
Submitted:
02 February 2024
Posted:
05 February 2024
You are already at the latest version
Abstract
Background: Advancement of next-generation sequencing (NGS) technologies provides opportunities for large-scale Pharmacogenetic (PGx) studies and pre-emptive PGx testing to cover a wide range of genotypes present in diverse populations. However, NGS-based PGx testing is limited by the lack of comprehensive computational tools to support genetic data analysis and clinical decisions.
Methods: Bioinformatics utilities specialized for human genomics and the latest cloud-based technologies are used for developing a bioinformatics pipeline for analyzing the genomic sequence data and reporting PGx genotypes. A database was created and integrated in the pipeline for filtering the actionable PGx variants and clinical interpretations. Strict quality verification procedures were conducted on variant calls with whole genome sequencing (WGS) dataset of the 1000 Genomes Project (G1K). The accuracy of PGx allele identification was validated using the whole genome sequencing dataset of the Pharmacogenetics Reference Materials from the Centers for Disease Control and Prevention (CDC).
Results: The newly created bioinformatics pipeline, Pgxtools, can analyze genomic sequence data, identify actionable variants in 13 PGx relevant genes, and generate reports annotated with specific interpretations and recommendations based on clinical practice guidelines. Verified with two independent methods, we have found that Pgxtools consistently identifies variants more accurately than the results in the G1K dataset on GRCh37 and GRCh38.
Conclusions: Pgxtools provides an integrated workflow for large-scale genomic data analysis and PGx clinical decision support. Implemented with cloud-native technologies, it is highly portable in a wide variety of environments from a single laptop to high performance computing clusters and cloud platforms for different production scales and requirements.
Keywords:
Pharmacogenetics
; bioinformatics pipeline
; cloud‐native technologies
; workflow
; genomic data analysis
1. Introduction
Genetic variations cause different drug responses in patients. The same dosage, effective in some patients, may inevitably be ineffective and may even cause adverse drug reactions (ADRs) in others. Studies have indicated that ADRs have been an important cause of hospital admissions and in-hospital mortality (1)(2). PGx tests translate germ-line genotypes into actionable phenotypes and provide recommendations on dosing of medications. Aiming to optimize drug therapy, prevent ADRs and improve patient safety, some PGx tests have been implemented clinically in the single-gene-drug-pair approach successfully (3)(4)(5)(6). This reactive testing approach has limited capacity, fixed coverage, bias in variant selection and may delay treatment while waiting for the PGx test result. The US FDA Table of Pharmacogenetic Associations lists more than 100 gene-drug pairs and their interactions (7). The Clinical Pharmacogenetics Implementation Consortium (CPIC), a shared project between Pharmacogenomics Knowledge Base (PharmGKB) and the National Institute of Health (NIH) Pharmacogenomics Research Network (PGRN), and the Dutch Working Group on Pharmacogenetics have developed guidelines on genotype guided drug therapy (8), which contains 517 gene-drug pairs in the database in v 1.36.0 in December 2023. To meet the increasing clinical demand, pre-emptive testing would be an ideal strategy to generate variant data for multiple genes before prescribing any target drugs (9). This strategy is under evaluation in some large international clinical trials and implemented in some institutions in the US and Europe (9)(10). Recently, a large-scale multicentre implementation study, PREPARE, has demonstrated that genotype-guided drug prescription using a 12-gene pharmacogenetic panel can reduce the incidence of clinically relevant ADRs significantly and improve the safety of drug therapy (11).
With the advancement of next-generation sequencing (NGS) technologies, the volume of genomic data has increased dramatically. Bioinformatics pipelines become essential tools to use genomic data to its full potential and to interpret it for clinical applications (12)(13). The availability of genomic data provides opportunities for large-scale PGx studies and pre-emptive PGx testing. However, there are barriers and challenges on how to analyze the genomic sequencing data to report the actionable PGx variants efficiently and reliably, and how to integrate the PGx results into electronic health records to deliver the clinical decision support at the point of prescribing (14)(15)(16). In this study, we aimed to develop a cloud-based bioinformatics pipeline for pharmacogenetic testing covering the entire workflow including NGS data analysis, variant allele assignment, genotype interpretation and clinical decision support.
2. Methods
2.1. Create PGx database:
The purpose of the PGx database is to provide a filter system for the pipeline to narrow the sequence analysis to the PGx relevant gene variants as well as the corresponding interpretations for these variants, such as clinical significance and dosing recommendations. The database contains the following attributes: gene name, drug name, genotype alias, variant position relative to the latest human reference genomes: GRCh37 and GRCh38, variant cDNA, amino acid change, enzyme activity or variant effect, rs number (a.k.a. reference SNP ID), genotype code, variants result, interpretation, and dosing recommendations. Each row of the PGx database represents a diplotype. Information regarding clinical significance is drawn mainly from the CPIC guidelines (17).
2.2. Design bioinformatics pipeline:
Genomic sequence analysis can be divided into three phases: primary analysis, secondary analysis and tertiary analysis (Figure 1). The primary analysis is usually completed on the sequencer. Classical bioinformatics pipelines focus on secondary analysis to create sequence alignments, and to identify variants. Our pipeline is designed to cover both secondary and tertiary analysis. The starting point is either Binary Alignment Map (BAM) / Compressed Reference-oriented Alignment Map (CRAM) or Variant Calling Format (VCF) input files from targeted sequencing panels or from whole genome sequencing (WGS).
With our pipeline, 20 - 100 thousand variants per exome or 3 - 4 million variants per genome can be prioritized, filtered to be comprehensible by healthcare providers. Variants with known clinical significance are annotated with suggestions and reference to PGx guidelines. The final clinical report contains tertiary analysis results specific to PGx testing. Our PGx database and pipeline form an automated workflow to provide PGx recommendations specific to patient genetic profiles.
2.3. Validate the variant calls and variant allele assignment
To verify the quality of the variant calls by Pgxtools, we used Pgxtools to analyze the WGS alignments (BAM/CRAM) in the G1K dataset (https://www.internationalgenome.org/1000-genomes-summary/), and compared the variants reported by Pgxtools with the variants documented in the G1K dataset. To further investigate the discordance in variant calls between our pipeline and the G1K datasets, we employed two independent methods: Samtools view command and EnsEMBL genome browser to identify which results are correct.
To validate the accuracy of variant to genotype mapping, we used PGx reference materials with “Consensus genotypes for 28 PGx genes” (18), archived on CDC (19) as a gold standard. We used Pgxtools to analyze all 70 WGS alignments used in the PGx reference materials archived in European Nucleotide Archive (ENA) (20) and reported the genotypes of the PGx genes. There are 3 genes (COMT, NUDT15 and IL28B) not included in the CDC 28 PGx gene panel. We compared the genotype calling by Pgxtools on the rest of 10 genes with the consensus genotypes published on this CDC dataset.
3. Results
3.1. System architecture
Our newly developed pipeline, named Pgxtools, is designed with the latest technologies to be completely cloud-native, and highly portable. The pipeline is containerized so it can run on Kubernetes clusters in different clouds without changes. We have used Kubernetes on MacBook Pro for development, and Google Cloud Platform (GCP) and HPC cluster at Genomics England for production.
The pipeline consists of a front-end of Graphical User Interface (GUI) on Jupyter notebook server augmented with IPywidgets and Pandas. It has a back-end runtime with Samtools, Bcftools based on Htslib. The Docker container is stateless by design. The data is either stored on a persistent volume managed by Kubernetes or from the various sources in the cloud or on storage volumes in HPC as described below (Figure 2).
The input of genomic alignment maps or variants, the output of PGx reports and the intermediate results are stored on a persistent volume outside of the container. They can survive the events of the container upgrade, shutdown, eviction, etc. The two human reference genomes: GRCh37 and the latest GRCh38 are downloaded from a public FTP site at European Bioinformatics Institute, European Molecular Biology Laboratory (EMBL-EBI). The 1000 Genomes Project (G1K) variants and alignments for GRCh37 and GRCh38 are accessed directly from an S3 bucket for public data on Amazon Web Services (AWS). The public API of the Variant Effect Predictor (VEP) at EMBL-EBI is used to get the details of PGx variants when the Docker image of the pipeline is built. Datashim is used to bridge the cloud object store and Linux POSIX filesystem.
We created a database including thirteen genes and most common drugs with ADRs caused by the variants in these genes. (Table 1). The genes and drugs were selected based on information in the CPIC guidelines. The genes include COMT, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A5, CYP4F2, DPYD, IL28B, NUDT15, SLCO1B1, TPMT and VKORC1. Detailed information of the variants can be found in the Supplemental Table 1. This database is stored in the Docker Image and used by the pipeline to perform targeted analysis. The Pgxtools references the database of the genes and drugs with the variant-specific interpretation and compares them against the genes and variants identified from the input sequence data. The tool generates specific PGx recommendations for the given genetic information.
3.2. Accuracy of variant calls, from alignments to variants
We used the variants in the G1K dataset as the gold standard to verify the quality of the variants called by Pgxtools from the sequence alignments (BAM / CRAM). We have run Pgxtools on 2504 WGS sample alignments on GRCh37 and 3200 WGS sample alignments on GRCh38 in the G1K dataset. There are at least 92.69% of samples with variants reported correctly on GRCh37, among all the 13 genes in our study except for CYP2D6 (See the GRCh37 row in Table 2). On GRCh38, Pgxtools reported 100% of variants matching the results in the G1K dataset except for CYP2D6. For CYP2D6, the concordance improved from 68.17% on GRCh37 to 98.59% on GRCh38 (Table 2).
We suspected that the discordance in variants reported by the Pgxtools and G1K project among the genes of CYP2D6, NUDT15, DPYD, TPMT and CYP3A5 were most likely caused by the quality of BAM files on GRCh37 in the G1K dataset. To test this hypothesis, we created the synthetic data by backporting the VCF and CRAM files with these genes from GRCh38 in the G1K dataset to GRCh37 coordinate with CrossMap (21). We ran Pgxtools on the backported CRAM files to compare the variants of the synthetic alignments. The concordance improved to 97.84% - 100% from 68.17% - 98.80% (See the row of GRCh38 to 37 in Table 2), proving that Pgxtools identifies variants correctly with both reference genomes of GRCh37 and GRCh38. The sample alignments (BAMs/CRAMs) for these genes on GRCh37 is the major limiting factor causing the discordance in variants reported.
We conducted detailed analysis on discordance in the variant calling by Pgxtools and G1K dataset. We employed two independent methods: Samtools view command and EnsEMBL genome browser to identify which variant calling results are correct.
On GRCh38, there are a total of 45 samples with 83 variant calls in CYP2D6 showing significant discordances between Pgxtools and G1K. With visual inspection of the 45 samples in EnsEMBL at EMBL-EBI, we conclude that 39 of the 83 variants (47.0%) are called correctly by Pgxtools alone, 6 variants (7.2%) called correctly by G1K alone and 31 variants (37.3%) called correctly by both Pgxtools and G1K. There are also 4 variants (4.8%) without contigs for variant calling and 3 variants (3.6%) called incorrectly by both Pgxtools and G1K (Figure 3). Overall, Pgxtools made the correct variant calls most of the time, performing significantly better than the variant calling in the G1K dataset (84.3% vs. 44.5%, respectively).
As a side note, CYP2D6 gene is known to be difficult for sequencing with NGS. The coverage can vary significantly from contig to contig, and some contigs contain very ambiguous base calls (e.g. G1K sample alignments NA19210.final.cram around 42130692 as shown in Supplemental Figure 1), making variant calling difficult and less accurate. Our data show that Pgxtools produces much better results for CYP2D6 gene variant calling.
There were 1132 samples with 1484 variants showing significant discordances between G1K and Pgxtools results on CYP2D6, NUDT15, DPYD, TPMT and CYP3A5 on GRCh37. We conducted manual inspection on sample alignment files with Samtools view command. We concluded that Pgxtools is reporting variant existence with 70.3%, 87.7%, 77.4%, 84.5% and 89.1% accuracy in CYP2D6, NUDT15, DPYD, TPMT and CYP3A5, respectively. However, G1K is reporting variants with 36.2%, 36.8%, 29.0%, 29.6% and 26.1% accuracy in CYP2D6, NUDT15, DPYD, TPMT and CYP3A5, respectively (Figure 4). However, there are significant numbers of sample files without alignments in variant positions on GRCh37 especially in CYP2D6 (9.6%) that led to reduced accuracy of variant findings.
As shown in the detailed analysis above, Pgxtools demonstrates much higher accuracy in variant calling for these genes. The G1K dataset on GRCh38 can be used as the gold standard but the dataset on GRCh37 is suboptimal for some genes.
3.3. From variants to genotypes
We use a consensus-based community standard “Consensus genotypes for 28 PGx genes” (18), archived on CDC (19) to verify the variant to genotype mapping by Pgxtools. There are 3 genes not included in the CDC 28 PGx gene panel: COMT, NUDT15 and IL28B. We analyzed the concordance of the genotype calling by Pgxtools and the CDC dataset on the rest of 10 genes. We used WGS BAM alignments from all 70 PGx reference materials by the original study archived in ENA (20). The result shows remarkable concordance rate with the community consensus.
There is 100% concordance of genotype calling for the genes of DPYD and TPMT between Pgxtools and the CDC dataset. For the rest of the 8 genes, the Pgxtools reports the genotypes 100% correctly in CYP2D6, CYP3A5, CYP4F2 and SLCO1B1 genes according to the available WGS alignment. However, there are 15.71%, 10.00%, 5.71% and 20% discordant genotype calls in these genes compared with the consensus genotype assignment. For CYP2C19 and CYP2B6, the Pgxtools has reported the genotypes 91.43% and 98.58% correctly after manual verification with the WGS alignment; however, the remaining 8.57% and 1.42% of the genotypes were assigned incorrectly by PGxtools with manual verification. The reason is still under investigation. For CYP2C9 and VKORC1, Pgxtools are 97.14% and 90% correct whereas the community consensus is 100% correct, indicating the need to further optimize the Pgxtools analysis on these two genes to improve concordance.
3.4. PGx interpretation and reports
The Pgxtools analyzes the whole genome or the targeted sequences the same way via the same user interfaces, command line interface (CLI) or graphic user interface (GUI) with alignments or variant calls as input. It can generate reports including gene-drug pairs, their corresponding PGx phenotype prediction and clinical decision support, such as dosing recommendations. On the user interface, the operator can choose gene(s) and drug(s) from their drop-down lists, and then create a customized report. The accuracy of the tertiary analysis and reporting was manually verified and cross checked for the consistency with clinical practice guidelines used for building our database.
In a report for a particular single-gene-drug-pair format (Supplemental Figure 2), it provides detailed analysis to support the PGx decisions. It lists all variants with clinical significance in the gene. For each variant, it reports nucleotide change, amino acid change and genotype. It also reports the actionable PGx interpretation of such changes. The comments of the gene variants and reference documentation are also included for further investigation.
The Pgxtools places more emphasis on the large-scale pre-emptive PGx testing. To provide a complete overview of potential PGx implications, Pgxtools can generate a report format of high-level summary of all genes and variants identified from the input sequencing data. This format doesn’t include PGx interpretation or dosing recommendations (Supplemental Figure 3). If detailed PGx decision support is needed, the most comprehensive all-gene-all-drug report format can be selected from the user interface. This report organizes the details by genes. For each gene, all the relevant variants are listed with the same level of details as described in the above single-gene-drug-pair report format.
4. Discussion
In this article, we report creation of a PGx database and a pipeline to perform both secondary and tertiary analysis of genomic sequences from targeted sequencing panels or whole genome sequences on both human reference genomes GRCh37 and GRCh38. The database and the pipeline are fully integrated to form a PGx clinical decision support workflow, which is implemented with cloud-native technologies to be highly portable in a wide variety of environments from a single laptop, to Google Cloud Platform, to high performance computing clusters for different production scales and requirements.
We conducted strict quality verification analysis against our pipeline with 5704 human genome sequences on both GRCh37 and GRCh38. We have demonstrated that the Pgxtools secondary analyses are highly reliable and accurate. In addition, we confirmed that the G1K dataset on GRCh38 can be used as the gold standard for PGx studies, while the G1K dataset on GRCh37 has lower accuracy and is not suitable for this purpose. With two independent methods, we have demonstrated that the Pgxtools reports more accurate results in the 13 PGx genes in the G1K dataset on GRCh37 and GRCh38.
During this investigation, we have encountered a number of challenges. First, each CPIC guideline is updated periodically on the CPIC website, and the latest publication may not reflect the most updated changes. Our database was created based on the original published guidelines. To follow the most updated therapeutic recommendations and allele definitions, we checked the CPIC and Pharmvar website periodically and amended our database accordingly.
Second, the therapeutic recommendations provided by the CPIC guidelines are based on different strengths of evidence. To indicate the strengths of evidence for therapeutic recommendations by CPIC, we kept the CPIC classification system in the Pgx database. The CPIC guidelines determined therapeutic recommendations based on evidence from functional and clinical data and/or other existing guidelines (22). Based on the strength of evidence, the CPIC guidelines assign “strong”, “moderate”, “optional” or “no recommendation” to their recommendations (22). We indicated this classification system in a bracket in front of each interpretation in the Pgx database.
Third, there are inconsistencies translating genotype to phenotype among CPIC guidelines for different target drugs. For example, for CYP2C19, we observed that in the guideline for selective serotonin reuptake inhibitors published in 2015, both the *17/*17 and *1/*17 genotypes are defined as ultrarapid metabolizers (23); However, in the newer guidelines for clopidogrel, proton pump inhibitors, tricyclic antidepressants and voriconazole, the *1/*17 is defined as a rapid metabolizer (24)(25)(23)(26). According to the Dutch Pharmacogenetics Working Group (DPWG), *1/*17 is defined as a normal metabolizer because of the relatively small increase in enzyme activity effect of the *17 allele (27). We defined *1/*17 as rapid metabolizer in the Pgx database because *17 and *1 alleles do have statistical differences in terms of pharmacokinetic parameters (26). Also since therapeutic recommendations for normal and rapid metabolizers of CYP2C19 in CPIC guidelines are the same, the phenotypic definition of *1/*17 does not affect its recommendation.
For CYP2D6, we observed that in the guidelines for selective serotonin reuptake inhibitors, tamoxifen, atomoxetine, tricyclic antidepressants and ondansetron/tropisetron, the allele *10 is given an activity score of 0.5 and genotype *10/*10 is defined as a normal metabolizer (28)(23)(29)(30)(31). However, in the guideline for opioid, *10 is given an activity score of 0.25 and *10/*10 is defined as an intermediate metabolizer (32). Due to inconsistencies in translating CYP2D6 genotype to phenotype across different laboratories and guidelines, CPIC used a modified Delphi method to obtain a consensus for translating CYP2D6 genotype to phenotype among a panel of international experts (33). As a result of the consensus, CPIC modified the activity score of *10 from 0.5 to 0.25 and changed the definition of metabolizers based on activity score as follows: ultrarapid metabolizer was changed from over 2 to over 2.25; normal metabolizer was changed from between 1 and 2 to between 1.25 to 2.25; intermediate metabolizer was changed from 0.5 to between 0 and 1.25 (33). Thus, the phenotype assignment for *10/*10 is now an intermediate metabolizer, as the genotype has an activity score of 0.5 (33). We adjusted interpretations in the Pgx database based on the latest consensus on the translation of CYP2D6 genotype to phenotype.
CYP2D6 is a polymorphic gene with over 100 known allelic variants (29). We noticed that the CPIC guidelines does not categorize CYP2D6 alleles based on their frequency, clinical relevance or the amount of evidence. On the other hand, the AMP guideline provides a “two tier” system for the allelic variants based on a set of criteria (34). Our PGx database includes all the variants covered in the “two tier system” described in the AMP guideline to reflect the importance of these variants.
The majority of clinically implemented PGx tests and commercially available PGx panels are targeted genotype methods covering a limited number of variants in each gene. The coverage of alleles may not be sufficient in diverse populations. In addition, low frequency no-function alleles not included in the testing panel will give false classification if the patient happens to be a carrier of that variant. The NGS-based PGx test gives opportunities to fill the gaps by analyzing all PGx relevant variants in the gene. The 1000 Genomes Project describes common human genetic variants in a diverse set of thousands of individuals from 26 populations (35). Our new pipeline, Pgxtools analyzes a total of 5704 individual whole-genome sequences (2504 on GRCh37 and 3200 on GRCh38) in the G1K dataset efficiently and accurately. For future development, we plan to expand the capacity of our pipeline to be able to report all actionable variants currently well characterized in the CPIC database regardless of their allele frequencies and population background.
Currently our pipeline is limited to analyzing the variants with rs ID in the SNP database. The next stage will be to develop another branch of tools to analyze gene structural change and variants without rs ID, such as CYP2D6 gene duplication/deletion and HLA typing.
In conclusion, we created a PGx database and pipeline for secondary and tertiary analysis of genomic sequence data that are fully integrated to form the basis of a PGx clinical decision support workflow. The workflow is implemented with cloud-native technologies to be highly portable. Our study reported here demonstrate that it is not only possible but also feasible to support the NGS-based large-scale pre-emptive PGx testing.
Acknowledgments
This project is supported partially by Sunnybrook LMMD Strategic Innovation Fund.
References
- Pirmohamed M, James S, Meakin S, Green C, Scott AK, Walley TJ, et al. Adverse drug reactions as cause of admission to hospital: prospective analysis of 18 820 patients. BMJ. 2004 Jul 3;329(7456):15–9. [CrossRef]
- Lazarou J, Pomeranz BH, Corey PN. Incidence of adverse drug reactions in hospitalized patients: a meta-analysis of prospective studies. JAMA. 1998 Apr 15;279(15):1200–5. [CrossRef]
- Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008 Feb 7;358(6):568–79. [CrossRef]
- Schaeffeler E, Fischer C, Brockmeier D, Wernet D, Moerike K, Eichelbaum M, et al. Comprehensive analysis of thiopurine S-methyltransferase phenotype-genotype correlation in a large population of German-Caucasians and identification of novel TPMT variants. Pharmacogenetics. 2004 Jul;14(7):407–17. [CrossRef]
- Morel A, Boisdron-Celle M, Fey L, Soulie P, Craipeau MC, Traore S, et al. Clinical relevance of different dihydropyrimidine dehydrogenase gene single nucleotide polymorphisms on 5-fluorouracil tolerance. Mol Cancer Ther. 2006 Nov;5(11):2895–904. [CrossRef]
- Wong BYL, Li Z, Raphael MJ, De Angelis C, Hwang DM, Fu L. Developing DPYD Genotyping Method for Personalized Fluoropyrimidines Therapy. J Appl Lab Med. 2023 Dec 12;jfad092. [CrossRef]
- Health C for D and R. Table of Pharmacogenetic Associations. FDA [Internet]. 2022 Oct 26 [cited 2024 Jan 15]; Available from: https://www.fda.gov/medical-devices/precision-medicine/table-pharmacogenetic-associations.
- Abdullah-Koolmees H, van Keulen AM, Nijenhuis M, Deneer VHM. Pharmacogenetics Guidelines: Overview and Comparison of the DPWG, CPIC, CPNDS, and RNPGx Guidelines. Front Pharmacol. 2021 Jan 25;11:595219. [CrossRef]
- Cavallari LH, Beitelshees AL, Blake KV, Dressler LG, Duarte JD, Elsey A, et al. The IGNITE Pharmacogenetics Working Group: An Opportunity for Building Evidence with Pharmacogenetic Implementation in a Real-World Setting. Clin Transl Sci. 2017 May;10(3):143–6. [CrossRef]
- van der Wouden CH, Böhringer S, Cecchin E, Cheung KC, Dávila-Fajardo CL, Deneer VHM, et al. Generating evidence for precision medicine: considerations made by the Ubiquitous Pharmacogenomics Consortium when designing and operationalizing the PREPARE study. Pharmacogenet Genomics. 2020 Aug;30(6):131–44. [CrossRef]
- Swen JJ, van der Wouden CH, Manson LE, Abdullah-Koolmees H, Blagec K, Blagus T, et al. A 12-gene pharmacogenetic panel to prevent adverse drug reactions: an open-label, multicentre, controlled, cluster-randomised crossover implementation study. Lancet Lond Engl. 2023 Feb 4;401(10374):347–56. [CrossRef]
- Yuan DY, Wildish T. Bioinformatics Application with Kubeflow for Batch Processing in Clouds. In: Jagode H, Anzt H, Juckeland G, Ltaief H, editors. High Performance Computing. Cham: Springer International Publishing; 2020. p. 355–67. [CrossRef]
- Pereira R, Oliveira J, Sousa M. Bioinformatics and Computational Tools for Next-Generation Sequencing Analysis in Clinical Genetics. J Clin Med. 2020 Jan 3;9(1):132. [CrossRef]
- Keeling NJ, Rosenthal MM, West-Strum D, Patel AS, Haidar CE, Hoffman JM. Preemptive pharmacogenetic testing: exploring the knowledge and perspectives of US payers. Genet Med Off J Am Coll Med Genet. 2019 May;21(5):1224–32. [CrossRef]
- Roden DM, McLeod HL, Relling MV, Williams MS, Mensah GA, Peterson JF, et al. Pharmacogenomics. Lancet Lond Engl. 2019 Aug 10;394(10197):521–32.
- Zhu Y, Swanson KM, Rojas RL, Wang Z, St Sauver JL, Visscher SL, et al. Systematic review of the evidence on the cost-effectiveness of pharmacogenomics-guided treatment for cardiovascular diseases. Genet Med Off J Am Coll Med Genet. 2020 Mar;22(3):475–86. [CrossRef]
- Clinical Pharmacogenetics Implementation Consortium Guidelines – CPIC [Internet]. [cited 2024 Jan 15]. Available from: https://cpicpgx.org/guidelines/.
- Pratt VM, Everts RE, Aggarwal P, Beyer BN, Broeckel U, Epstein-Baak R, et al. Characterization of 137 Genomic DNA Reference Materials for 28 Pharmacogenetic Genes: A GeT-RM Collaborative Project. J Mol Diagn JMD. 2016 Jan;18(1):109–23. [CrossRef]
- Reference Materials for Pharmacogenetics | CDC. Consensus genotypes for 28 PGx genes archived on CDC as part of the Reference Materials for Pharmacogenetics [Internet]. 2023 [cited 2024 Jan 15]. Available from: https://www.cdc.gov/labquality/get-rm/inherited-genetic-diseases-pharmacogenetics/pharmacogenetics.html.
- ENA Browser. The WGS samples for the Consensus genotypes for 28 PGx genes as PRJEB19931 archived by ENA [Internet]. [cited 2024 Jan 15]. Available from: https://www.ebi.ac.uk/ena/browser/view/PRJEB19931.
- What is CrossMap ? — CrossMap 0.6.4 documentation [Internet]. [cited 2024 Jan 15]. Available from: https://crossmap.sourceforge.net/.
- CPIC® Guideline for Thiopurines and TPMT and NUDT15 – CPIC [Internet]. [cited 2024 Jan 15]. Available from: https://cpicpgx.org/guidelines/guideline-for-thiopurines-and-tpmt/.
- CPIC® Guideline for Tricyclic Antidepressants and CYP2D6 and CYP2C19 – CPIC [Internet]. [cited 2024 Jan 15]. Available from: https://cpicpgx.org/guidelines/guideline-for-tricyclic-antidepressants-and-cyp2d6-and-cyp2c19/.
- CPIC® Guideline for Clopidogrel and CYP2C19 – CPIC [Internet]. [cited 2024 Jan 15]. Available from: https://cpicpgx.org/guidelines/guideline-for-clopidogrel-and-cyp2c19/.
- CPIC® Guideline for Proton Pump Inhibitors and CYP2C19 – CPIC [Internet]. [cited 2024 Jan 15]. Available from: https://cpicpgx.org/guidelines/cpic-guideline-for-proton-pump-inhibitors-and-cyp2c19/.
- CPIC® Guideline for Voriconazole and CYP2C19 – CPIC [Internet]. [cited 2024 Jan 15]. Available from: https://cpicpgx.org/guidelines/guideline-for-voriconazole-and-cyp2c19/.
- Brouwer JMJL, Nijenhuis M, Soree B, Guchelaar HJ, Swen JJ, van Schaik RHN, et al. Dutch Pharmacogenetics Working Group (DPWG) guideline for the gene-drug interaction between CYP2C19 and CYP2D6 and SSRIs. Eur J Hum Genet EJHG. 2022 Oct;30(10):1114–20. [CrossRef]
- Hicks JK, Bishop JR, Sangkuhl K, Müller DJ, Ji Y, Leckband SG, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for CYP2D6 and CYP2C19 Genotypes and Dosing of Selective Serotonin Reuptake Inhibitors. Clin Pharmacol Ther. 2015 Aug;98(2):127–34. [CrossRef]
- CPIC® Guideline for Ondansetron and Tropisetron based on CYP2D6 genotype – CPIC [Internet]. [cited 2024 Jan 15]. Available from: https://cpicpgx.org/guidelines/guideline-for-ondansetron-and-tropisetron-and-cyp2d6-genotype/.
- CPIC® Guideline for Tamoxifen based on CYP2D6 genotype – CPIC [Internet]. [cited 2024 Jan 15]. Available from: https://cpicpgx.org/guidelines/cpic-guideline-for-tamoxifen-based-on-cyp2d6-genotype/.
- Brown JT, Bishop JR, Sangkuhl K, Nurmi EL, Mueller DJ, Dinh JC, et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Cytochrome P450 (CYP)2D6 Genotype and Atomoxetine Therapy. Clin Pharmacol Ther. 2019 Jul;106(1):94–102. [CrossRef]
- CPIC® Guideline for Opioids and CYP2D6, OPRM1, and COMT – CPIC [Internet]. [cited 2024 Jan 15]. Available from: https://cpicpgx.org/guidelines/guideline-for-codeine-and-cyp2d6/.
- Caudle KE, Sangkuhl K, Whirl-Carrillo M, Swen JJ, Haidar CE, Klein TE, et al. Standardizing CYP2D6 Genotype to Phenotype Translation: Consensus Recommendations from the Clinical Pharmacogenetics Implementation Consortium and Dutch Pharmacogenetics Working Group. Clin Transl Sci. 2020 Jan;13(1):116–24. [CrossRef]
- Pratt VM, Cavallari LH, Del Tredici AL, Gaedigk A, Hachad H, Ji Y, et al. Recommendations for Clinical CYP2D6 Genotyping Allele Selection: A Joint Consensus Recommendation of the Association for Molecular Pathology, College of American Pathologists, Dutch Pharmacogenetics Working Group of the Royal Dutch Pharmacists Association, and the European Society for Pharmacogenomics and Personalized Therapy. J Mol Diagn JMD. 2021 Sep;23(9):1047–64. [CrossRef]
- Auton A, Abecasis GR, Altshuler DM, Durbin RM, Abecasis GR, Bentley DR, et al. A global reference for human genetic variation. Nature. 2015 Oct;526(7571):68–74. [CrossRef]
Figure 1.
Genomic sequence analysis workflow.
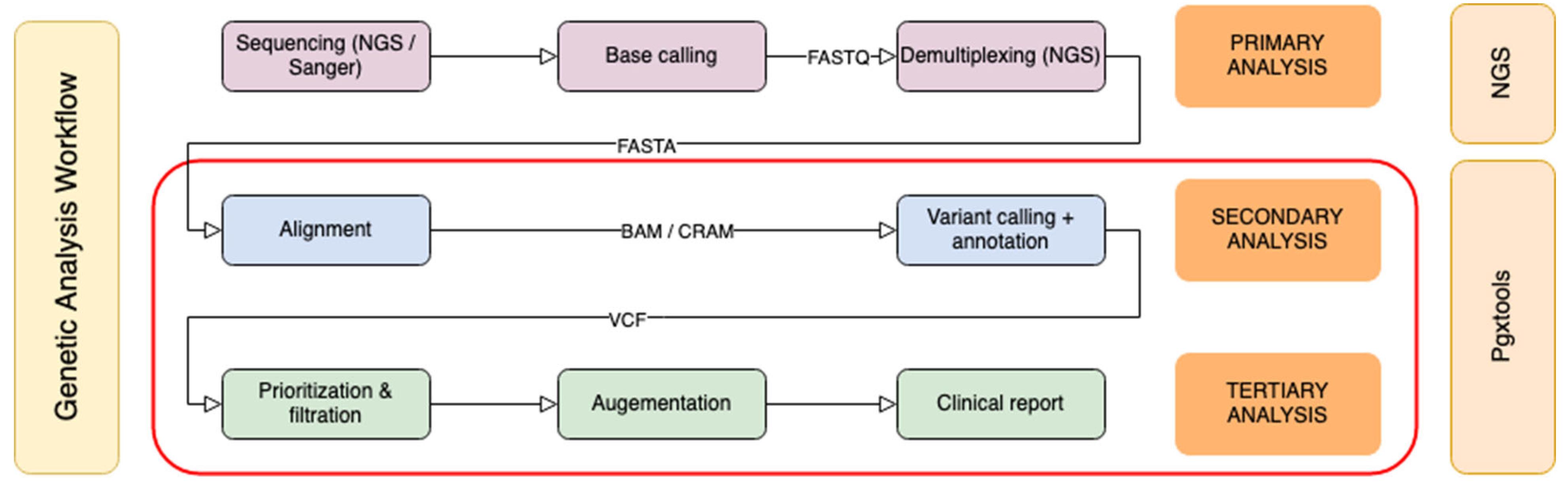
Figure 2.
Components and architecture of Pgxtools.

Figure 3.
Comparison of Pgxtools and G1K results in 45 samples with 83 variants in CYP2D6 gene on GRCh38. Percentages of variants correctly called by Pgxtools alone (green), G1K alone (orange), both Pgxtools and G1K (blue), or neither Pgxtools nor G1K (purple) are depicted. Variants without contigs for variant calling are depicted in red.
Figure 3.
Comparison of Pgxtools and G1K results in 45 samples with 83 variants in CYP2D6 gene on GRCh38. Percentages of variants correctly called by Pgxtools alone (green), G1K alone (orange), both Pgxtools and G1K (blue), or neither Pgxtools nor G1K (purple) are depicted. Variants without contigs for variant calling are depicted in red.
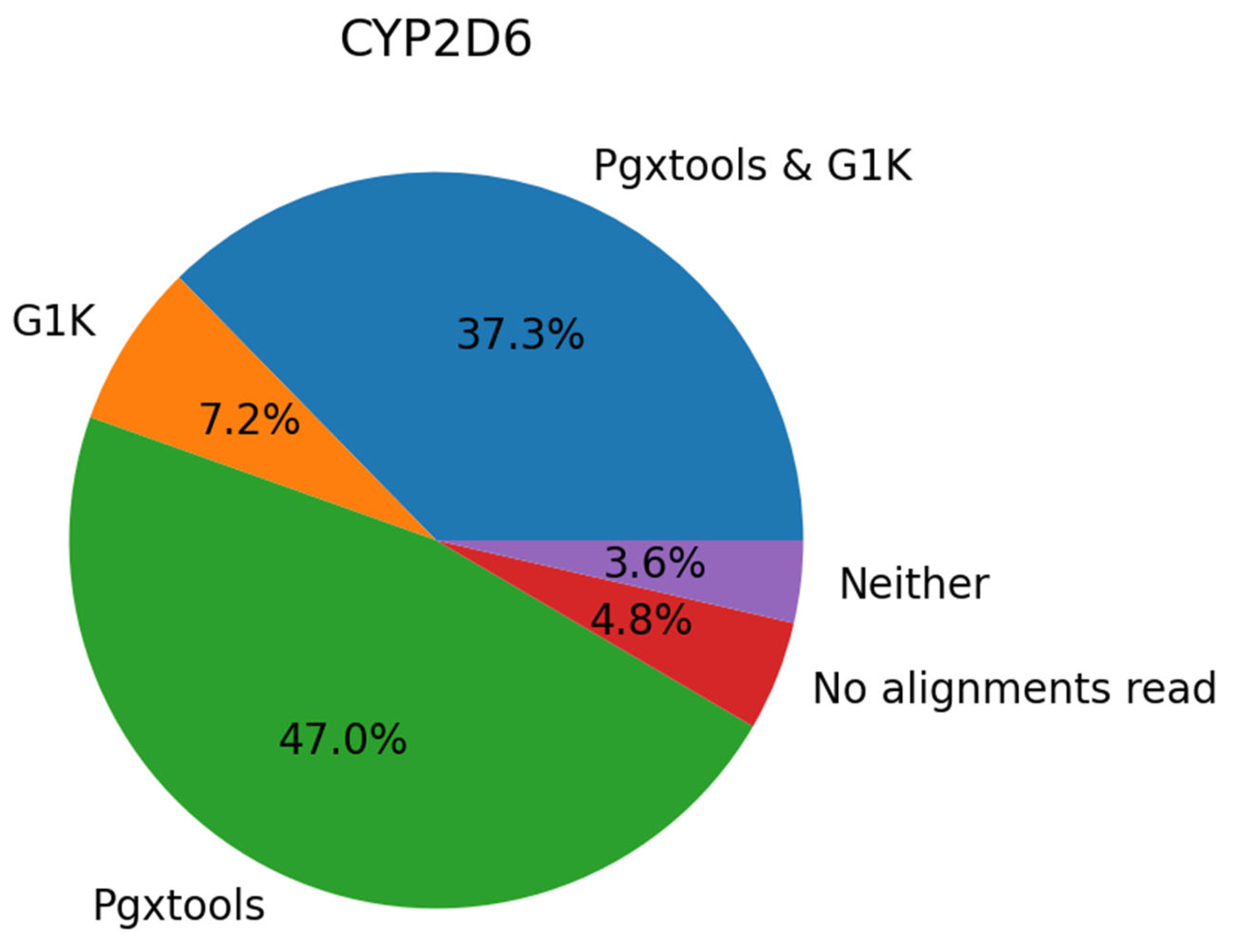
Figure 4.
Comparison of Pgxtools and G1K results in WGS samples with 1484 variants within CYP2D6, NUDT15, DPYD, TPMT and CYP3A5 genes on GRCh37.
Figure 4.
Comparison of Pgxtools and G1K results in WGS samples with 1484 variants within CYP2D6, NUDT15, DPYD, TPMT and CYP3A5 genes on GRCh37.
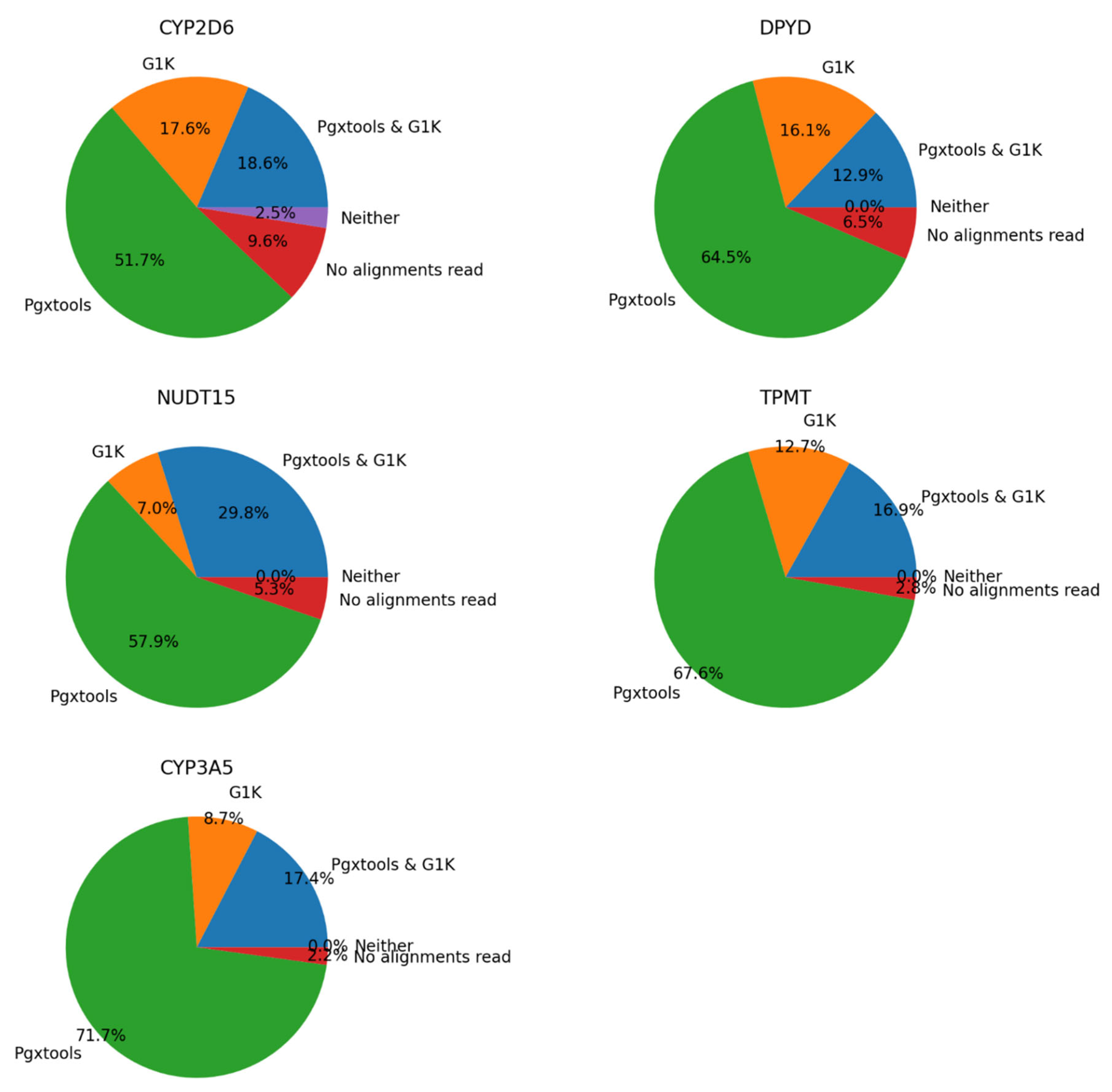
Figure 5.
The concordance between Pgxtools and the community consensus genotypes for the selected PGx genes with the CDC PGx reference materials.
Figure 5.
The concordance between Pgxtools and the community consensus genotypes for the selected PGx genes with the CDC PGx reference materials.
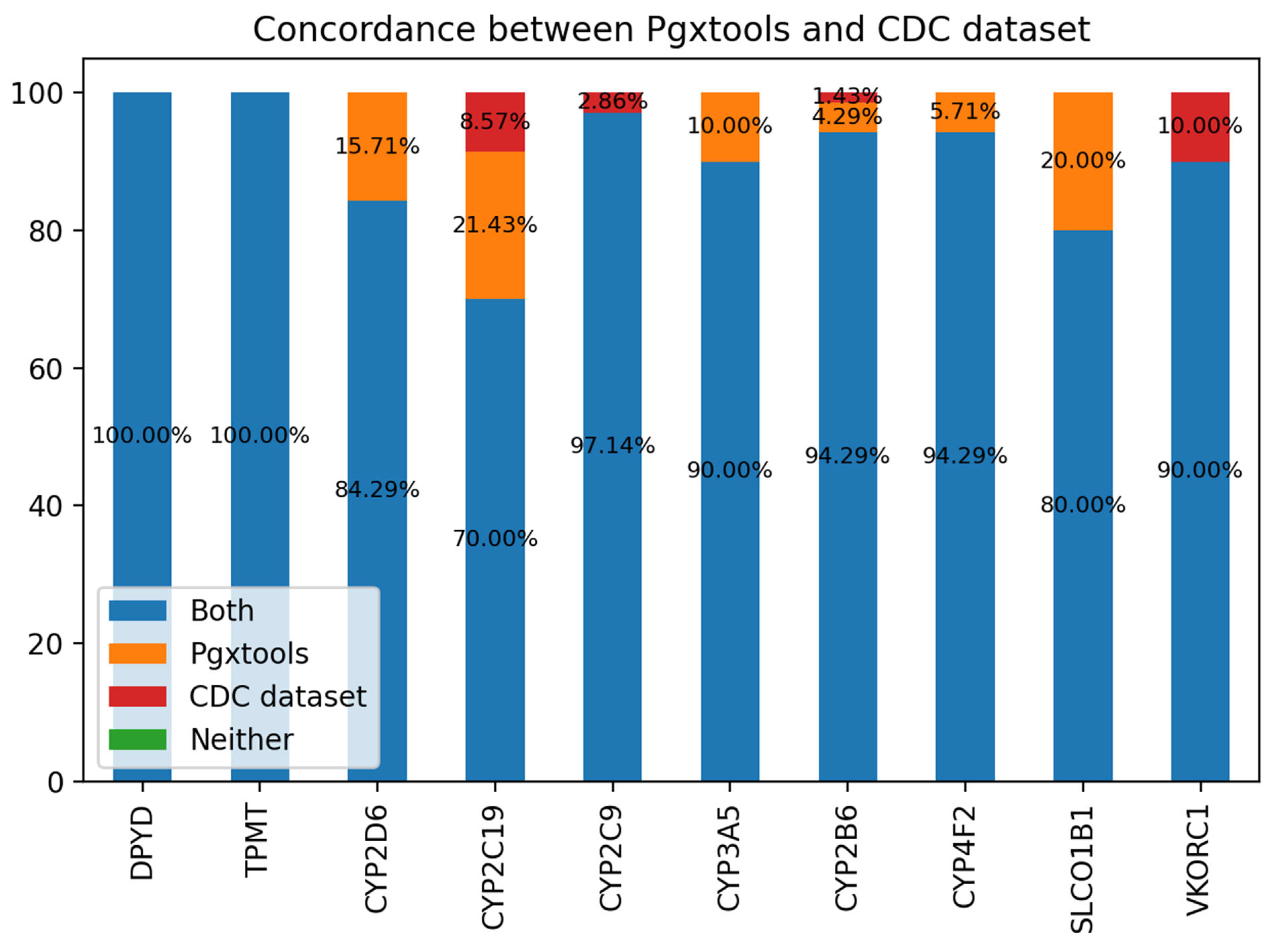

Table 1.
Genes and drugs for PGx analysis.
| Gene | Drug(s) |
|---|---|
| COMT | Opioid |
| CYP2B6 | Efavirenz |
| CYP2C9 | Warfarin |
| CYP2C19 | Clopidogrel, Proton Pump Inhibitors (Omeprazole, Lansoprazole, Pantoprazole and Dexlansoprazole), Selective Serotonin Reuptake Inhibitors (Citalopram, Escitalopram and Sertraline), Tricyclic Antidepressants (Tertiary Amines Amitriptyline, Clomipramine, Doxepin, Imipramine and Trimipramine), Voriconazole |
| CYP2D6 | Ondansetron and Tropisetron, Selective Serotonin Reuptake Inhibitors (Paroxetine and Fluvoxamine), Opioid (Codeine, Tramadol and Hydrocodone), Atomoxetine, Tricyclic Antidepressants, Tamoxifen |
| CYP3A5 | Tacrolimus |
| CYP4F2 | Warfarin |
| DPYD | Fluoropyrimidines 5-fluorouracil |
| IL28B | PEG Interferon-Alpha-Based Regimens |
| NUDT15 | Thiopurine |
| SLCO1B1 | Simvastatin |
| TPMT | Thiopurine (thioguanine, mercaptopurine and azathioprine) |
| VKORC1 | Warfarin |
Table 2.
Concordance of the variant calls by Pgxtools compared against the G1K dataset.
| CYP2D6 | CYP3A5 | DPYD | NUDT15 | TPMT | |
|---|---|---|---|---|---|
| GRCh37 | 68.17% | 92.69% | 98.80% | 97.72% | 97.40% |
| GRCh38 | 98.59% | 100% | 100% | 100% | 100% |
| GRCh38 to GRCh37 backported | 97.84% | 100% | 99.84% | 99.81% | 99.56% |
* There were 100% concordance in the genes, COMT, CYP2B6, CYP2C9, CYP2C19, CYP4F2, IL28B, SLCO1B1, and VKORC1 on both GRCh37 and GRCh38 in the 13 genes analyzed. Therefore, they are not listed in the table.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



