
Submitted:
06 February 2024
Posted:
06 February 2024
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
The expression of non-coding RNAs varies greatly depending on the cell type and physiological state. Changes in expression affect the regulatory properties of these molecules. We investigated RNA-seq data from sorted blood cells of healthy control donors and severe COVID-19 patients. Our aim was to display a distinguishable piRNA differential expression portrait for each type of cell and to study the behavior of piRNA in the blood cells of COVID-19 patients. Overall piRNA expression in sorted control cells (erythrocytes, monocytes, lymphocytes, eosinophils, basophils, neutrophils) differs depending on cell type. Four of the most abundant piRNA were expressed relatively equally in all cell types, they included piR-49145, piR-49144, piR-49143 and piR-33151. By reviewing piRNA expression in each group of sorted cells from severe COVID-19 patients we observed 3 upregulated and 10 downregulated piRNAs in erythrocytes. Monocytes were presented with a larger amount of statistically significant piRNA, 4 upregulated and 35 downregulated. In lymphocytes, all 19 piRNAs were upregulated.
Keywords:
piRNA
; blood cells
; severe COVID-19
; erythrocytes
; monocytes
; lymphocytes
; next generation sequencing of smallRNAs
; eosinophils
; basophils
; neutrophils
1. Introduction
Non-coding RNAs (ncRNA) are functional RNA molecules that are not translated into a protein. There are various types of ncRNAs, including transfer RNAs (tRNAs), microRNAs, small interfering RNAs (siRNAs), PIWI-interacting RNAs (piRNAs), small nucleolar RNAs (snoRNAs), small nuclear RNAs (snRNAs), and long ncRNAs such as Xist and HOTAIR [1]. The functional roles of ncRNAs in gene expression regulation have been widely recognized. They are known to modulate chromatin structure, regulate the assembly and function of nuclear bodies, alter the stability and translation of mRNAs, and interfere with signaling pathways in various ways [2].
The study of ncRNAs is an active area of research, and there is ongoing debate about the functional significance of various ncRNAs [3].
piRNAs are a large subclass of small non-coding RNA molecules [4]. piRNA molecules primarily function in the epigenetic and post-transcriptional silencing of genetic elements in germ line cells by forming complexes with piwi-subfamily Argonaute proteins [5,6]. They are created from loci that function as transposon traps, providing a kind of RNA-based immune system against transposons, which are DNA sequences that can change their position within the genome [6,7,8].
In addition to their role in transposon silencing, piRNAs are also involved in the regulation of other genetic elements in germ line cells. They are primarily expressed in the testes and ovaries of mammals and are essential for germline development and fertility [9]. The dysregulation of piRNAs has been associated with various diseases [10,11], including cancer [12], where they can influence gene expression in somatic cells [13]. It is also known that many piRNAs are dysregulated in tumor tissues, where they can either promote or suppress tumor growth [14]. For instance, piR-54265/PIWIL2 recruits STAT3 and p-SRC through the PIWIL2 PAZ domain to form the PIWIL2/STAT3/p-SRC complex, which facilitates the activation of signal pathways and phosphorylation of STAT3 by p-SRC, thereby promoting tumorigenesis in colorectal adenocarcinoma [15].
To date, there is insufficient data describing the behavior of piRNA in separate cell populations. Despite being previously associated with germinal cell lines [16,17], recent studies show that human piRNA may be related to a wider range of mechanisms. The evidence of an ancestral piRNA machinery that developed into a more complex and sophisticated form in multicellular eukaryotes could be reflected in the existence of PIWI-like machinery in Trypanosomatids and Apicomplexa. According to Horjales et al., ancient PIWI proteins were widely expressed and served purposes apart from germline preservation [18].
In a recent study of peripheral leukocytes of rheumatoid arthritis patients it has been shown that two immunoregulation piRNAs (piR-hsa-27620 and piR-hsa-27124) were significantly elevated in patients with rheumatoid arthritis [19]. Latest advancements in sequencing and molecular technologies, along with in silico studies have improved the annotation and functional validation of ncRNAs, enabling a better understanding of their roles in specific cell types and diseases [20]. However, the functional significance of ncRNAs, especially in poorly conserved species, remains a subject of ongoing research.
Given the fact how significantly COVID-19 alters the biochemical and morphologic features of blood cells [21,22,23], and taking in account the promising research on interrelations between ncRNA immune regulatory features and COVID-19 [24,25,26], it is necessary to state the importance of studying piRNA expression in blood cells of SARS-CoV-2 infected patients. The aim of this research was to analyze and compare the expression of piRNA in sorted blood cells in healthy control donors and in COVID-19 patients.
2. Material and methods
2.1. Patients and Data Collection
Eleven research participants in total were split up into two groups: six healthy donors and five COVID-19 patients. The six healthy donors in our control group had a mean age of 51 ± 22. The five COVID-19 patients, with an average age of 67 ± 11, arrived at our institution between April 1, 2022, and August 23, 2022. Real-time RT-PCR was used in the laboratory to confirm that all patients had SARS-CoV-2 infection. Two patients were transferred to the infectious disease unit and three patients were admitted to the intensive care unit. The following criteria were met by severe patients for ICU admission: body temperature ≥ 39 °C, respiration rate ≥ 30/min, and oxygen saturation (SpO2) ≤ 93%. These criteria are outlined in our national clinical recommendations.
2.2. Cell sorting and scanning electron microscopy
Sample staining for flow cytometry and subsequent sorting was carried out according to a standardized technique [23].
Erythrocyte sorting was carried out by the following technique:
10 µl whole blood was added to 100 µl PBS, 2 µl anti cd235 (IM2212U) was added, 2 µl cd41 (A07781), cd45 (A79392). 20 minutes incubated in dark, dissolved to 1 ml phosphate-buffered saline (PBS), next, fluorescent sorting was carried out up to 5 million events on a MoFlo Astrios EQ flow cytometer sorter (equipped with 405, 488, 645).
Leukocyte sorting was carried out by 2 different methods as follows:
100 µl of heparinized peripheral blood was taken, 1 ml of VersaLyse lysing solution (Beckman Coulter, USA) was added to each tube, stirred and incubated for 10 min. 10 µl of appropriate monoclonal antibodies were added: CD45-APC-AlexaFluor®750+ (A79392 Beckman Coulter, USA), CD16-PC7+(6607118 Beckman Coulter, USA), CD14-PC5.5+(A70204 Beckman Coulter, USA) was used to distinguish lymphocytes and monocytes.
The samples were stirred by vortex mixing and incubated for 15-20 minutes at room temperature in the dark. Afterwards, 1 mL of phosphate buffer (PBS) was added and centrifuged for 5 min at 1500 rpm, the supernatant was removed and 1 mL of buffer solution was added to each tube. Resuspended precipitant was analyzed on a MoFlo Astrios EQ flow cytometer sorter (equipped with 405, 488, 645).
For isolating subpopulations of granulocytes - eosinophils, basophils and neutrophils the duraclone IM granulocyte antibody panel (B88651) (Beckman Coulter, USA) was used.
Sorting was performed through 70 μm diameter nozzles, 50 000-100 000 events for the leukocyte cell population, into sterile 12x75 eppendorfs.
A standardized protocol for sample preparation for scanning electron microscopy (SEM) was followed [27]. Further analysis was performed using a Zeiss Merlin scanning electron microscope in high resolution mode with EHT 0.400 kV and a magnification of 1.00 KX.
2.3. RNA separation, library preparation and next generation sequencing
RNA was isolated\extracted using ExtractRNA reagent (Evrogen) according to the provided protocol. After extraction, RNA was dissolved in 10 µl RNAse free water. The quality of leukocyte RNA was checked on a TapeStation instrument (Agillent). Only samples with RIN>5 were used for the preparation of leukocyte libraries. 15-90 ng of RNA was taken in each reaction. Reversion, preparation of short RNA libraries, and circularization were performed using the Small RNA Library Prep Kit (BGI, 1000006383) according to the manufacturer’s recommendations. Libraries were purified by electrophoresis in a polyacrylamide 6% gel, cutting out the target band corresponding to the length of the target fragment 18-50b (library length 105-140b). Sequencing was performed on a DNBSEQ-G400 (BGI) instrument using the DNBSEQ-G400RS High-throughput Sequencing Set (Small RNA FCL SE50*) (BGI, 1000016998). After sequencing, the amount of information obtained from each sample was 400-1200MB.
2.4. Bioinformatics and statistical analysis
Bioinformatics analysis was performed in miRMaster 2 version 2.0.0 [28], a comprehensive analysis framework for small RNA-seq data. Current version of miRmaster used Ensembl [29], RNACentral piRNA [30], NCBI RefSeq [31], ENA [32], GeneCards [33], PirBase [34] databases for analyzing RNAseq FASTQ data. Statistical analysis was performed in RStudio (version 2023.09.1+494.pro2) [35]. Reads per million (RPM) was used as a normalized value of expression. Statistical analysis for the results was executed by applying the Wilcoxon–Mann–Whitney test. A p-value < 0.05 was considered statistically significant. For RNAseq volcano plot the p-value was log transformed to log10 (1.31) for data presentation. Heatmap raw expression data was preliminarily log transformed. Data visualization, images, and charts were made with RStudio ggplot2 [36] and tidyverse [37] open-source collection of packages. All raw data used for statistical analysis and visualization is presented in the supplementary materials.
3. Results
3.1. piRNA expression of sorted healthy donor blood cells
In order to compare piRNA expression in blood cells we sorted them by distinguishing the following cell types: erythrocytes (n=5), monocytes (n=5), lymphocytes (n=4), eosinophils (n=2), basophils (n=2), neutrophils (n=6). The cell sorting procedure is described in materials and methods. For accuracy, we checked the quality of sorting with SEM. As can be seen in the presented images (Figure 1 panel A) morphological features of cells correspond with sorted cell types [22,23]. We then isolated RNA from sorted cells, prepared small RNA libraries, performed NGS of small RNA and subjected the raw RNAseq data to bioinformatic analysis in miRMaster 2, details are described in materials and methods. All annotated piRNA data is presented in the supplementary materials. We compared the major RNAs overexpressed in different blood cells of healthy control donors. The results are presented in Figure 1 panel A (piechart). Additionally, log transformed raw expression data is presented in the heatmap (Figure 1 panel B), where 5 (red) indicates maximum expression values, and <1 (green) minimum. 4 of the most abundant piRNA were expressed relatively equally in all cell types, they included piR-49145, piR-49144, piR-49143 and piR-33151.
3.2. piRNA differential expression in blood cells of patients with COVID-19
Next, we analyzed the piRNA differential expression in healthy donors and patients with severe COVID-19 in three distinguished sorted cell types erythrocytes (n=5), monocytes (n=5) and lymphocytes (n=4), the results of the comparison are presented as a volcano plot (Figure 2) where positive increasing log2 fold change corresponds with gene downregulation, and negative values correspond with gene upregulation. All raw data used for analysis is presented in the supplementary materials section.
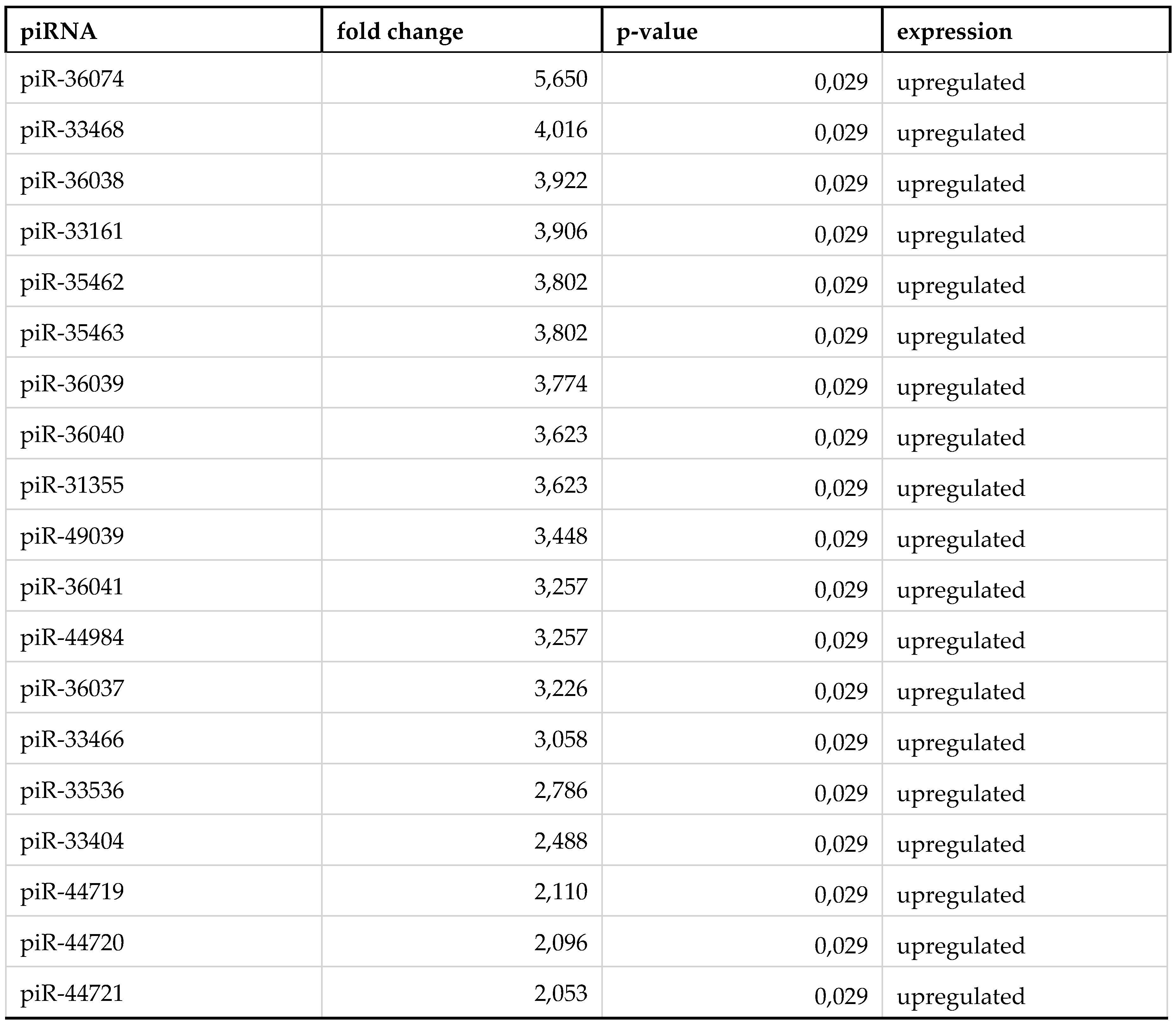
By reviewing piRNA expression in each group of sorted cells from severe COVID-19 patients we observed 3 upregulated and 10 downregulated piRNAs in erythrocytes (Table 1). Monocytes were presented with a larger amount of statistically significant piRNA, 4 upregulated and 35 downregulated (Table 2). In lymphocytes, each piRNA was upregulated (Table 3).
4. Discussion
Many ncRNAs exhibit cell type, tissue, and cancer specificity, making them valuable for understanding the molecular basis of various diseases [38,39].
By presenting piRNA expression profiles of sorted healthy control donor cells we attempted to portray cell type-specific piRNA, clusterization of which could compose a distinct and reliable piRNA expression portrait for healthy erythrocytes, lymphocytes, monocytes, eosinophils, basophils, neutrophils. For accuracy we additionally examined our sorted cells using SEM. The images that we observed confirmed that each sorted sample corresponded with the sought-for cell type. We observed mild cell shrinkage possibly due to the cell sorting procedure. (Figure 1 panel A). Such an approach would potentially allow us to distinguish piRNA peculiarities in sorted blood cells.
To date it is known that a highly specific ncRNA - mir-451 is abundant in red blood cells [40]. Other specific miRNA include miR-223-3p which is linked to the inhibition of erythrocyte differentiation and is recognized to be crucial in promoting granulocytic differentiation [41]. According to research, miR-223 is a potent granulopoiesis regulator that is mostly expressed in granulocytes [41]. However, we did not observe any similarly specific pattern of piRNA expression in either of the cell populations, therefore, it was not possible to state the prevalence of any distinct piRNA to a specific cell type. It is worth noting that despite the absence of a vivid piRNA expression pattern for erythrocytes, on our heatmap (Figure 1 panel C) we demonstrate that the top 20 most abundant erythrocyte piRNA show a mildly significant distinguishable expression demarcation when compared to other cell types. This could occur mainly due to the lack of cell nucleus [42] and ribosomes in mature erythrocytes [43]. Thus, the origin of changes in piRNA expression in erythrocytes remains a matter of debate. The overall top 4 expressed piRNA in healthy control donors, as we demonstrate in our heatmap (Figure 1 panel C) and percentages pie-chart (Figure 1 panel b), does not significantly differ depending on cell type. These piRNA include piR-49144, piR-49143, piR-33151 and piR-49145. Despite having a similar id, their sequence and length differ and each aforementioned piRNA is distinguished separately by RNACentral piRNA [30], ENA [32], GeneCards [33], PirBase [34].
Analyzing COVID-19 patients’ piRNA expression we observed several key features. The absolute majority of lymphocytic piRNA showed to be upregulated, while most erythrocytic and monocytic piRNA demonstrated a significant downregulation. (Figure 2 panel A volcano). In an attempt to interpret these findings we encountered difficulties due to the lack of data on piRNA and its potential roles in immune mediated mechanisms and viral infections as demonstrated with lncRNA [44].
It is important to give an overview on the involvement of piRNA in various biological processes known to date. piRNAs play diverse roles in gene regulation. They are best known for their functions in transposon silencing, fertility, and regulation of germline mRNAs and lncRNAs [6,7,8,9,16,17]. Recent studies have also highlighted their involvement in various neuronal processes, including neuronal differentiation and the development of neurodegenerative diseases. In the brain, piRNAs interact with a group of small RNAs and function as a complex to regulate cellular activities. Additionally, piRNAs have been implicated in the self-renewal of stem cells and are abundant in spermatogenic cells [6,45,46,47]. The precise mechanisms of piRNA-mediated gene regulation in the brain and their associations with neurodegenerative diseases are areas of active research.
We would like to emphasize on piRNAs role in transposon silencing. This process occurs in the cytoplasm, where a PIWI-piRNA complex binds to a piRNA-complementary transposon RNA and cleaves the RNA [6]. There are two main types of silencing: transcriptional silencing - mediated by nuclear PIWI proteins such as PIWI in Drosophila and MIWI2 PIWIL4 in mice and post-transcriptional silencing - mediated by cytoplasmic PIWI proteins such as Aubergine (PIWIL1) and MILI (a.k.a. PIWIL2) in mice [6,48].
The piRNA pathway guards the germline genome against transposable elements (TEs) by targeting two steps required for all transposons. Regulation of chromatin structure - In the nucleus, piRNAs are implicated in the regulation of chromatin structure, which can affect transcription of transposable elements [9].
Direct targeting and destruction of RNAs - In the cytoplasm, the piRNA pathway directly targets and destroys RNAs of transposons that escaped transcriptional silencing [9,49]. The piRNA pathway is essential for maintaining genome integrity, as transposable elements can cause DNA breaks, illegitimate recombination, and other genomic damage [9]. Mutations in genes involved in the piRNA pathway, such as Rhino and Armi, can lead to the activation of transposons and female sterility [48].
To date there are several significant reports of viral impact on transposable element activity [50,51]. It is explicitly stated in a study by Macchietto et al. that early up-regulated transposons are a component of the first wave response during virus infection and that transposon up-regulation is a common phenomena during virus infection in humans and mice [52]. According to Ivancevic et al. [53] immune cells show the highest enrichment of transposable element-derived enhancers. This allows us to propose that the expression of transposable elements could possibly correlate with the expression of certain piRNA, and elucidate our findings of the piRNA upregulation tendency in lymphocytes of COVID-19 patients. It has also been shown on a A549 cell culture that SARS-CoV-2 induces a great number of differentially expressed transposable element loci, which additionally supports our assumptions [54].
To better understand the nature of piRNA upregulation in lymphocytes during COVID-19 it is necessary to consider some individual piRNAs: piR-35462, piR-36074, piR-36038 have been found to be associated with eosinophil count and total serum IgE in large, independent childhood asthma cohorts [55].
Other piRNAs expressed in lymphocytes are mentioned by different authors in papers on germ cells [56], oocytes, embryos [57], human neuroblastoma [58], differentiation of human mesenchymal stromal cells [59], bladder cancer [60] and gastric cancer, where piR-31355, another piRNA upregulated in COVID-19 patients’ lymphocytes was pointed out as a potential biomarker of gastric cancer [61]. The function and role of aforementioned piRNAs remains a matter of debate, as the present piRNA dataset is not available for KEGG mapping or functional enrichment analysis [62].
When analyzing differential expression in COVID-19 patients’ monocytes our attention was drawn by 1 of 4 highly upregulated piRNAs’ - piR-31623. In a research dedicated to studying exosomes derived from lung basal epithelial cells infected by respiratory syncytial virus (RSV), Chahar et al. presented a table of top 15 highly upregulated exosome-derived piRNA, where piR-31626 demonstrates an average read count of 48.19 in RSV exosomes, and 0.62 in mock exosomes [63]. Additionally, in our research, we observed downregulation of piR-36169, whereas the same piRNA in RSV infected exosomes was highly upregulated [63]. Both RSV and SARS-CoV-2 can induce syncytia formation in infected cells, but the mechanisms and consequences of syncytia formation may differ between the two viruses [64]. In summary, RSV and SARS-CoV-2 have different genome sizes, polarities, protein encoding, and cellular receptors, however, both viruses can induce syncytia formation, but the mechanisms and consequences of syncytia formation may differ between the two viruses. Furthermore, exosomes isolated from RSV-infected cells were able to trigger the release of chemokines from A549 alveolar basal epithelial cells and human monocytes, according to Chahar et al. This suggests that exosomes released during infection may change cellular responses, either activating or suppressing the innate immune system [63]. Given the fact that both viruses cause syncytia formation, it is possible that piR-31626 may be circumstantial evidence of syncytium remodeling.
In our study we attempted to portray distinguishable expression profiles for erythrocytes of healthy control donors and COVID-19 patients. Erythrocytes express their genetic material and proteins during their development in the bone marrow [65]. There is currently limited information available on how piRNAs are expressed in erythrocytes, especially given the fact that they lack mitochondria, ribosomes, nuclei, and other organelles [43]. Supposedly, the piRNA content in red blood cells that we observed remains from proerythroblast stages.
Conclusion
Overall piRNA expression differs depending on cell type. Four of the most abundant piRNA were expressed relatively equally in all cell types. By reviewing piRNA expression in each group of sorted cells from severe COVID-19 patients we observed 3 upregulated and 10 downregulated piRNAs in erythrocytes. In lymphocytes, each of 19 piRNA were upregulated. Monocytes were presented with a larger amount of statistically significant piRNA, 4 upregulated and 35 downregulated. piRNA was upregulated in lymphocytes of all COVID-19 patients.
Funding
This work was supported by St. Petersburg State University, project ID: 95412780. The investigation of the structure by means of low-voltage scanning electron microscopy was carried out at the IRC for Nanotechnology of the Science Park of St. Petersburg State University within the framework of project No. AAAA-A19-119091190094.
Institutional Review Board Statement
This study was conducted according to the guidelines of the declaration of Helsinki and approved by the ethics committee.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hombach, S.; Kretz, M. Non-Coding RNAs: Classification, Biology and Functioning. Adv Exp Med Biol 2016, 937, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Statello, L.; Guo, C.-J.; Chen, L.-L.; Huarte, M. Gene Regulation by Long Non-Coding RNAs and Its Biological Functions. Nat Rev Mol Cell Biol 2021, 22, 96–118. [Google Scholar] [CrossRef]
- Palazzo, A.F.; Lee, E.S. Non-Coding RNA: What Is Functional and What Is Junk? Frontiers in Genetics 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Yoshitake, K.; Asakawa, S. A Review of Discovery Profiling of PIWI-Interacting RNAs and Their Diverse Functions in Metazoans. International Journal of Molecular Sciences 2021, 22, 11166. [Google Scholar] [CrossRef] [PubMed]
- Darricarrère, N.; Liu, N.; Watanabe, T.; Lin, H. Function of Piwi, a Nuclear Piwi/Argonaute Protein, Is Independent of Its Slicer Activity. Proc Natl Acad Sci U S A 2013, 110, 1297–1302. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lin, H. Roles of piRNAs in Transposon and Pseudogene Regulation of Germline mRNAs and lncRNAs. Genome Biology 2021, 22, 27. [Google Scholar] [CrossRef]
- Litwack, G. Chapter 10 - Nucleic Acids and Molecular Genetics. In Human Biochemistry; Litwack, G., Ed.; Academic Press: Boston, 2018; pp. 257–317. ISBN 978-0-12-383864-3. [Google Scholar]
- Xiao, Y.; Ke, A. PIWI Takes a Giant Step. Cell 2016, 167, 310–312. [Google Scholar] [CrossRef]
- Tóth, K.F.; Pezic, D.; Stuwe, E.; Webster, A. The piRNA Pathway Guards the Germline Genome Against Transposable Elements. Adv Exp Med Biol 2016, 886, 51–77. [Google Scholar] [CrossRef]
- Zhang, T.; Wong, G. Dysregulation of Human Somatic piRNA Expression in Parkinson’s Disease Subtypes and Stages. Int J Mol Sci 2022, 23, 2469. [Google Scholar] [CrossRef]
- Rayford, K.J.; Cooley, A.; Strode, A.W.; Osi, I.; Arun, A.; Lima, M.F.; Misra, S.; Pratap, S.; Nde, P.N. Trypanosoma Cruzi Dysregulates Expression Profile of piRNAs in Primary Human Cardiac Fibroblasts during Early Infection Phase. Frontiers in Cellular and Infection Microbiology 2023, 13. [Google Scholar] [CrossRef]
- Chattopadhyay, T.; Gupta, P.; Nayak, R.; Mallick, B. Genome-Wide Profiling of Dysregulated piRNAs and Their Target Genes Implicated in Oncogenicity of Tongue Squamous Cell Carcinoma. Gene 2023, 849, 146919. [Google Scholar] [CrossRef]
- Cheng, Y.; Wang, Q.; Jiang, W.; Bian, Y.; zhou, Y.; Gou, A.; Zhang, W.; Fu, K.; Shi, W. Emerging Roles of piRNAs in Cancer: Challenges and Prospects. Aging (Albany NY) 2019, 11, 9932–9946. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dou, M.; Song, X.; Dong, Y.; Liu, S.; Liu, H.; Tao, J.; Li, W.; Yin, X.; Xu, W. The Emerging Role of the piRNA/Piwi Complex in Cancer. Molecular Cancer 2019, 18, 123. [Google Scholar] [CrossRef] [PubMed]
- Mai, D.; Ding, P.; Tan, L.; Zhang, J.; Pan, Z.; Bai, R.; Li, C.; Li, M.; Zhou, Y.; Tan, W.; et al. PIWI-Interacting RNA-54265 Is Oncogenic and a Potential Therapeutic Target in Colorectal Adenocarcinoma. Theranostics 2018, 8, 5213–5230. [Google Scholar] [CrossRef] [PubMed]
- Kawase, M.; Ichiyanagi, K. The Expression Dynamics of piRNAs Derived From Male Germline piRNA Clusters and Retrotransposons. Frontiers in Cell and Developmental Biology 2022, 10. [Google Scholar] [CrossRef]
- Gainetdinov, I.V.; Skvortsova, Y.V.; Kondratieva, S.A.; Klimov, A.; Tryakin, A.A.; Azhikina, T.L. Assessment of piRNA Biogenesis and Function in Testicular Germ Cell Tumors and Their Precursor Germ Cell Neoplasia in Situ. BMC Cancer 2018, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Horjales, S.; Li Calzi, M.; Francia, M.E.; Cayota, A.; Garcia-Silva, M.R. piRNA Pathway Evolution beyond Gonad Context: Perspectives from Apicomplexa and Trypanosomatids. Frontiers in Genetics 2023, 14. [Google Scholar] [CrossRef] [PubMed]
- Ren, R.; Tan, H.; Huang, Z.; Wang, Y.; Yang, B. Differential Expression and Correlation of Immunoregulation Related piRNA in Rheumatoid Arthritis. Frontiers in Immunology 2023, 14. [Google Scholar] [CrossRef]
- Akimniyazova, A.; Yurikova, O.; Pyrkova, A.; Rakhmetullina, A.; Niyazova, T.; Ryskulova, A.-G.; Ivashchenko, A. In Silico Study of piRNA Interactions with the SARS-CoV-2 Genome. Int J Mol Sci 2022, 23, 9919. [Google Scholar] [CrossRef]
- Lucas, C.; Wong, P.; Klein, J.; Castro, T.B.R.; Silva, J.; Sundaram, M.; Ellingson, M.K.; Mao, T.; Oh, J.E.; Israelow, B.; et al. Longitudinal Analyses Reveal Immunological Misfiring in Severe COVID-19. Nature 2020, 584, 463–469. [Google Scholar] [CrossRef]
- Kondratov, K.A.; Artamonov, A.A.; Mikhailovskii, V.Y.; Velmiskina, A.A.; Mosenko, S.V.; Grigoryev, E.A.; Anisenkova, A.Y.; Nikitin, Y.V.; Apalko, S.V.; Sushentseva, N.N.; et al. SARS-CoV-2 Impact on Red Blood Cell Morphology. Biomedicines 2023, 11, 2902. [Google Scholar] [CrossRef] [PubMed]
- Velmiskina, A.A.; Nikitin, Yu.V.; Mikhailovskii, V.Yu.; Mosenko, S.V.; Anisenkova, A.Yu.; Apalko, S.V.; Sushentseva, N.N.; Scherbak, S.G.; Ivanov, A.M.; Galaktionov, N.K.; et al. Analysis of the Morphology of Monocytes and Lymphocytes from COVID-19 Patients Using Low-Voltage Scanning Electronic Microscopy. Bull Exp Biol Med 2023, 176, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Sun, Q.; Zhang, B.; Zhao, W.; Shen, C. The Regulation of lncRNAs and miRNAs in SARS-CoV-2 Infection. Front Cell Dev Biol 2023, 11, 1229393. [Google Scholar] [CrossRef] [PubMed]
- Ding, J.; Chen, J.; Yin, X.; zhou, J. Current Understanding on Long Non-Coding RNAs in Immune Response to COVID-19. Virus Res 2022, 323, 198956. [Google Scholar] [CrossRef]
- Arman, K.; Dalloul, Z.; Bozgeyik, E. Emerging Role of microRNAs and Long Non-Coding RNAs in COVID-19 with Implications to Therapeutics. Gene 2023, 861, 147232. [Google Scholar] [CrossRef]
- Fedorov, A.; Kondratov, K.; Kishenko, V.; Mikhailovskii, V.; Kudryavtsev, I.; Belyakova, M.; Sidorkevich, S.; Vavilova, T.; Kostareva, A.; Sirotkina, O.; et al. Application of High-Sensitivity Flow Cytometry in Combination with Low-Voltage Scanning Electron Microscopy for Characterization of Nanosized Objects during Platelet Concentrate Storage. Platelets 2020, 31, 226–235. [Google Scholar] [CrossRef]
- Fehlmann, T.; Kern, F.; Laham, O.; Backes, C.; Solomon, J.; Hirsch, P.; Volz, C.; Müller, R.; Keller, A. miRMaster 2.0: Multi-Species Non-Coding RNA Sequencing Analyses at Scale. Nucleic Acids Research 2021, 49, W397–W408. [Google Scholar] [CrossRef]
- Martin, F.J.; Amode, M.R.; Aneja, A.; Austine-Orimoloye, O.; Azov, A.G.; Barnes, I.; Becker, A.; Bennett, R.; Berry, A.; Bhai, J.; et al. Ensembl 2023. Nucleic Acids Research 2023, 51, D933–D941. [Google Scholar] [CrossRef]
- The RNAcentral Consortium RNAcentral: A Hub of Information for Non-Coding RNA Sequences. Nucleic Acids Research 2019, 47, D221–D229. [CrossRef] [PubMed]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res 2016, 44, D733–745. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Akhtar, R.; Birney, E.; Bower, L.; Cerdeno-Tárraga, A.; Cheng, Y.; Cleland, I.; Faruque, N.; Goodgame, N.; Gibson, R.; et al. The European Nucleotide Archive. Nucleic Acids Res 2011, 39, D28–D31. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Current Protocols in Bioinformatics 2016, 54, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Shi, Y.; Zhou, H.; Zhang, P.; Song, T.; Ying, Z.; Yu, H.; Li, Y.; Zhao, Y.; Zeng, X.; et al. piRBase: Integrating piRNA Annotation in All Aspects. Nucleic Acids Research 2022, 50, D265–D272. [Google Scholar] [CrossRef] [PubMed]
- RStudio Team (2020). RStudio: Integrated Development for R. 2020. http://www.rstudio.com/.
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag New York, 2016; ISBN 978-3-319-24277-4. https://ggplot2.tidyverse.org.
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; Hester, J.; et al. Welcome to the Tidyverse. Journal of Open Source Software 2019, 4, 1686. [Google Scholar] [CrossRef]
- Grillone, K.; Riillo, C.; Scionti, F.; Rocca, R.; Tradigo, G.; Guzzi, P.H.; Alcaro, S.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Non-Coding RNAs in Cancer: Platforms and Strategies for Investigating the Genomic “Dark Matter”. Journal of Experimental & Clinical Cancer Research 2020, 39, 117. [Google Scholar] [CrossRef]
- Lu, J.; Getz, G.; Miska, E.A.; Alvarez-Saavedra, E.; Lamb, J.; Peck, D.; Sweet-Cordero, A.; Ebert, B.L.; Mak, R.H.; Ferrando, A.A.; et al. MicroRNA Expression Profiles Classify Human Cancers. Nature 2005, 435, 834–838. [Google Scholar] [CrossRef]
- Sun, L.; Yu, Y.; Niu, B.; Wang, D. Red Blood Cells as Potential Repositories of MicroRNAs in the Circulatory System. Front Genet 2020, 11, 442. [Google Scholar] [CrossRef]
- Yuan, S.; Wu, Q.; Wang, Z.; Che, Y.; Zheng, S.; Chen, Y.; Zhong, X.; Shi, F. miR-223: An Immune Regulator in Infectious Disorders. Front Immunol 2021, 12, 781815. [Google Scholar] [CrossRef]
- Zhang, Z.-W.; Cheng, J.; Xu, F.; Chen, Y.-E.; Du, J.-B.; Yuan, M.; Zhu, F.; Xu, X.-C.; Yuan, S. Red Blood Cell Extrudes Nucleus and Mitochondria against Oxidative Stress. IUBMB Life 2011, 63, 560–565. [Google Scholar] [CrossRef]
- Pretini, V.; Koenen, M.H.; Kaestner, L.; Fens, M.H.A.M.; Schiffelers, R.M.; Bartels, M.; Van Wijk, R. Red Blood Cells: Chasing Interactions. Front Physiol 2019, 10, 945. [Google Scholar] [CrossRef]
- Li, Z.; Gao, J.; Xiang, X.; Deng, J.; Gao, D.; Sheng, X. Viral Long Non-Coding RNA Regulates Virus Life-Cycle and Pathogenicity. Mol Biol Rep 2022, 49, 6693–6700. [Google Scholar] [CrossRef]
- Sato, K.; Takayama, K.; Inoue, S. Role of piRNA Biogenesis and Its Neuronal Function in the Development of Neurodegenerative Diseases. Frontiers in Aging Neuroscience 2023, 15. [Google Scholar] [CrossRef]
- Zuo, L.; Wang, Z.; Tan, Y.; Chen, X.; Luo, X. piRNAs and Their Functions in the Brain. Int J Hum Genet 2016, 16, 53–60. [Google Scholar] [CrossRef]
- Ali, S.D.; Tayara, H.; Chong, K.T. Identification of piRNA Disease Associations Using Deep Learning. Comput Struct Biotechnol J 2022, 20, 1208–1217. [Google Scholar] [CrossRef] [PubMed]
- Halic, M.; Moazed, D. Transposon Silencing by piRNAs. Cell 2009, 138, 1058–1060. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Lu, J. Silencing of Transposable Elements by piRNAs in Drosophila: An Evolutionary Perspective. Genomics, Proteomics & Bioinformatics 2017, 15, 164–176. [Google Scholar] [CrossRef]
- Roy, M.; Viginier, B.; Saint-Michel, É.; Arnaud, F.; Ratinier, M.; Fablet, M. Viral Infection Impacts Transposable Element Transcript Amounts in Drosophila. Proc Natl Acad Sci U S A 2020, 117, 12249–12257. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.S.; Nanda, H.; Aliferis, C.; Langlois, R.A. Characterization of Influenza A Virus Induced Transposons Reveals a Subgroup of Transposons Likely Possessing the Regulatory Role as eRNAs. Sci Rep 2022, 12, 2188. [Google Scholar] [CrossRef] [PubMed]
- Macchietto, M.G.; Langlois, R.A.; Shen, S.S. Virus-Induced Transposable Element Expression up-Regulation in Human and Mouse Host Cells. Life Sci Alliance 2020, 3, e201900536. [Google Scholar] [CrossRef]
- Ivancevic, A.; Chuong, E.B. Transposable Elements Teach T Cells New Tricks. Proceedings of the National Academy of Sciences 2020, 117, 9145–9147. [Google Scholar] [CrossRef]
- Marston, J.L.; Greenig, M.; Singh, M.; Bendall, M.L.; Duarte, R.R.R.; Feschotte, C.; Iñiguez, L.P.; Nixon, D.F. SARS-CoV-2 Infection Mediates Differential Expression of Human Endogenous Retroviruses and Long Interspersed Nuclear Elements. JCI Insight 6, e147170. [CrossRef]
- Li, J.; Hong, X.; Jiang, M.; Kho, A.T.; Tiwari, A.; Wang, A.L.; Chase, R.P.; Celedón, J.C.; Weiss, S.T.; McGeachie, M.J.; et al. A Novel Piwi-Interacting RNA Associates with Type 2–High Asthma Phenotypes. Journal of Allergy and Clinical Immunology 2023. [Google Scholar] [CrossRef] [PubMed]
- Girard, A.; Sachidanandam, R.; Hannon, G.J.; Carmell, M.A. A Germline-Specific Class of Small RNAs Binds Mammalian Piwi Proteins. Nature 2006, 442, 199–202. [Google Scholar] [CrossRef] [PubMed]
- Roovers, E.F.; Rosenkranz, D.; Mahdipour, M.; Han, C.-T.; He, N.; Chuva de Sousa Lopes, S.M.; van der Westerlaken, L.A.J.; Zischler, H.; Butter, F.; Roelen, B.A.J.; et al. Piwi Proteins and piRNAs in Mammalian Oocytes and Early Embryos. Cell Rep 2015, 10, 2069–2082. [Google Scholar] [CrossRef] [PubMed]
- Roy, J.; Mallick, B. Investigating Piwi-Interacting RNA Regulome in Human Neuroblastoma. Genes Chromosomes Cancer 2018, 57, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Della Bella, E.; Menzel, U.; Basoli, V.; Tourbier, C.; Alini, M.; Stoddart, M.J. Differential Regulation of circRNA, miRNA, and piRNA during Early Osteogenic and Chondrogenic Differentiation of Human Mesenchymal Stromal Cells. Cells 2020, 9, 398. [Google Scholar] [CrossRef]
- Chu, H.; Hui, G.; Yuan, L.; Shi, D.; Wang, Y.; Du, M.; Zhong, D.; Ma, L.; Tong, N.; Qin, C.; et al. Identification of Novel piRNAs in Bladder Cancer. Cancer Lett 2015, 356, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Vinasco-Sandoval, T.; Moreira, F.C.; F. Vidal, A.; Pinto, P.; Ribeiro-dos-Santos, A.M.; Cruz, R.L.S.; Fonseca Cabral, G.; Anaissi, A.K.M.; Lopes, K. de P.; Ribeiro-dos-Santos, A.; et al. Global Analyses of Expressed Piwi-Interacting RNAs in Gastric Cancer. International Journal of Molecular Sciences 2020, 21, 7656. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A Web Server for Functional Enrichment Analysis and Functional Annotation of Gene Lists (2021 Update). Nucleic Acids Res 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Chahar, H.S.; Corsello, T.; Kudlicki, A.S.; Komaravelli, N.; Casola, A. Respiratory Syncytial Virus Infection Changes Cargo Composition of Exosome Released from Airway Epithelial Cells. Sci Rep 2018, 8, 387. [Google Scholar] [CrossRef]
- Lin, L.; Li, Q.; Wang, Y.; Shi, Y. Syncytia Formation during SARS-CoV-2 Lung Infection: A Disastrous Unity to Eliminate Lymphocytes. Cell Death Differ 2021, 28, 2019–2021. [Google Scholar] [CrossRef]
- Moras, M.; Lefevre, S.D.; Ostuni, M.A. From Erythroblasts to Mature Red Blood Cells: Organelle Clearance in Mammals. Front Physiol 2017, 8, 1076. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
piRNA expression in cells of healthy donors. Panel A. Percentages of different piRNA in each cell type are shown on the left. SEM images of sorted cells are displayed on the right. Scale bar is equal to 5 µm. Panel B. Log transformed raw expression data is presented in the heatmap (Figure 1 panel B), where 5 (red) indicates maximum expression values, and <1 (green) minimum. Erythrocytes (Er), Monocytes (Mon), Lymphocytes (Lym), Eosinophils (Eos), Basophils (Bas) Neutrophils (Neu).
Figure 1.
piRNA expression in cells of healthy donors. Panel A. Percentages of different piRNA in each cell type are shown on the left. SEM images of sorted cells are displayed on the right. Scale bar is equal to 5 µm. Panel B. Log transformed raw expression data is presented in the heatmap (Figure 1 panel B), where 5 (red) indicates maximum expression values, and <1 (green) minimum. Erythrocytes (Er), Monocytes (Mon), Lymphocytes (Lym), Eosinophils (Eos), Basophils (Bas) Neutrophils (Neu).
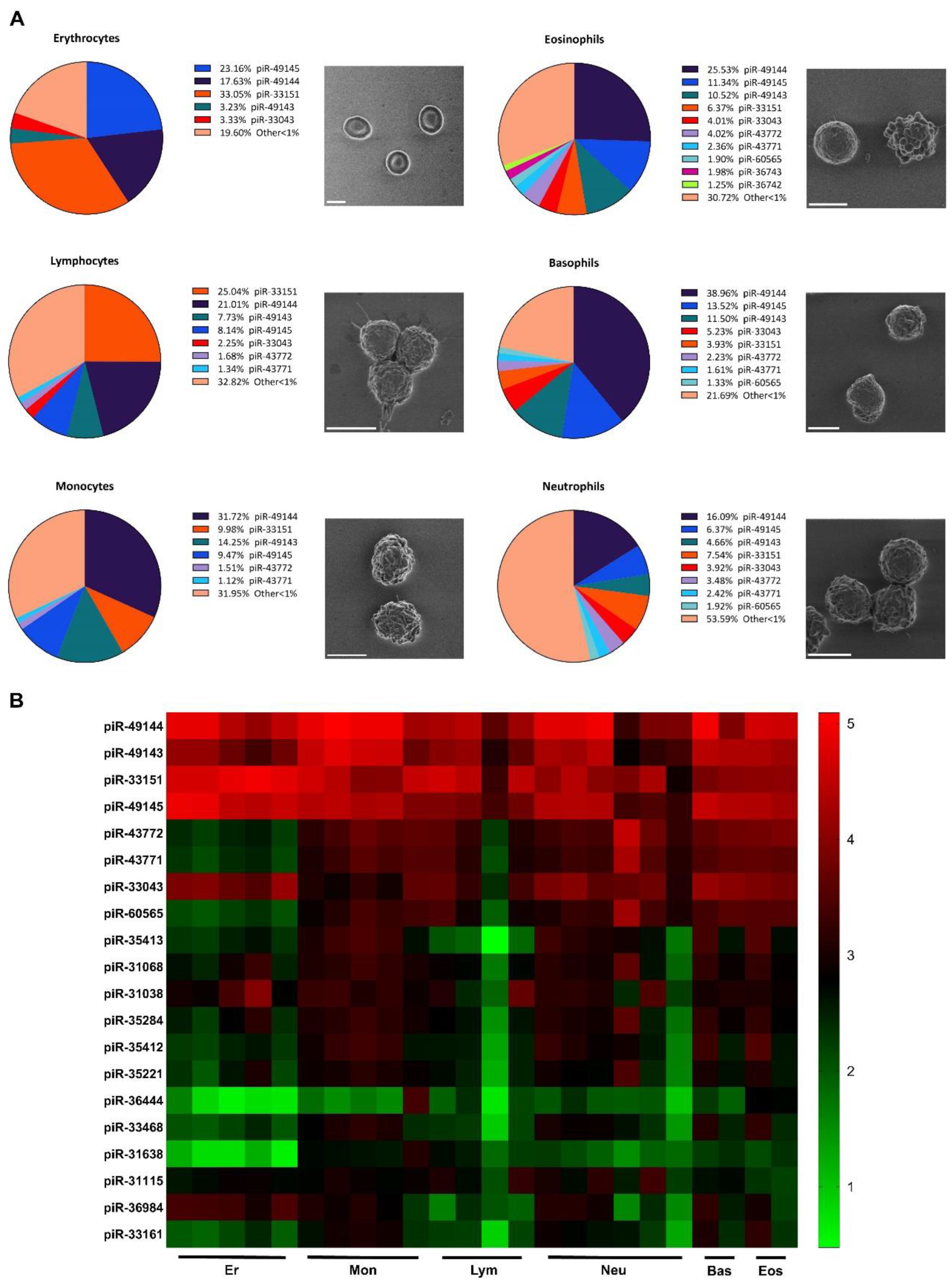
Figure 2.
Volcano plot representing differential expression of piRNA in erythrocytes, monocytes and lymphocytes of severe COVID-19 patients.
Figure 2.
Volcano plot representing differential expression of piRNA in erythrocytes, monocytes and lymphocytes of severe COVID-19 patients.
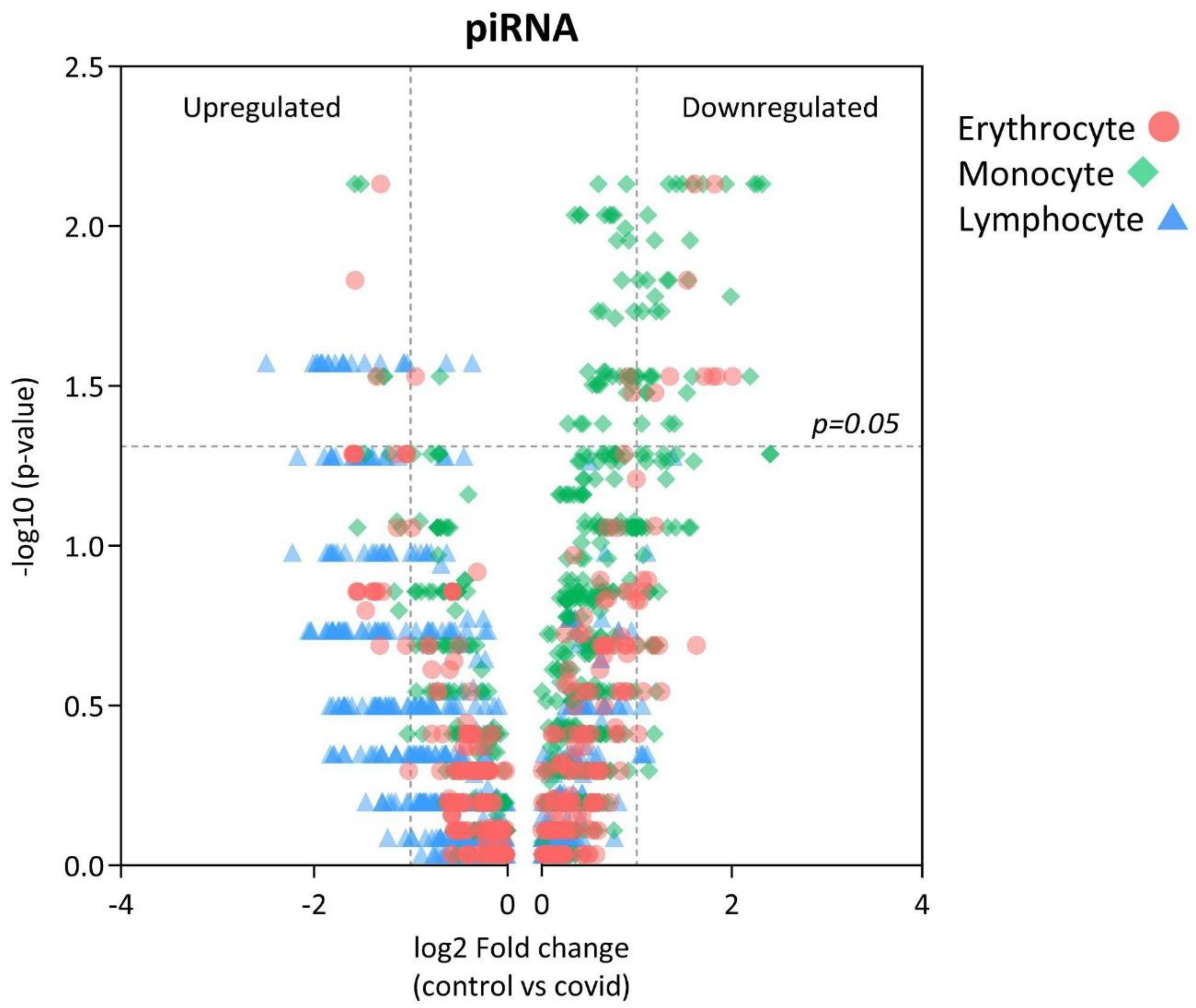

Table 1.
List of piRNAs significantly altering their expression in erythrocytes of severe COVID-19 patients.
Table 1.
List of piRNAs significantly altering their expression in erythrocytes of severe COVID-19 patients.
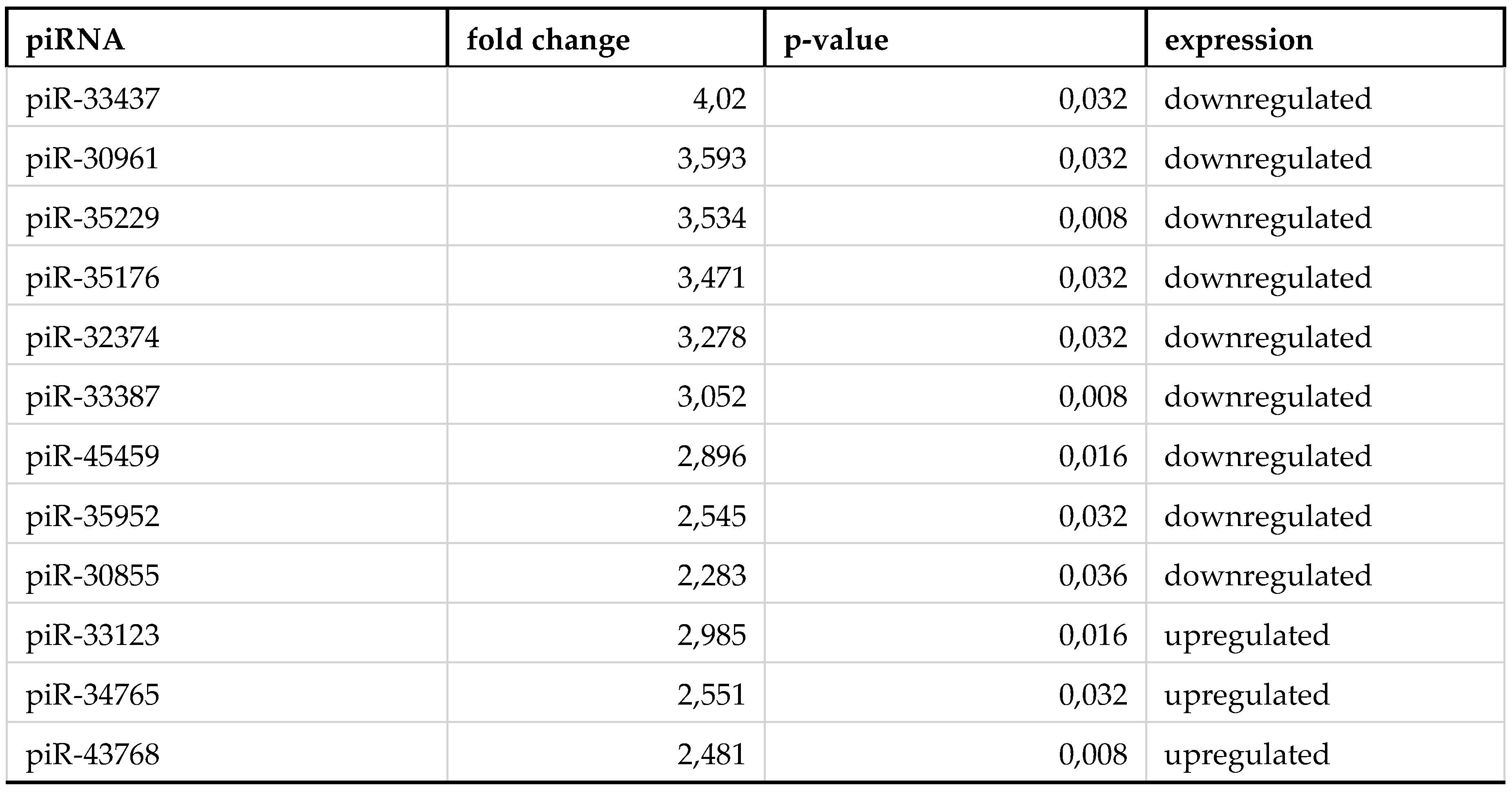
Table 2.
List of piRNAs significantly altering their expression in monocytes of severe COVID-19 patients.
Table 2.
List of piRNAs significantly altering their expression in monocytes of severe COVID-19 patients.
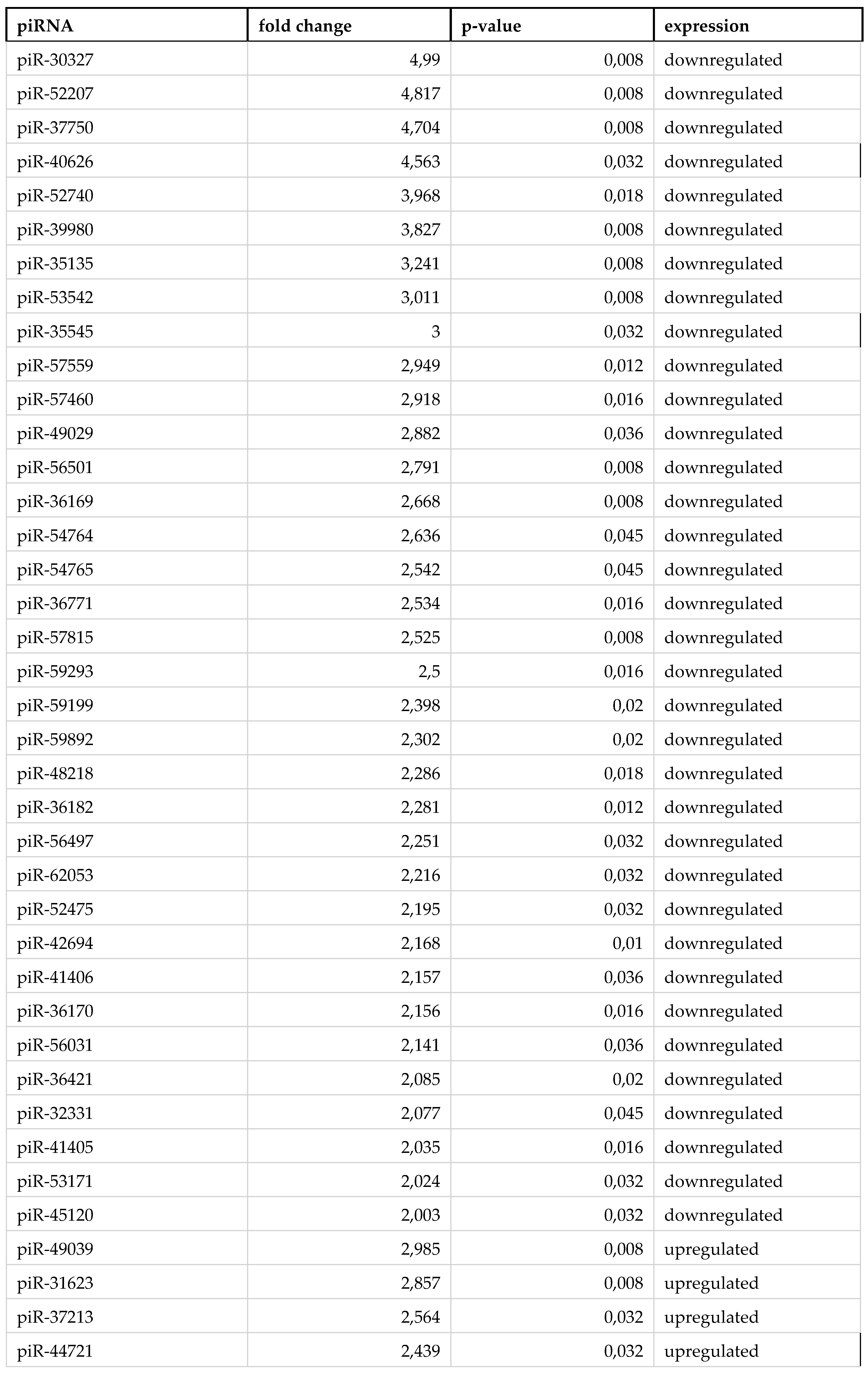

Table 3.
List of piRNAs significantly altering their expression in lymphocytes of severe COVID-19 patients.
Table 3.
List of piRNAs significantly altering their expression in lymphocytes of severe COVID-19 patients.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



