
Submitted:
07 February 2024
Posted:
08 February 2024
You are already at the latest version
Abstract
Glioblastoma, a deadly brain tumor, shows limited response to standard therapies like te-mozolomide (TMZ). Recent findings from the REGOMA trial underscore a significant survival improvement offered by regorafenib (REGO) in recurrent glioblastoma. Our study aimed to propose a 3D ex vivo drug response precision medicine approach to investigate recurrent glio-blastoma sensitivity to REGO and elucidate the underlying molecular mechanisms involved in tumor resistance or responsiveness to treatment. 3D glioblastoma organoids (GB-EXPs) obtained from 18 patients’ resected recurrent glioblastoma tumors were treated with TMZ and REGO. Drug responses were evaluated using NAD(P)H FLIM, stratifying tumors as responders (Resp) or non-responders (NR). Whole-exome sequencing was performed on 16 tissue samples, and whole-transcriptome analysis on 13 GB-EXPs treated and untreated. We found 35% (n=9) and 77% (n=20) of tumors responded to TMZ and REGO, respectively, with no instances of TMZ-Resp be-ing REGO-NR. Exome analysis revealed a unique mutational profile in REGO-Resp tumors compared to NR tumors. Transcriptome analysis identified distinct expression patterns in Resp and NR tumors, impacting Rho GTPase and NOTCH signaling, known to be involved in drug response. In conclusion, recurrent glioblastoma tumors were more responsive to REGO com-pared to TMZ treatment. Importantly, our approach enables a comprehensive longitudinal ex-ploration of the molecular changes induced by treatment, unveiling promising biomarkers in-dicative of drug response.
Keywords:
Glioblastoma
; NADP(H) FLIM
; Regorafenib
; drug response
; organoids
1. Introduction
Glioblastoma (GB) stands out as the most prevalent malignant primary brain tumor affecting adults [1]. The standard treatment protocol includes maximal surgery followed by temozolomide (TMZ) treatment and radiotherapy. However, the median survival is currently 14.6 months [2], with GB relapsing in nearly all patients around 6–9 months after the initial therapy[3,4]. Currently, managing disease recurrence poses a significant challenge.
Multikinase inhibitors, compounds designed to target a variety of kinases, have been examined in numerous studies for the treatment of recurrent [5,6]. The objective was to address diverse tumor-related pathways, encompassing invasion and metastasis, cell growth and survival, and neo-angiogenesis. Regorafenib (REGO), among the multikinase inhibitors, is presently utilized in clinical settings for the treatment of hepatocellular carcinoma, gastrointestinal stromal tumors and colorectal cancer [7,8,9]. Recently, in a phase II clinical trial focused on recurrent GB (REGOMA), this drug demonstrated promising and noteworthy results [10]. The study involved 119 patients with recurrent GB, revealing extended overall survival (OS) (7.4 months compared to 5.6 months with lomustine). Notably, the REGO arm exhibited a statistically significant 6-month improvement in progression-free survival (PFS) compared to the lomustine-treated group, as reported in the clinical trial. Based on these findings, the National Comprehensive Cancer Network (NCCN) 2020 Guidelines designated REGO as a preferred regimen for treating relapsed GB, and the Italian Agency of Medicine (AIFA) granted approval for its use in Italian patients with recurrent GB [11]. From a molecular perspective the angiogenic VEGFR 1-3, PDGFR-b, and the oncogenic c-KIT, RET, FGFR, Raf kinases represent REGO targets [12] REGO. However, despite REGO becoming a part of clinical practice as a treatment option for relapsed GB, our comprehension of the molecular mechanisms governing GBM patients’ sensitivity to REGO remains restricted [13]. The limited comprehension of the biological factors influencing a specific drug’s efficacy often results in trials that initially hold promise during early development but subsequently prove ineffective in later stages. Gaining insight into the biological underpinnings of these setbacks can be achieved by collecting tissue samples both before and after treatment. However, the routine practice of gathering such samples has yet to be established in the realms of neurosurgery and neuro-oncology drug development [14]. In pursuit of addressing this pressing need, we have recently pioneered an ex vivo drug response functional precision medicine approach. This innovative methodology allows us to assess how tumor samples respond to various cancer treatments, enabling us to analyze tissue samples both prior to and after treatment [15]. This approach involves the use of an in vitro 3D organoid model derived from vital patient glioblastoma tissue (referred to as GB-EXPs). These organoids are minimally manipulated and cultured briefly to maintain the tumor microenvironment. We leverage the fluorescence properties of NAD(P)H, a cellular enzyme cofactor, using Fluorescence Lifetime Imaging (FLIM). NAD(P)H may be present either bound to proteins or in a free state within cells, and these conditions impact its fluorescence lifetime decay. As previously shown [15], the shift of lifetimes distribution towards lower free-/bound-NADP(H) fractions is indicative of a responsive phenotype [15]. Our research primarily centers on GB-EXPs derived from patients’ recurrent tumors. We employ this model to investigate changes in the transcriptome of GB-EXPs after treatment through longitudinal in vitro tissue sampling. Additionally, we establish correlations between transcriptome or exome profile and response to REGO. Importantly, this study represents the first exploration of alterations in gene expression resulting from REGO drug treatments in patient-derived GB organoids. Our findings hold significant promise for advancing personalized precision medicine in the field.
2. Materials and Methods
2.1. GB tissue collection
The research was conducted in compliance with the principles outlined in the Declaration of Helsinki, and the protocol for sample collection received approval from the Ethics Committee of the University Hospital of Pisa (787/2015). Tumor specimens were sourced from 18 patients who had undergone surgical resection of histologically confirmed GBM after providing informed consent. Samples were acquired from either the Neurosurgery Department of the "Azienda Ospedaliero-Universitaria Pisana" or the Unit of Neurosurgery of Livorno Civil Hospital. All patients had a GB diagnosis without a prior history of brain neoplasia and did not exhibit R132 IDH1 or R172 IDH2 mutations. In three out of the 18 patients, neurosurgeons utilized neuronavigation-guided microsurgical techniques to collect both core and peripheral tumor samples. Peripheral tumor samples were obtained at the initial identification of GB during surgery, while core tumor samples were taken from the resected tumor mass. In cases where the tumor displayed a significant area of central necrosis, samples were collected from tumor regions outside the necrotic area. Patient clinical and demographic data are shown in Table 1. Resected tumors were put in MACS tissue storage solution (Miltenyi Biotec, Bergisch Gladbach, Germany) at 4°C for 2–4 hours. Each tumor specimen was rinsed with Dulbecco’s phosphate-buffered saline (DPBS) within a sterile dish and divided into ~ 0.5-1 mm2 pieces under a biological hood. In an effort to minimize variability due to sampling, we pooled 2-4 pieces of parental tumor tissue into one sample for the subsequent analysis.
Biopsies not immediately processed for GB-EXPs cultures were cryopreserved in 90% fetal bovine serum (FBS) and 1% dimethyl sulfoxide (DMSO) at -140°C. Tumor samples designated for histological analysis were promptly fixed in 10% formalin and embedded in paraffin, while portions allocated for additional analyses were stored at -80°C.
2.2. GB Explant Cultures (GB-EXPs)
The methodology employed in generating explant cultures for this study was previously described [15] but with some modifications. Briefly, Fresh GB tumors or frozen samples, following rapid thawing in a 37°C water bath, underwent washing with DPBS within a sterile dish and were finely sectioned using a scalpel. The resulting minced tissues were then passed through a 300-micron cell strainer to eliminate larger tissue fragments. Suspension was centrifuged at 1200 rpm for 5’. The pellet was resuspended in 3 ml eBiosceince 1X RBC Lysis Buffer (Invitrogen) for 5’ at room temperature. DPBS was then added to inactive reed blood lysis and suspension was centrifuged at 1200 rpm for 5’. Pellet was resuspended in 1200 ml of explant medium, composed by 89% DMEM:F12 without red phenol (Thermo Fisher Scientific), 10% FBS (Thermo Fisher Scientific) and 1% PenStrep (Thermo Fisher Scientific). Two volume of Vitrogel ORG-4 (TheWell Bioscience) was added. The solution so obtained was put in 8-well chamberslides coverglass (Nalge Nunc International), using 150 ul each well. Vitrogel was permitted to solidify for 20 minutes at 37°C, followed by the addition of 300 µl of medium. Subsequently, the cultures were placed within a sterile incubator maintained at 37°C, 5% CO2, and 90% humidity.
2.3. GB Cell Lines
The T98G, U118, and U87 GB cell lines were purchased from the American Type Culture Collection (ATCC, Rockville, MD). These cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) devoid of red phenol and supplemented with 10% FBS and 1% Penicillin-Streptomycin. For FLIM experiments, cells were cultivated in 35 mm Nunc Glass Bottom Dishes (Thermo Fisher Scientific).
2.4. Spheroid Cultures
Hanging Drop method was used to generated spheroids, using T98G, U118 and U87 GB commercial cell lines, as previously described [16].
2.5. Drug treatments
Regorafenib (Tebu-Bio, T1792) and TMZ (Sigma, St. Louis, MI, USA), were dissolved in DMSO to create stock concentrations of 20 mM and 50 mM, respectively.
2D cell lines were subjected to treatment when they reached 30% confluence, while spheroids were treated the following day after being cultured in vitrogel. The medium was replaced with fresh medium containing either 10, 50, or 100 mM of REGO for treated cells, or an equivalent volume of DMSO for control groups.
GB-EXPs were subjected to treatment 3 days after being cultured. The volume of the medium was substituted with fresh medium containing either TMZ at 600 μM, as detailed previously [15], or REGO at 50 μM for treated explants, with an equivalent volume of DMSO for control samples. Both cells and GB-EXPs underwent treatment for a duration of 72 hours.
2.6. Cell viability
For the assessment of cell viability in 2D cell lines, the WST-1 assay (Clontech Laboratories, Mountain View, CA, USA) was employed, while the CellTiter-Glo 3D Cell Viability Assay (Promega) was utilized for spheroids, following the respective manufacturer’s protocols.
In the 2D model, 5000 cells per well were seeded in a 96-well plate format. WST1 was added 72 hours following REGO treatment. The quantification of metabolically active cells was performed by assessing the absorbance at 450 nm using a multiwell plate reader (Tecan, Mannedorf, Switzerland). Optical density (OD) values were normalized to those of non-treated cells (controls).For 3D model, 2 spheroids for well were seeded in an ultra low attachment 96-well plate for luminescence. CellTiter-Glo 3D reagent was added 72 h after REGO treatment. Luminescence was recorded using Tecan multiwall plate reader. Each assay was conducted in triplicate.
2.7. Nucleic Acids Isolation
Genomic DNA was extracted from the original tissue sample, which was stored at -80 °C, using Maxwell 16 Tissue LEV DNA Purification Kit (Promega, Madison, WI, USA), following the manufacturer’s instructions. RNA extraction from GB-EXPs and 3D cell lines was performed with Maxwell 16 LEV Simply RNA Tissue Kit (Promega, Madison, WI, USA), following manufacturer’s protocol.
The concentrations of DNA and RNA were assessed utilizing the Qubit Fluorometer (Life Technologies, Carlsbad, CA), and their quality was evaluated using the Agilent 2200 Tapestation (Agilent Technologies, Santa Clara, CA) system.
2.8. KI67 Expression analysis
Complementary DNA (cDNA) was synthesized from 2 ng of total RNA using the iScript cDNA Synthesis Kit (Bio-Rad), following the manufacturer’s protocol, in a final volume of 20 μl. For the analysis of Ki67 expression in cell lines, semiquantitative real-time PCR was conducted in a 10 µL reaction mixture containing 5 µL of SsoAdvanced Universal SYBR Green supermix (Bio-Rad), 1 µL of primer Assay (Bio-Rad), 2.0 µL of cDNA, and 2 µL of nuclease-free water. We used Human Mki67 PrimePCR SYBR Green Assay (Bio-Rad) for Ki67 and Human ACTB PrimePCR™ SYBR® Green Assay (Bio-Rad), for B-actin housekeeping gene. PCR amplification was conducted using the CFX96 Touch Deep Well PCR system (Bio-Rad) with an initial template denaturation at 98°C for 30 seconds, followed by 40 cycles of denaturation at 98°C for 15 seconds and annealing at 60°C for 30 seconds. Every sample was evaluated in triplicate, with positive and negative controls run concurrently in each reaction. Following amplification, melting curve analysis was performed to evaluate PCR product specificity. Data analysis was carried out using the 2-ΔΔCT method for relative quantification [17].
2.9. Confocal Imaging
Images were captured using the Olympus Fluoview 3000 confocal microscope, which is furnished with four laser lines. (405/488/561/640 nm), 2 hybrid detectors and 2 standard detectors (Olympus, FV31-HSD and FV31-SD), using a quadriband 405/488/561/640 nm dichroic mirror (Chroma) and an UPLXAPO20X (20X magnification, N.A.=0.80) for brightfield acquisition or UPLXAPO60XO (60X magnification, N:A.=1.42) oil immersion objective for FLIM. Confocal pinhole diameter was set to 1 Airy.
2.10. Lifetime Imaging
Fluorescence lifetime imaging was performed using MultiHarp 150 (Picoquant) time-correlated single-photon counting (TCSPC) unit and a 405 nm LDH-P-C-375B (Picoquant) excitation laser, powered by a PDL 828 “Sepia II” laser driver (Picoquant) and interfaced through fiber port with the confocal setup previously described. Fluorescence was collected with two PMA hybrid detectors (Picoquant) using a dichroic filter (510 nm), and band pass filters 440/40 for NAD(P)H. Laser pulse frequency was set to 40 MHz, pixel dwell time was set to 10 us, and 240 cycles of acquisition were performed for each field. Images sizes were of 512X512 pixels. Temporal resolution was 80 ps. We obtained between 15 and 20 FLIM measurements for both control and treated samples.
FLIM data were analyzed according Phasor approach, using SimFCS suite (Globals for Images, Laboratory for Fluorescence Dynamics, Irvine). In brief, a mono-exponential lifetime standard (fluorescein (Sigma-Aldrich, 46955), 1 µM in NaOH 0.01M, pH12, t=4 ns) was first acquired in order to reference the other acquisitions and calibrate the universal circle. This was done on SimFCS 2. First the fluorescein phasor was auto-centered, and then, the acquisition files were all referenced. All data analysis of referenced files was performed using SimFCS 4 as previously described [15,18].
2.11. Histology and staining
Tissues were fixed for 24 hours in 10% neutral-buffered formalin (Sigma-Aldrich) at room temperature, processed through a graded-ethanol series followed by xylene, and embedded in paraffin. Explants, whether in vitrogel or in suspension, underwent the same fixation process before embedding in paraffin. Paraffin-embedded samples sections (5 μm) were stained with hematoxylin (Diapath C0303) for 40 s and with eosin (Diapath C0353) for 30 s.
For the immunohistochemical staining process, paraffin slides underwent deparaffinization, followed by antigen retrieval achieved through the use of Epitope Retrieval Solution (pH=8) (Leica Microsystems RE 716 CE). Samples were incubated with Ki67 monoclonal (SP6) (Thermo Fischer, MA5-14520) primary antibody using 1:50 diluition for 1 h at RT. Detection of bound antibody was accomplished with the Rabbit Specific HRP/DAP Detection IHC Kit (Abcam, ab64261). Immunohistological and H&E pictures were taken with microscope (CARL ZEISS Axio Observer Z1FLMot) after mounting with mounting medium (Fisher Scientific, 7 Miami, FL).
2.12. Whole Transcriptome RNA analysis (WTA) libraries
NextSeq 500 (Illumina, San Diego, CA, USA) was used for RNA-Seq. The libraries ware prepared using Illumina Stranded Total RNA Prep with Ribo-Zero Plus kit (Illumina), starting from 100 ng of total RNA, according manufacturer’s protocol. The libraries were quantified using Qubit reagents (Thermo Fisher Scientific, Waltham, MA, USA) and analyzed for validation through TapeStation (Agilent, Headquarters, Santa Clara, CA, USA). A maximum of 10 libraries were loaded on NextSeq High Output cartridge (150 cycles).
2.13. Whole Exome Analysis (WEA) libraries
Whole exome library preparation was performed using Illumina DNA Prep with Enrichment (Illumina, San Diego, CA, USA), following manufacturer procedure, starting from 500 ng of DNA. Paired-end sequencing was performed using NextSeq 500 (Illumina, San Diego, CA, USA) with 101 bp of read lenght. Up to 10 libraries for WET were loaded on NextSeq High Output cartridge (300 cycles, Illumina).
2.14. Data analysis
2.14.1. FLIM data analysis
FLIM data, referenced with SimFCS 2, were analyzed using SimFCS 4, according to the protocol described by Ranjiit, in the section “Two-component analysis of fractional NADH distribution” [18]. From this analysis and as previously described [15], we obtained a mean NAD(P)H fractional distribution curve for treated GB-EXPs and one for control GB-EXPS. Comparing these two curves as reported in Morelli et al. [15], we calculate a percentage of drug response (%DR). Using %DR samples were classified as: NR, non responder: %DR<5%; RESP, responder:DR≥5%
2.14.2. Next Generation Sequencing data analysis
As first step, RNA-Seq reads in FASTQ format were examined using the FASTQC program (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Subsequently, alignment against the Hg19 reference genome was carried out using the STAR aligner (version 2.5.3a). The quantification of read counts on known human genes was accomplished utilizing feature Counts version 1.5.1. Differential expression analysis was performed using EdgeR 2.6.12 and Cuffdiff 2.2.0 tools. Differentially expressed genes (DEGs) were identified by intersecting the lists of significant DEGs (p-value < 0.004) obtained from both Cuffdiff 2 and edgeR.A discriminant stepwise analysis was used to find genes discriminating our two groups: Resp and Non Resp. JMP 10.0.0 (SAS Institute).
To compare the prognostic significance of our discriminant gene set in predicting survival in glioblastoma patients, we evaluated it in TCGA Database with SurvExpress platform. Heat-maps and PCA plots were generated using ClustVis [19].
The initial analysis of exome data was conducted using the SeqMule pipeline [20]. For the detection of somatic single-nucleotide variants (SNVs) and indels within tumors, we employed a Panel of Normal (PoN) as recommended in the GATK best practices (https://console.cloud.google.com/storage/browser/gatk-best-practices/somatic-b37) along with the Mutect2 variant calling algorithms [21]. Rare variants were acquired by filtering out somatic variants cataloged in the non-cancer database gnomAD v3 [22], with a minor allele frequency (MAF) threshold of ≥0.01. The frequency and characteristics of mutations were analyzed using the R package MAFtools [23].
Copy number analysis was conducted utilizing CNVkitt [24]. CNApp was employed with default cutoffs to summarize copy number variations [25]. Comparative data for the CNV classifier were obtained from The Cancer Genome Atlas Glioblastoma Multiforme (TCGA-GB, https://www.cancer.gov/tcga) dataset (hg19 Legacy Database) via the TCGAbiolinks [26] and randomForest [27] R packages.
2.14.3. Statistical analysis
All presented summary data are expressed as means ± standard deviation (s.d.). Statistical analyses were conducted using R and GraphPad Prism software (GraphPad 7.0). Differences between two groups were assessed using Student’s two-tailed unpaired t-test or log–rank test, as specified in the figure legends. For t-tests, we assumed normality and equal distribution of variance among the different groups. No data points were excluded from the statistical analyses. Significance was defined as P < 0.05 (for all other experiments). Logistic regression analysis was performed with 5000 number of iteration and a learning rate of 0.005 with Statistic calculator DATAtab https://datatab.net/statistics.
3. Results
3.1. Tumor Samples Characteristics
Tumors were collected from 18 patients, for five of whom we had both primary and recurrent tumor samples. For the remaining patients, we only had recurrent samples, except for one patient for whom we only had the primary tumor. Additionally, for three patients, we obtained both core and peripheral portions from the same patient-derived tumor using neuronavigation-guided microsurgical techniques (Table 1). In total, we had 26 patient-derived surgical glioblastoma tissues. Patients included 13 men (72%) and 5 (28%) women in the age group of 52–79 years (mean age: 66 years). Informations on cerebral localization, MGMT methylation status are reported in Table 1. All tumors were wild-type for IDH1/2 genes. The patient-derived tumor sample Gb14_pr showed the deletion of CDKN2A and CDKN2B and Gb15_pr reported the deletion of 1p, while the Gb17

Table 1.
Tumor samples description. All tumors were recurrent, except those indicated with the suffix _pr, which denotes primary tumors. For recurrent tumors with both peripheral and core portions available (Y=yes), they are marked with the suffixes _pe and _co, respectively. In the column ‘Tumor Sample Portion Available,’ the type of sample (primary or recurrence) is specified, along with the portion of the recurrence (peripheral and core).
Table 1.
Tumor samples description. All tumors were recurrent, except those indicated with the suffix _pr, which denotes primary tumors. For recurrent tumors with both peripheral and core portions available (Y=yes), they are marked with the suffixes _pe and _co, respectively. In the column ‘Tumor Sample Portion Available,’ the type of sample (primary or recurrence) is specified, along with the portion of the recurrence (peripheral and core).
| Patient Nr | Sex | Age at intervention | Tumor Nr | Tumor sample portion available | Localizzation | |||
|---|---|---|---|---|---|---|---|---|
| Pr | Rec | Co | Pe | |||||
| Pt.01 | m | 57 | Gb1 | Y | right frontal | |||
| Pt.02 | m | 73 | Gb2 | Y | left temporal | |||
| Pt.03 | m | 52 | Gb3 | Y | left temporal | |||
| Pt.04 | m | 73 | Gb4 | y | left temporal-parietal | |||
| Pt.05 | m | 73 | Gb5 | Y | left occipital | |||
| Pt.06 | f | 57 | Gb6 | Y | right temporal | |||
| Pt.07 | f | 62 | Gb7 | Y | right parietal | |||
| Pt.08 | m | 61 | Gb8 | y | left fronto parieto-temporal | |||
| Pt.09 | m | 56 | Gb9 | y | ||||
| Pt.10 | m | 70 | Gb10_co | Y | Y | left temporal-parietal | ||
| Gb10_pe | Y | Y | ||||||
| Pt.11 | m | 70 | Gb11_co | y | Y | left fusiform and parahippocampal gyrus | ||
| Gb11_pe | y | Y | ||||||
| Pt.12 | f | 69 | Gb12_co | Y | Y | left fronto-parietal | ||
| Gb12_pe | Y | Y | ||||||
| Pt.13 | m | 55 | Gb13_pr | Y | right frontal | |||
| 56 | Gb13 | Y | right frontal | |||||
| Pt.14 | m | 72 | Gb14_pr | y | left frontal and insular | |||
| 73 | Gb14 | y | left temporal | |||||
| Pt.15 | f | 74 | Gb15_pr | Y | left temporal | |||
| 75 | Gb15 | Y | left fronto temporal | |||||
| Pt.16 | m | 78 | Gb16_pr | Y | left temporal | |||
| 79 | Gb16 | Y | left temporal | |||||
| Pt.17 | f | 56 | Gb17_pr | Y | left hemisphere multifocal | |||
| 58 | Gb17 | Y | left temporal | |||||
| Pt.18 | m | 76 | Gb18_pr | Y | left temporal | |||
3.2. FLIM imaging of patient-derived organoids
From the 26 GB tissues collected, patient-derived organoids named GB-EXPs were obtained following the protocol described previously [15] and summarized in Figure 1a. GB-EXPs were derived from patient-derived tissue surgery, cultured in vitrogel, and treated with either DMSO (controls) or REGO or TMZ for 72 hours. A minimum of 12 to a maximum of 20 FLIM images were collected for each experimental condition and analyzed using the phasor approach to obtain mean NAD(P)H fractional distribution curves for controls and one for treated GB-EXPs. By comparing the two curves, we calculated a %DR (see materials and methods), represented as the green area under the treated curve. A cutoff of 5%DR was used, as previously described [15], to stratify tumors into Non-Responders (NR) when DR≤5% and Responders (REGO) if DR>5%. Considering the data obtained from the 2D and 3D cell line models (see Supplementary Results), we opted for using a REGO concentration of 50 µM for GB-EXPs treatment. GB-EXPs were also treated with 600 µM of TMZ, as previously described [15]. All treatments lasted 72 hours. Representative images of a GB-EXP treated with 50 µM of REGO are shown in Figure 1b. The top row features a brightfield image, while the bottom row displays phasor-FLIM NAD(P)H lifetime maps. The coloration corresponds to the color bar defined on the side. Figure 1c shows the overlapping of the mean fractional NAD(P)H distribution curves of the control and Rego-treated patient-derived GB-EXPs, which is indicative of 0%DR, that means NR tumor. Conversely, in Figure 1d, we depict an instance of a GB-EXP, comprising a brightfield image (top row) and phasor-FLIM NAD(P)H lifetime maps (at the bottom). This demonstrates a statistically notable shift of the mean treated curve (red) towards a higher prevalence of bound NAD(P)H molecular species in REGO-treated explants compared to the control counterparts, with 87% drug response (Figure 1e). Increased levels of bound-state NAD(P)H indicate oxidative metabolism, characteristic of less proliferative cells, thereby signifying a responsive tumor [28,29].
The final annotation in Resp or NR, for each case and for both treatments, Rego and TMZ, leads us to stratify our tumor samples into 17 TMZ-NR (65%) and 9 TMZ-Resp (35%), 6 REGO-NR (23%) and 20 REGO-Resp (77%) (Figure 1f). These data show a higher percentage of responders in Rego-treated tumors (72%) compared to TMZ-treated ones (39%). Particular attention must be paid to the fact that all 6 REGO-NR were also TMZ-NR; on the contrary, 6 out of 17 TMZ-NR (35%) were REGO-Resp. Furthermore, in tumors treated with TMZ, both patient-derived samples from the peripheral (pe) and core (co) regions exhibited identical FLIM labels (NR), indicating a uniform metabolic response to TMZ. In the case of REGO-treated tumors, within the three pairs of pe/co samples, one (Gb12) displayed a distinct response, with the core portion showing an NR phenotype while the peripheral region exhibited a responsive phenotype (Resp). This divergence in response likely corresponds to the more aggressive phenotype typically observed in the core portion compared to the peripheral region in GB tumors. Regarding the difference in drug response among primary and recurrence of the same patient, heterogeneity was observed in both TMZ- and REGO-treated cases, with two patient-derived primary/recurrence tumor pairs having a different response to TMZ (Gb13 and Gb15), and one to REGO (Gb13). It is noteworthy that the Gb13 primary tumor is more responsive both to TMZ and to REGO than its relative recurrence.
3.3. Mutational genetic background in Regorafenib Resp and NR GB tumors
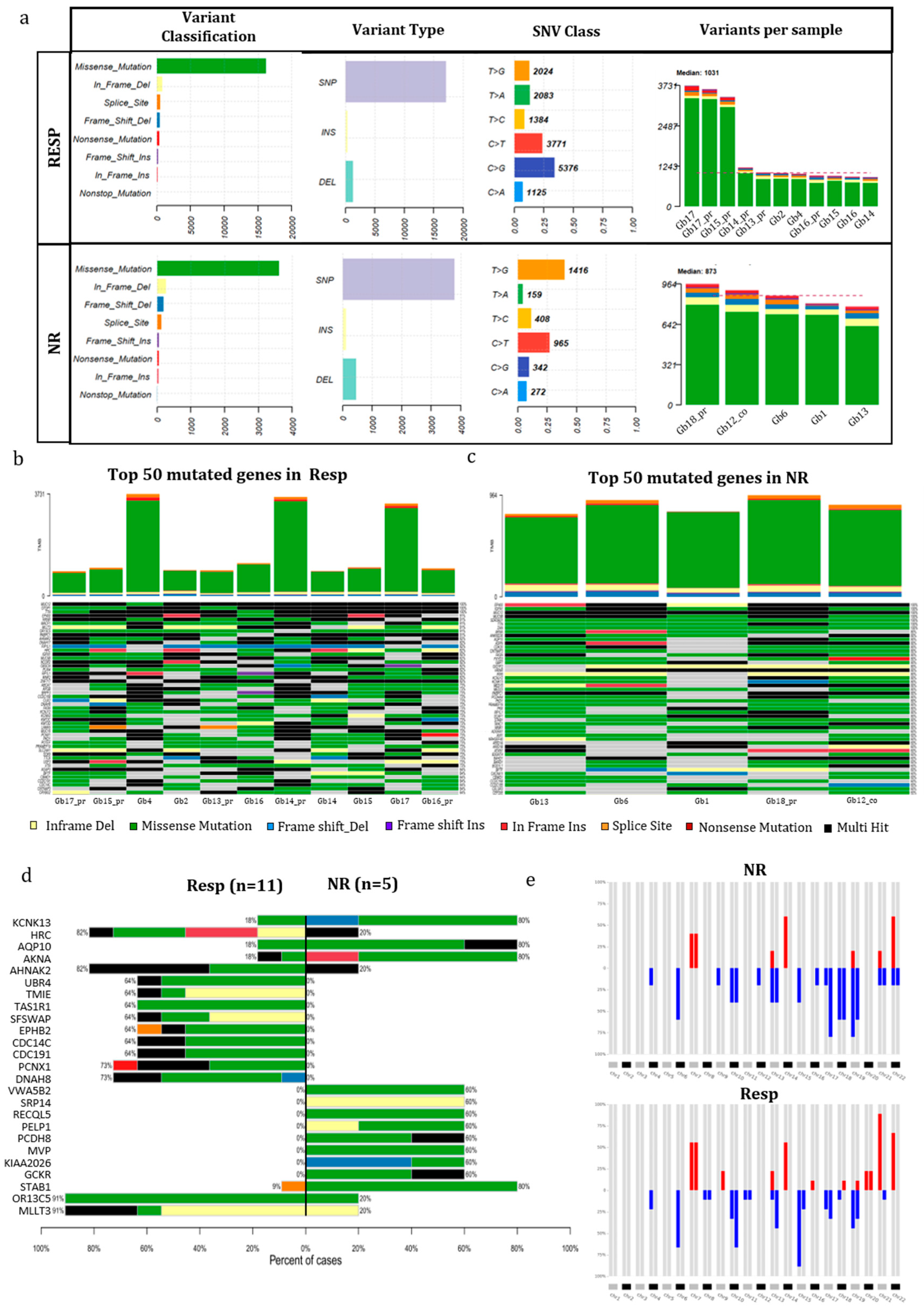
The mutational analysis conducted on 16 GB tumors before treatment, comprising 5 NR and 11 Resp cases, aimed to discern potential genetic distinctions between these two categories. We chose all mutations within the coding region known to affect protein function, along with all splicing mutations. Synonymous mutations with no predicted impact on splicing were excluded.
Figure 2a portrays the mutational landscape of both Resp and NR patients. The incidence of various types of mutations and the distribution of base substitutions exhibited remarkable parity between the two groups. However, a disparity emerged: C>G variations were notably more frequent in Resp (31%) compared to 10% in NR cases. Conversely, T>G variations prevailed in NR (36%) as opposed to Resp (12%). The number of variants per sample was, on average, greater in the Resp group (1032) compared to the NR group (837) (Figure 2a). Among the Resp tumors, Gb17 exhibited the highest number of variants in both recurrence and primary tumors, while Gb15 showed the highest number only in primary one, with approximately 3200 variants. In contrast, Gb14 had the lowest number of variants, falling below the median. In NR samples, the number of variants per sample was more evenly distributed (Figure 4a), with Gb18_pr having the highest count and Gb13 having the lowest.
Figure 4b and 4c highlight the top 50 mutated genes that harbored mutations in every single case of Resp and NR patients. In the Resp group, three genes exhibited mutations in all samples, while the NR group showed mutations in seven genes across all cases. Notably, two genes, MUC12 and TTN, displayed mutations in 100% of both Resp and NR samples (see Figure 2b and 2c). Further, analysis of Table 1S reveals that both genes carry multiple mutations in most of the patients. In Figure 2d, a co-bar plot illustrates the genes that significantly distinguish the Resp and NR tumors. Remarkably, four genes—KCNK13, AQP10, AKNA, and STAB1—have been identified with mutations prevalent in the majority of NR samples (4 out of 5). In contrast, only two out of the 11 Resp samples exhibit mutations in the KCNK13, AQP10, and AKNA genes, while one out of 11 Resp samples carries the mutated STAB1 gene. Concerning the KCNK3 gene, all samples, except for GB60, share a common mutation (Val391Gly), with the exceptional case of GB60 displaying a termination codon resulting from a frameshift starting at codon 88, as elucidated in Table 1S. The gene AQP10 also exhibits the same mutation (ASN54Ser) across all samples. The remaining genes display a variety of molecular variations distributed among the different samples, as detailed in Table 1S.
As depicted in Figure 2d, several genes exclusively exhibited mutations within the Resp group. Notably, two genes, PCNX1 and DNAH8, displayed mutations in 8 out of 11 Resp tumors, showcasing diverse mutations among samples (refer to Table 1S). Remarkably, 91% of the Resp group (10 out of 11) exhibited mutations in the OR13C5 and MLLT3 genes, in contrast to the NR group where only 20% (1 out of 5 tumors) showed mutations. It is noteworthy that both genes harbored the same mutation in all samples; specifically, the OR13C5 gene exhibited a consistent multinucleotide variation affecting Threonine in codon 81 (Thr81), while an in-frame deletion consistently affected codon 190 (Ser190) in the MLLT3 gene.
3.4. Transcriptional genetic background in Regorafenib Resp and NR GB tumors
WTA was performed on 13 GB-EXPs samples REGO-treated and on the 13 patient-corresponding GB-EXPs DMSO-treated (controls). Among these 13 cases, 9 were REGO-Resp and 4 NR. A p-value < 0.001 and a log2 fold change greater than 0.05 or less than -0.05 were considered to identify differentially expressed genes (DEGs).
To understand the background expression pattern’s involvement in the response to REGO, we conducted differential expression analysis between Resp and NR using the control samples. We identified 57 differentially expressed genes, (Figure 3a). In particular, among these genes we point out GRIK3, GSMT5, CXCL3 and SALL4 that result already involved in drug response mechanisms [30,31,32,33]. These four genes were significantly upregulated in the Resp tumors compared to the NR tumors except for GSTM5. Subsequently, we used the Reactome online tool to explore the molecular pathways associated with these DEGs. Our findings revealed that Rho GTPase and NOTCH signaling pathways were downregulated in Resp samples compared to NR samples (Figure 3b).
3.5. Early whole transcriptome changes induced by Regorafenib treatment in GB-EXPs
Whole transcriptome analysis was performed to explore early gene expression changes occurring after Regorafenib treatment in the Resp and NR samples. Even in this case, a p-value < 0.001 and a log2 fold change greater than 0.05 or less than -0.05 were considered to identify DEGs. Two classes of DEGs were obtained comparing control versus treated in Resp samples (DEGsA) and control versus treated in NR samples (DEGsB). Sixty and 33 DEGsA and DEGsB were found, respectively (Table 2S and 3S). In Figure 3c and Figure 3d heatmaps were run for Resp and non NR samples. The results show that DEGsA and DEGsB perfectly classify the ctrls and the treated GB-EXP samples among the Resp and NR groups. Only one control sample in Resp group (Gb14) was clustered among treated ones (Figure 3c). Upon analyzing the GLIOVIS database (http://gliovis.bioinfo.cnio.es/), we observed that most of the downregulated DEGsA in treated GB-EXPs, compared to GB-EXP controls, as DUSP6, displayed an increased expression pattern as tumor grade increased (Figure 3e). This finding suggests that REGO treatment in Resp samples could potentially restore the expression of these genes to a pattern more similar to that of less aggressive tumor tissue. On the contrary, the only upregulated gene in treated GB-EXPs compared to controls, RNF150, showed an opposite expression pattern (Figure 3e) when compared to the downregulated DEGsA. This might indicate that Resp samples increased the expression of this gene, making it more similar to the expression pattern observed in less aggressive tumor tissue. Of the significantly dysregulated genes in the Non-Resp group (DEGsB) only 1 gene, KI67, demonstrated a significant adjusted p-value of less than 0.05 (Figure 3f, Table 3S). KI67, a well-known marker for proliferation [34], shows a statistical significant downregulation at 72hr post REGO treatment compared to the non-treated sample. Among the genes typically involved in multidrug resistance [35] we found the drug transporter ABCA4 gene significantly downregulated after treatment in the NR group (Figure 3f) while no significant gene of the ABC family was found dysregulated in the Resp group.
Figure 3.
Gene expression analysis of GB-EXPs. (a) Heatmap of differentially expressed genes (DEGs) among Resp and NR samples in DMSO-treated GB-EXPs(controls). (b) Molecular pathways in which DEGsC are involved. (c) Heatmap of DEGs among controls and REGO-treated samples in Responders GB-EXPs. (d) Heatmap of DEGs among controls and REGO-treated samples in NR GB-EXPs. (e) DUSP6 and RNF150 gene expression in GB-EXPs of Resp samples, and in Gliovis online database (TCGA samples and Agilent platform) according tumor grade. (f) KI67 and ABCA4 genes expression in GB-EXPs of NR samples. Abbreviations. RESP, Responders; NR, Non Responders; CTRL, controls; TREAT, treated GB-EXPs.
Figure 3.
Gene expression analysis of GB-EXPs. (a) Heatmap of differentially expressed genes (DEGs) among Resp and NR samples in DMSO-treated GB-EXPs(controls). (b) Molecular pathways in which DEGsC are involved. (c) Heatmap of DEGs among controls and REGO-treated samples in Responders GB-EXPs. (d) Heatmap of DEGs among controls and REGO-treated samples in NR GB-EXPs. (e) DUSP6 and RNF150 gene expression in GB-EXPs of Resp samples, and in Gliovis online database (TCGA samples and Agilent platform) according tumor grade. (f) KI67 and ABCA4 genes expression in GB-EXPs of NR samples. Abbreviations. RESP, Responders; NR, Non Responders; CTRL, controls; TREAT, treated GB-EXPs.
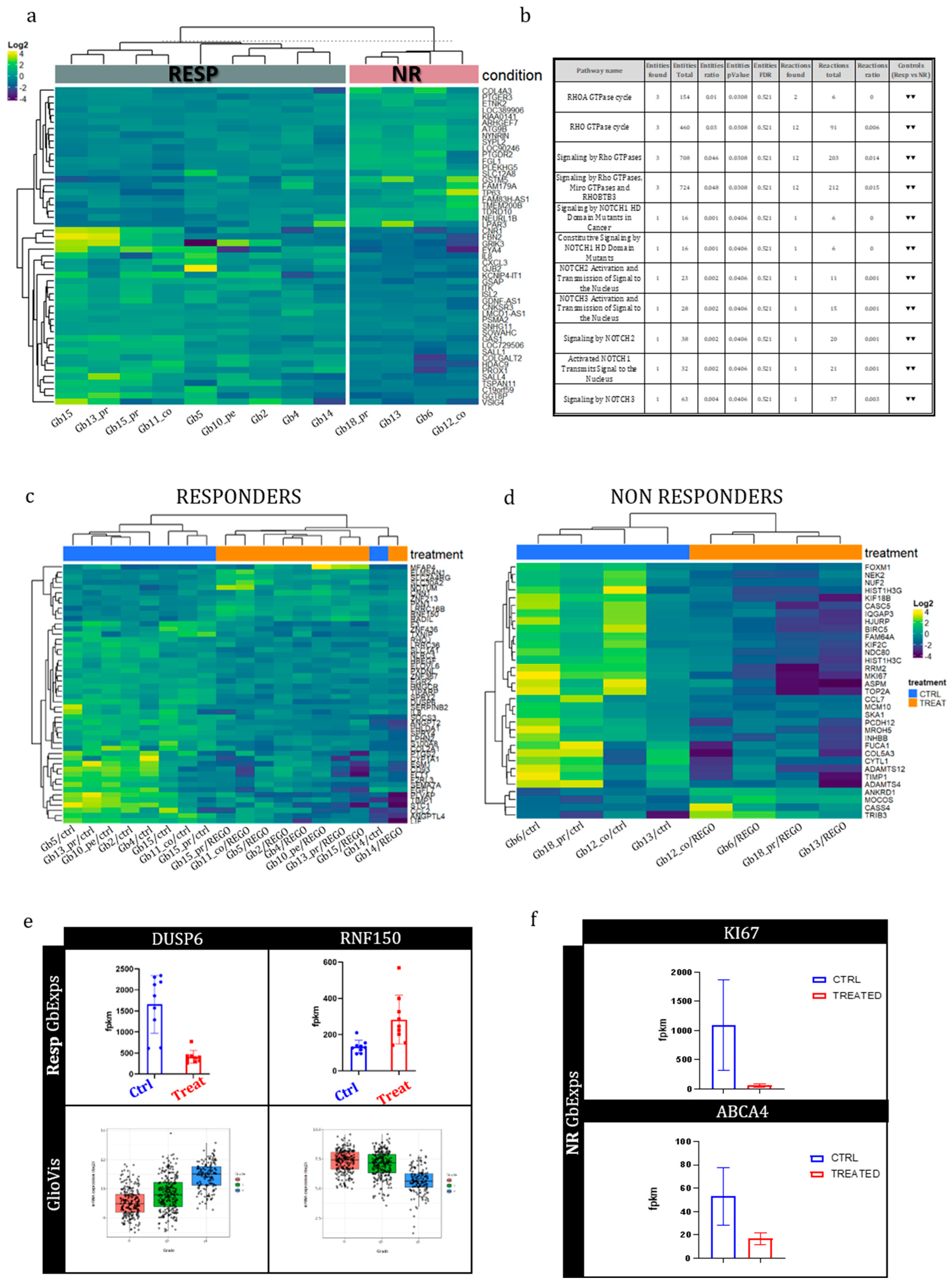
To understand pathways in which DEGsA and DEGsB were involved, we queried Reactome database (https://reactome.org/). We found that DEGsA, are primarily focused on MAP kinase (ERK/MAPK) signaling pathways inactivation/negative regulation in treated GB-EXPs, when compared to control samples (Table 4S), but also in interleukin-17 signaling, Toll-Like Receptor (TLR) cascades, and signaling by receptor tyrosine kinases (RTKs). On the other hand, DEGsB were centered around transcriptional regulation by E2F6, metabolic processes, membrane trafficking, and cell cycle regulation during the G2/M transition, underling the different response to REGO treatment in Resp and NR samples (Table 5S).
4. Discussion
Glioblastoma (GB) stands as the most prevalent malignant primary brain tumor among adults with an average survival period following standard treatment is just 14.6 months. This limited survival is largely attributed to the tumor’s inclination to reappear in nearly all patients within 6–9 months following initial therapy [36,37] Addressing disease recurrence remains a current challenge. To tackle recurrent GB, researchers have explored the potential of multikinase inhibitors, which are substances targeting various kinases. One such multikinase inhibitor is Regorafenib (REGO), already in use for conditions like hepatocellular carcinoma, gastrointestinal stromal tumors, and colorectal cancer. Both the National Comprehensive Cancer Network (NCCN) 2020 Guidelines and the Italian Agency of Medicine (AIFA) have endorsed REGO as a preferred treatment for Italian recurrent GB patients. From a molecular perspective, REGO targets various kinases, both angiogenic (VEGFR 1-3, PDGFR-b) and oncogenic (c-KIT, RET, FGFR, Raf). However, despite its adoption in clinical practice, the precise molecular mechanisms governing GB patients’ responsiveness to REGO remain poorly understood. Understanding these mechanisms is crucial, as it can prevent promising treatments from becoming ineffective in later stages. To delve into these biological factors, collecting tissue samples before and after treatment is imperative, yet this practice is not yet routine in the domains of neurosurgery and neuro-oncology drug development. In an effort to address this need, in this work a novel ex vivo drug response precision medicine approach has been used. This innovative method assesses how tumor samples react to different cancer treatments, enabling the analysis of tissue samples before and after treatment. The technique involves the use of in vitro 3D organoid models derived from patient glioblastoma tissue (referred to as GB-EXPs as previously described [15]). These organoids are minimally manipulated and briefly cultured to maintain the tumor microenvironment so that the whole process is conducted within 6 days from surgery to avoid extensive molecular changes due to in vitro culturing. Our research primarily focuses on GB-EXPs derived from patients with recurrent tumors. To evaluate REGO response and the difference in efficacy compared to TMZ of GB-EXPs, we employed NADH(P) Fluorescence Lifetime Imaging, as described in our previous work [15]. A substantial body of literature exists to corroborate the efficacy of the FLIM technique in detecting therapeutic response in cancer [38]. In our study, we observed a higher percentage of REGO responders (77%) compared to TMZ responders (35%). It is noteworthy that all REGO-NR (6 out of 6, 100%) were also TMZ-NR. Contrarily, 6 out of 17 TMZ-NR (35%) were REGO-Resp. These data are consistent with results obtained from Jang et al. [39] Nude mice received subcutaneous inoculation with U87 cells and were treated with REGO (20 mg/kg/day) alone or with or without TMZ (10 mg/kg/day) for 14 days. The study found that 20 μM Regorafenib induced a higher GB growth inhibition than 500 μM TMZ, and the cytotoxic impact of REGO was only minimally enhanced by TMZ. Additionally, the study confirmed these results in an orthotopic xenograft mouse model of human GB, showing that REGO significantly prolonged the survival of mice with orthotopic GB xenografts. Our findings, when considered alongside this evidence, may suggest that REGO has greater efficacy in reducing tumor aggressiveness than TMZ. However, clinical validation is required to confirm our data on Regorafenib response.
One of the most innovative approaches of this study was to analyze molecular changes of GB-EXPs after treatment through longitudinal in vitro tissue sampling, which facilitated a thorough understanding of the molecular effects of drugs in an ex vivo model, which closely mimics in vivo conditions. As an initial step, we conducted a comprehensive analysis of the mutational and transcriptome profiles of tumors stratified into REGO Responders (Resp) and Non-Responders (NR). In our examination, we pinpointed several genes exhibiting a preferential mutation pattern in the NR group, notably KCNK3, AQP10, AKNA, and STAB1. These mutations are deemed potentially pathogenetic, potentially impacting the functions of the corresponding proteins. KCNK3 belongs to the K+ channel protein family and plays a crucial role in regulating numerous biological processes. It is extensively expressed in the nervous system [40]. Recent studies have indicated a potential link between glioma and dysregulated expression of potassium ion channels. In particular, inhibitors of K+ channels have shown significant influence on glioma growth [41,42]. The selective expression of KCNK3 in glioma suggests its potential as a therapeutic target, holding substantial clinical application value [42]. Another focus of our analysis were Aquaporins (AQPs), a water channel proteins family distributed across various human tissues. They are accountable for transporting small solutes such as glycerol, gases, and ions. Overexpression of AQPs has been detected in gliomas and is significantly associated with the histological severity of tumors and the prognosis of cancer patients. Proposed as novel therapeutic strategies are inhibitors targeting AQPs in tumor cells and microvessels [43]. This underscores the potential significance of AQP10 mutations in the context of glioma response to REGO treatment, providing a basis for further exploration of targeted therapeutic interventions. The AKNA gene, encoding a transcription factor with an elusive role in cancer (https://www.genecards.org ), adds a layer of complexity to our understanding. Recently, the scavenger receptor stabilin-1 (STAB1), expressed in activated macrophages was identified as the first known receptor for SPARC which is a matricellular protein involved in cell migration in glioblastoma [44]. The discovery of STAB1 as a receptor for SPARC in the context of glioblastoma underscores its potential significance, warranting further exploration to unravel its role in cancer development and response to treatments like REGO.
In the REGO-Resp group, distinct molecular alterations emerged, notably in the PCNX1, DNAH8, OR13C5, and MLLT3 genes. PCNX1 was described hyper mutated in glioma, and some of its somatic variants were defined as predictors of chemotherapy response in breast cancer [45]. Axoneme Heavy Chain 8 (DNAH8) is involved in prostate cancer and lung cancer [46]. Furthermore, mutations in DNAH8 were found to be associated with increased tumor mutation burden (TMB), indicating their potential as predictive biomarkers for identifying cancer patients who may respond to immune checkpoint inhibitors inhibitors [47]. The OR13C5 receptor gene is part of the Odorant receptors (ORs) family, comprising approximately 60% of all human G protein-coupled receptors (GPCRs). ORs exhibit functions beyond odor perception when expressed outside of the nose, potentially playing a role in the pathogenesis of glioblastoma. Increasingly, ORs are being acknowledged as potential biomarkers and therapeutic targets for glioblastoma [48]. Finally, MLLT3, a DNA binding protein, has been frequently found fused with other partner genes in leukemia and more recently in glioblastoma [49]. In addition to its oncogenic role in osteosarcoma through partial regulation of the JNK signaling pathway, MLLT3 represents a promising treatment target of this tumor [50]. This diverse array of molecular alterations in the Resp group further underscores the intricate landscape of genetic factors influencing glioblastoma responses to REGO, highlighting potential avenues for targeted therapeutic interventions.
The comprehensive transcriptome analysis uncovered a distinct set of genes exhibiting differential expression between the genetic backgrounds of RESP and NR tumors, with a particular focus on GRIK3, GSMT5, and SALL4. The GRIK3 gene encodes the receptor for glutamate, serving as a ligand-gated ion channel in the central nervous system and playing a crucial role in excitatory synaptic transmission. The diverse spatial and temporal expression of ligand-gated ion channels implicates them in important cellular processes. Perturbations in the finely adjusted transcellular electrochemical gradient in eukaryotic cells can contribute to aberrancies, potentially altering the activity of pro-tumorigenic molecular pathways, including the MAPK/ERK, RAS, and mTOR pathways [31]. Thus, ligand-gated ion channels emerge as a promising targetable system for cancer therapeutics. Notably, similar ligand-gated ionotropic channel subtypes, such as the P2X7R, are highly expressed in various cancers, including breast, prostate, and pancreatic cancer. In breast cancer cell lines, REGO has been reported to modulate the P2X7R with demonstrated antitumor activity [51]. This insight into the modulation of ligand-gated ion channels by REGO provides a valuable perspective on potential therapeutic avenues, indicating that the effects observed in breast cancer cell lines may extend to glioblastoma through similar mechanisms. Building on the modulation of ligand-gated ion channels by REGO and its potential therapeutic implications, we propose that GRIK3, significantly more expressed in the Resp group, could serve as a promising therapeutic target for REGO action in glioblastoma. The findings reported by Sallahudin et al. [51], demonstrating a decrease in P2X7R gene expression in breast cancer cell lines following REGO treatment, align with our observations in RESP tumors. Although not reaching statistical significance, our data reveal a more than 2-fold decrease in GRIK3 expression after REGO treatment in RESP tumors. This parallel reduction in expression suggests a potential conserved mechanism of action for REGO across different cancer types, further supporting the exploration of GRIK3 as a viable therapeutic target in glioblastoma. The GSMT5 gene is part of the glutathione S-transferases (GSTs) family. They play a crucial role in metabolizing and/or detoxifying various endogenous and exogenous substances, including drugs. GSTs catalyze the conjugation of glutathione (GSH) to a diverse range of xenobiotics. This detoxification capability is vital for cellular protection against environmental factors and oxidative stress, but it is also associated with cellular resistance to certain chemotherapy drugs [32]. GSTM5 is closely associated with drug inactivation and multidrug resistance. Surprisingly, Cheng S.Y. et al. (2018) [52] reported increased levels of GSTM5 protein in TMZ-resistant cell lines, which is in line with our results. The higher statistically significant expression of the GSMT5 gene that we observe in the REGO resistant group could support the hypothesis that these tumors have developed a protection from the action of the REGO drug. SALL4, an embryonic stem cell regulator was observed to be significantly overexpressed in the NR tumor group, confirming previous findings of its overexpression in malignant cancers, including glioma. In breast cancer (BC) SALL4 has been associated with tumor progression and drug resistance. The development of therapies targeting SALL4 may provide new therapeutic strategies for aggressive BC and might address the problem of drug resistance [33]. The analysis of the Reactome database revealed that Rho GTPase and NOTCH signaling pathways were downregulated in Resp samples compared to NR samples. The Rho GTPase and NOTCH pathways are critical cell signaling pathways that regulate various cellular processes. NOTCH signaling can influence cell migration and invasion, potentially through the regulation of Rho GTPase activity or expression. Conversely, Rho GTPases are involved in cytoskeletal reorganization during processes such as Epithelial-Mesenchymal Transition (EMT). There is evidence of crosstalk between these two pathways, with NOTCH regulating Rho GTPase activity or expression and Rho GTPases influencing NOTCH signaling by affecting cell-cell adhesion or NOTCH ligand presentation. Therefore, the downregulation of these pathways is consistent with a higher response to Rego.
The utilization of a vital in-vitro model provided a valuable platform for studying gene expression changes induced by REGO drug treatment, with a specific focus on transcriptional alterations occurring 72 hours after treatment. Comparative analyses of gene expression profiles between treated and non-treated tumors within the Resp and NR groups revealed an overall trend of general gene expression downregulation, consistent with the direction of most statistically significant deregulated genes. However, It is noteworthy to point out that in the Resp group the expression DUSP6 and the RFN50 genes are statistically significantly modulated by the action of the REGO to reconstitute a genetic background typical of a lesser aggressive tumor, as reported in the Gliovis dataset (http://gliovis.bioinfo.cnio.es/). Unexpectedly, in the NR group, a striking observation was the statistically significant 20-fold downregulation of MKI67, a well-known proliferation marker, at 72 hours post REGO treatment compared to non-treated samples. This unexpected molecular alteration in the context of drug resistance challenges our understanding and warrants further investigation. Similar decreases in KI67 expression have been reported in the literature, such as the work of Mansy et al. (2020), where exogenous epidermal growth factor was examined in the submandibular salivary gland of albino rats receiving doxorubicin [53]. These findings emphasize the complexity of drug responses and the need for nuanced exploration of molecular pathways involved in resistance. In cancer cells, multi-drug resistance (MDR), whether intrinsic or acquired through various mechanisms, significantly undermines the effectiveness of therapeutic drugs. Often, the diminished therapeutic efficacy of drugs is primarily attributed to the inherent overexpression of ATP-binding cassette (ABC) transporter proteins on the cell membrane. This leads to reduced drug uptake, increased drug detoxification, and enhanced DNA repair mechanisms [35]. In our analyses, the ABCA4 gene was the only gene modulated by the REGO treatment. Significantly reduced expression of ABCA4 was observed in the NR group in response to treatment, suggesting the ability of these tumors to counteract REGO action by downregulating ABCA4, potentially depriving cells of drug uptake.
To elucidate the intricate pathways associated with DEGsA and DEGsB, we conducted a comprehensive analysis utilizing the Reactome database (https://reactome.org/). Our findings reveal that DEGsA primarily engage in the attenuation/negative regulation of MAP kinase (ERK/MAPK) signaling pathways within treated GB-EXPs, in contrast to control samples. Additionally, DEGsA exhibit involvement in interleukin-17 signaling, Toll-Like Receptor (TLR) cascades, and signaling by receptor tyrosine kinases (RTKs). These pathways potentially contribute to the broader landscape of drug response mechanisms to REGO. Notably, tyrosine kinase inhibitors (TKIs), such as REGO, target both MAP kinase (ERK/MAPK) signaling pathways and RTK signaling. However, establishing a direct correlation between DEGsA and responders to REGO necessitates further investigation through additional data and specific studies. Conversely, DEGsB demonstrate a distinct focus on transcriptional regulation by E2F6, metabolic processes, membrane trafficking, and cell cycle regulation during the G2/M transition.
These findings underscore the differential response to REGO treatment observed in Responders (Resp) and Non-Responders (NR) samples. As such, unraveling the nuanced molecular mechanisms associated sheds light on the multifaceted nature of cellular responses to REGO, emphasizing the need for a more targeted exploration of these intricate pathways in future studies. Notably, this study represents the first exploration of alterations in gene expression resulting from REGO drug treatments in patient-derived glioblastoma organoids. These findings need further validation and hold great potential for advancing personalized precision medicine in the field.
5. Conclusions
Our findings suggest that using our innovative approach, which allows for the study of longitudinal molecular alterations, it may be possible to identify patients who are more likely to respond to a treatment based on their tumor’s molecular profile. This could lead to the development of new therapies that target these pathways more effectively and to more personalized treatment decisions that could improve outcomes for patients with glioblastoma.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Viability and proliferation effects of Regorafenib on 2D and 3D cell lines models, T98G, U87 and U118.; Figure S2: FLIM analysis on 3D-T98G cell line using different REGO concentrations.; Supplementary Results: Sensitivity to Regorafenib in 2D and 3D commercial cell lines.
Author Contributions
Conceptualization, M.M. and C.M.M.; methodology, M.M, F.L, S.F and M.G.; software, P.A and A.M.; validation, M.Me, G.F. and F.L..; formal analysis, M.M.; investigation, M.M, G.F, F.L. and M.Me; resources, O.S.S, A.L.D, N.M., F.P, F.P. and C.G; data curation, M.M.; writing—original draft preparation, M.M and C.M.M.; writing—review and editing, M.M and C.M.M.; visualization, M.M.; supervision, C.M.M.; project administration, C.M.M.; funding acquisition, C.M.M. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki. The sample collection protocol was approved by the Ethics Committee of the University Hospital of Pisa (787/2015).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.”
Data Availability Statement
All data generated or analyzed during this study are included in this published article and its supplementary information files. The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Q. T. Ostrom, N. Patil, G. Cioffi, K. Waite, C. Kruchko, and J. S. Barnholtz-Sloan, “CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2013–2017,” Neuro. Oncol., vol. 22, no. Supplement_1, pp. iv1–iv96, Oct. 2020. [CrossRef]
- R. Stupp et al., “Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma,” N. Engl. J. Med., vol. 352, no. 10, pp. 987–996, Mar. 2005. [CrossRef]
- P. A. Rønning, E. Helseth, T. R. Meling, and T. B. Johannesen, “A population-based study on the effect of temozolomide in the treatment of glioblastoma multiforme,” Neuro. Oncol., vol. 14, no. 9, pp. 1178–1184, Sep. 2012. [CrossRef]
- R. Stupp et al., “Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial,” Lancet Oncol., vol. 10, no. 5, pp. 459–466, May 2009. [CrossRef]
- T. T. Batchelor et al., “Feasibility, phase I, and phase II studies of tandutinib, an oral platelet-derived growth factor receptor-β tyrosine kinase inhibitor, in patients with recurrent glioblastoma,” Neuro. Oncol., vol. 19, no. 4, pp. 567–575, Apr. 2017. [CrossRef]
- J. Kalpathy-Cramer et al., “Phase II study of tivozanib, an oral VEGFR inhibitor, in patients with recurrent glioblastoma,” J. Neurooncol., vol. 131, no. 3, pp. 603–610, Feb. 2017. [CrossRef]
- J. Bruix et al., “Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial,” Lancet, vol. 389, no. 10064, pp. 56–66, Jan. 2017. [CrossRef]
- A. Grothey et al., “Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): An international, multicentre, randomised, placebo-controlled, phase 3 trial,” Lancet, vol. 381, no. 9863, pp. 303–312, Jan. 2013. [CrossRef]
- G. D. Demetri et al., “Effi cacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): An international, multicentre, randomised, placebo-controlled, phase 3 trial,” Lancet, vol. 381, no. 9863, pp. 295–302, Jan. 2013. [CrossRef]
- G. Lombardi et al., “Regorafenib compared with lomustine in patients with relapsed glioblastoma (REGOMA): a multicentre, open-label, randomised, controlled, phase 2 trial,” Lancet Oncol., vol. 20, no. 1, pp. 110–119, Jan. 2019. [CrossRef]
- M. Sepúlveda-Sánchez et al., “Characterization of Glioblastoma Characterization of Glioblastoma Cells Response to Regorafenib,” 2022. [CrossRef]
- G. Lombardi et al., “Regorafenib in Recurrent Glioblastoma Patients: A Large and Monocentric Real-Life Study,” Cancers 2021, Vol. 13, Page 4731, vol. 13, no. 18, p. 4731, Sep. 2021. [CrossRef]
- M. P. Mongiardi et al., “Characterization of Glioblastoma Cells Response to Regorafenib,” Cancers (Basel)., vol. 14, no. 24, 2022. [CrossRef]
- K. Singh et al., “Correcting the drug development paradigm for glioblastoma requires serial tissue sampling,” Nat. Med. 2023 2910, vol. 29, no. 10, pp. 2402–2405, Jul. 2023. [CrossRef]
- M. Morelli et al., “Metabolic-imaging of human glioblastoma live tumors: A new precision-medicine approach to predict tumor treatment response early,” Front. Oncol., vol. 12, no. September, pp. 1–18, 2022. [CrossRef]
- R. Foty, “A simple hanging drop cell culture protocol for generation of 3D spheroids,” J. Vis. Exp., no. 51, 2011. [CrossRef]
- K. J. Livak and T. D. Schmittgen, “Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method,” Methods, vol. 25, no. 4, pp. 402–408, 2001. [CrossRef]
- S. Ranjit, L. Malacrida, D. M. Jameson, and E. Gratton, “Fit-free analysis of fluorescence lifetime imaging data using the phasor approach,” Nat. Protoc., vol. 13, no. 9, pp. 1979–2004, Sep. 2018. [CrossRef]
- T. Metsalu and J. Vilo, “ClustVis: a web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap,” Nucleic Acids Res., vol. 43, no. W1, pp. W566–W570, 2015. [CrossRef]
- Y. Guo, X. Ding, Y. Shen, G. J. Lyon, and K. Wang, “SeqMule: automated pipeline for analysis of human exome/genome sequencing data,” Sci. Reports 2015 51, vol. 5, no. 1, pp. 1–10, Sep. 2015. [CrossRef]
- D. Benjamin, T. Sato, K. Cibulskis, G. Getz, C. Stewart, and L. Lichtenstein, “Calling Somatic SNVs and Indels with Mutect2”. [CrossRef]
- K. J. Karczewski et al., “The mutational constraint spectrum quantified from variation in 141,456 humans,” Nature, vol. 581, no. 7809, pp. 434–443, 2020. [CrossRef]
- A. Mayakonda, D. C. Lin, Y. Assenov, C. Plass, and H. P. Koeffler, “Maftools: efficient and comprehensive analysis of somatic variants in cancer,” Genome Res., vol. 28, no. 11, pp. 1747–1756, Nov. 2018. [CrossRef]
- E. Talevich, A. H. Shain, T. Botton, and B. C. Bastian, “CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing,” PLoS Comput. Biol., vol. 12, no. 4, Apr. 2016. [CrossRef]
- S. Franch-Expósito et al., “CNApp, a tool for the quantification of copy number alterations and integrative analysis revealing clinical implications,” Elife, vol. 9, Jan. 2020. [CrossRef]
- A. Colaprico et al., “TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data,” Nucleic Acids Res., vol. 44, no. 8, p. e71, May 2016. [CrossRef]
- A. Liaw and M. Wiener, “Classification and Regression by randomForest,” vol. 2, no. 3, 2002, Accessed: Dec. 28, 2023. [Online]. Available: http://www.stat.berkeley.edu/.
- A. L. Trinh et al., “Tracking functional tumor cell subpopulations of malignant glioma by phasor fluorescence lifetime imaging microscopy of NADH,” Cancers (Basel)., vol. 9, no. 12, pp. 1–13, 2017. [CrossRef]
- M. M. Lukina et al., “Metabolic cofactors NAD(P)H and FAD as potential indicators of cancer cell response to chemotherapy with paclitaxel,” Biochim. Biophys. Acta - Gen. Subj., vol. 1862, no. 8, pp. 1693–1700, 2018. [CrossRef]
- B. Xiao et al., “Glutamate Ionotropic Receptor Kainate Type Subunit 3 (GRIK3) promotes epithelial-mesenchymal transition in breast cancer cells by regulating SPDEF/CDH1 signaling,” Mol. Carcinog., vol. 58, no. 7, pp. 1314–1323, Jul. 2019. [CrossRef]
- R. Rao et al., “Ligand-Gated Ion Channels as Targets for Treatment and Management of Cancers,” Front. Physiol., vol. 13, p. 839437, Mar. 2022. [CrossRef]
- Y. C. Jou et al., “Anti-Cancer Effects and Tumor Marker Role of Glutathione S-Transferase Mu 5 in Human Bladder Cancer,” Int. J. Mol. Sci. 2021, Vol. 22, Page 3056, vol. 22, no. 6, p. 3056, Mar. 2021. [CrossRef]
- B. Sun, L. Xu, W. Bi, and W. Bin Ou, “SALL4 Oncogenic Function in Cancers: Mechanisms and Therapeutic Relevance,” Int. J. Mol. Sci., vol. 23, no. 4, Feb. 2022. [CrossRef]
- C. Yang et al., “Ki67 targeted strategies for cancer therapy,” Clin. Transl. Oncol., vol. 20, no. 5, pp. 570–575, May 2018. [CrossRef]
- C. Duan, M. Yu, J. Xu, B. Y. Li, Y. Zhao, and R. K. Kankala, “Overcoming Cancer Multi-drug Resistance (MDR): Reasons, mechanisms, nanotherapeutic solutions, and challenges,” Biomed. Pharmacother., vol. 162, p. 114643, Jun. 2023. [CrossRef]
- F. Hanif, K. Muzaffar, K. Perveen, S. M. Malhi, and S. U. Simjee, “Glioblastoma Multiforme: A Review of its Epidemiology and Pathogenesis through Clinical Presentation and Treatment,” Asian Pac. J. Cancer Prev., vol. 18, no. 1, p. 3, 2017. [CrossRef]
- M. Ogawa, A. Tanaka, K. Namba, J. Shia, J. Y. Wang, and M. H. Roehrl, “Early-Stage Loss of GALNT6 Predicts Poor Clinical Outcome in Colorectal Cancer,” Front. Oncol., vol. 12, p. 2334, May 2022. [CrossRef]
- Y. Ouyang, Y. Liu, Z. M. Wang, Z. Liu, and M. Wu, “FLIM as a Promising Tool for Cancer Diagnosis and Treatment Monitoring,” vol. 1, p. 3. [CrossRef]
- J. Jiang et al., “Regorafenib induces lethal autophagy arrest by stabilizing PSAT1 in glioblastoma,” Autophagy, vol. 16, no. 1, pp. 106–122, Jan. 2020. [CrossRef]
- J. Liu, C. Qu, C. Han, M. M. Chen, L. J. An, and W. Zou, “Potassium channels and their role in glioma: A mini review,” Mol. Membr. Biol., vol. 35, no. 1, pp. 76–85, Jan. 2019. [CrossRef]
- A. Felipe et al., “Targeting the voltage-dependent K(+) channels Kv1.3 and Kv1.5 as tumor biomarkers for cancer detection and prevention,” Curr. Med. Chem., vol. 19, no. 5, pp. 661–674, Jan. 2012. [CrossRef]
- L. Huang, B. Li, W. Li, H. Guo, and F. Zou, “ATP-sensitive potassium channels control glioma cells proliferation by regulating ERK activity,” Carcinogenesis, vol. 30, no. 5, pp. 737–744, May 2009. [CrossRef]
- J. Wang et al., “Aquaporins as diagnostic and therapeutic targets in cancer: How far we are?,” J. Transl. Med., vol. 13, no. 1, pp. 1–11, Mar. 2015. [CrossRef]
- C. David, J. P. Nance, J. Hubbard, M. Hsu, D. Binder, and E. H. Wilson, “Stabilin-1 Expression in Tumor Associated Macrophages,” Brain Res., vol. 1481C, p. 71, Oct. 2012. [CrossRef]
- P. Daniel et al., “Detection of temozolomide-induced hypermutation and response to PD-1 checkpoint inhibitor in recurrent glioblastoma,” Neuro-Oncology Adv., vol. 4, no. 1, pp. 1–14, 2022. [CrossRef]
- Y. Wang, R. J. Ledet, K. Imberg-Kazdan, S. K. Logan, and M. J. Garabedian, “Dynein axonemal heavy chain 8 promotes androgen receptor activity and associates with prostate cancer progression,” vol. 7, no. 31, Accessed: Dec. 22, 2023. [Online]. Available: www.impactjournals.com/oncotarget.
- C. Zhang, “COL22A1 and DNAH8 mutations are associated with tumor mutation burden and prognosis of lung adenocarcinoma patients,” pp. 1–17, 2022.
- H. J. Cho and J. H. Koo, “Odorant G protein-coupled receptors as potential therapeutic targets for adult diffuse gliomas: a systematic analysis and review,” BMB Rep., vol. 54, no. 12, p. 601, Dec. 2021. [CrossRef]
- L. Wang et al., “Novel Gene Fusions in Glioblastoma Tumor Tissue and Matched Patient Plasma,” Cancers (Basel)., vol. 12, no. 5, May 2020. [CrossRef]
- J. Sun, P. Ren, L. Ye, N. Li, and D. Wang, “MLLT3 promotes proliferation of osteosarcoma cells by regulating JNK signaling,” Int. J. Clin. Exp. Pathol., vol. 10, no. 9, p. 9444, 2017, Accessed: Dec. 22, 2023. [Online]. Available: /pmc/articles/PMC6966015/.
- M. M. Salahuddin, G. A. Omran, M. W. Helmy, and M. E. Houssen, “Effect of Regorafenib on P2X7 Receptor Expression and Different Oncogenic Signaling Pathways in a Human Breast Cancer Cell Line: A Potential of New Insight of the Antitumor Effects of Regorafenib,” Curr. Issues Mol. Biol. 2021, Vol. 43, Pages 2199-2209, vol. 43, no. 3, pp. 2199–2209, Dec. 2021. [CrossRef]
- S. Y. Cheng, W.-F. Chen, and Z.-H. Wen, “Glutathione S-transferase M subfamily in TMZ-resistant glioblastoma cells,” Ann. Oncol., vol. 29, no. October, p. viii679, 2018. [CrossRef]
- M. Shamel, M. Mansy, M. Soliman, and R. Mubarak, “The role of exogenous epidermal growth factor on Ki-67 proliferation marker expression in the submandibular salivary gland of albino rats receiving doxorubicin,” F1000Research 2020 91393, vol. 9, p. 1393, Dec. 2020. [CrossRef]
Figure 1.
FLIM Analysis of Patient-Derived GB-EXPs. (a) Workflow: Patient-derived GB-EXPs were obtained from surgical tissue, cultured in vitrogel, and treated for 72 hours. FLIM analysis was performed on 12-20 patient GB-EXPs, including treated and control samples. Data were analyzed using the phasor approach, resulting in mean NAD(P)H fractional distribution curves for treated GB-EXPs (red) and control GB-EXPs (blue). The statistically significant leftward shift of the treated curve compared to the control curve is highlighted in green and indicates a more oxidative state in the treated GB-EXPs compared to the controls. The green area is indicative of the percentage of drug response (%DR). Tumors were stratified into non-responders (NR, %DR ≤ 5) and responders (Resp, %DR > 5) using a cutoff of 5%DR. (b-e) Exemplary instances of one NR and one Resp tumor-derived GB-EXP post-treatment, featuring a brightfield image (top) and the corresponding phasor map (bottom). In the case of NR, the NAD(P)H fractional mean distribution curves overlap between control (blue) and treated GB-EXPs (red), leading to a 0%DR (c). Conversely, in the case of Resp, the NAD(P)H fractional mean distribution curves exhibit a leftward shift of the red curves (treated GB-EXPs), resulting in a 74%DR (e). (f) Summary of %DR for all cases with both treatment modalities, TMZ (left) and REGO (right). The first column lists the sample identification number (Sample Nr), the second column displays the %DR, and the third column indicates the phenotype as NR or Resp. Cases are arranged from lower %DR at the top to higher %DR at the bottom, with color highlighting the %DR increase from white to red. Abbreviations: %DR, percentage of drug response; NR, Non Responder; Resp, Responder; cor, core portion of tumor; per, peripheral portion of the tumor; PR, primary tumor; REC, recurrent tumor.
Figure 1.
FLIM Analysis of Patient-Derived GB-EXPs. (a) Workflow: Patient-derived GB-EXPs were obtained from surgical tissue, cultured in vitrogel, and treated for 72 hours. FLIM analysis was performed on 12-20 patient GB-EXPs, including treated and control samples. Data were analyzed using the phasor approach, resulting in mean NAD(P)H fractional distribution curves for treated GB-EXPs (red) and control GB-EXPs (blue). The statistically significant leftward shift of the treated curve compared to the control curve is highlighted in green and indicates a more oxidative state in the treated GB-EXPs compared to the controls. The green area is indicative of the percentage of drug response (%DR). Tumors were stratified into non-responders (NR, %DR ≤ 5) and responders (Resp, %DR > 5) using a cutoff of 5%DR. (b-e) Exemplary instances of one NR and one Resp tumor-derived GB-EXP post-treatment, featuring a brightfield image (top) and the corresponding phasor map (bottom). In the case of NR, the NAD(P)H fractional mean distribution curves overlap between control (blue) and treated GB-EXPs (red), leading to a 0%DR (c). Conversely, in the case of Resp, the NAD(P)H fractional mean distribution curves exhibit a leftward shift of the red curves (treated GB-EXPs), resulting in a 74%DR (e). (f) Summary of %DR for all cases with both treatment modalities, TMZ (left) and REGO (right). The first column lists the sample identification number (Sample Nr), the second column displays the %DR, and the third column indicates the phenotype as NR or Resp. Cases are arranged from lower %DR at the top to higher %DR at the bottom, with color highlighting the %DR increase from white to red. Abbreviations: %DR, percentage of drug response; NR, Non Responder; Resp, Responder; cor, core portion of tumor; per, peripheral portion of the tumor; PR, primary tumor; REC, recurrent tumor.
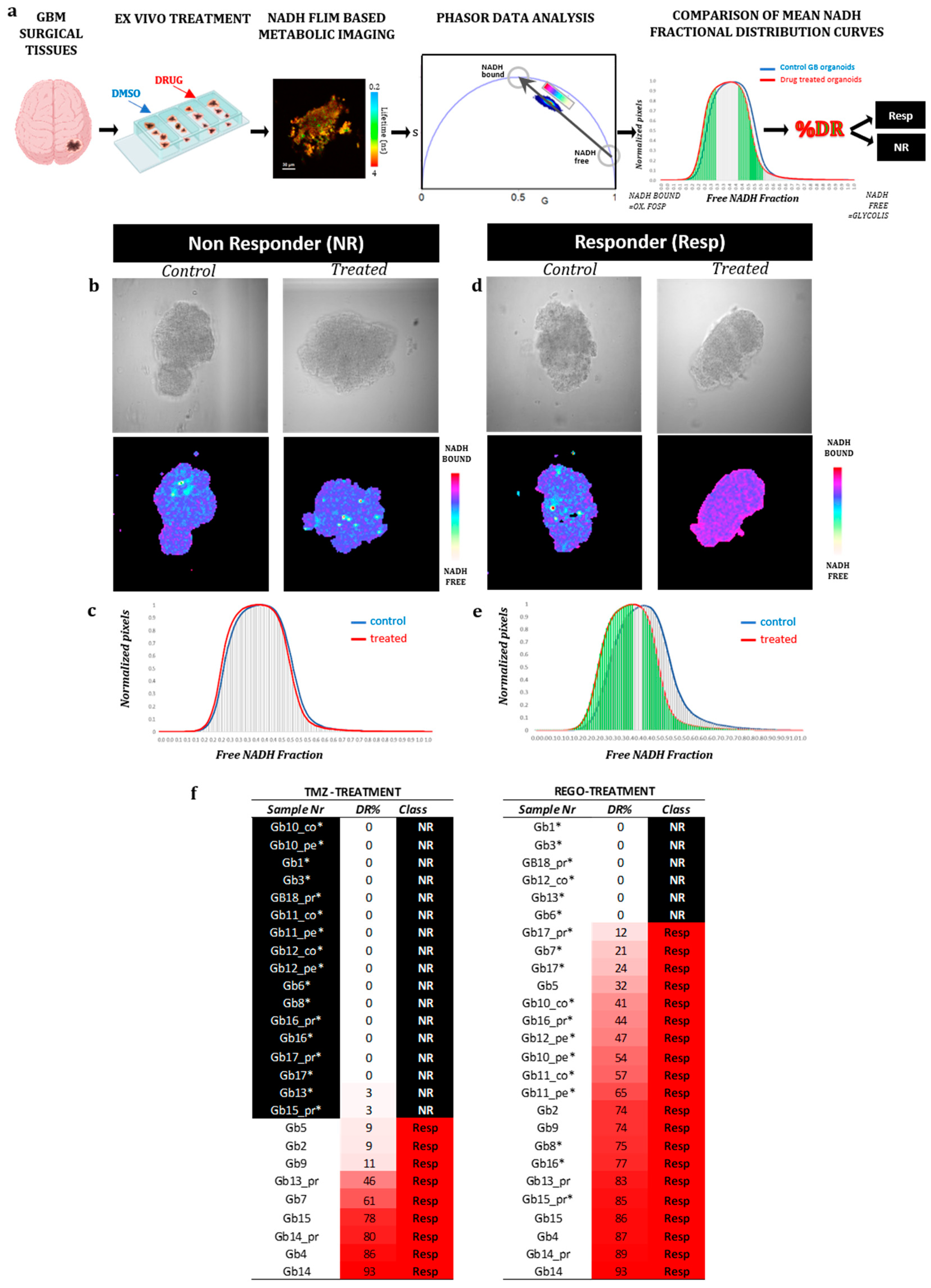
Figure 2.
Mutational analysis of GB patients. (a) The mutation landscape of GB patients categorized as Resp (n=11) and NR (n=5). This includes counts of each variant classification, variant type, single-nucleotide variant (SNV) classification, and the top 10 mutated genes. (b,c) Oncoplot illustrating the genes mutated in 100% (11/11) of Resp patients (b) and NR patients (c). (d) A co-bar plot indicating the genes significantly distinguishing between Resp and NR groups. Bars indicate the percentage of samples in which gene mutations were identified, with colors representing the types of mutation. (e) Heatmap displaying alterations in copy number of chromosome regions between Resp (n=11) and Non-Resp (n=5) groups (red indicates chromosome gains, and blue indicates losses). Abbreviations. Resp, Responders; NR, Non Responders.
Figure 2.
Mutational analysis of GB patients. (a) The mutation landscape of GB patients categorized as Resp (n=11) and NR (n=5). This includes counts of each variant classification, variant type, single-nucleotide variant (SNV) classification, and the top 10 mutated genes. (b,c) Oncoplot illustrating the genes mutated in 100% (11/11) of Resp patients (b) and NR patients (c). (d) A co-bar plot indicating the genes significantly distinguishing between Resp and NR groups. Bars indicate the percentage of samples in which gene mutations were identified, with colors representing the types of mutation. (e) Heatmap displaying alterations in copy number of chromosome regions between Resp (n=11) and Non-Resp (n=5) groups (red indicates chromosome gains, and blue indicates losses). Abbreviations. Resp, Responders; NR, Non Responders.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



