
Submitted:
09 February 2024
Posted:
12 February 2024
You are already at the latest version
Abstract
TNF-related apoptosis-inducing ligand (TRAIL or Apo2 or TNFSF10) belongs to the TNF superfamily. When bound to its agonistic receptors, TRAIL can induce apoptosis in tumour cells, while sparing healthy cells. This tumour selectivity prompted, over the last three decades, many studies aiming at evaluating the anti-tumoral potential of TRAIL or its derivatives. Although most of these attempts have failed, so far, novel formulations are still being evaluated. Yet, emerging evidence indicates that TRAIL can also trigger, on the other hand, a non-canonical signal transduction pathway that is likely to be detrimental for its use in oncology. Likewise, increasing studies suggest that TRAIL can induce, through Death receptor 5 (DR5) in some circumstances, tumour cell motility, potentially leading to and contributing to tumour metastasis. While the pro-apoptotic signal transduction machinery of TRAIL is well known from a mechanistic point of view, that of the non-canonical pathway is less understood. We are reviewing here the current state of knowledge of TRAIL non-canonical signalling.
Keywords:
Apoptosis
; Metastasis
; Migration
; EMT
; Cancer
; TNF
; TRAIL
; signalling
1. Introduction
TNF-Related Apoptosis Ligand (TRAIL) is known as a type II transmembrane protein belonging to the TNF ligands superfamily (TNFSF) and reported for the first time as a cytokine coded by a gene TNFSF10 in 3q26 position on human chromosome 3 [1,2]. TRAIL can bind to six receptors. Two agonist receptors have been reported to induce the canonical pro-apoptotic signal transduction, upon binding to TRAIL, namely DR4 (TRAIL-R1 encoded by TNFRSF10A gene) [3]and DR5 (two splice variants of TRAIL-R2 encoded by TNFRSF10B gene) [4,5,6,7,8]. The TRAIL agonist receptors, DR4 and DR5, are able to trigger apoptosis because they harbour a death domain (DD) in their c-terminal part (Figure 1), which is also found in TNF-R1 and Fas [9,10,11,12,13,14], and which is necessary and sufficient to engage the pro-apoptotic machinery [9,10].
In addition to these two agonists, TRAIL can also bind to four other receptors (Figure 2), but the latter are unable to induce apoptosis due either to the absence of a functional death domain (DD), in their intracellular c-terminal portion, or because these receptors are secreted to the extracellular compartment [15]. DcR1, DcR2 and OPG [16,17,18,19,20] solely interact with TRAIL, while DcR3, which has more recently been found to interact both with TRAIL [21], was originally found to interact with Fas ligand [18]. Both DcR1 (TRAIL-R3 encoded by TNFRSF10C) [22,23] and DcR2 [24] (TRAIL-R4 encoded by TNFRSF10D) are expressed at the cell surface. Albeit DcR1 lacks an intracellular domain, it is expressed on the cell surface, thanks to a GPI-anchor. DcR2, on the other hand is a transmembrane protein, but its truncated DD precludes the recruitment of the pro-apoptotic machinery and thus makes DcR2 unable to trigger apoptosis[16,25,26,27,28]. The two other antagonist receptors, OPG (osteoprotegerin) [29] and DcR3 [21] are secreted as soluble receptors in the extracellular compartment, and are thus unable to transduce cell death, including OPG which harbours two DD (Figure 2). All four antagonist receptors are capable of competing with TRAIL to inhibit apoptosis-induced by either DR4 or DR5 [16,17,30] (Figure 1 and Figure 2).
Like other members of the TNF superfamily, increasing evidence indicate that TRAIL can also, besides inducing cell death, display pleiotropic signalling activities ranging from cell differentiation [31,32,33,34], tumour progression, invasion to metastasis [35,36,37,38,39,40,41]. Although TNFRSF share both structural characteristics and signalling activation partners [42], and despite the fact that a large number of these receptors also share the ability to trigger similar signalling pathways, the modus operandi is not similar, involving distinct sequence of events, depending both on the considered receptor and ligand [43].
Likewise, and albeit much less efficiently than TNFR1, both TRAIL and Fas-ligand agonist receptors are able to trigger NF-kB signalling, leading in some cases to increased tumour growth and inflammation [44,45,46,47,48,49,50,51,52,53,54,55,56,57,58] (Figure 2). Moreover, increasing evidence indicate that soluble FasL or TRAIL may confer tumour cell resistance to apoptosis, contributing to pro-tumoral signalling or even inducing tumour cell growth[57,59,60,61]. For instance, seminal findings from Seamus Martin’s laboratory demonstrated that caspase-8, regardless of its proteolytic activity, serves as a scaffold for the formation of a FADD containing soluble complex recruiting RIPK1 and is necessary for NF-κB activation and pro-inflammatory proteins secretion, upon TRAIL stimulation [52,62,63]. The formation of this complex has been found to be directly controlled by the caspase-8 inhibitor c-FLIP [52] (Figure 2b).
Alternatively, although much less represented in the literature, other studies suggest that non-conventional ligand-to-receptor interactions may also exist, explaining how these agonist receptors may transduce non-apoptotic signalling pathways, such as the recently described soluble FasL/DR5 interaction, whose role during auto-antibody-induced arthritis has been associated with exacerbated inflammation in-vivo through regulation of NF-κB -mediated production of CX3XL1 [64].
While it is still unclear how these complexes are formed, these less studied, non-apoptotic signalling capabilities are likely to contribute to a large variety of human diseases. It is thus of utmost importance to study them, since only a better understanding of their mechanistic will allows us to develop novel therapeutic drugs to be tested in the clinic, to cure or at least alleviate patients suffering from autoimmune, inflammatory and cancer diseases.
We aim with this comprehensive review at discussing and at delineating the current understanding of the molecular events governing cell fate decision after TRAIL stimulation, with a special emphasis for non-apoptotic signal transduction.
2. The TRAIL System
2.1. TRAIL-Induced Cell Death
TRAIL was described for the first time as a pro-apoptotic ligand that induces apoptosis [1,2]. TRAIL is expressed as a cell surface protein, mostly by activated immune cells such as T and B cells [65], neutrophils [66,67,68], dendritic cells [69], monocytes and macrophages [70,71,72,73,74], natural killer and NKT cells (NK) [75,76,77,78,79,80,81,82,83,84,85]. TRAIL plays a crucial role both during viral clearance [86,87,88,89,90,91,92,93,94,95,96,97,98] and tumour immune surveillance [99,100,101,102,103,104]. Mechanistically, during innate immunity, NK cells and CTLs (cytotoxic T cells) promote apoptosis of target cells, either by releasing soluble factors such as the cytolytic granules [68,105,106,107], which contain perforin and granzymes, or by engaging membrane-bound death ligands like FasL or TRAIL [84,105,108,109,110,111,112,113].
Unlike FasL or TNFα [114,115], TRAIL induces apoptosis in tumour cells, selectively [116] and exhibits little to no cytotoxicity against normal human cells or mice cells [117,118,119,120,121,122,123]. Given that DR4 and DR5 are usually upregulated on cancer cells [124,125,126,127,128,129,130,131,132], and that TRAIL induces apoptosis in a p53-independent manner [133,134], contrary to most chemotherapeutic drugs [135], overcoming p53 escape [23], it has soon attracted major attention in oncology [136,137,138,139].
TRAIL binding to its two agonist receptors, DR4 and DR5, lead to the formation of homo or hetero multimeric complex on the cell surface, which in turn enable the recruitment of the adaptor protein FADD (Fas Associated via Death Domain) and the initiator pro-caspases-8 and/or -10, leading to the formation of the Death-Inducing Signalling Complex or DISC [48,140,141,142,143], in which the initiator caspase-8, like in the Fas DISC, is activated by mere proximity-induced dimerization [144,145,146]. Once activated this initiator caspase, self cleaves itself enabling it not only its free itself from the DISC, but also to reach and cleave substrates localized in the cytosol, such as the executioner caspases-3, -6 and -7 [147], which ultimately will concur in the execution of apoptosis, culminating in DNA fragmentation and the formation of apoptotic bodies [148].
Commitment to apoptosis upon TRAIL stimulation may further be regulated either by genetically regulated events, see below, or by cellular heterogeneity and stochasticity. Likewise, it has been demonstrated that random assembly of the receptors upon ligand stimulation [149], as well as intracellular or membrane-bound proteins stochastic distribution during cell division [150], may contribute to cell fate decision.
In the late 90′s two type of cells, found to rely or not on the activation of mitochondria, were described to transduce differentially apoptosis upon Fas ligand and TRAIL stimulation [151,152]. In type I cells, sufficient caspase-8 is activated to undergo apoptosis [153], regardless of mitochondria [154,155]. The intrinsic pathway is, however, required in type II cells, to fully transduce apoptosis upon TRAIL or FasL stimulation. Likewise, contrary to type I cells, mere loss of Bax expression [156] or overexpression of Bcl-2 anti-apoptotic members [153,157,158], is sufficient to abrogate the execution of apoptosis. Activation of the mitochondrial pathway by TRAIL receptors is mediated, in these cells, by a caspase-8-dependent cleavage of Bid [152,159], a BH3-only Bcl-2 family member, whose cleavage allows truncated Bid (tBid) insertion into mitochondrial membranes where it induces the translocation and oligomerization of Bax and Bak [160,161,162], inducing the release of cytochrome-c (Cyt-c). Once released from the outer membranes of mitochondria, cytochrome c forms, together with the initiator caspase-9 and APAF-1 (Apoptotic peptidase activating factor-1), the apoptosome complex [163,164,165], which allows the activation of the caspase-9 by mere dimerization [166] and which culminates in the activation of the executioner caspases (Figure 3).
Besides apoptosis, cell death induced by TRAIL may proceed through necrosis, in specific cell types or under certain conditions. Likewise, and similar to TNFα and FasL, TRAIL has been found, by a seminal work by the late Pr Jurg Tschopp [167], to induce necroptosis in the human jurkat T cell line, in a RIPK1-dependant manner, in the presence of a pan-caspase inhibitor or in the absence of FADD [167]. It was next found that at acidic extracellular pH (pHe), a condition that can be encountered in the tumour microenvironment (TME), TRAIL induced cell death proceeds through necroptosis. Likewise, mere acidification of the extracellular pH, in vitro, switches TRAIL-induced cell death from apoptosis to necroptosis [168], in a RIPK1-dependent manner [169]. The first inhibitor of this programmed inflammatory cell death, the necrostatine [170], was later found to inhibit RIPK1 [171]. RIPK1 is an integrator of cellular stimulation with protein kinase activity and scaffolding functions. RIPK1 is composed of a N-terminal kinase domain, an intermediary domain (ID), a C-terminal homology interaction motif (RHIM), and a DD. Owing to homotypic interactions, RIPK1 can be recruited to DD-containing receptors through its DD, and provided that it is not cleaved by the caspase-8 within the DISC [172,173], RIPK1 can recruit RIPK3 through the RHIM [174,175] and phosphorylate RIPK3 [176,177,178,179], forming the ripoptosome [180], which then phosphorylates and activates the pseudo kinase mixed lineage kinase domain-like protein (MLKL) [181,182]. Activation of MLKL leads to its oligomerization, translocation to the plasma membrane, forming large pores which engage ion channels to mediates ion influx, cell swelling, and plasma membrane rupture followed by the uncontrollable release of intracellular material [181,183,184,185] (Figure 4). Changes of pH naturally occur in the vicinity of tumour cells [186] as well as during ischemia [187]. The latter are, thus, likely to regulate TRAIL-induced cell death efficacy and modalities [188] and ultimately to affect immune antitumoral responses [189,190].
Last but not least, TRAIL agonist receptors have been found to induce cell death, in a ligand-independent manner, during unresolved unfolded-protein stress-induced response [191,192,193,194,195]. DR5 was found to serve as a receptor for misfolded proteins, explaining, at least in part, how apoptosis is transduced through this receptor during ER stress, in the absence of TRAIL [196]. Albeit it remains to be determined whether DR4 binds or not unfolded proteins, and despite the fact that most studies have focused on DR5, this second agonist TRAIL receptor has also been found to contribute to apoptosis during ER stress [63,193,194]. Moreover, although caspase-8 is involved during DR4- and DR5-mediated ER-stress-induced cell death [192], it has also been found, associated within an atypical platform devoid of DR4 and DR5, to be required for ER-induced apoptosis in an osteosarcoma cell line [197]. Yet, it has also been reported that TRAIL agonist receptors or caspase-8 are negligeable in some cases, such as in B-cell malignant cells or the colorectal cancer cell line HCT116 [198,199].
2.2. Comparison of the Proximal Regulatory Mechanisms Governing TRAIL-Induced Cell Death with Other TNFRSF Members
TNFα was the first ligand of the TNFSF superfamily tested for its anti-tumoral activity [200,201], followed by Fas-ligand [115,202]. While Fas-ligand [14,203,204] and to a much lesser extend TNFα, due to the requirement of protein synthesis or transcription inhibitors [205,206,207], are efficient in killing a variety of tumour cells, these ligands cause significant damage to normal tissues that result in life-threatening toxicities [116]. Despite the fact that TRAIL, TNFα and Fas share common pro-apoptotic partners and modalities, solely TRAIL displays tumour selective pro-apoptotic activity, sparing normal tissues or cells [116,123], including when administered to small animals or humans [122]. Administration of Fas or TNFα in rodents, on the other hand, is lethal [115,208,209,210]. Moreover, TNF is involved in sepsis-mediated organ failure due to cellular toxicity [200,211,212].
Unlike TNF-R1 [213], engagement of apoptosis induced by DR4, DR5 or Fas is primarily initiated directly from the plasma membrane, through the formation of a complex coined Death-inducing signalling complex (DISC) [140,141,143] after TRAIL or Fas ligand binding to their respective cognate agonist receptors (Figure 2). TNFR1 membrane-bound complex, on the other hands, triggers a NF-kB-dependant survival pathway on the first instance, without recruiting FADD nor the caspase-8, due essentially to the recruitment of the kinase RIPK1 [214,215,216] and the adaptor protein TRADD [217,218]. The group of David Goeddel in the late 90′s provided the first molecular demonstration that divergent signalling complexes could lead to distinct and antagonist signalling pathways [217]. Albeit FADD and caspase-8 have long been known to be required for TNF-induced apoptosis [219,220], the molecular comprehension of their temporal and spatial contribution was unveiled, almost a decade later, by the discovery that a secondary complex was required to initiate apoptosis. Complex II is a soluble scaffold multimeric protein complex which arises from complex I [213]. It contains, amongst others, the adaptor protein FADD, the cysteine protease caspase-8, as well as the post-translationally modified forms of RIPK1 and TRADD, whose modification is primarily initiated in complex I [213]. Transition from complex I to complex II, albeit still not fully understood, was later on found to involve two proteolytic steps, starting first with the shedding of TNFR1 extracellular domain by TACE (TNF-Alpha Converting Enzyme), also known as ADAM17 [221], and leading to the internalization of complex I through a clathrin-dependent mechanism, followed by an additional cleavage within TNFR1 transmembrane domain, by the γ-secretase, allowing the release of its intracellular domain, which contains bound TRADD, TRAF2 and RIPK1 amongst others proteins [221]. The release of complex I to the cytosol, in turn, subsequently allows the recruitment of FADD and caspase-8, forming the pro-apoptotic TNFR1-complex II (Figure 2).
Regardless of the modus operandi required for engaging cell death by these receptors, the latter have been found to form dimers or trimers, due to spontaneous self-association of their N-terminal extracellular domain, called pre-ligand assembly domain (PLAD) [222,223], which is generally present in the first cysteine-rich domain of some TNFSFRs (Figure 1). By favouring ligand-independent receptor multimerizations, the PLAD was both find to limit apoptosis induced by TRAIL due to the homodimerization of DR5 [224], or to the formation of heteromeric complexes DR4, DR5, DcR1 or DcR2 [16,30,225]. These self-association motifs have recently been demonstrated to be targetable. Interestingly it was found that, mere administration of a TNFR1 PLAD-Fc recombinant protein improves skin lesions in MRL/lpr [226], arthritis [227], as well as experimental autoimmune encephalomyelitis or diabetes [228], in experimental animal models.
Organization and arrangement of TNFRSF in homo- and heteromeric complexes into higher-order complexes has profound effect on their signalling capabilities [42,229,230] and is often required for efficient apoptosis triggering, as demonstrated with DR5 [231,232,233]. Likewise, it has been proposed that DR4, DR5 and Fas form, first of all, upon cognate ligand binding, trimer complexes whose multimerization or crosslinking with neighbouring trimers occurs via the dimerization between receptor interfaces, either located opposite the ligand-binding interfaces, resulting in a hexameric honeycomb-like structure [234]. A dimerization motif found in the transmembrane helix domain of the receptors is also suspected to play an important role for the assembly of the DISC, its stability and potency [231,234,235]. Moreover, as suggested for Fas, DISC stability may also be regulated at the level of the cytoplasmic domain of some agonist receptors by the adaptor protein FADD [236,237,238].
Furthermore, in line with the fact that most TNFSF receptors harbour putative glycosylation sites, it has been demonstrated that O- and N-glycosylations, post-translational modifications, also regulate TNFRSFs pro-apoptotic signalling transduction [239,240]. Likewise, based on the observation that TRAIL sensitivity in cancer cells was associated with high glycosylation profiles, the seminal work of Avi Ashenazi’s laboratory, provided the first molecular demonstration that DR5-mediated TRAIL-induced cell death could be regulated by the O-glycosylation [241]. While it remains to be determined whether O-glycosylation affects other receptors of the family [242], receptors such Fas, TNFR1 or DR4 were found, on the other hand, to be N-glycosylated [243,244,245,246,247]. This post-translational modification of DR4 or Fas increases cancer cell lines sensitivity to TRAIL- or FasL-induced cell death, respectively [243,245]. Similar gain of function associated with the fly tumour necrosis factor (TNF) receptor homolog glycosylation were demonstrated [248]. It shall be noted, however, N-glycosylation, on the other hand, was found to prevent TRAIL-induced cell death in normal mouse fibroblastic cells [244], suggesting that the increase in signal transduction induced by TNFRSFR mediated by their O- or N-glycosylation, maybe restricted to cancer cells. Regardless, it has been demonstrated that the gain of function associated with the O- or N-glycosylation of these agonist receptors, with the exception of one study [248], is not related to a change in ligand binding to its cognate receptor, but rather a stabilization of the membrane-bound primary complex, likely mediated by an increase in receptor aggregation, that ultimately leads to a better signalling activity, which in the case of Fas or TRAIL is associated with an increase in caspase-8 activation [241,243,245,249,250,251]. Consistent with this, glycan modifications or glycan-binding proteins were found to enhance or impair apoptosis induced both by TNFR1, FasL and TRAIL [242,250,252,253,254,255,256,257,258,259,260,261,262]. These post-translational modifications shall be distinguished from the O-GlcNAcylations or O-GlcNAc, as contrary to the O- or N-glycosylation, O-GlcNAc takes place within the cytosol, and shall thus affect the C-terminal cytosolic domains of TNFRSFs. Likewise, there have also been reports demonstrating that GlcNAcylation of both DR4 or DR5 C-termini, could be required for, or enhance, DISC formation and receptor clustering [249,263,264]. On the other hand, O-GlcNAc of death-domain containing proteins, has also been demonstrated to protect cells, infected by pathogens, from apoptosis induced by TNFRSF-death-containing receptors [265,266,267], and to protect erythrocytes from necroptosis by targeting RIPK1 [268]. Another intracellular post-translational modification may also affect death-domain containing receptor localization, aggregation and function. Likewise, it has been found that palmitoylation of DR4, Fas and TNFR1, but not DR5, enhances apoptosis induced by TRAIL [269] and Fas ligand [270,271,272] and is required for TNFR1 signal transduction [273].
3. Physiological and Physiopathological Functions of TRAIL
TRAIL exhibits pleiotropic physiological functions which are regulated by its cognate receptors due to their ability to trigger or not cell death. TRAIL and its receptors play an important role in maintaining tissue homeostasis [274,275,276,277,278]. Through transducing cell death, TRAIL and its agonist receptors are most notoriously known for their ability to kill cancerous cells and cells infected by viruses [91]. Yet, a tremendous amount of work also suggests that TRAIL and its receptors are also likely to play a role in several human diseases including, but not limited to, obesity and diabetes[279], associated with inflammation [32,63,280], neurological disorders [281] or cardiac diseases [282].
3.1. In immune System
In the immune system, TRAIL helps maintain lymphocyte homeostasis. Likewise, while activated CD8+ cells were described to be more sensitive than CD4+ T cells to TRAIL-induced cell death [283], CD8+ T cells can protect themselves from apoptosis induced by TRAIL by up-regulating both the antagonist receptors and c-FLIP [77,284]. Variation of TRAIL sensitivity, in CD8+ T cell blast, is both time- and stimuli-dependent, explaining TRAIL’s ability to actively contribute to CD8+ T cell AICD and to generate memory-like CD8+ T-cells [285,286,287,288,289,290,291]. Interestingly, using experimental animal models, TRAIL was found to inhibit autoimmune lymphoproliferative syndrome as well as spontaneous idiopathic thrombocytopenia purpura, due to its active contribution during activation induced cell-death (AICD) [290,292].
Besides its role in adaptative immunity, TRAIL plays an important role during in innate immunity [293], such as in anti-tumour immune surveillance [80,99,101,293,294]. TRAIL is often instrumental for the cytotoxic activity of immune cells. It is upregulated and contributes to the cytolytic activity of T cells, neutrophiles or monocytes stimulated by type I interferons [71,72,284], or after stimulation with IL-2 plus phytohemagglutinin [65], contributing to their anti-tumoral activity. TRAIL expression can also be induced in plasmacytoid dendritic cells by microbial or viral products such as LPS or Toll receptor agonists, contributing to their cytotoxic activity [295]. TRAIL is also thought to contribute to ocular [296] and placental immune privilege [297].
A recent study analysing the immune repertoire, in TRAIL-deficient mice, found organ-distribution differences of several types of immune cells, such as dendritic cells, in these animals as compared to parental mice [298]. Keeping in mind that CD8+ T cells were recently found to contribute to tissue remodelling [299] and that TRAIL can be expressed by a large number of immune cells, as mentioned above, including CD8+ cells, these studies collectively suggest that TRAIL may play a wider role in the immune system than expected. Indeed, growing evidence suggests that TRAIL non-apoptotic functions may also play a role in shaping and orchestrating the immune response to pathogens or cancer cells. TRAIL has for example recently been demonstrated to inhibit IL-15-induced cytotoxic granule granzyme B production in NK cells during viral infection, limiting viral clearance [91]. By regulating inflammation, in the absence of apoptosis, TRAIL can also contribute to the dysregulation of the immune system. Likewise, using TRAIL-R-deficient mice, it was found that TRAIL, by inhibiting T cell activation, supresses gut inflammation [300] or arthritis [301,302], in an apoptosis-independent manner [303]. Injection of TRAIL itself was also found to be beneficial in experimental animal models to inhibit autoimmune thyroiditis [304] or arthritis [305]. Suppression of auto-immunity by TRAIL, can proceed both through caspase-dependent and independent manner, as it was shown that TRAIL can on the one hand inhibit Th1 cells proliferation and on the other promote that of regulatory T cells, as demonstrated in TRAIL- [306] and TRAIL-R- deficient mice [301]. TRAIL deficient mice also unveiled the critical role of TRAIL in supressing experimental autoimmune encephalomyelitis [307]. In a remarkable way, TRAIL functions in autoimmune diseases by transducing both canonical and non-canonical signalling pathways, holding promises in autoimmune therapy [308,309]. Yet in other instances, TRAIL has also been found to trigger inflammation and/or to amplify other autoimmune diseases such as lupus erythematosus [310] and lupus nephritis [311].
3.2. In Diseases
TRAIL is associated with diseases beyond of the immune system. Likewise, TRAIL may play a physiological role in endothelial cell function [316], since it has been found to exhibit a pro-angiogenic activity [317,318] and to stimulate the proliferation of vascular smooth muscle cells [319]. In another study, TRAIL, on the contrary, was shown to inhibit angiogenesis-mediated by VEGF, through both a caspase-8-dependent and -independent manner [320]. In vivo, however, it was found, using Trail-/- mice, that TRAIL is able to promote angiogenesis and neovascularization after ischemia [321]. In the same line, an increasing number of studies also indicate that TRAIL could be involved during cell differentiation. Likewise, TRAIL induces the differentiation of intestinal cells [31], osteoblasts [322,323], skeletal muscle or myoblast cells [34,324] or keratinocytes [325], but appears to inhibit adipocyte differentiation [326].
TRAIL has also been described in lung and heart diseases. TRAIL induces survival, proliferation, and migration of human vascular smooth muscle cells (VSMC) in Pulmonary arterial hypertension (PAH) [327,328,329]. Its high expression levels in the serum of PAH patients correlates with the severity of the disease [329]. Through non-canonical signalling TRAIL promotes VSMC and fibroblasts proliferation and migration through ERK1/2 MAPK and the Serine/Threonine Kinase Akt activation, without affecting p38 MAPK or c-Jun N-terminal kinases (JNK) activation [330]. TRAIL stimulates proliferation of VSMC after Insulin-like growth factor-1 receptor (IGR1) regulation through NF-κB activation [319]. In addition to VSMC, TRAIL promotes survival and proliferation of primary human vascular endothelial cells, as well after Akt and ERK activation without affecting NF-κB pathway [331]. Activation of NF-κB in vascular smooth muscle cells by TRAIL has also been described to require the cleavage of protein kinase C-delta (PKC-δ) by caspases [332].
TRAIL and its three receptors, DR4, DR5 and DcR1, are highly expressed in human heart [127], and while cardiomyocytes express DR5, they are resistant to apoptosis, yet after TRAIL stimulation DR5 transduces the activation of the ERK1/2 pathway, in these cells, in a MMP-EGFR-dependent manner, as described by Panner et al. [333]. It has been proposed that TRAIL, by inducing the production of MMPs trigger the cleavage of the Epithelial Growth Factor Receptor ligand (HB-EGF) in the cell membrane to induce EGFR signalling, which promotes cardiomyocyte proliferation and ERK 1/2 signalling [333].
TRAIL pro-apoptotic or non-apoptotic signalling is also suspected to contribute at some extent to Alzheimer’s disease [334,335,336,337,338], and non-alcoholic fatty liver disease [339,340,341,342]. Like cancer cells [52,63,343], the molecular mechanisms driving TRAIL-induced non-apoptotic signalling, including cell motility in normal cells remain poorly understood.
4. Signalling Machinery Associated with TRAIL Non-Canonical Transduction
TRAIL, as reported in a growing number of studies, triggers the differentiation, proliferation or survival of normal cells, such as macrophages [32,322], intestinal mucosal cells [31], Skeletal myoblasts [324], keratinocytes, osteoclasts [323,344], vascular smooth muscle cells [34,330,331,345] or mouse fibroblasts [346].
In cancer cells, on the other hand, if apoptosis is not efficiently triggered, TRAIL can be detrimental to patients given that this cytokine also exhibits pro-tumoral properties, associated with TRAIL’s ability to induce inflammation, tumour cell motility and invasion, ultimately leading to metastasis [35,38,39,43,193,347,348,349,350]. Likewise, TRAIL was found to promote the proliferation in human glioma cells through ERK1/2 phosphorylation and the stabilization of the long form of c-FLIP(L) [351], in cholangiocarcinoma cells via NF-kB [40]. Migration and invasion were also promoted by TRAIL in NSCLC the A549 cell line in a RIPK1-dependent manner through phosphorylation of Src and STAT3 [39], in pancreatic ductal adenocarcinoma [38], in colorectal cancer cells, resistant [349] or not [193] to TRAIL-induced cell death, and in the triple negative breast cancer cell line MDA-MB-231 (TNBCs) [193]. In oesophageal squamous cell carcinomas (Figure 5), TRAIL induced epithelial-mesenchymal transition (EMT) and metastasis through ERK1/2 and stat3-dependent upregulation of PD-L1 [352]. PD-L1 regulation through ERK phosphorylation induced by TRAIL was also reported in TNBCs [294]. Using a TNBC xenograft model, TRAIL was also demonstrated to promote skeletal metastasis [350]. Consistent with these findings, deletion of murine TRAIL-R, in a non-small-cell lung cancer (NSCLC) and pancreatic ductal adenocarcinoma (PDAC) using a KRAS-driven experimental model, was found to drastically impair metastasis, and this effect was associated with a loss of cell migration, proliferation, and invasion [35].
Mechanistically, TRAIL was shown to induce NF-κB activation[48,353,354] and by analogy with TNFR1 signalling [213], albeit in a distinct manner, it was next found that TRAIL could lead to the formation of two main distinct molecular complexes, explaining, at least in part, how TRAIL receptors can transduce cell death or pro-inflammatory pathways [39,43]. The primary pro-apoptotic complex, known as TRAIL DISC, is mostly composed of the TRAIL receptors, FADD, caspase-8 or -10 and the inhibitor c-FLIP, and is localized at the level of cellular membranes [16,140,143,355,356]. RIPK1 is also present in this complex [52,357] as well as TRADD [48,353,358,359], albeit there might be some differences in TRADD binding to TRAIL receptors, given that TRADD seems to be preferentially recruited to DR4 [48,353,357]. In addition to these adaptor proteins and kinases, originally found to compose TRAIL membrane-Bound complex, kinases such as IKKα, IKKβ and IKKγ, recruited to complex I, explaining how NF-κB may be induced by TRAIL [360]. Native recruitment of ubiquitin ligases can also happen in the TRAIL DISC as demonstrated with the presence of the linear ubiquitin chain assembly complex LUBAC (Figure 2), whose components SHARPIN and HOIP limits TRAIL-induced cell death as well as NF-κB activation [360,361,362], due to RIPK1 and FADD linear ubiquitination [360]. Moreover, other proteins such as c-IAPs, A20 and TRAF-2 are also recruited in complex I [360].
The secondary non-apoptotic complex, on the other hand, is found in the cytosol, albeit it arises from complex I [361] (Figure 3). Complex II contains not only FADD and caspase-8, but also RIPK1, TNF receptor-associated factor 2 (TRAF2), TRADD, as well as a large number of apoptosis inhibitors, NF-κB regulators, including IKK and NEMO [43], not to mention LUBAC [360] (Figure 4). It must be stressed here that RIPK1 can not only be directly recruited to TRAIL receptors, as evidenced in native complex I [360,363,364], because it contains a death-domain [365], but that the latter is required for TRAIL-induced NF-κB activation [366]. Of interest, similar to Fas DISC [173], membrane-proximal localization of RIPK1 allows its cleavage by the initiator caspase-8 within its intermediary domain, abolishing TRAIL-induced NF-kΒ activation [363,364].
Given that RIPK1 is recruited to the TRAIL DISC and present in the cytosolic complex II, it is easy to understand how TRAIL triggers the NF-κB pathway. Yet, as demonstrated by Azijli and co-workers, more than 10 years ago, in the TRAIL-resistant cancer cell line A549, TRAIL also induces besides NF-κB, the phosphorylation of a large number of substrates associated with activation of the P38, ERK1/2, JNK1, Src, AKT, Raf1 and ROCK [367]. While the implication of TRADD for TRAIL signalling is less investigated, TRADD was found to afford protection against TRAIL-induced apoptosis [358,368,369], but more interestingly TRADD could play an important role in the secondary complex to induces IL-8 secretion in NSCLC, under TRAIL treatment [370]. Furthermore, TRADD and RIPK1 redundantly mediate pro inflammatory signalling in response to TRAIL in human ovarian HeLa metastatic cell line [357]. Despite the fact that several experimental evidence link for example ERK1/2 activation in glioma cells with c-FLIP [351] or JNK activation with RIPK1 [366], it remains unclear how upstream kinases are integrated and activated in the molecular platforms triggered by TRAIL, whether it be complex I or complex II.
Evidence accumulates demonstrating that TRAIL can be detrimental in oncology due to its ability to promote cell migration and metastasis, but it still remains unknown, however, whether both TRAIL agonist receptor trigger similar non canonical signalling activity. Contrary to rodents[8], primates express two TRAIL agonist receptors [1,4,6], and therefore findings obtained from genetically modified mice may not always transpose to primates. For instance, with the exception of one study [371], migration and metastasis promoting TRAIL’s activity seem to be mostly associated with DR5 [35,39,193,350]. While it remains unclear whether this peculiarity is due to DR5 splice variants or not [372], DR5 is found to be overexpressed in several cancer types and this overexpression is often associated with tumour aggressiveness and poor patient prognosis [373]. For example, DR5-positive staining is associated with increased risk of patient death in non-small cell lung cancer [126], breast [374] and renal cancer [375].
Activation of this non-canonical signalling pathway by DR5, which promotes tumour growth and metastasis through MAPK, PI3K/AKT or NF-κΒ signalling, is likely to be only visible in TRAIL-resistant cancer cells [39,349], including cell expressing TRAIL decoy receptors [27,376]. Alternatively, transition of the receptors once engaged with the ligand to membrane lipid rafts, may as demonstrated for TNF[377], contribute to induction of the pro-migratory signal. It has been suggested for example, that lipid rafts may provide an adequate membrane platform for aggregation for DR4/DR5 to transduce apoptosis [378]. Localization to lipid raft may be differentially occurring depending on the receptor and its potential palmitoylation status. Likewise, DR4 can be palmitoylated, translocating to lipid raft, where it was proposed to form and activate the pro-apoptotic complex I [269]. In B-cell hematologic malignant cells, DR4 was even proposed to be constitutively localized within lipid rafts [379]. Albeit DR5 was not found to be palmitoylated, it has also been described in lipid raft and described to recruit and activate the caspase-8 in these subcellular compartments [378,380,381,382,383]. However, while there is no doubt that TRAIL complex I may transit to lipid rafts, native TRAIL DISC formation in these lipid rich structures have never been demonstrated. On the contrary, it was found that TRAIL DISC-mediated activation of the initiator caspase-8, which is required for initiating apoptosis, rather occurs in non-lipid rich membranes [16,384]. Nonetheless, it cannot be excluded that transient translocation to lipid raft may account for TRAIL pro-tumoral properties.
4.1. Lessons from Fas/CD95 Induced Non-Canonical Signalling (Secondary Complex)
Non-canonical pro-motile and pro-metastatic signalling was also documented for Fas, a receptor of the TNF superfamily which like DR4 and DR5 is able to engage apoptosis from the membrane in a FADD- and caspase-8 dependent manner [385]. Fas ligand (FasL) was found to redistribute its agonist receptor Fas dynamically into lipid rafts, contributing to the elimination of activated T cells [386]. Lipid rafts were, thus, soon considered as possible check point controls for FasL-induced Fas signalling cellular outcome [387,388]. Like TRAIL, but to a much lesser extent than TNFα, FasL is also able to transduce NF-κB, regardless of its ability to trigger apoptosis [53,389]. NF-κΒ activation by FasL was associated with resistance to apoptosis in cancer cells [44], but also appeared to be associated, in addition, to cell motility and invasiveness [57]. It was also demonstrated that naturally cleaved FasL could induce cell migration [390,391,392]. Fas was found to induce proinflammatory cytokines in human monocytes [54,393]. In dendritic cells, Fas stimulation induce IL1β and IL-12 production and cell maturation [394].
Mechanistically, it remains unclear how Fas induce cytokine production or how it activates its pro-metastatic signalling pathway. FasL-induced cell motility and invasion has been associated with TRAF2 [395], PDGFR-β-mediated PLC-γ1 activation and PIP2 hydrolysis [396], activation of the kinase c-Yes and AKT and changes in cytosolic calcium [390], Rac1 [397], or through phosphorylation of Rock1 and involvement of the Na+/H+ exchanger NHE1 [392].
TRAF2 is recruited within the TRAIL DISC [360,398]. By allowing recruitment of ubiquitin ligases within the primary complex TRAF2 is able to limit caspase-8 activation [360,398,399]. TRAIL-induced JNK activation was found in cancer cell lines to require RIP and TRAF2 [400], suggesting that many of the non-canonical signalling pathways may be readily engaged from complex I. Alternatively, it has recently been proposed that NF-κΒ-mediated initiation of inflammation upon TRAIL stimulation may be induced, at least in part, through TRAF-2-mediated recruitment of cIAP1/2 and LUBAC into complex I, leading to the formation of a secondary complex coined ‘‘FADDosome’’ in which RIPK1 undergoes linear ubiquitination, allowing assembly of the NF-kB machinery and NF-κΒ-dependent regulation of inflammatory cytokines and chemokines [62] (Figure 5).
Linear ubiquitination and stabilization of the NF-κB signalling by LUBAC was first uncovered in TNFR1 complex I and found to rely on TRADD, whose absence precludes both TRAF2 and LUBAC recruitment to TNFR1 [401], consistent with the need of TRADD to induce NF-κB activation by TNFR1 [218] and to allow TRAF2 recruitment to TNFR1 [217]. Within the Fas DISC, the caspase-8 inhibitor c-FLIP was also found in the early days as a protein that could integrate at TRAF2, to induce both NF-κB and ERK signalling [402,403]. Keeping in mind that TRADD could be essential too, for TRAIL-mediated non-apoptotic signalling, including induction of NF-κB [357,369], it is worth mentioning that TRADD is found both associated with TRAIL receptors membrane complex I [353,360] and soluble complex II [43]. An alternative molecular circuitry may explain the biological activity of TRAF2 in driving TRAIL pro-tumoral effects. Likewise, it was described that NF-kB activation by TNFR1 requires sphingosine-1-phosphate (S1P). S1P interacted with TRAF2 as a co-factor to catalyze RIPK1 poly-ubiquitination and NF-κB activation [404]. Given that S1P may be critically linked to metastasis [405,406], it may be worth considering, in addition, the interesting work demonstrating that deletion of DR5 induce cell motility and promotes cell invasion in a TRAF2 and S1P-dependent manner, through activation of the JNK/AP-1 pathway in lung cancer cells [371,407] (Figure 4).
Direct recruitment of kinases associated with non-apoptotic Fas signal transduction as also been found, including Rac1 activation after binding to Fas membrane proximal domain (MPD), located in the intracellular part of the receptor, during neurite growth [397]. Albeit not characterized molecularly, TRAIL-induced cell motility was also associated with Rac1 activation in monocytes [408] and HeLa cells [409]. Interestingly, though, while Rac1 appears dispensable for the regulation of inflammatory proteins after TRAIL stimulation [410], Rac1 was required for DR5-mediated cancer cell motility and metastasis [35], and similar to Fas, the MPD of DR5 was also required to trigger this effect. (Figure 5). Rac1 was found to be directly recruited to DR5 [35], and consistent with mutated KRAS’s ability to inhibit ROCK1 [411], ROCK1 inhibitors allowed Rac1 recruitment to DR5 and transduction of a signalling pathway leading to invasion in non-mutated KRAS cells [35]. It is thus likely that direct recruitment of RAC1 into the TRAIL DISC may, due to its ability to promote filopodia and lamellipodia formation, lead to microtubules and cytoskeleton organization [412], accounting for the cell migration induced by DR5 [35] (Figure 5).
4.1. Calcium Signalling Inducing Cell Motility and Metastasis
Calcium signalling induced by ligands of the TNF family has initially been addressed with TNF [413] and FasL [414]. Increased cytosolic Ca2+ was found to occur almost immediately after stimulation, within the first 50 seconds. High calcium levels have been recorded after stimulation by FasL following activation of phospholipase C γ1 (PLCγ1), inositol 1,4,5-trisphosphate (IP3) generation, IP3 receptor (IP3R) calcium ionic channels stimulation and a late secondary Cytochrome-c-triggered activation of endoplasmic reticulum (ER)-resident calcium channels [415]. The role of Ca2+ in cancer cell proliferation, migration, and invasion has been well established [416]. Likewise, Ca2+ signalling is a potential key regulator for breast cancer bone metastasis and prostate cancer cells proliferation, angiogenesis, EMT, migration, and bone colonization [417]. Interestingly, both TRAIL- and FasL-induced pro-metastatic pathways are associated with an early increase in intracellular Ca2+ and tyrosine kinase signalling [193,418,419]. The use of isogenic stable cancer cells deficient for either DR4 or DR5 [193], demonstrated that TRAIL-induced pro-metastatic signalling was solely triggered by DR5 and correlated with a rapid Ca2+ flux [193,420,421]. Furthermore, early increased cytosolic Ca2+ was shown to be activated upon TRAIL exposure in both Jurkat and NB4 leukemia cells, protecting the latter from apoptosis [421]. It was found in these cells that recruitment of both p62 and ATG7 to complex I was required for calcium influx induced by TRAIL [421].
Like TRAIL, FasL also induces an increase of cytosolic Ca2+, associated with cell-motility and metastasis [390,418,422,423,424]. Intracellular increase in Ca2+ is generally induced by PLCγ1 and IP3R activation, due to ER Ca2+ release [425], but may also be triggered, as demonstrated in leukemia cells, after ORAI1 activation and CRAC channels opening [421]. Autophagy Related 7 (ATG7) [426] and Sequestosome 1 (p62/SQSTM1) [427], are two autophagic proteins related to ORAI1 and CRAC channels, whose recruitment to DR5 induce the release of Ca2+ from the ER [421]. Keeping in mind that DR5 is also involved during apoptosis induced during the ER stress and that this process is associated with Ca2+ release [191,196], while DR5, but not DR4, is able to induce a change in calcium flux after TRAIL stimulation, these findings suggest that calcium regulation is probably important for the triggering of TRAIL-mediated non-apoptotic signalling. Indeed, FasL also can induce high intracellular levels of Ca2+ ions to promote, depending on the context and cancer cell type, apoptosis or non-canonical signalling [415]. How Fas or DR5 trigger these changes in intracellular calcium remain unknown. However, in two studies performed using breast cancer models DR5 was proposed to directly interact with a protein which has a calcium dependent activity, the calmodulin (CaM) [428,429] (Figure 5 and Figure 6). CaM is a small Ca2+ binding protein that interacts with a large group of intracellular proteins and which participates in signalling pathways that regulate proliferation and motility [430,431]. In PDAC cells, CaM was also found to be recruited in the DR5 DISC together with c-FLIP and the proto-oncogene Src, contributing to cell resistance [432]. In NSCLC cells, CaM inhibition or Ca2+ deprivation inhibited the recruitment of Src and was associated with an increase in c-FLIP short degradation, sensitizing cells to DR5 agonist-induced apoptosis [433]. Src could play a role during TRAIL-induces non-canonical signalling [39], given that Src was described, in addition, to phosphorylate and, thus, to inhibit caspase-8 enzymatic activity [434]. Furthermore, CaM may allow recruitment and activation of the Src [435]. Interestingly, CaM has also been found to be recruited within the Fas DISC [424,436,437], and associated with the regulation of Src pro-tumoral activity [424,435]. Last, caspase-8, alone, was found to bind to the Focal Adhesion Kinase (FAK) and Calpain-2 Ca2+ dependent protease (CPN2), displaying, thus, pro-metastatic function properties in glioblastoma cell lines [438], (Figure 6).
4.2. Nuclear DR5 Regulates Both Proliferation and Metastasis
In other studies, regulation of TRAIL’s pro-tumoral signalling has been suggested to be due to the subcellular compartmentalization of DR5 in the nucleus [439,440]. It is not clear how DR5 goes to the nucleus, but it has been proposed that DR5 may undergo proteolytic cleavage or internalization upon ligand binding, allowing its translocation into the nucleus [441,442,443]. Interestingly, mostly DR5 but not DR4 is found in nuclear compartment in late cancer stage of NSCLC [440], pancreatic [444], and breast cancer [445]. DR5 harbours two nuclear localization signals (NLS) sequences which promote importin-β1 binding and nuclear translocation of the complex, limiting thus TRAIL-induced cell death sensitivity [442]. In the nucleus importin-β1/DR5 was found to regulates the micro-RNA let-7 maturation and to promote tumour cell proliferation [444].
Mature let-7 is known to control cell proliferation by inhibiting its targets, such as, the High mobility group AT-Hook protein-2 (HMGA2) and the Lin-28 homolog-B (Lin28B) protein expression. Upregulation of HMGA2 and Lin28B enhance cell proliferation and malignant progression [446,447,448,449] (Figure 4). HMGA2 and Lin28B are two proteins overexpressed in embryonic tissues and downregulated in differentiated tissues because of low expression of let-7. Let-7 overexpression prevent cell transformation in epithelial cells [450]. Furthermore, knockdown of DR5 using shRNA results in increased levels of mature let-7, consequently in reduced abundance of let-7 targets, which induce cell proliferation in pancreatic cancer cells [444]. Interestingly, knockdown of DR5 in metastatic breast cancer cells decrease bone homing and early colonization to the bone marrow and induce E-cadherin overexpression which contraries EMT in xenograft mice model [350]. Impaired cell migration was linked to decreased CXCR4 expression [350] and increased E-cadherin expression [451]. CXCR4 selectively binds the CXC chemokine stromal cell-derived factor-1 (SDF-1), also known as CXCL12, and plays a crucial role in several biological processes, including in cancer biology, where it was associated with tumour dissemination and metastasis [452]. CXCR4 is a marker of breast cancer cells poor prognosis. High CXCR4 expression is significantly correlated with lymph node status, distant metastasis, and poor survival [453]. Interestingly, nuclear DR5 regulates CXCR4 expression through inhibiting let-7 maturation [41,350], leading, as a consequence, to the expression of HMGA2 and CXCR4, and bone metastases formation of breast primary tumours [350,444,454] (Figure 4). All these findings suggest that nuclear DR5 may also play an important function in tumour aggressiveness. Yet, whether translocation of DR5 to the nucleus is fast enough to explain and concur to calcium-mediated pro-motile and metastatic signalling after TRAIL treatment, remains to be determined?
4.3. Caspase-8 Contribution in TRAIL Non-Canonical Signalling
Caspase-8 and FADD are required for TRAIL to induce apoptosis and are both recruited to TRAIL DISC upon TRAIL treatment [140,141], but recent evidence suggest that they may also contribute to TRAIL non-canonical signalling. Likewise, caspase-8 has been reported to be recruited to a FADDosome complex, whose formation after TRAIL stimulation is associated with cell proliferation and/or migration [62]. Interestingly, mutations of caspase-8 in head and neck squamous cell carcinomas represent almost 9% of the cases, and three out of the four mutations examined in Li’s study conferred caspase-8 with pro-motile and pro-invasive properties [455]. Moreover, phosphorylation of caspase-8 on tyrosine 380 by the Src kinase, which inhibits its aspartate protease activity and, thus, protect cells from TRAIL-induced cell death [434], was associated the likelihood of a regulation of caspase-8 functions, switching its pro-apoptotic activity to cell migration by SH2 kinases [456,457]. Caspase-8 Y380 residue was described to be essential for caspase-8 relocalization to lamella of migrating cells [458]. Src-induced phosphorylation of caspase-8 on Y380 was also found to drive the assembly of a soluble complex, containing IKKα, IKKβ and p65, that tiggers NF-kB activation in glioblastoma cells, leading to inflammation and angiogenesis [459].
Caspase-8 has been described to interact with p85α, subunit of PI3K to activate Rac1 through lipid products generation (PIP2 and PIP3) that activate guanine nucleotides-exchange factors (GEFs), [460] which are necessary to Rac1 activation [461]. In Neuroblastoma cell lines caspase-8 pro-migratory signalling capability was associated with its ability to interact with the focal adhesion kinase (FAK) and calpain 2 (CPN2) [462], two components of the focal adhesion complex (FAC) [438] (Figure 6). FAC is a signalling complex anchored by cell actin cytoskeleton, membrane integrins and extracellular matrix (ECM). This complex is known to contain many cytosolic proteases, phosphatases, and kinases, including the FAK, a key effector of metastasis [463]. Cytoplasmic phosphorylated FAK induce cell migration and invasion, cytoskeleton organization and EMT through FAC protein elements activation, like PI3K, Src and Rho [464]. Caspase-8 interacts with components of the FAC in a tyrosine-kinase dependent manner, promoting both cell migration and metastasis [456,464,465]. Of interest, it was also found that FADD, by inhibiting miR7a expression, is associated with an increase in FAK and spontaneous invasion and metastasis of the melanoma cell line B16 [466]. The increase in FAK overexpression, induced by a FADD-mediated downregulation of miR7a, leading to the expression of CCL5 and TGFβ expression, two cytokines involved in triggering metastasis [466,467] (Figure 6). Last, but not least, caspase-8 pro-motile and metastatic signalling has also been associated with its ability to promote Rab5-mediated internalization and recycling of β1 integrins [468,469].
Consistent with the findings described above and the work of Henry et al. [62], indicating that both FADD and caspase-8 may account for TRAIL non-apoptotic signalling, is the demonstration, in rheumatoid arthritis fibroblast-like synoviocytes, that caspase-8 is responsible for the cellular migration of these synoviocytes stimulated with PDGF, regardless of its enzymatic activity [470].
4.4. TRAIL Induce Cancer Metastasis after uPA and c-cbl Regulation
TRAIL was found to enhance inflammation and promote invasion of PDAC cells in vitro and metastasis in vivo by inducing the up-regulation of the urokinase-type plasminogen activator (uPA), IL-8 and CCL2 [38]. uPA is an agonist of the urokinase-type plasminogen activator receptor (uPAR) which can induce metastasis [471]. It has been found to be involved in triggering FasL-induced invasiveness [57]. uPA converts plasminogen to plasmin then activates MMPs under matrix extracellular degradation [472]. Activated uPAR can also, on the other hand, interact with other transmembrane receptors, including integrins and growth factor receptors [473,474,475]. These interactions trigger activation of the ERK1/2, FAK, Src and PI3K/Akt signalling pathways [476,477].
Besides regulating metastasis, uPAR was found to inhibit TRAIL-induced apoptosis, in glioma cells by regulating the expression of DR4 and DR5 [478], in colon cancer by the intrinsic mitochondrial pathway [477] or in TNBC through the regulation of miR-17 and miR-20, two miRNAs that were shown to impair DR4 expression [479]. Using a RAS-derived stepwise tumorigenesis models to recapitulate TRAIL selectivity, Pavet et al. demonstrated that PLAU mRNA levels, encoding uPA, increase with transformation, preventing TRAIL-induced apoptosis [480]. Depletion of uPA restored TRAIL sensitivity, through inhibiting ERK1/2 activation and DcR2 recruitment to the TRAIL DISC [480]. Mechanistically, how uPA/uPAR regulate TRAIL signalling and more specifically cell motility and metastasis is still unknow. Yet given that uPA is known to promote, not only cancer cell survival or proliferation, but also migration from primary tissues to distant organs [481], it remains an interesting potential TRAIL receptor complex partner to study.
In addition to uPA, the ubiquitin ligase Cbl proto-oncogene (c-Cbl) has also attracted attention as a potential TRAIL receptor partner for the triggering of TRAIL pro-metastatic signalling. This ubiquitin ligase was found to regulate both DR5 and DR4 expression levels [482,483,484].
c-Cbl was found to interact with the caspase-8 inhibitor c-FLIP and to induce its proteasomal degradation, sensitizing macrophages, infected by mycobacteria, to TNF-induced cell death [485]. A number of studies point to c-Cbl as a potential regulator of TRAIL non-canonical signalling pathways [486,487,488]. Likewise, after TRAIL stimulation, c-CBL appears to be involved in a complex involving Src and PI3K, which induces the phosphorylation of AKT [486]. CBL-b and c-CBL were found to interact with DR5, linking DR5 with TRAF2 and inducing ubiquitination of caspase-8 in TRAIL resistant gastric cancer cells [398]. CIN85 is an important c-Cbl binding protein which plays an essential role in cell survival (Dikic, 2002), such as for example in prostate adenocarcinoma cells, in which CIN85 was found to enhance the phosphorylation and activation of MAPKs during TRAIL treatment, leading to their survival [488].
Interestingly, and albeit only cell death was analysed in Xu and al.’s study, it was also found in these cells that deletion of CBL-b, restored TRAIL sensitivity, but also had an impact towards TRAIL receptor subcellular localization [487,489]. Besides TRAIL agonist receptors, it was found that activated c-Cbl induce EGFR redistribution into lipid rafts, facilitating its activation (L. Xu et al., 2012), which might ultimately promote metastasis in gastric cancer cells (Figure 5).
5. Conclusions and Perspectives
TRAIL has emerged as a promising anticancer agent, however, resistance to TRAIL is a major problem, not only because targeted tumours will likely survive to the treatment, but most of all because TRAIL may trigger, in resistant cells, a non-conventional signalling pathway that may ultimately lead to tumour spreading and metastasis.
While signalling pathways triggering cell death are well understood, non-canonical signalling pathways driving cell motility and leading to metastasis are still unclear. As discussed in this review, a number of molecular complexes have been described, explaining how TRAIL receptors may drive cell survival, proliferation, inflammation, and metastatic signal transduction. Yet it is still unclear whether NF-κB or MAP Kinase signal transduction requires a secondary complex or not, given that main kinases or adaptor proteins, including RIPK1, TRADD or TRAF2 can readily interact with complex I. Comprehension of both the temporality and the subcellular localization and composition of these complexes is still missing to provide a comprehensive view of the molecular circuitry which dictate pro-apoptotic or non-apoptotic signalling pathways triggered by TRAIL receptors.
Regardless, a better understanding of the molecular events involved during TRAIL-induced pro-metastatic signalling or non-apoptotic signalling pathways shall be beneficial for both cancer therapies and auto-immune diseases, as this will likely open interesting opportunities to prevent autoimmune diseases associated or not with inflammation or to inhibit or cure metastasis formation in patients.
Acknowledgements
O.M. was supported by grants from CCIR Est (Conférence de Coordination Interrégionale Est de la ligue contre le cancer, comité de Côte d’or), the INCA (Institut National du Cancer) PLBIO22-171, the ANR (Agence Nationale de la Recherche) program, “Investissements d’Avenir” Labcom IAM-IT (ANR22-LCV1-0005-01), Labex LipSTIC (ANR-11LABX-0021-01), ISITE-BFC (ANR-15-IDEX-0003) and the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No, 777995 (DISCOVER) and MSCA-2022-SE-01-01 (Chiron). A.G. was supported by a fellowship from the French Government.
References
- Wiley, S.R.; Schooley, K.; Smolak, P.J.; Din, W.S.; Huang, C.P.; Nicholl, J.K.; Sutherland, G.R.; Smith, T.D.; Rauch, C.; Smith, C.A. Identification and characterization of a new member of the TNF family that induces apoptosis. Immunity 1995, 3, 673–682. [Google Scholar] [CrossRef]
- Pitti, R.M.; Marsters, S.A.; Ruppert, S.; Donahue, C.J.; Moore, A.; Ashkenazi, A. Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor cytokine family. J. Biol. Chem. 1996, 271, 12687–12690. [Google Scholar] [CrossRef]
- Pan, G.; O’Rourke, K.; Chinnaiyan, A.M.; Gentz, R.; Ebner, R.; Ni, J.; Dixit, V.M. The receptor for the cytotoxic ligand TRAIL. Science 1997, 276, 111–113. [Google Scholar] [CrossRef]
- MacFarlane, M.; Ahmad, M.; Srinivasula, S.M.; Fernandes-Alnemri, T.; Cohen, G.M.; Alnemri, E.S. Identification and molecular cloning of two novel receptors for the cytotoxic ligand TRAIL. J. Biol. Chem. 1997, 272, 25417–25420. [Google Scholar] [CrossRef]
- Walczak, H.; Degli-Esposti, M.A.; Johnson, R.S.; Smolak, P.J.; Waugh, J.Y.; Boiani, N.; Timour, M.S.; Gerhart, M.J.; Schooley, K.A.; Smith, C.A.; et al. TRAIL-R2: a novel apoptosis-mediating receptor for TRAIL. Embo J. 1997, 16, 5386–5397. [Google Scholar] [CrossRef]
- Schneider, P.; Bodmer, J.L.; Thome, M.; Hofmann, K.; Holler, N.; Tschopp, J. Characterization of two receptors for TRAIL. FEBS Lett. 1997, 416, 329–334. [Google Scholar] [CrossRef]
- Schneider, P.; Olson, D.; Tardivel, A.; Browning, B.; Lugovskoy, A.; Gong, D.; Dobles, M.; Hertig, S.; Hofmann, K.; Van Vlijmen, H.; et al. Identification of a new murine tumor necrosis factor receptor locus that contains two novel murine receptors for tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Biol. Chem. 2003, 278, 5444–5454. [Google Scholar] [CrossRef]
- Wu, G.S.; Burns, T.F.; Zhan, Y.; Alnemri, E.S.; El-Deiry, W.S. Molecular cloning and functional analysis of the mouse homologue of the KILLER/DR5 tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) death receptor. Cancer Res. 1999, 59, 2770–2775. [Google Scholar]
- Boldin, M.P.; Mett, I.L.; Varfolomeev, E.E.; Chumakov, I.; Shemer-Avni, Y.; Camonis, J.H.; Wallach, D. Self-association of the “death domains” of the p55 tumor necrosis factor (TNF) receptor and Fas/APO1 prompts signaling for TNF and Fas/APO1 effects. J. Biol. Chem. 1995, 270, 387–391. [Google Scholar] [CrossRef]
- Boldin, M.P.; Varfolomeev, E.E.; Pancer, Z.; Mett, I.L.; Camonis, J.H.; Wallach, D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J. Biol. Chem. 1995, 270, 7795–7798. [Google Scholar] [CrossRef]
- Feinstein, E.; Kimchi, A.; Wallach, D.; Boldin, M.; Varfolomeev, E. The death domain: a module shared by proteins with diverse cellular functions. Trends Biochem. Sci. 1995, 20, 342–344. [Google Scholar]
- Hofmann, K. The modular nature of apoptotic signaling proteins. Cell Mol. Life Sci. 1999, 55, 1113–1128. [Google Scholar] [CrossRef]
- Tartaglia, L.A.; Ayres, T.M.; Wong, G.H.; Goeddel, D.V. A novel domain within the 55 kd TNF receptor signals cell death. Cell 1993, 74, 845–853. [Google Scholar]
- Itoh, N.; Nagata, S. A novel protein domain required for apoptosis. Mutational analysis of human Fas antigen. J. Biol. Chem. 1993, 268, 10932–10937. [Google Scholar] [CrossRef]
- Merino, D.; Lalaoui, N.; Morizot, A.; Solary, E.; Micheau, O. TRAIL in cancer therapy: present and future challenges. Expert. Opin. Ther. Targets 2007, 11, 1299–1314. [Google Scholar] [CrossRef]
- Merino, D.; Lalaoui, N.; Morizot, A.; Schneider, P.; Solary, E.; Micheau, O. Differential inhibition of TRAIL-mediated DR5-DISC formation by decoy receptors 1 and 2. Mol. Cell Biol. 2006, 26, 7046–7055. [Google Scholar] [CrossRef]
- Sheridan, J.P.; Marsters, S.A.; Pitti, R.M.; Gurney, A.; Skubatch, M.; Baldwin, D.; Ramakrishnan, L.; Gray, C.L.; Baker, K.; Wood, W.I.; et al. Control of TRAIL-induced apoptosis by a family of signaling and decoy receptors. Science 1997, 277, 818–821. [Google Scholar] [CrossRef]
- Pitti, R.M.; Marsters, S.A.; Lawrence, D.A.; Roy, M.; Kischkel, F.C.; Dowd, P.; Huang, A.; Donahue, C.J.; Sherwood, S.W.; Baldwin, D.T.; et al. Genomic amplification of a decoy receptor for Fas ligand in lung and colon cancer. Nature 1998, 396, 699–703. [Google Scholar] [CrossRef]
- Pan, G.; Ni, J.; Yu, G.; Wei, Y.F.; Dixit, V.M. TRUNDD, a new member of the TRAIL receptor family that antagonizes TRAIL signalling. FEBS Lett. 1998, 424, 41–45. [Google Scholar] [CrossRef]
- Pan, G.; Ni, J.; Wei, Y.F.; Yu, G.; Gentz, R.; Dixit, V.M. An antagonist decoy receptor and a death domain-containing receptor for TRAIL. Science 1997, 277, 815–818. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, M.; Sun, W.; Yang, S.; Su, Y.; Zhang, H.; Liu, C.; Li, X.; Lin, L.; Kim, S.; et al. Reduction of decoy receptor 3 enhances TRAIL-mediated apoptosis in pancreatic cancer. PLoS One 2013, 8, e74272. [Google Scholar] [CrossRef]
- Degli-Esposti, M.A.; Smolak, P.J.; Walczak, H.; Waugh, J.; Huang, C.P.; DuBose, R.F.; Goodwin, R.G.; Smith, C.A. Cloning and characterization of TRAIL-R3, a novel member of the emerging TRAIL receptor family. J. Exp. Med. 1997, 186, 1165–1170. [Google Scholar] [CrossRef]
- Sheikh, M.S.; Huang, Y.; Fernandez-Salas, E.A.; El-Deiry, W.S.; Friess, H.; Amundson, S.; Yin, J.; Meltzer, S.J.; Holbrook, N.J.; Fornace, A.J., Jr. The antiapoptotic decoy receptor TRID/TRAIL-R3 is a p53-regulated DNA damage-inducible gene that is overexpressed in primary tumors of the gastrointestinal tract. Oncogene 1999, 18, 4153–4159. [Google Scholar] [CrossRef]
- Degli-Esposti, M.A.; Dougall, W.C.; Smolak, P.J.; Waugh, J.Y.; Smith, C.A.; Goodwin, R.G. The novel receptor TRAIL-R4 induces NF-kappaB and protects against TRAIL-mediated apoptosis, yet retains an incomplete death domain. Immunity 1997, 7, 813–820. [Google Scholar] [CrossRef]
- Toscano, F.; Fajoui, Z.E.; Gay, F.; Lalaoui, N.; Parmentier, B.; Chayvialle, J.A.; Scoazec, J.Y.; Micheau, O.; Abello, J.; Saurin, J.C. P53-mediated upregulation of DcR1 impairs oxaliplatin/TRAIL-induced synergistic anti-tumour potential in colon cancer cells. Oncogene 2008, 27, 4161–4171. [Google Scholar] [CrossRef]
- Morizot, A.; Merino, D.; Lalaoui, N.; Jacquemin, G.; Granci, V.; Iessi, E.; Lanneau, D.; Bouyer, F.; Solary, E.; Chauffert, B.; et al. Chemotherapy overcomes TRAIL-R4-mediated TRAIL resistance at the DISC level. Cell Death Differ. 2011, 18, 700–711. [Google Scholar] [CrossRef]
- Lalaoui, N.; Morle, A.; Merino, D.; Jacquemin, G.; Iessi, E.; Morizot, A.; Shirley, S.; Robert, B.; Solary, E.; Garrido, C.; et al. TRAIL-R4 promotes tumor growth and resistance to apoptosis in cervical carcinoma HeLa cells through AKT. PLoS ONE 2011, 6, e19679. [Google Scholar] [CrossRef]
- Lalaoui, N.; Merino, D.; Morizot, A.; Jacquemin, G.; Granci, V.; Iessi, E.; Solary, E.; Micheau, O. DcR2 PROTECTS CANCER CELLS FROM TRAIL-INDUCED APOPTOSIS BY ACTIVATING Akt. Adv. Tnf Fam. Res. 2011, 691, 745–745. [Google Scholar]
- Emery, J.G.; McDonnell, P.; Burke, M.B.; Deen, K.C.; Lyn, S.; Silverman, C.; Dul, E.; Appelbaum, E.R.; Eichman, C.; DiPrinzio, R.; et al. Osteoprotegerin is a receptor for the cytotoxic ligand TRAIL. J. Biol. Chem. 1998, 273, 14363–14367. [Google Scholar] [CrossRef]
- Neumann, S.; Hasenauer, J.; Pollak, N.; Scheurich, P. Dominant negative effects of tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor 4 on TRAIL receptor 1 signaling by formation of heteromeric complexes. J. Biol. Chem. 2014, 289, 16576–16587. [Google Scholar] [CrossRef]
- Rimondi, E.; Secchiero, P.; Quaroni, A.; Zerbinati, C.; Capitani, S.; Zauli, G. Involvement of TRAIL/TRAIL-receptors in human intestinal cell differentiation. J. Cell. Physiol. 2006, 206, 647–654. [Google Scholar] [CrossRef] [PubMed]
- Gunalp, S.; Helvaci, D.G.; Oner, A.; Bursali, A.; Conforte, A.; Guner, H.; Karakulah, G.; Szegezdi, E.; Sag, D. TRAIL promotes the polarization of human macrophages toward a proinflammatory M1 phenotype and is associated with increased survival in cancer patients with high tumor macrophage content. Front. Immunol. 2023, 14, 1209249. [Google Scholar] [CrossRef]
- Loeuillard, E.; Li, B.; Stumpf, H.E.; Yang, J.; Willhite, J.; Tomlinson, J.L.; Wang, J.; Rohakhtar, F.R.; Simon, V.A.; Graham, R.P.; et al. Noncanonical TRAIL Signaling Promotes Myeloid-Derived Suppressor Cell Abundance and Tumor Progression in Cholangiocarcinoma. bioRxiv 2023. [Google Scholar] [CrossRef]
- Toffoli, B.; Tonon, F.; Tisato, V.; Zauli, G.; Secchiero, P.; Fabris, B.; Bernardi, S. TRAIL/DR5 pathway promotes AKT phosphorylation, skeletal muscle differentiation, and glucose uptake. Cell Death Dis. 2021, 12, 1089. [Google Scholar] [CrossRef]
- von Karstedt, S.; Conti, A.; Nobis, M.; Montinaro, A.; Hartwig, T.; Lemke, J.; Legler, K.; Annewanter, F.; Campbell, A.D.; Taraborrelli, L.; et al. Cancer cell-autonomous TRAIL-R signaling promotes KRAS-driven cancer progression, invasion, and metastasis. Cancer Cell 2015, 27, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Wilde, A.; Voloshanenko, O.; Bailey, S.L.; Longton, G.M.; Schaefer, U.; Csernok, A.I.; Schutz, G.; Greiner, E.F.; Kemp, C.J.; Walczak, H. TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J. Clin. Invest. 2008, 118, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Steitz, A.M.; Schroder, C.; Knuth, I.; Keber, C.U.; Sommerfeld, L.; Finkernagel, F.; Jansen, J.M.; Wagner, U.; Muller-Brusselbach, S.; Worzfeld, T.; et al. TRAIL-dependent apoptosis of peritoneal mesothelial cells by NK cells promotes ovarian cancer invasion. iScience 2023, 26, 108401. [Google Scholar] [CrossRef]
- Trauzold, A.; Siegmund, D.; Schniewind, B.; Sipos, B.; Egberts, J.; Zorenkov, D.; Emme, D.; Roder, C.; Kalthoff, H.; Wajant, H. TRAIL promotes metastasis of human pancreatic ductal adenocarcinoma. Oncogene 2006, 25, 7434–7439. [Google Scholar] [CrossRef]
- Azijli, K.; Yuvaraj, S.; Peppelenbosch, M.P.; Wurdinger, T.; Dekker, H.; Joore, J.; van Dijk, E.; Quax, W.J.; Peters, G.J.; de Jong, S.; et al. Kinome profiling of non-canonical TRAIL signaling reveals RIP1-Src-STAT3-dependent invasion in resistant non-small cell lung cancer cells. J. Cell Sci. 2012, 125 (Pt 19) Pt 19, 4651–4661. [Google Scholar] [CrossRef]
- Ishimura, N.; Isomoto, H.; Bronk, S.F.; Gores, G.J. Trail induces cell migration and invasion in apoptosis-resistant cholangiocarcinoma cells. Am. J. Physiol. 2006, 290, G129–136. [Google Scholar] [CrossRef]
- Xiao, C.; Rui, Y.; Zhou, S.; Huang, Y.; Wei, Y.; Wang, Z. TNF-related apoptosis-inducing ligand (TRAIL) promotes trophoblast cell invasion via miR-146a-EGFR/CXCR4 axis: A novel mechanism for preeclampsia? Placenta 2020, 93, 8–16. [Google Scholar] [CrossRef]
- Vanamee, E.S.; Faustman, D.L. On the TRAIL of Better Therapies: Understanding TNFRSF Structure-Function. Cells 2020, 9. [Google Scholar] [CrossRef]
- Varfolomeev, E.; Maecker, H.; Sharp, D.; Lawrence, D.; Renz, M.; Vucic, D.; Ashkenazi, A. Molecular determinants of kinase pathway activation by Apo2 ligand/tumor necrosis factor-related apoptosis-inducing ligand. J. Biol. Chem. 2005, 280, 40599–40608. [Google Scholar] [CrossRef]
- Trauzold, A.; Wermann, H.; Arlt, A.; Schutze, S.; Schafer, H.; Oestern, S.; Roder, C.; Ungefroren, H.; Lampe, E.; Heinrich, M.; et al. CD95 and TRAIL receptor-mediated activation of protein kinase C and NF-kappaB contributes to apoptosis resistance in ductal pancreatic adenocarcinoma cells. Oncogene 2001, 20, 4258–4269. [Google Scholar] [CrossRef] [PubMed]
- Wajant, H. TRAIL and NFkappaB signaling--a complex relationship. Vitam. Horm. 2004, 67, 101–132. [Google Scholar] [PubMed]
- Shetty, S.; Gladden, J.B.; Henson, E.S.; Hu, X.; Villanueva, J.; Haney, N.; Gibson, S.B. Tumor necrosis factor-related apoptosis inducing ligand (TRAIL) up-regulates death receptor 5 (DR5) mediated by NFkappaB activation in epithelial derived cell lines. Apoptosis 2002, 7, 413–420. [Google Scholar] [CrossRef]
- Zhang, L.; Dittmer, M.R.; Blackwell, K.; Workman, L.M.; Hostager, B.; Habelhah, H. TRAIL activates JNK and NF-kappaB through RIP1-dependent and -independent pathways. Cell Signal 2015, 27, 306–314. [Google Scholar] [CrossRef]
- Schneider, P.; Thome, M.; Burns, K.; Bodmer, J.L.; Hofmann, K.; Kataoka, T.; Holler, N.; Tschopp, J. TRAIL receptors 1 (DR4) and 2 (DR5) signal FADD-dependent apoptosis and activate NF-kappaB. Immunity 1997, 7, 831–836. [Google Scholar] [CrossRef]
- Luo, J.L.; Maeda, S.; Hsu, L.C.; Yagita, H.; Karin, M. Inhibition of NF-kappaB in cancer cells converts inflammation- induced tumor growth mediated by TNFalpha to TRAIL-mediated tumor regression. Cancer Cell 2004, 6, 297–305. [Google Scholar] [CrossRef]
- Tang, W.; Wang, W.; Zhang, Y.; Liu, S.; Liu, Y.; Zheng, D. TRAIL receptor mediates inflammatory cytokine release in an NF-kappaB-dependent manner. Cell Res. 2009, 19, 758–767. [Google Scholar] [CrossRef]
- Geismann, C.; Erhart, W.; Grohmann, F.; Schreiber, S.; Schneider, G.; Schafer, H.; Arlt, A. TRAIL/NF-kappaB/CX3CL1 Mediated Onco-Immuno Crosstalk Leading to TRAIL Resistance of Pancreatic Cancer Cell Lines. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Davidovich, P.; Higgins, C.A.; Najda, Z.; Longley, D.B.; Martin, S.J. cFLIP(L) acts as a suppressor of TRAIL- and Fas-initiated inflammation by inhibiting assembly of caspase-8/FADD/RIPK1 NF-kappaB-activating complexes. Cell Rep. 2023, 42, 113476. [Google Scholar] [CrossRef]
- Imamura, R.; Konaka, K.; Matsumoto, N.; Hasegawa, M.; Fukui, M.; Mukaida, N.; Kinoshita, T.; Suda, T. Fas ligand induces cell-autonomous NF-kappaB activation and interleukin-8 production by a mechanism distinct from that of tumor necrosis factor-alpha. J. Biol. Chem. 2004, 279, 46415–46423. [Google Scholar] [CrossRef]
- Lee, S.M.; Kim, E.J.; Suk, K.; Lee, W.H. Stimulation of Fas (CD95) induces production of pro-inflammatory mediators through ERK/JNK-dependent activation of NF-kappaB in THP-1 cells. Cell. Immunol. 2011, 271, 157–162. [Google Scholar] [CrossRef]
- Zhang, C.; Gao, F.; Teng, F.; Zhang, M. Fas/FasL Complex Promotes Proliferation and Migration of Brain Endothelial Cells Via FADD-FLIP-TRAF-NF-kappaB Pathway. Cell Biochem. Biophys. 2015, 71, 1319–1323. [Google Scholar] [CrossRef]
- Kreuz, S.; Siegmund, D.; Rumpf, J.J.; Samel, D.; Leverkus, M.; Janssen, O.; Hacker, G.; Dittrich-Breiholz, O.; Kracht, M.; Scheurich, P.; et al. NFkappaB activation by Fas is mediated through FADD, caspase-8, and RIP and is inhibited by FLIP. J. Cell Biol. 2004, 166, 369–380. [Google Scholar] [CrossRef]
- Barnhart, B.C.; Legembre, P.; Pietras, E.; Bubici, C.; Franzoso, G.; Peter, M.E. CD95 ligand induces motility and invasiveness of apoptosis-resistant tumor cells. EMBO J. 2004, 23, 3175–3185. [Google Scholar] [CrossRef]
- Legembre, P.; Barnhart, B.C.; Zheng, L.; Vijayan, S.; Straus, S.E.; Puck, J.; Dale, J.K.; Lenardo, M.; Peter, M.E. Induction of apoptosis and activation of NF-kappaB by CD95 require different signalling thresholds. EMBO Rep. 2004, 5, 1084–1089. [Google Scholar] [CrossRef]
- Kawakubo, T.; Okamoto, K.; Iwata, J.; Shin, M.; Okamoto, Y.; Yasukochi, A.; Nakayama, K.I.; Kadowaki, T.; Tsukuba, T.; Yamamoto, K. Cathepsin E prevents tumor growth and metastasis by catalyzing the proteolytic release of soluble TRAIL from tumor cell surface. Cancer Res. 2007, 67, 10869–10878. [Google Scholar] [CrossRef]
- Yagolovich, A.V.; Artykov, A.A.; Karmakova, T.A.; Vorontsova, M.S.; Pankratov, A.A.; Andreev-Andrievsky, A.A.; Dolgikh, D.A.; Kirpichnikov, M.P.; Gasparian, M.E. Genetically Modified DR5-Specific TRAIL Variant DR5-B Revealed Dual Antitumor and Protumoral Effect in Colon Cancer Xenografts and an Improved Pharmacokinetic Profile. Transl. Oncol. 2020, 13, 100762. [Google Scholar] [CrossRef]
- Chen, L.; Park, S.M.; Tumanov, A.V.; Hau, A.; Sawada, K.; Feig, C.; Turner, J.R.; Fu, Y.X.; Romero, I.L.; Lengyel, E.; et al. CD95 promotes tumour growth. Nature 2010, 465, 492–496. [Google Scholar] [CrossRef] [PubMed]
- Henry, C.M.; Martin, S.J. Caspase-8 Acts in a Non-enzymatic Role as a Scaffold for Assembly of a Pro-inflammatory “FADDosome” Complex upon TRAIL Stimulation. Mol. Cell 2017, 65, 715–729. [Google Scholar] [CrossRef]
- Sullivan, G.P.; O’Connor, H.; Henry, C.M.; Davidovich, P.; Clancy, D.M.; Albert, M.L.; Cullen, S.P.; Martin, S.J. TRAIL Receptors Serve as Stress-Associated Molecular Patterns to Promote ER-Stress-Induced Inflammation. Dev. Cell 2020, 52, 714–730 e715. [Google Scholar] [CrossRef]
- Jeong, D.; Kim, H.S.; Kim, H.Y.; Kang, M.J.; Jung, H.; Oh, Y.; Kim, D.; Koh, J.; Cho, S.Y.; Jeon, Y.K.; et al. Soluble Fas ligand drives autoantibody-induced arthritis by binding to DR5/TRAIL-R2. Elife 2021, 10. [Google Scholar] [CrossRef]
- Ehrlich, S.; Infante-Duarte, C.; Seeger, B.; Zipp, F. Regulation of soluble and surface-bound TRAIL in human T cells, B cells, and monocytes. Cytokine 2003, 24, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Kamohara, H.; Matsuyama, W.; Shimozato, O.; Abe, K.; Galligan, C.; Hashimoto, S.; Matsushima, K.; Yoshimura, T. Regulation of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptor expression in human neutrophils. Immunology 2004, 111, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Koga, Y.; Matsuzaki, A.; Suminoe, A.; Hattori, H.; Hara, T. Neutrophil-derived TNF-related apoptosis-inducing ligand (TRAIL): a novel mechanism of antitumor effect by neutrophils. Cancer Res. 2004, 64, 1037–1043. [Google Scholar] [CrossRef]
- Simons, M.P.; Leidal, K.G.; Nauseef, W.M.; Griffith, T.S. TNF-related apoptosis-inducing ligand (TRAIL) is expressed throughout myeloid development, resulting in a broad distribution among neutrophil granules. J. Leukoc. Biol. 2008, 83, 621–629. [Google Scholar] [CrossRef]
- Fanger, N.A.; Maliszewski, C.R.; Schooley, K.; Griffith, T.S. Human dendritic cells mediate cellular apoptosis via tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Exp. Med. 1999, 190, 1155–1164. [Google Scholar] [CrossRef]
- Cartland, S.P.; Genner, S.W.; Martinez, G.J.; Robertson, S.; Kockx, M.; Lin, R.C.; O’Sullivan, J.F.; Koay, Y.C.; Manuneedhi Cholan, P.; Kebede, M.A.; et al. TRAIL-Expressing Monocyte/Macrophages Are Critical for Reducing Inflammation and Atherosclerosis. iScience 2019, 12, 41–52. [Google Scholar] [CrossRef]
- Griffith, T.S.; Wiley, S.R.; Kubin, M.Z.; Sedger, L.M.; Maliszewski, C.R.; Fanger, N.A. Monocyte-mediated tumoricidal activity via the tumor necrosis factor-related cytokine, TRAIL. J. Exp. Med. 1999, 189, 1343–1354. [Google Scholar] [CrossRef]
- Tecchio, C.; Huber, V.; Scapini, P.; Calzetti, F.; Margotto, D.; Todeschini, G.; Pilla, L.; Martinelli, G.; Pizzolo, G.; Rivoltini, L.; et al. IFNalpha-stimulated neutrophils and monocytes release a soluble form of TNF-related apoptosis-inducing ligand (TRAIL/Apo-2 ligand) displaying apoptotic activity on leukemic cells. Blood 2004, 103, 3837–3844. [Google Scholar] [CrossRef]
- Halaas, O.; Vik, R.; Ashkenazi, A.; Espevik, T. Lipopolysaccharide induces expression of APO2 ligand/TRAIL in human monocytes and macrophages. Scand. J. Immunol. 2000, 51, 244–250. [Google Scholar] [CrossRef]
- Ho, T.C.; Chen, S.L.; Shih, S.C.; Chang, S.J.; Yang, S.L.; Hsieh, J.W.; Cheng, H.C.; Chen, L.J.; Tsao, Y.P. Pigment epithelium-derived factor (PEDF) promotes tumor cell death by inducing macrophage membrane tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Biol. Chem. 2011, 286, 35943–35954. [Google Scholar] [CrossRef]
- Johnsen, A.C.; Haux, J.; Steinkjer, B.; Nonstad, U.; Egeberg, K.; Sundan, A.; Ashkenazi, A.; Espevik, T. Regulation of APO-2 ligand/trail expression in NK cells-involvement in NK cell-mediated cytotoxicity. Cytokine 1999, 11, 664–672. [Google Scholar] [CrossRef]
- Zamai, L.; Ahmad, M.; Bennett, I.M.; Azzoni, L.; Alnemri, E.S.; Perussia, B. Natural killer (NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature primary human NK cells. J. Exp. Med. 1998, 188, 2375–2380. [Google Scholar] [CrossRef]
- Mirandola, P.; Ponti, C.; Gobbi, G.; Sponzilli, I.; Vaccarezza, M.; Cocco, L.; Zauli, G.; Secchiero, P.; Manzoli, F.A.; Vitale, M. Activated human NK and CD8+ T cells express both TNF-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors but are resistant to TRAIL-mediated cytotoxicity. Blood 2004, 104, 2418–2424. [Google Scholar] [CrossRef]
- Beraza, N.; Malato, Y.; Sander, L.E.; Al-Masaoudi, M.; Freimuth, J.; Riethmacher, D.; Gores, G.J.; Roskams, T.; Liedtke, C.; Trautwein, C. Hepatocyte-specific NEMO deletion promotes NK/NKT cell- and TRAIL-dependent liver damage. J. Exp. Med. 2009, 206, 1727–1737. [Google Scholar] [CrossRef]
- Nishihori, Y.; Kato, K.; Tanaka, M.; Okamoto, T.; Hagiwara, S.; Araki, N.; Kogawa, K.; Kuribayashi, K.; Nakamura, K.; Niitsu, Y. Interleukin-2 gene transfer potentiates the alpha-galactosylceramide-stimulated antitumor effect by the induction of TRAIL in NKT and NK cells in mouse models of subcutaneous and metastatic carcinoma. Cancer Biol. Ther. 2009, 8, 1763–1770. [Google Scholar] [CrossRef]
- Smyth, M.J.; Cretney, E.; Takeda, K.; Wiltrout, R.H.; Sedger, L.M.; Kayagaki, N.; Yagita, H.; Okumura, K. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) contributes to interferon gamma-dependent natural killer cell protection from tumor metastasis. J. Exp. Med. 2001, 193, 661–670. [Google Scholar] [CrossRef]
- Nieda, M.; Nicol, A.; Koezuka, Y.; Kikuchi, A.; Lapteva, N.; Tanaka, Y.; Tokunaga, K.; Suzuki, K.; Kayagaki, N.; Yagita, H.; et al. TRAIL expression by activated human CD4(+)V alpha 24NKT cells induces in vitro and in vivo apoptosis of human acute myeloid leukemia cells. Blood 2001, 97, 2067–2074. [Google Scholar] [CrossRef]
- Gomez-Santos, L.; Luka, Z.; Wagner, C.; Fernandez-Alvarez, S.; Lu, S.C.; Mato, J.M.; Martinez-Chantar, M.L.; Beraza, N. Inhibition of natural killer cells protects the liver against acute injury in the absence of glycine N-methyltransferase. Hepatol. Baltim. Md. 2012, 56, 747–759. [Google Scholar] [CrossRef]
- Kahraman, A.; Barreyro, F.J.; Bronk, S.F.; Werneburg, N.W.; Mott, J.L.; Akazawa, Y.; Masuoka, H.C.; Howe, C.L.; Gores, G.J. TRAIL mediates liver injury by the innate immune system in the bile duct-ligated mouse. Hepatol. Baltim. Md. 2008, 47, 1317–1330. [Google Scholar] [CrossRef] [PubMed]
- Metelitsa, L.S.; Weinberg, K.I.; Emanuel, P.D.; Seeger, R.C. Expression of CD1d by myelomonocytic leukemias provides a target for cytotoxic NKT cells. Leukemia 2003, 17, 1068–1077. [Google Scholar] [CrossRef]
- Teng, M.W.; Westwood, J.A.; Darcy, P.K.; Sharkey, J.; Tsuji, M.; Franck, R.W.; Porcelli, S.A.; Besra, G.S.; Takeda, K.; Yagita, H.; et al. Combined natural killer T-cell based immunotherapy eradicates established tumors in mice. Cancer Res. 2007, 67, 7495–7504. [Google Scholar] [CrossRef]
- Stelma, F.; de Niet, A.; Tempelmans Plat-Sinnige, M.J.; Jansen, L.; Takkenberg, R.B.; Reesink, H.W.; Kootstra, N.A.; van Leeuwen, E.M. Natural Killer Cell Characteristics in Patients With Chronic Hepatitis B Virus (HBV) Infection Are Associated With HBV Surface Antigen Clearance After Combination Treatment With Pegylated Interferon Alfa-2a and Adefovir. J. Infect. Dis. 2015, 212, 1042–1051. [Google Scholar] [CrossRef]
- Peteranderl, C.; Morales-Nebreda, L.; Selvakumar, B.; Lecuona, E.; Vadasz, I.; Morty, R.E.; Schmoldt, C.; Bespalowa, J.; Wolff, T.; Pleschka, S.; et al. Macrophage-epithelial paracrine crosstalk inhibits lung edema clearance during influenza infection. J. Clin. Invest. 2016, 126, 1566–1580. [Google Scholar] [CrossRef]
- Azam, S.; Manzoor, S.; Imran, M.; Ashraf, J.; Ashraf, S.; Resham, S.; Ghani, E. Role of interferon gamma and tumor necrosis factor-related apoptosis-inducing ligand receptor 1 single nucleotide polymorphism in natural clearance and treatment response of HCV infection. Viral Immunol. 2015, 28, 222–228. [Google Scholar] [CrossRef]
- Seyman, D.; Yalcin, A.D.; Oztoprak, N.; Genc, G.E.; Ozen, N.S.; Kizilates, F.; Berk, H.; Gumuslu, S. Soluble TRAIL levels decreased in chronic hepatitis C treatment with pegylated interferon alpha plus ribavirin: association with viral responses. Int. J. Clin. Exp. Med. 2014, 7, 5650–5656. [Google Scholar] [PubMed]
- Gyurkovska, V.; Ivanovska, N. Distinct roles of TNF-related apoptosis-inducing ligand (TRAIL) in viral and bacterial infections: from pathogenesis to pathogen clearance. Inflamm. Res. : Off. J. Eur. Histamine Res. Society... [Et Al.] 2016, 65, 427–437. [Google Scholar] [CrossRef]
- Cardoso Alves, L.; Berger, M.D.; Koutsandreas, T.; Kirschke, N.; Lauer, C.; Sporri, R.; Chatziioannou, A.; Corazza, N.; Krebs, P. Non-apoptotic TRAIL function modulates NK cell activity during viral infection. EMBO Rep. 2020, 21, e48789. [Google Scholar] [CrossRef]
- Sato, K.; Hida, S.; Takayanagi, H.; Yokochi, T.; Kayagaki, N.; Takeda, K.; Yagita, H.; Okumura, K.; Tanaka, N.; Taniguchi, T.; et al. Antiviral response by natural killer cells through TRAIL gene induction by IFN-alpha/beta. Eur. J. Immunol. 2001, 31, 3138–3146. [Google Scholar] [CrossRef]
- Warke, R.V.; Martin, K.J.; Giaya, K.; Shaw, S.K.; Rothman, A.L.; Bosch, I. TRAIL is a novel antiviral protein against dengue virus. J. Virol. 2008, 82, 555–564. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Loewendorf, A.; Wang, Q.; McDonald, B.; Redwood, A.; Benedict, C.A. Inhibition of the TRAIL death receptor by CMV reveals its importance in NK cell-mediated antiviral defense. PLoS Pathog. 2014, 10, e1004268. [Google Scholar] [CrossRef] [PubMed]
- Stacey, M.A.; Marsden, M.; Pham, N.T.; Clare, S.; Dolton, G.; Stack, G.; Jones, E.; Klenerman, P.; Gallimore, A.M.; Taylor, P.R.; et al. Neutrophils recruited by IL-22 in peripheral tissues function as TRAIL-dependent antiviral effectors against MCMV. Cell Host Microbe 2014, 15, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Smith, W.; Tomasec, P.; Aicheler, R.; Loewendorf, A.; Nemcovicova, I.; Wang, E.C.; Stanton, R.J.; Macauley, M.; Norris, P.; Willen, L.; et al. Human cytomegalovirus glycoprotein UL141 targets the TRAIL death receptors to thwart host innate antiviral defenses. Cell Host Microbe 2013, 13, 324–335. [Google Scholar] [CrossRef] [PubMed]
- Schuster, I.S.; Wikstrom, M.E.; Brizard, G.; Coudert, J.D.; Estcourt, M.J.; Manzur, M.; O’Reilly, L.A.; Smyth, M.J.; Trapani, J.A.; Hill, G.R.; et al. TRAIL+ NK cells control CD4+ T cell responses during chronic viral infection to limit autoimmunity. Immunity 2014, 41, 646–656. [Google Scholar] [CrossRef]
- Dunn, C.; Brunetto, M.; Reynolds, G.; Christophides, T.; Kennedy, P.T.; Lampertico, P.; Das, A.; Lopes, A.R.; Borrow, P.; Williams, K.; et al. Cytokines induced during chronic hepatitis B virus infection promote a pathway for NK cell-mediated liver damage. J. Exp. Med. 2007, 204, 667–680. [Google Scholar] [CrossRef]
- Takeda, K.; Smyth, M.J.; Cretney, E.; Hayakawa, Y.; Yamaguchi, N.; Yagita, H.; Okumura, K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in NK cell-mediated and IFN-gamma-dependent suppression of subcutaneous tumor growth. Cell. Immunol. 2001, 214, 194–200. [Google Scholar] [CrossRef]
- Wajant, H.; Pfizenmaier, K.; Scheurich, P. TNF-related apoptosis inducing ligand (TRAIL) and its receptors in tumor surveillance and cancer therapy. Apoptosis 2002, 7, 449–459. [Google Scholar] [CrossRef]
- Takeda, K.; Smyth, M.J.; Cretney, E.; Hayakawa, Y.; Kayagaki, N.; Yagita, H.; Okumura, K. Critical role for tumor necrosis factor-related apoptosis-inducing ligand in immune surveillance against tumor development. J. Exp. Med. 2002, 195, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Yamaguchi, N.; Akiba, H.; Kojima, Y.; Hayakawa, Y.; Tanner, J.E.; Sayers, T.J.; Seki, N.; Okumura, K.; Yagita, H.; et al. Induction of Tumor-specific T Cell Immunity by Anti-DR5 Antibody Therapy. J. Exp. Med. 2004, 199, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Cretney, E.; Hayakawa, Y.; Ota, T.; Akiba, H.; Ogasawara, K.; Yagita, H.; Kinoshita, K.; Okumura, K.; Smyth, M.J. TRAIL identifies immature natural killer cells in newborn mice and adult mouse liver. Blood 2005, 105, 2082–2089. [Google Scholar] [CrossRef] [PubMed]
- Anees, M.; Horak, P.; Schiefer, A.I.; Vanhara, P.; El-Gazzar, A.; Perco, P.; Kiesewetter, B.; Mullauer, L.; Streubel, B.; Raderer, M.; et al. The potential evasion of immune surveillance in mucosa associated lymphoid tissue lymphoma by DcR2-mediated up-regulation of nuclear factor-kappaB. Leuk. Lymphoma 2015, 56, 1440–1449. [Google Scholar] [CrossRef]
- Cassioli, C.; Baldari, C.T. The Expanding Arsenal of Cytotoxic T Cells. Front. Immunol. 2022, 13, 883010. [Google Scholar] [CrossRef]
- Kemp, T.J.; Ludwig, A.T.; Earel, J.K.; Moore, J.M.; Vanoosten, R.L.; Moses, B.; Leidal, K.; Nauseef, W.M.; Griffith, T.S. Neutrophil stimulation with Mycobacterium bovis bacillus Calmette-Guerin (BCG) results in the release of functional soluble TRAIL/Apo-2L. Blood 2005, 106, 3474–3482. [Google Scholar] [CrossRef]
- Shamili, F.H.; Bayegi, H.R.; Salmasi, Z.; Sadri, K.; Mahmoudi, M.; Kalantari, M.; Ramezani, M.; Abnous, K. Exosomes derived from TRAIL-engineered mesenchymal stem cells with effective anti-tumor activity in a mouse melanoma model. Int. J. Pharm. 2018, 549, 218–229. [Google Scholar] [CrossRef]
- Schmaltz, C.; Alpdogan, O.; Kappel, B.J.; Muriglan, S.J.; Rotolo, J.A.; Ongchin, J.; Willis, L.M.; Greenberg, A.S.; Eng, J.M.; Crawford, J.M.; et al. T cells require TRAIL for optimal graft-versus-tumor activity. Nat. Med. 2002, 8, 1433–1437. [Google Scholar] [CrossRef]
- Zamai, L.; Del Zotto, G.; Buccella, F.; Galeotti, L.; Canonico, B.; Luchetti, F.; Papa, S. Cytotoxic functions and susceptibility to apoptosis of human CD56(bright) NK cells differentiated in vitro from CD34(+) hematopoietic progenitors. Cytom. A 2012, 81, 294–302. [Google Scholar] [CrossRef]
- Sur, S.Y.; Lim, G.H.; Park, S.M.; Seo, K.W.; Youn, H.Y. Anti-tumor Effect of Activated Canine B Cells With Interleukin-21 and Anti-B Cell Receptor. Anticancer. Res. 2023, 43, 4007–4014. [Google Scholar] [CrossRef]
- van Vliet, A.A.; Peters, E.; Vodegel, D.; Steenmans, D.; Raimo, M.; Gibbs, S.; de Gruijl, T.D.; Duru, A.D.; Spanholtz, J.; Georgoudaki, A.M. Early TRAIL-engagement elicits potent multimodal targeting of melanoma by CD34(+) progenitor cell-derived NK cells. iScience 2023, 26, 107078. [Google Scholar] [CrossRef]
- Smyth, M.J.; Hayakawa, Y.; Takeda, K.; Yagita, H. New aspects of natural-killer-cell surveillance and therapy of cancer. Nat. Rev. Cancer 2002, 2, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Labrada, A.; Pesini, C.; Santiago, L.; Hidalgo, S.; Calvo-Perez, A.; Onate, C.; Andres-Tovar, A.; Garzon-Tituana, M.; Uranga-Murillo, I.; Arias, M.A.; et al. All About (NK Cell-Mediated) Death in Two Acts and an Unexpected Encore: Initiation, Execution and Activation of Adaptive Immunity. Front. Immunol. 2022, 13, 896228. [Google Scholar] [CrossRef]
- Asher, A.; Mule, J.J.; Reichert, C.M.; Shiloni, E.; Rosenberg, S.A. Studies on the anti-tumor efficacy of systemically administered recombinant tumor necrosis factor against several murine tumors in vivo. J. Immunol. 1987, 138, 963–974. [Google Scholar] [CrossRef] [PubMed]
- Ogasawara, J.; Watanabe-Fukunaga, R.; Adachi, M.; Matsuzawa, A.; Kasugai, T.; Kitamura, Y.; Itoh, N.; Suda, T.; Nagata, S. Lethal effect of the anti-Fas antibody in mice. Nature 1993, 364, 806–809. [Google Scholar] [CrossRef]
- Bonavida, B.; Ng, C.P.; Jazirehi, A.; Schiller, G.; Mizutani, Y. Selectivity of TRAIL-mediated apoptosis of cancer cells and synergy with drugs: the trail to non-toxic cancer therapeutics (review). Int. J. Oncol. 1999, 15, 793–802. [Google Scholar] [CrossRef]
- Pollack, I.F.; Erff, M.; Ashkenazi, A. Direct stimulation of apoptotic signaling by soluble Apo2l/tumor necrosis factor-related apoptosis-inducing ligand leads to selective killing of glioma cells. Clin. Cancer Res. 2001, 7, 1362–1369. [Google Scholar] [PubMed]
- Ashkenazi, A.; Pai, R.C.; Fong, S.; Leung, S.; Lawrence, D.A.; Marsters, S.A.; Blackie, C.; Chang, L.; McMurtrey, A.E.; Hebert, A.; et al. Safety and antitumor activity of recombinant soluble Apo2 ligand. J. Clin. Invest. 1999, 104, 155–162. [Google Scholar] [CrossRef] [PubMed]
- Kelley, S.K.; Harris, L.A.; Xie, D.; Deforge, L.; Totpal, K.; Bussiere, J.; Fox, J.A. Preclinical studies to predict the disposition of Apo2L/tumor necrosis factor-related apoptosis-inducing ligand in humans: characterization of in vivo efficacy, pharmacokinetics, and safety. J. Pharmacol. Exp. Ther. 2001, 299, 31–38. [Google Scholar]
- Herbst, R.S.; Mendolson, D.S.; Ebbinghaus, S.; Gordon, M.S.; O’Dwyer, M.; Lieberman, G.; Ing, J.; Kurzrock, R.; Novotny, W.; Eckhardt, S.G. A phase I safety and pharmacokinetic (PK) study of recombinant Apo2L/TRAIL, an apoptosis-inducing protein in patients with advanced cancer. J. Clin. Oncol. 2006, 2, abstr 3013. [Google Scholar]
- Ichikawa, K.; Liu, W.; Zhao, L.; Wang, Z.; Liu, D.; Ohtsuka, T.; Zhang, H.; Mountz, J.D.; Koopman, W.J.; Kimberly, R.P.; et al. Tumoricidal activity of a novel anti-human DR5 monoclonal antibody without hepatocyte cytotoxicity. Nat. Med. 2001, 7, 954–960. [Google Scholar] [CrossRef]
- Walczak, H.; Miller, R.E.; Ariail, K.; Gliniak, B.; Griffith, T.S.; Kubin, M.; Chin, W.; Jones, J.; Woodward, A.; Le, T.; et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat. Med. 1999, 5, 157–163. [Google Scholar] [CrossRef] [PubMed]
- French, L.E.; Tschopp, J. The TRAIL to selective tumor death. Nat. Med. 1999, 5, 146–147. [Google Scholar] [CrossRef] [PubMed]
- Kurbanov, B.M.; Geilen, C.C.; Fecker, L.F.; Orfanos, C.E.; Eberle, J. Efficient TRAIL-R1/DR4-mediated apoptosis in melanoma cells by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J. Invest. Dermatol. 2005, 125, 1010–1019. [Google Scholar] [CrossRef] [PubMed]
- Strater, J.; Hinz, U.; Walczak, H.; Mechtersheimer, G.; Koretz, K.; Herfarth, C.; Moller, P.; Lehnert, T. Expression of TRAIL and TRAIL receptors in colon carcinoma: TRAIL-R1 is an independent prognostic parameter. Clin. Cancer Res. 2002, 8, 3734–3740. [Google Scholar] [PubMed]
- Spierings, D.C.; de Vries, E.G.; Timens, W.; Groen, H.J.; Boezen, H.M.; de Jong, S. Expression of TRAIL and TRAIL death receptors in stage III non-small cell lung cancer tumors. Clin. Cancer Res. 2003, 9, 3397–3405. [Google Scholar] [PubMed]
- Spierings, D.C.; de Vries, E.G.; Vellenga, E.; van den Heuvel, F.A.; Koornstra, J.J.; Wesseling, J.; Hollema, H.; de Jong, S. Tissue distribution of the death ligand TRAIL and its receptors. J. Histochem. Cytochem. 2004, 52, 821–831. [Google Scholar] [CrossRef] [PubMed]
- Daniels, R.A.; Turley, H.; Kimberley, F.C.; Liu, X.S.; Mongkolsapaya, J.; Ch’En, P.; Xu, X.N.; Jin, B.Q.; Pezzella, F.; Screaton, G.R. Expression of TRAIL and TRAIL receptors in normal and malignant tissues. Cell Res. 2005, 15, 430–438. [Google Scholar] [CrossRef]
- Sanlioglu, A.D.; Korcum, A.F.; Pestereli, E.; Erdogan, G.; Karaveli, S.; Savas, B.; Griffith, T.S.; Sanlioglu, S. TRAIL death receptor-4 expression positively correlates with the tumor grade in breast cancer patients with invasive ductal carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2007, 69, 716–723. [Google Scholar] [CrossRef]
- Ganten, T.M.; Sykora, J.; Koschny, R.; Batke, E.; Aulmann, S.; Mansmann, U.; Stremmel, W.; Sinn, H.P.; Walczak, H. Prognostic significance of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in patients with breast cancer. J Mol Med (Berl) 2009, 87, 995–1007. [Google Scholar] [CrossRef]
- Chen, S.M.; Sun, H.; Liu, Y.F.; Ma, J.; Zhang, Q.T.; Zhu, J.; Li, T. Expression of TRAIL and its receptor DR5 and their significance in acute leukemia cells. Genet. Mol. Res. : GMR 2015, 14, 18562–18568. [Google Scholar] [CrossRef]
- Gaertner, F.; Kruger, S.; Roder, C.; Trauzold, A.; Rocken, C.; Kalthoff, H. The expression of death receptor systems TRAIL-R1/-R2/-R4, CD95 and TNF-R1 and their cognate ligands in pancreatic ductal adenocarcinoma. Histol. Histopathol. 2019, 34, 491–501. [Google Scholar] [CrossRef]
- Ravi, R.; Bedi, A. Requirement of BAX for TRAIL/Apo2L-induced apoptosis of colorectal cancers: synergism with sulindac-mediated inhibition of Bcl-x(L). Cancer Res. 2002, 62, 1583–1587. [Google Scholar] [PubMed]
- Willms, A.; Schittek, H.; Rahn, S.; Sosna, J.; Mert, U.; Adam, D.; Trauzold, A. Impact of p53 status on TRAIL-mediated apoptotic and non-apoptotic signaling in cancer cells. PLoS One 2019, 14, e0214847. [Google Scholar] [CrossRef]
- Micheau, O.; Shirley, S.; Dufour, F. Death receptors as targets in cancer. Br. J. Pharmacol. 2013, 169, 1723–1744. [Google Scholar] [CrossRef]
- Naoum, G.E.; Buchsbaum, D.J.; Tawadros, F.; Farooqi, A.; Arafat, W.O. Journey of TRAIL from Bench to Bedside and its Potential Role in Immuno-Oncology. Oncol. Rev. 2017, 11, 332. [Google Scholar] [CrossRef] [PubMed]
- Smyth, M.J.; Takeda, K.; Hayakawa, Y.; Peschon, J.J.; van den Brink, M.R.; Yagita, H. Nature’s TRAIL-On a Path to Cancer Immunotherapy. Immunity 2003, 18, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Stuckey, D.W.; Shah, K. TRAIL on trial: preclinical advances in cancer therapy. Trends Mol. Med. 2013, 19, 685–694. [Google Scholar] [CrossRef]
- Di Cristofano, F.; George, A.; Tajiknia, V.; Ghandali, M.; Wu, L.; Zhang, Y.; Srinivasan, P.; Strandberg, J.; Hahn, M.; Sanchez Sevilla Uruchurtu, A.; et al. Therapeutic targeting of TRAIL death receptors. Biochem. Soc. Trans. 2023, 51, 57–70. [Google Scholar] [CrossRef]
- Bodmer, J.L.; Holler, N.; Reynard, S.; Vinciguerra, P.; Schneider, P.; Juo, P.; Blenis, J.; Tschopp, J. TRAIL receptor-2 signals apoptosis through FADD and caspase-8. Nat. Cell Biol. 2000, 2, 241–243. [Google Scholar] [CrossRef]
- Kischkel, F.C.; Lawrence, D.A.; Chuntharapai, A.; Schow, P.; Kim, K.J.; Ashkenazi, A. Apo2L/TRAIL-dependent recruitment of endogenous FADD and caspase-8 to death receptors 4 and 5. Immunity 2000, 12, 611–620. [Google Scholar] [CrossRef]
- Werner, A.B.; de Vries, E.; Tait, S.W.; Bontjer, I.; Borst, J. TRAIL receptor and CD95 signal to mitochondria via FADD, caspase-8/10, Bid, and Bax but differentially regulate events downstream from truncated Bid. J. Biol. Chem. 2002, 277, 40760–40767. [Google Scholar] [CrossRef] [PubMed]
- Sprick, M.R.; Weigand, M.A.; Rieser, E.; Rauch, C.T.; Juo, P.; Blenis, J.; Krammer, P.H.; Walczak, H. FADD/MORT1 and caspase-8 are recruited to TRAIL receptors 1 and 2 and are essential for apoptosis mediated by TRAIL receptor 2. Immunity 2000, 12, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Muzio, M.; Stockwell, B.R.; Stennicke, H.R.; Salvesen, G.S.; Dixit, V.M. An induced proximity model for caspase-8 activation. J. Biol. Chem. 1998, 273, 2926–2930. [Google Scholar] [CrossRef] [PubMed]
- Boatright, K.M.; Deis, C.; Denault, J.B.; Sutherlin, D.P.; Salvesen, G.S. Activation of caspases-8 and -10 by FLIP(L). Biochem. J. 2004, 382 Pt 2, 651–657. [Google Scholar] [CrossRef]
- Boatright, K.M.; Renatus, M.; Scott, F.L.; Sperandio, S.; Shin, H.; Pedersen, I.M.; Ricci, J.E.; Edris, W.A.; Sutherlin, D.P.; Green, D.R.; et al. A unified model for apical caspase activation. Mol. Cell 2003, 11, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Stennicke, H.R.; Jurgensmeier, J.M.; Shin, H.; Deveraux, Q.; Wolf, B.B.; Yang, X.; Zhou, Q.; Ellerby, H.M.; Ellerby, L.M.; Bredesen, D.; et al. Pro-caspase-3 is a major physiologic target of caspase-8. J. Biol. Chem. 1998, 273, 27084–27090. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Green, D.R. Protease activation during apoptosis: death by a thousand cuts? Cell 1995, 82, 349–352. [Google Scholar] [CrossRef]
- Matveeva, A.; Fichtner, M.; McAllister, K.; McCann, C.; Sturrock, M.; Longley, D.B.; Prehn, J.H.M. Heterogeneous responses to low level death receptor activation are explained by random molecular assembly of the Caspase-8 activation platform. PLoS Comput. Biol. 2019, 15, e1007374. [Google Scholar] [CrossRef]
- Spencer, S.L.; Gaudet, S.; Albeck, J.G.; Burke, J.M.; Sorger, P.K. Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009, 459, 428–432. [Google Scholar] [CrossRef]
- Scaffidi, C.; Schmitz, I.; Zha, J.; Korsmeyer, S.J.; Krammer, P.H.; Peter, M.E. Differential modulation of apoptosis sensitivity in CD95 type I and type II cells. J. Biol. Chem. 1999, 274, 22532–22538. [Google Scholar] [CrossRef]
- Yamada, H.; Tada-Oikawa, S.; Uchida, A.; Kawanishi, S. TRAIL causes cleavage of bid by caspase-8 and loss of mitochondrial membrane potential resulting in apoptosis in BJAB cells. Biochem. Biophys. Res. Commun. 1999, 265, 130–133. [Google Scholar] [CrossRef]
- Walczak, H.; Bouchon, A.; Stahl, H.; Krammer, P.H. Tumor necrosis factor-related apoptosis-inducing ligand retains its apoptosis-inducing capacity on Bcl-2- or Bcl-xL-overexpressing chemotherapy-resistant tumor cells. Cancer Res. 2000, 60, 3051–3057. [Google Scholar]
- Gazitt, Y.; Shaughnessy, P.; Montgomery, W. Apoptosis-induced by TRAIL AND TNF-alpha in human multiple myeloma cells is not blocked by BCL-2. Cytokine 1999, 11, 1010–1019. [Google Scholar] [CrossRef]
- Keogh, S.A.; Walczak, H.; Bouchier-Hayes, L.; Martin, S.J. Failure of Bcl-2 to block cytochrome c redistribution during TRAIL-induced apoptosis. FEBS Lett. 2000, 471, 93–98. [Google Scholar] [CrossRef]
- LeBlanc, H.; Lawrence, D.; Varfolomeev, E.; Totpal, K.; Morlan, J.; Schow, P.; Fong, S.; Schwall, R.; Sinicropi, D.; Ashkenazi, A. Tumor-cell resistance to death receptor--induced apoptosis through mutational inactivation of the proapoptotic Bcl-2 homolog Bax. Nat. Med. 2002, 8, 274–281. [Google Scholar] [CrossRef]
- Hinz, S.; Trauzold, A.; Boenicke, L.; Sandberg, C.; Beckmann, S.; Bayer, E.; Walczak, H.; Kalthoff, H.; Ungefroren, H. Bcl-XL protects pancreatic adenocarcinoma cells against CD95- and TRAIL-receptor-mediated apoptosis. Oncogene 2000, 19, 5477–5486. [Google Scholar] [CrossRef]
- Guo, B.C.; Xu, Y.H. Bcl-2 over-expression and activation of protein kinase C suppress the trail-induced apoptosis in Jurkat T cells. Cell Res. 2001, 11, 101–106. [Google Scholar] [CrossRef]
- Huang, K.; Zhang, J.; O’Neill, K.L.; Gurumurthy, C.B.; Quadros, R.M.; Tu, Y.; Luo, X. Cleavage by Caspase 8 and Mitochondrial Membrane Association Activate the BH3-only Protein Bid during TRAIL-induced Apoptosis. J. Biol. Chem. 2016, 291, 11843–11851. [Google Scholar] [CrossRef]
- Desagher, S.; Osen-Sand, A.; Nichols, A.; Eskes, R.; Montessuit, S.; Lauper, S.; Maundrell, K.; Antonsson, B.; Martinou, J.C. Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis. J. Cell Biol. 1999, 144, 891–901. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.H.; Zhao, Y.; Barber, M.J.; Kuharsky, D.K.; Yin, X.M. Bid-induced cytochrome c release is mediated by a pathway independent of mitochondrial permeability transition pore and Bax. J. Biol. Chem. 2000, 275, 39474–39481. [Google Scholar] [CrossRef] [PubMed]
- Korsmeyer, S.J.; Wei, M.C.; Saito, M.; Weiler, S.; Oh, K.J.; Schlesinger, P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000, 7, 1166–1173. [Google Scholar] [CrossRef]
- Chinnaiyan, A.M. The apoptosome: heart and soul of the cell death machine. Neoplasia (New York N.Y 1999, 1, 5–15. [Google Scholar] [CrossRef]
- Zou, H.; Li, Y.; Liu, X.; Wang, X. An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J. Biol. Chem. 1999, 274, 11549–11556. [Google Scholar] [CrossRef]
- Acehan, D.; Jiang, X.; Morgan, D.G.; Heuser, J.E.; Wang, X.; Akey, C.W. Three-dimensional structure of the apoptosome: implications for assembly, procaspase-9 binding, and activation. Mol. Cell 2002, 9, 423–432. [Google Scholar] [CrossRef]
- Pop, C.; Timmer, J.; Sperandio, S.; Salvesen, G.S. The apoptosome activates caspase-9 by dimerization. Mol. Cell 2006, 22, 269–275. [Google Scholar] [CrossRef]
- Holler, N.; Zaru, R.; Micheau, O.; Thome, M.; Attinger, A.; Valitutti, S.; Bodmer, J.L.; Schneider, P.; Seed, B.; Tschopp, J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat. Immunol. 2000, 1, 489–495. [Google Scholar] [CrossRef]
- Meurette, O.; Rebillard, A.; Huc, L.; Le Moigne, G.; Merino, D.; Micheau, O.; Lagadic-Gossmann, D.; Dimanche-Boitrel, M.T. TRAIL induces receptor-interacting protein 1-dependent and caspase-dependent necrosis-like cell death under acidic extracellular conditions. Cancer Res. 2007, 67, 218–226. [Google Scholar] [CrossRef]
- Jouan-Lanhouet, S.; Arshad, M.I.; Piquet-Pellorce, C.; Martin-Chouly, C.; Le Moigne-Muller, G.; Van Herreweghe, F.; Takahashi, N.; Sergent, O.; Lagadic-Gossmann, D.; Vandenabeele, P.; et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ. 2012, 19, 2003–2014. [Google Scholar] [CrossRef]
- Degterev, A.; Huang, Z.; Boyce, M.; Li, Y.; Jagtap, P.; Mizushima, N.; Cuny, G.D.; Mitchison, T.J.; Moskowitz, M.A.; Yuan, J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 2005, 1, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Wickliffe, K.E.; Dugger, D.L.; Maltzman, A.; Roose-Girma, M.; Dohse, M.; Komuves, L.; Webster, J.D.; Dixit, V.M. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature 2019, 574, 428–431. [Google Scholar] [CrossRef]
- Micheau, O.; Thome, M.; Schneider, P.; Holler, N.; Tschopp, J.; Nicholson, D.W.; Briand, C.; Grutter, M.G. The long form of FLIP is an activator of caspase-8 at the Fas death-inducing signaling complex. J. Biol. Chem. 2002, 277, 45162–45171. [Google Scholar] [CrossRef] [PubMed]
- Mompean, M.; Li, W.; Li, J.; Laage, S.; Siemer, A.B.; Bozkurt, G.; Wu, H.; McDermott, A.E. The Structure of the Necrosome RIPK1-RIPK3 Core, a Human Hetero-Amyloid Signaling Complex. Cell 2018, 173, 1244–1253 e1210. [Google Scholar] [CrossRef]
- Wu, X.; Ma, Y.; Zhao, K.; Zhang, J.; Sun, Y.; Li, Y.; Dong, X.; Hu, H.; Liu, J.; Wang, J.; et al. The structure of a minimum amyloid fibril core formed by necroptosis-mediating RHIM of human RIPK3. Proc. Natl. Acad. Sci. U S A 2021, 118. [Google Scholar] [CrossRef]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef]
- Zhang, D.W.; Shao, J.; Lin, J.; Zhang, N.; Lu, B.J.; Lin, S.C.; Dong, M.Q.; Han, J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science 2009, 325, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Orozco, S.; Yatim, N.; Werner, M.R.; Tran, H.; Gunja, S.Y.; Tait, S.W.; Albert, M.L.; Green, D.R.; Oberst, A. RIPK1 both positively and negatively regulates RIPK3 oligomerization and necroptosis. Cell Death Differ. 2014, 21, 1511–1521. [Google Scholar] [CrossRef]
- Sun, L.; Wang, H.; Wang, Z.; He, S.; Chen, S.; Liao, D.; Wang, L.; Yan, J.; Liu, W.; Lei, X.; et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell 2012, 148, 213–227. [Google Scholar] [CrossRef]
- Bertrand, M.J.; Vandenabeele, P. The Ripoptosome: death decision in the cytosol. Mol. Cell 2011, 43, 323–325. [Google Scholar] [CrossRef]
- Wang, H.; Sun, L.; Su, L.; Rizo, J.; Liu, L.; Wang, L.F.; Wang, F.S.; Wang, X. Mixed lineage kinase domain-like protein MLKL causes necrotic membrane disruption upon phosphorylation by RIP3. Mol. Cell 2014, 54, 133–146. [Google Scholar] [CrossRef]
- Zhao, J.; Jitkaew, S.; Cai, Z.; Choksi, S.; Li, Q.; Luo, J.; Liu, Z.G. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc. Natl. Acad. Sci. U S A 2012, 109, 5322–5327. [Google Scholar] [CrossRef]
- Dondelinger, Y.; Declercq, W.; Montessuit, S.; Roelandt, R.; Goncalves, A.; Bruggeman, I.; Hulpiau, P.; Weber, K.; Sehon, C.A.; Marquis, R.W.; et al. MLKL compromises plasma membrane integrity by binding to phosphatidylinositol phosphates. Cell Rep. 2014, 7, 971–981. [Google Scholar] [CrossRef]
- Galluzzi, L.; Kepp, O.; Kroemer, G. MLKL regulates necrotic plasma membrane permeabilization. Cell Res. 2014, 24, 139–140. [Google Scholar] [CrossRef]
- Murphy, J.M.; Vince, J.E. Post-translational control of RIPK3 and MLKL mediated necroptotic cell death. F1000Research 2015, 4. [Google Scholar] [CrossRef]
- Wike-Hooley, J.L.; Haveman, J.; Reinhold, H.S. The relevance of tumour pH to the treatment of malignant disease. Radiother. Oncol. 1984, 2, 343–366. [Google Scholar] [CrossRef]
- Barja de Quiroga, G. Hypothesis that the acidification of a tissue which takes place during ischemia can lead to tissue hyperoxia during reperfusion due to the Bohr effect. Free Radic. Biol. Med. 1990, 8, 487–489. [Google Scholar] [CrossRef]
- Zhang, Z.X.; Gan, I.; Pavlosky, A.; Huang, X.; Fuhrmann, B.; Jevnikar, A.M. Intracellular pH Regulates TRAIL-Induced Apoptosis and Necroptosis in Endothelial Cells. J. Immunol. Res. 2017, 2017, 1503960. [Google Scholar] [CrossRef]
- Bogdanov, A.; Bogdanov, A.; Chubenko, V.; Volkov, N.; Moiseenko, F.; Moiseyenko, V. Tumor acidity: From hallmark of cancer to target of treatment. Front. Oncol. 2022, 12, 979154. [Google Scholar] [CrossRef]
- Huber, V.; Camisaschi, C.; Berzi, A.; Ferro, S.; Lugini, L.; Triulzi, T.; Tuccitto, A.; Tagliabue, E.; Castelli, C.; Rivoltini, L. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 2017, 43, 74–89. [Google Scholar] [CrossRef]
- Lu, M.; Lawrence, D.A.; Marsters, S.; Acosta-Alvear, D.; Kimmig, P.; Mendez, A.S.; Paton, A.W.; Paton, J.C.; Walter, P.; Ashkenazi, A. Opposing unfolded-protein-response signals converge on death receptor 5 to control apoptosis. Science 2014, 345, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Lam, M.; Lawrence, D.A.; Ashkenazi, A.; Walter, P. Confirming a critical role for death receptor 5 and caspase-8 in apoptosis induction by endoplasmic reticulum stress. Cell Death Differ. 2018, 25, 1530–1531. [Google Scholar] [CrossRef]
- Dufour, F.; Rattier, T.; Constantinescu, A.A.; Zischler, L.; Morle, A.; Ben Mabrouk, H.; Humblin, E.; Jacquemin, G.; Szegezdi, E.; Delacote, F.; et al. TRAIL receptor gene editing unveils TRAIL-R1 as a master player of apoptosis induced by TRAIL and ER stress. Oncotarget 2017, 8, 9974–9985. [Google Scholar] [CrossRef] [PubMed]
- Iurlaro, R.; Puschel, F.; Leon-Annicchiarico, C.L.; O’Connor, H.; Martin, S.J.; Palou-Gramon, D.; Lucendo, E.; Munoz-Pinedo, C. Glucose Deprivation Induces ATF4-Mediated Apoptosis through TRAIL Death Receptors. Mol. Cell Biol. 2017, 37. [Google Scholar] [CrossRef]
- Chang, T.K.; Lawrence, D.A.; Lu, M.; Tan, J.; Harnoss, J.M.; Marsters, S.A.; Liu, P.; Sandoval, W.; Martin, S.E.; Ashkenazi, A. Coordination between Two Branches of the Unfolded Protein Response Determines Apoptotic Cell Fate. Mol. Cell 2018, 71, 629–636. [Google Scholar] [CrossRef]
- Lam, M.; Marsters, S.A.; Ashkenazi, A.; Walter, P. Misfolded proteins bind and activate death receptor 5 to trigger apoptosis during unresolved endoplasmic reticulum stress. Elife 2020, 9. [Google Scholar] [CrossRef]
- Hattori, T.; Fundora, K.A.; Hamamoto, K.; Opozda, D.M.; Liang, X.; Liu, X.; Zhang, J.; Uzun, Y.; Takahashi, Y.; Wang, H.G. ER stress elicits non-canonical CASP8 (caspase 8) activation on autophagosomal membranes to induce apoptosis. Autophagy 2023, 1–16. [Google Scholar] [CrossRef]
- Favaro, F.; Both, D.; Derks, I.A.M.; Spaargaren, M.; Munoz-Pinedo, C.; Eldering, E. Negligible role of TRAIL death receptors in cell death upon endoplasmic reticulum stress in B-cell malignancies. Oncogenesis 2023, 12, 6. [Google Scholar] [CrossRef]
- Glab, J.A.; Doerflinger, M.; Nedeva, C.; Jose, I.; Mbogo, G.W.; Paton, J.C.; Paton, A.W.; Kueh, A.J.; Herold, M.J.; Huang, D.C.; et al. DR5 and caspase-8 are dispensable in ER stress-induced apoptosis. Cell Death Differ. 2017, 24, 944–950. [Google Scholar] [CrossRef]
- Havell, E.A.; Fiers, W.; North, R.J. The antitumor function of tumor necrosis factor (TNF), I. Therapeutic action of TNF against an established murine sarcoma is indirect, immunologically dependent, and limited by severe toxicity. J. Exp. Med. 1988, 167, 1067–1085. [Google Scholar] [CrossRef]
- North, R.J.; Havell, E.A. The antitumor function of tumor necrosis factor (TNF) II. Analysis of the role of endogenous TNF in endotoxin-induced hemorrhagic necrosis and regression of an established sarcoma. J. Exp. Med. 1988, 167, 1086–1099. [Google Scholar] [CrossRef]
- Rensing-Ehl, A.; Frei, K.; Flury, R.; Matiba, B.; Mariani, S.M.; Weller, M.; Aebischer, P.; Krammer, P.H.; Fontana, A. Local Fas/APO-1 (CD95) ligand-mediated tumor cell killing in vivo. Eur. J. Immunol. 1995, 25, 2253–2258. [Google Scholar] [CrossRef]
- Itoh, N.; Yonehara, S.; Ishii, A.; Yonehara, M.; Mizushima, S.; Sameshima, M.; Hase, A.; Seto, Y.; Nagata, S. The polypeptide encoded by the cDNA for human cell surface antigen Fas can mediate apoptosis. Cell 1991, 66, 233–243. [Google Scholar]
- Suda, T.; Takahashi, T.; Golstein, P.; Nagata, S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell 1993, 75, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Leist, M.; Gantner, F.; Bohlinger, I.; Germann, P.G.; Tiegs, G.; Wendel, A. Murine hepatocyte apoptosis induced in vitro and in vivo by TNF-alpha requires transcriptional arrest. J. Immunol. 1994, 153, 1778–1788. [Google Scholar] [CrossRef] [PubMed]
- Nio, Y.; Zighelboim, J.; Berek, J.; Bonavida, B. Cycloheximide-induced modulation of TNF-mediated cytotoxicity in sensitive and resistant ovarian tumor cells. Cancer Chemother. Pharmacol. 1990, 26, 1–8. [Google Scholar] [CrossRef]
- Wajant, H.; Haas, E.; Schwenzer, R.; Muhlenbeck, F.; Kreuz, S.; Schubert, G.; Grell, M.; Smith, C.; Scheurich, P. Inhibition of death receptor-mediated gene induction by a cycloheximide-sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD). J. Biol. Chem. 2000, 275, 24357–24366. [Google Scholar] [CrossRef]
- Tanaka, M.; Suda, T.; Yatomi, T.; Nakamura, N.; Nagata, S. Lethal effect of recombinant human Fas ligand in mice pretreated with Propionibacterium acnes. J. Immunol. 1997, 158, 2303–2309. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, V.; Freudenberg, M.A.; Galanos, C. Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J. Exp. Med. 1987, 165, 657–663. [Google Scholar] [CrossRef]
- Schuchmann, M.; Varfolomeev, E.E.; Hermann, F.; Rueckert, F.; Strand, D.; Koehler, H.; Strand, S.; Lohse, A.W.; Wallach, D.; Galle, P.R. Dominant negative MORT1/FADD rescues mice from CD95 and TNF-induced liver failure. Hepatol. (Baltim. Md. 2003, 37, 129–135. [Google Scholar] [CrossRef]
- Eichacker, P.Q.; Hoffman, W.D.; Farese, A.; Banks, S.M.; Kuo, G.C.; MacVittie, T.J.; Natanson, C. TNF but not IL-1 in dogs causes lethal lung injury and multiple organ dysfunction similar to human sepsis. J. Appl. Physiol. 1991, 71, 1979–1989. [Google Scholar] [CrossRef]
- Hinshaw, L.B.; Emerson, T.E., Jr.; Taylor, F.B., Jr.; Chang, A.C.; Duerr, M.; Peer, G.T.; Flournoy, D.J.; White, G.L.; Kosanke, S.D.; Murray, C.K.; et al. Lethal Staphylococcus aureus-induced shock in primates: prevention of death with anti-TNF antibody. J. Trauma. 1992, 33, 568–573. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.; Huang, J.; Shu, H.B.; Baichwal, V.; Goeddel, D.V. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity 1996, 4, 387–396. [Google Scholar]
- Kelliher, M.A.; Grimm, S.; Ishida, Y.; Kuo, F.; Stanger, B.Z.; Leder, P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity 1998, 8, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Ting, A.T.; Bertrand, M.J.M. More to Life than NF-kappaB in TNFR1 Signaling. Trends Immunol. 2016, 37, 535–545. [Google Scholar] [CrossRef]
- Hsu, H.; Shu, H.B.; Pan, M.G.; Goeddel, D.V. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell 1996, 84, 299–308. [Google Scholar]
- Hsu, H.; Xiong, J.; Goeddel, D.V. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell 1995, 81, 495–504. [Google Scholar]
- Varfolomeev, E.E.; Schuchmann, M.; Luria, V.; Chiannilkulchai, N.; Beckmann, J.S.; Mett, I.L.; Rebrikov, D.; Brodianski, V.M.; Kemper, O.C.; Kollet, O.; et al. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity 1998, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Boldin, M.P.; Goncharov, T.M.; Goltsev, Y.V.; Wallach, D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell 1996, 85, 803–815. [Google Scholar] [CrossRef]
- Chhibber-Goel, J.; Coleman-Vaughan, C.; Agrawal, V.; Sawhney, N.; Hickey, E.; Powell, J.C.; McCarthy, J.V. gamma-Secretase Activity Is Required for Regulated Intramembrane Proteolysis of Tumor Necrosis Factor (TNF) Receptor 1 and TNF-mediated Pro-apoptotic Signaling. J. Biol. Chem. 2016, 291, 5971–5985. [Google Scholar] [CrossRef]
- Chan, F.K.; Chun, H.J.; Zheng, L.; Siegel, R.M.; Bui, K.L.; Lenardo, M.J. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 2000, 288, 2351–2354. [Google Scholar] [CrossRef] [PubMed]
- Albogami, S.; Todd, I.; Negm, O.; Fairclough, L.C.; Tighe, P.J. Mutations in the binding site of TNFR1 PLAD reduce homologous interactions but can enhance antagonism of wild-type TNFR1 activity. Immunology 2021, 164, 637–654. [Google Scholar] [CrossRef]
- Du, G.; Zhao, L.; Zheng, Y.; Belfetmi, A.; Cai, T.; Xu, B.; Heyninck, K.; Van Den Heede, K.; Buyse, M.A.; Fontana, P.; et al. Autoinhibitory structure of preligand association state implicates a new strategy to attain effective DR5 receptor activation. Cell Res. 2023, 33, 131–146. [Google Scholar] [CrossRef]
- Clancy, L.; Mruk, K.; Archer, K.; Woelfel, M.; Mongkolsapaya, J.; Screaton, G.; Lenardo, M.J.; Chan, F.K. Preligand assembly domain-mediated ligand- independent association between TRAIL receptor 4 (TR4) and TR2 regulates TRAIL-induced apoptosis. Proc. Natl. Acad. Sci. U S A 2005, 102, 18099–18104. [Google Scholar] [CrossRef]
- Deng, G.M.; Liu, L.; Tsokos, G.C. Targeted tumor necrosis factor receptor I preligand assembly domain improves skin lesions in MRL/lpr mice. Arthritis Rheum. 2010, 62, 2424–2431. [Google Scholar] [CrossRef]
- Deng, G.M.; Zheng, L.; Chan, F.K.; Lenardo, M. Amelioration of inflammatory arthritis by targeting the pre-ligand assembly domain of tumor necrosis factor receptors. Nat. Med. 2005, 11, 1066–1072. [Google Scholar] [CrossRef]
- Wang, Y.L.; Chou, F.C.; Chen, S.J.; Lin, S.H.; Chang, D.M.; Sytwu, H.K. Targeting pre-ligand assembly domain of TNFR1 ameliorates autoimmune diseases - an unrevealed role in downregulation of Th17 cells. J. Autoimmun. 2011, 37, 160–170. [Google Scholar] [CrossRef]
- Micheau, O.; Rizzi, M.; Smulski, C.R. Editorial: TNFR Superfamily Oligomerization and Signaling. Front. Cell Dev. Biol. 2021, 9, 682472. [Google Scholar] [CrossRef]
- Vanamee, E.S.; Faustman, D.L. The benefits of clustering in TNF receptor superfamily signaling. Front. Immunol. 2023, 14, 1225704. [Google Scholar] [CrossRef]
- Pan, L.; Fu, T.M.; Zhao, W.; Zhao, L.; Chen, W.; Qiu, C.; Liu, W.; Liu, Z.; Piai, A.; Fu, Q.; et al. Higher-Order Clustering of the Transmembrane Anchor of DR5 Drives Signaling. Cell 2019, 176, 1477–1489. [Google Scholar] [CrossRef]
- Valley, C.C.; Lewis, A.K.; Mudaliar, D.J.; Perlmutter, J.D.; Braun, A.R.; Karim, C.B.; Thomas, D.D.; Brody, J.R.; Sachs, J.N. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) induces death receptor 5 networks that are highly organized. J. Biol. Chem. 2012, 287, 21265–21278. [Google Scholar] [CrossRef] [PubMed]
- Lewis, A.K.; Valley, C.C.; Peery, S.L.; Brummel, B.; Braun, A.R.; Karim, C.B.; Sachs, J.N. Death Receptor 5 Networks Require Membrane Cholesterol for Proper Structure and Function. J. Mol. Biol. 2016, 428 (24 Pt A), 4843–4855. [Google Scholar] [CrossRef]
- Zhao, L.; Fu, Q.; Pan, L.; Piai, A.; Chou, J.J. The Diversity and Similarity of Transmembrane Trimerization of TNF Receptors. Front. Cell Dev. Biol. 2020, 8, 569684. [Google Scholar] [CrossRef]
- Frazzette, N.; Cruz, A.C.; Wu, X.; Hammer, J.A.; Lippincott-Schwartz, J.; Siegel, R.M.; Sengupta, P. Super-Resolution Imaging of Fas/CD95 Reorganization Induced by Membrane-Bound Fas Ligand Reveals Nanoscale Clustering Upstream of FADD Recruitment. Cells 2022, 11. [Google Scholar] [CrossRef]
- Scott, F.L.; Stec, B.; Pop, C.; Dobaczewska, M.K.; Lee, J.J.; Monosov, E.; Robinson, H.; Salvesen, G.S.; Schwarzenbacher, R.; Riedl, S.J. The Fas-FADD death domain complex structure unravels signalling by receptor clustering. Nature 2009, 457, 1019–1022. [Google Scholar] [CrossRef]
- Salvesen, G.S.; Riedl, S.J. Structure of the Fas/FADD complex: a conditional death domain complex mediating signaling by receptor clustering. Cell Cycle (Georget. Tex. 2009, 8, 2723–2727. [Google Scholar] [CrossRef]
- Ho, K.L.; Harrington, H.A. Bistability in apoptosis by receptor clustering. PLoS Comput. Biol. 2010, 6, e1000956. [Google Scholar] [CrossRef] [PubMed]
- Micheau, O. Posttranslational Modifications and Death Receptor Signalling. In TRAIL, Fas Ligand, TNF and TLR3 in Cancer, Micheau, O. Ed.; Springer International Publishing, 2017; pp 247-290.
- Micheau, O. Regulation of TNF-Related Apoptosis-Inducing Ligand Signaling by Glycosylation. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Wagner, K.W.; Punnoose, E.A.; Januario, T.; Lawrence, D.A.; Pitti, R.M.; Lancaster, K.; Lee, D.; von Goetz, M.; Yee, S.F.; Totpal, K.; et al. Death-receptor O-glycosylation controls tumor-cell sensitivity to the proapoptotic ligand Apo2L/TRAIL. Nat. Med. 2007, 13, 1070–1077. [Google Scholar] [CrossRef]
- Jiang, Y.; Wen, T.; Yan, R.; Kim, S.R.; Stowell, S.R.; Wang, W.; Wang, Y.; An, G.; Cummings, R.D.; Ju, T. O-glycans on death receptors in cells modulate their sensitivity to TRAIL-induced apoptosis through affecting on their stability and oligomerization. FASEB J. 2020, 34, 11786–11801. [Google Scholar] [CrossRef]
- Dufour, F.; Rattier, T.; Shirley, S.; Picarda, G.; Constantinescu, A.A.; Morle, A.; Zakaria, A.B.; Marcion, G.; Causse, S.; Szegezdi, E.; et al. N-glycosylation of mouse TRAIL-R and human TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ. 2017, 24, 500–510. [Google Scholar] [CrossRef] [PubMed]
- Estornes, Y.; Dondelinger, Y.; Weber, K.; Bruggeman, I.; Peall, A.; MacFarlane, M.; Lebecque, S.; Vandenabeele, P.; Bertrand, M.J.M. N-glycosylation of mouse TRAIL-R restrains TRAIL-induced apoptosis. Cell Death Dis. 2018, 9, 494. [Google Scholar] [CrossRef]
- Shatnyeva, O.M.; Kubarenko, A.V.; Weber, C.E.; Pappa, A.; Schwartz-Albiez, R.; Weber, A.N.; Krammer, P.H.; Lavrik, I.N. Modulation of the CD95-induced apoptosis: the role of CD95 N-glycosylation. PLoS One 2011, 6, e19927. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Shiraishi, T.; Horinaka, M.; Wakada, M.; Sakai, T. Glycosylation modulates TRAIL-R1/death receptor 4 protein: different regulations of two pro-apoptotic receptors for TRAIL by tunicamycin. Oncol. Rep. 2007, 18, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Corti, A.; Merli, S.; Bagnasco, L.; D’Ambrosio, F.; Marino, M.; Cassani, G. Identification of two forms (31-33 and 48 kD) of the urinary soluble p55 tumor necrosis factor receptor that are differentially N- and O-glycosylated. J. Interferon Cytokine Res. : Off. J. Int. Soc. Interferon Cytokine Res. 1995, 15, 143–152. [Google Scholar] [CrossRef]
- de Vreede, G.; Morrison, H.A.; Houser, A.M.; Boileau, R.M.; Andersen, D.; Colombani, J.; Bilder, D. A Drosophila Tumor Suppressor Gene Prevents Tonic TNF Signaling through Receptor N-Glycosylation. Dev. Cell 2018, 45, 595–605. [Google Scholar] [CrossRef]
- Liang, Y.; Xu, W.; Liu, S.; Chi, J.; Zhang, J.; Sui, A.; Wang, L.; Liang, Z.; Li, D.; Chen, Y.; et al. N-Acetyl-Glucosamine Sensitizes Non-Small Cell Lung Cancer Cells to TRAIL-Induced Apoptosis by Activating Death Receptor 5. Cell Physiol. Biochem. 2018, 45, 2054–2070. [Google Scholar] [CrossRef]
- Zhang, B.; van Roosmalen, I.A.M.; Reis, C.R.; Setroikromo, R.; Quax, W.J. Death receptor 5 is activated by fucosylation in colon cancer cells. FEBS J. 2019, 286, 555–571. [Google Scholar] [CrossRef]
- Jeon, M.Y.; Seo, S.U.; Woo, S.M.; Min, K.J.; Byun, H.S.; Hur, G.M.; Kang, S.C.; Kwon, T.K. Oridonin enhances TRAIL-induced apoptosis through GALNT14-mediated DR5 glycosylation. Biochimie 2019, 165, 108–114. [Google Scholar] [CrossRef]
- Peter, M.E.; Hellbardt, S.; Schwartz-Albiez, R.; Westendorp, M.O.; Walczak, H.; Moldenhauer, G.; Grell, M.; Krammer, P.H. Cell surface sialylation plays a role in modulating sensitivity towards APO-1-mediated apoptotic cell death. Cell Death Differ. 1995, 2, 163–171. [Google Scholar]
- Liu, Z.; Swindall, A.F.; Kesterson, R.A.; Schoeb, T.R.; Bullard, D.C.; Bellis, S.L. ST6Gal-I regulates macrophage apoptosis via alpha2-6 sialylation of the TNFR1 death receptor. J. Biol. Chem. 2011, 286, 39654–39662. [Google Scholar] [CrossRef] [PubMed]
- Swindall, A.F.; Bellis, S.L. Sialylation of the Fas death receptor by ST6Gal-I provides protection against Fas-mediated apoptosis in colon carcinoma cells. J. Biol. Chem. 2011, 286, 22982–22990. [Google Scholar] [CrossRef] [PubMed]
- Holdbrooks, A.T.; Britain, C.M.; Bellis, S.L. ST6Gal-I sialyltransferase promotes tumor necrosis factor (TNF)-mediated cancer cell survival via sialylation of the TNF receptor 1 (TNFR1) death receptor. J. Biol. Chem. 2018, 293, 1610–1622. [Google Scholar] [CrossRef]
- Lee, Y.J.; Song, Y.K.; Song, J.J.; Siervo-Sassi, R.R.; Kim, H.R.; Li, L.; Spitz, D.R.; Lokshin, A.; Kim, J.H. Reconstitution of galectin-3 alters glutathione content and potentiates TRAIL-induced cytotoxicity by dephosphorylation of Akt. Exp. Cell Res. 2003, 288, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Oka, N.; Nakahara, S.; Takenaka, Y.; Fukumori, T.; Hogan, V.; Kanayama, H.O.; Yanagawa, T.; Raz, A. Galectin-3 inhibits tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis by activating Akt in human bladder carcinoma cells. Cancer Res. 2005, 65, 7546–7553. [Google Scholar] [CrossRef]
- Lin, C.I.; Whang, E.E.; Abramson, M.A.; Donner, D.B.; Bertagnolli, M.M.; Moore, F.D., Jr.; Ruan, D.T. Galectin-3 regulates apoptosis and doxorubicin chemoresistance in papillary thyroid cancer cells. Biochem. Biophys. Res. Commun. 2009, 379, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Mazurek, N.; Byrd, J.C.; Sun, Y.; Ueno, S.; Bresalier, R.S. A galectin-3 sequence polymorphism confers TRAIL sensitivity to human breast cancer cells. Cancer 2011, 117, 4375–4380. [Google Scholar] [CrossRef]
- Mazurek, N.; Byrd, J.C.; Sun, Y.; Hafley, M.; Ramirez, K.; Burks, J.; Bresalier, R.S. Cell-surface galectin-3 confers resistance to TRAIL by impeding trafficking of death receptors in metastatic colon adenocarcinoma cells. Cell Death Differ. 2012, 19, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Saksida, T.; Nikolic, I.; Vujicic, M.; Nilsson, U.J.; Leffler, H.; Lukic, M.L.; Stojanovic, I.; Stosic-Grujicic, S. Galectin-3 deficiency protects pancreatic islet cells from cytokine-triggered apoptosis in vitro. J. Cell. Physiol. 2013, 228, 1568–1576. [Google Scholar] [CrossRef]
- Li, J.; Sun, R.R.; Yu, Z.J.; Liang, H.; Shen, S.; Kan, Q. Galectin-1 Modulates the Survival and Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Sensitivity in Human Hepatocellular Carcinoma Cells. Cancer Biother. Radiopharm. 2015, 30, 336–341. [Google Scholar] [CrossRef]
- Lee, H.; Oh, Y.; Jeon, Y.J.; Lee, S.Y.; Kim, H.; Lee, H.J.; Jung, Y.K. DR4-Ser424 O-GlcNAcylation Promotes Sensitization of TRAIL-Tolerant Persisters and TRAIL-Resistant Cancer Cells to Death. Cancer Res. 2019, 79, 2839–2852. [Google Scholar] [CrossRef]
- Yang, S.Z.; Xu, F.; Yuan, K.; Sun, Y.; Zhou, T.; Zhao, X.; McDonald, J.M.; Chen, Y. Regulation of pancreatic cancer TRAIL resistance by protein O-GlcNAcylation. Lab. Investig. ; A J. Tech. Methods Pathol. 2020, 100, 777–785. [Google Scholar] [CrossRef]
- Xue, J.; Pan, X.; Peng, T.; Duan, M.; Du, L.; Zhuang, X.; Cai, X.; Yi, X.; Fu, Y.; Li, S. Auto Arginine-GlcNAcylation Is Crucial for Bacterial Pathogens in Regulating Host Cell Death. Front. Cell Infect. Microbiol. 2020, 10, 197. [Google Scholar] [CrossRef]
- Xue, J.; Hu, S.; Huang, Y.; Zhang, Q.; Yi, X.; Pan, X.; Li, S. Arg-GlcNAcylation on TRADD by NleB and SseK1 Is Crucial for Bacterial Pathogenesis. Front. Cell Dev. Biol. 2020, 8, 641. [Google Scholar] [CrossRef]
- Li, S.; Zhang, L.; Yao, Q.; Li, L.; Dong, N.; Rong, J.; Gao, W.; Ding, X.; Sun, L.; Chen, X.; et al. Pathogen blocks host death receptor signalling by arginine GlcNAcylation of death domains. Nature 2013, 501, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Kim, Y.; Ji, S.; Kim, H.B.; Jung, H.; Yi, E.C.; Lee, Y.H.; Shin, I.; Yang, W.H.; Cho, J.W. O-GlcNAcylation of RIPK1 rescues red blood cells from necroptosis. Front. Immunol. 2023, 14, 1160490. [Google Scholar] [CrossRef]
- Rossin, A.; Derouet, M.; Abdel-Sater, F.; Hueber, A.O. Palmitoylation of the TRAIL receptor DR4 confers an efficient TRAIL-induced cell death signalling. Biochem. J. 2009, 419, 182, 185–192. [Google Scholar] [CrossRef]
- Chakrabandhu, K.; Herincs, Z.; Huault, S.; Dost, B.; Peng, L.; Conchonaud, F.; Marguet, D.; He, H.T.; Hueber, A.O. Palmitoylation is required for efficient Fas cell death signaling. EMBO J. 2007, 26, 209–220. [Google Scholar] [CrossRef]
- Feig, C.; Tchikov, V.; Schutze, S.; Peter, M.E. Palmitoylation of CD95 facilitates formation of SDS-stable receptor aggregates that initiate apoptosis signaling. EMBO J. 2007, 26, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Rossin, A.; Durivault, J.; Chakhtoura-Feghali, T.; Lounnas, N.; Gagnoux-Palacios, L.; Hueber, A.O. Fas palmitoylation by the palmitoyl acyltransferase DHHC7 regulates Fas stability. Cell Death Differ. 2015, 22, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Zingler, P.; Sarchen, V.; Glatter, T.; Caning, L.; Saggau, C.; Kathayat, R.S.; Dickinson, B.C.; Adam, D.; Schneider-Brachert, W.; Schutze, S.; et al. Palmitoylation is required for TNF-R1 signaling. Cell Commun. Signal 2019, 17, 90. [Google Scholar] [CrossRef]
- Anel, A.; Bosque, A.; Naval, J.; Pineiro, A.; Larrad, L.; Alava, M.A.; Martinez-Lorenzo, M.J. Apo2L/TRAIL and immune regulation. Front. Biosci. 2007, 12, 2074–2084. [Google Scholar] [CrossRef]
- Bossi, F.; Bernardi, S.; Zauli, G.; Secchiero, P.; Fabris, B. TRAIL modulates the immune system and protects against the development of diabetes. J. Immunol. Res. 2015, 2015, 680749. [Google Scholar] [CrossRef]
- Sag, D.; Ayyildiz, Z.O.; Gunalp, S.; Wingender, G. The Role of TRAIL/DRs in the Modulation of Immune Cells and Responses. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Burgaletto, C.; Munafo, A.; Di Benedetto, G.; De Francisci, C.; Caraci, F.; Di Mauro, R.; Bucolo, C.; Bernardini, R.; Cantarella, G. The immune system on the TRAIL of Alzheimer’s disease. J. Neuroinflammation 2020, 17, 298. [Google Scholar] [CrossRef]
- Cardoso Alves, L.; Corazza, N.; Micheau, O.; Krebs, P. The multifaceted role of TRAIL signaling in cancer and immunity. FEBS J. 2021, 288, 5530–5554. [Google Scholar] [CrossRef]
- Harith, H.H.; Morris, M.J.; Kavurma, M.M. On the TRAIL of obesity and diabetes. Trends Endocrinol. Metab. 2013, 24, 578–587. [Google Scholar] [CrossRef]
- Remuzgo-Martinez, S.; Genre, F.; Lopez-Mejias, R.; Ubilla, B.; Mijares, V.; Pina, T.; Corrales, A.; Blanco, R.; Martin, J.; Llorca, J.; et al. Expression of osteoprotegerin and its ligands, RANKL and TRAIL, in rheumatoid arthritis. Sci. Rep. 2016, 6, 29713. [Google Scholar] [CrossRef]
- Gao, S.; Fang, Y.; Tu, S.; Chen, H.; Shao, A. Insight into the divergent role of TRAIL in non-neoplastic neurological diseases. J. Cell Mol. Med. 2020, 24, 11070–11083. [Google Scholar] [CrossRef]
- Kelland, E.; Patil, M.S.; Patel, S.; Cartland, S.P.; Kavurma, M.M. The Prognostic, Diagnostic, and Therapeutic Potential of TRAIL Signalling in Cardiovascular Diseases. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef]
- Bosque, A.; Pardo, J.; Martinez-Lorenzo, M.J.; Lasierra, P.; Larrad, L.; Marzo, I.; Naval, J.; Anel, A. Human CD8+ T cell blasts are more sensitive than CD4+ T cell blasts to regulation by APO2L/TRAIL. Eur. J. Immunol. 2005, 35, 1812–1821. [Google Scholar] [CrossRef]
- Kayagaki, N.; Yamaguchi, N.; Nakayama, M.; Eto, H.; Okumura, K.; Yagita, H. Type I interferons (IFNs) regulate tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) expression on human T cells: A novel mechanism for the antitumor effects of type I IFNs. J. Exp. Med. 1999, 189, 1451–1460. [Google Scholar] [CrossRef]
- Badovinac, V.P.; Messingham, K.A.; Griffith, T.S.; Harty, J.T. TRAIL deficiency delays, but does not prevent, erosion in the quality of “helpless” memory CD8 T cells. J. Immunol. 2006, 177, 999–1006. [Google Scholar] [CrossRef]
- Janssen, E.M.; Droin, N.M.; Lemmens, E.E.; Pinkoski, M.J.; Bensinger, S.J.; Ehst, B.D.; Griffith, T.S.; Green, D.R.; Schoenberger, S.P. CD4+ T-cell help controls CD8+ T-cell memory via TRAIL-mediated activation-induced cell death. Nature 2005, 434, 88–93. [Google Scholar] [CrossRef] [PubMed]
- Sacks, J.A.; Bevan, M.J. TRAIL deficiency does not rescue impaired CD8+ T cell memory generated in the absence of CD4+ T cell help. J. Immunol. 2008, 180, 4570–4576. [Google Scholar] [CrossRef]
- Wolkers, M.C.; Gerlach, C.; Arens, R.; Janssen, E.M.; Fitzgerald, P.; Schumacher, T.N.; Medema, J.P.; Green, D.R.; Schoenberger, S.P. Nab2 regulates secondary CD8+ T-cell responses through control of TRAIL expression. Blood 2012, 119, 798–804. [Google Scholar] [CrossRef]
- Zhang, X.R.; Zhang, L.Y.; Devadas, S.; Li, L.; Keegan, A.D.; Shi, Y.F. Reciprocal expression of TRAIL and CD95L in Th1 and Th2 cells: role of apoptosis in T helper subset differentiation. Cell Death Differ. 2003, 10, 203–210. [Google Scholar] [CrossRef]
- Martinez-Lorenzo, M.J.; Alava, M.A.; Gamen, S.; Kim, K.J.; Chuntharapai, A.; Pineiro, A.; Naval, J.; Anel, A. Involvement of APO2 ligand/TRAIL in activation-induced death of Jurkat and human peripheral blood T cells. Eur. J. Immunol. 1998, 28, 2714–2725. [Google Scholar] [CrossRef]
- Hamilton, S.E.; Wolkers, M.C.; Schoenberger, S.P.; Jameson, S.C. The generation of protective memory-like CD8+ T cells during homeostatic proliferation requires CD4+ T cells. Nat. Immunol. 2006, 7, 475–481. [Google Scholar] [CrossRef]
- Sedger, L.M.; Katewa, A.; Pettersen, A.K.; Osvath, S.R.; Farrell, G.C.; Stewart, G.J.; Bendall, L.J.; Alexander, S.I. Extreme lymphoproliferative disease and fatal autoimmune thrombocytopenia in FasL- and TRAIL-double deficient mice. Blood 2010. [Google Scholar] [CrossRef]
- Takeda, K.; Hayakawa, Y.; Smyth, M.J.; Kayagaki, N.; Yamaguchi, N.; Kakuta, S.; Iwakura, Y.; Yagita, H.; Okumura, K. Involvement of tumor necrosis factor-related apoptosis-inducing ligand in surveillance of tumor metastasis by liver natural killer cells. Nat. Med. 2001, 7, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Pimentel, J.M.; Zhou, J.Y.; Wu, G.S. The Role of TRAIL in Apoptosis and Immunosurveillance in Cancer. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Chaperot, L.; Blum, A.; Manches, O.; Lui, G.; Angel, J.; Molens, J.P.; Plumas, J. Virus or TLR agonists induce TRAIL-mediated cytotoxic activity of plasmacytoid dendritic cells. J. Immunol. 2006, 176, 248–255. [Google Scholar] [CrossRef]
- Griffith, T.S.; Brincks, E.L.; Gurung, P.; Kucaba, T.A.; Ferguson, T.A. Systemic immunological tolerance to ocular antigens is mediated by TRAIL-expressing CD8+ T cells. J. Immunol. 2011, 186, 791–798. [Google Scholar] [CrossRef]
- Phillips, T.A.; Ni, J.; Pan, G.; Ruben, S.M.; Wei, Y.F.; Pace, J.L.; Hunt, J.S. TRAIL (Apo-2L) and TRAIL receptors in human placentas: implications for immune privilege. J. Immunol. 1999, 162, 6053–6059. [Google Scholar] [CrossRef] [PubMed]
- Stoyanova, A.K.; Sattler, A.; Hahn, E.M.; Hering, N.A.; Arndt, M.; Lauscher, J.C.; Speichinger-Hillenberg, F.; Kotsch, K.; Berg, A.K.; Beyer, K. Immune Phenotypic Characterization of a TRAIL-Knockout Mouse. Cancers (Basel) 2023, 15. [Google Scholar] [CrossRef]
- Delacher, M.; Schmidleithner, L.; Simon, M.; Stuve, P.; Sanderink, L.; Hotz-Wagenblatt, A.; Wuttke, M.; Schambeck, K.; Ruhland, B.; Hofmann, V.; et al. The effector program of human CD8 T cells supports tissue remodeling. J. Exp. Med. 2024, 221. [Google Scholar] [CrossRef]
- Chyuan, I.T.; Tsai, H.F.; Wu, C.S.; Hsu, P.N. TRAIL suppresses gut inflammation and inhibits colitogeic T-cell activation in experimental colitis via an apoptosis-independent pathway. Mucosal Immunol. 2019, 12, 980–989. [Google Scholar] [CrossRef]
- Chyuan, I.T.; Tsai, H.F.; Liao, H.J.; Wu, C.S.; Hsu, P.N. An apoptosis-independent role of TRAIL in suppressing joint inflammation and inhibiting T-cell activation in inflammatory arthritis. Cell Mol. Immunol. 2018, 15, 846–857. [Google Scholar] [CrossRef]
- Song, K.; Chen, Y.; Goke, R.; Wilmen, A.; Seidel, C.; Goke, A.; Hilliard, B.; Chen, Y. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) is an inhibitor of autoimmune inflammation and cell cycle progression. J. Exp. Med. 2000, 191, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Chyuan, I.T.; Hsu, P.N. TRAIL regulates T cell activation and suppresses inflammation in autoimmune diseases. Cell Mol. Immunol. 2020, 17, 1281–1283. [Google Scholar] [CrossRef]
- Wang, S.H.; Cao, Z.; Wolf, J.M.; Van Antwerp, M.; Baker, J.R., Jr. Death ligand tumor necrosis factor-related apoptosis-inducing ligand inhibits experimental autoimmune thyroiditis. Endocrinology 2005, 146, 4721–4726. [Google Scholar] [CrossRef]
- Yao, Q.; Seol, D.W.; Mi, Z.; Robbins, P.D. Intra-articular injection of recombinant TRAIL induces synovial apoptosis and reduces inflammation in a rabbit knee model of arthritis. Arthritis Res. Ther. 2006, 8, R16. [Google Scholar] [CrossRef]
- Ikeda, T.; Hirata, S.; Fukushima, S.; Matsunaga, Y.; Ito, T.; Uchino, M.; Nishimura, Y.; Senju, S. Dual effects of TRAIL in suppression of autoimmunity: the inhibition of Th1 cells and the promotion of regulatory T cells. J. Immunol. 2010, 185, 5259–5267. [Google Scholar] [CrossRef]
- Cretney, E.; McQualter, J.L.; Kayagaki, N.; Yagita, H.; Bernard, C.C.; Grewal, I.S.; Ashkenazi, A.; Smyth, M.J. TNF-related apoptosis-inducing ligand (TRAIL)/Apo2L suppresses experimental autoimmune encephalomyelitis in mice. Immunol. Cell Biol. 2005, 83, 511–519. [Google Scholar] [CrossRef]
- Annibaldi, A.; Walczak, H. Death Receptors and Their Ligands in Inflammatory Disease and Cancer. Cold Spring Harb. Perspect. Biol. 2020, 12. [Google Scholar] [CrossRef]
- McGrath, E.E.; Marriott, H.M.; Lawrie, A.; Francis, S.E.; Sabroe, I.; Renshaw, S.A.; Dockrell, D.H.; Whyte, M.K. TNF-related apoptosis-inducing ligand (TRAIL) regulates inflammatory neutrophil apoptosis and enhances resolution of inflammation. J. Leukoc. Biol. 2011, 90, 855–865. [Google Scholar] [CrossRef]
- Zahn, S.; Rehkamper, C.; Ferring-Schmitt, S.; Bieber, T.; Tuting, T.; Wenzel, J. Interferon-alpha stimulates TRAIL expression in human keratinocytes and peripheral blood mononuclear cells: implications for the pathogenesis of cutaneous lupus erythematosus. Br. J. Dermatol. 2011, 165, 1118–1123. [Google Scholar] [CrossRef]
- Nguyen, V.; Cudrici, C.; Zernetkina, V.; Niculescu, F.; Rus, H.; Drachenberg, C.; Rus, V. TRAIL, DR4 and DR5 are upregulated in kidneys from patients with lupus nephritis and exert proliferative and proinflammatory effects. Clin. Immunol. 2009, 132, 32–42. [Google Scholar] [CrossRef]
- Daigle, I.; Simon, H.U. Alternative functions for TRAIL receptors in eosinophils and neutrophils. Swiss Med. Wkly. 2001, 131, 231–237. [Google Scholar]
- Robertson, N.M.; Zangrilli, J.G.; Steplewski, A.; Hastie, A.; Lindemeyer, R.G.; Planeta, M.A.; Smith, M.K.; Innocent, N.; Musani, A.; Pascual, R.; et al. Differential expression of TRAIL and TRAIL receptors in allergic asthmatics following segmental antigen challenge: evidence for a role of TRAIL in eosinophil survival. J. Immunol. 2002, 169, 5986–5996. [Google Scholar] [CrossRef] [PubMed]
- Weckmann, M.; Collison, A.; Simpson, J.L.; Kopp, M.V.; Wark, P.A.; Smyth, M.J.; Yagita, H.; Matthaei, K.I.; Hansbro, N.; Whitehead, B.; et al. Critical link between TRAIL and CCL20 for the activation of TH2 cells and the expression of allergic airway disease. Nat. Med. 2007, 13, 1308–1315. [Google Scholar] [CrossRef]
- Weckmann, M.; Kopp, M.V.; Heinzmann, A.; Mattes, J. Haplotypes covering the TNFSF10 gene are associated with bronchial asthma. Pediatr. Allergy Immunol. 2011, 22 Pt 1, 25–30. [Google Scholar] [CrossRef]
- Zauli, G.; Pandolfi, A.; Gonelli, A.; Di Pietro, R.; Guarnieri, S.; Ciabattoni, G.; Rana, R.; Vitale, M.; Secchiero, P. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) sequentially upregulates nitric oxide and prostanoid production in primary human endothelial cells. Circ. Res. 2003, 92, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Gonelli, A.; Carnevale, E.; Corallini, F.; Rizzardi, C.; Zacchigna, S.; Melato, M.; Zauli, G. Evidence for a proangiogenic activity of TNF-related apoptosis-inducing ligand. Neoplasia (New York N.Y 2004, 6, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Cartland, S.P.; Genner, S.W.; Zahoor, A.; Kavurma, M.M. Comparative Evaluation of TRAIL, FGF-2 and VEGF-A-Induced Angiogenesis In Vitro and In Vivo. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef]
- Kavurma, M.M.; Schoppet, M.; Bobryshev, Y.V.; Khachigian, L.M.; Bennett, M.R. TRAIL stimulates proliferation of vascular smooth muscle cells via activation of NF-kappaB and induction of insulin-like growth factor-1 receptor. J. Biol. Chem. 2008, 283, 7754–7762. [Google Scholar] [CrossRef]
- Na, H.J.; Hwang, J.Y.; Lee, K.S.; Choi, Y.K.; Choe, J.; Kim, J.Y.; Moon, H.E.; Kim, K.W.; Koh, G.Y.; Lee, H.; et al. TRAIL negatively regulates VEGF-induced angiogenesis via caspase-8-mediated enzymatic and non-enzymatic functions. Angiogenesis 2014, 17, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Di Bartolo, B.A.; Cartland, S.P.; Prado-Lourenco, L.; Griffith, T.S.; Gentile, C.; Ravindran, J.; Azahri, N.S.; Thai, T.; Yeung, A.W.; Thomas, S.R.; et al. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Promotes Angiogenesis and Ischemia-Induced Neovascularization Via NADPH Oxidase 4 (NOX4) and Nitric Oxide-Dependent Mechanisms. J. Am. Heart Assoc. 2015, 4. [Google Scholar] [CrossRef]
- Yen, M.L.; Tsai, H.F.; Wu, Y.Y.; Hwa, H.L.; Lee, B.H.; Hsu, P.N. TNF-related apoptosis-inducing ligand (TRAIL) induces osteoclast differentiation from monocyte/macrophage lineage precursor cells. Mol. Immunol. 2008, 45, 2205–2213. [Google Scholar] [CrossRef]
- Sambandam, Y.; Baird, K.L.; Stroebel, M.; Kowal, E.; Balasubramanian, S.; Reddy, S.V. Microgravity Induction of TRAIL Expression in Preosteoclast Cells Enhances Osteoclast Differentiation. Sci. Rep. 2016, 6, 25143. [Google Scholar] [CrossRef]
- Freer-Prokop, M.; O’Flaherty, J.; Ross, J.A.; Weyman, C.M. Non-canonical role for the TRAIL receptor DR5/FADD/caspase pathway in the regulation of MyoD expression and skeletal myoblast differentiation. Differentiation 2009, 78, 205–212. [Google Scholar] [CrossRef]
- Wu, N.L.; Lee, T.A.; Tsai, T.L.; Lin, W.W. TRAIL-induced keratinocyte differentiation requires caspase activation and p63 expression. J. Invest. Dermatol. 2011, 131, 874–883. [Google Scholar] [CrossRef]
- Zoller, V.; Funcke, J.B.; Keuper, M.; Abd El Hay, M.; Debatin, K.M.; Wabitsch, M.; Fischer-Posovszky, P. TRAIL (TNF-related apoptosis-inducing ligand) inhibits human adipocyte differentiation via caspase-mediated downregulation of adipogenic transcription factors. Cell Death Dis. 2016, 7, e2412. [Google Scholar] [CrossRef]
- Dawson, S.H.; Arnold, N.D.; Pickworth, J.A.; Francis, S.E.; Lawrie, A. TRAIL Deficient Mice Are Protected from Sugen/Hypoxia Induced Pulmonary Arterial Hypertension. Diseases 2014, 2, 260–273. [Google Scholar] [CrossRef]
- Hameed, A.G.; Arnold, N.D.; Chamberlain, J.; Pickworth, J.A.; Paiva, C.; Dawson, S.; Cross, S.; Long, L.; Zhao, L.; Morrell, N.W.; et al. Inhibition of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) reverses experimental pulmonary hypertension. J. Exp. Med. 2012, 209, 1919–1935. [Google Scholar] [CrossRef]
- Liu, H.; Yang, E.; Lu, X.; Zuo, C.; He, Y.; Jia, D.; Zhu, Q.; Yu, Y.; Lv, A. Serum Levels of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Correlate with the Severity of Pulmonary Hypertension. Pulm. Pharmacol. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Zerbinati, C.; Rimondi, E.; Corallini, F.; Milani, D.; Grill, V.; Forti, G.; Capitani, S.; Zauli, G. TRAIL promotes the survival, migration and proliferation of vascular smooth muscle cells. Cell Mol. Life Sci. 2004, 61, 1965–1974. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Gonelli, A.; Carnevale, E.; Milani, D.; Pandolfi, A.; Zella, D.; Zauli, G. TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation 2003, 107, 2250–2256. [Google Scholar] [CrossRef]
- Song, S.; Choi, K.; Ryu, S.W.; Kang, S.W.; Choi, C. TRAIL promotes caspase-dependent pro-inflammatory responses via PKCdelta activation by vascular smooth muscle cells. Cell Death Dis. 2011, 2, e223. [Google Scholar] [CrossRef]
- Tanner, M.A.; Thomas, T.P.; Grisanti, L.A. Death receptor 5 contributes to cardiomyocyte hypertrophy through epidermal growth factor receptor transactivation. J. Mol. Cell. Cardiol. 2019, 136, 1–14. [Google Scholar] [CrossRef]
- Wu, Y.Y.; Hsu, J.L.; Wang, H.C.; Wu, S.J.; Hong, C.J.; Cheng, I.H. Alterations of the Neuroinflammatory Markers IL-6 and TRAIL in Alzheimer’s Disease. Dement. Geriatr. Cogn. Dis. Extra 2015, 5, 424–434. [Google Scholar] [CrossRef]
- Uberti, D.; Ferrari-Toninelli, G.; Bonini, S.A.; Sarnico, I.; Benarese, M.; Pizzi, M.; Benussi, L.; Ghidoni, R.; Binetti, G.; Spano, P.; et al. Blockade of the tumor necrosis factor-related apoptosis inducing ligand death receptor DR5 prevents beta-amyloid neurotoxicity. Neuropsychopharmacol. : Off. Publ. Am. Coll. Neuropsychopharmacol. 2007, 32, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, D. A new TRAIL in Alzheimer’s disease therapy. Brain 2015, 138 Pt 1, 8–10. [Google Scholar] [CrossRef]
- Fossati, S.; Ghiso, J.; Rostagno, A. TRAIL death receptors DR4 and DR5 mediate cerebral microvascular endothelial cell apoptosis induced by oligomeric Alzheimer’s Abeta. Cell Death Dis. 2012, 3, e321. [Google Scholar] [CrossRef] [PubMed]
- Cantarella, G.; Di Benedetto, G.; Puzzo, D.; Privitera, L.; Loreto, C.; Saccone, S.; Giunta, S.; Palmeri, A.; Bernardini, R. Neutralization of TNFSF10 ameliorates functional outcome in a murine model of Alzheimer’s disease. Brain 2015, 138 Pt 1, 203–216. [Google Scholar] [CrossRef]
- Cartland, S.P.; Harith, H.H.; Genner, S.W.; Dang, L.; Cogger, V.C.; Vellozzi, M.; Di Bartolo, B.A.; Thomas, S.R.; Adams, L.A.; Kavurma, M.M. Non-alcoholic fatty liver disease, vascular inflammation and insulin resistance are exacerbated by TRAIL deletion in mice. Sci. Rep. 2017, 7, 1898. [Google Scholar] [CrossRef]
- Lee, M.; Shin, E.; Bae, J.; Cho, Y.; Lee, J.Y.; Lee, Y.H.; Lee, B.W.; Kang, E.S.; Cha, B.S. Dipeptidyl peptidase-4 inhibitor protects against non-alcoholic steatohepatitis in mice by targeting TRAIL receptor-mediated lipoapoptosis via modulating hepatic dipeptidyl peptidase-4 expression. Sci. Rep. 2020, 10, 19429. [Google Scholar] [CrossRef]
- Hirsova, P.; Weng, P.; Salim, W.; Bronk, S.F.; Griffith, T.S.; Ibrahim, S.H.; Gores, G.J. TRAIL Deletion Prevents Liver, but Not Adipose Tissue, Inflammation during Murine Diet-Induced Obesity. Hepatol. Commun. 2017, 1, 648–662. [Google Scholar] [CrossRef]
- Zheng, S.J.; Wang, P.; Tsabary, G.; Chen, Y.H. Critical roles of TRAIL in hepatic cell death and hepatic inflammation. J. Clin. Invest. 2004, 113, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Walczak, H. Death receptor-ligand systems in cancer, cell death, and inflammation. Cold Spring Harb. Perspect. Biol. 2013, 5, a008698. [Google Scholar] [CrossRef] [PubMed]
- Yen, M.L.; Hsu, P.N.; Liao, H.J.; Lee, B.H.; Tsai, H.F. TRAF-6 dependent signaling pathway is essential for TNF-related apoptosis-inducing ligand (TRAIL) induces osteoclast differentiation. PLoS One 2012, 7, e38048. [Google Scholar] [CrossRef] [PubMed]
- Chan, J.; Prado-Lourenco, L.; Khachigian, L.M.; Bennett, M.R.; Di Bartolo, B.A.; Kavurma, M.M. TRAIL promotes VSMC proliferation and neointima formation in a FGF-2-, Sp1 phosphorylation-, and NFkappaB-dependent manner. Circ. Res. 2010, 106, 1061–1071. [Google Scholar] [CrossRef]
- Lluis, J.M.; Nachbur, U.; Cook, W.D.; Gentle, I.E.; Moujalled, D.; Moulin, M.; Wong, W.W.; Khan, N.; Chau, D.; Callus, B.A.; et al. TAK1 is required for survival of mouse fibroblasts treated with TRAIL, and does so by NF-kappaB dependent induction of cFLIPL. PLoS One 2010, 5, e8620. [Google Scholar] [CrossRef] [PubMed]
- Azijli, K.; Weyhenmeyer, B.; Peters, G.J.; de Jong, S.; Kruyt, F.A. Non-canonical kinase signaling by the death ligand TRAIL in cancer cells: discord in the death receptor family. Cell Death Differ. 2013. [Google Scholar] [CrossRef]
- Chekkat, N.; Lombardo, C.M.; Seguin, C.; Lechner, M.C.; Dufour, F.; Nomine, Y.; De Giorgi, M.; Frisch, B.; Micheau, O.; Guichard, G.; et al. Relationship between the agonist activity of synthetic ligands of TRAIL-R2 and their cell surface binding modes. Oncotarget 2018, 9, 15566–15578. [Google Scholar] [CrossRef]
- Somasekharan, S.P.; Koc, M.; Morizot, A.; Micheau, O.; Sorensen, P.H.; Gaide, O.; Andera, L.; Martinou, J.C. TRAIL promotes membrane blebbing, detachment and migration of cells displaying a dysfunctional intrinsic pathway of apoptosis. Apoptosis 2013, 18, 324–336. [Google Scholar] [CrossRef] [PubMed]
- Fritsche, H.; Heilmann, T.; Tower, R.J.; Hauser, C.; von Au, A.; El-Sheikh, D.; Campbell, G.M.; Alp, G.; Schewe, D.; Hubner, S.; et al. TRAIL-R2 promotes skeletal metastasis in a breast cancer xenograft mouse model. Oncotarget 2015, 6, 9502–9516. [Google Scholar] [CrossRef]
- Vilimanovich, U.; Bumbasirevic, V. TRAIL induces proliferation of human glioma cells by c-FLIPL-mediated activation of ERK1/2. Cell Mol. Life Sci. 2008, 65, 814–826. [Google Scholar] [CrossRef]
- Zhang, H.; Qin, G.; Zhang, C.; Yang, H.; Liu, J.; Hu, H.; Wu, P.; Liu, S.; Yang, L.; Chen, X.; et al. TRAIL promotes epithelial-to-mesenchymal transition by inducing PD-L1 expression in esophageal squamous cell carcinomas. J. Exp. Clin. Cancer Res. : CR 2021, 40, 209. [Google Scholar] [CrossRef]
- Chaudhary, P.M.; Eby, M.; Jasmin, A.; Bookwalter, A.; Murray, J.; Hood, L. Death receptor 5, a new member of the TNFR family, and DR4 induce FADD-dependent apoptosis and activate the NF-kappaB pathway. Immunity 1997, 7, 821–830. [Google Scholar] [CrossRef]
- Jeremias, I.; Debatin, K.M. TRAIL induces apoptosis and activation of NFkappaB. Eur. Cytokine Netw. 1998, 9, 687–688. [Google Scholar] [PubMed]
- Humphreys, L.M.; Fox, J.P.; Higgins, C.A.; Majkut, J.; Sessler, T.; McLaughlin, K.; McCann, C.; Roberts, J.Z.; Crawford, N.T.; McDade, S.S.; et al. A revised model of TRAIL-R2 DISC assembly explains how FLIP(L) can inhibit or promote apoptosis. EMBO Rep. 2020, 21, e49254. [Google Scholar] [CrossRef]
- MacFarlane, M.; Harper, N.; Snowden, R.T.; Dyer, M.J.; Barnett, G.A.; Pringle, J.H.; Cohen, G.M. Mechanisms of resistance to TRAIL-induced apoptosis in primary B cell chronic lymphocytic leukaemia. Oncogene 2002, 21, 6809–6818. [Google Scholar] [CrossRef] [PubMed]
- Fullsack, S.; Rosenthal, A.; Wajant, H.; Siegmund, D. Redundant and receptor-specific activities of TRADD, RIPK1 and FADD in death receptor signaling. Cell Death Dis. 2019, 10, 122. [Google Scholar] [CrossRef]
- Chang, Z.; Dang, T.; Che, N.; Yu, H.; Chai, J.; Chen, W. Esophageal cancer cells convert the death signal from TRAIL into a stimulus for survival during acid/bile exposure. Dig. Liver Dis. 2020, 52, 1195–1200. [Google Scholar] [CrossRef]
- Cao, X.; Pobezinskaya, Y.L.; Morgan, M.J.; Liu, Z.G. The role of TRADD in TRAIL-induced apoptosis and signaling. FASEB J. 2011, 25, 1353–1358. [Google Scholar] [CrossRef]
- Lafont, E.; Kantari-Mimoun, C.; Draber, P.; De Miguel, D.; Hartwig, T.; Reichert, M.; Kupka, S.; Shimizu, Y.; Taraborrelli, L.; Spit, M.; et al. The linear ubiquitin chain assembly complex regulates TRAIL-induced gene activation and cell death. EMBO J. 2017, 36, 1147–1166. [Google Scholar] [CrossRef]
- Wajant, H. TRAIL- and TNF-induced signaling complexes-so similar yet so different. EMBO J. 2017, 36, 1117–1119. [Google Scholar] [CrossRef]
- Dorn, S.; Schoergenhofer, C.; Krainer, M.; Muller, M.; Jilma, B. LUBAC and ABIN-1 Modulate TRAIL-Based NF-kappaB Induction in Human Embryonic Kidney 293 Cells. Biores Open Access 2018, 7, 81–89. [Google Scholar] [CrossRef]
- Zhang, L.; Blackwell, K.; Workman, L.M.; Chen, S.; Pope, M.R.; Janz, S.; Habelhah, H. RIP1 Cleavage in the Kinase Domain Regulates TRAIL-Induced NF-kappaB Activation and Lymphoma Survival. Mol. Cell Biol. 2015, 35, 3324–3338. [Google Scholar] [CrossRef]
- Harper, N.; Farrow, S.N.; Kaptein, A.; Cohen, G.M.; MacFarlane, M. Modulation of tumor necrosis factor apoptosis-inducing ligand- induced NF-kappa B activation by inhibition of apical caspases. J. Biol. Chem. 2001, 276, 34743–34752. [Google Scholar] [CrossRef] [PubMed]
- Grimm, S.; Stanger, B.Z.; Leder, P. RIP and FADD: two “death domain”-containing proteins can induce apoptosis by convergent, but dissociable, pathways. Proc. Natl. Acad. Sci. U S A 1996, 93, 10923–10927. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Devin, A.; Cook, A.; Keane, M.M.; Kelliher, M.; Lipkowitz, S.; Liu, Z.G. The death domain kinase RIP is essential for TRAIL (Apo2L)-induced activation of IkappaB kinase and c-Jun N-terminal kinase. Mol. Cell Biol. 2000, 20, 6638–6645. [Google Scholar] [CrossRef]
- Azijli, K.; Yuvaraj, S.; van Roosmalen, I.; Flach, K.; Giovannetti, E.; Peters, G.J.; de Jong, S.; Kruyt, F.A. MAPK p38 and JNK have opposing activities on TRAIL-induced apoptosis activation in NSCLC H460 cells that involves RIP1 and caspase-8 and is mediated by Mcl-1. Apoptosis 2013, 18, 851–860. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Wang, W.; Zhang, Y.; Liu, S.; Liu, Y.; Zheng, D. Tumour necrosis factor-related apoptosis-inducing ligand (TRAIL)-induced chemokine release in both TRAIL-resistant and TRAIL-sensitive cells via nuclear factor kappa B. FEBS J. 2009, 276, 581–593. [Google Scholar] [CrossRef]
- Kim, J.Y.; Lee, J.Y.; Kim, D.G.; Koo, G.B.; Yu, J.W.; Kim, Y.S. TRADD is critical for resistance to TRAIL-induced cell death through NF-kappaB activation. FEBS Lett. 2011, 585, 2144–2150. [Google Scholar] [CrossRef]
- Favaro, F.; Luciano-Mateo, F.; Moreno-Caceres, J.; Hernandez-Madrigal, M.; Both, D.; Montironi, C.; Puschel, F.; Nadal, E.; Eldering, E.; Munoz-Pinedo, C. TRAIL receptors promote constitutive and inducible IL-8 secretion in non-small cell lung carcinoma. Cell Death Dis. 2022, 13, 1046. [Google Scholar] [CrossRef]
- Oh, Y.T.; Yue, P.; Wang, D.; Tong, J.S.; Chen, Z.G.; Khuri, F.R.; Sun, S.Y. Suppression of death receptor 5 enhances cancer cell invasion and metastasis through activation of caspase-8/TRAF2-mediated signaling. Oncotarget 2015, 6, 41324–41338. [Google Scholar] [CrossRef]
- Wang, T.T.; Jeng, J. Coordinated regulation of two TRAIL-R2/KILLER/DR5 mRNA isoforms by DNA damaging agents, serum and 17beta-estradiol in human breast cancer cells. Breast Cancer Res. Treat. 2000, 61, 87–96. [Google Scholar] [CrossRef]
- Sun, S.Y. Understanding the Role of the Death Receptor 5/FADD/caspase-8 Death Signaling in Cancer Metastasis. Mol. Cell Pharmacol. 2011, 3, 31–34. [Google Scholar]
- Ganten, T.M.; Sykora, J.; Koschny, R.; Batke, E.; Aulmann, S.; Mansmann, U.; Stremmel, W.; Sinn, H.P.; Walczak, H. Prognostic significance of tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) receptor expression in patients with breast cancer. J. Mol. Med. 2009, 87, 995–1007. [Google Scholar] [CrossRef] [PubMed]
- Macher-Goeppinger, S.; Aulmann, S.; Tagscherer, K.E.; Wagener, N.; Haferkamp, A.; Penzel, R.; Brauckhoff, A.; Hohenfellner, M.; Sykora, J.; Walczak, H.; et al. Prognostic value of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and TRAIL receptors in renal cell cancer. Clin. Cancer Res. 2009, 15, 650–659. [Google Scholar] [CrossRef] [PubMed]
- Shlyakhtina, Y.; Pavet, V.; Gronemeyer, H. Dual role of DR5 in death and survival signaling leads to TRAIL resistance in cancer cells. Cell Death Dis. 2017, 8, e3025. [Google Scholar] [CrossRef]
- Legler, D.F.; Micheau, O.; Doucey, M.A.; Tschopp, J.; Bron, C. Recruitment of TNF receptor 1 to lipid rafts is essential for TNFalpha-mediated NF-kappaB activation. Immunity 2003, 18, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Yang, C.; Liu, Y.; Xiong, J.; Zhang, J.; Zhong, Y.; Zhang, G.; Zhou, F.; Zhou, Y.; Xie, C. Redistribution of DR4 and DR5 in lipid rafts accounts for the sensitivity to TRAIL in NSCLC cells. Int. J. Oncol. 2011, 39, 1577–1586. [Google Scholar] [CrossRef]
- Marconi, M.; Ascione, B.; Ciarlo, L.; Vona, R.; Garofalo, T.; Sorice, M.; Gianni, A.M.; Locatelli, S.L.; Carlo-Stella, C.; Malorni, W.; et al. Constitutive localization of DR4 in lipid rafts is mandatory for TRAIL-induced apoptosis in B-cell hematologic malignancies. Cell Death Dis. 2013, 4, e863. [Google Scholar] [CrossRef] [PubMed]
- Bellail, A.C.; Tse, M.C.; Song, J.H.; Phuphanich, S.; Olson, J.J.; Sun, S.Y.; Hao, C. DR5-mediated DISC controls caspase-8 cleavage and initiation of apoptosis in human glioblastomas. J. Cell Mol. Med. 2010, 14 (6A), 1303–1317. [Google Scholar] [CrossRef]
- Lim, S.C.; Duong, H.Q.; Choi, J.E.; Lee, T.B.; Kang, J.H.; Oh, S.H.; Han, S.I. Lipid raft-dependent death receptor 5 (DR5) expression and activation are critical for ursodeoxycholic acid-induced apoptosis in gastric cancer cells. Carcinogenesis 2011, 32, 723–731. [Google Scholar] [CrossRef]
- Delmas, D.; Rébé, C.; Micheau, O.; Athias, A.; Gambert, P.; Grazide, S.; laurent, G.; Latruffe, N.; Solary, E. Redistribution of CD95, DR4 and DR5 in rafts accounts for the synergistic toxicity of resveratrol and death receptor ligands in colon carcinoma cells. Oncogene 2004, 23, 8979–8986. [Google Scholar] [CrossRef]
- Song, J.H.; Tse, M.C.; Bellail, A.; Phuphanich, S.; Khuri, F.; Kneteman, N.M.; Hao, C. Lipid rafts and nonrafts mediate tumor necrosis factor related apoptosis-inducing ligand induced apoptotic and nonapoptotic signals in non small cell lung carcinoma cells. Cancer Res. 2007, 67, 6946–6955. [Google Scholar] [CrossRef]
- Dickens, L.S.; Boyd, R.S.; Jukes-Jones, R.; Hughes, M.A.; Robinson, G.L.; Fairall, L.; Schwabe, J.W.; Cain, K.; Macfarlane, M. A death effector domain chain DISC model reveals a crucial role for caspase-8 chain assembly in mediating apoptotic cell death. Mol. Cell 2012, 47, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Guegan, J.P.; Ginestier, C.; Charafe-Jauffret, E.; Ducret, T.; Quignard, J.F.; Vacher, P.; Legembre, P. CD95/Fas and metastatic disease: What does not kill you makes you stronger. Semin. Cancer Biol. 2020, 60, 121–131. [Google Scholar] [CrossRef]
- Muppidi, J.R.; Siegel, R.M. Ligand-independent redistribution of Fas (CD95) into lipid rafts mediates clonotypic T cell death. Nat. Immunol. 2004, 5, 182–189. [Google Scholar] [CrossRef]
- Muppidi, J.R.; Tschopp, J.; Siegel, R.M. Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity 2004, 21, 461–465. [Google Scholar] [CrossRef]
- Siegel, R.M.; Muppidi, J.R.; Sarker, M.; Lobito, A.; Jen, M.; Martin, D.; Straus, S.E.; Lenardo, M.J. SPOTS: signaling protein oligomeric transduction structures are early mediators of death receptor-induced apoptosis at the plasma membrane. J. Cell Biol. 2004, 167, 735–744. [Google Scholar] [CrossRef] [PubMed]
- Ponton, A.; Clement, M.V.; Stamenkovic, I. The CD95 (APO-1/Fas) receptor activates NF-kappaB independently of its cytotoxic function. J. Biol. Chem. 1996, 271, 8991–8995. [Google Scholar] [CrossRef]
- Tauzin, S.; Chaigne-Delalande, B.; Selva, E.; Khadra, N.; Daburon, S.; Contin-Bordes, C.; Blanco, P.; Le Seyec, J.; Ducret, T.; Counillon, L.; et al. The naturally processed CD95L elicits a c-yes/calcium/PI3K-driven cell migration pathway. PLoS Biol. 2011, 9, e1001090. [Google Scholar] [CrossRef] [PubMed]
- Malleter, M.; Tauzin, S.; Bessede, A.; Castellano, R.; Goubard, A.; Godey, F.; Leveque, J.; Jezequel, P.; Campion, L.; Campone, M.; et al. CD95L cell surface cleavage triggers a prometastatic signaling pathway in triple-negative breast cancer. Cancer Res. 2013, 73, 6711–6721. [Google Scholar] [CrossRef]
- Monet, M.; Poet, M.; Tauzin, S.; Fouque, A.; Cophignon, A.; Lagadic-Gossmann, D.; Vacher, P.; Legembre, P.; Counillon, L. The cleaved FAS ligand activates the Na(+)/H(+) exchanger NHE1 through Akt/ROCK1 to stimulate cell motility. Sci. Rep. 2016, 6, 28008. [Google Scholar] [CrossRef]
- Park, D.R.; Thomsen, A.R.; Frevert, C.W.; Pham, U.; Skerrett, S.J.; Kiener, P.A.; Liles, W.C. Fas (CD95) induces proinflammatory cytokine responses by human monocytes and monocyte-derived macrophages. J. Immunol. 2003, 170, 6209–6216. [Google Scholar] [CrossRef]
- Rescigno, M.; Piguet, V.; Valzasina, B.; Lens, S.; Zubler, R.; French, L.; Kindler, V.; Tschopp, J.; Ricciardi-Castagnoli, P. Fas engagement induces the maturation of dendritic cells (DCs), the release of interleukin (IL)-1beta, and the production of interferon gamma in the absence of IL-12 during DC-T cell cognate interaction: a new role for Fas ligand in inflammatory responses. J. Exp. Med. 2000, 192, 1661–1668. [Google Scholar] [CrossRef] [PubMed]
- Trauzold, A.; Roder, C.; Sipos, B.; Karsten, K.; Arlt, A.; Jiang, P.; Martin-Subero, J.I.; Siegmund, D.; Muerkoster, S.; Pagerols-Raluy, L.; et al. CD95 and TRAF2 promote invasiveness of pancreatic cancer cells. FASEB J. 2005, 19, 620–622. [Google Scholar] [CrossRef] [PubMed]
- Steller, E.J.; Ritsma, L.; Raats, D.A.; Hoogwater, F.J.; Emmink, B.L.; Govaert, K.M.; Laoukili, J.; Rinkes, I.H.; van Rheenen, J.; Kranenburg, O. The death receptor CD95 activates the cofilin pathway to stimulate tumour cell invasion. EMBO Rep. 2011, 12, 931–937. [Google Scholar] [CrossRef] [PubMed]
- Ruan, W.; Lee, C.T.; Desbarats, J. A novel juxtamembrane domain in tumor necrosis factor receptor superfamily molecules activates Rac1 and controls neurite growth. Mol. Biol. Cell 2008, 19, 3192–3202. [Google Scholar] [CrossRef]
- Xu, L.; Zhang, Y.; Qu, X.; Che, X.; Guo, T.; Li, C.; Ma, R.; Fan, Y.; Ma, Y.; Hou, K.; et al. DR5-Cbl-b/c-Cbl-TRAF2 complex inhibits TRAIL-induced apoptosis by promoting TRAF2-mediated polyubiquitination of caspase-8 in gastric cancer cells. Mol. Oncol. 2017, 11, 1733–1751. [Google Scholar] [CrossRef]
- Gonzalvez, F.; Lawrence, D.; Yang, B.; Yee, S.; Pitti, R.; Marsters, S.; Pham, V.C.; Stephan, J.P.; Lill, J.; Ashkenazi, A. TRAF2 Sets a threshold for extrinsic apoptosis by tagging caspase-8 with a ubiquitin shutoff timer. Mol. Cell 2012, 48, 888–899. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Wang, Q.; Xu, J.; Xu, X.; Padilla, M.T.; Ren, G.; Gou, X.; Lin, Y. Attenuation of TNFSF10/TRAIL-induced apoptosis by an autophagic survival pathway involving TRAF2- and RIPK1/RIP1-mediated MAPK8/JNK activation. Autophagy 2012, 8, 1811–1821. [Google Scholar] [CrossRef]
- Haas, T.L.; Emmerich, C.H.; Gerlach, B.; Schmukle, A.C.; Cordier, S.M.; Rieser, E.; Feltham, R.; Vince, J.; Warnken, U.; Wenger, T.; et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol. Cell 2009, 36, 831–844. [Google Scholar] [CrossRef]
- Kataoka, T.; Budd, R.C.; Holler, N.; Thome, M.; Martinon, F.; Irmler, M.; Burns, K.; Hahne, M.; Kennedy, N.; Kovacsovics, M.; et al. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr. Biol. 2000, 10, 640–648. [Google Scholar] [CrossRef]
- Kataoka, T.; Tschopp, J. N-terminal fragment of c-FLIP(L) processed by caspase 8 specifically interacts with TRAF2 and induces activation of the NF-kappaB signaling pathway. Mol. Cell Biol. 2004, 24, 2627–2636. [Google Scholar] [CrossRef]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef]
- Nagahashi, M.; Yamada, A.; Katsuta, E.; Aoyagi, T.; Huang, W.C.; Terracina, K.P.; Hait, N.C.; Allegood, J.C.; Tsuchida, J.; Yuza, K.; et al. Targeting the SphK1/S1P/S1PR1 Axis That Links Obesity, Chronic Inflammation, and Breast Cancer Metastasis. Cancer Res. 2018, 78, 1713–1725. [Google Scholar] [CrossRef]
- Noujarede, J.; Carrie, L.; Garcia, V.; Grimont, M.; Eberhardt, A.; Mucher, E.; Genais, M.; Schreuder, A.; Carpentier, S.; Segui, B.; et al. Sphingolipid paracrine signaling impairs keratinocyte adhesion to promote melanoma invasion. Cell Rep. 2023, 42, 113586. [Google Scholar] [CrossRef]
- Oh, Y.T.; Yue, P.; Sun, S.Y. DR5 suppression induces sphingosine-1-phosphate-dependent TRAF2 polyubiquitination, leading to activation of JNK/AP-1 and promotion of cancer cell invasion. Cell Commun. Signal 2017, 15, 18. [Google Scholar] [CrossRef]
- Wei, W.; Wang, D.; Shi, J.; Xiang, Y.; Zhang, Y.; Liu, S.; Liu, Y.; Zheng, D. Tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) induces chemotactic migration of monocytes via a death receptor 4-mediated RhoGTPase pathway. Mol. Immunol. 2010, 47, 2475–2484. [Google Scholar] [CrossRef]
- Park, K.J.; Lee, C.H.; Kim, A.; Jeong, K.J.; Kim, C.H.; Kim, Y.S. Death receptors 4 and 5 activate Nox1 NADPH oxidase through riboflavin kinase to induce reactive oxygen species-mediated apoptotic cell death. J. Biol. Chem. 2012, 287, 3313–3325. [Google Scholar] [CrossRef]
- Hartwig, T.; Montinaro, A.; von Karstedt, S.; Sevko, A.; Surinova, S.; Chakravarthy, A.; Taraborrelli, L.; Draber, P.; Lafont, E.; Arce Vargas, F.; et al. The TRAIL-Induced Cancer Secretome Promotes a Tumor-Supportive Immune Microenvironment via CCR2. Mol. Cell 2017, 65, 730–742. [Google Scholar] [CrossRef]
- Salhia, B.; Rutten, F.; Nakada, M.; Beaudry, C.; Berens, M.; Kwan, A.; Rutka, J.T. Inhibition of Rho-kinase affects astrocytoma morphology, motility, and invasion through activation of Rac1. Cancer Res. 2005, 65, 8792–8800. [Google Scholar] [CrossRef]
- Bustelo, X.R.; Ojeda, V.; Barreira, M.; Sauzeau, V.; Castro-Castro, A. Rac-ing to the plasma membrane: the long and complex work commute of Rac1 during cell signaling. Small GTPases 2012, 3, 60–66. [Google Scholar] [CrossRef]
- Miloszewska, J.; Janik, P.; Ostrowski, J. The effect of tumor necrosis factor (TNF-alpha) on calcium (Ca2+) level. Arch Immunol Ther Exp (Warsz), 1991; 39, 99–102. [Google Scholar]
- Boehning, D.; van Rossum, D.B.; Patterson, R.L.; Snyder, S.H. A peptide inhibitor of cytochrome c/inositol 1,4,5-trisphosphate receptor binding blocks intrinsic and extrinsic cell death pathways. Proc. Natl. Acad. Sci. U S A 2005, 102, 1466–1471. [Google Scholar] [CrossRef]
- Wozniak, A.L.; Wang, X.; Stieren, E.S.; Scarbrough, S.G.; Elferink, C.J.; Boehning, D. Requirement of biphasic calcium release from the endoplasmic reticulum for Fas-mediated apoptosis. J. Cell Biol. 2006, 175, 709–714. [Google Scholar] [CrossRef]
- Prevarskaya, N.; Ouadid-Ahidouch, H.; Skryma, R.; Shuba, Y. Remodelling of Ca2+ transport in cancer: how it contributes to cancer hallmarks? Philos. Trans. R. Soc. London. Ser. B Biol. Sci. 2014, 369, 20130097. [Google Scholar] [CrossRef]
- Xie, T.; Chen, S.; Hao, J.; Wu, P.; Gu, X.; Wei, H.; Li, Z.; Xiao, J. Roles of calcium signaling in cancer metastasis to bone. Explor. Target. Antitumor Ther. 2022, 3, 445–462. [Google Scholar] [CrossRef]
- Khadra, N.; Bresson-Bepoldin, L.; Penna, A.; Chaigne-Delalande, B.; Segui, B.; Levade, T.; Vacher, A.M.; Reiffers, J.; Ducret, T.; Moreau, J.F.; et al. CD95 triggers Orai1-mediated localized Ca2+ entry, regulates recruitment of protein kinase C (PKC) beta2, and prevents death-inducing signaling complex formation. Proc. Natl. Acad. Sci. U S A 2011, 108, 19072–19077. [Google Scholar] [CrossRef]
- Siegmund, D.; Lang, I.; Wajant, H. Cell death-independent activities of the death receptors CD95, TRAILR1, and TRAILR2. FEBS J. 2017, 284, 1131–1159. [Google Scholar] [CrossRef]
- Reis, C.R.; Chen, P.H.; Bendris, N.; Schmid, S.L. TRAIL-death receptor endocytosis and apoptosis are selectively regulated by dynamin-1 activation. Proc. Natl. Acad. Sci. U S A 2017, 114, 504–509. [Google Scholar] [CrossRef]
- Airiau, K.; Vacher, P.; Micheau, O.; Prouzet-Mauleon, V.; Kroemer, G.; Moosavi, M.A.; Djavaheri-Mergny, M. TRAIL Triggers CRAC-Dependent Calcium Influx and Apoptosis through the Recruitment of Autophagy Proteins to Death-Inducing Signaling Complex. Cells 2021, 11. [Google Scholar] [CrossRef]
- Ahn, E.Y.; Lim, S.T.; Cook, W.J.; McDonald, J.M. Calmodulin binding to the Fas death domain. Regulation by Fas activation. J. Biol. Chem. 2004, 279, 5661–5666. [Google Scholar] [CrossRef]
- Chen, J.J.; Sun, Y.; Nabel, G.J. Regulation of the proinflammatory effects of Fas ligand (CD95L). Science 1998, 282, 1714–1717. [Google Scholar] [CrossRef] [PubMed]
- Yuan, K.; Jing, G.; Chen, J.; Liu, H.; Zhang, K.; Li, Y.; Wu, H.; McDonald, J.M.; Chen, Y. Calmodulin mediates Fas-induced FADD-independent survival signaling in pancreatic cancer cells via activation of Src-extracellular signal-regulated kinase (ERK). J. Biol. Chem. 2011, 286, 24776–24784. [Google Scholar] [CrossRef]
- Krebs, J.; Agellon, L.B.; Michalak, M. Ca(2+) homeostasis and endoplasmic reticulum (ER) stress: An integrated view of calcium signaling. Biochem. Biophys. Res. Commun. 2015, 460, 114–121. [Google Scholar] [CrossRef]
- Gong, K.; Chen, C.; Zhan, Y.; Chen, Y.; Huang, Z.; Li, W. Autophagy-related gene 7 (ATG7) and reactive oxygen species/extracellular signal-regulated kinase regulate tetrandrine-induced autophagy in human hepatocellular carcinoma. J. Biol. Chem. 2012, 287, 35576–35588. [Google Scholar] [CrossRef]
- Nihira, K.; Miki, Y.; Ono, K.; Suzuki, T.; Sasano, H. An inhibition of p62/SQSTM1 caused autophagic cell death of several human carcinoma cells. Cancer Sci. 2014, 105, 568–575. [Google Scholar] [CrossRef]
- Fancy, R.M.; Wang, L.; Schmid, T.; Zeng, Q.; Wang, H.; Zhou, T.; Buchsbaum, D.J.; Song, Y. Characterization of the interactions between calmodulin and death receptor 5 in triple-negative and estrogen receptor-positive breast cancer cells. AN INTEGRATED EXPERIMENTAL AND COMPUTATIONAL STUDY. J. Biol. Chem. 2016, 291, 23489. [Google Scholar] [CrossRef]
- Fancy, R.M.; Kim, H.; Zhou, T.; Zinn, K.R.; Buchsbaum, D.J.; Song, Y. Calmodulin Binding to Death Receptor 5-mediated Death-Inducing Signaling Complex in Breast Cancer Cells. J. Cell Biochem. 2017, 118, 2285–2294. [Google Scholar] [CrossRef] [PubMed]
- Chin, D.; Means, A.R. Calmodulin: a prototypical calcium sensor. Trends Cell Biol. 2000, 10, 322–328. [Google Scholar] [CrossRef]
- Villalobo, A. The multifunctional role of phospho-calmodulin in pathophysiological processes. Biochem. J. 2018, 475, 4011–4023. [Google Scholar] [CrossRef]
- Yuan, K.; Yong, S.; Xu, F.; Zhou, T.; McDonald, J.M.; Chen, Y. Calmodulin antagonists promote TRA-8 therapy of resistant pancreatic cancer. Oncotarget 2015, 6, 25308–25319. [Google Scholar] [CrossRef] [PubMed]
- Kaminskyy, V.O.; Surova, O.V.; Piskunova, T.; Zborovskaya, I.B.; Tchevkina, E.M.; Andera, L.; Zhivotovsky, B. Upregulation of c-FLIP-short in response to TRAIL promotes survival of NSCLC cells, which could be suppressed by inhibition of Ca2+/calmodulin signaling. Cell Death Dis. 2013, 4, e522. [Google Scholar] [CrossRef]
- Cursi, S.; Rufini, A.; Stagni, V.; Condo, I.; Matafora, V.; Bachi, A.; Bonifazi, A.P.; Coppola, L.; Superti-Furga, G.; Testi, R.; et al. Src kinase phosphorylates Caspase-8 on Tyr380: a novel mechanism of apoptosis suppression. EMBO J. 2006, 25, 1895–1905. [Google Scholar] [CrossRef]
- Stateva, S.R.; Salas, V.; Anguita, E.; Benaim, G.; Villalobo, A. Ca2+/Calmodulin and Apo-Calmodulin Both Bind to and Enhance the Tyrosine Kinase Activity of c-Src. PLoS One 2015, 10, e0128783. [Google Scholar] [CrossRef]
- Chen, Y.; Pawar, P.; Pan, G.; Ma, L.; Liu, H.; McDonald, J.M. Calmodulin binding to the Fas-mediated death-inducing signaling complex in cholangiocarcinoma cells. J. Cell Biochem. 2008, 103, 788–799. [Google Scholar] [CrossRef]
- Fernandez, T.F.; Samal, A.B.; Bedwell, G.J.; Chen, Y.; Saad, J.S. Structural and biophysical characterization of the interactions between the death domain of Fas receptor and calmodulin. J. Biol. Chem. 2013, 288, 21898–21908. [Google Scholar] [CrossRef]
- Barbero, S.; Mielgo, A.; Torres, V.; Teitz, T.; Shields, D.J.; Mikolon, D.; Bogyo, M.; Barila, D.; Lahti, J.M.; Schlaepfer, D.; et al. Caspase-8 association with the focal adhesion complex promotes tumor cell migration and metastasis. Cancer Res. 2009, 69, 3755–3763. [Google Scholar] [CrossRef]
- Chen, J.; Li, L.; Huangfu, L.; Du, H.; Ji, X.; Xing, X.; Ji, J. Death receptor 5 promotes tumor progression in gastric cancer. FEBS Open Bio 2023, 13, 2375–2388. [Google Scholar] [CrossRef]
- Leithner, K.; Stacher, E.; Wurm, R.; Ploner, F.; Quehenberger, F.; Wohlkoenig, C.; Balint, Z.; Polachova, J.; Olschewski, A.; Samonigg, H.; et al. Nuclear and cytoplasmic death receptor 5 as prognostic factors in patients with non-small cell lung cancer treated with chemotherapy. Lung Cancer 2009, 65, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Bertsch, U.; Roder, C.; Kalthoff, H.; Trauzold, A. Compartmentalization of TNF-related apoptosis-inducing ligand (TRAIL) death receptor functions: emerging role of nuclear TRAIL-R2. Cell Death Dis. 2014, 5, e1390. [Google Scholar] [CrossRef]
- Kojima, Y.; Nakayama, M.; Nishina, T.; Nakano, H.; Koyanagi, M.; Takeda, K.; Okumura, K.; Yagita, H. Importin beta1 protein-mediated nuclear localization of death receptor 5 (DR5) limits DR5/tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL)-induced cell death of human tumor cells. J. Biol. Chem. 2011, 286, 43383–43393. [Google Scholar] [CrossRef]
- Mert, U.; Adawy, A.; Scharff, E.; Teichmann, P.; Willms, A.; Haselmann, V.; Colmorgen, C.; Lemke, J.; von Karstedt, S.; Fritsch, J.; et al. TRAIL Induces Nuclear Translocation and Chromatin Localization of TRAIL Death Receptors. Cancers (Basel) 2019, 11. [Google Scholar] [CrossRef]
- Haselmann, V.; Kurz, A.; Bertsch, U.; Hubner, S.; Olempska-Muller, M.; Fritsch, J.; Hasler, R.; Pickl, A.; Fritsche, H.; Annewanter, F.; et al. Nuclear death receptor TRAIL-R2 inhibits maturation of let-7 and promotes proliferation of pancreatic and other tumor cells. Gastroenterology 2014, 146, 278–290. [Google Scholar] [CrossRef]
- Chen, J.J.; Shen, H.C.; Rivera Rosado, L.A.; Zhang, Y.; Di, X.; Zhang, B. Mislocalization of death receptors correlates with cellular resistance to their cognate ligands in human breast cancer cells. Oncotarget 2012, 3, 833–842. [Google Scholar] [CrossRef]
- Alam, M.; Ahmad, R.; Rajabi, H.; Kufe, D. MUC1-C Induces the LIN28B-->LET-7-->HMGA2 Axis to Regulate Self-Renewal in NSCLC. Mol. Cancer Res. 2015, 13, 449–460. [Google Scholar] [CrossRef]
- Song, H.; Xu, W.; Song, J.; Liang, Y.; Fu, W.; Zhu, X.C.; Li, C.; Peng, J.S.; Zheng, J.N. Overexpression of Lin28 inhibits the proliferation, migration and cell cycle progression and induces apoptosis of BGC-823 gastric cancer cells. Oncol. Rep. 2015, 33, 997–1003. [Google Scholar] [CrossRef]
- Unachukwu, U.; Chada, K.; D’Armiento, J. High Mobility Group AT-Hook 2 (HMGA2) Oncogenicity in Mesenchymal and Epithelial Neoplasia. Int. J. Mol. Sci. 2020, 21. [Google Scholar] [CrossRef]
- Viswanathan, S.R.; Powers, J.T.; Einhorn, W.; Hoshida, Y.; Ng, T.L.; Toffanin, S.; O’Sullivan, M.; Lu, J.; Phillips, L.A.; Lockhart, V.L.; et al. Lin28 promotes transformation and is associated with advanced human malignancies. Nat. Genet. 2009, 41, 843–848. [Google Scholar] [CrossRef]
- Park, S.M.; Shell, S.; Radjabi, A.R.; Schickel, R.; Feig, C.; Boyerinas, B.; Dinulescu, D.M.; Lengyel, E.; Peter, M.E. Let-7 prevents early cancer progression by suppressing expression of the embryonic gene HMGA2. Cell Cycle (Georget. Tex. 2007, 6, 2585–2590. [Google Scholar] [CrossRef]
- Mbalaviele, G.; Dunstan, C.R.; Sasaki, A.; Williams, P.J.; Mundy, G.R.; Yoneda, T. E-cadherin expression in human breast cancer cells suppresses the development of osteolytic bone metastases in an experimental metastasis model. Cancer Res. 1996, 56, 4063–4070. [Google Scholar] [PubMed]
- Chatterjee, S.; Behnam Azad, B.; Nimmagadda, S. The intricate role of CXCR4 in cancer. Adv. Cancer Res. 2014, 124, 31–82. [Google Scholar] [CrossRef]
- Zhang, Z.; Ni, C.; Chen, W.; Wu, P.; Wang, Z.; Yin, J.; Huang, J.; Qiu, F. Expression of CXCR4 and breast cancer prognosis: a systematic review and meta-analysis. BMC Cancer 2014, 14, 49. [Google Scholar] [CrossRef]
- Yun, J.; Frankenberger, C.A.; Kuo, W.L.; Boelens, M.C.; Eves, E.M.; Cheng, N.; Liang, H.; Li, W.H.; Ishwaran, H.; Minn, A.J.; et al. Signalling pathway for RKIP and Let-7 regulates and predicts metastatic breast cancer. EMBO J. 2011, 30, 4500–4514. [Google Scholar] [CrossRef]
- Li, C.; Egloff, A.M.; Sen, M.; Grandis, J.R.; Johnson, D.E. Caspase-8 mutations in head and neck cancer confer resistance to death receptor-mediated apoptosis and enhance migration, invasion, and tumor growth. Mol. Oncol. 2014, 8, 1220–1230. [Google Scholar] [CrossRef]
- Graf, R.P.; Keller, N.; Barbero, S.; Stupack, D. Caspase-8 as a regulator of tumor cell motility. Curr. Mol. Med. 2014, 14, 246–254. [Google Scholar] [CrossRef]
- Keller, N.; Ozmadenci, D.; Ichim, G.; Stupack, D. Caspase-8 function, and phosphorylation, in cell migration. Semin. Cell Dev. Biol. 2018, 82, 105–117. [Google Scholar] [CrossRef]
- Barbero, S.; Barila, D.; Mielgo, A.; Stagni, V.; Clair, K.; Stupack, D. Identification of a critical tyrosine residue in caspase 8 that promotes cell migration. J. Biol. Chem. 2008, 283, 13031–13034. [Google Scholar] [CrossRef]
- Contadini, C.; Ferri, A.; Di Martile, M.; Cirotti, C.; Del Bufalo, D.; De Nicola, F.; Pallocca, M.; Fanciulli, M.; Sacco, F.; Donninelli, G.; et al. Caspase-8 as a novel mediator linking Src kinase signaling to enhanced glioblastoma malignancy. Cell Death Differ. 2023, 30, 417–428. [Google Scholar] [CrossRef]
- Senft, J.; Helfer, B.; Frisch, S.M. Caspase-8 interacts with the p85 subunit of phosphatidylinositol 3-kinase to regulate cell adhesion and motility. Cancer Res. 2007, 67, 11505–11509. [Google Scholar] [CrossRef] [PubMed]
- Rossman, K.L.; Der, C.J.; Sondek, J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 2005, 6, 167–180. [Google Scholar] [CrossRef]
- Helfer, B.; Boswell, B.C.; Finlay, D.; Cipres, A.; Vuori, K.; Bong Kang, T.; Wallach, D.; Dorfleutner, A.; Lahti, J.M.; Flynn, D.C.; et al. Caspase-8 promotes cell motility and calpain activity under nonapoptotic conditions. Cancer Res. 2006, 66, 4273–4278. [Google Scholar] [CrossRef]
- Mishra, Y.G.; Manavathi, B. Focal adhesion dynamics in cellular function and disease. Cell Signal 2021, 85, 110046. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in cancer: mechanistic findings and clinical applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Mandal, R.; Barron, J.C.; Kostova, I.; Becker, S.; Strebhardt, K. Caspase-8: The double-edged sword. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188357. [Google Scholar] [CrossRef]
- Liu, Y.; Cui, H.; Huang, X.; Zhu, B.; Guan, S.; Cheng, W.; Lai, Y.; Zhang, X.; Hua, Z.C. MiR-7a is an important mediator in Fas-associated protein with death domain (FADD)-regulated expression of focal adhesion kinase (FAK). Oncotarget 2016, 7, 51393–51407. [Google Scholar] [CrossRef]
- Murphy, J.M.; Rodriguez, Y.A.R.; Jeong, K.; Ahn, E.E.; Lim, S.S. Targeting focal adhesion kinase in cancer cells and the tumor microenvironment. Exp. Mol. Med. 2020, 52, 877–886. [Google Scholar] [CrossRef]
- Torres, V.A.; Mielgo, A.; Barbero, S.; Hsiao, R.; Wilkins, J.A.; Stupack, D.G. Rab5 mediates caspase-8-promoted cell motility and metastasis. Mol. Biol. Cell 2010, 21, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.A.; Mielgo, A.; Barila, D.; Anderson, D.H.; Stupack, D. Caspase 8 promotes peripheral localization and activation of Rab5. J. Biol. Chem. 2008, 283, 36280–36289. [Google Scholar] [CrossRef] [PubMed]
- Ansalone, C.; Ainsworth, R.I.; Nygaard, G.; Ai, R.; Prideaux, E.B.; Hammaker, D.; Perumal, N.B.; Weichert, K.; Tung, F.; Kodandapani, L.; et al. Caspase-8 Variant G Regulates Rheumatoid Arthritis Fibroblast-Like Synoviocyte Aggressive Behavior. ACR Open Rheumatol. 2022, 4, 288–299. [Google Scholar] [CrossRef]
- Mauro, C.D.; Pesapane, A.; Formisano, L.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Monteleone, F.; et al. Urokinase-type plasminogen activator receptor (uPAR) expression enhances invasion and metastasis in RAS mutated tumors. Sci. Rep. 2017, 7, 9388. [Google Scholar] [CrossRef]
- de Vries, T.J.; van Muijen, G.N.; Ruiter, D.J. The plasminogen activation system in tumour invasion and metastasis. Pathol. Res. Pract. 1996, 192, 718–733. [Google Scholar] [CrossRef]
- Chabot, V.; Dromard, C.; Rico, A.; Langonne, A.; Gaillard, J.; Guilloton, F.; Casteilla, L.; Sensebe, L. Urokinase-type plasminogen activator receptor interaction with beta1 integrin is required for platelet-derived growth factor-AB-induced human mesenchymal stem/stromal cell migration. Stem Cell Res. Ther. 2015, 6, 188. [Google Scholar] [CrossRef]
- Tarui, T.; Mazar, A.P.; Cines, D.B.; Takada, Y. Urokinase-type plasminogen activator receptor (CD87) is a ligand for integrins and mediates cell-cell interaction. J. Biol. Chem. 2001, 276, 3983–3990. [Google Scholar] [CrossRef]
- Kreiling, J.L.; Byrd, J.C.; Deisz, R.J.; Mizukami, I.F.; Todd, R.F., 3rd; MacDonald, R.G. Binding of urokinase-type plasminogen activator receptor (uPAR) to the mannose 6-phosphate/insulin-like growth factor II receptor: contrasting interactions of full-length and soluble forms of uPAR. J. Biol. Chem. 2003, 278, 20628–20637. [Google Scholar] [CrossRef]
- Gondi, C.S.; Kandhukuri, N.; Kondraganti, S.; Gujrati, M.; Olivero, W.C.; Dinh, D.H.; Rao, J.S. RNA interference-mediated simultaneous down-regulation of urokinase-type plasminogen activator receptor and cathepsin B induces caspase-8-mediated apoptosis in SNB19 human glioma cells. Mol. Cancer Ther. 2006, 5, 3197–3208. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Qiu, F.; Liu, Z.; Lan, Y.; Wang, K.; Zhou, P.K.; Wang, Y.; Hua, Z.C. Urokinase-type plasminogen activator receptor regulates apoptotic sensitivity of colon cancer HCT116 cell line to TRAIL via JNK-p53 pathway. Apoptosis 2014, 19, 1532–1544. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, B.; Darnay, B.; Aggarwal, B.; Dinh, D.H.; Kouraklis, G.; Olivero, W.C.; Gujrati, M.; Rao, J.S. Glioma cells deficient in urokinase plaminogen activator receptor expression are susceptible to tumor necrosis factor-alpha-related apoptosis-inducing ligand-induced apoptosis. Clin. Cancer Res. 2001, 7, 4195–4201. [Google Scholar] [PubMed]
- Li, X.; Wu, B.; Chen, L.; Ju, Y.; Li, C.; Meng, S. Urokinase-type plasminogen activator receptor inhibits apoptosis in triple-negative breast cancer through miR-17/20a suppression of death receptors 4 and 5. Oncotarget 2017, 8, 88645–88657. [Google Scholar] [CrossRef]
- Pavet, V.; Shlyakhtina, Y.; He, T.; Ceschin, D.G.; Kohonen, P.; Perala, M.; Kallioniemi, O.; Gronemeyer, H. Plasminogen activator urokinase expression reveals TRAIL responsiveness and supports fractional survival of cancer cells. Cell Death Dis. 2014, 5, e1043. [Google Scholar] [CrossRef]
- Mahmood, N.; Mihalcioiu, C.; Rabbani, S.A. Multifaceted Role of the Urokinase-Type Plasminogen Activator (uPA) and Its Receptor (uPAR): Diagnostic, Prognostic, and Therapeutic Applications. Front. Oncol. 2018, 8, 24. [Google Scholar] [CrossRef]
- Kim, S.Y.; Kim, J.H.; Song, J.J. c-Cbl shRNA-expressing adenovirus sensitizes TRAIL-induced apoptosis in prostate cancer DU-145 through increases of DR4/5. Cancer Gene Ther. 2013, 20, 82–87. [Google Scholar] [CrossRef]
- Song, J.J.; Szczepanski, M.J.; Kim, S.Y.; Kim, J.H.; An, J.Y.; Kwon, Y.T.; Alcala, M.A., Jr.; Bartlett, D.L.; Lee, Y.J. c-Cbl-mediated degradation of TRAIL receptors is responsible for the development of the early phase of TRAIL resistance. Cell Signal 2010, 22, 553–563. [Google Scholar] [CrossRef]
- Park, E.J.; Min, K.J.; Choi, K.S.; Kubatka, P.; Kruzliak, P.; Kim, D.E.; Kwon, T.K. Chloroquine enhances TRAIL-mediated apoptosis through up-regulation of DR5 by stabilization of mRNA and protein in cancer cells. Sci. Rep. 2016, 6, 22921. [Google Scholar] [CrossRef]
- Kundu, M.; Pathak, S.K.; Kumawat, K.; Basu, S.; Chatterjee, G.; Pathak, S.; Noguchi, T.; Takeda, K.; Ichijo, H.; Thien, C.B.; et al. A TNF- and c-Cbl-dependent FLIP(S)-degradation pathway and its function in Mycobacterium tuberculosis-induced macrophage apoptosis. Nat. Immunol. 2009, 10, 918–926. [Google Scholar] [CrossRef]
- Song, J.J.; Kim, J.H.; Sun, B.K.; Alcala, M.A., Jr.; Bartlett, D.L.; Lee, Y.J. c-Cbl acts as a mediator of Src-induced activation of the PI3K-Akt signal transduction pathway during TRAIL treatment. Cell Signal 2010, 22, 377–385. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhang, Y.; Liu, J.; Qu, J.; Hu, X.; Zhang, F.; Zheng, H.; Qu, X.; Liu, Y. TRAIL-activated EGFR by Cbl-b-regulated EGFR redistribution in lipid rafts antagonises TRAIL-induced apoptosis in gastric cancer cells. Eur. J. Cancer 2012, 48, 3288–3299. [Google Scholar] [CrossRef]
- Kim, J.; Kang, D.; Sun, B.K.; Kim, J.H.; Song, J.J. TRAIL/MEKK4/p38/HSP27/Akt survival network is biphasically modulated by the Src/CIN85/c-Cbl complex. Cell Signal 2013, 25, 372–379. [Google Scholar] [CrossRef]
- Xu, L.; Qu, X.; Zhang, Y.; Hu, X.; Yang, X.; Hou, K.; Teng, Y.; Zhang, J.; Sada, K.; Liu, Y. Oxaliplatin enhances TRAIL-induced apoptosis in gastric cancer cells by CBL-regulated death receptor redistribution in lipid rafts. FEBS Lett. 2009, 583, 943–948. [Google Scholar] [CrossRef]
Figure 1.
Schematic representation of TNFRSF sub-family receptors binding to TNFα, FasL and TRAIL. Receptors are depicted with their three main functional domains. The extracellular domain of these receptors is composed of Cystein Rich Domains (CRD), Orange for TNFR1/2; blue for Fas and DcR3 and a panel of greens for DR4, DR5, DcR1, DcR2 and OPG. Their TM (Transmembrane Domain) is represented in red, whereas their Intracellular domains, with the exception of DcR3 and OPG which are secreted, is represented either by a bar (solid or not). Some of these receptors harbour in addition a Death Domain (DD), represented as a yellow box. Note that the DD of DcR2 is truncated. The solid bar underneath each ligand encompasses the receptors with which a physical interaction has been demonstrated experimentally.
Figure 1.
Schematic representation of TNFRSF sub-family receptors binding to TNFα, FasL and TRAIL. Receptors are depicted with their three main functional domains. The extracellular domain of these receptors is composed of Cystein Rich Domains (CRD), Orange for TNFR1/2; blue for Fas and DcR3 and a panel of greens for DR4, DR5, DcR1, DcR2 and OPG. Their TM (Transmembrane Domain) is represented in red, whereas their Intracellular domains, with the exception of DcR3 and OPG which are secreted, is represented either by a bar (solid or not). Some of these receptors harbour in addition a Death Domain (DD), represented as a yellow box. Note that the DD of DcR2 is truncated. The solid bar underneath each ligand encompasses the receptors with which a physical interaction has been demonstrated experimentally.
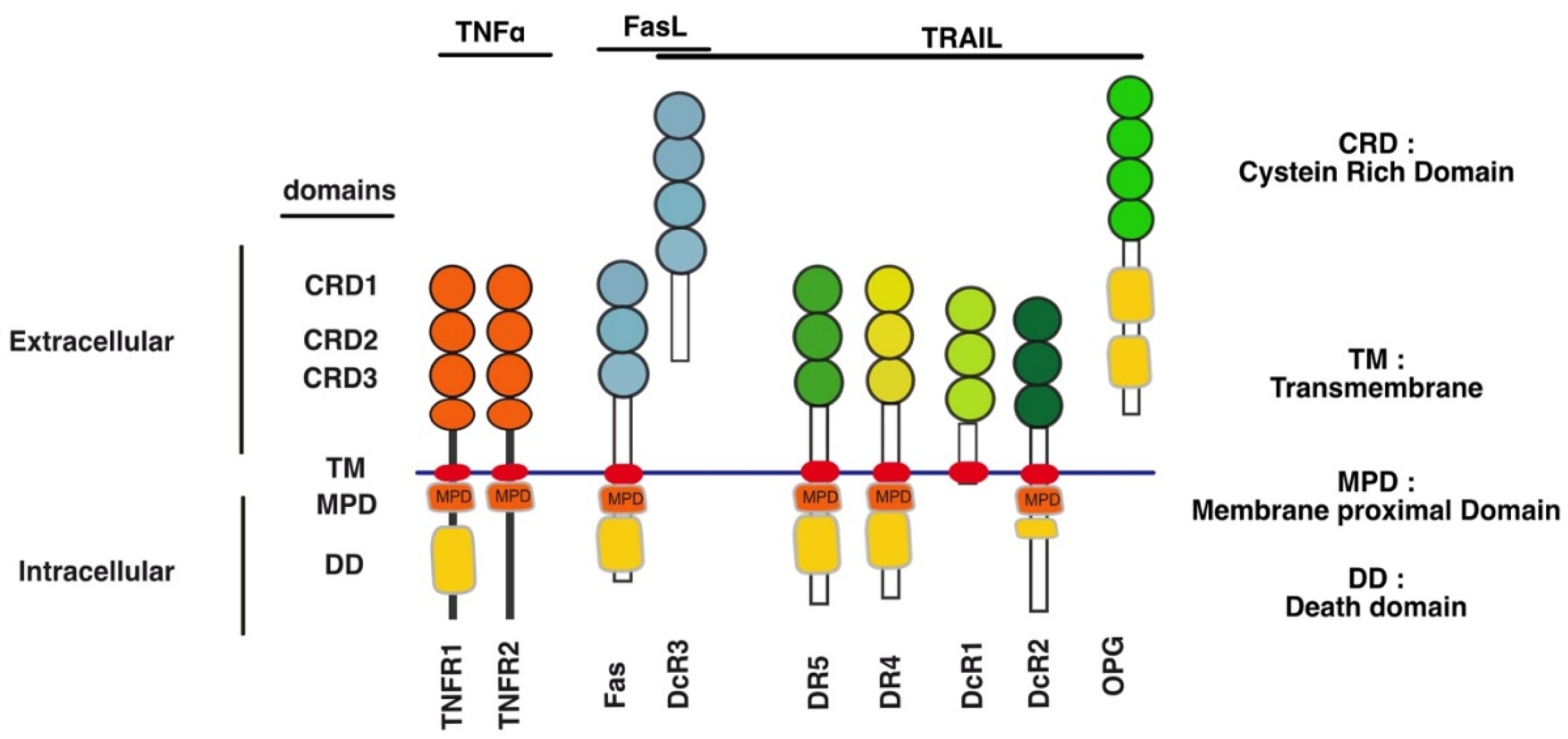
Figure 2.
Comparison of the signalling pathways triggered by TNFR1, Fas and TRAIL agonist receptors. a) TNFR1 signalling complexes upon TNFα stimulation are depicted in this panel (see also the text). TNFR1 engages first of all the formation of complex I, a survival membrane platform which leads to the activation of the NF-κB pathway, leading, in most cases to cell survival and inflammation. Regardless of the outcome, complex I is processed during activation to give rise to a secondary soluble complex (complex II), that recruits pro-apoptotic components such as the adaptor protein FADD and the caspase-8 to induce apoptosis. Cell death is usually never happening, unless activation of the NF-kB pathway fails, because the latter induce the transcriptional regulation of cellular FLIP (c-FLIP), the main inhibitor of caspase-8. b) Engagement of Fas, DR4 or DR5, contrary to TNFR1 enable direct recruitment of FADD and caspase-8 at the membrane complex I, and are thus more prone in triggering apoptosis than TNFR1. Non-apoptotic signal transduction, however, is thought to proceed from a secondary complex coined complex II, which has been described as the FADDosome or the MISC (Migration signalling complex). The latter leads to the activation of the NF-κB pathway to induce survival, pro-inflammatory and pro-tumoral effects (see text for more details). Proteins indicates in the rectangles have been described to be recruited in the distinct complexes depicted in this figure.
Figure 2.
Comparison of the signalling pathways triggered by TNFR1, Fas and TRAIL agonist receptors. a) TNFR1 signalling complexes upon TNFα stimulation are depicted in this panel (see also the text). TNFR1 engages first of all the formation of complex I, a survival membrane platform which leads to the activation of the NF-κB pathway, leading, in most cases to cell survival and inflammation. Regardless of the outcome, complex I is processed during activation to give rise to a secondary soluble complex (complex II), that recruits pro-apoptotic components such as the adaptor protein FADD and the caspase-8 to induce apoptosis. Cell death is usually never happening, unless activation of the NF-kB pathway fails, because the latter induce the transcriptional regulation of cellular FLIP (c-FLIP), the main inhibitor of caspase-8. b) Engagement of Fas, DR4 or DR5, contrary to TNFR1 enable direct recruitment of FADD and caspase-8 at the membrane complex I, and are thus more prone in triggering apoptosis than TNFR1. Non-apoptotic signal transduction, however, is thought to proceed from a secondary complex coined complex II, which has been described as the FADDosome or the MISC (Migration signalling complex). The latter leads to the activation of the NF-κB pathway to induce survival, pro-inflammatory and pro-tumoral effects (see text for more details). Proteins indicates in the rectangles have been described to be recruited in the distinct complexes depicted in this figure.
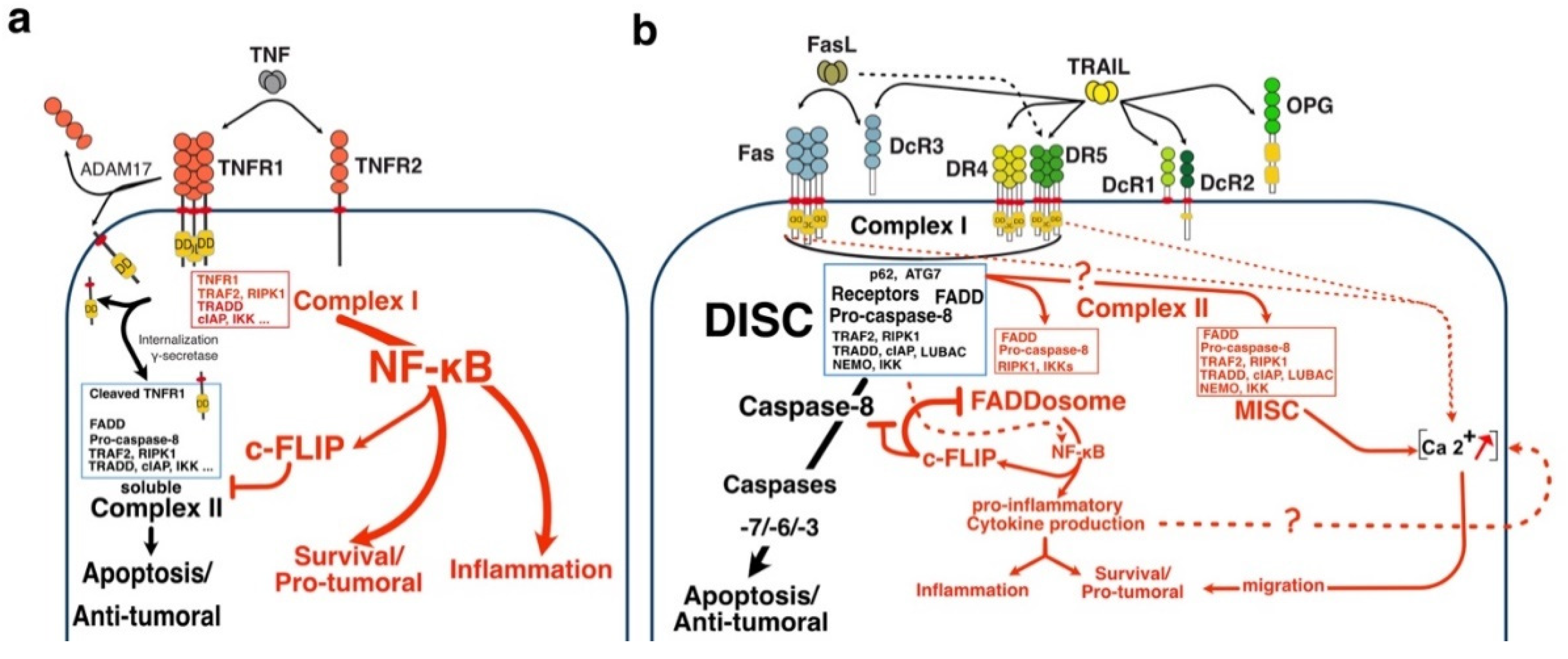
Figure 3.
Schematic representation of TRAIL canonical signalling pro-apoptotic pathway. Membrane-bound TRAIL, expressed by cytolytic immune cells such as NK cells induces apoptosis in cancer cells. TRAIL binding to DR4 and/or DR5 agonist receptors, induce their aggregation and the recruitment of FADD and caspase-8/10 forming the DISC (Death-Inducing Signalling Complex), or complex I, which ultimately will lead to the activation of the effector caspases 3/6/7, whose activation by enzymatic cleavage is either triggered directly by the active caspase-8 or indirectly through caspase-8-mediated Bid cleavage, allowing Bax translocation to mitochondria and the release of cytochrome c, whose binding with Apaf-1, amplifies apoptosis-induced by TRAIL receptors (extrinsic pathway), through the formation of a soluble pro-apoptotic complex coined apoptosome, that allows activation of the initiator caspase-9, that in turn will amplify the signal by cleaving and activating the effector caspases 3/6/7. The main inhibitors of this signalling pathway are represented in red, including the antagonist receptors (DcR1/2/3 and OPG) which compete for TRAIL binding or c-FLIP and XIAP the main caspase-8 and effector caspases inhibitor inhibitors, respectively. In addition, a schematic representation of the non-canonical signalling associated with complex I is shown, mainly describing potential activation of NF-κΒ which besides protecting the cells from TRAIL-induced apoptosis is involved in promoting TRAIL’s pro-tumoral activity. Main TRAIL-induced apoptosis inhibitors are shown in red.
Figure 3.
Schematic representation of TRAIL canonical signalling pro-apoptotic pathway. Membrane-bound TRAIL, expressed by cytolytic immune cells such as NK cells induces apoptosis in cancer cells. TRAIL binding to DR4 and/or DR5 agonist receptors, induce their aggregation and the recruitment of FADD and caspase-8/10 forming the DISC (Death-Inducing Signalling Complex), or complex I, which ultimately will lead to the activation of the effector caspases 3/6/7, whose activation by enzymatic cleavage is either triggered directly by the active caspase-8 or indirectly through caspase-8-mediated Bid cleavage, allowing Bax translocation to mitochondria and the release of cytochrome c, whose binding with Apaf-1, amplifies apoptosis-induced by TRAIL receptors (extrinsic pathway), through the formation of a soluble pro-apoptotic complex coined apoptosome, that allows activation of the initiator caspase-9, that in turn will amplify the signal by cleaving and activating the effector caspases 3/6/7. The main inhibitors of this signalling pathway are represented in red, including the antagonist receptors (DcR1/2/3 and OPG) which compete for TRAIL binding or c-FLIP and XIAP the main caspase-8 and effector caspases inhibitor inhibitors, respectively. In addition, a schematic representation of the non-canonical signalling associated with complex I is shown, mainly describing potential activation of NF-κΒ which besides protecting the cells from TRAIL-induced apoptosis is involved in promoting TRAIL’s pro-tumoral activity. Main TRAIL-induced apoptosis inhibitors are shown in red.
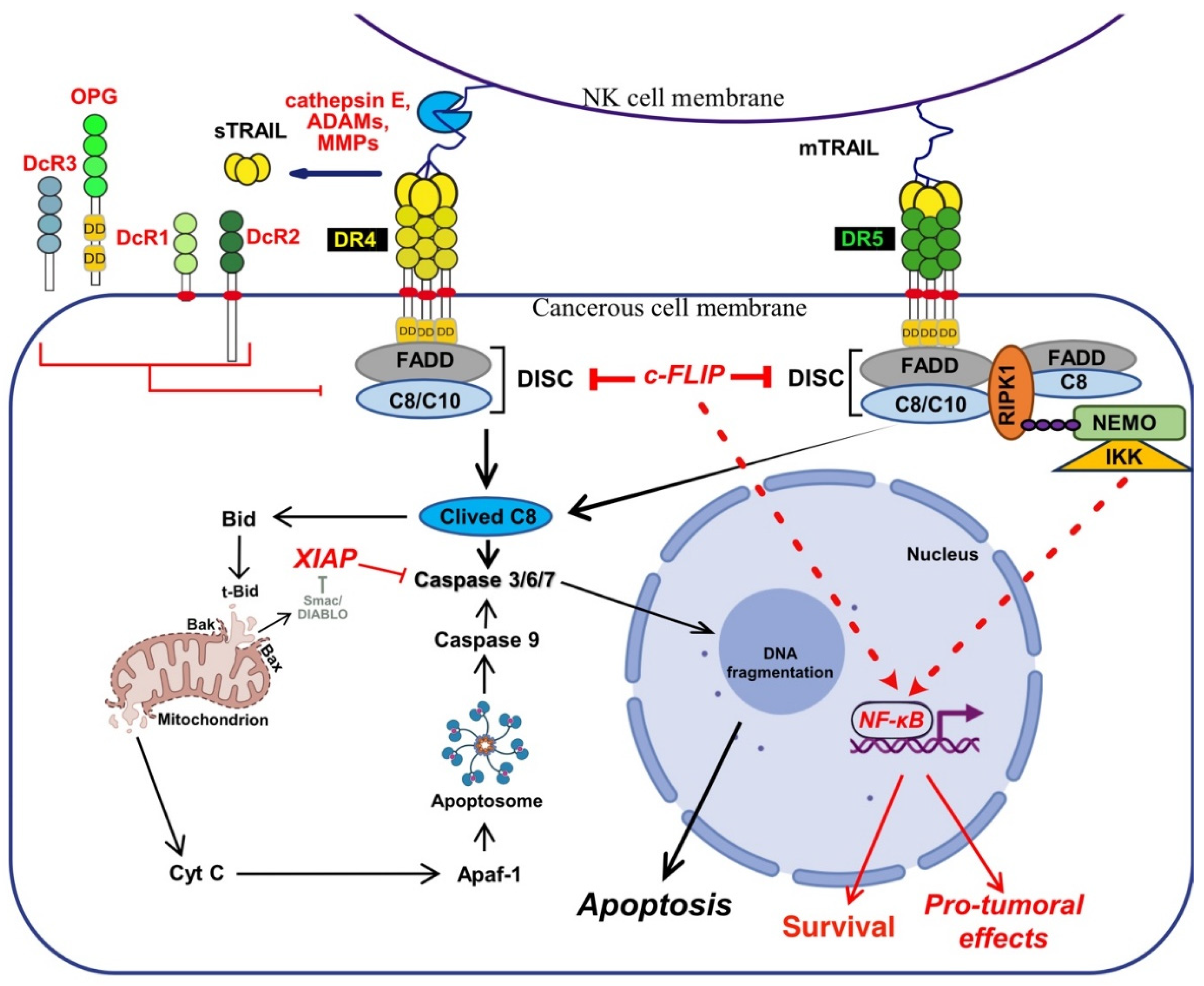
Figure 4.
Schematic representation of DR5 non-apoptotic signalling pathways. Illustration of a) DR5-mediated RIPoptosome and FADDosome secondary complexes and b) nuclear translocation of DR5 in the nucleus, potentially mediating cell migration. See text for explanation.
Figure 4.
Schematic representation of DR5 non-apoptotic signalling pathways. Illustration of a) DR5-mediated RIPoptosome and FADDosome secondary complexes and b) nuclear translocation of DR5 in the nucleus, potentially mediating cell migration. See text for explanation.
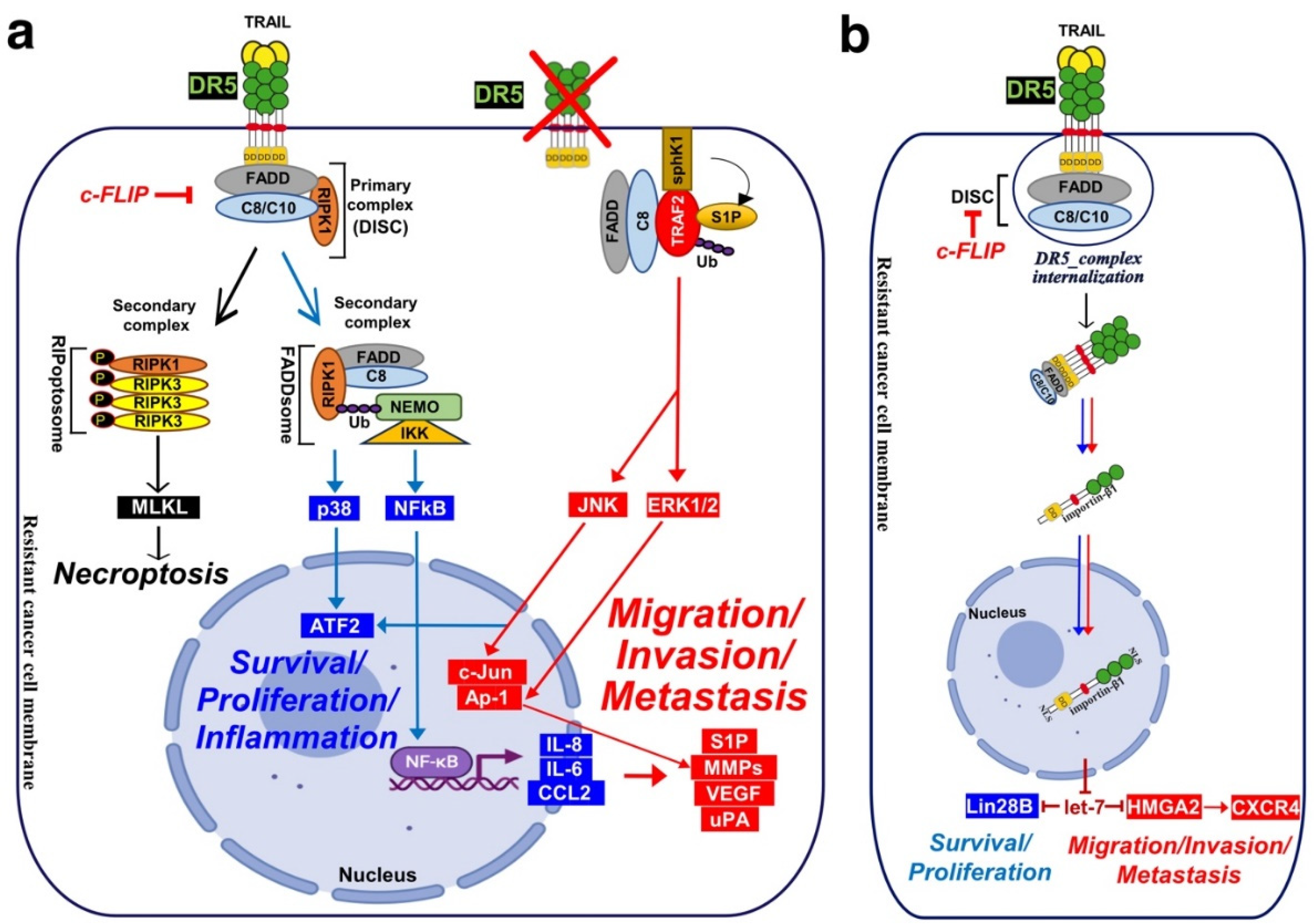
Figure 5.
TRAIL-induced non-canonical pro-tumoral signalling via DR5 secondary complex formation. TRAIL agonist receptors, especially DR5, depending on cancer type and stage, can promote tumour growth and metastasis either through complex I or through a soluble secondary complex. Complex II arises from complex I and contains amongst other FADD, caspase-8, RIPK1, TRAF2, TRADD, cIAP, LUBAC, NEMO and IKKs. While complex I. is associated with survival and proliferation through p38, JNK and NF-kB activation, complex II appears in addition able to activate ERK1/2 pathway and Src leading to metastasis in vivo (See text for explanations). DR5 can directly activate signalling proteins involved in metastasis, thanks to its membrane-proximal domain (MPD), represented in orange, which directly recruits a Ca2+ binding protein, the CaM whose recruitment, in the presence of calcium, induce the activation of the proto-oncogene Src and the ubiquitin ligase c-Cbl, leading to PI3K, JUN, STAT3 and Rac1 activation. Activation of Rac1 promotes microtubules and cytoskeleton organization to activate cell migration. Rac1 was also found, as illustrated here to be activated by direct recruitment to DR5 MPD, in a ligand independent manner. See text for additional details. Colours: writing highlights and arrows illustrate TRAIL-induced proliferation and inflammation (in blue), or TRAIL-induced metastasis (in red).
Figure 5.
TRAIL-induced non-canonical pro-tumoral signalling via DR5 secondary complex formation. TRAIL agonist receptors, especially DR5, depending on cancer type and stage, can promote tumour growth and metastasis either through complex I or through a soluble secondary complex. Complex II arises from complex I and contains amongst other FADD, caspase-8, RIPK1, TRAF2, TRADD, cIAP, LUBAC, NEMO and IKKs. While complex I. is associated with survival and proliferation through p38, JNK and NF-kB activation, complex II appears in addition able to activate ERK1/2 pathway and Src leading to metastasis in vivo (See text for explanations). DR5 can directly activate signalling proteins involved in metastasis, thanks to its membrane-proximal domain (MPD), represented in orange, which directly recruits a Ca2+ binding protein, the CaM whose recruitment, in the presence of calcium, induce the activation of the proto-oncogene Src and the ubiquitin ligase c-Cbl, leading to PI3K, JUN, STAT3 and Rac1 activation. Activation of Rac1 promotes microtubules and cytoskeleton organization to activate cell migration. Rac1 was also found, as illustrated here to be activated by direct recruitment to DR5 MPD, in a ligand independent manner. See text for additional details. Colours: writing highlights and arrows illustrate TRAIL-induced proliferation and inflammation (in blue), or TRAIL-induced metastasis (in red).
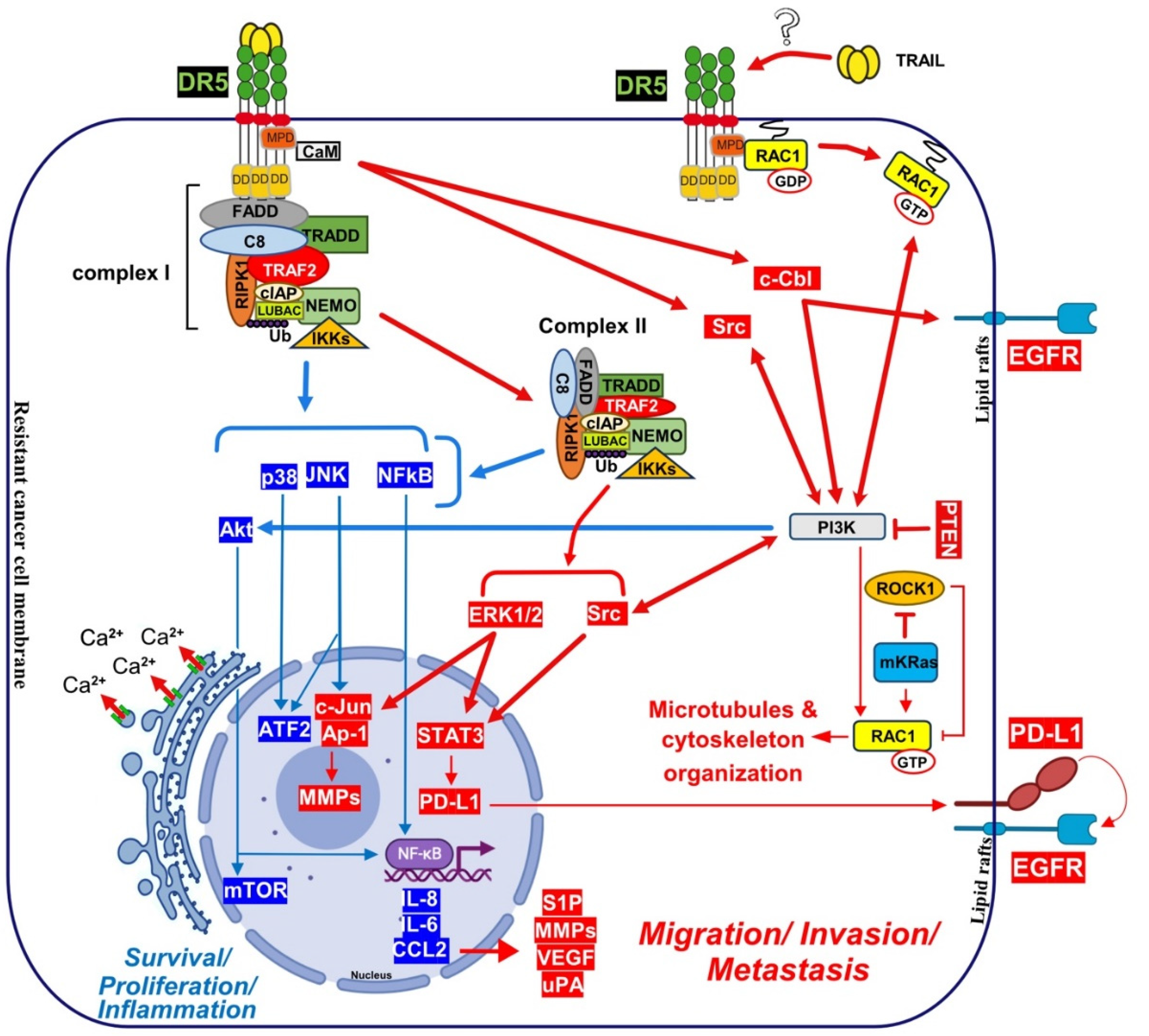
Figure 6.
TRAIL non-canonical signalling pathway associated with caspase-8 non-enzymatic functions. Caspase-8 is a pro-apoptotic protein that, in the presence of c-FLIP, can activate TRAIL non-canonical signalling in an enzymatic-independent manner. If an increase in Ca2+ ions from the ER is initiated at some point of the stimulation, the CaM can be recruited to DR5 MPD. Activated CaM, by inducing Src activation induces next the phosphorylation of the caspase-8, see text for details, enabling PI3K activation and subsequent activation of Rac1, leading to cell migration and invasion. Caspase-8 phosphorylation can also inhibit adhesion complex through complex elements FAC and CPN2 interaction. This interaction inhibits cell adhesion and allow complex elements activation. Then, cell migration and invasion can be induced by activating FAK and additional adhesion complex elements like PLCy, FAK, Rho, PI3K, and Src. Curiously, FADD seems to have the ability to trigger FAK-inhibited miR7a expression through unknown mechanisms. This is linked to the expression of pro-metastatic cytokines TGFβ and CCL5. Colours: writing highlights and arrows illustrate TRAIL-induced proliferation and inflammation (in blue), or TRAIL-induced metastasis (in red).
Figure 6.
TRAIL non-canonical signalling pathway associated with caspase-8 non-enzymatic functions. Caspase-8 is a pro-apoptotic protein that, in the presence of c-FLIP, can activate TRAIL non-canonical signalling in an enzymatic-independent manner. If an increase in Ca2+ ions from the ER is initiated at some point of the stimulation, the CaM can be recruited to DR5 MPD. Activated CaM, by inducing Src activation induces next the phosphorylation of the caspase-8, see text for details, enabling PI3K activation and subsequent activation of Rac1, leading to cell migration and invasion. Caspase-8 phosphorylation can also inhibit adhesion complex through complex elements FAC and CPN2 interaction. This interaction inhibits cell adhesion and allow complex elements activation. Then, cell migration and invasion can be induced by activating FAK and additional adhesion complex elements like PLCy, FAK, Rho, PI3K, and Src. Curiously, FADD seems to have the ability to trigger FAK-inhibited miR7a expression through unknown mechanisms. This is linked to the expression of pro-metastatic cytokines TGFβ and CCL5. Colours: writing highlights and arrows illustrate TRAIL-induced proliferation and inflammation (in blue), or TRAIL-induced metastasis (in red).
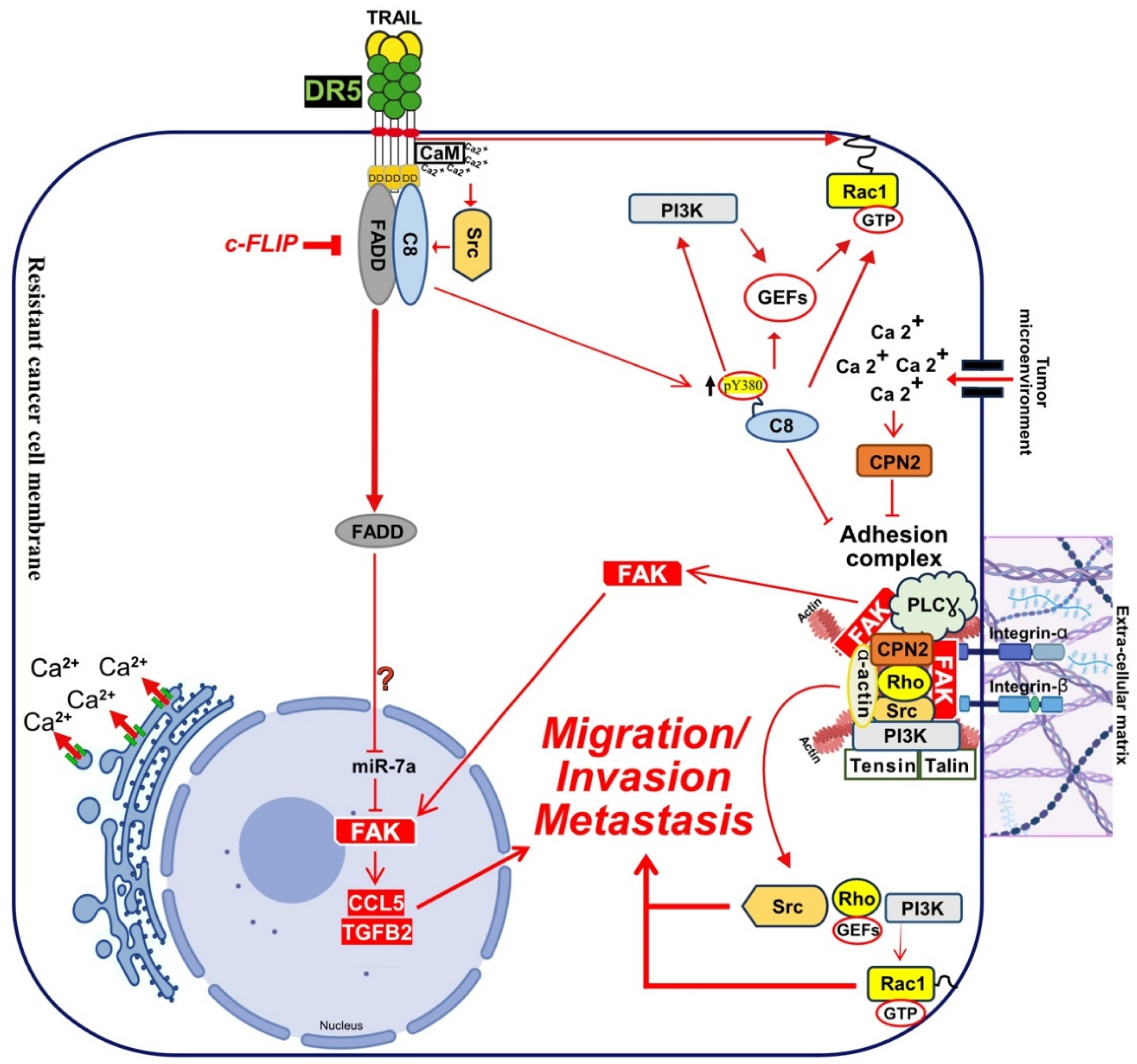
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



