-purpose solution addressing both waste reduction and sustainable packaging is evaluated. Expired milk and yogurt were selected as the primary raw materials due to their protein and carbohydrate content. The extracted casein was combined with various concentrations of chitosan while in some cases glycerol was used. The materials were characterized with attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy and X-ray analysis (XRD) to examine the interactions between the components of the formed binary blend. Scanning electron microscopy (SEM) was used to evaluate the surface morphology of the blends, while dynamic mechanical analysis (DMA) and tensile experiments were conducted to study the thermomechanical properties of the materials. Barrier properties, including water vapor transmission (WVTR) and oxygen permeability (OTR), were measured to assess the materials' potential to preserve food freshness. The findings reveal that expired dairy-based edible packaging materials exhibit promising mechanical properties, comparable to conventional plastic packaging. The barrier properties and especially the zero oxygen permeability of the membranes, indicate that these materials have the potential to effectively protect food products from external factors that could compromise quality and shelf life. The materials show encouraging signs of eco-friendliness, with a notable reduction in environmental persistence compared to conventional plastic packaging.
Keywords:
chitosan
; casein
; glycerol
; dairy wastes
; edible membranes
; food packaging
; sustainability
1. Introduction
Up-to-date food cycle, except from the food itself, consists of various other important tasks which are necessary such as processing, post-treatment storage, packaging, distribution, retail and consumption [1]. The transition from linear to circular bio-based economy is of utmost importance and new technologies that transform biological feedstock and/or resources into valuable products are required. The newly designed products should be renewable and cost effective to address the depletion of natural resources, the escalating global food demand and consumption as well as the severe climate change [2]. In recent scientific studies, the development of bio-based materials, showcasing desired properties such as sustainability, resource efficiency and low carbon dioxide emissions is reported [3]. The global output of plastics has reached an unprecedented value of 407 MMT (million metric tons), as already reported in the literature [4], and the pandemic COVID-19 has contributed significantly to an increase of the aforementioned value. Petroleum-based plastics utilized in the packaging sector alone are accountable of approximately 44% of the overall value, indicating their major contribution to the environmental pollution [4]. It should be mentioned though that various inherent properties including low cost, permeability, transparency, enhanced tensile and thermal performance and the ability to be easily sterilized has enabled the wide use of plastic materials in packaging applications [5] so far, despite the great environmental concerns the last couple of decades. The combination of carbon-carbon bonds in the polymeric materials; which does not allow their ease treatment and disintegration after the end-life cycle; with their excessive use, has led to devastating effects on the environment, causing contamination and putting in danger all living organisms. The inability to be disintegrated in reasonable time in nature from factors such as UV irradiation leads to an accumulation of plastics into the environment as well as oceans. Approximately 8 MMT of plastics end up in the oceans each year [6]. Currently, polymeric materials comprised of polyethylene, polypropylene, and poly(ethylene terephthalate) are extensively used in food packaging. These petroleum polymers despite their low weight and the ability to be easily transformed by extrusion in variable shapes do not showcase any sustainable characteristics. Their non-biodegradable nature contributes to the environmental pollution as has already been mentioned. Furthermore, the utilization of synthetic polymeric materials in food packaging has detrimental effects due to the release of carbon dioxide and other toxicants during the incineration process. Hazardous interactions between food and recycled or reused plastics have also been reported in the literature [7].
The development of new packaging materials targeting to food-packaging applications requires in depth analysis in terms of biodegradability. Plastics can be classified in two distinct categories strongly dependent on their components as bio- or fossil-based. In both types, biodegradable, non-biodegradable components and their combination can be found. It should be mentioned that the final chemical structure of the materials determines their biodegradability and not the initial resource used [8]. Bioplastics are therefore categorized as bio-based and nonbiodegradable, bio-based and biodegradable, and fossil-based and biodegradable respectively. Through this classification it is easily understood that not all bioplastics can be completely biodegradable and misinterpretations due to commercial purposes often occur.
The design and development of sustainable materials which are harmonized with the current environmental concerns and exhibit high quality, is vital for both industry and consumers. The scientific community has shifted its interest towards the production of edible and biodegradable materials that adhere to food quality and safety standards [9]. Environmental contamination related to expired dairy products including milk, cheese whey, colostrum and additional dairy industry by-products derived from the processing procedures has not gained tremendous attention yet, and only limited reports make use of these by-products to form sustainable materials [10]. The dairy byproducts may possibly lead to high-quality products further contributing to the circular economy.
In the case of food-grade components [11], bio-based, bio-degradable and edible products can be made. Edible packaging is rapidly evolving as a sustainable/biodegradable substitute of conventional packaging, demonstrating advantageous characteristics. The shelf life of edible membranes can be further extended using various additives such as lipids, chitosan/chitin, gums, cellulose derivatives, animal or plant-based proteins and starches. Valuable characteristics involving bio-compatibility, non-toxicity, non-polluting, gas and moisture barrier properties, render the specific materials quite important for packaging-based applications [12]. Furthermore, edible materials can be designed using proteins, polysaccharides and oils which are derived from feedstock and active chemicals such as antioxidants and/or antimicrobial agents. These reagents are used to further enhance their final properties making them ideal candidates in food science. The dual application of the above-mentioned materials, meaning packaging and consumption, without posing any threat on the human health is important, while by tuning the thickness of the films, different applications can be induced. The appropriate choice of edible packaging materials is related to the potential content/food to be packed and the processing technique [13].
Substances isolated from blood and glandular fluid, namely proteins, that vary in terms of molecular weight, concentration, and function are used in such applications. The use of dairy products has been extended beyond consumption and can be utilized in different fields as packaging [14]. The expired dairy products contribute significantly to the environmental pollution but still contain a variety of proteins that either offer defense against enteropathogens or are necessary to produce new dairy products [15]. Large amounts of proteinaceous waste, particularly whey and caseins, are produced from the dairy wastes. In bovine milk, caseins constitute approximately 80% of the total protein, concluding for it being the most abundant type of proteins [16]. Even though almost half of the whey generated globally is recovered and used in various products, including meals, supplements, and medications huge quantities are discarded without any prior processing [17]. Inherent characteristics of milk proteins, such as high barrier and film properties, make them ideal for biomaterial synthesis [18]. In recent years, the preparation of protein membranes from dairy waste has attracted the interest of several scientific groups. Laetitia M. Bonnaillie et al. [19] synthesized casein/glycerol/citric pectin membranes to study the structure and mechanical properties by adding a polysaccharide and a plasticizer. Muhammad Rehan Khan et al. [20] highlighted the impact of active ingredients on the composition of materials and lifespan of products, considering the presence of active ingredients in the casein matrix.
In the present study chitosan, casein, glycerol and squid ink were utilized for the development of the films. Chitosan was selected because of the attractive properties such as, antimicrobial properties, biodegradability, film forming properties it has [21]. Casein was used because of its excellent barrier and film forming properties while it is also biodegradable and non-toxic [22]. Τhe combination of the two biopolymers was carried out both to combine the excellent properties of the materials and to overcome the brittle nature of casein membrane. The squid ink was used to impart antimicrobial and antioxidant properties to the prepared films [23]. Casein protein was isolated from expired cow milk, by the precipitation method followed by the preparation of the relative membranes under different ratios of casein. For comparison reasons membranes of pure chitosan, chitosan and casein as well chitosan, casein and glycerol were prepared. For the final membranes according to the reagents used abbreviations of the type AxByCzDw are used where A, B, C and D are the compounds (chitosan, casein, glycerol and squid ink respectively) and x, y, z and w the relative %wt ratios.
2. Materials and Methods
2.1. Materials
Sigma–Aldrich (St. Louis, MO, USA) was the supplier of low molecular weight chitosan (75–85% deacetylated), glycerol, acetic acid (99,8%), methanol (99,8%), ethanol (99,8%), sodium hydroxide (98%) and diethyl ether. The expired milk was supplied from the national dairy industry DODONI S.A. and the squid ink was purchased from the local market.
2.2. Extraction of Casein
For the casein extraction from expired dairy product, a beaker containing 500 ml of expired milk, was heated till to reach 55 oC. In another beaker, 500 ml of acetic acid solution (approximately 10 % v/v), was heated under the same conditions (temperature and time). The foam formed on the surface of the milk was carefully removed and the acetic acid solution was added in beaker, dropwise, to adjust the pH at approximately 4.6, which is the isoelectric point of casein. To obtain the solid, the mixture was filtrated with filter paper. The solid sample was washed several times with distilled water and was placed in another beaker which contained enough amount of ethanol able to cover the solid. Filtration took place again with filter paper to collect the solid sample. The protein sample was washed with 250 ml solution of ethanol/diethyl ether in a ratio of 1:1 and once more with 100 ml of diethyl ether. The extracted sample was left to dry at room temperature.
2.3. Membrane Synthesis
For the Chi32Cas32Gly20SqInk16 membrane synthesis, in a beaker containing 50 mL of distilled water, 2 % (w/v) of extracted casein and 0.4 % (w/v) of sodium hydroxide were added. The mixture was transferred in an ice bath and it was sonicated using and ultrasonicator (UP100H, 100W, 30kHz, Hielscher Ultrasonics) for 5 min and then allowed to stir until completely dissolved. While stirring, 2 % (w/v) of chitosan was added in the solution followed by the addition of 2 % (v/v) acetic acid. The new solution was left to stir for approximately 10 min. In the same beaker 0.5 ml of glycerol was added followed by the addition of 1 % (w/v) squid ink. The final solution contains 1 g (2 % w/v) extracted casein, 1 g (2 % w/v) chitosan, 0.63 g (1.26 % w/v) glycerol and 0.5 g (1 % w/v) squid ink. It is then transferred to polystyrene dishes to evaporate the solvent and form the hydrogel membrane. For the preparation of all other materials, pure chitosan, pure casein, Chi50Cas50, Chi33Cas67 and Chi38Cas38Gly24, similar was followed by not including the additional step or steps on the aforementioned method. The abbreviations and quantities used in all the experiments are listed in Table 1.
2.4. Attenuated Total Reflectance-Fourier Transform Infrared Spectroscopy (ATR-FTIR)
ATR-FTIR analysis was conducted with a SHIMADZU IRSpirit fourier transform infrared spectrophotometer (1, Nishinokyo Kuwabara-cho, Nakagyo-ku, Kyoto 604-8511, Japan). The ATR objective featured a ZnSe prism with a 250 μm contact area on the studied samples. The prism allowed for a penetration depth of around 2.0 µm (@1000 cm−1) and enabled measurements starting from 650 cm−1.
2.5. X-Ray Diffraction (XRD)
The samples' crystallinity was examined using a PANalytical X'PertPRO diffractometer (Enigma Business Park, Grovewood Rd, Malvern WR14 1XZ, United Kingdom) using Cu/Kα radiation. The diffractometer is equipped with an X'Celerator detector running at 40 kV voltage and 40 mA current. The membranes underwent scanning within the 2θ range from 2° to 60°.
2.6. Thermogravimetric Analysis (TGA)
TGA analysis was carried out utilizing a Setsys Evolution-Setaram (7, rue de l'Oratoire 69300 Caluire-et-Cuire France) TGA, TG-DSC, and TG-DTA analyzer. The procedure involved placing roughly 30 mg of the sample into a platinum crucible, adjusting the heating and nitrogen (N2) flow rates, and then conducting the test. Throughout all experiments, the heating rate remained constant at 10 K/min, and the N2 flow rate at 25 ml/min within the temperature range from room temperature to 700 °C.
2.7. Dynamic Mechanical Analysis (DMA)
The films’ dynamic mechanical behavior was examined using a dynamic mechanical analyzer (DMA Q800, TA Instruments, 159 Lukens Drive New Castle, DE 19720, USA) in film tension mode. To evaluate the storage modulus (E′) and the loss factor (tan δ), a temperature range from -70 °C to 120 °C, at a rate of 3 K/min, along with a frequency of 1 Hz, was applied.
2.8. Mechanical Properties
The membranes' tensile characteristics were assessed following ASTM D638 standards, employing a custom horizontal tensile testing stage manufactured by ADMET (51 Morgan Drive | Norwood, MA 02062, USA). Specimens in type V dumbbell shapes were prepared and subjected to testing at a strain rate of 0.1 min−1 until failure. Each membrane type underwent testing at least three times by making the required type V dumbbell shaped specimens. The elongation of these specimens was tracked using a linear variable differential transformer (LVDT), while the load was measured through a 44.5 N load cell (or a 445 N load cell for the pure specimens). The elongation values were transformed into engineering strain by dividing each specimen's initial effective length, while the load values were transformed into engineering stress by dividing by the specimen's cross-sectional area.
2.9. Scanning Electron Microscopy (SEM)
The surface morphology of the samples was observed using a JEOL JSM-6510 LV SEM Microscope (Ltd., Tokyo, Japan) equipped with an X-Act EDS detector from Oxford Instruments, Abingdon, Oxford shire,UK (an acceleration voltage of 20 kV was applied) with possibility to function under low vacuum conditions. Before examination, all membranes were sputter-coated with gold/palladium for 45 s to prevent sample charging during observation with SEM.
2.10. Water Vapor Transmission Rate Measurements-Water Diffusion Coefficient Calculation
The water vapor transmission rate (WVTR [g/(cm2*s1)]) for all the obtained membranes was calculated according to the ASTM E96/E 96M-05 method at 38 °C and 95% RH, using a custom-made apparatus. The calculated WVTR values were converted into water vapor diffusivity (Dwv) values according to theory and relative equations which are described in detail in a previous publication by our group [24].
2.11. Oxygen Transmission Rate Measurements-Oxygen Permeability Calculation
The oxygen transmission rate (OTR) values (cc O2/m2/day) for each membrane was assessed following ASTM D 3985 standards (23 °C and 0% RH). An oxygen permeation analyzer (O.P.A., 8001, Systech Illinois Instruments Co., Johnsburg, IL, USA) was utilized for these measurements. Subsequently, using the derived OTR values, the oxygen permeability coefficient values (PeO2) were calculated, employing the theoretical framework and equations described thoroughly in a prior publication by our group [24].
3. Results
3.1. ATR-FTIR
ATR-FTIR spectroscopy was employed to evaluate the presence of electrostatic interactions between casein and chitosan. In Figure 1 the ATR-FTIR spectra of pure casein, Chi50Cas50 and Chi33Cas67 (chitosan/casein blend), Chi38Cas38Gly24 (chitosan/casein/glycerol blend) and Chi32Cas32Gly20SqInk16 (chitosan/casein/glycerol/squid ink blend) are presented. For pure casein, the FTIR spectra is shown in Figure 1a where the broad peak observed at ~ 3280 cm−1 is attributed to -OH and -NH- stretching (the peaks are overlapping). The peaks at 2923 cm−1 and 2856 cm−1 are attributed to -CH- stretching vibrations while the peaks observed at 1635 cm−1 (amide I), 1524 cm−1 (amide II), 1457 cm−1, 1388 cm−1 and 1236 cm−1 (amide III) are attributed to -C=O stretching vibrations, -NH- bending vibrations, -CH- bending vibrations (both 1457 and 1388 cm−1) and -NH- bending vibrations, respectively [25,26,27]. Figure 1b is referred to the pure chitosan membrane, where the broad peak between 3500-3000 cm−1 is attributed to stretching vibrations of the -OH groups and -NH- groups (overlapping as mentioned above). Peaks observed at 2923 and 2856 cm−1 are representative of -CH- stretching vibrations. The characteristic peaks at 1643 cm−1, 1554 cm−1, 1065 cm−1 and 1019 cm−1 are ascribed to -C=O stretching vibrations, -NH- bending vibrations and -C=O stretching vibrations, respectively [28,29]. Figure 1c,d show the spectra of the Chi50Cas50 and Chi33Cas67 membranes respectively, which are different in composition and are compared to the pure chitosan and pure casein membranes. The amide I peak observed at 1643 cm−1 in pure chitosan, moved to 1628 cm−1 indicating secondary interactions between the components of the blend [30]. Figure 1e shows the spectra of the glycerol containing material, Chi38Cas38Gly24. The characteristic peak at 1028 cm−1 can be attributed to the addition of glycerol and indicates its successful integration in the blend [28]. The addition of squid ink in the blend, concluded to no relative and drastic changes to the final FTIR spectra (Figure 1f).
3.2. XRD Analysis
XRD analysis was utilized to examine the crystallinity of pure casein (Figure 2a), pure chitosan (Figure 2b), Ch50Cas50 (Figure 2c), Ch33Cas67 (Figure 2d), Chi38Cas38Gly24 (Figure 2e) and Chi32Cas32Gly20SqInk16 (Figure 2f). Proteins do not exhibit a crystalline structure in general, which means that when they are subjected to XRD analysis, normal crystalline effects do not appear [31]. The pure casein diffraction pattern is typical of nonfibrous proteins (Figure 2a). The attraction between the polar groups that most likely occur in a complex protein structure is leading to the absence of structural order in casein and other similar proteins [32]. At diffraction angles 2θ of ~9.5◦ and 19.3◦, casein displayed two flat peaks, indicating the amorphous nature of the protein. In Figure 2b, the pure chitosan diffractogram is shown and provides information of the semicrystalline nature of chitosan, while the data have been already analyzed in our previous work [28]. In the diffractograms of Ch50Cas50 (Figure 2c) and Ch33s67 (Figure 2d), new peaks appeared, providing more evidence that intermolecular interactions between chitosan and casein are evident. The glycerol containing materials (Figure 2e,f) were amorphous due to the high plasticizer (glycerol) concentration used [28,33].
3.3. TGA
In Figure 3 the TGA thermograms of the membranes pure chitosan (Figure 3a), Ch50Cas50 (Figure 3b), Ch33Cas67 (Figure 3c), Chi38Cas38Gly24 (Figure 3d) and Chi32Cas32Gly20SI16 (Figure 3e) are given in the temperature range between 20–700 °C. The initial mass loss observed from ~80–120 °C is attributed to the water loss from the membranes. The incorporation of casein in the blend Ch50Cas50, did not affect the thermal stability of the membrane while the increase of its concentration (Ch33Cas67) seems to destabilize the material at ~180 °C. The major mass loss in the samples: pure chitosan, Ch50Cas50 and Ch33Cas67 occurs from 220–480 °C and is ascribed to the materials’ functional groups decomposition. The major loss observed in the glycerol containing materials, starts at lower temperature, phenomenon that has already been observed and has been thoroughly analyzed in our previous work [28,33]. The remaining ~20-30% of the mass is attributed to remaining ash that has been carbonized and cannot decompose any further [28,34].
3.4. DMA
Dynamic mechanical analysis (DMA) was utilized to study the thermomechanical properties of the final hydrogel membranes. In Figure 4 the results of the storage modulus as a function of temperature are illustrated for pure chitosan (Figure 4a), Ch50Cas50 (Figure 4b), Ch33Cas67 (Figure 4c), Chi38Cas38Gly24 (Figure 4d) and Chi32Cas32Gly20SqInk16 (Figure 4e). The thermomechanical properties of the materials were examined in the range between −70 to 120 °C. The addition of casein in the membranes Ch50Cas50 and Ch33s67 significantly increased the storage modulus of the materials in comparison to the pure chitosan membrane. On the other hand, the membranes Chi38Cas38Gly24 and Chi32Cas32Gly20SqInk16 showed decreased storage modulus due to the plasticizing effect of glycerol, while squid ink, did not affect the mechanical properties.
In Figure 5 the tan delta graphs as a function of temperature are shown. The glass transition temperature (Tg) of pure chitosan membrane Figure 5a was found to be approximately 90 °C. The addition of casein in the blends led to increased Tgs (98 °C and 108 °C respectively) of the hydrogel membranes Ch50Cas50 (Figure 5b) and Ch33Cas67 (Figure 5c). The increased Tg in the blends may be attributed to the Tg value of casein, which is higher than that of chitosan according to the literature [35] and from 1/1 ratio of chitosan/casein in the first blend has increased to 1/2 in the second case explaining the 10 °C increase. The glycerol addition in the hydrogel membranes Chi38Cas38Gly24 (Figure 5d) and Chi32Cas32Gly20SqInk16 (Figure 5e) respectively led to a significant overall decrease (~80 ◦C) in the Tg values of the blends due to the functioning role of glycerol as a plasticizer [28,33]. The Tg value of the glycerol containing materials were found to be 16 °C and 22 °C, for Chi38Cas38Gly24 and Chi32Cas32Gly20SqInk16 respectively. It is evident that the addition of the squid ink has led to a decreased ratio of the glycerol in the final membrane and therefore the increase of 6° C in the observed Tg can be explained as a decrease of the plasticizer content.
3.5. Tensile Properties
The tensile properties of the studied membranes are depicted in the stress vs strain plots of Figure 6 and the average stress and strain values for all samples are summarized in Table 2. The pure chitosan reference samples exhibited the highest strength of 102.82 MPa with an average strain of 6.8 %. The addition of casein in the membrane at a ratio of 50% caused a reduction of the strength by 49.8% and a small increase of the strain by 13.2%, while the addition of a higher amount of 67% casein led to a further embrittlement of the specimens with a reduction in both the strength and the strain values, by 64.2% and 61.5%, respectively, in comparison to the reference sample (pure chitosan). The combined addition of casein and glycerol caused a significant deterioration of the strength by 85.1% with a parallel significant enhancement of the strain by 467.5% in comparison to the pure chitosan. Finally, when chitosan was mixed with casein, glycerol and squid ink the resulting strength was substantially reduced by 88.2% but at the same time the strain was increased by 250.7% compared to the pure chitosan samples.
3.6. SEM Measurements
SEM was used to examine the surface morphology of the prepared hydrogel membranes. Indicative SEM images of pure casein and the hydrogel membranes pure chitosan, Chi50Cas50, Chi33Cas67, Chi38Cas38Gly24 and Chi38Cas38Gly24SqInk16 are shown in Figure 7. In Figure 7a, the formation of a thick network is evident, composed of casein aggregates while in Figure 7b, the dense morphology of the pure chitosan membrane is shown. In the case of Chi50Cas50 membrane (Figure 7c) a rough surface with voids is observed while in the case of Chi33Cas67 membrane (Figure 7d) the surface remained rough, but the voids significantly decreased. The integration of glycerol in the Chi38Cas38Gly24 (Figure 7e), further decreased the voids on the membrane surface and the introduction of the squid ink in the hydrogel membrane Chi32Cas32Gly20SqInk16 (Figure 7f), led to a smoother and continuous dense surface morphology.
3.7. WVTR-Water Diffusion Coefficient Calculation
The obtained water vapor transmission rate (WVTR) values for pure chitosan, Ch50Cas50, Chi33Cas67, Chi38Cas38Gly24 and Chi32Cas32Gly20SqInk16 membranes are summarised in Table 3. From these values, the water diffusivity (Dw) values are calculated and are listed in Table 3 for comparison. As is seen from Table 3, the incorporation of casein in the membranes led to an increase of the Dw (water diffusion coefficient) values of obtained Ch50Cas50 film. Increase of casein content in the Chi33Cas67 film led to higher Dw value. Furthermore, the addition of glycerol and squid ink further increased the Dw values of the Chi38Cas38Gly24 and Chi32Cas32Gly20SqInk16 films respectively.
3.8. OTR-Oxygen Permeability Calculation
The oxygen permeability studies revealed that all the prepared membranes showed zero oxygen permeability which is one of the most significant factors about packaging materials. That result significantly strengthens the initial assumption that these materials can be used as food packaging materials.
4. Conclusions
The prepared hydrogel membranes were synthesized through a solution casting-evaporation method and characterized through all the aforementioned analytical measurements. Casein was extracted from expired milk and to overcome its brittle nature, chitosan was incorporated in the blend. From the physicochemical characterizations it was proved that chitosan and casein interact via secondary interactions while glycerol successfully integrated in the blend. Glycerol’s incorporation further improved the strain property of the films while its low glass transition temperature allows flexibility both at room temperature and at lower temperatures. Squid ink was utilized to impart antimicrobial properties to the film. The zero-oxygen permeability of the prepared films indicates that they can provide effective food protection, quality assurance and shelf-life extension. Thus, all the results strongly support that the prepared novel materials can be used as sustainable packaging applications.
Author Contributions
Synthesis experiment design, Andreas Karydis-Messinis, Christina Kyriakaki , Eleni Triantafyllou, Dimitrios Moschovas and Apostolos Avgeropoulos; characterization measurements and interpretation, Andreas Karydis-Messinis, Christina Kyriakaki , Eleni Triantafyllou , Kyriaki Tsirka , Christina Gioti, Dimitris Gkikas, Konstantinos Nesseris , Dimitrios A. Exarchos, Spyridoula Farmaki, Aris Giannakas , Constantinos E. Salmas, Theodore E. Matikas, Dimitrios Moschovas and Apostolos Avgeropoulos; experimental data analysis and interpretation Andreas Karydis-Messinis, Christina Kyriakaki, Eleni Triantafyllou, Dimitrios Moschovas and Apostolos Avgeropoulos; overall evaluation of this work, Apostolos Avgeropoulos, Andreas Karydis-Messinis and Dimitrios Moschovas ; TGA, Christina Gioti; Tensile measurements, Kyriaki Tsirka ATR-FTIR Theodore E. Matikas, Dimitrios A. Exarchos and Spyridoula Farmaki SEM measurements, Dimitrios Moschovas and Apostolos Avgeropoulos ; WVTR and OTR Aris Giannakas and Constantinos E. Salmas.
Funding
This research received no funding.
Data Availability Statement
The datasets generated for this study are available on request to the corresponding author.
Acknowledgments
The expired dairy products were supplied by DODONI S.A..
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kummu, M.; de Moel, H.; Porkka, M.; Siebert, S.; Varis, O.; Ward, P.J. Lost food, wasted resources: Global food supply chain losses and their impacts on freshwater, cropland, and fertiliser use. Science of The Total Environment 2012, 438, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, R.; Jaiswal, A.K. Exploitation of Food Industry Waste for High-Value Products. Trends in Biotechnology 2016, 34, 58–69. [Google Scholar] [CrossRef]
- Rosenboom, J.-G.; Langer, R.; Traverso, G. Bioplastics for a circular economy. Nature Reviews Materials 2022, 7, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Rabnawaz, M.; Wyman, I.; Auras, R.; Cheng, S. A roadmap towards green packaging: the current status and future outlook for polyesters in the packaging industry. Green Chemistry 2017, 19, 4737–4753. [Google Scholar] [CrossRef]
- Luzi, F.; Torre, L.; Kenny, J.M.; Puglia, D. Bio- and Fossil-Based Polymeric Blends and Nanocomposites for Packaging: Structure⁻Property Relationship. Materials (Basel, Switzerland) 2019, 12. [Google Scholar] [CrossRef]
- Guillard, V.; Gaucel, S.; Fornaciari, C.; Angellier-Coussy, H.; Buche, P.; Gontard, N. The Next Generation of Sustainable Food Packaging to Preserve Our Environment in a Circular Economy Context. 2018, 5. [CrossRef]
- Aguirre-Joya, J.A.; De Leon-Zapata, M.A.; Alvarez-Perez, O.B.; Torres-León, C.; Nieto-Oropeza, D.E.; Ventura-Sobrevilla, J.M.; Aguilar, M.A.; Ruelas-Chacón, X.; Rojas, R.; Ramos-Aguiñaga, M.E.; et al. Chapter 1 - Basic and Applied Concepts of Edible Packaging for Foods. In Food Packaging and Preservation, Grumezescu, A.M., Holban, A.M., Eds.; Academic Press: 2018; pp. 1-61.
- Hamed, I.; Jakobsen, A.N.; Lerfall, J. Sustainable edible packaging systems based on active compounds from food processing byproducts: A review. 2022, 21, 198-226. [CrossRef]
- Trajkovska Petkoska, A.; Daniloski, D.; D'Cunha, N.M.; Naumovski, N.; Broach, A.T. Edible packaging: Sustainable solutions and novel trends in food packaging. Food Research International 2021, 140, 109981. [Google Scholar] [CrossRef]
- Sar, T.; Harirchi, S.; Ramezani, M.; Bulkan, G.; Akbas, M.Y.; Pandey, A.; Taherzadeh, M.J. Potential utilization of dairy industries by-products and wastes through microbial processes: A critical review. Science of The Total Environment 2022, 810, 152253. [Google Scholar] [CrossRef]
- Gheorghita, R.; Gutt, G.; Amariei, S.J.C. The Use of Edible Films Based on Sodium Alginate in Meat Product Packaging: An Eco-Friendly Alternative to Conventional Plastic Materials. 2020.
- Mellinas, C.; Valdés, A.; Ramos, M.; Burgos, N.; Garrigós, M.d.C.; Jiménez, A. Active edible films: Current state and future trends. 2016, 133. [CrossRef]
- Restrepo, A.E.; Rojas, J.D.; García, O.R.; Sánchez, L.T.; Pinzón, M.I.; Villa, C.C. Mechanical, barrier, and color properties of banana starch edible films incorporated with nanoemulsions of lemongrass ( Cymbopogon citratus) and rosemary ( Rosmarinus officinalis) essential oils. Food science and technology international = Ciencia y tecnologia de los alimentos internacional 2018, 24, 705–712. [Google Scholar] [CrossRef]
- Gobbetti, M.; Minervini, F.; Rizzello, C.G. Angiotensin I-converting-enzyme-inhibitory and antimicrobial bioactive peptides. 2004, 57, 173-188. [CrossRef]
- Korhonen, H.; Marnila, P.; Gill, H.S. Bovine milk antibodies for health. The British journal of nutrition 2000, 84 Suppl 1, S135–146. [Google Scholar] [CrossRef]
- Ryder, K.; Ali, M.A.; Carne, A.; Billakanti, J. The potential use of dairy by-products for the production of nonfood biomaterials. Critical Reviews in Environmental Science and Technology 2017, 47, 621–642. [Google Scholar] [CrossRef]
- Mazorra-Manzano, M.A.; Robles-Porchas, G.R.; González-Velázquez, D.A.; Torres-Llanez, M.J.; Martínez-Porchas, M.; García-Sifuentes, C.O.; González-Córdova, A.F.; Vallejo-Córdoba, B. Cheese Whey Fermentation by Its Native Microbiota: Proteolysis and Bioactive Peptides Release with ACE-Inhibitory Activity. 2020, 6, 19.
- Campos, C.A.; Gerschenson, L.N.; Flores, S.K. Development of Edible Films and Coatings with Antimicrobial Activity. Food and Bioprocess Technology 2011, 4, 849–875. [Google Scholar] [CrossRef]
- Bonnaillie, L.M.; Zhang, H.; Akkurt, S.; Yam, K.L.; Tomasula, P.M. Casein Films: The Effects of Formulation, Environmental Conditions and the Addition of Citric Pectin on the Structure and Mechanical Properties. 2014, 6, 2018-2036.
- Khan, M.R.; Volpe, S.; Valentino, M.; Miele, N.A.; Cavella, S.; Torrieri, E. Active Casein Coatings and Films for Perishable Foods: Structural Properties and Shelf-Life Extension. 2021, 11, 899.
- Babaei-Ghazvini, A.; Acharya, B.; Korber, D.R. Antimicrobial Biodegradable Food Packaging Based on Chitosan and Metal/Metal-Oxide Bio-Nanocomposites: A Review. Polymers (Basel) 2021, 13. [Google Scholar] [CrossRef]
- Chaudhary, V.; Kajla, P.; Kumari, P.; Bangar, S.P.; Rusu, A.; Trif, M.; Lorenzo, J.M. Milk protein-based active edible packaging for food applications: An eco-friendly approach. 2022, 9. [CrossRef]
- Nadarajah, S.K.; Vijayaraj, R.; Mani, J. Therapeutic Significance of Loligo vulgaris (Lamarck, 1798) ink Extract: A Biomedical Approach. Pharmacognosy research 2017, 9, S105–s109. [Google Scholar] [CrossRef]
- Giannakas, A.E.; Salmas, C.E.; Moschovas, D.; Zaharioudakis, K.; Georgopoulos, S.; Asimakopoulos, G.; Aktypis, A.; Proestos, C.; Karakassides, A.; Avgeropoulos, A.; et al. The Increase of Soft Cheese Shelf-Life Packaged with Edible Films Based on Novel Hybrid Nanostructures. 2022, 8, 539.
- Celli, G.B.; Ravanfar, R.; Kaliappan, S.; Kapoor, R.; Abbaspourrad, A. Annatto-entrapped casein-chitosan complexes improve whey color quality after acid coagulation of milk. Food Chemistry 2018, 255, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Chakrapani, V.; Ayaz Ahmed, K.B.; Kumar, V.V.; Ganapathy, V.; Anthony, S.P.; Anbazhagan, V. A facile route to synthesize casein capped copper nanoparticles: an effective antibacterial agent and selective colorimetric sensor for mercury and tryptophan. RSC Advances 2014, 4, 33215–33221. [Google Scholar] [CrossRef]
- Gebhardt, R.; Takeda, N.; Kulozik, U.; Doster, W. Structure and Stabilizing Interactions of Casein Micelles Probed by High-Pressure Light Scattering and FTIR. The Journal of Physical Chemistry B 2011, 115, 2349–2359. [Google Scholar] [CrossRef] [PubMed]
- Karydis-Messinis, A.; Moschovas, D.; Markou, M.; Gkantzou, E.; Vasileiadis, A.; Tsirka, K.; Gioti, C.; Vasilopoulos, K.C.; Bagli, E.; Murphy, C.; et al. Development, physicochemical characterization and in vitro evaluation of chitosan-fish gelatin-glycerol hydrogel membranes for wound treatment applications. Carbohydrate Polymer Technologies and Applications 2023, 6, 100338. [Google Scholar] [CrossRef]
- Ma, Y.; Xin, L.; Tan, H.; Fan, M.; Li, J.; Jia, Y.; Ling, Z.; Chen, Y.; Hu, X. Chitosan membrane dressings toughened by glycerol to load antibacterial drugs for wound healing. Materials Science and Engineering: C 2017, 81, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Fernández, C.; Ausar, S.F.; Badini, R.G.; Castagna, L.F.; Bianco, I.D.; Beltramo, D.M. An FTIR spectroscopy study of the interaction between αs-casein-bound phosphoryl groups and chitosan. International Dairy Journal 2003, 13, 897–901. [Google Scholar] [CrossRef]
- Astbury, W.T.; Lomax, R. X-Ray Photographs of Crystalline Pepsin. Nature 1934, 133, 795–795. [Google Scholar] [CrossRef]
- Clark, G.L.; Schaad, J.A. X-ray Diffraction Studies of Tendon and Intestinal Wall Collagen. 1936, 27, 339-356. [CrossRef]
- Arvanitoyannis, I.S.; Nakayama, A.; Aiba, S.-i. Chitosan and gelatin based edible films: state diagrams, mechanical and permeation properties. Carbohydrate Polymers 1998, 37, 371–382. [Google Scholar] [CrossRef]
- Karydis-Messinis, A.; Moschovas, D.; Markou, M.; Tsirka, K.; Gioti, C.; Bagli, E.; Murphy, C.; Giannakas, A.E.; Paipetis, A.; Karakassides, M.A.; et al. Hydrogel Membranes from Chitosan-Fish Gelatin-Glycerol for Biomedical Applications: Chondroitin Sulfate Incorporation Effect in Membrane Properties. 2023, 9, 844.
- Bengoechea, C.; Arrachid, A.; Guerrero, A.; Hill, S.E.; Mitchell, J.R. Relationship between the glass transition temperature and the melt flow behavior for gluten, casein and soya. Journal of Cereal Science 2007, 45, 275–284. [Google Scholar] [CrossRef]
Figure 1.
FTIR spectra of (a) pure casein, (b) pure chitosan, (c) Ch50Cas50, (d) Ch33s67, (e) Chi38Cas38Gly24 and (f) Chi32Cas32Gly20SqInk16.
Figure 1.
FTIR spectra of (a) pure casein, (b) pure chitosan, (c) Ch50Cas50, (d) Ch33s67, (e) Chi38Cas38Gly24 and (f) Chi32Cas32Gly20SqInk16.
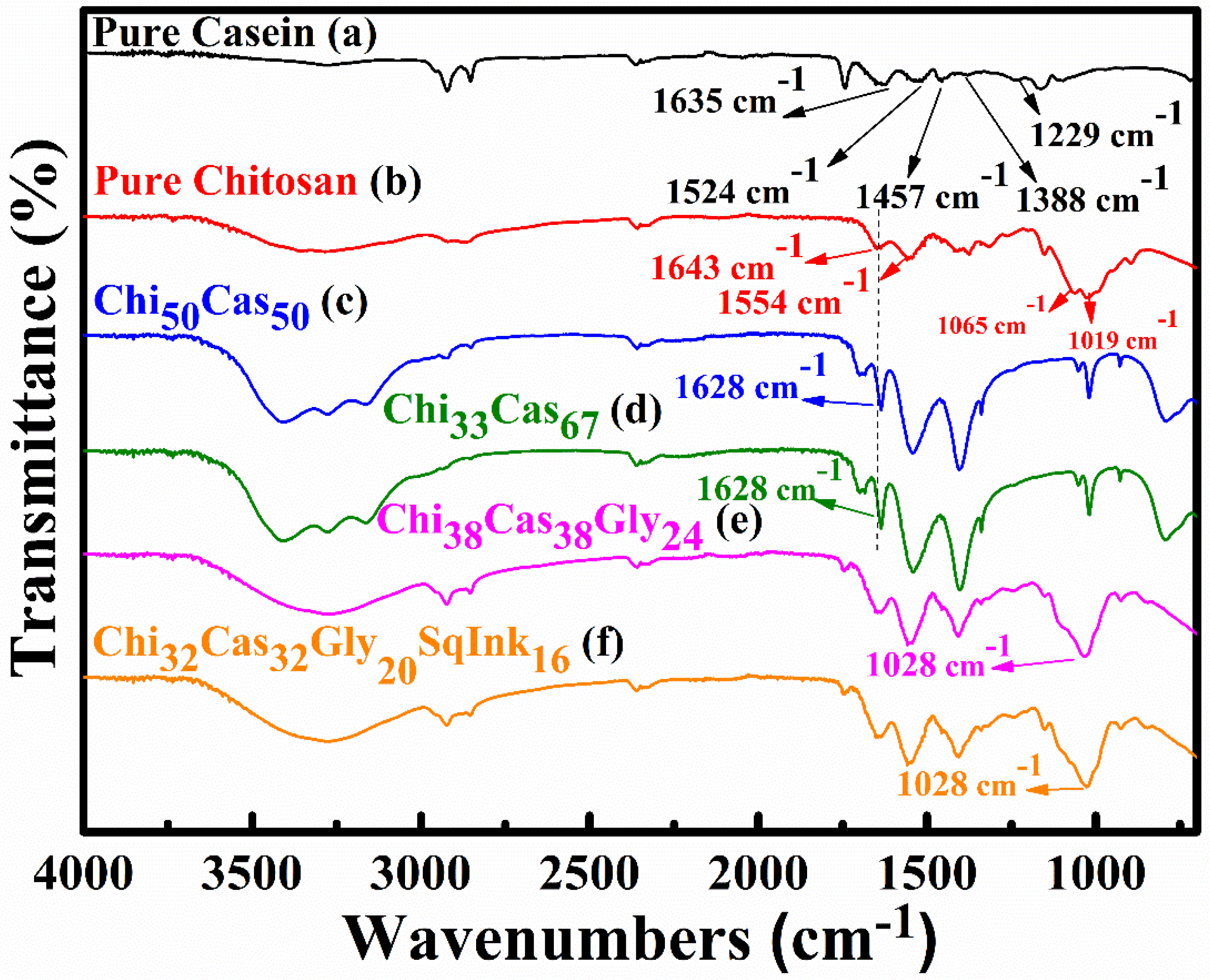
Figure 2.
XRD diffractograms of (a) pure casein, (b) pure chitosan, (c) Ch50Cas50, (d) Ch33Cas67, Chi38Cas38Gly24 and (f) Chi32Cas32Gly20SqInk16.
Figure 2.
XRD diffractograms of (a) pure casein, (b) pure chitosan, (c) Ch50Cas50, (d) Ch33Cas67, Chi38Cas38Gly24 and (f) Chi32Cas32Gly20SqInk16.
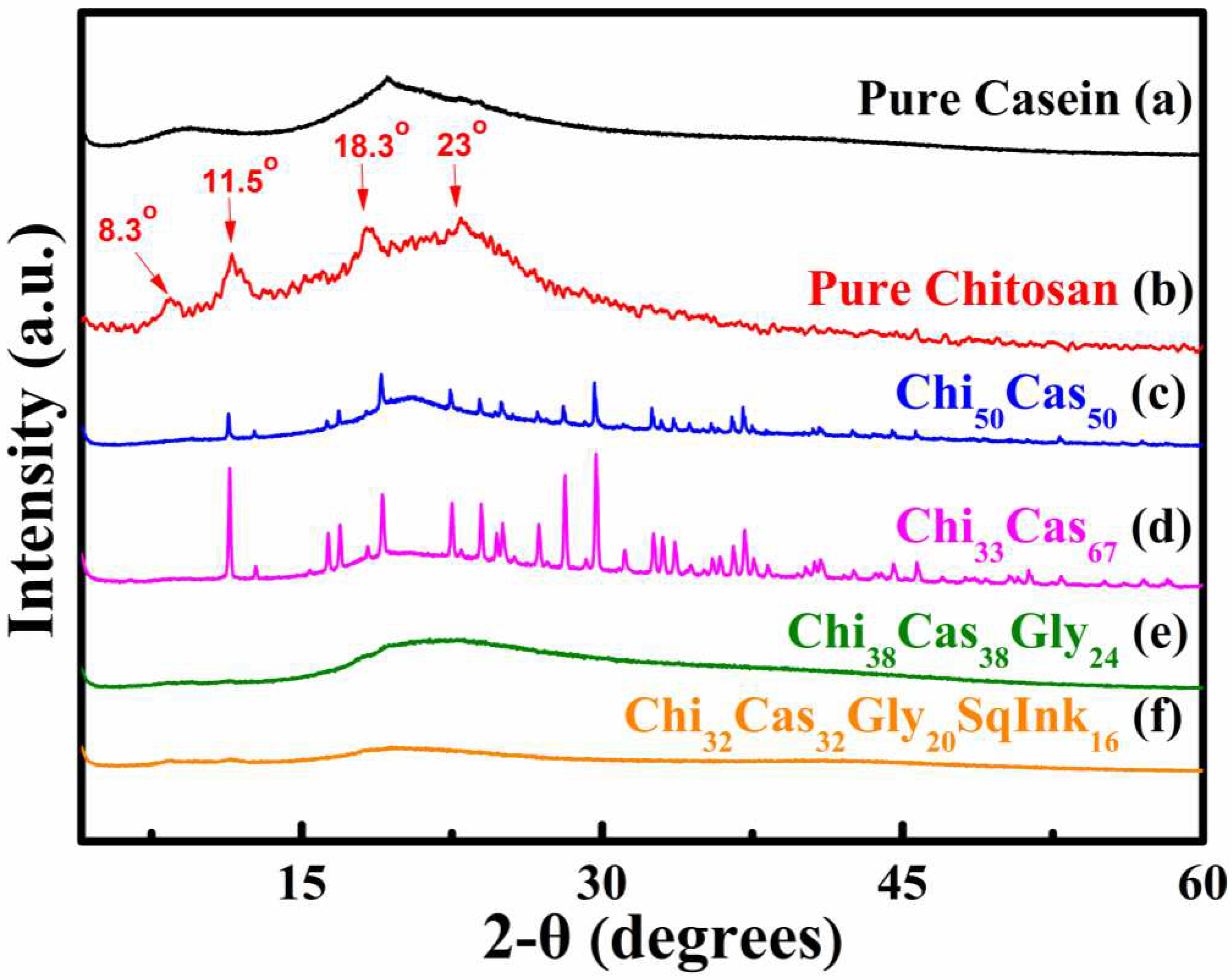
Figure 3.
TGA thermograms of (a) pure chitosan, (b) Ch50Cas50, (c) Ch33Cas67, (d) Chi38Cas38Gly24 and Chi32Cas32Gly20SqInk16.
Figure 3.
TGA thermograms of (a) pure chitosan, (b) Ch50Cas50, (c) Ch33Cas67, (d) Chi38Cas38Gly24 and Chi32Cas32Gly20SqInk16.
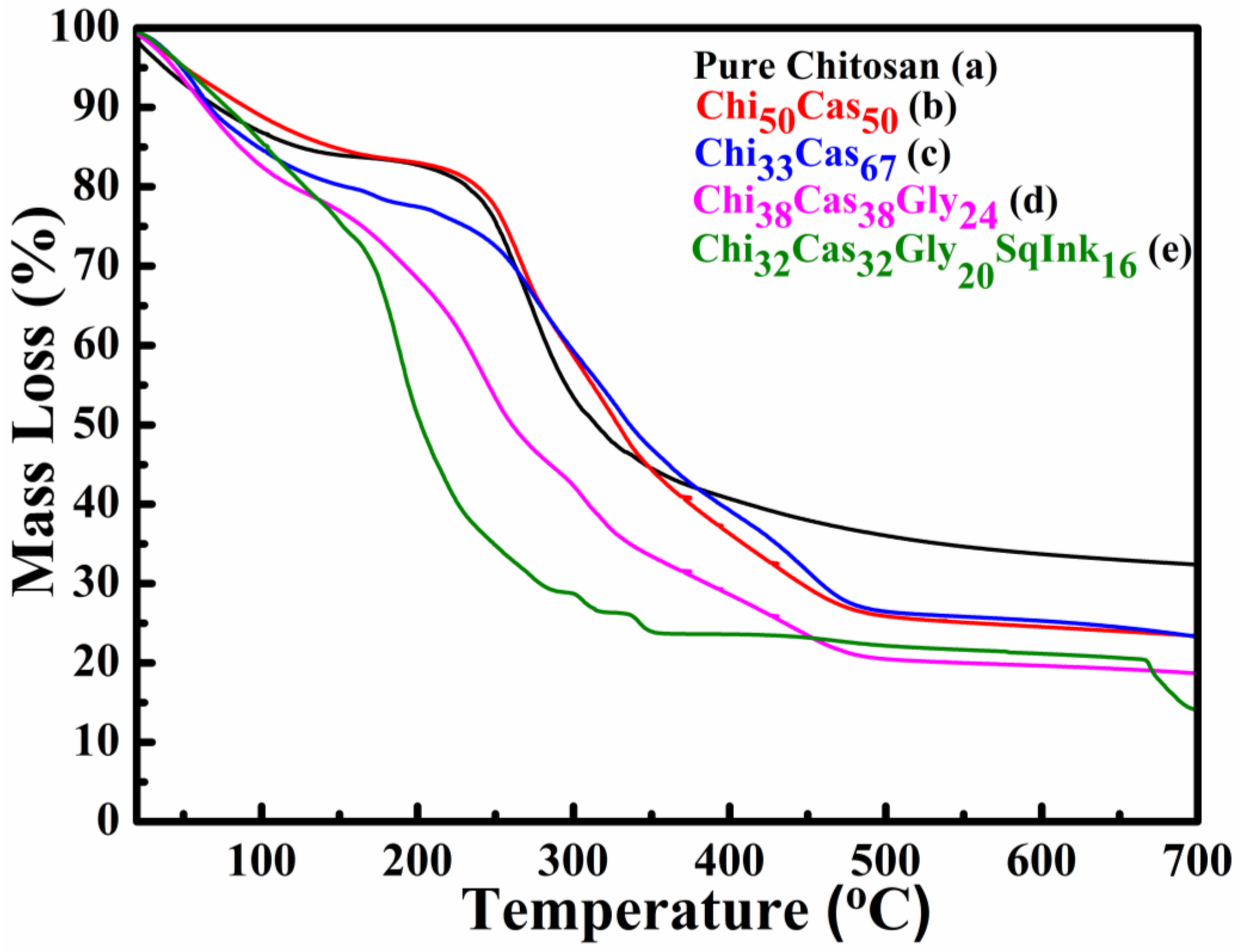
Figure 4.
Storage modulus plots of (a) pure chitosan, (b) Ch50Cas50, (c) Ch33Cas67, (d) Chi38Cas38Gly24 and Chi32Cas32Gly20SqInk16.
Figure 4.
Storage modulus plots of (a) pure chitosan, (b) Ch50Cas50, (c) Ch33Cas67, (d) Chi38Cas38Gly24 and Chi32Cas32Gly20SqInk16.
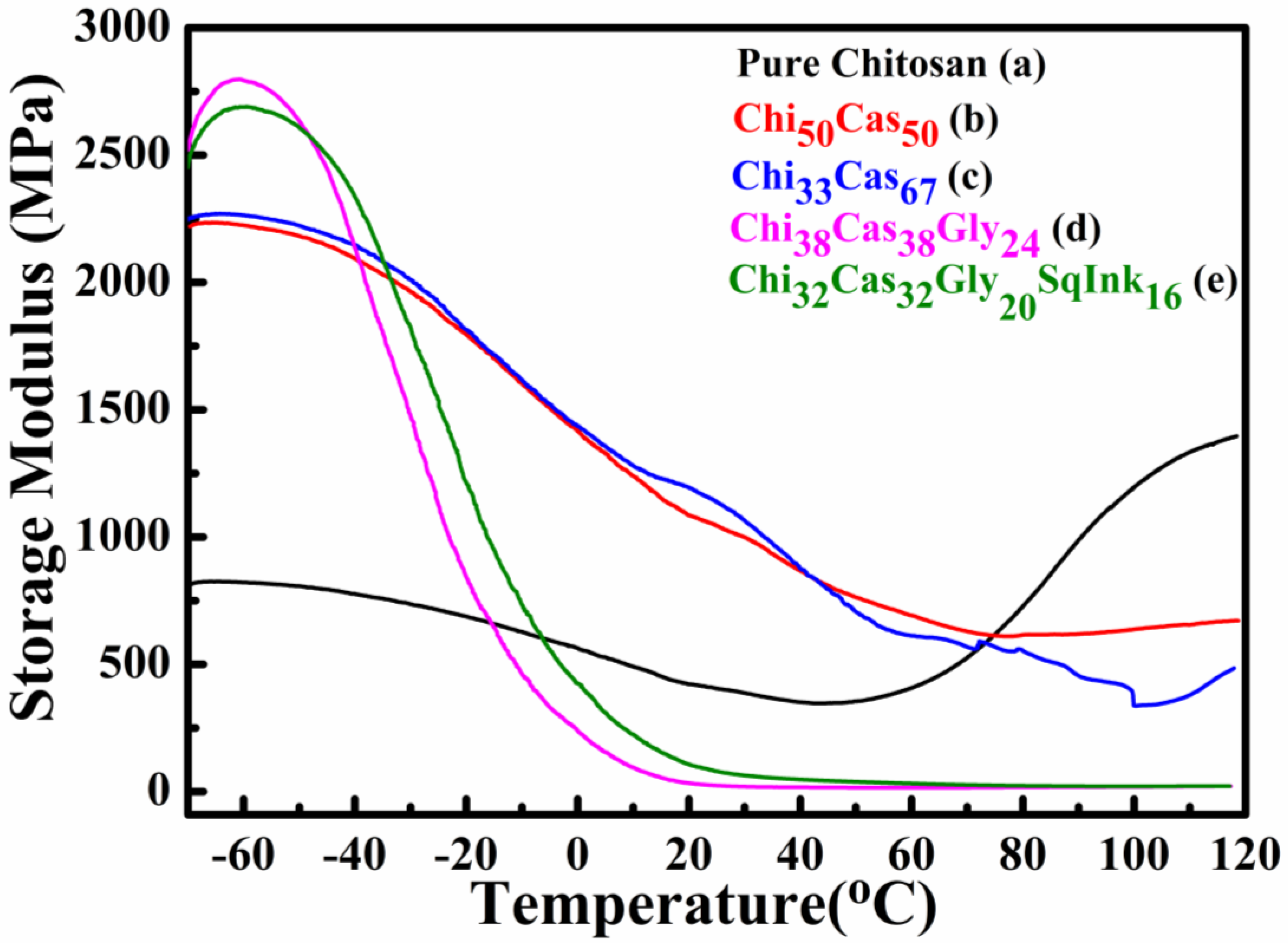
Figure 5.
Tan delta plots of (a) pure chitosan, (b) Ch50Cas50, (c) Ch33Cas67, (d) Chi38Cas38Gly24 and (e) Chi32Cas32Gly20SqInk16.
Figure 5.
Tan delta plots of (a) pure chitosan, (b) Ch50Cas50, (c) Ch33Cas67, (d) Chi38Cas38Gly24 and (e) Chi32Cas32Gly20SqInk16.
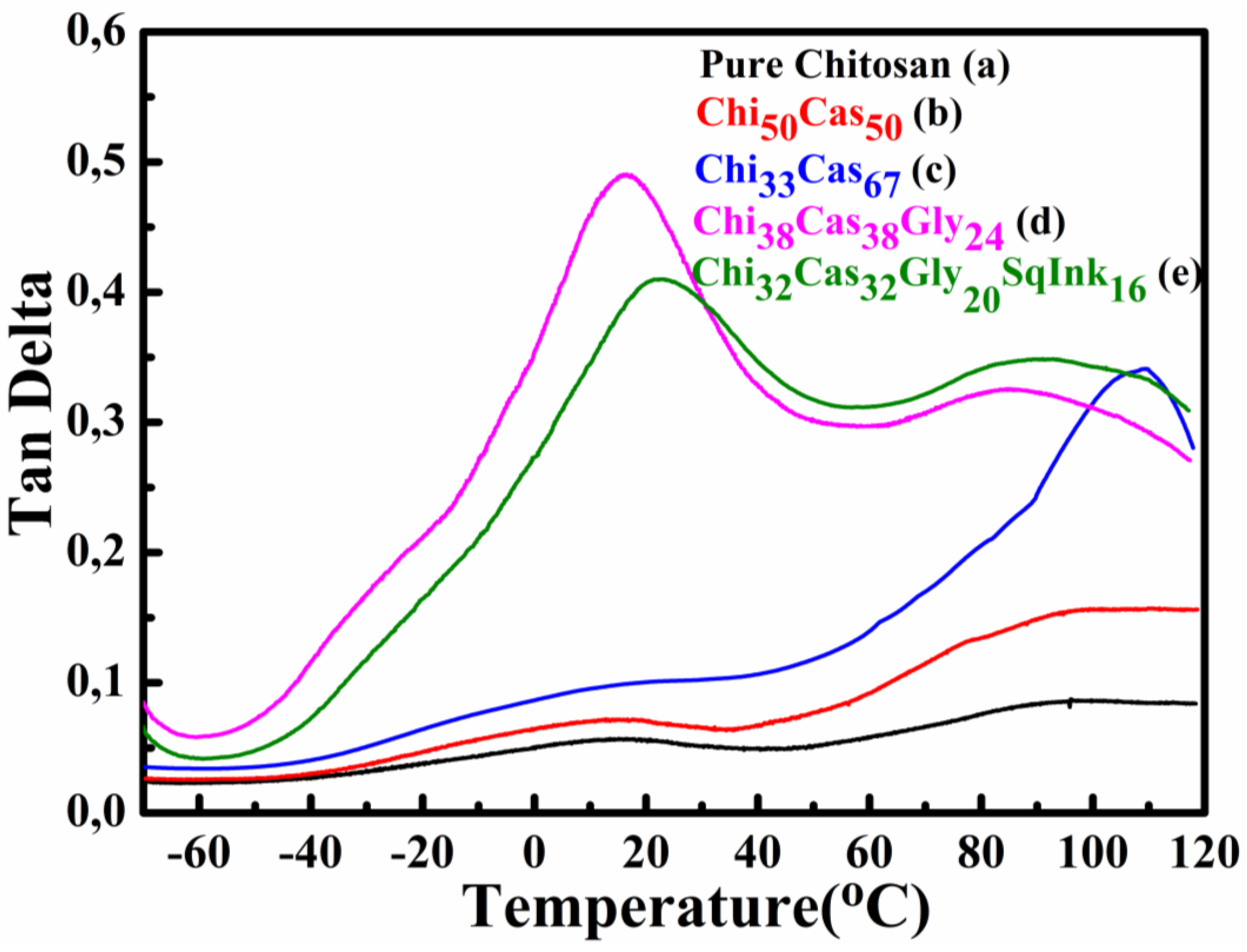
Figure 6.
Indicative stress vs strain plots of (a) pure chitosan, (b) Ch50Cas50, (c) Ch33Cas67, (d) Chi38Cas38Gly24 and Chi32Cas32Gly20SqInk16.
Figure 6.
Indicative stress vs strain plots of (a) pure chitosan, (b) Ch50Cas50, (c) Ch33Cas67, (d) Chi38Cas38Gly24 and Chi32Cas32Gly20SqInk16.
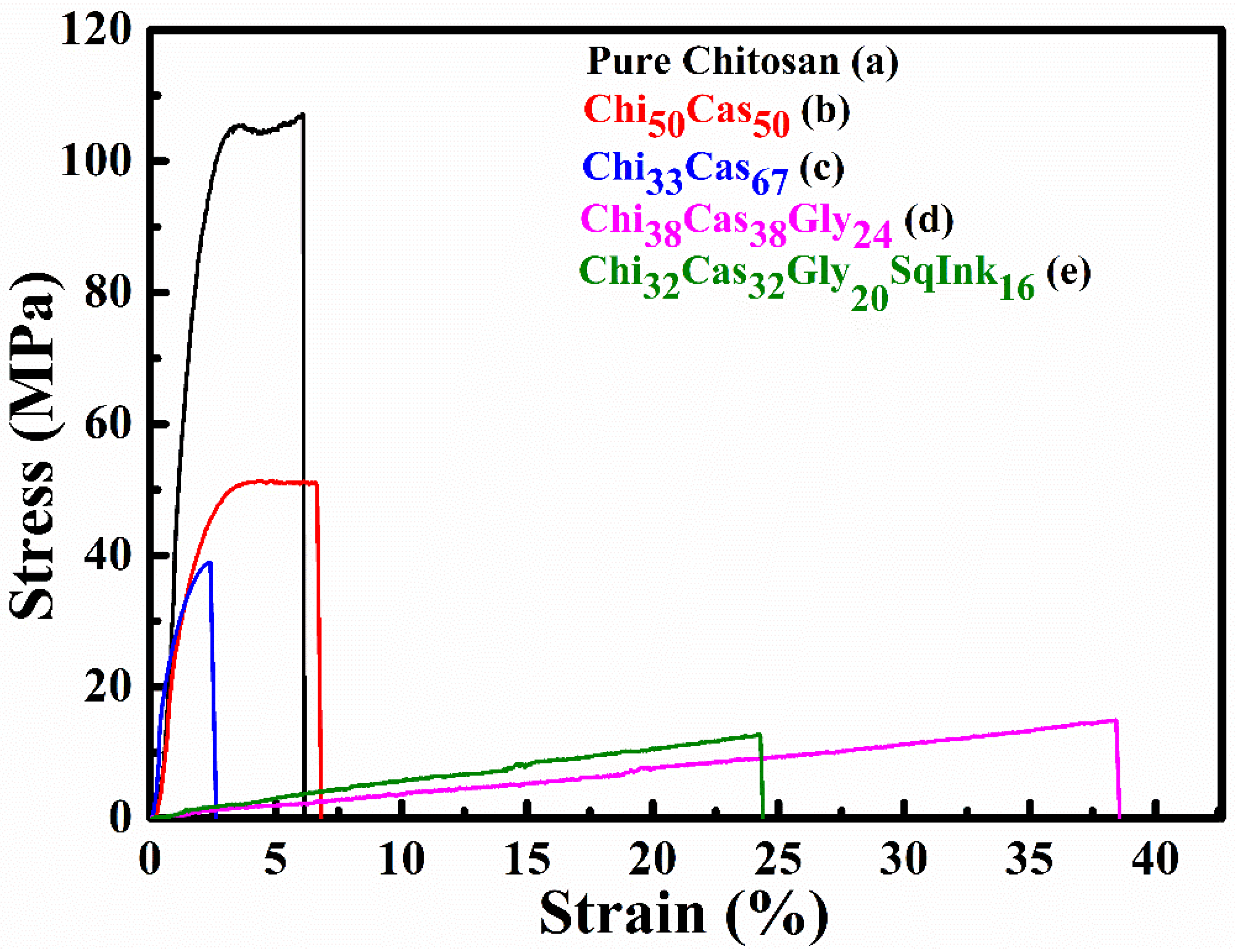
Figure 7.
SEM images of (a) pure casein, (b) pure chitosan, (c) Ch50Cas50, (d) Chi33Cas67, (e) Chi38Cas38Gly24 and (f) Chi32Cas32Gly20SqInk16.
Figure 7.
SEM images of (a) pure casein, (b) pure chitosan, (c) Ch50Cas50, (d) Chi33Cas67, (e) Chi38Cas38Gly24 and (f) Chi32Cas32Gly20SqInk16.


Table 1.
Abbreviations and compositions of the prepared hydrogel membranes.
| Sample code | Chitosan (w/v %) |
Casein (w/v %) |
Glycerol (w/v %) |
Squid Ink (w/v %) |
|---|---|---|---|---|
|
Chi32Cas32Gly20SqInk16 (%wt:32/32/20/16) |
2 | 2 | 1.26 | 1 |
|
Chi38Cas38Gly24 (%wt:38/38/24) |
2 | 2 | 1.26 | - |
|
Chi33Cas67 (%wt:33/67) |
2 | 4 | - | - |
|
Ch50Cas50 (%wt:50/50) |
2 | 2 | - | - |
|
Pure Chitosan (%wt:100) |
2 | - | - | - |
|
Pure Casein (powder) |
- | 100 | - | - |
Table 2.
Mechanical properties of the prepared hydrogel membranes.
| Specimen | Stress (MPa) | Strain (%) | % change in stress* | % change in strain* |
|---|---|---|---|---|
|
Pure Chitosan Reference system |
102.82 ± 6.97 | 6.77 ± 3.01 | Reference system | |
| Chi50Cas50 | 51.60 ± 2.00 | 7.66 ± 1.70 | -49.82 | +13.15 |
| Chi33Cas67 | 36.84 ± 2.02 | 2.61 ± 0.07 | -64.17 | -61.45 |
| Chi38Cas38Gly24 | 15.36 ± 0.63 | 38.42 ± 0.22 | -85.06 | +467.50 |
| Chi32Cas32Gly20SqInk16 | 12.18 ± 1.85 | 23.74 ± 3.56 | -88.15 | +250.66 |
*with respect to the reference system Pure Chitosan.
Table 3.
Water vapor transmission rate (WVTR) and water diffusivity (D) of the hydrogel membranes Pure Chitosan (a), Ch50Cas50 (b), Ch33s67 (c), Chi38Cas38Gly24 (d) and Chi32Cas32Gly20SqInk16 (e).
Table 3.
Water vapor transmission rate (WVTR) and water diffusivity (D) of the hydrogel membranes Pure Chitosan (a), Ch50Cas50 (b), Ch33s67 (c), Chi38Cas38Gly24 (d) and Chi32Cas32Gly20SqInk16 (e).
| Samples | WVTR [gr/(cm2*s)] | Dwv (cm2/s) |
|---|---|---|
| Pure Chitosan | 7.69367E-07(6.48778E-08) | 1.10E-04(1.98E-05) |
| Ch50Cas50 | 9.55637E-07(4.27094E-07) | 3.33E-04(0.79E-04) |
| Chi33Cas67 | 7.79506E-07(5.03817E-07) | 5.16E-04(1.04E-04) |
| Chi38Cas38Gly24 | 1.92141E-06(3.3557E-07) | 8.20E-04(1.69E-04) |
| Chi32Cas32Gly20SqInk16 | 1.53178E-06(3.217612E-07) | 8.51E-04(8.76E-05) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



