
Submitted:
07 February 2024
Posted:
08 February 2024
You are already at the latest version
Abstract
Chitosan (CS) nanoparticles are under investigation for a wide range of applications in nanomedicine. The structural, molecular and physicochemical properties of chitosan nanoparticles have a direct bearing on their efficacy in drug conjugation and delivery, gene delivery, and tissue engineering. Herein we report on the structural, morphological and molecular properties of chitosan nanoparticles synthesized using the ionic gelation method with the intended purpose of being used for drug delivery. The chitosan nanoparticles (CS-NPs) were prepared in the 0.25 to 1.0% w/v concentration range, whereas separate samples of the 1.0% w/v CS-NPs were also functionalized with polyethylene oxide (PEO) alone and with a diblock copolymer of PEO and polypropylene glycol (PPG). The average size of the CS-NPs was determined from TEM imaging to be in the range of 11.3 to 14.8 nm. The XRD and TEM analyses show that the CS-NPs have semi-crystalline structure with the degree of crystallinity depending upon the nature of synthesis. Our results show that functionalizing the 1.0% w/v CS-NPs with PEO increases the degree of crystallinity whereas functionalizing with the diblock copolymer PEO - PPG results in a significant increase in their degree crystallinity. The CS/TPP concentration (CS:TPP fixed at 1:1 ratio) had no apparent effect on the degree of crystallinity of the CS-NPs. The results from the FTIR analysis confirm incorporation of TPP with CS in the CS-NPs. Additionally, FTIR analysis shows an enhancement of hydrogen bonding that is consistent with the increase in the degree of crystallinity of the CS-NPs.
Keywords:
chitosan nanoparticles
; structure properties
; polymer functionalization
; x-ray diffraction
; transmission electron microscopy
; Fourier transform infrared spectroscopy
1. Introduction
Chitosan nanoparticles are currently under vigorous investigation as suitable agents for in vivo delivery of drugs and genetic material [1,2,3,4,5,6]. Current research has shown that their small size and selective functionalization enables chitosan nanoparticles (CS-NPs) to enter human tissue in a targeted manner [2,3,4,5,6,7,8]. This targeted modality of treatment has distinct advantages over many conventional treatment approaches, such as chemotherapy in the treatment of cancer. Furthermore, the fact that CS-NPs are derived from chitosan, which is a natural polysaccharide material, provides a distinct biocompatibility advantage over inorganic nanoparticles. The physicochemical properties that make functionalized CS-NPs highly suitable for drug delivery include biodegradability, biocompatibility, nano size, hydrophilicity, and high drug conjugation efficacy.
Chitosan (CS) is a polysaccharide derived by the N-deacetylation of chitin. The molecular structure of chitosan is a linear copolymer consisting of glucosamine and N-acetyl-glucosamine units: The repeatability of the units is determined by the degree of deacetylation (DD) [9,10]. The level of deacetylation directly affects the efficacy of CS in a wide range of applications [11,12,13,14]. Chitosan’s solubility is directly impacted by the DD level, pH, temperature, degree of crystallinity, and solvent type. The amount of protonated NH2 groups determines the solubility of chitosan in aqueous media: For instance, chitosan dissolves in aqueous solutions when 50% of the amino groups are protonated [9]. Chitosan is soluble in acidic solutions at relatively low DD values. Therefore, assuming all other variables remain constant, the degree of deacetylation directly affects the solubility, crystal structure and the size of glucosamine units in chitosan [9]. As a result, the degree of crystallinity and crystal size potentially plays a direct role in the biochemical, nanomedicinal, and pharmacological applications of CS-NPs [15,16].
A number of methodologies are employed to synthesize CS-NPs from chitosan [17,18]. These include ionic gelation, microemulsion, emulsion-based solvent evaporation, and emulsification solvent diffusion [19]. The molecular weight, the amount of chitosan employed, and the degree of chitosan deacetylation are the three key factors that directly affect the particle size, crystallinity, and surface charge of the CS-NPs obtained utilizing these preparation processes. Chitosan is classified as a semi-crystalline biopolymer since it consists of both a crystalline and an amorphous portion [20]. Rinaudo has made an extensive review of the structural properties of chitosan. Chitosan can exhibit diverse crystalline structural forms based on the various raw material sources: α-chitin with two anti-parallel molecules in the unit cell, β-chitin with two parallel molecules in the unit cell, and γ-chitin in a mixed arrangement of the two allomorphs [21]. Both the α-chitin and β-chitin structures have the polymeric chains ordered and held together by hydrogen bonding in sheets. In contrast to the normal profile of the β-chitin allomorph, which exhibits a monoclinic P21 symmetry with parallel displacement and decreased intersheet interaction, the pattern of α-chitin maintains the orthorhombic P212121 symmetry with antiparallel displacement of the polymeric chains [22]. The XRD patterns of the chitin allomorphs generally exhibit two high-intensity diffraction peaks occurring within the 8 – 11° and 19 – 21° ranges in 2θ, which coincide with the (110) and (120) reflections, respectively. On the other hand, secondary peaks are primarily observed in XRD patterns from α-chitin [21,22,23]. Depending upon procedures used in processing, chitin and chitosan can occur in hydrated form. Fachinatto et al. made a detailed structure study using XRD and NMR of CS showing that increasing the DD increases the degree of crystallinity of chitin [23]. In addition, this study showed that the degree of crystallinity does not differ significantly as a function of molecular weight, particularly when it comes to the molar mass changes in the samples with lower DD. This finding is consistent with the study carried out by Savitri et al. [24] showing that decreasing the molar mass from chitin with DA > 20% has a direct bearing on the degree of crystallinity of the CS. The authors purport that the crystallinity of CS depends on various factors including the source of extraction, deacetylation, and molecular weight.
In this study we have prepared CS-NPs using the ionic gelation method. Furthermore, we have functionalized the CS-NPs using PEO and a combined diblock copolymer PEO-PPG. The incorporation of hydrophilic polymers and diblock copolymers substantially improves the solubility, hemocompatibility, and bodily-fluid stability, while reducing cytotoxicity of CS-NPs [25]. For these reasons, the drug delivery capacity of CS-NPs modified by incorporating hydrophilic polymers and diblock copolymers is improved. However, studies of the structural characteristics of CS-NPs modified using polymers are exceedingly rare. The resulting CS-NPs were fully characterized using X-Ray diffraction (XRD), Transmission Electron Microscopy (TEM), and Fourier Transmission Infrared Spectroscopy (FTIR) to determine the structure and bonding properties of functionalized and non-functionalized CS-NPs.
2. Materials and Methods
2.1. Preparation of CS Nanoparticles
The polyethylene oxide (PEO) of molecular weight (MW) 300,000, polypropylene glycol (PPG) of MW of 4000, sodium tripolyphosphate (TPP), and 85% deacetylated chitosan (CS) that were used in this study were purchased from Thermo Fisher Scientific Chemicals. The CS-NPs were synthesized using the ionic gelation method, where aqueous solution of CS is mixed with an aqueous solution of sodium tripolyphosphate (TPP), whereby their opposing charges cause inter- and intra-molecular cross-linking, resulting in the formation of chitosan nanoparticles. High-purity liquid chromatography (HPLC) water was used to dissolve the CS and TPP to make the aqueous solution. CS was dissolved in HPLC water with acetic acid using a magnetic stirrer set to stir vigorously for approximately 2-3 hours at room temperature. Similarly, TPP was dissolved in HPLC water using a magnetic stirrer set to stir just as vigorously for 2-3 minutes at room temperature.
Bulk chitosan and TPP (in equal proportions) were prepared in concentrations of 0.25%, 0.5% and 1.0% w/v in acetic acid. In every instance, the volume in mL of glacial acetic acid concentration was 1.75 times the number of grams of CS added [26]. The CS and TPP were dissolved in two separate beakers of HPLC water at identical concentrations. The TPP was added to the CS-solution under magnetic stirring dropwise using a pipette. In this process, three different stages were identified as TPP solution was progressively added to the CS solution: clear, opalescent and aggregated solution of CS-NPs. At low CS concentrations, the opalescence observed is very faint. Subsequently, each aggregated sample was centrifuged using 8:1 ethanol: toluene solution 5 times to remove any impurities. The centrifuged sample was then dried and ground using mortar and pestle to the powdered form for characterization.
Functionalized CS-NPs were formed spontaneously upon incorporation of 8mL of 1% w/v of TPP in 20mL of 1% w/v CS solution, with the CS solution also containing 50 mg of PEO or PEO-PPG so that the surfactant was 1.75 times higher in concentration than CS. The procedure used was the same as outlined above for synthesis of the functionalized CS-NPs. The samples used in this study are shown in the table below.
2.2. Characterization of Chitosan Nanoparticles
X-ray diffraction (XRD), transmission electron microscopy (TEM), and Fourier transform infrared spectroscopy (FTIR) were employed for the structural and morphological characterization of the samples. The CS-NP samples were fixed on glass slides using ethanol for XRD measurements. The characterization was made using a Bruker D8 Discover diffractometer with Cu Kα (λ = 1.5405 Å) radiation. During the data collection, the same geometry had been followed to compare multiple samples in the same condition. Care was taken for proper alignment of the sample on the sample mount of the diffractometer for optimal data collection. Several XRD spectra were collected and averaged for measurements from each sample.
For TEM imaging, the samples were dissolved in hexane and a drop of a sample was placed on a carbon TEM grid. The TEM and HRTEM (high resolution transmission electron microscopy) imaging of the samples was performed on an FEI Titan 80−300 instrument at the University of Arkansas Nano-Bio Materials Characterization Facility, with the field emission gun set to 300 keV. The fast Fourier transform (FFT) analysis and size determination were made using imageJ software.
FTIR analyses of the CS-NPs samples were made using a Bruker Alpha II spectrometer. The Alpha II spectrometer is equipped with a Diamond Crystal ATR (Attenuated Total internal Reflectance) accessory. The FTIR spectra were measured from our CS-NP samples in powder form.
3. Results
3.1. XRD and TEM Analyses
Figure 1 a) Figure 1 a) shows the XRD data measured from the CS-NPs samples without and functionalized with PEO and with PEO-PPG, along with that measured from bulk CS, whereas the XRD patterns measured from the CS-NPs as a function of CS concentration are shown in Figure 1 b). As shown in Figure 1 a), the XRD pattern measured from bulk CS consists of two predominant peaks, one occurring at 10.1°, with a planar d spacing of 0.875 nm, and a wider peak occurring at 19.9° in 2θ stemming from planar d spacing of 0.446 nm. As mentioned above, peaks occurring in the 8 – 11° and 19 – 21° ranges are typically attributed to reflections from the (020) and (110) planes in the crystalline SC structure, respectively. The variability in the reported positions is typically attributed to the source of chitin used in the preparation of the chitosan [21]. The d spacings associated with the (020) and (110) reflections and the lack of additional notable reflections that occur for the α allomorph, including the (120) and the (130), makes our bulk CS sample most likely the β allomorph of chitosan [21,22,23]. Interestingly, the XRD pattern measured from the bulk CS sample also shows two lower intensity peaks, one at 14.9° and a smaller one at 29.2°. As seen in Figures 1 a) and b), the diffraction patterns measured from our CS-NP samples are characterized by a convolution of a very broad peak-like feature centered near 23° and broad diffraction peaks of smaller intensities. The extensive peak-like feature centered near 23° is indicative of a significant amorphous contribution to the overall structure of the CS-NPs whereas the smaller-intensity diffraction peaks are indicative of a crystalline contribution. The (110) peak is seen to be shifted in the XRD patterns measured from the CS-NPs to 18.6°, indicating an increased level of hydration in comparison to that of the bulk CS. This is consistent with the use of the aqueous solvent in the ionic gelation method to prepare the CS-NPs. Additionally, a lower intensity peak is found to occur in the 11.3 – 11.6° range for all CS-NPs as indicated in Figure 1. This peak may coincide with the crystalline plane reflections indicated by the 14.9° peak observed in the pattern measured from bulk CS but similarly shifted as the (110) peak due to hydration. There is also a prominent shoulder observed near 30° in all XRD patterns measured the CS-NPs which most likely is correlated with the 29.2° peak observed from the pattern measured from bulk CS.
Figure 1a) clearly indicates that the addition of PEO does not have much of a bearing in affecting the degree of crystallinity of the CS-NPs, in comparison to that of the 1.0% w/v CS-NP sample. This is in contradiction with the findings made by Darbasizadeh et al. [26] indicating that the addition of and functionalization with PEO increases the crystallinity of CS-NPs. These authors claim that the formation of hydrogen bonds between the amine group from chitosan and the polyether oxygen from PEO increased chain entanglement and solution spinnability when PEO was added to a chitosan solution. We do not find evidence for such action from PEO in the synthesis of our CS-NPs. However, we find that the addition of PEO-PPG has a significant impact on increasing the degree of crystallinity of the 1.0% w/v CS-NPs. We conjecture that this is due to the tendency of the PEO-PPG block copolymer to self-organize into ordered structures, such as micelles, in aqueous solutions. Mortensen et al. determined that the polystyrene (PS)-PEO diblock copolymer forms spherical micelles at up to 20% concentration in aqueous solutions [27]. The PEO-PPG block copolymer may first envelop the embryonic phase of the CS-NPs allowing for more controlled diffusive motion and greater organization of CS polymeric strands into crystalline units. Nevertheless, this hypothesis needs to be tested with additional structure studies of CS-NP formation in PEO-PPG bearing aqueous solution using small-angle neutron scattering and similar techniques. Figure 1 b) shows that the XRD patterns measured from the 0.25 – 1.0% w/v CS-NP samples (where CS:TPP ratio is fixed at 1:1) are qualitatively similar in appearance indicating that the CS/TPP concentration values used in the preparation have a negligible effect on the degree of crystallinity of the CS nanoparticles. A small peak is evident in the XRD spectra of the CS-NP samples lacking either PEO or PEO-PPG in the 8.6 – 8.9° range, which is consistent with a weak reflection from the chitosan (020) planes.
The morphology and size of CS-NPs was studied using TEM imaging whereas the structural aspects were studied using high resolution TEM (HRTEM). The CS-NPs are generally pseudo-spherical and prismatic shaped. The size and crystallinity of the CS-NPs was analyzed by processing the TEM images using imageJ software. As shown in Table 1, the size of the CS-NPs as determined from the histograms was found to be similar for all samples and in the 11 – 15 nm range. This is attributed to the fixed ratio of CS:TPP (1:1) used in the ionic gelation of the CS-NP samples. The semi-crystalline nature of the samples was verified using the fast Fourier transform (FFT) analysis of HRTEM images, which is shown in Figures 2 and S1. We also observe a variability in the extent of crystallinity exhibited by the CS-NPs: Some are predominantly amorphous with negligible crystallinity whereas others show varying degrees of crystallinity. The amorphous nature is partially exhibited in the FFTs by the wide and diffuse ring feature (hexane residue provides an additional contribution) whereas the spot-like pattern is indicative of reflections from specific crystalline planes. The FFTs of select nanoparticles in Figures 1c and 1f are consistent with a lower degree of crystallinity exhibited by the CNP-1% sample as compared to that of the CNP-PEPG sample, respectively. The overall TEM results are consistent with those from the analysis of the XRD patterns measured from the CS-NP samples.
3.2. FTIR Spectroscopy Analysis
FTIR spectra of bulk CS, CNP-1%, CNP-PEO, and CNP-PEPG samples are shown in Figure 3. The broad peak occurring in the ~3000 – 3500 cm-1 range results from N-H and O-H stretching vibrations in the polymer CS chains [27,28]. This peak is more pronounced in the spectrum measured from the CNP-PEPG sample compared to that of the CNP-PEO and CNP-1% samples, indicating that the hydrogen bonding in the former is more enhanced than in the latter samples. This result is consistent with the structure results obtained from the XRD and TEM analyses of these samples. The peak located at 2861cm-1 is due to the C-H asymmetric vibration [28,29] whereas the peak at 1630 cm-1 is due to C==O stretching of the -CONH2 group (amide I) mode, which is consistent with the results from Pighnelli et al. [30]. Additionally, the peaks at 1535 cm-1 and 1380 cm-1 result from N-H bending amide (-CONH2) II and acetyl CH3 modes, respectively. The hypsochromic shift of the peaks from 1650 cm-1 and 1567 cm-1 of CS to 1630 and 1535 cm-1 in the spectra measured from the CS-NPs are a result of the interaction between the NH3+ groups of chitosan and the phosphate groups of TPP, as was previously reported by Lustriane et al. [31]. We also observe the asymmetric stretching mode of the C-O-C bridge of glucose-β-1-4 at 1154 cm-1, which is consistent with the results of Zaman et al. [32]. In addition, a relatively intense peak at 1064 cm-1 stemming from the C-O stretching and 1025 cm-1 from the C-O bending mode is found in all spectra measured from the CS-NP samples. FTIR spectra measured from TPP show a PO4-2 group mode peak occurring at 888 cm-1, as previously reported by Agrawal et al. [33]. This feature is more enhanced and occurs at 890 cm-1 in the spectra measured from our CS-NPs, indicating incorporation of TPP upon CS nanoparticle formation.
Our next objective is to load our CS-NPs with known cancer treatment drugs and test their drug delivery potential in HeLa cell culture studies. The results of these studies will be published in the future.
4. Conclusions
The ionic gelation method was used to prepare CS-NPs at concentrations varying from 0.25 to 1.0% w/v with the CS:TPP ratio fixed at 1:1 and with the highest concentration sample functionalized with PEO alone and with a combination of PEO+PPG. Our XRD and HRTEM results show that the CS-NPs are semi-crystalline in that they are typically comprised of both amorphous and crystalline components. The CS-NP sample functionalized with PEO-PPG is found to have a significant increase in the degree of crystallinity whereas the CS-NP sample functionalized with PEO alone had no increase in the degree of crystallinity. In addition, the CS/TPP concentration had no apparent bearing on the degree of crystallinity of the CS-NPs. The results from the FTIR analysis confirm the cross-linkage of TPP with CS to form nanoparticles. The broad peak in the ~3000 – 3500 cm-1 range, that is attributed to O-H and N-H stretching vibrational modes, are found to intensify in spectra measured from CS-NPs functionalized with PEO-PPG but not CS-NPs or CS-NPs functionalized with POE alone, indicating the enhancement of hydrogen bonding in the former. This is consistent with the increased degree of crystallinity of the CS-NP sample functionalized with PEO-PPG. The CONH2 and NH2 peaks at 1650 cm-1 and 1567 cm-1 in the FTIR spectrum of CS shift hypsochromically to 1629 and 1535 cm-1, respectively, because of the interaction between the NH3+ groups of chitosan and the phosphate groups of TPP. The 1138 and 888 cm-1 peaks of PO4-2 groups are more enhanced and are shifted to 1154 and 891 cm-1 in case of CS-NPs, indicating the incorporation of TPP upon CS nanoparticle formation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: a) Size distribution histogram calculated from TEM data (inset shows a TEM image), b) a HRTEM image and c) a FFT of the region delineated by the rectangle shown in b) of sample CNP-0.5%; d) size distribution histogram and a corresponding a TEM image shown in the inset, e) a HRTEM image and f) a FFT of the region outlined by the rectangle in e) of sample 1% CNP-PEO. ; Table S2: title: FTIR spectra measured from the CNP-1.0% and CNP-0.5% samples indicating negligible concentration dependence.
Author Contributions
Author Contributions: Conceptualization, R.J. and R.A.M.; methodology, R.J., M.B., and R.A.M.; validation, R.J. M.B., and R.A.M.; formal analysis, R.J., H.H., M.B., and R.A.M.; data curation, R.J., H.H., M.B, and R.A.M.; writing—original draft preparation, R.J. and R.A.M.; writing—review and editing, R.J., H.H., M.B, and R.A.M.; visualization, R.J., H.H., and R.A.M.; supervision, R.J. and R.A.M.; project administration, R.A.M.; funding acquisition, R.A.M. All authors have read and agreed to the published version of the manuscript. .
Funding
Not Applicable.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data available on request.
Acknowledgments
We would like to thank Dr. Erich Steinle and Dr. Reza Sedaghat Herati for assistance with the FTIR measurements of the samples. .
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jha, R.; Mayanovic, R. A. A Review of the Preparation, Characterization, and Applications of Chitosan Nanoparticles in Nanomedicine. Nanomaterials 2023, 13(8), 1302. [Google Scholar] [CrossRef]
- Nagpal, K.; Singh, S. K.; Mishra, D. N. Chitosan Nanoparticles: A Promising System in Novel Drug Delivery. Chem. Pharm. Bull. 2010, 58(11), 1423–1430. [Google Scholar] [CrossRef]
- Mohammed, M.; Syeda, J.; Wasan, K.; Wasan, E. An Overview of Chitosan Nanoparticles and Its Application in Non-Parenteral Drug Delivery. Pharmaceutics 2017, 9(4), 53. [Google Scholar] [CrossRef] [PubMed]
- Garg, U.; Chauhan, S.; Nagaich, U.; Jain, N. Current Advances in Chitosan Nanoparticles Based Drug Delivery and Targeting. Adv. Pharm. Bull. 2019, 9(2), 195–204. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z. Recent Advances of Chitosan Nanoparticles as Drug Carriers. Int. J. Nanomedicine 2011, 765. [Google Scholar] [CrossRef] [PubMed]
- Naskar, S.; Koutsu, K.; Sharma, S. Chitosan-Based Nanoparticles as Drug Delivery Systems: A Review on Two Decades of Research. J. Drug Target. 2019, 27(4), 379–393. [Google Scholar] [CrossRef] [PubMed]
- De Campos, A. M.; Sánchez, A.; Alonso, M. J. Chitosan Nanoparticles: A New Vehicle for the Improvement of the Delivery of Drugs to the Ocular Surface. Application to Cyclosporin A. Int. J. Pharm. 2001, 224(1–2), 159–168. [Google Scholar] [CrossRef]
- Luo, Y.; Wang, Q. Recent Development of Chitosan-Based Polyelectrolyte Complexes with Natural Polysaccharides for Drug Delivery. Int. J. Biol. Macromol. 2014, 64, 353–367. [Google Scholar] [CrossRef]
- Kumirska, J.; Weinhold, M. X.; Thöming, J.; Stepnowski, P. Biomedical Activity of Chitin/Chitosan Based Materials- Influence of Physicochemical Properties Apart from Molecular Weight and Degree of N-Acetylation. Polymers (Basel). 2011, 3(4), 1875–1901. [Google Scholar] [CrossRef]
- Aranaz, I.; Mengibar, M.; Harris, R.; Panos, I.; Miralles, B.; Acosta, N.; Galed, G.; Heras, A. Functional Characterization of Chitin and Chitosan. Curr. Chem. Biol. 2012, 3(2), 203–230. [Google Scholar] [CrossRef]
- Kumirska, J.; Czerwicka, M.; Kaczyński, Z.; Bychowska, A.; Brzozowski, K.; Thöming, J.; Stepnowski, P. Application of Spectroscopic Methods for Structural Analysis of Chitin and Chitosan. Mar. Drugs 2010, 8(5), 1567–1636. [Google Scholar] [CrossRef]
- Hussain, R.; Maji, T. K.; Maji, T. K. Determination of Degree of Deacetylation of Chitosan and Their Effect on the Release Behavior of Essential Oil from Chitosan and Chitosan-Gelatin Complex Microcapsules. Int. J. Adv. Eng. Appl. 2013, 2(4), 4–12. [Google Scholar]
- Kasaai, M. R. Various Methods for Determination of the Degree of N-Acetylation of Chitin and Chitosan: A Review. J. Agric. Food Chem. 2009, 57(5), 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Ravi Kumar, M. N. V. Chitin and Chitosan Fibres: A Review. Bull. Mater. Sci. 1999, 22(5), 905–915. [Google Scholar] [CrossRef]
- Gan, Q.; Wang, T.; Cochrane, C.; McCarron, P. Modulation of Surface Charge, Particle Size and Morphological Properties of Chitosan-TPP Nanoparticles Intended for Gene Delivery. Colloids Surfaces B Biointerfaces 2005, 44(2–3), 65–73. [Google Scholar] [CrossRef]
- Badawy, M. E. I.; Rabea, E. I. A Biopolymer Chitosan and Its Derivatives as Promising Antimicrobial Agents against Plant Pathogens and Their Applications in Crop Protection. Int. J. Carbohydr. Chem. 2011, 2011, 1–29. [Google Scholar] [CrossRef]
- Divya, K.; Jisha, M. S. Chitosan Nanoparticles Preparation and Applications. Environ. Chem. Lett. 2018, 16(1), 101–112. [Google Scholar] [CrossRef]
- Aranaz, I.; Alcántara, A. R.; Civera, M. C.; Arias, C.; Elorza, B.; Caballero, A. H.; Acosta, N. Chitosan: An Overview of Its Properties and Applications. Polymers (Basel) 2021, 13(19). [Google Scholar] [CrossRef]
- Tamer, T. M.; Hassan, M. A.; Omer, A. M.; Valachová, K.; Eldin, M. S. M.; Collins, M. N.; Šoltés, L. Antibacterial and Antioxidative Activity of O-Amine Functionalized Chitosan. Carbohydr. Polym. 2017, 169, 441–450. [Google Scholar] [CrossRef]
- de Farias, B. S.; Sant’Anna Cadaval Junior, T. R.; de Almeida Pinto, L. A. Chitosan-Functionalized Nanofibers: A Comprehensive Review on Challenges and Prospects for Food Applications. Int. J. Biol. Macromol. 2019, 123, 210–220. [Google Scholar] [CrossRef]
- Rinaudo, M. Chitin and Chitosan: Properties and Applications. Prog. Polym. Sci. 2006, 31(7), 603–632. [Google Scholar] [CrossRef]
- Minke, R.; Blackwell, J. The Structure of α-Chitin. J. Mol. Biol. 1978, 120(2), 167–181. [Google Scholar] [CrossRef]
- Facchinatto, W. M.; Santos, D. M. dos; Fiamingo, A.; Bernardes-Filho, R.; Campana-Filho, S. P.; Azevedo, E. R. de; Colnago, L. A. Evaluation of Chitosan Crystallinity: A High-Resolution Solid-State NMR Spectroscopy Approach. Carbohydr. Polym. 2020, 250(July). [Google Scholar] [CrossRef]
- Savitri, E.; Juliastuti, S. R.; Handaratri, A.; Sumarno; Roesyadi, A. Degradation of Chitosan by Sonication in Very-Low-Concentration Acetic Acid. Polym. Degrad. Stab. 2014, 110, 344–352. [Google Scholar] [CrossRef]
- Mikušová, V.; Mikuš, P. Advances in Chitosan-Based Nanoparticles for Drug Delivery. Int. J. Mol. Sci. 2021, 22(17), 9652. [Google Scholar] [CrossRef]
- Alonso, M. J.; Calvo, P.; Remun, C. Novel Hydrophilic Chitosan-polyethylene Oxide Nanoparticles as Protein Carriers. J. Appl. Polym. Sci. 1997, 63(1), 125–132. [Google Scholar] [CrossRef]
- Mortensen, K.; Brown, W.; Almdal, K.; Alami, E.; Jada, A. Structure of PS-PEO Diblock Copolymers in Solution and the Bulk State Probed Using Dynamic Light-Scattering and Small-Angle Neutron-Scattering and Dynamic Mechanical Measurements. Langmuir 1997, 13(14), 3635–3645. [Google Scholar] [CrossRef]
- Darbasizadeh, B.; Motasadizadeh, H.; Foroughi-Nia, B.; Farhadnejad, H. Tripolyphosphate-Crosslinked Chitosan/Poly (Ethylene Oxide) Electrospun Nanofibrous Mats as a Floating Gastro-Retentive Delivery System for Ranitidine Hydrochloride. J. Pharm. Biomed. Anal. 2018, 153, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Sazak, C.; Attar, A.; Yilmaz, A.; Altikatoglu Yapaoz, M. Biofabrication of Acer Palmatum-Mediated Multifunctional CuO Nanoparticles for Dye Removal, Antibacterial-Antifungal Activity, and Molecular Docking. ACS Omega 2023. [CrossRef]
- Zaman, M.; Butt, M. H.; Siddique, W.; Iqbal, M. O.; Nisar, N.; Mumtaz, A.; Nazeer, H. Y.; Alshammari, A.; Riaz, M. S. Fabrication of PEGylated Chitosan Nanoparticles Containing Tenofovir Alafenamide: Synthesis and Characterization. Molecules 2022, 27(23). [Google Scholar] [CrossRef]
- L, P.; MF, G.; CM, B.; G, Z.; MG, R.; DS, C.; RL, P.; BG, Z.; M, K.; MF, T.; V, R.; MM, S.; CT, F.; CC, F. Structure and Properties of Nanocrystalline Chitosan. J. Appl. Biotechnol. Bioeng. 2016, 1(1), 13–20. [Google Scholar] [CrossRef]
- Lustriane, C.; Dwivany, F. M.; Suendo, V.; Reza, M. Effect of Chitosan and Chitosan-Nanoparticles on Post Harvest Quality of Banana Fruits. J. Plant Biotechnol. 2018, 45(1), 36–44. [Google Scholar] [CrossRef]
- Agarwal, M.; Agarwal, M. K.; Shrivastav, N.; Pandey, S.; Das, R.; Gaur, P. Preparation of Chitosan Nanoparticles and Their In-Vitro Characterization. Int. J. Life-Sciences Sci. Res. 2018, 4(2), 1713–1720. [Google Scholar] [CrossRef]
Figure 1.
a) XRD spectra measured from the CS-NP samples without and functionalized with PEO and with PEO+PPG compared to that of bulk CS and b) XRD spectra measured from the CS-NP samples as a function of CS concentration.
Figure 1.
a) XRD spectra measured from the CS-NP samples without and functionalized with PEO and with PEO+PPG compared to that of bulk CS and b) XRD spectra measured from the CS-NP samples as a function of CS concentration.
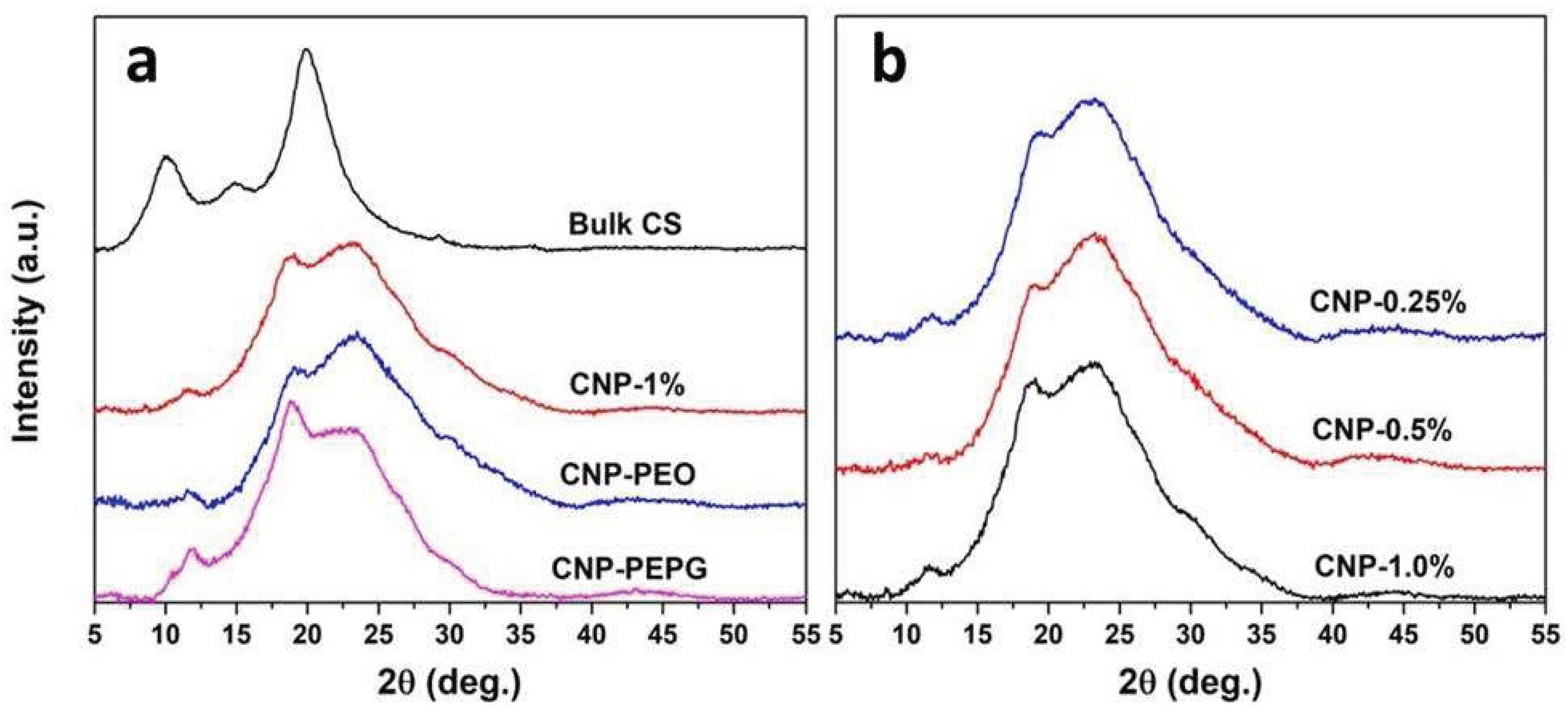
Figure 2.
a) Size distribution histogram with inset showing a TEM image, b) a high-resolution TEM image and c) a FFT of the region delineated by the rectangle in b) of sample CNP-1.0%; d) size distribution histogram with inset displaying a corresponding a TEM image, e) a high-resolution TEM image and f) a FFT of the region outlined by the rectangle in e) of sample CNP-PEPG.
Figure 2.
a) Size distribution histogram with inset showing a TEM image, b) a high-resolution TEM image and c) a FFT of the region delineated by the rectangle in b) of sample CNP-1.0%; d) size distribution histogram with inset displaying a corresponding a TEM image, e) a high-resolution TEM image and f) a FFT of the region outlined by the rectangle in e) of sample CNP-PEPG.
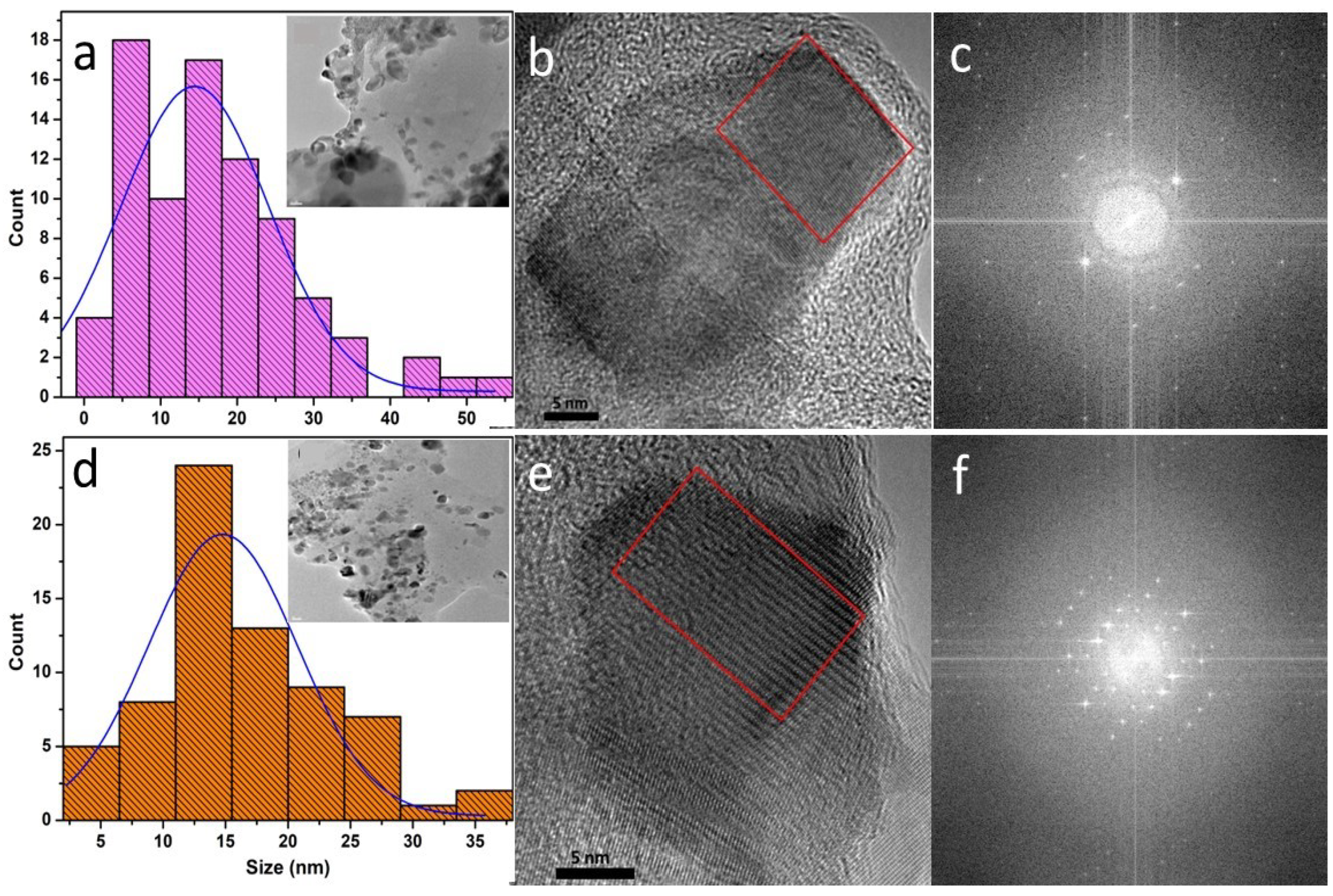
Figure 3.
FTIR spectra measured from the samples of this study.
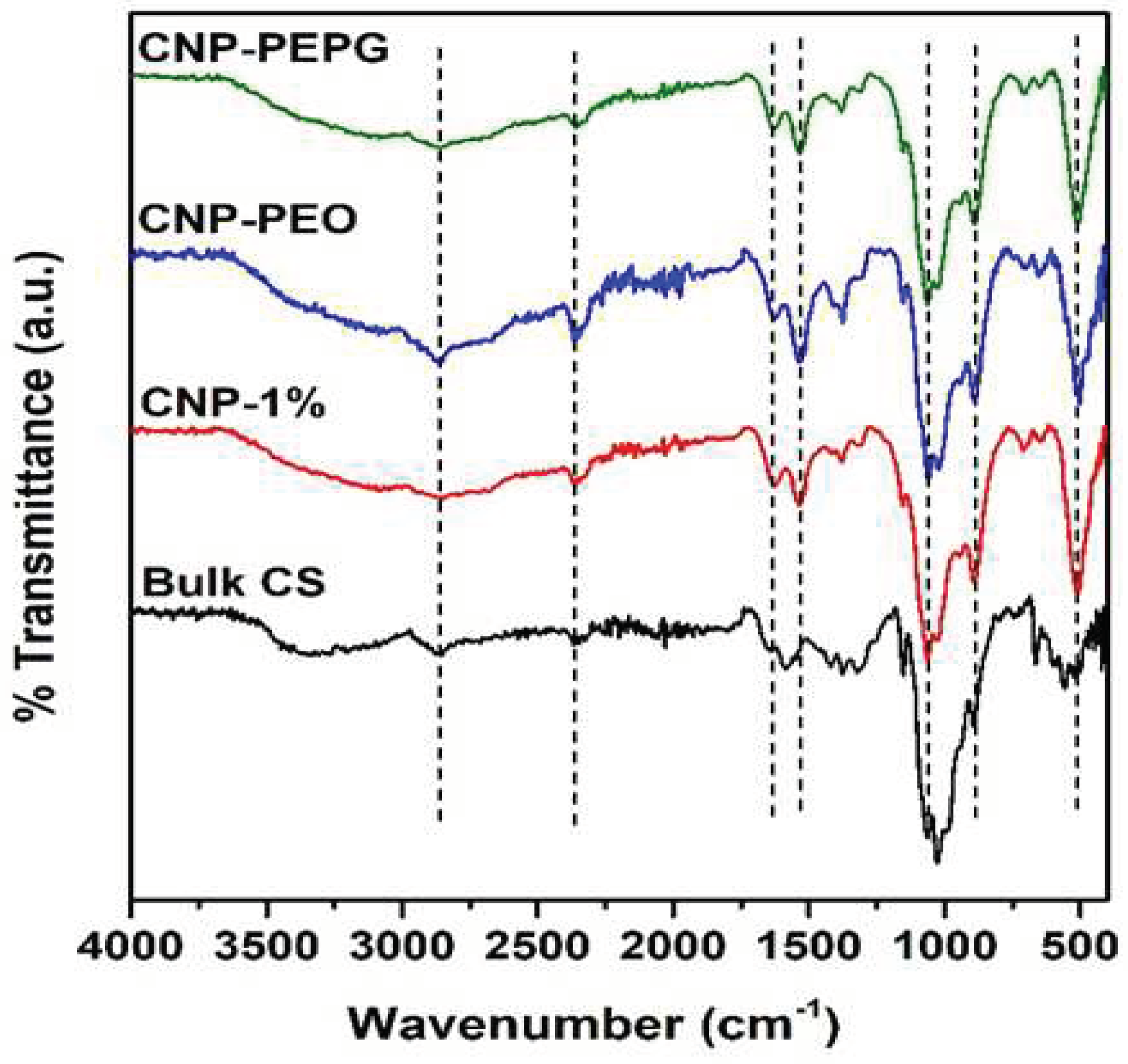

Table 1.
CS-NP samples of this study and their mean size as determined from TEM measurements.
| Sample | Description | Mean size (nm) | FWHM (nm) |
|---|---|---|---|
| CNP-0.25% | 0.25% w/v CS-NPs | - | - |
| CNP-0.5% | 0.5% w/v CS-NPs | 13.1 | 15.4 |
| CNP-1.0% | 1.0% w/v CS-NPs | 14.4 | 22.7 |
| CNP-PEO | 1.0% w/v CS-NPs+PEO | 11.3 | 13.0 |
| CNP-PEPG | 1.0% w/v CS-NPs+PEO+PPG | 14.8 | 14.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



