
Submitted:
05 February 2024
Posted:
06 February 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
Before the emergence of SARS-CoV-1, MERS-CoV, and most recently SARS-CoV-2, al-ready four other coronaviruses have been circulating in the human population. The alpha coronaviruses NL63 and 229E and the beta coronaviruses OC34 and HKU1. These circulating coronaviruses all cause mild respiratory illness during the winter seasons and most people are already infected in early life. Could antibodies and/or T cells, es-pecially against the beta coronaviruses have offered some form of protection against SARS-CoV-2? Related is the question whether survivors of SARS-CoV-1 or MERS-CoV would be relatively protected against SARS-CoV-2. More importantly, would humoral and cellular immunological memory, generated during the SARS-CoV-2 pandemic, ei-ther by infection or vaccination offer protection against future coronaviruses? Or rather than protection, could antibody dependent enhancement have taken place, a mecha-nism by which circulating corona antibodies enhance the severity of COVID-19? An-other related phenomenon, the original antigen sin, would also predict that the effec-tivity of the immune response to future coronaviruses would be impaired because of reactivation of memory against irrelevant epitopes. Current available evidence indicates that latter scenarios are highly unlikely and that especially cytotoxic memory T cells directed against conserved epitopes of human coronaviruses could at least offer partial protection against future coronaviruses.
Keywords:
SARS-CoV-2
; COVID-19
; human coronaviruses
; conserved T cell epitopes
; antibody dependent enhancement
; original antigenic sin.
1. Introduction
The COVID-19 pandemic pneumonia outbreak has brought attention to a critical global public health issue that has affected millions of people. The virus which causes COVID-19 is SARS-CoV-2, with a high degree of similarity with the SARS-CoV species which caused the 2003 coronavirus outbreak. This has greatly aided in identifying and full genetic characterization of SARS-CoV-2 [1]. The rapid spread of the illness necessitated the development of preventative and therapeutic measures for the care of SARS-CoV-2 infected individuals. Thanks mainly to the development and implementation of effective vaccines, the pandemic could be brought under control. On March 17, 2023, the Director-General of the WHO indicated that he was confident that in 2023 COVID-19 would be over as a public health emergency of international concern [2] and the official end of the pandemic was declared on May 23, 2023 [3]. SARS-CoV-2 however has not disappeared from the world, and still causes severe disease in the most vulnerable. In an optimistic scenario it can be envisioned that within time SARS-CoV-2 can be added to the so-called cold cousins, i.e., the circulating coronaviruses which cause the common cold during winter seasons (229E, OC43, NL63, and HKU1 [4]. In a pessimistic scenario immunological memory would not protect against novel coronaviruses, but due to antibody dependent and related mechanisms rather enhance severity of disease. In this paper the current available evidence will be discussed, which suggests that T cell memory, directed against conserved epitopes on the spike protein and nucleocapsid protein could protect against future coronaviruses.
Coronaviruses (CoVs) are positive-sense, single-stranded RNA viruses, they are members of the family Coronaviridae, subfamily Coronavirinae, and order Nidovirales. CoVs possess one of the largest genomes among all RNA viruses. The subfamily of Coronavirinae further can be divided into three major groups, distinguished by differences in their genetic makeup and antigenic cross-reactivity: the alpha-CoVs, beta-CoVs, and gamma-CoVs (also named group 1, 2 and 3 respectively) [5]. Originally, they were categorized based on serology, however, the categories named above are based on phylogenetic clustering [6].
The spike, envelope, membrane, and nucleocapsid are the four main structural proteins that make up the coronavirus virion structure (Figure 1). Although the number and location of accessory ORFs present in different coronaviruses species vary, distinct coronavirus strains share a common genetic organization for the coding region encoding for a canonical set of genes in the order 5′ end–ORF1a/b replicase, spike, envelope, membrane, nucleocapsid–3′ end. The subgenomic mRNAs that carry out gene translation combine with the viral genome to form a 5′ and 3′ co-terminal nested set. Subgenomic mRNAs are present along with a common 5′ leader sequence and a 3′ terminal sequence. The genome contains small untranslated regions at both the 3′ and 5′ ends. In addition, the viral genome encodes a number of nonstructural proteins (NSPs), such as the papain-like protease (PLpro), coronavirus main protease (3CLpro), and RNA-dependent RNA polymerase (RdRp) [7].
Before the emergence of SARS-CoV-1, Middle Eastern respiratory syndrome coronavirus (MERS-CoV), and most recently SARS-CoV-2, already four other coronaviruses have been circulating in the human population. The alpha coronaviruses NL63 and 229E and the beta coronaviruses OC34 and HKU1 (SARS-CoV-1, MERS-CoV, and SARS-CoV-2 also are beta coronaviruses) [8]. The circulating coronaviruses all cause mild respiratory illness during the winter seasons (which is the reason why they sometimes are called the cold cousins of SARS-CoV-2) and most people are already infected in early life [9,10]. Could antibodies or T cells, (especially against the beta coronaviruses) which have originally been generated after exposure to a circulating coronavirus have offered some form of protection against SARS-CoV-2? Related is the question whether survivors of SARS-CoV-1 or MERS-CoV would be relatively protected against SARS-CoV-2.
2. The immune response to SARS-CoV-2 infection and vaccination
Upon infection with SARS-CoV-2, an innate immune response is initiated in the respiratory tract. This involves massive activation of mainly proinflammatory cytokines [11,12]. The size of the initial viral load and the effectiveness of the innate immune response—especially the type I interferon-mediated response—appears to be crucial in determining the course of the subsequent adaptive response and the final clinical outcome [13]. Effective interferon signaling plays a critical role in acute infection, as demonstrated by both acquired and genetic factors. The indicators of severe clinical outcomes include early and persistent inflammation with elevated interferon (IFN)-α, TNF, and IFN-γ, as well as a slow decline in the viral load [14].
Like other viral respiratory infections, SARS-CoV-2 infection promotes the fast production of IgM, IgG, and IgA antibodies. These antibodies, including those that bind to the spike protein and nucleocapsid, are detectable in the sera as early as one week post-infection. The speed at which these reactions occur suggests that the antibodies originate from extrafollicular differentiation of naive B cells into short-lived antibody secreting cells, independent of the traditional germinal center reaction [15]. These antibodies also show neutralizing activity against live or pseudotyped SARS-CoV-2; latter activity can be readily detected in convalescent sera, though the degree of neutralization that can be achieved varies widely between people. The inconsistent outcomes of plasma therapy, which was tried early in the pandemic, could be partially explained by this variability [16].
2.1. The humoral immune response to SARS-CoV-2 infection
Hashem et al. studied the relationship between the severity of disease and the antibody titers in COVID-19 patients [17]. It was found that COVID-19 patients who experienced severe or moderate infections exhibited noticeably higher total neutralizing antibody (nAbs) levels. Furthermore, compared to mild cases, severe cases had significantly higher levels of S1-IgG antibodies. Notably, anti-N IgG and IgM levels were induced at higher levels in moderate and severe cases compared to mild infections, and these levels were significantly correlated with the severity of the disease. Anti-S1 and N antibodies (IgG and IgM) as well as nAbs were found to be significantly higher in patients who required ICU admission or had fatal outcomes when patients were stratified based on their need for ICU admission or infection outcome. Similar findings were also published by Liu et al. and Rijkers et al. who reported that nAbs were higher in hospitalized COVID-19 patients as compared to health care workers with mild clinical symptoms, not requiring hospitalization [18,19]. These findings unequivocally demonstrate the relationship between the severity of the COVID-19 infection and the activation state of the humoral immune system as a whole.
2.1.1. COVID-19 and response to vaccination in patients with humoral immunodeficiency or B cell depletion therapy.
The importance of the role played by humoral immunity in COVID-19 as well as a correlation of protection after vaccination remains uncertain, because SARS-CoV-2 infection or vaccination induce both a humoral and cellular immune response. The immune response of agammaglobulinemia patients during SARS-CoV-2 infection and the outcome of COVID-19 could serve as a model to further investigate the role of humoral immunity and thus help predict future outcomes.
Soresina et al. describe the cases of two X-linked agammaglobulinemia (XLA) patients who developed pneumonia as a clinical manifestation of SARS-CoV-2 infection, but did recover without requiring oxygen ventilation or intensive care treatment [20]. The cases of two adolescent male XLA patients were discussed by Devassikutty et al. where, except for a delayed recovery, both patients had successful outcomes [21]. In a paper on seven patients (2 with agammaglobulinemia and 5 with common variable immunodeficiency (CVID)) , it was noted that a milder course of COVID-19 was observed in agammaglobulinemia as compared to the CVID patients. Furthermore their COVID-19 course had a shorter duration and required no need for anti-inflammatory treatment with an IL-6 blocking drug [22]. These data, on an admittedly low number of patients, indicate that in the complete absence of B cells, such as is the case in XLA, COVID-19 does not necessarily takes a severe course. It could be argued that XLA patients are substituted with IVIG, and that antibodies contained within the preparation confer protection. The above mentioned studies however all were published in the first year of the pandemic and at that time IVIG preparations did not (yet) contain SARS-CoV-2 IgG antibodies.
From these findings in patients with B cell deficiencies it can be speculated that B cell-depletion may not have a major detrimental effect on COVID-19 recovery. In B cell lymphoma patients, treated with rituximab and bendamustine, the humoral immune response (but not the T cell response) to SARS-CoV-2 vaccination is impaired, as would be expected from a B cell depleting treatment [23]. Similar results were published by Candon et al. and Ishio et al. [24,25]. Patients with B cell lymphoma who were successfully treated with rituximab and have recovered, show normal antibody responses after (mRNA) SARS-CoV-2 vaccines [26]. Finally, in a group of patients with childhood-onset nephrotic syndrome who received rituximab for its steroid/calcineurin-inhibitor sparing effect, SARS-CoV-2 antibodies largely persisted. This indicates that long-lived plasma cells play a major role in maintaining antibody levels [27].
Most of the patients with multiple sclerosis who were treated with rituximab had a mild course of COVID-19 [28]. In a French cohort study on COVID-19 outcomes in patients with inflammatory rheumatic and musculoskeletal diseases treated with rituximab, severe disease occurred more frequently than in non-rituximab treated patients [29]. For other rituximab indications variable effects on incidence and severity of COVID-19 have been reported, but an extensive discussion of these data is beyond the scope of this paper.
2.1.2. Clinical effect of intravenous immunoglobulin (IVIG) administration on outcome of (severe) COVID-19
Another approach to estimate the relative importance of humoral immunity in COVID-19 is to analyze the effect of administration of antibodies, i.e., passive vaccination. A study in China that was already initiated in December 2019 found that administration of IVIG prolonged the length of hospital stay and duration of the disease. After correction for age and comorbidities, the treatment did reduce mortality [30] An Iranian study did find that administration of IVIG in patients with severe COVID-19 infection could improve their clinical outcome and significantly reduce mortality rate [31]. In a large multicenter randomized placebo controlled trial in France, patients with COVID-19 admitted to the ICU and on mechanical ventilation for moderate-to-severe ARDS, IVIG did not improve clinical outcomes [32]. Anti-SARS-CoV-2 IgG antibodies in the IVIG preparation or in the patients was not measured. In a meta-analysis, the pooled results from 4 retrospective studies shown no effects of IVIG use on COVID-19 mortality [33].
While the IVIG preparations used in above studies did not contain SARS-CoV-2 antibodies, a beneficial effect could be obtained via targeting the cytokine storm by influences on complement, innate immune cells, effector T-cells, and Tregs in patients with severe and critical COVID-19. The results of early administration of high-doses IVIG in severe or critical COVID-19 regarding mortality reduction are inconclusive [34].
2.1.3. Passive vaccination with monoclonal antibodies.
One of the treatment options for patients with severe COVID-19 disease is passive vaccination. Pooled sera from recovered patients have been used for this purpose, however with limited or no success [35,36,37]. Monoclonal antibodies have been developed, the first one was bamlanivimab, a monoclonal directed against the RBD of the spike protein of the original Wuhan strain of SARS-CoV-2 [38]. Administration of the antibody, especially in combination with another monoclonal antibody against the RBD of SARS-CoV-2 (etesvimab) to high-risk outpatients recently diagnosed with COVID-19 led to reduced viral load and decreased the risk of hospitalization and death [39]. When the omicron variants became the dominant strains, the effectivity of these antibodies was lost [40].
2.2. T cell mediated immunity against coronaviruses.
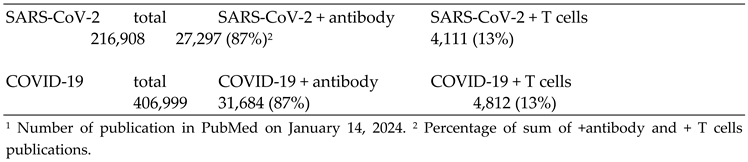
Probably fueled by the massive reports in the popular press on vaccination and immunity, the general conception is that mainly antibodies directed against the SARS-CoV-2 Spike protein offer protection against disease. Furthermore, antibody levels should remain high in order to stay protected. The role of T cell mediated immunity against SARS-CoV-2, and viruses in general, has received relative little attention. PubMed yields over 200,000 papers on SARS-CoV-2 and T cells (January 14, 2024) and over 400,000 papers on COVID-19. In the papers dealing with immunity, 87% concern humoral immunity and only 13% address T cell immunity (Table 1).
2.2.1. Importance of T-cell immunity
T-cells, in principle , can respond to any viral peptides, including those of more conserved regions. SARS-CoV-2, as well as SARS-CoV-1 for that matter, can enter the host cells through binding to the ACE2 receptor (see also Figure 5) . Upon entry, viral capsid proteins are broken down by enzymatic degradation, setting free the genetic material and exploiting the host cellular machinery for viral replication purposes [41]. Simultaneously, viral proteins are further processed by the proteasome after which any peptide that fits can be bound in the groove of an MHC class I molecule (HLA-A, -B, -C) and upon cell surface expression be presented to CD8+ T-cells or bind to MHC class II (HLA-DR, -DP, DQ) [42] molecules on antigen-presenting cells (APCs) to CD4+ T-helper cells. The selective interaction with the TCR elicits either a cytotoxic response by CD8+ T-cells or the activation of CD4+ T-helper cells that are necessary for proper stimulation of B-lymphocytes and antibody production [43]. Because all viral peptides, including those derived from non-structural regions, have the potential to elicit a T-cell response, they play a determining role in adaptive cellular immunity.
The receptor binding domain of the spike protein of SARS-CoV-2 is localized in the N-terminal region of the protein and contains 17 amino acids which directly interact with the ACE2 receptor on human host cells (Figure 2). Within the conserved regions of the Spike proteins of the other human coronaviruses there is little sequence homology. Homologies are more prominent in the C-terminal regions of the Spike proteins, which is the part where most CD8+ T cell epitopes are found (Figure 2)[44].
The Spike protein contains 3 major T cell epitopes, as defined by Ferretti et al., termed YLQ, KCY and QYI [44]. All three epitopes are located outside the RBD of SARS-CoV-2. The QYI epitope is highly conserved in SARS-CoV-1 and MERS-CoV, as well as in the other hCoVs. Furthermore, in the variants of SARS-CoV-2, from alpha to omicron, including the omicron subvariants, the QYI epitope remains unchanged. However, the epitope closest to the RBD (KCY) has been mutated in the omicron variants [45].
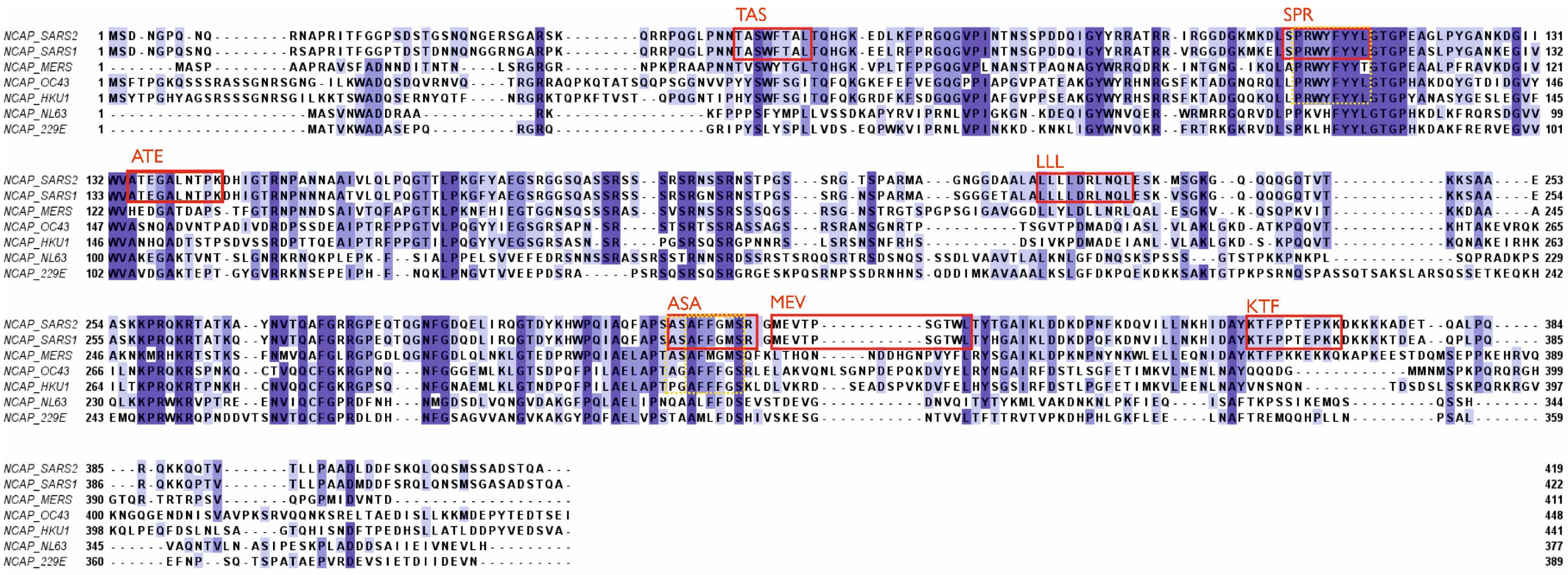
The nucleocapsid (N) protein is well conserved and has been shown to display a high degree of homology between different coronavirus strains [46]. N proteins are associated with the viral RNA (see Figure 1) and are not accessible for antibodies in an intact virus particle. Therefore, anti-N antibodies, which are produced in large quantities following infection, cannot prevent spread of the virus within the body. N proteins can serve however as important T-cell epitopes [47]. Six major epitopes have been identified in SARS-CoV-2, as indicated in Figure 3. Furthermore, all six epitopes are identical in SARS-CoV-1 and SARS-CoV-2 (including the delta and omicron variants of SARS-CoV-2), plus they are located within conserved regions of the different beta coronavirus strains. More specifically, the ASA epitope is conserved in all known human beta coronaviruses and SPR to a lesser degree (Figure 3). The MEV epitope is identical in SARS-CoV-1 and SARS-CoV-2, but not found in other human beta coronaviruses [48]. SARS-CoV-1 survivors showed a robust T cell response when activated in vitro with N protein epitopes, including ASA and SPR [48]. This is evidence supporting the suggestion that memory T-cells from previous infections with circulating coronaviruses could have been reactive to SARS-CoV-2 and thus have aided in the cellular protection against severe disease. Any possible existing cross-reactive T-cell immunity against SARS-CoV-2 could therefore have resulted from prior infection with another coronavirus i.e., SARS-CoV-1, MERS-CoV [49] or, and this would have had a much bigger impact, one of the common cold viruses OC43, HKU1, NL63 or 229E10 [50]. In a similar scenario, long-lasting cellular immunity could be provided through cross-reactivity against homologous epitopes for new coronaviruses in the future.
Figure 3.
Major T cell epitopes within the nucleocapsid protein. All epitopes are indicated by their first three letters. TAS = TASWFTAL (49-56); SPR = SPRWYFYYL (105-113); ATE = ATEGALNTPK (134-143); LLL = LLLDRLNQL ( 222-230); ASA = ASAFFGMSR (311-319); KTF = KTFPPTEPKK (361-369): MEV = MEVTPSGTWL (322-331). Further details in legend of Figure 2). Based on data from [44,47,48].
Figure 3.
Major T cell epitopes within the nucleocapsid protein. All epitopes are indicated by their first three letters. TAS = TASWFTAL (49-56); SPR = SPRWYFYYL (105-113); ATE = ATEGALNTPK (134-143); LLL = LLLDRLNQL ( 222-230); ASA = ASAFFGMSR (311-319); KTF = KTFPPTEPKK (361-369): MEV = MEVTPSGTWL (322-331). Further details in legend of Figure 2). Based on data from [44,47,48].
When targeting cellular immunity, the focus should be on MHC class I epitopes which are present on all nucleated cells [51] including those with ACE2 receptors. Displaying viral peptides via MHC I elicits a direct response of cytotoxic CD8+ T-cells, leading to programmed cell death of the virally infected cell [52]. This immediately halts viral replication as the host cell is necessary for the production of the virus. In COVID-19, an early cytotoxic T-cell response has been observed to correlate with efficient viral clearance and a mild disease course [14]. In addition, CD8+ memory T-cells generated by previous infection with coronaviruses facilitate a quick response to reinfection by the same strain, and might aid in fighting infection by a new strain or entirely new virus, provided there would be sufficient sequence homology between relevant epitopes. Therefore, T-cell immunity and especially MHC I epitopes should receive careful consideration when designing vaccines in the future.
Furthermore, Verma et al. found that peptides of the N protein can bind with high affinity to TLR4, expressed on monocytes and macrophages [47]. Triggering of TLR4 could be beneficial as this improves antigen uptake and presentation to both CD4+ T-cells, stimulating the B-cell response and antibody production as well as to CD8+ T cells [53]. Hence, identifying and utilizing epitopes that are recognized by APCs additionally to binding to MHC I could thus promote both cellular and humoral immunity. Apart from Spike and N protein, many T-cell epitopes in SARS-CoV-2 are also found in non-structural parts (NSP) like ORF1ab or ORF3a [54]. However, immunodominant are epitopes of the N protein, giving rise to the highest frequency of specific T-cells [14,54]. Overall, the indicated (immuno-)dominant HLA alleles in response to SARS-CoV-2 are HLA-A*01:01, HLA-A*02:01, HLA-A*03:01, HLA-A*11:01, HLA-B*07:02 [44]. These HLA types specifically have been shown to interact with a wide variety of different structural and non-structural SARS-CoV-2 peptides and at least one of the six HLA-alleles is present in about 85% of the world’s population [44]. All SARS-CoV-2 T cell epitopes discussed in this paper can interact with at least one of these aforementioned alleles.
3. Potential negative impact of existing antibodies and memory B cells on future viruses.
When, as discussed above, a novel emerging coronavirus would have considerable antigenic differences with circulating coronaviruses, existing antibodies and memory B-cells would offer no protection. It is even possible that existing antibodies and memory B-cells would have a negative effect and cause more severe clinical expression of the infection. Two different, but partly overlapping, mechanisms could be involved. The first one is antibody dependent enhancement, the second is the original antigenic sin.
3.1. Antibody Dependent Enhancement (ADE)
Antibody Dependent Enhancement (ADE) refers to the enhancement of disease due to the presence of antibodies to the pathogen which has caused the disease. These antibodies can have been induced by previous exposure to the given pathogen which caused a primary immune response, or previous vaccination which has induced the production of antibodies, or passive administration of (monoclonal) antibodies as a form of treatment. As far as coronaviruses are concerned, there has been a precedent of ADE after vaccination. Cats, vaccinated against feline infectious peritonitis virus, a corona virus, did produce anti-viral antibodies but after challenge with live virus were more susceptible than the non-vaccinated control animals [55,56]. ADE can lead to the facilitated entry of the virus into the cells that express FcRγs, mostly leukocytes, and depending on the cellular tropism of the virus in question, expand the number of target cells (see below, 3.1.3). While ADE is of concern to vaccine development in general, it especially was relevant for corona virus vaccines (see above). Thus, although the precise pathways of ADE remain elusive, and especially how it may enhance viral infections, the interest in ADE rose during the SARS-CoV-2 pandemic (Figure 4).
Figure 4.
Number of publications per year retrieved from PubMed (https://pubmed.ncbi.nlm.nih.gov/) using the search terms “antibody dependent enhancement” or “original antigenic sin” during the past 20 years.
Figure 4.
Number of publications per year retrieved from PubMed (https://pubmed.ncbi.nlm.nih.gov/) using the search terms “antibody dependent enhancement” or “original antigenic sin” during the past 20 years.

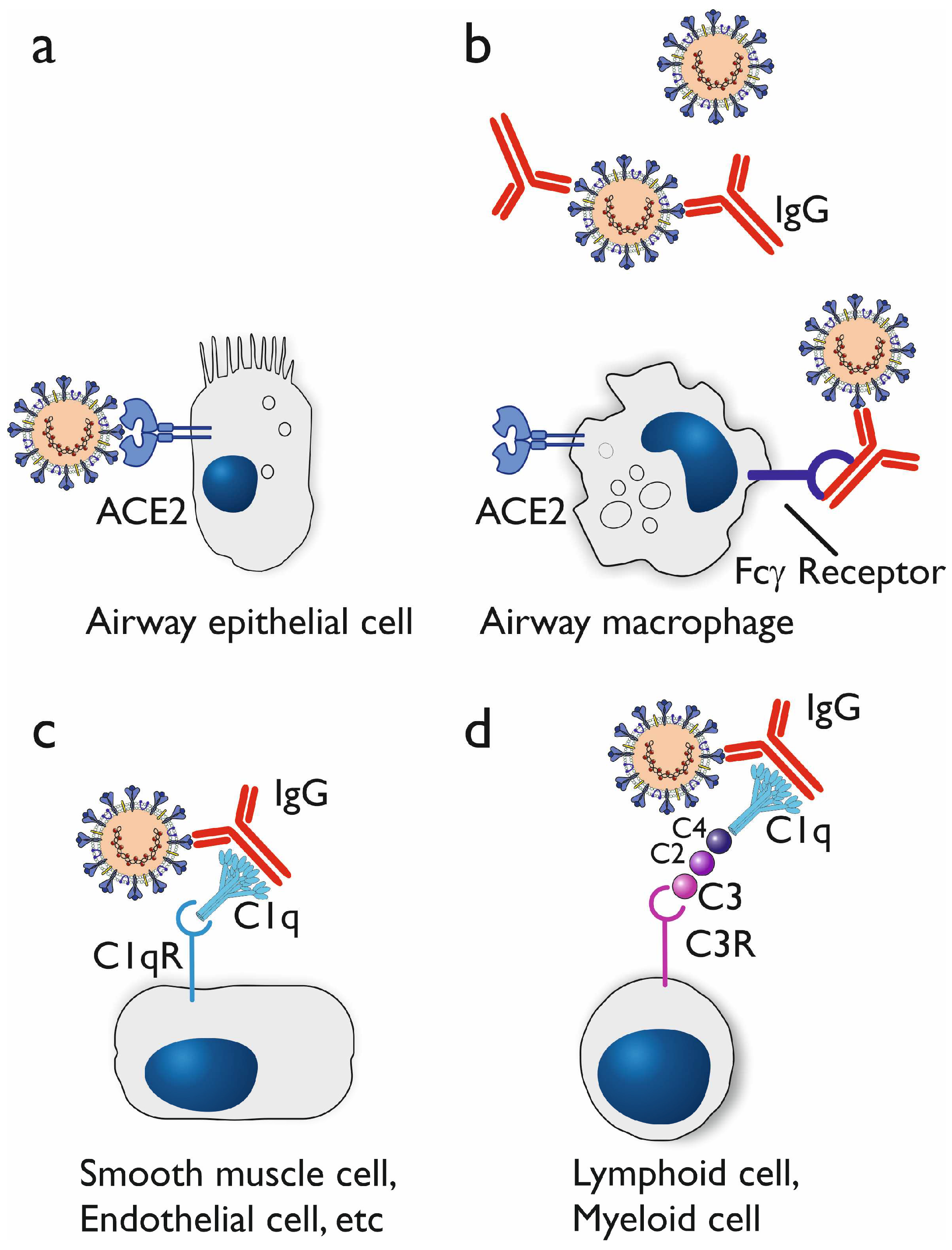
Figure 5.
Cellular tropism of SARS-CoV-2 and the potential mechanism of antibody dependent enhancement. Human cells expressing the angiotensin converting enzyme 2 (ACE2), such as airway epithelial cells (panel a) and airway macrophages (panel b) can be infected by SARS-CoV-2. When IgG antibodies (against viral surface proteins such as the Spike protein) are generated, these can form immune complexes with the virus. The Fc part of the IgG can be bound by cellular Fcγ receptors, which enables the virus to enter the cell via this route (panel b). When the complement system is activated, C1q binds to the IgG and the complex can be captured by C1q receptors (C1qR) (panel c). Ongoing complement activation leads to binding and activation of C4, C2, and C3 (C1r and C1s are not shown). C3 receptors (C3R), which include Complement Receptor 1 (CR1), CR2 and CR3, also are able to bind the complex of virus, antibody, and complement proteins.
Figure 5.
Cellular tropism of SARS-CoV-2 and the potential mechanism of antibody dependent enhancement. Human cells expressing the angiotensin converting enzyme 2 (ACE2), such as airway epithelial cells (panel a) and airway macrophages (panel b) can be infected by SARS-CoV-2. When IgG antibodies (against viral surface proteins such as the Spike protein) are generated, these can form immune complexes with the virus. The Fc part of the IgG can be bound by cellular Fcγ receptors, which enables the virus to enter the cell via this route (panel b). When the complement system is activated, C1q binds to the IgG and the complex can be captured by C1q receptors (C1qR) (panel c). Ongoing complement activation leads to binding and activation of C4, C2, and C3 (C1r and C1s are not shown). C3 receptors (C3R), which include Complement Receptor 1 (CR1), CR2 and CR3, also are able to bind the complex of virus, antibody, and complement proteins.
3.1.1. In vitro studies on antibody dependent enhancement
Wan et al. mention the mechanism by which previous coronaviruses have undergone ADE, specifically MERS-CoV and SARS-CoV [57]. This includes how cellular entry is mediated, and thus how MERS-CoV and SARS-CoV serve as excellent models for the study of ADE in vitro. Specifically of interest to this paper is the mechanism they discussed for SARS-CoV as SARS-CoV-2 shares the same viral receptor, namely angiotensin-converting enzyme 2 (ACE2). Using monoclonal antibodies specifically targeted to the receptor binding domain (RBD) of SARS-CoV, Wan et al. found that entry into CD32A expressing cells was mediated by this MAb, however it blocked entry into cells that expressed the ACE2 receptor [57]. This inhibitory effect serves as a possible mechanism by which ADE could occur with blood lymphocytes. since they show poor ACE2 expression when compared to tissue resident macrophages..
3.1.2. In vivo indications for antibody dependent enhancement
Evidence of ADE in flaviviruses, specifically dengue virus, has been observed and studied. The proposed mechanism for the ADE of dengue virus entails a primary infection of a specific serotype during which antibodies are produced that neutralize the serotype of the primary infection [58]. ADE then occurs during a secondary infection of a different serotype the same antibodies no longer function to the same extent, and instead of neutralizing the viral particles they potentiate the entry into leukocytes [58]. This is achieved as the Fab part of these antibodies will bind to the viral particles leaving the Fc part open to bind with Fcγ receptors on cells of the immune system. This will then allow leukocytes to recognize and neutralize the virus in normal function, however in ADE the Fc part of the antibody will mediate the entry of the viral particles into leukocytes, particularly the macrophages, which will lead to viral replication inside of the leukocytes and ultimately enhancement of the disease. This mechanism of disease enhancement also has been discussed in the case of coronaviruses (see above)[59]. The specific glycosylation states of IgG antibodies generated during a SARS-CoV-2 infection has been linked to how the course of the disease will progress ultimately. Afucosylation of the IgG antibodies leads to an increase in the affinity to FcγRIIIA (CD16a) and thus an increase in antibody dependent cellular cytotoxity (ADCC) [60,61]. Other modifications to antibodies and different antibody classes need to be researched to elucidate the role galactosylation and sialylation has on complement activation and disease progression which could play a role in ADE through the aforementioned mechanism.
3.1.3. Proposed mechanisms of antibody dependent enhancement
Fc gamma receptors (FcγR) are involved in the specific signaling pathways that purportedly cause ADE and are primarily expressed on the surface of leukocytes. These receptors, including FcγRI (CD64), FcγRIIa (CD32a), FcγRIIb (CD32b), FcγRIIc (CD32c), and FcγRIIIa (CD16a), play distinct roles in mediating immune responses upon binding to the Fc parts of antibodies, particularly IgG, the antibody class that dominates the humoral immune response to SARS-CoV-2 infection and vaccination. If binding of the Fc part of IgG would mediate viral entry into the leukocytes, on top of the capacity of the virus to enter target cells expressing ACE2 receptors, this could lead to ADE (Figure 5).
Upon engagement with immune complexes formed by antibodies and their corresponding antigens, FcγRs initiate intracellular signaling cascades. The pathways involved often include phosphorylation events mediated by tyrosine kinases, such as Lyn and Syk, leading to the activation of downstream signaling molecules. Ultimately this will lead to various cellular responses, including phagocytosis: antibody dependent cellular phagocytosis, ADCP [62]. Activation of monocytes and macrophages via FcγRs also leads to production of pro-inflammatory cytokines and cellular activation [63]. In the context of ADE, these signaling pathways may inadvertently contribute to increased infection by promoting the uptake of virus containing immune complexes. Therefore, in ADE the phagocytic cells could serve as a (additional) host for the virus.
Also the complement system may play a critical role in ADE. There are two main mechanisms involving complement that have been exploited by viruses, namely a C1q mediated mechanism and a C3 mediated mechanism. The former involves the formation of complexes composed of virus, antibody, and C1q [64,65]. Ultimately the binding of C1q to the C1q receptor leads to the increased viral fusion with the cellular membrane (Figure 5, panel c). As Wen et al. mention this serves as a possible explanation as to why ADE can occur in cells other than monocytes, due to the presence of C1q receptor on other cells such as endothelial cells, smooth muscle cells [66,67]. B cells, monocytes and macrophages also can be involved when C3 becomes activated through the classical pathway of the complement system after binding of antibodies to the viral surface proteins. The activated C3 will bind to C3 receptors and thus again lead to increased viral fusion with the cellular membrane due to the complexes of virus, antibody, and complement C3 (Figure 5, panel d) [66].
3.2. The original antigenic sin
Apart from ADE, another potential negative effect of immunological memory for future coronaviruses would be the so-called original antigenic sin [68]. This phenomenon was first described by Thomas Francis Jr. in 1960, and states that a new strain of an existing virus (in his case influenza virus) could activate memory B cells specific for a previous strain. Those antibodies could bind to the new strain, but not neutralize the virus [69]. This phenomenon needs to be considered when designing and evaluating current and future coronavirus vaccines [70]. Current data do not point towards a role for “original antigenic sin” in the immune response to SARS-CoV-2 infection or vaccination Does not negatively impact the efficacy of vaccination [71,72], if anything activation of existing memory to related coronaviruses could contribute to control of SARS-CoV-2. The sin therefore could also turn out to be a virtue [71,72].
Within the T cell system, an equivalent of ADE or original antigenic sin has not been described. Any negative effect of existing T cell memory on protection against future coronavirus infections therefore is not to be expected.
4. Concluding remarks and outlook for the future
The best model to assess the protective effect of existing immunological memory for future coronaviruses is to compare COVID-19 incidence and severity in survivors of SARS-CoV-1 or MERS-CoV infection. In SARS-CoV-1 survivors, cross-reactive SARS-CoV-2 IgG antibodies can be detected and these SARS survivors also generate a much stronger antibody response after SARS-CoV-2 vaccination [73]. In a retrospective cohort study it was found that symptomatic MERS-CoV patients were at a lower risk for COVID-19 [74]. Care must be taken when interpreting these data, because SARS-CoV-1 and MERS-CoV patients survived without being vaccinated or specifically treated. They therefore can have an immune system which was, and remained, a priori better equipped to combat coronaviruses.
Our analysis of the literature shows and confirms that cytotoxic T cells against conserved epitopes on the SARS-CoV-2 spike protein and nucleocapsid protein are expanded during infection and do limit the severity of COVID-19 [14,75]. It is possible that existing memory T cells, generated during exposure to circulating hCoVs, including SARS-CoV-1 and MERS, have offered partial protection against SARS-CoV-2, which would explain the lower case fatality rate of SARS-CoV-2 as compared to SARS and MERS. It therefore should be considered to include conserved epitopes of both the spike protein and nucleocapsid protein in future vaccines, in order to induce a robust and lasting T cell response against these relevant epitopes [75]. Appelberg et al. have shown that in their prototype universal SARS-CoV-2 vaccine, inclusion of only the relevant nucleoprotein T cell epitopes provided 60% protection against lethal infection in mice [76].
Infection with SARS-CoV-2 or vaccination for that matter, induces a strong response of both the humoral and cellular immune system. In assessment of the immune status of patients with COVID-19, as well as in the analysis of the immune response to vaccination, emphasis mostly is put on quantitation of (neutralizing) antibodies. Although no protective antibody titers have been established, the magnitude of the antibody response is taken as a correlate of protection. T cell immunity appears to be more important, and is directed to epitopes of viral proteins which are largely conserved among the hCoVs, including the variants of SARS-CoV-2. It therefore would be appropriate to (also) implement T cell based diagnostic assays. Development and implementation of these assays will be part of preparing for the future.
In their search for the origin of SARS-CoV-2, Temmam et al. have sampled Rhinolophus bats in Northern Laos [77]. Many previously unknown coronaviruses were found, including five sarbecoviruses. Three of those (BANAL-52, BANAL-103 and BANAL-236) are close relatives of SARS-CoV-2 and can bind to human ACE-2 [77]. These data show that bats, as well as many other animal species for that matter, are a large reservoir from which new coronaviruses can emerge with the potential to set off a new pandemic.
Thanks to the advancement of medical sciences, the original Darwinian principle struggle for life, survival of the fittest no longer holds completely true. Yet, despite vaccines, diagnostics, advanced treatment, the burden of infectious diseases, including SARS-CoV-2 for that matter, weighs on those with an impaired immune system. The “fitness” of the immune system, however, is not as easy to determine as for instance that of the cardiac or respiratory system. Unfortunately there is no simple litmus test for the immune system.
The research team of Amy Huei-Yi Lee has developed a multi-omic data integration tool which has the potential to create a kind of immune signature which could predict response to vaccination [78]. On a much larger scale, an initiative has been taken to collect thousands of immune parameters (cells, proteins, genes) of hundreds of thousands of people across the globe in a project called the Human Immunome Project (https://www.humanimmunomeproject.org/ ) [79]. Advancements like these will bring us in a position making it possible to predict whether or not a new virus, or any other micro-organism, would have the potential to give rise to endemic or pandemic outgrowth.
Author Contributions
Conceptualization, G.T.R, M.B. E.S.S., and L.M.S.; methodology, M.B. and G.T.R.; formal analysis, M.B.; writing—original draft preparation, G.T.R, M.B. E.S.S., and L.M.S.; writing—review and editing, G.T.R.; visualization, M.B., E.S.S., L.M.S., and G.T.R; supervision, G.T.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We apologize to all colleagues who have contributed immensely to our current understanding of the immunobiology of SARS-CoV-2 and other coronaviruses, but who’s work we could not include or cite due to space restrictions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, et al. China Novel Coronavirus Investigating and Research Team. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [CrossRef]
- World Health Organization. Virtual Press Conference on COVID-19 & Other Global Health Emergencies. Available online: https://www.who.int/publications/m/item/virtual-press-conference-on-covid-19---other-global-health-emergencies (accessed on January 14 2024).
- Harris E. WHO Declares End of COVID-19 Global Health Emergency. JAMA. 2023 Jun 6;329(21):1817. [CrossRef]
- Cohen J. COVID’s cold cousins. Four largely ignored coronaviruses circulate in humans without causing great harm and may portend the future for SARS-CoV-2. Science 383, 2024, 141-145. [CrossRef]
- Kaur N, Singh R, Dar Z, Bijarnia RK, Dhingra N, Kaur T. Genetic comparison among various coronavirus strains for the identification of potential vaccine targets of SARS-CoV2. Infect Genet Evol. 2021 Apr;89:104490. [CrossRef]
- Al-Qahtani WS, Alneghery LM, Alqahtani AQS, Al-Kahtani MD, Alkahtani S. A review of comparison study between corona viruses (Sars-cov, mers-cov) and novel corona virus (COVID-19). Revista Mexicana de Ingeniería Química 2020, 19(Sup. 1), 201-212. [CrossRef]
- Yang H, Rao Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat Rev Microbiol. 2021 Nov;19(11):685-700. [CrossRef]
- Tang G, Liu Z, Chen D. Human coronaviruses: Origin, host and receptor. J Clin Virol. 2022 Oct;155:105246. [CrossRef]
- Rajapakse N, Dixit D. Human and novel coronavirus infections in children: a review. Paediatr Int Child Health. 2021 Feb;41(1):36-55. [CrossRef]
- van der Hoek L, Pyrc K, Jebbink MF, Vermeulen-Oost W, Berkhout RJ, Wolthers KC, Wertheim-van Dillen PM, Kaandorp J, Spaargaren J, Berkhout B. Identification of a new human coronavirus. Nat Med. 2004 Apr;10(4):368-73. [CrossRef]
- Hu B, Huang S, Yin L. The cytokine storm and COVID-19. J Med Virol. 2021 Jan;93(1):250-256. [CrossRef]
- Fajgenbaum DC, June CH. Cytokine Storm. N Engl J Med. 2020 Dec 3;383(23):2255-2273. [CrossRef]
- Lei X, Dong X, Ma R, Wang W, Xiao X, Tian Z, Wang C, Wang Y, Li L, Ren L, et al. Activation and evasion of type I interferon responses by SARS-CoV-2. Nat Commun. 2020 Jul 30;11(1):3810. [CrossRef]
- Moss P. The T cell immune response against SARS-CoV-2. Nat Immunol. 2022 Feb;23(2):186-193. [CrossRef]
- Woodruff MC, Ramonell RP, Nguyen DC, Cashman KS, Saini AS, Haddad NS, Ley AM, Kyu S, Howell JC, Ozturk T, et al. Extrafollicular B cell responses correlate with neutralizing antibodies and morbidity in COVID-19. Nat Immunol. 2020 Dec;21(12):1506-1516. [CrossRef]
- Zhang Q, Wang Y, Qi C, Shen L, Li J. Clinical trial analysis of 2019-nCoV therapy registered in China. J Med Virol. 2020 Jun;92(6):540-545. [CrossRef]
- Hashem AM, Algaissi A, Almahboub SA, Alfaleh MA, Abujamel TS, Alamri SS, Alluhaybi KA, Hobani HI, AlHarbi RH, Alsulaiman RM, et al. Early Humoral Response Correlates with Disease Severity and Outcomes in COVID-19 Patients. Viruses. 2020 Dec 4;12(12):1390. [CrossRef]
- Liu X, Wang J, Xu X, Liao G, Chen Y, Hu CH. Patterns of IgG and IgM antibody response in COVID-19 patients. Emerg Microbes Infect. 2020 Dec;9(1):1269-1274. [CrossRef]
- Rijkers G, Murk JL, Wintermans B, van Looy B, van den Berge M, Veenemans J, Stohr J, Reusken C, van der Pol P, Reimerink J. Differences in Antibody Kinetics and Functionality Between Severe and Mild Severe Acute Respiratory Syndrome Coronavirus 2 Infections. J Infect Dis. 2020 Sep 14;222(8):1265-1269. [CrossRef]
- Soresina A, Moratto D, Chiarini M, Paolillo C, Baresi G, Focà E, Bezzi M, Baronio B, Giacomelli M, Badolato R. Two X-linked agammaglobulinemia patients develop pneumonia as COVID-19 manifestation but recover. Pediatr Allergy Immunol. 2020 Jul;31(5):565-569. [CrossRef]
- Devassikutty FM, Jain A, Edavazhippurath A, Joseph MC, Peedikayil MMT, Scaria V, Sandhya P, Govindaraj GM. X-Linked Agammaglobulinemia and COVID-19: Two Case Reports and Review of Literature. Pediatr Allergy Immunol Pulmonol. 2021 Sep;34(3):115-118. [CrossRef]
- Quinti I, Locatelli F, Carsetti R. The Immune Response to SARS-CoV-2 Vaccination: Insights Learned From Adult Patients With Common Variable Immune Deficiency. Front Immunol. 2022 Jan 19;12:815404. [CrossRef]
- Vanni A, Salvati L, Mazzoni A, Lamacchia G, Capone M, Francalanci S, Kiros ST, Cosmi L, Puccini B, Ciceri M, et al. Bendamustine impairs humoral but not cellular immunity to SARS-CoV-2 vaccination in rituximab-treated B-cell lymphoma-affected patients. Front Immunol. 2023 Dec 1;14:1322594. [CrossRef]
- Candon S, Lemee V, Leveque E, Etancelin P, Paquin C, Carette M, Contentin N, Bobee V, Alani M, Cardinael N, et al. Dissociated humoral and cellular immune responses after a three-dose schema of BNT162b2 vaccine in patients receiving anti-CD20 monoclonal antibody maintenance treatment for B-cell lymphomas. Haematologica. 2022 Mar 1;107(3):755-758. [CrossRef]
- Ishio T, Tsukamoto S, Yokoyama E, Izumiyama K, Saito M, Muraki H, et al. Anti-CD20 antibodies and bendamustine attenuate humoral immunity to COVID-19 vaccination in patients with B-cell non-Hodgkin lymphoma. Ann Hematol 2023; 12:1–11. [CrossRef]
- Perry C, Luttwak E, Balaban R, Shefer G, Morales MM, Aharon A, Tabib Y, Cohen YC, Benyamini N, Beyar-Katz, et al. Efficacy of the BNT162b2 mRNA COVID-19 vaccine in patients with B-cell non-Hodgkin lymphoma. Blood Adv. 2021 Aug 24;5(16):3053-3061. [CrossRef]
- Lu L, Chan CY, Lim YY, Than M, Teo S, Lau PYW, Ng KH, Yap HK. SARS-CoV-2 Humoral Immunity Persists Following Rituximab Therapy. Vaccines (Basel). 2023 Dec 18;11(12):1864. [CrossRef]
- Bsteh G, Assar H, Hegen H, Heschl B, Leutmezer F, Di Pauli F, Gradl C, Traxler G, Zulehner G, AUT-MuSC investigators. COVID-19 severity and mortality in multiple sclerosis are not associated with immunotherapy: Insights from a nation-wide Austrian registry. PLoS One. 2021 Jul 27;16(7):e0255316. [CrossRef]
- Avouac J, Drumez E, Hachulla E, Seror R, Georgin-Lavialle S, El Mahou S, Pertuiset E, Pham T, Marotte H, FAIR/SFR/SNFMI/SOFREMIP/CRI/IMIDIATE consortium and contributors. COVID-19 outcomes in patients with inflammatory rheumatic and musculoskeletal diseases treated with rituximab: a cohort study. Lancet Rheumatol. 2021 Jun;3(6):e419-e426. [CrossRef]
- Shao Z, Feng Y, Zhong L, Xie Q, Lei M, Liu Z, Wang C, Ji J, Liu H, Gu Z, Hu Z, Su L, Wu M, Liu Z. Clinical efficacy of intravenous immunoglobulin therapy in critical ill patients with COVID-19: a multicenter retrospective cohort study. Clin Transl Immunology. 2020 Oct 14;9(10):e1192. [CrossRef]
- Gharebaghi N, Nejadrahim R, Mousavi SJ, Sadat-Ebrahimi SR, Hajizadeh R. The use of intravenous immunoglobulin gamma for the treatment of severe coronavirus disease 2019: a randomized placebo-controlled double-blind clinical trial. BMC Infect Dis. 2020 Oct 21;20(1):786. [CrossRef]
- Mazeraud A, Jamme M, Mancusi RL, Latroche C, Megarbane B, Siami S, Zarka J, Moneger G, Santoli F, Argaud L, Chillet P, et al. Intravenous immunoglobulins in patients with COVID-19-associated moderate-to-severe acute respiratory distress syndrome (ICAR): multicentre, double-blind, placebo-controlled, phase 3 trial. Lancet Respir Med. 2022 Feb;10(2):158-166. [CrossRef]
- Pei L, Zhang S, Huang L, Geng X, Ma L, Jiang W, Li W, Chen D. Antiviral agents, glucocorticoids, antibiotics, and intravenous immunoglobulin in 1142 patients with coronavirus disease 2019: a systematic review and meta-analysis. Pol Arch Intern Med. 2020 Sep 30;130(9):726-733. [CrossRef]
- Kwapisz D, Bogusławska J. Intravenous immunoglobulins (IVIG) in severe/critical COVID-19 adult patients. Biomed Pharmacother. 2023 Jul;163:114851. [CrossRef]
- Piechotta V, Iannizzi C, Chai KL, Valk SJ, Kimber C, Dorando E, Monsef I, Wood EM, Lamikanra AA, Roberts DJ, et al. Convalescent plasma or hyperimmune immunoglobulin for people with COVID-19: a living systematic review. Cochrane Database Syst Rev. 2021 May 20;5(5):CD013600. Update in: Cochrane Database Syst Rev. 2023 Feb 1;2:CD013600. PMID: 34013969; PMCID: PMC8135693. [CrossRef]
- RECOVERY Collaborative Group. Convalescent plasma in patients admitted to hospital with COVID-19 (RECOVERY): a randomised controlled, open-label, platform trial. Lancet. 2021 May 29;397(10289):2049-2059. [CrossRef]
- Writing Committee for the REMAP-CAP Investigators; Estcourt LJ, Turgeon AF, McQuilten ZK, McVerry BJ, Al-Beidh F, Annane D, Arabi YM, Arnold DM, Beane A, Bégin P, et al. Effect of Convalescent Plasma on Organ Support-Free Days in Critically Ill Patients With COVID-19: A Randomized Clinical Trial. JAMA. 2021 Nov 2;326(17):1690-1702. [CrossRef]
- Chen P, Nirula A, Heller B, Gottlieb RL, Boscia J, Morris J, Huhn G, Cardona J, Mocherla B, Stosor V, et al., BLAZE-1 Investigators. SARS-CoV-2 Neutralizing Antibody LY-CoV555 in Outpatients with Covid-19. N Engl J Med. 2021 Jan 21;384(3):229-237. [CrossRef]
- Dougan M, Nirula A, Azizad M, Mocherla B, Gottlieb RL, Chen P, Hebert C, Perry R, Boscia J, Heller B, et al., BLAZE-1 Investigators. Bamlanivimab plus Etesevimab in Mild or Moderate Covid-19. N Engl J Med. 2021 Oct 7;385(15):1382-1392. [CrossRef]
- Li M, Lou F, Fan H. SARS-CoV-2 variant Omicron: currently the most complete “escapee” from neutralization by antibodies and vaccines. Signal Transduct Target Ther. 2022 Jan 28;7(1):28. [CrossRef]
- Anand P, Puranik A, Aravamudan M, Venkatakrishnan AJ, Soundararajan V. SARS-CoV-2 strategically mimics proteolytic activation of human ENaC. Elife. 2020 May 26;9:e58603. [CrossRef]
- Kotsias F, Cebrian I, Alloatti A. Antigen processing and presentation. Int Rev Cell Mol Biol. 2019;348:69-121. [CrossRef]
- Nagler A, Kalaora S, Barbolin C, Gangaev A, Ketelaars SLC, Alon M, Pai J, Benedek G, Yahalom-Ronen Y, Erez N, Greenberg P, et al. Identification of presented SARS-CoV-2 HLA class I and HLA class II peptides using HLA peptidomics. Cell Rep. 2021 Jun 29;35(13):109305. [CrossRef]
- Ferretti AP, Kula T, Wang Y, Nguyen DMV, Weinheimer A, Dunlap GS, Xu Q, Nabilsi N, Perullo CR, Cristofaro AW, et al. Unbiased Screens Show CD8+ T Cells of COVID-19 Patients Recognize Shared Epitopes in SARS-CoV-2 that Largely Reside outside the Spike Protein. Immunity. 2020 Nov 17;53(5):1095-1107.e3. [CrossRef]
- Suardana IBK, Mahardika BK, Pharmawati M, Sudipa PH, Sari TK, Mahendra NB, Mahardika GN. Whole-Genome Comparison of Representatives of All Variants of SARS-CoV-2, Including Subvariant BA.2 and the GKA Clade. Adv Virol. 2023 Mar 9;2023:6476626. [CrossRef]
- Bai Z, Cao Y, Liu W, Li J. The SARS-CoV-2 Nucleocapsid Protein and Its Role in Viral Structure, Biological Functions, and a Potential Target for Drug or Vaccine Mitigation. Viruses. 2021 Jun 10;13(6):1115. [CrossRef]
- Verma J, Kaushal N, Manish M, Subbarao N, Shakirova V, Martynova E, Liu R, Hamza S, Rizvanov AA, Khaiboullina SF, Baranwal M. Identification of conserved immunogenic peptides of SARS-CoV-2 nucleocapsid protein. J Biomol Struct Dyn. 2023 Sep 26:1-17. [CrossRef]
- Le Bert N, Tan AT, Kunasegaran K, Tham CYL, Hafezi M, Chia A, Chng MHY, Lin M, Tan N, Linster M, et al. SARS-CoV-2-specific T cell immunity in cases of COVID-19 and SARS, and uninfected controls. Nature. 2020 Aug;584(7821):457-462. [CrossRef]
- AlKhalifah JM, Seddiq W, Alshehri MA, Alhetheel A, Albarrag A, Meo SA, Al-Tawfiq JA, Barry M. Impact of MERS-CoV and SARS-CoV-2 Viral Infection on Immunoglobulin-IgG Cross-Reactivity. Vaccines (Basel). 2023 Feb 26;11(3):552. [CrossRef]
- Kesheh MM, Hosseini P, Soltani S, Zandi M. An overview on the seven pathogenic human coronaviruses. Rev Med Virol. 2022 Mar;32(2):e2282. [CrossRef]
- Hewitt EW. The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology. 2003 Oct;110(2):163-9. [CrossRef]
- Cassioli C, Baldari CT. The Expanding Arsenal of Cytotoxic T Cells. Front Immunol. 2022 Apr 20;13:883010. [CrossRef]
- Tay C, Kanellakis P, Hosseini H, Cao A, Toh BH, Bobik A, Kyaw T. B Cell and CD4 T Cell Interactions Promote Development of Atherosclerosis. Front Immunol. 2020 Jan 10;10:3046. [CrossRef]
- Kared H, Redd AD, Bloch EM, Bonny TS, Sumatoh H, Kairi F, Carbajo D, Abel B, Newell EW, Bettinotti MP, et al. SARS-CoV-2-specific CD8+ T cell responses in convalescent COVID-19 individuals. J Clin Invest. 2021 Mar 1;131(5):e145476. [CrossRef]
- Vennema H, de Groot RJ, Harbour DA, Dalderup M, Gruffydd-Jones T, Horzinek MC, Spaan WJ. Early death after feline infectious peritonitis virus challenge due to recombinant vaccinia virus immunization. J Virol. 1990 Mar;64(3):1407-9. [CrossRef]
- Olsen CW, Corapi WV, Ngichabe CK, Baines JD, Scott FW. Monoclonal antibodies to the spike protein of feline infectious peritonitis virus mediate antibody-dependent enhancement of infection of feline macrophages. J Virol. 1992 Feb;66(2):956-65. [CrossRef]
- Wan Y, Shang J, Sun S, Tai W, Chen J, Geng Q, He L, Chen Y, Wu J, Shi Z, et al. Molecular Mechanism for Antibody-Dependent Enhancement of Coronavirus Entry. J Virol. 2020 Feb 14;94(5):e02015-19. [CrossRef]
- Takada A, Kawaoka Y. Antibody-dependent enhancement of viral infection: molecular mechanisms and in vivo implications. Rev Med Virol. 2003 Nov-Dec;13(6):387-98. [CrossRef]
- Arvin AM, Fink K, Schmid MA, Cathcart A, Spreafico R, Havenar-Daughton C, Lanzavecchia A, Corti D, Virgin HW. A perspective on potential antibody-dependent enhancement of SARS-CoV-2. Nature. 2020 Aug;584(7821):353-363. [CrossRef]
- Pongracz T, Vidarsson G, Wuhrer M. Antibody glycosylation in COVID-19. Glycoconjugate Journal. 2022 Jun;39(3):335-44.
- van Osch, T.L.J., Nouta, J., Derksen, N.I.L., van Mierlo, G., van der Schoot, C.E., Wuhrer, M., Rispens, T., Vidarsson, G.: Fc galactosylation promotes hexamerization of human IgG1, leading to enhanced classical complement activation. J. Immunol. 207(6), 1545–1554 (2021). [CrossRef]
- Weiskopf K, Weissman IL. Macrophages are critical effectors of antibody therapies for cancer. MAbs. 2015;7(2):303-10. [CrossRef]
- Junker F, Gordon J, Qureshi O. Fc Gamma Receptors and Their Role in Antigen Uptake, Presentation, and T Cell Activation. Front Immunol. 2020 Jul 3;11:1393. [CrossRef]
- von Kietzell K, Pozzuto T, Heilbronn R, Grössl T, Fechner H, Weger S. Antibody-mediated enhancement of parvovirus B19 uptake into endothelial cells mediated by a receptor for complement factor C1q. J Virol. 2014 Jul;88(14):8102-15. [CrossRef]
- Eggleton P, Tenner AJ, Reid KB. C1q receptors. Clin Exp Immunol. 2000 Jun;120(3):406-12. [CrossRef]
- Wen J, Cheng Y, Ling R, Dai Y, Huang B, Huang W, Zhang S, Jiang Y. Antibody-dependent enhancement of coronavirus. Int J Infect Dis. 2020 Nov;100:483-489. [CrossRef]
- Thomas S, Smatti MK, Ouhtit A, Cyprian FS, Almaslamani MA, Thani AA, Yassine HM. Antibody-Dependent Enhancement (ADE) and the role of complement system in disease pathogenesis. Mol Immunol. 2022 Dec;152:172-182. [CrossRef]
- Monto AS, Malosh RE, Petrie JG, Martin ET. The Doctrine of Original Antigenic Sin: Separating Good From Evil. J Infect Dis. 2017 Jun 15;215(12):1782-1788. [CrossRef]
- Francis T. On the doctrine of original antigenic sin. Proc Am Philos Soc 1960; 104:572–578.
- Petráš M, Králová Lesná I. SARS-CoV-2 vaccination in the context of original antigenic sin. Hum Vaccin Immunother. 2022 Dec 31;18(1):1949953. [CrossRef]
- Pillai S. SARS-CoV-2 vaccination washes away original antigenic sin. Trends Immunol. 2022 Apr;43(4):271-273. [CrossRef]
- Rijkers GT, van Overveld FJ. The “original antigenic sin” and its relevance for SARS-CoV-2 (COVID-19) vaccination. Clin Immunol Communications. 2021; 1: 13-16. [CrossRef]
- Xia CS, Zhan M, Liu Y, Yue ZH, Song Y, Zhang F, Wang H. SARS-CoV-2 antibody response in SARS survivors with and without the COVID-19 vaccine. Int J Antimicrob Agents. 2023 Oct;62(4):106947. [CrossRef]
- El-Saed A, Othman F, Baffoe-Bonnie H, Almulhem R, Matalqah M, Alshammari L, Alshamrani MM. Symptomatic MERS-CoV infection reduces the risk of future COVID-19 disease; a retrospective cohort study. BMC Infect Dis. 2023 Nov 3;23(1):757. [CrossRef]
- Sette A, Sidney J, Crotty S. T Cell Responses to SARS-CoV-2. Annu Rev Immunol. 2023 Apr 26;41:343-373. Epub 2023 Feb 7. PMID: 36750314. [CrossRef]
- Appelberg S, Ahlén G, Yan J, Nikouyan N, Weber S, Larsson O, Höglund U, Aleman S, Weber F, Perlhamre E, et al. A universal SARS-CoV DNA vaccine inducing highly cross-reactive neutralizing antibodies and T cells. EMBO Mol Med. 2022 Oct 10;14(10):e15821. [CrossRef]
- Temmam S, Vongphayloth K, Baquero E, Munier S, Bonomi M, Regnault B, Douangboubpha B, Karami Y, Chrétien D, Sanamxay D, et al. Bat coronaviruses related to SARS-CoV-2 and infectious for human cells. Nature. 2022 Apr;604(7905):330-336. [CrossRef]
- Shannon CP, Blimkie TM, Ben-Othman R, Gladish N, Amenyogbe N, Drissler S, Edgar RD, Chan Q, Krajden M, Foster LJ, et al. Multi-Omic Data Integration Allows Baseline Immune Signatures to Predict Hepatitis B Vaccine Response in a Small Cohort. Front Immunol. 2020 Nov 30;11:578801. [CrossRef]
- Leslie M. Giant project will chart human immune diversity to improve drugs and vaccines. Science 2024 Jan 5; 383y: 13-14.
Figure 1.
Structure of SARS-CoV-2 with major structural protein. The immunological relevant domains of the Spike protein (S1, S2, and RBD) are indicated.
Figure 1.
Structure of SARS-CoV-2 with major structural protein. The immunological relevant domains of the Spike protein (S1, S2, and RBD) are indicated.
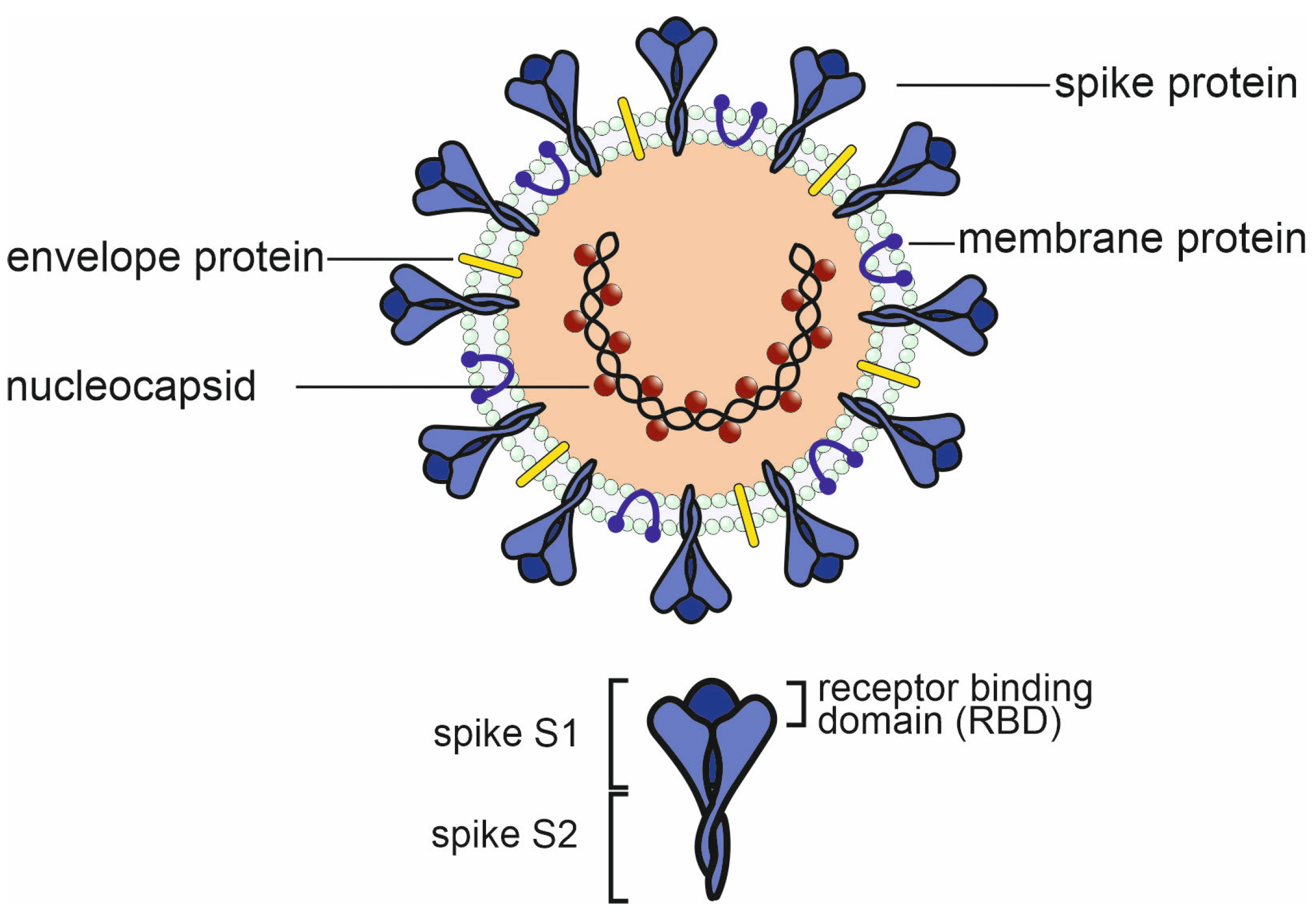
Figure 2.
Homology between spike proteins of the human coronaviruses. The amino acids which constitute the ACE2 receptor binding domain of SARS-CoV-2 are encircled in red. Major CD8 T cell epitopes are indicated by red rectangles and their first three amino acids according to the SARS-CoV-2 sequence. YLQ = YLQPRTFLL (269-277); KCY = KCYGVSPTKL (378-386); QYI = QYIKWPWYI (1208-1216) [44]. Figure 2 and Figure 3 were created using Clustal Omega (http://www.clustal.org/omega/ ) and Jalview sequence alignment software (https://www.jalview.org/ ). All viral peptide sequences were retrieved from the Uniprot data base (https://www.uniprot.org/ ).
Figure 2.
Homology between spike proteins of the human coronaviruses. The amino acids which constitute the ACE2 receptor binding domain of SARS-CoV-2 are encircled in red. Major CD8 T cell epitopes are indicated by red rectangles and their first three amino acids according to the SARS-CoV-2 sequence. YLQ = YLQPRTFLL (269-277); KCY = KCYGVSPTKL (378-386); QYI = QYIKWPWYI (1208-1216) [44]. Figure 2 and Figure 3 were created using Clustal Omega (http://www.clustal.org/omega/ ) and Jalview sequence alignment software (https://www.jalview.org/ ). All viral peptide sequences were retrieved from the Uniprot data base (https://www.uniprot.org/ ).
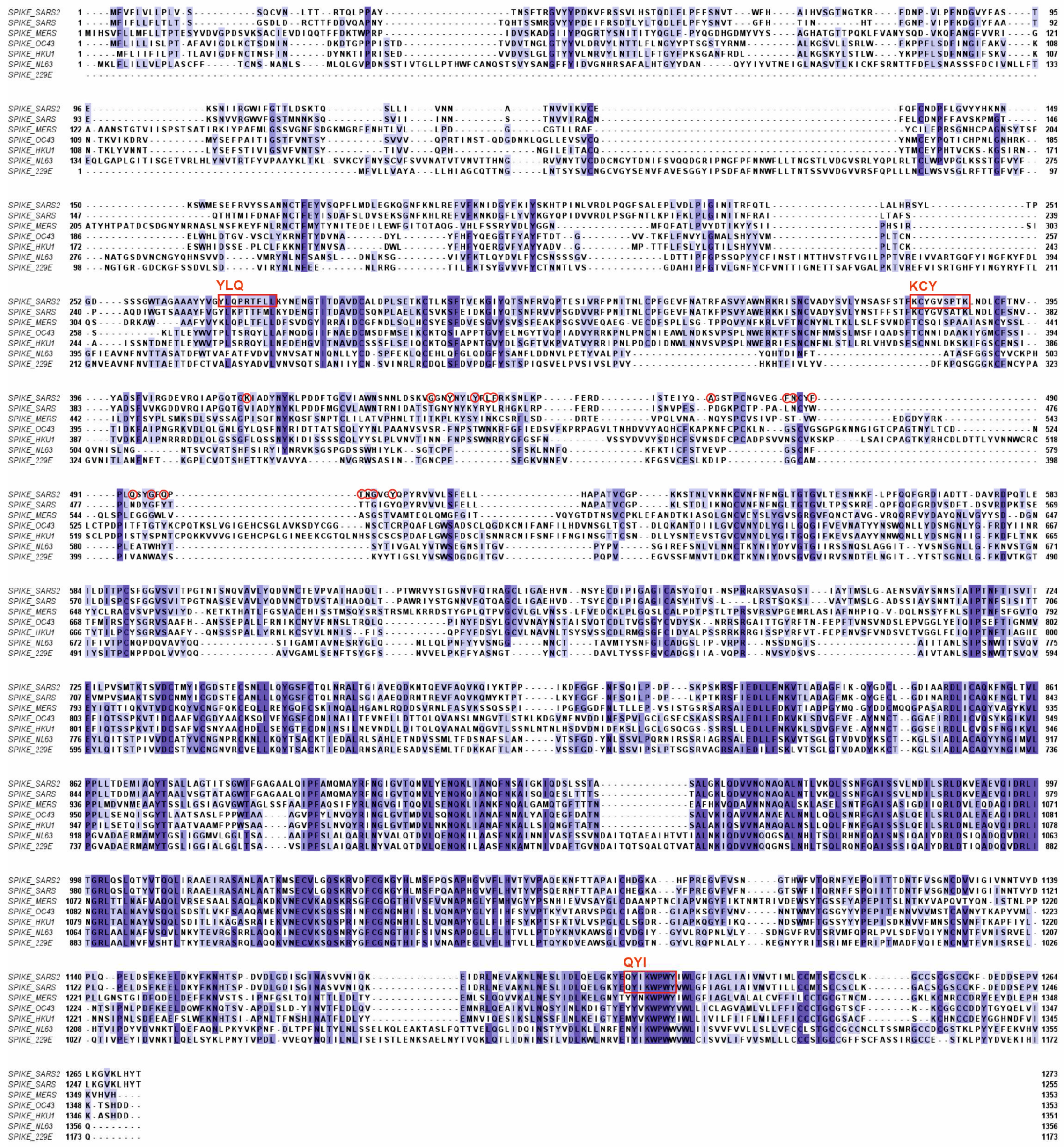

Table 1.
The humoral and cellular immune response to SARS-CoV-2 in COVID-191.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



