
Submitted:
07 February 2024
Posted:
08 February 2024
You are already at the latest version
Abstract
Colorectal cancer (CRC) is the second cause of cancer-related death where the CpG-island methylation pathways (CIMP) is associated to KRAS/BRAF mutations, two oncogenes rewiring cell metabolism, worst prognosis and resistance to classical chemotherapies. Despite this, the question of a possible metabolic rewiring in CIMPs has never been asked. Here we asked if metabolic dysregulations were associated to the methylation status of tumours by evaluating the transcriptome of CRC tumours. CIMP-high patients were found to present a hyper-metabolism activating mainly carbohydrates, folates, sphingolipids and arachidonic acid metabolic pathways. A third of these genes had epigenetic targets of Myc in their proximal promoter, activating carboxylic acid, tetrahydrofolate interconversion, nucleobase, and oxoacid metabolisms. Amongst the Myc signature, the expression of GAPDH, TYMS, DHFR, TK1 was enough to predict methylation levels, microsatellite instability (MSI) and mutations in the mismatch repair (MMR) machinery, which are strong indicators of responsiveness to immunotherapies. Finally, we discovered that CIMP patients harboured an increase in genes involved in the one-carbon metabolism, a pathway critical to provide nucleotides for cancer growth and methyl donors for DNA methylation which was associated to worse prognosis. Transcriptomics could hence become a tool to help clinicians stratify their patients better.
Keywords:
Myc
; metabolism
; colorectal cancer
; CIMP
; immunotherapy
; one-carbon metabolism
1. Introduction
Colorectal cancer (CRC) is the second cause of cancer-related death worldwide, responsible for nearly a million death in 2020 [1]. CRC tumours can arise from two main mechanisms of carcinogenesis: the conventional pathway, associated with mutations in APC and p53, and the serrated pathway that is associated with mutations in KRAS or BRAF and a hypermethylated phenotype (CpG island methylator phenotype or CIMP). Serrated tumours, although less frequent, have been shown to be the most aggressive subtype of CRC [2]. Nevertheless, the strong heterogeneity in their pathological presentation and mutational pattern makes them very hard to diagnose. Moreover, multiple methylation panels are used to diagnose the CIMP phenotype, leading to heterogeneity in the diagnosis and new means of identifying these higher risk tumours are needed.
The CIMP subtype of CRC represent around 20% of all CRC patients and are the ones with the worst survival and resistance to classical treatment like 5-Fluorouracil [3]. Moreover, several meta-analysis have highlighted the disparities in the diagnosis of CIMP patients: there is no consensus on the gene panels to be used or in the experimental method of choice to test the methylation of these genes leading to 16 different definitions of CIMP in the literature [4]. There is hence a need to homogenize our definition of CIMPs and find reliable phenotypic markers for this group of patients.
Since KRAS and BRAF are known regulators of metabolism in multiple cancer, driving its rewiring in tumour cells, CIMP tumours could harbor a particular metabolic signature differentiating them from other types of CRCs. Indeed, it has previously been shown that CIMP have upregulation of enzymes in glycolysis and related processes [5]. Moreover, since CIMP tumours are characterized by a hypermethylation phenotype, it is very likely that the metabolism of these tumours also needs to adapt to accommodate for the higher demand for methyl groups.
Methylation is one of the main epigenetic ways to regulate gene expression: it consists on the addition of a methyl group to the cysteine in position 5 of histones and usually results in the downregulation of the methylated gene. The one-carbon (1-C) metabolism is the central provider of methyl groups needed for methylation processes and has been shown to be associated with poorer prognosis in colorectal and other cancers [6,7,8,9,10,11]. Indeed, 5-Fluorouracil, an anti-metabolite chemotherapy targeting 1-C metabolism, is the first line of treatment for CRC patients [12] and one of the most widely used chemotherapy [13], indicating the importance of this pathway for cancer cell growth in many different types of cancers. Finally, it has recently been shown that the mitochondrial part of the 1-C metabolism is not required for cancer growth but fosters migration of cancer cells [14]. Altogether these data suggest that 1-C metabolism is important for both cancer growth and dissemination. Nevertheless, its contribution to CIMP specifically and its value as a prognostic factor have never been investigated. Furthermore, targeting of the 1-C metabolism pathway is difficult because it is so essential to normal cells and hence generates a lot of resistance to treatment. To date, most of the targeting is done through targeting the Dihydrofolate reductase (DHFR) or the Thymidylate synthase (TYMS) but more targets for therapeutic intervention are required.
The aim of the present study is to evaluate if CIMP tumours present a distinct metabolic signature, if their levels of methylation could be predicted using transcriptomic data and if new therapeutic targets can be identified.
2. Materials and Methods
2.1. TCGA RNA-Sequencing Dataset
TCGA consortium RNA-sequencing data matrices of Z-scores from the CRC-2012 cohort [15] were downloaded with their corresponding clinical data on Cbioportal website [16]. Supplemental biological information was also provided for these patients such as: methylation group status, MLH1 status, hyper-mutation and MSI status. This cohort is composed of 223 patients and stratification on methylation status showed 80 patients with positive methylation status (CIMP low and CIMP high) versus 143 patients with methylation status negative (cluster3 and cluster4) (Table 1).
The main clinical parameters provided in this study such as primary site of tumour localization, oncotree code, cancer type were found significant on this methylation stratification (Table 1). Metabolism pathway enrichment was performed with Geneset enrichment Analysis software version 4.0.1 [17] with KEGG and REACTOME databases implemented in MSigDB version 7.0 [18]. Bioinformatics analyses were performed in software environment version 3.5.3. Unsupervised principal component analysis was performed with Factominer R-package [19]. Boxplot were drawn with ggplot2 graph definition [20] and expression heatmap were done with pheatmap R-package with the option of Euclidean distances. Machine learning random forest was performed on selected transcriptome features during unsupervised analysis. This learning was perfomed with Randomforest R-package [21] and the model was built with 150 trees and optimized mtry parameter after tuning with rftune function. Variables importance were kept in the model to interpret gene priority on their accuracy.
2.2. Overall Survival Analysis on TCGA RNA-Sequencing with Multi-Omics Integration in Colorectal Cancer
Using the Cbioportal web application [16], RNA-seq data from the TCGA 2018 colorectal cancer cohort (our validation cohort) was analyzed with Z-score diploid V2 matrix normalization. This dataset composed of 379 patients also had overall survival information [22]. This interface was used to investigate one carbon metabolism gene candidates, defined by AMIGO web database of gene ontology information [23] and available in Supplementary Table S3, on patient survival.
2.3. ChIP-Sequencing Analysis
Results from ChIP sequencing experiments performed in the colorectal cell line LoVo [24] were downloaded on the Cistrome project website. Gene promoter annotation of MYC epigenetics intervals were performed with BETA minus algorithm version 1.0.7 on human genome version HG38. Proximal peaks were filtrated around Transcription starting sites with 5kb upstream and 0.5kb downstream. Functional enrichment on MYC epigenetics genomic intervals (HG38) was predicted with GREAT website application with Gene Ontology Biological Process database [25]. Functional enrichment network of GREAT metabolism connections was drawn with Cytoscape software version 3.6.0 [26]. Motif prediction was performed with RSAT genomic application on human HG38 with Jaspar vertebrae 2018 motif collection [27]. Promoter heatmap were performed with deeptools version 3.3.1 in Linux MINT 19 operating system with custom wrapper in BASH script (https://github.com/cdesterke/chip2heat). Full results are available on Supplementary Table S2.
2.4. Integrative Analysis
In order to integrate Myc epigenetics analyses with RNA-sequencing transcriptome of CRC patients, genomic coordinates of CHIP-sequencing initially aligned on HG38 human transcriptome were liftover to HG19 genomic coordinates. This operation was done with liftover algorithm on UCSC website (https://genome.ucsc.edu/cgi-bin/hgLiftOver). Circosplot was drawn with OmicCircos R bioconductor package for multi-omics integration [28].
2.5. Deep Learning
Keras version 2.7.0, Scikit learn version 1.0.1, tensorflow 2.7.0, pandas 1.3.5 and matplotlib version 3.5.1 python libraries (python version 3.7.6) were used with Jupyter notebook version 6.4.6 to implement deep-learning neuronal network for validations of MYC targets predicting clinical data: methylation status, hyper-mutated phenotype, MLH1 silencing and MSI status. Neuronal network was built with three sequential neuron dense layers: first one with 12 neurons and “Rectified Linear Unit” (ReLU) activation function, second one with 8 neurons with ReLU activation function and third one with one neuron and sigmoid activation function. The model was compiled with “adam” optimizer and loss function based on binary cross entropy and fit with 150 epochs and 10 batch size. Corresponding python code was deployed at the following web address: https://condescending-saha-2dfa16.netlify.app/ (accessed on 2021, December 20th).
2.6. Multivariable Model Built on Methylation Status Outcome
In order to test dependency between qualitative variables of the study “chi2loop” R-package was developed to perform iteration chi.square test between character variables from the cohort dataset. This package is available at the following address: https://github.com/cdesterke/chi2loop (accessed on 2021, nov 30th). This package takes as input a dataset of qualitative variables. To run iterative chi.square tests with “cltest” function, the dataset need to be imported with parameter “stringsAsFactors = FALSE” because input qualitative variables need to be in character format and not as factors. On dataset “cltest” function could be applied to perform chi.square iteration between character columns and output results could be graphically represented with the “nlpplot” function (NLP: negative log10 of chi.squre test p-values). Subsequently “chinet” function could be applied also on “cltest” results to detect variable communities with a Louvain classification algorithm. After selection of associated clinical and expression markers, a multivariable model was built with generalized linear model (GLM) R base function for a logistic regression using binomial family as parameters (methylation status as binary outcome).
Graphical output of multivariable logistic regression model censored on binomial status of methylation was drawn with the R-packages combination: broom version 0.7.10, broom.helpers version 1.4.0 and GGally version 2.1.2. In the multivariable logistic model, a p-value p<0.0.5 for included variables in the model was taken as threshold of significance for their independency.
2.7. Statistical Analyses
Statistical analyses were performed in R software environment version 4.1.0. With Z-scores from RNA-seq data (Gaussian transformation), multi-group comparison was performed with one Way ANOVA Fisher statistical test and dual group comparison was performed with bilateral ttest with Welch correction. For both approach, statistical significance was assessed for a p-value inferior to 0.05.
3. Results
3.1. Hypermetabolism in CIMP CRC Transcriptome
To investigate the metabolic signature of CIMP tumours, we used the CRC-2012 TCGA cohort [15], comprising 222 patients, where methylation levels were measured and CIMP statuses determined. Patients were hence divided into unmethylated (clusters C3 and C4) versus CIMP patients (clusters CIMP_low and CIMP_high). REACTOME and KEGG databases were used to perform Geneset enrichment analysis (GSEA) to compare the transcriptome of methylated and unmethylated patients, with an emphasis on metabolic pathways. Surprisingly, we found that all metabolic pathway significantly modulated in CIMP patients were up-regulated. This hypermetabolism associated to the CIMP phenotype comprised 169 enzymes (Supplementary Table S1), involved in pathways of the biochemistry of carbohydrates, nucleotides and sphingolipids (Figure 1A). We then performed an unsupervised principal component analysis (PCA) using this metabolic signature that confirmed its ability to separate methylated and unmethylated patients (p-value = 3.35e-19, Figure 1B and Supplementary Table S2) but was also able to show a progressive stratification of methylation axis of the unsupervised analysis (p-value = 6.48e-25, Figure 1C). These results suggest a close association between the CIMP phenotype and this hypermetabolism in CRC patients. Indeed, feeding our 169 metabolic markers to a supervised machine learning algorithm was able to discriminate methylated from unmethylated tumors with a global efficiency of 77.58% after 150 trees of random forest learning (Figure 1D). This efficiency was confirmed by unsupervised classification, performed with Euclidean distances, showing on the expression heatmap a smaller cluster comprising a majority of CIMP tumor and a larger cluster comprising a majority of unmethylated patients (Figure 1E). A random Forest variable importance study further confirmed the importance of glycolysis, nucleotide- and one carbon- metabolism to predict CIMP status, with GAPDH being the best metabolic marker (Figure 1F, Supplementary Table S2).
3.2. Myc Regulates one Third of the CIMP-CRC Metabolic Program
We next wondered if there could be a master regulator orchestrating this hypermetabolism, characteristic of CIMP tumours. Myc is a strong promoter of metabolism to foster cell proliferation and cell fitness [29] but also to control stem cell fate decisions [30]. It was shown that its expression is changed in nearly all colorectal tumours, suggesting a strong role in CRC pathogenesis [15]. We asked if the metabolic rewiring observed in CIMP could be driven by Myc by using publicly available ChIP-seq data from the colorectal cell line LoVo [24]. Promoter heatmap of the 38663 MYC peaks confirmed that the signal is well centered on Transcription Starting sites (TSS) (Figure 2A). A phast-conservation study for Myc confirmed that its chromatin binding events were found well conserved on the core mammalian promoter database (Figure 2B). Myc chromatin bindings were then mapped on HG38 human genome promoters and proximal events were filtered around the TSS (5kb upstream and 0.5kb downstream). The resulting 5000 gene promoter prediction was crossed, in the LoVo cell line, with genes from the CIMP metabolic signature and we found 53 genes with Myc binding sites in their promoters (Figure 2C, Supplementary Table S2). These results indicate that at least a third of the metabolic signature observed in CIMP patients could be driven by Myc. A genomic Circosplot on the Myc signature revealed that sexual chromosomes, chromosomes 6, 13, 16 and 20 were not involved in the CIMP-specific metabolic program (Figure 2D). Chromosome 12 contained the most gene from the signature with eight Myc targets, especially GAPDH that we found as best predictive marker of the CIMP phenotype (Figures 1F and 2D).
3.3. Genes from the Myc Transcriptional Program Also Have Binding Sites for Other Transcription Factors
The observed mean size of Myc chromatin binding peaks in the CIMP-specific metabolic program was 651 pb (SD: 259 pb, n=54 peaks for 53 genes) and the identified peaks were narrow and centered on Transcription Starting Sites (TSS) (Figure 3A). This proximal transcriptional program was compatible to perform motif prediction with the RSAT web application, using the JASPAR core promoter motif database. After picking corresponding nucleotide sequences on HG38 human genome, we performed a transcription factor binding motif identification and confirmed MAX binding sites, amongst other Myc known co-factors (Figure 3B). Interestingly, we also found 182 compatible sites for homeobox transcription factors like POU6F1, DLX1 and EMX2 – 142, 156 binding sites for Kruppel like factors KLF16 and KLF5 respectively and finally 111, 73 and 78 binding sites for HOX family homeobox transcription factors HOXA13, HOXA1913, HOXB13 (Figure 3B). Myc metabolic chromatin binding marks and found activated in transcriptome of CIMP-CRC, were conjointly analyzed with LOVO CHIP sequencing of DLX1, KLF5 and HOXA13. The resulting proximal promoter heatmap analyses revealed that myc metabolic program found CIMP CRC shared chromatin proximal binding sites with these transcription factors previously predict by RSAT: KLF5, DLX1 and HOXA13 (Figure 3C). Also, at transcriptome level, MAX, DLX1 and SP8 transcription factors were confirmed as being progressively overexpressed with level of methylation. These results suggested that myc metabolic program found CIMP CRC could be active in a chromatin context implicating homebox and Kruppel like factors.
Functional enrichment, performed on genomic intervals from the Myc signature on Gene Ontology Biological Process database (GO-BP), showed that the principally altered pathways were the carboxylic acid, tetrahydrofolate interconversion, nucleobase containing small molecule and oxoacid pathways (Figure 4A). This metabolism enrichment highlighted enzymes at the interface between glycolysis (GAPDH, ENO1, LDHA) and one-carbon metabolism (TYMS, SHMT2, MTHFR, MTHFD1), further confirming the importance of these two pathways in CIMP tumours (Figure 4B).
3.4. Metabolism Targets in the Myc Signature Are Associated to Worst Clinical Group in CRC
Several clinical parameters are available in the CRC-2012 cohort: methylation status, expression subtype (clustering into 3 subtypes, according to their mRNA profile), hypermutation and MLH1 silencing. We next evaluated the correlation between these parameters and our CIMP-specific metabolic signature. Based on the classification from the Random Forest prediction factor analysis (Figure 1F), the Myc epigenetic score from the LoVo ChIP-seq (Supplementary Table S2) and also the methylation predictive score, the best candidates to discriminate the pathological parameters from our TCGA cohort were GAPDH, TYMS, DHFR and TK1. All these markers had a specific increased expression in CIMP (Figure 4C, first two left panels) and were correlated to hypermutation and MLH1 silencing (Figure 4C, last two right panels). To go further, we built a multivariate deep-learning model by neural network using keras tensorflow Python libraries and these four Myc-specific targets. We found that this model was very efficient in predicting the Hypermutation and MLH1 silencing status of tumours (95 and 96% respectively) but also the CIMP and MSI status (82 and 85% respectively, Table 2). Predicting the MSI and MLH1 status is of particular interest, since it is one of the only parameters used to stratify patient care and decide on the use of immunotherapies [31]. Very interestingly, three of our four markers (TYMS, DHFR and TK1) are also part of the one-carbon metabolism pathway.
3.5. Overexpression of One Carbon Metabolism Enzymes Are Independent Markers of Methylation Status, MLH1 Silencing, Hypermutation and MSI in Colorectal Cancer
To study in more detail the prognostic value of one carbon metabolism in CRC, we selected the enzymes significantly associated to the methylation status (TYMS, TK1, SHMT2, MTHFD1, MTHFD2, DHFR, Figure 4B) and their normalized Z-score were transformed in quantile five (Q5) classes. This allowed us to perform iterative Chi-square tests with the different clinical parameters (for details see Supplementary Table S1) inputted as character string for Natural Language Processing (NLP) (see Materials and methods for more details, Figure 5A). Amongst one carbon metabolism genes, the expression of TYMS had the highest association to the hypermutation status, iCluster classification, methylation subtypes, MLH1 silencing and MSI status (Figure 5A). Consistent with these proteins being part of the same pathway, the same associations, although with less significance, were found for TK1, SHMT2 and MTHFD2 (Figure 5A). MTHDF1 did not show significant association with MSI status and was not able to discriminate between CIN/CIMP (Expression_subtype variable on Figure 5A). DHFR showed the weakest association with clinical parameters and was not associated with methylation or iCluster variables (Figure 5A). Interestingly, none of the one-carbon metabolism genes were able to discriminate for tumour stage (“Tumor stage” variable). This was consistent with the fact that stratifying patients per methylation status gave similar distribution of tumour stages (Table 1). We next performed a variable community detection using a Louvain algorithm on the results from the Chi.square tests (Figure 5A) This analysis confirmed the association of TYMS, TK1, SHMT2, MTHFD2 and DHFR expressions with the main prognosis markers (hypermutation, MSI, iCluster, expression subtype and tumor stage) while MTHFD1, and not MTHFD2, was more strongly associated to the methylation status (Figure 5B). Based on these observed associations, we finally built a methylation status multivariable model with minimum confounding parameters based on the expression of these one-carbon metabolism genes and clinical parameters. This logistic multivariate model particularly confirmed that overexpression of TYMS and MTHFD1 are strong, independent, one-carbon metabolism markers able to discriminate for the CIMP phenotype (Figure 5C). It also confirmed that CIMP tumours are strongly associated to MSI, as previously published [15].
3.6. Activation of 1-C Metabolism Genes Predicts Worst Prognosis Colorectal Cancer Patients
In order to validate the association of one-carbon metabolism and the hypermethylation status as well as bad prognosis for colorectal cancer patients, a multi-omics investigation was performed in the 2018 augmented CRC dataset (TCGA-COAD, our validation cohort) from the TCGA consortium [22]. Using the Gene Ontology database, through the AMIGO website, genes from the one-carbon metabolism were selected (Supplementary Table S3). We looked at the overexpression of these one-carbon metabolism genes in the CRC tumour RNAseq data and selected the most changed to establish a gene signature (Figure 6A).
Overall survival analysis revealed that patients presenting alteration in this one-carbon signature (i.e., increased expression of the genes in the signature) had worsen survival (Figure 6B). As previously reported [2], tumours from the right colon (comprising Ascending, Transverse colon, hepatic flexure and cecum) showed a significant association with increased one-carbon metabolism (Figure 6C) and were associated to a higher age at diagnosis (data not shown). Looking at HM450 methylation profiles, we found that the presence of this altered one-carbon signature was associated to an increase in the global methylation of the genome (Figure 6D and Supplemental Table S5 for a detailed list of genes with differentially methylated promoters).
We then performed a functional enrichment analysis on the differentially methylated genes using the KEGG or Gene Ontology database (Figure 6E,F). This revealed that most changes were seen in primary bile acid biosynthesis, but also affected vitamin absorption, PPAR signaling as well as stem cell functions (WNT signaling, pathways of pluripotency regulation). We also used the Gene Ontology database to perform the functional enrichment and found the most changes in gene implicated in the regulation of SMAD protein phosphorylation.
Altogether these results confirmed that the activation of one-carbon metabolism in CRC is associated to a worst prognosis for patients and a hypermethylation of gene promoters that could impact important functionalities in intestinal cells and have causative effects in cancer progression and dissemination. This strongly suggests that targeting one-carbon metabolism is of high interest for CRC patients.
4. Discussion
Despite the role of KRAS and BRAF in driving the CIMP phenotype and rewiring cancer metabolism [2,32,33,34], few studies have aimed at establishing if CIMP tumours have a different metabolism [5]. Using transcriptomics data and focusing only on metabolic pathways, we have found that CIMP tumours indeed present a hypermetabolism, with surprisingly no down-regulated but several up-regulated metabolic pathways. We were also able to determine a metabolic signature specific to CIMP tumours, able to discriminate hypermethylated tumours and even separate CIMP-L from CIMP-H patients with a high accuracy. This suggests the possibility of identifying these higher risk tumours, with high accuracy, by using transcriptomics data. This provides an alternative to the current techniques using the methylation of a panel of genes, with several gene panels being used over the world and no consensus and which panel and which technique to test for methylation [4]. Furthermore, we were also able to show that one-carbon metabolism enzymes were amongst the most efficient predictors of the CIMP phenotype, confirming the importance of this pathway, most likely in providing methyl groups to support the high methylation of the genome. Indeed, a background of high genome methylation dramatically fosters the ability of mutant BRAF to drive carcinogenesis, since high expression of mutated BRAF on its own rather triggers senescence and death [35]. This is what is thought to foster the development of BRAF-driven CIMP tumours [36].
Myc’s expression is changed in almost all colorectal cancer, underlying its role in tumorigenesis [15]. Amongst the genes in our metabolic signature, one third could be under the control of Myc in colorectal patients. Indeed we confirmed using ChIP-seq data that Myc physically binds to the promoter of these genes in the LoVo colorectal cancer cell line. Pardoxically, Myc expression is lowest in CIMP tumours, compared to the other tumour subtypes, where the signature is observed. We also found that binding sites for Max, one of the best characterized co-factors of Myc, are present within the promoters of the metabolic signature and that Max physically binds to the promoter of these genes in the LoVo Chip-seq data. This suggests that the specificity of the signature could be coming from the expression of co-factors rather than of Myc itself. Such a phenomenon has already been observed in Small Cell Lung Cancer, where it has been shown that Max expression regulates the 1C-metabolism pathway in a context-dependent manner [37].
Thanks to several meta-analysis of CRC cohorts, it is now becoming clear that the CIMP phenotype is associated to a worsen prognosis and that the benefit of an adjuvant fluorouracil (5-FU, one of the classical chemotherapy protocol for CRC patients) treatment after surgery is limited for CIMP-High patients, especially those with advanced stage III-IV tumours [3,38,39]. Given the important toxicity of these treatments, identifying those patients is very important to improve their quality of life and prevent their exposure to a medication with limited interest and high toxicity for them. Counter-intuitively we found that one of the gene most associated and predictive of the CIMP phenotype is TYMS (thymidylate synthase), which has been shown to be up-regulated by Myc and to drive sensitivity to 5-FU treatments [40]. This means that other mechanisms are at play and suggests that other targets in the 1C-metabolism pathway should be explored. Indeed, our results suggest that MTHFD1, 2 could be very interesting targets for the treatment of colorectal cancer. MTHFD2 is of particular interest since it has been shown to be expressed only in embryonic tissues but becomes re-activated in cancer tissues [41,42]. It is hence a tumour-specific marker that has been shown to be overexpressed and associated with a worse prognosis in many cancer tissues [42,43,44]. It has also been recently proposed to play a role in the regulation of the immune system, fostering cancer immune evasion [45]. Interestingly, in this study the authors showed that MTHFD2 promotes O-glycosylation which increases Myc’s stability and PD-L1 transcription. A recent study has shown that inhibitors of MTHFD2 lead to the specific death of MTHFD2-expressing cells, suggesting that these therapeutic approaches could be very specific to cancer cells and have minimal impact on normal cells.
5. Conclusions
Our study shows that transcriptomics holds great power towards unbiasedly identifying hypermethylated tumours. This approach could provide an alternative to the actual disparities in the methods used to assess methylation and allow more consistency and robustness. Additionally, we show that transcriptomics identified tumours with high MLH1 silencing, which is an indication of good responders to immunotherapy and these patients should receive this treatment rather than 5-FU [31]. This is in accordance with recent studies showing that transcriptomics is becoming more and more relevant in the clinical practice and provides a great clinical benefit to orient treatment protocols, especially for patients without tractable DNA mutations in their tumours [46].
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, CD and ED; methodology, software, CD; writing—review and editing, CD and ED; funding acquisition, ED. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Fondation Gustave Roussy, grant number DRE029-X75951 and the APC was funded by Fondation ARC pour la recherche sur le cancer.
Data Availability Statement
Links to all algorithms used in this study are listed in Materials and Methods
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Xi, Y.; Xu, P. Global Colorectal Cancer Burden in 2020 and Projections to 2040. Translational Oncology 2021, 14, 101174. [Google Scholar] [CrossRef] [PubMed]
- De Palma, F.D.E.; D’Argenio, V.; Pol, J.; Kroemer, G.; Maiuri, M.C.; Salvatore, F. The Molecular Hallmarks of the Serrated Pathway in Colorectal Cancer. Cancers 2019, 11, 1017. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Deng, Z.; Lang, X.; Jiang, J.; Xie, K.; Lu, S.; Hu, Q.; Huo, Y.; Xiong, X.; Zhu, N.; et al. Meta-Analysis of the Prognostic and Predictive Role of the CpG Island Methylator Phenotype in Colorectal Cancer. Dis Markers 2022, 2022, 4254862. [Google Scholar] [CrossRef] [PubMed]
- Jia, M.; Gao, X.; Zhang, Y.; Hoffmeister, M.; Brenner, H. Different Definitions of CpG Island Methylator Phenotype and Outcomes of Colorectal Cancer: A Systematic Review. Clinical Epigenetics 2016, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Fedorova, M.S.; Krasnov, G.S.; Lukyanova, E.N.; Zaretsky, A.R.; Dmitriev, A.A.; Melnikova, N.V.; Moskalev, A.A.; Kharitonov, S.L.; Pudova, E.A.; Guvatova, Z.G.; et al. The CIMP-High Phenotype Is Associated with Energy Metabolism Alterations in Colon Adenocarcinoma. BMC Medical Genetics 2019, 20, 52. [Google Scholar] [CrossRef]
- Ducker, G.S.; Rabinowitz, J.D. One-Carbon Metabolism in Health and Disease. Cell Metabolism 2017, 25, 27–42. [Google Scholar] [CrossRef]
- Newman, A.C.; Maddocks, O.D.K. One-Carbon Metabolism in Cancer. British Journal of Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef]
- Asai, A.; Konno, M.; Koseki, J.; Taniguchi, M.; Vecchione, A.; Ishii, H. One-Carbon Metabolism for Cancer Diagnostic and Therapeutic Approaches. Cancer Letters 2020, 470, 141–148. [Google Scholar] [CrossRef]
- Myte, R.; Gylling, B.; Häggström, J.; Schneede, J.; Löfgren-Burström, A.; Huyghe, J.R.; Hallmans, G.; Meyer, K.; Johansson, I.; Ueland, P.M.; et al. One-Carbon Metabolism Biomarkers and Genetic Variants in Relation to Colorectal Cancer Risk by KRAS and BRAF Mutation Status. PLOS ONE 2018, 13, e0196233. [Google Scholar] [CrossRef]
- Mentch, S.J.; Locasale, J.W. One-Carbon Metabolism and Epigenetics: Understanding the Specificity. Annals of the New York Academy of Sciences 2016, 1363, 91–98. [Google Scholar] [CrossRef]
- Hanley, M.P.; Rosenberg, D.W. One-Carbon Metabolism and Colorectal Cancer: Potential Mechanisms of Chemoprevention. Curr Pharmacol Rep 2015, 1, 197–205. [Google Scholar] [CrossRef]
- Heidelberger, C.; Chaudhuri, N.K.; Danneberg, P.; Mooren, D.; Griesbach, L.; Duschinsky, R.; Schnitzer, R.J.; Pleven, E.; Scheiner, J. Fluorinated Pyrimidines, A New Class of Tumour-Inhibitory Compounds. Nature 1957, 179, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Chalabi-Dchar, M.; Fenouil, T.; Machon, C.; Vincent, A.; Catez, F.; Marcel, V.; Mertani, H.C.; Saurin, J.-C.; Bouvet, P.; Guitton, J.; et al. A Novel View on an Old Drug, 5-Fluorouracil: An Unexpected RNA Modifier with Intriguing Impact on Cancer Cell Fate. NAR Cancer 2021, 3, zcab032. [Google Scholar] [CrossRef] [PubMed]
- Kiweler, N.; Delbrouck, C.; Pozdeev, V.I.; Neises, L.; Soriano-Baguet, L.; Eiden, K.; Xian, F.; Benzarti, M.; Haase, L.; Koncina, E.; et al. Mitochondria Preserve an Autarkic One-Carbon Cycle to Confer Growth-Independent Cancer Cell Migration and Metastasis. Nat Commun 2022, 13, 2699. [Google Scholar] [CrossRef]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Liberzon, A.; Birger, C.; Thorvaldsdóttir, H.; Ghandi, M.; Mesirov, J.P.; Tamayo, P. The Molecular Signatures Database (MSigDB) Hallmark Gene Set Collection. Cell Syst 2015, 1, 417–425. [Google Scholar] [CrossRef]
- Lê, S.; Josse, J.; Husson, F. FactoMineR: An R Package for Multivariate Analysis. Journal of Statistical Software 2008, 25, 1–18. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York 2009. [CrossRef]
- Breiman, L. Random Forests. Machine Learning 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Liu, J.; Lichtenberg, T.; Hoadley, K.A.; Poisson, L.M.; Lazar, A.J.; Cherniack, A.D.; Kovatich, A.J.; Benz, C.C.; Levine, D.A.; Lee, A.V.; et al. An Integrated TCGA Pan-Cancer Clinical Data Resource to Drive High-Quality Survival Outcome Analytics. Cell 2018, 173, 400–416.e11. [Google Scholar] [CrossRef] [PubMed]
- Carbon, S.; Ireland, A.; Mungall, C.J.; Shu, S.; Marshall, B.; Lewis, S. AmiGO Hub; Web Presence Working Group AmiGO: Online Access to Ontology and Annotation Data. Bioinformatics 2009, 25, 288–289. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Enge, M.; Whitington, T.; Dave, K.; Liu, J.; Sur, I.; Schmierer, B.; Jolma, A.; Kivioja, T.; Taipale, M.; et al. Transcription Factor Binding in Human Cells Occurs in Dense Clusters Formed around Cohesin Anchor Sites. Cell 2013, 154, 801–813. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT Improves Functional Interpretation of Cis-Regulatory Regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Cline, M.S.; Smoot, M.; Cerami, E.; Kuchinsky, A.; Landys, N.; Workman, C.; Christmas, R.; Avila-Campilo, I.; Creech, M.; Gross, B.; et al. Integration of Biological Networks and Gene Expression Data Using Cytoscape. Nat Protoc 2007, 2, 2366–2382. [Google Scholar] [CrossRef] [PubMed]
- Turatsinze, J.-V.; Thomas-Chollier, M.; Defrance, M.; van Helden, J. Using RSAT to Scan Genome Sequences for Transcription Factor Binding Sites and Cis-Regulatory Modules. Nat Protoc 2008, 3, 1578–1588. [Google Scholar] [CrossRef]
- Hu, Y.; Yan, C.; Hsu, C.-H.; Chen, Q.-R.; Niu, K.; Komatsoulis, G.A.; Meerzaman, D. OmicCircos: A Simple-to-Use R Package for the Circular Visualization of Multidimensional Omics Data. Cancer Inform 2014, 13, 13–20. [Google Scholar] [CrossRef]
- Grifoni, D.; Bellosta, P. Drosophila Myc: A Master Regulator of Cellular Performance. Biochimica et Biophysica Acta (BBA) - Gene Regulatory Mechanisms 2015, 1849, 570–581. [Google Scholar] [CrossRef]
- Cliff, T.S.; Wu, T.; Boward, B.R.; Yin, A.; Yin, H.; Glushka, J.N.; Prestegaard, J.H.; Dalton, S. MYC Controls Human Pluripotent Stem Cell Fate Decisions through Regulation of Metabolic Flux. Cell Stem Cell 2017, 21, 502–516.e9. [Google Scholar] [CrossRef]
- Motta, R.; Cabezas-Camarero, S.; Torres-Mattos, C.; Riquelme, A.; Calle, A.; Figueroa, A.; Sotelo, M.J. Immunotherapy in Microsatellite Instability Metastatic Colorectal Cancer: Current Status and Future Perspectives. J Clin Transl Res 2021, 7, 511–522. [Google Scholar]
- Kundu, S.; Ali, M.A.; Handin, N.; Conway, L.P.; Rendo, V.; Artursson, P.; He, L.; Globisch, D.; Sjöblom, T. Common and Mutation Specific Phenotypes of KRAS and BRAF Mutations in Colorectal Cancer Cells Revealed by Integrative -Omics Analysis. Journal of Experimental & Clinical Cancer Research 2021, 40, 225. [Google Scholar] [CrossRef]
- Charitou, T.; Srihari, S.; Lynn, M.A.; Jarboui, M.-A.; Fasterius, E.; Moldovan, M.; Shirasawa, S.; Tsunoda, T.; Ueffing, M.; Xie, J.; et al. Transcriptional and Metabolic Rewiring of Colorectal Cancer Cells Expressing the Oncogenic KRASG13D Mutation. Br J Cancer 2019, 121, 37–50. [Google Scholar] [CrossRef]
- Hutton, J.E.; Wang, X.; Zimmerman, L.J.; Slebos, R.J.C.; Trenary, I.A.; Young, J.D.; Li, M.; Liebler, D.C. Oncogenic KRAS and BRAF Drive Metabolic Reprogramming in Colorectal Cancer. Mol Cell Proteomics 2016, 15, 2924–2938. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Kang, B.; Petkovich, D.A.; Bhandari, Y.R.; In, J.; Stein-O’Brien, G.; Kong, X.; Xie, W.; Zachos, N.; Maegawa, S.; et al. Aging-like Spontaneous Epigenetic Silencing Facilitates Wnt Activation, Stemness, and BrafV600E-Induced Tumorigenesis. Cancer Cell 2019, 35, 315–328.e6. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, T.; Suzuki, H. The Origin of CIMP, At Last. Cancer Cell 2019, 35, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Augert, A.; Mathsyaraja, H.; Ibrahim, A.H.; Freie, B.; Geuenich, M.J.; Cheng, P.-F.; Alibeckoff, S.P.; Wu, N.; Hiatt, J.B.; Basom, R.; et al. MAX Functions as a Tumor Suppressor and Rewires Metabolism in Small Cell Lung Cancer. Cancer Cell 2020, 38, 97–114.e7. [Google Scholar] [CrossRef] [PubMed]
- Juo, Y.Y.; Johnston, F.M.; Zhang, D.Y.; Juo, H.H.; Wang, H.; Pappou, E.P.; Yu, T.; Easwaran, H.; Baylin, S.; van Engeland, M.; et al. Prognostic Value of CpG Island Methylator Phenotype among Colorectal Cancer Patients: A Systematic Review and Meta-Analysis. Annals of Oncology 2014, 25, 2314–2327. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, W.; Cao, P. Advances in CpG Island Methylator Phenotype Colorectal Cancer Therapies. Front Oncol 2021, 11, 629390. [Google Scholar] [CrossRef] [PubMed]
- Azwar, S.; Seow, H.F.; Abdullah, M.; Faisal Jabar, M.; Mohtarrudin, N. Recent Updates on Mechanisms of Resistance to 5-Fluorouracil and Reversal Strategies in Colon Cancer Treatment. Biology 2021, 10, 854. [Google Scholar] [CrossRef]
- Bolusani, S.; Young, B.A.; Cole, N.A.; Tibbetts, A.S.; Momb, J.; Bryant, J.D.; Solmonson, A.; Appling, D.R. Mammalian MTHFD2L Encodes a Mitochondrial Methylenetetrahydrofolate Dehydrogenase Isozyme Expressed in Adult Tissues*. Journal of Biological Chemistry 2011, 286, 5166–5174. [Google Scholar] [CrossRef]
- Nilsson, R.; Jain, M.; Madhusudhan, N.; Sheppard, N.G.; Strittmatter, L.; Kampf, C.; Huang, J.; Asplund, A.; Mootha, V.K. Metabolic Enzyme Expression Highlights a Key Role for MTHFD2 and the Mitochondrial Folate Pathway in Cancer. Nat Commun 2014, 5, 3128. [Google Scholar] [CrossRef]
- Koseki, J.; Konno, M.; Asai, A.; Colvin, H.; Kawamoto, K.; Nishida, N.; Sakai, D.; Kudo, T.; Satoh, T.; Doki, Y.; et al. Enzymes of the One-Carbon Folate Metabolism as Anticancer Targets Predicted by Survival Rate Analysis. Sci Rep 2018, 8, 303. [Google Scholar] [CrossRef]
- Noguchi, K.; Konno, M.; Koseki, J.; Nishida, N.; Kawamoto, K.; Yamada, D.; Asaoka, T.; Noda, T.; Wada, H.; Gotoh, K.; et al. The Mitochondrial One-Carbon Metabolic Pathway Is Associated with Patient Survival in Pancreatic Cancer. Oncol Lett 2018, 16, 1827–1834. [Google Scholar] [CrossRef] [PubMed]
- Shang, M.; Yang, H.; Yang, R.; Chen, T.; Fu, Y.; Li, Y.; Fang, X.; Zhang, K.; Zhang, J.; Li, H.; et al. The Folate Cycle Enzyme MTHFD2 Induces Cancer Immune Evasion through PD-L1 up-Regulation. Nat Commun 2021, 12, 1940. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Soria, J.-C.; Berger, R.; Miller, W.H.; Rubin, E.; Kugel, A.; Tsimberidou, A.; Saintigny, P.; Ackerstein, A.; Braña, I.; et al. Genomic and Transcriptomic Profiling Expands Precision Cancer Medicine: The WINTHER Trial. Nat Med 2019, 25, 751–758. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Hyper-metabolism in CIMP CRC transcriptome. (a) Barplot of metabolic pathway found over expressed in CIMP CRC as compared to unmethylated ones (NES: normalized enrichment scores); (b) Unsupervised PCA on CRC transcriptome separates unmethylated and CIMP CRCs using the 169 metabolic marker signature; (c) Unsupervised PCA on CRC transcriptome can stratify tumours into four clusters C3, C4, CIMP-low, CIMP-high using the 169 metabolic marker signature; (d) Machine learning misclassification error rate estimation by Random forest analysis to classify methylation status using the metabolic expression profile; (e) Unsupervised classification of CRC tumours with the metabolic signature (Euclidean distance and Ward.D2 method); (f) Variable importance plot of the best metabolic markers to stratify the methylation status with methylation status (clusters c3 and c4, CIMP_low and CIMP_high).
Figure 1.
Hyper-metabolism in CIMP CRC transcriptome. (a) Barplot of metabolic pathway found over expressed in CIMP CRC as compared to unmethylated ones (NES: normalized enrichment scores); (b) Unsupervised PCA on CRC transcriptome separates unmethylated and CIMP CRCs using the 169 metabolic marker signature; (c) Unsupervised PCA on CRC transcriptome can stratify tumours into four clusters C3, C4, CIMP-low, CIMP-high using the 169 metabolic marker signature; (d) Machine learning misclassification error rate estimation by Random forest analysis to classify methylation status using the metabolic expression profile; (e) Unsupervised classification of CRC tumours with the metabolic signature (Euclidean distance and Ward.D2 method); (f) Variable importance plot of the best metabolic markers to stratify the methylation status with methylation status (clusters c3 and c4, CIMP_low and CIMP_high).
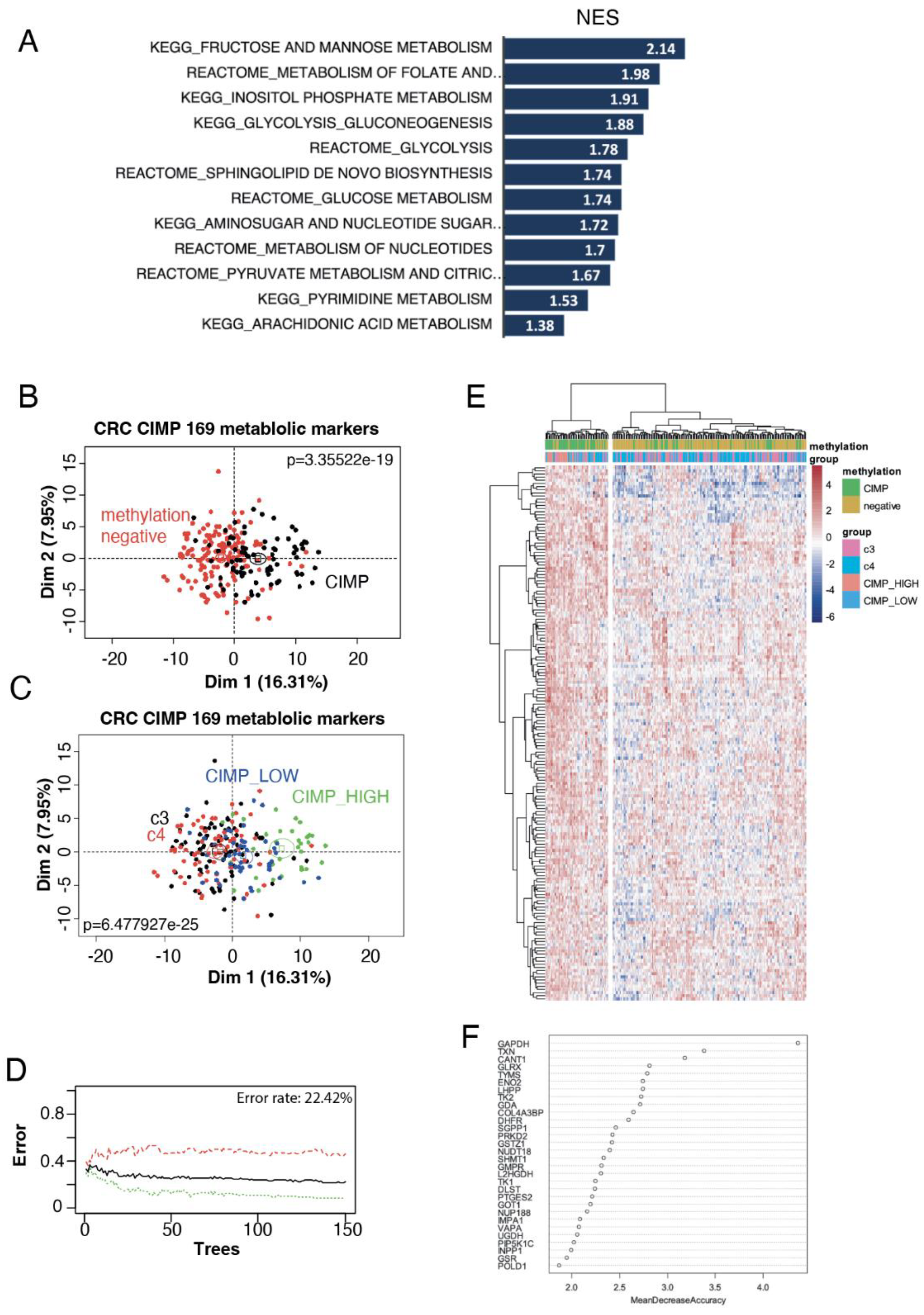
Figure 2.
Myc targets in CIMP CRC metabolism activated program. (a) Promoter heatmap for Myc chromatin binding in the whole genome of the LOVO colorectal cell line (matrix compute on proximal promoter regions: minus 5kb and plus 2kb around TSS); (b) Phastconservation plot of LOVO Myc CHIP-seq around mammalian core promoter database; (c) Venn diagram showing genes at the intersection between the LOVO Myc CHIP-seq dataset and the metabolic signature in CIMPs; (d) Circosplot presenting the results of the Myc CHIP-seq epigenetics analysis (purple: peak scores) and the mRNA levels in the transcriptome of CIMP CRC (blue: delta between CIMP and unmethylated CRC). Genes present in the metabolic signature are indicated in green. HG19 was used as referent genome.
Figure 2.
Myc targets in CIMP CRC metabolism activated program. (a) Promoter heatmap for Myc chromatin binding in the whole genome of the LOVO colorectal cell line (matrix compute on proximal promoter regions: minus 5kb and plus 2kb around TSS); (b) Phastconservation plot of LOVO Myc CHIP-seq around mammalian core promoter database; (c) Venn diagram showing genes at the intersection between the LOVO Myc CHIP-seq dataset and the metabolic signature in CIMPs; (d) Circosplot presenting the results of the Myc CHIP-seq epigenetics analysis (purple: peak scores) and the mRNA levels in the transcriptome of CIMP CRC (blue: delta between CIMP and unmethylated CRC). Genes present in the metabolic signature are indicated in green. HG19 was used as referent genome.
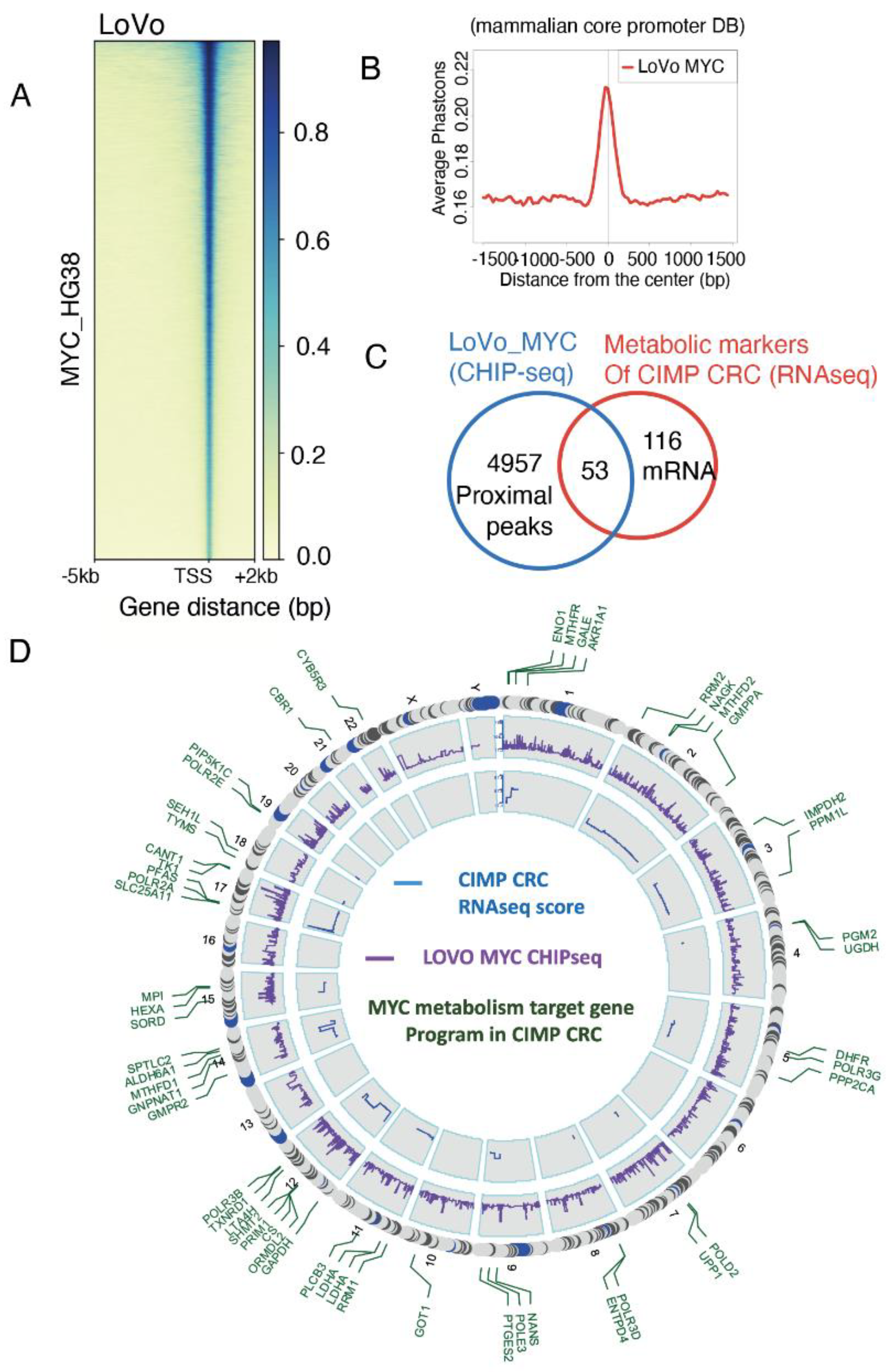
Figure 3.
Myc-driven hypermetabolism gene signature present binding sites for transcription co-factors. (a) Promoter barplot of all the Myc targets in the CIMP metabolic signature; (b) Motif prediction for binding on Myc intervals in the promoter of genes from the metabolic signature, based on Jaspar vertebrae 2018 motif prediction database; (c) Promoter heatmaps using CHIP-seq data from LoVo cells for Myc, DLX1, KLF5 and HOXA13 binding on the promoters of the 169 genes found in the CIMP metabolic signature; (d) Expression boxplot of Myc, Max, Dlx1 and SP8 transcription factors in the transcriptome of CRC patients, stratified by methylation status.
Figure 3.
Myc-driven hypermetabolism gene signature present binding sites for transcription co-factors. (a) Promoter barplot of all the Myc targets in the CIMP metabolic signature; (b) Motif prediction for binding on Myc intervals in the promoter of genes from the metabolic signature, based on Jaspar vertebrae 2018 motif prediction database; (c) Promoter heatmaps using CHIP-seq data from LoVo cells for Myc, DLX1, KLF5 and HOXA13 binding on the promoters of the 169 genes found in the CIMP metabolic signature; (d) Expression boxplot of Myc, Max, Dlx1 and SP8 transcription factors in the transcriptome of CRC patients, stratified by methylation status.
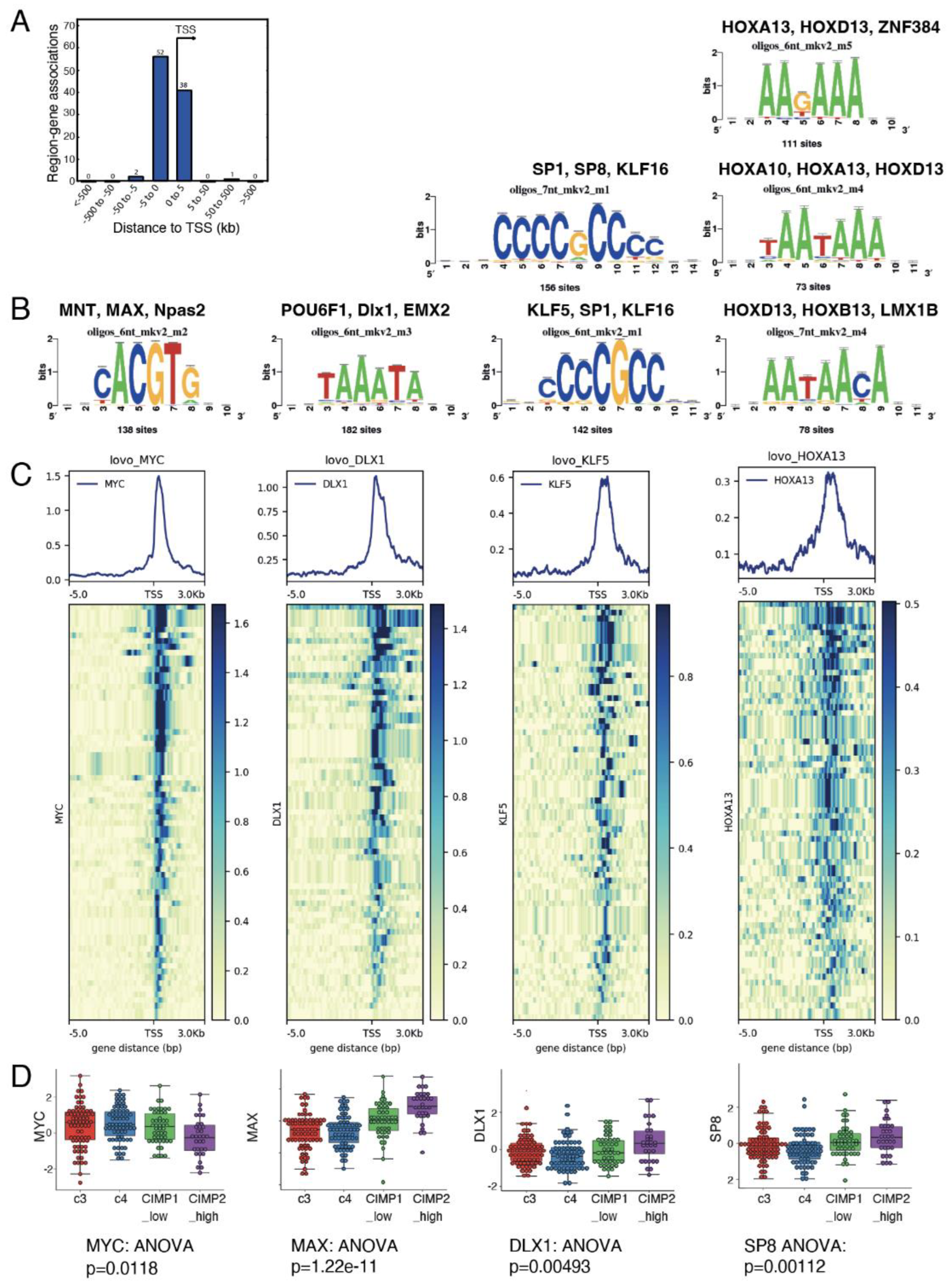
Figure 4.
Genes in the CIMP-specific metabolic signature are associated to worst clinical outcome. (a) Functional enrichment (Biological Process of Gene Ontology database: GO-BP) performed on Myc metabolic targets overexpressed in the CIMP signature. Barplots represent logarithm 10 of negative binomial p-values; (b) Functional enrichment network performed on the main metabolic functions enriched for Myc targets from the CIMP signature < over-expressed in CIMP-CRC; C/ Selection of four discriminant metabolic myc targets dysregulated in CIMP-CRC: for each candidate myc LOVO CHIP-seq and expression boxplot stratified on clinical data (methylation subgroups, expression subgroups, hypermutation status and MLH1 silencing) are represented.
Figure 4.
Genes in the CIMP-specific metabolic signature are associated to worst clinical outcome. (a) Functional enrichment (Biological Process of Gene Ontology database: GO-BP) performed on Myc metabolic targets overexpressed in the CIMP signature. Barplots represent logarithm 10 of negative binomial p-values; (b) Functional enrichment network performed on the main metabolic functions enriched for Myc targets from the CIMP signature < over-expressed in CIMP-CRC; C/ Selection of four discriminant metabolic myc targets dysregulated in CIMP-CRC: for each candidate myc LOVO CHIP-seq and expression boxplot stratified on clinical data (methylation subgroups, expression subgroups, hypermutation status and MLH1 silencing) are represented.
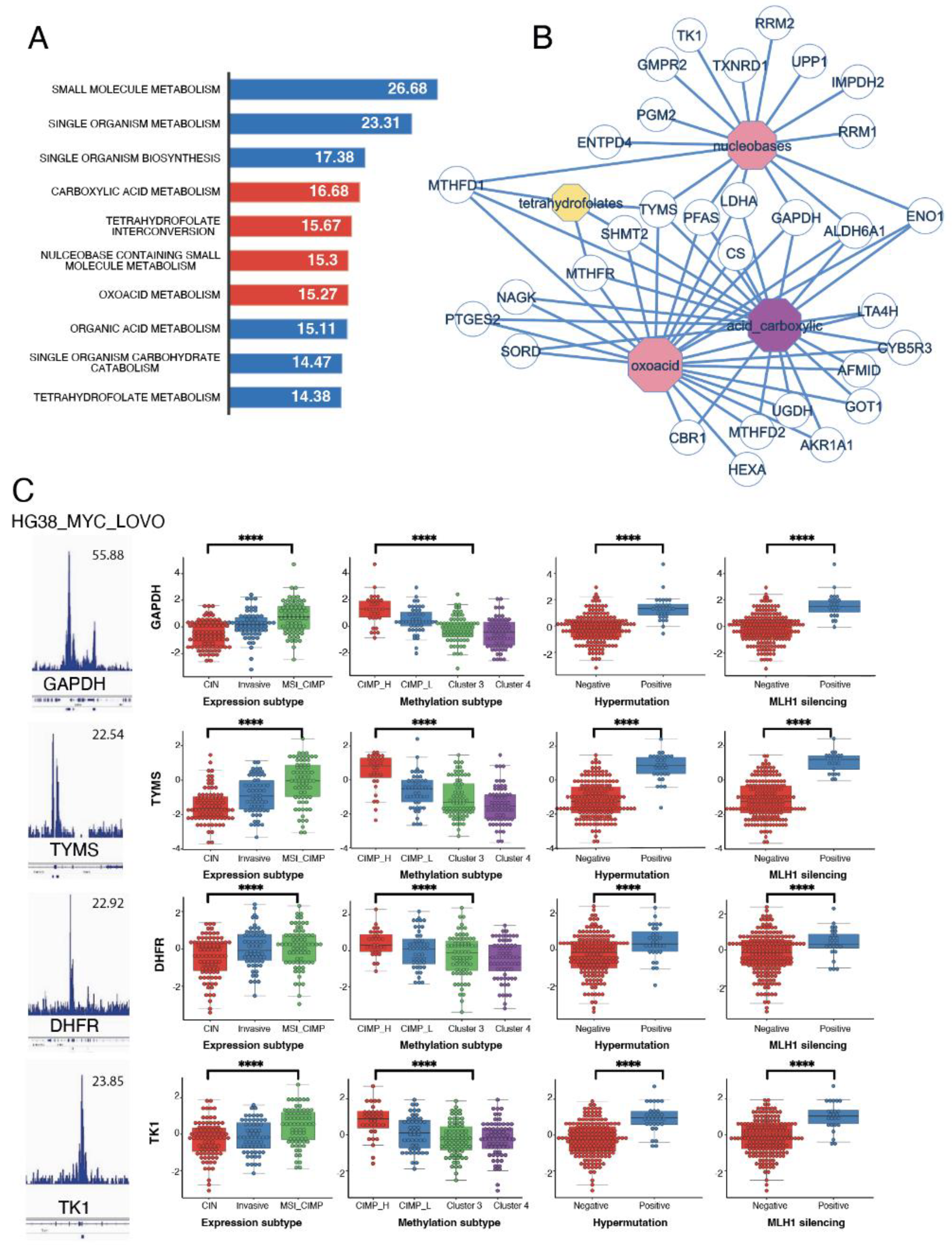
Figure 5.
One carbon metabolism multivariable model stratified on methylation status. (a) Negative log10 p-values (NLP) of iterative chi.square tests performed between parameters; (b) Network performed on the detection of communities between parameters; (c) Multivariable model showing the association between one carbon metabolism gene expression and clinical parameters to tumour methylation status, tested as logistic regression.
Figure 5.
One carbon metabolism multivariable model stratified on methylation status. (a) Negative log10 p-values (NLP) of iterative chi.square tests performed between parameters; (b) Network performed on the detection of communities between parameters; (c) Multivariable model showing the association between one carbon metabolism gene expression and clinical parameters to tumour methylation status, tested as logistic regression.
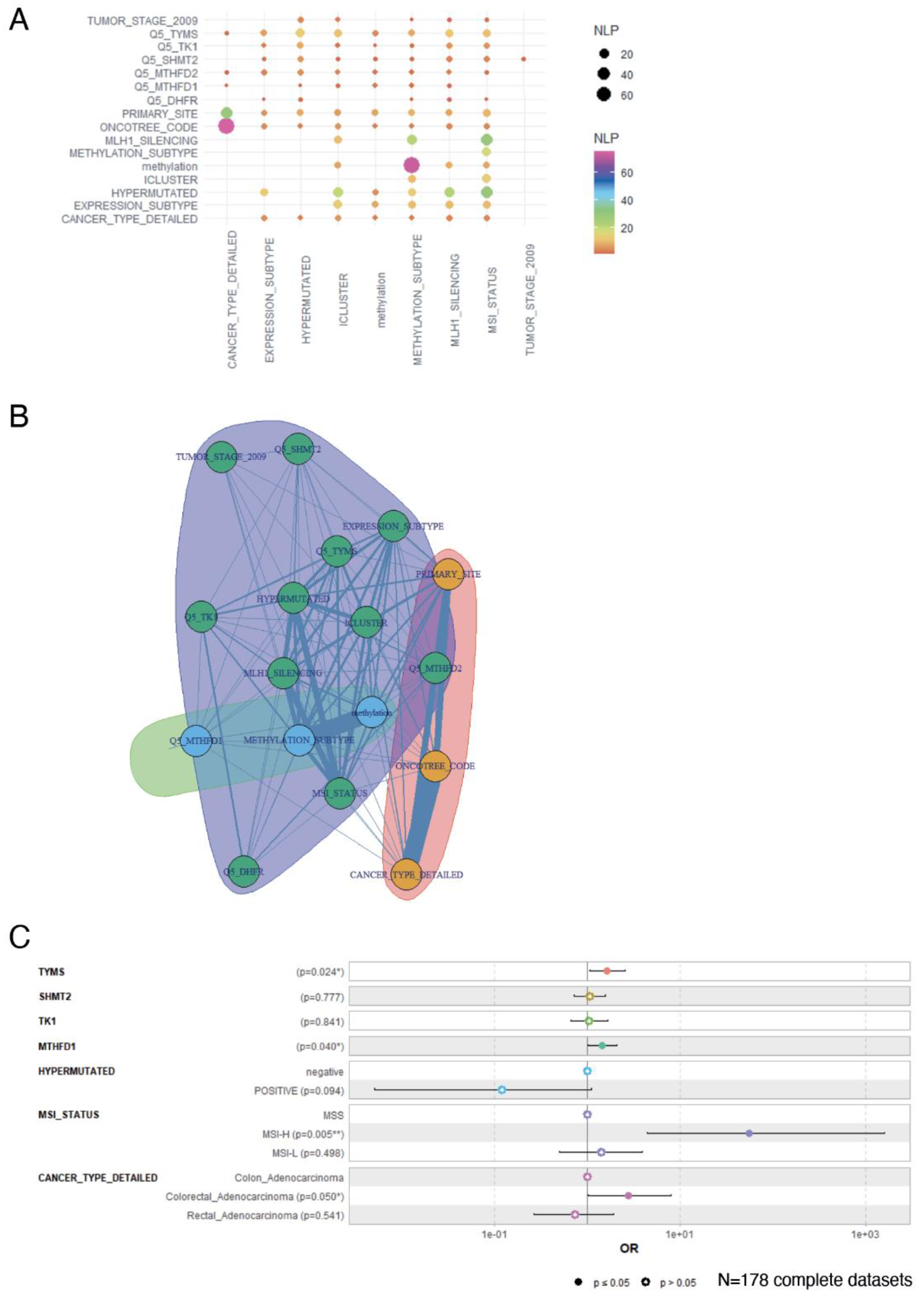
Figure 6.
Assessment of one carbon metabolism activation on overall survival prognosis in colorectal cancer. (a) Oncoprint analysis on one carbon metabolism gene candidates found to be strictly overexpressed in CRC tumours; (b) Kaplan Meier and log rank test performed between altered and unaltered groups of CRC patients focus on one carbon metabolism overexpression in RNA-sequencing; (c) barplot of tumor localization stratified on carbon metabolism alteration status; (d) Volcanoplot performed on differential methylation HM450 quantification between altered and unaltered one carbon metabolism CRC patients; (e) KEGG (Kyoto encyclopedia of Genes and Genomes) functional enrichment performed on hypermethylated gene promoters of patients presenting overexpression of one carbon metabolism genes in their transcriptomes; (f) Gene-ontology biological process enrichment performed on hypermethylated gene promoters of patients presenting overexpression of one carbon metabolism genes in their transcriptomes.
Figure 6.
Assessment of one carbon metabolism activation on overall survival prognosis in colorectal cancer. (a) Oncoprint analysis on one carbon metabolism gene candidates found to be strictly overexpressed in CRC tumours; (b) Kaplan Meier and log rank test performed between altered and unaltered groups of CRC patients focus on one carbon metabolism overexpression in RNA-sequencing; (c) barplot of tumor localization stratified on carbon metabolism alteration status; (d) Volcanoplot performed on differential methylation HM450 quantification between altered and unaltered one carbon metabolism CRC patients; (e) KEGG (Kyoto encyclopedia of Genes and Genomes) functional enrichment performed on hypermethylated gene promoters of patients presenting overexpression of one carbon metabolism genes in their transcriptomes; (f) Gene-ontology biological process enrichment performed on hypermethylated gene promoters of patients presenting overexpression of one carbon metabolism genes in their transcriptomes.
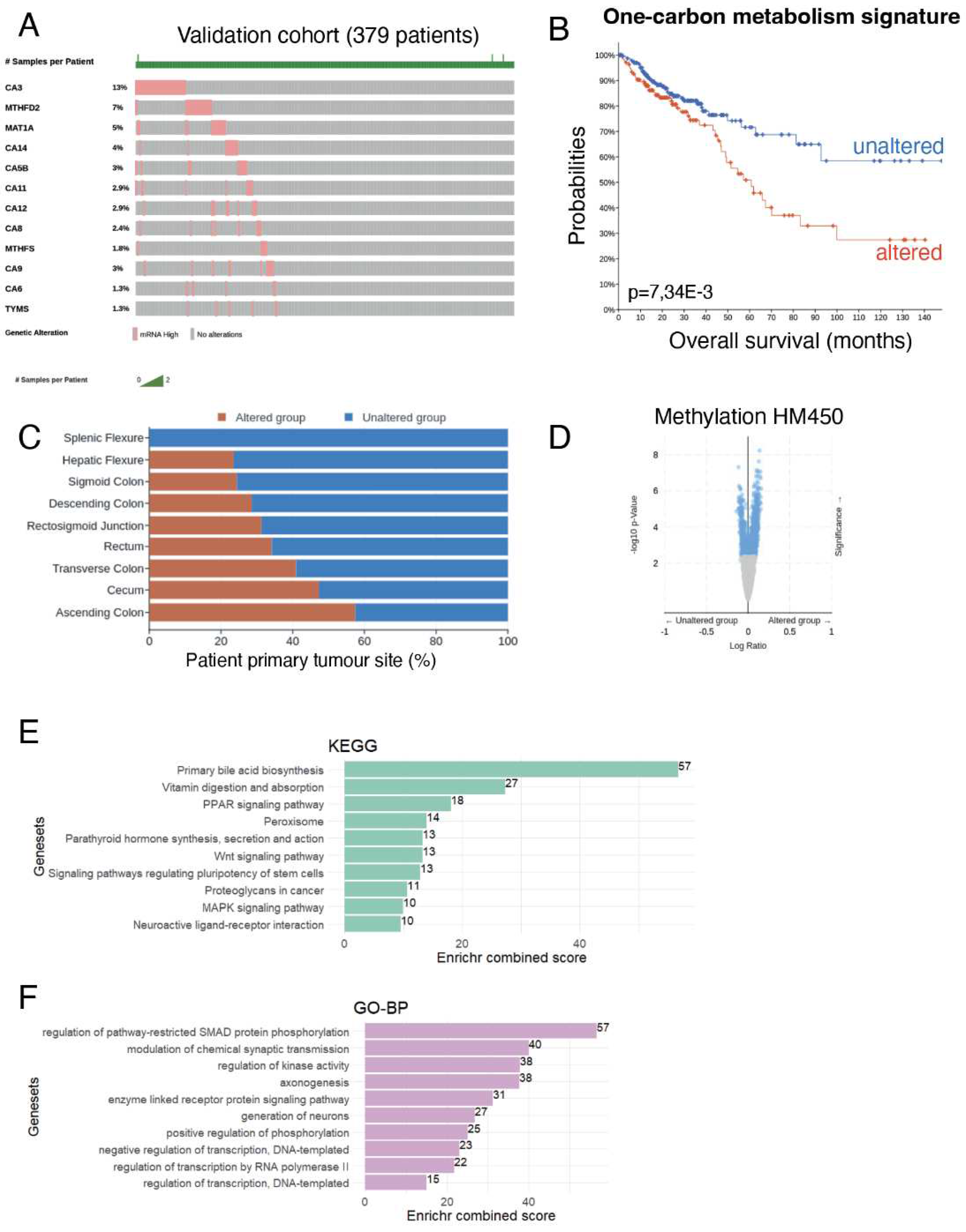

Table 1.
Clinical information of colorectal cancer RNA-seq cohort stratified on methylation status: univariate logistic regression was performed on available clinical parameters present in TCGA CRC cohort (2012[15]) and stratified on methylation status.
Table 1.
Clinical information of colorectal cancer RNA-seq cohort stratified on methylation status: univariate logistic regression was performed on available clinical parameters present in TCGA CRC cohort (2012[15]) and stratified on methylation status.
| Variable | Level | negative (n=143) | POSITIVE (n=80) | Total (n=223) | p-value |
|---|---|---|---|---|---|
| MSI_STATUS | MSS | 116 (81.1) | 41 (51.9) | 157 (70.7) | |
| MSI-L | 24 (16.8) | 13 (16.5) | 37 (16.7) | ||
| MSI-H | 3 (2.1) | 25 (31.6) | 28 (12.6) | < 1e-04 | |
| missing | 0 | 1 | 1 | ||
| METHYLATION_SUBTYPE | Cluster3 | 74 (51.7) | 0 (0.0) | 74 (33.2) | |
| Cluster4 | 69 (48.3) | 0 (0.0) | 69 (30.9) | ||
| CIMP_H | 0 (0.0) | 32 (40.0) | 32 (14.3) | ||
| CIMP_L | 0 (0.0) | 48 (60.0) | 48 (21.5) | < 1e-04 | |
| ICLUSTER | c1 | 43 (36.8) | 11 (16.7) | 54 (29.5) | |
| c2b | 16 (13.7) | 22 (33.3) | 38 (20.8) | ||
| c3 | 48 (41.0) | 9 (13.6) | 57 (31.1) | ||
| c2a | 10 (8.5) | 24 (36.4) | 34 (18.6) | < 1e-04 | |
| missing | 26 | 14 | 40 | ||
| MLH1_SILENCING | negative | 142 (99.3) | 56 (70.0) | 198 (88.8) | |
| POSITIVE | 1 (0.7) | 24 (30.0) | 25 (11.2) | < 1e-04 | |
| EXPRESSION_SUBTYPE | CIN | 77 (54.6) | 11 (13.9) | 88 (40.0) | |
| Invasive | 36 (25.5) | 25 (31.6) | 61 (27.7) | ||
| MSI_CIMP | 28 (19.9) | 43 (54.4) | 71 (32.3) | < 1e-04 | |
| missing | 2 | 1 | 3 | ||
| HYPERMUTATED | negative | 125 (93.3) | 51 (69.9) | 176 (85.0) | |
| POSITIVE | 9 (6.7) | 22 (30.1) | 31 (15.0) | < 1e-04 | |
| missing | 9 | 7 | 16 | ||
| CANCER_TYPE | Colorectal_Adenocarcinoma | 143 (100) | 80 (100) | 223 (100) | < 1e-04 |
| CANCER_TYPE_DETAILED | Colon_Adenocarcinoma | 84 (58.7) | 43 (53.8) | 127 (57.0) | |
| Colorectal_Adenocarcinoma | 15 (10.5) | 23 (28.8) | 38 (17.0) | ||
| Rectal_Adenocarcinoma | 44 (30.8) | 14 (17.5) | 58 (26.0) | 0.001041 | |
| ONCOTREE_CODE | COAD | 84 (58.7) | 43 (53.8) | 127 (57.0) | |
| COADREAD | 15 (10.5) | 23 (28.8) | 38 (17.0) | ||
| READ | 44 (30.8) | 14 (17.5) | 58 (26.0) | 0.001041 | |
| PRIMARY_SITE | 3_-_left_colon | 59 (41.5) | 13 (16.2) | 72 (32.4) | |
| 1_-_right_colon | 24 (16.9) | 42 (52.5) | 66 (29.7) | ||
| 2_-_transverse_colon | 5 (3.5) | 9 (11.2) | 14 (6.3) | ||
| 4_-_rectum | 54 (38.0) | 16 (20.0) | 70 (31.5) | < 1e-04 | |
| missing | 1 | 0 | 1 | ||
| TUMOR_STAGE_2009 | Stage_IIA | 46 (32.6) | 33 (41.8) | 79 (35.9) | |
| Stage_IIIC | 17 (12.1) | 3 (3.8) | 20 (9.1) | ||
| Stage_IIIB | 20 (14.2) | 11 (13.9) | 31 (14.1) | ||
| Stage_I | 31 (22.0) | 15 (19.0) | 46 (20.9) | ||
| Stage_IIIA | 3 (2.1) | 1 (1.3) | 4 (1.8) | ||
| Stage_IV | 22 (15.6) | 12 (15.2) | 34 (15.5) | ||
| Stage_IIB | 2 (1.4) | 3 (3.8) | 5 (2.3) | ||
| Stage_IVA | 0 (0.0) | 1 (1.3) | 1 (0.5) | 0.29398 | |
| missing | 2 | 1 | 3 |
Table 2.
Four MYC metabolic targets were used for deep learning predictions of prognostic status in patients with colorectal cancer. The neural network is based on the RNA quantification of 4 Myc metabolic targets: GAPDH + TYMS + TK1 + DHFR to predict each prognostic parameter.
Table 2.
Four MYC metabolic targets were used for deep learning predictions of prognostic status in patients with colorectal cancer. The neural network is based on the RNA quantification of 4 Myc metabolic targets: GAPDH + TYMS + TK1 + DHFR to predict each prognostic parameter.
| CRC status | Number of patients | Accuracy | Precision | Recall | F1 score | Cohen Kappa score | AUC: area under curve |
|---|---|---|---|---|---|---|---|
| methylation CIMP | 223 | 0.82 | 0.78 | 0.72 | 0.75 | 0.62 | 0.90 |
| Hypermutation | 207 | 0.95 | 0.87 | 0.83 | 0.85 | 0.82 | 0.98 |
| MLH1 silencing | 223 | 0.96 | 0.86 | 0.80 | 0.83 | 0.81 | 0.99 |
| MSI | 222 | 0.85 | 0.83 | 0.63 | 0.72 | 0.62 | 0.94 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



