
Submitted:
05 February 2024
Posted:
06 February 2024
You are already at the latest version
Abstract
We developed an ovarian cancer-targeted drug delivery system based on a follicle-stimulating hormone receptor (FSHR) peptide. The lipophilic chemotherapeutic drug SN38 and the photosensitizer IR820 were loaded into the phospholipid bilayer of liposomes. Near-infrared laser irradiation stimulated the generation of reactive oxygen species, causing short-term reactive molecular species and spatial-temporal restricted photodamage to the biological targets. The reactive molecular species and spatial-temporal restricted photodamage enhanced the release and uptake of chemotherapy drugs and increased the sensitivity to chemotherapy drugs in ovarian cancer cells. FSH liposomes loaded with SN38 and IR820 (SN38/IR820-Lipo@FSH) were prepared using thin-film hydration-sonication. FSH peptide binding was analyzed using 1H NMR spectrum and Maldi-Tof. The average size and zeta potential of SN38/IR820-Lipo@FSH were 105.1 ± 1.15 nm (PDI: 0.204 ± 0.03) and −27.8 ± 0.42 mV, respectively. SN38/IR820-Lipo@FSH exhibited good drug-loading properties and the in vitro release was slow. FSH significantly increased the uptake of liposomes, inhibited cell proliferation, and induced apoptosis in A2780 cells. Moreover, SN38/IR820-Lipo@FSH exhibited good tumor-targeting ability and anti-ovarian cancer activity in vivo. The SN38/IR820-Lipo@FSH exhibited specific targeting to A2780 ovarian cancer cells. The combination of chemotherapy and photodynamic treatment based on an FSH peptide-targeted delivery system may be an effective approach to treating ovarian cancer.
Keywords:
FSHβ (33-53) peptide
; Ovarian cancer
; Targeting therapy
; PDT
; SN38
1. Introduction
Ovarian cancer, known as the “silent killer,” has the highest mortality rate of all gynecological tumors [1], and over 90% of ovarian cancers are epithelial carcinomas [2]. Over 60% of ovarian cancers are diagnosed at advanced stages due to ineffective early detection strategies, leading to a high mortality rate [3]. The five-year survival rate is above 50% [4,5]. Ovarian cancer originates from the ovarian surface epithelial cells or the fallopian tube secretory epithelial cells, and malignant cells can spread directly to the peritoneum, resulting in metastasis and poor treatment outcomes [6]. Improvements in ovarian cancer treatments are urgently needed. Current research is focused on accurately targeting cancer cells and enhancing the efficacy of therapeutic drugs while reducing side effects. Chemotherapy remains the most important treatment modality for ovarian cancer.
SN38 (7-ethyl-10-hydroxythectothecin) is an active metabolite of irinotecan (CPT-11) and a derivative of camptothecin obtained by chemical structure modification. It has strong anti-tumor effect and high anti-tumor activity [7]. In vitro cytotoxicity test results showed that the anti-tumor activity of SN38 was 100-1000 times that of irinotecan for some tumor cell lines. However, due to the extremely poor physiological and pharmaceutical compatibility, and low tumour targeting ability, SN38 cannot be administered directly as a chemotherapeutic agent [8]. Clinical use of SN-38 is limited by poor water solubility and instability at physiological pH. The lactone ring of SN-38 is hydrolyzed at pH > 6 resulting in the inactive carboxylate form [9]. Recently, nanomedicines improved the pharmacological and therapeutic properties of SN-38. Nanosized delivery systems can improve anticancer efficacy, reduce side effects, and reverse drug resistance by targeting tumor tissues through enhanced permeability and retention and active targeting strategies [10]. Many attempts have been made to develop intravenous or oral preparations of SN-38 with better antitumor effects. Liposomes are small spherical vesicles derived from naturally occurring non-toxic phospholipids and are biodegradable nanosystems. Liposomes have been extensively utilized as drug delivery systems. Onivyde (Irinotecan liposome injection), a pioneer in the development of liposomes loaded with camptothecin and its derivatives, is used to treat patients with tumors. Although SN38-based chemotherapy improved the efficacy of ovarian cancer treatment, drug resistance develops in cancer cells and serious side effects occur. To overcome the inherent limitations of single chemotherapy drugs, chemotherapy has been combined with phototherapy induced by photosensitizers as a practical clinical cancer treatment platform. Combining chemotherapy and phototherapy increases the sensitivity of cancer cells to chemotherapy and improves the uptake efficiency of chemotherapeutic drugs by increasing cancer cell membrane permeability [11].
Photodynamic therapy (PDT) involves light activation of photosensitizers and the generation of short-lived reactive molecular species that induce spatially and temporally controlled photodamage to cellular organelles and biological targets [12]. PDT induces subcellular toxicity, which sensitizes target cells to subsequent therapies, including chemotherapy and direct cell killing [13]. PDT can be combined with chemotherapy, immunotherapy, gene therapy, or other treatment modalities to enhance antitumor efficacy [14,15]. By irradiating lesion sites at specific wavelengths, photosensitizers selectively accumulate and become activated in diseased tissue, triggering a photochemical reaction that destroys the lesion. Photosensitizers in PDT transfer energy to the surrounding oxygen, generating highly reactive singlet oxygen. Singlet oxygen can react with nearby biomolecules, producing cytotoxic effects that kill cancer cells [16,17]. Indocyanine green (IR820) is a clinical infrared fluorescent dye that efficiently absorbs laser light for photothermal and photodynamic therapy. IR820 is non-toxic until exposed to laser irradiation. The limitations of IR820 include rapid clearance (blood half-life of 2–4 min) and low cellular uptake. These limitations restrict the diagnostic and treatment applications of IR820. Enhancing the intracellular release of chemotherapeutic drugs during the PDT process is essential to amplifying the synergistic therapeutic effects of chemo-photodynamic therapy.
The follicle-stimulating hormone receptor (FSHR), a G protein-coupled receptor with seven transmembrane domains, is found on the surfaces of cells in the ovaries, testes, and other reproductive organs [18,19]. Follicle-stimulating hormone (FSH) is a heterodimeric glycoprotein composed of non-covalently bound α and β subunits [20]. The α subunit is encoded by a single gene shared by FSH, luteinizing hormone (LH), human chorionic gonadotropin (hCG), and thyroid-stimulating hormone (TSH). The β subunit is specific to FSH. The association of FSH β with the FSHR in the cell membrane affects target cells, such as granulosa cells and Sertoli cells. FSHβ 33–53 and FSHβ 81–95 peptides promote paclitaxel-loaded NP entry into FSH receptor (FSHR)-positive ovarian tissue to achieve excellent antitumor effects [21]. Thus, targeting therapy mediated by FSHR has a high potential for ovarian cancer treatment [18,20].
We developed an ovarian cancer-targeting drug delivery system based on the FSHβ (33–53) peptide. The lipophilic chemotherapeutic drug SN38 and the photosensitizer IR820 were loaded into the phospholipid bilayer of the liposomes. Near-infrared laser irradiation generates reactive oxygen species via IR820 to induce short-term reactive molecular species and spatial-temporal restricted photodamage in biological targets. These effects induce the release and uptake of chemotherapy drugs and increase sensitivity to chemotherapy drugs in ovarian cancer cells, as illustrated in Figure 1. Chemotherapy and phototherapy act synergistically and this dual-delivery liposome may be a promising strategy for tumor treatment.
2. Materials and Methods
2.1. Materials
SN38 was purchased from Dalian Meilun Biotechnology Co., Ltd. (Dalian, P. R. China). The FSH β (FSH) peptide (YTRDLVYKDPARPKIQKTCTF) was custom-synthesized by Nanjing TGpeptide Biotechnology Co., Ltd. (Nanjing, China). New indocyanine green (IR820) and Coumarin-6 (98%) were obtained from J&K Scientific Co., Ltd. (Beijing, China). DSPE-PEG2000, DSPE-PEG2000-Mal, 2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), and cholesterol were purchased from AVT (Shanghai) Pharmaceutical Technology Co., Ltd. (Shanghai, China). The Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Laboratories (Kumamoto, Japan). The Fluorescein Isothiocyanate (FITC)-Annexin V/Propidium Iodide (PI) apoptosis detection kit was acquired from Beyotime Biotechnology Co., Ltd. (Shanghai, China). The ROS Detection Kit was purchased from Enzo Life Sciences Co., Ltd. (Beijing, China). The Calcineurin-AM/PI Staining Kit and 4′, 6-diamidino-2-phenylindole (DAPI) were purchased from Solarbio Life Sciences Co., Ltd. (Beijing, China). The 1,1-dioctadecyltetramethyl indotricarbocyanine iodide (DIR) was obtained from Biotium Inc. (Hayward, CA, USA). Chloroform and methanol (HPLC grade) were purchased from Sigma-Aldrich Co. (St Louis, MO, USA). Fetal bovine serum (FBS) was purchased from GIBCO LLC. (Grand Island, NY, USA). RPMI 1640 medium, DMEM medium, and phosphate-buffered saline (PBS) were obtained from Thermo Fisher Scientific Co., Ltd. (Beijing, China).
2.2. Cells and Animals
High FSHR-expressing mouse A2780 cells and low FSHR-expressing HepG2 and C26 cells were purchased from the Department of Pathology at Peking Union Medical College Institute of Medicinal Biotechnology. A2780 cells and C26 cells were cultured in RPMI-1640 medium with 10% FBS at 37°C and 5% CO2. HepG2 cells were cultured in DMEM media with 10% FBS at 37°C and 5% CO2 in a humidified atmosphere. All experiments were performed in cells at the logarithmic growth stage.
Female BALB/c Nude mice (initial weight 18-20 g) were obtained from Vital River Laboratory Animal Technology Co., Ltd. (Beijing, PR China) and housed under standard conditions. All animal experiments were conducted following the guidelines prepared and approved by the Laboratory Animal Ethics Committee of the Institute of Materia Medica in Chinese Academy of Medical Sciences and Peking Union Medical College.
2.3. DSPE-PEG2000-FSH Synthesis and Characterization
The synthesis of DSPE-PEG2000-FSH was based on the method by Hong et al. [21], with slight modifications. The FSH-Cys cysteine residue was coupled to DSPE-PEG2000-MAL to make DSPE-PEG2000-FSH. FSH-Cys and DSPE-PEG2000-MAL (molar ratio 1:1) were dissolved in HEPES buffer at pH 8.0, and nitrogen was stirred at room temperature for 48 h. The reaction was conducted in dialysis bags in water. The unreacted FSH peptide was removed by aeration in water through a dialysis bag (MWCO: 2500) for 48 h. The final product was obtained by freeze-drying. The DSPE-PEG2000-FSH synthesis was confirmed by 1H nuclear magnetic resonance spectroscopy (400 MHz, Varian Medical Systems, Inc., Palo Alto, CA, USA) and matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF-MS) (4800 Plus, Applied Biosystems Inc., Waltham, MA, USA).
2.4. Liposome Preparation with or without FSH Peptide Modification
SN38/IR820-Lipo@FSH liposomes were prepared using thin-film hydration. After dissolving DOPC, cholesterol, DSPE-PEG2000, DSPE-PEG2000-FSH, SN38, and IR820 in chloroform, the organic solvents were removed by rotary evaporation at 37°C. The lipid membranes were hydrated with deionized water under spinning for 30 min. The whole mixture was sonicated at 65 W for 10 min with 2 s of sonication and 2 s of a break in an ice bath using an ultrasonic cell pulverizer (Scientz 950E; Ningbo Scientz Biotechnology Co., Ltd., Zhejiang, P. R. China). The unbound SN38 and IR820 were removed by filtration through a 0.22 μm nitrocellulose membrane. Samples were stored at 4°C. Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, and SN38/IR820-Lipo (without FSH peptide) were prepared using the same method.
2.5. Characterization of the liposomes
The average particle size, polydispersity index, and zeta potential of SN38/IR820-Lipo@FSH were measured using dynamic light scattering (DLS) and electrophoretic light scattering (Zetasizer Nano ZS90, Malvern Instruments, Malvern, UK). The structure and morphology were examined using transmission electron microscopy (JEM-1400PLUS, JEOL Ltd., Tokyo, Japan). The SN38 and IR820 concentrations were determined using high-performance liquid chromatography (HPLC, Agilent 1200 infinity; Agilent Technologies, Santa Clara, CA, USA) and UV-VIS spectrophotometry (TU-1810, Pulse Analyzer General Instrument Ltd., Beijing, China), respectively. The detection wavelength of SN38 was 265 nm, and methanol:water (70:30) was used as the mobile phase. Free SN38 and IR820 were separated from the liposomes using minicolumn centrifugation [22]. The entrapment efficiency (EE%) and drug loading capacity (DL%) were calculated as follows:
EE (%)=(weight of encapsulated drug)/(weight of total drug)× 100%
DL(%)=(weight of encapsulated drug)/(weight of total drug and lipid)× 100%
To determine the stability of SN38/IR820-Lipo@FSH, the prepared solution was stored at 4°C for 7 days in PBS or PBS containing 50% of serum. The color, transparency, particle size, zeta potential, and PDI of SN38/IR820-Lipo@FSH were recorded during the days.
The in vitro release of drugs from SN38/IR820-Lipo@FSH in a simulated physiological environment was evaluated using dialysis. SN38/IR820-Lipo@FSH solution (0.5 mL) was placed in a dialysis bag (MW: 10 kDa; MYM Biological Technology Co., Ltd., Hyderabad, India), and the dialysis bag was immersed in 10 mL of PBS (pH 7.4) containing 0.5% (v/v) Tween 80. The dialysis solution was stirred at 100 rpm for 12 h at 37°C, and 0.5 ml aliquots were removed at 0.5, 1, 2, 4, 6, 8, 10, 12, 24, 48, and 72 h. The aliquot volume was replaced with fresh medium. The SN38 and IR820 concentrations in the samples were determined using HPLC and UV-VIS spectrophotometry respectively. The cumulative release of SN38 and IR820 over time was calculated.
2.6. Interaction of SN38/IR820-Lipo@FSH with FSHR in vitro
SN38/IR820-Lipo and SN38/IR820-Lipo@FSH were added to A2780 cells and incubated at 37°C for 24 h to measure binding to the FSHR. Cells were washed twice with cold PBS and lysed (lysis buffer: 50 nM Tris-HCl pH = 6.8, 2% SDS, 6% Glycerol, 1% β-mercaptoethanol, 0.04% bromophenol blue). The cell lysates were incubated for 30 min at 4°C and centrifuged for 15 min at 12000 × g. The protein concentration was determined using a BCA Protein Assay Kit (Lot: PA115-01, Tiangen Biotech (Beijing) CO., LTD). The protein lysates were separated on sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene difluoride membranes. The membranes were incubated with ACTIN (Servicebio, GB12001) and FSHR antibodies (1:1000; Cell Signaling, 2808s) overnight at 4°C. After washing, the membranes were incubated with goat anti-rat IgG antibody for 1 h, and bands were detected using enhanced chemiluminescence (ImageQuant LAS 4000 mini, Fuji, Japan) [23].
2.7. Cellular Uptake Analysis
As a drug delivery system, the efficiency of liposome internalization by target cells is crucial [24]. Cou-6, a fluorescent probe and laser dye, was used to track liposome uptake [25]. Cou-6-labeled liposomes with or without FSH modifications (Cou-6-Lipo and Cou-6-Lipo@FSH) were prepared. A2780 cells, HepG2 cells, and C26 cells (1.5 × 105 cells/well) were seeded on circular glass slides in 12-well plates. After incubation at 37°C for 24 h, the cells were treated with free Cou-6, Cou-6-Lipo, or Cou-6-Lipo@FSH (Cou-6, 5 µg/mL) for 4 h. After washing three times with cold PBS buffer, the cells were fixed with 4% paraformaldehyde. Cells were stained with DAPI to visualize the cell nuclei. Finally, cells were imaged with a confocal laser scanning microscope (Carl Zeiss LSM 710; Carl Zeiss Microscope, Jena, Germany).
Free Cou-6, Cou-6-Lipo, and Cou-6-Lipo@FSH uptake efficiencies in A2780 cells were quantified using a flow cytometer (Becton Dickinson, Franklin Lake, NJ, USA). A2780 cells were seeded in a 12-well plates (1.5 × 105 cells/well). After 24 h, the cells were treated with free Cou-6, Cou-6-Lipo, or Cou-6-Lipo@FSH (Cou-6, 1 µg/mL) in serum-free culture medium at 37°C for 4 h. Cells were washed three times with cold PBS, treated with 0.25% trypsin, centrifuged, and resuspended in 0.5 mL PBS. The fluorescence intensity of the cells was measured by flow cytometry.
2.8. ROS Generation
To measure ROS production, A2780 cells (1.5 × 105 cells/well) were seeded into 12-well plates and cultured for 24 h at 37°C and 5% CO2. After washing three times with cold PBS, the cells were treated with Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, SN38/IR820-Lipo, SN38/IR820-Lipo@FSH (SN38: 1 µg/mL; IR820: 1 µg/mL), or physiological saline (control) for 4 h. The cells were treated with 1 mL of the ROS detection probe DCFH-DA (25 μM) and incubated for 10 min. The photic group was exposed to 5 min of 808 nm laser irradiation (0.5 W/cm2). Cells were treated following the kit instructions, and the fluorescence resulting from ROS production was observed using an inverted fluorescence microscope (CKX41, Olympus, Japan).
2.9. Cell Apoptosis Assay
Cell apoptosis was qualitatively evaluated using Calcein-AM/PI staining. A2780 cells were seeded into 12-well plates and incubated for 24 h. Cells were treated with Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, SN38/IR820-Lipo, or SN38/IR820-Lipo@FSH (SN38 and IR820 both 1 μg/mL) diluted in fresh culture medium. After 4 h of treatment, the cells were irradiated with an 808 nm laser (0.5 W/cm2) for 3 min. After 24 h, Calcein-AM/PI live/dead cell staining was performed. Cell apoptosis was observed and photographed using an inverted fluorescence microscope.
The apoptosis rate of A2780 cells was quantified using an Annexin V-FITC/PI apoptosis detection kit. A2780 cells (1.5 × 105 cells/well) were inoculated in 6-well plates and cultured for 24 h. Cells were treated as previously described, and irradiated for 3 min using an 808 nm laser (0.5 W/cm2). After 24 h, the cells were treated with 0.25% EDTA-free trypsin, washed three times with cold PBS, centrifuged, and resuspended in binding buffer. Annexin V-FITC/PI apoptosis detection was performed according to the manufacturer’s protocol. Cells were immediately analyzed using flow cytometry.
2.10. Wound healing Assay
The migratory ability of tumor cells after treatment with different formulations were evaluated. A2780 cells were plated in 12-well plates and cultured until they formed a monolayer. A scratch wound was created in the middle of the well using a sterile 200-μL pipette tip. The cells were washed with PBS to remove detached cells. The cells were treated with free Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH (Laser +), SN38/IR820-Lipo (Laser +) or SN38/IR820-Lipo@FSH (Laser +) (SN38 and IR820 both 0.2 μg/mL). Fresh medium was used as control. Migrating cells in the wound area were observed and imaged at 0 and 24 h using an inverted light microscope (Olympus, Hamburg, Germany).
2.11. Cell Viability Assay
The cytotoxic effects of the blank vectors on A2780 cells were measured using the CCK-8 assay. A2780 cells (4 × 103 cells/well) were seeded in a 96-well plates. After 24 h, the cells were treated with 0.001–50 μg/mL Blank-Lipo in fresh media for 24 or 48 h. After replacing the micelle with CCK-8 solution, the cells were incubated for 3 h at 37°C. The optical density was measured at 450 nm using a Synergy H1 Microplate Reader (BioTek Instruments, Inc., Winooski, VT, USA). Cells treated with culture media only were used as the control. Each group consisted of three parallel samples.
The inhibitory effects of SN38/IR820-Lipo and SN38/IR820-Lipo@FSH on A2780 cell proliferation were determined using the CCK-8 assay. A2780 cells (4 × 103 cells/well) were seeded into 96-well plates. After 24 h, cells were treated with SN38/IR820-Lipo and SN38/IR820-Lipo@FSH at SN38 concentrations of 0.001, 0.01, 0.1, 1, 10, and 50 μg/mL for 2 h. Control and Blank groups were included in the analysis. Cells were irradiated with an 808 nm laser (0.5 W/cm2) for 3 min per well, and the cells were incubated at 37°C for another 24 or 48 h. CCK-8 reagent was added to each well, and the optical density was measured at 450 nm. Cell viability (%) was calculated as follows:
Cell viability (%) = [(Atest − Ablank)/(Acontrol − Ablank)] ×100%
2.12. In Vivo Imaging
BALB/c Nude mice were injected in the right flank with 200 μL of A2780 cells (1 × 108 cells/mL). After tumor volumes reached 100 mm3, mice were randomly divided into three groups (n = 3/group). Free DIR, DIR-loaded liposomes (DIR-Lipo), or DIR-loaded liposomes with FSH (DIR-Lipo@FSH) were injected intravenously at a dose of 0.1 mg/kg of DIR. Mice were anesthetized and imaged using an in vivo imaging system (Caliper Life Sciences Inc., Mountain View, CA, USA) 1, 4, 8, and 24 h after injection. Hearts, livers, spleens, lungs, kidneys, and tumor tissues were collected from the mice 24 h after injection to assess the distribution of the different formulations in the major organs. Regions of interest were quantitatively analyzed using Living Image software (Version 4.3.1; Caliper Life Sciences Inc.).
2.13. In Vivo US-induced ROS generation
To evaluate US-induced ROS generation at the tumor sites, the tumor-bearing BALB/c nude mice (n = 3/group) were intravenously injected with PBS, Free IR820, SN38/IR820-Lipo, and SN38/IR820-Lipo@FSH (IR820 dose of 10 mg/kg). After 24 h, the mice were anesthetized with chloralic hydras and injected with 50 µg DCFH-DA intra-tumorally. After 10 min, the tumor sites of the mice were irradiated (808 nm, 0.5 W/cm2) for 10 min and then, the mice were sacrificed. The tumors were collected and flash frozen, conterstained with DAPI, and observed by CLSM.
2.14. Antitumor Efficacy and Safety Assessment
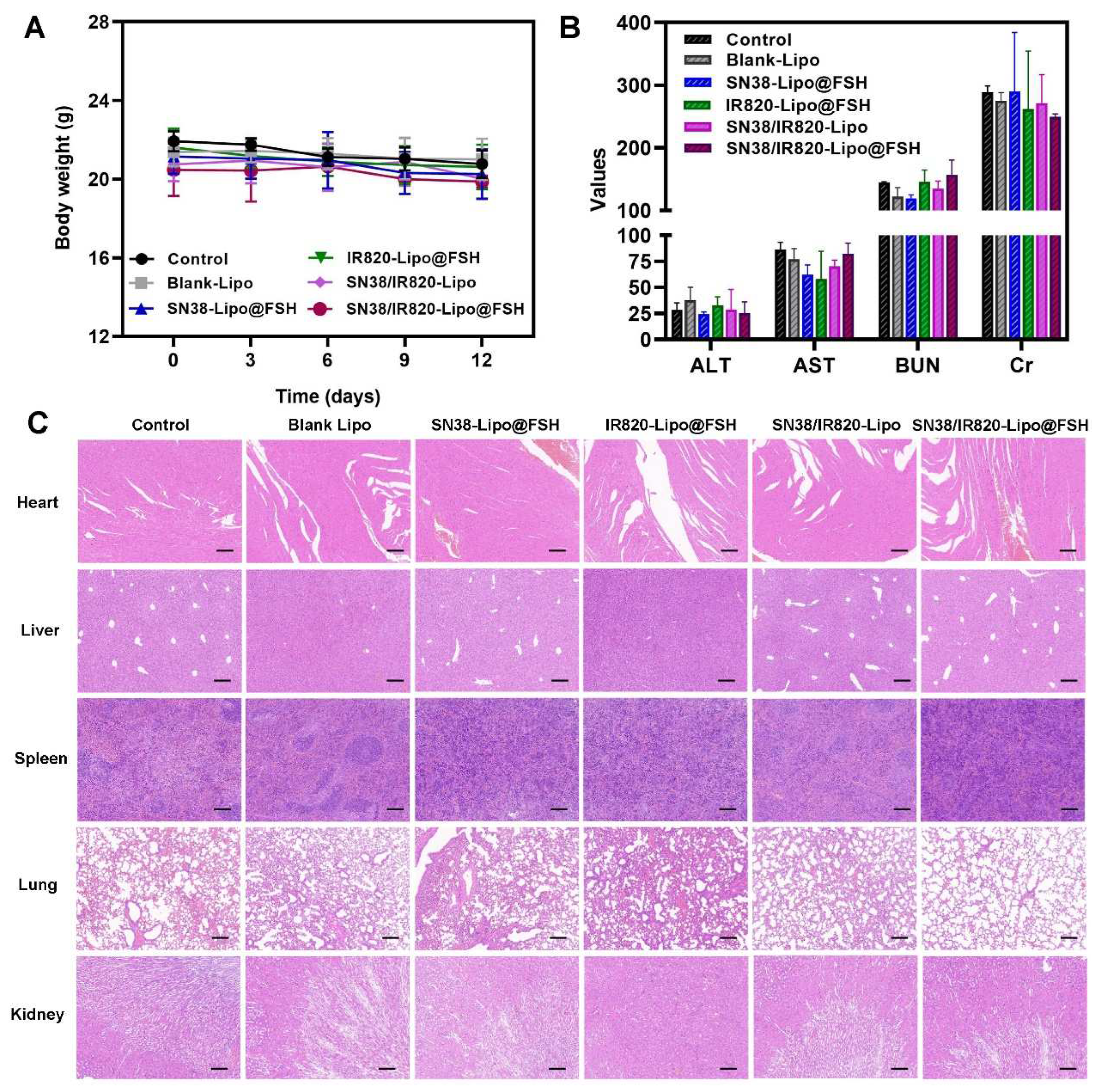
BALB/c nude mice were injected in the right flank with 200 μL of A2780 cell suspension (1 × 108 cells/mL). After the tumor volumes reached 100 mm3, mice were randomly divided into six groups (n = 4/group). Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, SN38/IR820-Lipo, SN38/IR820-Lipo@FSH (SN38, 10 mg/kg; IR820, 10 mg/kg), or saline (Negative Control) were injected intravenously. All the formulations were given to mice via the tail vein every 4 days for five times. Mice in the SN38/IR820-Lipo@FSH group were subjected to 808 nm laser irradiation (0.5 W/cm2) for 10 min on the second day after the injection. The irradiation treatment was repeated once every three days for a total of five treatments. Body weights and tumor dimensions (measured with a caliper) were recorded after each treatment.
Three days after the final treatment, mice were euthanized by cervical dislocation after blood collection from the orbital sinus. Hearts, livers, spleens, lungs, and kidneys were collected, fixed with 4% paraformaldehyde, embedded in paraffin, and stained with hematoxylin and eosin for histopathological analysis. Apoptotic cells were detected in the tumor tissues using terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL). Lung tissues were fixed in Bouin’s solution for 24 h and immersed in 50% ethanol for 2 h to detect tumor lung metastases. Blood samples were centrifuged at 3000 rpm at 4°C for 10 min to collect serum for measuring blood urea nitrogen, creatinine, aspartate transaminase, and alanine transaminase levels.
2.15. Statistical Analysis
All data are presented as the means ± standard deviations. GraphPad Prism software was used for data analysis. Groups were compared using two-tailed Student’s t-tests or one-way analysis of variance. Differences between groups were considered statistically significant at *P < 0.05, **P < 0.01, and ***P < 0.001.
3. Results
3.1. Synthesis and Characterization of DSPE-PEG2000-FSH
The synthesis of DSPE-PEG2000-FSH is depicted in Figure 2A. The FSH peptide was coupled with DSPE-PEG2000-MAL through an addition reaction. The maleimide group on DSPE-PEG2000-MAL reacted with the thiol group on the FSH peptide with nitrogen protection, resulting in the formation of DSPE-PEG2000-FSH. The 1H-NMR spectrum of DSPE-PEG2000-MAL and DSPE-PEG2000-FSH are shown in Figure 2B. The maleimide double bond was observed at 6.74 ppm in DSPE-PEG2000-MAL but not in DSPE-PEG2000-FSH, confirming the successful conjugation. The MALDI-TOF-MS spectrum showed that DSPE-PEG2000-MAL had an average relative molecular weight of about 2000~2200 Da, and the synthesized DSPE-PEG2000-FSH had an average relative molecular weight of about 4600~4700 Da. Considering that the molecular weight of FSH is 2543 Da, the increased molecular weight coincides with the molecular weight of FSH, indicating that FSH has successfully connected to DSPE-PEG. (Figure 2C, 2D).
3.2. Characterization of Liposomes
DLS revealed that the Blank-Lipo had an average particle size of 97.06 ± 3.12 nm and a PDI of 0.286 ± 0.053 (Figure 3A). SN38/IR820-Lipo@FSH, encapsulating SN38 and IR820, formed stable nanoparticles with an average particle size of 108.9 ± 2.88 nm and an average PDI of 0.249 ± 0.064 (Figure 3C). The zeta potentials of Blank-Lipo and SN38/IR820-Lipo@FSH were −28.9 ± 2.51 mV and −25.6 ± 2.74 mV, respectively. The particle size distribution was relatively narrow, which may reduce non-specific organ uptake and enhance accumulation at the tumor site. Negatively charged nanoparticles on the surface increase the stability of liposomes in the circulation. SN38/IR820-Lipo@FSH morphology was also examined using transmission electron microscopy. As shown in Figure 3B and 3D, the images revealed that both the Blank-Lipo and SN38/IR820-Lipo@FSH exhibited a smooth, spherical structure with uniform dispersion, consistent with the results obtained from DLS analysis. SN38/IR820-Lipo@FSH is stable, as shown in Figure 3E and 3F; minimal changes in the size and zeta potential were detected after storing the liposomes for 7 days at room temperature in PBS or PBS containing 50% serum. The EE percentages for SN38 and IR820 in SN38/IR820-Lipo@FSH were 90.2 and 91.5%, respectively. The DL percentages of SN38 and IR820 in SN38/IR820-Lipo@FSH were 1.42 and 1.79%, respectively. Moreover, the successful encapsulation of IR820 was confirmed by the characteristic UV-VIS absorption peaks at 820 nm (Figure 3G).
The FSHR content in vitro was confirmed and analyzed by western blotting analysis. Control, SN38/IR820-Lipo and SN38/IR820-Lipo@FSH had FSHR (71~78 kDa) protein bands (Figure 3H). However, the normalized gray value of FSHR absorbed on the SN38/IR820-Lipo@FSH was lower than the FSHR absorbed on the SN38/IR820-Lipo. These results suggest that SN38/IR820-Lipo@FSH binds to the FSHR receptor due to the FSH peptide modification, decreasing the FSH peptide concentration and increasing the specific capture of FSHR receptor-targeted apolipoproteins.
Sustained extracellular release of SN38 and IR820 by SN38/IR820-Lipo@FSH was observed for 12 h at 37°C in PBS (pH 7.4) containing 0.5% Tween 80, as shown in Figure 3I and 3J. The cumulative release of SN38 and IR820 was less than 20%, indicating excellent in vivo sustained release functionality of the liposomes.
3.3. Cellular Uptake Studies
Cou-6-Lipo and Cou-6-Lipo@FSH were prepared to track liposome uptake in A2780, HepG2, and C26 cells. The uptake and efficiency of SN38/IR820-Lipo@FSH were determined using flow cytometry and confocal microscopy. As shown in Figure 4A, the green fluorescence signal of Cou-6 increased in A2780 cells after incubation with Cou-6-Lipo@FSH for 4 h. In contrast, the fluorescent signal increased only slightly in HepG2 and C26 cells after treatment with Cou-6-Lipo@FSH. Furthermore, the uptake of liposomes with the FSH peptide was significantly higher than the uptake of liposomes without the FSH peptide in A2780 cells. These results indicate that the FSH peptide can accurately target the FSHR receptor in A2780 cells to enhance specificity. The results obtained from flow cytometry were consistent with the confocal microscopy results. As shown in Figure 4B,C, the uptake of Cou-6-Lipo@FSH was 4.04 times higher than the uptake of Cou-6-Lipo in A2780 cells. These results indicate that liposomes with the FSH peptide enhance uptake, selectivity, and efficiency in tumor cells with high FSHR expression.
3.4. ROS Generation
DCFH-DA is a cell-permeable, intracellular reactive oxygen species (ROS) detection probe (Ex/Em = 488/525 nm). ROS generation in A2780 cells in response to near-infrared laser irradiation was detected using this probe and fluorescence microscopy. After incubating A2780 cells with different liposomes for 4 h, ROS production increased significantly in the groups that underwent 808 nm laser irradiation compared with ROS production in the non-irradiated groups (Figure 4D). Furthermore, the SN38/IR820-Lipo@FSH group exhibited higher ROS generation in response to 808 nm laser irradiation than the Control, Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH and SN38/IR820-Lipo groups. These results show that IR820 PDT activity was activated in response to 808 nm laser irradiation, resulting in the generation of ROS.
3.5. In Vitro Cell Apoptosis Study
The apoptotic rate of cells was measured using annexin V-FITC/PI double staining and flow cytometry. Apoptotic rates were low in the Control and Blank-Lipo groups. Apoptosis after SN38/IR820-Lipo@FSH treatment was significantly higher than apoptosis after SN38-Lipo@FSH, IR820-Lipo@FSH, or SN38/IR820-Lipo treatments (Figure 5A). The apoptosis rate after light exposure in the SN38/IR820-Lipo@FSH treatment group was 52.7%, which was significantly higher than the other groups.
Calcein-AM staining of live cells and PI staining of dead or late apoptotic cells were visualized with an inverted fluorescence microscope. As shown in Figure 5B and 5F, SN38-Lipo@FSH, SN38/IR820-Lipo, and SN38/IR820-Lipo@FSH exhibited weak red signals in the absence of light. After light exposure, SN38/IR820-Lipo@FSH exhibited a significant red fluorescent signal, indicating significantly lower cell viability compared with the other groups. These results show that the strong cellular uptake induced by FSH peptide targeting induced more apoptosis in the SN38/IR820-Lipo@FSH treatment group.
3.6. Wound healing Assay
To further investigate the anti-metastatic effect of SN38/IR820-Lipo@FSH, a wound-healing assay was used to evaluate random cell migration and invasion. As shown in Figure 5C and 5G, scratches in the PBS and Blank-Lipo groups healed gradually. After treatment with SN38-Lipo@FSH or IR820-Lipo@FSH (Laser +), the scratches remained visible. However, scratches in the SN38/IR820-Lipo (Laser +) or SN38/IR820-Lipo@FSH (Laser +) group remained clearly visible owing to the greater cytotoxicity and distinct inhibition of A2780 cell migration compared to the other groups. Importantly, SN38/IR820-Lipo@FSH under irradiation exhibited a stronger anti-metastatic effect than the SN38/IR820-Lipo. The antimetastatic effect of SN38/IR820-Lipo@FSH was attributed to the high cellular internalization in A2780 tumor cells via FSH-mediated active targeting.
3.7. Cell Viability Assay
We assessed the viability of A2780 cells treated with Blank-Lipo using the CCK-8 assay. No significant cytotoxic effects were observed in A2780 cells after treatment with Blank-Lipo@; cell survival rates exceeded 90% after 24 or 48 h incubation (Figure 5D). Thus, Blank-Lipo is safe and biocompatible for in vivo applications.
The CCK-8 assay was used to assess the inhibitory effects of SN38/IR820-Lipo and SN38/IR820-Lipo@FSH on A2780 cell proliferation. As shown in Figure 5E, SN38/IR820-Lipo and SN38/IR820-Lipo@FSH significantly inhibited A2780 cell proliferation in a time- and concentration-dependent manner. SN38/IR820-Lipo@FSH inhibited cell proliferation more than SN38/IR820-Lipo, at a concentration range of 0.001–50 µg/mL after 24 or 48 h of incubation. The IC50 values for SN38/IR820-Lipo and SN38/IR820-Lipo@FSH were 2.485 and 0.5112 µg/mL, respectively, after 24 h of incubation. The IC50 of SN38/IR820-Lipo@FSH was significantly lower than the IC50 of SN38/IR820-Lipo. Consistent with the increased uptake resulting from FSH peptide targeting, the high cellular uptake of SN38/IR820-Lipo@FSH enhances its anti-proliferative effects in A2780 cells.
3.8. In Vivo Biodistribution
An in vivo imaging system was employed to determine the biodistribution of DIR-Lipo and DIR-Lipo@FSH in mice bearing A2780 subcutaneous tumors. Real-time in vivo images and corresponding quantitative fluorescence results were captured at different time points after the injection of DIR liposomes in ovarian cancer-bearing mice. As shown in Figure 6A, DIR-Lipo was rapidly eliminated after injection. DIR-Lipo@FSH significantly accumulated at the tumor site at various time points following systemic administration; the fluorescence signal lasted 24 h, indicating the crucial role of FSH peptide in tumor targeting. The fluorescence distribution in organs and tumors was determined 24 h after the last liposome administration. The fluorescence intensity was significantly higher in tumors from mice treated with DIR-Lipo@FSH compared with the fluorescence intensity in tumors from mice treated with DIR-Lipo (Figure 6B,C). Additionally, DIR-Lipo and DIR-Lipo@FSH were primarily distributed in macrophage-associated organs, including the liver and spleen, with limited distribution in other organs. This result suggests that the liposomes are metabolized in the liver and spleen. Overall, these findings indicate that DIR-Lipo@FSH exhibits excellent targeting properties.
3.9. In Vivo ROS generation efficiency
In order to determine the effect of ROS generated by IR820, we observed the levels of DCFH-DA in vivo. As can be seen in Figure 7, strong green fluorescence of DCF was detected throughout the tumor sections of the SN38/IR820-Lipo@FSH group under irradiation, indicating ROS generation in vivo. However, negligible levels of DCF green fluorescence was observed in the PBS group. Compared with the PBS control group, although the DCF fluorescence intensity of the Free IR820 and SN38/IR820-Lipo groups was strong, it was significantly lower than that of the SN38/IR820-Lipo@FSH, indicating that the IR820 photosensitizer in the SN38/IR820-Lipo@FSH was successfully excited under infrared irradiation and produced ROS in vivo.
3.10. In Vivo Antitumor Efficacy
An ovarian cancer subcutaneous tumor mouse model was established to evaluate liposome antitumor effects. Mice were separated into six groups (n = 4/group), which were intravenously injected with Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, SN38/IR820-Lipo, SN38/IR820-Lipo@FSH, or saline and exposed to 808 nm laser irradiation the following day (As shown in Figure 8A). The mice were euthanized three days after the final injection, and the hearts, livers, spleens, lungs, kidneys, and tumors were collected. Tumor suppression was enhanced after light exposure in mice treated with FSH peptide-modified liposomes; tumor volumes were lower in the SN38/IR820-Lipo@FSH group compared with tumor volumes in the other groups (Figure 8B, 8C, and 8D). Tumor volumes in the SN38-Lipo@FSH and IR820-Lipo@FSH groups were not significantly lower than tumor volumes in the saline group. These results suggested that the SN38/IR820-Lipo@FSH delivery system enhanced the in vivo antitumor activity of the drug.
To confirm the effects of SN38/IR820-Lipo@FSH on tumor apoptosis in vivo, tumor tissues were analyzed using TUNEL assays. More TUNEL-positive cells were detected in tumors from mice treated with SN38/IR820-Lipo@FSH than in mice from the other groups (Figure 8E), indicating widespread necrosis and apoptosis in the tumor tissue. This result suggests that SN38/IR820-Lipo@FSH can effectively penetrate the tumor tissue and induce cell death. In contrast, the SN38/IR820-Lipo group showed a lower cell apoptosis rate compared with the SN38/IR820-Lipo@FSH group. Furthermore, tumor localization of SN38-Lipo@FSH and IR820-Lipo@FSH was limited compared with SN38/IR820-Lipo@FSH. Thus, mice treated with SN38/IR820-Lipo@FSH exhibited the best anti-ovarian cancer activity among all the groups.
3.11. Safety Assessment
Weight loss reflects cancer proliferation and systemic drug toxicity [24], and preventing weight loss is essential for cancer survival [26]. Changes in mouse body weight after treatment with the various formulations are shown in Figure 9A. Significant changes in body weight did not occur in any treatment group, suggesting that the liposomes reduced the toxicity and side effects of the drugs and were safe in mice. Hematological analysis of blood samples from mice treated with the different formulations showed that serum alanine aminotransferase, aspartate aminotransferase, creatinine, and blood urea nitrogen were stable, and all the parameters fell within the safe range for each group (Figure 9B). These findings imply that SN38/IR820-Lipo@FSH may lower SN38 toxicity and improve chemotherapy drug safety, making it a valuable potential delivery strategy.
To assess the ability of SN38/IR820-Lipo@FSH to inhibit metastasis, the major organs were stained with hematoxylin and eosin. No visible inflammatory damage was detected in the hearts, livers, spleens, and kidneys of any group, indicating that the agents were safe and biocompatible (Figure 9C). Moreover, metastatic foci appeared in the lungs of the saline, SN38-Lipo@FSH, IR820-Lipo@FSH, and SN38/IR820-Lipo groups, but no tumor metastases were detected in mice treated with SN38/IR820-Lipo@FSH, suggesting that SN38/IR820-Lipo@FSH inhibited tumor metastasis.
4. Conclusions
In this study we developed SN38/IR820-Lipo@FSH, which combined photodynamic therapy and chemotherapy, to treat cancer. FSH-modified liposomes were used to target ovarian cancer; tumor cell targeting was demonstrated in vitro and in vivo. SN38 and IR820 were efficiently encapsulated within the liposomes, thereby enhancing the water solubility and stability of SN38 in aqueous solutions and significantly reducing toxicity in mice. Moreover, SN38/IR820-Lipo@FSH generated ROS after laser irradiation, leading to cell death and enhanced anticancer effects. In vitro studies demonstrated that SN38/IR820-Lipo@FSH effectively promoted drug uptake by tumor cells, leading to decreased proliferation and increased apoptosis of ovarian cancer cells. Importantly, in vivo experiments revealed that SN38/IR820-Lipo@FSH efficiently delivered targeted SN38 and IR820 to the tumor site, resulting in the suppression of tumor growth and metastasis. Furthermore, the novel carrier was simple to prepare, and encapsulation efficiency was high. This novel drug delivery system combined photodynamic therapy and other chemotherapeutic drugs to treat ovarian cancer and demonstrated clinical potential.
Author Contributions
Conceptualization, Z.G., D.J. and M.J.; methodology, L.P. and B.Z.; software, N.W., S.W. and H.W.; validation, H.W.; formal analysis, L.C.; investigation, W.H.; resources, L.C.; data curation, H.W.; writing—original draft preparation, L.P. and B.Z.; writing—review and editing, Z.G., D.J. and M.J.; visualization, M.J.; supervision, M.J.; project administration, M.J.; funding acquisition, Z.G. and M.J. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financially supported by National Natural Science Fund of China (82104106, 82073778). This work was supported by Beijing Key Laboratory of Drug Delivery Technology and Novel Formulations, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College.
Institutional Review Board Statement
The study was conducted according to the guidelines of the Laboratory Animal Ethics Committee in the Institute of Materia Medica at the Chinese Academy of Medical Sciences (CAMS) and Peking Union Medical College (PUMC). All the experimental procedures were performed in conformity with the institutional guidelines and protocols for the care and use of laboratory animals.
Data Availability Statement
All data available are reported in the article.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J Clin 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Epithelial Ovarian Cancer - PubMed.
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking Ovarian Cancer: Recommendations for Improving Outcomes. Nat Rev Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed]
- A Brief Review of the Management of Platinum-Resistant-Platinum-Refractory Ovarian Cancer - PubMed.
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian Cancer Statistics, 2018. CA Cancer J Clin 2018, 68, 284–296. [Google Scholar] [CrossRef] [PubMed]
- Rickard, B.P.; Conrad, C.; Sorrin, A.J.; Ruhi, M.K.; Reader, J.C.; Huang, S.A.; Franco, W.; Scarcelli, G.; Polacheck, W.J.; Roque, D.M.; et al. Malignant Ascites in Ovarian Cancer: Cellular, Acellular, and Biophysical Determinants of Molecular Characteristics and Therapy Response. Cancers (Basel) 2021, 13, 4318. [Google Scholar] [CrossRef] [PubMed]
- SN38-Based Albumin-Binding Prodrug for Efficient Targeted Cancer Chemotherapy - PubMed.
- In Situ Activation of STING Pathway with Polymeric SN38 for Cancer Chemoimmunotherapy - PubMed.
- Novel Prodrugs of SN38 Using Multiarm Poly(Ethylene Glycol) Linkers - PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/18370417/ (accessed on 2 February 2024).
- Si, J.; Zhao, X.; Gao, S.; Huang, D.; Sui, M. Advances in Delivery of Irinotecan (CPT-11) Active Metabolite 7-Ethyl-10-Hydroxycamptothecin. Int J Pharm 2019, 568, 118499. [Google Scholar] [CrossRef] [PubMed]
- Advances in Indocyanine Green-Based Codelivery Nanoplatforms for Combinatorial Therapy - PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/33539071/ (accessed on 2 February 2024).
- Castano, A.P.; Mroz, P.; Hamblin, M.R. Photodynamic Therapy and Anti-Tumour Immunity. Nat Rev Cancer 2006, 6, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Rickard, B.P.; Overchuk, M.; Obaid, G.; Ruhi, M.K.; Demirci, U.; Fenton, S.E.; Santos, J.H.; Kessel, D.; Rizvi, I. Photochemical Targeting of Mitochondria to Overcome Chemoresistance in Ovarian Cancer †. Photochem Photobiol 2023, 99, 448–468. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Huan, W.; Liu, X.; Zhang, Z.; Zhang, Y.; Wu, J.; Hu, Y. NIR Light-Activated Drug Release for Synergetic Chemo-Photothermal Therapy. Mol Pharm 2017, 14, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Phase-Change Material Packaged within Hollow Copper Sulfide Nanoparticles Carrying Doxorubicin and Chlorin E6 for Fluorescence-Guided Trimodal Therapy of Cancer - PubMed.
- Dobson, J.; de Queiroz, G.F.; Golding, J.P. Photodynamic Therapy and Diagnosis: Principles and Comparative Aspects. Vet J 2018, 233, 8–18. [Google Scholar] [CrossRef]
- Self-Assembled IR780-Loaded Transferrin Nanoparticles as an Imaging, Targeting and PDT/PTT Agent for Cancer Therapy - PubMed.
- Zhang, X.; Chen, J.; Kang, Y.; Hong, S.; Zheng, Y.; Sun, H.; Xu, C. Targeted Paclitaxel Nanoparticles Modified with Follicle-Stimulating Hormone β 81-95 Peptide Show Effective Antitumor Activity against Ovarian Carcinoma. Int J Pharm 2013, 453, 498–505. [Google Scholar] [CrossRef] [PubMed]
- Follicle-Stimulating Hormone Peptide Can Facilitate Paclitaxel Nanoparticles to Target Ovarian Carcinoma in Vivo - PubMed.
- Enhanced Antitumor Effects of Follicle-Stimulating Hormone Receptor-Mediated Hexokinase-2 Depletion on Ovarian Cancer Mediated by a Shift in Glucose Metabolism - PubMed.
- Hong, S.; Zhang, X.; Chen, J.; Zhou, J.; Zheng, Y.; Xu, C. Targeted Gene Silencing Using a Follicle-Stimulating Hormone Peptide-Conjugated Nanoparticle System Improves Its Specificity and Efficacy in Ovarian Clear Cell Carcinoma in Vitro. J Ovarian Res 2013, 6, 80. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Meng, S.; Zhou, X.; Gao, Z.; Piao, M.G. pH-Responsive and Mucoadhesive Nanoparticles for Enhanced Oral Insulin Delivery: The Effect of Hyaluronic Acid with Different Molecular Weights. Pharmaceutics 2023, 15, 820. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, Z.; Liu, L.; Li, M.; Zuo, A.; Guo, J. Preparation, in Vitro and in Vivo Evaluation of Chitosan-Sodium Alginate-Ethyl Cellulose Polyelectrolyte Film as a Novel Buccal Mucosal Delivery Vehicle. Eur J Pharm Sci 2022, 168, 106085. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Zeng, B.; Liu, Y.; Jin, L.; Hou, Y.; Liu, C.; Liu, W.; Wu, H.; Chen, L.; Gao, Z.; et al. Co-Delivery of Repurposing Itraconazole and VEGF siRNA by Composite Nanoparticulate System for Collaborative Anti-Angiogenesis and Anti-Tumor Efficacy against Breast Cancer. Pharmaceutics 2022, 14, 1369. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Jiang, L.; Meng, S.; Liu, C.; Wang, H.; Gao, Z.; Guo, J. Hollow Mesoporous Silica Nanoparticles-Loaded Ion-Crosslinked Bilayer Films with Excellent Mechanical Properties and High Bioavailability for Buccal Delivery. Int J Pharm 2022, 624, 122056. [Google Scholar] [CrossRef] [PubMed]
- Mehra, K.; Berkowitz, A.; Sanft, T. Diet, Physical Activity, and Body Weight in Cancer Survivorship. Med Clin North Am 2017, 101, 1151–1165. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Preparation of SN38/IR820-Lipo@FSH and synergistic effects of chemotherapy and phototherapy in ovarian cancer.
Figure 1.
Preparation of SN38/IR820-Lipo@FSH and synergistic effects of chemotherapy and phototherapy in ovarian cancer.
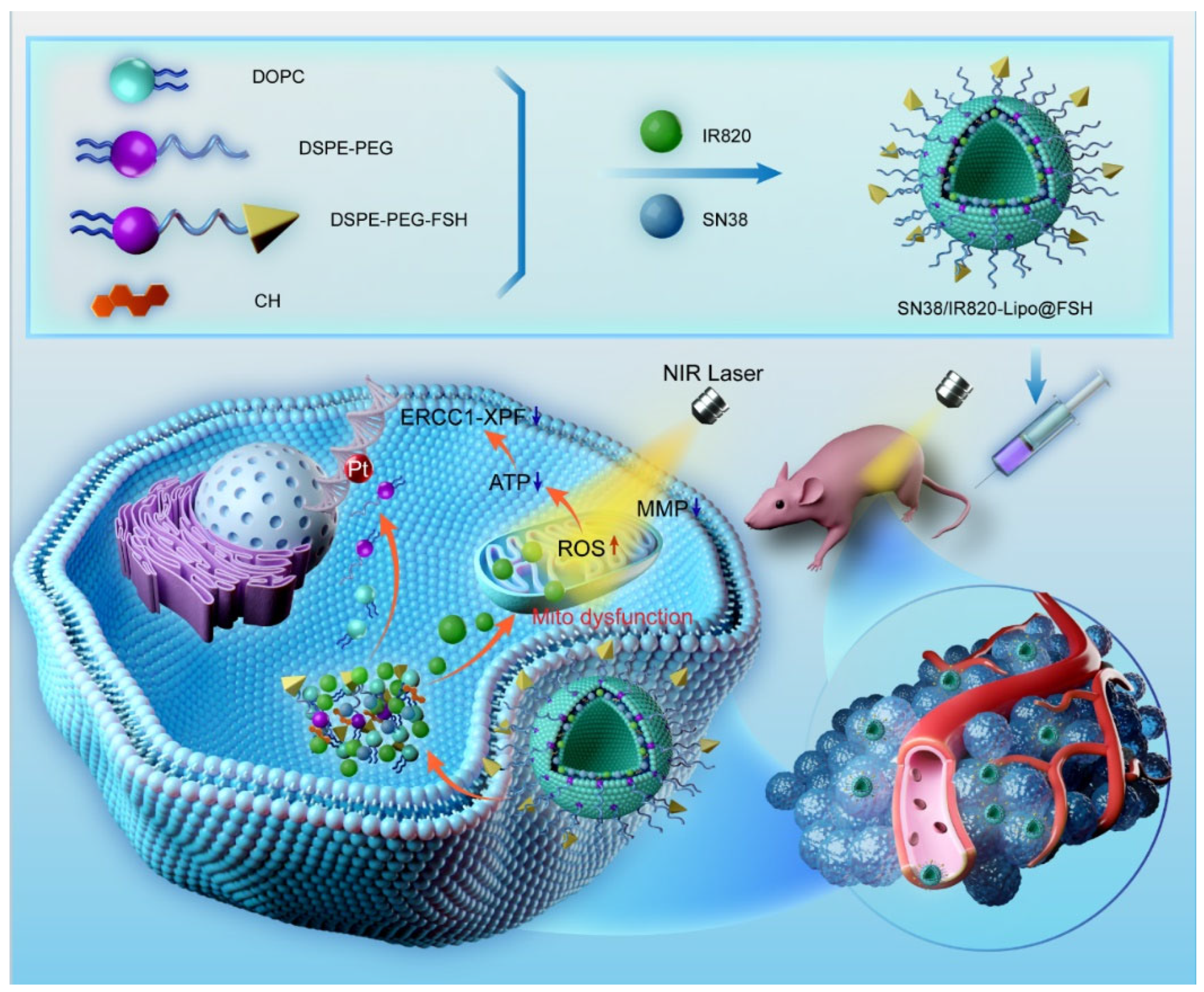
Figure 2.
(a) Synthesis route of DSPE-PEG2000-FSH; (b) 1H NMR spectra of DSPE-PEG2000-Mal and DSPE-PEG2000-FSH; (c). (d) MALDI-TOF-MS spectra of DSPE-PEG2000-FSH.
Figure 2.
(a) Synthesis route of DSPE-PEG2000-FSH; (b) 1H NMR spectra of DSPE-PEG2000-Mal and DSPE-PEG2000-FSH; (c). (d) MALDI-TOF-MS spectra of DSPE-PEG2000-FSH.
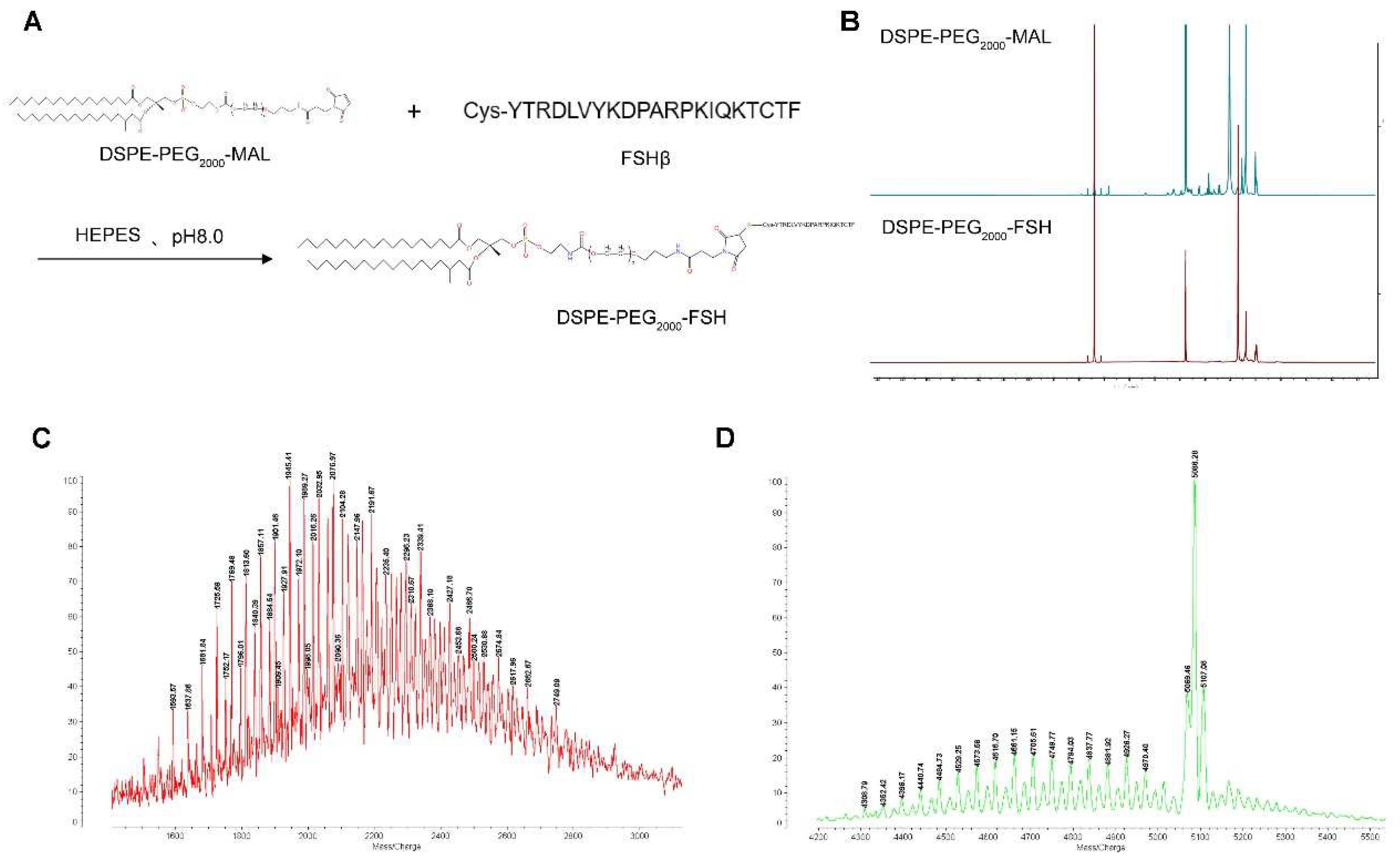
Figure 3.
Characterization of Blank Lipo and SN38/IR820-Lipo@FSH. (a). (b) The average size and zeta of Blank Lipo and TEM images of Blank Lipo (scale bar: 100 μm); (c). (d) The average size and zeta of SN38/IR820-Lipo@FSH and TEM images of SN38/IR820-Lipo@FSH (scale bar: 100 μm); (e). (f) Stability of SN38/IR820-Lipo@FSH in 4 ◦C within 7 days; (g) UV/VIS absorption spectra; (i). (j) The drug release behavior of SN38/IR820-Lipo@FSH within 12 h.
Figure 3.
Characterization of Blank Lipo and SN38/IR820-Lipo@FSH. (a). (b) The average size and zeta of Blank Lipo and TEM images of Blank Lipo (scale bar: 100 μm); (c). (d) The average size and zeta of SN38/IR820-Lipo@FSH and TEM images of SN38/IR820-Lipo@FSH (scale bar: 100 μm); (e). (f) Stability of SN38/IR820-Lipo@FSH in 4 ◦C within 7 days; (g) UV/VIS absorption spectra; (i). (j) The drug release behavior of SN38/IR820-Lipo@FSH within 12 h.
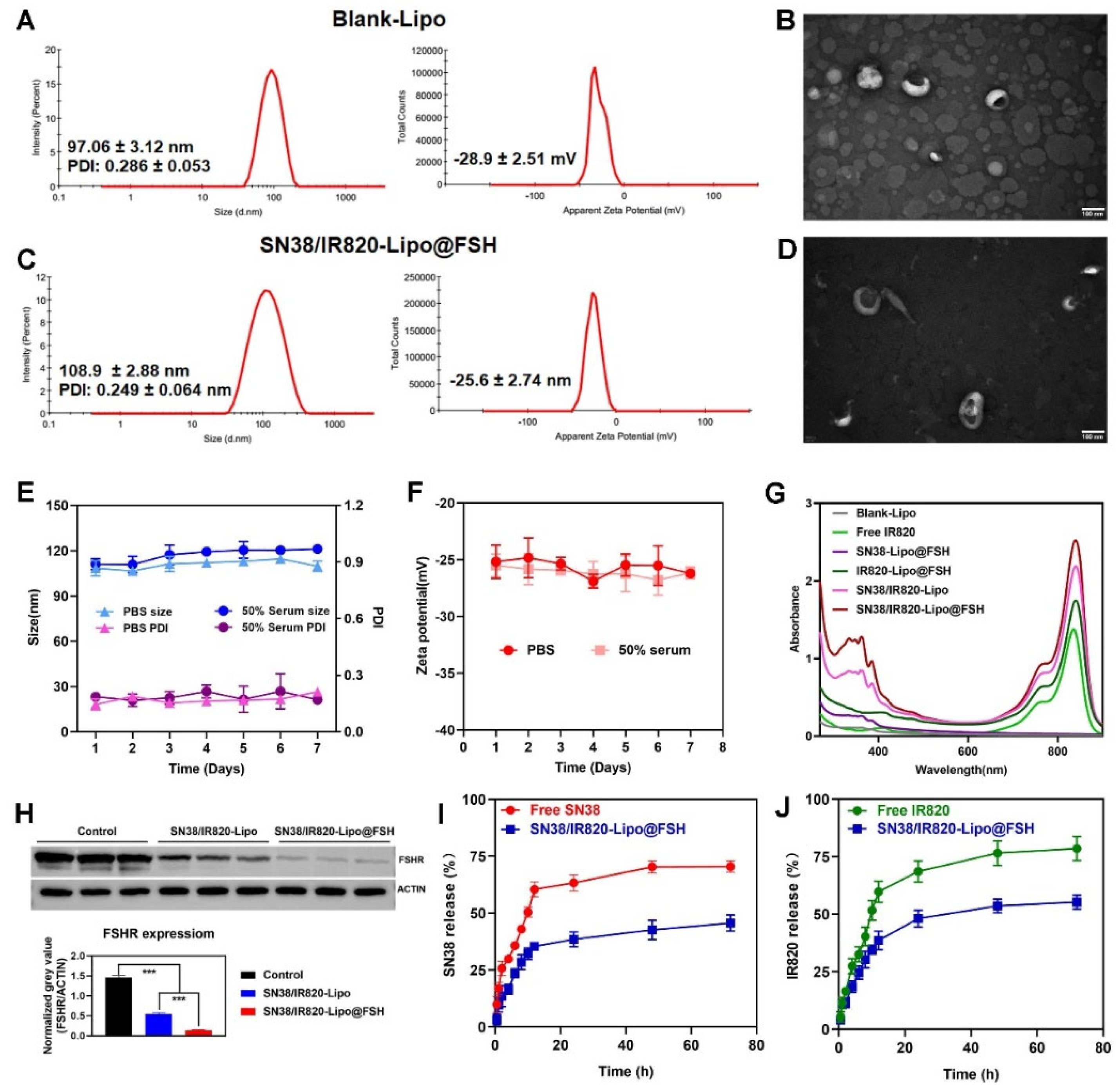
Figure 4.
Cellular uptake of SN38/IR820-Lipo@FSH. (a) Confocal images of cellular uptake in A2780, HepG2 and C26 cells (Scale bar: 50 μm); (b) Analysis of cellular uptake of the control, free Cou-6, Cou-6-Lipo and Cou-6-Lipo@FSH by flow cytometry and mean fluorescence intensity; (c). (d) Inverted fluorescence microscope image of ROS (Scale bar: 200 μm). Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 4.
Cellular uptake of SN38/IR820-Lipo@FSH. (a) Confocal images of cellular uptake in A2780, HepG2 and C26 cells (Scale bar: 50 μm); (b) Analysis of cellular uptake of the control, free Cou-6, Cou-6-Lipo and Cou-6-Lipo@FSH by flow cytometry and mean fluorescence intensity; (c). (d) Inverted fluorescence microscope image of ROS (Scale bar: 200 μm). Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 5.
(a). (f) The flow cytometry results of A2780 cell apoptosis and the percentage of early and late apoptosis after the 48 h treatment and near-infrared laser irradiation with Control, Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, SN/IR-Lipo and SN/IR-Lipo@FSH; (b). (g) Live/dead examination of A2780 cells subjected to different treatments was conducted using calcein-acetoxymethyl ester/propidium iodide (Calcein-AM/PI) double staining and observing by inverted fluorescence microscope. (Scale bar: 200 μm); (c) Wound healing Assay; (c) Wound healing Assay; (d) In vitro cytotoxicity of Blank-Lipo on A2780 cells; (e) Inhibitory capacity of SN38/IR820-Lipo and SN38/IR820-Lipo@FSH against A2780 cells proliferation. Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 5.
(a). (f) The flow cytometry results of A2780 cell apoptosis and the percentage of early and late apoptosis after the 48 h treatment and near-infrared laser irradiation with Control, Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, SN/IR-Lipo and SN/IR-Lipo@FSH; (b). (g) Live/dead examination of A2780 cells subjected to different treatments was conducted using calcein-acetoxymethyl ester/propidium iodide (Calcein-AM/PI) double staining and observing by inverted fluorescence microscope. (Scale bar: 200 μm); (c) Wound healing Assay; (c) Wound healing Assay; (d) In vitro cytotoxicity of Blank-Lipo on A2780 cells; (e) Inhibitory capacity of SN38/IR820-Lipo and SN38/IR820-Lipo@FSH against A2780 cells proliferation. Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.

Figure 6.
Biodistribution of SN38/IR820-Lipo@FSH in vivo. (a) In vivo fluorescence images of A2780-bearing BALB/c Nude mice treated with DIR-Lipo and DiR-Lipo@FSH. Images were taken at 1, 2, 3, 6, and 24 h after injection; (b) The fluorescence images of the excised tumors and major organs at 24 h after injection; (c) The quantitative ROI analysis of the excised tumors and major organs at 24 h after injection. Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 6.
Biodistribution of SN38/IR820-Lipo@FSH in vivo. (a) In vivo fluorescence images of A2780-bearing BALB/c Nude mice treated with DIR-Lipo and DiR-Lipo@FSH. Images were taken at 1, 2, 3, 6, and 24 h after injection; (b) The fluorescence images of the excised tumors and major organs at 24 h after injection; (c) The quantitative ROI analysis of the excised tumors and major organs at 24 h after injection. Data are shown as mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001.
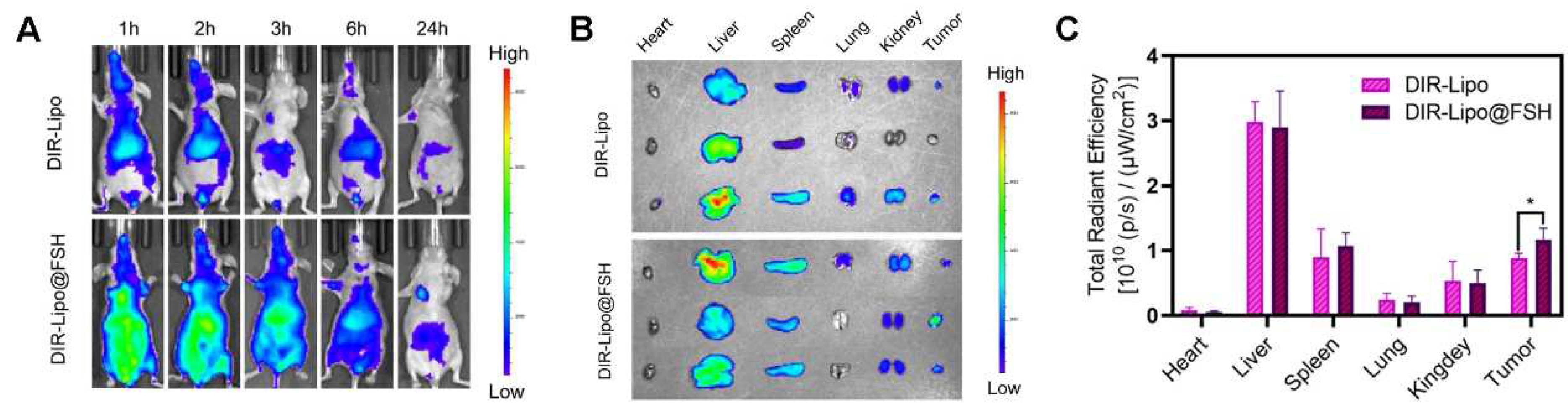
Figure 7.
In vivo ROS generation using CLSM (Scale bar: 2.5 mm).
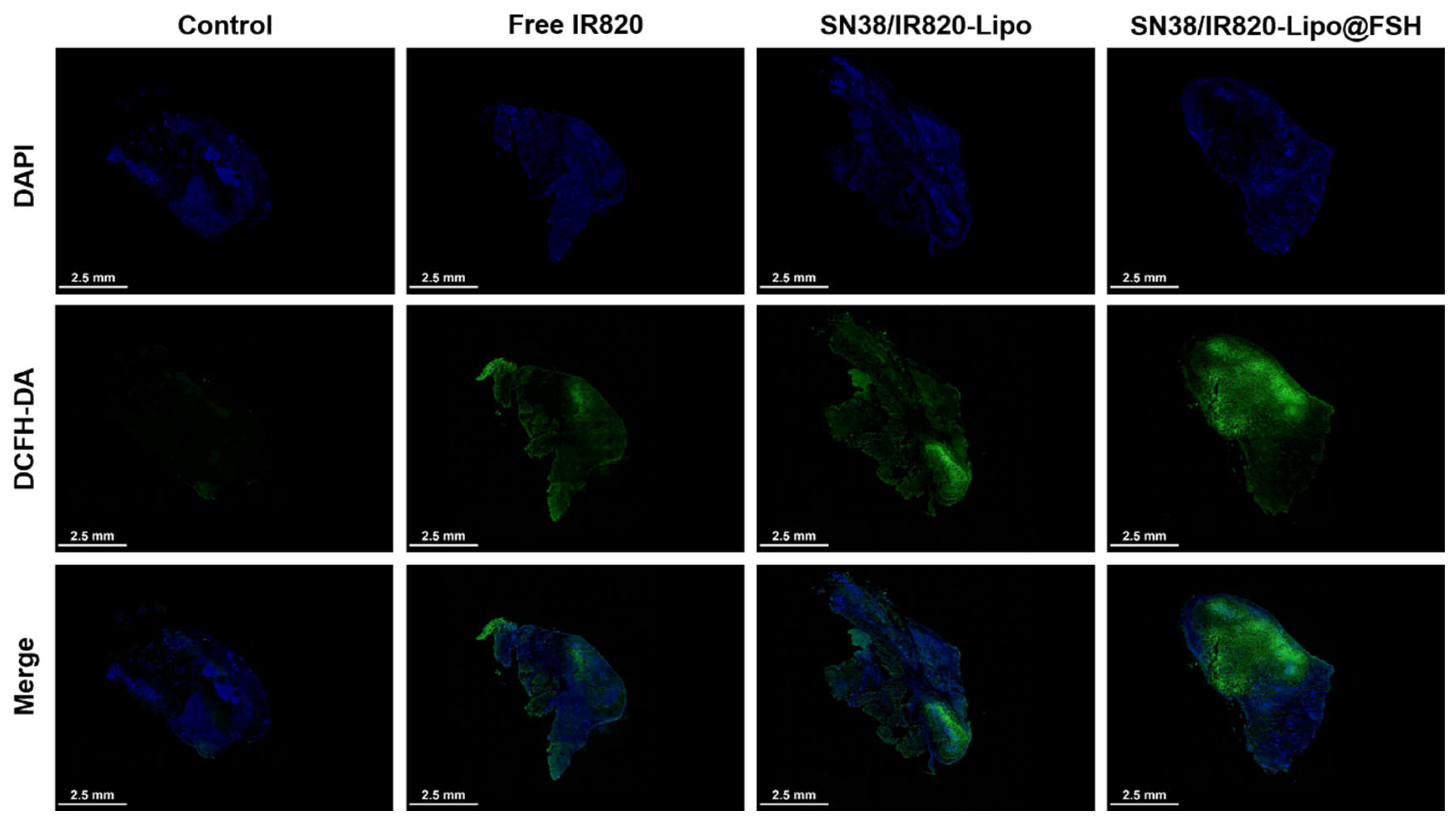
Figure 8.
Antitumor Evaluation of SN38/IR820-Lipo@FSH in vivo. The A2780-bearing BALB/c Nude mice were treated with Saline, Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, SN38/IR820-Lipo and SN38/IR820-Lipo@FSH. (a) Experimental procedure; (b) Image of isolated tumor tissues of mice; (c) Tumor volume of mice; (d) Weight of isolated tumors; (e) H&E and TUNEL assay of tumor tissues isolated from mice (Scale bar: 50 μm). Data are shown as mean ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 8.
Antitumor Evaluation of SN38/IR820-Lipo@FSH in vivo. The A2780-bearing BALB/c Nude mice were treated with Saline, Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, SN38/IR820-Lipo and SN38/IR820-Lipo@FSH. (a) Experimental procedure; (b) Image of isolated tumor tissues of mice; (c) Tumor volume of mice; (d) Weight of isolated tumors; (e) H&E and TUNEL assay of tumor tissues isolated from mice (Scale bar: 50 μm). Data are shown as mean ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001.
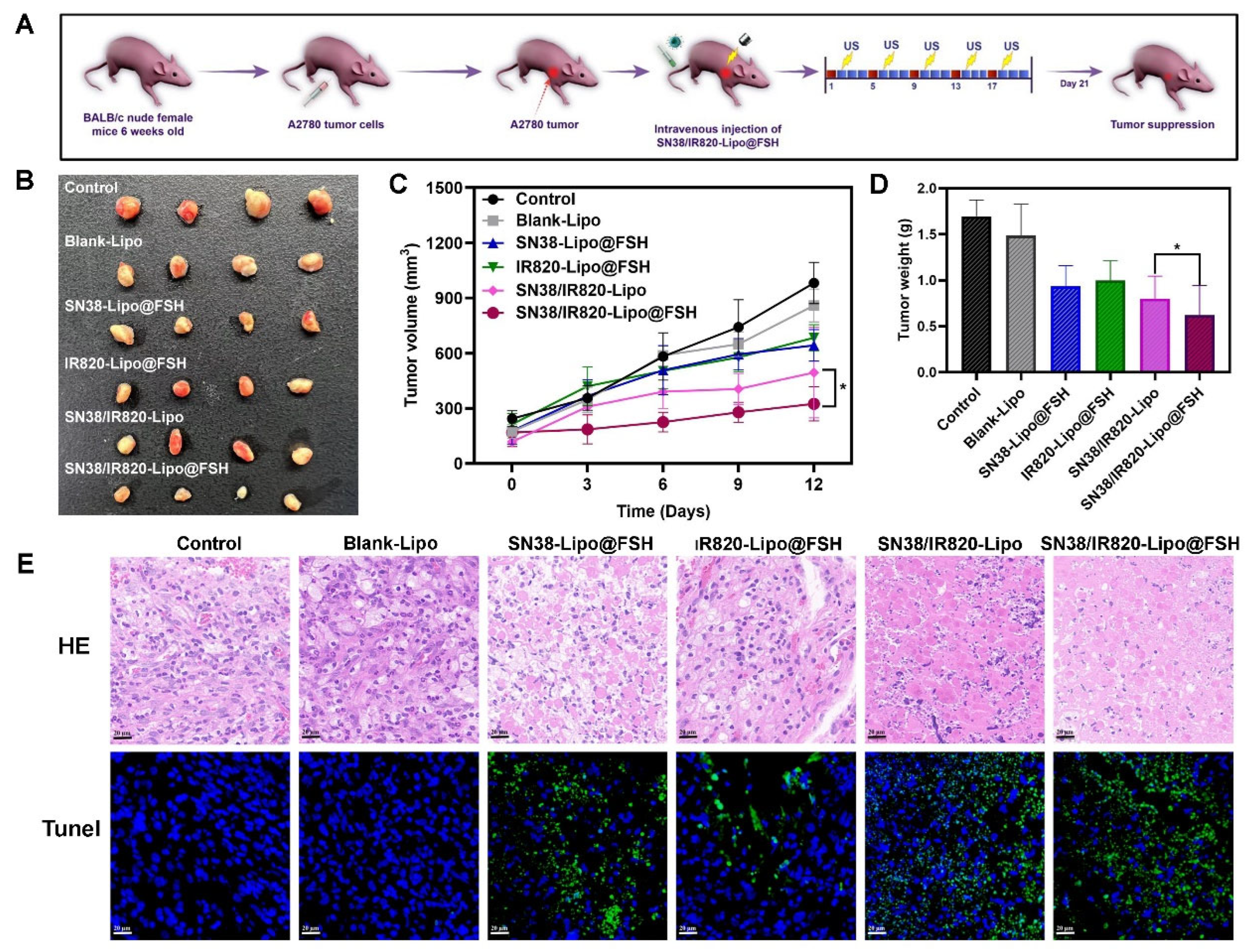
Figure 9.
Anti-metastasis evaluation of SN38/IR820-Lipo@FSH in vivo. The A2780-bearing BALB/c Nude mice were treated with Saline, Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, SN38/IR820-Lipo and SN38/IR820-Lipo@FSH. (a) Body weight of mice; (b) ALT, AST, BUN and Cr from the bloodsamples isolated from mice; (c) H&E staining of tumor and organs isolated from mice (Scale bar: 200 μm). Data are shown as mean ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001.
Figure 9.
Anti-metastasis evaluation of SN38/IR820-Lipo@FSH in vivo. The A2780-bearing BALB/c Nude mice were treated with Saline, Blank-Lipo, SN38-Lipo@FSH, IR820-Lipo@FSH, SN38/IR820-Lipo and SN38/IR820-Lipo@FSH. (a) Body weight of mice; (b) ALT, AST, BUN and Cr from the bloodsamples isolated from mice; (c) H&E staining of tumor and organs isolated from mice (Scale bar: 200 μm). Data are shown as mean ± SD (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



